
Differential transcript abundance and genotypic variation of ...
Upload
uniklinik-duesseldorfCategory
view
0download
0
Brain (2000), 123, 1612–1623
Phenotypic and genotypic heterogeneity inhereditary motor neuronopathy type VA clinical, electrophysiological and genetic study
Michaela Auer-Grumbach,1 Wolfgang N. Loscher,1 Klaus Wagner,2 Erwin Petek,2 Eva Korner,1
Hans Offenbacher3 and Hans-Peter Hartung1
1Department of Neurology, Karl-Franzens University, Correspondence to: Michaela Auer-Grumbach, MD,2Institute of Medical Biology and Human Genetics, Department of Neurology, Auenbruggerplatz 22, A-8036Karl-Franzens University, Graz and 3Department of Graz, AustriaNeurology, Landeskrankenhaus, Knittelfeld, Austria E-mail: [email protected]
SummaryWe report on a large four-generation Austrian family abnormal in older or severely affected persons.
Electromyography showed high-amplitude motor unitwith autosomal dominant distal hereditary motorpotentials and reduced recruitment compatible withneuronopathy type V (distal HMN V). Forty-seven at-anterior horn cell degeneration. Central motor conductionrisk family members, of whom 21 were definitely affected,times were prolonged in two-thirds of the patients.underwent detailed clinical, electrophysiological andMolecular genetic studies excluded Charcot–Marie–Toothgenetic studies. The age at onset was in the second1A syndrome and proximal spinal muscular atrophydecade of life in most affected individuals, but clinicallinked to chromosome 5q as well as the known gene locipresentation was rather variable. While the majority offor distal HMN II on chromosome 12q, HMN V onpatients were primarily disabled by progressivechromosome 7p and juvenile amyotrophic lateral sclerosisasymmetrical wasting of the thenar and the first dorsalon chromosome 9q. The findings in this family thusinterosseus muscles, others had marked foot deformityprovide detailed clinical and electrophysiologicaland gait disturbance with the occasional absence of handinformation on HMN V and demonstrate broad
involvement. Sensation sense was normal except for the phenotypic variability in this disorder. Hallmark featuresreduced response to vibration. Many individuals showed are discussed that appear to be most reliable tobrisk tendon reflexes and some elevated muscle tone in differentiate this type of HMN V from other variants ofthe lower limbs, but extensor plantar responses were hereditary neuropathies, and a set of diagnostic criteriararely observed. Electrophysiological evaluation revealed is proposed. Furthermore, this is the first report ofnormal or reduced motor nerve conduction velocities, prolonged central motor conduction times in HMN V,normal or prolonged distal motor latencies, and low which indicates additional involvement of the centralcompound motor action potentials, depending on the motor pathways in this disease. Finally, molecular geneticdegree of muscle wasting. Sensory nerve studies were studies demonstrate genetic heterogeneity, suggesting the
existence of at least a second genetic subtype in HMN V.usually within the normal range or slightly to moderately
Keywords: hereditary motor neuronopathy; HMN V; distal spinal muscular atrophy; Charcot–Marie–Tooth; chromosome 7p
Abbreviations: CMAP � compound motor action potential; CMCT � central motor conduction time; FDI � first dorsalinterosseus muscle; HMN � hereditary motor neuronopathy; HMSN � hereditary motor and sensory neuropathy; NCV �nerve conduction velocity
IntroductionCharcot–Marie–Tooth syndrome (hereditary motor and disorder can be divided into three main subtypes: a
demyelinating, an axonal and a spinal form. Charcot–Marie–sensory neuropathy, HMSN) is a genetically determinedcommon disorder of the peripheral nervous system with an Tooth type 1, the demyelinating form, and Charcot–Marie–
Tooth type 2, the axonal variant, are characterized clinicallyestimated incidence of 40/100 000 (Skre, 1974). Based onclinical, electrophysiological and morphological criteria, this by distal muscle weakness and wasting of the upper and
© Oxford University Press 2000

Heterogeneity in HMN V 1613
lower limbs, foot deformity, gait disturbance and reduced or Consequently, the disorder was classified as an axonalCharcot–Marie–Tooth type 2 and was subcategorizedabsent tendon reflexes (Harding and Thomas, 1980). A
variable degree of sensory loss is usually present and the genetically as Charcot–Marie–Tooth type 2D. Subsequently,a disease locus on chromosome 7p was confirmed in adisease is occasionally accompanied by additional features
(Kwon et al., 1995; Thomas et al., 1997; Auer-Grumbach large Mongolian Charcot–Marie–Tooth 2D kinship in whichpatients with and without sensory loss were observed withinet al., 1998). Until molecular genetic testing became available
for individual Charcot–Marie–Tooth 1 cases, the two subtypes the same family. Genetic linkage studies in this familysuggested that Charcot–Marie–Tooth 2D and HMN V arecould be distinguished only by electrophysiological and
histopathological methods (Harding and Thomas, 1980). The caused by mutations in a single gene (Sambuughin et al.,1998). A refined genetic analysis narrowed the critical regionspinal form of Charcot–Marie–Tooth syndrome [also called
distal spinal muscular atrophy; in the recent gene mapping for Charcot–Marie–Tooth 2D/HMN V to ~1.5 cM(centiMorgan) and a BAC/PAC contig map has beenliterature the preferred term is distal hereditary motor
neuronopathy (HMN)] is phenotypically different from constructed containing the whole of the region of interest(Ellsworth et al., 1999).classical Charcot–Marie–Tooth types 1 and 2 as there is no
clinically apparent sensory loss and the sensory nerves are We report here a large Austrian family presenting withsigns and symptoms of HMN V and provide detailed clinicalelectrophysiologically and morphologically normal (Harding,
1993). It has been emphasized that distal HMN is caused by and electrophysiological data in 21 affected and 26 at-riskindividuals. Genetic linkage analysis did not confirm thedegeneration of the spinal motor neurons (Harding, 1993).
