
Activation of Nrf2 in Endothelial Cells Protects Arteries From Exhibiting a Proinflammatory State
33
ISSN: 1524-4636 Copyright © 2009 American Heart Association. All rights reserved. Print ISSN: 1079-5642. Online 7272 Greenville Avenue, Dallas, TX 72514 Arteriosclerosis, Thrombosis, and Vascular Biology is published by the American Heart Association. DOI: 10.1161/ATVBAHA.109.193375 published online Sep 3, 2009; Arterioscler Thromb Vasc Biol Harald Carlsen, Dorian O. Haskard, Justin C. Mason and Paul C. Evans Cuhlmann, Shahir S. Hamdulay, Rob Krams, Indika Edirisinghe, Irfan Rahman, Mustafa Zakkar, Kim Van der Heiden, Le Anh Luong, Hera Chaudhury, Simon Proinflammatory State Activation of Nrf2 in Endothelial Cells Protects Arteries From Exhibiting a http://atvb.ahajournals.org/cgi/content/full/ATVBAHA.109.193375/DC2 http://atvb.ahajournals.org/cgi/content/full/ATVBAHA.109.193375/DC1 Data Supplement (unedited) at: http://atvb.ahajournals.org located on the World Wide Web at: The online version of this article, along with updated information and services, is http://www.lww.com/reprints Reprints: Information about reprints can be found online at [email protected] 410-528-8550. E-mail: Fax: Kluwer Health, 351 West Camden Street, Baltimore, MD 21202-2436. Phone: 410-528-4050. Permissions: Permissions & Rights Desk, Lippincott Williams & Wilkins, a division of Wolters http://atvb.ahajournals.org/subscriptions/ Biology is online at Subscriptions: Information about subscribing to Arteriosclerosis, Thrombosis, and Vascular at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.org Downloaded from
Transcript of Activation of Nrf2 in Endothelial Cells Protects Arteries From Exhibiting a Proinflammatory State

ISSN: 1524-4636 Copyright © 2009 American Heart Association. All rights reserved. Print ISSN: 1079-5642. Online
7272 Greenville Avenue, Dallas, TX 72514Arteriosclerosis, Thrombosis, and Vascular Biology is published by the American Heart Association.
DOI: 10.1161/ATVBAHA.109.193375 published online Sep 3, 2009; Arterioscler Thromb Vasc Biol
Harald Carlsen, Dorian O. Haskard, Justin C. Mason and Paul C. Evans Cuhlmann, Shahir S. Hamdulay, Rob Krams, Indika Edirisinghe, Irfan Rahman, Mustafa Zakkar, Kim Van der Heiden, Le Anh Luong, Hera Chaudhury, Simon
Proinflammatory State Activation of Nrf2 in Endothelial Cells Protects Arteries From Exhibiting a
http://atvb.ahajournals.org/cgi/content/full/ATVBAHA.109.193375/DC2 http://atvb.ahajournals.org/cgi/content/full/ATVBAHA.109.193375/DC1
Data Supplement (unedited) at:
http://atvb.ahajournals.orglocated on the World Wide Web at:
The online version of this article, along with updated information and services, is
http://www.lww.com/reprintsReprints: Information about reprints can be found online at
[email protected]. E-mail:
Fax:Kluwer Health, 351 West Camden Street, Baltimore, MD 21202-2436. Phone: 410-528-4050. Permissions: Permissions & Rights Desk, Lippincott Williams & Wilkins, a division of Wolters
http://atvb.ahajournals.org/subscriptions/Biology is online at Subscriptions: Information about subscribing to Arteriosclerosis, Thrombosis, and Vascular
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

Activation of Nrf2 in Endothelial Cells Protects ArteriesFrom Exhibiting a Proinflammatory State
Mustafa Zakkar, Kim Van der Heiden, Le Anh Luong, Hera Chaudhury, Simon Cuhlmann,Shahir S. Hamdulay, Rob Krams, Indika Edirisinghe, Irfan Rahman, Harald Carlsen,
Dorian O. Haskard, Justin C. Mason, Paul C. Evans
Objective—Proinflammatory mediators influence atherosclerosis by inducing adhesion molecules (eg, VCAM-1) onendothelial cells (ECs) via signaling intermediaries including p38 MAP kinase. Regions of arteries exposed to high shearstress are protected from inflammation and atherosclerosis, whereas low-shear regions are susceptible. Here weinvestigated whether the transcription factor Nrf2 regulates EC activation in arteries.
Methods and Results—En face staining revealed that Nrf2 was activated in ECs at an atheroprotected region of the murineaorta where it negatively regulated p38–VCAM-1 signaling, but was expressed in an inactive form in ECs at anatherosusceptible site. Treatment with sulforaphane, a dietary antioxidant, activated Nrf2 and suppressed p38–VCAM-1signaling at the susceptible site in wild-type but not Nrf2�/� animals, indicating that it suppresses EC activation viaNrf2. Studies of cultured ECs revealed that Nrf2 inactivates p38 by suppressing an upstream activator MKK3/6 and byenhancing the activity of the negative regulator MKP-1.
Conclusions—Nrf2 prevents ECs at the atheroprotected site from exhibiting a proinflammatory state via the suppressionof p38–VCAM-1 signaling. Pharmacological activation of Nrf2 reduces EC activation at atherosusceptible sites andmay provide a novel therapeutic strategy to prevent or reduce atherosclerosis. (Arterioscler Thromb Vasc Biol. 2009;29:00-00.)
Key Words: Nrf2 � arterial endothelium � shear stress � sulforaphane � proinflammatory activation � p38� MKK3/6 � MKP-1
Early atherosclerotic lesions contain monocytes andT-lymphocytes which are recruited from the circulation
by adhesion to activated vascular endothelial cells (ECs).1
This process is triggered by proinflammatory mediators (eg,TNF�) which induce cellular adhesion molecules (eg,VCAM-1) via signaling intermediaries including p38mitogen-activated protein (MAP) kinase, which is activatedby phosphorylation by MAP kinase kinases 3 and 6 (MKK3/6).2,3 Vascular inflammation and atherosclerosis developpredominantly at distinct sites of the arterial tree located nearbranches and bends which are exposed to nonuniform bloodflow, which exerts relatively low shear stress on vascularendothelium, whereas regions of arteries that are exposed tounidirectional high shear stress are protected.4–6 Proinflam-matory activation of ECs is reduced at high-shear sitescompared to low-shear regions, thus providing a potentialexplanation for the distinct spatial localization of vascular
inflammation and lesion formation.5,7–10 Similarly, the appli-cation of unidirectional high shear stress can suppress proin-flammatory activation of cultured ECs, whereas low oroscillatory shear can act as a positive regulator of ECactivation.5,10–15
The molecular mechanisms underlying the antiinflamma-tory effects of shear stress are uncertain, but previous studiesof cultured cells have suggested a role for the transcriptionfactor Nrf2.16–20 In unstimulated cells, Nrf2 is suppressed bykelch-like ECH-associated protein 1 (Keap1) which targets itfor ubiquitination and proteasomal processing. Nrf2 can beactivated by shear stress, dietary antioxidants (eg, sulfora-phane) and other physiological stimuli which disrupt Keap1-Nrf2 interactions leading to stabilization and nuclear translo-cation of Nrf2.16–22 A previous study of cultured ECsrevealed that activated Nrf2 can induce numerousantioxidant-defense genes (eg, heme oxygenase-1 [HO-1])
Received February 16, 2009; revision accepted August 18, 2009.From the British Heart Foundation Cardiovascular Sciences Unit (M.Z., K.V.d.H., L.A.L., H.C., S.C., S.S.H., D.O.H., J.C.M., P.C.E.), National Heart
and Lung Institute, Imperial College London, UK; the Department of Bioengineering (S.C., R.K.), Imperial College London, UK; University of RochesterMedical Center (I.E., I.R.), Rochester, NY; and the Department of Nutrition (H.C.), University of Oslo, Norway.
M.Z. and K.V.d.H. contributed equally to this study.Correspondence to Dr Paul C. Evans, Senior Lecturer, British Heart Foundation Cardiovascular Sciences Unit, National Heart and Lung Institute,
Imperial College London, Hammersmith Campus, Du Cane Road, London W12 ONN, UK. E-mail [email protected]© 2009 American Heart Association, Inc.
Arterioscler Thromb Vasc Biol is available at http://atvb.ahajournals.org DOI: 10.1161/ATVBAHA.109.193375
1 at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

and suppress adhesion molecule expression in ECs by inhib-iting phosphorylation/activation of p38.20 However, the ef-fects of Nrf2 on EC activation have not previously beenassessed in vivo.
Materials and MethodsFull details of all methods can be found in the supplemental materials(available online at http://atvb.ahajournals.org).
In Vivo StudiesMale C57BL/6 or Nrf2�/� (C57BL/6)23 mice were treated withsulforaphane (5 mg/kg) 24 hours and 4 hours before experimenta-tion. Mice were treated with LPS (4 mg/kg) for 1 hour or 6 hoursbefore assessment of p38 phosphorylation or VCAM-1 expression,respectively. Expression levels of specific proteins were assessed inECs at regions of the inner curvature (susceptible site) and outercurvature (protected site) of aortae by en face staining asdescribed.9,10
Cell Culture StudiesHuman umbilical vein endothelial cells (HUVECs) were collectedand cultured as described previously.14 Confluent cultures wereexposed to unidirectional laminar shear (12 dynes/cm2) for 24 hoursusing a parallel-plate flow chamber as described previously14 or weretreated with sulforaphane (1 �mol/L). Gene-specific small interfer-ing (si)RNAs were used to silence Nrf2, MAP kinase phosphatase-1(MKP-1), or HO-1. Transcript levels were quantified by real-timePCR using gene-specific primers (supplemental Table) as describedpreviously.14 MKP-1 redox state was assessed by nonreducingSDS-PAGE using total cell lysates, as described.24 The expressionand intracellular localization of Nrf2 was assessed by immunostain-ing using anti-Nrf2 antibodies and Alexafluor 568-conjugated sec-ondary antibodies followed by confocal microscopy. IntracellularROS were measured in HUVECs loaded with the redox-sensitivefluorescent probe 5-(and-6)-carboxy-2�7�-dichlorodihydrofluoresce-in diacetate (H2DCFDA) by quantification of fluorescence byconfocal microscopy.
