
Proinflammatory and Oxidative Stress Markers in Patients with Periodontal Disease
Upload
univ-lille2Category
view
1download
0
PHAGOCYTES, GRANULOCYTES, AND MYELOPOIESIS
p16INK4a deficiency promotes IL-4–induced polarization and inhibitsproinflammatory signaling in macrophages*Celine Cudejko,1-4 *Kristiaan Wouters,1-4 Lucía Fuentes,1-4 Sarah Anissa Hannou,1-4 Charlotte Paquet,1-4
Kadiombo Bantubungi,1-4 Emmanuel Bouchaert,1-4 Jonathan Vanhoutte,1-4 Sebastien Fleury,1-4 Patrick Remy,4,5
Anne Tailleux,1-4 Giulia Chinetti-Gbaguidi,1-4 David Dombrowicz,1-4 Bart Staels,1-4 and Rejane Paumelle1-4
1Universite Lille Nord de France, Lille, France; 2Inserm, U1011, Lille, France; 3Universite de Lille 2, Lille, France; 4Institut Pasteur de Lille, Lille, France; and5Service de Production des antigenes, Institut Pasteur de Lille, Lille, France
The CDKN2A locus, which contains thetumor suppressor gene p16INK4a, is asso-ciated with an increased risk of age-related inflammatory diseases, such ascardiovascular disease and type 2 diabe-tes, in which macrophages play a crucialrole. Monocytes can polarize toward clas-sically (CAM�) or alternatively (AAM�)activated macrophages. However, the mo-lecular mechanisms underlying the acqui-sition of these phenotypes are not welldefined. Here, we show that p16INK4a defi-ciency (p16�/�) modulates the macro-phage phenotype. Transcriptome analy-
sis revealed that p16�/� BM-derivedmacrophages (BMDMs) exhibit a pheno-type resembling IL-4–induced macro-phage polarization. In line with thisobservation, p16�/� BMDMs displayed adecreased response to classically polariz-ing IFN� and LPS and an increased sensi-tivity to alternative polarization by IL-4.Furthermore, mice transplanted withp16�/� BM displayed higher hepatic AAM�
marker expression levels on Schisto-soma mansoni infection, an in vivo modelof AAM� phenotype skewing. Surpris-ingly, p16�/� BMDMs did not display in-
creased IL-4–induced STAT6 signaling,but decreased IFN�-induced STAT1 andlipopolysaccharide (LPS)–induced IKK�,�phosphorylation. This decrease corre-lated with decreased JAK2 phosphoryla-tion and with higher levels of inhibitoryacetylation of STAT1 and IKK�,�. Thesefindings identify p16INK4a as a modulatorof macrophage activation and polariza-tion via the JAK2-STAT1 pathway withpossible roles in inflammatory diseases.(Blood. 2011;118(9):2556-2566)
Introduction
The tumor suppressor p16INK4a is encoded by the CDKN2A locuson the human chromosome 9p21 and on the murine chromosome 4.p16INK4a belongs to the INK4 family of cyclin-dependent kinase(CDK) inhibitors, also including p15INK4b, p18INK4c, and p19INK4d.1-5
p16INK4a inhibits cell-cycle progression by preventing cyclin D–CDK4/6 complex formation. As a consequence, pRb hyperphosphorylationand its association with E2F, which induces transcription of S-phasegenes, are inhibited. p16INK4a inactivation by deletion, point mutation, orpromoter methylation, occurs frequently in most tumors.6
Besides its role in cancer as an inhibitor of cell-cycle progres-sion, p16INK4a plays a crucial role in senescence and aging.7,8
Indeed, expression of p16INK4a increases with age in various tissuesfrom several species.9-11 A genome-wide association study hasshown association of the CDKN2A locus with an increased risk ofthe age-related frailty syndrome.12 In addition, increased p16INK4a
expression causes the age-dependent decline in proliferation ofself-renewing cellular compartments such as HSCs,13 which giverise to immune cells.
Although the role of p16INK4a in mature immune cells has notyet been investigated, several studies has shown that the CDKN2Alocus is associated with an increased risk for coronary heartdisease,14 atherosclerosis,15 and type 2 diabetes (T2D).16 In thesepathologies, immune cells, such as macrophages, play a crucialrole. Besides their pleiotropic immune functions, macrophages also
play a role in the development and homeostasis of several tissues,such as adipose tissue17 and liver.18 Depending on the cytokineenvironment, macrophages differentiate into distinct subclasseswith specific characteristics. Classically activated macrophages(CAM�) differentiate in presence of Th1 cytokines, such as IFN�,or in presence of bacterial products such as lipopolysaccharide(LPS). CAM� trigger proinflammatory responses required to killintracellular pathogens.19 Alternatively activated macrophages(AAM�), induced by Th2 cytokines such as IL-4 and IL-13, areassociated with Th2-type immune responses as seen in helminthparasite infections.19 During inflammation, AAM� play a key rolein protecting the organism against tissue damage.20 However, littleis known about the mechanisms underlying the acquisition of theAAM� phenotype.
In the present study, we investigated whether p16INK4a defi-ciency influences macrophage activation in vitro, using BM-derived macrophages (BMDMs), and in vivo by infection with theparasite Schistosoma mansoni. BMDMs from p16INK4a-deficient(p16�/�) mice exhibit a phenotype resembling IL-4–induced mac-rophage polarization and an enhanced response to the Th2 cytokineIL-4 compared with BMDMs from wild-type (p16�/�) mice. Bycontrast, their response to Th1 stimuli is diminished. Moreover,S mansoni infection of mice transplanted with p16�/� BM resultedin an increased hepatic AAM� signature in vivo. Finally, we show
Submitted October 14, 2010; accepted May 17, 2011. Prepublished online asBlood First Edition paper, June 2, 2011; DOI 10.1182/blood-2010-10-313106.
*C.C. and K.W. contributed equally to this work.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page chargepayment. Therefore, and solely to indicate this fact, this article is herebymarked ‘‘advertisement’’ in accordance with 18 USC section 1734.
© 2011 by The American Society of Hematology
2556 BLOOD, 1 SEPTEMBER 2011 � VOLUME 118, NUMBER 9
For personal use only.on March 27, 2016. by guest www.bloodjournal.orgFrom

that p16INK4a deficiency does not influence the IL-4–inducedSTAT6 pathway. By contrast, the lower response to classicactivation stimuli likely occurs through an inhibition of thephosphorylation of both STAT1 and IKK�,�, which correlatedwith decreased JAK2 phosphorylation and increased acetylation ofSTAT1 and IKK�,�.
Methods
Mice
C57BL/6J p16INK4a-deficient and littermate control mice on a C67BL/6Jbackground (� 97%) were kindly provided by P. Krimpenfort (NetherlandsCancer Institute, Amsterdam). p16�/� mice showed neither abnormalities inbiochemical parameters nor spontaneous tumor development at the studiedage. Flow cytometric analysis of splenic T and B cells or macrophagesshowed no differences between p16�/� and p16�/� mice (data not shown).All protocols were conducted with the approval of the Pasteur Instituteethical review board (Lille, France).
BM chimeras
Eight-week-old C57BL/6J mice were lethally irradiated (8 Gy) and injectedin the tail vein the next day with 10 � 106 BM cells isolated from p16�/� orp16�/� donor mice. Mice were supplied with autoclaved acidified water(pH 2) supplemented with neomycin 100 mg/L (Sigma-Aldrich), poly-myxin B sulfate 60 000 U/L (Invitrogen) 1 week before and 4 weeks aftertransplantation. Mice were studied 6 weeks posttransplantation to allowrepopulation by the donor BM. To ensure that donor BM replaced theresident blood cell population, DNA was extracted from whole blood withan Illustra blood kit (GE Healthcare). PCR was performed with forward5-GCA GTG TTG CAG TTT GAA CCC-3 and reverse 5-TGT GGCAAC TGA TTC AGT TTG-3 primer sets yielding products of differentlengths, which were separated on a 1.5% agarose gel and quantified with theGel Doc XR system (Bio-Rad). More than 95% of host blood cells werefrom donor origin.
