
Small molecule inhibitors of Ago2 decrease Venezuelan equine encephalitis virus replication
12
Small molecule inhibitors of Ago2 decrease Venezuelan equine encephalitis virus replication Cathaleen Madsen a,1 , Idris Hooper a,1 , Lindsay Lundberg a , Nazly Shafagati a , Alexandra Johnson a , Svetlana Senina a , Cynthia de la Fuente a , Lisa I. Hoover b,c , Brenda L. Fredricksen b,c,2 , Jonathan Dinman c , Jonathan L. Jacobs d , Kylene Kehn-Hall a,⇑ a National Center for Biodefense and Infectious Diseases, School of Systems Biology, George Mason University, Manassas, VA, USA b Maryland Pathogen Research Institute, College Park, MD, 20742, USA c Department of Cell Biology and Molecular Genetics, University of Maryland, College Park, MD, 20742, USA d MRI Global, 1330 Piccard Drive #101, Rockville, MD, 20850, USA article info Article history: Received 19 May 2014 Revised 1 October 2014 Accepted 7 October 2014 Available online 18 October 2014 Keywords: Venezuelan equine encephalitis virus Ago2 miRNA Acriflavine Therapeutic Alphavirus abstract Venezuelan equine encephalitis virus (VEEV) is classified as a Category B Select Agent and potential bio- terror weapon for its severe disease course in humans and equines and its potential for aerosol transmis- sion. There are no current FDA licensed vaccines or specific therapies against VEEV, making identification of potential therapeutic targets a priority. With this aim, our research focuses on the interactions of VEEV with host microRNA (miRNA) machinery. miRNAs are small non-coding RNAs that act as master regula- tors of gene expression by downregulating or degrading messenger RNA, thus suppressing production of the resultant proteins. Recent publications implicate miRNA interactions in the pathogenesis of various viral diseases. To test the importance of miRNA processing for VEEV replication, cells deficient in Ago2, an important component of the RNA-induced silencing complex (RISC), and cells treated with known Ago2 inhibitors, notably acriflavine (ACF), were utilized. Both conditions caused decreased viral replica- tion and capsid expression. ACF treatment promoted increased survival of neuronal cells over a non-trea- ted, infected control and reduced viral titers of fully virulent VEEV as well as Eastern and Western Equine Encephalitis Viruses and West Nile Virus, but not Vesicular Stomatitis Virus. ACF treatment of VEEV TC- 83 infected mice resulted in increased in vivo survival, but did not affect survival or viral loads when mice were challenged with fully virulent VEEV TrD. These results suggest that inhibition of Ago2 results in decreased replication of encephalitic alphaviruses in vitro and this pathway may be an avenue to explore for future therapeutic development. Ó 2014 Elsevier B.V. All rights reserved. 1. Introduction Venezuelan equine encephalitis virus (VEEV) is a new-world alphavirus that is a significant pathogen of humans and equines. The virus is normally spread by an arthropod vector, but can also be transmitted by aerosol exposure. The disease course in humans ranges from an acutely febrile yet mild illness (Aguilar et al., 2011) to a debilitating condition clinically indistinguishable from Dengue (Casals et al., 1943), and in rare cases can progress to coma or death (Zacks and Paessler, 2010). The disease course is more severe and frequently fatal in equines. Due to its severity and ability to be spread by aerosol exposure, VEEV is classified as a Category B Select Agent and potential bioterror weapon. There is currently no FDA-approved vaccine or specific treatment against VEEV, mak- ing identification of therapeutic targets a priority. Recent research has identified multiple microRNAs (miRNA) that are differentially regulated during VEEV infection of mouse brain (Bhomia et al., 2010), suggesting that such interactions could be important in a human-cell model of VEEV pathogenesis. miRNA are small, non-coding strands of RNA approximately 22 nucleotides in length, which can serve as master regulators of the cell by directly interacting with, and causing downregulation or degradation of target messenger RNA (mRNA). miRNA regulation was first described in Caenorhabditis elegansin 2001 http://dx.doi.org/10.1016/j.antiviral.2014.10.002 0166-3542/Ó 2014 Elsevier B.V. All rights reserved. ⇑ Corresponding author at: National Center for Biodefense and Infectious Diseases, School of Systems Biology, George Mason University, Biomedical Research Laboratory, 10650 Pyramid Place, Manassas, VA 20110, USA. Tel.: +1 703 993 8869; fax: +1 703 993 4280. E-mail address: [email protected] (K. Kehn-Hall). 1 The two first authors contributed equally to this research. 2 Current address: AIDS Review Branch, National Institute of Allergy and Infectious Diseases, National Institutes of Health, DHHS, Bethesda, MD 20817, USA. Antiviral Research 112 (2014) 26–37 Contents lists available at ScienceDirect Antiviral Research journal homepage: www.elsevier.com/locate/antiviral
Transcript of Small molecule inhibitors of Ago2 decrease Venezuelan equine encephalitis virus replication

Antiviral Research 112 (2014) 26–37
Contents lists available at ScienceDirect
Antiviral Research
journal homepage: www.elsevier .com/locate /ant iv i ra l
Small molecule inhibitors of Ago2 decrease Venezuelan equineencephalitis virus replication
http://dx.doi.org/10.1016/j.antiviral.2014.10.0020166-3542/� 2014 Elsevier B.V. All rights reserved.
⇑ Corresponding author at: National Center for Biodefense and InfectiousDiseases, School of Systems Biology, George Mason University, Biomedical ResearchLaboratory, 10650 Pyramid Place, Manassas, VA 20110, USA. Tel.: +1 703 993 8869;fax: +1 703 993 4280.
E-mail address: [email protected] (K. Kehn-Hall).1 The two first authors contributed equally to this research.2 Current address: AIDS Review Branch, National Institute of Allergy and Infectious
Diseases, National Institutes of Health, DHHS, Bethesda, MD 20817, USA.
Cathaleen Madsen a,1, Idris Hooper a,1, Lindsay Lundberg a, Nazly Shafagati a, Alexandra Johnson a,Svetlana Senina a, Cynthia de la Fuente a, Lisa I. Hoover b,c, Brenda L. Fredricksen b,c,2, Jonathan Dinman c,Jonathan L. Jacobs d, Kylene Kehn-Hall a,⇑a National Center for Biodefense and Infectious Diseases, School of Systems Biology, George Mason University, Manassas, VA, USAb Maryland Pathogen Research Institute, College Park, MD, 20742, USAc Department of Cell Biology and Molecular Genetics, University of Maryland, College Park, MD, 20742, USAd MRI Global, 1330 Piccard Drive #101, Rockville, MD, 20850, USA
a r t i c l e i n f o a b s t r a c t
Article history:Received 19 May 2014Revised 1 October 2014Accepted 7 October 2014Available online 18 October 2014
Keywords:Venezuelan equine encephalitis virusAgo2miRNAAcriflavineTherapeuticAlphavirus
Venezuelan equine encephalitis virus (VEEV) is classified as a Category B Select Agent and potential bio-terror weapon for its severe disease course in humans and equines and its potential for aerosol transmis-sion. There are no current FDA licensed vaccines or specific therapies against VEEV, making identificationof potential therapeutic targets a priority. With this aim, our research focuses on the interactions of VEEVwith host microRNA (miRNA) machinery. miRNAs are small non-coding RNAs that act as master regula-tors of gene expression by downregulating or degrading messenger RNA, thus suppressing production ofthe resultant proteins. Recent publications implicate miRNA interactions in the pathogenesis of variousviral diseases. To test the importance of miRNA processing for VEEV replication, cells deficient in Ago2,an important component of the RNA-induced silencing complex (RISC), and cells treated with knownAgo2 inhibitors, notably acriflavine (ACF), were utilized. Both conditions caused decreased viral replica-tion and capsid expression. ACF treatment promoted increased survival of neuronal cells over a non-trea-ted, infected control and reduced viral titers of fully virulent VEEV as well as Eastern and Western EquineEncephalitis Viruses and West Nile Virus, but not Vesicular Stomatitis Virus. ACF treatment of VEEV TC-83 infected mice resulted in increased in vivo survival, but did not affect survival or viral loads when micewere challenged with fully virulent VEEV TrD. These results suggest that inhibition of Ago2 results indecreased replication of encephalitic alphaviruses in vitro and this pathway may be an avenue to explorefor future therapeutic development.
� 2014 Elsevier B.V. All rights reserved.
