
GPCR Heteromers and their Allosteric Receptor-Receptor Interactions
Upload
independentCategory
view
0download
0
Cellular Signalling 18 (2006) 1169–1181www.elsevier.com/locate/cellsig
P2Y12 receptor signalling towards PKB proceeds through IGF-I receptorcross-talk and requires activation of Src, Pyk2 and Rap1
Kristof Van Kolen a, Kambiz Gilany b, Luc Moens b, Eddy L. Esmans c, Herman Slegers a,⁎
a Laboratory of Cellular Biochemistry, Department of Biomedical Sciences, University of Antwerp, Universiteitsplein 1, B-2610 Wilrijk-Antwerpen, Belgiumb Laboratory of Protein Chemistry, Department of Biomedical Sciences, University of Antwerp, B-2610 Wilrijk-Antwerpen, Belgiumc Nucleoside Research and Mass Spectrometry Unit, Department of Chemistry, University of Antwerp, B-2020 Antwerpen, Belgium
Received 26 August 2005; accepted 9 September 2005Available online 19 October 2005
Abstract
Previously it was shown that stimulation of the P2Y12 receptor activates PKB signalling in C6 glioma cells [K. Van Kolen and H. Slegers, J.Neurochem. 89, 442.]. In the present study, the mechanisms involved in this response were further elucidated. In cells transfected with the Gβγ-scavenger β-ARK1/GRK2 or Rap1GAPII, stimulation with 2MeSADP failed to enhance PKB phosphorylation demonstrating that the signallingproceeds through Gβγ-subunits and Rap1. Moreover, Rap1-GTP pull-down assays revealed that P2Y12 receptor stimulation induced a rapidactivation of Rap1. Treatment of cells with the Ca2+ chelator BAPTA-AM and inhibition of Src and PLD2 with PP2 or 1-butanol, respectively,abrogated P2Y12 receptor-mediated activation of Rap1 and PKB. In addition inhibition of PKCζ decreased basal and 2MeSADP-stimulatedphosphorylation of PKB indicating a role for this PKC isoform in PKB signalling. Although the increased PKB phosphorylation was abolished inthe presence of the IGF-I receptor tyrosine kinase inhibitor AG 1024, 2MeSADP did not significantly increase receptor phosphorylation.Nevertheless, phosphorylation of a 120 kDa IGF-I receptor-associated protein was observed. The latter protein was identified by MALDI-TOF/TOF-MS as the proline-rich tyrosine kinase 2 (Pyk2) that co-operates with Src in a PLD2-dependent manner. Consistent with the signallingtowards Rap1 and PKB, activation of Pyk2 was abrogated by Ca2+ chelation, inhibition of PLD2 and IGF-I receptor tyrosine kinase activity. Inconclusion, the data reveal a novel type of cross-talk between P2Y12 and IGF-I receptors that proceeds through Gβγ-, Ca2+-and PLD2-dependentactivation of the Pyk2/Src pathway resulting in GTP-loading of Rap1 required for an increased PKB phosphorylation.© 2005 Elsevier Inc. All rights reserved.
Keywords: IGF-I receptor; P2Y12 receptor; PKB; Pyk2; Rap1; Receptor cross-talk
Abbreviations:AC, adenylate cyclase; β-ARK, β-adrenergic receptor kinase;AG 1024, 3-bromo-5-t-butyl-4-hydroxybenzylidenemalonitrile; AG 1296, 6,7-dimethoxy-2-phenylquino-xaline; BAPTA-AM, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis-(aceto-methyl)ester; ERK, extracellularsignal-regulated kinase; IGFR, insulin-like growth factor-I receptor; MALDI-TOF/TOF-MS, matrix-assisted laser desorption-time of flight/time of flight-massspectrometry; 2MeSADP, 2-methylthioadenosine-5′-diphosphate; MRS 2395, 2-dimethyl-propionic acid-3-(2-chloro-6-methylaminopurin-9-yl)-2-(2,2-dimethyl-propionyl oxy-methyl)-propylester; PDGFR, platelet-derived growth factorreceptor; PI 3-K, phosphatidylinositol 3-kinase; PLD, phospholipase D; PP2, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4]pyrimidine; PPADS, pyridox-al-phosphate-6-azophenyl-2′,4′-disulfonic acid; Pyk2, proline-rich tyrosinekinase 2; RapGAP, Rap GTPase activating protein; RTK, receptor tyrosinekinase; SU 1498, (E)-3-(3,5-diisopropyl-4-hydroxyphenyl)-2-[(3-phenyl-n-propyl)amino-carbonyl]acrylonitrile; VEGFR, vascular endothelial growth factorreceptor.⁎ Corresponding author. Tel.: +32 3 8202306; fax: +32 3 8202248.E-mail address: [email protected] (H. Slegers).
0898-6568/$ - see front matter © 2005 Elsevier Inc. All rights reserved.doi:10.1016/j.cellsig.2005.09.005
1. Introduction
It is established that nucleotides initiate and regulate a varietyof biological processes including neurotransmission, inflam-mation, regulation of blood pressure, platelet aggregation, cellgrowth and differentiation [1–4]. Nucleosides and nucleotidesaffect these processes by binding to adenosine and AMP specificreceptors (P1 receptors) that are divided in A1, A2a, A2b and A3
receptors, and to P2 receptors responding to nucleoside di- andtri-phosphates. P2 receptors are subdivided into ionotropic P2Xand G protein-coupled P2Y receptors. In the nervous system,purine nucleotides released from different sources includingneurons, endothelial and glial cells, are reported to exertpresynaptic and postsynaptic actions but also regulate neuriteoutgrowth, neuronal cell proliferation and secretion of neuronalfactors [5].

1170 K. Van Kolen et al. / Cellular Signalling 18 (2006) 1169–1181
During brain injury, rapid hydrolysis to adenosine protectsneurons from excitatory cell death by adenosine receptorstimulation while ATP protects astrocytes from oxidative stressby a P2Y1 receptor-mediated upregulation of oxido-reductasegene products [6]. Recent reports also point to a role ofextracellular nucleotides in the proliferation and differentiationof cells including rat C6 glioma [7–10]. The latter cells arePTEN deficient resulting in a constitutively active PI 3-K/PKBpathway that contributes to their proliferative and invasiveproperties [11–13]. In addition, proliferation of these cells isalso controlled by autocrine stimulation of growth factorreceptors [14,15].
C6 cells express P2Y1, P2Y2, P2Y4, P2Y6, P2Y12 and P2Y13
receptors [17–21]. When nucleotide hydrolysis is prevented,agonists of the P2Y12 receptor increase proliferation by RhoA-and PKC-dependent activation of ERK and inhibit β-adrenergicreceptor (β-AR)-induced astrocytic differentiation by a PI 3-K/PKB-dependent mechanism [7,8,10,22].
Modulation of PKB activity is reported for a variety of Gprotein-coupled receptor (GPCR) ligands including adrener-gics, cannabinoids, carbachol, glutamate, histamine, nucleo-tides and thrombin [23–27].
In HEK293 cells, stimulation of β-AR with (−)-isopro-terenol acts independently of adenylate cyclase (AC) to activatePKB via Gβγ, Src, Ras and PI 3-K [28,29] while activation ofAC by Gsα exerts cell specific effects on PKB activity. In cellsexpressing Epac, cAMP activates PI 3-K/PKB via Rap1 whilein other cells cAMP activates PKA that exerts a negative actionon PI 3-K and PKB [30,31].
Gi protein-mediated activation of PKB can occur throughcoupling of Gβγ-subunits to activation of PI 3-K or by growthfactor receptor transactivation [32]. Stimulation of Gq protein-coupled muscarinic M3 receptors also triggers PI 3-K activationby ErbB3 transactivation that requires Ca2+ mobilization [33].In contrast, some reports showed an inhibitory pathway from Gq
protein-coupled receptors towards PI 3-K through interactionbetween Gα-subunits and p110α or by a selective inhibition ofIRS-1-associated PI 3-K activity [34–36].
Modulation of PI 3-K/PKB signalling is also reported for afew P2Y receptors. In bovine adventitial fibroblasts ATP isshown to induce proliferation through parallel but independentERK and PI 3-K signalling pathways that contribute to mTORand p70S6K phosphorylation [37]. In rat mesangial cells,stimulation of the P2Y2 receptor with ATP or UTP activatesPKB by a PDK-1-dependent mechanism while in C6 cells ADPactivates PI 3-K/PKB by the Gi protein-coupled P2Y12 receptorbut inhibits PI 3-K by a Gq/G11/12 protein-coupled P2Y1 receptorstimulation [9,10,38]. Although a P2Y12 receptor-inducedincrease in PI 3-K/PKB activity is observed in some cell types,different mechanisms seem to be involved. Overexpression ofthe P2Y12 receptor in CHO cells revealed that PDGFRtransactivation is at least partially involved in PI 3-K/PKB andERK signalling [32]. On the other hand, PI 3-K could also beactivated by the GTPase Rap1. In platelets, stimulation of theP2Y12 receptor activates Rap1 in a PI 3-K-dependent mannerwhile PI 3-K is positioned downstream of Rap1 in C6 cells[31,39,40]. In the latter cells, an increase in cAMP is shown to
abrogate PKB and ERK signalling by inhibition of Rap1.Therefore, it can not be excluded that activation of Rap1 by Giαprotein-dependent inhibition of AC is responsible for theenhanced PI 3-K activity upon P2Y12 receptor stimulation.