Primary cell isolation and culture
LPS-elicited neutrophils from air pouches were isolated as described.21
Splenic CD4� T cells and CD43� B cells were isolated from female mice,respectively using Dynal (Invitrogen), biotin-conjugated anti-CD43 (BDBiosciences) and streptavidin conjugated to magnetic beads (MiltenyiBiotec) according to the manufacturer’s instructions. BM-derived dendriticcells (DCs) were prepared as described.22 BMDMs were obtained fromfemoral and tibial BM suspensions, plated at 10 � 106 cells in 10-cm platesand differentiated in BMDM medium (RPMI 1640 containing HEPES25mM supplemented with 10% low endotoxin FBS, 15% L929-conditionedmedium, 2mM L-glutamine, 1mM gentamycine). Stimulations and microar-ray analysis were carried out at day 8. IL-4 (15 ng/mL) and IFN�(2.5 ng/mL and 10 ng/mL) were obtained from PeproTech. LPS (100 ng/mL) from Escherichia coli serotype 0111B4 was from Sigma-Aldrich.CINK4 (no. 219492) was obtained from Merck. For indirect cocultureexperiments, conditioned medium of alternatively differentiated p16�/�
and p16�/� macrophages was added to cultures of BMDMs 24 hours beforeLPS (100 ng/mL) stimulation. After 4 hours, CAM� were washed andcultured for 24 hours in fresh medium. The resulting media were assayedfor cytokine secretion by ELISA.
Flow cytometry
During in vitro differentiation, asynchronously growing cultures of BM-DMs were gently scraped every 2 days with 1� PBS/10mM EDTA/2%trypsine, washed with PBS and collected by centrifugation (5 minutes,146g) . For protein analysis, nonspecific staining was avoided by incubatingcells with purified rat anti–mouse CD16/CD32 (BD Pharmingen) for20 minutes at 4°C and labeled with anti–mouse F4/80-FITC (CaltagLaboratories) for 30 minutes at 4°C. Cells were analyzed using a Coulter
EPICS XL4-MCL Flow cytometer (Beckman Coulter) and Expo32 soft-ware (Applied Cytometry Systems). For cell sorting, cells were incubatedwith anti–mouse F4/80-FITC Abs (11-4801; eBioscience) and rat IgG2aK isotype (11-4321; eBioscience) and sorted on an EPICS-ALTRA cellsorter (Beckman Coulter). For FACS-based cell-cycle analysis, cells werefixed with 70% ethanol for 1 hour at �20°C and labeled with 50 �g/mLpropidium iodide containing 50 �g/mL RNase A for 30 minutes at roomtemperature. The percentage of cell-cycle distribution was obtained usingthe Multicycle software (Phoenix Flow Systems).
Microarray studies
Mouse genome MOE 430 2.0 gene chips (Affymetrix) were used intriplicate for each condition. Sample preparation, hybridization, andscanning were performed according the one-cycle protocol (Affymetrix).Only genes significantly regulated (P � .05) were analyzed further withGenomatix Bibliosphere. Microarray data have been deposited to http://www.ebi.ac.uk/arrayexpress under accession number E-MEXP-3216.
RNA extraction and analysis
Total RNA was obtained from BMDMs using EXTRACT-ALL, DNase-Itreated, and cDNA was generated using cDNA reverse transcription kit (AppliedBiosystems). Real-time quantitative PCR (QPCR) detection was conducted on aStratagene Mx3005P (Agilent technologies) using Brilliant II SYBR Greenreagent (Agilent Technologies) and specific primers (supplemental Table 1,available on the Blood Web site; see the Supplemental Materials link at the top ofthe online article). mRNA levels were normalized to 36B4 mRNA and foldinduction was calculated using the 2� Ct method.
Cytokine production measurement
IL-6, TNF, IL-1Rn, IL-4, IL-10, and IFN� levels in culture supernatantswere measured with ELISA kits according to the manufacturer’s instruc-tions (R&D Systems). Plasma IgE from infected animals was determinedby ELISA using purified rat anti–mouse IgE (BD Pharmingen) as a captureAb, and biotin rat anti–mouse IgE as a detection Ab (BD Pharmingen).Standards were prepared using purified mouse IgE � isotype control(BD Pharmingen).
Protein extraction and Western blotting
BMDM lysates were prepared using cell lysis buffer (Cell SignalingTechnology) supplemented with 1mM PMSF. Lysates were resolved in 16%or 10% SDS-PAGE and analyzed by Western blot. Anti-p16 (sc-1207),-actin (sc-1616), and -p38 (sc-535) Abs were purchased from Santa CruzBiotechnology. Anti–phospho-STAT6 and -STAT6 (nos. 9361 and 9362),anti–phospho-STAT1 and -STAT1 (nos. 9171 and 9172), anti–phospho-p38(no. 9211), anti–phospho-IKK�,� (no. 2681), anti-I�B� (no. 9242), andanti–phospho-JAK2 and -JAK2 (nos. 3771 and 3230) Abs were purchasedfrom Cell Signaling Technology. Hybridizations were done 1:1000 in 5%BSA, 1� TBS, 0.1% Tween 20 overnight at 4°C. Anti–rabbit IgGperoxidase-conjugate secondary Ab 1:10 000 (Sigma-Aldrich) was addedfor 1 hour at room temperature. Signals were visualized using the enhancedchemiluminescence Western blot detection kit (Amersham ECL plusWestern blotting Detection System; GE Healthcare). Membranes werestripped with Reblot Plus Strong Antibody Stripping Solution (Millipore)and reblotted with the indicated Abs.
Immunoprecipitation assay
Five hundred micrograms of BMDM lysates were prepared using lysisbuffer supplemented with trichostatin A 1�M (Sigma-Aldrich). Lysateswere mixed with 100 �L of protein G/protein A agarose (Millipore) androtated for 1 hour at 4°C. After centrifugation for 2 minutes at 900g,anti-acetyl-Lysine Ab 1:100 (ab21623; Abcam) was added. Samples wererotated overnight at 4°C. The protein G/protein A agarose mix (100 �L)was added to the samples and incubated for 1 hour at 4°C. The beads werewashed 3 times and the pellets dissolved in 40 �L of 2� Laemmli buffer.Proteins were analyzed by Western blotting as previously described.
p16INK4a FUNCTION IN MACROPHAGES 2557BLOOD, 1 SEPTEMBER 2011 � VOLUME 118, NUMBER 9
For personal use only.on March 27, 2016. by guest www.bloodjournal.orgFrom
S mansoni infection
Chimeric mice were anesthetized and percutaneously infected with40 S mansoni cercariae and killed 9 weeks later. The liver was perfused andadult worms were recovered and quantified. A piece of liver was weighedand dissolved in a KOH solution (5%, 37°C during 16 hours) and eggs werecounted. Organs were snap-frozen or fixed in paraformaldehyde for furtheranalysis. Resin-embedded livers were cut at 5 �m and Mallory trichromestaining was performed to determine granuloma size and hepatic collagen.Immunostaining was performed using Abs directed to Moma-2 (sc59332;Santa Cruz Biotechnology), Arginase1 (Arg-I; ab60176-100), Fizz1(ab39626-50), and Ym1 (ab93034) from Abcam. May-Grunwald-Giemsastaining was performed and eosinophils were counted per granuloma area.Granulomas were isolated using an Arcturus laser capture microdissectionsystem from 14-�m sections of snap-frozen liver samples. For ex vivorestimulation, spleens were homogenized, filtered (70 �m) and erythrocyteswere lysed. Cells were cultured in RPMI 1640 (Invitrogen) supplementedwith 10% low endotoxin FBS and 1mM gentamycin. Cells (5 � 105) werestimulated with 10 �g/mL soluble egg Ag (SEA) or anti-CD3 (no. 555274;BD Pharmingen) Abs during 4 days after which medium was collected forcytokine determination by ELISA.
Statistical analysis
Groups were compared using 2-tailed nonpaired t tests using GraphPadPrism software. In vitro data are expressed as means � SD and in vivo dataare expressed as means � SEM.
