
Mitochondrial calcium signalling in cell death
10
THE THEODOR BU ¨ CHER LECTURE Mitochondrial calcium signalling in cell death Delivered on 1 July 2004 at the 29th FEBS Congress in Warsaw Sara Leo 1 , Katiuscia Bianchi 1 , Marisa Brini 2 and Rosario Rizzuto 1 1 Department of Experimental and Diagnostic Medicine, Section of General Pathology, and Interdisciplinary Center for the Study of Inflammation (ICSI), University of Ferrara, Italy 2 Department of Biochemistry, University of Padova, Italy In the last few decades, much information has been obtained on the role of calcium ions as ubiquitous sec- ond messengers that translate the binding of signalling molecules to plasmamembrane receptors into defined cell activities [1]. Thanks to the development of highly efficient probes (the intracellularly trappable fluores- cent indicators developed by Tsien and coworkers) [2], it was possible to investigate the calcium signals elici- ted in a wide variety of cell types, either in culture (pri- mary cultures and immortalized cell lines) or in situ (organotypic slices or even the intact tissue within a living organism) by the opening of plasmamembrane Ca 2+ channels or Ca 2+ channels of intracellular reser- voirs, cytologically identifiable with the endoplasmic reticulum (ER) and, more recently, with the Golgi apparatus [3,4]. The use of Ca 2+ as a second messenger rests on the maintenance of a low cytosolic Ca 2+ concentration, through the energy-consuming pumping activity of Ca 2+ ATPases located in ER ⁄ SR (SERCA) or plasmamembrane (PMCA). As to the triggering mech- anism of the [Ca 2+ ] rise, a route involves either the stimulation of G-protein coupled receptors (specifically those coupled to a G(aq) protein, that activate phos- pholipase Cb and thus produce inositol 1,4,5-trisphos- phate (IP3) from the hydrolysis of the lipid phosphatidyl-inositol 4,5-diphosphate) or growth fac- tors receptors (also causing the production of IP3 through the activation of phospholipase Cc, containing an SH2 domain that recruits it to the activated GF-R) [1]. An alternative route for raising cytosolic Ca 2+ concentration ([Ca 2+ ] c ) depends on the opening of Keywords apoptosis; calcium; mitochondria; organelles; photoproteins; signal transduction Correspondence R. Rizzuto, Department of Experimental and Diagnostic Medicine, General Pathology Section, University of Ferrara, Via L. Borsari 46, 44100 Ferrara, Italy Fax: +39 0532247278 Tel: +39 0532291361 E-mail: [email protected] (Received 1 June 2005, accepted 11 July 2005) doi:10.1111/j.1742-4658.2005.04855.x The development of targeted probes (based on the molecular engineering of luminescent or fluorescent proteins) has allowed the specific measure- ment of [Ca 2+ ] in intracellular organelles or cytoplasmic subdomains. This approach gave novel information on different aspects of cellular Ca 2+ homeostasis. Regarding mitochondria, it was possible to demonstrate that, upon physiological stimulation of cells, Ca 2+ is rapidly accumulated in the matrix. We will discuss the basic characteristics of this process, its role in modulating physiological and pathological events, such as the regulation of aerobic metabolism and the induction of cell death, and new insight into the regulatory mechanisms operating in vivo. Abbreviations AGC, aspartate ⁄ glutamate metabolite carrier; COX8, cytochrome c oxidase; CRAC, Ca 2+ release-activated current; ER, endoplasmic reticulum; HBx, x protein of the hepatitis B virus; IP3, inositol 1,4,5-trisphosphate; PKC, protein kinase C; PMCA, plasmamembrane Ca 2+ ATPase; SERCA, sarcoplamic reticulum ⁄ ER Ca 2+ ATPase. FEBS Journal 272 (2005) 4013–4022 ª 2005 FEBS 4013
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Mitochondrial calcium signalling in cell death

THE THEODOR BUCHER LECTURE
Mitochondrial calcium signalling in cell death
Delivered on 1 July 2004 at the 29th FEBS Congress in Warsaw
Sara Leo1, Katiuscia Bianchi1, Marisa Brini2 and Rosario Rizzuto1
1 Department of Experimental and Diagnostic Medicine, Section of General Pathology, and Interdisciplinary Center for the Study of
Inflammation (ICSI), University of Ferrara, Italy
2 Department of Biochemistry, University of Padova, Italy
In the last few decades, much information has been
obtained on the role of calcium ions as ubiquitous sec-
ond messengers that translate the binding of signalling
molecules to plasmamembrane receptors into defined
cell activities [1]. Thanks to the development of highly
efficient probes (the intracellularly trappable fluores-
cent indicators developed by Tsien and coworkers) [2],
it was possible to investigate the calcium signals elici-
ted in a wide variety of cell types, either in culture (pri-
mary cultures and immortalized cell lines) or in situ
(organotypic slices or even the intact tissue within a
living organism) by the opening of plasmamembrane
Ca2+ channels or Ca2+ channels of intracellular reser-
voirs, cytologically identifiable with the endoplasmic
reticulum (ER) and, more recently, with the Golgi
apparatus [3,4].
The use of Ca2+ as a second messenger rests on the
maintenance of a low cytosolic Ca2+ concentration,
through the energy-consuming pumping activity of
Ca2+ ATPases located in ER ⁄SR (SERCA) or
plasmamembrane (PMCA). As to the triggering mech-
anism of the [Ca2+] rise, a route involves either the
stimulation of G-protein coupled receptors (specifically
those coupled to a G(aq) protein, that activate phos-
pholipase Cb and thus produce inositol 1,4,5-trisphos-
phate (IP3) from the hydrolysis of the lipid
phosphatidyl-inositol 4,5-diphosphate) or growth fac-
tors receptors (also causing the production of IP3
through the activation of phospholipase Cc, containingan SH2 domain that recruits it to the activated GF-R)
[1]. An alternative route for raising cytosolic Ca2+
concentration ([Ca2+]c) depends on the opening of
Keywords
apoptosis; calcium; mitochondria;
organelles; photoproteins; signal
transduction
Correspondence
R. Rizzuto, Department of Experimental and
Diagnostic Medicine, General Pathology
Section, University of Ferrara,
Via L. Borsari 46, 44100 Ferrara, Italy
Fax: +39 0532247278
Tel: +39 0532291361
E-mail: [email protected]
(Received 1 June 2005, accepted 11 July 2005)
doi:10.1111/j.1742-4658.2005.04855.x
The development of targeted probes (based on the molecular engineering
of luminescent or fluorescent proteins) has allowed the specific measure-
ment of [Ca2+] in intracellular organelles or cytoplasmic subdomains. This
approach gave novel information on different aspects of cellular Ca2+
homeostasis. Regarding mitochondria, it was possible to demonstrate that,
upon physiological stimulation of cells, Ca2+ is rapidly accumulated in the
matrix. We will discuss the basic characteristics of this process, its role in
modulating physiological and pathological events, such as the regulation of
aerobic metabolism and the induction of cell death, and new insight into
the regulatory mechanisms operating in vivo.
Abbreviations
AGC, aspartate ⁄ glutamate metabolite carrier; COX8, cytochrome c oxidase; CRAC, Ca2+ release-activated current; ER, endoplasmic
reticulum; HBx, x protein of the hepatitis B virus; IP3, inositol 1,4,5-trisphosphate; PKC, protein kinase C; PMCA, plasmamembrane Ca2+
ATPase; SERCA, sarcoplamic reticulum ⁄ ER Ca2+ ATPase.