1. Introduction
Venezuelan equine encephalitis virus (VEEV) is a new-worldalphavirus that is a significant pathogen of humans and equines.The virus is normally spread by an arthropod vector, but can alsobe transmitted by aerosol exposure. The disease course in humansranges from an acutely febrile yet mild illness (Aguilar et al., 2011)to a debilitating condition clinically indistinguishable from Dengue
(Casals et al., 1943), and in rare cases can progress to coma or death(Zacks and Paessler, 2010). The disease course is more severe andfrequently fatal in equines. Due to its severity and ability to bespread by aerosol exposure, VEEV is classified as a Category BSelect Agent and potential bioterror weapon. There is currentlyno FDA-approved vaccine or specific treatment against VEEV, mak-ing identification of therapeutic targets a priority. Recent researchhas identified multiple microRNAs (miRNA) that are differentiallyregulated during VEEV infection of mouse brain (Bhomia et al.,2010), suggesting that such interactions could be important in ahuman-cell model of VEEV pathogenesis.
miRNA are small, non-coding strands of RNA approximately 22nucleotides in length, which can serve as master regulators of thecell by directly interacting with, and causing downregulationor degradation of target messenger RNA (mRNA). miRNAregulation was first described in Caenorhabditis elegansin 2001

C. Madsen et al. / Antiviral Research 112 (2014) 26–37 27
(Lau et al., 2001; Lee and Ambros, 2001), and appears to be evolu-tionarily conserved among eukaryotes. Canonical processing ofmiRNA begins with transcription from the host genome andassembly of a primary miRNA containing a stem-loop or hairpinstructure, which is processed from the primary transcript by a ser-ies of enzymes to produce an asymmetrical miRNA duplex (Cullen,2005). One strand associates with Argonaute (Ago) proteins and isincorporated into the RNA-induced silencing complex (RISC),which in turn associates with a target mRNA. The other strand isdegraded. Perfect complementarity targets the mRNA for degrada-tion, while imperfect complementarity has inhibitory activity,reducing production of the encoded protein (Cullen, 2005). Manyviruses have shown significant interaction with host miRNA,including Epstein–Barr Virus (Lin et al., 2010), Pseudorabies Virus(Wu et al., 2012), and Rabies Virus (Zhao et al., 2012); the last ofwhich has been shown, as with VEEV, to affect host miRNA expres-sion in the brains of infected mice.
Viruses can also make use of miRNA for positive regulation, ashas been well-studied with Hepatitis C virus (HCV), which uses aliver-specific miRNA called miR-122 to enhance replication byenabling a more favorable association between viral RNA and thehost cell ribosomes (Henke et al., 2008; Jopling et al., 2005,2006). More recent research showed a complex interactionbetween uncapped RNA, miR-122, and the viral internal ribosomeentry site (IRES), in which translation activation is mediated by thepresence of Ago2, a component of the RISC (Roberts et al., 2011).While the complete mechanism remains unknown, it is clear thatmiR-122 provides both a valuable biomarker and a viable thera-peutic target for the treatment of HCV infection.
Based on these studies, it was hypothesized that VEEV interactswith cellular miRNA and miRNA processing machinery duringinfection of mammalian cell lines. To test this hypothesis, cellsdeficient in Ago2, a key miRNA processing enzyme, were infectedwith VEEV. The absence of Ago2 produced a decrease in viral geno-mic copies as monitored by qRT-PCR, as well as a decrease in rep-lication as measured by plaque assay. Treatment with a knownAgo2 inhibitor, acriflavine (ACF), inhibited viral replication. Theeffects of ACF on viral transcription, protein expression, and path-ogenicity during VEEV infection, as well as its inhibitory effects onother viruses are presented herein.
2. Materials and methods
2.1. Cell culture
Mouse embryonic fibroblasts (MEFs), U87MG, and Vero cellswere cultured in Dulbecco’s modified Eagle’s media (DMEM), sup-plemented and maintained as described in Lundberg et al. (2013).MEFs deficient in Ago2 (Ago2�/� MEFs) were obtained through Dr.G. Hannon of Cold Spring Harbor (Liu et al., 2004). AP-7 olfactoryneuronal cells, a generous gift of Dr. D.E. Griffin (Johns HopkinsUniversity), were cultured as described by Amaya et al. (2014).
2.2. Viruses
VEEV strains Trinidad Donkey (TrD), Mena II (Mena), 3880, andTC-83 were obtained from BEI Resources (Manassas, VA). The TrDstrain, considered the model organism for the fully virulent strains,is serological subtype IAB and is associated with periodic epidemicand epizootic outbreaks. The Mena and 3880 strains are subtypesID and IE, respectively, and, although fully virulent, are not associ-ated with such outbreaks (Powers et al., 1997). The TC-83 virus is alive attenuated vaccine derivative of the TrD strain propagated by83 serial passages in guinea pig heart cells (Berge et al., 1961),resulting in 12 nucleotide substitutions which confer attenuation
principally through changes within the 50-noncoding region andE2 envelope glycoprotein (Kinney et al., 1993). The TC-83 reportervirus, VEEV-GFP, was provided by Dr. I. Frolov (Atasheva et al.,2010) at the University of Alabama. Working stocks of WNV-NYstrain 3356 were generated as previously described (Hussmannet al., 2013) All work with VEEV-TrD, VEEV-Mena, VEEV-3880,WEEV, EEEV, and WNV-NY was performed at BSL-3.
2.3. Protein extraction
Cell pellets were lysed with 50 ll clear lysis buffer [50 mM Tris–HCl (pH 7.5), 120 mM NaCl, 5 mM EDTA, 0.5% NP-40, 50 mM NaF,0.2 mM Na3VO4, 1 mM DTT, and one complete protease cocktailtablet/50 mL]. Protein concentration was determined colorimetri-cally by a standard curve, using Bradford reagent (Sigma) and aBeckman-Coulter DTX-800 Multimode Detector/Multimode Analy-sis Software. For the ACF treatment assays, cell lysates were col-lected in blue lysis buffer as described in Lundberg et al. (2013).
2.4. Cell viability
Following treatment, cells were treated with the Celltiter Gloreagent (Promega) and viability was determined via fluorescenceas compared to the control sample, using a Beckman-CoulterDTX-800 Multimode Detector with Multimode Analysis Softwareplatform.
2.5. Western blot
Protein samples of 50 lg (clear lysis buffer) or 20 ll (blue lysisbuffer) were separated, transferred, and subsequently processed asdescribed in Austin et al. (2012), using a 5% bovine serum albuminsolution for loss of Ago2 assays, and 5% milk for ACF treatmentassays. Both the polyclonal Anti-Venezuelan equine encephalitisvirus, TC-83 (Subtype IA/B) Capsid Protein (antiserum, Goat), NR-9403 and polyclonal Anti-Venezuelan equine encephalitis virus,TC-83 (Subtype IA/B) Glycoprotein (antiserum, Goat) antibodieswere obtained through the NIH Biodefense and Emerging Infec-tions Research Resources Repository. Rabbit monoclonal antibod-ies to Ago2 were obtained from Abcam and Cell SignalingTechnologies. Beta actin HRP conjugated monoclonal antibodywas used as a loading control and was obtained from Abcam.
2.6. Plaque assays
Viral supernatants were serially diluted, and applied at 400 llin duplicate to confluent cultures of Vero cells in 6-well plates,or 200 ll to confluent cultures of Vero cells in 12-well plates, for1 h prior to immobilization. Immobilization and staining were per-formed as described in Narayanan et al. (2012) for neutral redstaining, or Kehn-Hall et al. (2012) for crystal violet staining. Pla-ques were assessed at two days post infection. For WNV-NY, mon-olayers of Vero cells in six-well plates were washed once withDulbecco’s phosphate buffered saline (DPBS) (Hyclone) followedby the addition of serial dilutions of viral samples (200 ll). Thecells were incubated in a 5% CO2 incubator for 1 h at 37 �C withrocking, the inocula removed, and a 0.9% agarose-complete DMEMoverlay added. Cell monolayers were incubated for 48 h and a sec-ond overlay of agarose-complete DMEM containing 0.003% neutralred (MP Biomedicals) was added. Plaques were counted at days 3and 4.
2.7. qRT-PCR
U87MG cells were infected with VEEV at an MOI of 0.1 andsupernatants collected after 24 h. Viral RNA was extracted using

28 C. Madsen et al. / Antiviral Research 112 (2014) 26–37
Ambion’s MagMax viral RNA extraction kit or Qiagen’s RNeasy kitand quantitated using q-RT-PCR with primers and probe for nucle-otides 7931–8005 of VEEV TC-83, as previously described (Kehn-Hall et al., 2012). qRT-PCR assays were performed using Invitro-gen’s RNA UltraSense™ One-Step Quantitative RT-PCR System.TC-83 RNA was amplified using the using an ABI Prism 7000. Theabsolute quantification was calculated based on the thresholdcycle (Ct) relative to the standard curve.
2.8. Flow cytometry
For the Ago2 assay, wild-type or Ago2�/� MEF cells were platedat 2.5 � 105 to 5.0 � 105 cells/well in 12-well plates and incubatedovernight prior to infection of triplicate samples of with VEEV-GFPat an MOI of 0.1 or 1.0. Samples were collected at 18 or 24 h postinfection. For the ACF assay, U87MG cells were plated at 5 � 105
in 12-well plates and treated with 2.5 lM or 1.25 lM of ACF orPLL. DMSO was included as a control. Cells were infected withVEEV-GFP, at an MOI of 1.0 and collected at 24 h post infection.All above samples were processed as described in Lundberg et al.(2013). Cells were analyzed on an Accuri C6 Flow Cytometer usinga CFlow Plus analysis platform to detect GFP expression.
