
285 Ans. Ans. UT = (744.4 cos 15°)(25) = 18.0 A103B ft # lb T ...

Molecular analysis of the cos region of the Lactobacilluscasei bacteriophage A2. Gene product 3, gp3,specifically binds to its downstream cos region
Pilar Garcı´a,1 Juan Carlos Alonso 2 and Juan EvaristoSuarez1*1Area de Microbiologıa and Instituto Universitario deBiotecnologıa de Asturias, Facultad de Medicina,Universidad de Oviedo, 33006 Oviedo, Spain.2Centro Nacional de Biotecnologıa. CSIC, Campus deCanto Blanco, 28034 Madrid, Spain.
Summary
The terminal nucleotide sequence of the Lactobacilluscasei bacteriophage A2 DNA revealed a single-stran-ded extension 13 bases in length (5 8-AACGGTCGGC-CTC-38) at its 3 8 termini that defines the packaginginitiation nicking site ( cosN ). The cosN sequence isbisected by an axis of hyphenated twofold rotationalsymmetry. Directly and inverted repeated sequenceslocated to the left ( cosL ) and the right ( cosR ) of thecosN site were observed. Analysis of the 3.4 kb Eco RIDNA sequence surrounding the cos region revealedfour complete and one incomplete open reading frames(orf s). Northern blots indicated that all were cotran-scribed in a single mRNA molecule in excess of10 kb that appeared late during infection. Minicellstudies indicated that the four orf s were translatedinto protein. From the ORF3 amino acid sequenceDNA-binding and NTP-binding domains can be pre-dicted. The purified ORF3 (predicted molecular mass16.8 kDa) shows specific binding to the A2 cos region,so it was renamed gp3. Gp3 forms a specific complexwith a 369 bp cos DNA segment in the presence ofATP. Gp3 interaction with the intrinsically bent cosDNA segment induces intramolecular ligation in thepresence of T4 DNA ligase. The data presented heresuggest that gp3 is the small subunit of the terminaseenzyme.
Introduction
Mesophilic lactobacilli constitute the vast majority of the sec-ondary microbiota involved in the ripening of many dairy
products. Furthermore, they are crucial in fermentationprocesses involving meats, vegetables, artisan cheeses,yakult, etc. (Buckenhuskes, 1993; McKay and Baldwin,1990). Finally, the interest in them is reinforced by theirprobiotic role (Bottazzi and Mercenier, 1994). By analogywith Lactococcus-driven industrial fermentations it is assu-med that bacteriophage attack will be recognized as themain cause of fermentation failure, once these processesbecome as highly standardized as is the manufacture ofdairy products (Accolas et al., 1994). This is promotingthe study of phages that infect lactobacilli and of mechan-isms of defence that impair their development. Further-more, phages are a source of useful genetic tools thatmay help to improve the manipulation of these industriallyand health relevant bacteria.
A2 is a temperate bacteriophage that infects strains ofLactobacillus casei and Lactobacillus paracasei. Its gen-ome is contained in a double-stranded DNA (dsDNA)molecule 44 kb long with 38-protruding single-strandedcohesive ends (Herrero et al., 1994). The 38-protrudingsingle-stranded DNA (ssDNA) ends are of complementarybase sequence and it is likely that they result from theaction of a phage-encoded terminase. Terminases are het-ero-oligomeric proteins, made of two subunits, which med-iate the specific interaction between the prohead of thevirus and its DNA. In addition, they are endonucleasesthat direct packaging of the DNA into a preformed shellin an ATP-consuming reaction. ATP also acts as an allos-teric ligand which enhances the specificity of bindingbetween terminase and phage DNA (reviewed by Black,1989). Two general principles for packaging of concate-meric DNA into a virus head have been proposed. Oneimplies a ‘headful’ packaging, in which the packaging initi-ates at a specific site, termed pac (e.g. in phages P22, P1,SPP1), but with the capacity of the prohead playing apredominant role in the termination step. In this case theterminase initiates DNA packaging by binding at the pacregion and cleaving at the pacC site. DNA packaging ter-minates when the DNA inside the head is separatedfrom the concatemer by a cutting process (headful cut)that occurs in a sequence-independent manner. Theheadful cleavage generates a new end that serves asstarting point for the second round of DNA packaging.Headful cleavage generates a heterogeneous population
Molecular Microbiology (1997) 23(3), 505–514
Q 1997 Blackwell Science Ltd
Received 10 May, 1996; revised 25 October, 1996; accepted 31 Octo-ber, 1996. *For correspondence. E-mail [email protected]; Tel. (8) 5103559; Fax (8) 5103148.
m
of terminally redundant and circularly permuted DNA mole-cules (reviewed by Black, 1989). The second principleimplies a site-specific packaging that shows some constraintin DNA size, in which the recognition sequence (termedcos ) plays an important role in initiation and termination ofpackaging. In the case of phage l the cos region is subdi-vided into three discrete sites (cosQ, cosN, and cosB).The terminase, with the help of accessory proteins (specific(IHF) or non-specific (HU) DNA-binding and DNA-bendingproteins (Shinder et al., 1988; Xin and Feiss, 1988;Mendelson et al., 1991)), forms a nucleoprotein complexat cosB (the DNA end to be encapsidated) and introducesstaggered nicks at the adjacent cosN site. After DNA trans-location into the prohead the encapsidation cycle is endedby binding of the terminase to cosQ (the non-encapsidatedend in the first event of the packaging series) and introdu-cing staggered nicks at cosN, generating the cohesiveends found in the virion DNA (Cue and Feiss, 1993). Clea-vage at cosN, which serves as the starting point for a secondround of DNA packaging, generates unit-length virion DNA.
Here we report the characterization of the cos-contain-ing region of bacteriophage A2 DNA and the identificationof cosN. We include data that may indicate that a proteinencoded in its vicinity (gp3) is the small subunit of thephage terminase.