Results
p16INK4a expression increases during macrophage maturation,but p16INK4a deficiency does not modulate macrophagecell-cycle progression
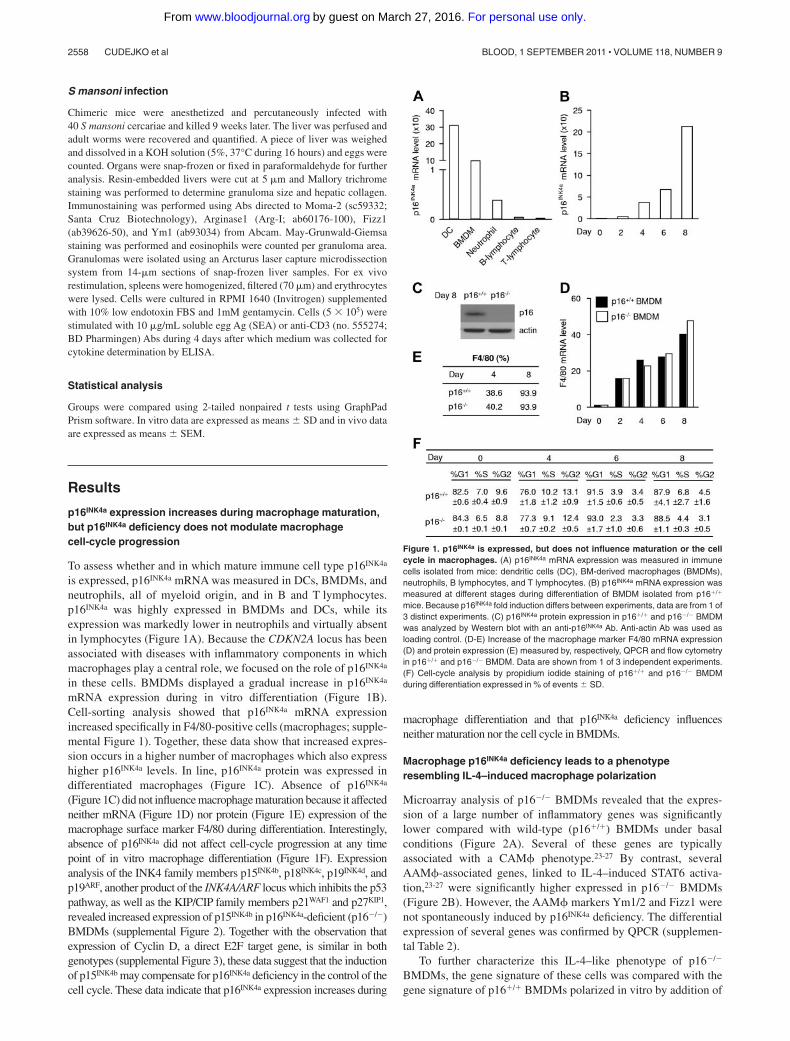
To assess whether and in which mature immune cell type p16INK4a
is expressed, p16INK4a mRNA was measured in DCs, BMDMs, andneutrophils, all of myeloid origin, and in B and T lymphocytes.p16INK4a was highly expressed in BMDMs and DCs, while itsexpression was markedly lower in neutrophils and virtually absentin lymphocytes (Figure 1A). Because the CDKN2A locus has beenassociated with diseases with inflammatory components in whichmacrophages play a central role, we focused on the role of p16INK4a
in these cells. BMDMs displayed a gradual increase in p16INK4a
mRNA expression during in vitro differentiation (Figure 1B).Cell-sorting analysis showed that p16INK4a mRNA expressionincreased specifically in F4/80-positive cells (macrophages; supple-mental Figure 1). Together, these data show that increased expres-sion occurs in a higher number of macrophages which also expresshigher p16INK4a levels. In line, p16INK4a protein was expressed indifferentiated macrophages (Figure 1C). Absence of p16INK4a
(Figure 1C) did not influence macrophage maturation because it affectedneither mRNA (Figure 1D) nor protein (Figure 1E) expression of themacrophage surface marker F4/80 during differentiation. Interestingly,absence of p16INK4a did not affect cell-cycle progression at any timepoint of in vitro macrophage differentiation (Figure 1F). Expressionanalysis of the INK4 family members p15INK4b, p18INK4c, p19INK4d, andp19ARF, another product of the INK4A/ARF locus which inhibits the p53pathway, as well as the KIP/CIP family members p21WAF1 and p27KIP1,revealed increased expression of p15INK4b in p16INK4a-deficient (p16�/�)BMDMs (supplemental Figure 2). Together with the observation thatexpression of Cyclin D, a direct E2F target gene, is similar in bothgenotypes (supplemental Figure 3), these data suggest that the inductionof p15INK4b may compensate for p16INK4a deficiency in the control of thecell cycle. These data indicate that p16INK4a expression increases during
macrophage differentiation and that p16INK4a deficiency influencesneither maturation nor the cell cycle in BMDMs.
Macrophage p16INK4a deficiency leads to a phenotyperesembling IL-4–induced macrophage polarization
Microarray analysis of p16�/� BMDMs revealed that the expres-sion of a large number of inflammatory genes was significantlylower compared with wild-type (p16�/�) BMDMs under basalconditions (Figure 2A). Several of these genes are typicallyassociated with a CAM� phenotype.23-27 By contrast, severalAAM�-associated genes, linked to IL-4–induced STAT6 activa-tion,23-27 were significantly higher expressed in p16�/� BMDMs(Figure 2B). However, the AAM� markers Ym1/2 and Fizz1 werenot spontaneously induced by p16INK4a deficiency. The differentialexpression of several genes was confirmed by QPCR (supplemen-tal Table 2).
To further characterize this IL-4–like phenotype of p16�/�
BMDMs, the gene signature of these cells was compared with thegene signature of p16�/� BMDMs polarized in vitro by addition of
Figure 1. p16INK4a is expressed, but does not influence maturation or the cellcycle in macrophages. (A) p16INK4a mRNA expression was measured in immunecells isolated from mice: dendritic cells (DC), BM-derived macrophages (BMDMs),neutrophils, B lymphocytes, and T lymphocytes. (B) p16INK4a mRNA expression wasmeasured at different stages during differentiation of BMDM isolated from p16�/�
mice. Because p16INK4a fold induction differs between experiments, data are from 1 of3 distinct experiments. (C) p16INK4a protein expression in p16�/� and p16�/� BMDMwas analyzed by Western blot with an anti-p16INK4a Ab. Anti-actin Ab was used asloading control. (D-E) Increase of the macrophage marker F4/80 mRNA expression(D) and protein expression (E) measured by, respectively, QPCR and flow cytometryin p16�/� and p16�/� BMDM. Data are shown from 1 of 3 independent experiments.(F) Cell-cycle analysis by propidium iodide staining of p16�/� and p16�/� BMDMduring differentiation expressed in % of events � SD.
2558 CUDEJKO et al BLOOD, 1 SEPTEMBER 2011 � VOLUME 118, NUMBER 9
For personal use only.on March 27, 2016. by guest www.bloodjournal.orgFrom

IL-4 from day 0 of differentiation, generating p16�/� AAM�.28
Compared with p16�/� BMDMs, 14 311 probesets were differen-tially expressed in p16�/� AAM� (Figure 2C green dots), whereas7605 probesets were regulated in p16�/� BMDMs (Figure 2C reddots), of which the majority (78%) was also differentially ex-pressed in p16�/� AAM� (Figure 2C blue dots). In addition, astrong correlation (P � .0001; Pearson R 0.7; R2 0.5) wasfound between the 2 experimental conditions (Figure 2C), as alsoillustrated by a heat map representation of a selection of AAM�and CAM� marker genes (Figure 2D, supplemental Table 3).
Because p16�/� BMDMs displayed lower expression levelsof inflammatory genes (Figure 2A,D), the secretion of inflamma-tory cytokines produced in p16�/� BMDMs compared withp16�/� BMDMs was measured. Under basal conditions, secre-tion of the pro-inflammatory cytokines IL-6 and TNF was lower(Figure 3A-B) in p16�/� BMDMs. By contrast, secretion ofanti-inflammatory IL-1Rn was, albeit modestly, higher (Figure3C). These observations are in line with the microarray data and
suggest that p16�/� BMDMs are functionally different fromtheir wild-type counterparts.
Next, we compared the response of p16�/� vs p16�/� BMDMsto polarization into AAM� induced by IL-4 added at the start (day0) of differentiation. Induction of the AAM� marker genes Arg-I(Figure 3D) and Ym1/2 (Figure 3E) was higher in p16�/� AAM�.However, induction of Mgl2 (Figure 3F) was not different in thesecells, suggesting that the IL-4 responsiveness of certain, but not all,genes is dependent on p16INK4a. This heterogeneity in regulation ofAAM� marker genes in IL-4–polarized p16�/� AAM� confirmsthe microarray results (Figure 2D).