2.9. Drug information and viability assays
ACF (acriflavine, A8126), PLL (poly-L-lysine, P6516), ATA (aurin-tricarboxylic acid, A1895), OXD (oxidopamine hydrochloride,H4381) and SUR (suramin, S2671) were obtained from Sigma. Allcompounds were dissolved in DMSO, with the exception of SUR,which was dissolved in deionized water. U87MG cells were seededat 5 � 104 in a 96-well white clear bottom plate, pre-treated withappropriate compounds for 24 h and then assayed with CelltiterGlo (Promega) as previously described. Results were confirmedthrough the MTT assay (Life Technologies) and evaluated colori-metrically at 570 nm using a Beckman-Coulter DTX-800 Multi-mode Detector/Multimode Analysis Software. All drugs weretested in triplicate, with concentrations of 10 lM, 5 lM, 2.5 lMand 1.25 lM for ACF and PLL, and 50 lM, 25 lM, 12.5 lM,6.25 lM and 3.125 lM for ATA, OXD, and SUR.
2.10. Time of addition analysis
U87MG cells were plated at 5 � 104 in a 96-well plate and con-trols pretreated with ACF (2.5 lM) for 2 h with DMSO used as anegative control. Afterwards, cells were infected with TC-83 (MOI0.1) for 1 h, and washed with PBS. Drug media was reapplied tocontrols and fresh media applied to experimental cultures. ACFand DMSO were added at desired time points. Viral supernatantswere collected 24 h post-infection and plaque assays performedto determine viral titer.
2.11. IC50 assay
U87MG cells were plated at 2.5 � 104 in a 96-well plate andincubated overnight at standard conditions prior to treatment.ACF was prepared at a top dose of 2.5 lM and serial dilutions of1:5. Cells were pre-treated with DMSO or ACF for 2 h prior to infec-tion at MOI 0.1 for 1 h as described above. After infection, cellswere washed with PBS and re-fed with DMSO or appropriate con-centration of drug media. Viral supernatants were collected at 24 hpost infection, and titers determined by plaque assay.
2.12. Animal experiments
For the VEEV TC-83 experiments, six to eight week old femaleC3H/HeN mice were obtained from Harlan Laboratories. Mice were
infected intranasally with a 90% lethal dose (2 � 107 pfu) of VEEVTC-83. Groups of 10 mice were treated by oral gavage with PBSor with ACF (10 mg/kg) daily and were monitored for survival for14 days. Mice were weighed daily and monitored for morbidityand mortality, including lethargy and ruffled fur.
For TrD infection experiments, six to eight week old femaleBALB/c mice were obtained from Harlan Laboratories. Groups of16 mice were infected with VEEV-TrD using Biaera’s AereoPm Sys-tem, whole body chamber and a three jet Collison nebulizer. Theywere exposed to 1 � 105 pfu/ml of VEEV-TrD for 10 min. To deter-mine the approximate number of virus aerosolized by the nebu-lizer during 10 min run, a plaque assay was performed on the AllGlass Impinger (AGI). Hank’s Balanced Salt Solution (HBSS) plus1% FBS was used for viral aerosol. Mice were pretreated 2 h priorto infection, and each day post infection, with 5 mg/kg of ACF viagavage. Control animals were treated intraperitoneally with PBS.Three animals from each group were euthanized Day 4 and Day7 post infection to determine the kinetics of disease in the mousesystem. Serum, lungs, spleen, and brain were collected from eachanimal. Organs were homogenized using IKA DT-20 dispersing sys-tem, and viral replication assessed by plaque assay.
The toxicity study was conducted using female six to eightweek old Balb/c mice treated with 5 mg/kg ACF via gavage, andcontrol animals treated with PBS by intraperitoneal injection.These animals were not infected but were monitored for weightloss for 14 days. All toxicity and TC-83 experiments were carriedout in animal bio-safety level 2 (BSL-2) facilities, and TrD experi-ments in bio-safety level 3 (BSL-3) facilities, in accordance withthe National Research Council’s Guide for the Care and Use of Lab-oratory Animals and under GMU IACUC protocol #0211 and #0248.
2.13. Statistics
Statistical significance was determined using Grubb’s test foroutliers and Student’s unpaired t test to compare the means of testvs. control data. Differences between test and control data weredeemed statistically significant if the two-tailed p value was 60.05.
3. Results
3.1. VEEV protein expression is decreased in the absence of Ago2
Wild-type and Ago2�/� MEFs were mock-infected or infectedwith VEEV-GFP at 0.1 or 1.0 MOI. The VEEV-GFP used for the studycontains the sequence for GFP with its own subgenomic promoterinserted into the VEEV genome at a position between the NSP4protein and the viral subgenomic promoter (Fig. 1A) (Atashevaet al., 2010). The infected cells were incubated for 16 h, and pro-cessed by flow cytometry to measure GFP expression. At an MOIof 0.1, findings were not significantly different between wild typeand Ago2�/� cells; however, at an MOI of 1.0, Ago2�/� cells showedan approximately 3-fold decrease in GFP expression compared towild type cells (Fig. 1A). Levels of viral capsid protein wereassessed by western blot analysis following infection at MOI of1.0. Both wild-type and Ago2�/� samples showed time-dependentincreased expression of VEEV capsid; however, the band intensitiesfor Ago2�/� samples were decreased to approximately 24% of thewild-type value at 8 h and 77% at 16 h, compared to wild-typesamples (Fig. 1B). In a third experiment (Fig. 1C), wild-type andAgo2�/�MEFs were infected at an MOI of 0.1 and resultant sampleswere analyzed by qRT-PCR for presence of VEEV genomic RNA. TheAgo2�/� samples showed a trend toward decreased levels of geno-mic RNA at 8 and 16 h post infection. Comparison of viral replica-tion through plaque assays revealed an approximately 1-log10
decrease in viral replication in Ago2�/� MEFs compared to wild

(A) (B)
(D)(C)
Fig. 1. Ablation of Ago2 decreases VEEV replication. (A) Top panel: diagram of VEEV-GFP genome, showing location of subgenomic promoter (Atasheva et al., 2010). Bottompanel: WT and Ago2�/� MEFs were plated in triplicate and mock-infected or infected with VEEV TC-83-GFP for 1 h at MOI of 0.1 or 1.0. Cells were harvested at 16 h post-infection and assessed by flow cytometry for the expression of GFP. ⁄p-value 60.05. (B) WT and Ago2�/� MEFs were mock-infected or infected with VEEV at an MOI of 0.1.Cells were harvested at 8 or 16 h post infection and extracted proteins were assessed by Western blot for the presence of VEEV capsid protein. Actin was used as a loadingcontrol. Values shown are percentage of normalized band intensity compared to WT at the same time point. Results shown are representative of two independentexperiments. (C) WT and Ago2�/�MEFs were infected with VEEV at an MOI of 0.1, and samples were collected at 8, 16, and 24 h post-infection. Viral RNA levels were assessedby qRT-PCR. ⁄p-value60.05 (compared to WT VEEV infected cells at the corresponding time point). (D) WT and Ago2�/�MEFs were infected at an MOI of 0.1 and supernatantscollected at 24 hpi. Viral replication was assessed by plaque assay. ⁄p-value 60.05 (compared to WT VEEV infected cells).
C. Madsen et al. / Antiviral Research 112 (2014) 26–37 29
type (Fig. 1D). Cumulatively, these results indicate that Ago2 isrequired for optimum VEEV production.
3.2. Inhibition of Ago2 reduces VEEV infection
We next compared the effects of known RISC inhibitors on viralreplication. The inhibitors included: aurintricarboxylic acid (ATA),thought to block the miRNA binding site of Ago2, oxidopaminehydrochloride (OXD) and suramin (SUR), shown to inhibit loadingof miRNA onto Ago2 (Tan et al., 2012), acriflavine (ACF), shown tointerfere with small molecules binding to the RISC, and Poly-L-Lysine (PLL), shown to interfere with the action of Dicer (Watashiet al., 2010). Dicer is a miRNA processing enzyme which acts inthe cytoplasm, but does not interact with the RISC; therefore PLLserves as a control for this assay. Cell viability assays were per-formed to determine the concentrations at which each compoundinduces 50% cytotoxicity (CC50) in U87MG astrocytoma cells. ACFand PLL had CC50 values of 7.22 lM and 6.04 lM, respectively(Fig. 2A). ATA and SUR showed no toxicity at concentrations ashigh as 500 lM, while OXD produced a CC50 value of 419.44 lM(Fig. 2B). Based on these findings, we used ACF and PLL concentra-tions of 2.5 lM and 1.25 lM, ATA and SUR at 500 lM, and OXD at125 lM for antiviral testing.
