
Spectrum of disease-causing mutations in protein secondary structures
Upload
independentCategory
view
4download
0
A R T I C L E S
1366 VOLUME 10 | NUMBER 12 | DECEMBER 2004 NATURE MEDICINE
WNV, a mosquito-borne single-stranded RNA virus, has been thecause of recent outbreaks of viral encephalitis in North America1.Though human infection is usually asymptomatic, life-threateningneurological disease, including encephalitis or meningitis, can ensuein the elderly and immunocompromised1. Viral pathogenesis is notcompletely understood, and specific therapies have not yet beenapproved for use in humans. In mice, WNV induces a systemic infec-tion and crosses the blood-brain barrier (BBB), resulting inencephalitis and death2,3. Type-1 interferons (IFNs)4–6 and humoralimmunity7,8 provide immediate defense against the dissemination ofWNV. Cellular immunity, including γδ T cells9, CD4+ (ref. 10) andCD8+ αβ T-cell11 responses also participate in recovery of the hostfrom WNV infection.
The Toll-like receptors (TLRs) have an essential role in the initiationof innate immunity. In mammals, the TLR family is composed of atleast 11–12 members and each TLR acts as a primary sensor of con-served microbial components and drives the induction of specific bio-logical responses12–15. It is well established that peptidoglycan,lipopolysaccharide and flagellin are recognized by TLR2, TLR4 andTLR5, respectively13. Specific recognition of viral proteins by TLRs hasbeen previously documented16–18. We have shown that TLR3 isinvolved in the recognition of viral components, such as poly(I:C) anddsRNA from Lang reovirus19,20. TLR7 and TLR8 are vital for therecognition of ssRNA viruses and ssRNA21–23. Furthermore, TLR9,which recognizes DNA unmethylated at CpG motifs, and TLR3 areboth involved in defense against mouse cytomegalovirus infection15.Hence, certain TLRs, including TLR3, are involved in viral recognition.
The functional role of TLR3 in recognizing viral infections in vivostill remains unclear. WNV is a positive ssRNA virus that produces
dsRNA in its life cycle and may be detected by TLR3 in the host24.Therefore, to elucidate the role of TLR3 in the control of viral infec-tion, we examined the responses of Tlr3–/– mice upon WNV infection.
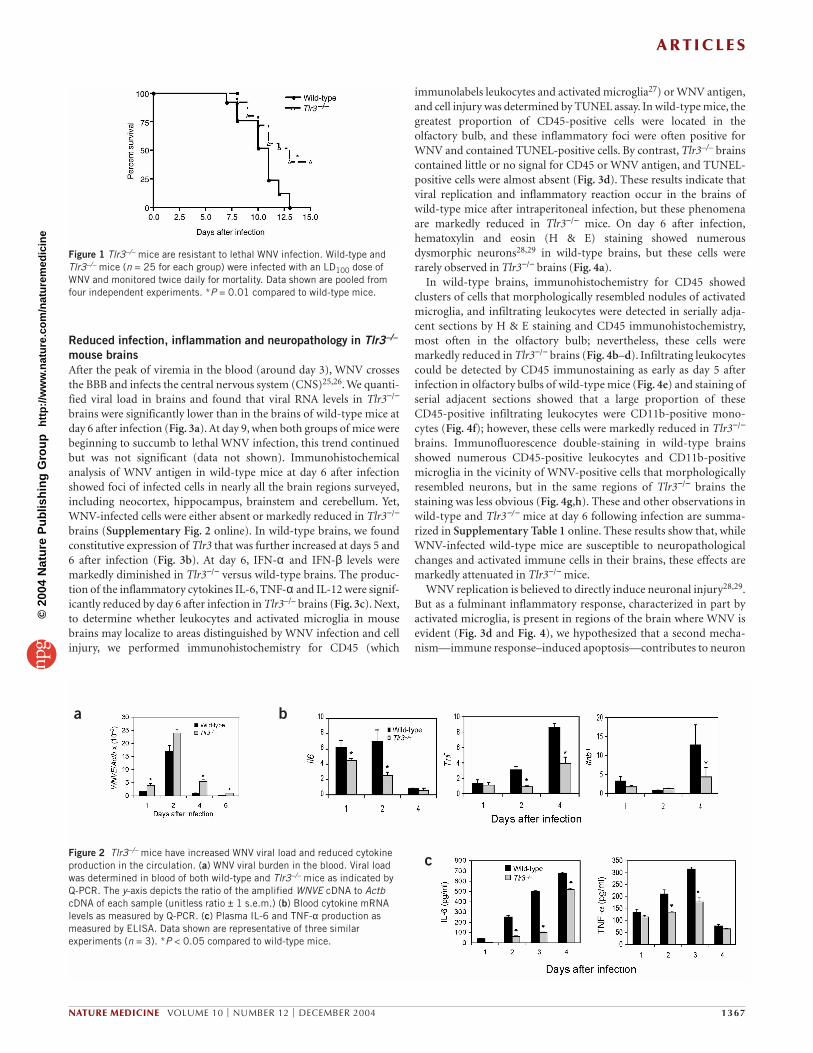
RESULTSTlr3–/– mice are more resistant to lethal WNV infectionTo examine the role of TLR3 in viral recognition in vivo, we used themouse model of WNV encephalitis. Wild-type and Tlr3−/− micewere challenged intraperitoneally with 1 × 103 plaque-forming units(p.f.u.) of WNV and monitored daily for survival. Tlr3−/− mice weremore resistant (40% survival) to lethal WNV infection than wild-type controls (0% survival) (Fig. 1). Next, we measured the viralload in the blood and spleen. In the blood, Tlr3−/− mice had signifi-cantly elevated viral burden at days 1, 4 and 6 compared to wild-type mice (Fig. 2a). WNV antigen was generally detected in thecytoplasm of marginal zone splenocytes near blood vessels.Quantitative PCR (Q-PCR) data measuring WNV envelope gene(WNVE) gene mRNA (see Supplementary Methods online) showeda very modest but significant increase of splenic viral load in wild-type versus Tlr3−/− mice at days 1 and 6 (Supplementary Fig. 1online); however, no significant differences were detected versusTlr3−/− by immunohistochemistry or plaque assay (data not shown).Moreover, we measured cytokine levels in the blood. The RNA levelsof interleukin (IL)-6 at days 1 and 2, tumor necrosis factor (TNF)-αat days 2 and 4, and IFN-β levels at day 4 were significantly higher inwild-type than in Tlr3−/− mice (Fig. 2b); however, these differencesdisappeared by day 6 (data not shown). The blood plasma proteinlevels of IL-6 and TNF-α were also clearly reduced in Tlr3−/− mice atdays 2 and 3 following infection (Fig. 2c).
