
Structures of magnetic null points in reconnection diffusion region: Cluster observations
Upload
independentCategory
view
0download
0
Phenotypic Effects of Null and Haploinsufficiency ofAcid-Labile Subunit in a Family with Two Novel IGFALSGene Mutations
Horacio M. Domene, Paula A. Scaglia, Aida Lteif, Farid H. Mahmud, Salman Kirmani, Jan Frystyk,Patricia Bedecarras, Mariana Gutierrez, and Hector G. Jasper
Centro de Investigaciones Endocrinologicas (H.M.D., P.A.S., P.B., M.G., H.G.J.), Hospital de Ninos “R. Gutierrez,” 1425Buenos Aires, Argentina; Pediatric Endocrinology and Metabolism (A.L., S.K.), Mayo Clinic, Rochester, Minnesota 55095;Endocrinology (F.H.M.), Children’s Hospital of Western Ontario, London, Canada N6A 4G5; and Medical ResearchLaboratories (J.F.), Clinical Institute, Aarhus University Hospital, DK-8000 Aarhus, Denmark
Context: IGF-I deficiency may result from impairment of GH secretionor action, or from defects in IGF-I synthesis, transport, or action. Com-plete deficiency of the acid-labile subunit (ALS), previously described intwo male patients, the only known inherited alteration in IGF-I trans-port, is characterized by severe circulating IGF-I and IGF binding pro-tein (IGFBP)-3 deficiency with only mild growth retardation.
Objective: Our objective was to study the characterization, at bio-chemical and molecular levels, of the cause for severe circulatingIGF-I and IGFBP-3 deficiency in a male patient with mild growthretardation.
Patients: We report an adolescent male with delayed growth andpubertal development (Tanner stage I, �2.00 SD score for height at theage of 15.3 yr), profound circulating IGF-I and IGFBP-3 deficiency,and poor response to GH treatment.
Results: The index case, as well as one of his brothers, and his sisterwere found to be compound heterozygotes for two novel IGFALS gene
mutations: C540R, a missense point mutation; and S195_197Rdup, a9-bp duplication. The parents and youngest brother were found to becarriers for one of these two mutations. The three affected siblings hadmarked reduction of IGF-I and IGFBP-3 levels, undetectable serumlevels of ALS, inability to form ternary complexes, and moderateinsulin resistance. All of them attained a normal near-adult height(between �1.0 and �0.5 SD score), which was nonetheless lower thanthat of their heterozygous brother. The IGF system was only modestlyaffected in the heterozygous carriers.
Conclusions: This study confirms the critical role of ALS in formingternary complexes and the maintenance of normal levels of IGF-I andIGFBP-3. Insulin resistance, pubertal delay in male patients, andpoor GH responsiveness seem to be frequent findings in ALS defi-ciency. However, haploinsufficiency of the IGFALS gene has no dis-cernible clinical effects with only modest impact on the IGF system.(J Clin Endocrinol Metab 92: 4444–4450, 2007)
THE ACID-LABILE SUBUNIT (ALS), a liver-derived,GH-dependent, 85-kD glycoprotein, is a key compo-
nent of the 150-kD IGF circulating ternary complex (1). Morethan 80% of the IGFs circulate as ternary complexes formedby one molecule each of IGF, IGF binding protein (IGFBP)-3or -5, and ALS (2, 3). When carried in ternary complexes, thehalf-life of IGF-I is more than 12 h, which is much longer thanthat of binary bound IGF-I (30–90 min) and free, unboundIGF-I (10 min) (4, 5).
The disruption of the IGFALS gene by targeted inactiva-tion in the mouse (6) as well as mutational inactivation of thegene in humans (7, 8) result in a marked reduction in cir-culating levels of both IGF-I and IGFBP-3, probably the resultof a more rapid clearance, due to either an increased effluxor an accelerated degradation. Despite the profound reduc-
tion in circulating IGF-I and IGFBP-3 levels, postnatal growthis only mildly affected if at all. Pubertal delay and insulinresistance were found in one ALS-deficient patient (7). How-ever, because that was the first reported case, it was not clearif these abnormalities were related to ALS deficiency per se,or merely coincidental findings. Thus, a recently reportednew ALS-deficient patient presented with pubertal devel-opment at an appropriate age (8).
We now report a family that was found to have threeaffected siblings with complete ALS deficiency, in associa-tion with two novel IGFALS gene mutations. The parents andone other sibling were found to be heterozygous carriers forone of the mutations. Thus, the effects of complete and hap-loinsufficiency of the IGFALS gene are described.
Subjects and Methods
Written informed consent was obtained from the parents. The indexcase (B2) and his siblings gave their assent to participate in this study.The study was conducted in accordance with the Helsinki Declaration.
The B2 was initially examined elsewhere in the United States at theage of 15.3 yr for delayed growth and pubertal development. He was thesecond of four siblings from nonconsanguineous, American, white par-ents of primarily Norwegian and German descent. Both parents hadnormal stature [father (F) 188.0 cm, �1.50 sd score (SDS); mother (M)157.5 cm, �0.93 SDS]. At the initial examination, his height was 156.2 cm
First Published Online August 28, 2007Abbreviations: ALS, Acid-labile subunit; B1, older brother; B2, index
case; B3, youngest brother; F, father; G1, sister; HOMA, homeostasismodel of assessment; IGFBP, IGF binding protein; LRR, leucine-richrepeat; M, mother; MPTH, midparental target height; rhGH, recombi-nant human GH; SDS, sd score.JCEM is published monthly by The Endocrine Society (http://www.endo-society.org), the foremost professional society serving the en-docrine community.