To test the effects of these inhibitors on VEEV replication,U87MG cells were pre-treated with inhibitors for 2 h prior to VEEV
infection, and the treatment media replaced following the expo-sure period. Viral replication was assessed by plaque assay. Cellstreated with both concentrations of ACF showed a dramatic reduc-tion in viral titers, with 2.5 lM causing an approximately 7-log10
drop in viral titer (Fig. 2C). PLL treatment did not reduce VEEVtiters and actually caused a statistically significant increase at bothconcentrations. Treatment with ATA and SUR caused 3- to 4-log10
decreases in viral titer. OXD treatment had no effect on VEEV titers(Fig. 2D), consistent with previous work (Tan et al., 2012) showingthat OXD does not affect RISC in cell-based assays. These findingssuggest that inhibition of miRNA processing inhibits VEEV replica-tion, with ACF being the most potent inhibitor tested.
3.3. Characterization of ACF treatment on VEEV replication
The kinetics of viral growth in U87MGs pre- and post-treatedwith ACF was assayed to further elucidate the effects of ACF onVEEV replication. In cultures treated with 1.25 lM of ACF, infec-tious particle production was below the level of detection for upto 8 h post-infection and was reduced by approximately 3.5-log10
compared to the DMSO control (Fig. 3A) at 16 h post-infection.Infectious particles production was also below the level of detec-tion for the first 16 h in cultures treated with 2.5 uM ACF. Viraltiters for both ACF-treated and DMSO-treated cultures increasedover time; however, the addition of 1.25 lM or 2.5 lM ACF

(A) (B)
(D)(C)
Fig. 2. Inhibition of Ago2 reduces VEEV infection. (A) U87MGs were treated with ACF or PLL for 24 h at indicated concentrations. Cell viability was determined by Cell TiterGloassay (Promega). (B) U87MGs were treated with ATA, OXD or SUR for 24 h at indicated concentrations. Cell viability was determined by Cell TiterGlo assay. (C) U87MGs werepre-treated for 2 h with ACF and PLL at concentrations indicated in the graph and infected with VEEV TC-83 (MOI 0.1) for 1 h. Viral supernatants were collected at 24 h post-infection and plaque assays were performed to obtain viral titers. ⁄p-value 60.05 (compared to DMSO treated VEEV infected cells). (D) U87MGs were pre-treated for 2 h withATA, OXD or SUR at 500 lM, 125 lM or 500 lM respectively, and infected afterwards with VEEV TC-83 (MOI 0.1) for 1 h. Viral media was removed, cells were washed withPBS, and ATA, OXO, or SUR treatment media was replaced. Viral supernatants were collected at 24 h post-infection and plaque assays were performed to obtain viral titers. ⁄p-value 60.001 (compared to DMSO treated VEEV infected cells).
30 C. Madsen et al. / Antiviral Research 112 (2014) 26–37
reduced viral titer by 2- or 4-log10 respectively, compared to theDMSO value at the 48-h time point.
To determine whether ACF was virucidal, VEEV viral stockswere pre-treated with ACF, PLL, or DMSO prior to infectingU87MGs at an MOI of 0.1 (Fig. 3B). ACF pre- and post-treatedU87MGs infected with untreated VEEV, as in Fig. 3A, were includedfor a direct comparison. Viral supernatants were collected after24 h and viral titers were determined by plaque assays. Cellsinfected with ACF-pre-treated virus showed no reduction in viraltiters while cells pre- and post-treated with ACF showed a reduc-tion in viral titer of approximately 5-log10 compared to DMSO con-trol. Treatment with PLL showed no effect on VEEV titer in eithercase. This suggests that ACF affects VEEV at the replication stage,rather than by damaging the virus directly.
To determine whether the addition of ACF at later times post-infection had an effect on VEEV replication, U87MG cells were trea-ted with DMSO or 2.5 lM ACF at 9 h post-infection, or were pre-and post-treated with DMSO or 2.5 lM ACF as controls. Viralsupernatants were collected 24 h after infection and titers mea-sured by plaque assays. Post-treatment with ACF at 9 h post infec-tion resulted in an approximately 6.5 log10 decrease compared tothe DMSO control, statistically similar to the effects observed withpre- and post-treated cells (Fig. 3C). This demonstrates that post-treatment alone is sufficient for ACF-induced inhibition of VEEVreplication.
To determine the IC50, the level at which ACF caused 50% inhi-bition of viral growth compared to a DMSO control, U87MG cellswere pre- and post-treated with DMSO or varying concentrationsof ACF as described above, and infected at MOI = 0.1. Supernatantswere collected at 24 hpi and titers measured by plaque assay.Interpolation of the data demonstrated that the IC50 of ACF underthese conditions was approximately 0.20 lM (Fig. 3D), or approx-imately 8% of the highest dose used in this assay. The selectiveindex, representing the ratio by which toxic concentration exceedsthe therapeutic concentration, is determined by dividing the CC50by the IC50 to give a value of approximately 36.1.
3.4. ACF inhibits VEEV protein expression and RNA production
In order to determine the effect of ACF on VEEV protein expres-sion, cells were pre-treated with ACF, PLL, or DMSO, infected withVEEV TC-83 GFP, and GFP expression was analyzed by flow cytom-etry. Mock infection had no effect on GFP expression, but reporterexpression was dramatically reduced in ACF treated cells while PLLincreased GFP expression up to 4-fold above the DMSO control(Fig. 4A). Western blot analysis revealed decreased levels of VEEVcapsid when cells were treated with ACF (Fig. 4B), consistent withthe GFP data. Collectively, these results indicate that ACF treatmentresults in a reduction in VEEV protein synthesis.

(A) (B)
(D)(C)
Fig. 3. Characterization of the effect of ACF treatment of VEEV. (A) U87MGs were pre-treated with ACF (2.5 or 1.25 lM) or DMSO 2 h before infection with VEEV TC-83,followed by post-treatment as previously described. Viral supernatants were collected at 8, 16, 24, 36 and 48 h post infection and plaque assays were performed to determineviral titer. ⁄p-value 60.05 (compared to DMSO treated VEEV infected cells at the corresponding time point). ND = none detected (limit of detection was 10 pfu/ml). (B)Aliquots of the VEEV TC-83 viral stock were pre-treated with 2.5 lM of ACF or PLL, or DMSO control, for 1 h at room temperature. U87MG controls were pre-treated with thesame concentrations of ACF or PLL, or with a DMSO control, for 2 h. Infections were performed at an MOI of 0.1 for 1 h. Following infection, all cultures were washed with PBS.Cell growth media was added to cultures containing pre-treated virus, while ACF, PLL, or DMSO media was added to cultures containing the pre-treated cells. Viralsupernatants were collected at 24 h post infection. Plaque assays were used to determine viral titers. ⁄p-value 60.05 (compared to DMSO treated VEEV infected cells). (C)U87MG pre-treated controls were treated with ACF (2.5 lM) or DMSO 2 h before infection with VEEV TC-83 (MOI 0.1), followed by post-treatment as previously described. ⁄p-value 60.05 (compared to DMSO treated VEEV infected cells). (D) U87MG cells were pre- and post-treated as described above, with DMSO control or with serial dilutions ofACF, and infected as described above. Viral supernatants were collected at 24 h post infection and titers determined by plaque assay. IC50 was interpolated from the resultinggraph to determine the concentration at which ACF induced 50% reduction of plaques compared to DMSO control.
C. Madsen et al. / Antiviral Research 112 (2014) 26–37 31
To determine if the decrease in protein levels corresponded tolower levels of viral RNA production, U87MG cells were first pre-treated with ACF for 2 h, infected with VEEV for 1 h, and thenpost-treated with ACF and incubated for 24 h. Samples were quan-tified by q-RT-PCR using primers targeting the capsid region. Asshown in Fig. 4C, levels of VEEV RNA in ACF treated cells were sub-stantially reduced at 24 h post-infection, with a decrease ofapproximately 10-fold at 2.5 lM ACF and approximately 2.5-foldat 1.25 lM ACF. This suggests that the effects of ACF on viral pro-tein production may be due to an overall reduction in viral RNAsynthesis.
3.5. ACF is effective in inhibiting VEEV in neuronal cells
Neuronal damage, which can be attributed to both necrosis andapoptosis, is a critical aspect of the brain lesions of VEEV infectionin mice (Jackson and Rossiter, 1997; Schoneboom et al., 2000;Steele et al., 1998) and is clinically significant as neurons do notnormally regenerate following injury. Therefore, the ability ofACF to protect against cell death was assayed using differentiatedAp7 (dAp7) rat neurons. dAp7 cells were not as sensitive to ACFas U87MG cells, as 5 lM of ACF did not dramatically inhibit cellviability (Fig. 5A). Cell viability was assessed 72 h post-infection,because VEEV replicates more slowly in neurons compared to
other commonly used cell lines, e.g. Vero and U87MG cells (datanot shown). Untreated VEEV-infected dAp7 cells displayed an aver-age viability of 33% at 72 h, whereas average viabilities were 70%and 52% in cells treated with 2.5 or 1.25 lM ACF respectively(Fig. 5B), demonstrating that ACF treatment is protective. Adecrease in viral RNA (to less than 0.1-fold the control value at2.5 lM ACF treatment) and a dramatic reduction in viral titers(nearly 8-log10 reduction at 2.5 lM and 6.5-log10 at 1.25 lM ACF)were also observed following ACF treatment (Fig. 5C and D). Theseresults suggest that ACF treatment can inhibit VEEV replication inneurons and is protective against VEEV induced death of neuronalcells.