Results
Determination of the terminal sequence ofbacteriophage A2 DNA
The DNA of bacteriophage A2 has 38-protruding comple-mentary cohesive ends which allows it to form circles or
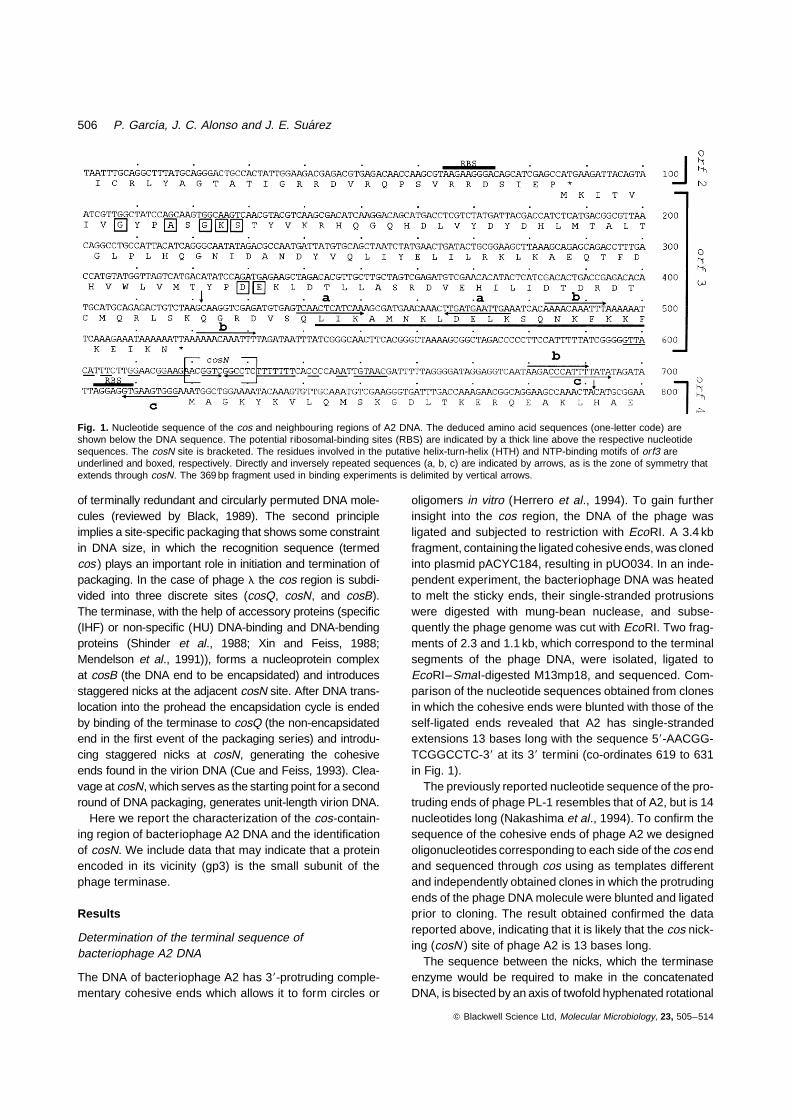
oligomers in vitro (Herrero et al., 1994). To gain furtherinsight into the cos region, the DNA of the phage wasligated and subjected to restriction with EcoRI. A 3.4 kbfragment, containing the ligated cohesive ends, was clonedinto plasmid pACYC184, resulting in pUO034. In an inde-pendent experiment, the bacteriophage DNA was heatedto melt the sticky ends, their single-stranded protrusionswere digested with mung-bean nuclease, and subse-quently the phage genome was cut with EcoRI. Two frag-ments of 2.3 and 1.1 kb, which correspond to the terminalsegments of the phage DNA, were isolated, ligated toEcoRI–SmaI-digested M13mp18, and sequenced. Com-parison of the nucleotide sequences obtained from clonesin which the cohesive ends were blunted with those of theself-ligated ends revealed that A2 has single-strandedextensions 13 bases long with the sequence 58-AACGG-TCGGCCTC-38 at its 38 termini (co-ordinates 619 to 631in Fig. 1).
The previously reported nucleotide sequence of the pro-truding ends of phage PL-1 resembles that of A2, but is 14nucleotides long (Nakashima et al., 1994). To confirm thesequence of the cohesive ends of phage A2 we designedoligonucleotides corresponding to each side of the cos endand sequenced through cos using as templates differentand independently obtained clones in which the protrudingends of the phage DNA molecule were blunted and ligatedprior to cloning. The result obtained confirmed the datareported above, indicating that it is likely that the cos nick-ing (cosN ) site of phage A2 is 13 bases long.
The sequence between the nicks, which the terminaseenzyme would be required to make in the concatenatedDNA, is bisected by an axis of twofold hyphenated rotational
Q Blackwell Science Ltd, Molecular Microbiology, 23, 505–514
Fig. 1. Nucleotide sequence of the cos and neighbouring regions of A2 DNA. The deduced amino acid sequences (one-letter code) areshown below the DNA sequence. The potential ribosomal-binding sites (RBS) are indicated by a thick line above the respective nucleotidesequences. The cosN site is bracketed. The residues involved in the putative helix-turn-helix (HTH) and NTP-binding motifs of orf3 areunderlined and boxed, respectively. Directly and inversely repeated sequences (a, b, c) are indicated by arrows, as is the zone of symmetry thatextends through cosN. The 369 bp fragment used in binding experiments is delimited by vertical arrows.
506 P. Garcıa, J. C. Alonso and J. E. Suarez

symmetry which extends to at least 29 bases on each sideof the axis (co-ordinates 597 to 654 in Fig. 1).
The region adjacent to cosN contains three directrepeats of 11 bp (labelled b in Fig. 1), two of them at theleft-hand side in our arbitrary drawing (tentatively termedcosL ) and the third, which shows two mismatches, at theright arm (cosR ). Hyphenated inversely repeated sequen-ces of 12 bp and 10 bp could be predicted within cosL andcosR, respectively (labelled a and c, Fig. 1). In addition,several other direct repeats, seven to nine nucleotideslong, were detected (data not shown).
Nucleotide sequence of the A2 cos region
The 3454 bp EcoRI DNA segment of the A2 genome whichcontains the cos region was sequenced. Part of the nuc-leotide sequence of the nonsense strand, which containsmost of the features discussed in this paper, is shown inFig. 1. The overall dG+dC content of the fragment was43.4%, which is well within the range of the average con-tent of lactobacilli (Pouwels and Leer, 1993; Pouwels andLeunissen, 1994). However, in the cosN region the dG+dC content is about 69%. This high dG+dC content ofcosN has also been reported for the Lactococcus phagefLC3 (Lillehaug et al., 1991) and possibly reflects thenecessity of a stable base-pairing of the cohesive endsof the phage DNA to become circularized shortly aftercell infection.
Computer-assisted analysis of the 3.4 kb DNA sequ-ence allowed the prediction of four potential protein codingframes and the 38-end of a fifth on the basis of the length
of the orfs, and the presence of a ribosome-binding site(RBS). By using the above parameters, no orfs could bepredicted from the reading of the complementary strand(data not shown). orf1 to orf3 were located to the left ofcosN. orf2 and orf3 predicted polypeptides of 123 and144 amino acids, respectively. The start codon of eachpolypeptide overlapped with the stop codon of the pre-vious one. orf3 and orf4 are separated by a 200 bp non-coding segment, which includes the cosN site (see above).orf4 is separated from orf5 by an intergenic region of 46nucleotides and predicted polypeptides of 151 and 563amino acids, respectively. It seemed likely that orf1 toorf3 or even all orfs described here formed a single tran-scriptional unit, since no evident rho-independent termina-tor was observed between them and all are in the sameorientation in circularized phage DNA. To test this hypoth-esis total RNA was isolated from L. casei 393 cells at var-ious times after infection with phage A2. This RNA washybridized with DNA probes that included orf3 (co-ordi-nates 574 to 1070; Fig. 1) or the 38 and 58 ends of orf4and orf5, respectively (co-ordinates 1603 to 2431; Fig. 1).In both cases, a major mRNA molecule in excess of 10 kbbecame clearly detectable 100 min after infection (Fig. 2).This indicated that all orfs were cotranscribed and thattheir expression occurred late during infection (A2 one-step growth curves, under the conditions of the experi-ment, show eclipse and latent periods of 110 and140 min, respectively; Herrero et al., 1994).