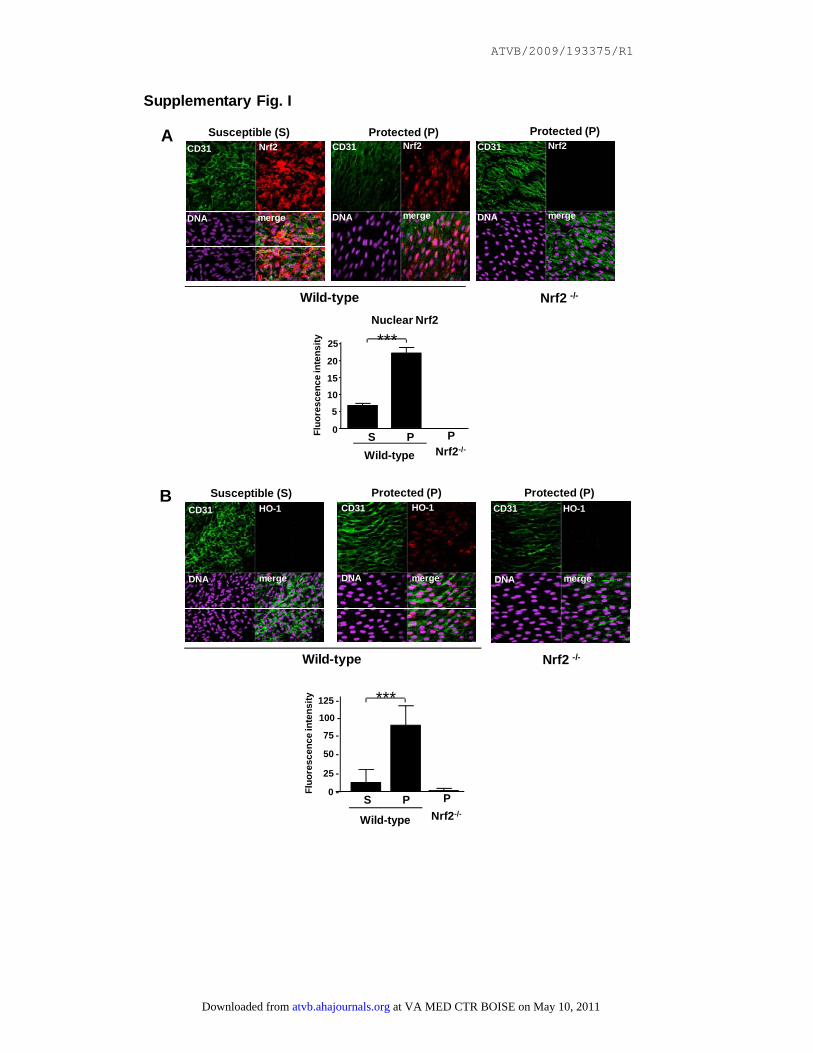
ResultsHigh Shear Stress Suppressed EC Activation at anAtheroprotected Region by Inducing Nrf2We examined whether the known spatial distribution ofshear stress in the murine aorta4 correlated with Nrf2activation in ECs. Nrf2 was localized predominantly in thenucleus of ECs in the outer curvature which is exposed tohigh shear stress and is protected from inflammation andatherosclerosis, but was localized predominantly in thecytoplasm of ECs in the inner curvature which is exposedto low shear and is atherosusceptible (supplemental FigureIA). Staining was not observed in parallel experimentsusing Nrf2�/� animals, indicating that staining was spe-cific for Nrf2. We confirmed that nuclear Nrf2 was activein the protected region because HO-1 (an Nrf2-targetmolecule) was expressed by ECs in the protected region inwild-type mice but not in Nrf2�/� mice (supplementalFigure IB). HO-1 was not detected in the susceptibleregion of wild-type mice, suggesting that cytoplasmic Nrf2is inactive at this site.
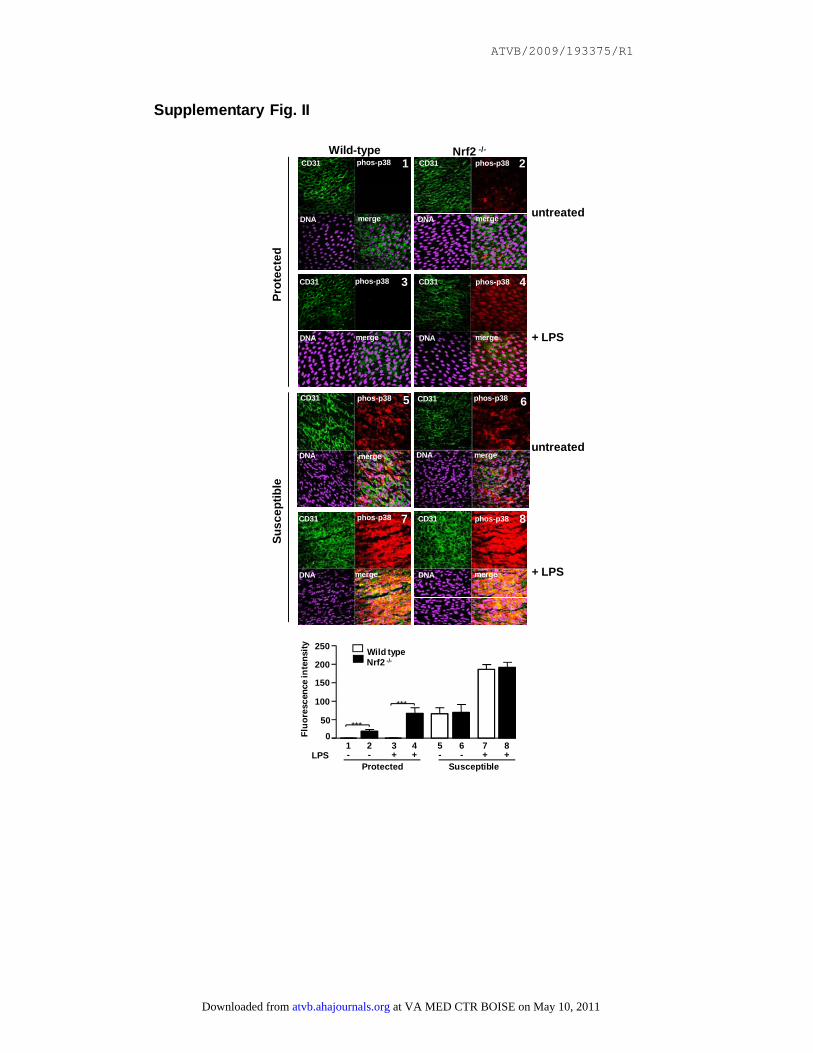
We determined whether Nrf2 regulates p38 signaling toVCAM-1 in protected regions of the aorta by performing
en face staining using antibodies that recognize activephosphorylated p38 or VCAM-1. In wild-type mice, phos-phorylated p38 and VCAM-1 were expressed at signifi-cantly lower levels in the protected site compared to thesusceptible site in both untreated and LPS-treated mice(supplemental Figures II and III). The suppression ofphosphorylated p38 in the protected region was not causedby reduced protein expression of p38 which was similarin susceptible and protected sites (supplemental FigureIV). We used Nrf2�/� mice to examine whether Nrf2 isnecessary for suppression of p38 activation and VCAM-1expression in the protected region. Genetic deletion ofNrf2 significantly enhanced p38 phosphorylation andVCAM-1 expression at the protected site in both untreatedand LPS-treated animals (supplemental Figures II and III),indicating that Nrf2 suppresses EC activation at the pro-tected region. In contrast, genetic deletion of Nrf2 hadlittle or no effect on EC activation at the susceptible site,which is consistent with our finding that Nrf2 is inactive atthis region in wild-type mice. Interestingly, levels ofphosphorylated p38 and VCAM-1 were higher in thesusceptible region compared to the protected region inNrf2�/� mice suggesting that other negative regulatorscooperate with Nrf2 to suppress EC activation at theprotected site.
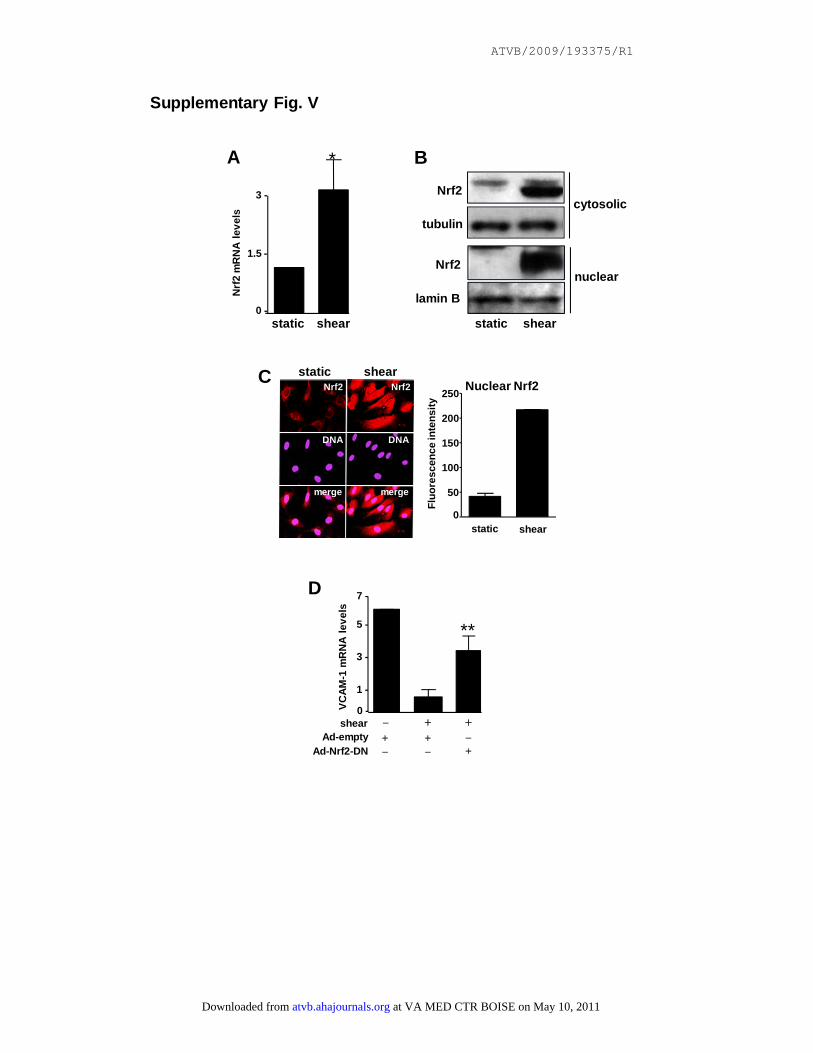
It is likely that Nrf2 suppresses p38–VCAM-1 signaling atthe protected site as a consequence of shear stress becauseparallel studies performed under well defined conditions invitro indicated that high shear stress (12 dynes/cm2 for 24hours) significantly enhanced expression and nuclear local-ization of Nrf2 and simultaneously reduced expression ofVCAM-1 in HUVECs (supplemental Figure VA throughVD). Inhibition of Nrf2 activity using an adenovirus contain-ing a dominant negative version (Ad-Nrf2-DN) restoredVCAM-1 expression in sheared HUVECs (supplementalFigure VD) indicating that endogenous Nrf2 is essential forsuppression of EC activation by shear stress. Taken together,our in vitro and in vivo studies suggest that Nrf2 activation byshear stress suppresses p38 activation and VCAM-1 expres-sion at protected sites, which may be a key molecularmechanism that governs the spatial distribution of atheroscle-rotic plaques.