FEBS Journal 272 (2005) 4013–4022 ª 2005 FEBS 4013

various classes of plasma membrane Ca2+ channels,
such as those directly opened by ligand binding (e.g.
the ionotropic glutamate receptors of neurons), those
opened by the depolarization of the plasmamembrane
(the wide number of voltage-dependent Ca2+ channels)
or those opened by other intracellular signals (e.g. sec-
ond messengers or the depletion of intracellular Ca2+
stores) [5,6].
The concerted action of channels with distinct spa-
tial distribution and kinetics of opening determines a
high spatio-temporal specificity of the signals elicited
by different agonists, which in turn are decoded into
radically different intracellular effects. This adds fur-
ther interest and complexity to the signalling properties
of Ca2+: not only tissue-specific functions (e.g. endo-
crine and neuro-secretion, muscle contraction and fer-
tilization), but also decisions on cell fate (proliferation,
cell death by necrosis or apoptosis) are controlled by
Ca2+ [7]. Thus, not surprisingly deregulations in intra-
cellular Ca2+ homeostasis have been implicated in the
pathogenesis of genetic (e.g. familial migraine and skin
disorders, such as Darier’s and Hailey-Hailey diseases)
and multifactorial (e.g. hypertension and diabetes) dis-
eases [8,9].
The recognition of the spatio-temporal complexity
of calcium signals and of their multiple signalling roles
has ignited interest in clarifying the molecular mecha-
nisms that allow to specifically decode different signal-
ling patterns. Extensive work in the past decades has
revealed the broad repertoire of Ca2+ effectors, i.e.
enzymes, channels or structural proteins that modify
their activity upon binding of Ca2+. At first, these
included cytosolic proteins, such as the Ca2+-depend-
ent kinases [protein kinase C (PKC), CamK] or phos-
phatases (calcineurin) and their targets [1]. In recent
years, however, it became clear that also processes
occurring within intracellular organelles (gene tran-
scription, post-translational modification of proteins
and aerobic metabolism) are modulated by [Ca2+]
changes [10,11]. Thus, Ca2+-dependent effects within
organelles are now considered a significant component
of the ‘Ca2+ symphony’, initiated by physiological or
pathological stimuli, which may influence its final out-
come.
In this context, measuring Ca2+ concentrations
within organelles with accuracy and specificity has
become an important experimental task. This task has
largely been accomplished, thanks to the development
of a new class of probes that are based on Ca2+-sensi-
tive reporter proteins and take advantage of the highly
selective mechanisms that target cellular proteins to
the correct location. The first successful example has
been that of aequorin. Aequorin is a Ca2+-sensitive
photoprotein of the jellyfish Aequorea victoria, which
emits light upon binding of Ca2+ to three high-affinity
sites present in the protein sequence. The protein can
be purified from jellyfish extracts and microinjected in
cells. Using this classical indicator, seminal observa-
tions were made, such as the repetitive [Ca2+]c spiking
induced by agonist stimulation [12]. More recently, we
have taken advantage of molecular biology techniques
for developing a series of specifically targeted Ca2+
probes. The rationale was that of fusing the aequorin
cDNA with DNA sequences encoding specific target-
ing signals, i.e. the protein sequences that are necessary
and sufficient for localizing a mammalian protein to
the correct subcellular location. Figure 1A shows an
example, mtAEQ, which is the recombinant protein
developed for measuring [Ca2+] within the mitochond-
rial matrix and was instrumental in gaining new insight
into mitochondrial Ca2+ handling, i.e. the topic of this
review [13]. In the chimeric cDNA, an aequorin moiety
including an HA1 tag was fused in frame with the
N-terminal portion (including the 25 amino acids
cleavable presequence and the first eight amino acids
of the mature polypeptide) of subunit VIII of cyto-
chrome c oxidase (COX8). The fusion protein, when
expressed in mammalian cells, is entirely distributed
to the mitochondria (Fig. 1B). The localization of
mtAEQ is revealed by immunofluorescence using an
antibody that recognizes the HA1 domain.
In general, targeted aequorins (also developed for
other intracellular compartments using similar strat-
egies) have proved to be extremely valuable, and
allowed many new data and novel concepts in Ca2+
signalling to be obtained. The most important ones
A
B
Fig. 1. Schematic map of the mtAEQ construct (aequorin targeted
to the mitochondrial matrix) and its localization by immunofluores-
cence.
Mitochondrial calcium signalling in cell death S. Leo et al.
4014 FEBS Journal 272 (2005) 4013–4022 ª 2005 FEBS

not covered in this review are the estimates of ER
[Ca2+] in the near-millimolar range (� 0.5 mm)
[14,15], the role of the agonist-sensitive Ca2+ store
played by the Golgi apparatus (endowed with a resting
[Ca2+] of � 0.3 mm and rapidly emptying after agonist
stimulation) [16], the rapid equilibration of cytosolic
and nuclear [Ca2+] [17], the estimates of resting and
stimulated [Ca2+]c under the plasmamembrane, well
above those of the bulk cytosol [18]. This paper
focuses on mitochondria, as Ca2+ handling by these
organelles was not only significantly different from
that expected, but also identified them as critical
checkpoints, in which radically different effects can be
triggered by a rise in [Ca2+].
Mechanism and role of mitochondrialCa2+ homeostasis
The participation of mitochondria in Ca2+ homeostasis
is a concept that alternated periods of glory and com-
plete dismissal. Indeed, as soon as the chemiosmotic
theory was accepted as the basis of energy conservation
in mitochondria, it became obvious that these organ-
elles could, at least potentially, efficiently accumulate
Ca2+ down the electrochemical gradient established
across the inner membrane by the activity of respirat-
ory complexes. This possibility was actually directly
demonstrated by an extensive body of work carried out
with isolated mitochondria. Respiring mitochondria
can rapidly accumulate Ca2+ through an electrogenic
pathway, termed the ‘mitochondrial Ca2+ uniporter’
(MCU) [19]. This route was (and still is) undefined at
the molecular level, although very recent work by Clap-
ham and coworkers demonstrated that it is a bona fide
Ca2+ channel [20]. Ca2+ is then re-extruded by electro-
neutral exchangers (Na2+ ⁄Ca2+ and H+ ⁄Ca2+
exchangers, mostly expressed in nonexcitable and excit-
able cells, respectively) [21]. Based on this evidence,
mitochondria were thought to dynamically change the
matrix Ca2+ concentration ([Ca2+]m) in living cells
challenged with Ca2+-mobilizing agonists.
This possibility was severely questioned in the 1980s,
when the signalling pathways downstream of receptor
stimulation were clarified. It was then demonstrated
that G-protein coupled and growth factor receptors
mobilize Ca2+ from an intracellular store that proved
be the endoplasmic reticulum, not mitochondria [22].
Moreover, the accurate measurement of [Ca2+]c levels
with fluorescent indicators suggested that mitochondria
did not receive the Ca2+ released either. Indeed, both
at rest and after stimulation the [Ca2+]c values were
well below those necessary for rapid Ca2+ uptake
through the MCU. Thus, the general consensus
became that mitochondria can accumulate a significant
amount of Ca2+ only when large and sustained
[Ca2+]c increases occurred, such as those postulated to
occur in various pathological derangements (e.g. the
Ca2+ overload during neuronal excitotoxicity).
