
Complexation of a four-strand tetraalcohol with labile metal ions probed by electrospray mass...
10
Inorganica Chimica Acta 328 (2002) 159 – 168 www.elsevier.com/locate/ica Complexation of a four-strand tetraalcohol with labile metal ions probed by electrospray mass spectrometry Geoffrey A. Lawrance *, Mark J. Robertson, Sutrisno, Ellak I. von Nagy-Felsobuki Discipline of Chemistry, School of Biological and Chemical Sciences, The Uniersity of Newcastle, Callaghan 2308, Australia Received 11 July 2001; accepted 4 October 2001 Abstract The saturated, stereodefined tetraalcohol 2,3,5,6-endo,endo,endo,endo -tetrakis(hydroxymethyl)bicyclo[2.2.1]heptane (tetol, L 1 ) and the simple alcohol butane-1,3-diol (L 2 ) form complexes with alkali metal ions (lithium, sodium, potassium, rubidium and caesium), alkali earth cations (magnesium, calcium, strontium and barium) and Ga(III) and Ce(IV) in aqueous solution, characterised by electrospray ionisation mass spectrometry (ESMS). Metal ion exchange between the Li + complex of L 1 and the other metal ions is rapid, with a range of M(L 1 ) n m+ species detected, in addition to solvated species. With the alkal metal ions, M(L 1 ) + and M(L 1 ) 2 + are dominant, although speciation varies with metal ion size. For the alkaline earth ions, a range of complex ions up to n =8 are observed, although n =1–3 dominate. A preference for M(L 1 ) 2 2 + with Mg 2 + versus M(L 1 ) 3 2 + with Ca 2 + may again relate to a larger ion size. For the higher-charged Ga(III) and Ce(IV), hydroxo species M(OH)(L 1 ) n (m−1) + are dominant reflecting bulk solution behaviour, which the ESMS studies appear to map generally. © 2002 Elsevier Science B.V. All rights reserved. Keywords: Polyalcohol; Alkali metal ions; Alkaline earth metal ions; Metal complexes; Electrospray ionisation mass spectrometry 1. Introduction Polyalcohols (polyols), as potential ligands, offer a set of hydroxy groups for binding metal ions. The interaction with metal ions is generally weak, and the complex formation by a neutral polyalcohol may not be significant in aqueous solution, although deprotonated polyalcohols represent strong and efficient metal bind- ing agents [1], as, since polyols are weak acids (pK a 12), the deprotonated polyols are even stronger bases than polyamines. Due to the high tendency of the deprotonated hydroxy group to bind to more than one metal ion, polyalcohols are particularly suitable for the stabilisation of polynuclear complexes, with bridging by -alkoxo groups a basic structural element in such complexes [2]. For metal ions in alkaline media, com- plex formation with polyols competes with hydrolytic polymerisation reactions. Moreover, partially hy- drolysed species (polynuclear polyolato complexes with additional oxo or hydroxo bridges) are also known. Such aggregates can be regarded to be composed of an inorganic metal-oxide or metal-hydroxide core that is protected by the peripherally coordinated polyolato ligand, thereby preventing complete hydrolysis and yielding species that are intermediate between well- defined low molecular weight complexes and solid phases. This intermediate state is currently of particular interest with respect to applications in materials science (e.g. the sol – gel technique) [3]. The wide structural diversity found in the naturally occurring polyols gives rise to a variety of different geometries for donor sets, each with characteristic coordination properties. This diversity could possibly be used to control complex stability in terms of metal ion size [4]. Further, selective metal complex formation could be of interest in terms of a controlled protection or activation of individual hydroxy groups providing opportunities for devising new routes in synthetic organic chemistry [5]. The ability of a variety of natural occurring polyol ligands, such as glycerol or sorbitol, to act as sequester- * Corresponding author. Tel.: +61-2-492 15471; fax: +61-2-492 15472. E-mail address: [email protected] (G.A. Lawrance). 0020-1693/02/$ - see front matter © 2002 Elsevier Science B.V. All rights reserved. PII:S0020-1693(01)00728-9
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Complexation of a four-strand tetraalcohol with labile metal ions probed by electrospray mass...

Inorganica Chimica Acta 328 (2002) 159–168
www.elsevier.com/locate/ica
Complexation of a four-strand tetraalcohol with labile metal ionsprobed by electrospray mass spectrometry
Geoffrey A. Lawrance *, Mark J. Robertson, Sutrisno, Ellak I. von Nagy-FelsobukiDiscipline of Chemistry, School of Biological and Chemical Sciences, The Uni�ersity of Newcastle, Callaghan 2308, Australia
Received 11 July 2001; accepted 4 October 2001
Abstract
The saturated, stereodefined tetraalcohol 2,3,5,6-endo,endo,endo,endo-tetrakis(hydroxymethyl)bicyclo[2.2.1]heptane (tetol, L1)and the simple alcohol butane-1,3-diol (L2) form complexes with alkali metal ions (lithium, sodium, potassium, rubidium andcaesium), alkali earth cations (magnesium, calcium, strontium and barium) and Ga(III) and Ce(IV) in aqueous solution,characterised by electrospray ionisation mass spectrometry (ESMS). Metal ion exchange between the Li+ complex of L1 and theother metal ions is rapid, with a range of M(L1)n
m+ species detected, in addition to solvated species. With the alkal metal ions,M(L1)+ and M(L1)2
+ are dominant, although speciation varies with metal ion size. For the alkaline earth ions, a range of complexions up to n=8 are observed, although n=1–3 dominate. A preference for M(L1)2
2+ with Mg2+ versus M(L1)32+ with Ca2+
may again relate to a larger ion size. For the higher-charged Ga(III) and Ce(IV), hydroxo species M(OH)(L1)n(m−1)+ are
dominant reflecting bulk solution behaviour, which the ESMS studies appear to map generally. © 2002 Elsevier Science B.V. Allrights reserved.
