
Identification and characterization of a heat-labile type I glutamine synthetase fromStreptomyces...
10
Folia Microbiol. 42 (5), 431-440 (1997) Identification and Characterization of a Heat-Labile Type I Glutamine Synthetase from Streptomyces cinnamonensis K.T. NGUYEN**, L.T. NGUYEN, O. BENADA and V. BI~HAL* Institute of Microbiology, Academy of Sciences of the Czech Republic, 142 20 Prague 4, Czech Republic Received May 15, 1997 ABSTRACT. Streptomycetes have two distinct glutamine synthetases (GS): a heat-stable dodecameric GSI and a heat-labile octamerie GSII. A heat-inactivated GS activity was detected in crude extracts of Streptomyces cinnaraonensis cells grown with nitrate or glutamate as the nitrogen source. The purified enzyme obtained from crude extracts of the nitrate-grown cells after affinity and anion-exchange chromatography was also heat-labile; it was inactivated by 80 % when incubated at 50 ~ for 1 h. However, the enzyme has properties typical of GSI and similar with those of the heat-stable GSI purified from S. aureofaciens: It is composed of twelve subunits, each of M55 kDa, and has a native molar mass of 625 kDa and an isoelectrie point at pH 4.2. In addition, its activity is regulated by reversible adenylylation. Mg2+ and NaCI but not Mn2+ protected the purified enzyme from thermal inactivation, and both NaCI and Mn 2+ or Mg 2+ stabilized its activity at 4-8 ~ As compared with GSI from S. aureofaciens, the S. cinnamonensis enzyme was cleaved more extensively during SDS-PAGE, was less sensitive to feedback inhibitors, and similarly affected by divalent cations. The Km values were 12_5 mmol/L for L-glutamate, 0.1 for NH +, 1.25 for ATP, 18_5 for L-glutamine, 3.3 for hydroxylamine and 0.087 for ADP. To our best knowledge, this is the first report of a heat- labile GSI from any source. Streptomycetes are commercially important Gram-positive bacteria that produce numerous antibiotics and other bioactive secondary metabolites. The biosynthesis of secondary metabolites usu- ally occurs after the depletion of nitrogen or other essential nutrients, such as carbon and phosphate (Vining and Stuttard 1995). Under nitrogen-limited growth conditions, glutamine synthetase (GS) activity that catalyzes the ATP-dependent synthesis of glutamine from ammonium and glutamate is activated (Tyler 1978). Since glutamine is the preferred donor of the amino group in structures of purines, pyrimidines, amino acids and amino sugars, it has been suggested that GS might play a role in processes that occur under nutritional limitation, such as antibiotic biosynthesis, by participating in the quantitative maintenance of cell components, coenzymes and enzymes, particularly of those directly involved in the antibiotic biosynthetic pathways (Nguyen et al. 1994). GS has been found in several distinct isoforms (Woods and Reid 1993). The prokaryote-asso- ciated GSI (product of the glnA gene) is composed of twelve subunits, each of M 50 kDa. GSII (product of the glnH gene) found in eukaryotes and several bacteria is an octamer with subunits of M 40 kDa. GSI and GSII share only 15 % similarity in amino acid sequence and only GSI is modified post-translationally by reversible adenylylation (Edmands et al. 1987; Wray and Fisher 1988; Hillemznn et al. 1993). GSI is heat-stable and distinguished from the heat-labile GSII by thermal inactivation (Hil- lemann et al. 1993; Kumada et al. 1990). GSIII found in Bacteriodes fragilis is composed of six larger subunits (M 75 kDa), and GST recently purified from Rhizobium meliloti is an octamer with subunits of M 46 kDa (Shatters et al. 1993). Although these GS isoforms are quite different in primary as well as quaternary structures, they contain identical amino acid residues in five conserved regions associated with putative active sites, and require Mn 2+ or Mg 2§ for catalytic activity and structural stability; removal of Mn 2§ from E. coli GSI molecules by EDTA treatment causes a reversible inactivation and structure unfolding (Almassy et al. 1986; Rhee et al. 1989). Since glutamine is an important nitrogen-contzining intermediate, the activ- ity of the bacterial GS has been found to be tightly regulated by a number of mechanisms: covalent modifications by adenylylation and ADP-ribosylation, feedback inhibitions by end products of the glu- tamine metabolism, proteolysis, divalent cations and metal-mediated oxidation in the presence of reducing reagents, such as ascorbate or 2-mercaptoethanol (Rhee et al. 1989; Penyige et al. 1994). GSII activity is also regulated by feedback inhibitions and an unknown post-translational modification (Hille- mznn et aL 1993; Tate and Meister 1971). *Corresponding author. **Present address: Biozentrum, Department of Microbiology, University of Basel, Switzerland.
-
Upload
independent -
Category
Documents
-
view
5 -
download
0
Transcript of Identification and characterization of a heat-labile type I glutamine synthetase fromStreptomyces...

