
Pharmacology of Palmitoylethanolamide and ... - DiVA-Portal
82
Pharmacology of Palmitoylethanolamide and Related Compounds by Kent-Olov Jonsson Umeå 2005 Department of Pharmacology and Clinical Neuroscience, Pharmacology, Umeå University
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of Pharmacology of Palmitoylethanolamide and ... - DiVA-Portal

Pharmacology of
Palmitoylethanolamide and Related
Compounds
by Kent-Olov Jonsson
Umeå 2005
Department of Pharmacology and Clinical Neuroscience, Pharmacology, Umeå University

Printed by Solfjädern Offset AB, Umeå, Sweden Copyright © Kent-Olov Jonsson, 2005
ISBN 91-7305-821-1
ii

ABSTRACT
Anandamide (AEA) is an endogenous fatty acid which activates the same cannabinoid receptors as ∆9-tetrahydrocannabinol, the psychoactive substance in marijuana. In vivo, AEA exerts a number of actions including effects upon pain and inflammation. However, AEA has a short duration of action since it is rapidly metabolised, primarily by the intracellular enzyme fatty acid amide hydrolase (FAAH).
The general aim of this thesis has been to identify and characterize compounds capable of preventing the metabolism of AEA. The chemical approach was based on the endogenous anti-inflammatory compound palmitoylethanolamide (PEA), a compound related to AEA with the ability to inhibit AEA degradation by substrate competition, but without the ability to directly activate cannabinoid receptors.
A number of compounds were identified as inhibitors of rat brain FAAH in the initial in vitro studies, without having major affinity for the cannabinoid receptors. In particular, palmitoylisopropylamide (PIA) was found to reduce the metabolism of AEA in intact C6 glioma cells with potency similar to the prototypical AEA reuptake inhibitor AM404. This compound was in addition found to exert less effect upon C6 glioma cell proliferation than either AM404 or the closely related uptake inhibitor VDM11. To evaluate if PIA was effective in vivo, a model of mast cell dependent inflammation, oedema of the ear following local injection of compound 48/80, was set up using anaesthetised mice. Initially, a CB2 cannabinoid receptor-selective agonist was used to probe the model and demonstrated to produce an anti-oedema effect. In contrast, the compound was inactive in vitro in skin slice preparations. PIA showed a similar pattern, although there was a large variation in responses which affected the significance of the result obtained, as did the vehicle used to dissolve the compound.
Taken together, the present data would suggest that PIA can inhibit the degradation of AEA without having deleterious effects upon cell proliferation or affinity for the cannabinoid receptors. Further experimentation is necessary to elucidate the usef
Keywords: anandamide, cannabinoid, FAA mitoylethanolamide, palmitoylisopropylamide, inflam
ulness of this compound in vivo.
H, palmation, mast cells
iii

CONTENTS
CO
eral Background 9
18
34
37 β-Hexosaminidase release 38
38 40
NTENTS i
ORIGINAL PAPERS 6
EXPLANATIONS OF ABBREVIATIONS 7
INTRODUCTION 9 GenThe endocannabinoid system 9 Cannabinoid receptor ligands 13
In vivo effects of cannabinoid receptor activation 15 Anandamide (AEA) 17 N-acylethanolamines (NAEs) Synthesis and release of anandamide 19 Anandamide uptake 21 Anandamide degradation 23
FAAH inhibitors 24 Therapeutic potential of FAAH inhibitors 26
AIMS OF THE STUDY 28
METHODS AND METHODOLOGICAL CONSIDERATIONS 29 FAAH assay 29 Anandamide uptake in RBL-2H3 and C6 cells 31 Radioligand binding experiments 32 Cell cultures 33 Cell proliferation assay 34
Analysis of cell density Analysis of caspase activity 34
Animals 35 Plasma extravasation in mouse ear pinna 36 Mice paw skin release
Immunohistochemistry and PCR Data analyses and statistics
4

RESULTS AND DISCUSSION 41 Inhibition of FAAH catalysed anandamide hydrolysis 41 Interaction with CB1 and CB2 cannabinoid receptors 47 Inhibition of anandamide uptake 47 Inhibition of anandamide uptake and hydrolysis in intact C6 cells 49 Effects of PIA, AM404 and VDM11 upon cell proliferation 51 Effects of PIA and JWH133 upon inflammation in vivo and in vitro 53
GENERAL CONCLUSIONS AND FUTURE PERSPECTIVES 57
ACKNOWLEDGEMENTS 59
REFERENCES 61
APPENDIX 81
5

ORIG S
erals.
DM, Tiger G, Fowler CJ. Effects
oid anandamide. British Journal of Pharmacology
II. Vand M. Modifications
in N-palmitoylethanolamine: synthesis and evaluat the metabolism of anandamide. Journal
1448.
III. , Jacobsson SOP, Vandevoorde S, Lambert DFowler CJ. AM404 and VDM 11 non-specifically inhibit C6 glioma cprolif cellular accumulation of t
de. Archives of Toxicology (2003), 77: 201-207.
eptor-select33 reduces mast cell oedema in response to compound 48/
se from skin slices in vi
V. J. Characterisation of palmitoylisopropylamide
mast and in vivo. In manuscript.
pers were made with permission from the respective publishers.
INAL PAPER
The present thesis is based on the following papers which will be referred to in text by their roman num
I. Jonsson KO, Vandevoorde S, Lambert of
homologues and analogues of palmitoylethanolamide upon the inactivation of the endocannabin(2001), 133: 1263-1275.
evoorde S, Jonsson KO, Fowler CJ, Lambert D of the ethanolamine head ion of new agents interfering with of Medicinal Chemistry (2003), 46: 1440-
Jonsson KO, Andersson A M, ell
eration at concentrations used to block the he endocannabinoid anandami
IV. Jonsson KO, Persson E, Fowler CJ. The cannabinoid CB2 rec ive
agonist JWH1 80 in vivo but not the release of ß-hexosaminida tro. Submitted for publication.
Jonsson KO, Fowler C in cell dependent systems in vitro
Reprints of the published pa
6

EXPLANATIONS OF ABBREVIATIONS
trahydrocannabinol 2-AG AC ACEA yl-2-chloroethylamide AEA AM12AM25
agonist
CB1 cannabinoid receptor type 1 CB2 cannabinoid receptor type 2 Compound 48/80 mast cell degranulator CP55,940 non-selective cannbinoid receptor agonist FAAH fatty acid amide hydrolase (EC 3.5.1.4) FAAH-NS neurospecific FAAH knockout mice HU210 non-selective cannabinoid receptor agonist HU308 CB2 receptor-selective agonist IC50 concentration producing 50 % of the maximal
attainable inhibition JWH133 CB2 receptor-selective agonist MAFP methylarachidonylfluorophosphonate,
FAAH inhibitor NAEs N-acylethanolamines NAPE N-acylphosphatidylethanolamine NAPE-PLD N-acylphosphatidylethanolamine specific
phospholipase D NAT N-acyltransferase O-1812 CB1 receptor-selective agonist OEA oleoylethanolamide
∆9-THC ∆9-te 2-arachidonylglycerol adenylyl cyclase
arachidon anandamide (arachidonylethanolamide)
41 CB2 receptor-selective agonist 1 CB1 receptor-selective antagonist/inverse agonist
AM404 AEA uptake inhibitor AM630 CB receptor-selective antagonist/inverse 2
ATMK or ATFMK arachidonoyltrifluoromethylketone, FAAH inhibitor
BSA bovine serum albumine cAMP cyclic adenosine monophosophate
7

OL-135
palmitoylisopropylamide PMSF P RRBL-RT-PCSEA oylethanolamide SR141716A, rimonabant CB reS 4TASKTNF- TRPV tential, vanilloid receptor 1 URB532 UVDMWIN5
FAAH inhibitor PEA palmitoylethanolamide pI50 negative logarithmic value of IC50
PIA phenylmethylsulphonylfluoride, FAAH inhibitor
PA -α peroxisome proliferator-activated receptor alpha 2H3 rat basophilic leukaemia cells
R reverse transcriptase polymerase chain reaction stear
1 ceptor-selective antagonist/inverse agonist R14 528 CB2 receptor-selective antagonist/inverse agonist
-1 background membrane K+ channel α tumor necrosis factor alpha 1 transient receptor po
FAAH inhibitor RB597 FAAH inhibitor
11 AEA uptake inhibitor 5,212-2 non-selective cannabinoid receptor agonist
8

Pharmacology of Palmitoylethanolamide and Related Compounds
INTRODUCTION
General Background
t, Cacts t ut
ent of pain, inflammation, and m ar
bis, ∆9 etrahydrocannabinol n 1964 , a ∆9-TH d the UK for
treatment of chemotherapy-in d vomiting. Dronabinol, ∆9-THC e ouse of
ittee of K House of Lords conc f anecdotal evidence as e therapeutic efficacy o osis and chronic pain
”as a ma ation in proper clinical trials are now
dertaken for these in et al., n al., 2004). N utic agents of
HC i e same s as ∆ y their psychotropic
s. A major challenge fo es to harness the therapeutic pot abinoids without producing
The endocannabinoid sy
s and assumed that the anna unspecific
membranes du he mid activity was highly stereospecific
r and its endogenous ence erisation of the first made in ne et al., 1988).
The cannabis plan nnabis sativa, has been used for millennia as a recreational drug and affe he body in several ways. It has been used throughohistory for the treatm a number of diseases such asmuscle spasms, nausea any more (Reynolds 1890; Baker et al., 2003; Kumet al., 2001). The major active constituent of canna -t(∆9-THC) was isolated i (Gaoni & Mechoulam, 1964). Currently nabilonestructural analogue of C, is used in parts of the USA an
duced nausea anitself, is used as an appetit stimulator in AIDS patients in the USA (HLords, 1998). In their hearing in 1998, the Science and Technology Commthe U luded that the large amount oto th f cannabis in multiple sclerconditions warranted tter of urgency” investig(House of Lords, 1998). Clinical trials of standardised cannabis extractsbeing un dications (see e.g. Wade et al., 2003; Zajicek2004; Berma et onetheless, the usefulness as therapesuch extracts, of ∆9-T tself, or of synthetic compounds with thpharmacological action 9-THC, is greatly hampered beffect r many researchers is to find new approach
ential of these cannunwanted effects.
stem
Back in the 1970’ early 1980’s it was generally effects of cpsychotropic bis and synthetic cannabinoids was rather
on nerve cell e to their high lipophilicity. However, in t1980’s, several groups showed that cannabinoid(Razdan, 1986) which led to the search for a specific recepto
tmediators. As a consequ , the pharmacological characcannabinoid receptor was rat brain in the late 80’s (Deva
9

Kent-Olov Jonsson
10
ept brain by Matsuda and co-e a mbrane G-protein
led receptor superfamily e first endogenous ty mide, a lipid
ng to a group of fatty s), ted from pig brain rit liss, (Devan
located cytic ., 1993). In the same
epor A, as fatt ,
in 1994 the first da EA uptake was (Di Marzo et al., 199 molecular proof of this protein. In 1995, th the
, 2-arac ), belonging to a group of oacylgly tracted from rat brain and
t (Sugiura et al., 199 995). The knowledge of the system is co sed that
other endogenous compounds, including dihomo-γ-linolenoylethanolamide and docosatetraenoylethanolamide (Hanus et al., 1993), 2-arachidonyl-glycerylether (noladin ether, 2-AGE; Hanus et al., 2001), O-arachidonoyl-ethanolamine (virhodamine; Porter et al., 2002), N-arachidonoyl-dopamine (NADA; Bisogno et al., 2000; Huang et al., 2002) and oleamide (Legget et al., 2004), also function as cannabinoid receptor ligands, but its not clear as to whether they are true endocannabinoids.
NH
OOH
Shortly after, the CB1 rec or was cloned from ratworkers and found to b member of the seven transmecoup (Matsuda et al., 1990). In 1992, thcompound to exert activi at CB1 receptors, arachidonylethanolabelongi acid amines called the N-acylethanolamines (NAEwas extrac and named anandamide (AEA) after the Sanskword for b ananda e et al., 1992; structure, see Fig. 1). Around the same time, a peripherally receptor was cloned from human promyeloleukemia HL-60 cells and named the CB2 receptor (Munro et alyear, Deutsch and Chin r ted an enzyme responsible for hydrolysis of AEnowadays referred to y acid amide hydrolase (FAAH; Deutsch & Chin1993), and ta suggesting a transport system for Areported 4), although there is still no putative e second endogenous compound to activate cannabinoid receptors hidonoylglycerol (2-AGlipids called the mon cerols (MAGs), was excanine gu 5; Mechoulam et al., 1endocannabinoid ntinuously growing and it has been propo
NH
OOH
O
NH
OHO
NH
OH
Fig. 1. Structures of anandamide (AEA) and the related N-acylethanolamine palmitoylethanolamide (PEA).

Pharmacology of Palmitoylethanolamide and Related Compounds
In respect of the human C reports of two different splice variants, hCB1a and hCB1b, showing a slightly different pharmacological
l-length hCB1 (Shire et al., 1995; Ryberg et al., 2005). ns of different types of receptors, including a CB2-
like
B1 receptor, there have been
profile compared to the fulThere have also been indicatio
receptor, a mesenteric non-CB1 non-CB2 receptor and a non-CB1 non-CB2 brain receptor, although they have not been cloned so far (Howlett et al., 2002).
The identification of cannabinoid receptors and FAAH, the key enzyme in AEA degradation, as well as a putative transport protein has lead to the development and search for selective compounds revealing more information about the endogenous cannabinoid system as well as developing new therapeutic agents that act through the cannabinoid system.
So what is the physiological function of the endocannabinoid system (shown schematically in Fig. 2 below)? Even if this of course is a complex question, the dramatic expansion of studies concerning endocannabinoids and related compounds, as well as the well known effects of cannabis, have revealed a number of possible physiological roles. These include pain processing, memory functions, cognition, food intake, gastrointestinal and cardiovascular function, regulation of the immune system and control of motor function (reviews, see Piomelli, 2003; Gerdeman & Lovinger, 2003; De Petrocellis et al., 2004; Di Marzo et al., 2004; Fowler et al., 2005). This range of actions means that there is, at least in theory, considerable therapeutic potential for compounds affecting the endocannabinoid system. See figure 2 below for a schematic overview of the synthesis, release, action and inactivation of the endocannabinoid AEA.
11

Kent-Olov Jonsson
AEA
mGluR+
Ca2+, Na+
+
AEA
Ethanolamine
Ca2+
FAAH
NAPE-PLD
NAT
CB1
-
NAPE
AEA AEA?
GABA
?
TRPV1
Arachidonicacid
AEA
mGluR++
Ca2+, Na+
++
AEA
Ethanolamine
Ca2+
FAAHFAAH
NAPE-PLD
NAT
CB1
--
NAPE
AEA AEA?
GABA
?
TRPV1
Arachidonicacid
Arachidonicacid
Fig.2. Schematic figure of the synthesis, action and metabolism of AEA in neurons (uptake and metabolism has also been shown in other cell types). AEA is synthesised in a two-step process via intracellularly membrane bound enzymes. In the first step, a lipid precursor is converted into NAPE by NAT. Secondly; NAPE is further transformed into AEA by NAPE-PLD. The synthesis is initialised “on demand” by a rise in intracellular calcium or by neurotransmitters such as glutamate. After synthesis, AEA is released directly out in the synapse by an unclear mechanism after which it can activate the G-protein-coupled CB
arachidonic acid and membrane of intracell
receptors aptically in the brain, le c neuron, such as GAB alised into the ce protein. Inside
e ion channel, which responds to h of capsaicin, the “hot” co hydrolysed into
ethanolamine by the enzyme FAAH, which is located in the ular organelles such as mitochondria. Abbreviations in the figure:
idylethanolamine phospholipase D; CB, cannabinoid receptor; TRPV1, transient receptor potential vanilloid 1; GABA, γ-aminobutyric acid; mGluR, metabotropic glutamate receptor; FAAH, fatty acid amide hydrolase.
. The activation of CB1 receptors, which are mainly located presynads to a decreased release of neurotransmitters located in the presynapti
A or glutamate. After activation of the CB receptors, AEA is internll by a process which may (or may not) involve a surface membrane
the cell, AEA can also activate the TRPV1 receptor, a non-selectiveat and low pH and is in addition responsible for the actions
mponent of red chili pepper. Inside the cell, AEA is also
AEA, anandamide; NAPE, N-acylphosphatidylethanolamine; NAT, N-acyltransferase; NAPE-PLD, N-acylphosphat
12

Pharmacology of Palmitoylethanolamide and Related Compounds
Ca
1
phase III clinical trials under the name rimonabant, and the CB2 antagonist/inverse agonist SR144528 (Rinaldi-Carmona et al., 1994; 1998). Another example in this group is the CB1 receptor antagonists/inverse agonist AM251. Among other chemical series, AM630, which is an aminoalkylindole, is also a commonly used CB2 antagonist/inverse agonist.
nnabinoid receptor ligands
Since the isolation of ∆9-THC (Gaoni & Mechaoulam, 1964), a number of different agonists and antagonists at the cannabinoid receptors have been developed. See figure 3 below for a selection of different CB ligands. The cannabinoid receptor agonists can be divided into several groups based on their chemical structure. They are divided into classical cannabinoids, non-classical cannabinoids, aminoalkylindoles and eicosanoids (Howlett et al., 2002). The classical group includes the plant derived agonist of C. sativa, including ∆9-THC, as well as synthetic analogues such as HU210. Both of these bind and activate CB1 and CB2 receptors without showing any particular selectivity between the receptors. Among the non-classical cannabinoids, CP55,940 is an important member, showing similar selectivity for both cannabinoid receptors, and the use of which led to the pharmacological characterization of the CB1 receptor (Devane et al., 1988). In the third group, the aminoalkylindoles, WIN55,212-2 is a compound showing similar affinity for both the CB1 and the CB2 receptor. The endocannabinoids, AEA and 2-AG, are important members of the eicosanoids.
Examples of potent and selective agonists at the CB1 receptor are the AEA analogues arachidonoyl-2-ethylchloride (ACEA) (Hillard et al., 1999) and O-1812 (Di Marzo et al., 2001b). Examples of CB2-selective agonists include HU-308, JWH133 and AM1241 (Hanus et al., 1999; Huffman et al., 1999; Malan et al., 2001b).
Concerning the antagonists/inverse agonists, the diarylpyrazoles is a major group, including the CB antagonist/inverse agonist SR141716A nowadays in
13

Kent-Olov Jonsson
Agonists
ACEA
O-1812
HU-308
JWH133
AM1241
NH
OCl O
OH
O
O
OH
O
OOO
NO2O
N
I
N
NO2O
N
I
NN
Fig. 3. Structures of selected cannabinoid receptor ligands.
CP55940
Rimonabant(SR141716A)
SR144528
(-)-∆9-THC
HU-210
R-(+)-WIN55212
NH
OCl
NH
OOH
CN
NH
OOH
CN
OH
O
OH
O
OH
HO
O
OH
HO
O
O
NO
O
N
O
NO
O
N
OH
OH
OH
OH
OH
OH
Cl
Cl
Cl
N
N
O
NH
N
Cl
Cl
Cl
Cl
Cl
N
N
O
NH
N N
O
NH
NN
O
NH
N
CB1 selective Non-selective CB2 selective
Agonists
(-)-∆9-THC
HU-210
R-(+)-WIN55212
ACEA
O-1812
HU-308
JWH133
AM1241
NH
OCl O
OH
O
O
OH
O
OOO
NO2O
N
I
N
NO2O
N
I
NN
NH
OCl
NH
OOH
CN
NH
OOH
CN
OH
O
OH
O
OH
HO
O
OH
HO
O
O
NO
O
N
O
NO
O
N
OH
OH
OH
OH
OH
OH
Antagonists
CB1 selective CB2 selective
CP55940
Rimonabant(SR141716A)
SR144528
Cl
Cl
Cl
N
N
O
NH
N
Cl
Cl
Cl
Cl
Cl
N
N
O
NH
N N
O
NH
NN
O
NH
N
CB1 selective Non-selective CB2 selective
Antagonists
CB1 selective CB2 selective
14