Sulforaphane Inhibits p38–VCAM-1 Signaling viaActivation of Nrf2We reasoned that sites exposed to low shear may besusceptible to endothelial activation and vascular inflam-mation because of the absence of Nrf2 activation in ECs.Therefore, strategies to activate Nrf2 in susceptible sitesmay suppress vascular inflammation. To address thishypothesis we used sulforaphane, an isothiocyanate de-rived from cruciferous vegetables (eg, broccoli) that canactivate Nrf2 by dissociating it from Keap1 and can induceantioxidants in cultured human ECs21 and in murine tissuesin vivo.22 We observed that sulforaphane treatment ofHUVECs did not influence Nrf2 mRNA expression (sup-plemental Table) but significantly enhanced expressionand nuclear localization of Nrf2 protein (Figure 1A and
2 Arterioscler Thromb Vasc Biol November 2009
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

1B) and simultaneously reduced the expression ofVCAM-1 at both the mRNA (Figure 1C and 1D) andprotein levels (Figure 1E). We concluded that endogenousNrf2 is essential for suppression of VCAM-1 expressionby sulforaphane because VCAM-1 expression insulforaphane-treated cells was restored by treatment withAd-Nrf2-DN (Figure 1C), and by gene silencing usingNrf2-specific siRNA (Figure 1D and 1E). Pulse-chaseexperiments revealed that sulforaphane did not influencethe stability of VCAM-1 mRNA in activated HUVECs(supplemental Figure VI), suggesting that sulforaphaneinhibits VCAM-1 at a transcriptional level. To furtherstudy the molecular mechanism underlying VCAM-1 sup-pression by Nrf2 we examined whether sulforaphaneregulates p38 or NF-�B which are essential for VCAM-1expression. We observed that pretreatment of ECs withsulforaphane suppressed activation of p38 and MKK3/6 in
response to TNF� (Figure 1F and 1G) but did not influenceNF-�B activation (Figure 1G). Ad-Nrf2-DN restored p38activation in sulforaphane-treated HUVECs (Figure 1F),indicating that sulforaphane inhibits p38 activity via Nrf2.Similarly, overexpression of Nrf2 in HUVECs (supple-mental Figure VII) using an adenovirus (A; Ad-Nrf2)suppressed induction of VCAM-1 (B) and activation ofp38 and MKK3/6 (C and D) by TNF� but did not influenceNF-�B (E).
Given that sulforaphane induces antioxidants, we hy-pothesized that it suppresses p38 activation by alteringcellular ROS. Consistent with this idea, treatment ofHUVECs with sulforaphane (Figure 1H) or Ad-Nrf2 (sup-plemental Figure VIIF) suppressed the induction of cellu-lar ROS. Moreover, screening of a panel of antioxidants byquantitative PCR revealed that sulforaphane inducesHO-1, aldo-keto reductase family 1 members C1 and C3,
Figure 1. Sulforaphane suppresses p38–VCAM-1 signaling in cultured ECs viaNrf2. HUVECs were transduced withAd-Nrf2-DN or with an empty adenovirus(Ad-empty; C and F), or were transfectedwith Nrf2-specific (D and E) or MKP-1–specific siRNA (I) or with a scrambledcontrol (Scr), or remained untreated. Cellswere then exposed to SFN or vehiclealone for 4 hours. A, Cytosolic or nuclearlysates were tested by Western blottingusing specific antibodies. Representativeof 3 experiments. B, Immunofluorescencestaining using anti-Nrf2 antibodies andnuclear counterstain (DNA). Representa-tive images and quantitation of nuclearNrf2 levels from multiple ECs (mean�SD)are shown. C through G, Cells were stim-ulated with TNF� for 15 to 30 minutes (G),1 hour (F), 4 hours (C and D), or 6 hours(E) or remained untreated. C and D, Levelsof VCAM-1 or Nrf2 transcripts were quan-tified by real-time PCR. Mean values(�SD) calculated from triplicate measure-ments are shown. Cell lysates were testedby Western blotting using anti-Nrf2 orantitubulin antibodies. Representative of 3independent experiments. E, VCAM-1 sur-face expression was assessed by immu-nostaining followed by flow cytometry.Data were pooled from 2 independentexperiments and mean RFI values (�SD)are shown. F and G, Lysates were testedby Western blotting using specific anti-bodies. phos-p38 indicates Phosphorylat-ed-p38; phos-MKK3/6, PhosphorylatedMKK3/6. Representative of 2 independentexperiments. H, HUVECs were loadedwith the redox-sensitive probe H2DCFDA,treated with H2O2 (50 �mol/L), and fluo-rescence was assessed at varying timesby confocal microscopy. Representativeimages and quantitation of fluorescentcells after 5 minutes stimulation(mean�SD; pooled from 3 experiments)are shown. I, Cells were stimulated withTNF� (4 hours) or remained untreated.Levels of MKP-1 or VCAM-1 transcriptswere quantified by real-time PCR. Datawere pooled from 2 independent experi-ments and mean values (�SD) are shown.
Zakkar et al Nrf2 Suppresses Endothelial Activation in Arteries 3
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

and thioredoxin reductase 1 (supplemental Table). As theinduction of HO-1 was most prominent, we examined itsrole in EC responses to SFN. Inhibition of HO-1 activityusing zinc protoporphyrin or silencing of HO-1 expressionusing prevalidated siRNA sequences25 did not influenceVCAM-1 suppression by sulforaphane (supplemental Fig-ure VIII), suggesting that HO-1 is not essential for theantiinflammatory effects of sulforaphane. It is plausiblethat the other Nrf2-induced antioxidants can compensatefor the absence of HO-1 activity in sulforaphane-treatedcells. We next examined whether sulforaphane suppressesp38 activation via MKP-1, an antiinflammatory negativeregulator of p38 that is expressed by ECs in atheropro-tected regions.10 Sulforaphane did not induce MKP-1expression (supplemental Table). However, silencing ofMKP-1 significantly elevated VCAM-1 expression insulforaphane-treated HUVECs (Figure 1I), indicating thatendogenous MKP-1 is involved in suppression ofVCAM-1 expression by sulforaphane. A previous studydemonstrated that ROS can inactivate MKP-1 by oxidizingcatalytic cysteine residues to sulfenic acid, thus promotingthe formation of disulfide bonds between MKP-1 andcellular proteins.24 Given that Nrf2 reduces ROS, weexamined whether Nrf2 can enhance MKP-1 activity bypromoting the reduced form. Analysis of cell lysates bySDS-PAGE in the absence of reducing agents revealedboth oxidized and reduced forms of MKP-1, which dis-played a marked difference in electrophoretic mobility(supplemental Figure VIIG), as described previously.24
Treatment of HUVECs with H2O2 induced a high-molecular-weight disulfide-linked form of MKP-1 (oxi-dized; compare 1 and 2) that could be reduced by incuba-tion of cell lysates with DTT (compare 2 and 4). Thereduced form of MKP-1 was promoted by overexpressionof Nrf2 using Ad-Nrf2 (compare 2 and 3), suggesting thatNrf2 can enhance MKP-1 catalytic activity by influencingits redox state.
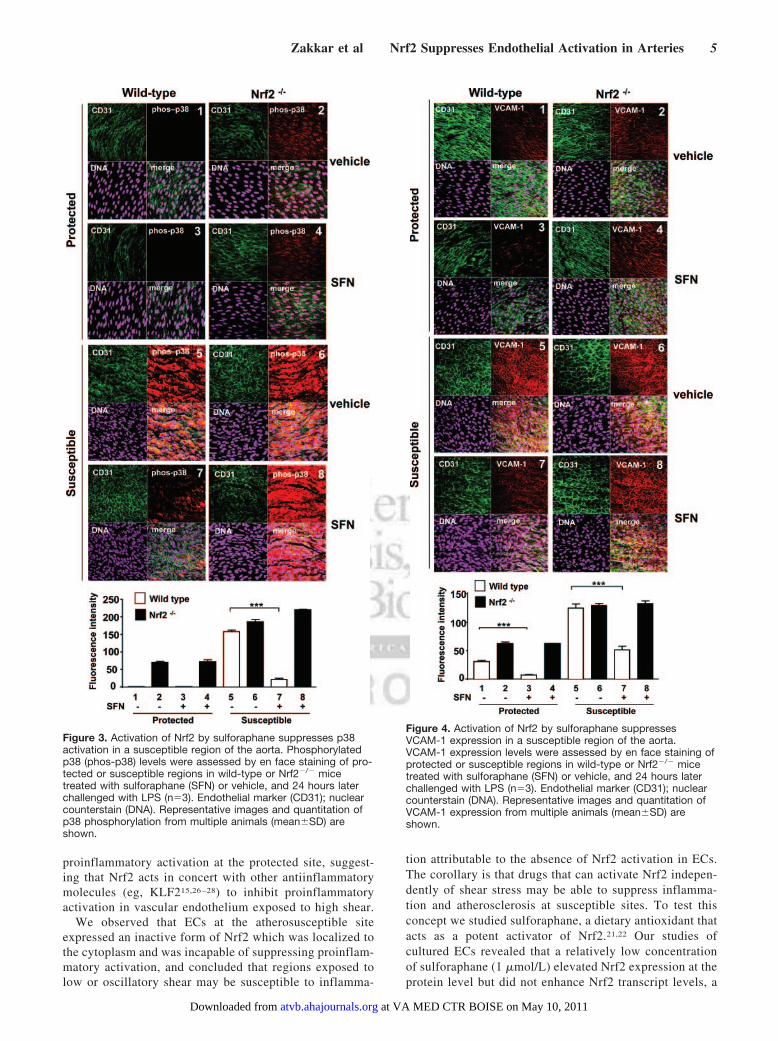
Pharmacological Activation of Nrf2 Suppresses ECActivation in a Susceptible Region of theMurine AortaWe examined whether pharmacological activation of Nrf2using sulforaphane can reduce proinflammatory activation atatherosusceptible sites. Treatment of mice with sulforaphaneactivated Nrf2 in a susceptible region of the aorta (Figure 2).Sulforaphane treatment also reduced p38 activation andVCAM-1 expression at the susceptible site in wild-type mice(Figures 3 and 4, compare 5 with 7) but had no effect inNrf2�/� mice (compare 6 with 8), indicating that sulfora-phane suppresses p38–VCAM-1 signaling at the susceptiblesite by activating Nrf2. At the protected region of wild-typeanimals, Nrf2 was constitutively active and sulforaphanetreatment had relatively modest effects on Nrf2 activity(Figure 2), p38 phosphorylation (Figure 3), and VCAM-1expression (Figure 4).