In this communication, we demonstrated that the P2Y12
receptor-induced increase in PKB activity is due to a Ca2+- andSrc-dependent Rap1 activation. Although P2Y12 receptorstimulation did not induce detectable phosphorylation ofIGF-IR or PDGFR, a novel type of receptor cross-talk wasdemonstrated in which interaction between IGF-IR and Pyk2is involved in signalling towards Rap1 and PKBphosphorylation.
2. Materials and methods
2.1. Materials
Rat C6 glioma cells (ATCC no. CCL 107) were obtainedfromATCC (Manassas, VA, USA) and maintained in monolayerculture as described previously [41]. Nucleotides, LY294002,BAPTA-AM, MRS2395, PPADS and methyl-β-cyclodextrinwere from RBI (Bornem, Belgium). PKC inhibitors (Bisin-dolylmaleimide I, Bisindolylmaleimide IX, Gö6976, Rottlerin,PKCζ pseudosubstrate inhibitor), tyrphostins (AG 1024, AG1296, SU 1498), KN62 and PP2 were obtained from Cal-biochem (La Jolla, CA, USA). Human recombinant IGF-I wasfrom R&D Systems (Minneapolis, USA). The pMT2HA-Rap1GAPII construct, the pRK-βARKI/GRK2 construct andToxin B were kind gifts from Dr. J. Bos (Department ofPhysiological Chemistry, University Medical Center Utrecht,The Netherlands), Dr. R.J. Lefkowitz (Duke University MedicalCenter, Durham, NC, USA) and Dr. T. Giesemann (Institute forExperimental and Clinical Pharmacology and Toxicology,Freiburg, Germany), respectively.
2.2. Immunoblotting
Cells were cultivated in 96-well plates in chemically definedmedium up to a density of 105 cells/cm2. After stimulation ofthe cells as described in the figure legends, cellular proteinswere separated by SDS PAGE [22] and electroblotted overnightonto a nitrocellulose membrane (Hybond-C pure, AmershamBiosciences, NJ, USA). Phosphorylated PKB and total PKBwere detected with anti-phospho Ser473 PKB (Cell SignallingTechnology, Hertfordshire, UK) and anti-PKB (Santa CruzBiotechnologies, CA, USA) antibodies, respectively. Phos-phorylated Pyk2 was detected with anti-phospho Tyr402 (CellSignalling Technology, Hertfordshire, UK) and anti-phosphoTyr579 (Santa Cruz Biotechnologies, CA, USA) antibodies.Primary antibodies were detected with horseradish peroxidase-conjugated antibody (DAKOCytomation, Glostrup, Denmark)and visualized by enhanced chemiluminescence (LumiGlo®Cell Signalling Technology, Hertfordshire, UK) according tothe manufacturer's instructions. Luminograms were recordedwith the Gel Doc 2000 system (Bio-Rad, Hercules, CA, USA)and analyzed with Image Quant (Molecular Dynamics,Amersham Biosciences, NJ, USA).

1171K. Van Kolen et al. / Cellular Signalling 18 (2006) 1169–1181
2.3. Transient transfection of C6 cells
C6 glioma cells were plated in 96-well plates and cultivatedin serum-free chemically defined medium up to a density of8×104 cells/cm2. Cells were transfected in serum-free mediumcontaining Lipofectamine 2000 (Invitrogen) transfection rea-gent and DNA at a ratio of 3 :1 (μL:μg). After 18 h, cells werestimulated with PBS or 2MeSADP (1 μM, 10 min) as indicatedand lysed in SDS PAGE sample buffer. PKB phosphorylationwas quantified by immunoblotting. Expression of pMT2HA-Rap1GAPII was detected with an anti-HA-Tag antibody (SantaCruz Biotechnologies, CA, USA) while the βARK1-(495–689)peptide was detected with an anti-βARK1/GRK2 antibody(Santa Cruz Biotechnologies, CA, USA) that reacts withendogenous βARK1 (90 kDa) and overexpressed βARK1-(495–689) peptide (27 kDa).
2.4. Rap1-GTP pull-down assay
C6 cells were plated in 100 mm dishes and cultivated inserum-free chemically defined medium up to a density of 105
cells/cm2. Upon stimulation, cells were washed with 5 mL ice-cold PBS and lysed in 400 μLTLB [50 mM Tris/HCl (pH 7.4),500 mM NaCl, 10% (v/v) glycerol, 5 mMMgCl2, 1 mM PMSF,1 mM NaVO3, 1% (v/v) NP-40, 10 mM NaF and a protease-inhibitor cocktail (Roche, Mannheim, Germany)]. The lysatewas collected by scraping, passed through a syringe andcentrifuged for 5 min at 12000 g. Small aliquots (20 μL) wereanalyzed for total Rap1 while pull-down of GTP-bound Rap1was performed by incubation with Ral GDS-RBDagarose(Upstate, Charlottesville, Virginia, USA) for 45 min. Sub-sequently, beads were collected by centrifugation at 12000 gfor 20 s, washed three times with TLB and suspended indouble concentrated Laemmli buffer. Rap1 was immuno-detected using an anti-Rap1 antibody (Upstate, Charlottesville,Virginia, USA).
2.5. Growth factor receptor immunoprecipitation
C6 cells were plated in 100 mm dishes and cultivated inserum-free chemically defined medium up to a density of 105
cells/cm2. After stimulation as indicated in the figure legends,cells were washed with 5 mL ice-cold PBS and lysis buffer [20mM Tris/HCl (pH 7.4), 100 mM NaCl, 1 mM PMSF, 1 mMNaVO3, 1% (v/v) NP-40, 10 mM NaF and a protease-inhibitorcocktail (Roche, Mannheim, Germany)] was added. After 5 minincubation on ice, the lysate was collected by scraping, passedthrough a syringe and centrifuged for 5 min at 12000 g. Thesupernatant was pre-cleared with protein G beads (Pierce,Rockford, Illinois, USA). Subsequently the supernatant wasincubated for 2 h with anti-IGF-IR β-subunit or anti-PDGFRβantibody (Santa Cruz Biotechnology, CA, USA). Protein Gbeads were added and collected after 1 h by centrifugation.Beads were washed five times with lysis buffer and suspendedin double concentrated Laemlli buffer. Proteins were separatedby SDS PAGE (7.5% w/v) and blotted overnight as describedabove. Phosphorylation of the immunoprecipitated receptors
was determined by immunoblotting using mouse anti-phospho-Tyr (Cell Signalling Technology, Hertfordshire, UK) andperoxidase-conjugated goat anti-mouse antibody (DAKOCy-tomation, Glostrup, Denmark). Subsequently, blots were strip-ped and probed with anti-growth factor receptor antibody as aninternal control. For co-immunoprecipitation and detection oftyrosine phosphorylated IGFR-associated proteins, the sameprocedure was used but final washing was once with lysis bufferand three times with PBS.
2.6. Peptide mass fingerprinting of IGFR-associated protein
Isolated immunocomplexes were solubilized in Laemllibuffer and subjected to a one dimensional SDS PAGE (10% (w/v)). Proteins were stained with “Colloidal Coomassie” stain.Bands were excised and subjected to an ‘in gel’ tryptic digest[42]. Subsequently, peptides were eluted from the gel anddesalted on a home made capillary reversed phase column.Peptides were eluted with matrix solution [2 mg/ml α-cyano-4-hydroxycinnamic acid (Sigma) dissolved in acetonitrile, 0.1%trifluoroacetic acid (70 :30, v/v)], directly applied on theMALDI plate and their mass spectrum measured by MALDI-TOF/TOF-MS (Applied Biosystems, Foster City, CA, USA).Mono-isotopic masses were assigned to individual peaks andused for database searches with the MASCOT search engine.This software uses the peptide mass values of the enzymaticdigest for protein identification. The data were compared withthe NCBI non-redundant protein sequence database.
2.7. Statistical analysis
Results are represented as the mean±SEM calculated from atleast three independent experiments. Statistically significantdifferences were calculated using the Student's t-test.