Pharmacology of Palmitoylethanolamide and Related Compounds
In vivo effects of cannabinoid receptor activation
rt-term
or the CB1 receptor. Long term effects include impairment of the immune and
involvement of the CB1 receptor in this effect (Huestis et al., 2001).
As stated in the introduction, the current use of cannabinoid ligands as pharmaceuticals is for the treatment of nausea and for stimulation of appetite. The involvement in the regulation of food intake is shown by the use of rimonabant, which is now in phase III clinical trials for weight reduction (also in phase III trials for aid to smoking cessation; Le Fur, 2004). Another major therapeutical indication
The hitherto discovered cannabinoid receptors include the central CB1 receptor and the peripheral CB2 receptor. Agonist activation of these G-protein coupled receptors leads to several signal transduction pathways through Gi/o proteins. The most examined pathway is the inhibition of adenylyl cyclase (AC), leading to a decrease in cAMP production and inhibition of protein kinase A (PKA). In the case of CB1, inhibition of voltage-gated Ca2+ channels and activation of inwardly rectifying K+ channels, also seem to be important (Howlett et al., 2002; Pertwee, 1997). Cannabinoid receptor activation also modulates other signalling pathways via G-proteins, including the extracellular signal-regulated kinase pathway and stimulation of ceramide synthesis, a lipid that can activate apoptosis (Guzman et al., 2001).
The biological consequences of cannabinoid receptor activation are numerous.
The main adverse psychotropic effects in human seen with both cannabis and ∆9-THC include euphoria and relaxation, time distortion and impairment of sho
memory and motor skills. Acute effects also include anxiety, paranoia and panic reactions as well as tachycardia and lowering of blood pressure by vasodilation (Carlini, 2004; Kumar et al., 2001; Baker et al., 2003). The effects seen with ∆9-THC are also seen with both synthetic and endogenous ligands with affinity f
reproductive system (Kumar et al., 2001). In experimental animals, the use of cannabinoid ligands, as well as the use of genetically modified mice, has identified a tetrad of behavioural effects that point to activation of central CB1 receptors. These are hypothermia, hypoactivity, antinociception and catalepsy (see Martin et al., 1999) and correlate well with affinity for the CB1 receptor (Howlett et al., 2002). In respect of the “high” produced by smoked cannabis, clinical studies using rimonabant have confirmed the
15

Kent-Olov Jonsson
is in the treatment of pain and several studies have been conducted in this respect. Cannabinoids modulate pain at supraspinal, spinal and peripheral levels (see Pertwee, 2001; Rice, 2001). The involvement of the cannabinoid system in this respect seems to be broad as shown by the fact that both CB1 and CB2 receptor agonists as well as palmitoylethanolamide (PEA), a fatty acid with no activity at the cannabinoid receptors, and other non-CB receptor cannabinoid related substances such as ajulemic acid can produce an antinociceptive response (Burstein et al., 2004). In the case of CB1 receptors, there is always the potential problem of separation between the wanted and unwanted (psychotropic) effects, unless the compounds are given locally (Dogrul et al., 2003) or designed so as not to cross the blood brain barrier (Fride et al., 2004). In the case of CB2 receptors it was initially believed that they did not participate in nociception, based on their restriction to immune cells. However, studies showing that PEA could be effective against carrageenan-induced hyperalgesia (Mazzari et al., 1996), suggested an involvement of the CB2 receptor. Since PEA was believed to be an endogenous ligand for the CB2 receptor (Facci et al., 1995) the involvement of CB2 receptors was examined. It has later been shown that PEA does not interact with the cannabinoid receptors in vitro, unless very high concentrations are used (Lambert et al., 1999; Sugiura et al., 2000; see also Paper I of this thesis).
There have also been reports showing effects of CB2-selective agonists. The first came in 1999, showing that HU-308 could reduce pain responses in the formalin test, which was reversed by SR144528 but not by rimonabant (Hanus et al., 1999). HU-308 was also effective in decreasing arachidonic acid-induced ear oedema in a SR144528, but not rimonabant, -sensitive manner. Similar results were seen using AM1241, which reversed carrageenan-induced hyperalgesia and this effect could be blocked by AM630 or SR144528 but not by AM251 or rimonabant (Quartilho et al., 2003; Nackley et al., 2003). AM1241 has also been shown to be effective against substance P-induced inflammation (Malan et al., 2001a). This compound has also been shown to suppress capsaicin-induced hyperalgesia in a SR144528 sensitive manner and to be effective in a model of neuropathic pain in an AM630 sensitive way (Hohmann et al., 2004; Ibrahim et al., 2003). A recent study has also shown the effect of the CB2 agonist JWH133 in models of inflammatory and neuropathic SR144528 but not pain, with the effects blocked by by rimonabant (Elmes et al., 2004).
16

Pharmacology of Palmitoylethanolamide and Related Compounds
Cannabinoids have also been shown to produce beneficial effects in animal mo
h a shorter duration (Fride & Mechoulam, 1993; Smith et al., 1994). It is a
dels of multiple sclerosis. For example, WIN55212 and JWH133 were shown to be effective against tremor and spasticity in CREAE mice (Baker et al., 2000).
Based on the findings that natural plant cannabinoids and synthetic cannabinoids exert a number of effects it is not surprising that the endogenous compounds produce similar effects. Indeed, this has been found in numerous studies and in the case of AEA some examples will be given below.
Anandamide (AEA)
AEA is the most studied of the endocannabinoids. It exerts the same effects as ∆9-THC and other cannabinoids in the tetrad test when exogenously administered, although wit
partial agonist at both CB1 and CB2 receptors in vitro (Mackie et al., 1993). AEA has been shown to produce a number of effects including antinociceptive and anti-inflammatory effects (Calignano et al., 1998; Jaggar et al., 1998; Farquhar-Smith & Rice, 2001; Richardson et al., 1998), stimulation of food intake (Williams & Kirkham, 1999; Hao et al., 2000; Di Marzo et al., 2001c), amelioration of spasticity in mice (Baker et al., 2001) and reduction of blood pressure (Varga et al., 1995). In addition to its interaction with the cannabinoid receptors, AEA has been reported to interact directly with other targets including L-type Ca2+ and background TASK-1 K+ channels as well as G-protein coupled non-CB1 and non-CB2 receptors (see Di Marzo et al., 2002b; Howlett et al., 2002; Maingret et al., 2001). Better investigated is the interaction between AEA and TRPV1 (vanilloid) receptors, a non-selective ion channel which also is activated by capsaicin (the pungent component of chili pepper), heat and protons (Zygmunt et al., 1999; Smart et al., 2000 see Di Marzo et al., 2002b; Ross et al., 2003). It has been proposed that AEA functions as an endovanilloid, although the concentration required for activation of the vanilloid receptor is much higher than for the cannabinoid receptors. Nevertheless, some of the effects of exogenously applied AEA upon hyperalgesia and vasodilation are mediated by the TRPV1 receptor (Di Marzo et al., 2001a, 2002a; Ross et al., 2003). This binding site for AEA, in contrast to the cannabinoid receptors, is intracellularly located (De Petrocellis et al., 2001; Jordt & Julius, 2002).
17

Kent-Olov Jonsson
N-acylethanolamines (NAEs)
Long chain NAEs are fatty acid amides that are normally found in low levels in almost all mammalian cells and tissues as well as in fish, invertebrates, plants and microorganisms (Schmid et al., 2002). In addition to AEA, NAEs such as palmitoylethanolamide (PEA), stearoylethanolamide (SEA) and oleoylethanolamide (OEA) produce interesting pharmacological effects both in vitro and in vivo. In most tissues, AEA is found at low levels compared to other NAEs. For example, in pig brain AEA content, corresponding to 1 % of total NAE content, was 6±1 ng/g wet tissue. In comparison, the levels of PEA, OEA and SEA were 205±49, 96±17 and 227±43 ng/g wet tissue, respectively (Schmid et al., 1995). In contrast, 200-2000 times higher levels of AEA has been found in mouse uterus and in that tissue, the relative amount of AEA was 75-95 % of total NAE content (Schmid et al., 1997). NAE levels are elevated in response to different types of stimuli, particularly those related to cellular stress and damage. For example, cadmium chloride-induced inflammation in rat testis results in a 5-fold increase in AEA amount and a 39-fold PEA increase (Kondo et al., 1998). Neurodegeneration is also associated with a large increase in NAE content (see below). However, cell damage rather than the inflammatory stimulus may be a prerequisite for such increases, since in two models of inflammation (intraplantar formalin and inhalation of lipopolysaccharide), no obvious increase in NAE content was seen (Beaulieu et al., 2000; Holt et al., 2004).
As mentioned above, AEA is active at CB receptors. The other “non-cannabinoid” fatty acid amines, although not being active at CB receptors, have been found to be endogenous signalling lipids and exert a number of effects. For example, OEA has been shown to reduce food consumption in rats and mice via activation of the nuclear peroxisome proliferator-activated receptor-α (PPAR-α) (Rodríguez de Fonseca et al., 2001; Fu et al., 2003) and exert weak antinociceptive effects (Calignano et al., 2001). SEA, the saturated homologue of OEA, has been found to produce the same effects as AEA on catalepsy, motility, analgesia, and body temperature of mice (Maccarone et al., 2002) and reduce food consumption in mice independent of an increased PPAR-α gene expression (Terrazzino et al., 2004 but see Fu et al., 2003). On the basis of a finding that a phospholipid fraction from egg yolk showed anti-allergic activity (Coburn et al., 1954), Kuehl and colleagues identified PEA as a constituent of egg yolk and reported further that it
18

Pharmacology of Palmitoylethanolamide and Related Compounds
affe
duce sub
synergistically with AEA (Jaggar et al., 1998; Calignano et al., 1998). Since PEA was believed at
ous ligand for the CB2 receptor (Facci et al., 1995), the involvement of CB receptors was examined. The anti-nociceptive actions of PEA was
tep enzymatic pathway (Schmid & Berdyshev, 2002). In the first step, a calcium-dependent membrane bound N-acyltransferase (NAT) catalyses the
cted joint anaphylaxis in the guinea pig (Kuehl et al., 1957). Following the discovery of the CB receptors, the effects of PEA saw new interest. In 1993, Aloe et al. reported that PEA, as well as other N-acylethanolamines, could re
stance P-induced mast cell degranulation in rat ear pinna (Aloe et al., 1993). Mazzari et al. (1996) showed a few years later a spectrum of actions of PEA in different models of inflammation. In 1998, PEA was also shown to be effective in two different models of inflammatory pain and in addition to act
that time to be an endogen2
found to be sensitive to SR144528, but not to rimonabant, which is consistent with a CB2 sensitive pathway (Calignano et al., 1998, 2001; Farquar-Smith et al., 2001).
Newer studies have investigated the possible involvement of CB2 receptors in the anti-inflammatory effect of PEA. In this respect, pre-treatment with PEA decreased carrageenan-induced oedema, in a SR144528 sensitive manner (Conti et al., 2002). The same effect was seen when PEA was administered after induction of inflammation, however, in this case, the effect could not be reversed by SR144528 (Costa et al., 2002). In a recent study, it has been shown that PEA binds and activates the nuclear PPAR-α. The authors attributed the anti-inflammatory effects of PEA solely to the activation of the PPAR-α, and indicated that this effect was independent of the CB2 receptor shown by the lack of effect of SR144528 (Lo Verme et al., 2005). In addition to the anti-inflammatory abilities, PEA has also shown other effects including alleviation of spasticity in a mouse model of multiple sclerosis (Baker et al., 2000, 2001) and suppression of convulsions in rats and mice (Lambert et al., 2001; Sheerin et al., 2004). The mechanisms responsible for these effects have not been elucidated, although the former is presumably related to the ability of PEA to reduce the rate of AEA metabolism by acting as an alternative FAAH substrate (see below).
Synthesis and release of anandamide
The synthesis of NAEs, including AEA and PEA, seems to be performed by the same two-s
19

Kent-Olov Jonsson
transfer of an acyl group from the sn-1 position of glycerophospholipid to the amino group of the phospolipid membrane precursor, phosphatidylethanolamine (PE), producing N-acylphosphatidylethanolamine (NAPE) (Schmid & Berdyshev, 2002). In the second step, a specific phospholipase D (NAPE-PLD), which recently has been cloned (Okamoto et al., 2004), hydrolyses the conversion into the corresponding NAE, although others have reported an alternative second pathway (Sun et al, 2004). With respect to AEA and PEA, synthesis has been shown in different tissues and cells, including cells of both neuronal and immune origin (Di Marzo et al., 1994; Di Marzo et al., 1996; Bisogno et al., 1996; Bisogno et al., 1997).
Although the mechanisms behind the release of NAEs are far from evident, it is established that they are synthesised and released “on demand” in neurons, at least in the case of the endocannabinoid AEA, under physiological and pathological conditions (reviews see Piomelli, 2003; Fowler, 2003). The calcium-dependency of the NAE synthesizing enzymes (see above) mean that conditions that result in an increased intracellular calcium level would be expected to result in an increased NAE synthesis. Consistent with this, depolarising stimuli, calcium ionophores and stimulation of receptors such as metabotropic glutamate receptors result in a stimulation of synthesis (see Di Marzo et al., 1994; Alger, 2002; Wilson & Nicoll, 2002; Piomelli, 2003). Calcium dysregulation is an important excitotoxic event (see Rang et al., 2004), and it has been shown that the levels of the endocannabinoids are elevated in certain brain areas in different animal models of diseases including stroke, multiple sclerosis and Parkinson’s disease (Di Marzo et al., 2004). For example, in rats with permanent middle cerebral artery occlusion, a model of stroke, different N-acylethanolamines was increased 15-44 times with an 18-fold increase of AEA levels in the striatum (Berger et al., 2004). In the central nervous system, the main function seems to be regulation of synaptic transmission and AEA has been shown to function as a retrograde messenger. In this role, endocannabinoids are released from the postsynaptic membrane and travel backwards into the synapse to activate presynaptic CB1 receptors and thereby decrease the presynaptic release of other neurotransmitters such as GABA and glutamate (Alger, 2002; Wilson & Nicoll, 2002). Indeed, the fact that endocannabinoids affect the activity of many neurotransmitters indicates a modulatory role for the endocannabinoid system in the brain (Baker et al., 2003). In the periphery, endocannabinoid systems are involved in reproduction, cardiovascular control and the gastrointestinal system (see Di Marzo et al., 2004).
20

Pharmacology of Palmitoylethanolamide and Related Compounds
For example, low AEA degrading activity has been shown to be related to miscarriage in humans (Maccarrone et al., 2000b). With respect to cardiovascular control, AEA levels were found to be elevated in rats during haemorrhagic shock and the blood from these rats induced vasodilation, which could be antagonised by rimonabant, in normal rats (Wagner et al., 1997).
Anandamide uptake
Since the degradation of AEA is brought about intracellularly (see below), released AEA needs to be internalized into the cell before this can occur. How this process works is not fully elucidated and is a matter of current debate. The accumulation of AEA has been found in a number of different cell lines, including cells of neuronal and immune origin from mouse, rat and man (see McFarland & Bar
bstrate specificity and selective inhibitability by structurally related compounds (see e.g. Di Marzo et al., 1994; Hillard et al., 1997;
xistence of a membrane protein is e inhibitors enhance extracellularly
med
ker, 2004; Fowler & Jacobsson, 2002 for review). Following early suggestions that the uptake of AEA was mediated by an energy
and sodium-independent process of facilitated diffusion (Di Marzo et al., 1994; Hillard et al., 1997), much effort has been made toward revealing this mechanism. There is today no clear consensus in this matter, although a number of different models has been proposed (Deutsch et al., 2001; Glaser et al., 2003; Hillard & Jarrahian, 2003; McFarland & Barker, 2004; Fowler et al., 2004; Ortega-Gutiérrez et al., 2004). These include: a) facilitated diffusion mediated by a membrane carrier protein, b) simple diffusion driven by intracellular FAAH, c) simple diffusion driven by intracellular sequestration of AEA or binding of AEA to other intracellular targets, d) endocytotic internalisation of AEA (review see McFarland & Barker, 2004).
In support of a surface membrane protein, it has been argued that the accumulation show all characteristics of this, including saturability, exhibiting comparable Km values with brain amines and amino acids transporters, temperature dependence, su
Beltramo et al., 1997; Fegley et al., 2004). The efurther supported by the fact that AEA uptak
iated receptor effects (CB1 receptors) and decrease intracellularly mediated receptor effects (TRPV1) (De Petrocellis et al., 2001; see also Paper III in this thesis). However, many different cells show AEA accumulation, which would
21

Kent-Olov Jonsson
suggest a rather unspecific process. The fact that AEA is highly lipophilic, makes diffusion through the lipid layer a distinct possibility.
The importance of FAAH in driving accumulation was suggested by Deutsch et al. (2001) who showed that FAAH transfected cells accumulated AEA to a larger extent than wild type cells with low FAAH activity, although it was still present in untransfected cells. Evidence against a separate membrane protein responsible for AEA accumulation was presented by Glaser et al. (2003). They showed that AEA transport inhibitors were not able to inhibit AEA uptake when using short incubation times (<1 min), but instead a saturable uptake was seen using longer incubation times. These authors indicated FAAH as the protein responsible for the saturable inhibition of uptake, and attributed the crossing over the plasma membrane as simple diffusion driven by the concentration gradient. In contrast to these results, Fegley et al. (2004) showed that there was no difference upon AEA uptake between neurons from FAAH (-/-) and FAAH (+/+) mice, indicating a lack of effect of FAAH. These results are in line with other studies identifying compounds that lack effects on FAAH but still can inhibit AEA uptake (Ortar et al., 2003; López-Rodríguez et al., 2003; Fegley et al., 2004). Furthermore, Fegley et al. (2004) reported that the prototypical AEA uptake inhibitor AM404 was still pharmacologically active in vivo when given to FAAH (-/-) mice.
Hillard & Jarrahian (2003) proposed that AEA is taken over the cell membrane by simple diffusion, and thereafter intracellularly sequestered or bound to proteins, in which the second process is the saturable part in AEA uptake. This model was proposed by the finding that radiolabelled AEA reaches higher intracellular concentrations than found extracellularly. An alternative to sequestration is the presence of intracellular shuttling protein(s), which take the AEA from the inside of the plasma membrane to the endoplasmic reticulum, where the FAAH is located (Fowler et al., 2004; Ortega-Gutiérrez et al., 2004). Since uptake inhibitors like AM404 and the closely related VDM11 can inhibit AEA binding to plastic wells (Karlsson et al., 2004; Fowler et al., 2004; Ortega-Gutiérrez et al., 2004; see also Paper I of this thesis), the structural requirements of such binding proteins may not need to be that stringent. In the fourth model, McFarland & Barker (2004) proposed that the sequestration of AEA was due to a caveolae-related endocytic process. Caveolae are flask shaped invaginations of the plasma membrane, and their model involves the binding of AEA to a protein located on
22

Pharmacology of Palmitoylethanolamide and Related Compounds
the extracellular part of the surface membrane in the caveolae, followed by an endocytic process where the membrane is pinched off and released into the cell.
Even if the exact process of AEA internalization is unclear, it is a fact that compounds that are identified as putative transport inhibitors enhances the effects of AEA in vitro and in vivo. For example, AM404 increased latency in the hot plate test when administered together with AEA (Beltramo et al., 1997). No effect was
ed alone. Another example is VDM11, which reduced spasticity in mice using a model of multiple sclerosis (Baker et al., 2001). Although, the
se AEA was reported for the first time in 1993 by Deutsch and Chi
seen when AM404 was us
specificity of these compounds have been questioned (e.g. Zygmunt et al., 2000; see Paper III of this thesis) there have recently been studies conducted using more selective uptake inhibitors, also showing similar in vivo effects (De Lago et al., 2004). For a recent compilation of the in vivo effects of putative AEA uptake blockers, see Fowler et al. (2005).
Anandamide degradation
The main enzyme responsible for the inactivation of AEA, in both peripheral tissue and the central nervous system, is fatty acid amide hydrolase, FAAH. This enzyme hydrolyses AEA into arachidonic acid and ethanolamine. Although it has been shown that other enzymes also are capable of inactivating AEA in vitro, such as cyclooxygenase-2 and lipoxygenase (see Kozak & Marnett, 2002; Maccarone et al., 2004), FAAH seems to be the key enzyme since FAAH (-/-) mice show a 15-fold increase in brain levels of AEA as well as a 50-100 fold decrease of catalytic activity in brain and liver compared to wild type (Cravatt et al., 2001). The ability of FAAH to metaboli
n (Deutsch & Chin, 1993). After the cloning of FAAH (Cravatt et al., 1996; Giang et al., 1997), it was confirmed that this enzyme was the same as the enzyme responsible of hydrolysing other NAEs including PEA and OEA (Bachur & Udenfriend, 1966; Schmid et al., 1983; Natarajan et al., 1984; Schmid et al., 1985).
FAAH is a membrane bound enzyme, and subcellular fractionation and
immunohistochemical studies have indicated that it is primarily localised to mitochondria and microsomal fractions (Hillard et al., 1995; Watanabe et al., 1998; Tiger et al., 2000; Glaser et al., 2003). FAAH shows an alkaline pH optimum ranging from pH 8.5 to 10, depending on source of enzyme (Deutsch et al., 2002).
23

