
Transcription factor IRF5 drives P2X4R+-reactive microglia gating neuropathic pain
Upload
independentCategory
view
1download
0
Proinflammatory Mediators Released by ActivatedMicroglia Induces Neuronal Death in JapaneseEncephalitisAYAN GHOSHAL,� SULAGNA DAS,� SOUMYA GHOSH, MANOJ KUMAR MISHRA, VIVEK SHARMA,PREETI KOLI, ELLORA SEN, AND ANIRBAN BASU*
National Brain Research Centre, Manesar, Haryana, India
KEY WORDScytokines; virus; iNOS; Cox-2; cortex; hippocampus; lectin
ABSTRACTWhile a number of studies have documented the importanceof microglia in central nervous system (CNS) response toinjury, infection and disease, little is known regarding itsrole in viral encephalitis. We therefore, exploited an experi-mental model of Japanese Encephalitis, to better under-stand the role played by microglia in Japanese EncephalitisVirus (JEV) infection. Lectin staining performed to assessmicroglial activation indicated a robust increase in reactivemicroglia following infection. A difference in the topographicdistribution of activated, resting, and phagocytic microgliawas also observed. The levels of various proinflammatorymediators, such as inducible nitric oxide synthase (iNOS),cyclooxygenase-2 (Cox-2), IL-6, IL-1b, TNF-a, and MCP-1that have been implicated in microglial response to an acti-vational state was significantly elevated following infection.These cytokines exhibited region selective expression in thebrains of infected animals, with the highest expressionobserved in the hippocampus. Moreover, the expression ofneuronal specific nuclear protein NeuN was markedly down-regulated during progressive infection indicating neuronalloss. In vitro studies further confirmed that microglial acti-vation and subsequent release of various proinflammatorymediators induces neuronal death following JEV infection.Although initiation of immune responses by microglial cellsis an important protective mechanism in the CNS, unrest-rained inflammatory responses may result in irreparablebrain damage. Our findings suggest that the increasedmicroglial activation following JEV infection influences theoutcome of viral pathogenesis. It is likely that the increasedmicroglial activation triggers bystander damage, as the ani-mals eventually succumb to infection. VVC 2007 Wiley-Liss, Inc.
INTRODUCTION
Microglia are the resident immune cells of the centralnervous system (CNS) and have a critical role in hostdefense against invading microorganisms. Microglial ac-tivation is viewed as an adaptive response whereby micro-glia release neuroprotective factors to facilitate the recov-ery of injured neurons and they also phagocytose dying ordamaged neurons, before they lyse and release toxicagents into surrounding areas. Activated microglia secretecytokines, such as interleukin-1 (IL-1) and tumor necrosisfactor alpha (TNF-a), which can cause toxic effects in the
brain (Quagliarello et al., 1991). Additionally, other solu-ble factors such as neurotoxins (Giulian et al., 1990), exci-tatory neurotransmitters (Stone and Connick, 1985), pros-taglandin (Zhang et al., 2006), reactive oxygen, and nitro-gen species (Mhatre et al., 2004) are secreted by activatedmicroglia.
Japanese encephalitis virus (JEV) is an acute zoonoticinfection that commonly affects children and is a majorcause of acute encephalopathy (Chen et al., 2002). JEVtargets CNS, clinically manifesting with fever, headache,vomiting, signs of meningeal irritation, and altered con-sciousness leading to high mortality and neurologicalsequel in some of those who survive (Kumar et al., 1990).Though neurological disorders caused by conventionalviruses are often characterized by evidence of immunesystem recognition and the presence of inflammatory com-ponents among the neuropathological changes (Wolinsky,1990), the mechanisms by which these viruses cause neu-rological disease are not fully understood. In many cases,the virus is probably not directly involved in the destruc-tion of brain tissue but may cause damage indirectly bytriggering cell mediated immune response by activatingmicroglia. Previous studies using the Reovirus infectionmodel have demonstrated increased inducible nitric oxidesynthase (iNOS) expression (Goody et al., 2005). Nitric ox-ide (NO) has been implicated as a contributor to the host’sinnate defense against viral infections including thoseaffecting the CNS. Infection of immunocompetent adultrats with Borna disease virus (BDV) results in severeencephalitis and neural dysfunction. The expression ofCyclooxygenase-2 (Cox-2) previously implicated in CNSdisease and peripheral inflammation is dramatically upre-gulated in the cortical neurons of BDV-infected rats. Thisupregulation in neuronal Cox-2 predominantly occurs inbrain areas where macrophages/microglia accumulate(Rohrenbeck et al., 1999).
Clinically, the infection of JEV results in increased levelsof inflammatory mediators in the serum and cerebrospinal
Grant sponsor: Department of Biotechnology; Grant number: BT/PR/5799/MED/14/698/2005.
*Correspondence to: Anirban Basu, National Brain Research Centre, Manesar,Haryana 122050, India. E-mail: [email protected]@nbrc.res.in
�AG and SD contributed equally to this work.
Received 19 July 2006; Accepted 22 November 2006
DOI 10.1002/glia.20474
Published online 3 January 2007 in Wiley InterScience (www.interscience.wiley.com).
GLIA 55:483–496 (2007)
VVC 2007 Wiley-Liss, Inc.

fluid (CSF) (Ravi et al., 1997). Accumulating evidenceshows that the mortality rate increases with increasingconcentration of cytokine in serum and CSF in patients(Winter et al., 2004). Despite the pathological importanceof JE, the immunological mechanisms occurring during vi-ral infection and role played by microglia in the pathoge-nesis of JE is still not well defined. In this communication,we have evaluated the functional status of microglia fol-lowing JE. We have demonstrated for the first time thatactivation of microglia and subsequent production of mul-tiple proinflammatory mediators contributes to neuronalloss following JEV infection.
MATERIALS AND METHODSViruses and Cell
The JaOArS982 strain of JEV was initially obtainedfrom Dr. Sudhanshu Vrati (National Institute of Immunol-ogy, New Delhi, India) and further propagated in sucklingBALB/c mice. The mouse brain derived virus was storedat 270�C and was used as the source of virus for all theexperiments (Appaiahgari et al., 2006). Virus inactivationwas done by placing viruses in boiled water for 15 min(Chen et al., 1996). Porcine stable kidney (PS) cell lineand mouse neuroblastoma Neuro2a (N2a) were obtainedfrom National Centre for Cell Science, Pune, India. Mousemicroglial cell line BV-2 was a kind gift from Dr. SteveLevison, University of Medicine and Dentistry, NJ. All thecell lines were grown at 37�C in Dulbecco’s modifiedeagle’s medium (DMEM) supplemented with 7.5% sodiumbi-carbonate (NaHCO3), 10% fetal bovine serum, and peni-cillin/streptomycin. All the reagents related to cell culturewere obtained from Sigma, St. Louis, MO, unless other-wise stated.
Virus Titration
JEV was titrated by plaque formation on the mono-layers of PS cell line. Monolayers of PS cells were inocu-lated with 10-fold dilutions of virus sample made in MEMcontaining 1% FCS and incubated for 1 h at 37�C withoccasional shaking. The inoculums was removed byaspiration and the monolayers were further overlaid withMEM containing 4% FCS, 1% low melting point agarose,and a cocktail of antibiotic-antimyotic solution (Gibco, CA)containing penicillin, streptomycin and amphotericin-B.Plates were incubated at 37�C for 3–7 days until plaqueswere visible. To allow counting of the plaques, the cellmonolayer was stained with crystal violet after fixing thecells with 10% formaldehyde (Vrati et al., 1999).
Coculturing BV-2 Cell Line with Virus
BV-2 mouse microglial cell line was plated at 5 3105 cells/well in a six-well plate. After 24 h in DMEMwith 10% serum, the cells were switched to serum freemedia for 12 h. BV-2 cell line was then adsorbed with ei-
ther live JEV (multiplicity of infection 5 5) or boiled virusfor 1 h. After adsorption, unbound viruses were removedby gentle washing with phosphate-buffered saline (PBS).Fresh serum free medium was added to each well for fur-ther incubation for 24 h at 37�C. Following incubation, thesupernatant was collected and stored at 230�C until fur-ther use, and cells were processed for immunoblot analy-sis as described later in materials and methods. Virus ti-tration was performed with this supernatant to furtherconfirm the presence or absence of live viruses.