DiscussionStudies using cultured ECs suggest that the molecularmechanism underlying the antiinflammatory effects of
shear stress involves the transcription factors KLF215,26,27
and Nrf2,16 –20 both of which can be activated by shearstress and can limit proinflammatory activation whenoverexpressed in ECs. Indeed KLF2 and Nrf2 can actsynergistically to modulate the transcriptome in culturedECs exposed to shear stress.28 The involvement of KLF2has been confirmed by studies of human, murine andzebrafish arteries which have demonstrated that KLF2 isexpressed at high shear regions.26,29,30 In contrast, theregulation and physiological role of Nrf2 in the vasculaturein vivo has thus far received little attention. Althoughprevious studies have revealed that Nrf2 is expressed in thedescending thoracic aorta19 and can be activated by oxi-dized phospholipids in the carotid artery,31 the capacity ofNrf2 to regulate proinflammatory activation of ECs inarteries has not been previously investigated. Here wedemonstrate that Nrf2 is constitutively active in ECs at ahigh-shear atheroprotected region of the murine aorta andthat it reduces proinflammatory activation at this site byinactivating p38 MAP kinase and suppressing VCAM-1expression. Thus Nrf2 activation by shear stress may be akey molecular mechanism that governs the spatial distri-bution of atherosclerotic plaques. Interestingly, we ob-served that genetic deletion of Nrf2 did not entirely restore
Figure 2. Sulforaphane activates Nrf2 in ECs at a susceptibleregion of the aorta. Nrf2 nuclear localization was assessed byen face staining in protected or susceptible regions in wild-typemice (treated with sulforaphane [SFN] or vehicle alone as a con-trol (n�3 per group). Endothelial marker (CD31); nuclear coun-terstain (DNA). Representative images and the proportion ofnuclear Nrf2 calculated from multiple animals (mean�SD) areshown.
4 Arterioscler Thromb Vasc Biol November 2009
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from
proinflammatory activation at the protected site, suggest-ing that Nrf2 acts in concert with other antiinflammatorymolecules (eg, KLF215,26 –28) to inhibit proinflammatoryactivation in vascular endothelium exposed to high shear.
We observed that ECs at the atherosusceptible siteexpressed an inactive form of Nrf2 which was localized tothe cytoplasm and was incapable of suppressing proinflam-matory activation, and concluded that regions exposed tolow or oscillatory shear may be susceptible to inflamma-
tion attributable to the absence of Nrf2 activation in ECs.The corollary is that drugs that can activate Nrf2 indepen-dently of shear stress may be able to suppress inflamma-tion and atherosclerosis at susceptible sites. To test thisconcept we studied sulforaphane, a dietary antioxidant thatacts as a potent activator of Nrf2.21,22 Our studies ofcultured ECs revealed that a relatively low concentrationof sulforaphane (1 �mol/L) elevated Nrf2 expression at theprotein level but did not enhance Nrf2 transcript levels, a
Figure 3. Activation of Nrf2 by sulforaphane suppresses p38activation in a susceptible region of the aorta. Phosphorylatedp38 (phos-p38) levels were assessed by en face staining of pro-tected or susceptible regions in wild-type or Nrf2�/� micetreated with sulforaphane (SFN) or vehicle, and 24 hours laterchallenged with LPS (n�3). Endothelial marker (CD31); nuclearcounterstain (DNA). Representative images and quantitation ofp38 phosphorylation from multiple animals (mean�SD) areshown.
Figure 4. Activation of Nrf2 by sulforaphane suppressesVCAM-1 expression in a susceptible region of the aorta.VCAM-1 expression levels were assessed by en face staining ofprotected or susceptible regions in wild-type or Nrf2�/� micetreated with sulforaphane (SFN) or vehicle, and 24 hours laterchallenged with LPS (n�3). Endothelial marker (CD31); nuclearcounterstain (DNA). Representative images and quantitation ofVCAM-1 expression from multiple animals (mean�SD) areshown.
Zakkar et al Nrf2 Suppresses Endothelial Activation in Arteries 5
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

finding that is consistent with the observation that sulfora-phane can stabilize Nrf2 by inhibiting Keap1.32 We ob-served that sulforaphane suppressed p38 activation andVCAM-1 expression in cultured ECs and subsequentstudies, in which Nrf2 was either silenced or inactivated,revealed that Nrf2 was required for these antiinflammatoryeffects. These data are consistent with our observation thatsulforaphane suppressed endothelial activation at a suscep-tible site in wild-type mice but not in Nrf2�/� animals. Arecent study demonstrated that relatively high concentra-tions of sulforaphane can influence EC activation throughNrf2-independent mechanisms,33 and we therefore con-clude that sulforaphane may have concentration-dependenteffects on proinflammatory signaling pathways.
Our study demonstrates that Nrf2 suppresses endothelialactivation by targeting the MAP kinase signaling pathwayat 2 levels (ie, by reducing MKK3/6 signaling to p38 andby enhancing the activity of MKP-1, an antiinflammatorymolecule that inactivates p38 by dephosphorylation). Nrf2did not regulate MKP-1 at the level of expression. Instead,we provide evidence that Nrf2 can enhance the catalyticactivity of MKP-1 by promoting a reducing environmentvia the induction of multiple antioxidants. We have previ-ously demonstrated that MKP-1 is induced by shear stressand is preferentially expressed by ECs in a protectedregion of the murine aorta where it inhibits p38 –VCAM-1signaling.10 Thus we propose that shear stress suppressesEC activation at atheroprotected sites by inducing MKP-1and by simultaneously enhancing MKP-1 activity viaactivation of Nrf2. Our studies revealed that Nrf2 alsosuppresses the activation of MKK3/6, a signaling interme-diary that activates p38 by phosphorylation. The underly-ing mechanism for MKK3/6 suppression by Nrf2 mayinvolve redox regulation as well because ASK1, a MAPkinase kinase kinase that acts upstream from MKK3/6, isknown to be inhibited by reduced forms of glutathione andthioredoxin.34,35
We believe that sulforaphane is the first example of atherapeutic intervention that enhances the activity of anantiinflammatory transcription factor at atherosusceptibleregions, and that this compound may have clinical utilityfor the prevention or treatment of vascular inflammation.In addition, given that cruciferous vegetables are a sourceof sulforaphane it is plausible that their known beneficialeffects on cardiovascular health36 involve activation ofNrf2 at atherosusceptible regions. We administered sul-foraphane to mice i.p. because dosing this route can becontrolled more accurately than oral administration. Thedose used in our study was based in the efficacy ofsulforaphane established in previous studies.37,38 Furtheranimal studies are now required to assess directly whetherthe consumption of green vegetables can influence Nrf2activation and inflammation at susceptible sites. A recentstudy revealed that genetic deletion of Nrf2 reducesatherosclerosis in ApoE�/� mice, and demonstrated thatNrf2 exerts proatherogenic effects in cultured macro-phages by positively regulating the expression of CD36which is a scavenger receptor for modified LDL.39 How-ever, our current findings indicate that Nrf2 may exert
antiatherogenic effects in vascular endothelium by sup-pressing inflammation. Thus, activation of Nrf2 in endo-thelial cells and macrophages may exert opposing effectson lesion development. Studies using conditional knock-outs of Nrf2 are now required to assess the functionsof Nrf2 in specific vascular cells in relation toatherosclerosis.
Sources of FundingThe study was funded by the British Heart Foundation.
DisclosuresNone.
References1. Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874.2. Ahmad M, Theofanidis P, Medford RM. Role of activating protein-1
in the regulation of the vascular cell adhesion molecule-1 geneexpression by tumor necrosis factor-alpha. J Biol Chem. 1998;273:4616 – 4621.
3. Pietersma A, Tilly BC, Gaestel N, deJong N, Lee JC, Koster JF, SluiterW. P38 mitogen activated protein kinase regulates endothelial VCAM-1expression at the post-transcriptional level. Biochem Biophys Res Comm.1997;230:44–48.
4. Suo J, Ferrara DE, Sorescu D, Guldberg RE, Taylor WR, Giddens DP.Hemodynamic shear stresses in mouse aortas - Implications for athero-genesis. Arterioscler Thromb Vasc Biol. 2007;27:346–351.
5. Dai GH, Kaazempur-Mofrad MR, Natarajan S, Zhang YZ, Vaughn S,Blackman BR, Kamm RD, Garcia-Cardena G, Gimbrone MA. Distinctendothelial phenotypes evoked by arterial waveforms derived from ath-erosclerosis-susceptible and -resistant regions of human vasculature. ProcNatl Acad Sci U S A. 2004;101:14871–14876.
6. Caro CG. Discovery of the role of wall shear in atherosclerosis.Arterioscler Thromb Vasc Biol. 2009;29:158 –161.
7. Passerini AG, Polacek DC, Shi CZ, Francesco NM, Manduchi E, GrantGR, Pritchard WF, Powell S, Chang GY, Stoeckert CJ, Davies PF.Coexisting proinflammatory and antioxidative endothelial transcriptionprofiles in a disturbed flow region of the adult porcine aorta. Proc NatlAcad Sci U S A. 2004;101:2482–2487.
8. Iiyama K, Hajra L, Iiyama M, Li HM, DiChiara M, Medoff BD, CybulskyMI. Patterns of vascular cell adhesion molecule-1 and intercellularadhesion molecule-1 expression in rabbit and mouse atheroscleroticlesions and at sites predisposed to lesion formation. Circ Res. 1999;85:199–207.
9. Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. TheNF-kappa B signal transduction pathway in aortic endothelial cells isprimed for activation in regions predisposed to atherosclerotic lesionformation. Proc Natl Acad Sci U S A. 2000;97:9052–9057.
10. Zakkar M, Chaudhury H, Sandvik G, Enesa K, Luong LA, Cuhlmann S,Mason JC, Krams R, Clark AR, Haskard DO, Evans PC. Increasedendothelial mitogen-activated protein kinase phosphatase-1 expressionsuppresses proinflammatory activation at sites that are resistant to ath-erosclerosis. Circ Res. 2008;103:726–732.
11. Yamawaki H, Lehoux S, Berk BC. Chronic physiological shear stressinhibits tumor necrosis factor-induced proinflammatory responses inrabbit aorta perfused ex vivo. Circulation. 2003;108:1619–1625.
12. Sheikh S, Rainger GE, Gale Z, Rahman M, Nash GB. Exposure tofluid shear stress modulates the ability of endothelial cells to recruitneutrophils in response to tumor necrosis factor-alpha: a basis forlocal variations in vascular sensitivity to inflammation. Blood. 2003;102:2828 –2834.
13. Chiu JJ, Lee PL, Chen CN, Lee CI, Chang SF, Chen LJ, Lien SC, Ko YC,Usami S, Chien S. Shear stress increases ICAM-1 and decreasesVCAM-1 and E-selectin expressions induced by tumor necrosisfactor-alpha in endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24:73–79.