This situation was completely reversed when the tar-
geted recombinant indicators (first aequorin, then the
more recent GFP-based fluorescent probes) clearly
demonstrated that a [Ca2+]c rise elicited by a physiolo-
gical stimulation is almost invariably paralleled by
a robust [Ca2+]m increase [23], that usually largely
exceeds the values observed in the bulk cytosol and
reached values as high as 500 lm [24]. The apparent
discrepancy with the sluggish rate of Ca2+ uptake
observed in isolated mitochondria upon exposure to
Ca2+ concentrations similar to those measured in the
bulk cytosol was reconciled by postulating that mito-
chondria upon opening of the IP3-sensitive channels
are not exposed to those low [Ca2+], but rather to the
much higher values generated in the proximity of the
channel (the microdomain hypothesis) [25]. In support
of this notion, organelle labelling of the ER and mito-
chondria showed closed appositions and an aequorin
chimera located on the mitochondrial membrane detec-
ted [Ca2+] values well above those of the bulk cytosol
[26].
Numerous studies then followed that demonstrated,
both in cell lines and in intact tissues, the occurrence
of rapid [Ca2+]m transients in cells as diverse as HeLa,
hepatocytes, cardiac and skeletal muscle and neurons
[27]. A striking example is that of cardiac muscle, in
which a [Ca2+]m transient was detected at every con-
tractile cycle [28]. This implies that both the uptake
and the release mechanism are highly efficient in situ,
and allow the completion of a Ca2+ cycle within the
short time frame of a single contraction.
What is the role of mitochondrial Ca2+ homeosta-
sis? A first obvious function stems from long-standing
biochemical evidence, i.e. the demonstration by Den-
ton, McCormack and Hansford in the 1960s that three
key metabolic enzymes (the pyruvate, a-ketoglutarateand isocitrate dehydrogenases) are activated by Ca2+,
by different mechanisms. In the case of pyruvate dehy-
drogenase, this is through a Ca2+-dependent dephos-
phorylation step, whereas in the latter two cases this is
through the direct binding of Ca2+ to the enzyme
complex [29,30]. Thus, a [Ca2+] rise in the matrix may
allow the up-regulation of aerobic metabolism and
tuning of ATP production to the increased needs of
stimulated cells. This could be demonstrated by the
direct measurement of mitochondrial ATP levels with
a targeted chimera of the ATP-sensitive photoprotein
luciferase. Parallel measurements of Ca2+ and ATP
S. Leo et al. Mitochondrial calcium signalling in cell death
FEBS Journal 272 (2005) 4013–4022 ª 2005 FEBS 4015
levels showed that the Ca2+ signal within the mito-
chondria is responsible for the enhanced ATP produc-
tion, an effect that lasts longer than the Ca2+ signal
itself, highlighting a novel form of cellular ‘metabolic
memory’ [31].
Interestingly, recent work indicates that other Ca2+-
dependent metabolic checkpoints are operative.
Namely, the aspartate ⁄ glutamate metabolite carriers
(AGCs) were shown to include EF-hand domains, and
Ca2+ binding to these sites was shown to increase their
activity [32]. In turn, recombinant expression of wild
type AGCs enhanced ATP production upon cell stimu-
lation, an effect that was not observed with truncated
mutants lacking the Ca2+-binding domain [33].
Substantial evidence has built up in recent years
indicating that metabolic regulation is only one of the
roles of the mitochondrial Ca2+ signal. It now appears
evident that massive Ca2+ loading (as in the case of
glutamate excitotoxicity of neurons) and ⁄or the com-
bined action of apoptotic agents or pathophysiological
conditions (e.g. oxidative stress) can induce a profound
alteration of organelle structure and function [34–36].
As a consequence, bioenergetic dysfunction and ⁄orrelease into the cytosol of proteins acting as caspase
cofactors, such as cytochrome c [37,38], AIF [39] and
Smac ⁄Diablo [40], may lead the cell to necrotic or
apoptotic cell death. In relation to this effect, the anti-
oncogene Bcl-2 was shown to reduce the steady state
Ca2+ levels in the ER (and thus dampen the pro-apop-
totic Ca2+ signal) [41,42].
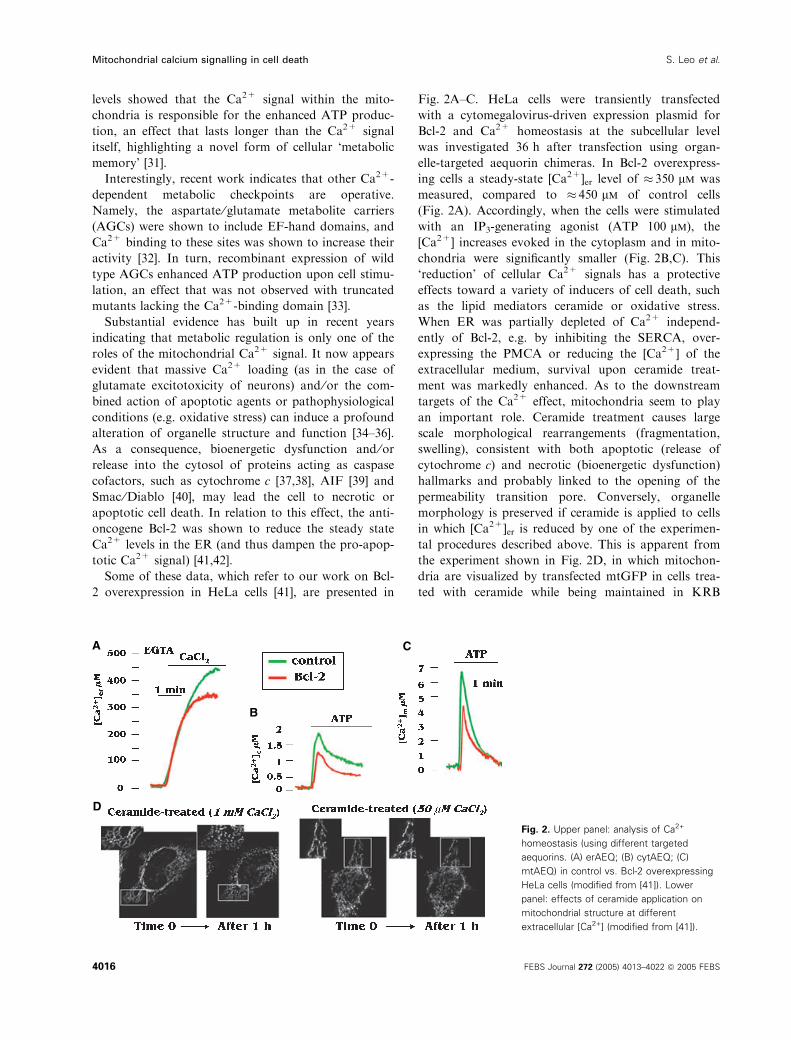
Some of these data, which refer to our work on Bcl-
2 overexpression in HeLa cells [41], are presented in
Fig. 2A–C. HeLa cells were transiently transfected
with a cytomegalovirus-driven expression plasmid for
Bcl-2 and Ca2+ homeostasis at the subcellular level
was investigated 36 h after transfection using organ-
elle-targeted aequorin chimeras. In Bcl-2 overexpress-
ing cells a steady-state [Ca2+]er level of � 350 lm was
measured, compared to � 450 lm of control cells
(Fig. 2A). Accordingly, when the cells were stimulated
with an IP3-generating agonist (ATP 100 lm), the
[Ca2+] increases evoked in the cytoplasm and in mito-
chondria were significantly smaller (Fig. 2B,C). This
‘reduction’ of cellular Ca2+ signals has a protective
effects toward a variety of inducers of cell death, such
as the lipid mediators ceramide or oxidative stress.