1Section of Rheumatology, Department of Internal Medicine, 2Section of Immunobiology and 3the Howard Hughes Medical Institute, Yale University School ofMedicine, 300 Cedar Street, New Haven, Connecticut 06520, USA. 4Department of Entomology, Connecticut Agricultural Experiment Station, P. O. Box 1106, NewHaven, Connecticut 06504, USA. 5Current address: Centre d’Immunologie de Marseille-Luminy, CNRS-INSERM, Campus de Luminy, Case 906, 13288 Marseille,Cedex 9, France. 6These authors contributed equally to this work. Correspondence should be addressed to E.F. ([email protected]).
Published online 21 November 2004; doi:10.1038/nm1140
Toll-like receptor 3 mediates West Nile virus entry intothe brain causing lethal encephalitisTian Wang1,6, Terrence Town2,3,6, Lena Alexopoulou2,5,6, John F Anderson4, Erol Fikrig1 & Richard A Flavell2,3
West Nile virus (WNV), a mosquito-borne single-stranded (ss)RNA flavivirus, causes human disease of variable severity. Weinvestigated the involvement of Toll-like receptor (Tlr) 3, which recognizes viral double-stranded (ds)RNA, on WNV infection.Tlr3-deficient (Tlr3–/–) mice were more resistant to lethal WNV infection and had impaired cytokine production and enhancedviral load in the periphery, whereas in the brain, viral load, inflammatory responses and neuropathology were reduced comparedto wild-type mice. Peripheral WNV infection led to a breakdown of the blood-brain barrier and enhanced brain infection in wild-type but not in Tlr3–/– mice, although both groups were equally susceptible upon intracerebroventricular administration of thevirus. Tumor necrosis factor-α receptor 1 signaling is vital for blood-brain barrier compromise upon Tlr3 stimulation by dsRNA orWNV. Collectively, WNV infection leads to a Tlr3-dependent inflammatory response, which is involved in brain penetration of thevirus and neuronal injury.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine
A R T I C L E S
NATURE MEDICINE VOLUME 10 | NUMBER 12 | DECEMBER 2004 1367
Reduced infection, inflammation and neuropathology in Tlr3–/–
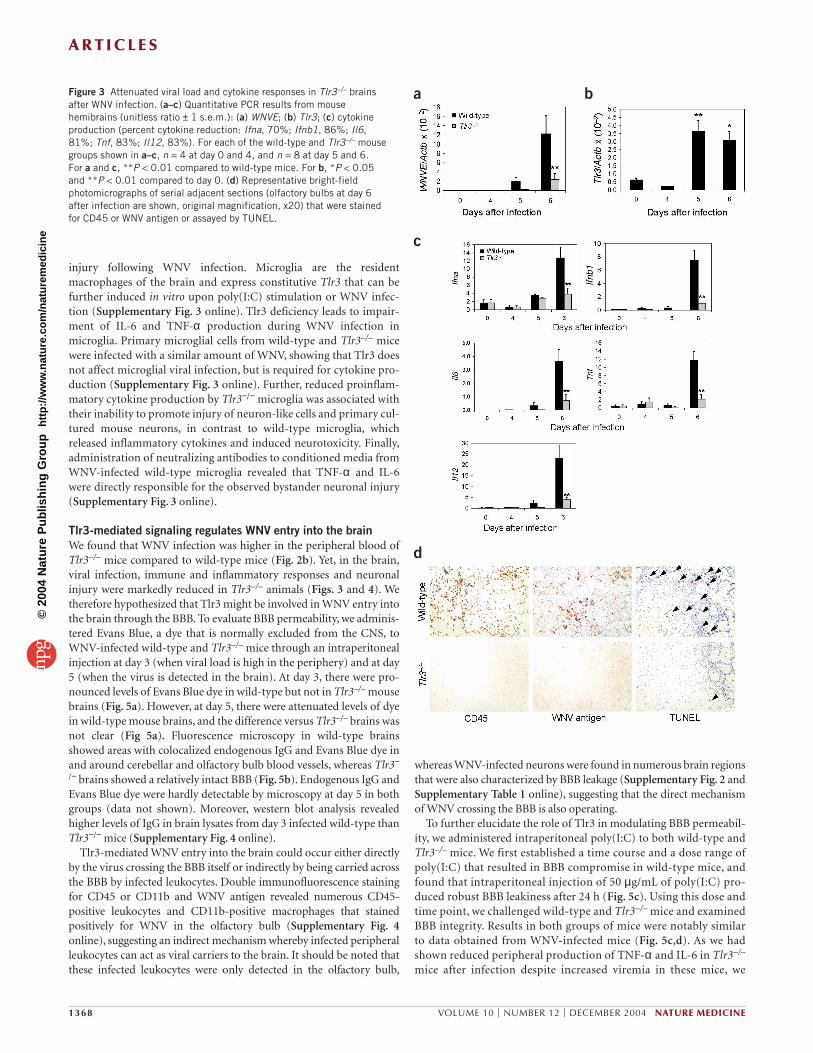
mouse brainsAfter the peak of viremia in the blood (around day 3), WNV crossesthe BBB and infects the central nervous system (CNS)25,26. We quanti-fied viral load in brains and found that viral RNA levels in Tlr3−/−
brains were significantly lower than in the brains of wild-type mice atday 6 after infection (Fig. 3a). At day 9, when both groups of mice werebeginning to succumb to lethal WNV infection, this trend continuedbut was not significant (data not shown). Immunohistochemicalanalysis of WNV antigen in wild-type mice at day 6 after infectionshowed foci of infected cells in nearly all the brain regions surveyed,including neocortex, hippocampus, brainstem and cerebellum. Yet,WNV-infected cells were either absent or markedly reduced in Tlr3−/−
brains (Supplementary Fig. 2 online). In wild-type brains, we foundconstitutive expression of Tlr3 that was further increased at days 5 and6 after infection (Fig. 3b). At day 6, IFN-α and IFN-β levels weremarkedly diminished in Tlr3−/− versus wild-type brains. The produc-tion of the inflammatory cytokines IL-6, TNF-α and IL-12 were signif-icantly reduced by day 6 after infection in Tlr3–/– brains (Fig. 3c). Next,to determine whether leukocytes and activated microglia in mousebrains may localize to areas distinguished by WNV infection and cellinjury, we performed immunohistochemistry for CD45 (which
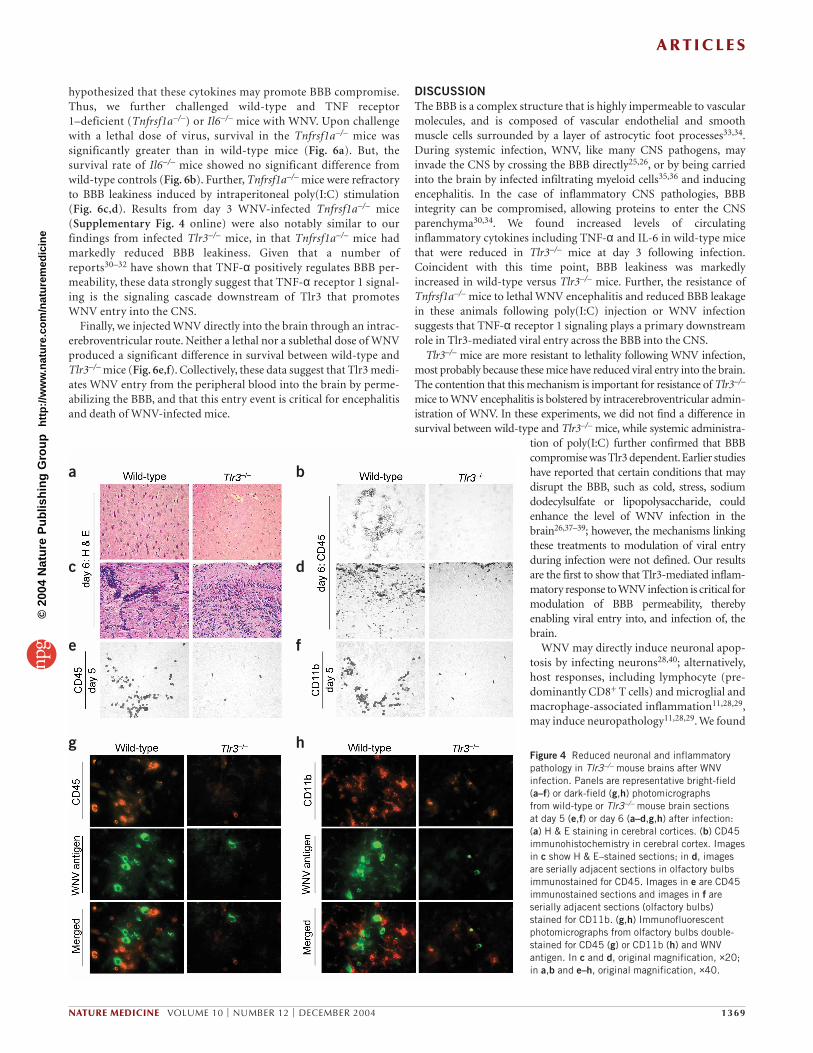
immunolabels leukocytes and activated microglia27) or WNV antigen,and cell injury was determined by TUNEL assay. In wild-type mice, thegreatest proportion of CD45-positive cells were located in theolfactory bulb, and these inflammatory foci were often positive forWNV and contained TUNEL-positive cells. By contrast, Tlr3–/– brainscontained little or no signal for CD45 or WNV antigen, and TUNEL-positive cells were almost absent (Fig. 3d). These results indicate thatviral replication and inflammatory reaction occur in the brains ofwild-type mice after intraperitoneal infection, but these phenomenaare markedly reduced in Tlr3−/− mice. On day 6 after infection,hematoxylin and eosin (H & E) staining showed numerousdysmorphic neurons28,29 in wild-type brains, but these cells wererarely observed in Tlr3−/− brains (Fig. 4a).