Folia Microbiol. 42 (5), 431-440 (1997)
Identification and Characterization of a Heat-Labile Type I Glutamine Synthetase from Streptomyces cinnamonensis K.T. NGUYEN**, L.T. NGUYEN, O. BENADA and V. BI~HAL*
Institute of Microbiology, Academy of Sciences of the Czech Republic, 142 20 Prague 4, Czech Republic
Received May 15, 1997
ABSTRACT. Streptomycetes have two distinct glutamine synthetases (GS): a heat-stable dodecameric GSI and a heat-labile octamerie GSII. A heat-inactivated GS activity was detected in crude extracts of Streptomyces cinnaraonensis cells grown with nitrate or glutamate as the nitrogen source. The purified enzyme obtained from crude extracts of the nitrate-grown cells after affinity and anion-exchange chromatography was also heat-labile; it was inactivated by 80 % when incubated at 50 ~ for 1 h. However, the enzyme has properties typical of GSI and similar with those of the heat-stable GSI purified from S. aureofaciens: It is composed of twelve subunits, each of M55 kDa, and has a native molar mass of 625 kDa and an isoelectrie point at pH 4.2. In addition, its activity is regulated by reversible adenylylation. Mg 2+ and NaCI but not Mn 2+ protected the purified enzyme from thermal inactivation, and both NaCI and Mn 2+ or Mg 2+ stabilized its activity at 4-8 ~ As compared with GSI from S. aureofaciens, the S. cinnamonensis enzyme was cleaved more extensively during SDS-PAGE, was less sensitive to feedback inhibitors, and similarly affected by divalent cations. The Km values were 12_5 mmol/L for L-glutamate, 0.1 for NH +, 1.25 for ATP, 18_5 for L-glutamine, 3.3 for hydroxylamine and 0.087 for ADP. To our best knowledge, this is the first report of a heat- labile GSI from any source.
Streptomycetes are commercially important Gram-positive bacteria that produce numerous antibiotics and other bioactive secondary metabolites. The biosynthesis of secondary metabolites usu- ally occurs after the depletion of nitrogen or other essential nutrients, such as carbon and phosphate (Vining and Stuttard 1995). Under nitrogen-limited growth conditions, glutamine synthetase (GS) activity that catalyzes the ATP-dependent synthesis of glutamine from ammonium and glutamate is activated (Tyler 1978). Since glutamine is the preferred donor of the amino group in structures of purines, pyrimidines, amino acids and amino sugars, it has been suggested that GS might play a role in processes that occur under nutritional limitation, such as antibiotic biosynthesis, by participating in the quantitative maintenance of cell components, coenzymes and enzymes, particularly of those directly involved in the antibiotic biosynthetic pathways (Nguyen et al. 1994).
GS has been found in several distinct isoforms (Woods and Reid 1993). The prokaryote-asso- ciated GSI (product of the glnA gene) is composed of twelve subunits, each of M 50 kDa. GSII (product of the glnH gene) found in eukaryotes and several bacteria is an octamer with subunits of M 40 kDa. GSI and GSII share only 15 % similarity in amino acid sequence and only GSI is modified post-translationally by reversible adenylylation (Edmands et al. 1987; Wray and Fisher 1988; Hillemznn et al. 1993). GSI is heat-stable and distinguished from the heat-labile GSII by thermal inactivation (Hil- lemann et al. 1993; Kumada et al. 1990). GSIII found in Bacteriodes fragilis is composed of six larger subunits (M 75 kDa), and GST recently purified from Rhizobium meliloti is an octamer with subunits of M 46 kDa (Shatters et al. 1993).
Although these GS isoforms are quite different in primary as well as quaternary structures, they contain identical amino acid residues in five conserved regions associated with putative active sites, and require Mn 2+ or Mg 2§ for catalytic activity and structural stability; removal of Mn 2§ from E. coli GSI molecules by EDTA treatment causes a reversible inactivation and structure unfolding (Almassy et al. 1986; Rhee et al. 1989). Since glutamine is an important nitrogen-contzining intermediate, the activ- ity of the bacterial GS has been found to be tightly regulated by a number of mechanisms: covalent modifications by adenylylation and ADP-ribosylation, feedback inhibitions by end products of the glu- tamine metabolism, proteolysis, divalent cations and metal-mediated oxidation in the presence of reducing reagents, such as ascorbate or 2-mercaptoethanol (Rhee et al. 1989; Penyige et al. 1994). GSII activity is also regulated by feedback inhibitions and an unknown post-translational modification (Hille- mznn et aL 1993; Tate and Meister 1971).
*Corresponding author. **Present address: Biozentrum, Department of Microbiology, University of Basel, Switzerland.

432 K.T. NGUYEN et aL Vol. 42
Several GS isoforms have been found in a single organism of Synechocystis (GSI and GSIII) (Reyes and Florencio 1994), in Rhizobiaceae (GSI, GSII, GST) (Shatters et aL 1993), in Frankia (GSI and GSII) (Edmands et al. 1987) and Streptomyces (GSI and GSII) (Kumada et aL 1990; Behrmann et al. 1990). In Rhizobiaceae and Frankia, GSI is constitutively expressed and GSII synthesis is regulated in response to the availability of the nitrogen source and under the control of a global nitrogen regula- tory system (Ntr). In contrast, GSI expression in Streptomyces is affected by the nitrogen source and activated by GInR, a transcription activator (Fisher and Wray 1989; Wray et al. 1991) and GSII is expressed constitutively at low levels in S. viridochromogenes grown either with or without a nitrogen source (HiUemann et al. 1993). The heat-labile GSII activity of S. coelicolor was also not inducible by nitrogen starvation (Nguyen et aL 1994). While a high GSII activity was found in a S. lividans strain car- rying the glnlI gene cloned from S. hygroscopicus, and a S. viridochromogenes mutant with a disrupted glnA gene (Hillemann et al. 1993; Kumada et al. 1990), suggesting that GSII expression is triggered by the absence of an active GSI rather than by nitrogen starvation.
So far, GSII has not been purified from any genetically nonmanipulated Streptomyces strain, and this prompted us to search for a heat-labile GS activity in S. aureofaciens and S. cinnamonensis cells grown with nitrate or glutamate as the nitrogen source. We detected and purified a highly expressed heat-inactivated GS activity from crude extracts of S. cinnamonensis; however, this enzyme has properties typical of GSI rather than GSII. Therefore, the enzyme appears to be the first described heat-labile GSI from any source.