Kent-Olov Jonsson
The enzyme was first cloned from rat liver and has since then been cloned from a number of species including human and mouse (Cravatt et al., 1996; Giang et al., 199
acylnitroanilides (Cr
7). The enzyme exists as a dimer and is believed to deliver AEA from the membrane to its active site (Bracey et al., 2002).
FAAH is widely distributed in mammalian tissue, and the distribution has been studied in detail. Many different types of tissues in rat and mouse, including liver, brain, small intestine, testis, eye and uterus have shown enzyme activity but activity was hardly detectable in heart and muscle (Deutsch & Chin, 1993; Desarnaud et al., 1995; Cravatt et al., 1996; Paria et al., 1996; Matsuda et al., 1997). This distribution correlates well with mRNA levels in rat (Cravatt et al., 1996; Katayama et al., 1997). The highest activities are found in liver and brain. In the brain, the localisation of FAAH is complementary to the CB1 receptor with FAAH located in neurons postsynaptic to CB1 receptors (Egertová et al., 1998, 2003). In contrast to the distribution in rat, FAAH mRNA in human organs is most abundant in pancreas, brain, kidney, and skeletal muscle with lower levels in liver and placenta (Giang et al., 1997).
The enzyme boasts broad substrate specificity and is capable of metabolising a number of different NAEs (see above), the endocannabinoid 2-AG (Lang et al., 1999; Goparaju et al., 1999) as well as N-acylamides and N-
avatt et al., 1996; Lang et al., 1999; Boger et al., 2000a; Patricelli et al., 2001). In view of the fact that the biological activity of AEA is offset by its short
lifespan, a possible pharmacological target would be to inhibit the degradation of this endocannabinoid. In this respect, the development of FAAH inhibitors has been undertaken by a number of groups.
FAAH inhibitors
Since the first inhibitor of FAAH, phenylmethylsulfonylfluoride (PMSF), was described in 1993 (Deutsch & Chin, 1993), a number of different FAAH inhibitors have been synthesised and tested for the ability to inhibit AEA degradation. The main groups of FAAH inhibitors can be divided into organophosphates and organosulphates, substrate analogues and carbamates (Fowler, 2004b). The non-specific serine hydrolase inhibitor PMSF belongs to the first group and inhibits FAAH irreversibly with an IC50 value in the low micromolar range at physiological pH (Deutsch & Chin, 1993; Desarnaud et al., 1995; Deutsch et al., 1997a, 1997b). In
24

Pharmacology of Palmitoylethanolamide and Related Compounds
vivo, PMSF has been shown to potentiate the effects of exogenous AEA in the cannabinoid tetrad response, without being active per se (Compton et al., 1997; Wil
inhibitor, and later studies have reported IC50 values ranging between high nanomolar and low micromolar concentrations (see
les are the oleoyl- and palmitoyltrifluoromethyl-ketones, showing similar IC values (Patterson et al, 1996; see Paper I of this the
d to inhibit FAAH in the low nanomolar range with URB597 and URB532 as the mo
ey et al., 2000). Other examples in this group include methyl-arachidonylfluorophosphonate (MAFP), which is a more potent irreversible inhibitor with IC50 values in the low nanomolar range (Deutsch et al., 1997b). The lack of specificity of these compounds, as well as potential toxicological effects including acetylcholinesterase inhibition (Quistad et al., 2002), however, limits their usefulness.
There have been a number of publications where FAAH inhibitors have been based on AEA or other fatty acids as a chemical template. In 1994 came the first report of a number of different substrate analogues (Koutek et al., 1994). Among these compounds, arachidonoyltrifluoromethylketone (ATMK or ATMFK) was found to be the most potent FAAH
Fowler et al., 2001). Other examp50
sis). With respect to their selectivity, the trifluoromethyl ketones are also inhibitors of phospholipase A2 and ATMK shows affinity for the CB1 receptor (Lehr, 1997; Koutek et al., 1994).
Another approach with substrate analogues has led to α-ketoheterocycle
inhibitors of FAAH, and this has given rise to highly potent compounds which have shown to be active in vivo, producing a decrease in locomotor activity (Boger et al., 2000b; Fedorova et al., 2001). In respect of their selectivity towards other components of the endocannabinoid system, not much have been done so far. However, in a very recent study, reversible potent and selective FAAH inhibitors belonging to this group were reported and among these, OL-135 was found to produce CB1 receptor mediated analgesia (Lichtman et al., 2004a).
In the last group, the carbamates, a number of compounds have been reporte
st potent (Kathuria et al., 2003). They showed no affinity for the CB receptors. In vivo, these compounds potentiated AEA-induced hypothermia without showing the same signs of CB1 receptor activation as AEA itself did on catalepsy, hypothermia and increased food intake (Kathuria et al., 2003; Fegley et al., 2005). In
25

Kent-Olov Jonsson
addition to these results, URB597 showed a CB1 receptor sensitive anxiolytic effect in a mouse model (Kathuria et al., 2003).
In addition to the above, compounds that have actions on other systems, have
also
1 sensitive antinociceptive response et al., 2004a). The same is seen in FAAH (-/-)
rence in locomotor activity and body temperature compared to FA
been shown to inhibit FAAH at pharmacologically relevant concentrations. These include propofol (Patel et al., 2003) and some acidic non-steroidal anti-inflammatory agents (Fowler et al., 1997; 2003a), and there is some evidence to suggest that this action may contribute to some of the pharmacological properties of these compounds in vivo (Patel et al., 2003; Gühring et al., 2002; Ates et al., 2003; review see Fowler, 2004a).
Therapeutic potential of FAAH inhibitors
Since AEA and other endogenous NAEs, such as PEA, possess a number of interesting effects in vivo, one can argue that inhibition of the metabolism of these compounds would be of therapeutic potential. In this respect, inhibition of FAAH have been suggested to possess a possible therapeutic value in a number of disorders such as pain, anxiety, inflammation and neuroprotection (Cravatt & Lichtman, 2004; Gaetani & Piomelli, 2003; Fowler et al., 2005).
A drawback in this respect could be the unwanted psychotropic effects, which
is mainly mediated by the CB1 receptor. However, inhibition of FAAH and, hence, enhanced levels of the NAEs, is not accompanied by catalepsy, hypothermia or increased food intake but instead by a CB(Kathuria et al., 2003; Lichtman mice, with no diffe
AH (+/+) mice (Cravatt et al., 2001). In contrast to the lack of these effects, FAAH (-/-) mice have shown a decreased sensitivity in several models of acute and inflammatory pain as well as an anti-inflammatory phenotype (Cravatt et al., 2001; Lichtman et al., 2004b; Massa et al., 2004). The relative involvement of central and peripheral FAAH in these effects was recently shown using a transgenic mouse model (FAAH-NS) where FAAH expression was restricted to the central nervous system. The levels of AEA, PEA and OEA were normal in the CNS and spinal cord but elevated in the periphery, in contrast to the FAAH (-/-) mice
26

Pharmacology of Palmitoylethanolamide and Related Compounds
where a general increase was seen throughout the body (Cravatt et al., 2001; 2004). The antinociceptive effects of the NAEs were attributed to the CNS and in contras the anti-inflammatory effects involved the periphery (Cravatt et al., 2004).
As stated earlier, AEA possesses a number of benificial effects and is
synthesised and released “on demand”. If this means that AEA is produced only when and where it is needed, FAAH inhibitors would enhance the levels of AEA mainly in the
t
areas where the production of AEA already is increased. This could enhance the beneficial effects, without causing the general CB1 receptor-mediated effects. Even if it can not be excluded that inhibition of FAAH would produce the psychotropic effects seen with cannabis, the results so far with FAAH inhibitors support the possibility that AEA-mediated effects can be separated.
In summary, it is concluded that modulation of the endocannabinoid system could be of therapeutic value and that the development of selective and potent FAAH inhibitors could be one approach in this respect.
27

Kent-Olov Jonsson
AIMS OF THE STUDY
ased on the discussion above, the deB velopment of compounds which can inte
having effects upon cannabinoid receptors.
ability of compounds structurally affect the uptake and hydrolysis of the
lamine head group f palmitoylethanolamide with respect to its interaction with fatty acid amide
ract with the degradation of anandamide and other fatty acids would be of interest. At the beginning of this study, there were few selective FAAH inhibitors available and those described were either non-selective (i.e. PMSF, ATMK) or poorly characterised apart from their effect on FAAH (α-heterocycle inhibitors). In consequence the present thesis was undertaken with the ultimate goal of identifying and characterising selective FAAH inhibitors. The endogenous N-acylethanolamine palmitoylethanolamide was chosen as a template upon its ability to inhibit FAAH without
Paper I: To evaluate pharmacologically the based on palmitoylethanolamide to endogenous cannabinoid anandamide, as well as their ability to interfere with cannabinoid receptors. The general aim was to identify compounds able to inhibit anandamide metabolism without having activity at cannabinoid receptors. Paper II: To explore further the importance of the ethanoohydrolase. Paper III: To test some compounds identified in Paper I for effects not related to FAAH inhibition or anandamide uptake in biological systems, and in this respect to compare them with two established anandamide uptake inhibitors. To compare the ability of these compounds to inhibit intracellularly TRPV1-mediated effects of anandamide. Paper IV: To establish models of inflammation in vitro and in vivo for further investigation of compounds identified in Paper I-III and to investigate the effects of a CB2 receptor agonist in these models. Paper V: To investigate the effects of palmitoylisopropylamide in models of inflammation in vivo and in vitro.
28

Pharmacology of Palmitoylethanolamide and Related Compounds
METHODS AND METHODOLOGICAL
CONSIDERATIONS
The methods used in this thesis are presented briefly together with comments abo
Fig. 4. Hydrolysis of [3H]-AEA into arachidonic acid and [3H]-ethanolamine. The asterisk indicates the position of the tritium labelled for the FAAH assay.
[3H]-labelled AEA, with the ethanolamine part labelled, was used as substrate (Fig. 4). The addition of chloroform and methanol to the homogenates after the incubation phase allows the separation of the water-soluble [3H]-ethanolamine from the lipid soluble [3H]-AEA. The ability of PEA and related compounds to inhibit this hydrolysis was examined. The method was originally described by Omeir et al. (1995) and modified by Fowler et al. (1997).
Frozen brains (minus cerebellum) from adult rats were thawed and
homogenized at 4°C in HEPES buffer, pH 7.0. The homogenates were centrifuged twice at 36,000 g at 4 °C after which the tissue pellets were re-suspended in homogenisation buffer and incubated at 37 °C. After centrifugation at 36,000 g at 4°C, membranes were re-suspended in Tris-HCl buffer, pH 7.4. The protein content in the membrane preparations were determined according to the method of Harrington (1990), using bovine serum albumin as standard.
Test compounds or ethanol carrier and assay buffer were preincubated for 0 - 180 min at 37 °C. [3H]-AEA and untritated AEA were added and the samples were incubated at 37 °C. Reactions were stopped by placing the tubes in ice and adding
ut the methods. For further details of each method, see the original papers.
FAAH assay (Paper I and II)
AEA is converted to arachidonic acid and ethanolamine by the intracellular enzyme fatty acid amide hydrolase, FAAH. This method was used to measure the ability of PEA-related compounds to inhibit this conversion.
O
OH
OO
OH*NH2
OH*NH2
OHNH
OHO
*NH
OHO
NH
OH* +
FAAH
H2O
Arachidonic acidAnandamide Ethanolamine
29

Kent-Olov Jonsson
chloroform: methanol (1:1 ixing of the tubes the phases wer
sch & Chin, 1993) or high-performance liquid chromatography (Maccarone et al., 1999). Thechrdispusinproperf y. Another possibility is to separate the labelled ethanolamine part from the arachidonic part is by using open bed column chromatography (Dealloreproducibility. The two main disadvantages are that the product is not unequivocally identified, and that the assay uses chloroform. In this respect, the metproprodessho FAAH inhibitors, it is in theory possible that additional hydrolytic enzymes contribute to the observed activity. Cerbeehom brain samples from mice lacking FAAH do not hydrolyse AEA (Cravatt et al., 2001). A second common factinvo y acid-free BSA (see e.g. Omeir et al., 1995) or
v/v) in the tubes. After vortex me separated by centrifugation. Aliquots of the methanol/buffer phase
were removed and analysed for radioactivity by liquid scintillation spectroscopy with quench correction. Blanks contained distilled water instead of the homogenate preparations.
Comments on the FAAH assay
A number of FAAH assays have been described in the literature. Some assays, for example, use substrate radiolabelled in the arachidonoyl part of the molecule and the products are then separated by thin-layer chromatography (Deut
re are also methods without radiolabelled AEA, using different types of omatographic separation (Lang et al., 1996; Watanabe et al., 1998), a fluorescent lacement assay (Thumser et al., 1997) as well as a spectrophotometric assay, g fatty acid p-nitroanilides (Patricelli et al., 2001). The assay used separates the ducts by simple phase separation with chloroform and methanol, which is ormed easil
sarnaud et al., 1995). The main advantage of the present assay is its simplicity, wing a good throughput of samples, and its robustness, in terms of its
hod has been further developed by separating the substrate from the labelled duct using charcoal which binds to the substrate, but not the ethanolamine duct (Wilson et al., 2003; Boldrup et al., 2004). Common to all the assays cribed above is that they measure the hydrolysis of AEA and, although they w the appropriate sensitivities to standard
tainly, a hydrolytic activity distinct from FAAH that can metabolise PEA has n identified in the lung (Ueda et al., 1999). However, the use of brain ogenates should eliminate this possibility, since
or for all assays is the lipophilic nature of the substrate (and test compounds) lved. Most assays use either fatt
30

Pharmacology of Palmitoylethanolamide and Related Compounds
detergents (see ssay used here (but with a charcoal he inhibitory potency of indomethacin (Boldrup et ature of the compounds use
us, potencies of test compounds are best interpreted relative to standard compounds
e same method.
An
The hydrolysis of AEA by FAAH is performed inside the cell which makes it necessary for AEA to be transported across the cell membrane before this event can occur. The exact process of cellular uptake of AEA, however, is a matter of current debate, see introduction for further discussion.
In this model, cells are incubated with [3H]-labelled AEA. After the cells have ed and analysed in
regard t
the cells. The ability of PEA and related compounds to inhibit AEA uptake was exa
at 37 °C was allowed for 15 min. After the incubation, the plates were placed on ice after which the cells wer
ied version of the met
e.g. Boger et al., 2000b). Addition of detergent to the a extraction) does not affect tal., 2004), but the lipophilic n
d nevertheless means that inhibitory potencies of compounds can vary between laboratories (see e.g. Fowler et al., 2004 for an example with VDM11). Th
assayed in the same laboratory with th
andamide uptake in RBL-2H3 and C6 cells (Paper I)
been allowed to internalise AEA they are washed, solubilizo tritium content (Rakhshan et al., 2000). This method measures both
metabolised ([3H]-ethanolamine part) and non-metabolised [3H]-AEA taken up by
mined. RBL-2H3 or C6 cells were plated onto 24-well plates and incubated at 37 °C
for 18 hours. After the incubation period, the cells were washed once with warm assay buffer and pre-incubated with the test compounds or ethanol carrier at 37 °C for 10 min [3H]-AEA was added to the wells and uptake
e rinsed three times with ice-cold buffer containing 1 % BSA to terminate the transport. The cells were aspirated from the buffer and incubated at 75 °C in NaOH for 15 min. Aliquots of the solubilized cells were transferred to scintillation vials and assayed for tritium content by liquid scintillation spectroscopy with quench correction.
In Paper I some of the compounds were tested in a modifhod above with the major difference in analysing only the [3H]-ethanolamine
part. This was achieved by separating the radioactive substrate from the
31

Kent-Olov Jonsson
[3H]-ethanolamine product in a similar way as in the FAAH method, using chloroform:methanol. These results reflect the combined inhibition of both uptake and FAAH degradation of AEA.
Comments on the uptake method
These highly lipophilic compounds have the ability to adhere to plastic as well f the data somewhat troublesome. A possible way by using fatty acid-free bovine serum albumin,
whi
or WIN 55,212-2 (CHO-CB2). After incubation, the membrane suspension was
as glass, making interpretation oto decrease this interference is
ch binds to AEA and thereby prevent the binding of AEA to plastic (Karlsson et al., 2004). Indeed, some studies have utilized BSA when measuring the uptake (Glaser et al., 2003). Other authors, however, argue that BSA per se inhibits AEA uptake (Di Marzo et al, 1994). As a consequence, when the study concerning the compounds in Paper I was carried out, we chose following the prevailing view and to implement the experiments without BSA. Due to the problems with the method concerning plastic binding and use of BSA or not, we chose to not perform any uptake experiments with the compounds in Paper II awaiting a better method. Since then, later studies have shown 0.15 % fatty acid-free BSA may be a useful concentration to use in the study of AEA uptake (Sandberg & Fowler, 2005). Nonetheless, there is still considerable controversy as to the best way to assay AEA uptake (see Fowler et al., 2004), as indeed there is concerning the nature of the uptake process itself (Glaser et al., 2003; Hillard & Jarrahian, 2003; McFarland & Barker, 2004).
Radioligand binding experiments (Paper I and II)
The PEA-related compounds were all tested for their ability to interfere at human CB1 and CB2 receptors overexpressed in Chinese Hamster Ovary Cells. This method, which was undertaken by Dr Séverine Vandevoorde in Belgium in the studies reported here, measures the ability of the test compounds to displace a tritiated potent CB agonist from the cell membranes.
Membranes from CHO-CB1 and CHO-CB2 were incubated at 30 °C with [3H]-CP55,940 (CHO-CB1) or [3H]-WIN 55,212-2 (CHO-CB2) for 1 hour in Tris-HCl with MgCl2 and EDTA (pH 7.4) in the presence of PMSF and BSA. Non-specific binding was determined with high concentrations of HU-210 (CHO-CB1)
32

Pharmacology of Palmitoylethanolamide and Related Compounds
rapidly filtered through 0.5 % polyethyleneimine-pretreated GF/B glass fibre filters, and the radioactivity trapped on the filters was measured by liquid scintillation spectroscopy.
Comments on the radioligand method
Radioligand binding is nowadays considered a standard method for assessing affinity at receptors. In general, it is recommended that the non-specific binding is measured using a different compound than the radioligand. This was not the case for the CHO- CB2 cells. However, the high levels of receptor expression, and the
ic binding, mea
r CB2 (CHO- CB2) receptors (Paper I and II) were used. The different cells were cultured in: Eagle's minimum essential medium, 2 mM L-glut
Rakhshan et al., 2000; De Petrocellis et al., 2000; Jacobsson & Fowler, 2001; McFarland
correspondingly low level of non-specific binding relative to the specifn that the use of the same ligand to assess non-specific binding is unlikely to
introduce errors. Indeed, the same results were found when HU-210 was used at high concentrations instead of WIN55,212-2 (data not shown). By using transfected cells, overexpressing a specific receptor, compared to using tissue naturally expressing receptors, involvement of other receptors could by avoided.
Cell cultures (Paper I, II, III, IV and V)
Rat basophilic leukaemia (RBL-2H3) cells (Paper I, IV and V), rat C6 glioma cells (Paper I and III) and Chinese hamster ovary transfected with human CB1 (CHO- CB1) o
amine supplemented with 15 % FBS (RBL-2H3), Ham’s F10 medium supplemented with 10 % FBS (C6) or Ham’s F12 medium supplemented with 10 % FBS and 200 µg ml-1 G418 (CHO-CB1) and CHO-CB2). All cells were grown with 100 units ml-1 penicillin and 100 µg ml-1 streptomycin present.
Comments on the cells used
RBL-2H3 (rat basophilic leukemia) is a mastocyte tumour cell line which shows similar homology to rat mucosal mast cells (Seldin et al., 1985) and these cells have been thoroughly characterized in their ability to internalise [3H]-AEA (Bisogno et al., 1997;
et al, 2004). C6 rat glioma cells have also been characterized and shown to accumulate AEA (Deutsch & Chin, 1993; De
33