In wild-type brains, immunohistochemistry for CD45 showedclusters of cells that morphologically resembled nodules of activatedmicroglia, and infiltrating leukocytes were detected in serially adja-cent sections by H & E staining and CD45 immunohistochemistry,most often in the olfactory bulb; nevertheless, these cells weremarkedly reduced in Tlr3−/− brains (Fig. 4b–d). Infiltrating leukocytescould be detected by CD45 immunostaining as early as day 5 afterinfection in olfactory bulbs of wild-type mice (Fig. 4e) and staining ofserial adjacent sections showed that a large proportion of theseCD45-positive infiltrating leukocytes were CD11b-positive mono-cytes (Fig. 4f); however, these cells were markedly reduced in Tlr3−/−
brains. Immunofluorescence double-staining in wild-type brainsshowed numerous CD45-positive leukocytes and CD11b-positivemicroglia in the vicinity of WNV-positive cells that morphologicallyresembled neurons, but in the same regions of Tlr3−/− brains thestaining was less obvious (Fig. 4g,h). These and other observations inwild-type and Tlr3−/− mice at day 6 following infection are summa-rized in Supplementary Table 1 online. These results show that, whileWNV-infected wild-type mice are susceptible to neuropathologicalchanges and activated immune cells in their brains, these effects aremarkedly attenuated in Tlr3−/− mice.
WNV replication is believed to directly induce neuronal injury28,29.But as a fulminant inflammatory response, characterized in part byactivated microglia, is present in regions of the brain where WNV isevident (Fig. 3d and Fig. 4), we hypothesized that a second mecha-nism—immune response–induced apoptosis—contributes to neuron
Figure 1 Tlr3–/– mice are resistant to lethal WNV infection. Wild-type andTlr3–/– mice (n = 25 for each group) were infected with an LD100 dose ofWNV and monitored twice daily for mortality. Data shown are pooled fromfour independent experiments. *P = 0.01 compared to wild-type mice.
a b
cFigure 2 Tlr3–/– mice have increased WNV viral load and reduced cytokineproduction in the circulation. (a) WNV viral burden in the blood. Viral loadwas determined in blood of both wild-type and Tlr3–/– mice as indicated byQ-PCR. The y-axis depicts the ratio of the amplified WNVE cDNA to ActbcDNA of each sample (unitless ratio ± 1 s.e.m.) (b) Blood cytokine mRNAlevels as measured by Q-PCR. (c) Plasma IL-6 and TNF-α production asmeasured by ELISA. Data shown are representative of three similarexperiments (n = 3). *P < 0.05 compared to wild-type mice.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine
A R T I C L E S
1368 VOLUME 10 | NUMBER 12 | DECEMBER 2004 NATURE MEDICINE
injury following WNV infection. Microglia are the residentmacrophages of the brain and express constitutive Tlr3 that can befurther induced in vitro upon poly(I:C) stimulation or WNV infec-tion (Supplementary Fig. 3 online). Tlr3 deficiency leads to impair-ment of IL-6 and TNF-α production during WNV infection inmicroglia. Primary microglial cells from wild-type and Tlr3–/– micewere infected with a similar amount of WNV, showing that Tlr3 doesnot affect microglial viral infection, but is required for cytokine pro-duction (Supplementary Fig. 3 online). Further, reduced proinflam-matory cytokine production by Tlr3−/− microglia was associated withtheir inability to promote injury of neuron-like cells and primary cul-tured mouse neurons, in contrast to wild-type microglia, whichreleased inflammatory cytokines and induced neurotoxicity. Finally,administration of neutralizing antibodies to conditioned media fromWNV-infected wild-type microglia revealed that TNF-α and IL-6were directly responsible for the observed bystander neuronal injury(Supplementary Fig. 3 online).