Proteins encoded in the A2 cos region
The predicted molecular masses of the protein-codingframes under study (ORF2 to ORF5) are 14.5, 16.8, 17and 64.2 kDa in size, respectively. To determine whetherall the orfs were in fact translated into proteins, plasmidpUO034 was introduced into the minicell-producing strainE. coli AR1062 and the products of its expression wereseparated by SDS–PAGE and analysed by fluorography.
Radiolabelled polypeptides with Mr values of approxi-mately 15 500, 17 500, 18 500, and 68 000 were observed(data not shown). The 15.5 and the 68 kDa polypeptidescorrespond to ORF2 and ORF5, respectively; the 17.5 kDaband corresponds to a doublet made by ORF3 and ORF4,and the 18.5 kDa band corresponds to a hybrid proteinbetween the amino-proximal end of the polypeptideencodedby the chloramphenicol-resistance gene of the vector andthe cloned portion of ORF1, since its expected size wouldbe 17.3 kDa. Indirectly, it might be deduced that expres-sion of all proteins occurs from the chloramphenicol-resis-tance promoter, which favours the idea that the five orfsare part of a unique operon.
A computer-based comparison of the deduced aminoacid sequences of the five ORFs with primary proteinsequences currently available in the NBRF–PIR (Release
Q Blackwell Science Ltd, Molecular Microbiology, 23, 505–514
Fig. 2. Northern blot of the mRNA transcribed from the cos-surrounding region of bacteriophage A2 DNA at various times post-infection of L. casei ATCC 393 cells. Two probes encompassing,respectively, orf3 and the junction between orf4 and orf5 (i.e.segments of the DNA located at each side of cosN ) were used andsimilar pictures were obtained. Size markers indicated on the rightwere a commercial RNA ladder.
DNA–protein interaction at the cos region of phage A2 507
47) or the SWISSPROT (Release 32) protein sequencedatabases was performed and no significant identity wasdetected (data not shown). The analysis of ORF3 pre-dicted (i) an NTP-binding domain, with an A-type Walkermotif (G/AXXG/AXGKT/S) between residues 8 and 15,and a nucleotide-binding pocket, DE (motif B), corre-sponding to residues 82 and 83 (Walker et al., 1982),and (ii) a helix-turn-helix DNA-binding motif (Pabo andSauer, 1984; Brennan and Matthews, 1989) between resi-dues 120 and 140 (Fig. 1).
Overexpression and purification of ORF3
In general the genome organization and map position ofthe DNA-packaging genes are highly conserved amongthe different bacteriophages from Gram-positive andGram-negative bacteria, indicating that their location hasa strong functional advantage (reviewed by Black, 1989).Hence, we hypothesize that the product of orf3 might bethe small subunit of the terminase enzyme because ofits size (16.8 kDa), location (in the vicinity of cosN ) andfunctional motifs (ATP- and DNA-binding sites). To testthis hypothesis we overproduced and purified ORF3through expression of the corresponding gene under thecontrol of the P10 promoter of T7 in the presence of rifam-picin (see the Experimental procedures).
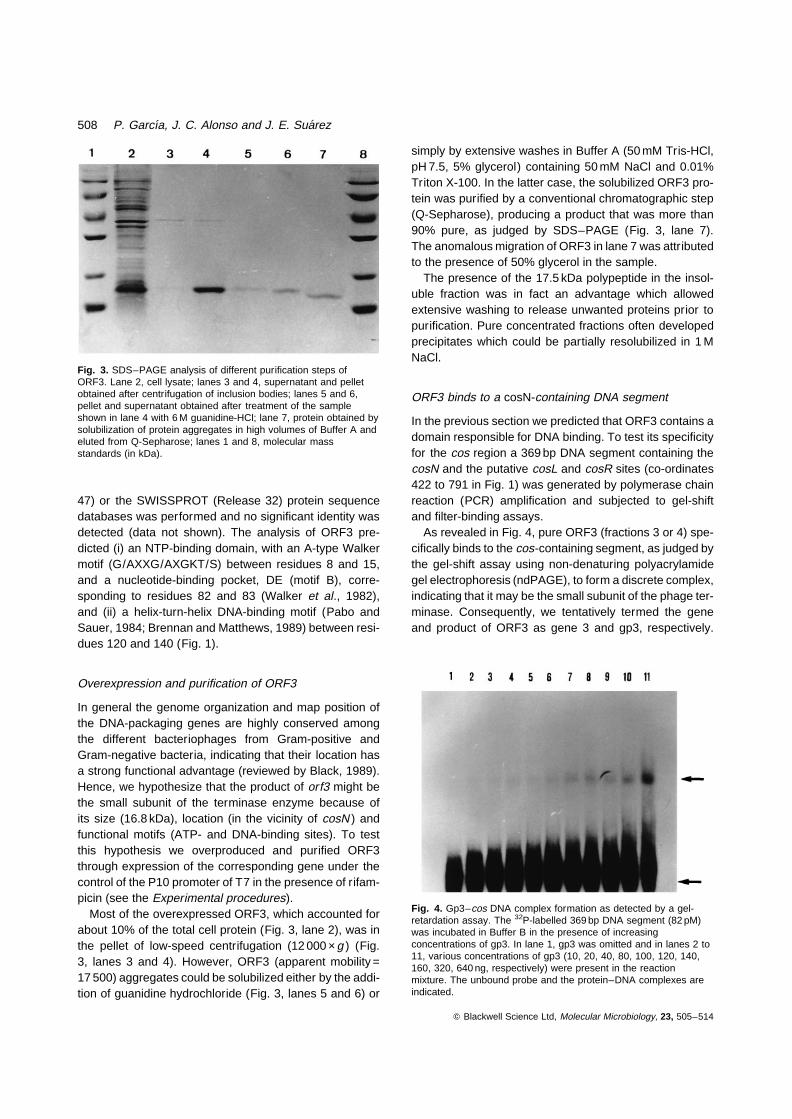
Most of the overexpressed ORF3, which accounted forabout 10% of the total cell protein (Fig. 3, lane 2), was inthe pellet of low-speed centrifugation (12 000 ×g ) (Fig.3, lanes 3 and 4). However, ORF3 (apparent mobility =17 500) aggregates could be solubilized either by the addi-tion of guanidine hydrochloride (Fig. 3, lanes 5 and 6) or
simply by extensive washes in Buffer A (50 mM Tris-HCl,pH 7.5, 5% glycerol) containing 50 mM NaCl and 0.01%Triton X-100. In the latter case, the solubilized ORF3 pro-tein was purified by a conventional chromatographic step(Q-Sepharose), producing a product that was more than90% pure, as judged by SDS–PAGE (Fig. 3, lane 7).The anomalous migration of ORF3 in lane 7 was attributedto the presence of 50% glycerol in the sample.