MATERIALS A N D METHODS
Microorganisms and cultivation conditions. Mycelium of S. cinnamonensis C-100-5 grown for 1 d in a complex medium (Pospf~il et aL 1983) was used as inoculum (5 %, V]V) for a chemically defined medium containing glucose (carbon source) and nitrate or glutamate (nitrogen source) (Nguyen et al. 1994). The cultivations were performed on a shaker (2.7 Hz) at 34 ~ S. aureofaciens BS-5 was cultivated in similar media at 28 *C with shaking (1.6 Hz) as previously described (Nguyen et al. 1995a).
Preparation of crude extracts and GS assay. Crude extracts were prepared by sonication with 20 mmol/L imidazole-HCl (pH 7.5) containing 1 mmol/L Mn 2+ (added in the form of MnC12) as the extraction buffer (Nguyen et al. 1994). GS transferase, semisynthetase and biosynthetase activities were determined at 40 *C for 15 rain in the assay mixtures (pH 7.2) described by Bender et al. (1977). Unless otherwise stated, GS activity was determined in the transferase assay system. Enzyme activity was expressed as nmol product formed per second (nkat). The Michaelis constant (Kin) for the purified GS was determined under standard assay conditions from double-reciprocal plots of reaction velocity vs. substrate concentrations (Nguyen et al. 1995b). In studies of feedback inhibition, to increase the sensi- tivity of S. cinnamonensis GS to inhibitors, biosynthetase assay mixtures containing lowered concentra- tions of glutamate (16 retool/L), and ammonium and ATP (5 mmol/L) were used.
Heat treatment. Diluted crude extracts (1.0-2.8 mg protein per mL) or purified GS samples (11 ~tg/mL) were incubated in a water bath at 50 ~ Just before the heat treatment, small molecules or NaC1 were removed from the enzyme samples by gel filtration on a G-25 column (bed volume 10 mL) pre-equilibrated with the extraction buffer.
Ammonium shock and phosphodiesterase digestion. NH4CI (37 mmol/L) was added into a 10- h-old S. cinnamonensis culture exponentially growing in a glucose-nitrate medium. Samples (100 ~tL) were taken at intervals and assayed for GS transferase activity in whole cells permeabilized by 100 lag/mL hexadecyltrimethylammonium bromide (Fisher and Wray 1989). For GS reactivation, crude extracts were incubated at 37 ~ with 0.1 mg snake venom phosphodiesterase per mg protein in 100 mmol/L Tris-HCl (pH 8.8) containing 100 mmol/L NaC1 and 5 mmol/L MgCI2. Transferase activity in samples taken at intervals was measured.
GS purification
Blue-Sepharose. Forty mL crude extract in buffer A (20 mmol/L imidazole-HCl at pH 7.5, containing 1 mmol/L Mn 2 § was transferred to buffer B (buffer A, pH 6.3) by ultrafiltration with an Amicon CF-25 membrane (M 25 kDa cut-off) and then recycled overnight at 10 mL/h to a Blue- Sepharose column (16 x 50 ram, bed volume 10 mL) pre-equilibrated with buffer B. The column was washed with 100 mL buffer B containing 1 mol/L NaCI, and then the absorbed proteins were eluted

1997 GLUTAMINE SYNTHETASE FROM S. cinnamonensis 433
with 60 mL buffer A. Fractions of 2 mL were collected at 30 mL/h. The active fractions were pooled and transferred to buffer A by ultrafiltration.
Q-Sepharose chromatography. GS from the previous step was loaded at 10 mL/h on a Q- Sepharose column (16 x 50 mm, bed volume 10 mL) pre-equilibrated with buffer A. The column was washed with 80 mL buffer A and eluted at 30 mL/h with a linear gradient of 0.2 to 0.7 mol/L NaCI in buffer A (total volume 80 mL). Active fractions of 0.5 mL were collected, pooled and stored at 4 ~ in the presence of 1 mol/L NaCI.
Analytical methods. SDS-PAGE on a 12 % acrylamide gel and native-PAGE on a linear gradi- ent 4-10 % acrylamide gel were done as previously described (Hames 1981). Isoelectric focusing (IEF) was performed on a 5 % agarose gel (0.5 ram-thick) calibrated with Pharmacia pl standard pro- teins according to the Pharmacia Guide Book. Two-dlmensional PAGE was done with a mixture of Servalyte 4-9 and 4-6 (4 : 1, V/V) (O'Farrell 1975). Native molar mass of GS was determined by gel ill- tration on a Sephacryl S-300 HR column with 20 mmol/L Tris-HC1 buffer (pH 7.4) containing 20 mmol/L MgCI2 and 200 mmol/L NaCI as the equilibration and elution buffer (Nguyen et al. 1995a). GS structure was viewed by electron microscopy (Benada and Pokorn~ 1990). Protein concentrations were determined by the dye-binding method (Bradford 1976).