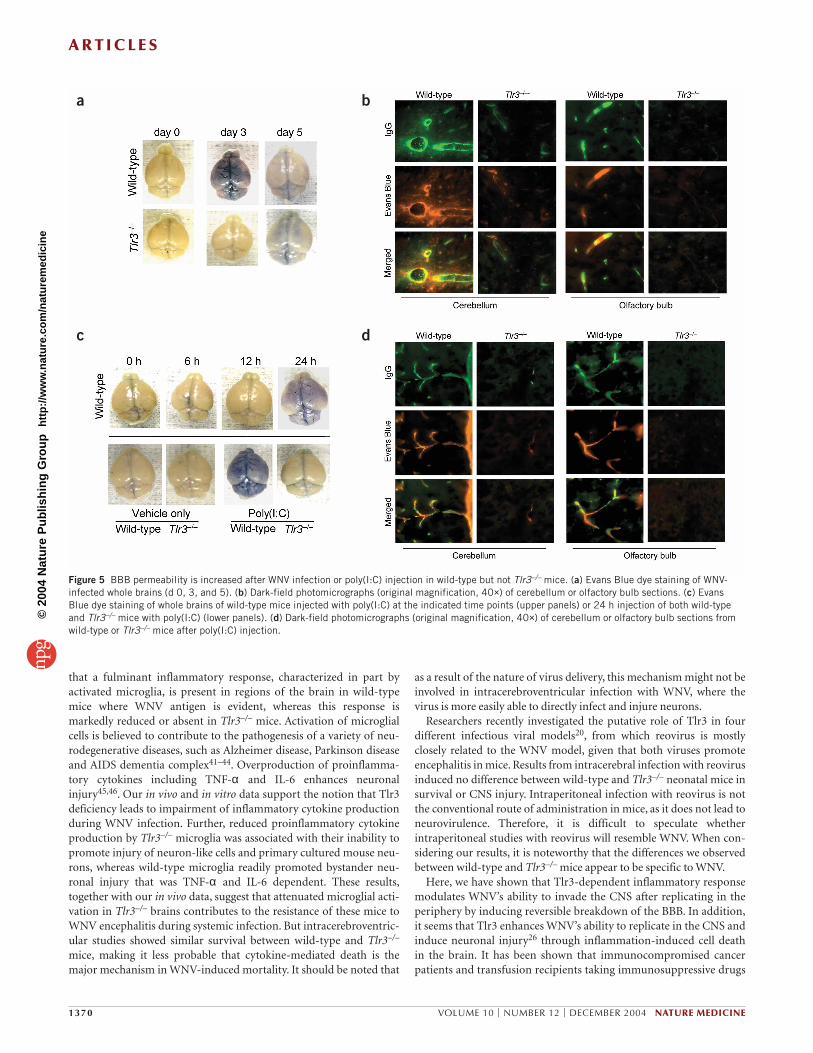
Tlr3-mediated signaling regulates WNV entry into the brainWe found that WNV infection was higher in the peripheral blood ofTlr3–/– mice compared to wild-type mice (Fig. 2b). Yet, in the brain,viral infection, immune and inflammatory responses and neuronalinjury were markedly reduced in Tlr3–/– animals (Figs. 3 and 4). Wetherefore hypothesized that Tlr3 might be involved in WNV entry intothe brain through the BBB. To evaluate BBB permeability, we adminis-tered Evans Blue, a dye that is normally excluded from the CNS, toWNV-infected wild-type and Tlr3–/– mice through an intraperitonealinjection at day 3 (when viral load is high in the periphery) and at day5 (when the virus is detected in the brain). At day 3, there were pro-nounced levels of Evans Blue dye in wild-type but not in Tlr3–/– mousebrains (Fig. 5a). However, at day 5, there were attenuated levels of dyein wild-type mouse brains, and the difference versus Tlr3–/– brains wasnot clear (Fig 5a). Fluorescence microscopy in wild-type brainsshowed areas with colocalized endogenous IgG and Evans Blue dye inand around cerebellar and olfactory bulb blood vessels, whereas Tlr3−
/− brains showed a relatively intact BBB (Fig. 5b). Endogenous IgG andEvans Blue dye were hardly detectable by microscopy at day 5 in bothgroups (data not shown). Moreover, western blot analysis revealedhigher levels of IgG in brain lysates from day 3 infected wild-type thanTlr3−/− mice (Supplementary Fig. 4 online).
Tlr3-mediated WNV entry into the brain could occur either directlyby the virus crossing the BBB itself or indirectly by being carried acrossthe BBB by infected leukocytes. Double immunofluorescence stainingfor CD45 or CD11b and WNV antigen revealed numerous CD45-positive leukocytes and CD11b-positive macrophages that stainedpositively for WNV in the olfactory bulb (Supplementary Fig. 4online), suggesting an indirect mechanism whereby infected peripheralleukocytes can act as viral carriers to the brain. It should be noted thatthese infected leukocytes were only detected in the olfactory bulb,
whereas WNV-infected neurons were found in numerous brain regionsthat were also characterized by BBB leakage (Supplementary Fig. 2 andSupplementary Table 1 online), suggesting that the direct mechanismof WNV crossing the BBB is also operating.
To further elucidate the role of Tlr3 in modulating BBB permeabil-ity, we administered intraperitoneal poly(I:C) to both wild-type andTlr3–/– mice. We first established a time course and a dose range ofpoly(I:C) that resulted in BBB compromise in wild-type mice, andfound that intraperitoneal injection of 50 µg/mL of poly(I:C) pro-duced robust BBB leakiness after 24 h (Fig. 5c). Using this dose andtime point, we challenged wild-type and Tlr3–/– mice and examinedBBB integrity. Results in both groups of mice were notably similarto data obtained from WNV-infected mice (Fig. 5c,d). As we hadshown reduced peripheral production of TNF-α and IL-6 in Tlr3–/–
mice after infection despite increased viremia in these mice, we
a
c
d
bFigure 3 Attenuated viral load and cytokine responses in Tlr3–/– brainsafter WNV infection. (a–c) Quantitative PCR results from mousehemibrains (unitless ratio ± 1 s.e.m.): (a) WNVE; (b) Tlr3; (c) cytokineproduction (percent cytokine reduction: Ifna, 70%; Ifnb1, 86%; Il6,81%; Tnf, 83%; Il12, 83%). For each of the wild-type and Tlr3–/– mousegroups shown in a–c, n = 4 at day 0 and 4, and n = 8 at day 5 and 6. For a and c, **P < 0.01 compared to wild-type mice. For b, *P < 0.05and **P < 0.01 compared to day 0. (d) Representative bright-fieldphotomicrographs of serial adjacent sections (olfactory bulbs at day 6after infection are shown, original magnification, x20) that were stainedfor CD45 or WNV antigen or assayed by TUNEL.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine
A R T I C L E S
NATURE MEDICINE VOLUME 10 | NUMBER 12 | DECEMBER 2004 1369
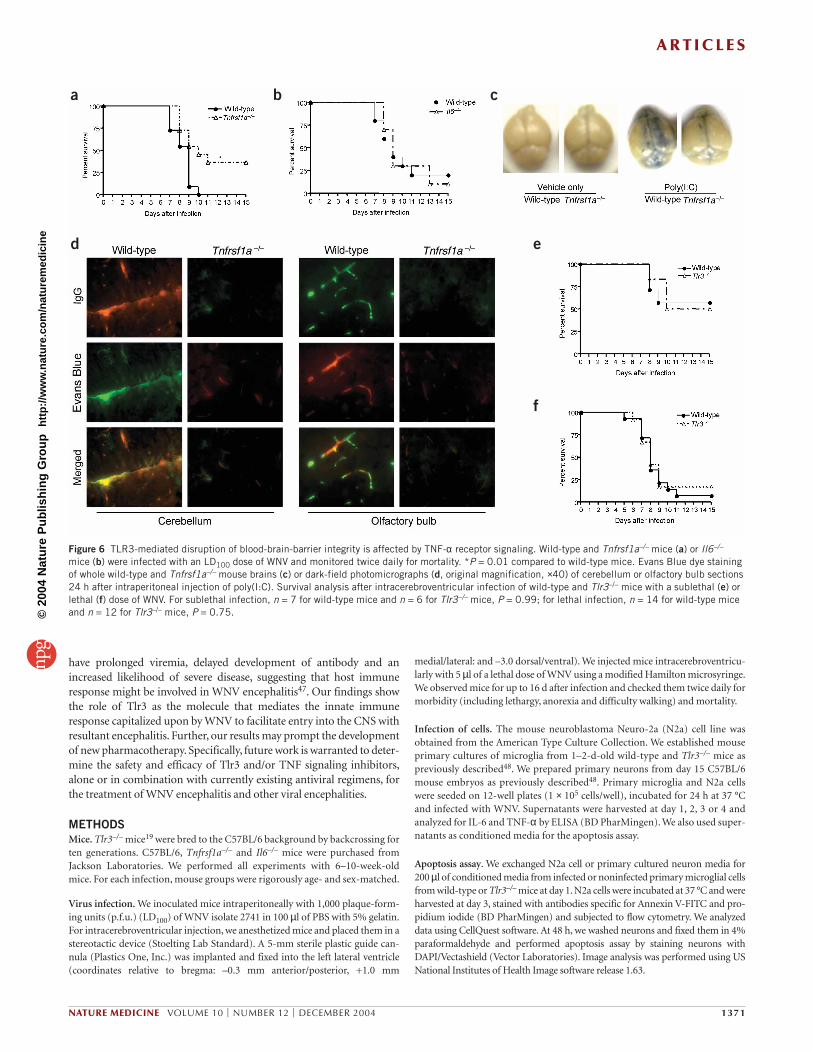
hypothesized that these cytokines may promote BBB compromise.Thus, we further challenged wild-type and TNF receptor 1–deficient (Tnfrsf1a–/–) or Il6–/– mice with WNV. Upon challengewith a lethal dose of virus, survival in the Tnfrsf1a–/– mice wassignificantly greater than in wild-type mice (Fig. 6a). But, thesurvival rate of Il6–/– mice showed no significant difference fromwild-type controls (Fig. 6b). Further, Tnfrsf1a–/– mice were refractoryto BBB leakiness induced by intraperitoneal poly(I:C) stimulation(Fig. 6c,d). Results from day 3 WNV-infected Tnfrsf1a–/– mice(Supplementary Fig. 4 online) were also notably similar to ourfindings from infected Tlr3–/– mice, in that Tnfrsf1a–/– mice hadmarkedly reduced BBB leakiness. Given that a number ofreports30–32 have shown that TNF-α positively regulates BBB per-meability, these data strongly suggest that TNF-α receptor 1 signal-ing is the signaling cascade downstream of Tlr3 that promotesWNV entry into the CNS.
Finally, we injected WNV directly into the brain through an intrac-erebroventricular route. Neither a lethal nor a sublethal dose of WNVproduced a significant difference in survival between wild-type andTlr3–/– mice (Fig. 6e,f). Collectively, these data suggest that Tlr3 medi-ates WNV entry from the peripheral blood into the brain by perme-abilizing the BBB, and that this entry event is critical for encephalitisand death of WNV-infected mice.
DISCUSSIONThe BBB is a complex structure that is highly impermeable to vascularmolecules, and is composed of vascular endothelial and smoothmuscle cells surrounded by a layer of astrocytic foot processes33,34.During systemic infection, WNV, like many CNS pathogens, mayinvade the CNS by crossing the BBB directly25,26, or by being carriedinto the brain by infected infiltrating myeloid cells35,36 and inducingencephalitis. In the case of inflammatory CNS pathologies, BBBintegrity can be compromised, allowing proteins to enter the CNSparenchyma30,34. We found increased levels of circulatinginflammatory cytokines including TNF-α and IL-6 in wild-type micethat were reduced in Tlr3–/– mice at day 3 following infection.Coincident with this time point, BBB leakiness was markedlyincreased in wild-type versus Tlr3–/– mice. Further, the resistance ofTnfrsf1a–/– mice to lethal WNV encephalitis and reduced BBB leakagein these animals following poly(I:C) injection or WNV infectionsuggests that TNF-α receptor 1 signaling plays a primary downstreamrole in Tlr3-mediated viral entry across the BBB into the CNS.
Tlr3–/– mice are more resistant to lethality following WNV infection,most probably because these mice have reduced viral entry into the brain.The contention that this mechanism is important for resistance of Tlr3–/–
mice to WNV encephalitis is bolstered by intracerebroventricular admin-istration of WNV. In these experiments, we did not find a difference insurvival between wild-type and Tlr3–/– mice, while systemic administra-
tion of poly(I:C) further confirmed that BBBcompromise was Tlr3 dependent.Earlier studieshave reported that certain conditions that maydisrupt the BBB, such as cold, stress, sodiumdodecylsulfate or lipopolysaccharide, couldenhance the level of WNV infection in thebrain26,37–39; however, the mechanisms linkingthese treatments to modulation of viral entryduring infection were not defined. Our resultsare the first to show that Tlr3-mediated inflam-matory response to WNV infection is critical formodulation of BBB permeability, therebyenabling viral entry into, and infection of, thebrain.