The presence of the 17.5 kDa polypeptide in the insol-uble fraction was in fact an advantage which allowedextensive washing to release unwanted proteins prior topurification. Pure concentrated fractions often developedprecipitates which could be partially resolubilized in 1 MNaCl.
ORF3 binds to a cosN-containing DNA segment
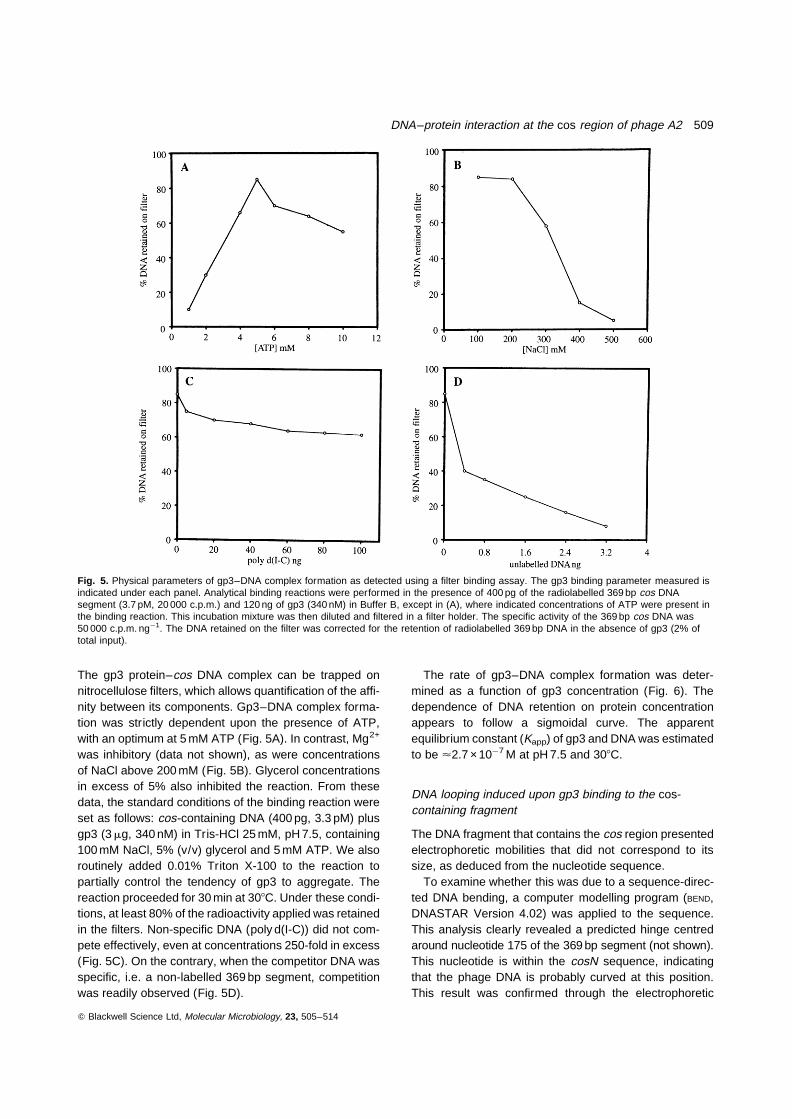
In the previous section we predicted that ORF3 contains adomain responsible for DNA binding. To test its specificityfor the cos region a 369 bp DNA segment containing thecosN and the putative cosL and cosR sites (co-ordinates422 to 791 in Fig. 1) was generated by polymerase chainreaction (PCR) amplification and subjected to gel-shiftand filter-binding assays.
As revealed in Fig. 4, pure ORF3 (fractions 3 or 4) spe-cifically binds to the cos-containing segment, as judged bythe gel-shift assay using non-denaturing polyacrylamidegel electrophoresis (ndPAGE), to form a discrete complex,indicating that it may be the small subunit of the phage ter-minase. Consequently, we tentatively termed the geneand product of ORF3 as gene 3 and gp3, respectively.
Q Blackwell Science Ltd, Molecular Microbiology, 23, 505–514
Fig. 3. SDS–PAGE analysis of different purification steps ofORF3. Lane 2, cell lysate; lanes 3 and 4, supernatant and pelletobtained after centrifugation of inclusion bodies; lanes 5 and 6,pellet and supernatant obtained after treatment of the sampleshown in lane 4 with 6 M guanidine-HCl; lane 7, protein obtained bysolubilization of protein aggregates in high volumes of Buffer A andeluted from Q-Sepharose; lanes 1 and 8, molecular massstandards (in kDa).
Fig. 4. Gp3–cos DNA complex formation as detected by a gel-retardation assay. The 32P-labelled 369 bp DNA segment (82 pM)was incubated in Buffer B in the presence of increasingconcentrations of gp3. In lane 1, gp3 was omitted and in lanes 2 to11, various concentrations of gp3 (10, 20, 40, 80, 100, 120, 140,160, 320, 640 ng, respectively) were present in the reactionmixture. The unbound probe and the protein–DNA complexes areindicated.
508 P. Garcıa, J. C. Alonso and J. E. Suarez
The gp3 protein–cos DNA complex can be trapped onnitrocellulose filters, which allows quantification of the affi-nity between its components. Gp3–DNA complex forma-tion was strictly dependent upon the presence of ATP,with an optimum at 5 mM ATP (Fig. 5A). In contrast, Mg2+
was inhibitory (data not shown), as were concentrationsof NaCl above 200 mM (Fig. 5B). Glycerol concentrationsin excess of 5% also inhibited the reaction. From thesedata, the standard conditions of the binding reaction wereset as follows: cos-containing DNA (400 pg, 3.3 pM) plusgp3 (3 mg, 340 nM) in Tris-HCl 25 mM, pH 7.5, containing100 mM NaCl, 5% (v/v) glycerol and 5 mM ATP. We alsoroutinely added 0.01% Triton X-100 to the reaction topartially control the tendency of gp3 to aggregate. Thereaction proceeded for 30 min at 308C. Under these condi-tions, at least 80% of the radioactivity applied was retainedin the filters. Non-specific DNA (poly d(I-C)) did not com-pete effectively, even at concentrations 250-fold in excess(Fig. 5C). On the contrary, when the competitor DNA wasspecific, i.e. a non-labelled 369 bp segment, competitionwas readily observed (Fig. 5D).