Keywords: Polyalcohol; Alkali metal ions; Alkaline earth metal ions; Metal complexes; Electrospray ionisation mass spectrometry
1. Introduction
Polyalcohols (polyols), as potential ligands, offer aset of hydroxy groups for binding metal ions. Theinteraction with metal ions is generally weak, and thecomplex formation by a neutral polyalcohol may not besignificant in aqueous solution, although deprotonatedpolyalcohols represent strong and efficient metal bind-ing agents [1], as, since polyols are weak acids (pKa�12), the deprotonated polyols are even stronger basesthan polyamines. Due to the high tendency of thedeprotonated hydroxy group to bind to more than onemetal ion, polyalcohols are particularly suitable for thestabilisation of polynuclear complexes, with bridging by�-alkoxo groups a basic structural element in suchcomplexes [2]. For metal ions in alkaline media, com-plex formation with polyols competes with hydrolyticpolymerisation reactions. Moreover, partially hy-
drolysed species (polynuclear polyolato complexes withadditional oxo or hydroxo bridges) are also known.Such aggregates can be regarded to be composed of aninorganic metal-oxide or metal-hydroxide core that isprotected by the peripherally coordinated polyolatoligand, thereby preventing complete hydrolysis andyielding species that are intermediate between well-defined low molecular weight complexes and solidphases. This intermediate state is currently of particularinterest with respect to applications in materials science(e.g. the sol–gel technique) [3]. The wide structuraldiversity found in the naturally occurring polyols givesrise to a variety of different geometries for donor sets,each with characteristic coordination properties. Thisdiversity could possibly be used to control complexstability in terms of metal ion size [4]. Further, selectivemetal complex formation could be of interest in termsof a controlled protection or activation of individualhydroxy groups providing opportunities for devisingnew routes in synthetic organic chemistry [5].
The ability of a variety of natural occurring polyolligands, such as glycerol or sorbitol, to act as sequester-
* Corresponding author. Tel.: +61-2-492 15471; fax: +61-2-49215472.
E-mail address: [email protected] (G.A.Lawrance).
0020-1693/02/$ - see front matter © 2002 Elsevier Science B.V. All rights reserved.
PII: S 0 0 2 0 -1693 (01 )00728 -9

G.A. Lawrance et al. / Inorganica Chimica Acta 328 (2002) 159–168160
ing agents—preventing formation of solid metal oxidesor hydroxides in alkaline aqueous solution—has beenwell known for many years [6]. However, the correctstructures of the complexes that were formed were notelucidated until a number of recent X-ray crystal struc-tures [7,8]. The use of polyols as selective receptors formetal ions has been reviewed [9]. However, the detailedsynthetic and structural studies of sugar–metal com-plexes have been limited due to their complicated stere-ochemistry, multi-functionality and the hygroscopicnature of species. Synthetic polyols, particularly thosewith pre-defined isomeric character, present themselvesas more practical molecules for study of metal ion–polyol interactions. We have sought ways of examiningthe relatively weak complexation between metal ionsand neutral polyalcohols in aqueous solution, and haveidentified electrospray mass spectrometry (ESMS) as apotentially revealing method [10]. Spectroscopic meth-ods have been used to investigate polyol and relatedpolyaminol ligand complexation previously [11], butwere not able to provide definitive answers regardingsolution structures.
Examination of polyol–metal ion interactions bymass spectrometry (MS) does not appear to have beenpursued previously, although complexation of weakinteractions of the polyethers 18-crown-6 andcryptand[2.2.2] with alkali metal cations using ESMShas been probed [12]. In the present study, we haveapplied ESMS to characterise the tetraalcohol 2,3,5,6-endo,endo,endo,endo-tetrakis(hydroxymethyl)bicyclo-[2.2.1]heptane (tetol, L1), a saturated, stereodefinedmolecule, interacting with alkali metal ions (lithium,sodium, potassium, rubidium and caesium), alkali earthcations (magnesium, calcium, strontium and barium)and higher-charged ions, gallium(III) and cerium(IV).The interaction of a simple chelating model ligand withalkali metal cations is also examined. Tetol is the firstexample of a four-strand ligand where four pendantgroups extend from a rigid core in one direction. Thisstudy involves the direct observation of metal ion ex-change in solution by means of ESMS influenced by therelative stability of complexes formed. The ‘soft’ ionisa-tion technique of ESMS, which has now been appliedto a range of both inert and labile complexes, appearsto reflect speciation in solution [13,14]. Thus this ESMSstudy offers the potential to provide information oncomplexation in bulk media.
2. Experimental
2.1. Materials
All reagents were commercially certified ACS gradeor better. Chloride salts of metal ions were usually
employed, except for cerium(IV), where the sulfate wasused. Butane-1,3-diol (L2) was obtained from Sigma(98%). Doubly deionised Milli-Q water (Millipore) wasused as solvent. All chemicals were used as purchasedwithout further purification. The tetraalcohol 2,3,5,6-endo,endo,endo,endo-tetrakis(hydroxymethyl) bicy-clo[2.2.1] heptane (tetol) was synthesised by a variation[10] of a reported method [15] isolated as the lithiumsalt LiL1Cl. Some was converted to KL1Cl salt by ionexchange on a column of Dowex 50W cation exchangeresin. The LiL1Cl has been characterised by an X-raycrystal structures analysis [10].
2.2. Electrospray ionisation mass spectrometry
For LiL1Cl and KL1Cl, sample solutions were pre-pared by dissolving the solid samples in deionised wa-ter. For butane-1,3-diol (L2), the solution was preparedby dissolution of a weighed amount of the alcohol inwater. The concentration of all sample solutions was10−4 M. For investigation of cation exchange oflithium complexes with other cations, aqueous solutionsof LiL1Cl with the cations in equimolar concentrationswere prepared. Similarly, for the simple model ligandbutane-1,3-diol (L2), 1:1 solutions of metal:ligand wereprepared. All analyte samples (in 1.8-ml vials) wereintroduced readily using an auto injector, which wasthoroughly rinsed with solvent between injections. Allthe ESMS experiments were performed on a VG Plat-form II single quadrupole mass spectrometer (Micro-mass Ltd, Altrincham, UK) coupled to a highperformance liquid chromatography (HPLC) binarypump system. For all spectral acquisitions, the tip ofthe capillary was at a potential of �3.5 kV relative toground. The source temperature was maintained at80 °C, and nitrogen was used as the bath and nebulis-ing gas. The mobile phase solution was the same as thesolvent of the analyte solution. An optimum flow rateof 10 �l min−1 was employed. The cone voltage (CV)was varied between +20 to +60 for positive-ion and−20 to −50 V for negative-ion mode. A Reodyneinjector fitted with a 10-�l loop was used to inject thesample solution into the flow of the mobile phase.