R E S U L T S
Detection of heat-labile GS activity in crude extracts
Although streptomycetes contain genes coding for the heat-stable GSI and the heat-labile GSII, only a S. lividans strain carrying an inserted glnlI gene cloned from S. hygroscopicus and a S. viri- dochromogenes strain with a disrupted glnA gene overexpress GSII (Hillemann et al. 1993; Kumada et al. 1990). To examine whether genetically nonmanipulated Streptomyces strains synthesize high GSII levels, we studied the heat-stability of GS activity in S. aureofaciens and S. cinnamonensis cells grown with nitrate or glutamate as the nitrogen source for 10, 16, and 20 h, corresponding, respectively, to mid-exponential, early and late stationary growth phases. These nitrogen sources were chosen because they supported high GS activity. Crude extracts were prepared in 20 mmol/L imidazole-HCl buffer (pH 7.5) containing 1 mmol/L Mn 2+. It has been reported that in this buffer GSI is relatively stable but GSII is inactivated after a 1-h incubation at 50 *C (Kumada et al. 1990). After similar incubations, GS in all S. aureofaciens crude extracts lost only 5-10 % activity; whereas GS in all S. cinnamonensis crude extracts was inactivated by 70-90 %. Dilution of S. cinnamonensis crude extracts three times or removal of small molecules from crude extracts by G-25 gel filtration prior to heat treatments did not increase the enzyme stability, suggesting that GS inactivation was caused neither by accelerated aggre- gation of GS protein present at high concentrations nor inhibition by metabolites during the heat treatment. GS activity in crude extracts of S. cinnamonensis cells grown at 28 ~ (instead of 34 ~ with lower shaking (1.6 instead of 2.7 Hz) was also heat-labile. We decided to purify and characterize the heat-labile GS from S. cinnamonensis.
GS purification
GS from crude extracts of S. cinnamonensis cells grown for 20 h in the glucose-nitrate medium was purified by affinity chromatography on Blue-Sepharose and anion-exchange chromatogra- phy on Q-Sepharose. Only one peak of GS activity was detected during the purification. The elution from Blue-Sepharose of GSIs from S. cattleya and S. aureofaciens required AMP or ADP and 1 mol/L NaC1 (Streicher and Tyler 1981; Nguyen 1995b). In contrast, the S. cinnamonensis enzyme was eluted by increasing the pH of the buffer from 6.3 to 7.4; addition of adenine nucleotides into the elution buffer did not increase GS activity recovery and the presence of 1 mol/L NaCI in this buffer reduced the recovery to 18 %. After Q-Sepharose chromatography, the S. cinnamonensis enzyme was purified 41.2-fold with a 56.7 % recovery (Table I) to homogeneity as judged by native-PAGE on a gradient acrylamide gel (Fig. 1B) and IEF (Fig. 1C). However, at least five protein bands were found on a SDS- PAGE gel (Fig. 1A). The purified enzyme exhibited a transferase specific activity of 3588 nkat/mg. The S. aureofaciens enzyme was cleaved to a lesser extent but has a lower specific activity (3083 nkat/mg) (Nguyen 1995b). A further purification step by gel filtration on a Sephacryl S-300 HR column did not increase the specific activity of these enzymes. The GS fragmentation on SDS-PAGE gels will be dis- cussed below.

K.T. NGUYEN eta/. Vol. 42
Table I. Purification of the heat-labile GSI from S. c/nnamonens/~
Total Total Specific Recovery Purification Purification s tep protein activity activity factor
mg nkat nkat/mg b %
Crude extract 80 6 962 87 100 1 Blue-Sepharose 2.25 4 386 1949 63 22.4 Q-Sepharose 1.1 3 947 3 588 56.7 41.2
aGSI was purified from 40 mL of crude extract of S. cinnamonensis grown for 20 h in the glucose-nitrate medium. The purification was repeated three times with similar results.
bThe transferase specific activity of 3 588 nkat/mg corresponds to a Mg2+-dependent biosynthetase specific activ- ity of 188 nkat/mg.
Stability at high and low temperatures
Activity of the purified GS from S. aureofaciens remained unchanged when incubated at 50 ~ for i h, indicating that it is a heat-stable GSI (Nguyen 1995). Whereas the purified GS from S. cinna- monensis was heat-labile as the enzyme in crude extracts; transferase and semibiosynthetase activities of desalted pure GS samples were reduced by 80 % (Fig. 2, top). Addition of 20 mmol/L Mg 2 + (as MgCl2) or 0.2-1 mol/L NaC1 protected GS activity at temperatures up to 65 *C (Fig. 2, bottom); how- ever, GS lost activity rapidly at 70 ~ regardless of the presence of Mg 2+ or NaC1. Surprisingly, 10 mmol/L Mn 2+ (added as MnCl2) offered no protection for GS incubated at 50 ~
The enzyme was irreversibly and completely inactivated at 4 - 8 ~ if Mn 2+ or Mg 2+ were removed from the gel filtration buffer. The enzyme also lost 50 % activity when incubated at 8 ~ for 2 h; addition of i mol/L NaCI provided a full protection. Under similar conditions, GSI activity from S. aureofaciens was not affected.
Molecular properties
The Streptomyces GSII is an octamer with eight subunits of M 40 kDa (Kumada et al. 1990). The heat-labile GS from S. cinnamonensis has a twelve-subunit structure (Fig. 1D), a native molar mass of 625 kDa as determined by gel filtration and an isoelectric point at pH 4.2 (Fig. 1C). From the determined native molar mass and structure, the M of GS subunits was calculated to be 52 kDa. These molecular properties are very similar to those of the heat-stable GS from S. aureofaciens (Nguyen 1995b) and other bacteria (Woods and Reid 1993), indicating that the S. cinnamonensis enzyme is a GSI rather than GSII.
To obtain further support for this condusion, we added ammonium ions to a 10-h S. cin- namonensis culture exponentially growing in a glucose-nitrate medium; it is well known that only GSI but not GSII is inactivated by adenylylation. As expected, ammonium shock reduced 60 % of GS activ- ity (Fig. 3A), perhaps by adenylylation, because the inactivated enzyme regained 90 % original activity after a 1-h treatment with snake venom phosphodiesterase. As detected on two-dimensional PAGE gels, the ammonium shock induced an increase of the 55 kDa, acidic protein at the expense of a closely located, basic protein with the same molar mass (Fig. 3B, C). These two protein spots might represent the adenylylated and nonadenylylated GS subunits, respectively (Edmands et al. 1987; Streicher and Tyler 1981). Taken together, these results show that the purified GS from S. cinnamonensis is a heat- labile GSI composed of twelve subunits, each of M 55 kDa. Our further experiments aimed to deter- mine whether the heat-labile GSI from S. cinnamonensis is markedly different in catalytic and regula- tory properties from the heat-stable GSI from S. aureofaciens.