The rate of gp3–DNA complex formation was deter-mined as a function of gp3 concentration (Fig. 6). Thedependence of DNA retention on protein concentrationappears to follow a sigmoidal curve. The apparentequilibrium constant (Kapp) of gp3 and DNA was estimatedto be <2.7 ×10¹7 M at pH 7.5 and 308C.
DNA looping induced upon gp3 binding to the cos-containing fragment
The DNA fragment that contains the cos region presentedelectrophoretic mobilities that did not correspond to itssize, as deduced from the nucleotide sequence.
To examine whether this was due to a sequence-direc-ted DNA bending, a computer modelling program (BEND,DNASTAR Version 4.02) was applied to the sequence.This analysis clearly revealed a predicted hinge centredaround nucleotide 175 of the 369 bp segment (not shown).This nucleotide is within the cosN sequence, indicatingthat the phage DNA is probably curved at this position.This result was confirmed through the electrophoretic
Q Blackwell Science Ltd, Molecular Microbiology, 23, 505–514
Fig. 5. Physical parameters of gp3–DNA complex formation as detected using a filter binding assay. The gp3 binding parameter measured isindicated under each panel. Analytical binding reactions were performed in the presence of 400 pg of the radiolabelled 369 bp cos DNAsegment (3.7 pM, 20 000 c.p.m.) and 120 ng of gp3 (340 nM) in Buffer B, except in (A), where indicated concentrations of ATP were present inthe binding reaction. This incubation mixture was then diluted and filtered in a filter holder. The specific activity of the 369 bp cos DNA was50 000 c.p.m. ng¹1. The DNA retained on the filter was corrected for the retention of radiolabelled 369 bp DNA in the absence of gp3 (2% oftotal input).
DNA–protein interaction at the cos region of phage A2 509

migration behaviour of the segment in 8% ndPAGE at 48Cand 658C (Fig. 7). At the higher temperature, the curvatureof the fragment was lost and consequently it migrated inaccordance with its size. In contrast, at 48C it migratedmuch more slowly (like a c. 600 bp segment) becausebending was maximized. To determine if gp3 induces abend in DNA upon binding we employed the cyclizationor ring-closure assay (Shore and Baldwin, 1983). The pro-cess of formation of a stable loop, which brings distantsegments into close proximity, resembles DNA cyclization.By using a DNA ligation reaction with sticky-ended restric-tion fragments, Shore and Baldwin (1983) showed that theprobability of interaction of two distant sites on an isolatedDNA molecule exhibits a relative maximum when they areseparated by about 500 bp. The intrinsic stiffness of theDNA double helix precludes the spontaneous contact ofDNA ends that are separated by distances significantlyless than the persistence length (in other words the statis-tical segment length determines the probability of cycliza-tion or loop formation). For helical DNA in aqueoussolution at 200 mM Na+ the persistence length is about140 bp (reviewed by Hagerman, 1990). In order to deter-mine if gp3 promoted further bending of the cos-contain-ing segment, the blunt-ended linear DNA fragment wasligated under very diluted conditions, which favour intra-molecular ligation, in the presence of increasing concen-trations of gp3 (10, 20, 40, 80, 160, 320 and 640 nM). Asrevealed in Fig. 8, the presence of 320 or 640 nM of gp3remarkably enhances the cyclization of the segmentwhen compared to the result obtained in its absence(lane 1) or with lower amounts of gp3 (lanes 2 to 6). This
is in good agreement with the Kapp of gp3–cos DNA com-plex formation (270 nM, see above).
Discussion
The importance of lactobacilli to the agro-food and feedindustrial sector and their relevance to human and animalhealth is promoting research that may help in the improve-ment and/or construction of biotechnologically relevantstrains (Kammerer, 1996; Mercenier et al., 1994). Phagesare a rich source of tools which, if properly used, mayincrease the versatility of genetical manipulation of bac-teria. Knowledge of the region surrounding the cohesiveends of the L. casei bacteriophage A2 may be used inthe design of cosmids and at the same time will help inthe elucidation of the packaging mechanism of the phage.
In the case of phage l, the site of action of the terminaseis called the cos site, and it includes the site nicked by theenzyme (cosN ) as well as the binding sites for the termi-nase and accessory protein(s) (cosB and cosQ ) (see theIntroduction ). To maintain the same nomenclature wedesignated the site nicked by the A2 terminase enzymeas cosN. The phage A2 cos region contains, in additionto the 13 nucleotides that form the cohesive ends, a fewneighbouring bases that could play an essential role interminase recognition. First, the cosN sequence isembedded into a 58 bp region of twofold hyphenated rota-tional symmetry. The phosphodiester bonds hydrolysed byterminase are symmetrically placed within this pseudodyad axis. Additionally, inversely and directly repeatedsequences were detected at both sides of cosN. The sig-nificance of these repeated regions, if any, requires furtheranalysis. Furthermore, the determination of the first seg-ment to be encapsidated into a preformed proheadremains to be determined.
Q Blackwell Science Ltd, Molecular Microbiology, 23, 505–514
Fig. 6. Binding of DNA to membrane filters in the presence of gp3.400 pg of 32P-labelled 369 bp cos DNA and increasingconcentrations of gp3 were incubated in Buffer B at 308C for30 min. The quantification of the binding products was as describedin Fig. 5.
Fig. 7. Non-denaturing PAGE run at 48C (A) and at 658C (B) of the399 bp BamHI–HindIII fragment containing cos. Lane 1, l DNAdigested with HindIII; lane 2, pUO035 digested with BamHI–HindIII; lane 3, l DNA digested with Pst I. The arrows indicate themigration of the 564 bp l HindIII DNA fragment.
510 P. Garcıa, J. C. Alonso and J. E. Suarez

Terminases are commonly composed of a small and alarge subunit. The small subunit, which is implicated inthe binding to a specific DNA site, appears to bind andhydrolyse ATP. The large subunit appears to bind to theproheads and may be involved in cutting the phage mole-cules (Black, 1989). Inspection of the primary structure ofgp3 revealed DNA- and ATP-binding motifs. A commonfeature of the packaging models is that the terminasesmall subunit forms a nucleoprotein structure, in a reactionthat is enhanced by ATP, that helps to position the largesubunit to cleave at the pac or cos site (Feiss, 1986; Black,1989; Murialdo, 1991). In the case of phage l or P1, the pur-ified terminase small subunit binds to the cos or pac regionin the presence of an accessory protein with either specific(IHF) or non-specific (HU) DNA-binding and DNA-bendingactivity (Shinder et al., 1988; Xin and Feiss, 1988; Mendel-son et al., 1991; Skorupski et al., 1994). Both IHF and HUbelong to a large family of proteins collectively termed his-tone-like or chromatin-associated proteins (reviewed byDrlica and Rouviere-Yaniv, 1987). However, gp3 of phageA2 binds to the a 369 bp segment that contains the cosregion in the absence of any accessory protein, althoughthe reaction is absolutely dependent upon the presence ofATP. Furthermore, the addition of Hbsu (the Bacillus subtiliscounterpart of E. coli HU protein) neither modified norenhanced the gp3–cos DNA complex formation (our unpub-lished results). Gp3 alone cannot promote cosN cleavage(data not shown). The Kapp for the gp3–cos DNA complexwas estimated to be 270 nM. However, the Kapp value calcu-lated here may be a gross underestimation, because wehave found large aggregates of free gp3 upon storage andalso under the experimental conditions used.