Kent-Olov Jonsson
Petrocellis et al., 2000; Bisogno et al., 2001; McFarland et al, 2004). They respond to AEA by enhanced production of apoptotic bodies (Maccarrone et al., 2000a) as well as decreased rate of proliferation (Jacobsson et al., 2001) and this effect seems to involve TRPV1 and CB1 receptors. The AEA binding site on TRPV1 receptors is intracellularly located (De Petrocellis et al., 2001; Jordt & Julius, 2002), and so it
en up into the cell before it can interact with this rece
ell proliferation assay kit was used, which quantifies the total nucleic acid content of the samples and is based on a dye which becomes
erature for 30 min bef
An
is necessary for the AEA to be takptor.
Cell proliferation assay (Paper III)
The method was used to examine the effects of two AEA uptake inhibitors and compare them with PIA from Paper I on cell function and to compare the ability of these compounds to affect AEA inhibition of cell proliferation. The method described by Jacobsson et al. (2001) was used. C6 glioma cells were plated on 96-well plates and incubated at 37 ºC. After 6 h, the cells were incubated with various concentrations of the compounds. Half of the media were replaced daily for four days with fresh substances dissolved in media. 24, 48, 72 and 96 h after the first addition of compounds, the plates were emptied of media and stored at -80 ºC.
Analysis of cell density
The assay was used to analyse the density of cells in the proliferation assay. The CyQuant® c
fluorescent when bound to nucleic acids. At the day of analysis, the plates were placed in room temp
ore addition of reagents and incubation for 5 min. Fluorescence was measured at excitation/emission of 485/520 nm. A series of calibration curves indicated that the relationship between cell density and fluorescence deviated very slightly from linear at high cell densities.
alysis of caspase activity
AEA has been shown to affect cell proliferation in C6 cells by induction of apoptosis (Maccarone et al., 2000) and the assay used, which can detect the
34

Pharmacology of Palmitoylethanolamide and Related Compounds
activities of several caspases, was employed to determine if the AEA uptake inhibitors effects upon cell proliferation was due to activation of caspases. Cells were cultured and treated with test compounds as described in the cell proliferation assay. After 48 or 96 h of incubation, the medium was removed and the plates were frozen. On the day of assay, the plates were thawed and aliquots of
-Glu-Val-Asp-Rhodamine 110) in lysate buffer was
h measures mitochondrial activity. We measured cell proliferation as an endpoint,
and a fluorescent marker. In addition, it was possible to evaluate the effect of the test compounds on the anti
totic bodies. , can not give a complete answer if the effects seen on ptosis, but will at least indicate if caspase activation is an
imp
were used and maintained under pathogen-free conditions at the animal facility at
medium were added. Substrate (Asp added and the samples were then incubated for 2 h at 37 °C. Free rhodamine
110 produced by the action of caspases upon this substrate was detected fluorometrically using excitation and emission wavelengths of 485 and 520 nm respectively. Lysate from apoptotic U937 cells treated with camptothecin was used as positive control.
Comments on cell proliferation assay
There are a large number of different assays that can be used to measure cell viability and proliferation. Examples of these include measurement of the release of LDH, a cytoplasmic enzyme present in all cells, or by using MTT assays, whic
which is easily performed using microtiter plates
proliferative effects of AEA, indicating an effect on AEA uptake. The method, although highly sensitive, does not give any direct answers on the mechanism behind the antiproliferative effects of the compounds.
To evaluate if apoptosis could be responsible for the antiproliferative effects seen, an assay that measures the activity of caspases, which is one of the initial biochemical changes in apoptosis, was assessed. This approach does not account for later events of apoptosis, and the most common characteristic, the fragmentation of DNA into 180 bp fragments, so called apopMeasuring caspase activitycell viability are due to apo
ortant step in inhibition of cell proliferation produced by these compounds.
Animals
For the in vivo experiments female BALB/cJBom mice (8-9 weeks, 18-23 g)
35

Kent-Olov Jonsson
Umeå University. The mice were fed with standard food and water ad libitum and kept at least a week at the animal facility before the experiments. All animal experiments carried out in this report were approved by the ethical committee at the Umeå University. The mice were anaesthetized with an intraperitoneal (i.p.) injection (0.1 ml/mouse) of “cocktail”, a mixture of 0.315 mg ml-1 fentanyl citrate and 10 mg ml-1 fluanisone (Hypnorm™, Janssen, Saunderton, UK), 5 mg ml-1 midazolam (Dormicum®, Roche, Basel, Switzerland) and sterile water at a ratio of 1:1:2. Anaesthesia was retained under the whole experiment with additional injections of anaesthetic mixture. In the experiments when only tissue was used,
xygen before cervical dislocation.
or vehicle. The mice were injected subcutaneously in one ear pinna with compound 48/80 and vehicle alone in the other ear pinna. Immediately after the
re given Evans blue intravenously and euthanized 2 h later. Eva
ks.
d formalin-induced (Mazzari et al., 1996) paw oedema, arachidonic acid-induced ear oedema (Hanus et al., 1999), LPS-induced pulmonary inflammation (Berdyshev et al., 1998) and turpentine-induced bladder inflammation (Jaggar et al., 1998). The different models assess effects of a variety of inflammatory mediators acting either alone or in concert, and of course the best
the animals were euthanized with CO2 and o
Plasma extravasation in mouse ear pinna
The method was used to examine the effects of JWH133, a CB2 agonist, as well as the PEA analogue PIA upon inflammation and a model of mast cell dependent inflammation was chosen. The method by Mazzari et al. (1996) was used with some modifications. Mice were injected i.p. with compounds or vehicle 30 min prior to induction of inflammation. In the experiments with SR144528, the compound (or vehicle) was administered i.p. additionally 45 min before PIA, JWH133
ear injections, mice wens blue was extracted by homogenising of the ear in formamide followed by a
2 h incubation at 50 °C. After centrifugation the supernatant was analysed by optical absorbance at 620 nm (Saria and Lundberg, 1983). Ears from mice not injected with Evans blue were used as blan
Comments on the plasma extravasation method
Models of inflammation that has been used to evaluate effects of PEA and CB2 receptor agonists includes: carrageenan-induced (Conti et al., 2002; Costa et al., 2002; Mazzari et al., 1996) an
36

Pharmacology of Palmitoylethanolamide and Related Compounds
information regarding the anti-inflammatory effect of a new compound would be to u
. All steps with drugs were mad
se a selection of models of inflammation. However, the limited time available precluded this approach, and we chose to use an apparently simple system involving mast cells, on the basis of previous findings that PEA and CB2 receptor agonists have been shown to be effective against mast cell dependent inflammation (Mazzari et al., 1996 and Malan et al., 2001a). Initially the method was assessed using substance P as a mast cell degranulator. The results using substance P did however not give any significant increase in plasma extravasation. This could be due to instability of this compound, or some other methodological errors. In consequence, compound 48/80, a chemically stable mast cell degranulator, was chosen as an inducer of inflammation. This compound produces oedema that is mast cell dependent, shown by lack of effect in mast cell deficient mice (Kim et al., 1999) and gave a significant increase in plasma extravasation in all experiments, although very variable between animals.
Mice paw skin release
The method was set up to examine the effects of JWH133, PIA and PEA upon effects of mast cells in vitro.
The skin on the upper side from both hind paws of the mice was separated from the connective tissue. The skin was gently loosened with a small forceps followed by additional incisions with a scissor and placed in an oxygenated super interstitial fluid (SIF) buffer. After weighing, the isolated skin was mounted on glass rods with the outside facing the rods and transferred into tubes containing SIF buffer continuously bubbled with 95 % O2 and 5 % CO2 at 37 ºC. The tissue was left to equilibrate for 90 min before drug treatment
e in bubbled SIF. After equilibration, the tissue was transferred to new tubes containing drugs or vehicle and incubated for 15 min before compound 48/80 or vehicle was added to the test tubes. Fluid was collected at 60 min after the addition of compound 48/80 and stored at -80 ºC until analysis. Following the collection of fluid, the tissue was weighed and placed on a paper and an outline of the area from each piece of tissue was made. The outline was weighed and an area was calculated from the known weight of 10 cm2 paper.
37

Kent-Olov Jonsson
Comments of the paw skin method
the release of ß-hexosaminidase. This enzyme is stored in granules and its release
(Schwartz & Austen, 1980). ompounds like CB2 receptor
ago
cular Devices).
The method was set up in Ruth Ross lab in Aberdeen and was based on the work by Kilo et al. (1997) and Ellington et al. (2002). By using whole skin tissue pieces a system with mast cells surrounded by their natural environment is utilized. This was chosen as a model system since mast cells seem to respond differently when separated from their environment (Robinson et al., 1989; Sweiter et al., 1993; Hamawy et al., 1994; Ra et al., 1994). Initially it was intended to study the release of TNF-α, an important inflammatory mediator that is synthesised and released in response to mast cell activation (Ackermann & Harvima, 1998; Gibbs et al., 2001). Initial experiments, conducted at the laboratory of Dr. Ruth Ross in Aberdeen, U.K., indicated however that the release of TNF-α in response to non-immunogenic stimulation of mast cells was under the limit of detection. In consequence, upon return to Umeå, a different end-point was used, namely
represents the immediate response to degranulationOf course, the possibility cannot be ruled out that c
nists and PIA differently affect the release of preformed and de-novo synthesised mediators.
β-Hexosaminidase release
β-Hexosaminidase content was analysed by using the method, with some modification, of Smith et al. (1997). Fluid samples from the paw skin experiments were allowed to reach room temperature and added to a 96-well microtiter plate. The reaction was started by adding p-nitrophenyl-N-acetyl-D-glucosaminide in a citrate-buffer as substrate. After 2 h of incubation at 37 °C, the reaction was stopped by addition of Tris-buffer, pH 9.0. Optical density was measured spectrophotometrically at 405 nm (subtraction at 570 nm), using a computer-operated ThermoMax microplate reader (Mole
Immunohistochemistry and PCR
Immunohistochemistry was assessed to determine the location of CB2 receptors in skin. Mouse skin tissue were cut into pieces, embedded in Tissue-Tek® and snap frozen on dry ice before storage at -80°C. Cryostat sections were made
38

Pharmacology of Palmitoylethanolamide and Related Compounds
and allowed to air dry for 1 h, followed by fixation in paraformaldehyde for 30 min. The sections were rinsed in phosphate buffered saline (PBS) between every step, except after blocking of unspecific binding with serum. Endogenous peroxidase activity was blocked by 3 % hydrogen peroxide in methanol for 5 min at room temperature (RT). Unspecific binding was blocked with normal goat serum for 5 min at RT. The primary antibody was directed against the first transmembrane domain of the human CB2 receptor. Sections were incubated with the primary antibody at 4°C O/N. Biotinylated goat anti-rabbit immunoglobulins were used as secondary antibody and incubated for 30 min. For detection, sections were incubated with horseradish peroxidase-labelled streptavidin and DAB. After detection, sections were washed in tap water, counterstained with haematoxylin and mounted in glycerol jelly. Control experiments were performed without the primary antibody as well as preabsorption with the blocking peptide used as antigen for production of the CB2 receptor antibody. In the preabsorption experiments the primary antibody and blocking peptide were incubated for one hour at RT.
To determine if CB2 receptor mRNA was present in skin tissue, pieces of skin
was prepared and analysed by using RT-PCR. Mouse spleen was used as a positive racted from skin and spleen with TRIzol LS Reagent
by following the manufacturer’s protocol. The RNA was quantified spe
nly RNA preparations showing intact species wer
control. Total RNA was ext
ctrophotometrically and the integrity of the RNA preparations was examined by agarose gel electrophoresis. O
e used for subsequent analysis. One microgram of total RNA was reverse transcribed into single-stranded cDNA. After incubation at, the AMV reverse transcriptase was denatured and the cDNA was stored at -20ºC until used for polymerase chain reaction (PCR).
The synthesized cDNA was amplified by PCR. The PCR analyses for mouse CB2 receptor and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were performed using PCR Core Kit standard protocol except for the use of HotStar Taq polymerase in the CB2 reactions. The expression was compared at the logarithmic phase of the PCR reaction. The PCR products were electrophoretically size fractionated in 1.5 % agarose gel and visualized using ethidium bromide. The estimated size of the PCR product was 479 bp for CB2 and 267 bp for GAPDH. The PCR products were purified using a QIAquick purification kit according to
39

Kent-Olov Jonsson
manufacturer’s protocol, and the sequence was confirmed using a DNA Sequencer.
Data analyses and statistics
test, or paired two-tailed t-test (Paper IV, Fig. 2A), using the
Km and Vmax values for inhibition of [3H]-AEA metabolism by PIA and OEA in Paper I were calculated from the mean data using the Direct Linear Plot analysis (Eisenthal & Cornish-Bowden, 1974) using the enzyme kinetics v 1.4 software package, Trinity Software, Campton, NH, USA. These values were used in secondary replots to determine Ki(slope) and Ki(intercept) values. For the analyses of pI50 values in Paper I and II non-linear regression analysis built into the GraphPad Prism computer programme (San Diego, CA, USA) were used (for details, see result and discussion below). The pI50 values for inhibition of cell proliferation in Paper III were calculated from the data expressed as % of control using the same computer programme with the same approach as in Paper I and II, and the top and bottom value was set to 100 % and 0 % respectively. The cell density data in Paper III was analysed either using one-way ANOVA followed by Bonferroni-Dunn post hoc tests the using Statview computer programme (SAS institute Inc. Cary, NC, USA) or two-tailed unpaired t-test using the GraphPad Prism programme. In Paper IV and V plasma extravasation and β-hexosaminidase release were analysed using one-way ANOVA followed by Dunnett’s multiple comparisonsGraphPad Prism software.
40

Pharmacology of Palmitoylethanolamide and Related Compounds
RESULTS AND DISCUSSION
gramme used (GraphPad Prism, GraphPad Software Inc., San Diego, CA, USA). When the bottom value gave a 95 % confidence interval value stra
ds. Although other possibilities cannot be ruled out
Inhibition of FAAH catalysed anandamide hydrolysis (Paper I
and II)
As the initial goal was to find new potent compounds which could affect the degradation of AEA, a number of compounds were tested for their ability to block FAAH-catalysed AEA hydrolysis by rat brain homogenates. The compounds were based on PEA as a chemical template, in view of the findings that this compound is inactive at CB receptors, but is able to inhibit AEA degradation by itself being a substrate for FAAH (Schmid et al., 1985; Tiger et al., 2000). Being a pharmacological study, the idea at the outset was to construct dose-response curves and thereby determine the pI50 values, i.e. the positive calculated logarithmic value of the concentration of the compound producing 50 % inhibition. However, as is apparent in Fig. 2 of Paper II, several of the compounds did not produce a maximal level of inhibition. Comparison of potencies between compounds with different maximal levels of effect is somewhat difficult. In Papers I and II, an approach was used whereby the data was fitted to sigmoidal curves of variable slope with an initial value of 100 % and the ”bottom” value determined by the computer pro
ddling zero (such as seen for compound 13 in Fig. 2 of Paper II), the bottom value was set to 0 and thereby the calculated maximum inhibition was 100 % for these compounds. In some cases not enough inhibition was seen to permit meaningful calculation of pI50 values and in these cases the inhibition seen at the highest concentration tested was given. In many cases, however, the 95 % confidence intervals of the compounds were both above zero. In these cases, the bottom value was used to calculate the maximum observable inhibition and the pI50 value represents the potency with respect to the inhibitable fraction. In the papers, it was suggested that the residual activity simply reflected a solubility issue for these highly lipophilic compoun
(for example, the presence of FAAH subtypes, presumably resulting from the posttranslational modification of a single gene product, can be suggested although there is as yet no evidence in support of such a suggestion), the compounds do
41

Kent-Olov Jonsson
have limited solubilities and this would seem to be the most rational explanation of the data.
While the use of pI50 and maximum observable inhibition allows for the
compact description of a large number of dose response curves, interpretation of the data is complicated. Is, for example, a compound with a pI50 value of 5.54 more useful than a compound with a pI50 value of 5.08 when the former only produces a maximum of 24 % inhibition whereas the latter produces a maximum
nd 37, respectively, of Paper II)? In terms of the ability to prevent AEA metabolism, it may be more useful simply to show to the
ty, and as pI50 values, reflecting potency. This value is the positive calculated logarithmic value of the concentration of the compound inhibiting 50 % of the measured response.
of 87 % inhibition (compounds 6 a
total inhibitory capability at a given concentration, such as 10 µM. In order to give the reader the opportunity to see such values, the original data for a series of compounds studied in Papers I and II have been redrawn to show the inhibition produced by 10 and 100 µM (Fig. 6). In Paper I, several homologues with fatty acid chain length varying from 3 (C3:0, propanoylethanolamide) to 18 carbons (C18:0, stearoylethanolamide, SEA) was tested against their ability to inhibit FAAH catalysed AEA hydrolysis. The mono-unsaturated homologue of SEA, where a double binding was introduced between carbon 9 and 10 in the fatty acid chain (C18:1, oleoylethanolamide, OEA), was also tested.
The chemical approach was to change the fatty acid chain (Fig. 5A), herein
termed homologues, or the ethanolamine head of PEA (Fig. 5B), herein termed analogues, and all chemical work was done at the Catholic University of Louvain in Brussels, Belgium, by Dr Lambert and co-workers. All these compounds show different degrees of solubility, which of course affect their ability and usefulness as potential pharmacological tools. In this respect, the data concerning the FAAH inhibition are presented and compared both at individual concentrations, somewhat reflecting solubili
42

Pharmacology of Palmitoylethanolamide and Related Compounds
For the homologues with chain lengths fhigher than 20 % of [3H]-AEA metabolism wlength instead contained 10 to 18 carbons a metabolism was seen at 100 µM of these hunsatured homologue OEA (C18:1) was thmetabolism by 70 % (Fig. 6A). This compoundinhibitor, which was somewhat unexpected (Schmid et al., 1985; Licthman et al., 2002) andas competitive inhibitors. However, in this resa concentration of 1 µM, inhibited rat brainmanner, whereas the inhibition produced by(Tiger et al., 2000). It was pointed out in thatmixed and competitive was rather small anparticularly since the blank values were relativactivities at high substrate concentrations”. ThAt 10 µM, PEA (C16) showed the highest degrhomologues with decreasing inhibition with adpattern was seen at 100 µM when instead theshowed the highest degree of inhibition tosaturated homologues containing 17 or 18 concentration, also showed lower inhibition cohighest degree of inhibition using PEA contahomologue with 12 carbons at 100 µM, is probthese compounds, limiting the observed
NH
O
OH
C3
C18C18:1
NH
O
OH
C3
C18C18:1
NH
O
RNH
O
R
A
B
43
Fig. 5. (A) Modificafatty acid chain
tions of the (broken lines)
creating homologues of PEA. C3, i.e 3 carbons in the acyl chain, is
(OEA) indicates the number of the double bounds in the molecule. (B)
the shortest and C18 the longest ofthe homologues. The :1 in C18:1
Modifications of the ethanolamine head (dotted line) creating analogues of PEA. In some cases, the hydrogen on the amine was replaced by an additional R-group.
rom 3 to 8 carbons, no inhibition as seen at 100 µM. When the chain 50 -100 % inhibition of [3H]-AEA omologues. At 10 µM, the mono-e most efficient, inhibiting AEA was also found to be mixed type of since it is a substrate for FAAH alternate substrates should interact pect it has been found that AEA, at PEA hydrolysis in a competitive 2 µM AEA was mixed in nature study that “the difference between d may reflect experimental error, ely high with respect to the specific e same conclusion may apply here. ee of inhibition among the saturated ditional carbons. A slightly different homologue containing 12 carbons gether with OEA (Fig. 6A). The carbon in the side chains, at this mpared to PEA. This shift, with the ining 16 carbons at 10 µM, to the ably a reflection of the solubility of inhibition at the highest tested