Based on the wide spectrum of clinical variation and different known HMN V gene locus on chromosome 7p in our family,therefore suggesting genetic heterogeneity of this disorder,modes of inheritance, distal HMN has been divided into
seven subtypes (distal HMN I–VII) (Harding, 1993). When and we attempted to establish diagnostic criteria for differentgenetic subtypes. The large number of definitely affectedmuscle weakness and wasting are confined predominantly to
the hands, the disease is termed HMN type V. Both sporadic persons in this family allowed us to demonstrate the broadspectrum of phenotypic variation of this disorder. We alsoand familial cases of HMN V have been observed (Meadows
and Marsden, 1969; McLeod and Prineas, 1971; O’Sullivan investigated whether additional electrophysiological findingscontribute to the diagnosis of the disease in subclinical casesand McLeod, 1978). To date, however, only a few families
have been described in the literature (Silver, 1966; Lander and whether they aid in distinguishing different geneticsubtypes.et al., 1976; van Gent et al., 1985; de Visser et al., 1988;
Christodoulou et al., 1995; Gross et al., 1998).While some of these kinships demonstrated symmetrical
wasting of all small hand muscles, others presented with aPatients and methodsstriking, asymmetrical distribution, usually affecting the
thenar and first dorsal interosseus (FDI) muscles Family data and clinical assessmentA part of the pedigree of the family comprising 134 memberspredominantly. Clinical sensory loss was rarely observed and
was only a minor feature when it was present, but foot in four generations is shown in Fig. 1. Several male-to-maletransmissions suggest autosomal dominant inheritance. Indeformity was a frequent sign. In addition, mild spastic
paraplegia was noted in some families and was sometimes total, 47 at-risk individuals were recruited after obtaininginformed consent for clinical, electrophysiological andprominent enough for the disorder to be classified as a
hereditary spastic paraplegia rather than an HMN (Silver, genetic studies, and a further eight spouses participated inthe genetic linkage studies. A full neurological and detailed1966; de Visser et al., 1988).
Recent molecular genetic studies have led to the definition electrophysiological examination was performed on all at-risk family members (aged 12–68 years, mean 35 years) byof two gene loci in distal HMN: HMN II on chromosome 12q
(Timmerman et al., 1996) and HMN V on chromosome 7p one of us (M.A.-G.). Information on deceased family memberswas obtained independently from several older relatives,(Christodoulou et al., 1995). The latter was first reported in
a large Bulgarian kinship in which affected individuals were when available.Motor strength was assessed using the standard MRCafflicted with prominent wasting of the thenar and FDI
muscles. Lower limb involvement was observed in 40% (Medical Research Council) scale (grades 0–5) and reflexeswere quantified as absent � 0, hypoactive � �1, normal �of cases. Brisk tendon reflexes and mild pyramidal tract
involvement were found in only one branch of the family. �2, brisk � �3 or �4, following the NINDS (NationalInstitute of Neurological Disorders and Stroke) scale. TouchSensation was clinically and electrophysiologically normal
except mildly reduced vibration. Interestingly, in 1996 sensation was tested with a monofilament and vibration sensewas quantitated with a graduated Rydel–Seiffer tuning forkIonasescu and colleagues described a similar phenotype in a
family from Iowa, and the gene locus also mapped to (grades 0–8). The ability to recognize written numbers in thedistal parts of the upper and lower limbs was assessed inchromosome 7p (Ionasescu et al., 1996). In contrast to the
Bulgarian family, in affected individuals of this family every individual.As there was considerable phenotypic variation in thetendon reflexes were reduced and sensory loss was common.

1614 M. Auer-Grumbach et al.
Fig
.1
Part
ial
pedi
gree
offa
mily
with
dist
alH
MN
type
V.
Defi
nite
lyaf
fect
edfa
mily
mem
bers
are
show
nin
blac
k;at
-ris
kin
divi
dual
san
dsp
ouse
sar
em
arke
das
norm
al.

Heterogeneity in HMN V 1615
expression of the disease and the family was ascertained cortex stimulations were performed with underlyingcontraction and stimulator output was increased to yieldfor genetic linkage studies, strict diagnostic criteria were
used for the phenotypic classification of the patients and the maximum response amplitude. At least two reproducibleresponses were recorded from each stimulation site. Corticalat-risk individuals. According to their medical history
and physical and electrophysiological evaluation, at-risk and spinal latencies were determined visually and thecentral motor conduction times were calculated.individuals were diagnosed as being either definitely
affected or probably affected. None was classified as notaffected, because the penetrance of the disease is notknown. A person was classified as definitely affected when
Statisticsall of the following criteria were met: (i) age at onsetThe distribution of electrophysiological data was analysedbefore the fourth decade of life; (ii) unilateral or bilateralusing the Shapiro–Wilks W-test and non–parametric statisticsweakness and wasting of thenar and/or FDI muscles and/were used accordingly. As no side differences were foundor marked foot deformity; (iii) clinical absence of sensoryfor nerve conduction studies (Wilcoxon matched pair test),loss except for impaired vibration sense; (iv) pathologicaldata for both sides were pooled to compare definitely andmotor nerve conduction speeds in at least two nerves butprobably affected groups. Differences between groups werenormal or only slightly abnormal sensory nerve conductionanalysed using the Mann–Whitney U-test.speeds. Individuals with polyneuropathy of known cause