Cata~tic and inhibitory properties
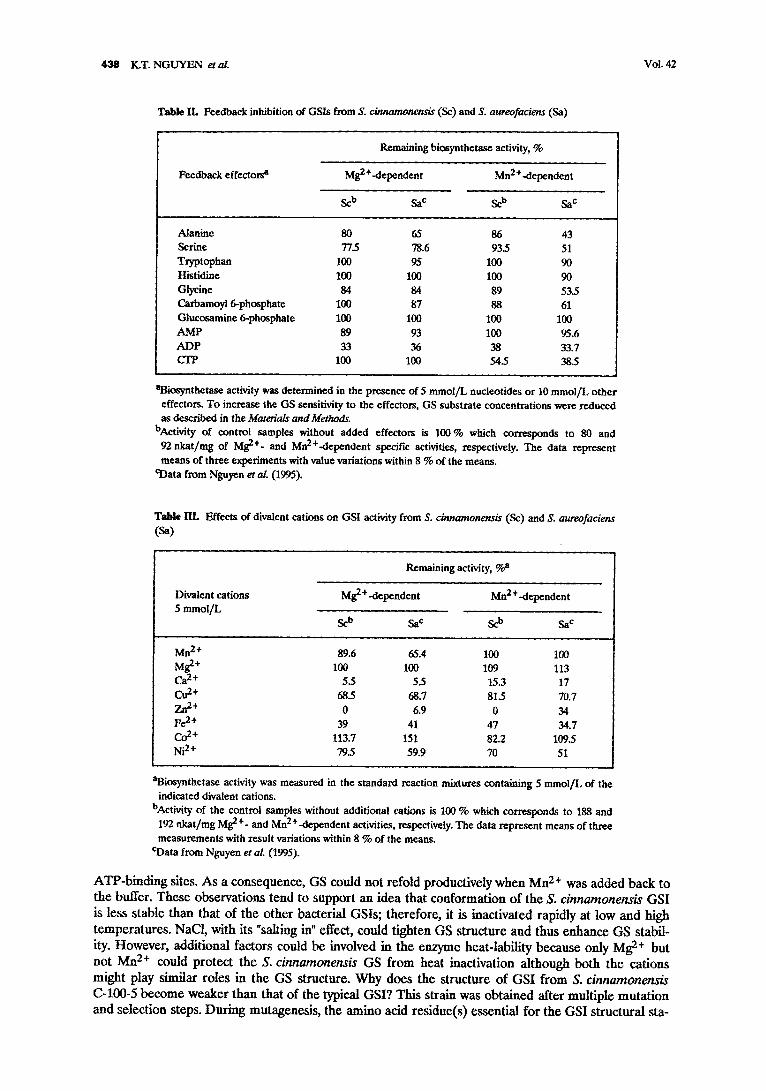
When assayed over a pH range from 6.3 to 7.8 and temperatures from 30 to 60 *C, the highest transferase and biosynthetase activities were found at pH 7.2 and 40-50 *C, respectively. Under opti- mum assay conditions, the Km values were (retool/L): glutamate 12.5, ammonium 0.1, ATP 1.25, glut- amine 18.5, hydroxylamine 3.3, ADP 87. These substrate affinities are similar with those of the heat- stable GSI from S. aureofaciens (Nguyen et al. 1995); however, the S. cinnamonensis enzyme was less sensitive to some feedback inhibitors (Table H). ADP was a potent inhibitor of both Mn 2+- and Mg 2+-

1997 GLLITAMINE SYNTHETASE FROM S. cinnamonensis 435
Fig. 1. Electrophoresis (A, B, C) and electron micrograph of GS (D); SDS-PAGE (A), native-PAGE (13) and IEF (C) of pure GSs from S. aureofaciens (lanes 1) and S. cinnamonensis (lanes 2). The positions marked by arrows correspond to M 55 kDa (A) and pI at pH 4.2 (C). Positions of the M and pI standard proteins run in the same gels are not shown. Electron micrograph of the pure GS from S. cinnamonensis (D). GS particles exhibit two specific views: the end-on view with hexagonal symme- try (arrow) and the side-on view.
dependent activities but CTP inhibited only the MnX+-dependent activity. Amino acids and amino sug- ars exerted partial inhibition. The effects of divalent cations on GSI activities from S. cinnamonensis and S. aureofaciens are very similar (Table III). Zn 2+, Ca 2+, Fe 2+, Cu 2 + and Ni 2 + are strong inhibitors and Co 2+ activated slightly the MgX+-dependent activity but inhibited the Mn2+-dependent activity by 18 %. Higher activation of Co 2§ was found with the S. aureofaciens enzyme.

436 K.T. NGUYEN et al. Vol. 42
100 %
8O:
60
~0
20
1 "" I i
I . . I 20 /.0 60
rain
120 - I ! | "" I %
80
~0
0 2Q /.0 60 80 *C
Fig. 2. Thermo6tability of purified GS from S. cinna- monensis; top: desalted GS samples (11 lag/mL; 30 ~tL) were incubated in a water bath at 50 ~ At the indi- cated time, samples were cooled in an ice bath and the remaining transferase (1) and semisynthetase (2) activi- ties were assayed; bottom: desalted GS samples were incubated at the indicated temperatures for 10 rain (I), or in the presence of 20 mmol/L Mg 2+ (2) or 1 mol/L NaCI (3). The remaining transferase activity was deter- mined. All data represent the means of three measure- ments with value variations within 5 % of the means.