0021-972X/07/$15.00/0 The Journal of Clinical Endocrinology & Metabolism 92(11):4444–4450Printed in U.S.A. Copyright © 2007 by The Endocrine Society
doi: 10.1210/jc.2007-1152
4444

(�2.00 SDS), weight 56 kg (body mass index 22.9 kg/m2, �1.23 SDS),bone age 14 yr, testicular volume 2 cm3, and pubic hair development wasat Tanner stage I. Preliminary studies showed a normal GH response of10 �g/liter to an arginine test and low IGF-I levels of 12 �g/liter (1.56nmol/liter) (reference values 109–485 �g/liter or 14.2–63.0 nmol/liter).With the diagnosis of non-GH deficient short stature, recombinant hu-man GH (rhGH) treatment was started at the age 15.5 yr at a dose of 0.3mg/kg�wk. The dose was gradually increased to 0.8 mg/kg�wk due tothe poor clinical and IGF-I responses to treatment. Pretreatment growthvelocity was 6.2 cm/yr, whereas posttreatment velocity was 8.0 cm/yr,and IGF-I level only increased to 54 �g/liter (7.02 nmol/liter). At the ageof 16.1 yr, while the patient was on GH treatment, he was admitted tothe Mayo Clinic, Rochester, Minnesota. At that time he had a height of163.4 cm (�1.0 SDS), weight 64.8 kg (0.0 SDS), genital and pubic hairTanner stage I, and testicular volume 2.1–2.4 cm3. Laboratory evaluationshowed: IGF-I, 57 �g/liter (7.41 nmol/liter) (237–601 �g/liter or 30.8–78.1 nmol/liter); IGFBP-3, 0.3 �g/liter (10.5 nmol/liter) (2.5–4.8 �g/literor 87.5–168 nmol/liter); GHBP, 504 pmol/liter (431–1892); negativeanti-GH antibodies; and prepubertal testosterone levels of 0.28 ng/ml(0.97 nmol/liter). The dose of rhGH was reduced to 0.5 mg/kg�wk, andim testosterone enanthate was added at 50 mg/month. Total length ofrhGH treatment was 17 months. The association of mild growth retar-dation with marked reduction of IGF-I and IGFBP-3 levels, and theinability of GH to improve growth and serum IGF-I levels suggested thediagnosis of ALS deficiency, which was confirmed by undetectableserum levels of ALS (�0.5 mg/liter, reference values 20–35 mg/liter).
Subsequently, blood samples were obtained from the B2 and all hisimmediate family members for the study of the GH-IGF axis and formolecular characterization of the IGFALS gene.
Serum levels of T4, free T4, T3, TSH, antithyroperoxidase antibodies,LH, FSH, estradiol, and prolactin were determined by electrochemolu-minescence (Elecsys; Roche, Indianapolis, IN), levels of GH and insulinby immunofluorometric assay (Immulite; Diagnostic Products Corp.,Los Angeles, CA), and testosterone by RIA (Diagnostic Systems Labo-ratories, Inc., Webster, TX). IGF-I levels were determined by RIA afterserum extraction by the acid-ethanol method followed by cryoprecip-itation (9), IGFBP-3 by immunoradiometric assay, and ALS by ELISA(10) using commercial kits (Diagnostic Systems Laboratories, Inc.).
Serum IGFBPs were evaluated by Western ligand blot (11), and ALSprotein by Western immunoblot using antibodies against the aminoterminal (hALS1–34) and the carboxy terminal regions (hALS551–578;Diagnostic Systems Laboratories, Inc.) of the ALS protein (7).
Ternary complex formation was evaluated by neutral size-exclusionchromatography (7). A total of 100 �l serum was incubated overnightat 22 C with 3.5 � 106 counts per minute of 125I-IGF-I, and then cross-linked with the addition of disuccinimidyl suberate (Sigma-Aldrich, St.Louis, MO). Samples were chromatographed on a HiPrep 16/60Sephacryl S-200HR column (Amersham Pharmacia Biotech AB, Upp-sala, Sweden). A total of 500-�l aliquots was loaded onto the column, and1-ml fractions were collected and counted.
Genomic DNA from all family members was isolated from peripheralleukocytes based on the use of cetyltrimethylammonium bromide lysisbuffer and isoamyl alcohol-chloroform extraction (12). Exons 1 and 2 andcontiguous intron sequences, corresponding to the IGFALS gene (Gen-Bank accession no. AF192554), were amplified by PCR using oligonu-cleotide primers flanking both exons (exon 1: F1 5�-ccgcagtggggaaatc-caga-3�; and R1 5�-gggtggaggacgggacagag-3�. Exon 2 was amplified intwo fragments: fragment A: F2A, 5�-gagccaagccctgagcgtctgt-3�; and R2A5�-ccttgaggaagagtcggcgg-3�; and fragment B: F2B 5�-aacgtgttcgtgcagct-gcc-3� and R2B 5�-tccccagcacaaggtgagcc-3�). PCR products were purifiedusing Wizard Clean up (Promega, Madison, WI). Afterward, the puri-fied products were sequenced in both directions in an ABI 3730xl au-tomatic sequencer (Macrogen, Seoul, South Korea) using the same oli-gonucleotides primers used for amplification and/or internaloligonucleotide primers (for exon 1: INT-1 reverse 5�-agcgggctcagggt-cacaga-3�; for exon 2 fragment A: INT-A1 reverse 5�-tcgtcatcgtagctgca-gac-3�; INT-A2 reverse 5�-ctgctcagacggttgttgct-3�; INT-A3 reverse 5�-gcagcgatgaggttgcggtcca-3�; and INT-A1 forward 5�-agcaacaaccgtctgagcag-3�; for fragment B: 2A reverse 5�-ccttgaggaagagtcggcgg-3�; INT-B forward5�-agctgcctgggacgcatccg-3�; and 2B reverse 5�-tccccagcacaaggtgagcc-3�).
The presence of a missense point mutation (c.1618T�C; C540R) anda 9-bp duplication (c.583_591dup9; p.S195_197Rdup) was examined inall family members by restriction fragment length polymorphism, taking
advantage of the fact that each mutation introduces a new restriction sitefor the Btg I and Fsp I enzymes (New England BioLabs, Inc., Ipswich,MA), respectively.
