
Antidepressant Augmentation Using the N -Methyl- d -Aspartate Antagonist Memantine

ORIGINAL ARTICLE
Altered calcium homeostasis in autism-spectrumdisorders: evidence from biochemical and genetic studiesof the mitochondrial aspartate/glutamate carrier AGC1L Palmieri1,2, V Papaleo3,4,16, V Porcelli1,16, P Scarcia1, L Gaita3,4, R Sacco3,4, J Hager5, F Rousseau5,
P Curatolo6, B Manzi6, R Militerni7, C Bravaccio7, S Trillo8, C Schneider9, R Melmed10, M Elia11,
C Lenti12, M Saccani12, T Pascucci13,14, S Puglisi-Allegra13,14, K-L Reichelt15 and AM Persico3,4
1Laboratory of Biochemistry and Molecular Biology, Department of Pharmaco-Biology, University of Bari, Bari, Italy; 2ConsiglioNazionale delle Ricerche, Institute of Biomembranes and Bioenergetics, Bari, Italy; 3Laboratory of Molecular Psychiatry andNeurogenetics, University ‘Campus Bio-Medico’, Rome, Italy; 4Laboratory of Molecular Psychiatry and Psychiatric Genetics,Department of Experimental Neurosciences, I.R.C.C.S. ‘Fondazione Santa Lucia’, Rome, Italy; 5IntegraGen SA. Genopole,Evry, France; 6Department of Child Neuropsychiatry, University ‘Tor Vergata’, Rome, Italy; 7Department of ChildNeuropsychiatry, University of Naples Federico II, Naples, Italy; 8ASL RM/B, Rome, Italy; 9Center for Autism Research andEducation, Phoenix, AZ, USA; 10Southwest Autism Research and Resource Center, Phoenix, AZ, USA; 11Unit of Neurologyand Clinical Neurophysiopathology, I.R.C.C.S. ‘Oasi Maria S.S.’, Enna, Italy; 12Department of Child Neuropsychiatry,University of Milan, Milan, Italy; 13Department of Psychology, University ‘La Sapienza’, Rome, Italy; 14Laboratory of BehavioralNeurobiology, Department of Experimental Neurosciences, I.R.C.C.S. ‘Fondazione Santa Lucia’, Rome, Italy and15Department of Pediatric Research, Rikshospitalet, University of Oslo, Oslo, Norway
Autism is a severe developmental disorder, whose pathogenetic underpinnings are still largelyunknown. Temporocortical gray matter from six matched patient–control pairs was used toperform post-mortem biochemical and genetic studies of the mitochondrial aspartate/glutamate carrier (AGC), which participates in the aspartate/malate reduced nicotinamideadenine dinucleotide shuttle and is physiologically activated by calcium (Ca2þ ). AGC transportrates were significantly higher in tissue homogenates from all six patients, including thosewith no history of seizures and with normal electroencephalograms prior to death. Thisincrease was consistently blunted by the Ca2þ chelator ethylene glycol tetraacetic acid;neocortical Ca2þ levels were significantly higher in all six patients; no difference in AGCtransport rates was found in isolated mitochondria from patients and controls followingremoval of the Ca2þ -containing postmitochondrial supernatant. Expression of AGC1, thepredominant AGC isoform in brain, and cytochrome c oxidase activity were both increased inautistic patients, indicating an activation of mitochondrial metabolism. Furthermore, oxidizedmitochondrial proteins were markedly increased in four of the six patients. Variants of theAGC1-encoding SLC25A12 gene were neither correlated with AGC activation nor associatedwith autism-spectrum disorders in 309 simplex and 17 multiplex families, whereas someunaffected siblings may carry a protective gene variant. Therefore, excessive Ca2þ levels areresponsible for boosting AGC activity, mitochondrial metabolism and, to a more variabledegree, oxidative stress in autistic brains. AGC and altered Ca2þ homeostasis play a keyinteractive role in the cascade of signaling events leading to autism: their modulation couldprovide new preventive and therapeutic strategies.Molecular Psychiatry (2010) 15, 38–52; doi:10.1038/mp.2008.63; published online 8 July 2008
Keywords: autistic disorder; calcium signaling; chromosome 2q; oxidative stress; pervasivedevelopmental disorders; SLC25A12
Introduction
Autism is a severe pervasive developmental disordercharacterized by impaired language, communicationand social skills, as well as by repetitive andstereotypic patterns of behavior.1 Its incidence hasdramatically risen from 2–5 to 15–60/10 000 childrenduring the past two decades: broader diagnosticcriteria and increased medical awareness have con-tributed to determine this trend, but the involvement
Received 20 July 2007; revised 7 April 2008; accepted 16 May2008; published online 8 July 2008
Correspondence: Professor AM Persico, Laboratory of MolecularPsychiatry and Neurogenetics, University ‘Campus Bio-Medico’,Via Alvaro del Portillo 21, I-00128 Rome, Italy.E-mail: [email protected] Professor L Palmieri, Laboratory of Biochemistry and Mole-cular Biology, Department of Pharmaco-Biology, University ofBari, Via Orabona 4, Bari 70125, Italy.E-mail: [email protected] authors contributed equally to this work.
Molecular Psychiatry (2010) 15, 38–52& 2010 Nature Publishing Group All rights reserved 1359-4184/10 $32.00
www.nature.com/mp

of environmental factors is also likely.2,3 Despitestrong familial components, clinical and geneticcomplexities have posed a major challenge to ourunderstanding of autism pathogenesis.3 Clinically, thebehavioral expression of autism-predisposing genescan range from minimal autistic traits to full-blownautism, identifying a broad clinical entity referred toas ‘autism-spectrum disorders’ (ASDs).3,4 In addition,comorbidity with seizures and mental retardationoccurs in up to 30% and in 80% of autistic patients,respectively.5 Genetically, the picture is complicatedby significant interindividual heterogeneity, num-erous contributing loci, incomplete penetrance,phenocopies, gene–gene and gene–environment inter-actions.3 Several lines of evidence strongly support aprenatal onset for developmental abnormalities laterleading to autism:6 post-mortem assessments of brainsof autistic patients have unveiled early neurodevelop-mental alterations including reduced programmedcell death and/or increased cell proliferation, alteredcell migration, abnormal cell differentiation withreduced neuronal size and altered synaptogenesis;3,7
many children later diagnosed with ASD displaymotor abnormalities8 and/or excessive body growth,9
already on the day of birth or in early neonatal life. Inaddition, systemic signs and symptoms includingmacrosomy,9 nonspecific enterocolitis,10,11 immunedysreactivity11 and renal oligopeptiduria,12 pose aut-ism as a multiorgan systemic disorder encompassingseveral developmental components, not restricted tothe central nervous system (CNS).