TUNEL Assay
Mouse neuroblastoma cell line N2a was plated at adensity of 5 3 104 cells/well of eight-well chamber slides(Nunc, Denmark) in medium containing 1% FBS. After24 h, the medium was replaced by 50% of culture super-natant obtained from previous experiment (BV-2 and vi-rus co culture) and 50% of fresh medium containing 1% se-rum. In two of the wells Celecoxib (50 ng/mL), a specificinhibitor for Cox-2, aminoguanidine bicarbonate (10 mM),and a specific inhibitor for iNOS was added. After 24 hdying N2a cells were identified using In situ Cell DeathDetection Kit, TMR red (Roche, Germany). Briefly, neuro-nal cultures (N2a) were fixed with 4% paraformaldehyde(PFA) in PBS and blocked with 4% BSA containing 0.02%Triton-X-100. The fixed cells were then incubated inTUNEL mix (terminal deoxynucleotidyl transferase in sto-rage buffer and TMR red labeled-nucleotide mixture inreaction buffer) for 1 h at room temperature. The slideswere mounted with Vectashield mounting media contain-ing DAPI (Vector Laboratories, CA).
Virus Infection of Animals
We have used a previously described animal model ofJapanese Encephalitis (Vrati et al., 1999). Suckling BALB/cmice of either sex were injected intra-cerebrally with�100 p.f.u (in 30 lL of PBS) of JEV of strain JaOArS982and control animals received the same amount of PBS.Groups of three mice were sacrificed at each time pointeither for tissue or protein or RNA. From third day post-infection, animals started to show symptoms of JE in-cluding limb paralysis, poor pain response, and whole bodytremor. On the fourth day postinfection, all animals suc-cumbed to infection. All experiments were performedaccording to the protocol approved by the InstitutionalAnimal Ethics Committee.
Immunohistochemistry
Animals at days 2 and 4 postinfection and age matchedcontrols used for immunohistochemistry were perfusedwith PBS containing 7 U/mL heparin, followed by a fixa-tive containing 2.5% PFA in PBS. The brains were pro-cessed for cryostat sectioning and the sections werestained for tomato lectin as described earlier (Basu et al.,
484 GHOSHAL ET AL.
GLIA DOI 10.1002/glia

2002b). Briefly, the cryostat sections from control and JEinfected animals were washed with PBS followed byquenching with 0.3% H2O2. The sections were then rinsedwith PBS containing CaCl2, MgCl2, MnCl2, and thenblocked with 1% BSA for 1 h. The sections were then incu-bated overnight in a humidified chamber at 4�C with bio-tinylated-tomato Lectin (10 lg/mL; Vector laboratories)diluted in PBS containing CaCl2, MgCl2, MnCl2, and 0.2%Triton-X-100. Following incubation in Streptavidin HRP(used as the secondary antibody at 1:500; Vector Laborato-ries), the sections were further incubated in inactivatedDAB for 5 min followed by incubation in activated DABfor 10 min. The sections were then dehydrated in gradedalcohol and xylene and mounted with DPX.
Fluorescence immunohistochemistry was performedfor the following antibodies- phagocytic microglia were la-beled with rat anti-CD11b antibodies (1:100; BD Phar-mingen; NJ), which were then double stained with rabbitanti-iNOS (1:1000, Chemicon, CA) and rabbit anti-Cox-2(1:100, Cell Signaling Tech., Boston, MA), respectively.The corresponding secondary antibodies were used- don-key anti-rat Alexa Fluor 594 (1:1000; Molecular Probes,OR) for CD11b, goat anti-rabbit Fluorescein Isothiocya-nate (FITC, 1:200; Vector Laboratories) for both iNOS andCox-2. For labeling mature neurons, NeuN staining wasdone by performing antigen retrieval on the sections. Sec-tions were blocked for 1 h with blocking solution and thenstained with mouse anti-NeuN (1:500; Chemicon), over-night at 4�C. After PBS wash, the sections were incubatedin horse anti-mouse FITC (1:200; Vector Laboratories).The stained slides were observed under the Zeiss Axioplan2 Fluorescence microscope.
Counting
The three different types of microglia as identified bylectin staining were differentially counted in four regionsof the mouse brain viz., cortex, hippocampus, striatum,and thalamus. Pictures were captured under 203 magnifi-cation and the different subtypes were morphologicallysorted and counted from five different fields from the sameregion by using the software IM50 (Leica). Each type wasthen expressed as the percentage of total number of micro-glia in a particular region and was plotted in a graph.
Immunoblot Analysis
The brain tissue from animals at days 2 and 4 postin-fection and controls were dissected and placed in micro-fuge tubes with PBS containing protease inhibitor cock-tail (Sigma). Samples were homogenized and centrifugedat 8000g for 20 min. Supernatant was further sonicatedand protein concentrations were determined using Brad-ford Method.
BV-2 cells were washed twice with ice-cold PBS, thenlysed in buffer containing 1% Triton-X-100, 10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.5% Nonidet P (NP-40),1 mM EDTA, 0.2% EGTA, 0.2% sodium orthovanadate,
and protease inhibitor cocktail. The lysate was incubatedon a rocking platform at 4�C for 30 min before centrifuga-tion at 10,000g for 15 min at 4�C. Protein levels weredetermined by Bradford method. To increase the intracel-lular accumulation of iNOS protein in BV-2 cells, Brefel-din A (BD Pharmingen, 1 lg/mL) was added to the cul-ture for the last 4 h of incubation (Basu et al., 2000).
Ten microgram of each sample was electrophoresed onpolyacrylamide gel and transferred onto a nitrocellulosemembrane. The membrane was then blocked in 2.5%skimmed milk in PBS-Tween-20 for 1 h at room tempera-ture with gentle agitation. After blocking, the blots wereincubated with rabbit anti-iNOS (Chemicon) diluted1:2,000 in 1% BSA in PBS-Tween-20 overnight at 4�Cwith gentle agitation. After extensive washes in PBS-Tween-20, blots were incubated with goat anti-rabbithorseradish peroxidase (Vector Laboratories) at a dilutionof 1:5,000 in 1% BSA diluent for 1 h, with agitation. Theblots were rinsed again in PBS-Tween-20. The chemilumi-nescence reagent from Roche (Basel, Switzerland) wasused according to the manufacturer’s instructions. Theblots were developed by exposing them to Chemigenius,Bioimaging System (Syngene, Cambridge, UK). The imageswere captured and analyzed using the GeneSnap andGeneTools software respectively, from Syngene. The blotswere stripped (30 min at 50�C in 62.5 mmol/L Tris-HClpH 6.8, 2% sodium dodecyl sulfate (SDS), 100 mmol/L2-mercaptoethanol) and reprobed with anti-b-tubulin (1:1,000;Santa Cruz Biotechnology; CA) to determine equivalentloading of samples. Similarly, blots were developed forrabbit anti-Cox-2 (1:1000, Cell Signaling Tech) using thesecondary goat anti-rabbit HRP (1:5000, Vector Laborato-ries). With in vivo protein samples blots for mouse anti-NeuN (1:1000, Chemicon) were developed using the sec-ondary antibody horse anti-mouse HRP (1:5000, VectorLaboratories).
RNA Isolation and Semi QuantitativeRT-PCR Analysis
Tissue samples were homogenized in Trizol reagent(Sigma) and total cellular RNA was isolated from 2 and4 days postinfected animals and from age matched con-trol as described previously (Basu et al., 2002b). IsolatedRNA was quantified using spectrophotometry, aliquoted,and stored at 270�C. Reverse-transcriptase polymerasechain reaction (RT-PCR) was performed using the onestepRT-PCR kit (Qiagen Biosciences; Hamburg, Germany) fol-lowing the manufacturer’s protocol and reactions werecarried out on Genius Techne thermal cycler. One micro-gram of the total RNAwas used as template in 25 lL PCRreactions, containing 53 PCR Buffer (5 lL), dNTPs (1 lL),Enzyme Mix (1 lL), specific forward and reverse primers(0.6 lM), and RNase free water. Oligonucleotide primerpairs against mouse IL-6, IL-1b, TNF-a, MCP-1, andcyclophilin mRNAs were prepared from Microsynth(Balgach, Switzerland) (Table 1). Amplification of themRNA sequences of IL-6, IL-1b, TNF-a, MCP-1, andCyclophilin produced single bands that were of the size
485MICROGLIAL ACTIVATION IN JAPANESE ENCEPHALITIS
GLIA DOI 10.1002/glia

predicted from the reported sequences for these mRNAs.PCR products were separated on 2% agarose gel, stainedwith ethidium bromide, and photographed using Gene-Snap software provided with Chemigenius BioimagingSystem, Syngene. Photographs were analyzed by Gene-Tools software provided with same Bioimaging system.
Cytokine Bead Array
Cytokine bead array (CBA, mouse inflammation kit; BDBiosciences) was used to quantitatively measure cytokineexpression levels in the control and infected mouse braintissue lysates and in the supernatant collected from mock-infected and JEV cocultured BV-2 cell line. CBA was alsoperformed to measure cytokines from different brainregions (cortex, hippocampus, striatum, and thalamus).The assay was performed according to the manufacturer’sinstructions and analyzed on the FACS Calibur (BectonDickinson). Analysis was performed using CBA softwarethat allows the calculation of cytokine concentrations inunknown samples (Soldan et al., 2004).