3. Results
3.1. P2Y12 receptor-mediated activation of PKB requires Src,Ca2+ and PLD2
Previous studies of our laboratory ruled out the involvementof tyrosine kinases in the P2Y12 receptor-dependent signallingtowards ERK [22]. In contrast, recruited tyrosine kinases areinvolved in the activation of PKB by this receptor. Treatment ofC6 cells with PP2 (10 μM), a specific inhibitor of the Src-familyof tyrosine kinases, decreased PKB phosphorylation in controlcells and abolished the P2Y12 receptor-mediated activation ofPKB (Fig. 1). In agreement with previous reports, a similarobservation was made when cells were treated with BAPTA-AM (50 μM, 1 h) [10,22]. These data indicated that, in contrastto ERK signalling, P2Y12 receptor-enhanced phosphorylationof PKB requires Src and Ca2+. Stimulation of the P2Y12
receptor with 2MeSADP induces a PLC-independent Ca2+
influx that is probably responsible for the increase in PKBphosphorylation [22]. In NG108 neuroblastoma cells NMDA-induced Ca2+ mobilization activates PKB by a Ca2+/calmo-dulin-dependent protein kinase kinase (CaM-KK)-mediated

0
50
100
150
200
250
300
C KN62 10 μM KN62 50 μM
Rel
ativ
e ph
osph
o-P
KB
/PK
B r
atio
(%
)
Phospho-PKB
Control 2MeSADP
Phospho-PKB
Total PKB
2MeSADP - + - + - +KN62 (μM) 0 10 50
++
+
++
*
* *° °
A
B
TotalPKB
C 1-But 2-But PP2 BAPTA
C 1-But 2-But PP2 BAPTA
C 1-But 2-But PP2 BAPTA
0
50
100
150
200
250
Rel
ativ
e ph
osph
o-P
KB
/PK
B r
atio
(%
)
Fig. 1. P2Y12 receptor-mediated activation of PKB requires Ca2+, Src and PLD.Cells were grown in chemically defined medium up to a density of 105 cells/cm2
and were pre-incubated with (A) 1-butanol (1-But, 0.3% v/v, 5 min), 2-butanol(2-But, 0.3% v/v, 5 min), PP2 (10 μM, 30 min) or BAPTA-AM (50 μM, 60 min)or (B) with KN62 for 30 min at the indicated concentrations. Subsequently, cellswere not stimulated (white bars) or stimulated with 2MeSADP (1 μM, 10 min,black bars) and the amount of Ser473-phosphorylated PKB and total PKB wasdetermined by immunoblotting. The detected phospho-PKB/ total PKB ratio innon-stimulated cells (control) was taken as 100%. Data are the mean±SEM ofthree independent experiments. Statistically significant differences from therespective controls (+), from control cells (°) and from 2MeSADP stimulatedcells (*) are indicated (p<0.05). The insert shows a representative blot of at leastthree independent experiments.
1172 K. Van Kolen et al. / Cellular Signalling 18 (2006) 1169–1181
mechanism [43]. In C6 cells, this mechanism could be rejectedby the fact that the CaM-KK inhibitor KN62 did not affect thephosphorylation of PKB after activation of the P2Y12 receptor(Fig. 1B).
It has been reported that in response to Ca2+ mobilization,Src can co-operate with the proline-rich tyrosine kinase 2(Pyk2) [44]. The latter interaction depends on PLD2,demonstrated to be constitutively active in C6 cells [45]. Toinvestigate a possible role of PLD2 in the signalling of theP2Y12 receptor towards PKB, we used 1-butanol (0.3% (v/v)),
an inhibitor of PLD, and 2-butanol (0.3% (v/v)) its inactiveanalogue as a control. Data from Fig. 1A demonstrated that theP2Y12 receptor-dependent increase in PKB phosphorylationwas abrogated by 1-butanol but not by 2-butanol suggesting theinvolvement of PLD2 in this mechanism.
3.2. PKCζ is involved in basal and P2Y12 receptor-enhancedPKB phosphorylation
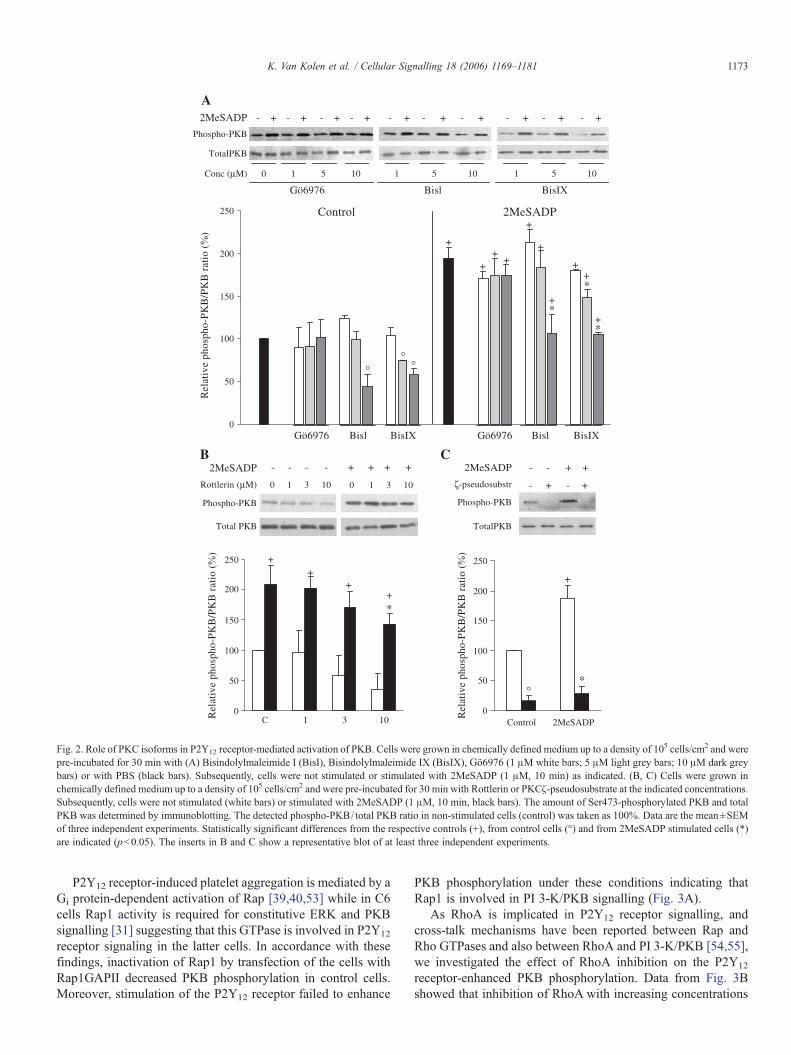
To determine the involvement of PKC in the P2Y12 receptor-activated signalling to PKB in C6 cells, we evaluated the effectof several PKC inhibitors (Fig. 2A). Treatment of the cells withBisindolylmaleimide (Bis) I (1 and 5 μM) did not affect PKBphosphorylation while addition of 10 μM of this compoundreduced the basal and P2Y12 receptor-enhanced PKB phos-phorylation suggesting the involvement of a PKC isoform in thelatter mechanism. Similar results were obtained when cells weretreated with BisIX but a significant inhibition was alreadyobserved at 5 μM (Fig. 2A). Since Ca2+ chelation inhibits PKBphosphorylation in C6 cells classical PKC isoforms (α, βI, βII,γ) might be involved in the signalling towards PKB. However,experiments with Gö6976 showed that inhibition of these PKCsdid not affect PKB phosphorylation and that only novel oratypical isoforms could be involved. Although novel PKCsrequire DAG formation, PKCδ is regulated independently ofDAG by several site specific tyrosine phosphorylations [46].Moreover, this isoform has been reported to have an importantrole in the proliferation, differentiation and apoptosis of C6 cells[47–49]. We treated the cells with increasing concentrations ofRottlerin, a compound often used to inhibit PKCδ. Althoughthis inhibitor reduced the basal PKB phosphorylation,stimulation of the P2Y12 receptor with 2MeSADP still increasedPKB phosphorylation more than twofold (Fig. 2B) in ac-cordance with studies performed in blood platelets [50]. Toevaluate the involvement of atypical PKCs, we determined theeffect of a PKCζ-pseudosubstrate inhibitor peptide on PKBphosphorylation (Fig. 2C). Treatment of the cells with 50 μM ofthis peptide significantly inhibited PKB phosphorylation incontrol cells and stimulation of the P2Y12 receptor with2MeSADP did not increase PKB phosphorylation. These datasuggest a role for PKCζ in the basal and P2Y12 receptor-induced increase in PKB phosphorylation.
3.3. PKB activation involves Gβγ and Rap1 signalling
Previous experiments with pertussis toxin revealed that PKBphosphorylation upon stimulation of the P2Y12 receptor is Gi
protein-dependent [10]. Since PKB activation by GPCRs can bemediated by Gα- or Gβγ-subunits [29,51], we investigated therole of Gβγ-subunits by transfection with the β-ARK1/GRK2-(495–689) peptide, a Gβγ-subunit scavenger [52]. Expressionof this construct was evaluated by probing immunoblots with ananti-GRK2 antibody that detects endogenous expressed GRK2(data not shown) and overexpressed β-ARK1 (27 kDa) intransfected cells (Fig. 3A). Data from Fig. 3A showed thatimpeding the Gβγ-subunit-activated signal transduction de-creased PKB phosphorylation by more than 40%.