Evidence linking altered energy metabolism toautistic disorder has been available for some time,including peripheral markers, such as increasedplasma lactate levels, and rare instances of associa-tion between respiratory chain disorders and aut-ism.13,14 Interest in assessing the role of mitochondriain this disorder has been revitalized by the associa-tion between autism and variants of the SLC25A12gene, which encodes the predominant isoform of themitochondrial aspartate (asp)/glutamate (glu) carrier(AGC) in brain.15,16 AGC belongs to a family of integralproteins that catalyze the transport of metabolites andcofactors across the inner mitochondrial membrane.17
In particular, AGC is important in energy metabolismby transporting glutamate into mitochondria inexchange for matrix aspartate, a key regulatory stepin the malate/aspartate reduced nicotinamideadenine dinucleotide (NADH) shuttle.18,19 Its twoisoforms, AGC1 and AGC2, also named aralar(1) andcitrin, are encoded by the SLC25A12 and SLC25A13genes, located on human chromosomes 2q24 and7q21.3, respectively.19 AGC1 and AGC2 expressionoverlaps during early prenatal life, but divergesbeginning in late gestation and into adulthood, withAGC1 predominantly expressed in the brain, heartand skeletal muscle, whereas AGC2 is mainly ex-pressed in liver and kidney.20,21 In the CNS, AGC1 ishighly expressed in neurons, whereas glial cellsexpress both isoforms at much lower levels.20,21
Importantly, AGC activity is regulated by intracellular
calcium (Ca2þ ) through four ‘EF-hand’ domains22
located at its N-terminus, hanging into the intermem-brane space.18,19 As Ca2þ concentrations in themitochondrial intermembrane space and cytosol arein equilibrium, cytosolic Ca2þ can rapidly activateAGC transport, thereby increasing the NADH/NADratio in the mitochondrial matrix and consequentlyboosting electron flow through the respiratorychain and adenosine triphosphate (ATP) generationby oxidative phosphorylation.18,19,23 Through thismechanism, AGC1 is important in the transductionof small Ca2þ signals to neuronal mitochondria.19 Anexcessive amplitude and/or duration of Ca2þ spikesleading to AGC activation can, however, contribute tothe formation of reactive oxygen species (ROS) and tooxidative stress.24 Genetic and/or environmentalfactors could thus interfere with neuronal ATPproduction and with oxidative stress by affecting theAGC1 carrier, either directly or through Ca2þ home-ostasis.
In this study, we have assessed AGC transport rates,SLC25A12 gene expression, Ca2þ concentrations andrespiratory chain activity in the post-mortem tempor-ocortical gray matter (Brodmann area 41/42 or 22) ofsix pairs of nonsyndromic ASD patients and sex-, age-and post-mortem interval (PMI)-matched controls(Table 1). Neocortex from the superior temporal gyruswas selected, as it hosts structural and functionalabnormalities well documented in autistic indivi-duals.25 Sequencing genomic DNA and cDNA fromthese same tissues, we have searched for polymorph-isms and/or mutations in the SLC25A12 gene corre-lated with biochemical parameters. Finally, we havesearched for SLC25A12 gene variants conferringautism vulnerability using a family-based associationapproach in 309 simplex and 17 multiplex familieswith a nonsyndromic autistic proband.
Materials and methods
Patient brain tissue informationRelevant clinical and demographic information wasobtained from the Autism Tissue Program (www.atpportal.org) and is summarized in Table 1. Patientswere selected for the absence of CNS malformationsor dysmorphology; controls were selected to matchpatients, based on sex, age (±2 years) and PMI. Thesame, or slightly larger paired patient–control sam-ples, have been the object of recent reports.26–28
Brain tissue processingFresh frozen brain tissue samples dissected from thesuperior temporal gyrus (BA 41/42 or 22) of patientsand controls were obtained through the AutismTissue Program from the NICHD Brain and TissueBank and the Harvard Brain Tissue Resource Center.Cortical gray matter (100–200 mg) was suspended in abuffer containing 250 mM sucrose and 10 mM TRIS,pH 7.4 and homogenated using a Teflon glass potter.Cell debris was pelleted by centrifugation at 800� gand discarded. Cytochrome c oxidase (COX) and
Altered calcium homeostasis in autismL Palmieri et al
39
Molecular Psychiatry

Table
1B
rain
tiss
ue
info
rmati
on
for
pati
en
tsan
dcon
trols
Pair
no.
Case
no.a
Dia
gn
osi
sA
ge
(years
)b
Sex
PM
I(h
)c
Cau
seof
death
Men
tal
reta
rdati
on
Ep
ilep
syO
ther
featu
res
Dru
gth
era
pie
sat
the
tim
eof
death
1B
5342
Au
tism
11
F13
Dro
wn
ing
(seiz
ure
?)Y
es
Yes
Recu
rren
toti
tis
Top
am
ax,
Lam
icta
l,A
dd
era
ll
2B
5569
PD
D-N
OS
5M
25.5
Dro
wn
ing
No
No
Recu
rren
toti
tis,
an
gio
ed
em
a,
food
all
erg
ies
Pro
zac,
mela
ton
ind
3U
MB
4721
Au
tism
8M
16
Dro
wn
ing
Un
kn
ow
nN
o—
Non
e
4B
6337
Au
tism
22
M25
Seiz
ure
Yes
Yes
Inte
stin
al
lym
ph
oad
en
op
ath
y,h
yp
ert
rop
hic
sple
en
,re
cu
rren
toti
tis
Lam
ecti
l,Z
on
egra
n,
Neu
ron
tin
,A
bil
ify,
flax
seed
oil
,O
-3,
mu
ltiv
itam
in
5B
6294
Au
tism
16
MU
nkn
ow
nS
eiz
ure
Un
kn
ow
nY
es
—T
op
am
ax,
Dep
akote
,A
llegra
,C
lari
tin
,N
uT
hera
mu
ltiv
itam
in
6B
5144
Au
tism
20
M23.7
Tra
um
aY
es
No
—N
on
e
1U
MB
1407
Con
trol
9F
20
Ast
hm
aN
oN
o—
Non
e2
UM
B1185
Con
trol
4M
17
Dro
wn
ing
No
No
—N
on
e3
UM
B1860
Con
trol
8M
5C
ard
iac
arr
hyth
mia
No
No
—N
on
e
4B
6221
Con
trol
22
M24
Un
kn
ow
nN
oN
o—
Non
e5
B6207
Con
trol
16
M26
Isch
em
ich
eart
att
ack
No
No
—N
on
e
6B
3829
Con
trol
22
M12
Cen
tral
hep
ati
cla
cera
tion
No
No
—N
on
e
aA
uti
smT
issu
eP
rogra
mid
en
tifi
er.