ResultsGrowth and pubertal development
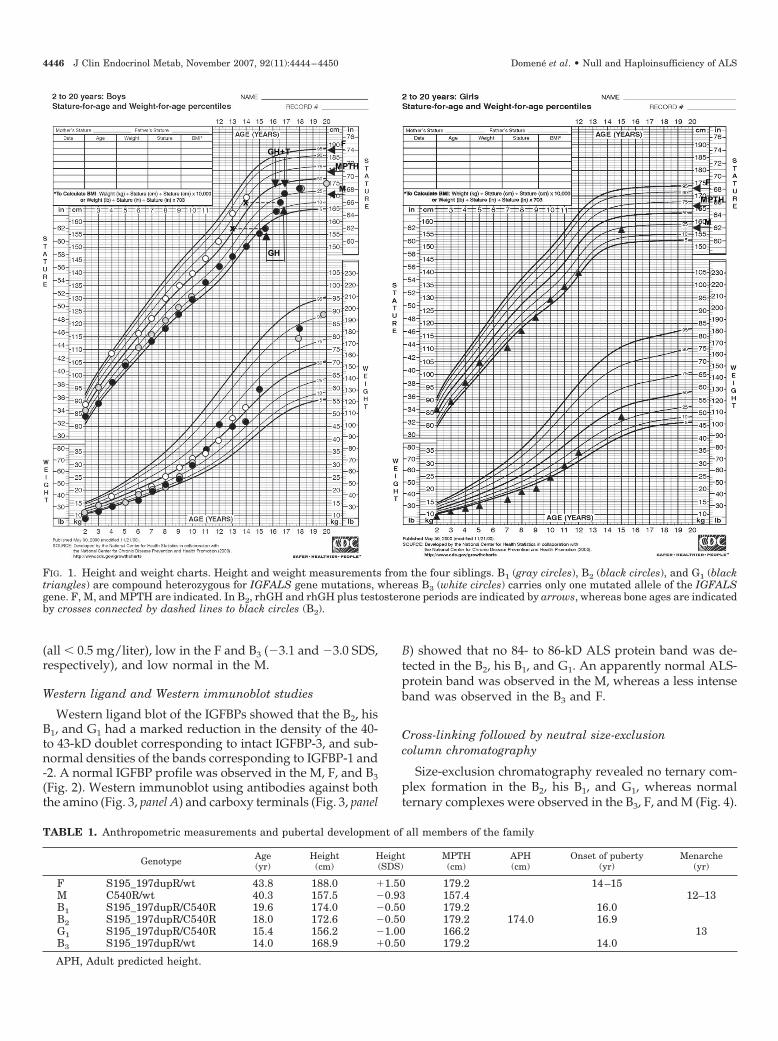
The B2 had delay of growth and pubertal development(Tanner stage I at 16.1 yr). After a 6-month course of testos-terone treatment, puberty developed normally, and hereached Tanner stage V for both sexual development andpubic hair at the age of 18.0 yr. Although his height wasbelow normal at 16 yr, he had a normal growth spurt whilereceiving rhGH plus testosterone, reaching a normal near-adult height of 172.6 cm (�0.5 SDS) at the age of 18.0 yr (Fig.1), close to his midparental target height (MPTH) of 179.2 cm(Table 1). Retrospectively collected data showed a similarpubertal development and growth history for the oldestbrother, who entered puberty at 16 yr of age. He was foundto be at Tanner stage V and had a normal adult height of 174.0cm (�0.5 SDS) at the age of 19.6 yr. In contrast, the pubertaldevelopment was normal in the youngest sister (G1), whohad menarche at the age of 13 yr, and a normal height of 156.2cm (�1.00 SDS) at 15.4 yr of age. There was no history ofpubertal delay in either of the parents, or in the youngestbrother (B3) (Table 1).
Biochemical profile
Total, high-density lipoprotein-, and low-density lipopro-tein-cholesterol, as well as triglycerides, were normal exceptfor the B2 [total cholesterol 205 mg/dl (5.3 mmol/liter), ref-erence values � 200 mg/dl (5.17 mmol/liter); and triglyc-erides 183 mg/dl (2.07 mmol/liter), reference values � 160mg/dl; (1.81 mmol/liter)]. Levels of calcium, phosphorous,and PTH were normal in all family members. Thyroid func-tion evaluated by TSH, and total, free T4 and T3 levels wasalso normal in all family members (data not shown).
Gonadal function evaluation
All subjects studied had normal LH, FSH, prolactin, andestradiol levels according to their sex and pubertal stage(data not shown). Testosterone levels were normal in themales (7.0, 5.1, 5.8, and 6.1 ng/ml, or 24.3, 17.7, 20.1, and 21.2nmol/liter for the F and three brothers, respectively).
Glucose–insulin
Although glucose levels were normal in all the childrenand their parents, insulin levels, and consequently thehomeostasis model of assessment (HOMA) index, wereelevated in the B2, his older brother (B1), and younger G1(Table 2).
GH-IGF axis
Basal GH levels were elevated only in the B2 (6.8 �g/liter).Circulating levels of IGF-I and IGFBP-3 were markedly de-creased in the B2, his B1, and G1, whereas IGF-I levels weresubnormal in the F, low normal in the M, and normal in theB3 (Table 2). ALS was undetectable in the B2, his B1, and G1
Domene et al. • Null and Haploinsufficiency of ALS J Clin Endocrinol Metab, November 2007, 92(11):4444–4450 4445
(all � 0.5 mg/liter), low in the F and B3 (�3.1 and �3.0 SDS,respectively), and low normal in the M.
Western ligand and Western immunoblot studies
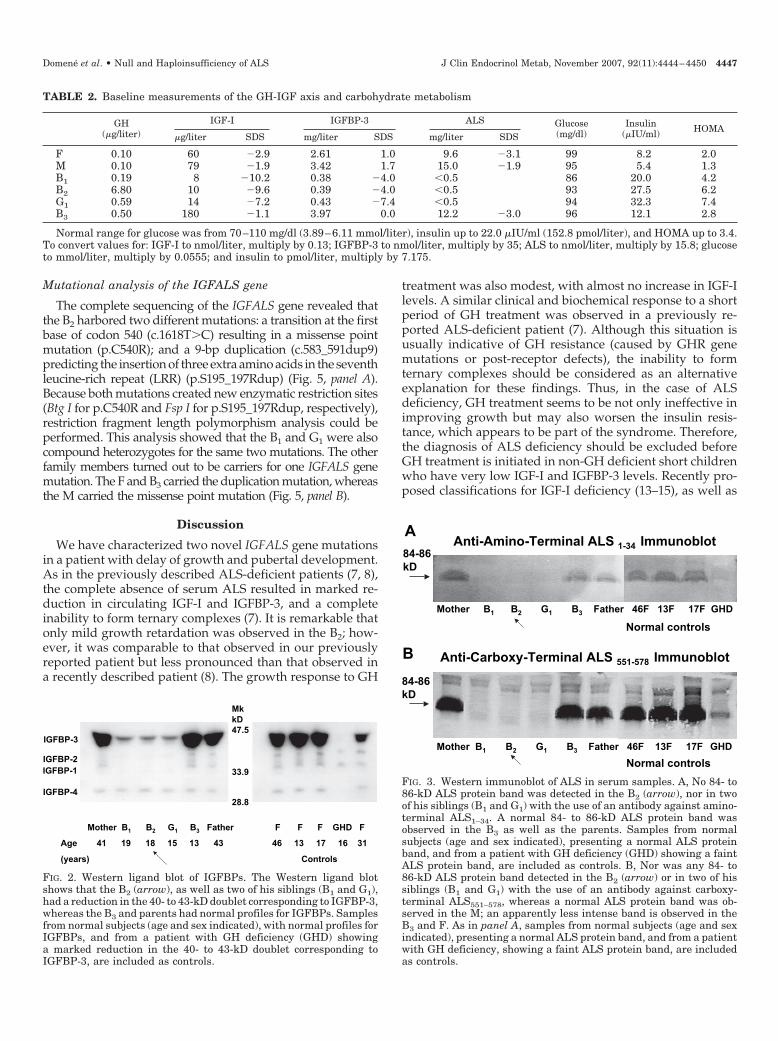
Western ligand blot of the IGFBPs showed that the B2, hisB1, and G1 had a marked reduction in the density of the 40-to 43-kD doublet corresponding to intact IGFBP-3, and sub-normal densities of the bands corresponding to IGFBP-1 and-2. A normal IGFBP profile was observed in the M, F, and B3(Fig. 2). Western immunoblot using antibodies against boththe amino (Fig. 3, panel A) and carboxy terminals (Fig. 3, panel
B) showed that no 84- to 86-kD ALS protein band was de-tected in the B2, his B1, and G1. An apparently normal ALS-protein band was observed in the M, whereas a less intenseband was observed in the B3 and F.