When ER was partially depleted of Ca2+ independ-
ently of Bcl-2, e.g. by inhibiting the SERCA, over-
expressing the PMCA or reducing the [Ca2+] of the
extracellular medium, survival upon ceramide treat-
ment was markedly enhanced. As to the downstream
targets of the Ca2+ effect, mitochondria seem to play
an important role. Ceramide treatment causes large
scale morphological rearrangements (fragmentation,
swelling), consistent with both apoptotic (release of
cytochrome c) and necrotic (bioenergetic dysfunction)
hallmarks and probably linked to the opening of the
permeability transition pore. Conversely, organelle
morphology is preserved if ceramide is applied to cells
in which [Ca2+]er is reduced by one of the experimen-
tal procedures described above. This is apparent from
the experiment shown in Fig. 2D, in which mitochon-
dria are visualized by transfected mtGFP in cells trea-
ted with ceramide while being maintained in KRB
A
B
C
D
Fig. 2. Upper panel: analysis of Ca2+
homeostasis (using different targeted
aequorins. (A) erAEQ; (B) cytAEQ; (C)
mtAEQ) in control vs. Bcl-2 overexpressing
HeLa cells (modified from [41]). Lower
panel: effects of ceramide application on
mitochondrial structure at different
extracellular [Ca2+] (modified from [41]).
Mitochondrial calcium signalling in cell death S. Leo et al.
4016 FEBS Journal 272 (2005) 4013–4022 ª 2005 FEBS
supplemented with physiological (1 mm; left) or a
lower (0.05 mm; right) Ca2+ concentration, which cau-
ses a partial Ca2+ depletion of the ER. Overall, the
data indicate that the anti-apoptotic protein Bcl-2, by
reducing Ca2+ signals evoked by physiological and
pathological stimuli, counteracts the efficacy of death
pathways acting through the mitochondrial check-
point.
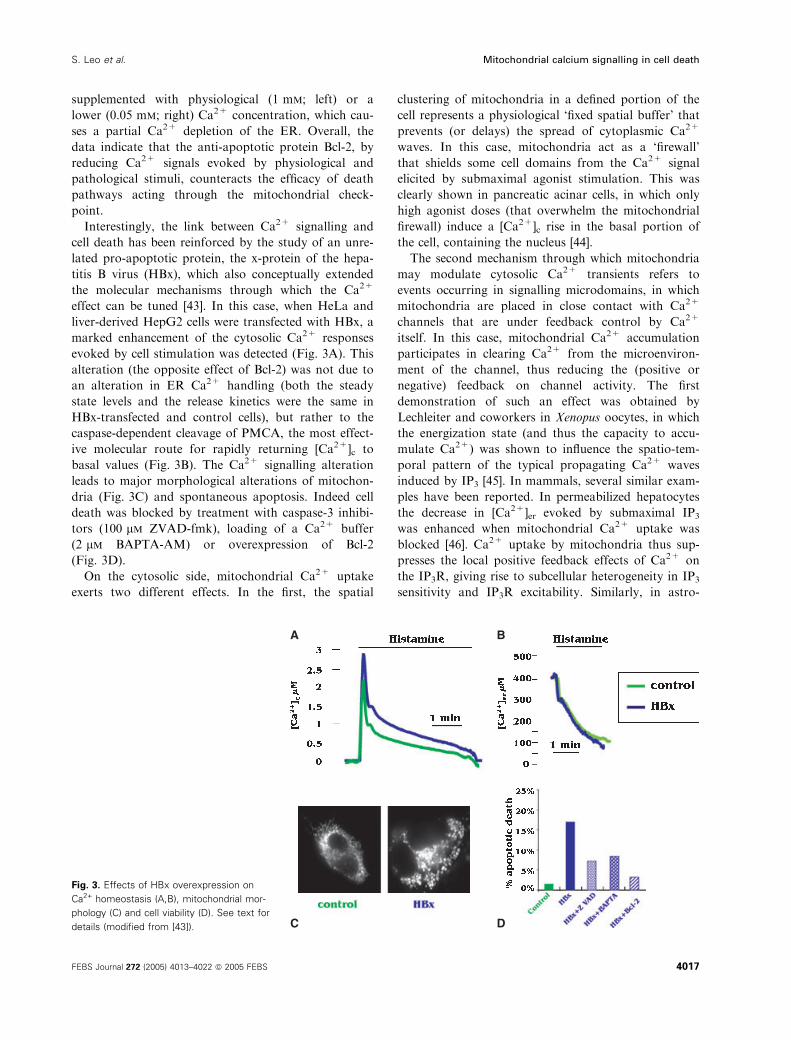
Interestingly, the link between Ca2+ signalling and
cell death has been reinforced by the study of an unre-
lated pro-apoptotic protein, the x-protein of the hepa-
titis B virus (HBx), which also conceptually extended
the molecular mechanisms through which the Ca2+
effect can be tuned [43]. In this case, when HeLa and
liver-derived HepG2 cells were transfected with HBx, a
marked enhancement of the cytosolic Ca2+ responses
evoked by cell stimulation was detected (Fig. 3A). This
alteration (the opposite effect of Bcl-2) was not due to
an alteration in ER Ca2+ handling (both the steady
state levels and the release kinetics were the same in
HBx-transfected and control cells), but rather to the
caspase-dependent cleavage of PMCA, the most effect-
ive molecular route for rapidly returning [Ca2+]c to
basal values (Fig. 3B). The Ca2+ signalling alteration
leads to major morphological alterations of mitochon-
dria (Fig. 3C) and spontaneous apoptosis. Indeed cell
death was blocked by treatment with caspase-3 inhibi-
tors (100 lm ZVAD-fmk), loading of a Ca2+ buffer
(2 lm BAPTA-AM) or overexpression of Bcl-2
(Fig. 3D).
On the cytosolic side, mitochondrial Ca2+ uptake
exerts two different effects. In the first, the spatial
clustering of mitochondria in a defined portion of the
cell represents a physiological ‘fixed spatial buffer’ that
prevents (or delays) the spread of cytoplasmic Ca2+
waves. In this case, mitochondria act as a ‘firewall’
that shields some cell domains from the Ca2+ signal
elicited by submaximal agonist stimulation. This was
clearly shown in pancreatic acinar cells, in which only
high agonist doses (that overwhelm the mitochondrial
firewall) induce a [Ca2+]c rise in the basal portion of
the cell, containing the nucleus [44].
The second mechanism through which mitochondria
may modulate cytosolic Ca2+ transients refers to
events occurring in signalling microdomains, in which
mitochondria are placed in close contact with Ca2+
channels that are under feedback control by Ca2+
itself. In this case, mitochondrial Ca2+ accumulation
participates in clearing Ca2+ from the microenviron-
ment of the channel, thus reducing the (positive or
negative) feedback on channel activity. The first
demonstration of such an effect was obtained by
Lechleiter and coworkers in Xenopus oocytes, in which
the energization state (and thus the capacity to accu-
mulate Ca2+) was shown to influence the spatio-tem-
poral pattern of the typical propagating Ca2+ waves
induced by IP3 [45]. In mammals, several similar exam-
ples have been reported. In permeabilized hepatocytes
the decrease in [Ca2+]er evoked by submaximal IP3
was enhanced when mitochondrial Ca2+ uptake was
blocked [46]. Ca2+ uptake by mitochondria thus sup-
presses the local positive feedback effects of Ca2+ on
the IP3R, giving rise to subcellular heterogeneity in IP3
sensitivity and IP3R excitability. Similarly, in astro-
A B
C D
Fig. 3. Effects of HBx overexpression on
Ca2+ homeostasis (A,B), mitochondrial mor-
phology (C) and cell viability (D). See text for
details (modified from [43]).