Statistical Analysis
All the experiments performed and the data generatedwere analyzed statistically by paired two-tailed Student’st-test.
RESULTSTopographic Distribution of Activated
Microglial Following JE
As quiescent microglia undergo a series of stereotypedmorphological, phenotypic, and functional changes inresponse to an activating stimulus, we examined the re-sponse of this resident immune cell of the CNS to JEV in-fection. Lectin staining gives a clear image about the mor-phology of the microglia. Using this histochemical stain,we have shown the presence of resting or quiescent cellsexhibiting long thin rod like processes in the control ani-mals (Fig. 1A). Following infection, the resting microgliadecrease in number, and numerous cells with thicker pro-cesses and star-shaped morphology are seen-a characteris-tic of reactive microglia (Fig. 1B). A further shorteningand thickening of the processes occurs as the microgliatransforms from activated to amoeboid or phagocytic formby attaining a rounded darkly stained cell morphology(Fig. 1B). In the infected brain, the number of star-shaped
‘‘reactive’’ or ‘‘activated’’ microglia (Fig. 1B) appeared to bemore frequent in the hippocampus and the adjoining areawhen compared with the cortex. A robust increase in thenumber of activated and phagocytic microglia was ob-served in the thalamus (Fig. 1D), hippocampus (Fig. 1E),and striatum (Fig. 1F), but to lesser extent in the cortex(Fig. 1C). Importantly, the overall number of microgliawas less in the control sections and most of them wererod-shaped and represented the resting state (Fig. 1A).These results clearly indicate that JEV infection is accom-panied by robust microglial activation and that an unequaldistribution of activated microglia exists across differentbrain regions. We therefore determined the topographicaldistribution of activated microglia in the infected brain, bycounting the number of resting, activated, and phagocyticmicroglia in the different regions. Five fields of lectinstained sections of infected brain were chosen for each re-gion under 203 magnification. Lightly stained rod-shapedcells were counted as resting cells (Fig. 1B), star-shapedcells as reactive cells (Fig. 1B), and darkly stained roundor triangular shaped cells (Fig. 1B) were considered asamoeboid cells. The different types of microglia were ex-pressed as a percentage of the total number of microglia.In the hippocampus (59.43 6 8.4)% were activated, (29.6 64.9)% were phagocytic, and (10.97 6 1.7)% were restingcells. The thalamus had (57.74 6 8.18)% activated, (28.73 63.78)% phagocytic, and (13.52 6 3.36)% resting microglia.While the striatum had (60.34 6 2.1)% activated, (24.14 68.5)% phagocytic and (15.52 6 3.5)% resting microglia, thecortex of the infected brain housed (36.81 6 9.5)% acti-vated, (22.9 6 3)% phagocytic microglia, and (40.29 64.9)% resting microglia.
In the hippocampus and the striatum, the number ofactivated cells was more than anywhere else in the brainclosely followed by that in the thalamus (Fig. 1G). In thecortex, number of activated microglia was significantlyless when compared with other regions of the mouse brain(P < 0.01) (Fig. 1G). The number of phagocytic microgliawas more or less similar in all areas of the brain (Fig. 1G).Interestingly, the cortex had a significantly higher per-centage of resting cells when compared with any otherregions (P < 0.01) of the infected brain (Fig. 1G).
The Heightened Release of ProinflammatoryCytokines Following JE is Brain Region Selective
Previous studies have indicated that activated microgliasecrete various proinflammatory cytokines. We, therefore,investigated the functional profile of activated microgliafollowing JEV infection, by determining mRNA transcripts
TABLE 1. Primers Used for RT-PCR Experiments and Expected Size of Amplified Products
Forward primer Reverse primer Product size (bp)
IL-6 50-GAGGATACCACTCCCAACAGACC-30 50-AAGTGCATCATCGTTGTTCATACA-30 117IL-1b 50-TGGAAAAGCGGTTTGTCT-30 50-ATAAATAGGTAAGTGGTTGCC-30 294TNF-a 50-GGCAGGTCTACTTTGGAGTCATTGC-30 50-ACATTCGAGGCTCCAGTGAATTCGG-30 275MCP-1 50-ACCAAGCTCAAGAGAGAGGT-30 50-CTGGATTCACAGAGAGGGAA-30 380Cyclophilin 50-CCATCGTGTCATCAAGGACTTCAT-30 50-TTGCCATCCAGCCAGGAGGTCT-30 192
IL, Interleukins; TNF-a, Tumor necrosis factor-a; RT-PCR, Reverse transcriptase-Polymerase chain reaction.
486 GHOSHAL ET AL.
GLIA DOI 10.1002/glia

Fig. 1. Activation of microglia and topographic distribution of acti-vated microglia following JEV infection. A & B: Cryostat sections fromcontrol (A) and infected mouse brains (B) were processed for lectin stain-ing. A robust increase in expression of lectin, a surface marker for micro-glia, was seen accompanied with a distinct change in morphology inresponse to JEV infection when compared with the control brain. Whilethe control sections exhibited only resting microglia (A; 1fi), the infectedbrains showed the presence of activated (B; 2fi) and amoeboid microglia(B; 3fi). A few resting microglia were also observed in the infected brain(B; 1fi). C–F: Activation of microglia in different regions of the brain.The thalamus (D), hippocampus (E), and striatum (F) were completely
infiltrated by activated and phagocytic microglia. The number of acti-vated microglia in the cortex (C) was less when compared with that ofthe other regions. The insets characterize the respective areas. Scale baris 20 lm. G: The percentage of different types of microglia in differentareas of the brain. The percentage of activated microglia in the hippo-campus and the striatum is the highest closely followed by that in thethalamus. The percentage of activated microglia is significantly lowerand that of resting microglia is significantly higher in the cortex thanother areas of the brain (*significant change P < 0.01). The percentageof phagocytic microglia is almost similar across different regions of theinfected brain, with the highest in the hippocampus.
487MICROGLIAL ACTIVATION IN JAPANESE ENCEPHALITIS
GLIA DOI 10.1002/glia

levels of various proinflammatory cytokines by RT-PCR.Included in the panel of proinflammatory cytokines wereIL-6, IL-1b, MCP-1, and TNF-a (Figs. 2A,B). A generaltrend towards an increase in the expression of proinflam-matory mRNA transcripts was observed during thecourse of a progressive infection when compared with theuninfected controls. The expression of IL-6 (117 bp), wasenhanced by 2-fold (P < 0.01) and 4-fold (P < 0.05) at 2and 4 days postinfection respectively, when comparedwith the normal controls (Figs. 2A,B). The levels of IL-1b(294 bp) band, was elevated significantly by 9-fold (P <0.001) and 8-fold (P < 0.05) at 2 and 4 days postinfectionrespectively, when compared with the uninfected controls(Figs. 2A,B). The chemokine MCP-1 (380 bp) also showeda significant 8-fold (P < 0.05) and 11-fold increase (P <0.005) on 2 and 4 days postinfection respectively, with re-spect to uninfected controls. However, amongst all theproinflammatory molecules investigated, the increase inthe expression of TNF-a was the maximum. The increasein TNF-a transcript (275 bp) was as great as 14-fold (P <0.001) and 12-fold (P < 0.05) on 2 and 4 days postinfec-tion respectively, when compared with the control sam-ples (Figs. 2A,B).
We also performed CBA (Fig. 2C) to observe the levelsof different proinflammatory cytokines in control and
infected mouse brain. Included in the list of proinflamma-tory cytokines examined were MCP-1, IL-6, and TNF-a.While the expression of IL-6 and TNF-a was upregulatedat 2 days postinfection, the increase seemed only slightwhen compared with the dramatic 180-fold increaseobserved in the level of MCP-1 (P < 0.001) at this stage ofinfection. However, once elevated this enhanced level ofMCP-1 was maintained at approximately 200-fold (P <0.001) over control, at 4 days postinfection (Fig. 2C). Onthe contrary, the levels of IL-6 and TNF-a increased mark-edly reaching levels greater than 700 and 100-fold respec-tively, at 4 days postinfection, when compared with thecontrol (P < 0.001) (Fig. 2C). Taken together, there was anoverall induction of proinflammatory cytokines and che-mokines at the protein level following JE infection.