Because p16�/� macrophages display increased AAM� markerexpression after IL-4–induced polarization and because AAM�exert paracrine anti-inflammatory effects,29 the functional effects ofp16INK4a deficiency on IL-4–induced macrophage polarization wereanalyzed by indirect coculture experiments. Hereto, conditionedmedium from IL-4–polarized p16�/� or p16�/� AAM� was addedto p16�/� BMDMs and their response to LPS was measured
Figure 2. p16INK4a deficiency induces a gene expres-sion profile resembling IL-4–induced macrophagepolarization. Microarray analysis using mRNA fromp16�/� BMDM compared with p16�/� BMDM showed(A) decreased mRNA expression of classically activatedmacrophage-associated genes and (B) increased mRNAexpression of alternatively activated macrophage-asso-ciated genes. Data are expressed as fold change relativeto p16�/� BMDM. (C) Differential gene expression inp16�/� BMDM relative to p16�/� BMDM was correlatedwith the changes induced in IL-4–induced p16�/� AAM�.The figure shows 2 log values of the probesets signifi-cantly (P � .05) regulated only in p16�/� BMDM (reddots), only in IL-4–polarized p16�/� AAM� (green dots)and by both conditions (blue dots), compared withp16�/� BMDM. The x-axis represents differences ingene expression induced by IL-4, whereas the y-axisrepresents the effect of p16INKa deficiency. These com-parisons are depicted in the schematic representation ofthe protocol in the corresponding colors. Pearson corre-lation analysis was done for probesets differentiallyexpressed by both conditions (blue). (D) Heat map ofp16�/� BMDM, p16�/� BMDM, IL-4–polarized p16�/�,and p16�/� AAM� gene expression profiles. Colorsfluctuate from blue (poorly expressed) to green (interme-diate expression) and yellow (high expression). Addi-tional information regarding gene description, fold induc-tion, and P value can be found in supplemental Table 3.
p16INK4a FUNCTION IN MACROPHAGES 2559BLOOD, 1 SEPTEMBER 2011 � VOLUME 118, NUMBER 9
For personal use only.on March 27, 2016. by guest www.bloodjournal.orgFrom

(Figure 3G). Conditioned medium from p16�/� AAM� resulted ina more pronounced inhibition of LPS-induced secretion of IL-6 andTNF (Figure 3H-I). Thus, medium from p16�/� AAM� morepotently inhibits proinflammatory responses.
Collectively, these results evidence that p16INK4a deficiencyresults in a phenotype partially resembling IL-4–induced macro-phage polarization. Moreover, IL-4–polarized p16�/� macrophagesdisplay a more pronounced AAM� phenotype.
p16INK4a-deficient macrophages are more responsive to IL-4,while they respond less to IFN� or LPS
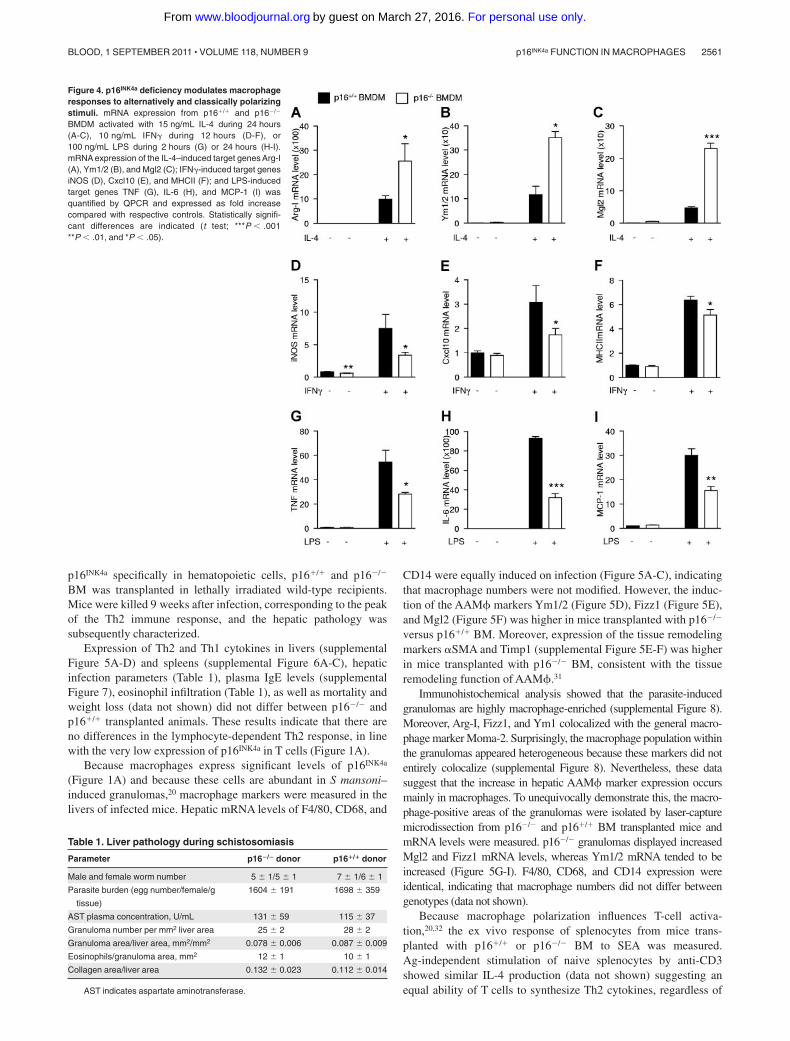
Because p16�/� BMDMs resemble IL-4–polarized macrophages andbecause p16�/� AAM� show more potent anti-inflammatory effectsthan wild-type macrophages, the response of p16�/� BMDMs to acute(24 hours) IL-4 activation was assessed. Interestingly, IL-4 treatmentresulted in a larger increase of Arg-I, Ym1/2, and Mgl2 mRNA inp16�/� compared with p16�/� BMDMs (Figure 4A-C).
AAM� are known to be less sensitive to inflammatory stimuli.19
Therefore, the effects of IFN� and LPS, 2 proinflammatory stimuli,
were tested on these cells. Interestingly, p16�/� BMDMs were lessresponsive to IFN�, shown by the lower induction of the IFN�target genes iNOS, Cxcl10, and MHCII (Figure 4D-F), as well as toLPS, shown by the decreased response of the LPS-regulated genesTNF, IL-6, and MCP-1 (Figure 4G-I). Expression levels of thecognate receptors for IL-4, IFN�, and LPS was similar in bothgenotypes (supplemental Figure 4A-C), suggesting that the alteredresponses were not because of differences in receptor expression.
Collectively, these data show that p16�/� BMDMs are moreresponsive to alternative activation by IL-4 and less responsive tothe classic macrophage activators IFN� and LPS.
Hematopoietic p16INK4a deficiency increases hepaticexpression of alternatively activated macrophage markers onS mansoni infection
Because helminth parasite infections induce a strong Th2 immuneresponse resulting in AAM� differentiation,20,30 the influence ofp16INK4a deficiency on macrophage polarization in vivo wasinvestigated during S mansoni infection. To assess the role of
Figure 3. p16INK4a-deficient macrophages phenotypically and functionally resemble IL-4–polarized alternatively activated macrophages. Protein secretion of IL-6(A), TNF (B), and IL-1Rn (C) was measured in the culture medium of p16�/� and p16�/� BMDM by ELISA. (D-F) QPCR analysis of Arg-I (D), Ym1/2 (E), and Mgl2 (F) of p16�/�
and p16�/� AAM� polarized by addition of 15 ng/mL IL-4 from day 0 of differentiation. (G) Schematic representation of the indirect coculture experiment. (H-I) LPS-inducedsecretion of IL-6 (H), and TNF (I) was determined by ELISA in p16�/� BMDM supernatants resulting from indirect coculture. Statistically significant differences are indicated(t test; ***P � .001, **P � .01, and *P � .05).
2560 CUDEJKO et al BLOOD, 1 SEPTEMBER 2011 � VOLUME 118, NUMBER 9
For personal use only.on March 27, 2016. by guest www.bloodjournal.orgFrom
p16INK4a specifically in hematopoietic cells, p16�/� and p16�/�
BM was transplanted in lethally irradiated wild-type recipients.Mice were killed 9 weeks after infection, corresponding to the peakof the Th2 immune response, and the hepatic pathology wassubsequently characterized.
Expression of Th2 and Th1 cytokines in livers (supplementalFigure 5A-D) and spleens (supplemental Figure 6A-C), hepaticinfection parameters (Table 1), plasma IgE levels (supplementalFigure 7), eosinophil infiltration (Table 1), as well as mortality andweight loss (data not shown) did not differ between p16�/� andp16�/� transplanted animals. These results indicate that there areno differences in the lymphocyte-dependent Th2 response, in linewith the very low expression of p16INK4a in T cells (Figure 1A).