Kent-Olov Jonsson
concentration. When comparing the pI50 values of the homologues against the inhibitable part of AEA hydrolysis, compounds with side chains from 10-18 carb ns show similar values, except for PEA and OEA showing slightly higher valu
hanolamine head were tested. The most important findings from Paper I and Paper II and examples are indicated her
h the highest activity in the ethylamide analogue of PEA (Fig. 6B, [4]), showing a lower obtainable inhibition with both lower and higher chain length at 10 µM. The same pattern was found with 100 µM, although in this case the methylamide analogue (Fig. 6B, [3]) showed the same obtainable inhibition as the ethylamide analogue (Fig. 6B). This is consistent with studies by Schmid et al. (1985) showing that the length of the chain of the amino
oes and the C10 homologue showing slightly lower potency. The homologues
with 3-8 carbons in the side chain were inactive, as mentioned earlier. Thus, a decrease in number of carbons enhances solubility, but too few carbon atoms in the side chain produce an inactive compound. The introduction of a double bound between carbon 9 and 10 seems to enhance solubility, although no major difference in potency is seen shown by the similar pI50 values between the saturated and the mono-unsaturated homologue. This lack of importance of the double binding concerning potency is in line with studies using the same experimental conditions, showing the oleoyl-analogue of PTMK exhibiting similar potency as PTMK used in Paper I (Fowler et al., 2000; Holt et al., 2001).
Instead of changing the fatty acid chain, another approach was to modify the
ethanolamine head of PEA. In Paper I, four analogues of PEA with a butylamide, isopropylamide, cyclohexamide or a (2-methyl)ethanolamide chain instead of the ethanolamide part was tested. In Paper II, this approach was taken further and a number of analogues with modifications in the et
e. For further details of all of the analogues investigated, see Paper II. By replacing the –OH group in the ethyl chain with a halogen atom, no major
difference in potency between the compounds compared to PEA was seen except when a fluoride atom was introduced (data not shown), and in this case all inhibitory activity was abolished. At both 10 and 100 µM, here shown with a chloride atom as replacement of the –OH group, the obtained inhibition seemed to decrease slightly compared to PEA (Fig. 6B). The same pattern, with no major difference in potency, was seen when hydrogen was introduced instead of the –OH group. In this case, the length of the head group seemed to affect the highest obtainable inhibition of AEA hydrolysis, wit
44

Pharmacology of Palmitoylethanolamide and Related Compounds
alcohol moiety is of importance upon interaction with the amidohydrolase in rat liver. These results, taken together, indicate tha is not of major importance in the ability of the hydrolysis. The length of the carbon chain see inhibition. The lack of importance of the r arachidonoyl-based compounds, where the et replaced by cyclopropyl, methylcyclopropyl or FAAH catalysed metabolism of [14C]-AEA in ra
The increase of steric groups seemed t introduction of one additional methyl group at cy as well as maximum obtained inhibition (Fig. 6B, PIA), and two additional methyl gro
t the –OH group in the ethyl chainse analogues to interfere with AEAms to affect maximum obtainable
–OH group is also the case fohanolamine head group of AEAcyclobutyl was reported to inhibit t brain (Jarrahian et al., 2000). o be of some importance, sincethe alpha-carbon decreased poten
ups gave an inactive compound (Fig. 6B, [9]). The first of these compounds (PIA) was found to be a mixed type of inhibitor (Ki(slope) 15 µM, Ki(intercept) 87 µM; but see discussion with respect to oleoylethanolamide and AEA concerning the difference between mixed and competitive inhibition) and is probably not a substrate itself for FAAH, since the arachidonyl isopropylamide analogue of AEA have no substrate activity (Lang et al., 1999). This effect of steric hindrance is not only due to a general bulky steric effect, since some aryl analogues were active (see e.g. compound 24), although the maximum inhibition of AEA hydrolysis was no more than around 30 % for these compounds. The steric parameters for these group was indeed tightly regulated, shown by difference in activity between compounds 23 and 24 (Fig. 6B).
45

Kent-Olov Jonsson
C3:
0
C4:
0
C6:
0
C8:
0
C10
:0
C12
:0
C14
:0
C16
:0
C17
:0
C18
:0
C18
:1
0
20
40
60
80
100
120
10 µM100 µM
A
4.26
4.79
4.77
5.30
4.71
4.95
5.54
no p
I 50
no p
I 50
no p
I 50
no p
I 50
% In
hibi
tion
of [3 H
]AEA
met
abol
ism
Fig. 6. Effects of homologues (A) and analogues (B) of PEA (C16:0) upon rat brain FAAH catalysed hydrolysis of 2 µM [3H]-AEA. In the homologues, the numbers of carbons in the fatty acid chain are indicated. In the analogues both the structure of the changed ethanolamine head group and the abbreviation or compound number (taken from Paper II) are indicated. The numbers above the columns in the figure are the pI50 values of the compounds, taken from Paper II.
[PEA
]
[34] [2
]
[3]
[4]
[5]
[6]
[19]
[PIA
]
[9]
[PC
A]
[24]
[23]
80
10 µM100 µM
B
5.01
4.5
5.4
5.56
5.54
5.58
4.89
no p
I 50
5.08
5.
no p
I 50no
pI 5
0
~3.6
7 530
% In
hibi
tion
of [3 H
]AE
met
abol
ism
A
0
20
40
60
NO
HN
OH
NN N NN NN NN NN NC
lN
Cl
NN NN NN NN NN
46

Pharmacology of Palmitoylethanolamide and Related Compounds
Interaction with CB1 and CB2 cannabinoid receptors (Paper I
and II)
All compounds in Paper I and Paper II were tested at 10 µM, as well as at 100 µM in some cases, upon their ability to interact with human CB1 and CB2 receptors. The analogues and homologues were assessed for their ability to inhibit specific binding of the synthetic cannabinoid receptor ligands [3H]-CP55,940 (CB1) or [3H]-WIN55,212-2 (CB2) on CHO cells transfected with the human CB1 or CB2 rece
seen. The analogue emanating as one of the most interesting from these studies, PIA, showed low affinity and inhibited specific binding at CB1 receptors with 25 % and 15 % and at CB2 receptors with 12 % and 21 % at 10 and 100 µM, respectively.
Inhibition of anandamide uptake (Paper I)
If inhibition of AEA hydrolysis and thereby prolongation of the lifespan of this compound is beneficiary, another approach is to reduce accumulation (and hence FAAH accessibility) of AEA. By identifying compounds inhibiting the process which accumulate AEA this is a way of avoiding, or at least delaying, AEA degradation. Indeed, putative AEA uptake inhibitors, such as AM404, have been shown to be active in vivo in a variety of models (see Fowler et al., 2005). All compounds in Paper I were screened for their ability to inhibit AEA uptake in
ptors. Concerning the homologues in Paper I, all except one produced <15 % at 10 µM or <25 % at 100 µM inhibition of specific binding with [3H]-WIN55,212-2 on CHO cells expressing CB2 receptors. In the case of the CB1 receptors, the data was less clear with inhibition of [3H]-CP55,940 between 19-32 % at 10 µM and between 8-41 % at 100 µM of the homologues. The only exception was the mono-saturated homologue oleoylethanolamide which gave 13-14 % inhibition at 10 µM and around 64 % at 100 µM at both CB1 and CB2 receptors. The analogues did not produce any inhibition of CB2 specific binding higher than 16 % at 10 µM or higher than 22 % at 100 µM for the compounds tested at that concentration in Paper I. Concerning the interaction with CB1 receptors, the analogues showed, to some extent, more affinity compared with the interaction at the CB2 receptor. None of the compounds were highly active with the most of these exhibiting an inhibition of specific binding less than 30 %. Even with the most active analogues, an inhibition less than 45 % was
47

Kent-Olov Jonsson
RBL-2H3 cells and were initially tested at 1, 10 and 100 µM. In this first screening, none of the compounds were active at 1 µM, although it was found that at concentrations of 10 or 100 µM the homologues containing more than 10 carbons in the fatty acid chain, exhibited some ability to inhibit [3H]-AEA uptake. The shorter homologues were inactive at all concentrations tested. As with the longer homologues, 10 or 100 µM of the analogues tested in this paper, showed ability to inhibit [3H]-AEA uptake.
As a result of the observation that AM404 can affect binding of [3H]-AEA to cell culture wells, which we became aware of after the initial screening, all compounds was tested retroactively for their ability to block [3H]-AEA binding to the wells. Neither the saturated homologues nor the analogues affected this binding to a major extent. In contrast, AM404 as well as the mono-saturated homologue OEA did indeed inhibit the binding of [3H]-AEA to the wells.
As a consequence of the effect of binding to plastic, the most promising compounds from the initial uptake data as well as AM404 and PEA were tested using RBL-2H3 and/or C6 cells with parallel assays using wells. In Fig. 7 the data are presented as cell specific, i.e. the [3H]-AEA uptake of each individual compound in wells with no cells is subtracted from the uptake for this compound when cells are present. The data indicated that 30 µM of PIA, as well as 100 µM AM404, inhibited cell specific [3H]-AEA uptake in both RBL-2H3 (Fig. 7A) and C6 (Fig. 7B) cells. In contrast to these compounds, 30 µM of PEA did not affect and 30 µM of OEA showed only a small effect on cell specific [3H]-AEA uptake in C6 cells (Fig. 7C). As mentioned above, both AM404 and OEA per se inhibited the [3H]-AEA uptake (see Paper I for figures) in wells without cells, making it troublesome to interpret the uptake data of these compounds. Among the compounds analysed, PIA was the substance that showed most potential in affecting AEA uptake, and in contrast to AM404 and OEA, this compound had little effect on the binding of [3H]-AEA to plastic.
48

Pharmacology of Palmitoylethanolamide and Related Compounds
RBL-2H3 C6 C6
Fig. 7. Effects of PEA and related compounds (µM) upon the uptake of 10 µM [3H]-AEA into RBL-2H3 basophilic leukaemia cells (A) and rat C6 glioma cells (B, C). Shown are means±s.e.mean of cell specific, i.e. the [3H]-AEA uptake of each individual compound in wells not containing cells is subtracted from the uptake for this compound when cells are present. Data are recalculated from figure 6 of Paper I.
Inhibition of anandamide uptake and hydrolysis in intact C6 cells
(Paper I)
Since the in vivo effects of AEA metabolism is dependent on both uptake and hydrolysis, a compound producing inhibition of both processes might be favourable compared to compounds only affecting each separate process. To investigate the combined effects upon these processes, the compounds in figure 7 were tested for their ability to inhibit [3H]-ethanolamine production in intact C6 cells. By separating this product from the added [3H]-AEA substrate, a measurement of the compounds effect on both AEA uptake and degradation can be accomplished. PIA and OEA together with AM404 was found to be the most potent in this respect, showing a concentration dependent inhibition of [3H]-
m 3-30 µM (Fig. 8A), with ion tested. PEA and PCA
exh
ethanolamine production, at concentrations ranging froPIA being the most effective at the highest concentrat
ibited the same concentration dependent pattern as the compounds above, although the latter did not inhibit [3H]-ethanolamine production to the same extent (Fig. 8B). The relative contribution of uptake or FAAH inhibition in this model is somewhat difficult to speculate on, since the FAAH inhibition also is dependent on the ability of the compounds to be taken up by the cells. In this respect, less is known about the cellular uptake of the examined compounds, although the lipophilicity indicates at least a possible passive diffusion over the cell membrane.
Non
e
PCA
10
PCA
30
PIA
10
PIA
30
AM
404
100
0
5
10
15A
ake
in.-1
)
B C
[3H
](p
mo
AEA
upt
l.wel
l-1.m
Non
e
PCA
10
PCA
30
PIA
10
PIA
30
AM
404
100
0.0
2.5
5.0
7.5
10.0
[3H
]AEA
u(p
mol
.wel
l-1pt
ake
.min
.-1)
Non
e
OEA
10
OEA
30
PEA
30
0
4
8
12
[3H
]AEA
u(p
mol
.wel
l-1pt
ake
.min
.-1)
49
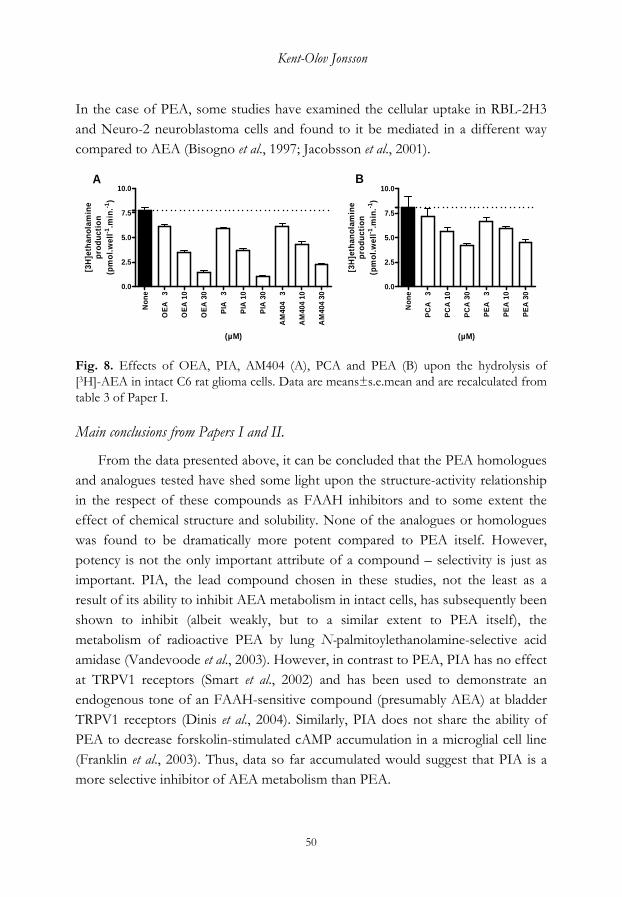
Kent-Olov Jonsson
In the case of PEA, some studies have examined the cellular uptake in RBL-2H3 and Neuro-2 neuroblastoma cells and found to it be mediated in a different way compared to AEA (Bisogno et al., 1997; Jacobsson et al., 2001).
Fig. 8. Effects of OEA, PIA, AM404 (A), PCA and PEA (B) upon the hydrolysis of [3H]-AEA in intact C6 rat glioma cells. Data are means±s.e.mean and are recalculated from table 3 of Paper I.
Main conclusions from Papers I and II.
From the data presented above, it can be concluded that the PEA homologues and analogues tested have shed some light upon the structure-activity relationship in the respect of these compounds as FAAH inhibitors and to some extent the effect of chemical structure and solubility. None of the analogues or homologues was found to be dramatically more potent compared to PEA itself. However, potency is not the only important attribute of a compound – selectivity is just as important. PIA, the lead compound chosen in these studies, not the least as a result of its ability to inhibit AEA metabolism in intact cells, has subsequently been shown to inhibit (albeit weakly, but to a similar extent to PEA itself), the metabolism of radioactive PEA by lung N-palmitoylethanolamine-selective acid amidase (Vandevoode et al., 2003). However, in contrast to PEA, PIA has no effect at TRPV1 receptors (Smart et al., 2002) and has been used to demonstrate an endogenous tone of an FAAH-sensitive compound (presumably AEA) at bladder TRPV1 receptors (Dinis et al., 2004). Similarly, PIA does not share the ability of PEA to decrease forskolin-stimulated cAMP accumulation in a microglial cell line (Franklin et al., 2003). Thus, data so far accumulated would suggest that PIA is a more selective inhibitor of AEA metabolism than PEA.
Non
e
OEA
3
OEA
10
OEA
30
PIA
3
PIA
10
PIA
30
AM
404
3
AM
404
10
AM
404
30
0.0
2.5
5.0
7.5
10.0A
hano
lam
ine
uctio
nll-1
.min
.-1)
(µM)
[3H
] p(p
mol
.we
et rod
Non
e
PCA
3
PCA
10
PCA
30
PEA
3
PEA
10
PEA
30
0.0
2.5
5.0
7.5
10.0B
min
en in
.-1)
(µM)[3
H]e
than
prod
uc(p
mol
.wel
lola tio -1.m
50

Pharmacology of Palmitoylethanolamide and Related Compounds
Effects of PIA, AM404 and VDM11 upon cell proliferation
(Paper III)
After the identification of compounds from Paper I and II, we further wanted to test if the most promising analogues from the initial studies affected cell proliferation. As a comparison, other presumptive AEA uptake inhibitors such as AM404 and VDM11 were also tested in this approach. The specificity of AM404 has been questioned since it has shown to cause an influx of calcium into a variety
at a concentration of 1 µM; this effect was not blocked by either the CB1 receptor
g a 96 h incubation, it was found that both AM404 and its structurally relat
of cells (Chen et al., 2001) at concentrations used for AEA uptake studies, which may be due to the fact that the compound is an agonist at TRPV1 receptors (Zygmunt et al., 1999; 2000; Jordt & Julius, 2002). More recently Kelley & Thayer (2004) reported that AM404 affected calcium spiking in rat hippocampal neurons
antagonist rimonabant, the TRPV1 antagonist capsazepine, or by treatment of pertussis toxin.
Followined analogue VDM11 inhibited, in a concentration dependent manner, the
proliferation of C6 glioma cells. This was also seen with PCA. In contrast, PIA did not inhibit cell proliferation at all at daily added concentrations of 1, 3 and 10 µM, and produced only a small effect at 30 µM. These data are summarised in Fig. 9A together with data performed at the same time with OEA. The dramatic inhibition seen at 30 µM of this compound may be related to the ability of this compound to inhibit ceramidase (Sugita et al., 1975).
AM404, VDM11 and PIA were also tested for their ability to inhibit cell
proliferation at different time points. The dose and time response data showed that both AM404 and VDM11 exerted a major effect upon cell proliferation at concentrations of 10 and 30 µM at all time points investigated (24, 48, 72 and 96 h after administration). At 3 µM, the same pattern was seen for VDM11 whereas AM404 had a very small (albeit significant) effect. At 1 µM, a small, but significant inhibition of cell proliferation could be seen after the 72 h treatment with both of these compounds. PIA did not affect cell proliferation at all when tested at 1, 3 and 10 µM at any time point. At the highest concentration tested, 30 µM, PIA showed a significant reduction after 24 h, although this effect was not as pronounced as with the other AEA uptake inhibitors. These results indicate that AM404 and
51

Kent-Olov Jonsson
VDM11 indeed affect cell proliferation at the same concentrations used for AEA uptake studies (De Petrocellis et al., 2000), and to a larger extent than is seen for PIA.
To evaluate further the mechanism behind the cell proliferation inhibiting effects found, AM404, VDM11, PCA and PIA were incubated at concentrations that affected cell proliferation with a mixture of cannabinoid (AM251+AM630) and TRPV1 (capsazepine) receptor antagonists or α-tocopherol, an antioxidant. The antiproliferative effects could not be antagonised by these treatments, except in the case of 30 µM PCA, where a partial block was seen with α-tocopherol. Since apoptotic cell death can be induced by uncontrolled calcium influx (see Kim et al., 2002 for example) and AM404 induces a rise in calcium influx (Chen et al., 2001), we also wanted to test if the antiproliferative effects could be a due to induction of apoptosis. In this respect, we tested AM404, VDM11 and PIA upon their ability to
positive control supplied with the kit (camptothecin treated U937 en the treated and untreated cells in
caspase ac
ition per se were included.
activate caspases, which is an initial event in apoptosis. Very low levels of caspase activity were seen after either 48 or 96 h treatment with the compounds (<2.5 %), compared to the cells). Although there was a difference betwe
tivity, this difference essentially reflected the cell content of the wells, which was measured in parallel plates using the same treatment.
In addition to the effects per se of the compounds, they were tested upon their
ability to reduce the effect of exogenously administered AEA in this system. AEA has previously been shown to produce antiproliferative effects in these cells in a TRPV1 mediated way (Jacobsson et al., 2001). Given by the fact that the AEA binding site for TRPV1 receptors is intracellularly located (De Petrocellis et al, 2001; Jordt & Julius, 2002), a hindering of AEA uptake would decrease the antiproliferative effects of AEA. Indeed, such an effect was seen when AM404, VDM11 or PIA was administered together with different concentrations of AEA and incubated for 96 h. The uptake inhibitors were tested at concentrations where they showed no or only a small effect on proliferation per se, and examples of the effects upon 2 µM AEA are given in Fig. 9B. The compounds showed a similar pattern in their way of inhibiting the antiproliferative effects of AEA. It should be noted that a large variability was seen upon the effects of AEA, and since the aim was to investigate whether AM404, VDM11 and PIA could decrease these effects, only those experiments where AEA produced an inhib
52