S. Leo et al. Mitochondrial calcium signalling in cell death
FEBS Journal 272 (2005) 4013–4022 ª 2005 FEBS 4017

cytes inhibition of mitochondrial Ca2+ uptake almost
doubled the rate of propagation of the calcium wave
across the cell [47]. In contrast, in BHK cells inhibition
of mitochondrial Ca2+ uptake resulted in reduction of
ER Ca2+ release [48], indicating that in this case
mitochondria play a major role in preventing the
Ca2+-depended inhibition of the InsP3 channel. This
effect is not limited to ER Ca2+ channels. Indeed, sev-
eral papers from the groups of Lewis and Parekh have
demonstrated that Ca2+ uptake by energized mito-
chondria relieves the Ca2+-dependent inhibition of
Ca2+ release-activated current (CRAC) channels, i.e.
those activated by the emptying of intracellular Ca2+
stores [49,50]. This notion explains the high buffering
(mimicking mitochondrial activity) required to observe
ICRAC in many experimental conditions. A complex
role, somewhat similar to that proposed for the modu-
lation of capacitative Ca2+ influx, has been proposed
by Nicholls and coworkers for the modulation of
Ca2+ activation-inhibition of glutamate and voltage
operated Ca2+ channels in cerebellar granule cells [51].
Specific regulatory pathways formitochondrial Ca2+ homeostasis
Mitochondria can thus be regarded as critical check-
points in Ca2+ signalling, acting as membrane-bound
Ca2+ buffers, in which Ca2+ itself plays a regulatory
role. In this situation, the possibility that their uptake
capacity (kinetics, amplitude) is tuned by converging
signalling pathways may add further complexity (and
option for regulation) to Ca2+-mediated signal trans-
duction.
We investigated this possibility, focusing on two
aspects: the role of the broad family of PKC kinases and
of the three-dimensional structure of mitochondria, in
turn controlled by the activity of large GTPases indu-
cing organelle fusion or fission (mitofusins, dynamin-
related protein-1).
PKC comprises a family of serine-threonine kinases
that are involved in the transduction of a wide number
of extracellular signals. Based on their biochemical
properties, they are divided into classical (e.g. a, bI,bII, c), activated by Ca2+ and diacylglycerol, novel
(e.g. d, e, g, h), activated by diacylglycerol, and atyp-
ical (e.g. f, k), insensitive to both Ca2+ and diacylglyc-
erol. Different isozymes are coexpressed in the various
cell types giving rise to a highly flexible molecular rep-
ertoire, which can mediate radically different intra-
cellular effects, e.g. isoforms belonging to the same
group, such as d and e, have been reported to play
opposite effects on apoptosis. To evaluate specific
effects of PKC isoforms on cellular Ca2+ homeostasis,
we overexpressed PKC–GFP chimeras in HeLa cells
and investigated agonist-dependent Ca2+ signals in
the cytosol and mitochondria, using organelle-targeted
aequorin chimeras [52]. An interesting scenario
emerged, with distinct roles for the various isoforms.
Overexpression of PKCe did not modify the amplitude
and the kinetics of either the cytosolic and mitochond-
rial Ca2+ transients evoked by histamine stimulation,
indicating that this PKC isoform does not influence
either Ca2+ signalling globally in the cell or mito-
chondrial Ca2+ accumulation. Conversely, PKCaoverexpression greatly reduces the agonist-evoked
[Ca2+] increase both in the cytosol and in the mito-
chondria. The monitoring of ER [Ca2+] demonstrated
that this is due to the sharp reduction of Ca2+ release
from the organelle. These data are in keeping with pre-
vious demonstration that phorbol esters inhibited ER
Ca2+ release [53], suggesting that PKCa is the isoform
responsible for this effect. As to the target, the demon-
stration of the phosphorylation of the IP3 receptor by
PKC [54] indicates that the ER Ca2+ release channel
itself is a plausible site of action for the PKCa effect.
Interestingly, some isoforms appear to have an
effect on mitochondrial, but not on cytosolic, Ca2+
handling. Indeed, in cells overexpressing PKCb (and
to a smaller extent PKCd), the mitochondrial but not
the cytosolic [Ca2+] rise is reduced. Conversely, over-
expression of PKCf enhances the mitochondrial Ca2+
responses to agonist stimulation, while still leaving
cytosolic [Ca2+] increases unaffected. These effects do
not depend on alterations of mitochondrial structure
(monitored by mtRFP labelling), an important deter-
minant of organelle responsiveness, nor to significant
changes in the driving force for Ca2+ accumulation
(the mitochondrial membrane potential, DYm). A pos-
sibility is that the Ca2+ uptake machinery of
the organelle is directly modulated, but the demonstra-
tion of this possibility awaits the molecular characteri-
zation of the uptake process. As to the functional
significance of this regulatory mechanism, ‘mitochond-
rial Ca2+ desensitization’, i.e. the sharp reduction of
[Ca2+]m peaks upon repetitive cell stimulation, was
proposed to be responsible for phenomena, such as
the down-regulation of insulin secretion in pancreatic
b-cells [55]. Interestingly, inhibition of PKCb greatly
reduces the [Ca2+]m reduction upon repetitive agonist
stimulation, indicating that it could represent the
molecular route for Ca2+-dependent inhibition of cel-
lular responses.
The other physiological regulation of mitochondria
that has been studied in relation to Ca2+ signalling is
the state of fusion and fission. The study of mito-
chondrial structure, and of the mechanisms that
Mitochondrial calcium signalling in cell death S. Leo et al.
4018 FEBS Journal 272 (2005) 4013–4022 ª 2005 FEBS
dynamically control it in living cells, is a field of study
that literally boomed in the past decade. Indeed,
although the idea that mitochondria can form an inter-
connected reticulum was shown by reconstructing elec-
tron microscopy images in the late 1970s [56] and
more recently by using fluorescent GFP-based probes
and high-resolution digital imaging systems [26], it was
only after the identification of the molecules involved
that it became clear that the morphology of mitochon-
dria is tightly controlled by a dedicated cellular
machinery. Large GTPases, such as dynamin-related
protein-1 (Drp-1; a mechanoenzyme involved in mem-
brane constriction and fission), OPA1 (a dynamin rela-
ted GTPase, involved in fusion and mostly located in
the inner mitochondrial membrane) and mitofusins
(also involved in fusion and homologous to the first
element described, the ‘fuzzy onion’, fzo, protein of
Drosophila melanogaster [57]), and docking proteins,
such as Fis-1 (a transmembrane protein of the outer
mitochondrial membrane that participates in recruiting
Drp-1 to the organelle during organelle fragmenta-
tion). It is beyond the scope of this review to describe
in detail this fascinating field, and we refer to excellent
reviews on this topic [58–60]. Recent work gives some
insight into the dynamic regulation of the process, and
suggests that Ca2+ could be involved: (a) Ca2+ release
from the ER promotes the translocation of Drp-1 from
the cytoplasm to the outer mitochondrial membrane
[61]; (b) treatment with the Ca2+ ionophore, A23187,
triggers mitochondrial fission [62]; and (c) a novel
group of rho-GTPases have been described (mitoch-
ondrial rho 1 and 2; miro-1 and miro-2), that promote
mitochondrial fusion and include EF-hand Ca2+ bind-
ing sites in their sequence [63].