were excluded. At-risk individuals were defined as probablyaffected if they were normal on clinical and electro-physiological examination or had only mild foot deformity
Molecular geneticsand/or brisk tendon reflexes and/or sweating disturbance orpathological sensory nerve conduction speeds or prolonged DNA marker analysis
Peripheral blood samples were obtained from patients andcentral motor conduction times.relatives after they had given informed consent. DNAisolation from leucocytes was performed according tostandard methods.Neurophysiology
After determination of the DNA concentration, ~100 ngElectromyography and nerve conductiongenomic DNA was used for PCR (polymerase chain
velocity studies reaction) to amplify short tandem repeats for the genotypeNerve conduction velocity (NCV) studies and EMG analysis. One oligonucleotide primer was labelled with afollowed standard techniques (Aminoff, 1998) using the fluorochrome (IRD700, IRD800; MWG–Biotech, Ebersberg,electromyograph MS60 (Medelec, Old Woking, UK) or a Germany) and the resulting amplification products wereMyohandy portable EMG (Micromed Neurodata, Mogliano analysed using a DNA sequencer with a dual laser systemVeneto, Italy). Responses for motor nerve conduction (DNA sequencer 4000; LI–COR, Lincoln, Nebr., USA).studies were recorded from distal muscles using surface For the exclusion of distal HMN II on chromosome 12,electrodes. Sensory nerve conduction studies were performed the DNA markers D12S1282, D12S1349, D12S2079,antidromically with the use of surface or ring electrodes. D12S340, D12S378 and D12S86 were used; for theMultiple motor nerves, i.e. median, ulnar, peroneal and exclusion of HMN V on chromosome 7 the markerstibial nerves, were measured bilaterally in most individuals D7S435, D7S1514, D7S2492, D7S632 were usedand sensory nerve conduction studies were performed on (Christodoulou et al., 1995; Timmerman et al., 1996;the median and sural nerve on at least one side. Sambuughin et al.,1998). Juvenile amyotrophic lateralSemiquantitative EMG was performed with concentric sclerosis, linked to chromosome 9q, was excluded usingneedle electrodes on distal muscles of the upper and the markers D9S1830 and D9S1863 (Chance et al., 1998),lower limbs. Charcot–Marie–Tooth 2B was excluded by the use of the
markers D3S1551 and DS31744, and markers D9S197 andD9S910 were used to exclude HSN I (Kwon et al., 1995;Nicholson et al., 1996). PCR was performed in a 15 µlMagnetic evoked potentials
Transcranial and nerve root magnetic stimuli were delivered reaction containing 1 pmol of each primer, 0.15 UDYNAZyme (Finzyme, Espoo, Finland) in a UNO IIthrough a Magstim 200 stimulator (Magstim, Whitland,
Dyfed, UK). A standard round coil with an inner diameter thermocycler (Biometra, Gottingen, Germany). Before gelanalysis, 1 µl of the PCR product was mixed with 4 µlof 9 cm was used. The motor cortex of the hand and foot
area and the spinal nerve roots C8 and L5 were stimulated loading dye, denatured for 3 min at 70°C and loaded ona 4% polyacrylamide sequencing gel for the LI–CORand magnetic evoked motor potentials were recorded from
the abductor digiti minimi and the tibialis anterior muscles sequencer. Data collection and analysis were performedusing BaseImagIR Data Collection V4.00 and RFLPScanbilaterally using surface electrodes and a commercial EMG
amplifier (Toennies–Jager, Wurzburg, Germany). Motor V3.00 as supplied by LI–COR.

1616 M. Auer-Grumbach et al.
Fig. 2 Foot deformity and hand muscle wasting in patient III/37. This 47-year-old patientremembered having had foot deformity since early childhood. At the age of 18 years he firstnoticed wasting of the thenar and FDI muscles of the right hand. At that time he becameaware of being afflicted with a hereditary disease. Foot deformity progressed to severe gaitdisturbance.
Charcot–Marie–Tooth 1A caused by a duplication on Linkage analysischromosome 17p11.2 and proximal spinal muscular atrophy Two-point linkage studies were performed using thelinked to chromosome 5q were tested by routine methods LINKAGE computer package version 5.1 obtained from the(Reiter et al., 1996) in one representative family member Laboratory of Statistical Genetics at Rockefeller University,
USA (http://linkage.rockefeller.edu). We used five age-(IV/58).
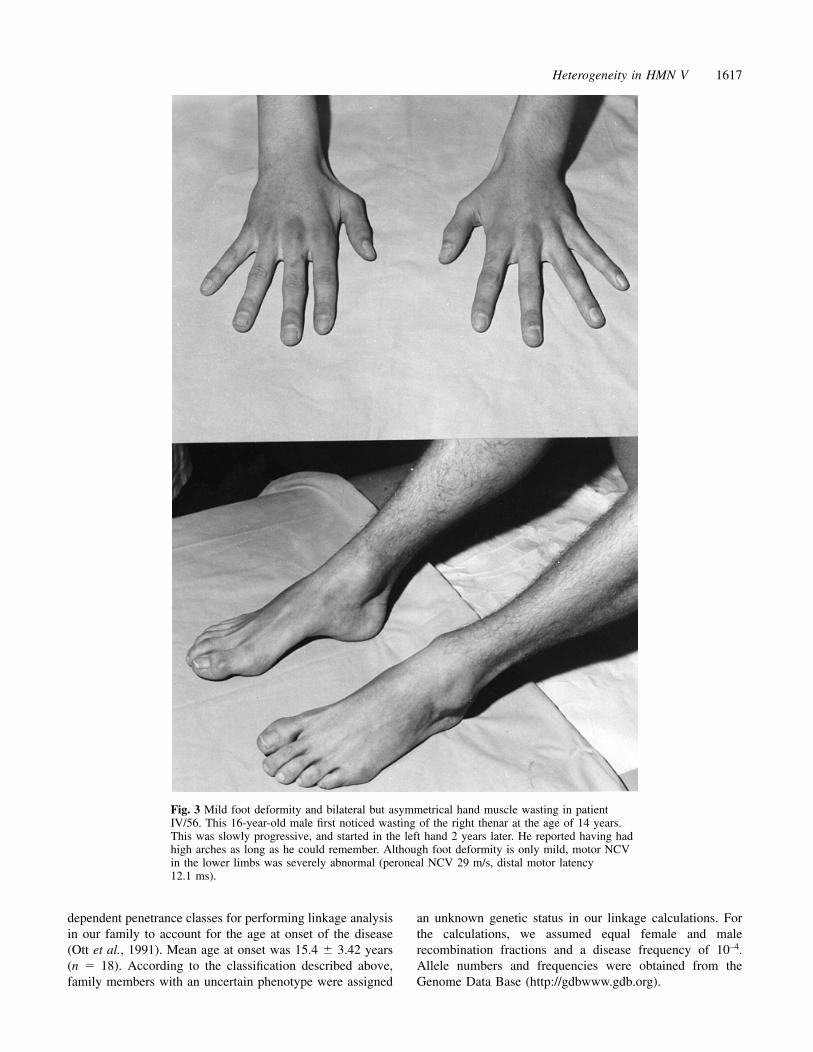
Heterogeneity in HMN V 1617
Fig. 3 Mild foot deformity and bilateral but asymmetrical hand muscle wasting in patientIV/56. This 16-year-old male first noticed wasting of the right thenar at the age of 14 years.This was slowly progressive, and started in the left hand 2 years later. He reported having hadhigh arches as long as he could remember. Although foot deformity is only mild, motor NCVin the lower limbs was severely abnormal (peroneal NCV 29 m/s, distal motor latency12.1 ms).
dependent penetrance classes for performing linkage analysis an unknown genetic status in our linkage calculations. Forthe calculations, we assumed equal female and malein our family to account for the age at onset of the disease
(Ott et al., 1991). Mean age at onset was 15.4 � 3.42 years recombination fractions and a disease frequency of 10–4.Allele numbers and frequencies were obtained from the(n � 18). According to the classification described above,
family members with an uncertain phenotype were assigned Genome Data Base (http://gdbwww.gdb.org).