Mass spectra were acquired by scanning the quadru-pole mass filter from m/z 2000 to 2, and ions weredetected by means of a scintillator detector. Approxi-mately 40 scans were summed to give a mass spectrum.All data were acquired and processed using the Micro-mass MassLynx system. Experimental peak valuesthroughout this study are identified by the m/z ratio ofthe most abundant peak in the parent group. Calcu-lated m/z values tabulated are those based on the mostabundant isotopes. Peak intensities are cited as percent-ages of the base (major) peak intensity (%BPI).

G.A. Lawrance et al. / Inorganica Chimica Acta 328 (2002) 159–168 161
3. Results and discussion
The molecule L1 is a stereochemically definedmolecule with four �CH2�OH arms pendant in onedirection to a stereorigid but saturated bicy-clo[2.2.1]heptane frame. The arms are sufficiently flex-ible to either wrap up a metal ion in a 1:1 mode, or elseto share binding of ions with other molecules in dimericor other polymeric motifs. The X-ray structure of theLiL1Cl complex displays a polymeric structure in thesolid state [10]. However, this is not anticipated topersist in solution. To pursue the solution structure ofthe labile and relatively spectroscopically silent alkaliand alkaline earth ion complexes, we have turned to thetechnique of ESMS, which has been used previously to
provide information about other labile systems, includ-ing inorganic clusters. Although not necessarily definingthe bulk solution structure strictly, speciation in this‘soft’ MS technique does appear to identify thermody-namically significant species.
3.1. Positi�e-ion ESMS of LiL1Cl, KL1Cl and H+L2
The positive-ion mode ESMS for the LiL1Cl complexreveals a series of lithium complexes and their solvatedions (Fig. 1(a)). The dominant peaks at a CV of 30 Varise from complexes with observed m/z of 223.0,(100% BPI) and 439.3 (78% BPI) assigned to (LiL1)+
(calc. m/z 223) and (Li(L1)2)+ (calc. m/z 439). Further,smaller peaks assigned to higher ratio Li:ligand speciesand solvated ions are observed. For KL1Cl (Fig. 1(b)),a series of potassium complexes with increasing num-bers of ligands (up to four) was observed [namely(KL1)+, (K(L1)2)+, (K(L1)3)+and (K(L1)4)+] as well asa series of solvated ions. For both lithium and potas-sium complexes, minor peaks at m/z 216.2, 198.2, 181.2and 164.2 were identified at a CV of 30 V assigned tomono-protonated ligands that have lost successive hy-droxy groups through dehydration reactions. Alcoholdehydration reactions to form alkenes can occur se-quentially for L1 (Scheme 1), with loss of a watermolecule in each step leading to formation of a doublebond. The final step to a tetraene leads to a species thatmay not undergo ready protonation or complexationand, therefore, is less likely to be detected.
The positive-ion ESMS spectra of the simple modelligand butane-1,3-diol exhibits similar behaviour. Thespectrum is simple, with a major peak assigned to themonoprotonated diol (L2+H)+ (m/z obsd. 91.1 (100%BPI), calc. 91). Dehydrated charged species are as-signed to two smaller peaks (m/z obsd. 73.1 (25% BPI),calc. 73 and m/z obsd. 55.1 (20% BPI), calc. 55). Thebehaviour is consistent with the scheme:
The behaviour of L2 in the absence of any metal ionconfirms the chemistry observed in the more complexmolecule L1. However, these reactions are minor at lowCV, although they increase as the CV is increased. Atthe cone voltages typically employed in this study, thischemistry does not occur at a significant level for L1 atleast.
Addition of a mixture of Li+, Na+, K+, Rb+ andCs+ produces a range of (ML)+ species and theirsolvated forms indicating that metal exchange is rapidas anticipated. This occurs with both L1 and the simplechelate L2; Fig. 2 illustrates the behaviour with L2. The
Fig. 1. Positive-ion ESMS of (a) LiL1Cl and (b) KL1Cl complexes inwater at neutral pH (cone voltage 30 V).
Scheme 1.
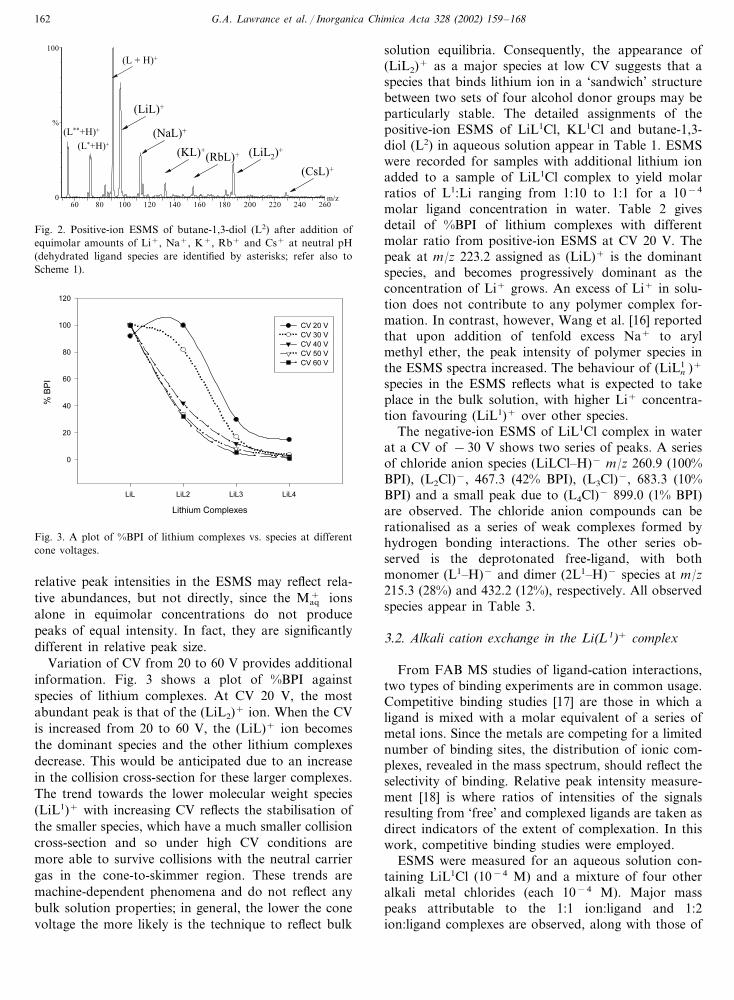
G.A. Lawrance et al. / Inorganica Chimica Acta 328 (2002) 159–168162
Fig. 2. Positive-ion ESMS of butane-1,3-diol (L2) after addition ofequimolar amounts of Li+, Na+, K+, Rb+ and Cs+ at neutral pH(dehydrated ligand species are identified by asterisks; refer also toScheme 1).