No good homologue to the large subunit of the termi-nase enzyme was found in the 3.4 kb DNA segmentsequenced. The best candidate might be ORF5 becauseof its size, but two points argue against it. First, no ATP-binding motif could be predicted in the orf5 product and,second, no cosN cleavage was observed when both orf3and orf5 were present in the same clone, in spite of thefact that expression of both genes was detected, at leastin Escherichia coli minicells.
The cos region contains an intrinsically bent sequenceand binding of gp3 greatly enhances the cyclization ofthe segment. Furthermore, it is likely that the centre ofthe bend falls in the cosN sequence. We hypothesizedthat the intrinsic bend at cosN is necessary for co-opera-tive interaction between gp3 molecules bound at their cog-nate sites at cosL and cosR. This is consistent with the factthat gp3 interacts co-operatively with a cos-containingDNA segment and that at a gp3 concentration midpointfor gp3–DNA complex formation the protein enhancesDNA ligation.
Taken together these results suggest that gp3 is thesmall subunit of the A2 terminase. The availability of puri-fied gp3 will allow a more detailed study of the gp3domains, the interaction with its target site, and the char-acterization of the large subunit of the terminase enzyme.
Experimental procedures
Bacterial strains, plasmids and bacteriophages
L. casei ATCC 393 was used to propagate bacteriophage A2(Herrero et al., 1994). E. coli HB101, XL-1 Blue, AR1062 andBL21(DE3) were used (Sambrook et al., 1989; Studier andMoffatt, 1986). Strain HB101 was used as recipient of the con-structions made in plasmids pACYC184 (Chang and Cohen,1978) and pUC18 (Yanisch-Perron et al., 1985). Strain XL-1Blue was used to propagate phage M13 derivatives (Yanisch-Perron et al., 1985). Strain AR1062, which was kindly pro-vided by C. Thompson (Universitat Basel, Switzerland), wasthe source of minicells. Strain BL21(DE3) was used in expres-sion studies of genes cloned into plasmid pAR3038 under thecontrol of the P10 promoter of bacteriophage T7 (Studier andMoffatt, 1986).
L. casei and E. coli were grown and maintained on MRS(Biokar) and Luria media (Sambrook et al., 1989), respec-tively. E. coli was made competent and transformed asdescribed by Inoue et al. (1990). Antibiotic-resistance trans-formants were selected using 100 mg ml¹1 ampicillin (Ap),50 mg ml¹1 chloramphenicol (Cm) and/or 50 mg ml¹1 tetracy-cline (Tc). All manipulations with bacteriophage A2 were per-formed according to the standard procedures described byHerrero et al. (1994).
Minicell techniques
E. coli AR1062 was a source of minicells in protein-labellingexperiments. The strain bearing the plasmids of interest wasgrown to mid-exponential phase and minicells were purified
Q Blackwell Science Ltd, Molecular Microbiology, 23, 505–514
Fig. 8. DNA looping induced by gp3 interaction with cos DNA. Theintramolecular interaction of the DNA ends of the blunt-ended399 bp segment that contains A2 cos DNA is revealed by itsenhancement of cyclization. The ligation was performed at a lowDNA concentration (200 ng ml¹1) in the presence of increasingconcentrations of gp3 and 0.3 units of T4 DNA ligase. Lane 1,control without gp3; lanes 2–8, 10, 20, 40, 80, 160, 320 and640 nM of gp3 were added, respectively.
DNA–protein interaction at the cos region of phage A2 511

by centrifugation through continuous sucrose gradients (10–30%), followed by incubation in LB containing 400 mg ml¹1
D-cycloserine. The synthesized proteins were labelled with[35S]-methionine (1103.2 Ci mmol¹1; Amersham) and detec-ted by fluorography as previously described (Villar et al.,1986).
DNA manipulations
Plasmid DNA was obtained by the alkaline method of Birn-boim and Doly (1979). Bacteriophage A2 DNA was preparedas described by Suarez and Chater (1981). Single-strandedclosed circular M13mp18 and M13mp19 DNAs were purifiedas described by Messing (1983). Purification of fragmentswas carried out by electroelution (Sambrook et al., 1989).
Analytical and preparative gel electrophoreses of plasmidDNAs and restriction fragments were carried out in 0.8%(w/v) agarose/Tris-Borate horizontal slab gels or 4% and8% (w/v) polyacrylamide/Tris-Borate gels.
Different DNA segments of phage A2 origin were clonedinto M13mp18 and M13mp19 and sequenced by the dideoxy-nucleotide chain termination method (Sanger et al., 1977)using [35S]-dATP (1200 Ci mmol¹1; Amersham) and a Seque-nase 2.0 DNA sequencing kit (US Biochemicals Corp., Amer-sham). Specific primers were used when no suitable restrictionsites were available. The nucleotide sequence reported herehas been submitted to the EMBL Nucleotide Sequence DataLibraries under Accession Number X97563. Computer-aideddatabase searching and sequence analyses were carriedout using the University of Wisconsin Genetics ComputerGroup programs package (UWGCG; Devereux et al., 1984).
The 369 bp DNA segment containing the cos region of A2was obtained by PCR amplification (see the Results section)and cloned into HindII-cleaved pUC18 to generate pUO035.A 399 bp BamHI–HindIII segment containing the 369 bp cossite was purified by electroelution and end-labelled by fillingin the staggered ends with T7 DNA polymerase in the pre-sence of [a- 32P]-dCTP (3000 Ci mmol¹1) and cold dTTP,dATP and dGTP. In gel retardation assays the 369 bp seg-ment was labelled with [g-32P]-dATP (3000 Ci mmol¹1) usingpolynucleotide kinase (Boehringer Mannheim).
Plasmid pAR3 was constructed as follows: primersupstream (58-CAGCATCGACATATG-38) and downstream(58-GCCGCTTGGATCCCGTG-38) of gene 3 were used toamplify the encompassing region. The primers were designedto introduce NdeI and BamHI restriction sites (underlined).The amplified DNA segment was purified, NdeI- andBamHI-cleaved, and ligated to pAR3038 digested with thesame enzymes to generate plasmid pAR3. The plasmid-borne gene 3 (pAR3) was then introduced into the E. coliBL21[DE3] strain.