1618 M. Auer-Grumbach et al.
Results Neurological examination: results for the 21Clinical findings definitely affected individualsClassification of family members Proximal muscle strength was normal in both the upper andForty-seven at-risk individuals underwent complete the lower limbs in all definitely affected individuals. In 15neurological examination. Mean age at investigation was 35 persons muscle power in the small hand muscles was reducedyears. Eighteen individuals expressed the disease fully at the depending on the degree of muscle wasting (MRC grade 0time of investigation and were aware of being affected. The to 4�). Fourteen individuals revealed diminished muscleremaining 29 at-risk individuals were screened systematically power in the distal lower limbs, which occurred particularlyfor HMN V by clinical and electrophysiological testing and in the toe and ankle extensors (MRC grade 1 to 4�). Thetwo further definitely affected individuals were detected. One tendon reflexes in the upper and lower limbs were normalindividual (III/51), who was clinically and electro- (NINDS �2), diminished (NINDS �1) or brisk (NINDS �3physiologically normal but gave birth to an affected child, or �4) or rarely absent (NINDS � 0). Muscle tone waswas also classified as affected, although she did not fulfil the usually normal in the upper limbs and increased in the lowerdiagnostic criteria mentioned above. The asymptomatic status limbs in three patients only. A Babinski sign was present inof this person might be the result of incomplete penetrance. only one patient. No individual had a deformity of the spinalAlternatively, the affected child IV/56 may even represent a column or an abnormality of the cranial nerves. Clinical andphenocopy. Thus, a total of 21 definitely affected and 26 neurological findings for all 21 definitely affected familyprobably affected family members were identified. Histories members are summarized in Table 1.of the deceased individuals of generation II and III revealedthat nine further persons had been definitely affected.
Main findings in 26 probably affected familymembersNine individuals had mild foot deformity, 20 had brisk tendonAge at onset and presenting features in the 21reflexes in the upper or lower limbs and 14 complained aboutdefinitely affected family membersexcessive sweating. None of them showed signs of muscleThe age at onset was taken to be the time when the patientweakness or wasting or other additional features. One 18-first noticed weakness in the hands and/or gait disturbance.year-old individual was clinically normal but had pathologicalAlthough this varied widely, from 10 to 38 years (mean �median motor nerve conduction speeds. A further 52-year-15.4 � 3.42 years), most patients (17/21) developedold person was clinically normal but had marked prolongedsymptoms during the second decade of life. Three individualscentral motor conduction times to the lower limbs, as didwere unable to define the time when the disease started. Inothers.12 cases, atrophy of the thenar and FDI muscles was the
initial and most prominent symptom, although on furtherquestioning most of them remembered that foot deformity
Main findings in nine definitely affected deceasedhad been present, but so far unnoticed, to a variable degreefamily memberssince early childhood. In others, amyotrophy of the handsHand muscle involvement had been observed in six patients,appeared later (3/9) or even remained absent (6/9).although this was the first sign of the disease in only two ofInterestingly, muscle atrophy in the hands was frequentlythem. Seven individuals had had marked foot deformity andasymmetrical in distribution (12/15), the right side beinggait problems. One of them had been unable to walk atmore severely affected than the left in most cases (9/12),advanced age; all others remained ambulatory. One personwhile the lower limbs were always involved to an equalhad been asymptomatic but had children who weredegree. Eight patients were brought to medical attentiondefinitely affected.primarily because of marked foot deformity and gait
problems. All patients except the clinically normal diseasecarrier had mild to very severe foot deformity, presenting as
Neurophysiological resultspes cavus, pes planus and/or hammer-toes. Less frequently,atrophy of the ankle extensors occurred. None of the patients Nerve conduction velocity studies
NCV studies were performed in 21 definitely affected andcomplained about sensory loss and only two individualsnoticed paraesthesia, and, in addition, muscle cramps in the 26 at-risk individuals. In general the median and the peroneal
nerves were more severely impaired than the ulnar and tibiallegs. Eight persons reported prominent hyperhidrosis in thehands and feet, and five frequently had cold feet. Additional nerves. The values of motor NCV, distal motor latencies and
compound motor action potentials (CMAPs) in the definitelyfeatures such as hypacusis, depression and hip dysplasia wereobserved rarely. The disease was slowly progressive in all affected individuals varied widely from normal to severely
reduced, and this was strongly correlated with the degree ofpatients, but none was severely handicapped. Figures 2 and3 demonstrate the variability of hand muscle involvement muscle wasting. Conduction block was never observed, but
pronounced dispersion of CMAPs was frequently present. Inand foot deformity.

Heterogeneity in HMN V 1619
Table 1 Clinical features in 21 patients with definite disease
Patient/ Age at Age at onset of Foot Peroneal Hand muscle Vibration Tendon reflexes Hyper-pedigree/ examination symptomatic deformity muscle atrophy wasting UE/LE (Radial/ knee/ankle) hidrosissex (years) disease (years) right/left
III/2/F 61 15 ��� � �/�� 8/7 2/2/1 �III/3/M 59 12 ��� � –/– 7/6 3/4/3 �III/18/M 40 17 �� �� ���/� 7/6 3/4/3 –III/19/F 40 14 �� – �/� 6/4 2/4/3 –III/28/M 55 10 � – –/– 7/5 2/0/0 ��III/34/M 52 14 ��� �� ���/� 6/4 2/3/2 �III/35/F 51 15 �� � –/– 7/5 2/4/3 �III/37/M 47 18 ��� ��� ���/� 6/5 3/3/0 ��III/43/M 68 ? � � –/– 6/6 2/3/0 –III/45/F 43 38 � – ��/� 7/6 3/3/3 –III/47/M 50 ? �� �� ���/�� 7/4 2/3/2 �III/51/F 40 ? – – –/– 8/7 2/2/2 –III/53/F 39 10 �� � ��/� 7/7 2/3/1 –IV/4/F 32 15 � � �/� 6/6 2/3/3 –IV/6/M 28 18 �� �� �/�� 8/7 1/3/1 –IV/26/M 19 15 � – �/� 8/7 2/3/3 –IV/ 27/M 12 12 � – ���/�� 8/7 2/3/3 –IV/33/M 28 14 �� �� –/– 7/5 2/3/3 ��IV/37/F 12 17 � – �/��� 6/4 1/2/2 –IV/56/M 16 14 � – ���/�� 7/4 1/2/2 –IV/58/M 19 10 �� � ���/�� 7/5 2/3/1 –
Reflex grading according to NINDS scale. UE � upper extremities; LE � lower extremities; � � mild; �� � moderate; ��� �severe; – � not present; ? � unknown.