solution equilibria. Consequently, the appearance of(LiL2)+ as a major species at low CV suggests that aspecies that binds lithium ion in a ‘sandwich’ structurebetween two sets of four alcohol donor groups may beparticularly stable. The detailed assignments of thepositive-ion ESMS of LiL1Cl, KL1Cl and butane-1,3-diol (L2) in aqueous solution appear in Table 1. ESMSwere recorded for samples with additional lithium ionadded to a sample of LiL1Cl complex to yield molarratios of L1:Li ranging from 1:10 to 1:1 for a 10−4
molar ligand concentration in water. Table 2 givesdetail of %BPI of lithium complexes with differentmolar ratio from positive-ion ESMS at CV 20 V. Thepeak at m/z 223.2 assigned as (LiL)+ is the dominantspecies, and becomes progressively dominant as theconcentration of Li+ grows. An excess of Li+ in solu-tion does not contribute to any polymer complex for-mation. In contrast, however, Wang et al. [16] reportedthat upon addition of tenfold excess Na+ to arylmethyl ether, the peak intensity of polymer species inthe ESMS spectra increased. The behaviour of (LiL1
n )+
species in the ESMS reflects what is expected to takeplace in the bulk solution, with higher Li+ concentra-tion favouring (LiL1)+ over other species.
The negative-ion ESMS of LiL1Cl complex in waterat a CV of −30 V shows two series of peaks. A seriesof chloride anion species (LiLCl�H)− m/z 260.9 (100%BPI), (L2Cl)−, 467.3 (42% BPI), (L3Cl)−, 683.3 (10%BPI) and a small peak due to (L4Cl)− 899.0 (1% BPI)are observed. The chloride anion compounds can berationalised as a series of weak complexes formed byhydrogen bonding interactions. The other series ob-served is the deprotonated free-ligand, with bothmonomer (L1�H)− and dimer (2L1�H)− species at m/z215.3 (28%) and 432.2 (12%), respectively. All observedspecies appear in Table 3.
3.2. Alkali cation exchange in the Li(L1)+ complex
From FAB MS studies of ligand-cation interactions,two types of binding experiments are in common usage.Competitive binding studies [17] are those in which aligand is mixed with a molar equivalent of a series ofmetal ions. Since the metals are competing for a limitednumber of binding sites, the distribution of ionic com-plexes, revealed in the mass spectrum, should reflect theselectivity of binding. Relative peak intensity measure-ment [18] is where ratios of intensities of the signalsresulting from ‘free’ and complexed ligands are taken asdirect indicators of the extent of complexation. In thiswork, competitive binding studies were employed.
ESMS were measured for an aqueous solution con-taining LiL1Cl (10−4 M) and a mixture of four otheralkali metal chlorides (each 10−4 M). Major masspeaks attributable to the 1:1 ion:ligand and 1:2ion:ligand complexes are observed, along with those of
Fig. 3. A plot of %BPI of lithium complexes vs. species at differentcone voltages.
relative peak intensities in the ESMS may reflect rela-tive abundances, but not directly, since the M+
aq ionsalone in equimolar concentrations do not producepeaks of equal intensity. In fact, they are significantlydifferent in relative peak size.
Variation of CV from 20 to 60 V provides additionalinformation. Fig. 3 shows a plot of %BPI againstspecies of lithium complexes. At CV 20 V, the mostabundant peak is that of the (LiL2)+ ion. When the CVis increased from 20 to 60 V, the (LiL)+ ion becomesthe dominant species and the other lithium complexesdecrease. This would be anticipated due to an increasein the collision cross-section for these larger complexes.The trend towards the lower molecular weight species(LiL1)+ with increasing CV reflects the stabilisation ofthe smaller species, which have a much smaller collisioncross-section and so under high CV conditions aremore able to survive collisions with the neutral carriergas in the cone-to-skimmer region. These trends aremachine-dependent phenomena and do not reflect anybulk solution properties; in general, the lower the conevoltage the more likely is the technique to reflect bulk

G.A. Lawrance et al. / Inorganica Chimica Acta 328 (2002) 159–168 163
Table 1Positive-ion ESMS of LiL1Cl and KL1Cl in water at CV 20 V
Species m/z (Theoretical)Major ions observed a m/z (Experimental), (%BPI)
164LiL1Cl 163.9, 5(L1***+H)+
(L1**+H)+ 181 181.1, 25(L1*+H)+ 198 198.2, 8
216(L1+H)+ 216.0, 11(LiL1)+ 223 223.0, 100
241 239.5, 30(LiL1(H2O))+
421(LiL1(H2O)11)+ 421.3, 11(LiL1
2 )+ 439 439.3, 88457 455.4, 6(LiL1
2 (H2O))+
655(LiL13 )+ 655.7, 18
871 871.5, 8(LiL14 )+
164(L1***+H)+ 164.3, 4KL1Cl(L1**+H)+ 181 181.5, 7
198(L1*+H)+ 198.3, 9(L1+H)+ 216 216.2, 9(KL1)+ 255 255.3, 100
327(KL1(H2O)4)+ 327.3, 10(KL1
2 )+ 471 471.2, 60543 544.0, 10(KL1
2 (H2O)4)+
687(KL13 )+ 687.2, 28
(KL13 (H2O)4)+ 759 760.1, 10
904(KL14 )+ 905.2, 4
a Refer to Scheme 1 for ligand species identification.