Cross-linking followed by neutral size-exclusioncolumn chromatography
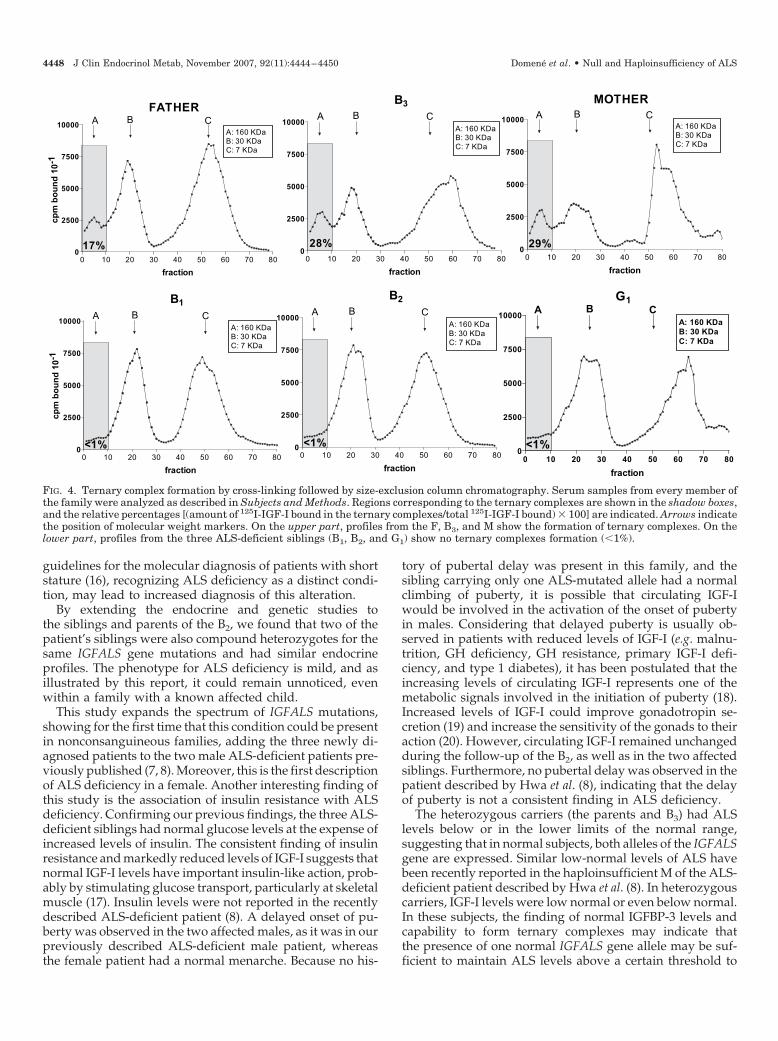
Size-exclusion chromatography revealed no ternary com-plex formation in the B2, his B1, and G1, whereas normalternary complexes were observed in the B3, F, and M (Fig. 4).
TABLE 1. Anthropometric measurements and pubertal development of all members of the family
Genotype Age(yr)
Height(cm)
Height(SDS)
MPTH(cm)
APH(cm)
Onset of puberty(yr)
Menarche(yr)
F S195_197dupR/wt 43.8 188.0 �1.50 179.2 14–15M C540R/wt 40.3 157.5 �0.93 157.4 12–13B1 S195_197dupR/C540R 19.6 174.0 �0.50 179.2 16.0B2 S195_197dupR/C540R 18.0 172.6 �0.50 179.2 174.0 16.9G1 S195_197dupR/C540R 15.4 156.2 �1.00 166.2 13B3 S195_197dupR/wt 14.0 168.9 �0.50 179.2 14.0
APH, Adult predicted height.
GH
GH+TF
M
MPTH
M
MPTH
F
X
X
FIG. 1. Height and weight charts. Height and weight measurements from the four siblings. B1 (gray circles), B2 (black circles), and G1 (blacktriangles) are compound heterozygous for IGFALS gene mutations, whereas B3 (white circles) carries only one mutated allele of the IGFALSgene. F, M, and MPTH are indicated. In B2, rhGH and rhGH plus testosterone periods are indicated by arrows, whereas bone ages are indicatedby crosses connected by dashed lines to black circles (B2).