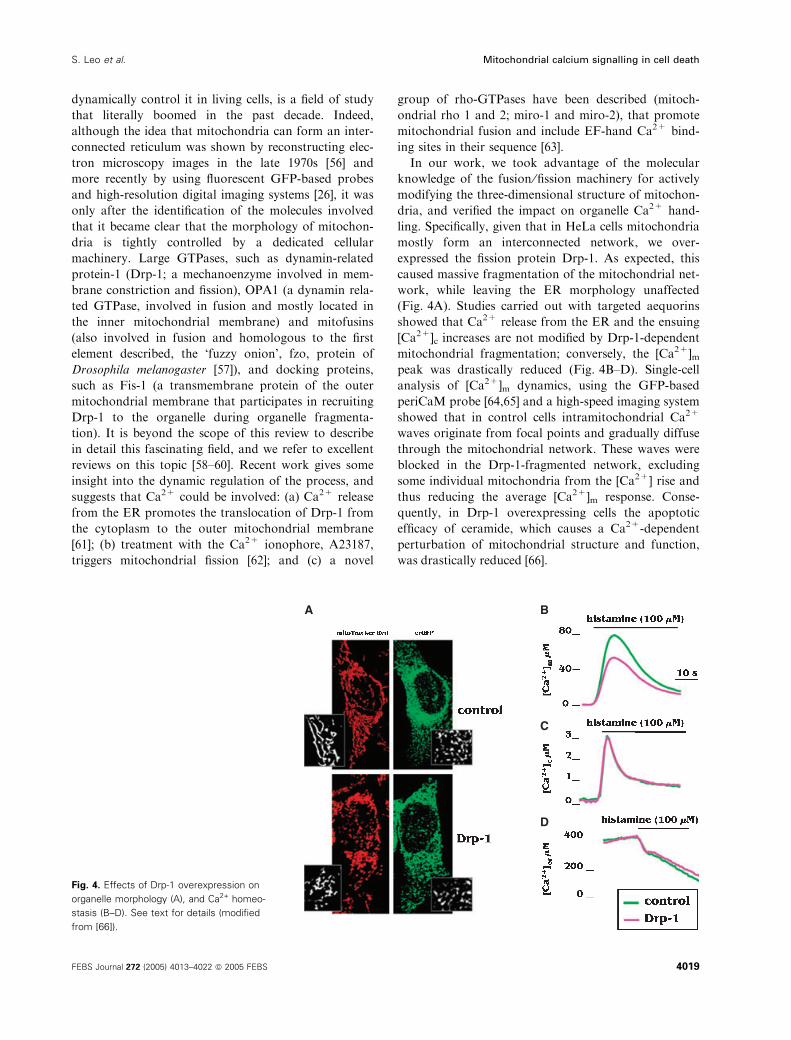
In our work, we took advantage of the molecular
knowledge of the fusion ⁄fission machinery for actively
modifying the three-dimensional structure of mitochon-
dria, and verified the impact on organelle Ca2+ hand-
ling. Specifically, given that in HeLa cells mitochondria
mostly form an interconnected network, we over-
expressed the fission protein Drp-1. As expected, this
caused massive fragmentation of the mitochondrial net-
work, while leaving the ER morphology unaffected
(Fig. 4A). Studies carried out with targeted aequorins
showed that Ca2+ release from the ER and the ensuing
[Ca2+]c increases are not modified by Drp-1-dependent
mitochondrial fragmentation; conversely, the [Ca2+]mpeak was drastically reduced (Fig. 4B–D). Single-cell
analysis of [Ca2+]m dynamics, using the GFP-based
periCaM probe [64,65] and a high-speed imaging system
showed that in control cells intramitochondrial Ca2+
waves originate from focal points and gradually diffuse
through the mitochondrial network. These waves were
blocked in the Drp-1-fragmented network, excluding
some individual mitochondria from the [Ca2+] rise and
thus reducing the average [Ca2+]m response. Conse-
quently, in Drp-1 overexpressing cells the apoptotic
efficacy of ceramide, which causes a Ca2+-dependent
perturbation of mitochondrial structure and function,
was drastically reduced [66].
A B
C
D
Fig. 4. Effects of Drp-1 overexpression on
organelle morphology (A), and Ca2+ homeo-
stasis (B–D). See text for details (modified
from [66]).
S. Leo et al. Mitochondrial calcium signalling in cell death
FEBS Journal 272 (2005) 4013–4022 ª 2005 FEBS 4019

Conclusions
A large body of experimental work shows that in vir-
tually every cell type the [Ca2+]c increases elicited in
the cytosol, by the opening of plasma membrane or
ER ⁄SR Ca2+ channels, is paralleled by a large
increase in the [Ca2+] of the mitochondrial matrix.
This process has two important functional conse-
quences. The first is that part of the Ca2+ entering the
cytoplasm is rapidly removed. Mitochondria thus act
as Ca2+ buffers, and this activity influences both the
local microdomain (hence affecting local inhibitory or
activatory effects of Ca2+ itself on the channel) or the
kinetics of the diffusion across the cytosol (thus acting
as a barrier limiting the [Ca2+]c rise to one portion of
the cell). Mitochondrial role is not limited, however, to
Ca2+ buffering. Indeed, within the organelle Ca2+ can
regulate functions as diverse as aerobic metabolism
(through Ca2+ sensitive enzymes and metabolite trans-
porters) and cell death (probably by activating the per-
meability transition pore). Given this high functional
plasticity, it is not surprising that we start to obtain
evidence that different mechanisms can finely tune
amplitude and kinetics of the mitochondrial Ca2+
responses. We have described a few intriguing exam-
ples (PKC, the fusion ⁄fission machinery), but it is rea-
sonable to predict that in the near future we will learn
much about these signalling routes and their cross-talk,
and get to know (finally!) the mitochondrial targets,
namely the channels, the regulatory elements and the
scaffolding proteins.
Acknowledgements
Experimental work in the authors’ laboratory was sup-
ported by grants from the Italian Ministry of Education
(FIRB, PRIN, local interest grants), the PRRIITT pro-
gram of the Emilia-Romagna region (ER-GenTech),
Telethon-Italy, the Italian Association for Cancer
Research (AIRC) and the Italian Space Agency (ASI).
References
1 Berridge MJ, Bootman MD & Roderick HL (2003) Cal-
cium signalling: dynamics, homeostasis and remodelling.
Nat Rev Mol Cell Biol 4, 517–529.
2 Tsien RY, Pozzan T & Rink TJ (1982) Calcium homeo-
stasis in intact lymphocytes: cytoplasmic free calcium
monitored with a new, intracellularly trapped fluores-
cent indicator. J Cell Biol 94, 325–334.
3 Pozzan T, Rizzuto R, Volpe P & Meldolesi J (1994)
Molecular and cellular physiology of intracellular cal-
cium stores. Physiol Rev 74, 595–636.