Table 2Variation of peak intensities of lithium complexes in water with excess Li+ at CV 20 V
(LiL2)+ (%BPI) (LiL3)+ (%BPI)Molar ratio Li:LiL1Cl (LiL4)+ (%BPI)(LiL)+ (%BPI)
1001 18.892.0 9.888.02 21.0100 6.573.4 12.6100 5.1467.4 11.16 4.210055.6 4.7100 1.18
10 100 34.5 0.74 0.5
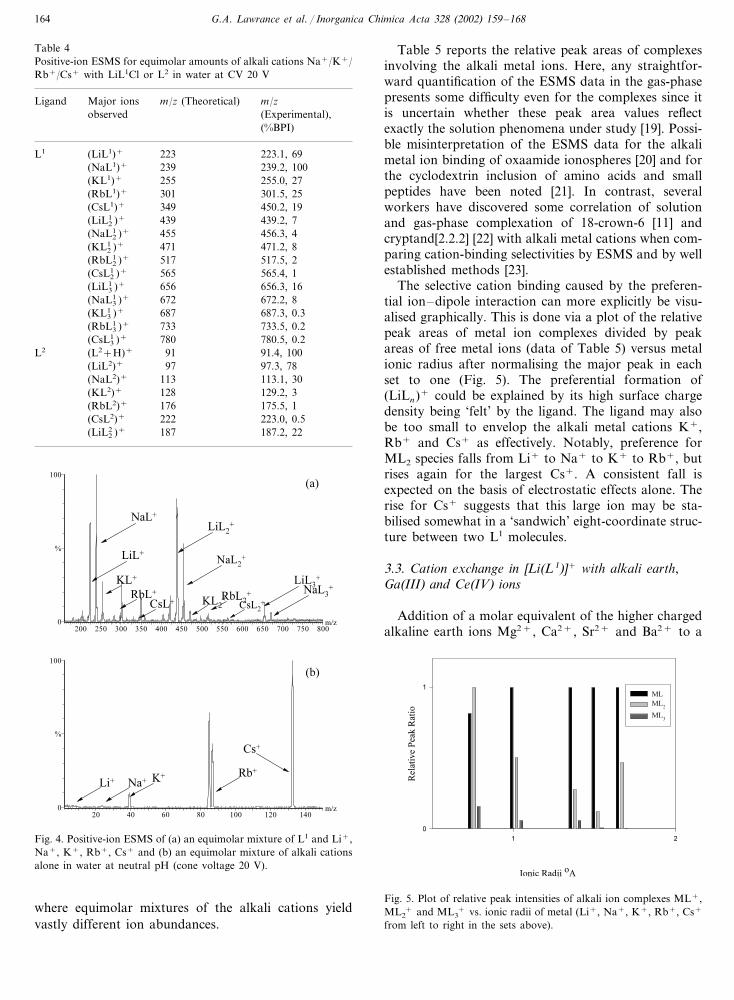
the 1:3 complexes (Table 4). Fig. 4(a) shows the spec-trum of the mixture of equimolar ratios of LiL1Cl withNa+, K+, Rb+ and Cs+, whereas Fig. 4(b) shows thespectrum of an equimolar mixture of free alkali ions.The total %BPI for all (ML1
n )+ complexes are 172 (Li),162 (Na), 35.3 (K), 27.2 (Rb) and 20.2 (Cs), whichequates with peak ratios of 1:0.94:0.21:0.16:0.12. It issuggested that the ligand prefers lithium ion (ionicradius 0.76 A� ) for complexation rather than otherscations, which have larger ionic radii. This behaviour isconsistent with a similar preference for lithium com-plexes in the mixture of alkali ions with the simplemodel ligand (L2), where %BPI of the lithium complexis the largest relative to the others (see Fig. 5).
To survey the selective cation binding, relative peakareas were obtained for each metal complex by dividingthe peak area of the ESMS of 1:1 metal complexes bythose of the corresponding metal ions alone. The needto normalise against the free metal ions is of paramount
importance due to the ESI process yielding differentconversion efficiencies for the alkali metal ions (i.e.conversion of bulk ions into the gas-phase are differentfor each alkali ion). This is clearly evident in Fig. 4(b),
Table 3Negative-ion ESMS of LiL1Cl in water at CV −30 V
m/z (Experimental), (%BPI)m/zMajor ions(Theoretical)
215(L1�H)− 214.8, 28251(L1Cl)− 251.1, 50260(LiL1Cl-H)− 260.9, 100313(LiL1Cl2(H2O))− 313.0, 38
432.2, 12432(L12 �H)−
467(L12 Cl)− 467.3, 42
(L13 Cl)− 683 683.3, 10
(L14 Cl)− 899 899.0, 1
G.A. Lawrance et al. / Inorganica Chimica Acta 328 (2002) 159–168164
Table 4Positive-ion ESMS for equimolar amounts of alkali cations Na+/K+/Rb+/Cs+ with LiL1Cl or L2 in water at CV 20 V
m/zMajor ions m/z (Theoretical)Ligandobserved (Experimental),
(%BPI)
L1 223(LiL1)+ 223.1, 69239(NaL1)+ 239.2, 100
(KL1)+ 255 255.0, 27(RbL1)+ 301 301.5, 25
349(CsL1)+ 450.2, 19(LiL1
2 )+ 439 439.2, 7455(NaL1
2 )+ 456.3, 4(KL1
2 )+ 471 471.2, 8(RbL1
2 )+ 517 517.5, 2565(CsL1
2 )+ 565.4, 1(LiL1
3 )+ 656 656.3, 16672(NaL1
3 )+ 672.2, 8687 687.3, 0.3(KL1
3 )+
733(RbL13 )+ 733.5, 0.2
780 780.5, 0.2(CsL13 )+
91(L2+H)+ 91.4, 100L2
97 97.3, 78(LiL2)+
113(NaL2)+ 113.1, 30(KL2)+ 128 129.2, 3
176(RbL2)+ 175.5, 1(CsL2)+ 222 223.0, 0.5(LiL2
2 )+ 187 187.2, 22
Table 5 reports the relative peak areas of complexesinvolving the alkali metal ions. Here, any straightfor-ward quantification of the ESMS data in the gas-phasepresents some difficulty even for the complexes since itis uncertain whether these peak area values reflectexactly the solution phenomena under study [19]. Possi-ble misinterpretation of the ESMS data for the alkalimetal ion binding of oxaamide ionospheres [20] and forthe cyclodextrin inclusion of amino acids and smallpeptides have been noted [21]. In contrast, severalworkers have discovered some correlation of solutionand gas-phase complexation of 18-crown-6 [11] andcryptand[2.2.2] [22] with alkali metal cations when com-paring cation-binding selectivities by ESMS and by wellestablished methods [23].