4446 J Clin Endocrinol Metab, November 2007, 92(11):4444–4450 Domene et al. • Null and Haploinsufficiency of ALS
Mutational analysis of the IGFALS gene
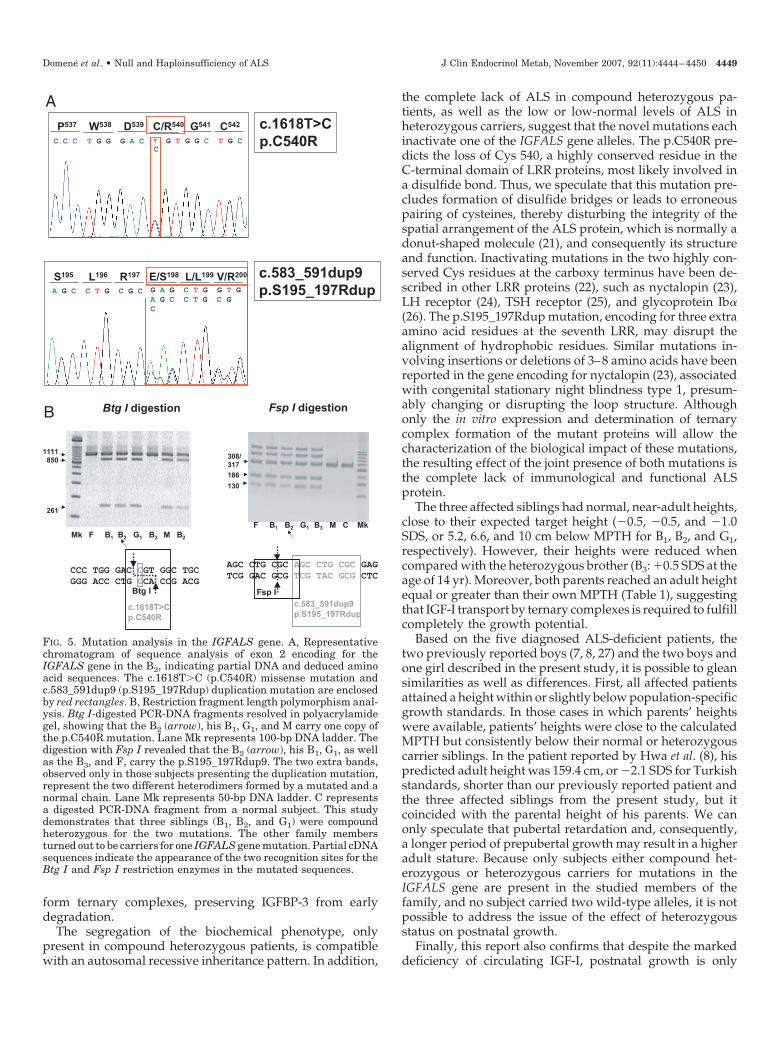
The complete sequencing of the IGFALS gene revealed thatthe B2 harbored two different mutations: a transition at the firstbase of codon 540 (c.1618T�C) resulting in a missense pointmutation (p.C540R); and a 9-bp duplication (c.583_591dup9)predicting the insertion of three extra amino acids in the seventhleucine-rich repeat (LRR) (p.S195_197Rdup) (Fig. 5, panel A).Because both mutations created new enzymatic restriction sites(Btg I for p.C540R and Fsp I for p.S195_197Rdup, respectively),restriction fragment length polymorphism analysis could beperformed. This analysis showed that the B1 and G1 were alsocompound heterozygotes for the same two mutations. The otherfamily members turned out to be carriers for one IGFALS genemutation. The F and B3 carried the duplication mutation, whereasthe M carried the missense point mutation (Fig. 5, panel B).
Discussion
We have characterized two novel IGFALS gene mutationsin a patient with delay of growth and pubertal development.As in the previously described ALS-deficient patients (7, 8),the complete absence of serum ALS resulted in marked re-duction in circulating IGF-I and IGFBP-3, and a completeinability to form ternary complexes (7). It is remarkable thatonly mild growth retardation was observed in the B2; how-ever, it was comparable to that observed in our previouslyreported patient but less pronounced than that observed ina recently described patient (8). The growth response to GH
treatment was also modest, with almost no increase in IGF-Ilevels. A similar clinical and biochemical response to a shortperiod of GH treatment was observed in a previously re-ported ALS-deficient patient (7). Although this situation isusually indicative of GH resistance (caused by GHR genemutations or post-receptor defects), the inability to formternary complexes should be considered as an alternativeexplanation for these findings. Thus, in the case of ALSdeficiency, GH treatment seems to be not only ineffective inimproving growth but may also worsen the insulin resis-tance, which appears to be part of the syndrome. Therefore,the diagnosis of ALS deficiency should be excluded beforeGH treatment is initiated in non-GH deficient short childrenwho have very low IGF-I and IGFBP-3 levels. Recently pro-posed classifications for IGF-I deficiency (13–15), as well as
Mother B1 B2 G1 B3 Father F F F GHD F
Age 41 19 18 15 13 43 46 13 17 16 31
(years) Controls
MkkD47.5
33.9
28.8
IGFBP-3
IGFBP-2IGFBP-1
IGFBP-4
FIG. 2. Western ligand blot of IGFBPs. The Western ligand blotshows that the B2 (arrow), as well as two of his siblings (B1 and G1),had a reduction in the 40- to 43-kD doublet corresponding to IGFBP-3,whereas the B3 and parents had normal profiles for IGFBPs. Samplesfrom normal subjects (age and sex indicated), with normal profiles forIGFBPs, and from a patient with GH deficiency (GHD) showinga marked reduction in the 40- to 43-kD doublet corresponding toIGFBP-3, are included as controls.
Anti-Carboxy-Terminal ALS 551-578 Immunoblot
Anti-Amino-Terminal ALS 1-34 Immunoblot
Mother B1 B2 G1 B3 Father 46F 13F 17F GHD
84-86kD
84-86kD
Mother B1 B2 G1 B3 Father 46F 13F 17F GHD
A
B
Normal controls
Normal controls
FIG. 3. Western immunoblot of ALS in serum samples. A, No 84- to86-kD ALS protein band was detected in the B2 (arrow), nor in twoof his siblings (B1 and G1) with the use of an antibody against amino-terminal ALS1–34. A normal 84- to 86-kD ALS protein band wasobserved in the B3 as well as the parents. Samples from normalsubjects (age and sex indicated), presenting a normal ALS proteinband, and from a patient with GH deficiency (GHD) showing a faintALS protein band, are included as controls. B, Nor was any 84- to86-kD ALS protein band detected in the B2 (arrow) or in two of hissiblings (B1 and G1) with the use of an antibody against carboxy-terminal ALS551–578, whereas a normal ALS protein band was ob-served in the M; an apparently less intense band is observed in theB3 and F. As in panel A, samples from normal subjects (age and sexindicated), presenting a normal ALS protein band, and from a patientwith GH deficiency, showing a faint ALS protein band, are includedas controls.
TABLE 2. Baseline measurements of the GH-IGF axis and carbohydrate metabolism
GH(�g/liter)
IGF-I IGFBP-3 ALS Glucose(mg/dl)
Insulin(�IU/ml) HOMA
�g/liter SDS mg/liter SDS mg/liter SDS
F 0.10 60 �2.9 2.61 1.0 9.6 �3.1 99 8.2 2.0M 0.10 79 �1.9 3.42 1.7 15.0 �1.9 95 5.4 1.3B1 0.19 8 �10.2 0.38 �4.0 �0.5 86 20.0 4.2B2 6.80 10 �9.6 0.39 �4.0 �0.5 93 27.5 6.2G1 0.59 14 �7.2 0.43 �7.4 �0.5 94 32.3 7.4B3 0.50 180 �1.1 3.97 0.0 12.2 �3.0 96 12.1 2.8
Normal range for glucose was from 70–110 mg/dl (3.89–6.11 mmol/liter), insulin up to 22.0 �IU/ml (152.8 pmol/liter), and HOMA up to 3.4.To convert values for: IGF-I to nmol/liter, multiply by 0.13; IGFBP-3 to nmol/liter, multiply by 35; ALS to nmol/liter, multiply by 15.8; glucoseto mmol/liter, multiply by 0.0555; and insulin to pmol/liter, multiply by 7.175.