Because macrophages express significant levels of p16INK4a
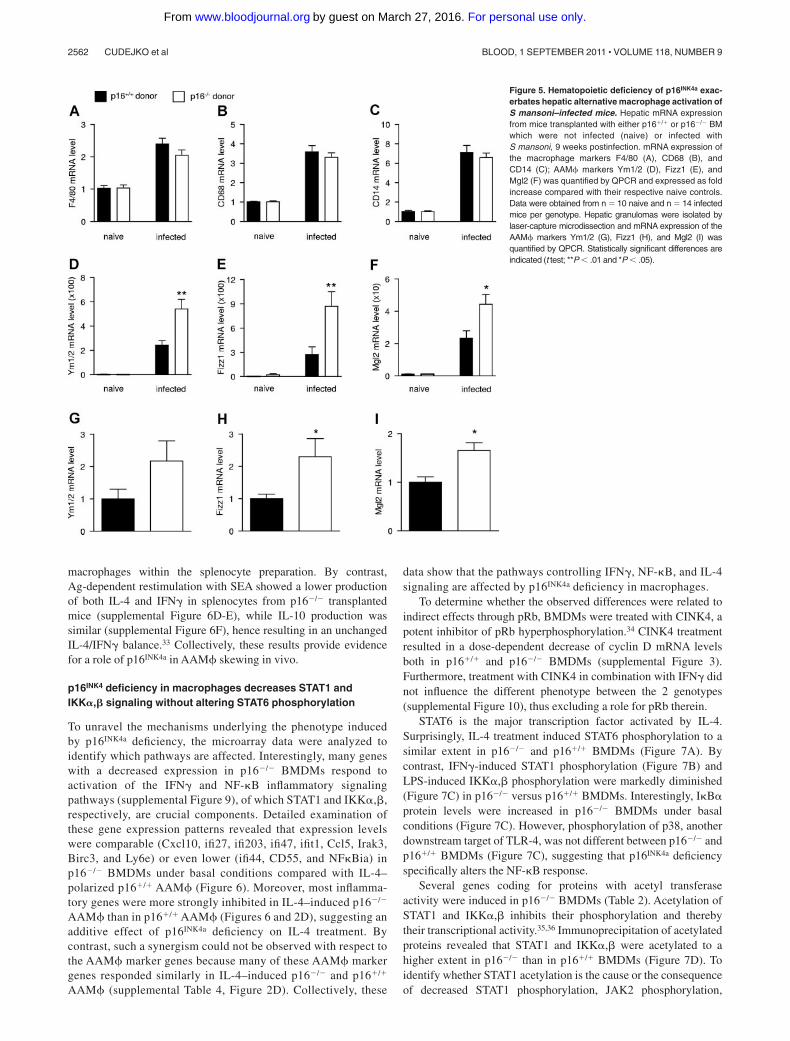
(Figure 1A) and because these cells are abundant in S mansoni–induced granulomas,20 macrophage markers were measured in thelivers of infected mice. Hepatic mRNA levels of F4/80, CD68, and
CD14 were equally induced on infection (Figure 5A-C), indicatingthat macrophage numbers were not modified. However, the induc-tion of the AAM� markers Ym1/2 (Figure 5D), Fizz1 (Figure 5E),and Mgl2 (Figure 5F) was higher in mice transplanted with p16�/�
versus p16�/� BM. Moreover, expression of the tissue remodelingmarkers �SMA and Timp1 (supplemental Figure 5E-F) was higherin mice transplanted with p16�/� BM, consistent with the tissueremodeling function of AAM�.31
Immunohistochemical analysis showed that the parasite-inducedgranulomas are highly macrophage-enriched (supplemental Figure 8).Moreover, Arg-I, Fizz1, and Ym1 colocalized with the general macro-phage marker Moma-2. Surprisingly, the macrophage population withinthe granulomas appeared heterogeneous because these markers did notentirely colocalize (supplemental Figure 8). Nevertheless, these datasuggest that the increase in hepatic AAM� marker expression occursmainly in macrophages. To unequivocally demonstrate this, the macro-phage-positive areas of the granulomas were isolated by laser-capturemicrodissection from p16�/� and p16�/� BM transplanted mice andmRNA levels were measured. p16�/� granulomas displayed increasedMgl2 and Fizz1 mRNA levels, whereas Ym1/2 mRNA tended to beincreased (Figure 5G-I). F4/80, CD68, and CD14 expression wereidentical, indicating that macrophage numbers did not differ betweengenotypes (data not shown).
Because macrophage polarization influences T-cell activa-tion,20,32 the ex vivo response of splenocytes from mice trans-planted with p16�/� or p16�/� BM to SEA was measured.Ag-independent stimulation of naive splenocytes by anti-CD3showed similar IL-4 production (data not shown) suggesting anequal ability of T cells to synthesize Th2 cytokines, regardless of
Figure 4. p16INK4a deficiency modulates macrophageresponses to alternatively and classically polarizingstimuli. mRNA expression from p16�/� and p16�/�
BMDM activated with 15 ng/mL IL-4 during 24 hours(A-C), 10 ng/mL IFN� during 12 hours (D-F), or100 ng/mL LPS during 2 hours (G) or 24 hours (H-I).mRNA expression of the IL-4–induced target genes Arg-I(A), Ym1/2 (B), and Mgl2 (C); IFN�-induced target genesiNOS (D), Cxcl10 (E), and MHCII (F); and LPS-inducedtarget genes TNF (G), IL-6 (H), and MCP-1 (I) wasquantified by QPCR and expressed as fold increasecompared with respective controls. Statistically signifi-cant differences are indicated (t test; ***P � .001**P � .01, and *P � .05).
Table 1. Liver pathology during schistosomiasis
Parameter p16�/� donor p16�/� donor
Male and female worm number 5 � 1/5 � 1 7 � 1/6 � 1
Parasite burden (egg number/female/g
tissue)
1604 � 191 1698 � 359
AST plasma concentration, U/mL 131 � 59 115 � 37
Granuloma number per mm2 liver area 25 � 2 28 � 2
Granuloma area/liver area, mm2/mm2 0.078 � 0.006 0.087 � 0.009
Eosinophils/granuloma area, mm2 12 � 1 10 � 1
Collagen area/liver area 0.132 � 0.023 0.112 � 0.014
AST indicates aspartate aminotransferase.
p16INK4a FUNCTION IN MACROPHAGES 2561BLOOD, 1 SEPTEMBER 2011 � VOLUME 118, NUMBER 9
For personal use only.on March 27, 2016. by guest www.bloodjournal.orgFrom
macrophages within the splenocyte preparation. By contrast,Ag-dependent restimulation with SEA showed a lower productionof both IL-4 and IFN� in splenocytes from p16�/� transplantedmice (supplemental Figure 6D-E), while IL-10 production wassimilar (supplemental Figure 6F), hence resulting in an unchangedIL-4/IFN� balance.33 Collectively, these results provide evidencefor a role of p16INK4a in AAM� skewing in vivo.
p16INK4 deficiency in macrophages decreases STAT1 andIKK�,� signaling without altering STAT6 phosphorylation
To unravel the mechanisms underlying the phenotype inducedby p16INK4a deficiency, the microarray data were analyzed toidentify which pathways are affected. Interestingly, many geneswith a decreased expression in p16�/� BMDMs respond toactivation of the IFN� and NF-�B inflammatory signalingpathways (supplemental Figure 9), of which STAT1 and IKK�,�,respectively, are crucial components. Detailed examination ofthese gene expression patterns revealed that expression levelswere comparable (Cxcl10, ifi27, ifi203, ifi47, ifit1, Ccl5, Irak3,Birc3, and Ly6e) or even lower (ifi44, CD55, and NF�Bia) inp16�/� BMDMs under basal conditions compared with IL-4–polarized p16�/� AAM� (Figure 6). Moreover, most inflamma-tory genes were more strongly inhibited in IL-4–induced p16�/�
AAM� than in p16�/� AAM� (Figures 6 and 2D), suggesting anadditive effect of p16INK4a deficiency on IL-4 treatment. Bycontrast, such a synergism could not be observed with respect tothe AAM� marker genes because many of these AAM� markergenes responded similarly in IL-4–induced p16�/� and p16�/�
AAM� (supplemental Table 4, Figure 2D). Collectively, these
data show that the pathways controlling IFN�, NF-�B, and IL-4signaling are affected by p16INK4a deficiency in macrophages.
To determine whether the observed differences were related toindirect effects through pRb, BMDMs were treated with CINK4, apotent inhibitor of pRb hyperphosphorylation.34 CINK4 treatmentresulted in a dose-dependent decrease of cyclin D mRNA levelsboth in p16�/� and p16�/� BMDMs (supplemental Figure 3).Furthermore, treatment with CINK4 in combination with IFN� didnot influence the different phenotype between the 2 genotypes(supplemental Figure 10), thus excluding a role for pRb therein.