bM
ean
age
(±s.
d)
for
the
au
tism
gro
up
=13.2
±7.5
,fo
rcon
trols
=13.0
±8.4
;p
air
ed
-t=
0.3
07,
5d
.f.,
P=
0.7
71.
cP
MI,
post
-mort
em
inte
rval;
mean
PM
I(±
s.d
)fo
rth
eau
tism
gro
up
=20.6
±5.7
,fo
rcon
trols
=15.6
±7.4
,p
air
ed
-t=
1.4
17,
4d
.f.,
P=
0.2
30.
dF
or
B5569,
the
last
rep
ort
sum
mari
zin
gth
ep
harm
acolo
gic
al
thera
py
date
sto
less
than
ayear
pri
or
tod
eath
.
Altered calcium homeostasis in autismL Palmieri et al
40
Molecular Psychiatry

citrate synthase (CS) activities were measured, ac-cording to the standard procedures.29 Mitochondriafrom brain homogenates were harvested by centrifu-gation at 13 000� g for 12 min at 4 1C. Mitochondria-enriched pellets and postmitochondrial supernatantswere then stored at �80 1C until further processing.Total RNA was extracted using the TRIzol reagent(Invitrogen, Carlsbad, CA, USA) according to stan-dard methods, and RNA quality was checked using aBioanalyzer (Agilent, Santa Clara, CA, USA); all RNAsamples used in this study displayed RNA integritynumber (RIN) values > 6.9. DNA was recoveredby phenol/chloroform extraction and ethanol preci-pitation, following tissue digestion in proteinase Kovernight at 55 1C.
Solubilization and functional reconstitution of AGC1Proteins from tissue homogenates or isolated mito-chondria were solubilized in 0.4% Triton X-114(w/v), 20 mM Na2SO4, 10 mM MOPS, pH 7.0, at a finalconcentration of 1.9–3.5 mg protein per ml (0.5–1.5 mUCS ml�1); 5 mM ethylene glycol tetraacetic acid(EGTA) was added 10 min prior to solubilization,where specified. After incubation for 20 min at 4 1C,the mixture was spun at 138 000� g for 12 min.Triton-solubilized proteins were immediately incor-porated into phospholipid vesicles by cyclic removalof the detergent using a hydrophobic column.30 Thecomposition of the initial mixture use for reconstitu-tion was: 25 ml of Triton-solubilized extract, 70 ml of10% Triton X-114, 110ml of 10% phospholipids in theform of sonicated liposomes, 20 mM glutamate, 10 mM
MOPS (pH 7.0), 0.4 mg cardiolipin and water to a finalvolume of 700ml. Deionized Milli-Q water was usedfor preparation of all solutions. Transport at 25 1C wasstarted by adding 40 mM [14C]-aspartate to the proteo-liposomes and terminated at each time interval byadding 30 mM pyridoxal 50-phosphate and 10 mM
bathophenanthroline.30 Entrapped radioactivity wascounted. The initial rate of transport was calculatedfrom the time course of isotope equilibration.30
Expression analysis by real-time PCRTotal RNA was reverse-transcribed with the GeneAmpRNA PCR Core kit (Applied Biosystems, Foster City,CA, USA). Probes and primers for AGC1, AGC2 andPiC were purchased from Applied Biosystems. Theamount of each cDNA was measured using thecomparative method (2�DDCt ; User Bulletin 2 P/N4303859, Applied Biosystems), and the humanb-actin was used as standard normalizer.
Western blottingWestern blotting of brain homogenates was performedusing polyclonal antibodies raised in rabbit againstrecombinant AGC123 or mouse monoclonal antibodiesagainst the b subunit of human F1-ATPase (cod.612519; BD Biosciences, San Jose, CA, USA).Antigen–antibody complexes were detected usinganti-rabbit or anti-mouse IgG-coupled horseradishperoxidase (cod. 31460 or 31430; Pierce, Rockford,
IL, USA), in combination with the Amersham ECLsystem (GE Healthcare, Piscataway, NJ, USA).
Fluorimetric measurement of Ca2þ concentrationsFree Ca2þ levels were measured in postmitochondrialsupernatants by the fluorescent indicator Fura-2.Following incubation of each postmitochondrialsupernatant (20 mg protein) with 1 mM Fura-2 penta-potassium salt (F6799; Molecular Probes, Eugene, OR,USA), the ratio of fluorescence intensities detected at510 nm at two different excitation wavelengths (340and 380 nm) was determined using a PerkinElmer LS50 spectrofluorimeter. The indicator calibration andCa2þ measurements were performed at room tem-perature, as described.31
Quantification of mitochondrial protein oxidationCarbonylated mitochondrial proteins were detectedusing the OxyBlot Protein Oxidation Detection Kit(Chemicon, Temecula, CA, USA). 2,4 dinitrophenyl-hydrazine-derivatized crude protein extracts were dotblotted, incubated with a primary antibody againstDNP-hydrazone followed by a horseradish peroxi-dase-conjugated secondary antibody, visualized usingthe ECL chemiluminescent detection method andquantified by densitometry.
SLC25A12 cDNA and genomic DNA sequencingThe SLC25A12 coding sequence was screened be-tween bp þ 13 and þ2941, relative to the ATGtranslation start site, using cDNA and genomic DNAobtained from the same six ASD–control pairsassessed for transport and gene expression studies.Primer sequences and PCR conditions are summar-ized in Supplementary Table S1. DNA sequencingwas performed using a CEQ8000 DNA sequencer(Beckman-Coulter, Fullerton, CA, USA). All single-nucleotide polymorphisms (SNPs) identified by DNAsequencing were confirmed by restriction digest.