4 Petersen OH, Tepikin A & Park MK (2001) The endo-
plasmic reticulum: one continuous or several separate
Ca(2+) stores? Trends Neurosci 24, 271–276.
5 Montell C (2001) Physiology, phylogeny, and functions
of the TRP superfamily of cation channels. Sci. STKE
2001, re1.
6 Parekh AB & Putney JW Jr (2005) Store-operated cal-
cium channels. Physiol Rev 85, 757–810.
7 Orrenius S, Zhivotovsky B & Nicotera P (2003) Regula-
tion of cell death: the calcium-apoptosis link. Nat Rev
Mol Cell Biol 4, 552–565.
8 Carafoli E (2004) Calcium-mediated cellular signals: a
story of failures. Trends Biochem Sci 29, 371–379.
9 Rizzuto R & Pozzan T (2003) When calcium goes
wrong: genetic alterations of a ubiquitous signaling
route. Nat Genet 34, 135–141.
10 Duchen MR (2000) Mitochondria and calcium: from
cell signalling to cell death. J Physiol 529 Part 1, 57–68.
11 Dolmetsch R (2003) Excitation-transcription coupling:
signaling by ion channels to the nucleus. Sci STKE
2003, pe4.
12 Woods NM, Cuthbertson KS & Cobbold PH (1986)
Repetitive transient rises in cytoplasmic free calcium in
hormone-stimulated hepatocytes. Nature 319, 600–602.
13 Rizzuto R, Simpson AW, Brini M & Pozzan T (1992)
Rapid changes of mitochondrial Ca2+ revealed by spe-
cifically targeted recombinant aequorin. Nature 358,
325–327.
14 Montero M, Brini M, Marsault R, Alvarez J, Sitia R,
Pozzan T & Rizzuto R (1995) Monitoring dynamic
changes in free Ca2+ concentration in the endoplasmic
reticulum of intact cells. EMBO J 14, 5467–5475.
15 Barrero MJ, Montero M & Alvarez J (1997) Dynamics
of [Ca2+] in the endoplasmic reticulum and cytoplasm
of intact HeLa cells: a comparative study. J Biol Chem
272, 27694–27699.
16 Pinton P, Pozzan T & Rizzuto R (1998) The Golgi
apparatus is an inositol 1,4,5-trisphosphate-sensitive
Ca2+ store, with functional properties distinct from
those of the endoplasmic reticulum. EMBO J 17, 5298–
5308.
17 Brini M, Marsault R, Bastianutto C, Pozzan T &
Rizzuto R (1994) Nuclear targeting of aequorin: a new
approach for measuring nuclear Ca2+ concentration in
intact cells. Cell Calcium 16, 259–268.
18 Marsault R, Murgia M, Pozzan T & Rizzuto R (1997)
Domains of high Ca2+ beneath the plasma membrane
of living A7r5 cells. EMBO J 16, 1575–1581.
19 Carafoli E (2003) Historical review: mitochondria and
calcium: ups and downs of an unusual relationship.
Trends Biochem Sci 28, 175–181.
20 Kirichok Y, Krapivinsky G & Clapham DE (2004) The
mitochondrial calcium uniporter is a highly selective ion
channel. Nature 427, 360–364.
Mitochondrial calcium signalling in cell death S. Leo et al.
4020 FEBS Journal 272 (2005) 4013–4022 ª 2005 FEBS

21 Cox DA & Matlib MA (1993) Modulation of intramito-
chondrial free Ca2+ concentration by antagonists of
Na+–Ca2+ exchange. Trends Pharmacol Sci 14, 408–
413.
22 Streb H, Irvine RF, Berridge MJ & Schulz I (1983)
Release of Ca2+ from a nonmitochondrial intracellular
store in pancreatic acinar cells by inositol-1,4,5-trisphos-
phate. Nature 306, 67–69.
23 Rizzuto R, Bastianutto C, Brini M, Murgia M &
Pozzan T (1994) Mitochondrial Ca2+ homeostasis in
intact cells. J Cell Biol 126, 1183–1194.
24 Montero M, Alonso MT, Carnicero E, Cuchillo-Ibanez
I, Albillos A, Garcia AG, Garcia-Sancho J & Alvarez J
(2000) Chromaffin-cell stimulation triggers fast millimo-
lar mitochondrial Ca2+ transients that modulate secre-
tion. Nat Cell Biol 2, 57–61.
25 Rizzuto R, Brini M, Murgia M & Pozzan T (1993)
Microdomains with high Ca2+ close to IP3-sensitive
channels that are sensed by neighboring mitochondria.
Science 262, 744–747.
26 Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty
KE, Lifshitz LM, Tuft RA & Pozzan T (1998) Close
contacts with the endoplasmic reticulum as determinants
of mitochondrial Ca2+ responses. Science 280, 1763–
1766.
27 Brini M, De Giorgi F, Murgia M, Marsault R,
Massimino ML, Cantini M, Rizzuto R & Pozzan T
(1997) Subcellular analysis of Ca2+ homeostasis in
primary cultures of skeletal muscle myotubes. Mol Biol
Cell 8, 129–143.
28 Rudolf R, Mongillo M, Magalhaes PJ & Pozzan T
(2004) In vivo monitoring of Ca2+ uptake into mito-
chondria of mouse skeletal muscle during contraction.
J Cell Biol 166, 527–536.
29 McCormack JG, Halestrap AP & Denton RM (1990)
Role of calcium ions in regulation of mammalian
intramitochondrial metabolism. Physiol Rev 70, 391–
425.
30 Hansford RG (1994) Physiological role of mitochondrial
Ca2+ transport. J Bioenerg Biomembr 26, 495–508.
31 Jouaville LS, Pinton P, Bastianutto C, Rutter GA &
Rizzuto R (1999) Regulation of mitochondrial ATP
synthesis by calcium: evidence for a long-term metabolic
priming. Proc Natl Acad Sci USA 96, 13807–13812.
32 Palmieri L, Pardo B, Lasorsa FM, del Arco A,
Kobayashi K, Iijima M, Runswick MJ, Walker JE,
Saheki T, Satrustegui J et al. (2001) Citrin and aralar1
are Ca2+-stimulated aspartate ⁄ glutamate transporters in
mitochondria. EMBO J 20, 5060–5069.
33 Lasorsa FM, Pinton P, Palmieri L, Fiermonte G,
Rizzuto R & Palmieri F (2003) Recombinant expression
of the Ca2+-sensitive aspartate ⁄ glutamate carrier increa-
ses mitochondrial ATP production in agonist-stimulated
Chinese hamster ovary cells. J Biol Chem 278, 38686–
38692.
34 Szalai G, Krishnamurthy R & Hajnoczky G (1999)
Apoptosis driven by IP(3)-linked mitochondrial calcium
signals. EMBO J 18, 6349–6361.
35 Pinton P, Ferrari D, Di Virgilio F, Pozzan T & Rizzuto
R (2001) Molecular machinery and signalling events in
apoptosis. Drug Dev Res 52, 558–570.
36 Pinton P, Ferrari D, Rapizzi E, Di Virgilio FD, Pozzan
T & Rizzuto R (2001) The Ca2+ concentration of the
endoplasmic reticulum is a key determinant of cera-
mide-induced apoptosis: significance for the molecular
mechanism of Bcl-2 action. EMBO J 20, 2690–2701.
37 Kluck RM, Bossy-Wetzel E, Green DR & Newmeyer
DD (1997) The release of cytochrome c from mitochon-
dria: a primary site for Bcl-2 regulation of apoptosis.