The selective cation binding caused by the preferen-tial ion–dipole interaction can more explicitly be visu-alised graphically. This is done via a plot of the relativepeak areas of metal ion complexes divided by peakareas of free metal ions (data of Table 5) versus metalionic radius after normalising the major peak in eachset to one (Fig. 5). The preferential formation of(LiLn)+ could be explained by its high surface chargedensity being ‘felt’ by the ligand. The ligand may alsobe too small to envelop the alkali metal cations K+,Rb+ and Cs+ as effectively. Notably, preference forML2 species falls from Li+ to Na+ to K+ to Rb+, butrises again for the largest Cs+. A consistent fall isexpected on the basis of electrostatic effects alone. Therise for Cs+ suggests that this large ion may be sta-bilised somewhat in a ‘sandwich’ eight-coordinate struc-ture between two L1 molecules.
3.3. Cation exchange in [Li(L1)]+ with alkali earth,Ga(III) and Ce(IV) ions
Addition of a molar equivalent of the higher chargedalkaline earth ions Mg2+, Ca2+, Sr2+ and Ba2+ to a
Fig. 4. Positive-ion ESMS of (a) an equimolar mixture of L1 and Li+,Na+, K+, Rb+, Cs+ and (b) an equimolar mixture of alkali cationsalone in water at neutral pH (cone voltage 20 V).
Fig. 5. Plot of relative peak intensities of alkali ion complexes ML+,ML2
+ and ML3+ vs. ionic radii of metal (Li+, Na+, K+, Rb+, Cs+
from left to right in the sets above).
where equimolar mixtures of the alkali cations yieldvastly different ion abundances.
G.A. Lawrance et al. / Inorganica Chimica Acta 328 (2002) 159–168 165
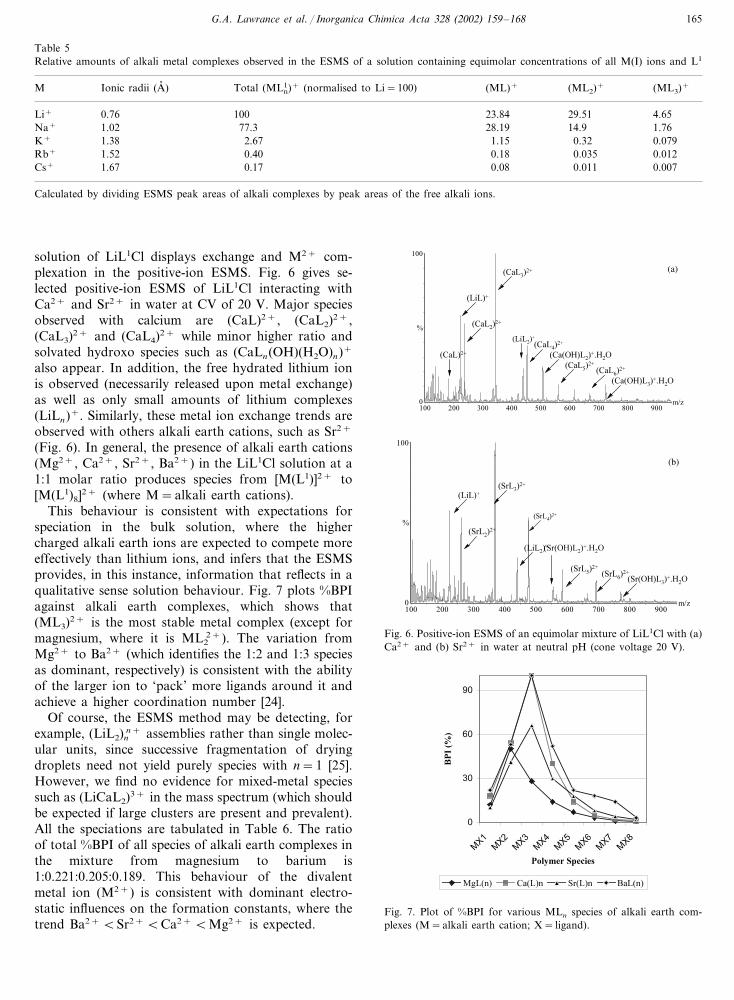
Table 5Relative amounts of alkali metal complexes observed in the ESMS of a solution containing equimolar concentrations of all M(I) ions and L1
M Total (ML1n)+ (normalised to Li=100)Ionic radii (A� ) (ML)+ (ML2)+ (ML3)+
100 23.84Li+ 29.510.76 4.65Na+ 77.31.02 28.19 14.9 1.76
2.67 1.151.38 0.32K+ 0.0790.40 0.18 0.035 0.012Rb+ 1.520.17 0.08 0.011 0.0071.67Cs+
Calculated by dividing ESMS peak areas of alkali complexes by peak areas of the free alkali ions.
Fig. 6. Positive-ion ESMS of an equimolar mixture of LiL1Cl with (a)Ca2+ and (b) Sr2+ in water at neutral pH (cone voltage 20 V).
solution of LiL1Cl displays exchange and M2+ com-plexation in the positive-ion ESMS. Fig. 6 gives se-lected positive-ion ESMS of LiL1Cl interacting withCa2+ and Sr2+ in water at CV of 20 V. Major speciesobserved with calcium are (CaL)2+, (CaL2)2+,(CaL3)2+ and (CaL4)2+ while minor higher ratio andsolvated hydroxo species such as (CaLn(OH)(H2O)n)+
also appear. In addition, the free hydrated lithium ionis observed (necessarily released upon metal exchange)as well as only small amounts of lithium complexes(LiLn)+. Similarly, these metal ion exchange trends areobserved with others alkali earth cations, such as Sr2+
(Fig. 6). In general, the presence of alkali earth cations(Mg2+, Ca2+, Sr2+, Ba2+) in the LiL1Cl solution at a1:1 molar ratio produces species from [M(L1)]2+ to[M(L1)8]2+ (where M=alkali earth cations).
This behaviour is consistent with expectations forspeciation in the bulk solution, where the highercharged alkali earth ions are expected to compete moreeffectively than lithium ions, and infers that the ESMSprovides, in this instance, information that reflects in aqualitative sense solution behaviour. Fig. 7 plots %BPIagainst alkali earth complexes, which shows that(ML3)2+ is the most stable metal complex (except formagnesium, where it is ML2
2+). The variation fromMg2+ to Ba2+ (which identifies the 1:2 and 1:3 speciesas dominant, respectively) is consistent with the abilityof the larger ion to ‘pack’ more ligands around it andachieve a higher coordination number [24].