DISCUSSION
The presence of GSI and GSII in several groups of soil bacteria including Rhizobiaceae, Fran- Ida and Streptomyces raises questions about regulation and physiological function of each GS isoform. In Rhizobiaceae and Franida, GSII appears to play the main role in ammonium assimilation under nitrogen-starvation conditions. In contrast, GSII activity from S. coelicolor and S. viridochromogenes is not induced by nitrogen starvation (Nguyen et al. 1994; Hillemann et al. 1993). Low heat-labile GSII activity was found in a wild-type S. viridochrornogenes strain and much higher GSII activity was detected in a mutant defective in GSI activity (Hillemann et al. 1993). So far, the Streptomyces GSII has been purified only from a S. lividans strain carrying and overexpressing the glnlI gene cloned from S. hygroscopicus (Kumada et al. 1990).
In S. coelicolor and S. aureofaciens, less than 10 % GS activity was found to be heat-inacti- vated, suggesting that these species express only the heat-stable GSI (Nguyen et al. 1994; Nguyen 1995b). Unexpectedly, the monensin-producing S. cinnamonensis cells grown with nitrate or glutamate as nitrogen source contained high heat-inactivated GS activity. The enzyme activity in crude extracts of cells growing in several growth phases under two temperature and shaking regimes was similarly inacti- vated by 80 % when incubated for 1 h at 50 ~ The possibility that this heat-inactivated activity repre- sented GSII activity prompted us to purify and characterize the enzyme. The results showed that the purified enzyme is also heat-labile but it has a dodecameric structure, an pI of 4.2, and an activity being regulated by adenylylation similar with properties of the heat-stable GSIs from S. aureofaciens and the other bacteria. Using diluted and desalted crude extracts or pure GS samples from S. cinnamonensis did not enhance GS heat stability, excluding the possibility that this enzyme is heat-stable but was inac- tivated rapidly due to accelerated aggregation and inhibition by metabolites during heat treatment. It was also unlikely that proteolysis was responsible for the GS heat inactivation because the enzyme in crude extracts or pure samples incubated at high temperatures up to 65 ~ in the presence of Mg 2+ or NaCI remained fully active; Mg 2 + and NaCI are not proteinase inhibitors.

1997 GLUTAMINE SYNTHETASE FROM S. cinnamonensis 437
Fig. 3. Effect of ammonium shock on GS activity (A) and GS modification (B, C); A: NH4CI was added to one half of a 10-h- old culture growing in glucose-nitrate medium to a final concentration of 37 mmol/L at the indicated time (arrow). The other half was used as control. Samples (100 laL) were taken at intervals and GS transferase activity in the permeabilized cells of the control (1) and the ammonium-shocked (2) cultures was determined as previously described (Fisher and Wray 1989); B, C: two-dimensional gels of total proteins in the control (B) and the ammonium-shocked (B) cells. Only parts of the gels containing most of the proteins are shown. Ammonium shock caused an increase of the modified GS subunits (arrow) at the expense of the closely located, unmodified proteins (arrow). The M values (kDa) are indicated.
The molecular basis of the heat lability of S. cinnamonensis GSI is currently unknown. High temperatures might cause the unfolding of GS molecules and thus promote the formation of inactive aggregates (Merkler et al. 1988). Therefore, a weaker structural conformation may lead to a lower sta- bility of GS. Bacterial GSIs bind to the Cibacron Blue dye at nucleotide-binding sites, and therefore could be eluted from Blue-Sepharose by adenine nucleotides. The elution of S. cinnamonensis GSI by simply increasing the pH of the buffer from 6.3 to 7.4 suggests that the nucleotide-binding site of this enzyme is not conformationally stable and is readily altered by pH change. Two ions of Mn 2+ (or Mg 2+) are usually found in each active site located in the subunit interfaces of GS (Almassy et al. 1986). The less tightly bound n2 ion has a role in ATP binding and the tightly bound nl stabilizes GS conformation (Liaw et al. 1993). Removal of Mn 2§ or Mg 2§ from the E. coli GSI by EDTA resulted in reversible inactivation and unfolding of the enzyme. The S. cinnamonensis GSI was readily inactivated just by omitting Mn 2§ or Mg 2§ from the buffer, suggesting that this enzyme is very sensitive to the lack of the specific cations. As shown by gel filtration the inactivated enzyme still remained in a dodeca- meric structure but addition of Mn 2 § did not reactivate it. It is possible that when Mn 2 § was omitted from the buffer, the nl Mn 2§ might still tightly bind to the GS molecules and stabilize the GS quater- nary structure but the n2 ion was lost, resulting in a marked conformational change, particularly at
438 ICT. NGUYEN et al. Vol. 42
Table IL Feedback inhibition of GSIs from S. c/nnamonens/s (Sc) and S. aureofaciens (Sa)
Feedback effectors a
Remaining biosynthetase activity, %
Mg 2 +-dependent Mn2+-dependent
Scb Sac Scb Sac
Alanine 80 65 86 43 Serine 77_5 78.6 93.5 51 Tryptophan 100 95 100 90 Histidine 100 100 100 90 Glycine 84 84 89 53.5 Carbamoyl 6-phosphate 100 87 88 61 Glucosamine 6-phosphate 1130 100 100 100 AMP 89 93 I00 95.6 ADP 33 36 38 33.7 CTP 100 100 54.5 383
aBiosynthetnse activity was determined in the presence of 5 mmol/L nucleotides or 10 mmol/L other effectors. To increase the GS sensitivity to the effectors, GS substrate concentrations were reduced as described in the Materials and Methods.
bAetivity of control samples without added effectors is 100 % which corresponds to 80 and 92 nkat/mg of Mg 2+- and Mn2+-dependent specific activities, respectively. The data represent means of three experiments with value variations within 8 % of the means.
eData from Nguyen et al. (1995).