only one case was it impossible to obtain a CMAP from the conduction times (CMCTs) were observed in 9/15 definitelyaffected and 6/15 probably affected (χ2 test, n.s.). However,extensor digitorum brevis muscle. Median and sural sensory
NCVs and sensory nerve action potentials were usually within when comparing upper and lower extremities separately,prolonged CMCTs were more frequently observed in thethe normal range, but occasionally sensory NCVs were
slightly to moderately slowed and sensory nerve action definitely than the probably affected group (Fisher’s exacttest, P � 0.05). Thus, CMCTs were prolonged in 2/30 upperpotentials were reduced, corresponding to a severe clinical
phenotype or a long duration of the disease. In 14/26 probably and 9/29 lower limbs in definitely affected patients whileonly 3/30 upper and 5/28 lower limbs showed prolongedaffected individuals, NCVs were slightly abnormal and
frequently consisted of prolonged median distal motor CMCTs in probably affected persons. The means and standarddeviations of CMCTs are shown in Table 3.latencies, whereas the NCVs of the ulnar, peroneal and tibial
nerves were within the normal range in most cases, as werethose of the sensory median and the sural nerves. In themajority of these people, pathological values were obtained Results of molecular genetic studiesin only one motor nerve and therefore cannot be associated Genotyping of the definitely affected and probably affectedunequivocally with the disease. Table 2 summarizes the individuals revealed recombinants for the HMN II and HMN Vresults of motor and sensory nerve conduction studies. loci on chromosomes 12q and 7p, respectively. Two-point
linkage studies resulted in significant negative LOD (log10
odds ratio) scores for almost all markers analysed (Table 4)EMGexcept for the markers D7S2492 and D12S2079, whichEMG revealed severely reduced recruitment, indicatingyielded inconclusive LOD scores (–2 � z � �3) because ofpronounced loss of motor neurons. Individual motor unituninformative alleles in the family. These results indicatepotentials were often polyphasic and very high in amplitudethat the disease in our family is not linked to the two hitherto(up to 15 mV). Spontaneous activity was rarely observeddescribed HMN loci on chromosomes 7p (distal HMN V)and, when present, consisted of low-frequency fibrillation orand 12q (distal HMN II). Furthermore, juvenile amyotrophicfasciculation potentials. These findings are consistent withlateral sclerosis on chromosome 9q, Charcot–Marie–Tooth 2Bchronic neurogenic disorder.and HSNI on chromosomes 3q and 9q were excluded by ourlinkage studies (data not shown). In addition, we calculatedtwo-point LOD scores with individuals III/51 and IV/56,Magnetic evoked potentials
Motor evoked potentials were recorded in 15 definitely and who had been classified as unknown because the clinicallynormal mother had an affected child and a phenocopy could15 probably affected subjects and prolonged central motor

1620 M. Auer-Grumbach et al.
Table 2 Results of electrophysiological studies
Probably affected individuals Definitely affected individuals P
Mean � SD 95% confidence Mean � SD 95% confidence(n) limits (n) limits
M–DML 3.9 � 0.66 (49) 3.7–4.1 4.5 � 0.80 (40) 4.2–4.8 0.001M–CV 57.7 � 7.42 (49) 55.5–59.8 53.5 � 5.70 (40) 51.7–55.3 0.005M–CMAP 10.4 � 3.46 (49) 9.4–11.4 5.9 � 4.55 (40) 4.5–7.4 0.000
U–DML 3.1 � 0.74 (23) 2.8–3.5 3.4 � 0.77 (20) 3.1–3.8 0.209U–CV 61.1 � 3.79 (19) 59.2–62.9 55.3 � 12.87 (18) 48.9–61.7 0.070U–CMAP 14.2 � 3.63 (23) 12.6–15.7 12.2 � 4.23 (19) 10.2–14.3 0.121
P–DML 4.6 � 0.79 (47) 4.4–4.8 7.3 � 3.68 (35) 60.0–8.5 0.000P–CV 49.7 � 4.28 (47) 48.4–51.0 38.3 � 13.31 (35) 33.8–42.9 0.000P–CMAP 9.9 � 4.14 (47) 8.6–11.1 3.2 � 4.65 (36) 1.6–4.7 0.000
1.6–4.4T–DML 4.8 � 0.73 (47) 4.6–5.0 6.5 � 2.34 (36) 5.7–7.3 0.000T–CV 50.5 � 3.88 (47) 49.4–51.7 42.4 � 5.94 (35) 40.4–44.5 0.000T–CMAP 22.9 � 8.17 (47) 20.5–25.3 9.1 � 8.75 (35) 6.1–12.1 0.000
M–S–CV 55.4 � 7.31 (24) 52.3–58.5 48.4 � 6.50 (27) 45.9–51.0 0.001M–S–A 42.5 � 23.84 (23) 32.2–52.9 22.6 � 16.86 (27) 16.0–29.3 0.001SU–CV 45.7 � 4.38 (32) 44.2–47.3 40.0 � 9.39 (27) 36.3–43.7 0.003SU–A 21.6 � 12.15 (32) 17.3–26.0 11.3 � 7.88 (15) 6.9–15.7 0.004
Family members were classified as definitely affected or probably affected as described in Patients and methods. As there was no sidedifference for any parameter, data for the two sides are pooled. M � median nerve; U � ulnar nerve; P � peroneal nerve; T � tibialnerve; SU � sural nerve; DML � distal motor latency; CV � conduction velocity; CMAP � compound motor action potential; S �sensory; A � amplitude.