Subjects recruited for the family-based associationstudyOur clinical sample includes 309 simplex and 17multiplex families with a nonsyndromic autisticproband (Supplementary Table S2), encompassing atotal of 346 patients and 120 unaffected siblings.Demographic and clinical characteristics of ASDpatients are summarized in Supplementary TableS3. Inclusion criteria and diagnostic screening meth-ods used to exclude probands with syndromic autismhave been previously reported.32 Briefly, patientsfulfilling DSM-IV diagnostic criteria for autisticdisorder1 were screened for nonsyndromic autismusing MRI, electroencephalogram (EEG), audiometry,urinary amino acid and organic acid measurements,cytogenetic and fragile-X testing. Patients with dys-morphic features were excluded even in the absenceof detectable cytogenetic alterations. Patients withsporadic seizures (that is, < 1 every 6 months) wereincluded; patients with frequent seizures or focalneurological deficits were excluded. Large subsets of
Altered calcium homeostasis in autismL Palmieri et al
41
Molecular Psychiatry

patients were assessed for adaptive functioning usingthe Vineland Adaptive Behavior Scales, for autisticbehaviors using the official Italian version of theAutism Diagnostic Observation Schedule and theAutism Diagnostic Interview—Revised,33,34 as soonas these were made publicly available in 2006, forintelligence quotient (IQ) using either the GriffithMental Developmental Scales, the Coloured RavenMatrices, the Bayley Developmental Scales or theLeiter International Performance Scale. Given the useof different scales in different clinical centers and thegenerally low reliability of IQ measurements inautistic patients, IQ scores were dichotomized as< 70 or X70 (that is, presence/absence of mentalretardation) and are presented in SupplementaryTable S3 according to this criterion. In addition, oursample encompasses a total of 120 unaffected sib-lings, from 92 families with one unaffected sibling, 9families with 2, 2 families with 3 and another with 4unaffected siblings, whereas 222 families with anautistic proband include no unaffected brothers andsisters. The definition of ‘unaffected sibling’ is basedupon the clinical judgement of the same cliniciansresponsible for the DSM-IV diagnosis of autisticdisorder in the affected sibling. All parents gavewritten informed consent for themselves and for theirchildren, using the consent form approved by theInstitutional Review Board of University ‘CampusBio-Medico’ (Rome, Italy).
GenotypingA total of 11 SNPs spanning the SLC25A12 locuswere genotyped either by TaqMan or by SNPlex(Supplementary Table S4). Probes for TaqMan andSNPlex were synthesized by the manufacturerand used according to manufacturer’s guidelines(Applied Biosystems).
Biochemical and morphological endophenotypesHead circumference, serotonin (5-HT) blood levels,and urinary peptide excretion rates were measured, aspreviously described.32,35
Statistical analysesCase–control contrasts were performed using theWilcoxon’s signed-ranks test for enzymatic and asp/glu transport activity data, and the paired Student’st-test for Ca2þ levels, depending upon normality andvariance homogeneity which were tested with theLevene and Kolmogorov–Smirnov tests, respectively,as implemented by the SPSS software package (ver.14.0). Hardy–Weinberg equilibrium was tested usingthe w2 statistic, as implemented by the Haploviewsoftware (available at http://www.broad.mit.edu/mpg/haploview/index.php)36 which extracts a max-imal set of unrelated individuals from the totalsample, and by the HWE software (available athttp://linkage.rockefeller.edu/soft/linkutil/), used toanalyze separately mothers, fathers, autistic patients,and unaffected siblings (including only one autisticand unaffected sibling per family). Family-based
single-marker and haplotype association tests wereperformed using the family-based association test(FBAT) statistic (S =ST�X, where T is the phenoty-pic trait and X is the marker value), as implementedby the FBAT software package (available at http://www.biostat.harvard.edu/~fbat/fbat.htm), under anadditive model.37 The FBAT statistic stems from thetransmission/disequilibrium test (TDT), where pre-ferential allelic transmission from heterozygous par-ents to affected offspring is tested by applying the(b�c)2/(bþ c) statistics and the w2 (‘McNemar test’).38
Maternal and paternal transmissions were also ana-lyzed separately using the same FBAT approach. Tofurther ensure reliability, despite lower statisticalpower, we also performed single-marker TDTs oncomplete trios only and randomly choosing one trioper multiplex family, by using TDTPHASE (http://www.rfcgr.mrc.ac.uk/~fdudbrid/software/unphased/),39
which implements likelihood ratio tests based on log-linear models. Quantitative traits were analyzed by(1) quantitative transmission/disequilibrium test(qTDT), as implemented by the same FBAT statistic,where T is the quantitative trait of interest (instead ofa dichotomic affected/unaffected status as in theTDT),37 and by (2) parametric analysis of variance(ANOVA) based on genotype distributions, or non-parametric Kruskal–Wallis ANOVA, implementedwhenever the assumptions of normality and/orvariance homogeneity could not be satisfied despitedata transformation. Patients treated with selective 5-HT-reuptake inhibitors were excluded from analysesof 5-HT blood levels. Linkage disequilibrium (LD)was tested using Haploview,36 which calculates D0
coefficients reported here and constructs haplotypeblocks by implementing the approach of Gabrielet al.40 The consistency of these D0 coefficientswas also verified using the HBAT command inFBAT on pedigree data. Analyses were carried outmerging together 290 Italian and 36 Caucasian-American families (Supplementary Table S2), afterpopulation structure analyses (see SupplementaryMethods in ref.27 for a detailed description) providedno evidence of major genetic dyshomogeneityin a subgroup of 155 Italians and 24 Caucasian-Americans autistic patients randomly chosen one perfamily, genotyped at 90 unlinked SNPs distributedgenome wide and analyzed using the Structuresoftware.41
Data are expressed as mean±s.e.m., except for headcircumference and urinary peptide excretion rates,expressed as median percentile±interquartilic range.Nominal two-tail P-values are reported. Statisticalsignificance is set by applying a Bonferroni correctionfor multiple testing as follows: (1) Hardy–Weinberganalyses: P < 0.05/55 (11 SNPs � 5 sets of analyses bystatus) = 0.00091; (2) haplotype analyses: P < 0.05/2(two haplotype blocks) = 0.025; single-marker ana-lyses: P < 0.05/5 (two haplotype blocksþ three SNPsoutside the blocks) = 0.01. We did not apply aBonferroni correction considering all 11 SNPs asindependent observations, because the existence of
Altered calcium homeostasis in autismL Palmieri et al
42
Molecular Psychiatry

two haplotype blocks implies by definition thenonindependence of single-marker and endopheno-typic analyses involving SNPs contained within eachblock. No correction for multiple testing was adoptedfor the two ethnic groups, given the outcome ofpopulation structure analyses described above.