WNV may directly induce neuronal apop-tosis by infecting neurons28,40; alternatively,host responses, including lymphocyte (pre-dominantly CD8+ T cells) and microglial andmacrophage-associated inflammation11,28,29,may induce neuropathology11,28,29. We found
a b
c d
e f
g hFigure 4 Reduced neuronal and inflammatorypathology in Tlr3–/– mouse brains after WNVinfection. Panels are representative bright-field(a–f) or dark-field (g,h) photomicrographs from wild-type or Tlr3–/– mouse brain sections at day 5 (e,f) or day 6 (a–d,g,h) after infection: (a) H & E staining in cerebral cortices. (b) CD45immunohistochemistry in cerebral cortex. Imagesin c show H & E–stained sections; in d, imagesare serially adjacent sections in olfactory bulbsimmunostained for CD45. Images in e are CD45immunostained sections and images in f areserially adjacent sections (olfactory bulbs)stained for CD11b. (g,h) Immunofluorescentphotomicrographs from olfactory bulbs double-stained for CD45 (g) or CD11b (h) and WNVantigen. In c and d, original magnification, ×20;in a,b and e–h, original magnification, ×40.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine
A R T I C L E S
1370 VOLUME 10 | NUMBER 12 | DECEMBER 2004 NATURE MEDICINE
that a fulminant inflammatory response, characterized in part byactivated microglia, is present in regions of the brain in wild-typemice where WNV antigen is evident, whereas this response ismarkedly reduced or absent in Tlr3–/– mice. Activation of microglialcells is believed to contribute to the pathogenesis of a variety of neu-rodegenerative diseases, such as Alzheimer disease, Parkinson diseaseand AIDS dementia complex41–44. Overproduction of proinflamma-tory cytokines including TNF-α and IL-6 enhances neuronalinjury45,46. Our in vivo and in vitro data support the notion that Tlr3deficiency leads to impairment of inflammatory cytokine productionduring WNV infection. Further, reduced proinflammatory cytokineproduction by Tlr3–/– microglia was associated with their inability topromote injury of neuron-like cells and primary cultured mouse neu-rons, whereas wild-type microglia readily promoted bystander neu-ronal injury that was TNF-α and IL-6 dependent. These results,together with our in vivo data, suggest that attenuated microglial acti-vation in Tlr3–/– brains contributes to the resistance of these mice toWNV encephalitis during systemic infection. But intracerebroventric-ular studies showed similar survival between wild-type and Tlr3–/–
mice, making it less probable that cytokine-mediated death is themajor mechanism in WNV-induced mortality. It should be noted that
as a result of the nature of virus delivery, this mechanism might not beinvolved in intracerebroventricular infection with WNV, where thevirus is more easily able to directly infect and injure neurons.
Researchers recently investigated the putative role of Tlr3 in fourdifferent infectious viral models20, from which reovirus is mostlyclosely related to the WNV model, given that both viruses promoteencephalitis in mice. Results from intracerebral infection with reovirusinduced no difference between wild-type and Tlr3–/– neonatal mice insurvival or CNS injury. Intraperitoneal infection with reovirus is notthe conventional route of administration in mice, as it does not lead toneurovirulence. Therefore, it is difficult to speculate whetherintraperitoneal studies with reovirus will resemble WNV. When con-sidering our results, it is noteworthy that the differences we observedbetween wild-type and Tlr3–/– mice appear to be specific to WNV.
Here, we have shown that Tlr3-dependent inflammatory responsemodulates WNV’s ability to invade the CNS after replicating in theperiphery by inducing reversible breakdown of the BBB. In addition,it seems that Tlr3 enhances WNV’s ability to replicate in the CNS andinduce neuronal injury26 through inflammation-induced cell deathin the brain. It has been shown that immunocompromised cancerpatients and transfusion recipients taking immunosuppressive drugs
a b
c d
Figure 5 BBB permeability is increased after WNV infection or poly(I:C) injection in wild-type but not Tlr3–/– mice. (a) Evans Blue dye staining of WNV-infected whole brains (d 0, 3, and 5). (b) Dark-field photomicrographs (original magnification, 40×) of cerebellum or olfactory bulb sections. (c) EvansBlue dye staining of whole brains of wild-type mice injected with poly(I:C) at the indicated time points (upper panels) or 24 h injection of both wild-typeand Tlr3–/– mice with poly(I:C) (lower panels). (d) Dark-field photomicrographs (original magnification, 40×) of cerebellum or olfactory bulb sections fromwild-type or Tlr3–/– mice after poly(I:C) injection.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine
A R T I C L E S
NATURE MEDICINE VOLUME 10 | NUMBER 12 | DECEMBER 2004 1371
have prolonged viremia, delayed development of antibody and anincreased likelihood of severe disease, suggesting that host immuneresponse might be involved in WNV encephalitis47. Our findings showthe role of Tlr3 as the molecule that mediates the innate immuneresponse capitalized upon by WNV to facilitate entry into the CNS withresultant encephalitis. Further, our results may prompt the developmentof new pharmacotherapy. Specifically, future work is warranted to deter-mine the safety and efficacy of Tlr3 and/or TNF signaling inhibitors,alone or in combination with currently existing antiviral regimens, forthe treatment of WNV encephalitis and other viral encephalities.
METHODSMice. Tlr3–/– mice19 were bred to the C57BL/6 background by backcrossing forten generations. C57BL/6, Tnfrsf1a–/– and Il6–/– mice were purchased fromJackson Laboratories. We performed all experiments with 6–10-week-oldmice. For each infection, mouse groups were rigorously age- and sex-matched.
Virus infection. We inoculated mice intraperitoneally with 1,000 plaque-form-ing units (p.f.u.) (LD100) of WNV isolate 2741 in 100 µl of PBS with 5% gelatin.For intracerebroventricular injection, we anesthetized mice and placed them in astereotactic device (Stoelting Lab Standard). A 5-mm sterile plastic guide can-nula (Plastics One, Inc.) was implanted and fixed into the left lateral ventricle(coordinates relative to bregma: –0.3 mm anterior/posterior, +1.0 mm
medial/lateral: and –3.0 dorsal/ventral). We injected mice intracerebroventricu-larly with 5 µl of a lethal dose of WNV using a modified Hamilton microsyringe.We observed mice for up to 16 d after infection and checked them twice daily formorbidity (including lethargy, anorexia and difficulty walking) and mortality.
Infection of cells. The mouse neuroblastoma Neuro-2a (N2a) cell line wasobtained from the American Type Culture Collection. We established mouseprimary cultures of microglia from 1–2-d-old wild-type and Tlr3–/– mice aspreviously described48. We prepared primary neurons from day 15 C57BL/6mouse embryos as previously described48. Primary microglia and N2a cellswere seeded on 12-well plates (1 × 105 cells/well), incubated for 24 h at 37 °Cand infected with WNV. Supernatants were harvested at day 1, 2, 3 or 4 andanalyzed for IL-6 and TNF-α by ELISA (BD PharMingen). We also used super-natants as conditioned media for the apoptosis assay.
Apoptosis assay. We exchanged N2a cell or primary cultured neuron media for200 µl of conditioned media from infected or noninfected primary microglial cellsfrom wild-type or Tlr3–/– mice at day 1. N2a cells were incubated at 37 °C and wereharvested at day 3, stained with antibodies specific for Annexin V-FITC and pro-pidium iodide (BD PharMingen) and subjected to flow cytometry. We analyzeddata using CellQuest software. At 48 h, we washed neurons and fixed them in 4%paraformaldehyde and performed apoptosis assay by staining neurons withDAPI/Vectashield (Vector Laboratories). Image analysis was performed using USNational Institutes of Health Image software release 1.63.