14. Partridge J, Carlsen H, Enesa K, Chaudhury H, Zakkar M, Luong L,Kinderlerer A, Johns M, Blomhoff R, Mason JC, Haskard DO, Evans
6 Arterioscler Thromb Vasc Biol November 2009
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

PC. Laminar shear stress acts as a switch to regulate divergentfunctions of NF-kappa B in endothelial cells. FASEB J. 2007;21:3553–3561.
15. Fledderus JO, van Thienen JV, Boon RA, Dekker RJ, Rohlena J, VolgerOL, Bijnens APJJ, Daemen MJAP, Kuiper J, van Berkel TJC, PannekoekH, Horrevoets AJG. Prolonged shear stress and KLF2 suppress consti-tutive proinflarnmatory transcription through inhibition of ATF2. Blood.2007;109:4249–4257.
16. Chen XL, Varner SE, Rao AS, Grey JY, Thomas S, Cook CK, Was-serman MA, Medford RM, Jaiswal AK, Kunsch C. Laminar flowinduction of antioxidant response element-mediated genes in endothelialcells - A novel anti-inflammatory mechanism. J Biol Chem. 2003;278:703–711.
17. Hosoya T, Maruyama A, Kang MI, Kawatani Y, Shibata T, Uchida K,Itoh K, Yamamoto M. Differential responses of the Nrf2-Keap1 system tolaminar and oscillatory shear stresses in endothelial cells. J Biol Chem.2005;280:27244–27250.
18. Warabi E, Takabe W, Minami T, Inoue K, Itoh K, Yamamoto M, Ishii T,Kodama T, Noguchi N. Shear stress stabilizes NF-E2-related factor 2 andinduces antioxidant genes in endothelial cells: Role of reactive oxygen/nitrogen species. Free Radic Biol Med. 2007;42:260–269.
19. Dai G, Vaughn S, Zhang Y, Wang ET, Garcia-Cardena G, Gimbrone MA.Biomechanical forces in atherosclerosis-resistant vascular regionsregulate endothelial redox balance via phosphoinositol 3-kinase/akt-dependent activation of Nrf2. Circ Res. 2007;101:723–733.
20. Chen XL, Dodd G, Thomas S, Zhang XL, Wasserman MA, Rovin BH,Kunsch C. Activation of Nrf2/ARE pathway protects endothelial cellsfrom oxidant injury and inhibits inflammatory gene expression. Am JPhysiol Heart Circ Physiol. 2006;290:H1862–H1870.
21. Xue MZ, Qian QW, Adaikalakoteswari A, Rabbani N, Babaei-Jadidi R,Thornalley PJ. Activation of NF-E2-related factor-2 reverses biochemicaldysfunction of endothelial cells induced by hyperglycemia linked tovascular disease. Diabetes. 2008;57:2809–2817.
22. Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamato M,Biswal S. Identification of Nrf2-regulated genes induced by the chemo-preventive agent sulforaphane by oligonucleotide microarray. CancerRes. 2002;62:5196–5203.
23. Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T,Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2small Maf heterodimer mediates the induction of phase II detoxifyingenzyme genes through antioxidant response elements. Biochem BiophysRes Comm. 1997;236:313–322.
24. Kamata H, Honda S, Maeda S, Chang LF, Hirata H, Karin M. Reactiveoxygen species promote TNF alpha-induced death and sustained JNKactivation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661.
25. Ali F, Zakkar M, Karu K, Lidington EA, Hamdulay SS, Boyle JJ, Zloh M,Bauer A, Haskard DO, Evans PC, Mason JC. Induction of the cytopro-tective enzyme heme oxygenase-1 by statins is enhanced in vascularendothelium exposed to laminar shear stress and impaired by disturbedflow. J Biol Chem. 2009;284:18882–18892.
26. Dekker RJ, van Soest S, Fontijn RD, Salamanca S, de Groot PG,VanBavel E, Pannekoek H, Horrevoets AJG. Prolonged fluid shear stress
induces a distinct set of endothelial cell genes, most specifically lungKruppel-like factor (KLF2). Blood. 2002;100:1689–1698.
27. Senbanerjee S, Lin ZY, Atkins GB, Greif DM, Rao RM, Kumar A,Feinberg MW, Chen ZP, Simon DI, Luscinskas FW, Michel TM,Gimbrone MA, Garcia-Cardena G, Jain MK. KLF2 is a novel transcrip-tional regulator of endothelial proinflammatory activation. J Exp Med.2004;199:1305–1315.
28. Fledderus JO, Boon RA, Volger OL, Hurttila H, Yla-Herttuala S,Pannekoek H, Levonen AL, Horrevoets AJ. KLF2 primes the antioxidanttranscription factor Nrf2 for activation in endothelial cells. ArteriosclerThromb Vasc Biol. 2008;28:1339–1346.
29. Parmar KM, Larman HB, Dai GH, Zhang YH, Wang ET, Moorthy SN,Kratz JR, Lin ZY, Jain MK, Gimbrone MA, Garcia-Cardena G. Inte-gration of flow-dependent endothelial phenotypes by Kruppel-like factor2. J Clin Invest. 2006;116:49–58.
30. Lee JS, Yu C, Shin JT, Sebzda E, Bertozzi C, Chen M, Mericko P,Stadtfeld M, Zhou D, Cheng L, Graf T, Macrae CA, Lepore JJ, Lo CW,Kahn ML. KIf2 is an essential regulator of vascular hemodynamic forcesin vivo. Dev Cell. 2006;11:845–857.
31. Jyrkkanen HK, Kansanen E, Inkala M, Kivela AM, Hurttila H, HeinonenSE, Goldsteins G, Jauhiainen S, Tiainen S, Makkonen H, Oskolkova O,Afonyushkin T, Koistinaho J, Yamamoto M, Bochkov VN, Yla-HerttualaS, Levonen AL. Nrf2 regulates antioxidant gene expression evoked byoxidized phospholipids in endothelial cells and murine arteries in vivo.Circ Res. 2008;103:e1–e9.
32. Eggler AL, Liu G, Pezzuto JM, van Breemen RB, Mesecar AD. Mod-ifying specific cysteines of the electrophile-sensing human Keap1 proteinis insufficient to disrupt binding to the Nrf2 domain Neh2. Proc NatlAcad Sci U S A. 2005;102:10070–10075.
33. Chen X-L, Dodd G, Kunsch C. Sulforaphane inhibits TNF-alpha-inducedactivation of p38 MAP kinase and VCAM-1 and MCP-1 expression inendothelial cells. Inflamm Res. 2009;58:513–521.
34. Hojo Y, Saito Y, Tanimoto T, Hoefen RJ, Baines CP, Yamamoto K,Haendeler J, Asmis R, Berk BC. Fluid shear stress attenuates hydrogenperoxide-induced c-jun NH2-terminal kinase activation via a glutathionereductase-mediated mechanism. Circ Res. 2002;91:712–718.
35. Liu YM, Yin GY, Surapisitchat J, Berk BC, Min W. Laminar flowinhibits TNF-induced ASK1 activation by preventing dissociation ofASK1 from its inhibitor 14-3-3. J Clin Invest. 2001;107:917–923.
36. Yochum L, Kushi LH, Meyer K, Folsom AR. Dietary flavonoid intakeand risk of cardiovascular disease in postmenopausal women. Am JEpidem. 1999;149:943–949.
37. Zhao X, Sun G, Zhang J, Strong R, Dash PK, Kan YW, Grotta JC,Aronowski J. Transcription factor Nrf2 protects the brain fromdamage produced by intracerebral hemorrhage. Stroke. 2007;38:3280 –3286.
38. Tanito M, Masutani H, Kim Y-C, Nishikawa M, Ohira A, Yodoi J.Sulforaphane induces thioredoxin through the antioxidant-responsiveelement and attenuates retinal light damage in mice. Invest Opthalmol VisSci. 2005;46:979–987.
39. Sussan TE, Jun J, Thimmulappa R, Bedja D, Antero M, Gabrielson KL ,.Polotsky VY, Biswal S. Disruption of Nrf2, a key inducer of antioxidantdefenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS ONE.2008;3:e3791.
Zakkar et al Nrf2 Suppresses Endothelial Activation in Arteries 7
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
SUPPLEMENT MATERIAL
MATERIALS AND METHODS
Animals Male C57BL/6 mice between 2 and 3 months of age were used. The Nrf2
knockout mouse strain (Nrf2-/- (C57BL/6))1 was generously supplied by Prof. Masayuki
Yamamoto, University of Tsukuba, Japan via the RIKEN BioResource Center,
Tsukuba, Japan. Mice were treated with sulforaphane (5 mg/kg in 10% corn-oil/ 90%
PBS) by intraperitoneal injections 24h and 4h prior to experimentation. Mice were
treated with LPS (4mg/kg) by intraperitoneal injection for 1 hour or 6 hours prior to
assessment of p38 phosphorylation or VCAM-1 expression, respectively. All
experiments were performed within guidelines set out by the Federation of European
Laboratory Animal Science Associations.
Reagents and antibodies Human recombinant TNFα and LPS (R&D) and anti-
phosphorylated-MKK3/6 (Cell signalling Technology), anti-phosphorylated-p38
Tyr180/Thr182 (Cell signalling Technology), anti-p38 (Cell Signaling Technology),
anti-RelA (Santa Cruz), anti-CD31-FITC (BD Biosciences Pharmingen), anti-Nrf2
(Santa Cruz Biotechnology), anti-HO-1 (Santa Cruz Biotechnology), anti-tubulin
(Sigma Aldrich) and anti-lamin B (Affinity BioReagents) antibodies were obtained
commercially. TNFα was used at a final concentration of 10ng/ml. The generation of
anti-VCAM-1 (1.G11) antibodies was described previously2. Anti-MKK3/6 antibodies3
were generously supplied by Dr Jonathan Dean, Imperial College London, UK.
Adenoviruses containing wild-type Nrf2 (Ad-Nrf2) or a dominant negative version that
lacks the transactivation domain (Ad-Nrf2-DN) were generously provided by Prof.
Jeffrey A. Johnson, University of Wisconsin-Madison, USA and have been described
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
previously4. Other reagents were purchased from Sigma Aldrich unless otherwise
stated.