0
50
100
150
200
250
C 31 100
50
100
150
200
250
Control 2MeSADP
Phospho-PKB
2MeSADP
ζ-pseudosubstr - + - +
- - + +
+
Control 2MeSADP
°
*
°
*
+
++
+
+
+
+
+
°
+
+
*
*°
++
++*
TotalPKB
A
C
0 1 5 10 1 5 10 1 5 10
Phospho-PKB
TotalPKB
2MeSADP - + - + - + - + - + - + - + - + - + - +
Gö6976 Bisl BisIX
Gö6976 Bisl BisIX Gö6976 Bisl BisIX
Conc (μM)
0 1 3 10 0 1 3 10Rottlerin (μM)
Phospho-PKB
2MeSADP - - - - + + + +B
Total PKB
0
50
100
150
200
250R
elat
ive
phos
pho-
PK
B/P
KB
rat
io (
%)
Rel
ativ
e ph
osph
o-P
KB
/PK
B r
atio
(%
)
Rel
ativ
e ph
osph
o-P
KB
/PK
B r
atio
(%
)
Fig. 2. Role of PKC isoforms in P2Y12 receptor-mediated activation of PKB. Cells were grown in chemically defined medium up to a density of 105 cells/cm2 and werepre-incubated for 30 min with (A) Bisindolylmaleimide I (BisI), Bisindolylmaleimide IX (BisIX), Gö6976 (1 μM white bars; 5 μM light grey bars; 10 μM dark greybars) or with PBS (black bars). Subsequently, cells were not stimulated or stimulated with 2MeSADP (1 μM, 10 min) as indicated. (B, C) Cells were grown inchemically defined medium up to a density of 105 cells/cm2 and were pre-incubated for 30 min with Rottlerin or PKCζ-pseudosubstrate at the indicated concentrations.Subsequently, cells were not stimulated (white bars) or stimulated with 2MeSADP (1 μM, 10 min, black bars). The amount of Ser473-phosphorylated PKB and totalPKB was determined by immunoblotting. The detected phospho-PKB/ total PKB ratio in non-stimulated cells (control) was taken as 100%. Data are the mean±SEMof three independent experiments. Statistically significant differences from the respective controls (+), from control cells (°) and from 2MeSADP stimulated cells (*)are indicated (p<0.05). The inserts in B and C show a representative blot of at least three independent experiments.
1173K. Van Kolen et al. / Cellular Signalling 18 (2006) 1169–1181
P2Y12 receptor-induced platelet aggregation is mediated by aGi protein-dependent activation of Rap [39,40,53] while in C6cells Rap1 activity is required for constitutive ERK and PKBsignalling [31] suggesting that this GTPase is involved in P2Y12
receptor signaling in the latter cells. In accordance with thesefindings, inactivation of Rap1 by transfection of the cells withRap1GAPII decreased PKB phosphorylation in control cells.Moreover, stimulation of the P2Y12 receptor failed to enhance
PKB phosphorylation under these conditions indicating thatRap1 is involved in PI 3-K/PKB signalling (Fig. 3A).
As RhoA is implicated in P2Y12 receptor signalling, andcross-talk mechanisms have been reported between Rap andRho GTPases and also between RhoA and PI 3-K/PKB [54,55],we investigated the effect of RhoA inhibition on the P2Y12
receptor-enhanced PKB phosphorylation. Data from Fig. 3Bshowed that inhibition of RhoA with increasing concentrations

0
50
100
150
200
250
V β-ARK1 Rap1GAPII
Rel
ativ
e ph
osph
o-P
KB
/PK
B r
atio
(%
)
0
50
100
150
200
250
Rel
ativ
e ph
osph
o-P
KB
/PK
B r
atio
(%
)
+ + + ++
*+
*
Phospho-PKB
Total PKB
β-ARK1 27 kDa
HA-Tag
V β-ARK1 Rap1GAPII
2MeSADP - + - + - +
C 2MeSADP
0 1 10 50 0 1 10 50Tox B (ng/mL)
Phospho-PKB
Total PKB
°
A B
C 1 10 50 ng/mL Tox B
Fig. 3. PKB activation requires Gβγ and Rap1 signalling. (A) Cells grown in chemically defined medium up to a density of 0.8×105 cells/cm2 were transfected with anempty vector (V), βARK1-(495–689) or pMT2HA-Rap1GAPII as described in Materials and methods. Expression of βARK1/GRK2 and Rap1GAPII was detectedwith an anti-GRK2 and an anti-HA-Tag antibody, respectively, as shown in the insert. (B) Cells were grown in chemically defined medium up to a density of 105 cells/cm2 and were pre-incubated for 60 min with the indicated concentrations of Toxin B (Tox B). Subsequently, cells were not stimulated (white bars) or stimulated with2MeSADP (1 μM, 10 min, black bars). The amount of Ser473-phosphorylated PKB and total PKB was determined by immunoblotting. The detected phospho-PKB/total PKB ratio in non-stimulated cells (control) was taken as 100%. Data are the mean±SEM of three independent experiments. Statistically significant differencesfrom the respective controls (+), from control cells (°) and from 2MeSADP stimulated cells (*) are indicated (p<0.05). The insert shows the representative blots of atleast three independent experiments.
1174 K. Van Kolen et al. / Cellular Signalling 18 (2006) 1169–1181
of Toxin B did not affect the P2Y12 receptor-dependentactivation of PKB indicating that Rap1 modulates PI 3-K/PKBsignalling independent of Rho GTPases.
To confirm a role of Rap1 in P2Y12 receptor-mediatedsignalling, we performed Rap1-GTP pull-down assays atdifferent time points after stimulation of C6 cells with2MeSADP. Data of Fig. 4A showed that stimulation with2MeSADP induced a rapid GTP loading of Rap1 (1 min) thatwas still maintained after 10 min. Pre-incubation of the cellswith the P2Y12 receptor antagonist 2-dimethyl-propionic acid-3-(2-chloro-6-methylaminopurin-9-yl)-2-(2,2-dimethyl-propio-nyloxymethyl)-propylester (MRS 2395) [56] abolished Rap1activation indicating that 2MeSADP exerts this effect bystimulation of the P2Y12 receptor. In blood platelets, P2Y12
receptor-induced Rap1 activation requires activation of PI 3-K[40]. As a consequence, the observed activation of Rap1 in thisstudy might be due to an increased PI 3-K activity. However, inthe presence of the PI 3-K inhibitor LY294002 (10 μM), P2Y12
receptor stimulation still activated Rap1 while Ca2+ chelationwith BAPTA-AM (50 μM) and inhibition of Src and PLD2 withPP2 (10 μM) and 1-butanol (0.3% (v/v)), respectively,abolished this response (Fig. 4B, C). These data, together withthe transfection experiments presented in Fig. 3, indicated thatP2Y12 receptor-induced GTP loading of Rap1 acts upstream ofPI 3-K but downstream of Ca2+ mobilization and activation ofSrc. PLD2 activity is also required for the activation of Rap1.
These observations are consistent with the data of Fig. 1Awhere P2Y12 receptor-dependent PI 3-K/PKB signalling isshown to require activation of the same components.
3.4. Effect of RTK inhibitors on P2Y12 receptor signalling
Implication of growth factor receptor transactivation isreported for P2Y12 receptor-dependent signalling in CHO cells[32]. Therefore, the possibility of IGF-IR, PDGFR or VEGFRtransactivation upon stimulation of the P2Y12 receptor wasinvestigated in C6 cells. We used AG1024, AG1296 andSU1498 to inhibit the intrinsic tyrosine kinase activity of IGF-IR, PDGFR and VEGFR, respectively.
Treatment of the cells with AG1024 (20 μM) and AG1296(10, 20 μM) inhibited PKB phosphorylation in control cellsindicating that basal PKB phosphorylation is due to autocrineIGF-I and PDGF signalling (Fig. 5A, B) in accordance withprevious studies performed in these cells [14–16]. While P2Y12
receptor-enhanced phosphorylation of PKB is inhibited by 20μM AG1024, stimulation of the cells with 2MeSADP in thepresence of AG1296 and SU1498 still significantly increasedthe phosphorylation level of PKB (Fig. 5A, B). These dataindicate that the observed attenuation of PKB phosphorylationby AG1296 and SU1498 can be explained by inhibition ofautocrine growth factor receptor signalling. Only in thepresence of the IGF-IR tyrosine kinase inhibitor AG1024 (20

Phospho-PKB
Total PKB
Control 2MeSADP (1 μM)
°° °
°
++
+ +
+
*
* *
C AG1024 AG1296 SU
- 10 20 10 20 20
C AG1024 AG1296 SU
- 10 20 10 20 20
0
50
100
150
200
250
300
C AG1024(10μM)
AG 1024(20μM)
AG 1296(10μM)
AG 1296(20μM)
SU 1498(20μM)
Rel
ativ
e ph
osph
o-P
KB
/PK
B r
atio
(%
)
Fig. 5. P2Y12 receptor-mediated PKB activation requires IGFR. Cells were grown in chemically defined medium up to a density of 105 cells/cm2 and were pre-incubated with the indicated inhibitors. Subsequently, cells were not stimulated (white bars) or stimulated with 2MeSADP (1 μM, 10 min, black bars) and the amountof Ser473-phosphorylated PKB and total PKB was determined by immunoblotting. The detected phospho-PKB/ total PKB ratio in non-stimulated cells (control) wastaken as 100%. Data are the mean±SEM of three independent experiments. Statistically significant differences from the respective controls (+), from control cells (°)and from 2MeSADP stimulated cells (*) are indicated (p<0.05). The insert shows a representative blot of at least three independent experiments.