STAT6 is the major transcription factor activated by IL-4.Surprisingly, IL-4 treatment induced STAT6 phosphorylation to asimilar extent in p16�/� and p16�/� BMDMs (Figure 7A). Bycontrast, IFN�-induced STAT1 phosphorylation (Figure 7B) andLPS-induced IKK�,� phosphorylation were markedly diminished(Figure 7C) in p16�/� versus p16�/� BMDMs. Interestingly, I�B�protein levels were increased in p16�/� BMDMs under basalconditions (Figure 7C). However, phosphorylation of p38, anotherdownstream target of TLR-4, was not different between p16�/� andp16�/� BMDMs (Figure 7C), suggesting that p16INK4a deficiencyspecifically alters the NF-�B response.
Several genes coding for proteins with acetyl transferaseactivity were induced in p16�/� BMDMs (Table 2). Acetylation ofSTAT1 and IKK�,� inhibits their phosphorylation and therebytheir transcriptional activity.35,36 Immunoprecipitation of acetylatedproteins revealed that STAT1 and IKK�,� were acetylated to ahigher extent in p16�/� than in p16�/� BMDMs (Figure 7D). Toidentify whether STAT1 acetylation is the cause or the consequenceof decreased STAT1 phosphorylation, JAK2 phosphorylation,
Figure 5. Hematopoietic deficiency of p16INK4a exac-erbates hepatic alternative macrophage activation ofS mansoni–infected mice. Hepatic mRNA expressionfrom mice transplanted with either p16�/� or p16�/� BMwhich were not infected (naive) or infected withS mansoni, 9 weeks postinfection. mRNA expression ofthe macrophage markers F4/80 (A), CD68 (B), andCD14 (C); AAM� markers Ym1/2 (D), Fizz1 (E), andMgl2 (F) was quantified by QPCR and expressed as foldincrease compared with their respective naive controls.Data were obtained from n 10 naive and n 14 infectedmice per genotype. Hepatic granulomas were isolated bylaser-capture microdissection and mRNA expression of theAAM� markers Ym1/2 (G), Fizz1 (H), and Mgl2 (I) wasquantified by QPCR. Statistically significant differences areindicated (t test; **P � .01 and *P � .05).
2562 CUDEJKO et al BLOOD, 1 SEPTEMBER 2011 � VOLUME 118, NUMBER 9
For personal use only.on March 27, 2016. by guest www.bloodjournal.orgFrom

which is upstream in this pathway, was assessed. JAK2 phosphory-lation was less pronounced under basal conditions and afteractivation with IFN� in p16�/� BMDMs (Figure 7E). Becauseacetylation of STAT1 can interfere with NF-�B signaling36 andboth pathways act synergistically,37 the decreased STAT1 andNF-�B signaling and their increased acetylation probably resultsfrom decreased JAK2 phosphorylation.
Discussion
We identify a novel role for the tumor suppressor proteinp16INK4a in the regulation of the macrophage phenotype in vitroand in vivo. p16INK4a deficiency promotes a STAT6-independentIL-4–induced macrophage polarization, whereas LPS and IFN�responses are lower. These actions of p16INK4a deficiency likelyoccur through modification of the JAK2-STAT1 and NF-�Bpathways.
Although p16INK4a is mainly known as a cell-cycle inhibitor, cellcycle and macrophage maturation were unaffected in the absenceof p16INK4a, and an E2F target gene signature,38,39 which would beexpected to be increased by p16INK4a deficiency, could not beidentified in p16�/� BMDMs. Because p15INK4b is a critical tumorsuppressor in the absence of p16INK4a 40 and because an increase ofp15INK4b expression was observed in p16�/� BMDMs, a compensa-tory mechanism between these CDKI might have taken place withrespect to cell-cycle control.
Interestingly, p16INK4a has been previously shown to modulatetranscriptional activities of the proinflammatory transcription fac-tors NF-�B and AP-1. Via its ankyrin motifs, p16INK4a binds c-jun41
and NF-�B,42 inhibiting their activity in several immortalized celllines. However, this mechanism does not appear to play a role inprimary murine macrophages, where the loss of p16INK4a hasanti-inflammatory effects. Another CDKI, p21WAF1/CIP1, was alsoreported to modulate inflammation without altering cell cycle ordifferentiation. However, in contrast with p16INK4a deficiency,p21WAF1/CIP1 deficiency in macrophages resulted in a higher sensitiv-ity to inflammatory stimuli.43,44 In addition, p21WAF1/CIP1 expressionwas unchanged in p16�/� BMDMs.
In a model of AAM� differentiation in vivo, infection with thehelminth S mansoni resulted in equal Th2-associated responses inp16�/� and p16�/� BM transplanted mice. These observationsindicate unaltered T-lymphocyte function, in line with the lowexpression levels of p16INK4a in these cells. By contrast, AAM�markers were significantly increased in the livers and in hepaticgranulomas of p16�/� BM transplanted animals, probably becauseof a cell-autonomous activity of p16INK4a in macrophages. Becausetotal BM was transplanted, a contribution of other hematopoietic celltypes to this cell-autonomous macrophage response cannot be excluded.However, because expression of the measured markers was alsoup-regulated in the macrophage-rich granulomas and because AAM�markers colocalized with macrophages in the granuloma, the effects are,at least in part, because of p16INK4a deficiency in the macrophages.Surprisingly, different AAM� markers did not strictly colocalize,suggesting the existence of different macrophage populations in definedregions of the granuloma, which could be because of different gradientsof stimuli within the granuloma.
Microarray analysis showed that p16�/� BMDMs underbasal conditions display a gene signature resembling that ofIL-4–induced macrophage polarization,27 with the exception of
Figure 6. p16INK4a deficiency results in an alteration of STAT1 and NF-�B signaling. Representation of the relative microarray intensity values from a selection ofdown-regulated genes in p16�/� and p16�/� BMDM with or without polarization (AAM�) by 15 ng/mL IL-4 from day 0 of differentiation. Statistically significant differences areindicated (a: P � .05 compared with p16�/� BMDM; b: P � .05 compared with p16�/� BMDM; c: P � .05 compared with p16�/� AAM�).
p16INK4a FUNCTION IN MACROPHAGES 2563BLOOD, 1 SEPTEMBER 2011 � VOLUME 118, NUMBER 9
For personal use only.on March 27, 2016. by guest www.bloodjournal.orgFrom

the AAM� marker genes Ym1/2 and Fizz1. Moreover, p16�/�
BMDMs were more responsive to IL-4 activation and displayedweaker proinflammatory responses to IFN� and LPS. Impor-tantly, our data show that p16INK4a deficiency does not influencepRb-mediated activation of the E2F transcription factor. Thedecreased IFN�-induced phosphorylation of STAT1 and theLPS-induced IKK�,� phosphorylation in p16�/� BMDMs prob-ably account for the attenuated inflammatory responses. De-creased IKK�,� phosphorylation combined with increasedI�B� protein levels correlated with the profound decrease ofNF-�B target gene expression in p16�/� BMDMs. Because acluster of genes coding for proteins with acetyl transferase
activity was induced in p16�/� BMDMs and because STAT136
and IKK�,�35 acetylation reduces their ability to be phosphory-lated, the decrease in STAT1 and IKK�,� phosphorylation mayresult from increased acetylation of these proteins in p16�/�
BMDMs. The acetylation-phosphorylation balance may beinfluenced by alterations upstream in the signaling pathway.Because phosphorylation of JAK2, the first component to bephosphorylated on receptor dimerization,45 was decreased, itappears that the p16INK4a deficiency-induced defect in JAK2phosphorylation may be implicated in the reduced Stat1 phos-phorylation. The increased acetylation status may be a conse-quence of this mechanism, contributing to the phenotype of
Figure 7. p16INK4a deficiency diminishes JAK2-STAT1 and NF-�Bsignaling and increases acetylation of STAT1 and IKK�,�. Total proteinextracts of p16�/� and p16�/� BMDM treated with 15 ng/mL IL-4(A), 2.5 ng/mL IFN� (B) and (E), and 100 ng/mL LPS (C) for the timesindicated. Western blots were performed with the indicated Abs. (D) Totalprotein extracts from p16�/� and p16�/� BMDM treated with 2.5 ng/mLIFN� for 5 hours were then immunoprecipitated (IP) with an anti-Acetyl-LysAb and then immunoblotted with an anti-STAT1 Ab or with an anti-IKK�,�Ab, as indicated. Input corresponds to 5% of total protein extract used forimmunoprecipitation.