We next investigated whether the expression profile ofproinflammatory cytokines in different brain regions hasany correlation with the region selective microglial acti-vation that follows JEV infection. Cortex, hippocampus,striatum, and thalamus were dissected from infectedbrains and age matched controls. The protein extractedfrom each of these brain regions were analyzed using CBA.A dramatic increase in the levels MCP-1, IL-6, and TNF-awas observed in the hippocampus with respect to the otherbrain regions (Figs. 3A–C). There was a significant 650
Fig. 2. Induction of pro-inflammatorycytokines in the mouse brain at differentdays post JEV infection. A & B: TotalmRNA was isolated from control and 2and 4 days postinfected mice brains andreverse transcribed. The levels of IL-6, IL-1b, TNF-a, andMCP-1 were assessed usingsemi quantitative RT-PCR and quantifiedusing the GeneTools software. Expressionprofiles of IL-6, IL-1b, TNF-a, and MCP-1at 2 and 4 days postinfection with JEV(A). Data shown is a representative of oneindividual animal from each group. Thegraph indicates fold change in IL-6, IL-1b, TNF-a, and MCP-1 mRNA levelsover the control at different days postin-fection (B). Values represent means 6SEM from six mice in each group. *Sig-nificant change from control P < 0.001(IL-1b, TNF-a), P < 0.01 (IL-6), P < 0.05(MCP-1); **significant change from andcontrol 2 days infected samples respec-tively, P < 0.001 and P < 0.05 (IL-1b),P < 0.01 and P < 0.05 (IL-6, TNF-a), andfor MCP-1 P < 0.005 from both controland 2 days postinfection. C: Expression ofMCP-1, IL-6, and TNF-a as observed byCBA at 2 and 4 days postinfection. Levelof MCP-1 was significantly high at both 2and 4 days postinfection when comparedwith the control. Levels of IL-6 and TNF-a were significantly enhanced at 4 dayspostinfection when compared with 2 dayspostinfection and control group. Valuesrepresent means 6 SEM from 6 mice ineach group. *Significant change from con-trol P < 0.001; **significant change fromcontrol and 2 days infected samples P <0.001.
488 GHOSHAL ET AL.
GLIA DOI 10.1002/glia

and 750-fold increase in the expression of MCP-1, in thecortex and hippocampus respectively, at 2 days postinfec-tion when compared with the control. The difference in thelevels of MCP-1 between these two regions was significant
(P < 0.005). The level of MCP-1 declined in the cortex by 8-fold at 4 days postinfection when compared with 2 dayspostinfection (Fig. 3A). On the contrary, the enhanced levelof MCP-1 was not only maintained in the hippocampus butalso showed a further elevation (1000-fold over control) at4 days postinfection, thereby maintaining it at a level fargreater than that observed in the cortex at similar timepoint (P < 0.001). MCP-1 levels were also induced in thestriatum and thalamus at days 2 and 4 postinfection overuninfected animals, but the increase was negligible whencompared with its level in the hippocampus (at 2 and 4days postinfection) and cortex (at 2 days postinfection). A9-fold and 12 fold increase in the level of IL-6 was observedin cortex and hippocampus respectively, at 2 days postin-fection over control, indicating a significant difference inits level between the two regions (P < 0.05). While thelevel of IL-6 released from the cortex at 4 days postinfec-tion was comparable to that seen on day 2 postinfection, adramatic 490-fold increase in expression was observed inthe hippocampus on day 4 postinfection when comparedwith the control. The difference in the levels of IL-6 be-tween the two regions was significant (P < 0.001) (Fig. 3B).While there was no significant difference in the expressionlevel of TNF-a between cortex and hippocampus at 2 dayspostinfection, a significant 40-fold increase was seen inthe hippocampus when compared with only 6-fold increasein the cortex at 4 days postinfection when compared withthe control (P < 0.005) (Fig. 3C). While an induction of IL-6 and TNF-a in the striatum and thalamus was observedat 2 and 4 days postinfection when compared with unin-fected animal, the increase was negligible when comparedwith the level of those two cytokines released from thehippocampus at 4 days postinfection. These results indi-cate differential expression of proinflammatory cytokinesfrom different brain regions during a progressive JEVinfection.
Induction of iNOS Following JE
As inducible nitric oxide synthase (iNOS) is an impor-tant proinflammatory molecule secreted by activatedmicroglia, we compared the levels of iNOS in mouse brainsat 2 and 4 days postinfection with controls. A significant2-fold increase in the expression of iNOS (which migratedas single band of 130 kDa) was observed at day 2 post-infection when compared with the control (P < 0.05)(Figs. 4A,B). Interestingly, this enhanced level of iNOSdeclined significantly at 4 days postinfection (P < 0.05),attaining levels comparable to that observed in unin-fected animals (Figs. 4A,B). Therefore, the peak in iNOSexpression at 2 days postinfection was followed by a sig-nificant decline as infection progressed.
Immunohistochemistry performed on control and 2 dayspostinfected brain sections not only confirmed an increasein iNOS expression following JE infection but also demon-strated that the release of iNOS occurs from the activatedmicroglia, as evident from the double staining of iNOS withCD11b (a marker for activated microglia/macrophages)(Figs. 4C–F). While the presence of double positive cells in
Fig. 3. Differential expression of proinflammatory cytokines acrossspecific brain regions: Protein extracted from cortex, hippocampus, stria-tum, and thalamus dissected from controls, 2 and 4 days postinfectedbrains were analyzed using CBA. A–C: The graphs depict the fold changein TNF-a, MCP-1, and IL-6 levels at days 2 and 4 postinfection when com-pared with the control. There was a dramatic increase in the level ofMCP-1 in the hippocampus (both at 2 and 4 days postinfection) and cortex(only at 2 days postinfection) when compared with the control as well asstriatum and thalamus of infected animal. The increase in IL-6 and TNF-a levels was confined to the hippocampus alone, and the levels of thesetwo cytokines in the cortex, striatum, and thalamus of infected animalwas almost comparable to that of the control level. Values representmeans 6 SEM from five mice in each group. *Significant change of hippo-campal cytokine level from cortex P < 0.005 and P < 0.001 (MCP-1),P < 0.05 and P < 0.001 (IL-6) at 2 and 4 days postinfection respectively,and P < 0.005 (TNF-a) for 4 days postinfection.
489MICROGLIAL ACTIVATION IN JAPANESE ENCEPHALITIS
GLIA DOI 10.1002/glia

Fig. 4. Induction of iNOS in mouse brain following JE. A: Protein iso-lated from control, 2 and 4 days postinfected mice brains were analyzedby immunoblot. A significant increase in the level of iNOS at 2 days post-infection was followed by a significant decrease at 4 days postinfection.Data shown is a representative of two individual animals from eachgroup. B: The graph represents the fold change in the expression levelsof iNOS in infected animals over the control. Values represent the means6 SEM from six animals in each group (*significant change from controlP < 0.05; ** significant change from 2 days infected samples P < 0.05).C–F: Cryostat sections from control and 2 days postinfected BALB/cmice brain were double-stained for CD11b and iNOS and mounted using
Vectashield containing DAPI. The control sections did not exhibit anystaining for both CD11b (C) and iNOS (D). The inset picture in (C)depicts the representative hippocampal field. The infected brain sections(E-F) show marked presence of activated microglia, as evident from theco-localization of CD11b (Alexa Fluor, red) and DAPI (blue) shown by thearrows (E) in a representative hippocampal field (inset at E). The releaseof proinflammatory mediator iNOS from these activated microglia isdemonstrated by the co-localization of iNOS (FITC, green) with CD11b(Alexa Fluor, red) indicated by the arrows (F). The inset depicts (F) ahigher power view of CD11b and iNOS co- expressing cells (yellow).Scale bar: 20 lm.
Fig. 5. Induction of Cox-2 in mouse brain following JEV infection. A:Protein isolated from control, 2 and 4 days postinfected mice brains wereanalyzed by immunoblot. A significant increase in the level of Cox-2 at 2days postinfection was followed by a significant decrease at 4 days post-infection. Data shown is a representative of two individual animals fromeach group. B: The graph represents the fold change in the expressionlevels of Cox-2 in infected animals over the control. Values represent themeans 6 SEM from 6 animals in each group (*significant change fromcontrol P < 0.05; **significant change from 2 days infected samples P<0.05). C–F: Cryostat sections from control and 2 days postinfectedBALB/c mice brain were double-stained for CD11b and Cox-2 and
mounted using Vectashield containing DAPI. The control brain showednegligible presence of activated microglia (C) as well as lack of expres-sion of Cox-2 (D) as observed by the absence of staining for both CD11b(Alexa Fluor, red) and Cox-2 (FITC, green). The inset picture depicts therepresentative hippocampal field. However, a robust activation of micro-glia was seen in the infected brain sections (co-localization of CD11bwith DAPI) (E). Co-localization of CD11b and Cox-2 as shown by thearrows in the same hippocampal field indicates the release of Cox-2 bythe activated microglia (F). Inset The inset depicts (Fig F) a higherpower view of CD11b and Cox-2 co- expressing cells (yellow). Scale bar:20 lm.

the infected brain (Figs. 4E,F) was clearly evident, nosuch cells were observed in the control brain (Figs. 4C,D).