Results
AGC activity is boosted by excessive Ca2þ levelsin autistic brainsAGC activity, normalized by CS activity to adjust fordifferences in absolute mitochondria tissue content,displays a prominent threefold increase in neocorticalhomogenates from nonsyndromic autistic patientscompared to matched controls in all six pairs(Wilcoxon’s test: Z =�2.201, P < 0.05; Figure 1a, left).AGC1 protein levels only show a very modest,nonsignificant increase (Supplementary Figure S1).Instead, Ca2þ chelation by EGTA reduces asp/gluexchange rates to a much larger extent in patientsthan in controls (2.1- vs 0.35-fold, respectively),reducing case–control differences to only 36.1%(Figure 1a, middle). No difference between patientsand controls is anymore detectable upon reconstitu-tion of protein extracts from isolated mitochondria(P = 0.17; Figure 1a, right). These results stronglypoint toward excessive Ca2þ concentrations as mostlikely responsible for increased asp/glu exchangerates in the brains of autistic patients.
To further test this hypothesis, two aliquots ofisolated mitochondria from each control have beenexposed in parallel experiments either to his/her ownpostmitochondrial supernatant, or to the postmito-chondrial supernatant of his/her matched autisticpatient (Figure 1b, left). Remarkably, the patientsupernatant activates asp/glu exchange rates to asignificantly larger extent than the control super-natant in all six pairs, yielding mean 3.2- vs 1.7-foldincreases, respectively (P < 0.05). This differencebetween case and control supernatants is no longersignificant following Ca2þ chelation by EGTA(þ119.8 vs þ 82.3%, respectively; P=0.074; Figure 1b,
right). These results confirm the primary role ofexcessive Ca2þ levels in boosting AGC activity inthe brains of autistic patients. Experiments employingmitochondria from autistic patients provide super-imposable results (Figure 1c), except for a statisticallysignificant elevation of asp/glu transport rates overcontrol levels retained also following exposure of thesupernatants to EGTA (Figures 1b vs 1c, right panels).
Finally, direct measurements of Ca2þ concentra-tions in the postmitochondrial supernatant revealedconsistently and significantly increased Ca2þ levelsin all six ASD patients compared to matchedcontrols (paired Student’s t-test: t =�5.259, 5 degrees
Figure 1 Aspartate/glutamate exchange rates are activatedby enhanced calcium levels in neocortical tissue of autisticpatients. (a) Aspartate/glutamate carrier (AGC) activity inneocortical tissue homogenate, in the absence (left) or in thepresence (middle) of 5 mM ethylene glycol tetraacetic acid(EGTA), and in mitochondria isolated from the same tissuespecimens (right); (b) AGC activity in mitochondria isolatedfrom the neocortical tissue of each of six controls exposed tothe postmitochondrial supernatant from his/her matchedautistic patient (autism-spectrum disorder, ‘ASD’ label) orto his/her own supernatant (‘controls’ label), preincubatedin the absence (left) or in the presence (right) of 5 mM EGTA;(c) AGC activity in mitochondria isolated from the neocor-tical tissue of each of six ASD patients, exposed to thehis/her own postmitochondrial supernatant (‘ASD’ label) orto the supernatant of his/her matched control (‘controls’label), preincubated in the absence (left) or in the presence(right) of 5 mM EGTA.
Altered calcium homeostasis in autismL Palmieri et al
43
Molecular Psychiatry

of freedom (d.f.), P < 0.01; Figure 2a). This increase isentirely independent of a positive or negative history ofseizures, antiepileptic medications or seizure-relateddeath (patients N=3 positive vs 3 negative history,Ca2þ levels: t=0.288, 4 d.f., P=0.788; autistic/controlCa2þ ratios: t=�0.699, 4 d.f., P=0.523; Figure 2b).
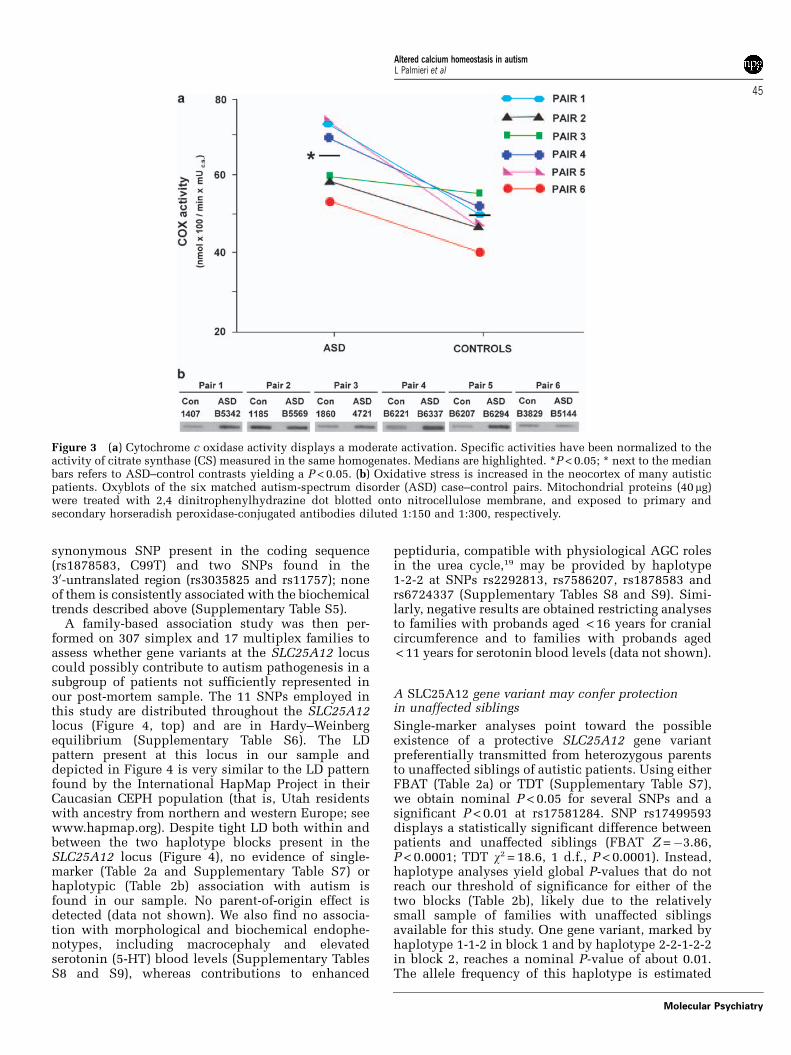
AGC activation increases mitochondrial metabolismand oxidative stressBy increasing the availability of reducing equivalentsin the mitochondrial matrix through AGC, enhancedcytosolic Ca2þ can be predicted to steadily boostmitochondrial metabolism and oxidative phosphor-ylation.18,19,23 Indeed, transcript amounts of themitochondrial phosphate carrier (PiC), AGC1, andAGC2 (expressed at much lower levels compared toAGC1), are all increased (Supplementary Figure S2).Also COX activity is elevated to a similar extent in allsix autistic patients compared to their matchedcontrols (P < 0.05; Figure 3a).