3.6. ACF treatment inhibits replication of virulent alphavirus strains inculture
To assess whether ACF treatment is effective against the fully-virulent Trinidad Donkey (TrD) strain of VEEV in culture, Vero cellswere pre- and post-treated with ACF at 2.5 lM, 1.25 lM, or DMSOcontrol, infected at an MOI of 0.1 for 1 h as described previously,and inhibition assessed by plaque assay. ACF treatment at1.25 lM resulted in approximately 1.5-log10 reduction in VEEV-TrD titers at 8 hpi, and approximately 1-log10 reduction at 18 and24 hpi, compared to DMSO alone. Treatment at 2.5 lM resulted

(A)
(C)
(B)
Fig. 4. ACF Inhibits VEEV Protein Expression and RNA Production. (A) U87MGs were pre-treated with ACF or PLL at the specified concentrations for 2 h, and then infected withVEEV TC-83 GFP (MOI 4.0) for 1 h. Drug media was reapplied after infection and cells were collected 24 h post-infection. Cells were analyzed by flow cytometry to determineGFP expression levels (⁄p-value 60.05). (B) U87MGs were pre-treated with ACF or PLL at the specified concentrations for 2 h, infected with VEEV TC-83 GFP (MOI 1.0) for 1 h,and then post-treated. Western blot analysis was performed using anti-capsid antibody. Actin was used as a loading control. Values shown are percentage of normalized bandintensity compared to DMSO control at the same time point. Figure is representative of two replicate trials. (C) U87MGs were pre/post treated with ACF at the specifiedconcentrations or with DMSO as a control, and infected with VEEV TC-83 (MOI 0.1) for 1 h. Cell lysates were collected 24 h post-infection and viral RNA extracted. RNA wasquantified using absolute q-RT-PCR. ⁄p-value 60.05 (compared to DMSO treated VEEV infected cells at the corresponding time point).
32 C. Madsen et al. / Antiviral Research 112 (2014) 26–37
in an approximately 4-log10 reduction compared to DMSO (Fig. 6A),consistent with the TC-83 data. The responses of two additionalVEEV strains, Mena II (Mena) and 3880, were also compared toACF at 2.5 lM, with MOI of 0.1 in Vero cells and at a single 18-htimepoint (Fig. 6B). ACF-induced inhibition at the 18-h timepointwas similar across all strains, approximately 4- to 4.5-log10 com-pared to the DMSO control, again consistent with previous resultsin TrD and in TC-83.
EEEV and WEEV replication were also measured in the presenceof ACF. EEEV exhibited a greater sensitivity to ACF at 8 hpi, in thatEEEV titers were below the level of detection in the presence of2.5 lM ACF. EEEV titers were also reduced at later time points,with the level of reduction being similar to that observed forVEEV-TrD (Fig. 6C). By comparison, WEEV showed slightly less sen-sitivity to ACF, with a decrease of 2- to 3-log10 at the 2.5 lM con-centration for each time point (Fig. 6D). Taken together, thesefindings demonstrate that ACF affects pathways which arerequired for optimal viral replication in encephalitic alphaviruses,and suggests that Ago2 plays a role in these pathways.
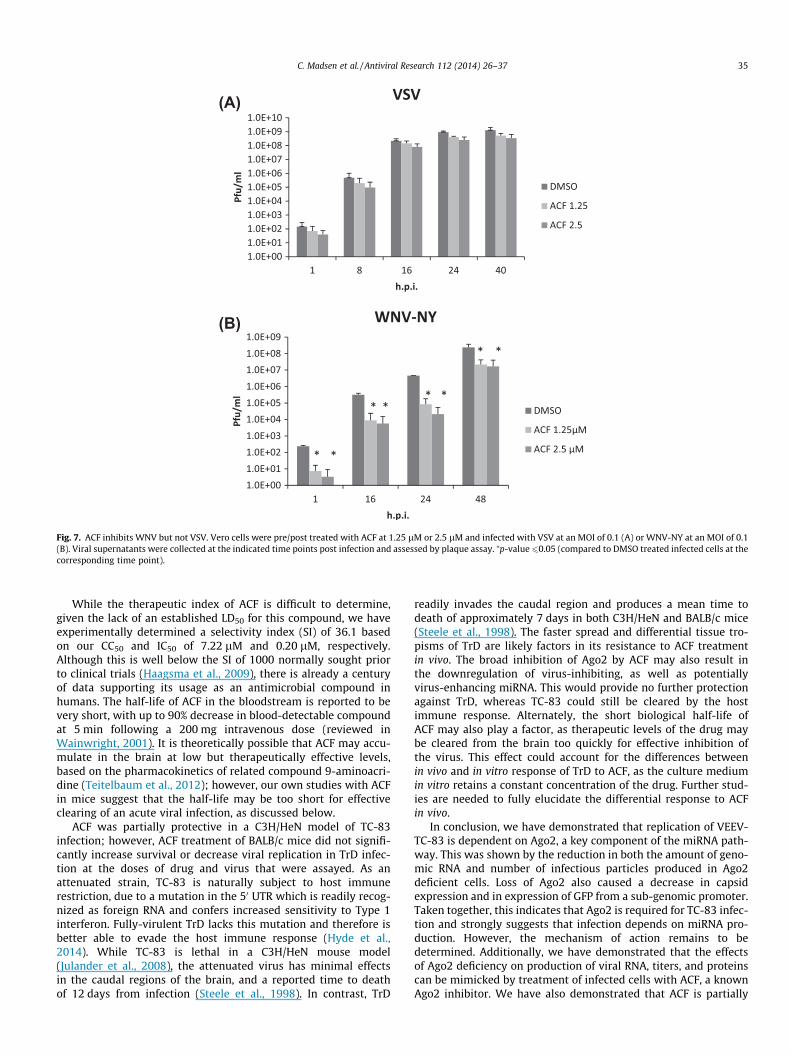
3.7. ACF inhibits WNV but not VSV
To determine if the effect of ACF was limited to Alphaviruses,replication of vesicular stomatitis virus (VSV), a member of the(�) ssRNA stranded Rhabdoviridae, and the New York strain of WestNile Virus (WNV-NY), a representative of the (+) ssRNA Flaviviridaewere assayed in the presence of ACF. Vero cells were pre- and
post-treated with 1.25 or 2.5 lM of ACF. The effect of ACF on viralreplication was assessed by plaque assay. ACF treatment had nosignificant effect on VSV (Fig. 7A). However, WNV-NY titers werereduced by approximately 2- to 4-log10 in the presence of1.25 lM and 2.5 lM of ACF (Fig. 7B). These results demonstratethat ACF is also capable of inhibiting at least one virus outsidethe Togaviridae family, but suggest that it may be less effectiveagainst (�) ssRNA viruses.
3.8. ACF treatment is partially protective in a VEEV TC-83 C3H/HeNmouse model, but not in a TrD BALB/c mouse model
In order to assess the ability of ACF to mitigate VEEV infectionin vivo, C3H/HeN mice were infected intranasally with a 90% lethaldose (2 � 107 pfu) of VEEV TC-83 (Julander et al., 2008). Groups of10 mice were treated by oral gavage daily with PBS or with ACF at10 mg/kg, and were monitored for survival for 16 days. Sixty per-cent of the ACF-treated mice survived VEEV infection, comparedto 10% of mock-treated mice (Fig. 8A). Even though the ACF-treatedgroup showed an overall increased survival, some mice succumbedto disease earlier than then PBS treated control group, suggestingsome toxicity when combined with viral infection.
Next, the ability of ACF to mitigate infection of the fully-virulentTrD strain was assessed. For these studies, a lower dose of ACF(5 mg/kg) was utilized based on the results observed with theVEEV BSL-2 model. Groups of 16 BALB/c mice were treated withACF or PBS prior to infection with 105 pfu/ml by the aerosol route.