Northern blot hybridizations
Cultures of L. casei 393 were grown to A580 = 0.5 and infectedwith A2 at a multiplicity of infection of 5 (time zero). After10 min at 48C to allow adsorption, incubation was continueduntil lysis of the culture was observed (about 150 min). Sam-ples were taken at various time intervals and total RNA waspurified as previously described by Chomczynski and Sacchi
(1987), except that breakage of the cells was achieved by vor-texing with glass beads in denaturing solution. Northern blotanalysis was performed by fractionation of RNA samples(20 mg per lane) on 1.5% agarose gels containing 2% formal-dehyde (Sambrook et al., 1989), followed by transfer to nylonmembrane filters. RNA was fixed to the filter by UV light. RNAsize markers were those of Boehringer Manheim and werestained with ethidium bromide.
The DNA segments used as probes were obtained by PCR,purified by electroelution, and labelled with [a-32P]-dCTP(3000 Ci mmol¹1; Rediprime, Amersham). Hybridization con-ditions were as described by Sambrook et al. (1989). The fil-ters were blot-dried and exposed to a Kodak X-Omat film at¹708C.
Purification of gene product 3
Gp3 was overproduced by the addition of 1 mM IPTG (Boeh-ringer Mannheim) to exponentially growing cultures ofBL21[DE3] cells carrying pAR3. After 30 min, rifampicin (to200 mg ml¹1 final concentration) was added and the cellswere further incubated for 90 min. The cells were harvested,resuspended in Buffer A (50 mM Tris-HCl, pH 7.5, 5% (v/v)glycerol) containing 50 mM NaCl and 1 mg ml¹1 lysozymeand broken by the freeze and thaw procedure. Most of thegp3 protein was in the pellet, which was washed three timeswith 10 vols of Buffer A containing 50 mM NaCl. The pelletwas repeatedly incubated with 10 vols of Buffer A plus50 mM NaCl for 30 min at 48C and the supernatants of the suc-cessive centrifugations pooled (Fraction 1). Fraction 1 wasloaded onto a Q-Sepharose (Pharmacia) column equilibratedwith Buffer A. The column was washed with 10 column vols ofthe same buffer and eluted with a gradient of increasing saltconcentration (50 mM to 1 M NaCl). The fractions containingsoluble gp3 were pooled (Fraction 3), and then diluted in gly-cerol (50% final concentration) and stored at ¹208C.
Alternatively, the pellet, containing more than 80% of gp3,was resuspended in one volume of Buffer A containing 6 Mguanidine hydrochloride (Sigma) and stirred for 60 min at48C. The solution was then centrifuged at 12 000 ×g for 30 minand the supernatant was retained (Fraction 2). Refolding con-ditions were chosen to minimize re-formation of aggregates.In short, guanidine hydrochloride was slowly removed by dia-lysis against equal volumes of Buffer A containing 1M NaCl at48C. Afterwards, the gp3 protein was diluted in glycerol (50%final concentration) and stored at ¹208C (Fraction 4).
Gel mobility shift assays
The end-labelled 369 bp DNA fragment (400 pg, 82 pM)expanding at both sides of cosN (co-ordinates 422 to 791,Fig. 1), was incubated with increasing amounts of gp3 pro-tein in 20 ml of Buffer B (25 mM Tris-HCl, pH 7.5, 100 mMNaCl, 5% (v/v) glycerol, 0.01% Triton X-100) containing5 mM ATP at 308C for 30 min. Then 3 ml of loading buffer(30% (v/v) glycerol, 0.25% (w/v) bromophenol blue and0.25% (w/v) xylene cyanol) were added and the reactionmixture was loaded on a 4% (w/v) non-denaturing polyacry-lamide gel. Gels were run at 15 mA at 48C and dried prior toautoradiography.
Q Blackwell Science Ltd, Molecular Microbiology, 23, 505–514
512 P. Garcıa, J. C. Alonso and J. E. Suarez

Filter binding assays
The formation of DNA complexes was measured as the radio-activity retained on nitrocellulose filters (Millipore, type HAWP,pore size of 0.45 mm) as previously described (McEnte et al.,1980). The standard reaction was performed for 30 min at308C and contained 400 pg of the [32P]-labelled 369 bp cosDNA segment (obtained as a 399 bp BamHI–HindIII frag-ment), 5 mM ATP and 3 mg of gp3 (340 nM) in a total volumeof 500 ml of Buffer B, unless stated otherwise. Ice-cold BufferB (10 ml) was added to stop the reaction, and the mixture wasfiltered and extensively washed with cold Buffer B. Filterswere dried and the radioactivity bound to them was deter-mined by scintillation counting. The DNA retained on the filterwas corrected for the retention of radioactively labelled DNAin the absence of protein. The specific activity of the labelledDNA was measured as trichloroacetic-precipitable material.All reactions were performed in duplicate.
Cyclization assay
The DNA concentration was kept at 200 ng ml¹1 so that theformation of monomolecular reaction products (circles) by T4DNA ligase was favoured. A typical 20 ml reaction mix con-tained 400 pg of the 369 bp cos DNA segment (82 pM)(obtained as a blunt-ended 399 bp HindIII–BamHI DNA frag-ment), 100 mM Tris-HCl, pH 7.5, 50 mM NaCl, 1 mM DTT,1 mM ATP, 5% (v/v) glycerol and increasing concentrationsof gp3 (10, 20, 40, 80, 160, 320 and 640 nM). Protein bindingwas allowed for 30 min at 308C. Then 5 mM MgCl2 (final con-centration) and 0.3 units of T4 DNA ligase were added and thetemperature of the reaction mixture was slowly lowered from308C to 48C. Deproteinized ligation products were analysedusing 4% ndPAGE. Autoradiographs of the dried gels weresubsequently obtained.
Acknowledgements
We would like to thank Sunghee Chai for help and encourage-ment during part of this work, and Manuel Espinosa for com-puter studies on the structure of the sequences reported. Thiswork was partially supported by Grant BIOT-CT94-3055 fromthe European Union and grants BIO94-0189 and PB93-0116from DGICYT of the Spanish Ministry of Education to J.E.Sand J.C.A., respectively. P.G. was the recipient of a Predoc-toral Fellowship from FICYT (Fondo de Investigacion Cientı-fica y Tecnica de Asturias).
References
Accolas, J.P., Peigney, C., Limsowtin, G.K.Y., Cluzel, P.J.,and Sechaud, L. (1994) Lutte contre les bacteriophagesdans l’industrie laitiere. In Bacteries Lactiques. de Rois-sart, H., and Luquet, F.M. (eds). Uriage, France: Lorica,pp. 473–492.
Birboim, H.C., and Doly, J. (1979) A rapid alkaline extractionprocedure for screening recombinant plasmid DNA. NuclAcids Res 7: 1513–1523.
Black, L.W. (1989) DNA packaging in dsDNA bacterio-phages. Annu Rev Microbiol 43: 267–292.
Bottazzi, V., and Mercenier, A. (1994) Proprietes prophylac-tiques et therapeutiques des bacteries lactiques. In Bac-teries Lactiques. de Roissart, H., and Luquet, F.M. (eds).Uriage, France: Lorica, pp. 409–418.