Ca2þ -triggered increases of the NADH/NAD ratio inthe mitochondrial matrix and of the electron flowthrough the respiratory chain could potentially foster
oxidative stress in genetically liable individuals.24
Markers of oxidative damage show an impressivethreefold increase in autistic brains compared tocontrols, with mean carbonylated mitochondrialprotein content up by 202.3% (Figure 3b). However,this increase does not reach statistical significance inour sample (P = 0.12) due to four patients displayinglarge increases in markers of oxidative damage(þ147, þ204, þ 393 and þ 492%), whereas patientsin pairs 2 and 6 show levels comparable to thoserecorded in their matched controls (�7 and �15.5%,respectively; Figure 3b). The latter results likelyreflect elevated oxidative stress in one control(UMB1185 in pair 2), and normal levels in oneautistic subject (B5144 in pair 6).
No evidence of SLC25A12 gene contributions to autismvulnerability
Sequencing of the AGC1-encoding SLC25A12 cDNAand genomic DNA in these same six case–controlpairs does not detect any nonsynonymous codingmutation. Only five known SNPs are identified, twolocated in intron 1 (rs3821092 and rs12991441), one
Figure 2 Calcium levels measured directly in the postmitochondrial supernatant by fluorimetry. (a) Calcium concentrationsare significantly higher in the neocortical tissue of autism-spectrum disorder (ASD) patients compared to matched controls.Data are presented as mean±s.d. of three measures per sample; paired Student’s t-test: t =�5.259, 5 degrees of freedom (d.f.),P < 0.01; (b) calcium concentrations do not differ between patients with a positive or negative history of seizures,antiepileptic medications or seizure-related death (patients N = 3 positive vs 3 negative history, calcium levels: t = 0.288,4 d.f., P = 0.788; autistic/control calcium ratios: t =�0.699, 4 d.f., P = 0.523).
Altered calcium homeostasis in autismL Palmieri et al
44
Molecular Psychiatry
synonymous SNP present in the coding sequence(rs1878583, C99T) and two SNPs found in the30-untranslated region (rs3035825 and rs11757); noneof them is consistently associated with the biochemicaltrends described above (Supplementary Table S5).
A family-based association study was then per-formed on 307 simplex and 17 multiplex families toassess whether gene variants at the SLC25A12 locuscould possibly contribute to autism pathogenesis in asubgroup of patients not sufficiently represented inour post-mortem sample. The 11 SNPs employed inthis study are distributed throughout the SLC25A12locus (Figure 4, top) and are in Hardy–Weinbergequilibrium (Supplementary Table S6). The LDpattern present at this locus in our sample anddepicted in Figure 4 is very similar to the LD patternfound by the International HapMap Project in theirCaucasian CEPH population (that is, Utah residentswith ancestry from northern and western Europe; seewww.hapmap.org). Despite tight LD both within andbetween the two haplotype blocks present in theSLC25A12 locus (Figure 4), no evidence of single-marker (Table 2a and Supplementary Table S7) orhaplotypic (Table 2b) association with autism isfound in our sample. No parent-of-origin effect isdetected (data not shown). We also find no associa-tion with morphological and biochemical endophe-notypes, including macrocephaly and elevatedserotonin (5-HT) blood levels (Supplementary TablesS8 and S9), whereas contributions to enhanced
peptiduria, compatible with physiological AGC rolesin the urea cycle,19 may be provided by haplotype1-2-2 at SNPs rs2292813, rs7586207, rs1878583 andrs6724337 (Supplementary Tables S8 and S9). Simi-larly, negative results are obtained restricting analysesto families with probands aged < 16 years for cranialcircumference and to families with probands aged< 11 years for serotonin blood levels (data not shown).
A SLC25A12 gene variant may confer protectionin unaffected siblings
Single-marker analyses point toward the possibleexistence of a protective SLC25A12 gene variantpreferentially transmitted from heterozygous parentsto unaffected siblings of autistic patients. Using eitherFBAT (Table 2a) or TDT (Supplementary Table S7),we obtain nominal P < 0.05 for several SNPs and asignificant P < 0.01 at rs17581284. SNP rs17499593displays a statistically significant difference betweenpatients and unaffected siblings (FBAT Z =�3.86,P < 0.0001; TDT w2 = 18.6, 1 d.f., P < 0.0001). Instead,haplotype analyses yield global P-values that do notreach our threshold of significance for either of thetwo blocks (Table 2b), likely due to the relativelysmall sample of families with unaffected siblingsavailable for this study. One gene variant, marked byhaplotype 1-1-2 in block 1 and by haplotype 2-2-1-2-2in block 2, reaches a nominal P-value of about 0.01.The allele frequency of this haplotype is estimated
Figure 3 (a) Cytochrome c oxidase activity displays a moderate activation. Specific activities have been normalized to theactivity of citrate synthase (CS) measured in the same homogenates. Medians are highlighted. *P < 0.05; * next to the medianbars refers to ASD–control contrasts yielding a P < 0.05. (b) Oxidative stress is increased in the neocortex of many autisticpatients. Oxyblots of the six matched autism-spectrum disorder (ASD) case–control pairs. Mitochondrial proteins (40 mg)were treated with 2,4 dinitrophenylhydrazine dot blotted onto nitrocellulose membrane, and exposed to primary andsecondary horseradish peroxidase-conjugated antibodies diluted 1:150 and 1:300, respectively.
Altered calcium homeostasis in autismL Palmieri et al
45
Molecular Psychiatry

at approximately 23–26% (Table 2b), making it arelatively common variant among unaffected siblings.
Discussion
This study reports increased asp/glu exchange ratesand significantly higher Ca2þ concentrations in post-mortem neocortical tissue specimens of six nonsyn-dromic autistic patients compared to age-, sex- andPMI-matched controls. Altogether, our resultsstrongly support excessive Ca2þ levels as primarilyresponsible for the observed activation of asp/gluexchange rates, whereas genetic contributions appearneither widespread nor necessary, at least in our post-mortem and genetic samples.