En face staining The expression levels of specific proteins were assessed in EC at
regions of the inner curvature (susceptible site) and outer curvature (protected site) of
murine aortae by en face staining as described previously5,6. Animals were sacrificed by
CO2 inhalation. Aortae were perfused in situ with PBS (at a pressure of approximately
100mm Hg) and then perfusion-fixed with 2% formalin prior to harvesting. Fixed
aortae were tested by immunostaining using specific primary antibodies and
Alexafluor568-conjugated secondary antibodies (red). EC were identified by co-
staining using anti-CD31 antibodies conjugated to the fluorophore FITC (green). Nuclei
were identified using a DNA-binding probe with far-red emission (To-Pro-3;
Invitrogen). Stained vessels were mounted prior to visualization of endothelial surfaces
en face using confocal laser-scanning microscopy (Zeiss LSM 510 META). Isotype-
matched monoclonal antibodies raised against irrelevant antigens or pre-immune rabbit
sera were used as experimental controls for specific staining (data not shown). The
expression of particular proteins at each site was assessed by quantification of
fluorescence intensity for multiple cells (at least 100 per site) using LSM 510 software
(Zeiss) and calculation of mean fluorescence intensities with standard deviations.
Endothelial cell culture Human umbilical vein endothelial cells (HUVEC) were
collected using collagenase and cultured as described previously7. Confluent HUVEC
cultures were exposed to high unidirectional laminar shear (12 dynes/cm2) for 24h using
a parallel-plate flow chamber (Cytodyne) as described previously7. A stock solution of
sulforaphane (1 mM in DMSO) was used to treat HUVEC to achieve a final
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
concentration of 1 μM sulforaphane. Alternatively, HUVEC were treated with an
equivalent volume of DMSO alone as a control.
Gene silencing. RNA interference was carried out using small interfering (si)RNAs that
specifically target Nrf2 (5'-AAGGATTATTATGACTGTTAA-3'; Qiagen), MAP kinase
phosphatase-1 (MKP-1) (5’-AAGCUGGACGAGGCCUUUGAGUU-3’; Dharmacon)
or HO-1 (5’-UGCUGAGUUCAUGAGGAACUU-3’; Dharmacon). Alternatively, cells
were treated with non-targeting scrambled controls (Silencer Negative Control no. 1
siRNA; Ambion, Foster City, Calif, or non-targeting siRNA no.1; Thermo Fisher
Scientific). Cell cultures that were 80% to 90% confluent were transfected with siRNA
(5 μM final concentration) by microporation (Digital BioTechnology, Seoul, Korea)
following the manufacturer’s instructions and then incubated in growth medium without
antibiotics for 48h before analysis.
Comparative real time PCR Transcript levels were quantified by comparative real-
time PCR using gene-specific primers (Supplementary Table). Extraction and reverse
transcription of total RNA and real-time PCR were carried out as described previously7.
Reactions were performed in triplicate. Relative gene expression was calculated by
comparing the number of thermal cycles that were necessary to generate threshold
amounts of product as described previously7.
Flow cytometry HUVEC were harvested and incubated on ice for 1 hour with mouse
IgG antibodies specific for VCAM-1. After washing and application of fluorescein
isothiocyanate-conjugated secondary antibody, cells were analysed by flow cytometry
after gating out dead cells. The relative fluorescence index (RFI) was calculated after
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
subtracting background fluorescence measured using secondary-only controls.
Western blotting Cytosolic or nuclear lysates were prepared using the Nuclear
Extraction Kit (Active Motif). MKP-1 redox state was assessed by non-reducing SDS-
PAGE using total cell lysates, as described8. Western blotting was carried out using
specific primary antibodies, horse radish peroxidase-conjugated secondary antibodies
and chemiluminescent detection.
Assay of NF-κB DNA binding Binding of RelA NF-κB sub-units to consensus
oligonucleotides was assessed by DNA-binding ELISA (Active Motif) using nuclear
lysates prepared using the NucBuster kit (Novagen).
Immunofluorescent staining of Nrf2 in HUVEC The expression and intracellular
localization of Nrf2 in cultured HUVEC was assessed by immunostaining of methanol-
fixed cells using anti-Nrf2 antibodies and Alexafluor 568-conjugated secondary
antibodies followed by laser-scanning confocal microscopy (LSM 510 META; Zeiss).
Nuclei were identified using the DNA-binding probe To-Pro-3. Image analysis was
performed using Zeiss LSM 510 META software to calculate average fluorescence
values after subtracting background fluorescence values from cells stained with
secondary antibody alone.
Measurement of intracellular reactive oxygen species (ROS). Intracellular ROS were
measured in HUVEC cultured on glass slide chambers. They were incubated with the
redox-sensitive fluorescent probe 5-(and-6)-carboxy-2´7´-dichlorodihydrofluorescein
diacetate (H2DCFDA, 10 μM; Invitrogen) for 45 minutes and then washed with
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
phosphate-buffered saline prior to experimentation. Fluorescence was quantified in live
cells using a laser-scanning confocal microscope with a specialized stage that
maintained cultures at 37oC in a 5% CO2 environment (LSM 510 META; Zeiss).
Statistics Differences between samples were analysed using an unpaired Student’s t-
test or one-way ANOVA with Bonferroni adjustment (*p<0.05, **p<0.01,
***p<0.001).
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
REFERENCES FOR SUPPLEMENT MATERIAL
(1) Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N,
Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2 small Maf heterodimer
mediates the induction of phase II detoxifying enzyme genes through antioxidant
response elements. Biochem Biophys Res Comm. 1997;236:313--322.
(2) Thornhill MH, Wellicome SM, Mahiouz DL, Lanchbury JSS, Kyanaung U, Haskard
DO. Tumor-Necrosis-Factor Combines with Il-4 Or Ifn-Gamma to Selectively Enhance
Endothelial-Cell Adhesiveness for T-Cells - the Contribution of Vascular Cell-
Adhesion Molecule-1-Dependent and Molecule-1-Independent Binding Mechanisms. J
Immunol 1991; 146:592-8.
(3) Lasa M, Brook M, Saklatvala J, Clark AR. Dexamethasone destabilizes
cyclooxygenase 2 mRNA by inhibiting mitogen-activated protein kinase p38.
Molecular and Cellular Biology 2001; 21(3): 771-80.
(4) Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent
antioxidant response element activation by tert-butylhydroquinone and sulforaphane
occurring preferentially in astrocytes conditions neurons against oxidative insult. J
Neurosci 2004;24(5):1101-12.
(5) Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B
signal transduction pathway in aortic endothelial cells is primed for activation in
regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA.
2000;97:9052--9057.
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
(6) Zakkar M, Chaudhury H, Sandvik G, Enesa K, Luong LA, Cuhlmann S, Mason JC,
Krams R, Clark AR, Haskard DO, Evans PC. Increased endothelial mitogen-activated
protein kinase phosphatase-1 expression suppresses proinflammatory activation at sites
that are resistant to atherosclerosis. Circ Res. 2008;103:726--732.
(7) Partridge J, Carlsen H, Enesa K, Chaudhury H, Zakkar M, Luong L, Kinderlerer A,
Johns M, Blomhoff R, Mason JC, Haskard DO, Evans PC. Laminar shear stress acts as
a switch to regulate divergent functions of NF-kappa B in endothelial cells. FASEB J.
2007;21:3553--3561.
(8) Kamata H, Honda S, Maeda S, Chang LF, Hirata H, Karin M. Reactive oxygen
species promote TNF alpha-induced death and sustained JNK activation by inhibiting
MAP kinase phosphatases. Cell. 2005;120:649--661.
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
LEGENDS FOR SUPPLEMENTARY TABLE AND FIGURES
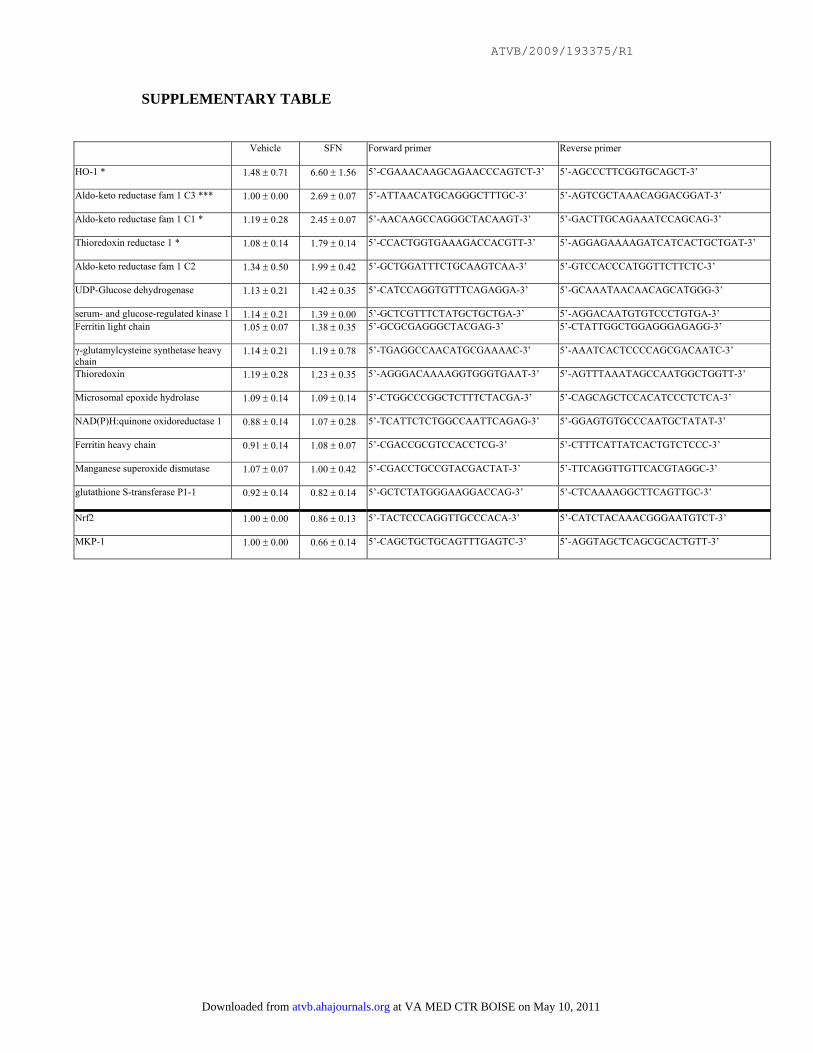
Supplementary Table
Effects of sulforaphane on gene expression. HUVEC were treated with SFN (1 μM)
or with vehicle alone and incubated for 4 hours. Levels of particular transcripts were
quantified by real-time PCR using gene-specific primers (sequences indicated) and
were normalised by measuring β-actin transcript levels. Mean values (+/- SD)
calculated from two independent experiments are shown.
Supplementary Fig. I
Nrf2 is activated in EC at a protected site in the aorta. Nrf2 expression levels and
intracellular localization (A) or HO-1 expression levels (B) were assessed in EC at
protected (P) or susceptible (S) regions of the aorta in wild-type or Nrf2-/- mice by en
face staining using anti-Nrf2 or anti-HO-1 antibodies. Endothelial marker (CD31);
nuclear counterstain (DNA). Representative images and quantitation of nuclear Nrf2
(A) and HO-1 (B) from multiple animals are shown.
Supplementary Fig. II
Nrf2 suppresses p38 activation in EC in a protected region of the aorta.