0
50
100
150
200
250
300
C MRS BAPTA LY PP2 1-But
Rel
ativ
e R
ap1-
GT
P/R
ap1
rati
o (%
)
Rap1-GTP
Total Rap1
2MeSADP
Rap1-GTP
Total Rap1
2MeSADP- 1’ 3’ 10’
+ +
*
*
**
°
C C MRS PP2 1-But LY BAPTA
A- + + + + + +
B
C
Fig. 4. Rap1 is rapidly activated upon P2Y12 receptor stimulation. (A) Cells were grown in chemically defined medium up to a density of 105 cells/cm2 and werestimulated with 2MeSADP (1 μM) for the indicated time. (B, C) Cells were pre-incubated with PBS,MRS 2395 (20 μM, 15 min), PP2 (10 μM, 30 min), LY294002 (10μM, 30 min), BAPTA-AM (50 μM, 60 min) or 1-butanol (1-But, 0.3% v/v, 5 min) and stimulated with PBS (white bars) or with 2MeSADP (1 μM, 2 min, black bars).Subsequently, cells were rinsed in ice-cold PBS and Rap1-GTP loading was determined as described in Materials and methods. The detected Rap1-GTP/ total Rap1ratio in non-stimulated cells (control) was taken as 100%. Statistically significant differences from the respective controls (+), from control cells (°) and from2MeSADP stimulated cells (*) are indicated (p<0.05). Panel B shows a representative blot of Rap1-GTP analysis of 2MeSADP-stimulated cells in the presence of theindicated inhibitors.
1175K. Van Kolen et al. / Cellular Signalling 18 (2006) 1169–1181

WB P-Tyr
2MeSADPPP2
IGF-I
- + - + -- - + + -- - - - +
175 kDa
82 kDa
Pro-IGF-IR
Pro-IGF-IR
IGFR
IGFR
IgG
175 kDa
82 kDa
IP: IGF-IRWB: P-Tyr
IP: IGF-IRWB: IGF-IR
WB PDGFR
x 120 kDa
A
B
WB IGF-IR
WB P-Tyr
Control 2MeSADP IGF-I
IP: IGFRIP: PDGFR
Control 2MeSADP FCS
IGF-IR
IgG
C 2MeS C 2MeS
Tryptic digest
IP: IGFRIB: P-Pyk2
MALDI-TOF/TOF-MS
Pyk2
175 kDa
82 kDa
x
C
Fig. 6. Identification of Pyk2 as a component of the P2Y12 receptor signalling.(A) Cells grown in chemically defined medium up to a density of 105 cells/cm2
were stimulated with PBS (control), 2MeSADP (1 μM, 1 min), IGF-I (100 ng/mL, 1min) or FCS (10% v/v, 1 min) as indicated. Subsequently cells were rinsedwith ice-cold PBS and IGF-IR or PDGFR was immunoprecipitated and tyrosinephosphorylation was determined with immunoblotting. Detection of IGF-IR orPDGFRwas used as internal control. The immunoblots shown are representativeof at least three independent experiments. (B) Cells grown in chemically definedmedium up to a density of 105 cells/cm2 were pre-incubated for 30 min in thepresence or absence of PP2 (10 μM) and were stimulated with 2MeSADP (1μM, 1 min) or IGF-I (100 ng/mL, 1 min) as indicated. Subsequently cells wererinsed with ice-cold PBS and IGF-IR-associated proteins were co-im-munoprecipitated as described in Materials and methods. Phosphorylation of co-immunoprecipitated proteins was determined with immunodetection of phosphotyrosine while reprobing the blot with anti-IGF-IR antibody was used as internalcontrol. Detection of phosphorylated IGF-IR and pro-IGF-IR and a receptor-associated protein (x) are indicated. (C) The immunoprecipitated proteins wereseparated on a one dimensional SDS PAGE and were stained with “ColloidalCoomassie”. An ‘in gel tryptic digest’ of the 120 kDa protein (x) was excisedand analysed by MALDI-TOF/TOF-MS. Based on peptide mass fingerprinting,the protein was identified as Pyk2 (GenBank accession no. P70600). Theidentification was confirmed by immunodetection of Tyr402-phosphorylatedPyk2 in the receptor immunoprecipitates of control cells and 2MeSADP (1 μM,1 min) stimulated cells.
1176 K. Van Kolen et al. / Cellular Signalling 18 (2006) 1169–1181
μM) PKB phosphorylation is not significantly different between2MeSADP-stimulated and non-stimulated cells. Although thesedata exclude PDGFR and VEGFR and point to a role of IGF-IR,other RTKs can not be excluded.
To further evaluate a possible RTK transactivation, westudied the tyrosine phosphorylation of immunoprecipitatedIGF-IR and PDGFR after stimulation of the P2Y12 receptor with2MeSADP (1 μM) for 1 min. Immunoblots presented in Fig. 6Aindicated no increase in PDGFR tyrosine phosphorylationconfirming that the inhibitory effect of AG1296 on PKBphosphorylation is due to a decrease of its basal phos-phorylation level. Unexpectedly, while experiments in thepresence of AG1024 suggested that IGF-IR transactivation wasinvolved in the P2Y12 receptor-dependent enhanced phos-phorylation of PKB, stimulation with 2MeSADP did notincrease the tyrosine phosphorylation of IGF-IR. Theobservation that stimulation with IGF-I and FCS (10% v/v)induced tyrosine phosphorylation of IGF-IR and PDGFR,respectively, indicated that phosphorylation of these growthfactor receptors can be measured in the used experimentalconditions (Fig. 6A). Besides activation of the kinase activityand tyrosine phosphorylation of growth factor receptors,transactivation can also proceed through phosphorylation ofdocking proteins or recruited tyrosine kinases that interact withactivated growth factor receptors [57]. Since inhibition of IGF-IR autophosphorylation attenuated the P2Y12 receptor-mediatedphosphorylation of PKB but stimulation of the latter receptorfailed to induce IGF-IR tyrosine phosphorylation, we assumedthat stimulation of the P2Y12 receptor might induce tyrosinephosphorylation of proteins that interact with IGF-IR.Therefore, we immunoprecipitated IGF-IR and analyzed thetyrosine phosphorylation pattern of the precipitated proteins byimmunoblotting. Fig. 6B revealed that only a 120 kDa protein(marked as x) was increased in phosphorylation after 1 minstimulation of the cells with 2MeSADP (1 μM). In addition,pretreatment of the cells with PP2 (10 μM) abolished this effectindicating that the latter phosphorylation is Src-dependent.Surprisingly, stimulation with IGF-I did not induce phos-phorylation of this protein indicating that an increased receptortyrosine kinase activity is not required for the phosphorylationof the 120 kDa protein. Re-probing the blot with an anti-IGF-IRantibody as internal control showed that the phosphorylatedprotein had a higher molecular weight as compared to thephosphorylated receptor.
3.5. Identification of Pyk2 as a component of the P2Y12receptor signalling
To identify the phosphorylated protein that associated withIGF-IR upon stimulation of the P2Y12 receptor, we performed apreparative co-immunoprecipitation and separated the proteinsby one dimensional SDS PAGE. After staining with “ColloidalCoomassie”, the protein was excised and identified with massspectrometry as Pyk2 (Fig. 6C). The identification wasconfirmed by immunodetection of phosphorylated Pyk2 in theIGF-IR immunoprecipitate of P2Y12 receptor stimulated cells.Data from Fig. 7 indicated that stimulation with 2MeSADP
induced a rapid increase in phosphorylation of Pyk2 at tyrosines402 and 579 that returned to the basal level in control cellswithin 10 and 60 min, respectively (Fig. 7A). Consistent withprevious observations, also PKB phosphorylation was tran-siently increased over a period of 60 min (Fig. 7A, [10]). Theincreased Pyk2 and PKB phosphorylation upon stimulation with

2MeSADP
2MeSADP
0’ 1’ 5’ 0’ 1’ 5’
BAPTA - - - + + +
- + + + + + +
C C LY 1-But 2-But CD AG1024
A
B
C
2MeSADP 0’ 1’ 5’ 10’ 30’ 60’
Phospho-Tyr402 Pyk2
Phospho-Tyr597 Pyk2
Phospho-SerPKB
Phospho-Tyr597 Pyk2
Phospho-Tyr597 Pyk2
Phospho-Tyr402 Pyk2
Phospho-Tyr402 Pyk2
Phospho-SerPKB
Phospho-SerPKB
Total PKB
Total PKB
Total PKB
- 5’ 5’ 5’
B B P MRS
Fig. 7. Pyk2 activation through Ca2+ mobilization and constitutive PLD2 activity. Cells were grown in chemically defined medium up to a density of 105 cells/cm2 andwere (A, left panel) stimulated with 2MeSADP (1 μM) for the indicated time, (A, right panel) pre-incubated with PBS (C, 10 min), PPADS (P, 50 μM, 10 min) or MRS2395 (MRS, 20 μM, 10 min) as indicated, (B) pre-incubated with BAPTA-AM (50 μM, 60 min), (C) pre-incubated with 1-Butanol (1-But, 0.3% v/v, 5 min), 2-Butanol(2-But, 0.3% v/v, 5 min), AG1024 (AG, 20 μM, 30 min), LY294002 (LY, 10 μM, 30 min) and methyl-β-cyclodextrin (CD, 10 mM, 30 min). Subsequently, cells werenot stimulated or stimulated with 2MeSADP and phosphorylation of PKB at Ser473 and Pyk2 at Tyr402 and Tyr597 was determined by immunoblotting. Detection oftotal PKB was used as internal control. The blots shown are representative of at least three independent experiments.