2564 CUDEJKO et al BLOOD, 1 SEPTEMBER 2011 � VOLUME 118, NUMBER 9
For personal use only.on March 27, 2016. by guest www.bloodjournal.orgFrom

p16�/� BMDMs. Moreover, JAK2 phosphorylation was lesspronounced already under basal culture conditions, in line withthe observation that p16INK4a deficiency confers a lower basalinflammatory phenotype, which is further exacerbated on stimu-lation. Because p16INK4a deficiency alters the IFN� responseearly in the pathway and TLR4 signaling only at the level ofNF-�B, but not, for example, at the level of p38, p16INK4a
deficiency thus likely acts directly on the IFN� and indirectly onthe NF-�B signaling pathway.
Surprisingly, STAT6 phosphorylation, the major transcriptionfactor for IL-4–mediated signaling response, was comparable inBMDMs of both genotypes, although IL-4–responsive genes wereincreased in p16�/� BMDMs, suggesting that other pathways areinvolved. It has been shown that STAT1 activation antagonizesexpression of STAT6-induced AAM� marker genes46,47 and thatdecreased STAT1 phosphorylation can result in an increase ofAAM� marker genes48 by several molecular mechanisms. First,IL-4 action is inhibited by STAT1 activation through inhibition ofSTAT6 phosphorylation and its subsequent nuclear translocation inhuman monocytes.49 However, p16INK4a deficiency did not modifySTAT6 phosphorylation. Second, IFN� inhibits STAT6 activationby inducing the expression of the suppressor of cytokine signaling1 (SOCS1) in macrophages.46 Although SOCS1 mRNA levels werenot different between p16�/� and p16�/� BMDMs (data notshown), we cannot exclude its involvement via posttranscriptionalmechanisms. Third, STAT1 and STAT6 have been shown tocompete for binding the same promoters47 because decreasedSTAT1 activity increases binding of STAT6 to the promoters ofseveral of its target genes. To our knowledge, an effect of NF-�Bsignaling on STAT6-dependent gene expression has not yet beendemonstrated. Therefore, it is possible that the AAM�-like pheno-type of p16�/� BMDMs is secondary to the decreased CAM�-associated response via JAK2-STAT1.
In conclusion, our results identify p16INK4a as a novelmodulator of macrophage polarization and show that p16INK4a
deficiency skews macrophages toward an IL-4–like phenotype.Because the CDKN2A locus is predictive for the risk of T2D andCVD,14,15 in which macrophage polarization plays a role,28,50 the
role of p16INK4a in the control of macrophage function could beone mechanism contributing to the association of this locus withthese diseases.
Acknowledgments
P. Krimpenfort provided p16INK4a-deficient mice. Discussions withG. Raes, J. Vanginderachter, and M. de Winther are greatly acknowl-edged. The authors thank E. Vallez for mouse breeding, J. Brozek(Genfit SA, Loos, France) for microarray raw data analysis, andT. Coevoet, N. Jouy, and A. Lucas for technical assistance.
This work was supported by the Fondation pour la RechercheMedicale (DCV20070409276, B.S.), the EFSD/GSK Program2009, the Cost Action (BM0602), and the Conseil regional NordPas-de-Calais and FEDER. C.C. was supported by a doctoralfellowship from the Nouvelle Societe Francaise d’Atherosclerose/Schering-Plough/MSD. K.W. was supported by a European FP7Marie Curie grant (PIEF-GA-2009-235221) and a European Athero-sclerosis Society grant.
The funders had no role in study design, data collection andanalysis, decision to publish, or preparation of the manuscript.
Authorship
Contribution: C.C. and K.W. designed and performed experiments,analyzed data, and wrote the manuscript. L.F., S.A.H., C.P., K.B.,E.B., J.V., S.F., and P.R. contributed to some of the experiments;A.T. and G.C. contributed to data interpretation; and D.D., B.S.,and R.P. contributed to data interpretation, manuscript preparation,and project supervision.
Conflict-of-interest disclosure: The authors declare no compet-ing financial interests.
Correspondence: Bart Staels, Inserm UR 1011, Institut Pasteurde Lille, 1 rue du Professeur Calmette, BP 245, Lille 59019,France; e-mail: [email protected].
References
1. Serrano M, Hannon GJ, Beach D. A new regula-tory motif in cell-cycle control causing specificinhibition of cyclin D/CDK4. Nature. 1993;366(6456):704-707.
2. Hannon GJ, Beach D. p15INK4B is a potentialeffector of TGF-beta-induced cell cycle arrest.Nature. 1994;371(6494):257-261.
3. Guan KL, Jenkins CW, Li Y, et al. Growth sup-pression by p18, a p16INK4/MTS1- andp14INK4B/MTS2-related CDK6 inhibitor, corre-lates with wild-type pRb function. Genes Dev.1994;8(24):2939-2952.
4. Chan FK, Zhang J, Cheng L, Shapiro DN, Winoto A.Identification of human and mouse p19, a novel
CDK4 and CDK6 inhibitor with homology to p16ink4.Mol Cell Biol. 1995;15(5):2682-268.
5. Hirai H, Roussel MF, Kato JY, Ashmun RA, Sherr CJ.Novel INK4 proteins, p19 and p18, are specific inhibi-tors of the cyclin D-dependent kinases CDK4 andCDK6. Mol Cell Biol. 1995;15(5):2672-2681.
6. Sharpless NE. INK4a/ARF: a multifunctional tu-mor suppressor locus. Mutat Res. 2005;576(1-2):22-38.
7. Alcorta DA, Xiong Y, Phelps D, Hannon G,Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in repli-cative senescence of normal human fibroblasts.Proc Natl Acad Sci U S A. 1996;93(24):13742-13747.
8. Hara E, Smith R, Parry D, Tahara H, Stone S,Peters G. Regulation of p16CDKN2 expressionand its implications for cell immortalization andsenescence. Mol Cell Biol. 1996;16(3):859-867.
9. Krishnamurthy J, Torrice C, Ramsey MR, et al.Ink4a/Arf expression is a biomarker of aging.J Clin Invest. 2004;114(9):1299-1307.
10. Nielsen GP, Stemmer-Rachamimov AO, Shaw J,Roy JE, Koh J, Louis DN. Immunohistochemicalsurvey of p16INK4A expression in normal humanadult and infant tissues. Lab Invest. 1999;79(9):1137-1143.
11. Zindy F, Quelle DE, Roussel MF, Sherr CJ. Ex-pression of the p16INK4a tumor suppressor ver-sus other INK4 family members during mouse
Table 2. Genes coding for proteins with acetyl transferase activity in p16INK4a-deficient macrophages
Probeset Symbol Description Fold change P
1435224_at CBP/p300 CREB binding protein 1.96 .0001
1426783_at Gcn5l2 GCN5 general control of amino acid synthesis-like 2 1.91 .00022
1448711_at Mcm3ap Mini chromosome maintenance-deficient 3–associated protein 1.55 .00890
1433692_at Nat10 N-acetyltransferase 10 1.65 .00018
1419213_at Nat6 N-acetyltransferase 6 2.11 .00008
1435842_at Nat8l N-acetyltransferase 8-like 1.32 .04600
1436580_at Hgsnat Heparan-alpha-glucosaminide N-acetyltransferase 1.38 .01800
p16INK4a FUNCTION IN MACROPHAGES 2565BLOOD, 1 SEPTEMBER 2011 � VOLUME 118, NUMBER 9
For personal use only.on March 27, 2016. by guest www.bloodjournal.orgFrom

development and aging. Oncogene. 1997;15(2):203-211.
12. Melzer D, Frayling TM, Murray A, et al. A commonvariant of the p16(INK4a) genetic region is asso-ciated with physical function in older people.Mech Ageing Dev. 2007;128(5-6):370-377.
13. Janzen V, Forkert R, Fleming HE, et al. Stem-cellageing modified by the cyclin-dependent kinaseinhibitor p16INK4a. Nature. 2006;443(7110):421-426.
14. McPherson R, Pertsemlidis A, Kavaslar N, et al. Acommon allele on chromosome 9 associated withcoronary heart disease. Science. 2007;316(5830):1488-1491.
15. Liu Y, Sanoff HK, Cho H, et al. INK4/ARF tran-script expression is associated with chromosome9p21 variants linked to atherosclerosis. PLoSONE. 2009;4(4):e5027.