Table 3 Central motor conduction times
Definitely affected individuals Probably affected individuals Upper limitof normal (ms)
Prolonged Normal Prolonged NormalCMCT (ms) CMCT (ms) CMCT (ms) CMCT (ms)
Upper limbs 10.4 � 0.3 6.6 � 0.9 9.9 � 1.1 6.2 � 1.2 8.3(n � 2/30) (n � 28/30) (n � 3/30) (n � 27/30)
Lower limbs 20.4 � 1.5 15.6 � 2.3 22.8 � 1.8 15.7 � 1.3 18.5(n � 10/29) (n � 19/29) (n � 5/28) (n � 23/28)
Means and standard deviations of central motor conduction times are shown for upper and lower limbs in patients classified as definitelyand probably affected by HMN V. Numbers in brackets denote the frequencies of normal and prolonged CMCT within each subgroup.The upper limits of normal are the reference values established in our laboratory, representing the mean � 2.5 SD derived from a normalpopulation of 40 healthy subjects. Recordings in the upper limb were taken from the abductor digiti minimi and in the lower limbs fromthe tibialis anterior muscle.
not be ruled out. As a result of this modification, the the enormous heterogeneity in the presenting signs, thedistribution of wasting and upper motor neuron involvement.LOD scores in our family changed only marginally and no
significant differences were obtained. The diagnosis of HMN type V in this family was basedprimarily on the findings of predominant wasting andCharcot–Marie–Tooth 1A and proximal spinal muscular
atrophy linked to chromosome 5q were excluded in one weakness restricted to the hands, which was the leading signin �50% of the patients, while gait disturbance was usuallyseverely affected patient (IV/58).less pronounced and sensory abnormalities were absent oronly minimally present. In 1966, Silver described two familiesafflicted with a neuropathy of autosomal dominantDiscussion
The present study provides the largest series of HMN V inheritance, in which wasting of the hands was the firstand most prominent manifestation of the disease, althoughpatients reported so far and focuses on the clinical
presentation, the neurophysiological characteristics and the differences were clearly present (Silver, 1966). While inthe first family (family K) hand muscle involvement wasgenetic background of this disorder. In principle, it shows

Heterogeneity in HMN V 1621
Table 4 Two-point LOD scores between HMN II and HMN V and various short tandem repeat markers
Recombination fractions
0 0.01 0.05 0.1 0.2 0.3 0.4
Chromosome 7D7S435 –3.65 –2.80 –1.66 –0.93 –0.22 0.02 0.05D7S2492 –0.76 –0.72 –0.29 0.07 0.30 0.26 0.14D7S632 –3.16 –1.53 –0.64 –0.11 0.35 0.41 0.27D7S1514 –9.16 –4.76 –2.75 –1.90 –0.94 –0.36 –0.06
Chromosome 12D12S86 –2.43 –1.04 –0.46 –0.25 0.11 0.25 0.18D12S2079 –0.35 –0.34 –0.17 0.01 0.13 0.10 0.02D12S1282 –2.37 –1.90 –0.51 0.19 0.58 0.49 0.25D12S1349 –3.09 –1.64 –0.93 –0.43 0.11 0.19 0.13D12S378 –3.72 –2.16 –1.42 –0.89 –0.26 –0.04 0.03D12S340 –3.47 –1.76 –0.66 –0.21 0.05 0.16 0.14
frequently distributed asymmetrically to the thumb and the the family history carefully and the examination of furtherfamily members to arrive at a correct clinical diagnosis.FDI muscles, patients of the second family (family A)
invariably had bilateral symmetrical wasting of all small Hyper-reflexia was most frequently associated withpredominant wasting of the small hand muscles, and canhand muscles, which developed at a later stage, but was
more severe and disabling. Similar phenotypes were observed therefore be considered as a typical, albeit inconstant, findingin HMN V. Occasionally, additional increased muscle tone,in the families reported by Lander and colleagues (Lander
et al.,1976), van Gent and colleagues (van Gent et al., 1985) extensor plantar responses and a spastic gait indicateinvolvement of the pyramidal tract in the disease. In theand de Visser and colleagues (de Visser et al.,1988), in which
hand muscle involvement particularly resembled that of families reported by Lander and colleagues (Lander et al.,1976), van Gent and colleagues (van Gent et al., 1985) andSilver’s family K, as was the case in our family. In contrast
to these families, however, all members of Gross’s kinship Gross and colleagues (Gross et al., 1998) and in our family,pyramidal signs were usually only slight, whereas they were(Gross et al., 1998) presented with symmetrical weakness of
the distal muscles of the upper limbs in the distribution of absent or barely present in the chromosome 7p-linked families(Christodoulou et al., 1995; Sambuughin et al., 1998).the radial, ulnar and median nerves, similarly to Silver’s
family A. It is unclear whether this striking asymmetrical However, in some of the affected family members of Silver(Silver, 1966) and de Visser and colleagues (de Visser et al.,atrophy of the thenar and FDI muscles, noted in some families
with HMN V, reflects the existence of a distinct genetic 1988), pyramidal tract involvement was so prominent thatthe disorder was classified as hereditary spastic paraplegia.entity. Unfortunately, genetic linkage studies in the above-
mentioned families have not yet been reported. In our Since both authors observed individuals with hand muscleinvolvement or gait and pyramidal disturbance only, it hasfamily the disease was not associated with a gene locus on
chromosome 7p, as found in the Bulgarian HMN V kinship been suggested that both lower and upper motor neuron signswere segregating as independent autosomal dominant traits.(Christodoulou et al., 1995), the family from Iowa with
Charcot–Marie–Tooth 2D (Ionasescu et al., 1996) and the This seems very unlikely in our family, as we foundindividuals displaying clinical features that resembledMongolian HMN V/Charcot–Marie–Tooth 2D family
(Sambuughin et al., 1998), in which asymmetrical wasting and peroneal muscular atrophy with mild pyramidal featureswithout hand muscle involvement, who, surprisingly had anweakness of the thenar and FDI muscles were not reported.
A further representative and common feature in our HMN V affected child with predominant hand muscle wasting. Thus,we hypothesize that both features are caused by only onepatients was foot deformity of a variable degree, which
occurred frequently in all the families with HMN V reported mutated gene, which, however, can give rise to markedphenotypic differences.to date. However, special attention should be paid to those
individuals in whom the disease starts in the feet and The absence or presence of sensory abnormalities has beenused as the most important feature to distinguish HMN fromprogresses to weakness and wasting of the ankle extensors,
whereas the hands are involved later or might even be spared, HMSN. Nevertheless, it has been demonstrated already thatboth phenotypes, with or without sensory loss, can beas found in eight of our patients. On the one hand, such
cases demonstrate the broad clinical variability of the disease. observed within one family (van Gent et al., 1985;Sambuughin et al., 1998). This, again, has implications forOn the other hand, in these individuals the disorder cannot be
distinguished from peroneal muscular atrophy with pyramidal the classification within this disease group. We, too, foundimpaired vibration and mildly to moderately slowed sensoryfeatures on purely clinical grounds (Harding and Thomas,
1984). This clearly emphasizes the importance of evaluating NCV and reduced sensory nerve amplitudes in older and

1622 M. Auer-Grumbach et al.
severely affected patients, indicating that the sensory nerves account for the slight slowing of CMCT noted in our patients.In support of a report by Schnider and colleagues, whomay become involved additionally with advanced disease.