Pharmacology of Palmitoylethanolamide and Related Compounds
This variability has been subsequently investigated in detail and been shown nbe related to analogue show
ot to the lipophilicity of AEA, since its water soluble phosphate ester s the same variability (Fowler et al., 2003b).
inding that CB2 receptor agonists, together with PEA, can affect mast cell dependent inflammation in different models which seem
Fig. 9. (A) Effects of PEA and AEA related compounds upon the proliferation of C6 rat glioma cells. The cell density was measured after 96 h. Data are expressed as % of cells treated with vehicle. Data are means±s.e.mean and are taken from Paper III. (B) Effects of AM404, VDM11 and PIA upon the antiproliferative effects of 2 µM AEA. Data are means±s.e.mean and are taken from figure 4 of Paper III.
Effects of PIA and JWH133 upon inflammation in vivo and in
vitro (Papers IV and V)
After the initial identification and characterisation of potential analogues from Paper I-III, we wanted to test if PIA could be effective in vivo. To establish an in vivo system for this purpose, we set up a model of mast cell dependent inflammation. This was based on the f
s, at least to some extent, to involve CB2 receptors (Malan et al., 2001a; Aloe et al., 1993; Mazzari et al., 1996; Hanus et al., 1999). As a reference the CB2 receptor agonist, JWH133 (Huffman et al., 1999) was used.
The first set of experiments showed that 20 or 200 µg but not 600 µg
(corresponding to 1, 10 and 30 mg/kg) JWH133 was significantly effective in reducing compound 48/80-induced plasma extravasation when the CB2 agonist was i.p. administered in an ethanol vehicle (Fig. 10A). To investigate if CB2 receptors were involved in this response, the CB2 receptor antagonist/inverse agonist SR144528 was administered i.p. prior to JWH133. In this set of experiment, however, the effect of JWH133 did not reach statistical significance
1 3 10 30 1 3 10 30 1 3 10 30 1 3 10 30 1 3 10 30
0
25
50
75
100
AAM 404 VDM 11 PCA PIA OEA
(µM)
Cel
l den
sity
(% o
f con
trol
)
0
25
50
75
100
AEA (2µM)
AEA + PIA (3µM)
AEA + VDM11 (1µM)AEA + AM404 (1µM)
B
Cel
l den
sity
(% o
f con
trol
)
53

Kent-Olov Jonsson
when injected alone at a dose of 200 µg, although the pattern was the same as in the initial experiment (Fig. 10B). Concerning the effects of SR144528, the results are hard to interpret in terms of antagonising the effects of JWH133, since the SR1
e of carrier. As a con quence, PIA and PEA were dissolved in a preparation of eth
44528 itself seemed to decrease the response of compound 48/80 (Fig. 10B). Although this is at first sight somewhat an unexpected finding, it is in line with data by Iwamura and co-workers, who found that SR144528 decreased carrageenan-induced paw oedema in mice (Iwamura et al., 2001). Similar results were also found in a recent study, were topical application of SR144528 reduced TPA (tetradecanoylphorbol acetate)-induced ear swelling as well as leukotriene B4 production and neutrophil infiltration in the mouse ear (Sugiura et al., 2004). Smith et al (2001) also reported that SR144528 reduced neutrophil migration in a mouse peritonitis model, and cited preliminary data that this was also seen in a CB1/ CB2 double knockout, indicating a CB1 and CB2 receptor independent effect.
In the case of PIA, as well as PEA (data not shown), initially no effects were
seen on plasma extravasation when the compounds were administered in an ethanol vehicle (Fig. 10C). Given the fact that these compounds are highly lipophilic, and that PEA has proven effective in other mast cell models, we wanted to test if the lack of effect in this model was due to the choic
seanol:Cremophor®:saline (1:1:18) instead. In this case, both treatments seemed
to decrease the compound 48/80-induced plasma extravasation, although this was not found to be statistically significant due to one animal in each PIA treated group which did not respond to the treatment (Fig. 10D). One explanation could be that the injection of PIA in these animals did not result in a systemic administration of the drug, although there is no proof for this concept. If this is a plausible explanation, the present data are in line with the finding by Mazzari et al. (1996) that PEA is effective in reducing mast cell dependent inflammation in vivo, and that PIA and JWH133 produces similar effects.
As a complement to the effects of the compounds in vivo, we wanted to evaluate if any effects could be seen using an in vitro system. Instead of using mast cell lines or isolated mast cells, in which the cells ability to respond is altered (Robinson et al., 1989; Sweiter et al., 1993; Hamawy et al., 1994; Ra et al., 1994), we chose to establish a skin tissue model where the cells are surrounded by a natural environment. When the mouse paw skin tissue was stimulated by compound
54

Pharmacology of Palmitoylethanolamide and Related Compounds
48/80 a significant release of the granular enzyme β-hexosaminidase was seen, although this was not followed by a detectable production of the cytokine TNF-α. Pre-treatment of the skin tissue with JWH133, PIA or PEA did not alter the basal release of β-hexosaminidase.
To investigate if CB2 receptors were present in skin tissue, PCR and
immunohistochemical experiments also were undertaken. The PCR experiments indicated that the skin samples expressed mRNA for the CB2 receptor. Immunoreactivity towards the CB2 receptor antibody used was detected in the epidermal rather than the dermal layers.
al values, taken from Paper IV and V, are shown. Statistically significant differences in one-way ANOVA followed by Dun
Fig. 10. Effects of JWH133, PIA, PEA and SR144528 upon compound 48/80-induced plasma extravasation in mouse ear pinna. The individu
nett’s multiple comparisons post-hoc test are indicated as **P<0.01 and *P<0.05 when compared with vehicle treatment.
vehicle 20 200 6000.0
0.5
1.0
1.5
2.0
2.5
* **
A
xtra
vasa
tion
s bl
ue, µ
g/m
l)
[JWH133] (µg)
E(E
van
vehicle JWH133 (200) vehicle JWH133 (200)0
1
2
3
4 Control SR144528Bxt
rava
satio
nva
ns b
lue,
µg/
ml)
Treatm ent (µg)
E(E
Vehicle 20 200 6000
1
2
3
[PIA] (µg)
Extr
avas
atio
n(E
vans
blu
e, µ
g/m
l)
C
Vehicle PIA (20) PIA (200) PEA (200)0
1
2
3
4
Pretreatm ent (µg)
Extr
avas
atio
n(E
vans
blu
e, µ
g/m
l)
D
55

Kent-Olov Jonsson
Main conclusions from Papers IV and V
Perhaps the most important conclusion to be made is that the ”apparently simple” (see above) model for inflammation chosen turned out to be far from simple, with both a large variation in response and the choice of vehicle producing problems of their own. The most robust findings of the studies are that JWH133 reduces the oedema response to compound 48/80 in vivo but does not affect the release of the granular enzyme ß-hexosaminidase in response to this agent in the skin slices, and that mRNA for the CB2 receptor is present in the skin samples. Our interpretation of the present data is that, at least for JWH133, the action of this compound is not directly upon mast cells, but rather an indirect one, presumably mediated by CB2 receptors. Our data would suggest that PIA is also able to modify mast cell function in vivo, although the reason for the non-responsiveness of two of the animals is not clear.
56

Pharmacology of Palmitoylethanolamide and Related Compounds
GENERAL CONCLUSIONS AND FUTURE
PERSPECTIVES
The present thesis has been undertaken with the main goal of identifying and cha terising selective FAAH inhibitors. The main conclusions that can be drawn are:
electivity for FAAH versus CB receptors, making them suitable as pharmacological research tools.
• The lead compound, PIA, shows ability to hinder cellular accumulation of AEA, and experimental data also suggest that the total effect upon AEA degradation equals other known AEA uptake inhibitors, with the advantage of showing less effects not related to FAAH inhibition or AEA uptake.
• JWH133, a CB2-selective agonist, can reduce plasma extravasation in a mouse model of mast cell dependent inflammation and PIA may be active in a similar way.
A number of additional observations can be made. For example, as stated in the introduction, it is not clear how AEA is accumulated intracellularly, which makes is somewhat difficult to interpret uptake data. Whatever mechanism is
Lago et al., 2002; 2004). This would suggest that compounds that can inhibit both AEA metabolism and accumulation may be more effective than compounds acting on each process per se. Some of the compounds studied in this thesis appear to be able to inhibit both processes, but further studies will have to be conducted before definitive conclusions can be made. The in vivo data in the thesis suggests that PIA could be effective against mast cell dependent
rac
• The compounds, which where chemically based on the endogenous anti-inflammatory fatty acid PEA, may not be the most potent FAAH inhibitors developed, but show s
responsible for AEA uptake into cells, there are data suggesting that compounds like URB707 and OMDM-2 that inhibit AEA uptake, but do not inhibit FAAH (López-Rodríguez et al., 2003; Ortar et al., 2003), are still effective in potentiating the effects of AEA in vivo (De
57

Kent-Olov Jonsson
inflammation, although, as noted earlieHowever, PEA, which is a well known
r, the results did not reach significance. anti-inflammatory compound, did not
pro
ed to CB1 agonists that they lack the psychotropic effects mediated by the CB1 receptor. Our data showed the presence of CB2 mRNA in skin and suggested that the location of the receptors was in the epidermis, although this conclusion is dependent upon the true specificity of the antibody used. Thus, more studies are needed to confirm the location of functional CB2 receptors present in skin tissue. Another approach to examine if the anti-inflammatory effects seen with JWH133 are due to CB2 receptor activation is to test the compound in CB2 (-/-) mice which, to my knowledge, has not been performed with any CB2 agonist so far.
What could be future approaches? A major drawback with the compounds
identified in this thesis is the limited solubility, hampering their usefulness both in vitro and in vivo. This is a general phenomenon, when dealing with lipophilic compounds such as the cannabinoids. A future approach would be to develop less lipophilic substances, to decrease these issues. If FAAH inhibition, AEA uptake inhibition and CB2 activation is beneficiary, a possible chemical approach would be to develop compounds with combined activity at these targets.
Although there are many unsolved questions, and whatever the future holds in
its hands, the present work has hopefully contributed to an increased knowledge in the continuously growing scientific field of cannabinoids.
duce a significant response either, indicating that the in vivo model itself was troublesome, due to large variability. More investigations are therefore needed to establish the usefulness of PIA in vivo. If, for the sake of argument, an effect was seen with PIA, what is the mechanism responsible for its actions in vivo? Even if the first data clearly showed a FAAH and AEA uptake inhibiting role of PIA in vitro it is possible that other mechanisms could be responsible for its actions in vivo. For example, a recent study attributed the anti-inflammatory effects of PEA to activation of PPAR-α, although this is the only study published so far in this respect (Lo Verme et al., 2005). The same mechanism could operate here as well, and this hypothesis should be tested.
A more clear-cut result of this study (and of others) is the effectiveness of CB2 agonists in vivo, which have the advantage compar
58

Pharmacology of Palmitoylethanolamide and Related Compounds
ACKNOWLEDGEMENTS
ainly performed at the DThe work in this thesis was m epartment of Pharmacology and Clini
reach this point. I wish to express my sincere gratitude to all those who have contributed during this journey. In particular, I wish to thank the following people: First I supervisendocan ent and ideas, and for keeping me off the streets! Dr Gupharmac Dr Stig son, for critical discussions concering science, teaching and technical issues, which have been very valuable during this work. Dr SéveBelgium peration, which have been a foundation for this thesis. I al
cal Neuroscience at Umeå University, Umeå, Sweden. During my years as a graduate student I have met several people who have helped me to
would like to express my gratitude to Professor Chris Fowler, my or, who has wakened my interest for the scientific field of nabinoids. Thank you for your never-ending encouragem
nnar Tiger, my co-supervisor, for fruitful discussions concering ology in general and for making the department a funnier place on earth.
Jacobs
rine Vandevoorde and Dr Didier Lambert, our co-workers in Brussels, , for fruitful co-o
so would like to thank Dr Ruth Ross in Aberdeen, for giving me the chance to explore Scottish science in her lab. Douglas McHugh, my friend from Aberdeen, who I had the opportunity to meet during my stay in Scotland. Thank you for helping me out in the lab as well as showing me around outside the University! Also thanks to all other people at the lab in Aberdeen for making my stay enjoyable. Professor Torbjörn Egelrud and Dr Annelii Ny for help with the immunohistochemistry experiments.
59

Kent-Olov Jonsson
Olov Nilsson and Sandra Holt, PhD students at the department, for scientific and teaching discussions. I also want to thank the rest of the members of the department, especially Britt Jacobsson and Ingrid Persson, who have helped me with some scientific work, and also Emma Söderström, Alf Olsson and Björn Carlsson for help with all non-scientific issues at the department. I also would like to thank all my friends who have, although not always in a direct way, contributed to this thesis. Especially Tobias Holmberg, a garrulous, singing friend, Dr Jörgen Kajberg, another “shy and quiet” friend and Dr Bengt Wikman, a friend who built an excellent mobile sauna. I am also very grateful to my family, for giving me support through life and always being helpful. Finally, Emma, thank you for being part of my life and helping me with everything, especially during the last year. I am indeed also grateful for your scientific contribution to this thesis.
60

Pharmacology of Palmitoylethanolamide and Related Compounds
REFERENCES
Ackermann L, Harvima IT (1998) Mast cells of psoriatic and atopic dermatitis skin are positive for TNF-alpha and their degranulation is associated with expression of ICAM-1 in the epidermis. Arch Dermatol Res 290: 353-359
mastocyte behaviour. Agents Actions 39: C145-147
nthesis of fatty acid amides. J Biol Chem 241: 1308–1313
) Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature 404:
A, Khanolkar A, Layward L, Fezza F, Bisogno T, Di Marzo V (2001) Endocannabinoids control
f cannabis. Lancet Neurol 2: 291-298
nous cannabinoid system in the formalin test of persistent pain in the rat. Eur J Pharmacol 396: 85-92
gente V (1998) Effects of cannabinoid receptor ligands on LPS-induced pulmonary inflammation in mice. Life Sci 63: PL125-
M, Schwab S, Schmid HH (2004) Massive accumulation of N-acylethanolamines after stroke. Cell signalling in acute cerebral ischemia? J Neurochem 88: 1159-1167
Alger BE (2002) Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol 68: 247-286
Aloe L, Leon A, Montalcini RL (1993) A proposed autacoid mechanism controlling
Ates M, Hamza M, Seidel K, Kotalla CE, Ledent C, Gühring H (2003) Intrathecally applied flurbiprofen produces an endocannabinoid-dependent antinociception in the rat formalin test. Eur J Neurosci 17: 597-604
achur NR, Udenfriend S (1966) Microsomal syB
Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Huffman JW, Layward L (2000
84-87
Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Makriyannis
spasticity in a multiple sclerosis model. FASEB J 15: 300-302
Baker D, Pryce G, Giovannoni G, Thompson AJ (2003) The therapeutic potential o
Beaulieu P, Bisogno T, Punwar S, Farquhar-Smith WP, Ambrosino G, Di Marzo V, Rice ASC (2000) Role of the endoge
Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D (1997) Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science 277: 1094-1097
Berdyshev E, Biochot E, Corbel M, Germain N, La
129
erger C, Schmid PC, Schabitz WR, Wolf B
61

Kent-Olov Jonsson
Berman JS, Symonds C, Birch R (2004) Efficacy of two cannabis based medicinal extracts for relief of central neuropathic pain from brachial plexus avulsion: results of a randomised controlled trial. Pain 112: 299-306
Bisogno T, Maccarrone M, De Petrocellis L, Jarrahian A, Finazzi-Agrò A, Hillard C, Di Marzo V (2001) The uptake by cells of 2-arachidonoylglycerol, an endogenous agonist of cannabinoid receptors. Eur J Biochem 268: 1982-1989
VV, De Petrocellis L, Di
Biochem J 351: 817-824
P, Fecik RA, Miyauchi H, Wilkie GD, Austin BJ, Patricelli MP, Cravatt BF (2000b) Exceptionally potent inhibitors of fatty acid amide hydrolase: the enzyme responsible for degradation of endogenous oleamide and anandamide. Proc Natl Acad Sci U S A 97: 5044-5049
Boldrup L, Wilson SJ, Barbier AJ, Fowler CJ (2004) A simple stopped assay for fatty acid amide hydrolase avoiding the use of a chloroform extraction phase. J Biochem Biophys Methods 60: 171-177
Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF (2002) Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science 298: 1793-1796
Burstein SH, Karst M, Schneider U, Zurier RB (2004) Ajulemic acid: A novel cannabinoid produces analgesia without a "high". Life Sci 75: 1513-1522
Calignano A, La Rana G, Giuffrida A, Piomelli D (1998) Control of pain initiation by endogenous cannabinoids. Nature 394: 277-281
Calignano A, La Rana G, Piomelli D (2001) Antinociceptive activity of the endogenous fatty acid amide, palmitylethanolamide. Eur J Pharmacol 419: 191-198
Carlini EA (2004) The good and the bad effects of (-) trans-delta-9-tetrahydrocannabinol (Delta 9-THC) on humans. Toxicon 44: 461-467
Bisogno T, Maurelli S, Melck D, De Petrocellis L, Di Marzo V (1997) Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J Biol Chem 2: 3315–3323
Bisogno T, Melck D, Bobrov MYu, Gretskaya NM, Bezuglov Marzo V (2000) N-acyl-dopamines: novel synthetic CB(1) cannabinoid-receptor ligands and inhibitors of anandamide inactivation with cannabimimetic activity in vitro and in vivo.
Boger DL, Fecik RA, Patterson JE, Miyauchi H, Patricelli MP, Cravatt BF (2000a) Fatty acid amide hydrolase substrate specificity. Bioorg Med Chem Lett 10: 2613–2616
Boger DL, Sato H, Lerner AE, Hedrick M
62

Pharmacology of Palmitoylethanolamide and Related Compounds
Chen WC, Huang JK, Cheng JS, T Liu CP, Jan CR (2001) 2+
amide in the mouse model of
Connd the synthetic cannabinoid nabilone in a
Cos agnoni G, Colleoni M (2002) Therapeutic effect of the endogenous
Crav Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH
t degrades neuromodulatory fatty-acid amides.
Crav
tl Acad Sci U S A 101: 10821-10826
tiates hypokinetic and antinociceptive effects of anandamide. Eur J Pharmacol
sai JCR, Chiang AJ, Chou KJ, AM-404 elevates renal intracellular Ca , questioning its selectivity as a pharmacological tool for investigating the anandamide transporter. J Pharmacol Toxicol Meth 45: 195-198
Coburn AF, Graham CE, Haninger J (1954) The effects of egg yolk in diets on anaphylactic arthritis (passive arthus phenomenon) in the guinea pig. J Exp Med 100: 425-435
Compton DR, Martin BR (1997) The effect of the enzyme inhibitor phenylmethylsulfonyl fluoride on the pharmacological effect of anandcannabimimetic activity. J Pharmacol Exp Ther 283: 1138-1143
ti S, Costa B, Colleoni M, Parolaro D, Giagnoni G (2002) Antiinflammatory action of endocannabinoid palmitoylethanolamide amodel of acute inflammation in the rat. Br J Pharmacol. 135: 181-187
ta B, Conti S, Gifatty acid amide, palmitoylethanolamide, in rat acute inflammation: inhibition of nitric oxide and cyclo-oxygenase systems. Br J Pharmacol 137: 413-420
att BF,(2001) Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A 98: 9371-9376
Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB (1996) Molecular characterization of an enzyme thaNature 384: 83–87
att BF, Lichtman AH (2004) The endogenous cannabinoid system and its role in nociceptive behavior. J Neurobiol 61: 149-160
Cravatt BF, Saghatelian A, Hawkins EG, Clement AB, Bracey MH, Lichtman AH (2004) Functional disassociation of the central and peripheral fatty acid amide signaling systems. Proc Na
de Lago E, Fernández-Ruiz J, Ortega-Gutiérrez S, Viso A, López-Rodríguez ML, Ramos JA (2002) UCM707, a potent and selective inhibitor of endocannabinoid uptake, poten449: 99-103
63

Kent-Olov Jonsson
de Lago E, Ligresti A, Ortar G, Morera E, Cabranes. A, Pryce G, Bifulco M, Baker D, Fernandez-Ruiz JJ, Di Marzo V (2004) In vivo pharmacological actions of two novel inhibitors of anandamide cellular uptake. Eur J Pharmacol 484: 249-257
capsaicin-like activity. FEBS
De anandamide at vanilloid VR1 receptors requires facilitated transport
De col 141: 765-774
5.
Deuto its breakdown by fatty-acid
Deuuorides inhibit anandamide metabolism and bind to the
Deuarachidonyl fluorophosphonate: a potent irreversible inhibitor of
Deu mide hydrolase (FAAH).
34: 605-13
De Petrocellis L, Bisogno T, Davis JB, Pertwee RG, Di Marzo V (2000) Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible Lett 483: 52-56
Petrocellis L, Bisogno T, Maccarrone M, Davis JB, Finazzi-Agrò A, Di Marzo V (2001) The activity ofacross the cell membrane and is limited by intracellular metabolism. J Biol Chem 276): 12856-12863
Petrocellis L, Cascio MG, Di Marzo V (2004) The endocannabinoid system: a general view and latest additions. Br J Pharma
Desarnaud F, Cadas H, Piomelli D (1995) Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. J Biol Chem 270: 6030–603
Deutsch DG, Chin SA (1993) Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem Pharmacol 46: 791–796
tsch DG, Glaser ST, Howell JM, Kunz JS, Puffenbarger RA, Hillard CJ, Abumrad N (2001) The cellular uptake of anandamide is coupled amide hydrolase. J Biol Chem 276: 6967-6973
tsch DG, Lin S, Hill WA, Morse KL, Salehani D, Arreaza G, Omeir RL, Makriyannis A (1997a) Fatty acid sulfonyl flcannabinoid receptor. Biochem Biophys Res Commun 231: 217–221
tsch DG, Omeir R, Arreaza G, Salehani D, Prestwich GD, Huang Z, Howlett A (1997b) Methyl anandamide amidase. Biochem Pharmacol 53: 255-260
tsch DG, Ueda N, Yamamoto S (2002) The fatty acid aProstaglandins Leukot Essent Fatty Acids 66: 201-210 Erratum in: Prostaglandins Leukot Essent Fatty Acids 2003 68: 69
Devane WA, Dysarz FA, Johnson RM, Melvin LS, Howlett AC (1988) Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol
64