Phosphorylated p38 (phos-p38) levels were assessed by en face staining of protected or
susceptible regions in wild-type or Nrf2-/- mice (treated with LPS or untreated; n=4 per
group). Endothelial marker (CD31); nuclear counterstain (DNA). Representative
images and quantitation of p38 phosphorylation from multiple animals are shown.
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
Supplementary Fig. III
Nrf2 suppresses VCAM-1 expression in EC in a protected region of the aorta.
VCAM-1 expression levels were assessed by en face staining of protected or
susceptible regions in wild-type or Nrf2-/- mice (treated with LPS or untreated; n=4 per
group). Endothelial marker (CD31); nuclear counterstain (DNA). Representative
images and quantitation of VCAM-1 expression from multiple animals are shown.
Supplementary Fig. IV
Expression levels of p38 are similar in protected and susceptible regions of wild-
type and Nrf2-/- mice. p38 expression levels were assessed by en face staining of
protected or susceptible regions in wild-type or Nrf2-/- mice. Endothelial marker
(CD31); nuclear counterstain (DNA). Representative images and quantitation of p38
expression from multiple animals are shown.
Supplementary Fig. V
Nrf2 is essential for suppression of pro-inflammatory activation by shear stress in
cultured EC. HUVEC were exposed to unidirectional shear stress for 24 hours or were
cultured under static conditions. (A) Levels of Nrf2 transcripts were quantified by real-
time PCR. Mean values (+/- SD) calculated from three independent experiments are
shown. (B) Cytosolic or nuclear lysates were tested by Western blotting using anti-
Nrf2, anti-tubulin or anti-lamin B antibodies. Representative of three independent
experiments. (C) Nrf2 expression and intra-cellular localization was assessed by
immunofluorescence staining using anti-Nrf2 antibodies and nuclear counterstaining
(DNA). Representative images and mean nuclear Nrf2 levels (+/- SD) calculated from
multiple EC are shown. (D) HUVEC were transduced with Ad-Nrf2-DN or with an
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
empty adenovirus (Ad-empty) or remained untreated and were incubated for 24 hours.
Cells were then either stimulated with TNFα for 4 hours or remained untreated. Levels
of VCAM-1 transcripts were quantified by real-time PCR. Mean values (+/- SD)
calculated from three independent experiments are shown.
Supplementary Fig. VI
Sulforaphane does not influence VCAM-1 mRNA stability
The effect of sulforaphane (SFN) on the stability of VCAM-1 mRNA was assessed by
pulse-chase experiments. HUVEC were stimulated with TNFα for 4 hours or remained
untreated. Activated cells were subsequently incubated with actinomycinD (50 μM) to
block transcription, either in the presence of SFN (1 μM) or vehicle alone. VCAM-1
transcript levels were then measured at various time points by real-time PCR. Mean
values (+/- SD) calculated from two independent experiments are shown.
Supplementary Fig. VII
Nrf2 suppresses p38-VCAM-1 signalling
(A) The effect of an Nrf2-containing adenovirus (Ad-Nrf2) on HO-1 expression levels
was determined. HUVEC were transduced with Ad-Nrf2 or with an empty adenovirus
(Ad-empty) and incubated for 24 hours. Cytosolic lysates were tested by Western
blotting using anti-Nrf2, anti-HO-1 or anti-tubulin antibodies. Representative of three
independent experiments. (B, C, D, E, F) The effect of Nrf2 overexpression on pro-
inflammatory signalling and on the induction of ROS was determined. HUVEC were
transduced with Ad-Nrf2, Ad-Nrf2-DN or Ad-empty and were incubated for 24 hours,
or were untreated. Cells were then stimulated with TNFα for 30 minutes (D), 4 hours
(B) or varying times (C, E). (B) Levels of VCAM-1 transcripts were quantified by real-
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
time PCR. Mean values (+/- SD) calculated from triplicate measurements are shown.
Representative of three independent experiments. (C, D) Cell lysates were tested by
Western blotting using antibodies that recognise phosphorylated p38 (phos-p38), total
p38, phosphorylated MKK3/6 or total MKK3/6. Representative of three independent
experiments. (E) NF-κB activation was assessed by DNA-binding ELISA. Mean optical
densities (450 nm) calculated from triplicate wells are shown with standard deviations.
Representative of three independent experiments. (F) HUVEC were loaded with the
redox-sensitive probe H2DCFDA, treated with H2O2 (50 μM) and fluorescence was
assessed at varying times by confocal microscopy. Representative images and
quantitation of fluorescent cells after 5 minutes stimulation (mean +/- SD; pooled from
3 experiments) are shown. (G) The effect of Nrf2 expression on MKP-1 redox state was
determined. Cells were treated with 0.3 μM H2O2 for 10 minutes or remained untreated
as a control. Total cell lysates were analysed by SDS-PAGE in the presence or absence
of DTT (as indicated) followed by Western blotting using anti-MKP-1 antibodies.
Oxidised and reduced forms of MKP-1 are indicated. NS, non-specific band.
Supplementary Fig. VIII
HO-1 activity is not required for suppression of VCAM-1 expression by
sulforaphane. The role of HO-1 in EC responses to SFN was assessed. (A) HUVEC
were treated with zinc protoporphyrin (ZnPP; 10 μM for 24 hours) which is a
pharmacological inhibitor of HO-1 or with vehicle alone. (B) Alternatively, HUVEC
were transfected with HO-1 specific siRNA or with a scrambled control (Scr). (A, B)
Cells were then exposed to SFN (1 μM) or vehicle alone for 4 hours, and then
stimulated with TNFα for 4 hours or remained untreated. Levels of HO-1 or VCAM-1
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
transcripts were quantified by real-time PCR. Mean values (+/- SD) calculated from
triplicate measurements are shown.
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from
ATVB/2009/193375/R1
SUPPLEMENTARY TABLE
Vehicle SFN Forward primer Reverse primer
HO-1 * 1.48 ± 0.71 6.60 ± 1.56 5’-CGAAACAAGCAGAACCCAGTCT-3’ 5’-AGCCCTTCGGTGCAGCT-3’
Aldo-keto reductase fam 1 C3 *** 1.00 ± 0.00 2.69 ± 0.07 5’-ATTAACATGCAGGGCTTTGC-3’ 5’-AGTCGCTAAACAGGACGGAT-3’
Aldo-keto reductase fam 1 C1 * 1.19 ± 0.28 2.45 ± 0.07 5’-AACAAGCCAGGGCTACAAGT-3’ 5’-GACTTGCAGAAATCCAGCAG-3’
Thioredoxin reductase 1 * 1.08 ± 0.14 1.79 ± 0.14 5’-CCACTGGTGAAAGACCACGTT-3’ 5’-AGGAGAAAAGATCATCACTGCTGAT-3’
Aldo-keto reductase fam 1 C2 1.34 ± 0.50 1.99 ± 0.42 5’-GCTGGATTTCTGCAAGTCAA-3’ 5’-GTCCACCCATGGTTCTTCTC-3’
UDP-Glucose dehydrogenase 1.13 ± 0.21 1.42 ± 0.35 5’-CATCCAGGTGTTTCAGAGGA-3’ 5’-GCAAATAACAACAGCATGGG-3’
serum- and glucose-regulated kinase 1 1.14 ± 0.21 1.39 ± 0.00 5’-GCTCGTTTCTATGCTGCTGA-3’ 5’-AGGACAATGTGTCCCTGTGA-3’ Ferritin light chain 1.05 ± 0.07 1.38 ± 0.35 5’-GCGCGAGGGCTACGAG-3’ 5’-CTATTGGCTGGAGGGAGAGG-3’