Domene et al. • Null and Haploinsufficiency of ALS J Clin Endocrinol Metab, November 2007, 92(11):4444–4450 4447
guidelines for the molecular diagnosis of patients with shortstature (16), recognizing ALS deficiency as a distinct condi-tion, may lead to increased diagnosis of this alteration.
By extending the endocrine and genetic studies tothe siblings and parents of the B2, we found that two of thepatient’s siblings were also compound heterozygotes for thesame IGFALS gene mutations and had similar endocrineprofiles. The phenotype for ALS deficiency is mild, and asillustrated by this report, it could remain unnoticed, evenwithin a family with a known affected child.
This study expands the spectrum of IGFALS mutations,showing for the first time that this condition could be presentin nonconsanguineous families, adding the three newly di-agnosed patients to the two male ALS-deficient patients pre-viously published (7, 8). Moreover, this is the first descriptionof ALS deficiency in a female. Another interesting finding ofthis study is the association of insulin resistance with ALSdeficiency. Confirming our previous findings, the three ALS-deficient siblings had normal glucose levels at the expense ofincreased levels of insulin. The consistent finding of insulinresistance and markedly reduced levels of IGF-I suggests thatnormal IGF-I levels have important insulin-like action, prob-ably by stimulating glucose transport, particularly at skeletalmuscle (17). Insulin levels were not reported in the recentlydescribed ALS-deficient patient (8). A delayed onset of pu-berty was observed in the two affected males, as it was in ourpreviously described ALS-deficient male patient, whereasthe female patient had a normal menarche. Because no his-
tory of pubertal delay was present in this family, and thesibling carrying only one ALS-mutated allele had a normalclimbing of puberty, it is possible that circulating IGF-Iwould be involved in the activation of the onset of pubertyin males. Considering that delayed puberty is usually ob-served in patients with reduced levels of IGF-I (e.g. malnu-trition, GH deficiency, GH resistance, primary IGF-I defi-ciency, and type 1 diabetes), it has been postulated that theincreasing levels of circulating IGF-I represents one of themetabolic signals involved in the initiation of puberty (18).Increased levels of IGF-I could improve gonadotropin se-cretion (19) and increase the sensitivity of the gonads to theiraction (20). However, circulating IGF-I remained unchangedduring the follow-up of the B2, as well as in the two affectedsiblings. Furthermore, no pubertal delay was observed in thepatient described by Hwa et al. (8), indicating that the delayof puberty is not a consistent finding in ALS deficiency.
The heterozygous carriers (the parents and B3) had ALSlevels below or in the lower limits of the normal range,suggesting that in normal subjects, both alleles of the IGFALSgene are expressed. Similar low-normal levels of ALS havebeen recently reported in the haploinsufficient M of the ALS-deficient patient described by Hwa et al. (8). In heterozygouscarriers, IGF-I levels were low normal or even below normal.In these subjects, the finding of normal IGFBP-3 levels andcapability to form ternary complexes may indicate thatthe presence of one normal IGFALS gene allele may be suf-ficient to maintain ALS levels above a certain threshold to
FATHER
0 10 20 30 40 50 60 70 800
2500
5000
7500
10000 A B CA: 160 KDaB: 30 KDaC: 7 KDa
17%
fraction
MOTHER
0 10 20 30 40 50 60 70 800
2500
5000
7500
10000 A B CA: 160 KDaB: 30 KDaC: 7 KDa
29%
fraction
B3
0 10 20 30 40 50 60 70 800
2500
5000
7500
10000A B C
A: 160 KDaB: 30 KDaC: 7 KDa
28%
fraction
B1
0 10 20 30 40 50 60 70 800
2500
5000
7500
10000A B C
A: 160 KDaB: 30 KDaC: 7 KDa
<1%
fraction
cpm
bo
un
d 1
0-1
cpm
bo
un
d 1
0-1
B2
0 10 20 30 40 50 60 70 800
2500
5000
7500
10000A B C
A: 160 KDaB: 30 KDaC: 7 KDa
<1%
fraction
G1
0 10 20 30 40 50 60 70 800
2500
5000
7500
10000A B C
A: 160 KDaB: 30 KDaC: 7 KDa
<1%
fraction
FIG. 4. Ternary complex formation by cross-linking followed by size-exclusion column chromatography. Serum samples from every member ofthe family were analyzed as described in Subjects and Methods. Regions corresponding to the ternary complexes are shown in the shadow boxes,and the relative percentages [(amount of 125I-IGF-I bound in the ternary complexes/total 125I-IGF-I bound) � 100] are indicated. Arrows indicatethe position of molecular weight markers. On the upper part, profiles from the F, B3, and M show the formation of ternary complexes. On thelower part, profiles from the three ALS-deficient siblings (B1, B2, and G1) show no ternary complexes formation (�1%).
4448 J Clin Endocrinol Metab, November 2007, 92(11):4444–4450 Domene et al. • Null and Haploinsufficiency of ALS
form ternary complexes, preserving IGFBP-3 from earlydegradation.
The segregation of the biochemical phenotype, onlypresent in compound heterozygous patients, is compatiblewith an autosomal recessive inheritance pattern. In addition,
the complete lack of ALS in compound heterozygous pa-tients, as well as the low or low-normal levels of ALS inheterozygous carriers, suggest that the novel mutations eachinactivate one of the IGFALS gene alleles. The p.C540R pre-dicts the loss of Cys 540, a highly conserved residue in theC-terminal domain of LRR proteins, most likely involved ina disulfide bond. Thus, we speculate that this mutation pre-cludes formation of disulfide bridges or leads to erroneouspairing of cysteines, thereby disturbing the integrity of thespatial arrangement of the ALS protein, which is normally adonut-shaped molecule (21), and consequently its structureand function. Inactivating mutations in the two highly con-served Cys residues at the carboxy terminus have been de-scribed in other LRR proteins (22), such as nyctalopin (23),LH receptor (24), TSH receptor (25), and glycoprotein Ib�(26). The p.S195_197Rdup mutation, encoding for three extraamino acid residues at the seventh LRR, may disrupt thealignment of hydrophobic residues. Similar mutations in-volving insertions or deletions of 3–8 amino acids have beenreported in the gene encoding for nyctalopin (23), associatedwith congenital stationary night blindness type 1, presum-ably changing or disrupting the loop structure. Althoughonly the in vitro expression and determination of ternarycomplex formation of the mutant proteins will allow thecharacterization of the biological impact of these mutations,the resulting effect of the joint presence of both mutations isthe complete lack of immunological and functional ALSprotein.