Therefore, with regard to the families of van Gent and observed prolonged CMCTs in clinically unaffected membersof a HMSN V family (Schnider et al., 1991), we occasionallycolleagues (van Gent et al., 1985) and Sambuughin and
colleagues (Sambuughin et al., 1998) and our family, we recorded prolonged CMCTs in probably affected individuals,even in the absence of clinical signs of the disease. Whetherbelieve that distinction between HMN and HMSN cannot be
based exclusively on the involvement of the sensory nerves. this reflects predominant involvement of the corticospinaltract in HMN V or earlier expression of disease-relatedOf more reliable value might be the relation between the
pathology of a motor nerve and its analogous sensory nerve; changes in long tracts cannot be decided at present.Two loci for HMN have been reported to date. HMN IIin HMN, motor nerves can be severely impaired or CMAPs
might even not be recordable, while the sensory nerves has been mapped to chromosome 12q and HMN V tochromosome 7p (Christodoulou et al., 1995; Timmermanremain normal for a long time, whereas in HMSN both motor
and sensory nerves are affected simultaneously. et al., 1996). Linkage and segregation analysis with sixchromosome 12 and four chromosome 7 markers yieldedWe have not undertaken studies to determine autonomic
nervous system involvement in our family. Hyperhidrosis, conclusively negative LOD scores, excluding these types ofHMN in our family. This strongly implies the existence of awhich was frequently reported by our patients and individuals
who were probably affected, may be a feature of additional further locus responsible for HMN and particularly highlightsthe genetic heterogeneity of HMN V.involvement of the autonomic nervous system.
Other findings in our kindred, such as depression, hip In conclusion, the findings in this family with HMN V(the largest such family ever reported) demonstrate clearlydysplasia and hypacusis, seem to occur by chance as they
have been observed only in single patients. that this disorder represents a phenotypically and geneticallyheterogeneous disease with autosomal dominant inheritance.The results of NCV and EMG studies were consistent with
a predominant chronic axonal motor neuropathy in the At least a second genetic subtype of HMN V must exist,which may be distinguished from HMN V linked tomajority of the definitely affected patients investigated. The
CMAPs were largely reduced in all but the ulnar nerves and chromosome 7p by the presence of asymmetrical weaknessand wasting of the thenar and FDI muscles and pyramidalthe EMG showed high-amplitude potentials and reduced
recruitment, whereas distal motor latencies and motor NCVs involvement, sometimes evidenced by brisk tendon reflexesand/or prolonged central motor conduction times. Footwere normal or mildly to severely abnormal. In the upper
limbs the median nerve was significantly more severely deformity and sensory disturbance are less reliable in thedifferential diagnosis of both types of HMN V. Due to itsdamaged than the ulnar nerve, suggesting that the median
nerve is most useful for screening of this type of HMN V. broad clinical variability, the disorder can mimic peronealmuscular atrophy with pyramidal features in individual cases.Our observation in one 18-year-old female with a normal
phenotype but mild abnormalities of the median motor NCV Electrophysiological studies might occasionally identifysubclinical involvement in clinically normal family members.might emphasize that the disease is sometimes recognized
only electrophysiologically, and underlines the importance Genetic linkage studies of further HMN V families areneeded to expand our knowledge of phenotype–genotypeof electrophysiological assessment in detecting gene carriers.
The sensory nerves were mildly to moderately involved correlations in different types of HMN V and to confirm thedifferential diagnostic considerations based on observationsin advanced disease and the changes were predominantly
axonal. At later stages of the disease, the electrophysiological in our HMN V patients. A genome-wide search in this largefamily is warranted to elucidate the second gene locusfeatures may be indistinguishable from the findings in
HMSN II (Chad, 1989; Emeryk–Szajewska et al., 1998; responsible for HMN V.Paraskevas et al., 1998), but the clinical characteristics aredistinctive enough to exclude the latter disease.
Transcranial magnetic stimulation revealed prolongedAcknowledgementsCMCTs in 50% of the family members studied, indicatingWe gratefully acknowledge the cooperation of the familyadditional CNS involvement in HMN V. Prolonged CMCTsmembers and thank Mrs Margit Schuster and Mrs Gerdahave been reported in hereditary neuropathies, but the resultsZmugg for their excellent technical assistance. This workso far remain conflicting. In HMSN I some authors havewas supported by the Austrian Science Fund (FWF), grantfound central motor pathway involvement independently ofP13563–BIO. We thank Professor K. V. Toyka, Wurzburg,clinical pyramidal signs (Mano et al., 1993; Sartucci et al.,Germany, for critical reading of the manuscript.1997), and in 1990 Claus and colleagues observed prolonged
CMCTs in the presence of only pyramidal signs (Claus et al.,1990). In the latter study, the slowing of central motorconduction was less pronounced in patients with the axonal Referencesvariant, HMSN II, suggesting a different pathology. Disease- Aminoff MJ. Electromyography in clinical practice. 3rd ed. New
York: Churchill Livingstone; 1998.related changes in motor neuron excitability might also

Heterogeneity in HMN V 1623
Auer-Grumbach M, Strasser-Fuchs S, Wagner K, Korner E, Mano Y, Nakamuro T, Ikoma K, Takayanagi T, Mayer RF. Aclinicophysiologic study of central and peripheral motor conductionFazekas F. Roussy-Levy syndrome is a phenotypic variant ofin hereditary demyelinating motor and sensory neuropathy.Charcot–Marie–Tooth syndrome 1A associated with a duplicationElectromyogr Clin Neurophysiol 1993; 33: 101–7.on chromosome 17p11.2. J Neurol Sci 1998; 154: 72–5.