1177K. Van Kolen et al. / Cellular Signalling 18 (2006) 1169–1181
2MeSADP was prevented by pretreatment of the cells withMRS2395 indicating that the observed effect is mediated bystimulation of the P2Y12 receptor. Involvement of P2Y1 orP2Y13 receptors could be excluded by the observation thatpretreatment with PPADS (50 μM) was without effect (Fig. 7A).In addition, Ca2+ chelation with BAPTA-AM (50 μM) abolishedP2Y12 receptor-mediated Pyk2 phosphorylation while inhibitionof PI 3-K with LY294002 inhibited PKB phosphorylation buthad no effect on the activation of Pyk2 which requires Ca2+
mobilization and takes place upstream of PI 3-K (Fig. 7B, C).Since inhibition of PLD2 abolished activation of Rap1 and
PKB (Figs. 1 and 4, respectively) we investigated whether thiseffect could be due to inhibition of Pyk2. Consistent with thedata mentioned above, treatment with 1-butanol (0.3% (v/v))decreased P2Y12 receptor-mediated Pyk2 phosphorylation atTyr579 to the basal level but not at Tyr402 (Fig. 7C). Theseobservations are in accordance with the study of Banno et al.[44], who reported that Ca2+ mobilization is sufficient forTyr402 phosphorylation while the Src-dependent phospho-rylation of Tyr597 requires PLD2 activity. A recent report
indicated that the constitutive PLD2 activity in C6 cells wassignificantly inhibited upon disruption of caveolae with methyl-β-cyclodextrin [45]. In addition, Banno et al. [58] demonstratedthat PLD is activated upon stimulation of IGF-IR in CHO cells.Consistent with these findings, Pyk2 phosphorylation at Tyr579was also diminished by treatment with methyl-β-cyclodextrin(10 mM) and AG1024. The observation that Tyr402 phos-phorylation was inhibited more potently by methyl-β-cyclodextrin pointed to involvement of other lipid raft-associated component (Fig. 7C).
4. Discussion
Previously we reported that stimulation of the P2Y12
receptor inhibits the cAMP-dependent induction of diff-erentiation by reactivation of PKB [10]. P2Y12 receptorstimulation is also reported to activate PKB in blood plateletsand CHO cells, although different signal transduction mecha-nisms are involved [59,32]. In platelets, activation of PKB ismediated by PI 3-Kγ, an isoform directly activated by Gβγ

1178 K. Van Kolen et al. / Cellular Signalling 18 (2006) 1169–1181
subunits [60,61] while in CHO cells PDGFR transactivationparticipates in the P2Y12 receptor-dependent PKB response[32]. In C6 cells, stimulation of the P2Y12 receptor induces arapid activation of PI 3-K [9,10].
In this communication we demonstrated that PKB phos-phorylation requires Ca2+ mobilization, activation of Src,Pyk2 and PLD2 and proceeds through a novel type of cross-talk between GPCRs and RTKs. Although Yano et al. [43]reported direct activation of PKB in vitro by CaM-KK,experiments with KN62 rejected this mechanism. Alter-natively, PKB can be phosphorylated on Ser473 by PKCα/βindependently of PI 3-K [62]. In C6 cells inhibition of PKCattenuates PI 3-K activity indicating that cross-talk betweenPKC and the PKB activating pathways can take place atdifferent levels [9]. In our study, BisI and BisIX inhibited thebasal and P2Y12 receptor-enhanced PKB phosphorylationwhile treatment of the cells with Gö6976 was without effectexcluding involvement of classical PKC isoforms. Further-more, experiments with Rottlerin suggested an involvement ofPKCδ in basal, but not in P2Y12 receptor-enhanced PKBsignalling. These data are consistent with observations madein platelets where inhibition of PKC with BisI attenuatedthrombin-induced PKB phosphorylation. This cascade pro-ceeds by Gq protein- and PKCδ-mediated release of ADPthat, on its turn, activates PKB upon P2Y12 receptor binding[50,59]. On the other hand, PKCδ might also contribute to thebasal PKB activity by interaction with Src and subsequentEGFR transactivation as reported in human U-1242 glio-blastoma cells [63].
Our observations that the PKCζ pseudosubstratesignificantly inhibited PKB phosphorylation in control and2MeSADP-stimulated cells indicated that atypical PKCs areimportant regulators of PKB signalling in C6 cells. In somecells PKCζ activity has been shown to attenuate PKB signallingthrough direct inhibition [64] or by Ser phosphorylation of IRS-I serving as a negative feedback for insulin-induced PI 3-K/PKB signalling [65]. However, our data are consistent with arecent study that revealed a role for PKCζ in astrocytomaprogression [66]. In C6 cells PKCζ and NF-κB contribute to IL-1- and TNFα-induced invasiveness by regulation of theexpression of MMP9 [67]. Since activation of PKB is involvedin the proliferative and invasive properties of glioma cells [68],these data suggest that PKCζ also exerts its pro-tumoral effectvia activation of PKB. It is still not clear whether PKCζ isactivated upon P2Y12 receptor stimulation or that itsconstitutive activity in C6 cells is sufficient for activation ofPKB.
Besides Ca2+ mobilization, PLD2 and Src activation,transfection experiments indicated that P2Y12 receptor-enhanced phosphorylation of PKB involves Rap1 and Gβγ-subunits. Moreover, activation of Rap1 was confirmed withGTP pull-down assays that revealed a rapid GTP-loadingupon stimulation with 2MeSADP. Involvement of the P2Y12
receptor in this effect was confirmed by the use of MRS2395,a specific P2Y12 receptor antagonist [56]. Rap signalling bythe P2Y12 receptor is also reported in platelets but in contrastto our results, this response is mediated by PI 3-K [39,40]. In
C6 cells, Rap1 inhibition by cAMP is shown to attenuate PI3-K/PKB signalling suggesting that Rap1 acts upstream of PI3-K in these cells [31]. Indeed, treatment of cells withLY294002 did not affect P2Y12 receptor-induced activation ofRap1 while PI 3-K/PKB signalling is abolished in RapGAP-transfected cells, indicating that P2Y12 receptor-mediatedPKB phosphorylation is mediated by Rap1. Since the P2Y12
receptor is negatively coupled to AC, Rap1 might beactivated by a Gi protein-dependent decrease in cAMP synthesis.Alternatively, Src is reported to activate Rap1 by phos-phorylation of the RapGEFC3G [28]. In our experiments, P2Y12
receptor-mediated activation of Rap1 was abolished by PP2supporting an involvement of Src. However, similar to PKBphosphorylation, Rap1 signalling was also inhibited by Ca2+
chelation and inhibition of PLD2, pointing to a more complexmechanism.
An important mechanism involved in Gi protein-mediatedactivation of PI 3-K/PKB is transactivation of growth factorreceptors. Stimulation of vascular smooth muscle cells withangiotensin II activates PI 3-K upon transactivation of IGF-IR while in CHO cells, stimulation of the P2Y12 receptorinduces PDGFR tyrosine phosphorylation leading to PKBand ERK activation [32,69]. Although experiments withAG1024 and AG1296 pointed to an involvement of IGF-IRinstead of PDGFR, analysis of receptor tyrosine phosphory-lation excluded a direct transactivation mechanism uponstimulation of the P2Y12 receptor. Interestingly, in IGF-IRimmunoprecipitates, we observed an increased phosphory-lation of Pyk2 upon P2Y12 receptor stimulation. Pyk2, amember of the focal adhesion protein kinase family, becomesactivated by stimuli that increase the intracellular Ca2+
concentration like LPA, carbachol and thrombin by a PKC-dependent mechanism [70]. It has been demonstrated thatPyk2 cooperates with Src to activate the Grb2/SOS/Ras/Raf/MEK/ERK cascade upon P2Y2 receptor stimulation [71]. Inastrocytes, stimulation of Gq protein-coupled protease activatedreceptor-1 induces phosphorylation of Pyk2 on Tyr402 thatserves as binding site for Src that in turn phosphorylates Tyr881to facilitate binding of Grb2 leading to ERK activation [72].Besides ERK signalling, cross-talk between Pyk2 and PI 3-K-dependent signalling is also reported. In platelets, Pyk2phosphorylation induced by procoagulant agonists like ADP andthrombin is mediated by PI 3-K while in PC12 cells H2O2
induces a PLD2-dependent association of Pyk2/Src that actsupstream of PI 3-K [44,73]. Our experiments reveal thatstimulation of the P2Y12 receptor induces a transientphosphorylation of Pyk2 at Tyr402 and Tyr597, that depends onCa2+ mobilization and PLD2. The latter enzyme is constitutivelyactive in C6 cells and is located in lipid microdomains(caveolae). Disruption of these structures by methyl-β-cyclo-dextrin induces inhibition of PLD2 [45]. In these conditions,P2Y12 receptor-increased Pyk2 phosphorylation at Tyr597 wasabolished while phosphorylation at Tyr402 was almostundetectable confirming the necessity of caveolae-associatedPLD2 activity in the P2Y12 receptor-mediated activation of thePyk2/Src complex. The inhibition of Pyk2 phosphorylation byAG1024 demonstrated the involvement of basal IGF-IR

PI 3-K*
P2Y12
Giα
ADPIGF-R
P
P
P
P
PI(4,5)P2
Ca2+
PLD
2
PKB*
PKCζSrc
Pyk2*
PDK
Rap1*
Gβγ
PI(3,4,5)P3
RapGAP PP2
BAPTA β-ARK1
1-butanol
AG1024
Lipid Rafts
methyl-β-CD
123
4
5
6
Ca2+influx*
COO-
+H3N
Fig. 8. Mechanism of P2Y12 receptor-mediated PKB activation. P2Y12 receptor stimulation induces Gβγ protein-mediated Ca2+ mobilization (1) leading to a rapidactivation of Pyk2/Src (2). Pyk2/Src signalling requires caveolae-associated constitutive PLD2 activity and autocrine IGF-IR signalling that is not enhanced uponP2Y12 receptor stimulation but serves as a docking site to facilitate interaction between signalling components (3). Coordinated Pyk2/Src/PLD2 signalling inducesactivation of Rap1 (4) which in turn activates the PI 3-K/PKB cascade (5, 6). PKCδ is involved in PKB phosphorylation in control cells, while PKCζ is implicated inP2Y12 receptor-mediated activation of PKB. The asterisks “*” are demonstrated activations upon P2Y12 receptor stimulation. The P2Y12-dependent Ca
2+ influx wasreported in Grobben et al. [22]. Activation of Rap1 and Pyk2 is shown in this study. Activation of PI 3-K and PKB was published by Czajkowski et al. [9] and by VanKolen and Slegers [10].