γ-glutamylcysteine synthetase heavy chain
1.14 ± 0.21 1.19 ± 0.78 5’-TGAGGCCAACATGCGAAAAC-3’ 5’-AAATCACTCCCCAGCGACAATC-3’
Thioredoxin 1.19 ± 0.28 1.23 ± 0.35 5’-AGGGACAAAAGGTGGGTGAAT-3’ 5’-AGTTTAAATAGCCAATGGCTGGTT-3’
Microsomal epoxide hydrolase 1.09 ± 0.14 1.09 ± 0.14 5’-CTGGCCCGGCTCTTTCTACGA-3’ 5’-CAGCAGCTCCACATCCCTCTCA-3’
NAD(P)H:quinone oxidoreductase 1 0.88 ± 0.14 1.07 ± 0.28 5’-TCATTCTCTGGCCAATTCAGAG-3’ 5’-GGAGTGTGCCCAATGCTATAT-3’
Ferritin heavy chain 0.91 ± 0.14 1.08 ± 0.07 5’-CGACCGCGTCCACCTCG-3’ 5’-CTTTCATTATCACTGTCTCCC-3’
Manganese superoxide dismutase 1.07 ± 0.07 1.00 ± 0.42 5’-CGACCTGCCGTACGACTAT-3’ 5’-TTCAGGTTGTTCACGTAGGC-3’
glutathione S-transferase P1-1 0.92 ± 0.14 0.82 ± 0.14 5’-GCTCTATGGGAAGGACCAG-3’ 5’-CTCAAAAGGCTTCAGTTGC-3’
Nrf2 1.00 ± 0.00 0.86 ± 0.13 5’-TACTCCCAGGTTGCCCACA-3’ 5’-CATCTACAAACGGGAATGTCT-3’
MKP-1 1.00 ± 0.00 0.66 ± 0.14 5’-CAGCTGCTGCAGTTTGAGTC-3’ 5’-AGGTAGCTCAGCGCACTGTT-3’
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from
ATVB/2009/193375/R1
Fluo
resc
ence
inte
nsity
Nuclear Nrf2
Supplementary Fig. I
Protected (P)Susceptible (S)A
mergeDNA
CD31 Nrf2
mergeDNA
CD31 Nrf2
mergeDNA
CD31 HO-1
Nrf2 -/-Wild-type
125 -
100 -
75 -
50 -
25 -
0 -
***
Wild-type Nrf2-/-S P P
Fluo
resc
ence
inte
nsity
mergeDNA
CD31 HO-1Protected (P)
mergeDNA
CD31 HO-1Susceptible (S) Protected (P)B
Nrf2 -/-Wild-type
Protected (P)
mergeDNA
CD31 Nrf2
20
15
10
0
5
25
Wild-type Nrf2-/-S P P
***
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from
ATVB/2009/193375/R1
mergeDNA
CD31 phos-p38 7
mergeDNA
CD31 phos-p38 3
mergeDNA
CD31 phos-p38 4
mergeDNA
CD31 phos-p38 8
+ LPS
mergeDNA
CD31 phos-p38 1
mergeDNA
CD31 phos-p38 2
mergeDNA
CD31 phos-p38 5
mergeDNA
CD31 phos-p38 6
Prot
ecte
d
Nrf2 -/-
untreated
Wild-type
Supplementary Fig. II
Susc
eptib
le
+ LPS
untreated
Fluo
resc
ence
inte
nsity
LPS
0
50
100
150
200
250 Wild typeNrf2 -/-
1 2 3 4 5 6 7 8
***
***
- - + + - - + +Protected Susceptible
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
mergeDNA
CD31 VCAM-1 1
5
mergeDNA
CD31 VCAM-1 7
mergeDNA
CD31 VCAM-1 2
mergeDNA
CD31 VCAM-1 3
mergeDNA
CD31 VCAM-1 4
mergeDNA
CD31 VCAM-1
mergeDNA
CD31 VCAM-1 6
mergeDNA
CD31 VCAM-1 8
Nrf2 -/-
+ LPS
untreated
Wild-type
Supplementary Fig. III
Fluo
resc
ence
inte
nsity
0
50
100
150
200
250 Wild typeNrf2 -/-
***
***
1 2 3 4 5 6 7 8LPS - - + + - - + +
Protected Susceptible
+ LPS
untreated
Prot
ecte
d Su
scep
tible
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
Supplementary Fig. IV
mergeDNA
CD31 p38 1
mergeDNA
CD31 p38 3
mergeDNA
CD31 p38 2
mergeDNA
CD31 p38 4
Susceptible
Protected
Nrf2 -/-Wild-type
Fluo
resc
ence
inte
nsity
0
10
20
30 Wild typeNrf2 -/-
Protected Susceptible1 2 3 4
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from
ATVB/2009/193375/R1
shear
Ad-Nrf2-DNAd-empty
0 -
1 -
3 -
5 -
7 -
**
++ −−− +
+− +
D
VCAM
-1 m
RN
A le
vels
A
lamin B
Nrf2
shear
B
nuclear
tubulin
Nrf2cytosolic
0 -
1.5 -
*
3 -
Supplementary Fig. V
CNrf2
DNA
merge
Nrf2
DNA
merge
shearstatic
0
50
100
150
200
250
static shear
Fluo
resc
ence
inte
nsity
Nuclear Nrf2
Nrf
2 m
RN
A le
vels
static shear static
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
Supplementary Fig. VI
0
200
400
600
800
1000
1200
DMSOSFN
VCA
M-1
mR
NA
leve
ls600
800
1000
400
200
0TNFα − + + + + +
Chase time (min) 0 30 45 60 90
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
Supplementary Fig. VII
C
TNFα (min)Ad-Nrf2-DNAd-empty
0 10 15 30 60 90 0 10 15 30 60 90 0 10 15 30 60 90 0 10 15 30 60 90
p38
phos-p38
Ad-Nrf2No adenovirus
HO-1
Nrf2
MOI 500 100 200 400 500
tubulin
Ad-empty Ad-Nrf2
A
TNFα + +− −
Ad-empty Ad-Nrf2
160 -
80 -
0 -
VCA
M-1
mR
NA
leve
ls
**
B
E
0
1
2
3
NF-
κB D
NA
bind
ing
TNFα (min)
Ad-empty
0 15 15
+ + -Ad-Nrf2 - - +
+ -- +
30 30F
Tim
e (m
in)
Ad-empty Ad-Nrf2
0
2
4
80
50
100
150
200
250
Ad empty Nrf2
**
Posi
tive
cells
TNFαAd-empty
phos-MKK3/6
- + ++ + -
Ad-Nrf2 - - +
MKK3/6
D
G97 kD
64 kD
51 kD
39 kD
Ad-emptyAd-Nrf2
H2O2
DTT
- oxidised
- reduced
- ns
- ns
+ +- -
- ++ -
- + + +- - - +1 2 3 4
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

ATVB/2009/193375/R1
Supplementary Fig. VIII
Data 3
vcam-1 data with ho-1 sirna
0
25
50
75
100
125
100
150
200
250
300
VCAM
-1 m
RN
A le
vels *A
B
0
25
50
75
100
125
0
50
TNFα − + + +SFN − − − +
ZnPP − − + +
TNFα
ScrHO-1
−+ + +
− − − +
+ ++−SFN + +−−
0
10
20
VCAM
-1 m
RN
A le
vels
HO
-1 m
RN
A le
vels
**
*
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

Nrf2
lamin B
SFNvehicle
A
nuclear
tubulin
Nrf2cytosolic
vcam-1-nrf2sirna data for paper
Scr-tnf
Scr+tnf
Scr+tnf+1
sfn
Sirna+
tnf+1sfn
010
20
30
40
50
60
VCA
M-1
mR
NA
lev
els
C
TNFα
Ad-Nrf2-DNAd-empty
- + +SFN - - +
- + +- - +
*
Scr -+- +Nrf2
0
0.5 -
1-
Nrf
-2 m
RN
A l
evel
s*
D
0
20
40
60
80
100
TNFαSFN
ScrNrf2
VCA
M-1
mR
NA
lev
els *
*
-+ + +- - - +
+ +--- + + +
E
0
1
2
3
4
5
*
*
TNFαSFN
Scr
+ +--- + + +
-+ + +
F
TNFα
Ad-Nrf2-DNAd-empty
p38
phos-p38
- + +SFN - - +
- + +- - +
G
TNFα
Scr
Data 2
0
6
9
MK
P-1
mR
NA
lev
els
2
4
8 *I
+ + -0
100
200
300
VCA
M-1
mR
NA
lev
els
*
*
TNFαSFN
ScrMKP-1
+ + -+- - +-+ + +-- + +-
MKP-1 - - +- + +
VCA
M-1
exp
ress
ion
(RFI
)
Tim
e (m
in)
vehicle SFN
0
2
4
8
H
0
20
40
60
80
100
vehicle SFN
Pos
itive
cel
ls
**
B SFNvehicle
0
1020
304050
6070
Vehicle SFN
Nrf2
DNA
merge
Nrf2
DNA
merge
*
Fluo
resc
ence
inte
nsity
Nuclear Nrf2
NF-κB
lamin B
TNFα (mins)SFN - + -
0 15 15 30 30- +
phos-MKK3/6
MKK3/6Nrf2 - - - +
Nrf2
Tubulin
nuclear
total
total
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

Fig. 2
mergeDNA
CD31 Nrf2
mergeDNA
CD31 Nrf2
mergeDNA
CD31 Nrf2
mergeDNA
CD31 Nrf2
vehicle
SFN
ProtectedSusceptible
Fluo
resc
ence
inte
nsity
Nuclear Nrf2***
010203040506070
SFN - + - +susceptible protected
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from
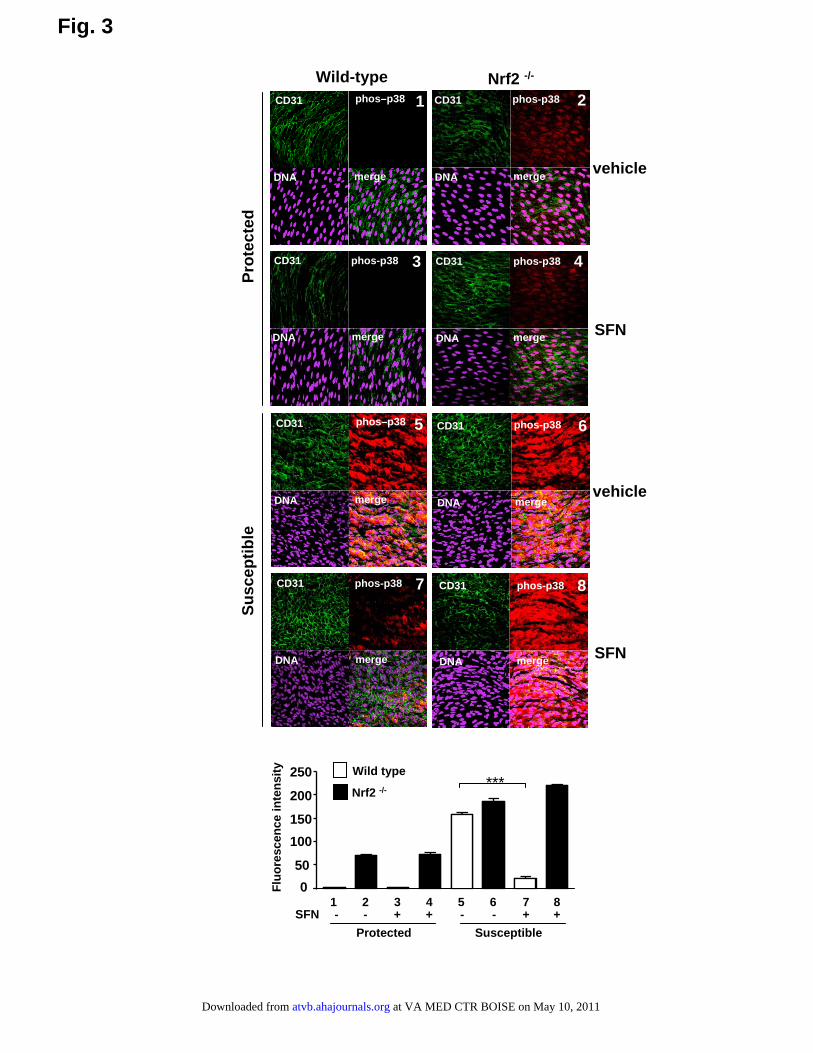
Fig. 3
mergeDNA
CD31 phos-p38 2
mergeDNA
CD31 phos–p38 1
mergeDNA
CD31 phos-p38 4
mergeDNA
CD31 phos-p38 3
mergeDNA
CD31 phos-p38 6
mergeDNA
CD31 phos–p38 5
mergeDNA
CD31 phos-p38 8
mergeDNA
CD31 phos-p38 7
Nrf2 -/-Wild-type
vehicle
SFN
vehicle
SFN
Fluo
resc
ence
inte
nsity
050
100150
200
250***
SFN
Wild type
Nrf2 -/-
1 2 3 4 5 6 7 8- - + + - - + +
Protected Susceptible
Prot
ecte
d Su
scep
tible
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from

Fig. 4
Nrf2 -/-Wild-type
vehicle
SFN
vehicle
SFN
mergeDNA
CD31 VCAM-1 1
mergeDNA
CD31 VCAM-1 3
mergeDNA
CD31 VCAM-1 2
mergeDNA
CD31 VCAM-1 4
mergeDNA
CD31 VCAM-1 5
mergeDNA
CD31 VCAM-1 7
mergeDNA
CD31 VCAM-1 6
mergeDNA
CD31 VCAM-1 8
Fluo
resc
ence
inte
nsity
***Wild type
Nrf2 -/-
***
0
50
100
150
SFN1 2 3 4 5 6 7 8- - + + - - + +
Protected Susceptible
Prot
ecte
d Su
scep
tible
at VA MED CTR BOISE on May 10, 2011 atvb.ahajournals.orgDownloaded from




