Induction of Cox-2 Following JEV Infection
As Cox-2 is another potent proinflammatory mediator(besides iNOS), its expression level following JEV infec-tion was also determined by immunoblot (a single band of80 kDa) in 2 and 4 days postinfected brain. A significant2.5-fold increase was noted at 2 days postinfection whencompared with control (P < 0.05) (Figs. 5A,B). However,the expression of Cox-2 declined along a progressive infec-tion, with levels becoming comparable to uninfected con-trol at 4 days postinfection (P < 0.05) (Figs. 5A,B).
We also performed immunohistochemistry to detect theexpression of Cox-2 in uninfected and 2 days postinfectedmouse brain sections. While the expression of Cox-2 wasundetectable in control animals as expected (Fig. 5D), asignificant increase in Cox-2 expression was observed fol-lowing infection (Fig. 5F). Importantly, co-localization ofCox-2 and CD11b in infected animals further establishedthat activated microglia releases Cox-2 following JE in-fection (Fig. 5F).
In Vitro Co-Culture of Microglia with JEVInduces Release of Proinflammatory
Mediators and Cytokines
To confirm the in vivo findings that proinflammatory med-iators and cytokines are released predominantly from micro-glia following JE, we infected mouse microglial cell line BV-2with JEV in vitro. Immunoblot analysis revealed a 4-foldincrease in iNOS and a 3-fold increase in the level of Cox-2in BV-2 cells treated with the virus, when compared withthe mock-infected BV-2 cells (P < 0.005) (Figs. 6A,B). Thisprovides clear evidence of the release of proinflammatorymediators (primarily iNOS and Cox-2) by the BV-2 micro-glial population upon JEV infection.
The CBA profile of the infected BV-2 supernatantdemonstrated an increase in the release of various proin-flammatory cytokines. A 8-fold and 6.5-fold increase inthe levels of TNF-a and IL-6 respectively, was observedin the infected supernatant when compared with themock infected control (P < 0.005) (Fig. 6C). Similarly,MCP-1 expression was elevated by 6-fold in BV-2 cellstreated with the virus when compared with the mock-infected cells (P < 0.005).
Microglial Activation Results in Neuronal DeathBoth In Vivo and In Vitro
To investigate whether microglial activation and theresulting release of proinflammatory cytokines followingJEV activation can result in a bystander damage of neu-rons, we determined the expression of NeuN–a marker of
mature neurons, during the course of a progressive infec-tion. Western blot analysis revealed two distinct bands(46 and 48 kDa) out of the three possible bands of theNeuN protein in all the samples examined (Fig. 7A).Although the levels of NeuN at 2 days postinfection wasslightly lower than the control, a significant decrease inexpression was observed in 4 days postinfected samples(P < 0.05) (Fig. 7B). To investigate whether this decreasein NeuN expression had resulted from (i) a decrease in thetotal number of neurons or (ii) a decrease in the expres-sion of NeuN in the existing neurons, we performed im-munostaining of control, 2 and 4 days JEV infected brainsections with NeuN.
In the control brain sections, numerous NeuN positivehealthy cells were observed in a representative corticalfield (Fig. 7C). In 2 days postinfected sections the numberof NeuN positive cells were slightly less than the control,in a similar cortical region under the same magnification(Fig. 7D). However, at 4 days postinfection there was a dra-matic decrease in the number of NeuN positive cells whencompared with a similar control cortical field (Fig. 7E).These results suggest that a loss in the total number ofneurons occurs during a progressive JE infection.
To investigate whether proinflammatory mediators re-leased from activated microglia mediate the neuronal lossobserved during a progressive JEV infection, in vitro stu-dies were performed. Mouse microglial cell line BV-2 wereeither mock infected or cocultured for 1 h with live JEV orboiled virus as described in materials and methods. Thesupernatant collected from these cells was then used totreat mouse N2a cell line in presence or absence of cele-coxib and aminoguanidine, respective inhibitors of Cox-2and iNOS. To confirm that the effect seen upon treatingN2a with culture supernatant is not the direct effect of vi-rus present in the supernatant on N2a, plaque assay wasperformed to determine the presence of virus in the super-natant. Plaque assay results confirmed that the superna-tant was devoid of any live virus (data not shown).
To determine whether microglia-produced cytotoxinsare involved in inducing increased neuronal killing viaapoptosis, TUNEL staining was performed on N2a celltreated with BV-2 supernatant collected from the above-mentioned conditions. There was no significant differencein the percentage of apoptotic cell in N2a cultured witheither the boiled-virus treated (Figs. 7H,K) or mock in-fected BV-2 supernatant (Figs. 7F,K). Interestingly, a dra-matic 25-fold increase in TUNEL positive apoptotic cellwas observed when N2a were cultured in supernatantobtained from BV-2 co-cultured with live JEV (P < 0.005)(Figs. 7G,K). Moreover, a significant decrease in the num-ber of TUNEL positive cells (P < 0.005) with respect tovirus-treated BV-2 supernatant was also noticed, whenthis supernatant was supplemented with either celecoxibor aminoguanidine (Figs. 7I–K). The results obtainedfrom TUNEL assay clearly indicate that proinflammatorymediators released from activated microglia adverselyaffect neuronal viability ultimately culminating in neuro-nal loss, and that preventing the action of these mediatorsby using selective inhibitors offers protection against neu-ronal death.
491MICROGLIAL ACTIVATION IN JAPANESE ENCEPHALITIS
GLIA DOI 10.1002/glia

DISCUSSION
As the primary immune effector cells of the CNS, micro-glia respond to injury, inflammation or the presence ofpathogens by becoming activated. When activated, micro-glia undergo proliferation, chemotaxis, and morphologicalalteration and generate numerous mediators involved inthe inflammatory and immunomodulatory response. Thesemediators not only coordinate tissue remodeling in re-sponse to damage but also possess several means by whichthey protect cells from infection. Though the central roleplayed by microglia in the CNS response to damage is welldocumented, few studies have investigated the role ofmicroglia in JE. We therefore investigated the role ofmicroglia in the pathogenesis of JE using an experimen-tal model. Our studies have demonstrated for the firsttime that (i) JEV infection activates microglia both mor-phologically and functionally, in vivo (ii) infection leads toan elevation of proinflammatory mediators; (iii) microgliaare the predominant source of proinflammatory mediators,and (iv) the cytotoxins released from activated microgliaare instrumental in inducing neuronal death that accom-panies JE. Our findings therefore clearly suggest thatmicroglial activation may be an important contributoryfactor in the pathogenesis of JE.
Although primary neurons are easily infected by JEV(Chen et al., 2000), glial cells could also be infectedin vitro (Chen et al., 2000; Liao et al., 2002). However,the functional state of microglial activation followingJEV infection remains uncertain. Though immunoreac-
tivity of JEV NS3 (Non-structural protein 3) has beenfound to be localized to microglia in JEV-infected mixedglial culture (Chen et al., 2004), the preference of micro-glia for JEV infection in vivo is unknown. We haveobserved a dramatic increase in activated microglia fol-lowing infection as demonstrated by the increased num-ber of lectin positive cells exhibiting shorter and thickerprocesses- a feature characteristic of activated microglia.Moreover, the number of activated microglia is greater inthe hippocampus when compared with other regions ofthe brain. Survivors of JE are often left with severe neu-rological sequel, including motor deficits, cognitive andlanguage impairment, learning difficulties, and behav-ioral problems (Desai et al., 1995). As the number of acti-vated microglia is greatest in the hippocampus- the brainregion best linked to learning and memory, these findingscould provide some explanations for the cognitive impair-ments observed in survivors of JE.
The response to cerebral infection with JEV in mice ischaracterized at the pathological level by significantrecruitment and extravasation of immunoinflammatorycells (Chaturvedi et al., 1979; Mathur et al., 1988, 1992).The inflammatory response observed within the CNS inviral encephalitis is partly mediated by chemokines,which are released by various cells of the CNS (Ishiguroet al., 1997). Neuroinflammation is thought to develop asa consequence of two processes that follow sequentially.The first process is the activation of microglia followedclosely by the mobilization and recruitment of peripheralinflammatory cells into the site of damage. The second
Fig. 6. JE virus induces the release ofproinflammatory mediators following co-cul-ture with mouse microglial cell line BV-2.Mouse microglial cell line BV-2 was co-cul-tured with (MOI 5 5) and without JE virusfor 1 h at 37�C. After 24 h, supernatant wascollected for CBA and protein was extractedfrom the cells for immunoblot. A: A signifi-cant increase in the expression of both iNOS(130 kDa) and Cox-2 (80 kDa) was noticedfollowing treatment of the BV-2 cells with vi-rus. The blots were then stripped and re-probed with b-tubulin to confirm equal pro-tein loading. B: The graph depicts the foldincrease in expression of iNOS and Cox-2 inBV-2 cells cocultured with the virus whencompared with mock-infected cells. Valuesrepresent the means 6 SEM from 3 inde-pendent experiments performed in duplicate(*, # significant change from mock-infectedBV-2 cells P < 0.005). C: The CBA profiledemonstrates the fold increase in MCP-1,TNF-a, IL-6 following co-culture of BV-2cells with JEV over mock-infected BV-2 cells(*significant change from mock-infected BV-2 cells P < 0.005).