Pharmacology of Palmitoylethanolamide and Related Compounds
Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LG, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258: 1946-1949
Marzo V, Bifulco M, De PDi etrocellis L (2004) The endocannabinoid system and its
Di Ml Sci 22: 346-349
ptor "hybrid" ligands.
Di M
Di M of anandamide and
Di Mand inactivation of endogenous cannabinoid anandamide in central
Di Mos G (2001c) Leptin-regulated endocannabinoids are involved in
Din S, Nagy I, Cruz F (2004) Anandamide-
Eger expression in the mouse brain: evidence of a
therapeutic exploitation. Nat Rev Drug Discov 3: 771-784
arzo V, Bisogno T, De Petrocellis L (2001a) Anandamide: some like it hot. Trends Pharmaco
Di Marzo V, Bisogno T, De Petrocellis L, Brandi I, Jefferson RG, Winckler RL, Davis JB, Dasse O, Mahadevan A, Razdan RK, Martin BR (2001b) Highly selective CB1 cannabinoid receptor ligands and novel CB1/VR1 vanilloid receBiochem Biophys Res Commun 281: 444-451
arzo V, Blumberg PM, Szallasi A (2002a) Endovanilloid signaling in pain. Curr Opin Neurobiol 12: 372-379
Di Marzo V, De Petrocellis L, Fezza F, Ligresti A, Bisogno T (2002b) Anandamide receptors. Prostaglandins Leukot Essent Fatty Acids 66: 377-391
arzo V, De Petrocellis L, Sepe N, Buono A (1996) Biosynthesisrelated acylethanolamides in mouse J774 macrophages and N18 neuroblastoma cells. Biochem J. 316: 977-984
arzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D (1994) Formation neurons. Nature 372: 686-691
arzo V, Goparaju SK, Wang L, Liu J, Batkai S, Jarai Z, Fezza F, Miura GI, Palmiter RD, Sugiura T, Kunmaintaining food intake. Nature 410: 822-825
is P, Charrua A, Avelino A, Yaqoob M, Bevan evoked activation of vanilloid receptor 1 contributes to the development of bladder hyperreflexia and nociceptive transmission to spinal dorsal horn neurons in cystitis. J Neurosci 24: 11253-11263
Dogrul A, Gul H, Akar A, Yildiz O, Bilgin F, Guzeldemir E (2003) Topical cannabinoid antinociception: synergy with spinal sites. Pain 105: 11-16
rtová M, Cravatt BF, Elphick MR (2003) Comparative analysis of fatty acid amide hydrolase and cb(1) cannabinoid recepto
65

Kent-Olov Jonsson
widespread role for fatty acid amide hydrolase in regulation of endocannabinoid signaling. Neuroscience 119: 481-496
rtová M, Giang DK, Cravatt BF, Elphick MR (1998) A new peEge rspective on
Ellin calcitonin gene-related peptide (CGRP) release from the isolated paw
Elm
urons in naïve rats and in rat models of inflammatory and neuropathic
Facc on A (1995) Mast cells express
he urinary bladder.
Fedth multiple
Fegl597: Effects on
Fegl kriyannis A, Piomelli D (2004)
N-acyl ethanolamine breakdown as therapeutic strategies to avoid pyschotropic effects. Brain Res Brain Res Rev 41: 26-43
cannabinoid signalling: complementary localization of fatty acid amide hydrolase, and the CB1 receptor in rat brain. Proc R Soc Lond B Biol Sci 265: 2081–2085
gton HC, Cotter MA, Cameron NE, Ross RA (2002) The effect of cannabinoids on capsaicin-evokedskin of diabetic and non-diabetic rats. Neuropharmacology 42: 966-975
es SJR, Jhaveri MD, Smart D, Kendall DA, Chapman V (2004) Cannabinoid CB2 receptor activation inhibits mechanically evoked responses of wide dynamic range dorsal horn nepain. Eur J Neurosci 20: 2311-2320
i L, Dal Toso R, Romanello S, Buriani A, Skaper SD, Lea peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc Natl Acad Sci U S A 92: 3376-3380
Farquhar-Smith WP, Rice ASC (2001) Administration of endocannabinoids prevents a referred hyperalgesia associated with inflammation of tAnesthesiology 94: 507-513
orova I, Hashimoto A, Fecik RA, Hedrick MP, Hanus LO, Boger DL, Rice KC, Basile AS (2001)Behavioral evidence for the interaction of oleamide wineurotransmitter systems.J Pharmacol Exp Ther 299: 332-342
ey D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D (2005) Characterization of the fatty-acid amide hydrolase inhibitor URBanandamide and oleoylethanolamide deactivation. J Pharmacol Exp Ther (Fast Forward. Published on December 3, 2004 as DOI:10.1124/jpet.104.078980)
ey D, Kathuria S, Mercier R, Li C, Goutopoulos A, MaAnandamide transport is independent of fatty-acid amide hydrolase activity and is blocked by the hydrolysis-resistant inhibitor AM1172. Proc Natl Acad Sci 101: 8756-8761
Fowler CJ (2003) Plant-derived, synthetic and endogenous cannabinoids as neuroprotective agents. Non-psychoactive cannabinoids, 'entourage' compounds and inhibitors of
66

Pharmacology of Palmitoylethanolamide and Related Compounds
Fowler CJ (2004a) Possible involvement of the endocannabinoid system in the actions of three clinically used drugs. Trends Pharmacol Sci 25: 59-61
ler CJ (2004b) Metabolism of the endocannabinoids anandamide and 2-arFow achidonoyl
seases and pain. Curr Med
Fow (2000) Differences in the pharmacological properties of
Fow
hydrolase in a pH-dependent manner. J Enzyme Inhib Med
Fow arachidonoylglycerol
Fow ersson A, Juntunen J, Jarvinen T, Vandevoorde S, Lambert
onse and involvement of arachidonic acid. Biochem
Fowyme hydrolyzing anandamide, 2-
Fowellular uptake versus enzymatic hydrolysis--a difficult issue
Fow s rat brain deamidation of
glycerol, a review, with emphasis on the pharmacology of fatty acid amide hydrolase, a possible target for the treatment of neurodegenerative diChem 4: 161-174
ler CJ, Börjesson M, Tiger Grat and chicken brain fatty acid amidohydrolase. Br J Pharmacol 131: 498-504
ler CJ, Holt S, Nilsson O, Jonsson KO, Tiger G, Jacobsson SOP (2005) The endocannabinoid signaling system: pharmacological and therapeutic aspects. Pharmacol Biochem Behav, in press
Fowler CJ, Holt S, Tiger G (2003) Acidic nonsteroidal anti-inflammatory drugs inhibit rat brain fatty acid amide Chem 18: 55-58
ler CJ, Jacobsson SOP (2002) Cellular transport of anandamide, 2-and palmitoylethanolamide--targets for drug development? Prostaglandins Leukot Essent Fatty Acids. 66: 193-200
ler CJ, Jonsson KO, AndDM, Jerman JC, Smart D (2003) Inhibition of C6 glioma cell proliferation by anandamide, 1-arachidonoylglycerol, and by a water soluble phosphate ester of anandamide: variability in respPharmacol 66: 757-767
ler CJ, Jonsson KO, Tiger G (2001) Fatty acid amide hydrolase: biochemistry, pharmacology, and therapeutic possibilities for an enzarachidonoylglycerol, palmitoylethanolamide, and oleamide. Biochem Pharmacol 62: 517-526
ler CJ, Tiger G, Ligresti A, López-Rodríguez ML, Di Marzo V (2004) Selective inhibition of anandamide cto handle. Eur J Pharmacol 492: 1-11
ler CJ, Tiger G, Stenström A (1997) Ibuprofen inhibitanandamide at pharmacologically relevant concentrations. Mode of inhibition and structure-activity relationship. J Pharmacol Exp Ther 283: 729-734
67

Kent-Olov Jonsson
Franer focal cerebral ischemia and potentiates
Fridal activity
Frid
Gao nthesis of an active
Geracol 140: 781-789
Gib 001) Human skin mast
Glas
Gop
em Pharmacol 57: 417–423
klin A, Parmentier-Batteur S, Walter L, Greenberg DA, Stella N (2003) Palmitoylethanolamide increases aftmicroglial cell motility. J Neurosci 23: 7767-7775
e E, Feigin C, Ponde DE, Breuer A, Hanus L, Arshavsky N, Mechoulam R (2004) (+)-Cannabidiol analogues which bind cannabinoid receptors but exert peripheronly. Eur J Pharmacol 506: 179-188
e E, Mechoulam R (1993) Pharmacological activity of the cannabinoid receptor agonist, anandamide, a brain constituent. Eur J Pharmacol 231:313-314
Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodríguez De Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D (2003) Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α. Nature 425: 90-93
Gaetani S, Cuomo V, Piomelli D (2003) Anandamide hydrolysis: a new target for anti-anxiety drugs? Trends Mol Med 9: 474-478
ni Y, Mechoulam R (1964) Isolation, structure, and partial syconstituent of hashish. J Am Chem Soc 86: 1646-1647
deman GL, Lovinger DM (2003) Emerging roles for endocannabinoids in long-term synaptic plasticity. Br J Pharm
Giang DK, Cravatt BF (1997) Molecular characterization of human and mouse fatty acid amide hydrolases. Proc Natl Acad Sci U S A 94: 2238-2242
bs BF, Wierecky J, Welker P, Henz BM, Wolff HH, Grabbe J (2cells rapidly release preformed and newly generated TNF-alpha and IL-8 following stimulation with anti-IgE and other secretagogues. Exp Dermatol 10: 312-320
er S, Abumrad N, Fatade F, Kaczocha M, Studholme K, Deutsch D (2003) Evidence against the presence of an anandamide transporter. Proc Natl Acad Sci U S A 100: 4269-4274
araju SK, Ueda N, Taniguchi K, Yamamoto S (1999) Enzymes of porcine brain hydrolyzing 2-arachidonoylglycerol, an endogenous ligand of cannabinoid receptors. Bioch
Gühring H, Hamza M, Sergejeva M, Ates M, Kotalla CE, Ledent C, Brune K (2002) A role for endocannabinoids in indomethacin-induced spinal antinociception. Eur J Pharmacol 454: 153-163
68

Pharmacology of Palmitoylethanolamide and Related Compounds
Guzmán M, Galve-Roperh I, Sanchez C (2001) Ceramide: a new second messenger of cannabinoid action. Trends Pharmacol Sci 22(: 19-22
Hamawy MM, Mergenhagen SE Siraganian RP (1994) Adhesion molecules as regulators of mast-cell and basophil function. Immunol Today 15: 62-66
us L, Abu-Lafi S, Fride E, Breuer A, Vogel Z, Shalev DE, Kustanovich I, Mechoulam R (2001) 2-arachid
Hanonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1
Han G, Ross
Han lam R (1993) Two new unsaturated fatty acid
Hao S, Avraham J, Mechoulam R, Berry EM (2000) Low dose anandamide affects food
Har assay containing sodium dodecyl sulfate in microtiter
Hill
urochem 69: 631-638
HillRG, Campbell WB (1999) Synthesis and characterization of potent and
Hill , Campbell WB (1995) Characterization of the
receptor. Proc Natl Acad Sci U S A 98: 3662-3665
us L, Breuer A, Tchilibon S, Shiloah S, Goldenberg D, Horowitz M, Pertwee RRA, Mechoulam R, Fride E (1999) HU-308: a specific agonist for CB2, a peripheral cannabinoid receptor. Proc Natl Acad Sci U S A 96: 14228-14233
us L, Gopher A, Almog S, Mechouethanolamides in brain that bind to the cannabinoid receptor. J Med Chem 36: 3032-3034
intake, cognitive function, neurotransmitter and corticosterone levels in diet-restricted mice. Eur J Pharmacol 392: 147-156
rington CR (1990) Lowry proteinplates for protein determination on fractions from brain tissue. Analyt Biochem 186: 285-287
ard CJ, Edgemond WS, Jarrahian A, Campbell WB (1997) Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J Ne
Hillard CJ, Jarrahian A (2003) Cellular accumulation of anandamide: consensus and controversy. Br J Pharmacol 140: 802-808
ard CJ, Manna S, Greenberg MJ, DiCamelli R, Ross RA, Stevenson LA, Murphy V, Pertwee selective agonists of the neuronal cannabinoid receptor (CB1). J Pharmacol Exp Ther 289: 1427-1433
ard CJ, Wilkison DM, Edgemond WSkinetics and distribution of N-arachidonylethanolamine (anandamide) hydrolysis by rat brain. Biochim Biophys Acta 1257: 249–256
69

Kent-Olov Jonsson
Hohmann AG, Farthing JN, Zvonok AM, Makriyannis A (2004) Selective activation of cannabinoid CB2 receptors suppresses hyperalgesia evoked by intradermal capsaicin. J Pharmacol Exp Ther 308: 446-453
Holt S, Nilsson J, Omeir R, Tiger G, Fowler CJ (2001) Effects of pH on the inhibition of fatty acid amidohydrolase by ibuprofen. Br J Pharmacol 133: 513-520. Erratum in: Br J Pharmacol 2001 134: 451
Holt S, Rocksen D, Bucht A, Petersen G, Hansen HS, Valenti M, Di Marzo V, Fowler CJ (2004) Lipopolysaccharide-induced pulmonary inflammation is not accompanied by a
Houffice.co.uk/pa/ld199798/ldselect/ldsctech/151/15101.htm]
oulam R, Pertwee RG (2002) International
N, Geppetti P, Walker JM, Di Marzo
99: 8400-8405
receptor. Bioorg Med Chem 7: 2905-2914
neuropathic pain: pain inhibition by receptors not
Iwamrization of JTE-907, a novel selective ligand for cannabinoid
CB2 receptor. J Pharmacol Exp Ther 296: 420-425
release of anandamide into the lavage fluid or a down-regulation of the activity of fatty acid amide hydrolase. Life Sci 76: 461-472
se of Lords, The United Kingdom Parliament (1998) [http://www.parliament.the-stationery-o
Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, MechUnion of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 54: 161-202
Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F, Tognetto M, Petros TJ, Krey JF, Chu CJ, Miller JD, Davies SV (2002) An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci U S A
Huestis MA, Gorelick DA, Heishman SJ, Preston KL, Nelson RA, Moolchan ET, Frank RA (2001) Blockade of effects of smoked marijuana by the CB1-selective cannabinoid receptor antagonist SR141716. Arch Gen Psychiat 58: 322-328
Huffman JW, Liddle J, Yu S, Aung MM, Abood ME, Wiley JL, Martin BR (1999) 3-(1',1'-dimethylbutyl)-1-deoxy-∆8-THC and related compounds: synthesis of selective ligands for the CB2
Ibrahim MM, Deng H, Zvonok A, Cockayne DA, Kwan J, Mata HP, Vanderah TW, Lai J, Porreca F, Makriyannis A, Malan TP (2003) Activation of CB2 cannabinoid receptors by AM1241 inhibits experimentalpresent in the CNS. Proc Natl Acad Sci U S A 100: 10529-10533
ura H, Suzuki H, Ueda Y, Kaya T, Inaba T (2001) In vitro and in vivo pharmacological characte
70

Pharmacology of Palmitoylethanolamide and Related Compounds
Jacobsson SOP, Fowler CJ (2001) Characterization of palmitoylethanolamide transport in mouse Neuro-2a neuroblastoma and rat RBL-2H3 basophilic leukaemia cells:
Jaco glioma cell proliferation by
Jagg he anti-hyperalgesic actions of the
Jarra 0) Structure-activity
Jordt SE, Julius D (2002) Molecular basis for species-specific sensitivity to “hot” chili
Karndamide to cell culture wells as a confounding
Katde amidohydrolase in rat tissues with special reference to small intestine.
Katlmery M, Cuomo V, Piomelli D (2003)
Kilo C, Hargreaves KM, Flores CM (1997) Peripheral CGRP release as a
Kim inhibits mast cell-dependent immediate-type allergic reactions. Pharmacol Res 39: 107-111
comparison with anandamide. Br J Pharmacol 132: 1743–1754.
bsson SOP, Wallin T, Fowler CJ (2001) Inhibition of rat C6 endogenous and synthetic cannabinoids. Relative involvement of cannabinoid and vanilloid receptors. J Pharmacol Exp Ther 299: 951-959
ar SI, Hasnie FS, Sellaturay S, Rice ASC (1998) Tcannabinoid anandamide and the putative CB2 receptor agonist palmitoylethanolamide in visceral and somatic inflammatory pain. Pain 76: 189-199
hian A, Manna S, Edgemond WS, Campbell WB, Hillard CJ (200relationships among N-arachidonoylethanolamine (anandamide) head group analogues for the anandamide transported. J Neurochem 74: 2597-2606
peppers. Cell 108: 421-430
lsson M, Påhlsson C, Fowler CJ (2004) Reversible, temperature-dependent, and AM404-inhibitable adsorption of anafactor in release experiments. Eur J Pharm Sci 22: 181-189
ayama K, Ueda N, Kurahashi Y, Suzuki H, Yamamoto S, Kato I (1997) Distribution of anandamiBiochim Biophys Acta 1347: 212-218
huria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, PaModulation of anxiety through blockade of anandamide hydrolysis. Nature Med 9: 76-81
Kelley BG, Thayer SA (2004) Anandamide transport inhibitor AM404 and structurally related compounds inhibit synaptic transmission between rat hippocampal neurons in culture independent of cannabinoid CB1 receptors. Eur J Pharmacol 96: 33-39
S, Harding-Rosemarker for neurogenic inflammation: a model system for the study of neuropeptide secretion in rat paw skin. Pain 73: 201-207
HM, Yi JM, Lim KS (1999) Magnoliae flos
71

Kent-Olov Jonsson
Kim M-J, Jo D-G, Hong G-S, Kim BJ, Lai BJ, Cho D-H, Kim K-W, Bandyopadhyay A, Hong Y-M, Kim DH, Cho C, Liu JO, Snyder SH, Jung Y-K (2002) Calpain-dependent cleavage of cain/cabin1 activates calcineurin to mediate calcium-triggered cell death.
Kones including N-arachidonoylethanolamine (anandamide) in
Kou
Kumar RN, Chambers WA, Pertwee RG (2001) Pharmacological actions and therapeutic
Lam
l ligands
Lame endocannabinoid, in
Lan amidase activity in rat brain
Lan
02
Proc Natl Acad Sci U S A 99: 9870-9875
do S, Sugiura T, Kodaka T, Kudo N, Waku K, Tokumura A (1998) Accumulation of various N-acylethanolamincadmium chloride-administered rat testis. Arch Biochem Biophys 354: 303-310
tek B, Prestwich GD, Howlett AC, Chin SA, Salehani D, Akhavan N, Deutsch DG (1994) Inhibitors of arachidonoyl ethanolamide hydrolysis. J Biol Chem 269: 22937-22940
Kozak KR, Marnett LJ (2002) Oxidative metabolism of endocannabinoids. Prostaglandins Leukotrienes Ess FattyAcids 66: 211-220
Kuehl FA, Jacob TA, Ganley OH, Ormond RE, Meisinger MAP (1957) The identification of N-(2-hydroxyethyl)-palmitamide as a naturally occurring anti-inflammatory agent. J Am Chem Soc 79: 5577-5578
uses of cannabis and cannabinoids. Anaesthesia 56: 1059-1068
bert DM, DiPaolo FG, Sonveaux P, Kanyonyo M, Govaerts SJ, Hermans E, Bueb J, Delzenne NM, Tschirhart EJ (1999) Analogues and homologues of N-palmitoylethanolamide, a putative endogenous CB2 cannabinoid, as potentiafor the cannabinoid receptors. Biochim Biophys Acta. 1440: 266-274
bert DM, Vandevoorde S, Diependaele G, Govaerts SJ, Robert AR (2001) Anticonvulsant activity of N-palmitoylethanolamide, a putativmice. Epilepsia 42: 321-327
g W, Qin C, Hill WA, Lin S, Khanolkar AD, Makriyannis A (1996) High-performance liquid chromatographic determination of anandamidemicrosomes. Anal Biochem 238: 40–45. Erratum in Anal Biochem (1996) 241: 274
g W, Qin C, Lin S, Khanolkar AD, Goutopoulos A, Fan P, Abouzid K, Meng Z, Biegel D, Makriyannis A (1999) Substrate specificity and stereoselectivity of rat brain microsomal anandamide amidohydrolase. J Med Chem 42: 896 –9
Le Fur G (2004) Clinical results with rimonabant in obesity. International Cannabinoid Research Society 2004 Symposium on the Cannabinoids June 22nd - June 27th, Paestum, Italy
72