Indeed the AGC activation documented here is (1)EGTA sensitive (Figures 1a–c), (2) recorded only inthe presence of the postmitochondrial supernatant,and absent in reconstituted protein extracts fromisolated mitochondria (Figure 1a), and (3) signifi-cantly more prominent when AGC reconstituted frommitochondrial membranes of each control is exposedto the postmitochondrial supernatant of his/hermatched ASD case, than to his/her own supernatant(Figure 1b). Measurements employing AGC reconsti-tuted from mitochondrial membranes of each ASDpatient exposed either to their own supernatant or tothe supernatant of his/her matched control, essen-tially confirm the latter result (Figure 1c). The only
difference between these two complementaryapproaches is that AGC reconstituted from mito-chondrial membranes of ASD patients retains astatistically significant elevation of asp/glu transportrates over control levels also following exposure toEGTA (Figure 1c). This result nicely parallels theresidual difference observed with homogenates (butnot with isolated mitochondria), indicating that thehigher Ca2þ levels present in the patient supernatantlikely render equal concentrations of EGTA lesseffective, as compared to control supernatant. Mostimportantly, direct measurements of neocortical Ca2þ
concentrations clearly demonstrate a significant ele-vation of neocortical Ca2þ levels in all autisticpatients compared to controls (Figure 2a). Finally,the thorough cDNA sequencing performed in ourpost-mortem cases, the highly favorable LD patternpresent at the SLC25A12 locus (Figure 4), the numberof informative SNPs assayed in our family-basedassociation study, the similar extent of asp/glutransport found in the protein extracts from isolatedmitochondria of patients and controls (Figure 1a,right panel), the nonspecific generalized trans-criptional activation of mitochondrial transporters(Supplementary Figure S1), which does not translateinto noticeable increases in AGC1 protein amounts(Supplementary Figure S2), all strongly argueagainst genetic mutations or functional polymorph-isms at the SLC25A12 locus as primarily responsible
Figure 4 High linkage disequilibrium (LD) at the SLC25A12 locus. Genomic structure, genotyping markers, D0 values� 100(that is, LD measure ranging 0–100) in our patients and first-degree relatives, and LD blocks at the SLC25A12 locus, accordingto the Haploview software.36
Altered calcium homeostasis in autismL Palmieri et al
46
Molecular Psychiatry

Table
2S
LC
25A
12
gen
evari
an
tsd
on
ot
con
fer
au
tism
vu
lnera
bil
ity;
acom
mon
pro
tecti
ve
vari
an
tcou
ldbe
pre
sen
tam
on
gu
naff
ecte
dsi
bli
ngs
(a)
Mark
er
All
ele
sA
uti
stic
pati
en
tsU
naff
ecte
dsi
bli
ngs
Nu
mber
of
fam
ilie
sS
E(S
)V
ar(
S)
ZP
-valu
eN
um
ber
of
fam
ilie
sS
E(S
)V
ar(
S)
ZP
-valu
e
rs2292813
C74
112.0
00
114.3
33
21.5
56
�0.5
03
0.6
15266
19
34.0
00
33.6
67
6.7
22
0.1
29
0.8
97702
T74
46.0
00
43.6
67
21.5
56
0.5
03
19
14.0
00
14.3
33
6.7
22
�0.1
29
rs17499593
C141
189.0
00
198.5
00
45.7
50
�1.4
05
0.1
60164
50
86.0
00
77.5
00
18.2
50
1.9
90
0.0
46624
G141
105.0
00
95.5
00
45.7
50
1.4
05
50
30.0
00
38.5
00
18.2
50
�1.9
90
rs17581284
C214
196.0
00
187.0
00
70.5
00
1.0
72
0.2
83772
70
81.0
00
67.0
00
27.0
00
2.6
94
0.0
07054
T214
254.0
00
263.0
00
70.5
00
�1.0
72
70
87.0
00
101.0
00
27.0
00
�2.6
94
rs7586207
A156
204.0
00
207.0
00
49.5
00
�0.4
26
0.6
69815
56
69.0
00
79.5
00
18.2
50
�2.4
58
0.0
13977
G156
124.0
00
121.0
00
49.5
00
0.4
26
56
57.0
00
46.5
00
18.2
50
2.4
58
rs12692976
A199
243.0
00
252.0
00
67.5
00
�1.0
95
0.2
73322
70
90.0
00
92.5
00
24.7
50
�0.5
03
0.6
15303
C199
177.0
00
168.0
00
67.5
00
1.0
95
70
68.0
00
65.5
00
24.7
50
0.5
03
rs2271758
G197
238.0
00
244.1
67
65.4
72
�0.7
62
0.4
45990
61
79.0
00
89.3
33
22.7
22
�2.1
68
0.0
30175
T197
176.0
00
169.8
33
65.4
72
0.7
62
61
69.0
00
58.6
67
22.7
22
2.1
68
rs3770445
A160
209.0
00
212.0
00
51.5
00
�0.4
18
0.6
75918
56
70.0
00
78.5
00
18.2
50
�1.9
90
0.0
46624
G160
127.0
00
124.0
00
51.5
00
0.4
18
56
54.0
00
45.5
00
18.2
50
1.9
90
rs2056202
C89
113.0
00
132.8
33
27.3
06
0.0
32
0.9
74556
25
44.0
00
46.1
67
7.9
72
�0.7
67
0.4
42964
T89
57.0
00
57.1
67
27.3
06
�0.0
32
25
20.0
00
17.8
33
7.9
72
0.7
67
rs1878583
A73
45.0
00
43.5
00
21.7
50
0.3
22
0.7
47730
19
15.0
00
14.0
00
7.2
50
0.1
86
0.8
52684
G73
111.0
00
112.5
00
21.7
50
�0.3
22
19
35.0
00
35.5
00
7.2
50
�0.1
86
rs6724337
C167
218.0
00
221.0
00
53.7
50
�0.4
77
0.6
33080
54
66.0
00
76.0
00
17.5
00
�2.3
90
0.0
16827
T167
132.0
00
128.0
00
53.7
50
0.4
77
54
54.0
00
44.0
00
17.5
00
2.3
90
rs7573003
A181
144.0
00
142.5
00
58.2
50
0.1
97
0.8
44190
62
56.0
00
48.5
00
19.7
50
1.6
88
0.0
91482
T181
234.0
00
235.5
00
58.2
50
�0.1
97
62
76.0
00
83.5
00
19.7
50
�1.6
88
Altered calcium homeostasis in autismL Palmieri et al
47
Molecular Psychiatry

for the increase in AGC activity observed in oursample.