492 GHOSHAL ET AL.
GLIA DOI 10.1002/glia
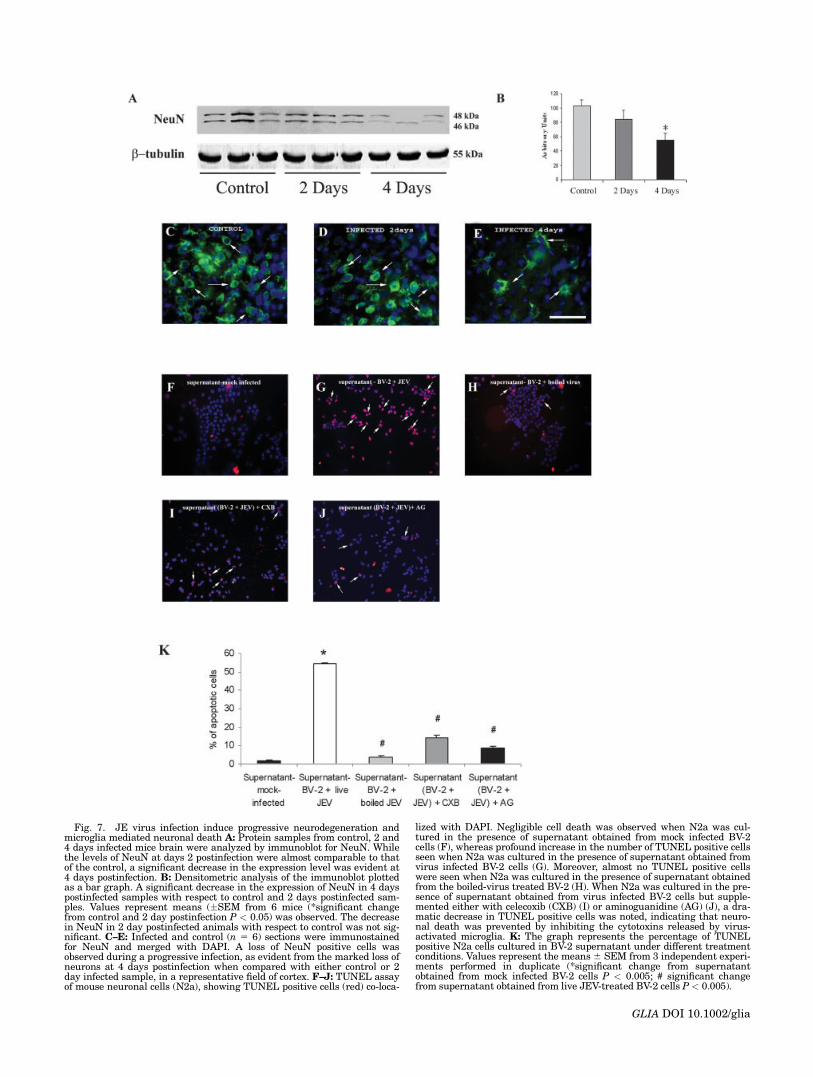
Fig. 7. JE virus infection induce progressive neurodegeneration andmicroglia mediated neuronal death A: Protein samples from control, 2 and4 days infected mice brain were analyzed by immunoblot for NeuN. Whilethe levels of NeuN at days 2 postinfection were almost comparable to thatof the control, a significant decrease in the expression level was evident at4 days postinfection. B: Densitometric analysis of the immunoblot plottedas a bar graph. A significant decrease in the expression of NeuN in 4 dayspostinfected samples with respect to control and 2 days postinfected sam-ples. Values represent means (�SEM from 6 mice (*significant changefrom control and 2 day postinfection P < 0.05) was observed. The decreasein NeuN in 2 day postinfected animals with respect to control was not sig-nificant. C–E: Infected and control (n 5 6) sections were immunostainedfor NeuN and merged with DAPI. A loss of NeuN positive cells wasobserved during a progressive infection, as evident from the marked loss ofneurons at 4 days postinfection when compared with either control or 2day infected sample, in a representative field of cortex. F–J: TUNEL assayof mouse neuronal cells (N2a), showing TUNEL positive cells (red) co-loca-
lized with DAPI. Negligible cell death was observed when N2a was cul-tured in the presence of supernatant obtained from mock infected BV-2cells (F), whereas profound increase in the number of TUNEL positive cellsseen when N2a was cultured in the presence of supernatant obtained fromvirus infected BV-2 cells (G). Moreover, almost no TUNEL positive cellswere seen when N2a was cultured in the presence of supernatant obtainedfrom the boiled-virus treated BV-2 (H). When N2a was cultured in the pre-sence of supernatant obtained from virus infected BV-2 cells but supple-mented either with celecoxib (CXB) (I) or aminoguanidine (AG) (J), a dra-matic decrease in TUNEL positive cells was noted, indicating that neuro-nal death was prevented by inhibiting the cytotoxins released by virus-activated microglia. K: The graph represents the percentage of TUNELpositive N2a cells cultured in BV-2 supernatant under different treatmentconditions. Values represent the means6 SEM from 3 independent experi-ments performed in duplicate (*significant change from supernatantobtained from mock infected BV-2 cells P < 0.005; # significant changefrom supernatant obtained from live JEV-treated BV-2 cells P < 0.005).
GLIA DOI 10.1002/glia

involves the production of a number of proinflammatorymediators from activated microglia, which further in-creases the competence of the cerebral endothelium torecruit peripheral immune cells, into the damaged brain(del Zoppo et al., 2000).
The set of mediators that are elevated following JEVinfection consists of proinflammatory cytokines, whichwould normally be associated with the recruitment ofinflammatory cells, to the site of infection. These cyto-kines could contribute to the disruption of the blood brainbarrier (BBB), which facilitates additional disseminationof the virus within the CNS. JEV infection induces theexpression of IL-1b, IL-6, and TNF-a- mediators that areassociated with microglial activation. As reported earlier,IL-1b is the upstream mediator of the innate immuneresponse and is essential for both basal and induced ex-pression of IL-6 (Basu et al., 2000b). Moreover, IL-6 is fre-quently induced by TNF-a and has been associated withneurotropic viral infections (Aloisi et al., 1992; Breenet al., 1990; Nakajima et al., 1989; Okuda et al., 1998;Olson et al., 2001; Perrin et al., 2000). TNF-a, an impor-tant mediator of proinflammatory functions in viral dis-eases (Arend, 2001; Barbara et al., 1996; Wesselinghet al., 1994) also induces iNOS, which can contribute tothe development of the pathological process (Li et al.,2004).
Our results indicate that JEV infection induces theexpression of iNOS, the key enzyme in nitric oxide (NO)synthesis. There are several consequences for aberrantproduction of NO: (i) NO from activated leukocytes andastrocytes leads to the production of peroxynitrite, whichcan irreversibly damage lipids and proteins causing celldeath (Iadecola and Zhang, 1996), (ii) NO can nega-tively regulate mammalian neurogenesis (Packer et al.,2003), and (iii) NO can enhance glutamate excitotoxicity(Hewett et al., 1996). The significant increase in iNOSlevel at 2 days postinfection followed by the declinein level (comparable to the control) at 4 days postinfec-tion, prompts us to speculate the existence of a compen-satory mechanism that attempts to reduce the level ofinflammation.
Our results also suggest that JEV infection induces theproduction of Cox-2, an inducible enzyme that catalyzesthe first step in the synthesis of prostanoids such as pros-taglandin E2 and prostaglandin I2. These lipid-derivedmediators of inflammation can induce edema, which isdeleterious to neuronal function and survival. In addi-tion, a byproduct of Cox-2 catalysis is the production ofreactive oxygen species (ROS), which multiple studieshave shown to contribute to brain damage. Additionalevidence that cyclooxygenases and prostaglandin produc-tion exacerbate brain damage comes from studies show-ing that Cox-2-overexpressing transgenic mice are morevulnerable to excitotoxic insult, and that mice lackingphospholipase A2 are less vulnerable to cerebral ischemia(Bonventre et al., 1997; Kelley et al., 1999).