McLeod JG, Prineas JW. Distal type of chronic spinal muscularChad DA. AAEE case report #20: hereditary motor and sensory
atrophy. Clinical, electrophysiological and pathological studies.neuropathy, type I. Muscle Nerve 1989; 12: 875–82.
Brain 1971; 94: 703–14.
Chance PF, Rabin BA, Ryan SG, Ding Y, Scavina M, Crain B, Meadows JC, Marsden CD. A distal form of chronic spinal muscularet al. Linkage of the gene for an autosomal dominant form of atrophy. Neurology 1969; 19: 53–8.juvenile amyotrophic lateral sclerosis to chromosome 9q34. Am J Nicholson GA, Dawkins JL, Blair IP, Kennerson ML, Gordon MJ,Hum Genet 1998; 62: 633–40. Cherryson AK, et al. The gene for hereditary sensory neuropathy
type I (HSN-I) maps to chromosome 9q22.1–q22.3. Nat GenetChristodoulou K, Kyriakides T, Hristova AH, Georgiou DM,1996; 13:101–14.
Kalaydjieva L, Yshpekova B, et al. Mapping of a distal formO’Sullivan DJ, McLeod JG. Distal chronic spinal muscular atrophyof spinal muscular atrophy with upper limb predominance toinvolving the hands. J Neurol Neurosurg Psychiatry 1978; 41: 653–8.chromosome 7p. Hum Mol Genet 1995; 4: 1629–32.
Ott J. Analysis of human genetic linkage. Baltimore: Johns HopkinsClaus D, Waddy HM, Harding AE, Murray NM, Thomas PK. University Press; 1991.Hereditary motor and sensory neuropathies and hereditary spastic
Paraskevas GP, Panousopoulou A, Karandreas N, Piperos P,paraplegia: a magnetic stimulation study. Ann Neurol 1990; 28: 43–9.Lygidakis C, Papageorgiou C. Correlation between denervationactivity and compound muscle action potential amplitude inde Visser M, Ongerboer de Visser BW, Verjaal M. Amyotrophy of thehereditary motor and sensory neuropathy I and II. Electromyogrhands and pyramidal features of predominantly the legs segregatingClin Neurophysiol 1998; 38: 343–7.within one large family. J Neurol Sci 1988; 88: 241–6.
Reiter LT, Murakami T, Koeuth T, Pentao L, Muzny DM, GibbsEllsworth RE, Ionasescu V, Searby C, Sheffield VC, Braden VV,
RA, et al. A recombination hotspot responsible for two inheritedKucaba TA, et al. The CMT2D locus: refined genetic position and
peripheral neuropathies is located near a mariner transposon-likeconstruction of a bacterial clone-based physical map. Genome Res element. Nat Genet 1996; 12: 288–97.1999; 9: 568–74.
Sambuughin N, Sivakumar K, Selenge B, Lee HS, Friedlich D,Emeryk-Szajewska B, Badurska B, Kostera-Pruszczyk A. Baasanjav D, et al. Autosomal dominant distal spinal muscular
atrophy type V (dSMA-V) and Charcot–Marie–Tooth disease typeElectrophysiological findings in hereditary motor and sensory2D (CMT2D) segregate within a single large kindred and map to aneuropathy type I and II—a conduction velocity study. Electromyogrrefined region on chromosome 7p15. J Neurol Sci 1998; 161: 23–8.Clin Neurophysiol 1998; 38: 95–101.
Sartucci F, Sagliocco L, Murri L. Central motor pathway evaluationGross DW, Rajput AH, Yeung M. Distal hereditary upper limb
using magnetic coil stimulation in hereditary motor and sensorymuscular atrophy. J Neurol Neurosurg Psychiatry 1998; 64: 217–20.
neuropathy type I (HMSN type I, Charcot–Marie–Tooth disease).Int J Neurosci 1997; 92: 145–59.Harding AE. Inherited neuronal atrophy and degeneration
predominantly of lower motor neurons. In: Dyck PJ, Thomas PK, Schnider A, Hess CW, Koppi S. Central motor conduction in aGriffin JW, Low PA, Poduslo JF, editors. Peripheral neuropathy. 3rd family with hereditary motor and sensory neuropathy with pyramidal
signs (HMSN V). J Neurol Neurosurg Psychiatry 1991; 54: 511–5.ed. Philadelphia: W.B. Saunders; 1993. p. 1051–64.
Silver JR. Familial spastic paraplegia with amyotrophy of the hands.Harding AE, Thomas PK. The clinical features of hereditary motorJ Neurol Neurosurg Psychiatry 1966; 29: 135–44.
and sensory neuropathy types I and II. Brain 1980; 103: 259–80.Skre H. Genetic and clinical aspects of Charcot–Marie–Tooth’s
Harding AE, Thomas PK. Peroneal muscular atrophy with pyramidal disease. Clin Genet 1974; 6: 98–118.features. J Neurol Neurosurg Psychiatry 1984; 47: 168–72. Thomas PK, Marques W Jr, Davis MB, Sweeney MG, King RH,
Bradley JL, et al. The phenotypic manifestations of chromosomeIonasescu V, Searby C, Sheffield VC, Roklina T, Nishimura D,17p11.2 duplication. Brain 1997; 120: 465–78.Ionasescu R. Autosomal dominant Charcot–Marie–Tooth axonalTimmerman V, De Jonghe P, Simokovic S, Lofgren A, Beuten J,neuropathy mapped on chromosome 7p (CMT2D). Hum Mol GenetNelis E, et al. Distal hereditary motor neuropathy type II (distal1996; 5: 1373–5.HMN II): mapping of a locus to chromosome 12q24. Hum Mol
Kwon JM, Elliott JL, Yee WC, Ivanovich J, Scavarda NJ, Genet 1996; 5: 1065–9.Moolsintong PJ, et al. Assignment of a second Charcot–Marie–
van Gent EM, Hoogland RA, Jennekens FG. Distal amyotrophy ofTooth type II locus to chromosome 3q. Am J Hum Genet 1995; 57: predominantly the upper limbs with pyramidal features in a large853–8. kinship. J Neurol Neurosurg Psychiatry 1985; 48: 266–9.
Lander CM, Eadie MJ, Tyrer JH. Hereditary motor peripheralneuropathy predominantly affecting the arms. J Neurol Sci 1976; Received December 6, 1999. Revised March 1, 2000.
Accepted March 20, 200028: 389–94.
Copyright © 2022 FDOKUMEN




