1179K. Van Kolen et al. / Cellular Signalling 18 (2006) 1169–1181
tyrosine kinase activity that is not significantly increased by2MeSADP.
The signal transduction cascade towards enhanced phos-phorylation of PKB upon activation of the P2Y12 receptor isschematically presented in Fig. 8. As mentioned above,activation of Pyk2 requires Ca2+ mobilization upon activationof the P2Y12 receptor. Since this response is mediated byGβγ-subunits [74,75] the inhibitory effect of β-ARK on PKBsignalling is probably due to inhibition of Ca2+ mobilization.The fact that PKB activation by the P2Y12 receptor is notcompletely abolished in the presence of βγ-subunit scav-engers, may be explained by partial Giα-mediated activationof Src [28]. It is interesting to mention that in plateletsstimulation of the P2Y12 receptor also activates Rap1 andPyk2, but these responses are positioned downstream of PI3-K indicating that activation of this module is cell typespecific [39,40,73]. Since activation of Pyk2 is often associatedwith ERK signalling one might expect that a similar mechanismis used by the P2Y12 receptor to activate ERK. Previously weexcluded the involvement of Ca2+ mobilization, activation oftyrosine kinases and PLD2 in P2Y12 receptor-mediatedactivation of ERK in C6 cells [22] indicating that the PKB andERK cascades proceed through separate mechanisms pointingto compartmentalization of these signalling components.
Since Pyk2 co-immunoprecipitated with IGF-IR and itsphosphorylation was attenuated by the tyrosine kinase inhibitorAG1024 but also by the PLD2 inhibitor 1-butanol, a concertedaction of IGF-IR and PLD2 can be suggested (Fig. 8). Such aphenomenon is already reported in CHO cells where PLD isinvolved in IGF-mediated ERK activation [58]. Since autocrineIGF stimulation is demonstrated in C6 cells [31], thismechanism may contribute to the constitutive activity ofcaveolae-associated PLD2 [45]. Therefore, it can be expected
that the effect of methyl-β-cyclodextrin on the signallingmechanism is due to inhibition of PLD2. On the other hand,cholesterol depletion might also affect IGF-IR signalling whichcan be activated by direct interaction with caveolin or can becompartmentalized in caveolae [76]. We also demonstrated thatinhibition of Pyk2/Src with 1-butanol abolished downstreamsignalling towards Rap1 and PKB signalling (Fig. 8). SincePyk2 and PLD2 activity are regulated by PKC, these findingsmay explain the inhibitory effect of BisI and BisIX on PKBphosphorylation.
From the presented data we may conclude thatstimulation of the P2Y12 receptor induces a Gβγ- and Ca2+-dependent activation of the Pyk2/Src/PLD2 complex thatassociates with IGF-IR in caveolae and induces GTP-loadingof Rap1 leading to PI 3-K/PKB signalling. The assembly ofthe Pyk2/Src/PLD2 complex may contribute to compart-mentalization of the signalling pathway. Further character-ization of signal transduction components will provide moreinsight in the cellular specificity of G protein-coupledreceptor responses.
Acknowledgements
We thank Dr. J. Bos (Department of PhysiologicalChemistry, University Medical Center Utrecht, the Nether-lands), Dr. R.J. Lefkowitz (Duke University Medical Center,Durham, NC, USA) and Dr. T. Giesemann (Institute forExperimental and Clinical Pharmacology and Toxicology,Freiburg, Germany) for kindly providing the pMT2HA-Rap1GAPII construct, the pRK-βARKI construct and Toxin B,respectively. This work was supported by grants from the Fundfor Scientific Research Flanders (HS/ELE), Concerted ResearchAction (ELE/LM/HS) and BOF-NOI (HS/LM) of the

1180 K. Van Kolen et al. / Cellular Signalling 18 (2006) 1169–1181
University of Antwerp. KVK is a fellow of the Institute ofScientific Technology (IWT). KG is a fellow of BOF-NOI. Dr.J. Goris (Department of Biochemistry, Faculty of Medicine,Catholic University of Leuven) is acknowledged for helpfuldiscussions.
References
[1] M.P. Abbracchio, M.J. Saffrey, V. Höpker, G. Burnstock, Neuroscience 59(1994) 67.
[2] G. Burnstock, M. Williams, J. Pharmacol. Exp. Ther. 295 (2000) 862.[3] G. Burnstock, Arterioscler. Thromb. Vasc. Biol. 22 (2002) 364.[4] V. Ralevic, G. Burnstock, Pharmacol. Rev. 50 (1998) 413.[5] M.P. Rathbone, P.J. Middlemiss, J.W. Gysbers, C. Andrew, M.A. Herman,
J.A. Reed, R. Cicarelli, P. Di Ioro, F. Caciagli, Prog. Neurobiol. 59 (1999)663.
[6] Y. Shinozaki, S. Koizumi, K. Ishida, J.I. Sawada, Y. Ohno, K. Inoue, Glia49 (2005) 288.
[7] P. Claes, B. Grobben, K. Van Kolen, D. Roymans, H. Slegers, Br. J.Pharmacol. 134 (2001) 402.
[8] P. Claes, K. Van Kolen, D. Roymans, D. Blero, K. Vissenberg, C. Erneux,J.P. Verbelen, E.L. Esmans, H. Slegers, Biochem. Pharmacol. 67 (2004)1489.
[9] R. Czajkowski, W. Banachewicz, O. Ilnytska, L.B. Drobot, J. Baranska,Br. J. Pharmacol. 141 (2004) 497.
[10] K. Van Kolen, H. Slegers, J. Neurochem. 89 (2004) 442.[11] T. Kubiatowski, T. Jang, M.B. Lachyankar, R. Salmonsen, R.R. Nabi, P.J.
Quesenberry, N.S. Litofski, A.H. Ross, L.D. Recht, J. Neurosurg. 95(2001) 480.
[12] D. Roymans, H. Slegers, Eur. J. Biochem. 268 (2001) 487.[13] B. Grobben, P.P. De Deyn, H. Slegers, Cell Tissue Res. 310 (2002) 257.[14] N. Okumura, K. Takimoto, M. Okada, H. Nakagawa, J. Biochem. 106
(1989) 904.[15] M. Resnicoff, C. Sell, M. Rubini, D. Coppola, D. Ambrose, R. Baserga, R.
Rubin, Cancer Res. 54 (1994) 2218.[16] L.M. Strawn, E. Mann, S.S. Elliger, L.M. Chu, L.L. Germain, G.
Niederfellner, A. Ullrich, L.K. Shawver, J. Biol. Chem. 269 (1994) 21215.[17] R. Czajkowski, L. Lei, P. Sabala, J. Baranska, FEBS Lett. 513 (2002) 179.[18] R.A. Nicholas, E.R. Lazarowski, W.C. Watt, Q. Li, J. Boyer, T.K. Harden,
J. Auton. Pharm. 16 (1996) 319.[19] M.T. Tu, S.F. Luo, C.C. Wang, C.S. Chien, C.T. Chiu, C.C. Lin, C.M.
Yang, Br. J. Pharmacol. 129 (2000) 1481.[20] P. Claes, H. Slegers, Curr. Neuropharmacol. 2 (2004) 207.[21] R. Czajkowski, J. Baranska, Acta Biochim. Pol. 49 (2002) 877.[22] B. Grobben, P. Claes, K. Van Kolen, D. Roymans, P. Fransen, S.U. Sys, H.