The three affected siblings had normal, near-adult heights,close to their expected target height (�0.5, �0.5, and �1.0SDS, or 5.2, 6.6, and 10 cm below MPTH for B1, B2, and G1,respectively). However, their heights were reduced whencompared with the heterozygous brother (B3: �0.5 SDS at theage of 14 yr). Moreover, both parents reached an adult heightequal or greater than their own MPTH (Table 1), suggestingthat IGF-I transport by ternary complexes is required to fulfillcompletely the growth potential.
Based on the five diagnosed ALS-deficient patients, thetwo previously reported boys (7, 8, 27) and the two boys andone girl described in the present study, it is possible to gleansimilarities as well as differences. First, all affected patientsattained a height within or slightly below population-specificgrowth standards. In those cases in which parents’ heightswere available, patients’ heights were close to the calculatedMPTH but consistently below their normal or heterozygouscarrier siblings. In the patient reported by Hwa et al. (8), hispredicted adult height was 159.4 cm, or �2.1 SDS for Turkishstandards, shorter than our previously reported patient andthe three affected siblings from the present study, but itcoincided with the parental height of his parents. We canonly speculate that pubertal retardation and, consequently,a longer period of prepubertal growth may result in a higheradult stature. Because only subjects either compound het-erozygous or heterozygous carriers for mutations in theIGFALS gene are present in the studied members of thefamily, and no subject carried two wild-type alleles, it is notpossible to address the issue of the effect of heterozygousstatus on postnatal growth.
Finally, this report also confirms that despite the markeddeficiency of circulating IGF-I, postnatal growth is only
P537 W538 D539 C/R540 G541 C542
S195 L196 R197 E/S198 L/L199 V/R200
G A G C T G G T GA G C C T G C GC
c.1618T>Cp.C540R
c.583_591dup9p.S195_197Rdup
C
A G C C T G C G C
C C C T G G G A C G T G G C T G C
308/317
186
130
Fsp I digestion
AGC CTG CGC AGC CTG CGC GAGTCG GAC GCG TCG TAC GCG CTC
c.583_591dup9p.S195_197Rdup
Fsp I
Btg I digestion
CCC TGG GAC CGT GGC TGCGGG ACC CTG GCA CCG ACG
c.1618T>Cp.C540R
Btg I
1111850
261
F B1 B2 G1 B3 M C MkMk F B1 B2 G1 B3 M B2
A
B
FIG. 5. Mutation analysis in the IGFALS gene. A, Representativechromatogram of sequence analysis of exon 2 encoding for theIGFALS gene in the B2, indicating partial DNA and deduced aminoacid sequences. The c.1618T�C (p.C540R) missense mutation andc.583_591dup9 (p.S195_197Rdup) duplication mutation are enclosedby red rectangles. B, Restriction fragment length polymorphism anal-ysis. Btg I-digested PCR-DNA fragments resolved in polyacrylamidegel, showing that the B2 (arrow), his B1, G1, and M carry one copy ofthe p.C540R mutation. Lane Mk represents 100-bp DNA ladder. Thedigestion with Fsp I revealed that the B2 (arrow), his B1, G1, as wellas the B3, and F, carry the p.S195_197Rdup9. The two extra bands,observed only in those subjects presenting the duplication mutation,represent the two different heterodimers formed by a mutated and anormal chain. Lane Mk represents 50-bp DNA ladder. C representsa digested PCR-DNA fragment from a normal subject. This studydemonstrates that three siblings (B1, B2, and G1) were compoundheterozygous for the two mutations. The other family membersturned out to be carriers for one IGFALS gene mutation. Partial cDNAsequences indicate the appearance of the two recognition sites for theBtg I and Fsp I restriction enzymes in the mutated sequences.
Domene et al. • Null and Haploinsufficiency of ALS J Clin Endocrinol Metab, November 2007, 92(11):4444–4450 4449

mildly affected. It is not known how almost normal growthis maintained in this condition. This may result from pre-served peripheral IGF-I production acting at the cartilagelevel. Increased IGF-I efflux to the peripheral tissues at theexpense of the circulating IGF-I pool and IGF-I-independentGH growth promoting effects may also have an effect.
In summary, this family illustrates the main phenotypicfeatures of ALS deficiency: 1) marked circulating IGF-I de-ficiency, insulin resistance, and delayed puberty in malepatients, with a mild effect on linear growth in patients withbiallelic deficiency; and 2) no clinical effects and only mod-erate impact on the IGF system in subjects carrying onemutated allele of the IGFALS gene.
Acknowledgments
We thank all members of this family who agreed to participate in thisstudy. We also thank Mrs. Perla Rossano for her invaluable technicalassistance.
Received May 24, 2007. Accepted August 17, 2007.Address all correspondence and requests for reprints to: Horacio M.
Domene, Centro de Investigaciones Endocrinologicas, Hospital de Ni-nos “R. Gutierrez,” Gallo 1330, 1425 Buenos Aires, Argentina. E-mail:[email protected].
This work was supported by grants from the Agencia Nacional dePromocion Cientıfica y Tecnologica (BID 1278/OC-AR PICT-2003 no.05-14354) and an Independent Research Grant from Pfizer Global Phar-maceutical. P.A.S. is supported by a Fellowship from Pfizer GlobalPharmaceutical. J.F. is supported by grants from the Danish MedicalResearch Council.
Disclosure Statement: F.H.M. has received lecture fees less than$10,000, and J.F. has received consulting fees less than $10,000. H.M.D.,P.A.S., A.L., S.K., P.B., M.G., and H.G.J. have nothing to declare.