Brennan, R.G., and Matthews, B. (1989) The helix-turn-helixDNA binding motif. J Mol Chem 264: 1903–1906.
Buckenhuskes, H.J. (1993) Selection criteria for lactic acidbacteria to be used as starter cultures for various foodcommodities. FEMS Microbiol Rev 12: 253–271.
Chang, A.C., and Cohen, S.N. (1978) Construction and char-acterization of amplifiable multicopy DNA cloning vehiclesderived from the p15 cryptic miniplasmid. J Bacteriol134: 1141–1156.
Chomczynski, P., and Sacchi, N. (1987) Single step methodof RNA isolation by acid guanidinum thiocyanate–phenol–chloroform extraction. Anal Biochem 162: 156–159.
Cue, D., and Feiss, M. (1993) A site required for terminationof packaging of the phage l chromosome. Proc Natl AcadSci USA 90: 9290–9294.
Devereux, J., Haeberli, P., and Smithies, O. (1984) A com-prehensive set of sequence analysis programs for theVAX. Nucl Acids Res 12: 387–395.
Drlica, K., and Rouviere-Yaniv, J. (1987) Histone-like pro-teins of bacteria. Microbiol Rev 51: 301–319.
Feiss, M. (1986) Terminase and the recognition, cuttingand packaging of chromosomes. Trends Genet 2: 100–104.
Hagerman, P.J. (1990) Sequence-directed curvature of DNA.Annu Rev Biochem 59: 755–781.
Herrero, M., de los Reyes-Gavilan, C.G., Caso, J.L., andSuarez, J.E. (1994) Characterization of 393-A2, a bacter-iophage that infects Lactobacillus casei. Microbiology140: 2585–2590.
Inoue, H., Nojima, H., and Okayama, H. (1990) High effi-ciency transformation of Escherichia coli with plasmids.Gene 96: 23–28.
Kammerer, B. (1996) Genetics of lactobacilli, leuconostocand pediococci: screening, selection and construction ofmutants. Le Lait 76: 51–65.
Lillehaug, D., Lindqvist, B.H., and Birkeland, N.K. (1991)Characterization of fLC3, a Lactoccocus lactis ssp. cre-moris temperate bacteriophage with cohesive single-stranded ends. Appl Environ Microbiol 57: 3206–3211.
McEntee, K., Weinstock, G.M., and Lehman, I.R. (1980) recAprotein-catalysed strand assimilation: stimulation byEscherichia coli single-stranded DNA-binding protein.Proc Natl Acad Sci USA 77: 857–861.
McKay, L.L., and Baldwin, K.A. (1990) Applications for bio-technology: present and future improvements in lacticacid bacteria. FEMS Microbiol Rev 87: 3–14.
Mendelson, I., Gottesman, M., and Oppenheim, A.B. (1991)HU and integration host factor function as auxiliary proteinsin cleavage of phage l cohesive ends by terminase. JBacteriol 173: 1670–1676.
Mercenier, A., Pouwels, P.H., and Chassy, B.M. (1994)Genetic engineering of lactobacilli, leuconostocs and Strep-tococcus thermophilus. In Genetics and Biotechnology ofLactic Acid Bacteria. Gasson, M.J., and De Vos, W.M.(eds). London: Blackie, pp. 252–293.
Messing, J. (1983) New M13 vectors for cloning genes. MethEnzymol 101: 20–78.
Q Blackwell Science Ltd, Molecular Microbiology, 23, 505–514
DNA–protein interaction at the cos region of phage A2 513

Murialdo, H. (1991) Bacteriophage lambda DNA maturationand DNA packaging. Annu Rev Biochem 60: 125–153.
Nakashima, Y., Ikeda, H., Kakita, Y., Miake, F., and Wata-nabe, K. (1994) Restriction map of the genomic DNA ofLactobacillus casei bacteriophage PL-1 and nucleotidesequence of its cohesive single-stranded ends. J GenVirol 75: 2537–2541.
Pabo, C.O., and Sauer, R.T. (1984) Protein–DNA recogni-tion. Annu Rev Biochem 53: 293–321.
Pouwels, P.H., and Leer, R.J. (1993) Genetics of lactobacilli:plasmids and gene expression. Antonie van Leeuwenhoek64: 85–107.
Pouwels., P.H., and Leunissen, J.A.M. (1994) Divergence incodon usage of Lactobacillus species. Nucl Acids Res 22:929–936.
Sambrook, J., Maniatis, T., and Fritsch, E.F. (1989) Molecu-lar Cloning: A Laboratory Manual. 2nd edn. Cold SpringHarbor, New York: Cold Spring Harbor Laboratory Press.
Sanger, F., Micklen, S., and Coulson, A.R. (1977) DNAsequencing with chain-terminating inhibitors. Proc NatlAcad Sci USA 74: 5463–5467.
Shinder, G., and Gold, M. (1988) The Nu1 subunit of bacter-iophage lambda terminase binds to specific sites in cosDNA. J Virol 62: 387–392.
Shore, D., and Baldwin, R.L. (1983) DNA flexibility studied bycovalent closure of short fragments into circles. J Mol Biol170: 957–981.
Skorupski, N., Sauer, B., and Sternberg, N. (1994) Faithfulcleavage of the P1 packaging site (pac ) requires twophage proteins, PacA and PacB, and two Escherichiacoli proteins, IHF and HU. J Mol Biol 243: 268–282.
Studier, N.F., and Moffat, B.A. (1986) Use of bacteriophageT7 RNA polymerase to direct selective high-level expres-sion of cloned genes. J Mol Biol 189: 113–130.
Suarez, J.E., and Chater, K.F. (1981) Development of a DNAcloning in Streptomyces using phage vectors. Cienc Biol 6:99–110.
Villar, C.J., Hardisson, C., and Suarez, J.E. (1986) Cloningand molecular epidemiology of plasmid determined fosfo-mycin resistance. Antimicrob Agents Chemother 29: 309–314.
Walker, J.E., Saraste, M., Runswick, M.J., and Gay, N.J.(1982) Distantly related sequences in the a- and b-sub-units of ATP synthase, myosin, kinase and other ATP-requiring enzymes and a common nucleotide bindingfold. EMBO J 1: 945–951.
Xin, W., and Feiss, M. (1988) The interaction of Escherichiacoli integration host factor with the cohesive end sites ofphage l and 21. Nucl Acids Res 16: 2015–2030.
Yanisch-Perron, C., Vieira, J., and Messing, J. (1985)Improved M13 phage cloning vectors and host strains:nucleotide sequences of the M13 mp18 and pUC19 vec-tors. Gene 33: 103–119.
Q Blackwell Science Ltd, Molecular Microbiology, 23, 505–514
514 P. Garcıa, J. C. Alonso and J. E. Suarez
Copyright © 2022 FDOKUMEN




