There are strong data implicating cytokines in micro-glial activation. Cytokines such as IL-1b, g-interferon,TNF-a, and IL-6 have been shown to directly activatemicroglia (Basu et al., 2002a; Loughlin et al., 1992; Suzu-
mura et al., 1990). Activated microglial cells release solu-ble effectors that can directly or indirectly cause damageto neuronal cells. Microglia release IL-1b, IL-3, IL-6,TNF-a, vascular endothelial growth factor (VEGF), lym-photoxin, macrophage inflammatory protein 1a (MIP-1a),matrix metalloproteinases (MMPs), NO, and reactive ox-ygen species (ROS) (Banati and Kreutzberg, 1993; Coltonand Gilbert, 1987; Gehrmann et al., 1995). The cytokinesIL-1b, IL-6, TNF-a, and lymphotoxin alter expression ofvascular cell adhesion molecules to recruit lymphocytesand macrophages to injury sites (Shima et al., 1995). Inaddition, lymphotoxin, TNF-a, NO, and ROS can directlykill cells. TNF-a has been shown to induce the death ofneurons via a direct apoptotic effect after receptor bind-ing and by directly interfering with intracellular sub-strates used by growth factors such as IGF-I and the in-sulin receptor (Shen et al., 2004; Venters et al., 1999).Elevated cytokines activate microglia, thereby stimulat-ing a cycle of inflammation that recruits leukocytes,causes vascular breakdown, and directly induces celldeath through the release of cytotoxic substances. Ourin vitro bioassay data clearly demonstrated the ability ofactivated microglia to release soluble factors that can in-duce neuronal death. Thus, microglial activation appearsto be intricately related with a number of pathologiesassociated with Japanese Encephalitis, including neuro-nal death and glial dysfunction.
It has been previously reported that JEV replicates inthe mouse brain and the viral load is detectable onlyaround 2 day postinfection followed by gradual increasein load with the titer reaching its peak at 4 day postinfec-tion (Vrati et al., 1999). Though JEV is predominantly aneurotropic virus (with its principal target of infectionin vivo being neuronal cells) (Hase et al., 1987), in vivoreplication of JEV in mouse mononuclear cells (Kelkarand Banerjee, 1977) as well as in vitro infection of humanmonocytes (Hasegawa et al., 1990; Kedarnath et al.,1986) and other mammalian cell lines such as Vero, BHK21, and PC 12 have also been reported. We have foundoccasional co-localization of viral protein with microglialspecific marker. However, it is difficult to concludewhether the co-localization is indicative of microglialinfection by the virus or the phagocytosis of viral particlealong with neuronal debris by microglia. Our findingsraises the possibility that besides its primary target neu-rons, JE virus also infects microglia, although in vivomicroglial activation could be a response to neuronaldamage with the subsequent inflammation resulting innegative consequences. Our study demonstrates for thefirst time that coculturing microglia in the presence ofJEV induces the release of mediators capable of killingneurons.
Although the net effect of the proinflammatory media-tors is to kill infectious organisms and infected cells aswell as to stimulate the production of molecules thatamplify the mounting response to damage, it is also evi-dent that in a nonregenerating organ such as brain, adysregulated innate immune response would be deleteri-ous. Our findings suggest that numerous proinflamma-tory mediators released by activated microglia have the
494 GHOSHAL ET AL.
GLIA DOI 10.1002/glia

ability to activate aspects of the microglial responsethemselves. One possible mechanism for such a destruc-tive cycle might be the result of a deregulated feed-forward stimulation of microglia by proinflammatorycytokines. In JE the tight regulation of microglial activa-tion appears to be disturbed, resulting in an autotoxicloop of microglial activation with dire consequences.Obviously, more research is required to determine theregulation and role of cytokines in microglial activationand the possible implication in progressive neurodegen-eration in JE.
ACKNOWLEDGMENTS
S.D. and M.K.M are recipient of Junior Research Fel-lowship from University Grants Commission, Govern-ment of India. The authors thank Mr Kanhaiya LalKumawat for excellent technical assistance. We thankProf. Vijayalakshmi Ravindranath, Director NBRC forher continuous support and encouragement. We thankDr Soumya Iyenger for her help with microscopy. Wethank Zydus Research Centre, Ahmedabad for providingus Celecoxib as a gift.
REFERENCES
Aloisi F, Care A, Borsellino G, Gallo P, Rosa S, Bassani A, Cabibbo A,Testa U, Levi G, Peschle C. 1992. Production of hemolymphopoieticcytokines (IL-6, IL-8, colony-stimulating factors) by normal humanastrocytes in response to IL-1b and tumor necrosis factor-a. J Immu-nol 149:2358–2366.
Appaiahgari MB, Saini M, Rauthan M, Jyoti, Vrati S. 2006. Immuniza-tion with recombinant adenovirus synthesizing the secretory form ofJapanese encephalitis virus envelope protein protects adenovirus-exposed mice against lethal encephalitis. Microbes Infect 8:92–104.
Arend WP. 2001. Cytokine imbalance in the pathogenesis of rheumatoidarthritis: The role of interleukin-1 receptor antagonist. Semin Arthri-tis Rheum 30(5, Suppl 2):1–6.
Banati RB, Kreutzberg GW. 1993. Flow cytometry: Measurement of pro-teolytic and cytotoxic activity of microglia. Clin Neuropathol 12:285–288.
Barbara JA, Van ostade X, Lopez A. 1996. Tumour necrosis factor-a(TNF-a): The good, the bad and potentially very effective. ImmunolCell Biol 74:434–443.
Basu A, Chakrabarti G, Saha A, Bandyopadhyay S. 2000. Modulation ofCD11C1 splenic dendritic cell functions in murine visceral leishmani-asis: Correlation with parasite replication in the spleen. Immunology99:305–313.
Basu A, Krady JK, Enterline JR, Levison SW. 2002a. Transforminggrowth factor b1 prevents IL-1b-induced microglial activation, whereasTNFa- and IL-6-stimulated activation are not antagonized. Glia 40:109–120.
Basu A, Krady JK, O’Malley M, Styren SD, DeKosky ST, Levison SW.2002b. The type 1 interleukin-1 receptor is essential for the efficientactivation of microglia and the induction of multiple proinflamma-tory mediators in response to brain injury. J Neurosci 22:6071–6082.
Bonventre JV, Huang Z, Taheri MR, O’Leary E, Li E, Moskowitz MA,Sapirstein A. 1997. Reduced fertility and postischaemic brain injury inmice deficient in cytosolic phospholipase A2. Nature 390:622–625.
Breen EC, Rezai AR, Nakajima K, Beall GN, Mitsuyasu RT, Hirano T,Kishimoto T, Martinez-Maza O. 1990. Infection with HIV is asso-ciated with elevated IL-6 levels and production. J Immunol 144:480–484.
Chaturvedi UC, Mathur A, Tandon P, Natu SM, Rajvanshi S, TandonHO. 1979. Variable effect on peripheral blood leucocytes during JEvirus infection of man. Clin Exp Immunol 38:492–498.
Chen CJ, Chen JH, Chen SY, Liao SL, Raung SL. 2004. Upregulation ofRANTES gene expression in neuroglia by Japanese encephalitis virusinfection. J Virol 78:12107–12119.
Chen CJ, Liao SL, Kuo MD, Wang YM. 2000. Astrocytic alteration in-duced by Japanese encephalitis virus infection. Neuroreport 11:1933–1937.
Chen CJ, Raung SL, Kuo MD, Wang YM. 2002. Suppression of Japaneseencephalitis virus infection by non-steroidal anti-inflammatory drugs.J Gen Virol 83 (Part 8):1897–1905.
Chen LK, Lin YL, Liao CL, Lin CG, Huang YL, Yeh CT, Lai SC, Jan JT,Chin C. 1996. Generation and characterization of organ-tropismmutants of Japanese encephalitis virus in vivo and in vitro. Virology223:79–88.
Colton CA, Gilbert DL. 1987. Production of superoxide anions by a CNSmacrophage, the microglia. FEBS Lett 223:284–288.
del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, FeuersteinGZ. 2000. Inflammation and stroke: Putative role for cytokines, adhe-sion molecules and iNOS in brain response to ischemia. Brain Pathol10:95–112.
Desai A, Shankar SK, Ravi V, Chandramuki A, Gourie-Devi M. 1995.Japanese encephalitis virus antigen in the human brain and its topo-graphic distribution. Acta Neuropathol (Berl) 89:368–373.
Gehrmann J, Banati RB, Wiessner C, Hossmann KA, Kreutzberg GW.1995. Reactive microglia in cerebral ischaemia: An early mediator oftissue damage? Neuropathol Appl Neurobiol 21:277–289.
Giulian D, Vaca K, Noonan CA. 1990. Secretion of neurotoxins by mono-nuclear phagocytes infected with HIV-1. Science 250:1593–1596.
Goody RJ, Hoyt CC, Tyler KL. 2005. Reovirus infection of the CNSenhances iNOS expression in areas of virus-induced injury. Exp Neu-rol 195:379–390.