Of course, the ESMS method may be detecting, forexample, (LiL2)n
n+ assemblies rather than single molec-ular units, since successive fragmentation of dryingdroplets need not yield purely species with n=1 [25].However, we find no evidence for mixed-metal speciessuch as (LiCaL2)3+ in the mass spectrum (which shouldbe expected if large clusters are present and prevalent).All the speciations are tabulated in Table 6. The ratioof total %BPI of all species of alkali earth complexes inthe mixture from magnesium to barium is1:0.221:0.205:0.189. This behaviour of the divalentmetal ion (M2+) is consistent with dominant electro-static influences on the formation constants, where thetrend Ba2+ �Sr2+ �Ca2+ �Mg2+ is expected.
Fig. 7. Plot of %BPI for various MLn species of alkali earth com-plexes (M=alkali earth cation; X= ligand).

G.A. Lawrance et al. / Inorganica Chimica Acta 328 (2002) 159–168166
Table 6ESMS for equimolar concentration of LiL1Cl with individual alkali earth cations in water at CV 20 V
Added M2+ Major ion observed m/z (Theoretical) m/z (Experimental), %BPI
120Mg2+ 119, 20(MgL1)2+
129(MgL1(H2O))2+ 128.9, 20138(MgL1(H2O)2)2+ 137.9, 6
(MgL1(H2O)3)2+ 147 147.2, 100(L1H(H2O)3)+ 164 164.1,38
181(L1H(H2O)2)+ 180.8, 25198(L1H(H2O))+ 198.2, 5223(LiL1)+ 223.0, 19
(MgL12 )2+ 228 226.8, 52
(Mg(OH)L1(H2O))+ 275 275.7, 13386(MgL1
3 )2+ 386.4, 30439 438.0, 3(LiL1
2 )+
444(MgL14 )2+ 439.4, 10
(Mg(OH)L12 (H2O))+ 491 491.0, 20
(MgL15 )2+ 552 552.2, 30
660(MgL16 )2+ 660.0, 3
707(Mg(OH)L13 (H2O))+ 706.7, 60
768(MgL17 )2+ 768.1, 6
(MgL18 )2+ 876 876.0, 0.4
(CaL1)2+Ca2+ 128 128.0, 19137(CaL1(H2O))2+ 137.1, 25146 145.9, 18(CaL1(H2O)2)2+
155(CaL1(H2O)3)2+ 155.2, 3(L1H(H2O)2)+ 181 181.0, 18(LiL1)+ 223 223.0, 54
236(CaL12 )2+ 236.0, 52
245 245.1, 3(CaL12 (H2O))2+
291(Ca(OH)L1(H2O))+ 291.2, 5(CaL1
3 )2+ 344 343.2, 100(LiL1
2 )+ 440 439.2, 39452(CaL1
4 )2+ 452.1, 22507 507.1, 11(Ca(OH)L1
2 (H2O))2+
560(CaL15 )2+ 561.5, 12
(CaL16 )2+ 668 668.0, 2
(Ca(OH)L13 (H2O))+ 723 723.1, 0.8
776(CaL17 )2+ 775.1, 0.5
(CaL18 )2+ 884 884.2, 0.2
152(SrL1)2+ 152.2, 28Sr2+
(SrL1(H2O))2+ 161 161.2, 30(SrL1(H2O)2)2+ 170 170.0, 20
180(SrL1(H2O)3)2+ 179.1, 12164 164.0, 5(L1H(H2O)3)+
182(L1H(H2O)2)+ 181.2, 25(L1H(H2O))+ 198 198.0, 8(LiL1)+ 223 223.2, 62
259(SrL12 )2+ 260.1, 58
338 338.0, 4(Sr(OH)L1(H2O))+
368(SrL13 )2+ 368.0, 100
(LiL12 )+ 440 440.3, 30
(SrL14 )2+ 476 476.2, 53
555(Sr(OH)L12 (H2O))+ 555.2, 5
584 584.2, 23(SrL15 )2+
692(SrL16 )2+ 692.2, 18
(Sr(OH)L13 (H2O))+ 772 772.1,7
(SrL17 )2+ 800 800.2, 2
908(SrL18 )2+ 908.3, 0.4
(BaL1)2+Ba2+ 177 176.2, 9186(BaL1(H2O))2+ 186.1, 12
(BaL1(H2O)2)2+ 195 195.1, 11(BaL1(H2O)3)2+ 204 204.1, 5
181(L1H(H2O)2)+ 181.1, 18223 223.2, 40(LiL1)+
285(BaL12 )2+ 285.0, 40
(BaL12 (H2O))2+ 294 294.2, 10
(Ba(OH)L1(H2O))+ 388 388.1, 2393(BaL1
3 )2+ 393.2, 68(LiL1
2 )+ 440 440.2, 18501(BaL1
4 )2+ 501.2, 33(Ba(OH)L1
2 (H2O))+ 406 406.2, 2(BaL1
5 )2+ 609 607.2, 11717(BaL1
6 )2+ 717.2, 5(Ba(OH)L1
3 (H2O))+ 622 622, 0.2825(BaL1
7 )2+ 825.2, 3833 833.2, 0.2(BaL1
8 )2+

G.A. Lawrance et al. / Inorganica Chimica Acta 328 (2002) 159–168 167
Table 7ESMS for equimolar concentration of LiL1Cl with Ga(III) and Ce(IV) cations in water at CV 20 V
m/z (Theoretical)Added M2+ m/z (Experimental), (%BPI)Major ion observed
160 160.5, 100Ga3+ (Ga(OH)L1(H2O))2+
178(Ga(OH)L1(H2O)3)2+ 179.0, 88(Ga(OH)L1(H2O)5)2+ 196 196.1, 49(Ga(OH)L1(H2O)7)2+ 214 214.2, 48
223(LiL1)+ 223.2, 15304 304.6, 9(Ga(OH)L1
2 (H2O)5)2+
320(Ga(OH)2L1)+ 320.3, 40(Ga(OH)2L1(H2O))+ 338 338.4, 9(Ga(OH)2L1(H2O)2)+ 356 356.2, 20
98(CeL1(H2O)2)4+ 98.2, 61Ce4+
108 108.2, 87(CeL1(H2O)4)4+
124(Ce(OH)L1)3+ 124.4, 100(Ce(OH)L1(H2O)2)3+ 136 136.3, 68(Ce(OH)L1
3 )3+ 268 268.4, 40274(Ce(OH)L1
3 (H2O))3+ 274.5, 28280 280.3, 38(Ce(OH)L1
3 (H2O)2)3+
303(Ce(OH)2L12 )2+ 304.3, 16
(Ce(OH)2L12 (H2O))2+ 312 312.3, 24
407 407.2, 28(Ce(OH)2L1)2+
425 425.3, 13(Ce(OH)3L1(H2O))2+
Positive-ion mode ESMS of an equimolar mixture ofLiL1Cl with Ga3+ in aqueous solution was also investi-gated. Two series of charge reduction gallium com-plexes, (Ga(OH)L(H2O)n)2+ and (Ga(OH)2L(H2O)n)+
were present in the spectra. Only a small residual(LiL)+ peak was observed (Fig. 8(a)). For Ce4+, undercomparable conditions, a series of cerium complexes
were observed, namely, (CeL(H2O)n)4+, (Ce(OH)L-(H2O)n)3+, (Ce(OH)L3(H2O)n)3+, (Ce(OH)2L2-(H2O)n)2+ and (Ce(OH)3L(H2O)n)+ (Fig. 8(b)). Bothgallium and cerium complex species are assigned inTable 7. Both a tendency of the Ga3+ and Ce4+ ionsto hydrolyse readily, and a general trend in ESMStowards favouring lower-charged species support theobservation of hydroxo complexes. With the highercharged ions, exchange equilibria lie strongly towardsthe multiply-charged ions and little if any Li+ com-plexes are observed.