Slegers, J. Neurochem. 78 (2001) 1325.[23] J.M. Dickenson, Br. J. Pharmacol. 135 (2002) 1967.[24] T.F. Franke, S.I. Yang, T.O. Chan, K. Datta, A. Kazlauskas, D.K.
Morrison, D.R. Kaplan, P.N. Tsichlis, Cell 81 (1995) 727.[25] L. Iacovelli, V. Bruno, L. Salvatore, D. Melchiorri, R. Gradini, A.
Caricasole, E. Barletta, A. De Blasi, F. Nicoletti, J. Neurochem. 82 (2002)216.
[26] C. Murga, L. Laguinge, R. Wetzker, A. Cuadrado, J.S. Gutkind, J. Biol.Chem. 273 (1998) 19080.
[27] M.G. Sanchez, L. Ruiz-Llorente, A.M. Sancez, I. Diaz-Laviada, Cell.Signal. 15 (2003) 851.
[28] J.M. Schmitt, P.J.S. Stork, J. Biol. Chem. 277 (2002) 43024.[29] R.K. Bommakanti, S. Vinayak, F. Simonds, J. Biol. Chem. 275 (2000)
38870.[30] F.C. Mei, J. Qiao, O.M. Tsygankova, J.L. Meinkoth, L.A. Quilliam, X.
Cheng, J. Biol. Chem. 277 (2002) 11497.[31] L. Wang, F. Liu, M.L. Adamo, J. Biol. Chem. 276 (2001) 37242.[32] C. Soulet, V. Sauzeau, M. Plantavid, J.M. Herbert, P. Pacaud, B. Payrastre,
P. Savi, J. Thromb. Haemost. 2 (2004) 135.[33] X. Tang, I.H. Batty, C.P. Downes, J. Biol. Chem. 277 (2002) 338.[34] L.M. Ballou, M.E. Cross, S. Huang, E.M. McReynolds, B.X. Zhang, R.Z.
Lin, J. Biol. Chem. 275 (2000) 4803.
[35] L.M. Ballou, H.-Y. Lin, G. Fan, Y.-P. Jiang, R.Z. Lin, J. Biol. Chem. 278(2003) 23472.
[36] I.H. Batty, I.N. Fleming, C.P. Downes, Biochem. J. 379 (2004) 641.[37] E.V. Gerasimovskaya, D.A. Tucker, M. Weiser-Evans, J.M. Wenzlau, D.J.
Klemm, M. Banks, K.R. Stenmark, J. Biol. Chem. 280 (2005) 1838.[38] A. Huwiler, J. Pfeilschifter, Br. J. Pharmacol. 113 (2002) 1455.
[39] P. Lova, S. Paganini, F. Sinigaglia, C. Balduini, M. Torti, J. Biol. Chem.277 (2002) 12009.
[40] P. Lova, S. Paganini, E. Hirsch, L. Barberis, M. Wymann, F. Sinigaglia, C.Balduini, M. Torti, J. Biol. Chem. 278 (2003) 131.
[41] H. Slegers, M. Joniau, J. Neurochem. 66 (1996) 466.[42] A. Shevchenko, I. Chernushevich, M. Wilm, M. Mann, Methods Mol.
Biol. 146 (2000) 1.[43] S. Yano, H. Tokumitsu, T.R. Soderling, Nature 396 (1998) 584.[44] Y. Banno, K. Ohguchi, N. Matsumoto, M. Koda, M. Ueda, A. Hara, I.
Dikic, Y. Nozawa, J. Biol. Chem. 280 (2005) 16319.[45] M. Bobeszko, P. Krzeminski, P. Pomorski, A. Dygas, J. Baranska,
Biochem. Biophys. Res. Commun. 317 (2004) 689.[46] S.F. Steinberg, Biochem. J. 384 (2004) 449.[47] Y. Ueda, S.I. Hirai, S.I. Osada, A. Suzuki, K. Mizuno, S. Ohno, J. Biol.
Chem. 271 (1996) 23512.[48] M. Blass, I. Kronfeld, G. Kazimirsky, P.M. Blumberg, C. Brodie, Mol.
Cell. Biol. 22 (2002) 182.[49] I. Kronfeld, G. Kazimirsky, P.S. Lorenzo, S.H. Garfield, P.M. Blumberg,
C. Brodie, J. Biol. Chem. 275 (2000) 35491.[50] S. Murugappan, F. Tuluc, R.T. Dorsam, H. Shankar, S.P. Kunapuli, J. Biol.
Chem. 279 (2004) 2360.[51] E.H.Wu, Y.H.Wong, Cell. Signal. 18 (2006) (Electronic publication ahead
of print), doi:10.1016/j.cellsig.2005.04.009.[52] W.J. Koch, B.E. Hawes, J. Inglese, L.M. Luttrett, R.J. Lefkowitz, J. Biol.
Chem. 269 (1994) 6193.[53] F. Greco, F. Sinigaglia, C. Balduini, M. Torti, J. Thromb. Haemost. 2
(2004) 2223.[54] J.L. Bos, Curr. Opin. Cell Biol. 17 (2005) 123.[55] M. Reuveny, H. Heller, E. Bengal, FEBS Lett. 569 (2004) 129.[56] B. Xu, A. Stephens, G. Kirschenheuter, A.F. Greslin, X. Cheng, J. Sennelo,
M. Cattaneo, M.L. Zighetti, A. Chen, S.A. Kim, H.S. Kim, N.Bischofberger, G. Cook, K.A. Jacobson, J. Med. Chem. 45 (2002) 5694.
[57] G.R. Guy, P. Yusoff, D. Bangarusamy, C.W. Fong, E.S. Wong, Cell.Signal. 14 (2002) 11.
[58] Y. Banno, Y. Takuwa, M. Yamada, N. Takuwa, K. Ohguchi, A. Hara, Y.Nozawa, Biochem. J. 369 (2003) 363.
[59] S. Kim, J. Jin, S.P. Kunapuli, J. Biol. Chem. 279 (2004) 4186.[60] E. Hirsch, O. Bosco, P. Tropel, M. Laffargue, R. Calvez, F. Altruda, M.
Wymann, G. Montrucchio, FASEB J. 15 (2001) 2019.[61] B. Stoyanov, S. Volinia, T. Hanck, I. Rubio, M. Loubtchenkov, D. Malek,
S. Stoyanova, B. Vanhaesebroeck, R. Dhand, B. Nürnberg, P. Gierschik, K.Seedorf, J.J. Hsuan, M.D. Waterfield, R. Wetzker, Science 269 (1995) 690.
[62] C. Kroner, K. Eybrechts, J.W.N. Akkerman, J. Biol. Chem. 275 (2000)27790.
[63] S. Amos, P.M. Martin, G.A. Polar, S.J. Parsons, I.M. Hussaini, J. Biol.Chem. 280 (2005) 7729.
[64] M. McConkey, H. Gillin, C.R. Webster, M.S. Anwer, J. Biol. Chem. 279(2004) 20882.
[65] Y.F. Liu, K. Paz, A. Herschkovitz, A. Alt, T. Tennenbaum, S.R. Sampson,M. Ohba, T. Kuroki, D. LeRoith, Y. Zick, J. Biol. Chem. 276 (2001)14459.
[66] A. Xiao, C. Yin, C. Yang, A. Di Cristofano, P.P. Pandolfi, T. Van Dyke,Cancer Res. 65 (2005) 5172.
[67] P.O. Estève, E. Chicoine, O. Robledo, F. Aoudjit, A. Descoteaux, E.F.Potworowski, Y. St-Pierre, J. Biol. Chem. 277 (2002) 35150.
[68] P. Pu, C. Kang, J. Li, H. Jiang, Tumour Biol. 25 (2004) 172.[69] P. Zahradka, B. Litchie, B. Storie, G. Helwer, Endocrinology 145 (2004)
2978.[70] H. Avraham, S.Y. Park, K. Shinkmann, S. Avraham, Cell. Signal. 12
(2000) 123.[71] S.P. Soltoff, H. Avraham, S. Avraham, L.C. Cantley, J. Biol. Chem. 273
(1998) 2653.

1181K. Van Kolen et al. / Cellular Signalling 18 (2006) 1169–1181
[72] H. Wang, G. Reiser, J. Neurochem. 84 (2003) 1349.[73] K. Koziak, E. Kaczmarek, S.Y. Park, Y. Fu, S. Avraham, H. Avraham, Br.
J. Haematol. 141 (2001) 134.[74] A.K. Filippov, J.M. Fernandez-Fernandez, S.J. Marsh, J. Simon, E.A.
Barnard, D.A. Brown, Mol. Pharmacol. 66 (2004) 468.
[75] J. Simon, A.K. Filippov, S. Göransson, Y.H. Wong, C. Frelin, A.D. Michel,D.A. Brown, E.A. Barnard, J. Biol. Chem. 277 (2002) 31390.
[76] Y. Ishikawa, K. Otsu, J. Oshikawa, Cell. Signal. 17 (2005) 1175.
Copyright © 2022 FDOKUMEN




