Table IlL Effects of divalent cations on GSI activity from S. cinnamonensis (Sc) and S. aureofaciens (Sa)
Remaining activity, %a
Divalent cations Mg 2 +-dependent Mn 2 +-dependent 5 mmol/L
Sc b SaC Sob Sa c
Mn 2§ 89.6 65.4 100 100 Mg 2+ 100 100 109 113 Ca 2+ 5.5 5.5 15.3 17 Cu 2+ 68.5 68.7 81.5 70.7 Zn 2+ 0 6.9 0 34 Fe 2+ 39 41 47 34.7 Co 2+ 113.7 151 82.2 109.5 Ni 2§ 79_5 59.9 70 51
aBiosynthetase activity was measured in the standard reaction mixtures containing 5 mmol/L of the indicated divalent cations.
bActivity of the control samples without additional cations is 100 % which corresponds to 188 and 192 nkat/mg Mg 2+- and Mn2+-dependent activities, respectively. The data represent means of three measurements with result variations within 8 % of the means.
eData from Nguyen et al. (1995).
ATP-binding sites. As a consequence, GS could not refold productively when M n 2 + was added back to the buffer. These observations tend to support an idea that conformation of the S. c innamonens i s GSI is less stable than that o f the other bacterial GSIs; therefore, it is inactivated rapidly at low and high temperatures. NaC1, with its "salting in" effect, could tighten GS structure and thus enhance GS stabil- ity. However , additional factors could be involved in the enzyme heat-lability because only M g 2+ but not M n 2+ could protect the S. c innamonens is GS from heat inactivation al though both the cations might play similar roles in the GS structure. Why does the structure o f GSI f rom S. c innamonens i s
C-100-5 become weaker than that of the typical GSI? This strain was obtained after multiple muta t ion and selection steps. Dur ing mutagenesis, the amino acid residue(s) essential for the GSI structural sta-

1997 GLUTAMINE SYNTHETASE FROM S. cinnamonensis 4:39
bility might be replaced. To test this hypothesis, further studies on the heat stability of GS activity f rom the wild type strain and other mutants of S. cinnamonensis are necessary.
GSs f rom streptomycetes appeared to be cleaved for unknown reasons as detected by SDS- P A G E . We found that the cleavage of S. aureofaciens GSI was dependent on type of the incubation buffer and the presence of 2-mercaptoethanol in the SDS-sample buffer (Nguyen 1995b). I f the Strepto- myces GSs were digested by proteinases they would be inactivated (Rhee et al. 1989), and the digestion products would be detected by na t ive-PAGE and IEF. However, the enzymes still possessed very high specific activities and appeared as sharp proteins on na t ive-PAGE and I E F gels. Unde r the milder conditions of two-dimensional P A G E , subunits of GSI from S. cinnamonensis were found to be intact with an M of 55 kDa after incubation at room temperature with urea (in the first dimension) and SDS- sample buffer (in the second dimension). Several proteins such as ~-amylase have been reported to be cleaved at A s p - P r o and A s p - C y s bonds when boiled with SDS (Rittenhouse and Marcus 1984, Kubo 1995). It is also possible that GS cleavage might be due to boiling of the GS proteins with the SDS- sample buffer. Thus, we assume that S. cinnamonensis GSI might contain more SDS-susceptible bonds than GSI f rom S. aureofaciens. However, this phenomenon might not have any relation to the heat sta- bility of these enzymes. Further analysis of amino acid sequence and conformational change during heating of GSI f rom S. cinnamonensis may illuminate more precisely the molecular basis of the heat lability of this enzyme.
We are grateful to Dr. S. Pospigil for providing the S. cinnamonensis strain and cultivation conditions. The first and second authors are participants of the Long-Term Post-Graduate Training Course UNESCO-Roste organized in the Institute of Microbiology, Academy of Sciences of the Czech Republic. This work was supported in part by the Grant Agency of the Czech Republic (Grant no. 203/95/1054).
REFERENCES
ALMASSY R.J., JANSON C.A., HAMLIN S., XUONG N.-H., E1SENBERG D.: Novel subunit-subunit interactions in the structure of glutamine synthetase. Nature 323, 304-309 (1986).
BEHRMANN I., HILLEMANN D., PUHLER A., STRAUCH E., WOHLLEBEN W.: Overexpression of a Streptomyces viridochromogenes gene (glnll) encoding a glutamine synthetase similar to those of eucaryotes confers resistance against the antibiotic phosphinothricyl-alanyl-alanine. ZBacteriol. 172, 5326-5334 (1990).
BENADA O., POKORN~ V.: Modification of the Polaron sputter-coater unit for glow-discharge activation of carbon support films. ZElectron.Micr.Tech. 16, 235-239 (1990).
BENDER R.A., JANSSEN K.A., RESNICK A.D., BLUMENBERG M., FOOR F., MAGASANIK B.: Biochemical parameter of glutamine synthetase from Klebsiella aerogenes. ZBacteriol. 129, 1001-1009 (1977).
BRADFORD M.M.: A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. AnaLBiochem. 72, 248-254 (1976).
EDMANDS J., NORIDGE N.A., BENSON D.R.Y.: The actinorhizal root-nodule symbiont Frankia sp. strain CpI 1 has two glutamine synthetases. Proc.Nat.Acad.Sci.USA 84, 6126-6130 (1987).