(A) (B)
(C) (D)
Fig. 5. Treatment with ACF protects neuronal cells against VEEV infection. (A) Differentiated Ap7 rat neurons were treated with ACF for 24 h and cell viability determined byCell TiterGlo assay (Promega). (B) Differentiated Ap7 rat neurons were pre-treated with ACF for 2 h, infected with VEEV TC-83 (MOI 5), and then post-treated. Cell viabilitywas determined by Cell TiterGlo assay (Promega) at 72 h post-infection (⁄p-value 60.05, ⁄⁄p-value 60.001). Uninfected cell viability is set to 100%. (C) Differentiated Ap7 ratneurons were pre-treated with ACF for 2 h, infected with VEEV TC-83 (MOI 5), and then post-treated. Viral supernatants were collected 72 h post-infection and analyzed by q-RT-PCR for the presence of capsid RNA. ⁄⁄p-value 60.001 (compared to DMSO treated VEEV infected cells). (D) Differentiated Ap7 rat neurons were pre-treated with ACF at1.25 or 2.5 lM for 2 h, infected with VEEV TC-83 (MOI 5), and then post-treated. Viral supernatants were collected 72 h post-infection and analyzed by plaque assay. ⁄⁄p-value60.001 (compared to DMSO treated VEEV infected cells). ND = none detected (limit of detection was 10 pfu/ml).
C. Madsen et al. / Antiviral Research 112 (2014) 26–37 33
Following infection, the mice were treated daily with PBS or withACF, and monitored for survival. Three mice from each group weresacrificed on days 4 and 7 for organ collection and assessment ofviral replication by plaque assay. Treatment with ACF did not sig-nificantly increase survival of VEEV TrD infected mice (Fig. 8B),nor did it induce a significant decrease in viral plaques at 4 days(Fig. 8C) or 7 days post infection (data not shown). Toxicity ofACF in uninfected BALB/c mice was assessed by daily dosing withPBS or with ACF (5 mg/kg), and monitoring for changes in bodyweight over a two-week period. Both PBS and ACF-treated miceshowed a slight increase in bodyweight during the treatment per-iod, with no significant difference in average weights between thetwo groups (Fig. 8D). Taken together, this indicates that ACF is notsufficient to induce inhibition of TrD in vivo, and that further stud-ies are needed to identify an effective inhibitor of the fully virulentstrain.
4. Discussion and conclusions
miRNA are a class of regulatory molecules that have most typ-ically been shown to downregulate gene expression. By virtue oftheir ability to participate in sequence specific interactions withother RNA molecules, they constitute a potentially important
new and poorly understood mode of host-pathogen interactionwhich may be used by the host to inhibit viral replication, or bythe virus to increase viral replication. The most well studied exam-ple of the latter is HCV, which, as discussed previously, actuallyrequires miR-122 for viral replication. Current treatments againstHCV, including pegylated interferon alpha and ribavirin, carry sig-nificant side effects and often fail to eradicate the virus (Schinaziet al., 2010). Inhibition of miR-122, as by the novel drug candidatemiravirsen, shows promise as a therapeutic in the treatment ofHCV (Janssen et al., 2013) suggesting that miRNA inhibitors maybe useful in the treatment of other viral infections such as VEEV.The marked decrease in VEEV replication in the absence of Ago2suggests that VEEV is also subject to miRNA-mediated positive reg-ulation, and may therefore be similarly controlled by miRNAinhibitors.
This study assessed the effects of several inhibitors known tointerfere with the miRNA pathway on VEEV. These included PLL,OXD, ATA, SUR, and ACF. PLL is known to inhibit Dicer, the enzymethat trims the loop from the hairpin of pre-miRNA (Tan et al., 2012;Watashi et al., 2010); OXD, ATA, and SUR have been shown to pre-vent formation of the RISC complex (Watashi et al., 2010). ACFinhibits small molecules binding to Ago2 (Watashi et al., 2010),and disrupts associations between Ago2 and its co-factors TRBP

(A) (B)
(C) (D)
Fig. 6. Effect of ACF on VEEV-TrD, virulent strains, EEEV, and WEEV. Vero cells were pre/post-treated with ACF or PLL (2.5 lM for each) or a DMSO control prior to infectionwith VEEV-TrD (A), fully-virulent strains TrD, Mena, or 3880 (B), or EEEV (C), or WEEV (D) at an MOI of 0.1 for 1 h. Supernatants were collected at 8, 18 and 24 h post infectionfor TC-83, and at 18 h post infection for other viruses. Plaque assays were used to determine viral titer. ⁄p-value 60.05 (compared to DMSO treated infected cells at thecorresponding time point). ND = none detected (limit of detection was 10 pfu/ml).
34 C. Madsen et al. / Antiviral Research 112 (2014) 26–37
and RHA, which assist in RNA loading onto the RISC (Chendrimadaet al., 2005; Robb and Rana, 2007; Watashi et al., 2010). Treatmentwith ATA or SUR reduced VEEV TC-83 infection by approximately3- to 4-log10 compared to a DMSO control, while ACF showed evengreater reduction, at approximately 5- to 6-log10. OXD had no sig-nificant effect, while PLL caused a slight increase (0.5- to 1-log10) inviral titer. It is notable that the effect of OXD on the RISC was pre-viously demonstrated to occur in in vitro assays, but not in cell-based assays (Tan et al., 2012). The failure of OXD to disrupt RISCduring viral infection could account for the lack of inhibitionobserved with this drug in the current study. Nonetheless, threeof the four drugs known to act on the end stage of miRNA process-ing significantly decreased viral titers in cell culture. Although thetest drugs can have pleiotropic effects within the cell, the observa-tion that viral RNA, expression of capsid protein, and expression ofGFP from a subgenomic promoter were similarly affected in ACF-treated and Ago2 deficient cells, and that three of the four RISC-inhibiting drugs decreased VEEV replication, suggests that a func-tional Ago2/RISC complex is required for optimal VEEV replication.
While it is possible that ACF may induce mutations within theviral genome, it has been shown that mutations occurred at con-centrations far above the 1.25–5 lM used in the current studies(Polat and Karakus, 2013). Thus, alterations in DNA or RNA areunlikely to be the source of the decrease in VEEV replicationobserved here. Additionally, ACF did not appear to damage thevirus itself, as pre-treated viral samples showed no change in infec-tivity compared to mock-treated controls. Full inhibition wasachieved only when the cells remained in contact with the drugmedia throughout the infection period, but was consistent acrossseveral strains of virus in culture.
The significant reduction in viral titer from ACF-treated cell cul-tures was also observed in fully-virulent strains of VEEV, as well asin the related Alphaviruses EEEV and WEEV, suggesting that ACFacts upon a replication pathway which is conserved amongencephalitic Alphaviruses. Interestingly, ACF also decreased repli-cation of WNV-NY, another positive-stranded RNA virus, but didnot significantly decrease replication of VSV, a negative-strandedRNA virus. This demonstrates that ACF is not a pan-viral inhibitor,although it does appear effective against at least two viral familiesin cultured cells.
ACF has a long history of usage in treating parasitic, viral, andbacterial infections. It was first investigated as a treatment forsleeping sickness in 1912 and was used extensively to treat battle-field wounds during World War II (Wainwright, 2001). In the pre-antibiotic era it was prescribed orally or intravenously to treatvenereal disease, and orally to treat throat infections(Wainwright, 2001). More recently, it has been combined withother virostatics to treat HIV infection, using oral dosages up to100 mg/day for seven days (Mathé et al., 1998). ACF is currentlybeing studied for its potential to treat malaria in a mouse model(Dana et al., 2014) using intraperitoneal dosages of 5 mg/kg/day,and to reduce vascularization of tumor cells in a cell-based assaythrough its mediation of HIF-1 (Lee et al., 2009). Specifically rele-vant for treatment of VEEV, acridine compounds are known tocross the blood–brain barrier readily (Cornford et al., 1992) andthere is evidence that ACF does so as well (Umschweif et al.,2013). Toxicity is relatively low; as reviewed by Wainwright(2001), a 1931 study involved 2500 men treated daily with 90–120 mg of ACF, with a single case of jaundice as the only seriousside effect.
(A)
(B)
Fig. 7. ACF inhibits WNV but not VSV. Vero cells were pre/post treated with ACF at 1.25 lM or 2.5 lM and infected with VSV at an MOI of 0.1 (A) or WNV-NY at an MOI of 0.1(B). Viral supernatants were collected at the indicated time points post infection and assessed by plaque assay. ⁄p-value60.05 (compared to DMSO treated infected cells at thecorresponding time point).
C. Madsen et al. / Antiviral Research 112 (2014) 26–37 35
While the therapeutic index of ACF is difficult to determine,given the lack of an established LD50 for this compound, we haveexperimentally determined a selectivity index (SI) of 36.1 basedon our CC50 and IC50 of 7.22 lM and 0.20 lM, respectively.Although this is well below the SI of 1000 normally sought priorto clinical trials (Haagsma et al., 2009), there is already a centuryof data supporting its usage as an antimicrobial compound inhumans. The half-life of ACF in the bloodstream is reported to bevery short, with up to 90% decrease in blood-detectable compoundat 5 min following a 200 mg intravenous dose (reviewed inWainwright, 2001). It is theoretically possible that ACF may accu-mulate in the brain at low but therapeutically effective levels,based on the pharmacokinetics of related compound 9-aminoacri-dine (Teitelbaum et al., 2012); however, our own studies with ACFin mice suggest that the half-life may be too short for effectiveclearing of an acute viral infection, as discussed below.