References
1. Baxter RC, Martin JL, Beniac VA 1989 High molecular weight insulin-likegrowth factor binding protein complex: purification and properties of theacid-labile subunit from human serum. J Biol Chem 264:11843–11848
2. Baxter RC 1990 Circulating levels and molecular distribution of the acid-labile(�)-subunit of the high molecular weight insulin-like growth factor-bindingprotein complex. J Clin Endocrinol Metab 70:1347–1353
3. Twigg SM, Baxter RC 1998 Insulin-like growth factor (IGF)-binding protein5 forms an alternative ternary complex with IGFs and the acid-labile subunit.J Biol Chem 273:6074–6079
4. Guler HP, Zapf J, Schmid C, Froesh ER 1989 Insulin-like growth factors I andII in healthy man. Estimations of half-lives and production rates. Acta Endo-crinol (Copenh) 121:753–758
5. Zapf J, Hauri C, Futo E, Hussain M, Rutishauser J, Maack CA, Froesch ER1995 Intravenously injected insulin-like growth factor (IGF) I/IGF bindingprotein-3 complex exerts insulin-like effects in hypophysectomized, but not innormal rats. J Clin Invest 95:179–186
6. Ueki I, Ooi GT, Tremblay ML, Hurst KR, Bach LA, Boisclair YR 2000 In-activation of the acid labile subunit gene in mice results in mild retardation ofpostnatal growth despite profound disruptions in the circulating insulin-likegrowth factor system. Proc Natl Acad Sci USA 97:6868–6873
7. Domene HM, Bengolea SV, Martınez AS, Ropelato MG, Pennisi P, ScagliaP, Heinrich JJ, Jasper HG 2004 Deficiency of the circulating insulin-like growthfactor system associated with inactivation of the acid-labile subunit gene.N Engl J Med 350:570–577
8. Hwa V, Haeusler G, Pratt KL, Little BM, Frisch H, Koller D, Rosenfeld RG2006 Total absence of functional acid labile subunit, resulting in severe insulin-like growth factor deficiency and moderate growth failure. J Clin EndocrinolMetab 91:1826–1831
9. Martınez AS, Domene HM, Ropelato MG, Jasper HG, Pennisi PA, EscobarMe, Heinrich JJ 2000 Estrogen priming effect on growth hormone (GH) pro-vocative test: a useful tool for the diagnosis of GH deficiency. J Clin EndocrinolMetab 85:4168–4172
10. Khosravi MJ, Diamandi A, Mistry J, Krishna RG, Khare A 1997 Acid-labilesubunit of human insulin-like growth factor-binding protein complex: Mea-surement, molecular, and clinical evaluation. J Clin Endocrinol Metab 82:3944–3951
11. Hossenlopp P, Seurin D, Segovia-Quinson B, Hardouin S, Binoux M 1986Analysis of serum insulin-like growth factor binding proteins using Westernblotting: use of the method for titration of the binding proteins and competitivebinding studies. Anal Biochem 154:138–143
12. Del Sal G, Manfioletti G, Schneider C 1989 The CTAB-DNA precipitationmethod: a common mini-scale preparation of template DNA from phagemids,phages or plasmids suitable for sequencing. Biotechniques 7:514–520
13. Rosenfeld RG 2006 Molecular mechanisms of IGF-I deficiency. Horm Res65(Suppl 1):15–20
14. Rosenbloom AL, Guevara-Aguirre J 2006 Controversy in clinical endocrinol-ogy: reclassification of insulin-like growth factor I production and actiondisorders. J Clin Endocrinol Metab 91:4232–4234
15. Cohen P 2006 Controversy in clinical endocrinology: problems with reclassi-fication of insulin-like growth factor I production and action disorders. J ClinEndocrinol Metab 91:4235–4236
16. Walenkamp MJ, Wit JM 2006 Genetic disorders in the growth hormone–insulin-like growth factor-I axis. Horm Res 66:221–230
17. Clemmons DR 2006 Involvement of insulin-like growth factor-I in the controlof glucose homeostasis. Curr Opin Pharmacol 6:620–625
18. Hiney JK, Ojeda SR, Dees WL 1991 Insulin-like growth factor I: a possiblemetabolic signal involved in the regulation of female puberty. Neuroendo-crinology 54:420–423
19. Hiney JK, Srivastava V, Nyberg CL, Ojeda SR, Dees WL 1996 Insulin-likegrowth factor I of peripheral origin acts centrally to accelerate the initiation offemale puberty. Endocrinology 137:3717–3728
20. Kasson BG, Hsueh AJ 1987 Insulin-like growth factor-I augments gonado-tropin-stimulated androgen biosynthesis by cultured rat testicular cells. MolCell Endocrinol 52:27–34
21. Janosi JB, Ramsland PA, Mott MR, Firth SM, Baxter RC, Delhanty PJ 1999The acid-labile subunit of the serum insulin-like growth factor-binding proteincomplexes. Structural determination by molecular modeling and electron mi-croscopy. J Biol Chem 274:23328–23332
22. Matsushima N, Tachi N, Kuroki Y, Enkhbayar P, Osaki M, Kamiya M,Kretsinger RH 2005 Structural analysis of leucine-rich-repeat variants in pro-teins associated with human diseases. Cell Mol Life Sci 62:2771–2791
23. Pusch CM, Zeitz C, Brandau O, Pesch K, Achatz H, Feil S, Scharfe C, MaurerJ, Jacobi FK, Pinckers A, Andreasson S, Hardcastle A, Wissinger B, BergerW, Meindl A 2000 The complete form of X-linked congenital stationary nightblindness is caused by mutations in a gene encoding a leucine-rich repeatprotein. Nat Genet 26:324–327
24. Laue L, Chan WY, Hsueh AJ, Kudo M, Hsu SY, Wu SM, Blomberg L, CutlerJr GB 1995 Genetic heterogeneity of constitutively activating mutations of thehuman luteinizing hormone receptor in familial male-limited precocious pu-berty. Proc Natl Acad Sci USA 92:1906–1910
25. Alberti L, Proverbio MC, Costagliola S, Romoli R, Boldrighini B, VigoneMC, Weber G, Chiumello G, Beck-Peccoz P, Persani L 2002 Germline mu-tations of TSH receptor gene as cause of nonautoimmune subclinical hypo-thyroidism. J Clin Endocrinol Metab 87:2549–2555
26. Simsek S, Noris P, Lozano M, Pico M, von dem Borne AE, Ribera A, GallardoD 1994 Cys209Ser mutation in the platelet membrane glycoprotein Ib alphagene is associated with Bernard-Soulier syndrome. Br J Haematol 88:839–844
27. Domene HM, Martınez AS, Frystyk J, Bengolea SV, Ropelato MG, ScagliaPA, Chen J-W, Heuck C, Wolthers OD, Heinrich JJ, Jasper HG 2007 Normalgrowth spurt and final height despite low levels of all forms of circulating IGF-Iin a patient with ALS deficiency. Horm Res 67:243–249
JCEM is published monthly by The Endocrine Society (http://www.endo-society.org), the foremost professional society serving theendocrine community.
4450 J Clin Endocrinol Metab, November 2007, 92(11):4444–4450 Domene et al. • Null and Haploinsufficiency of ALS
Copyright © 2022 FDOKUMEN




