4. Conclusion
The polyalcohol (L1) is capable of forming complexeswith mainly alkali and alkaline earth metal ions. Rapidmetal ion exchange is implied from ESMS experiments,consistent with expectations for these metal ions. Pref-erences for higher charged and smaller ions support adominant electrostatic character to the bonding. How-ever, observations such as higher than anticipatedamounts of (SrL1
2 )2+ in the ESMS infers some shape-based effect on complex stability. Given the tetrapodalshape of the ligand, this is not unreasonable. Forma-tion of species as high in ligand number as (ML1
8 )n+
infers that chelation does not operate in all species,although the dominant species have fewer L1 moleculesaround and are more likely to involve multidentatechelators. Some further structural studies in the solid-state could infer more fully some of the species thatcould also exist in solution. Elucidation of stereo-chemistries is not obtained from ESMS, although it is
Figure 8. Numerical solutions of the extended Boussinesqequations model for the propagation of a deep water wave(kh=3.14).

G.A. Lawrance et al. / Inorganica Chimica Acta 328 (2002) 159–168168
likely that coordination numbers can vary appreciablyand range up to at least 12, as found in the solid-stateand in solution under equilibrium conditions used formetal ions in this study. The value of ESMS to probelabile metal– ligand interactions evident in earlier stud-ies has been further defined in this study, the first ofstructurally defined synthetic polyalcohols.
Acknowledgements
Support of this research by the Australian ResearchCouncil is gratefully acknowledged.
References
[1] S. Yono, Coord. Chem. Rev. 92 (1988) 113.[2] D. Klaassan, P. Klufers, Inorg. Chem. 619 (1993) 661.[3] L.G. Hubert-Pfalzgraf, New J. Chem. 11 (1997) 663.[4] A. Appelt, A.C. Willis, S.B. Wild, J. Chem. Soc., Chem. Com-
mun. (1986) 1001.[5] T. Tanase, F. Shimizu, S. Yono, S. Yoshikawa, J. Chem. Soc.,
Chem. Commun. (1986) 1009.[6] P. Goldschmidt, E. Weinggardt, W. Bachmann, Koloid Z. 47
(1929) 49.[7] J. Burger, C. Gack, P. Klufers, Angew. Chem. 107 (1995) 2950.
[8] K. Hegetschweiler, L. Prino, H. Koppenol, V. Gramlich, W.Meyer, Angew. Chem. 107 (1995) 2421.
[9] K. Hegetschweiler, Chem. Soc. Rev. 28 (1999) 239.[10] P.V. Bernhardt, G.A. Lawrance, S.M. Luther, M. Maeder, M.J.
Robertson, Sutrisno, Inorg. Chem. Commun. 3 (2000) 410.[11] K. Hegetschweiler, Bol. Soc. Chilena Quim. 42 (1997) 257.[12] E. Leize, J. Jaffrezic, A. van Dorsselaer, J. Mass Spectrom. 31
(1996) 537.[13] D.K. Walanda, R.C. Burns, G.A. Lawrance, E.I. von Nagy-Fel-
sobuki, J. Chem. Soc., Dalton Trans. (1999) 311.[14] Sutrisno, Y. Baran, G.A. Lawrance, E.I. von Nagy-Felsobuki,
D.C. Richens, H. Xiao, Inorg. Chem. Commun. 2 (1999) 107.[15] D.N. Butler, T.J. Munshaw, Can. J. Chem. 59 (1981) 3365.[16] K. Wang, X. Han, R.W. Gross, G.W. Gokel, J. Am. Chem. Soc.
117 (1995) 7680.[17] R.A. Johnstone, M.E. Rose, J. Chem. Soc., Chem. Commun.
(1983) 1268.[18] A.E. Lewis, M.C. Grossel, Tetrahedron 39 (1993) 1597.[19] G.J. Angley, D.G. Hamilton, M.C. Grossel, J. Chem. Soc.,
Perkin Trans. 2 (1995) 929.[20] M. Goodall, P.M. Kelly, D. Parker, K. Gloe, H. Stephan, J.
Chem. Soc., Perkin Trans. 59 (1997) 442.[21] J.B. Cunniff, P. Vouras, J. Am. Soc. Mass Spectrom. 6 (1996)
4693.[22] K. Wang, G.W. Gokel, J. Org. Chem. 61 (1996) 4693.[23] R.M. Izatt, J.S. Bradshaw, S.A. Nielsen, J.D. Lamb, Chem. Rev.
85 (1995) 271.[24] F.A. Cotton, G. Wilkinson, A.C. Murillo, M. Bochmann, Ad-
vanced Inorganic Chemistry, sixth ed., Wiley, New York, 1999.[25] G. Horlick, G.R. Agnes, Appl. Spectrosc. 46 (1992) 401.




