ACF was partially protective in a C3H/HeN model of TC-83infection; however, ACF treatment of BALB/c mice did not signifi-cantly increase survival or decrease viral replication in TrD infec-tion at the doses of drug and virus that were assayed. As anattenuated strain, TC-83 is naturally subject to host immunerestriction, due to a mutation in the 50 UTR which is readily recog-nized as foreign RNA and confers increased sensitivity to Type 1interferon. Fully-virulent TrD lacks this mutation and therefore isbetter able to evade the host immune response (Hyde et al.,2014). While TC-83 is lethal in a C3H/HeN mouse model(Julander et al., 2008), the attenuated virus has minimal effectsin the caudal regions of the brain, and a reported time to deathof 12 days from infection (Steele et al., 1998). In contrast, TrD
readily invades the caudal region and produces a mean time todeath of approximately 7 days in both C3H/HeN and BALB/c mice(Steele et al., 1998). The faster spread and differential tissue tro-pisms of TrD are likely factors in its resistance to ACF treatmentin vivo. The broad inhibition of Ago2 by ACF may also result inthe downregulation of virus-inhibiting, as well as potentiallyvirus-enhancing miRNA. This would provide no further protectionagainst TrD, whereas TC-83 could still be cleared by the hostimmune response. Alternately, the short biological half-life ofACF may also play a factor, as therapeutic levels of the drug maybe cleared from the brain too quickly for effective inhibition ofthe virus. This effect could account for the differences betweenin vivo and in vitro response of TrD to ACF, as the culture mediumin vitro retains a constant concentration of the drug. Further stud-ies are needed to fully elucidate the differential response to ACFin vivo.
In conclusion, we have demonstrated that replication of VEEV-TC-83 is dependent on Ago2, a key component of the miRNA path-way. This was shown by the reduction in both the amount of geno-mic RNA and number of infectious particles produced in Ago2deficient cells. Loss of Ago2 also caused a decrease in capsidexpression and in expression of GFP from a sub-genomic promoter.Taken together, this indicates that Ago2 is required for TC-83 infec-tion and strongly suggests that infection depends on miRNA pro-duction. However, the mechanism of action remains to bedetermined. Additionally, we have demonstrated that the effectsof Ago2 deficiency on production of viral RNA, titers, and proteinscan be mimicked by treatment of infected cells with ACF, a knownAgo2 inhibitor. We have also demonstrated that ACF is partially

(A) (B)
(C) (D)
Fig. 8. ACF treatment is partially protective in a VEEV TC-83 C3H/HeN mouse model, but not a TrD BALB/c mouse model. (A) Six to eight week old female C3H/HeN mice wereinfected intranasally with 2 � 107 pfu/mouse of VEEV TC-83. Groups of 10 mice were treated by daily by oral gavage with PBS or with ACF at 10 mg/kg. Mice were monitoredfor 16 days for survival, weighed daily and monitored for signs of morbidity, including lethargy and ruffled fur. (B, C) Six to eight week old female BALB/c mice werepretreated with PBS control or ACF at 5 mg/kg, then infected by the aerosol route with 105 pfu/ml of VEEV-TrD. Groups of 16 mice were treated by daily intraperitonealinjection with PBS or by gavage with ACF at 5 mg/kg. (B) Serum, spleen, brain, and lung samples were collected on Day 4 from control-treated or ACF-treated TrD-infectedBALB/c mice. Viral replication in each tissue type was assessed by plaque assay. Error bars represent the standard error of mean; dotted line represents the lower limit ofdetection. (C) Mice were monitored for 10 days for survival and signs of morbidity as discussed above. (D) Uninfected six to eight week old female BALB/c mice were treateddaily with PBS or with ACF (5 mg/kg). Mice were monitored for survival and weighed daily.
36 C. Madsen et al. / Antiviral Research 112 (2014) 26–37
protective in a mouse model of TC-83 infection, though not of TrDinfection. In cell culture, ACF treatment induced decreased replica-tion of three fully-virulent subtypes of VEEV, as well as EEEV,WEEV, and WNV, but not VSV. These results demonstrate thatACF is not a pan-viral inhibitor, but is capable of inhibiting at leasttwo families of (+)-strand RNA viruses, Flaviviridae and Togaviridae.Taken together, these findings suggest that Ago2 is an importantfactor for efficient replication of encephalitic viruses. Further char-acterization of the role of Ago2 may contribute to a further under-standing of pathogenic processes, as well as provide potentialtargets for novel therapeutics.
Acknowledgments
The authors wish to thank the following persons for their gen-erous gifts of supplies: Dr. Ilya Frolov of the University of Alabamaat Birmingham for the VEEV TC83-GFP virus, Dr. Gregory Hannonof Cold Spring Harbor Laboratories for the Ago2�/� MEFs, Dr. DianeGriffin of Johns Hopkins University for the dAp-7 neuronal cells,and the late Dr. K.T. Jeang of National Institutes of Health for ACFand PLL. The authors also thank Dr. Fatah Kashanchi (GeorgeMason University) for helpful discussions. The following reagentswere obtained through the NIH Biodefense and Emerging Infec-tions Research Resources Repository, NIAID, NIH: polyclonal Anti-Venezuelan Equine Encephalitis Virus, TC-83 (Subtype IA/B) CapsidProtein (antiserum, Goat) antibody, NR-9403, Venezuelan EquineEncephalitis Virus, TC83 (Subtype IAB), NR63, and Venezuelan
equine encephalitis virus Trinidad Donkey (subtype IA/B), NR-332. This work was funded through a Defense Threat ReductionAgency (DTRA) - USA grant, HDTRA1-13-1-0005, to KK and JJ.
References
Aguilar, P.V., Estrada-Franco, J.G., Navarro-Lopez, R., Ferro, C., Haddow, A.D.,Weaver, S.C., 2011. Endemic Venezuelan equine encephalitis in the Americas:hidden under the dengue umbrella. Future Virol. 6, 721–740.
Amaya, M., Voss, K., Sampey, G., Senina, S., de la Fuente, C., Mueller, C., Calvert, V.,Kehn-Hall, K., Carpenter, C., Kashanchi, F., et al., 2014. The role of IKKb inVenezuelan equine encephalitis virus infection. PLoS ONE 9, e86745.
Atasheva, S., Krendelchtchikova, V., Liopo, A., Frolova, E., Frolov, I., 2010. Interplay ofacute and persistent infections caused by Venezuelan equine encephalitis virusencoding mutated capsid protein. J. Virol. 84, 10004–10015.
Austin, D., Baer, A., Lundberg, L., Shafagati, N., Schoonmaker, A., Narayanan, A.,Popova, T., Panthier, J.J., Kashanchi, F., Bailey, C., Kehn-Hall, K., 2012. p53Activation following Rift Valley fever virus infection contributes to cell deathand viral production. PLoS ONE 7, e36327.
Berge, T.O., Banks, I.S., Tigertt, W.D., 1961. Attenuation of Venezuelan equineencephalomyelitis virus by in vitro cultivation in guinea-pig heart cells. Am. J.Epidemiol. 73, 209–218.
Bhomia, M., Balakathiresan, N., Sharma, A., Gupta, P., Biswas, R., Maheshwari, R.,2010. Analysis of microRNAs induced by Venezuelan equine encephalitis virusinfection in mouse brain. Biochem. Biophys. Res. Commun. 395, 11–16.
Casals, J., Curnen, E.C., Thomas, L., 1943. Venezuelan equine encephalomyelitis inman. J. Exp. Med. 77, 521–530.
Chendrimada, T.P., Gregory, R.I., Kumaraswamy, E., Norman, J., Cooch, N., Nishikura,K., Shiekhattar, R., 2005. TRBP recruits the Dicer complex to Ago2 for microRNAprocessing and gene silencing. Nature 436, 740–744.
Cornford, E.M., Young, D., Paxton, J.W., 1992. Comparison of the blood–brain barrierand liver penetration of acridine antitumor drugs. Cancer Chemother.Pharmacol. 29, 439–444.
Cullen, B.R., 2005. RNAi the natural way. Nat. Genet. 37, 1163–1165.

C. Madsen et al. / Antiviral Research 112 (2014) 26–37 37
Dana, S., Prusty, D., Dhayal, D., Gupta, M.K., Dar, A., Sen, S., Mukhopadhyay, P., Adak,T., Dhar, S.K., 2014. The potent anti-malarial activity of acriflavine in vitro andin vivo. ACS Chem. Biol., 2366–2373.
Haagsma, A.C., Abdillahi-Ibrahim, R., Wagner, M.J., Krab, K., Vergauwen, K.,Guillemont, J., Andries, K., Lill, H., Koul, A., Bald, D., 2009. Selectivity ofTMC207 towards mycobacterial ATP synthase compared with that towards theeukaryotic homologue. Antimicrob. Agents Chemother. 53, 1290–1292.