FISHER S.H., WRAY L.V. Jr.: Regulation of glutamine synthetase in Streptomyces coelicolor. J.Bacteriol. 171, 2378-2383 (1989). IhMss B.D.: An introduction to polyacrylamide gel electrophoresis, pp. 1-91 in B.D. Hames, D. Rickwood (Eds): Gel Electro-
phoresis of Proteins: A Practical Approach. IRL Press, London-Washington (DC) 1981. HILLEMANN D., D~MmqN T., HILLEMANN A., WOHLLEREN W.: Genetic and biochemical characterization of the two glutamine
synthetases GSI and GSII of the phosphinothricyl-alanyt-alanine producer, Streptomyces viridochromogenes Tu 494. J.Gen.Microbiol. 139, 1773-1783 (1993).
Kuno K.: Variability in heat-induced fragmentation of a protein in the presence of dodecyl sulfate: the role of an intramolecu- lar sulfhydryl/disulfide exchange. J.Biochem. 118, 1112-1117 (1995).
KUMADA Y., TAg.'~O E., NAGAOIeA K., THOMPSON CJ.: Streptomyces hygroscopicus has two glutamine synthetase genes. J.Bac- teriol. 172, 5343-5351 (1990).
L~w S.H., V I S T A JJ., EISENnERG D.: A model for oxidative modification of glutamine synthetase, based on crystal structures of mutant H269N and the oxidized enzyme. Biochemistry 32, 7999-8003 (1993).
MERKLER D.J., SRIKUMAR K., ]VtARCHESE-RAGONA S.P., WEDLER F.C.: Aggregation and thermoinactivation of glutamine synthetase from an extreme thermophile Bacillus caiydoticus. Biochim.Biophys.Acta 952, 101-114 (1988).
NGUYE~ K.T., NGUYE~ L.T., B~.HAL V.: What type of glutamine synthetase is important for Streptomyces coelicolor A(3)2 under nitrogen-limited conditions. Biotechnol.Lett. 16, 1027-1030 (1994).
NGLrVEN K.T.: Enzymes of nitrogen metabolism of Streptomyces. Studies on glutamine synthetase and glutamate dehydroge- nase. PhD Thesis. Institute of Microbiology, Academy of Sciences of the Czech Republic, Prague 1995.
NGUYEN L.T., NGUYEN K.T, KOPECK~ J., NOVOTNA J., Nova P., Bi~h~L V.: Purification and characterization of a novel valine dehydrogenase from Streptomyces aureofaciens. Biochim.Biophys,4cta 1251, 186-190 (1995a).
NGLryEN K.T., NGLrVE~ L.T., B~p,L V.: How is glutamine synthetase 1 from Streptomyces aureofaciens regulated? Biotechnol. Lett. 17, 609-614 (1995b).
O'FmgRELL P.H.: High resolution twc~imensional electrophoresis of proteins. J.Biol.Chem. 250, 4007-4021 (1975).

440 K.T. NGUYEN et al. Voi. 42
PENYIGE A., KAI.MANCZHELYI A., SIPOS A., ENSXOS J.C., B ~ G.: Modification of glutamine synthetase in Streptomyces gr/seus by ADP-ribosylation and adenylylation. Biochem.Biophys.Res.Commun. 204, 598-605 (1994).
POSP[glL S., SEDMERA P., HAVRANEK M., KRUMPHAHZL V., VANS Z.: Biosynthesis of monensins A and B. J,4ntibiot. 36, 617- 619 (1983).
REVES J.C., FLORENOO FJ.: A new type of glutamine synthetase in cyanobacteria: the protein encoded by the glnN gene supports nitrogen assimilation in Synechocystis sp. strain PCC 6803. J.Bacteriol. 176, 1260-1267 (1994).
RHEE S.G., Chock P.B., STAtYrMA~ E.1L: Regulation of Escherichia coli glutamine synthetase.Adv.Enzymol. 62, 37-92 (1989). P-aTr~HOUSE J., MARO3S F.: Peptide mapping by polyacrylamide gel electrophoresis after cleavage at aspartyl-prolyl peptide
bonds in sodium dodecyl sulfate-containing buffers. Anal.Biochem. 138, 442-448 (1984). SnA~'ERS 1LG., LIu Y., KAnN M.L.: Isolation and characterization of a novel glutamine synthetase from Rhizobium meliloti.
ZBiol.CRem. 268, 469-475 (1993). STRFJCHER S L, TYLER B.: Regulation of glutamine synthetase activity by adenylylation in Gram-positive bacterium Strepto-
myces cattleya. Proc3Vat.Acad.Sci.USA 78, 229-233 (1981). TATE S.S., MElsa~ A.: Regulation of rat liver glutamine synthetase: activation by 0t-ketoglutarate and inhibition by glycine,
alanine and carbamyl phosphate. Proc.Nat.Acad.Scs 68, 781-785 (1971). TYLER B.: Regulation of the assimilation of nitrogen compounds. Ann.Rev.Biochem. 47, 1127-1162 (1978). VIN1NO L.C., SlxYrrARD C.: Genetics and Biochemisay of Antibiotic Production. Butterworth-Heinemann, Newton 1995. WooDs D.1L, REID SJ.: Recent developments on the regulation and structure of glutamine synthetase enzymes from selected
bacterial groups. FEMS Microbiol.Rev. 11, 273-284 (1993). WRAY L.V. Jr., FISHER S.H.: Cloning and nucleotide sequence of the Streptomyces coelicolor gene encoding glutamine synthet-
ase. Gene 71, 247-256 (1988). WRAY L.V. Jr., AT~NSON M.R., FISHER S.H.: Identification and cloning of the glnR locus, which is required for transcription of
the g/nA gene in Streptomyces coelicolor A3(2). J.Bacteriol. 173, 7351-7360 (1991).




