Pharmacology of Palmitoylethanolamide and Related Compounds
Leggett JD, Aspley S, Beckett SR, D'Antona AM, Kendall DA, Kendall DA (2004) Oleamide is a selective endogenous agonist of rat and human CB1 cannabinoid receptors. Br J Pharmacol 141: 253-262
Lehr M (1997) Synthesis, biological evaluation, and structure-activity relationships of 3-acylindole-2-carboxylic acids as inhibitors of the cytosolic phospholipase A2. J Med Chem 40: 2694-2705
Lichtman AH, Hawkins EG, Griffin G, Cravatt BF (2002) Pharmacological activity of fatty acid amides is regulated, but not mediated, by fatty acid amide hydrolase in vivo. J Pharmacol Exp Ther 302:73-79
Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, Cravatt BF (2004a) Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J Pharmacol
Lich ani T, Cravatt BF (2004b) Mice lacking fatty acid amide
Lo V A, Piomelli D (2005) The
Lóp utiérrez S, Fowler CJ, Tiger G, de Lago E,
Maccarrone M (2004) Inhibition of anandamide hydrolysis: cells also know how to do it.
Mac
tral nervous system. Mol Cell Neurosci 21:
Exp Ther 311: 441-448
tman AH, Shelton CC, Advhydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain 109: 319-327
erme J, Fu J, Astarita G, La Rana G, Russo R, Calignanonuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol 67: 15-19
ez-Rodríguez ML, Viso A, Ortega-GFernández-Ruiz J, Ramos JA (2003) Design, synthesis and biological evaluation of new endocannabinoid transporter inhibitors: comparison with effects upon fatty acid amidohydrolase. J Med Chem 46: 1512-1522
Trends Mol Med 10: 13-14
carrone M, Bari M, Agro AF (1999) A sensitive and specific radiochromatographic assay of fatty acid amide hydrolase activity. Anal Biochem 267: 314-318
Maccarrone M, Cartoni A, Parolaro D, Margonelli A, Massi P, Bari M, Battista N, Finazzi-Agrò A (2002) Cannabimimetic activity, binding, and degradation of stearoylethanolamide within the mouse cen126-40
73

Kent-Olov Jonsson
Maccarrone M, Lorenzon T, Bari M, Melino G, Finazzi-Agrò A (2000) Anandamide induces apoptosis in human cells via vanilloid receptors. Evidence for a protective role of cannabinoid receptors. J Biol Chem 275: 31938-31945
carrone M, Valensise H, Bari M, LazzarMac in N, Romanini C, Finazzi-Agrò A (2000)
Mac
Maingret F, Patel AJ, Lazdunski M, Honore E (2001) The endocannabinoid anandamide is
-mediated peripheral
Mar
9
Invest 113: 1202-1209
down-modulating mast cell activation. Eur J Pharmacol 300: 227–236
Relation between decreased anandamide hydrolase concentrations in human lymphocytes and miscarriage. Lancet 355: 1326-1329
kie K, Devane WA, Hille B (1993) Anandamide, an endogenous cannabinoid, inhibits calcium currents as a partial agonist in N18 neuroblastoma cells. Mol Pharmacol 44:498-503
a direct and selective blocker of the background K(+) channel TASK-1. EMBO J 20: 47-54
Malan TP, Ibrahim M, Deng H, Makriyannis A (2001) Anti-inflammatory effects of the CB2 cannabinoid receptor agonist AM1241. American Society of Anesthesiologists Annual Meeting Abstracts A-894
Malan, T.P., Ibrahim, M., Deng, H., Liu, Q., Mata, H.P., Vanderah, T., Porreca, F., Makriyannis, A. (2001) CB2 cannabinoid receptorantinociception. Pain 93: 239-245
tin BR, Jefferson R, Winckler R, Wiley JL, Huffman JW, Crocker PJ, Saha B, Razdan RK (1999) Manipulation of the tetrahydrocannabinol side chain delineates agonists, partial agonists, and antagonists. J Pharmacol Exp Ther 290; 1065-107
Massa F, Marsicano G, Hermann H, Cannich A, Monory K, Cravatt BF, Ferri GL, Sibaev A, Storr M, Lutz B (2004) The endogenous cannabinoid system protects against colonic inflammation. J Clin
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346: 561-564
Matsuda S, Kanemitsu N, Nakamura A, Mimura Y, Ueda N, Kurahashi Y, Yamamoto S (1997) Metabolism of anandamide, an endogenous cannabinoid receptor ligand, in porcine ocular tissues. Exp Eye Res 64: 707–711
Mazzari S, Canella R, Petrelli L, Marcolongo G, Leon A (1996) N-(2-Hydroxyethyl) hexadecamide is orally active in reducing edema formation, and inflammatory hyperalgesia by
74

Pharmacology of Palmitoylethanolamide and Related Compounds
McFarland MJ, Barker EL (2004) Anandamide transport. Pharmacol Ther 104: 117-135
houlam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, P
Mecertwee RG, Griffin G, Bayewitch M, Barg J,
Mun bu-Shaar M (1993) Molecular characterization of a peripheral
Nac fos protein expression and pain behavior in a rat
Nat
Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N (2004) Molecular characterization of
Om in S, Hong Y, Ahern DG, Deutsch DG (1995) Arachidonoyl ethanolamide-
Ortaarmacol 65:
Orte
e CB1 receptor to anandamide
Pari K (1996) The uterus is a potential site for anandamide
prod Dev 45:
Pate
s brain N-arachidonylethanolamine (anandamide) content and inhibits fatty acid amide hydrolase. Br J Pharmacol 139: 1005-1013
Vogel Z (1995) Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50: 83-90
ro S, Thomas KL, Areceptor for cannabinoids. Nature 365: 61-65
kley AG, Makriyannis A, Hohmann AG (2003) Selective activation of cannabinoid CB2 receptors suppresses spinalmodel of inflammation. Neuroscience 119: 747-757
arajan V, Schmid P, Reddy P, Schmid H (1984) Catabolism of Nacylethanolamine phospholipids by dog brain preparations. J Neurochem 42: 1613–1619
a phospholipase D generating anandamide and its congeners. J Biol Chem 279: 5298-5305
eir RL, Ch[1,2-14C] as a substrate for anandamide amidase. Life Sci 56: 1999-2005
r G, Ligresti A, De Petrocellis L, Morera E, Di Marzo V (2003) Novel selective and metabolically stable inhibitors of anandamide cellular uptake. Biochem Ph1473-1481
ga-Gutiérrez S, Hawkins EG, Viso A, López-Rodríguez ML, Cravatt BF (2004) Comparison of anandamide transport in FAAH wild-type and knockout neurons: evidence for contributions by both FAAH and thuptake. Biochemistry 43: 8184-8190
a BC, Deutsch DD, Dey Ssynthesis and hydrolysis: differential profiles of anandamide synthase and hydrolase activities in the mouse uterus during the periimplantation period. Mol Re183–192
l S, Wohlfeil ER, Rademacher DJ, Carrier EJ, Perry LJ, Kundu A, Falck JR, Nithipatikom K, Campbell WB, Hillard CJ (2003) The general anesthetic propofol increase
75

Kent-Olov Jonsson
Patricelli MP, Cravatt BF (2001) Characterization and manipulation of the acyl chain selectivity of fatty acid amide hydrolase. Biochemistry 40: 6107-6115
wee RG (1997) Pharmacology of cannabinoid CB1 andPert CB2 receptors. Pharmacol
Pert
Piom signalling. Nature Rev Neurosci
Portr FP, Leese AB, Felder CC (2002) Characterization of a novel
Quartilho A, Mata HP, Ibrahim MM, Vanderah TW, Porreca F, Makriyannis A, Malan TP
Quistad GB, Sparks SE, Segall Y, Nomura DK, Casida JE (2002) Selective inhibitors of
Ra C (1994) Fibronectin receptor integrins are involved
Rakxp Ther 292:
Ran
Rey 1890) Therapeutical uses and toxic effects of cannabis indica. Lancet. March
Rice
Rina
Ther 74: 129-180
wee RG (2001) Cannabinoid receptors and pain. Progr Neurobiol 63: 569-611
elli D (2003) The molecular logic of endocannabinoid4: 873-884
er AC, Sauer JM, Knierman MD, Becker GW, Berna MJ, Bao J, Nomikos GG, Carter P, Bymasteendocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J Pharmacol Exp Ther 301: 1020-1024
(2003) Inhibition of inflammatory hyperalgesia by activation of peripheral CB2 cannabinoid receptors. Anesthesiology 99: 955-960
fatty acid amide hydrolase relative to neuropathy target esterase and acetylcholinesterase: toxicological implications. Toxicol Appl Pharmacol 179: 57-63
, Yasuda M, Yagita H, Okumura Kin mast cell activation. J Allergy Clin Immunol 94: 625-628
hshan F, Day TA, Blakely RD, Barker EL (2000) Carrier-mediated uptake of the endogenous cannabinoid anandamide in RBL-2H3 cells. J Pharmacol E960-967
g HP, Dale MM, Ritter JM, Moore PK (2003) Pharmacology, 5th Edition, Churchill Livingstone, London 491-493
Razdan RK (1986) Structure-activity relationships in cannabinoids. Pharmacol Rev. 38: 75-149
nolds JR (22 637-368
ASC (2001) Cannabinoids and pain. Curr Opin Investig Drugs 2: 399-414
Richardson JD, Kilo S, Hargreaves KM (1998) Cannabinoids reduce hyperalgesia and inflammation via interaction with peripheral CB1 receptors. Pain 75: 111-119
ldi-Carmona M, Barth F, Héaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Néliat G, Caput D, Ferrara P, Soubrié P, Brelière J-C, Le Fur G (1994)
76

Pharmacology of Palmitoylethanolamide and Related Compounds
SR141716A, a potent and selective antagonist of the brain cannabinoid receptor.
Rina
id receptor. J
Rob 1989) The IgE- and calcium-dependent
Rod redo L, Nava F, Fu J, Murillo-
–212
Ryb Identification and characterisation of a novel splice variant of the human CB1
Sand mponents of
Sari erg JM (1983) Evans blue fluorescence: quantitative and morphological
Schm
Schm ria BC, Krebsbach RJ, Schmid HH, Dey SK (1997) Changes in anandamide
Schmn phosphodiesterase of the
phospholipase D type. J Biol Chem 258: 9302-9306
FEBS Letts 350: 240-244
ldi-Carmona M, Barth F, Millan J, Derocq J-M, Casellas P, Congy C, Oustric D, Sarran M, Boulaboula M, Calandra B, Portier M, Shire D, Brelière J-C, Le Fur G (1998) SR 144528, the first potent and selective antagonist of the CB2 cannabinoPharmacol Exp Ther 284: 644-650
inson C, Benyon C, Holgate ST, Church MK (release of eicosanoids and histamine from human purified cutaneous mast cells. J Invest Derm 93: 397-404
ríguez de Fonseca F, Navarro M, Gómez R, EscuRodríguez E, Giuffrida A, LoVerme J, Gaetani S, Kathuria S, Gall C, Piomelli, D (2001) An anorexic lipid mediator regulated by feeding. Nature 414: 209
Ross RA (2003) Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol 140: 790-801
erg E, Vu HK, Larsson N, Groblewski T, Hjorth S, Elebring T, Sjogren S, Greasley PJ (2005)receptor. FEBS Lett 579: 259-264
berg A, Fowler CJ (2005) Measurement of saturable and non-saturable coanandamide uptake into P19 embryonic carcinoma cells in the presence of fatty acid-free bovine serum albumin. Chem Phys Lipid, in press
a A, Lundbevaluation of vascular permeability in animal tissues. J Neurosci Methods 8: 41-49
id HH, Berdyshev EV (2002) Cannabinoid receptor-inactive N-acylethanolamines and other fatty acid amides: metabolism and function. Prostaglandins Leukot Essent Fatty Acids 66: 363-376
Schmid PC, Krebsbach RJ, Perry SR, Dettmer TM, Maasson JL, Schmid HH (1995) Occurrence and postmortem generation of anandamide and other long-chain N-acylethanolamines in mammalian brain. FEBS Lett 375: 117-120
id PC, Palevels in mouse uterus are associated with uterine receptivity for embryo implantation. Proc Natl Acad Sci U S A 94: 4188-4192
id PC, Reddy PV, Natarajan V, Schmid HHO (1983) Metabolism of N- acylethanolamine phospholipids by a mammalia
77

Kent-Olov Jonsson
Schmid PC, Zuzarte-Augustin ML, Schmid HHO (1985) Properties of rat liver N-acylethanolamine amidohydrolase. J Biol Chem 260: 14 145–14 149
Schwartz LB, Austen KF (1980) Enzymes of the mast cell granule. J Invest Dermatol 74: 349-353
Seldin DC, Adelman S, Austen KF, Stevens RL, Hein A, Caulfield JP, Woodbury RG
cad Sci U S A 82: 3871–3875
Shirl variant of the central cannabinoid receptor
Sma ir AI, Chambers JK, Randall AD,
Sma ts
anandamide metabolism.
Smi Solari R (1997) Rat basophilic leukaemia (RBL) cells overexpressing Rab3a
Smi P, Razdan RK, Mechoulam R, Martin BR (1994) The
rmacol Exp Ther 270: 219-27
receptor ligands in mouse peritonitis models. European Journal of
Biochim
(1985) Homology of the rat basophilic leukemia cell and the rat mucosal mast cell. Proc Natl A
Sheerin AH, Zhang X, Saucier DM, Corcoran ME (2004) Selective antiepileptic effects of N-palmitoylethanolamide, a putative endocannabinoid. Epilepsia 45: 1184-1188
e D, Carillon C, Kaghad M, Calandra B, Rinaldi-Carmona M, Le Fur G, Caput D, Ferrara P (1995) An amino-terminaresulting from alternative splicing. J Biol Chem 270: 3726-3731. Erratum in: J Biol Chem 1996 271: 33706
rt D, Gunthorpe MJ, Jerman JC, Nasir S, Gray J, MuDavis JB (2000) The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1). Br J Pharmacol 129: 227-230
rt D, Jonsson KO, Vandevoorde S, Lambert DM, Fowler CJ (2002) ‘Entourage’ effecof N-acyl ethanolamines at human vanilloid receptors. Comparison of effects upon anandamide-induced vanilloid receptor activation and uponBr J Pharmacol 136: 452-458
th J, Thompson N, Thompson J, Armstrong J, Hayes B, Crofts A, Squire J, Teahan C, Upton L,have a reversible block in antigen-stimulated exocytosis. Biochem J 323: 321-328
th PB, Compton DR, Welch Spharmacological activity of anandamide, a putative endogenous cannabinoid, in mice. J Pha
Smith SR, Denhardt G, Terminelli C (2001) The anti-inflammatory activities of cannabinoidPharmacology 432: 107-119
Sugita M, Williams M, Dulaney JT, Moser HW (1975) Ceramidase and ceramide synthesis in human kidney and cerebellum. Description of a new alkaline ceramidase. Biophys Acta 398: 125-31
78

Pharmacology of Palmitoylethanolamide and Related Compounds
SugiEvidence that 2-arachidonoylglycerol but not N-
Sugi ane S, Shinoda A, Itoh K, Yamashita A, Waku K
Sugi ishimoto S, Waku K (2004) New perspectives in the studies
Sun
Swieof fibronectin and fibroblasts on the functional
Terr mice via down-regulation of
Thu the
Tiger G, Stenström A, Fowler CJ (2000) Pharmacological properties of rat brain fatty acid
Ued terization of an acid
Van O, Fowler CJ, Lambert DM (2003) Esters,
ura T, Kondo S, Kishimoto S, Miyashita T, Nakane S, Kodaka T, Suhara Y, Takayama H, Waku K (2000) palmitoylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. J Biol Chem 275: 605-612
ura T, Kondo S, Sugukawa A, Nak(1995) 2-Arachidonylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun 215: 89-97
ura T, Oka S, Gokoh M, Kon endocannabinoid and cannabis: 2-arachidonoylglycerol as a possible novel mediator of inflammation. J Pharmacol Sci 96: 367-375
YX, Tsuboi K, Okamoto Y, Tonai T, Murakami M, Kudo I, Ueda N (2004) Biosynthesis of anandamide and N-palmitoylethanolamine by sequential actions of phospholipase A2 and lysophospholipase D. Biochem J 380: 749-756
ter M, Hamawy MM, Siraganian RP, Mergenhagen SE (1993) Mast cells and their microenvironment: the influence repertoire of rat basophilic leukemia cells. J Periodontol 64: 492-496
azzino S, Berto F, Dalle Carbonare M, Fabris M, Guiotto A, Bernardini D, Leon A (2004) Stearoylethanolamide exerts anorexic effects inliver stearoyl-coenzyme A desaturase-1 mRNA expression. FASEB J. 18: 1580-1582
mser AE, Voysey J, Wilton DC (1997) A fluorescence displacement assay for measurement of arachidonoyl ethanolamide (anandamide) and oleoyl amide (octadecenoamide) hydrolysis. Biochem Pharmacol 53: 433–435
amidohydrolase in different subcellular fractions using palmitoylethanolamide as substrate. Biochem Pharmacol 59: 647–653
a N, Yamanaka K, Yamamoto S (1999) Purification and characamidase selective for N-palmitoylethanolamine, a putative endogenous anti-inflammatory substance. J Biol Chem 276: 35552-35557
devoorde S, Tsuboi K, Ueda N, Jonsson Kretroesters, and a retroamide of palmitic acid: pool for the first selective inhibitors of N-palmitoylethanolamine-selective acid amidase. J Med Chem 46: 4373-4376
79

Kent-Olov Jonsson
Varga K, Lake K, Martin BR, Kunos G (1995) Novel antagonist implicates the CB I cannabinoid receptor in the hypotensive action of anandamide Eur J Pharmacol 278:
Wade DT, Robson P, House H, Makela P, Aram J (2003) A preliminary controlled study to
Wag , Martin BR, Kunos G (1997) Activation of
Wat
Wileenylmethylsulfonyl fluoride on anandamide brain levels and
Will315-317
Wils
Zaji t D, Vickery J, Nunn A, Thompson A on behalf of the
Zyg A, Chuang H, Sørgard M, Di Marzo V, Julius D,
279-283
determine whether whole-plant cannabis extracts can improve intractable neurogenic symptoms. Clin Rehab 17: 21-29
ner JA, Varga K, Ellis EF, Rzigalinski BAperipheral CB1 cannabinoid receptors in haemorrhagic shock. Nature. 390: 518-521
anabe K, Ogi H, Nakamura S, Kayano Y, Matsunaga T, Yoshimura H, Yamamoto I (1998) Distribution and characterization of anandamide amidohydrolase in mouse brain and liver. Life Sci 62: 1223–1229
y JL, Dewey MA Jefferson RG, Winckler RL, Bridgen DT, Willoughby KA, Martin BR (2002) Influence of phpharmacological effects. Life Sci 67: 1573-1583
iams CM, Kirkham TC (1999) Anandamide induces overeating: mediation by central cannabinoid (CB1) receptors. Psychopharmacology 143:
Wilson RI, Nicoll RA (2002) Endocannabinoid signaling in the brain. Science 296: 678-682
on SJ, Lovenberg TW, Barbier AJ (2003) A high-throughput-compatible assay for determining the activity of fatty acid amide hydrolase. Anal Biochem 318: 270–275
cek J, Fox P, Sanders H, WrighUK MS Research Group (2003) Cannabinoids for treatment of spasticity and other symptoms related to multiple sclerosis (CAMS study): multicentre randomised placebo-controlled trial. Lancet 362: 1517-1526
Zygmunt PM, Chuang H-h, Movahed P, Julius D, Högestätt ED (2000) The anandamide transport inhibitor AM404 activates vanilloid receptors. Eur J Pharmacol 396: 39-42
munt PM, Petersson J, Andersson DHögestätt ED (1999) Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400: 452-457
80


APPENDIX
Papers I-V




