16. Saxena R, Voight BF, Lyssenko V, et al. Genome-wide association analysis identifies loci for type 2diabetes and triglyceride levels. Science. 2007;316(5829):1331-1336.
17. Wei S, Lightwood D, Ladyman H, et al. Modula-tion of CSF-1-regulated post-natal developmentwith anti-CSF-1 antibody. Immunobiology. 2005;210(2-4):109-119.
18. Baffy G. Kupffer cells in non-alcoholic fatty liverdisease: the emerging view. J Hepatol. 2009;51(1):212-223.
19. Gordon S. Alternative activation of macrophages.Nat Rev Immunol. 2003;3(1):23-35.
20. Herbert DR, Holscher C, Mohrs M, et al. Alterna-tive macrophage activation is essential for sur-vival during schistosomiasis and downmodulatesT helper 1 responses and immunopathology. Im-munity. 2004;20(5):623-635.
21. Bellosta S, Via D, Canavesi M, et al. HMG-CoAreductase inhibitors reduce MMP-9 secretion bymacrophages. Arterioscler Thromb Vasc Biol.1998;18(11):1671-1678.
22. Dubois A, Deruytter N, Adams B, et al. Regulationof Th2 responses and allergic inflammationthrough bystander activation of CD8� T lympho-cytes in early life. J Immunol. 2010;185(2):884-891.
23. Van Ginderachter JA, Movahedi K, HassanzadehGhassabeh G, et al. Classical and alternative ac-tivation of mononuclear phagocytes: picking thebest of both worlds for tumor promotion. Immuno-biology. 2006;211(6-8):487-501.
24. Vats D, Mukundan L, Odegaard JI, et al. Oxida-tive metabolism and PGC-1beta attenuate mac-rophage-mediated inflammation. Cell Metab.2006;4(1):13-24.
25. Martinez FO, Gordon S, Locati M, Mantovani A.Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization:new molecules and patterns of gene expression.J Immunol. 2006;177(10):7303-7311.
26. Noel W, Raes G, Hassanzadeh Ghassabeh G,De Baetselier P, Beschin A. Alternatively acti-vated macrophages during parasite infections.Trends Parasitol. 2004;20(3):126-133.
27. Mantovani A, Sica A, Sozzani S, Allavena P,Vecchi A, Locati M. The chemokine system in di-verse forms of macrophage activation and polar-ization. Trends Immunol. 2004;25(12):677-686.
28. Bouhlel MA, Derudas B, Rigamonti E, et al.PPARgamma activation primes human mono-cytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6(2):137-143.
29. Katakura T, Miyazaki M, Kobayashi M, Herndon DN,Suzuki F. CCL17 and IL-10 as effectors that enablealternatively activated macrophages to inhibit thegeneration of classically activated macrophages.J Immunol. 2004;172(3):1407-1413.
30. Pearce EJ, M Kane C, Sun J, J Taylor J, McKee AS,Cervi L. Th2 response polarization during infectionwith the helminth parasite Schistosoma mansoni.Immunol Rev. 2004;201:117-126.
31. Wynn TA. Fibrotic disease and the T(H)1/T(H)2paradigm. Nat Rev Immunol. 2004;4(8):583-594.
32. Elliott DE, Boros DL. Schistosome egg antigen(s)presentation and regulatory activity by macro-phages isolated from vigorous or immunomodu-lated liver granulomas of Schistosoma mansoni-infected mice. J Immunol. 1984;132(3):1506-1510.
33. Mosmann TR, Coffman RL. TH1 and TH2 cells:different patterns of lymphokine secretion lead todifferent functional properties. Annu Rev Immu-nol. 1989;7:145-173.
34. Soni R, O’Reilly T, Furet P, et al. Selective in vivoand in vitro effects of a small molecule inhibitor ofcyclin-dependent kinase 4. J Natl Cancer Inst.2001;93(6):436-446.
35. Mittal R, Peak-Chew S, McMahon HT. Acetylationof MEK2 and I kappa B kinase (IKK) activationloop residues by YopJ inhibits signaling. Proc NatlAcad Sci U S A. 2006;103(49):18574-18579.
36. Kramer OH, Baus D, Knauer SK, et al. Acetyla-tion of Stat1 modulates NF-kappaB activity.Genes Dev. 2006;20(4):473-485.
37. Hiroi M, Ohmori Y. The transcriptional coactivatorCREB-binding protein cooperates with STAT1and NF-kappa B for synergistic transcriptionalactivation of the CXC ligand 9/monokine induced
by interferon-gamma gene. J Biol Chem. 2003;278(1):651-660.
38. Vernell R, Helin K, Muller H. Identification of tar-get genes of the p16INK4A-pRB-E2F pathway.J Biol Chem. 2003;278(46):46124-46137.
39. Stanelle J, Stiewe T, Theseling CC, Peter M,Putzer BM. Gene expression changes in re-sponse to E2F1 activation. Nucleic Acids Res.2002;30(8):1859-1867.
40. Krimpenfort P, Ijpenberg A, Song J, et al.p15Ink4b is a critical tumour suppressor in theabsence of p16Ink4a. Nature. 2007;448(7156):943-946.
41. Choi BY, Choi HS, Ko K, et al. The tumor sup-pressor p16(INK4a) prevents cell transformationthrough inhibition of c-Jun phosphorylation andAP-1 activity. Nat Struct Mol Biol. 2005;12(8):699-707.
42. Wolff B, Naumann M. INK4 cell cycle inhibitorsdirect transcriptional inactivation of NF-kappaB.Oncogene. 1999;18(16):2663-2666.
43. Scatizzi JC, Mavers M, Hutcheson J, et al. TheCDK domain of p21 is a suppressor of IL-1beta-mediated inflammation in activated macro-phages. Eur J Immunol. 2009;39(3):820-825.
44. Trakala M, Arias CF, García MI, et al. Regulationof macrophage activation and septic shock sus-ceptibility via p21(WAF1/CIP1). Eur J Immunol.2009;39(3):810-819.
45. Shuai K, Liu B. Regulation of JAK-STAT signal-ling in the immune system. Nat Rev Immunol.2003;3(11):900-911.
46. Dickensheets HL, Venkataraman C, Schindler U,Donnelly RP. Interferons inhibit activation ofSTAT6 by interleukin 4 in human monocytes byinducing SOCS-1 gene expression. Proc NatlAcad Sci U S A. 1999;96(19):10800-10805.
47. Ohmori Y, Hamilton TA. IL-4-induced STAT6 sup-presses IFN-gamma-stimulated STAT1-depen-dent transcription in mouse macrophages. J Im-munol. 1997;159(11):5474-5482.
48. Porta C, Rimoldi M, Raes G, et al. Tolerance andM2 (alternative) macrophage polarization are re-lated processes orchestrated by p50 nuclear fac-tor kappaB. Proc Natl Acad Sci U S A. 2009;106(35):14978-14983.
49. Dickensheets HL, Donnelly RP. Inhibition of IL-4-inducible gene expression in human monocytesby type I and type II interferons. J Leukoc Biol.1999;65(3):307-312.
50. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH,et al. Macrophage-specific PPARgamma controlsalternative activation and improves insulin resis-tance. Nature. 2007;447(7148):1116-1120.
2566 CUDEJKO et al BLOOD, 1 SEPTEMBER 2011 � VOLUME 118, NUMBER 9
For personal use only.on March 27, 2016. by guest www.bloodjournal.orgFrom

online June 2, 2011 originally publisheddoi:10.1182/blood-2010-10-313106
2011 118: 2556-2566
Anne Tailleux, Giulia Chinetti-Gbaguidi, David Dombrowicz, Bart Staels and Réjane PaumelleKadiombo Bantubungi, Emmanuel Bouchaert, Jonathan Vanhoutte, Sébastien Fleury, Patrick Remy, Céline Cudejko, Kristiaan Wouters, Lucía Fuentes, Sarah Anissa Hannou, Charlotte Paquet, proinflammatory signaling in macrophages
induced polarization and inhibits− deficiency promotes IL-4INK4ap16
http://www.bloodjournal.org/content/118/9/2556.full.htmlUpdated information and services can be found at:
(545 articles)Phagocytes, Granulocytes, and Myelopoiesis Articles on similar topics can be found in the following Blood collections
http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://www.bloodjournal.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://www.bloodjournal.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036.Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society
For personal use only.on March 27, 2016. by guest www.bloodjournal.orgFrom
Copyright © 2022 FDOKUMEN




