Science 275, 1132–1136.
38 Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J,
Peng TI, Jones DP & Wang X (1997) Prevention of
apoptosis by Bcl-2: release of cytochrome c from mito-
chondria blocked. Science 275, 1129–1132.
39 Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow
BE, Brothers GM, Mangion J, Jacotot E, Costantini P,
Loeffler M et al. (1999) Molecular characterization of
mitochondrial apoptosis-inducing factor. Nature 397,
441–446.
40 Du C, Fang M, Li Y, Li L & Wang X (2000) Smac,
a mitochondrial protein that promotes cytochrome
c-dependent caspase activation by eliminating IAP inhi-
bition. Cell 102, 33–42.
41 Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K,
Di Virgilio F, Pozzan T & Rizzuto R (2000) Reduced
loading of intracellular Ca2+ stores and downregulation
of capacitative Ca(2+) influx in Bcl-2-overexpressing
cells. J Cell Biol 148, 857–862.
42 Foyouzi-Youssefi R, Arnaudeau S, Borner C, Kelley
WL, Tschopp J, Lew DP, Demaurex N & Krause KH
(2000) Bcl-2 decreases the free Ca2+ concentration
within the endoplasmic reticulum. Proc Natl Acad Sci
USA 97, 5723–5728.
43 Chami M, Ferrari D, Nicotera P, Paterlini-Brechot P
& Rizzuto R (2003) Caspase-dependent alterations of
Ca2+ signaling in the induction of apoptosis by hepa-
titis B virus X protein. J Biol Chem 278, 31745–
31755.
44 Tinel H, Cancela JM, Mogami H, Gerasimenko JV,
Gerasimenko OV, Tepikin AV & Petersen OH (1999)
Active mitochondria surrounding the pancreatic acinar
granule region prevent spreading of inositol trispho-
sphate-evoked local cytosolic Ca(2+) signals. EMBO J
18, 4999–5008.
45 Jouaville LS, Ichas F, Holmuhamedov EL, Camacho P
& Lechleiter JD (1995) Synchronization of calcium
waves by mitochondrial substrates in Xenopus laevis
oocytes. Nature 377, 438–441.
46 Hajnoczky G, Hager R & Thomas AP (1999) Mito-
chondria suppress local feedback activation of inositol
S. Leo et al. Mitochondrial calcium signalling in cell death
FEBS Journal 272 (2005) 4013–4022 ª 2005 FEBS 4021

1,4, 5-trisphosphate receptors by Ca2+. J Biol Chem
274, 14157–14162.
47 Boitier E, Rea R & Duchen MR (1999) Mitochondria
exert a negative feedback on the propagation of intra-
cellular Ca2+ waves in rat cortical astrocytes. J Cell
Biol 145, 795–808.
48 Landolfi B, Curci S, Debellis L, Pozzan T & Hofer AM
(1998) Ca2+ homeostasis in the agonist-sensitive inter-
nal store: functional interactions between mitochondria
and the ER measured in situ in intact cells. J Cell Biol
142, 1235–1243.
49 Hoth M, Fanger CM & Lewis RS (1997) Mitochondrial
regulation of store-operated calcium signaling in T lym-
phocytes. J Cell Biol 137, 633–648.
50 Gilabert JA & Parekh AB (2000) Respiring mitochon-
dria determine the pattern of activation and inactivation
of the store-operated Ca2+ current I (CRAC). EMBO J
19, 6401–6407.
51 Budd SL & Nicholls DG (1996) Mitochondria, calcium
regulation, and acute glutamate excitotoxicity in cultured
cerebellar granule cells. J Neurochem 67, 2282–2291.
52 Pinton P, Leo S, Wieckowski MR, Di Benedetto G &
Rizzuto R (2004) Long-term modulation of mitochon-
drial Ca2+ signals by protein kinase C isozymes. J Cell
Biol 165, 223–232.
53 Montero M, Lobaton CD, Gutierrez-Fernandez S,
Moreno A & Alvarez J (2003) Modulation of histamine-
induced Ca2+ release by protein kinase C. Effects on
cytosolic and mitochondrial [Ca2+] peaks. J Biol Chem
278, 49972–49979.
54 Vermassen E, Fissore RA, Nadif KN, Vanderheyden V,
Callewaert G, Missiaen L, Parys JB & De Smedt H
(2004) Regulation of the phosphorylation of the inositol
1,4,5-trisphosphate receptor by protein kinase C. Bio-
chem Biophys Res Commun 319, 888–893.
55 Maechler P, Kennedy ED, Wang H & Wollheim CB
(1998) Desensitization of mitochondrial Ca2+ and insu-
lin secretion responses in the beta cell. J Biol Chem 273,
20770–20778.
56 Bakeeva LE, Chentsov Y & Skulachev VP (1978)
Mitochondrial framework (reticulum mitochondriale) in
rat diaphragm muscle. Biochim Biophys Acta 501, 349–
369.
57 Hales KG & Fuller MT (1997) Developmentally regu-
lated mitochondrial fusion mediated by a conserved,
novel, predicted GTPase. Cell 90, 121–129.
58 Bossy-Wetzel E, Barsoum MJ, Godzik A,
Schwarzenbacher R & Lipton SA (2003) Mitochondrial
fission in apoptosis, neurodegeneration and aging. Curr
Opin Cell Biol 15, 706–716.
59 Scorrano L (2003) Divide et impera: Ca2+ signals, mit-
ochondrial fission and sensitization to apoptosis. Cell
Death Differ 10, 1287–1289.
60 Perfettini JL, Roumier T & Kroemer G (2005) Mito-
chondrial fusion and fission in the control of apoptosis.
Trends Cell Biol 15, 179–183.
61 Breckenridge DG, Stojanovic M, Marcellus RC & Shore
GC (2003) Caspase cleavage product of BAP31 induces
mitochondrial fission through endoplasmic reticulum
calcium signals, enhancing cytochrome c release to the
cytosol. J Cell Biol 160, 1115–1127.
62 Duncan CJ, Greenaway HC, Publicover SJ, Rudge MF
& Smith JL (1980) Experimental production of
‘septa’ and apparent subdivision of muscle
mitochondria. J Bioenerg Biomembr 12, 13–33.
63 Fransson A, Ruusala A & Aspenstrom P (2003) Atypi-
cal Rho GTPases have roles in mitochondrial homeosta-
sis and apoptosis. J Biol Chem 278, 6495–6502.
64 Nagai T, Sawano A, Park ES & Miyawaki A (2001)
Circularly permuted green fluorescent proteins engi-
neered to sense Ca2+. Proc Natl Acad Sci USA 98,
3197–3202.
65 Filippin L, Magalhaes PJ, Di Benedetto G, Colella M
& Pozzan T (2003) Stable interactions between mito-
chondria and endoplasmic reticulum allow rapid accu-
mulation of calcium in a subpopulation of
mitochondria. J Biol Chem 278, 39224–39234.
66 Szabadkai G, Simoni AM, Chami M, Wieckowski MR,
Youle RJ & Rizzuto R (2004) Drp-1-dependent division
of the mitochondrial network blocks intraorganellar
Ca2+ waves and protects against Ca2+-mediated apop-
tosis. Mol Cell 16, 59–68.
Mitochondrial calcium signalling in cell death S. Leo et al.
4022 FEBS Journal 272 (2005) 4013–4022 ª 2005 FEBS




