Hase T, Summers PL, Eckels KH, Baze WB. 1987. Maturation process ofJapanese encephalitis virus in cultured mosquito cells in vitro andmouse brain cells in vivo. Arch Virol 96:135–151.
Hasegawa H, Satake Y, Kobayashi Y. 1990. Effect of cytokines on Japaneseencephalitis virus production by human monocytes. Microbiol Immunol34:459–466.
Hewett SJ, Muir JK, Lobner D, Symons A, Choi DW. 1996. Potentiationof oxygen-glucose deprivation-induced neuronal death after inductionof iNOS. Stroke 27:1586–1591.
Iadecola C, Zhang F. 1996. Permissive and obligatory roles of NO in cere-brovascular responses to hypercapnia and acetylcholine. Am J Physiol271(4, Part 2):R990–R1001.
Ishiguro A, Suzuki Y, Inaba Y, Fukushima K, Komiyama A, Koeffler HP,Shimbo T. 1997. The production of IL-8 in cerebrospinal fluid in asep-tic meningitis of children. Clin Exp Immunol 109:426–430.
Kedarnath N, Gore MM, Dayaraj C, Sathe PS, Ghosh SN. 1986. Effect ofvarious mitogens on the replication of Japanese encephalitis virus inhuman mononuclear leukocyte cultures. Indian J Med Res 84:231–238.
Kelkar SD, Banerjee K. 1977. Mononuclear cells in Japanese encephali-tis virus infection: Changes in cells counts and specific fluorescence.Acta Virol 21:417–421.
Kelley KA, Ho L, Winger D, Freire-Moar J, Borelli CB, Aisen PS,Pasinetti GM. 1999. Potentiation of excitotoxicity in transgenic mice over-expressing neuronal cyclooxygenase-2. Am J Pathol 155:995–1004.
Kumar R, Mathur A, Kumar A, Sharma S, Chakraborty S, Chaturvedi UC.1990. Clinical features and prognostic indicators of Japanese encephali-tis in children in Lucknow (India). Indian J Med Res 91:321–327.
Li Y, Fu L, Gonzales DM, Lavi E. 2004. Coronavirus neurovirulence cor-relates with the ability of the virus to induce proinflammatory cyto-kine signals from astrocytes and microglia. J Virol 78:3398–3406.
Liao SL, Raung SL, Chen CJ. 2002. Japanese encephalitis virus stimu-lates superoxide dismutase activity in rat glial cultures. Neurosci Lett324:133–136.
Loughlin AJ, Woodroofe MN, Cuzner ML. 1992. Regulation of Fc receptorand major histocompatibility complex antigen expression on isolated ratmicroglia by tumour necrosis factor, interleukin-1 and lipopolysaccha-ride: Effects on interferon-g induced activation. Immunology 75:170–175.
Mathur A, Bharadwaj M, Kulshreshtha R, Rawat S, Jain A, Chaturvedi UC.1988. Immunopathological study of spleen during Japanese encephali-tis virus infection in mice. Br J Exp Pathol 69:423–432.
Mathur A, Khanna N, Chaturvedi UC. 1992. Breakdown of blood-brainbarrier by virus-induced cytokine during Japanese encephalitis virusinfection. Int J Exp Pathol 73:603–611.
Mhatre M, Floyd RA, Hensley K. 2004. Oxidative stress and neuroin-flammation in Alzheimer’s disease and amyotrophic lateral sclerosis:Common links and potential therapeutic targets. J Alzheimers Dis 6:147–157.
Nakajima K, Martinez-Maza O, Hirano T, Breen EC, Nishanian PG,Salazar-Gonzalez JF, Fahey JL, Kishimoto T. 1989. Induction of IL-6(B cell stimulatory factor-2/IFN-b 2) production by HIV. J Immunol142:531–536.
Okuda Y, Sakoda S, Bernard CC, Fujimura H, Saeki Y, Kishimoto T,Yanagihara T. 1998. IL-6-deficient mice are resistant to the inductionof experimental autoimmune encephalomyelitis provoked by myelinoligodendrocyte glycoprotein. Int Immunol 10:703–708.
495MICROGLIAL ACTIVATION IN JAPANESE ENCEPHALITIS
GLIA DOI 10.1002/glia

Olson JK, Girvin AM, Miller SD. 2001. Direct activation of innate andantigen-presenting functions of microglia following infection withTheiler’s virus. J Virol 75:9780–9789.
Packer MA, Stasiv Y, Benraiss A, Chmielnicki E, Grinberg A, WestphalH, Goldman SA, Enikolopov G. 2003. Nitric oxide negatively regulatesmammalian adult neurogenesis. Proc Natl Acad Sci USA 100:9566–9571.
Perrin PJ, Rumbley CA, Beswick RL, Lavi E, Phillips SM. 2000. Differ-ential cytokine and chemokine production characterizes experimentalautoimmune meningitis and experimental autoimmune encephalomy-elitis. Clin Immunol 94:114–124.
Quagliarello VJ, Wispelwey B, Long WJ Jr, Scheld WM. 1991. Recombi-nant human interleukin-1 induces meningitis and blood-brain barrierinjury in the rat. Characterization and comparison with tumor necro-sis factor. J Clin Invest 87:1360–1366.
Ravi V, Parida S, Desai A, Chandramuki A, Gourie-Devi M, Grau GE.1997. Correlation of tumor necrosis factor levels in the serum and cer-ebrospinal fluid with clinical outcome in Japanese encephalitispatients. J Med Virol 51:132–136.
Rohrenbeck AM, Bette M, Hooper DC, Nyberg F, Eiden LE, DietzscholdB, Weihe E. 1999. Upregulation of COX-2 and CGRP expression inresident cells of the Borna disease virus-infected brain is dependentupon inflammation. Neurobiol Dis 6:15–34.
Shen WH, Jackson ST, Broussard SR, McCusker RH, Strle K, FreundGG, Johnson RW, Dantzer R, Kelley KW. 2004. IL-1b suppresses pro-longed Akt activation and expression of E2F-1 and cyclin A in breastcancer cells. J Immunol 172:7272–7281.
Shima DT, Adamis AP, Ferrara N, Yeo KT, Yeo TK, Allende R, FolkmanJ, D’Amore PA. 1995. Hypoxic induction of endothelial cell growth fac-tors in retinal cells: Identification and characterization of vascular en-dothelial growth factor (VEGF) as the mitogen. Mol Med 1:182–193.
Soldan SS, Alvarez Retuerto AI, Sicotte NL, Voskuhl RR. 2004. Dysregu-lation of IL-10 and IL-12p40 in secondary progressive multiple sclero-sis. J Neuroimmunol 146:209–215.
Stone TW, Connick JH. 1985. Quinolinic acid and other kynurenines inthe central nervous system. Neuroscience 15:597–617.
Suzumura A, Sawada M, Yamamoto H, Marunouchi T. 1990. Effects ofcolony stimulating factors on isolated microglia in vitro. J Neuroimmu-nol 30:111–120.
Venters HD, Tang Q, Liu Q, VanHoy RW, Dantzer R, Kelley KW. 1999.A new mechanism of neurodegeneration: A proinflammatory cytokineinhibits receptor signaling by a survival peptide. Proc Natl Acad SciUSA 96:9879–9884.
Vrati S, Agarwal V, Malik P, Wani SA, Saini M. 1999. Molecular charac-terization of an Indian isolate of Japanese encephalitis virus thatshows an extended lag phase during growth. J Gen Virol 80 (Part 7):1665–1671.
Wesselingh SL, Glass J, McArthur JC, Griffin JW, Griffin DE. 1994. Cyto-kine dysregulation in HIV-associated neurological disease. Adv Neu-roimmunol 4:199–206.
Winter PM, Dung NM, Loan HT, Kneen R, Wills B, Thu le T, House D,White NJ, Farrar JJ, Hart CA, Solomon T. 2004. Proinflammatorycytokines and chemokines in humans with Japanese encephalitis. JInfect Dis 190:1618–1626.
Wolinsky JS. 1990. Subacute sclerosing panencephalitis progressiverubella panencephalitis, and multifocal leukoencephelapathy. In: Waks-man BH, editor. Immunological mechanisms in neurologic and psychiat-ric disease. New York: Raven Press. pp 259–268.
Zhang J, Fujii S, Wu Z, Hashioka S, Tanaka Y, Shiratsuchi A, Nakanishi Y,Nakanishi H. 2006. Involvement of COX-1 and up-regulated prostaglan-din E synthases in phosphatidylserine liposome-induced prostaglandinE2 production by microglia. J Neuroimmunol 172:112–120.
496 GHOSHAL ET AL.
GLIA DOI 10.1002/glia
Copyright © 2022 FDOKUMEN




