a
e
f
b
d
c
Figure 6 TLR3-mediated disruption of blood-brain-barrier integrity is affected by TNF-α receptor signaling. Wild-type and Tnfrsf1a–/– mice (a) or Il6–/–
mice (b) were infected with an LD100 dose of WNV and monitored twice daily for mortality. *P = 0.01 compared to wild-type mice. Evans Blue dye stainingof whole wild-type and Tnfrsf1a–/– mouse brains (c) or dark-field photomicrographs (d, original magnification, ×40) of cerebellum or olfactory bulb sections24 h after intraperitoneal injection of poly(I:C). Survival analysis after intracerebroventricular infection of wild-type and Tlr3–/– mice with a sublethal (e) orlethal (f) dose of WNV. For sublethal infection, n = 7 for wild-type mice and n = 6 for Tlr3–/– mice, P = 0.99; for lethal infection, n = 14 for wild-type miceand n = 12 for Tlr3–/– mice, P = 0.75.©
2004
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
emed
icin
e

A R T I C L E S
1372 VOLUME 10 | NUMBER 12 | DECEMBER 2004 NATURE MEDICINE
Histology and immunohistochemistry. Mice were transcardially perfused withPBS. We removed brains and spleens and placed them in 4% paraformaldehydeovernight at 4 °C, and, in the case of cryoimmunohistochemistry, cryoprotectedthem with 10% (w/v) followed by 20% (w/v) sucrose in PBS for 24 h each beforeembedding them in Optimal Cutting Temperature compound. Subsequently,specimens were processed and histological slides were prepared for staining withvarious antibodies, TUNEL reaction or H & E staining. For paraffin sections, weprocessed antigen retrieval at 90 °C for 30 min in 10% (v/v) target retrieval solu-tion (Dakocytomation) or by treatment with 88% formic acid for 8 min. Theendogenous peroxidase activity was quenched in 0.3% H2O2 for 30 min at roomtemperature. We performed the staining using the Vectastain Elite ABC kit cou-pled to the 3′-3 diaminobenzadine substrate (Vector Laboratories). Rat antibodyspecific for mouse CD45 (clone YW 62.3, Serotec), CD11b (clone M1/70.15;CALTAG Laboratories), or mouse ascitic fluid WNV antibodies were used. Weperformed TdT-mediated dUTP nick-end labeling (TUNEL) on paraffin sec-tions using the ApopTag peroxidase in situ apoptosis detection kit (Serologicals).Sections were digitized with Kodak scientific imaging software (Eastman Kodak).
Immunofluorescence microscopy. We incubated frozen sections overnight at4 °C with various antibodies, washed and incubated the sections with goatantibody to rat IgG conjugated with Alexa Fluor 568 (Molecular Probes) andF(ab′)2 fragment of goat antibody to mouse IgG conjugated with Alexa Fluor488 (Molecular Probes) secondary antibodies. We mounted sections inVectashield fluorescence mounting medium with DAPI (Vector Laboratories).Immunofluorescence was observed using an Axioplan 2 microscope (KarlZeiss), and images were digitized using Openlab software (Improvision).
Evaluation of BBB permeability. Mice were injected intraperitoneally with 800 µlof 1% (w/v) Evans Blue dye, perfused with PBS 1 h later and evaluated for BBBleakage. We excised brains and macroscopically noted the extent of leakage ofEvans Blue dye. We processed hemibrains for 8–10-micron sagittal sections.Sections were stained with a F(ab′)2 fragment of goat antibody to mouse IgGconjugated with Alexa Fluor 488 for fluorescence microscopy as described above.We homogenized the other brain halves, lysed them with SDS-PAGE samplebuffer (Bio-Rad) and denatured proteins by boiling the halves. Equal amounts ofsamples were electrophoresed and subjected to western blotting with a mouseIgG peroxidase F(ab′)2 fragment antibody (Roche), and the expected band wasdetected using the Enhanced Chemiluminescence system (Amersham).
Statistical analysis. We calculated standard errors of the means (s.e.m.) andanalyzed data by nonpaired Student’s t-test for single mean comparisons orANOVA and post-hoc testing for multiple comparisons of the means. We per-formed survival curve comparisons using the log-rank test (equivalent to theMantel-Haenszel test, Prism software).
ACKNOWLEDGMENTSWe thank F. Manzo for assistance in preparing the manuscript and P. Cresswell for the use of the Zeiss Axioplan 2 fluorescence microscope. All experiments have been approved by the Yale University Animal Care Committee. These studieswere supported by grants from the United States National Institutes of Health(AIO55749) and the United States Department of Agriculture. R.A.F. is aninvestigator of the Howard Hughes Medical Institute. E.F. is the recipient ofa Burroughs Wellcome Clinical Scientist Award in Translational Research. T.W.is supported by a J. Kempner Postdoctoral Fellowship from University of TexasMedical Branch at Galveston. T.T. is supported by a Ruth L. Kirschstein NIH/NRSAPostdoctoral Fellowship.
COMPETING INTERESTS STATEMENTThe authors declare that they have no competing financial interests.
Received 24 May; accepted 1 November 2004Published online at http://www.nature.com/naturemedicine/
1. Campbell, G.L., Marfin, A.A., Lanciotti, R.S. & Gubler, D.J. West Nile virus. LancetInfect. Dis. 2, 519–529 (2002).
2. Wang, T. et al. Immunization of mice against West Nile virus with recombinant enve-lope protein. J. Immunol. 167, 5273–5277 (2001).
3. Lustig, S. et al. A live attenuated West Nile virus strain as a potential veterinary vac-cine. Viral Immunol. 13, 401–410 (2000).
4. Anderson, J.F. & Rahal, J.J. Efficacy of interferon alpha-2b and ribavirin againstWest Nile virus in vitro. Emerg. Infect. Dis. 8, 107–108 (2002).
5. Brinton, M.A. Host factors involved in West Nile virus replication. Ann. N Y Acad.Sci. 951, 207–219 (2001).
6. Lucas, M. et al. Infection of mouse neurones by West Nile virus is modulated by theinterferon-inducible 2’-5′ oligoadenylate synthetase 1b protein. Immunol. Cell Biol.81, 230–236 (2003).
7. Diamond, M.S., Shrestha, B., Marri, A., Mahan, D. & Engle, M. B cells and antibodyplay critical roles in the immediate defense of disseminated infection by West Nileencephalitis virus. J. Virol. 77, 2578–2586 (2003).
8. Diamond, M.S. et al. A critical role for induced IgM in the protection against WestNile virus infection. J. Exp. Med. 198, 1853–1862 (2003).
9. Wang, T. et al. IFN-gamma-producing gammadelta T cells help control murine WestNile virus infection. J. Immunol. 171, 2524–2531 (2003).
10. Kulkarni, A.B., Mullbacher, A. & Blanden, R.V. Functional analysis of macrophages,B cells and splenic dendritic cells as antigen-presenting cells in West Nile virus-spe-cific murine T lymphocyte proliferation. Immunol. Cell Biol. 69, 71–80 (1991).
11. Wang, Y., Lobigs, M., Lee, E. & Mullbacher, A. CD8+ T cells mediate recovery andimmunopathology in West Nile virus encephalitis. J. Virol. 77, 13323–13334(2003).
12. Qureshi, S.T. & Medzhitov, R. Toll-like receptors and their role in experimental mod-els of microbial infection. Genes Immun. 4, 87–94 (2003).