Henke, J.I., Goergen, D., Zheng, J., Song, Y., Schüttler, C.G., Fehr, C., Jünemann, C.,Niepmann, M., 2008. microRNA-122 stimulates translation of hepatitis C virusRNA. EMBO J. 27, 3300–3310.
Hussmann, K.L., Samuel, M.A., Kim, K.S., Diamond, M.S., Fredericksen, B.L., 2013.Differential replication of pathogenic and nonpathogenic strains of west nilevirus within astrocytes. J. Virol. 87, 2814–2822.
Hyde, J.L., Gardner, C.L., Kimura, T., White, J.P., Liu, G., Trobaugh, D.W., Huang, C.,Tonelli, M., Paessler, S., Takeda, K., et al., 2014. A viral RNA structural elementalters host recognition of nonself RNA. Science 343, 783–787.
Jackson, A.C., Rossiter, J.P., 1997. Apoptotic cell death is an important cause ofneuronal injury in experimental Venezuelan equine encephalitis virus infectionof mice. Acta Neuropathol. (Berl.) 93, 349–353.
Janssen, H.L.A., Reesink, H.W., Lawitz, E.J., Zeuzem, S., Rodriguez-Torres, M., Patel, K.,van der Meer, A.J., Patick, A.K., Chen, A., Zhou, Y., et al., 2013. Treatment of HCVinfection by targeting microRNA. N. Engl. J. Med. 368, 1685–1694.
Jopling, C.L., Yi, M., Lancaster, A.M., Lemon, S.M., Sarnow, P., 2005. Modulation ofhepatitis C virus RNA abundance by a liver-specific microRNA. Science 309,1577–1581.
Jopling, C.L., Norman, K.L., Sarnow, P., 2006. Positive and negative modulation ofviral and cellular mRNAs by liver-specific microRNA miR-122. Cold Spring Harb.Symp. Quant. Biol. 71, 369–376.
Julander, J.G., Skirpstunas, R., Siddharthan, V., Shafer, K., Hoopes, J.D., Smee, D.F.,Morrey, J.D., 2008. C3H/HeN mouse model for the evaluation of antiviral agentsfor the treatment of Venezuelan equine encephalitis virus infection. AntiviralRes. 78, 230–241.
Kehn-Hall, K., Narayanan, A., Lundberg, L., Sampey, G., Pinkham, C., Guendel, I., VanDuyne, R., Senina, S., Schultz, K.L., Stavale, E., et al., 2012. Modulation of GSK-3bactivity in Venezuelan equine encephalitis virus infection. PLoS ONE 7, e34761.
Kinney, R.M., Chang, G.J., Tsuchiya, K.R., Sneider, J.M., Roehrig, J.T., Woodward, T.M.,Trent, D.W., 1993. Attenuation of Venezuelan equine encephalitis virus strainTC-83 is encoded by the 50-noncoding region and the E2 envelope glycoprotein.J. Virol. 67, 1269–1277.
Lau, N.C., Lim, L.P., Weinstein, E.G., Bartel, D.P., 2001. An abundant class of tiny RNAswith probable regulatory roles in Caenorhabditis elegans. Science 294, 858–862.
Lee, R.C., Ambros, V., 2001. An extensive class of small RNAs in Caenorhabditiselegans. Science 294, 862–864.
Lee, K., Zhang, H., Qian, D.Z., Rey, S., Liu, J.O., Semenza, G.L., 2009. Acriflavine inhibitsHIF-1 dimerization, tumor growth, and vascularization. Proc. Natl. Acad. Sci.U.S.A. 106, 17910–17915.
Lin, Z., Wang, X., Fewell, C., Cameron, J., Yin, Q., Flemington, E.K., 2010. Differentialexpression of the miR-200 family microRNAs in epithelial and B cells andregulation of epstein–barr virus reactivation by the miR-200 family membermiR-429. J. Virol. 84, 7892–7897.
Liu, J., Carmell, M.A., Rivas, F.V., Marsden, C.G., Thomson, J.M., Song, J.J., Hammond,S.M., Joshua-Tor, L., Hannon, G.J., 2004. Argonaute2 is the catalytic engine ofmammalian RNAi. Science 305, 1437–1441.
Lundberg, L., Pinkham, C., Baer, A., Amaya, M., Narayanan, A., Wagstaff, K.M., Jans,D.A., Kehn-Hall, K., 2013. Nuclear import and export inhibitors alter capsid
protein distribution in mammalian cells and reduce Venezuelan equineencephalitis virus replication. Antiviral Res. 100, 662–672.
Mathé, G., Triana, K., Pontiggia, P., Blanquet, D., Hallard, M., Morette, C., 1998. Dataof pre-clinical and early clinical trials of acriflavine and hydroxy-methyl-ellipticine reviewed, enriched by the experience of their use for 18 months to6 years in combinations with other HIV1 virostatics. Biomed. Pharmacother.[Bioméd. Pharmacothér.] 52, 391–396.
Narayanan, A., Kehn-Hall, K., Senina, S., Lundberg, L., Duyne, R.V., Guendel, I., Das, R.,Baer, A., Bethel, L., Turell, M., et al., 2012. Curcumin inhibits rift valley fevervirus replication in human cells. J. Biol. Chem. 287, 33198–33214.
Polat, Z.A., Karakus, G., 2013. Cytotoxic effect of acriflavine against clinical isolatesof Acanthamoeba spp.. Parasitol. Res. 112, 529–533.
Powers, A.M., Oberste, M.S., Brault, A.C., Rico-Hesse, R., Schmura, S.M., Smith, J.F.,Kang, W., Sweeney, W.P., Weaver, S.C., 1997. Repeated emergence of epidemic/epizootic Venezuelan equine encephalitis from a single genotype of enzooticsubtype ID virus. J. Virol. 71, 6697–6705.
Robb, G.B., Rana, T.M., 2007. RNA helicase a interacts with RISC in human cells andfunctions in RISC loading. Mol. Cell 26, 523–537.
Roberts, A.P.E., Lewis, A.P., Jopling, C.L., 2011. MiR-122 activates hepatitis C virustranslation by a specialized mechanism requiring particular RNA components.Nucleic Acids Res. 39, 7716–7729.
Schinazi, R.F., Bassit, L., Gavegnano, C., 2010. HCV drug discovery aimed at viraleradication. J. Viral Hepat. 17, 77–90.
Schoneboom, B.A., Catlin, K.M.K., Marty, A.M., Grieder, F.B., 2000. Inflammation is acomponent of neurodegeneration in response to Venezuelan equineencephalitis virus infection in mice. J. Neuroimmunol. 109, 132–146.
Steele, K.E., Davis, K.J., Stephan, K., Kell, W., Vogel, P., Hart, M.K., 1998. Comparativeneurovirulence and tissue tropism of wild-type and attenuated strains ofVenezuelan equine encephalitis virus administered by aerosol in C3H/HeN andBALB/c mice. Vet. Pathol. 35, 386–397.
Tan, G.S., Chiu, C.-H., Garchow, B.G., Metzler, D., Diamond, S.L., Kiriakidou, M., 2012.Small molecule inhibition of RISC loading. ACS Chem. Biol. 7, 403–410.
Teitelbaum, A.M., Gallardo, J.L., Bedi, J., Giri, R., Benoit, A.R., Olin, M.R., Morizio, K.M.,Ohlfest, J.R., Remmel, R.P., Ferguson, D.M., 2012. 9-Amino acridinepharmacokinetics, brain distribution, and in vitro/in vivo efficacy againstmalignant glioma. Cancer Chemother. Pharmacol. 69, 1519–1527.
Umschweif, G., Alexandrovich, A.G., Trembovler, V., Horowitz, M., Shohami, E., 2013.Hypoxia-inducible factor 1 is essential for spontaneous recovery from traumaticbrain injury and is a key mediator of heat acclimation induced neuroprotection.J. Cereb. Blood Flow Metab. 33, 524–531.
Wainwright, M., 2001. Acridine—a neglected antibacterial chromophore. J.Antimicrob. Chemother. 47, 1–13.
Watashi, K., Yeung, M.L., Starost, M.F., Hosmane, R.S., Jeang, K.-T., 2010.Identification of small molecules that suppress MicroRNA function andreverse tumorigenesis. J. Biol. Chem. 285, 24707–24716.
Wu, Y.-Q., Chen, D.-J., He, H.-B., Chen, D.-S., Chen, L.-L., Chen, H.-C., Liu, Z.-F., 2012.Pseudorabies virus infected porcine epithelial cell line generates a diverseset of host microRNAs and a special cluster of viral microRNAs. PLoS ONE 7,e30988.
Zacks, M.A., Paessler, S., 2010. Encephalitic alphaviruses. Vet. Microbiol. 140, 281–286.
Zhao, P., Zhao, L., Zhang, T., Wang, H., Qin, C., Yang, S., Xia, X., 2012. Changes inmicroRNA expression induced by rabies virus infection in mouse brains. Microb.Pathog. 52, 47–54.




