13. Yamamoto, M., Takeda, K. & Akira, S. TIR domain-containing adaptors define thespecificity of TLR signaling. Mol. Immunol. 40, 861–868 (2004).
14. Zhang, D. et al. A toll-like receptor that prevents infection by uropathogenic bacte-ria. Science 303, 1522–1526 (2004).
15. Tabeta, K. et al. Toll-like receptors 9 and 3 as essential components of innateimmune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci.USA 101, 3516–3521 (2004).
16. Rassa, J.C., Meyers, J.L., Zhang, Y., Kudaravalli, R. & Ross, S.R. Murine retrovirusesactivate B cells via interaction with toll-like receptor 4. Proc. Natl. Acad. Sci. USA99, 2281–2286 (2002).
17. Kurt-Jones, E.A. et al. Pattern recognition receptors TLR4 and CD14 mediateresponse to respiratory syncytial virus. Nat. Immunol. 1, 398–401 (2000).
18. Bieback, K. et al. Hemagglutinin protein of wild-type measles virus activates toll-likereceptor 2 signaling. J. Virol. 76, 8729–8736 (2002).
19. Alexopoulou, L., Holt, A.C., Medzhitov, R. & Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413,732–738 (2001).
20. Edelmann, K.H. et al. Does Toll-like receptor 3 play a biological role in virus infec-tions? Virology 322, 231–238 (2004).
21. Lund, J.M. et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7.Proc. Natl. Acad. Sci. USA 101, 5598–5603 (2004).
22. Diebold, S.S., Kaisho, T., Hemmi, H., Akira, S. & Reis, E.S.C. Innate antiviralresponses by means of TLR7-mediated recognition of single-stranded RNA. Science303, 1529–1531 (2004).
23. Heil, F. et al. Species-specific recognition of single-stranded RNA via toll-like recep-tor 7 and 8. Science 303, 1526–1529 (2004).
24. Samuel, C.E. Host genetic variability and West Nile virus susceptibility. Proc. Natl.Acad. Sci. USA 99, 11555–11557 (2002).
25. Halevy, M. et al. Loss of active neuroinvasiveness in attenuated strains of West Nilevirus: pathogenicity in immunocompetent and SCID mice. Arch. Virol. 137,355–370 (1994).
26. Lustig, S., Danenberg, H.D., Kafri, Y., Kobiler, D. & Ben-Nathan, D. Viral neuroinva-sion and encephalitis induced by lipopolysaccharide and its mediators. J. Exp. Med.176, 707–712 (1992).
27. Bush, T.G. et al. Leukocyte infiltration, neuronal degeneration, and neurite out-growth after ablation of scar-forming, reactive astrocytes in adult transgenic mice.Neuron 23, 297–308 (1999).
28. Xiao, S.Y., Guzman, H., Zhang, H., Travassos da Rosa, A.P. & Tesh, R.B. West Nilevirus infection in the golden hamster (Mesocricetus auratus): a model for West Nileencephalitis. Emerg. Infect. Dis. 7, 714–721 (2001).
29. Shrestha, B., Gottlieb, D. & Diamond, M.S. Infection and injury of neurons by WestNile encephalitis virus. J. Virol. 77, 13203–13213 (2003).
30. de Vries, H.E. et al. The influence of cytokines on the integrity of the blood-brainbarrier in vitro. J. Neuroimmunol. 64, 37–43 (1996).
31. Fiala, M. et al. TNF-alpha opens a paracellular route for HIV-1 invasion across theblood-brain barrier. Mol. Med. 3, 553–564 (1997).
32. Mayhan, W.G. Cellular mechanisms by which tumor necrosis factor-alpha producesdisruption of the blood-brain barrier. Brain Res. 927, 144–152 (2002).
33. Kakinuma, Y. et al. Impaired blood-brain barrier function in angiotensinogen-defi-cient mice. Nat. Med. 4, 1078–1080 (1998).
34. Paul, R. et al. Lack of IL-6 augments inflammatory response but decreases vascularpermeability in bacterial meningitis. Brain 126, 1873–1882 (2003).
35. Gollins, S.W. & Porterfield, J.S. Flavivirus infection enhancement in macrophages:an electron microscopic study of viral cellular entry. J. Gen. Virol. 66, 1969–1982(1985).
36. Cardosa, M.J., Porterfield, J.S. & Gordon, S. Complement receptor mediatesenhanced flavivirus replication in macrophages. J. Exp. Med. 158, 258–263(1983).
37. Ben-Nathan, D., Lustig, S. & Feuerstein, G. The influence of cold or isolation stresson neuroinvasiveness and virulence of an attenuated variant of West Nile virus. Arch.Virol. 109, 1–10 (1989).
38. Ben-Nathan, D., Huitinga, I., Lustig, S., van Rooijen, N. & Kobiler, D. West Nilevirus neuroinvasion and encephalitis induced by macrophage depletion in mice.Arch. Virol. 141, 459–469 (1996).
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine

A R T I C L E S
NATURE MEDICINE VOLUME 10 | NUMBER 12 | DECEMBER 2004 1373
39. Kobiler, D., Lustig, S., Gozes, Y., Ben-Nathan, D. & Akov, Y. Sodium dodecylsul-phate induces a breach in the blood-brain barrier and enables a West Nile virus vari-ant to penetrate into mouse brain. Brain Res. 496, 314–316 (1989).
40. Yang, J.S. et al. Induction of inflammation by West Nile virus capsid through thecaspase-9 apoptotic pathway. Emerg. Infect. Dis. 8, 1379–1384 (2002).
41. Michel, P.P., Hirsch, E.C. & Agid, Y. Parkinson disease: mechanisms of cell death.Rev. Neurol. (Paris) 158, S24–S32 (2002).
42. Garden, G.A. Microglia in human immunodeficiency virus-associated neurodegener-ation. Glia 40, 240–251 (2002).
43. Rogove, A.D., Lu, W. & Tsirka, S.E. Microglial activation and recruitment, but notproliferation, suffice to mediate neurodegeneration. Cell Death Differ. 9, 801–806(2002).
44. Ferencik, M., Novak, M., Rovensky, J. & Rybar, I. Alzheimer’s disease, inflammationand non-steroidal anti-inflammatory drugs. Bratisl. Lek. Listy 102, 123–132(2001).
45. Jeohn, G.H., Kong, L.Y., Wilson, B., Hudson, P. & Hong, J.S. Synergistic neurotoxiceffects of combined treatments with cytokines in murine primary mixed neuron/gliacultures. J. Neuroimmunol. 85, 1–10 (1998).
46. Meda, L. et al. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature 374, 647–650 (1995).
47. Pealer, L.N. et al. Transmission of West Nile virus through blood transfusion in theUnited States in 2002. N. Engl. J. Med. 349, 1236–1245 (2003).
48. Tan, J. et al. Microglial activation resulting from CD40–CD40L interaction after beta-amyloid stimulation. Science 286, 2352–2355 (1999).
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine
Copyright © 2022 FDOKUMEN




