Increased AGC transport rates, COX activities andCa2þ levels consistently recorded in all six neocor-tical specimens from ASD patients crossvalidate eachother, confirming the reliability and biological sig-nificance of these findings. This consistency arguesagainst spurious effects exclusively due to seizures ordrug treatment at the time of death. Potentialconfounding factors are inevitably intermingled withautism in our post-mortem sample, as they are at thephenotypic level in real life. Approximately 80% ofautistic patients indeed have mental retardation andup to 30% a positive history of seizures, for whichthey often times receive pharmacological treatment.5
Our post-mortem sample, though small, faithfullyreflects the ASD population in terms of comorbidity.Indeed, three out of six ASD patients having apositive history for seizures, were receiving antiepi-leptic drugs at the time of death, and some of them
may have undergone a seizure-related death, assummarized in Table 1. Furthermore, several anti-epileptic drugs taken at the time of death eitherdirectly or indirectly modulate Ca2þ levels. Inprinciple, we cannot exclude that epilepsy couldpartly contribute to generate oxidative stress in theneocortex of autistic patients. However, our threeseizure-positive and three seizure-negative ASD pa-tients display absolutely no difference in supernatantCa2þ concentrations and in matched patient/controlCa2þ ratios (Figure 2b), nor do they differ in any AGC-related measure (Figure 1). For example, patientsB5342 and B5144 display the largest elevation in Ca2þ
levels: the former is seizure-positive, the latterseizure-negative (Figure 2a). Patients B5569 andUMB4721 display some of the largest increases inasp/glu exchange rates (see pairs 2 and 3 in Figures 1aand b) despite normal EEG reports, no history ofseizure, and causes of death unrelated to seizures(Table 1). Similarly, at least two patients, namely
Table 2 Continued
(b)Estimatedfrequency
Number offamilies
S E(S) Var(S) Z P-value
Autistic patientsHaplotype block 1
1-2-1 0.446 180.0 190.994 205.738 70.739 �1.753 0.0795941-1-2 0.257 138.0 128.972 125.583 47.914 0.490 0.6244182-2-1 0.183 115.9 104.978 96.195 37.964 1.425 0.1540131-1-1 0.108 79.0 58.006 54.423 24.313 0.727 0.467441
Global haplotype test: allele n = 7, d.f. 4, w2 = 4.785, P = 0.310079
Haplotype block 21-1-1-2-1 0.634 148.0 214.000 216.905 57.858 �0.382 0.7025492-2-1-2-2 0.232 122.0 102.000 99.333 43.222 0.406 0.6850252-1-2-1-1 0.077 54.0 34.000 31.762 16.300 �0.554 0.5793442-1-2-2-1 0.035 26.0 15.000 16.500 7.750 �0.539 0.590014
Global haplotype test: allele n = 13, d.f. 4, w2 = 0.805, P = 0.937744
Unaffected siblingsHaplotype block 1
1-2-1 0.446 66.0 77.000 83.694 27.401 �1.279 0.2009411-1-2 0.257 50.0 55.000 44.583 16.414 2.571 0.0101352-2-1 0.183 46.0 31.500 38.239 17.581 �1.726 0.0842781-1-1 0.108 28.0 25.000 21.467 10.334 1.099 0.271742
Global haplotype test: allele n = 7, d.f. 4, w2 = 9.118, P = 0.058224
Haplotype block 21-1-1-2-1 0.634 53.0 80.000 90.476 21.636 �2.252 0.0243082-2-1-2-2 0.232 45.0 46.000 35.833 15.496 2.583 0.0098042-1-2-1-1 0.077 17.0 12.000 11.690 6.047 0.126 0.8998352-1-2-2-1 0.035 10.0 10.000 8.500 4.083 0.742 0.457901
Global haplotype test: allele n = 13, d.f. 4, w2 = 8.503, P = 0.074782
(a) Single-marker family-based association test (FBAT), performed using the FBAT software under an additive model;37 (b)haplotype-based intrafamilial association test for autistic patients and unaffected siblings, performed using the HBATcommand in the FBAT software, under an additive model.37 The two haplotype blocks depicted in Figure 4 are analyzedseparately. Only haplotypes with estimated frequencies > 1% are listed. FBAT output variables: N, number of informativenuclear families; S, test statistic (that is, genotypic distribution in the offspring conditioned on affection status and parentalgenotypes); E(S), expected value for S; Var(S), variance of S under the null hypothesis of no linkage and no association.P-values < 0.05 and < 0.01 are highlighted in bold and gray, respectively.
Altered calcium homeostasis in autismL Palmieri et al
48
Molecular Psychiatry

UMB4721 and B5144 (see pairs 3 and 6 in Figures 1aand b), were not taking psychoactive drugs at the timeof death; pharmacological treatments were also quitedifferent from patient to patient (Table 1). Alsodifferences in PMI cannot explain our results. MeanPMI values are neither significantly different betweenpatients and controls (see note c in Table 1), nor arethey correlated with AGC activation, as pair 1displays the strongest AGC activation induced bythe supernatant of the autistic patient (Figure 1b),whereas her PMI is shorter than the PMI of hermatched control (Table 1). In summary, altered Ca2þ
homeostasis is the only factor shared by all autisticcortical tissue samples (Figure 2a), able to boost AGCactivity,18,19,23 and previously linked to the pathogen-esis of autism per se.42
The existence of altered Ca2þ signaling in autismhas been suggested in recent years by several lines ofresearch.42 Gain-of-function mutations in the L-typevoltage-gated Ca2þ channel Cav1.2 (CACNA1C) causeTimothy syndrome, a multisystem disorder includingmental retardation and autism.43 Similarly, mutationsin the L-type voltage-gated Ca2þ channel Cav1.4(CACNA1F) cause the incomplete form of X-linkedcongenital stationary night blindness (CSNB2):gain-of-function mutations cause CSNB2 frequentlyaccompanied by cognitive impairment and eitherautism or epilepsy, whereas CSNB2 due to loss-of-function mutations is not accompanied bythese symptoms.44 All of these gain-of-functionmutations prevent voltage-dependent channel inacti-vation leading to excessive Ca2þ influx. Also muta-tions indirectly yielding increased cytosolic Ca2þ
levels or amplifying intracellular Ca2þ signaling byhampering Ca2þ -activated negative feedback mechan-isms have been found associated with autism.42,45
The bioelectrical instability resulting from thesemutations nicely parallels the high prevalence ofseizures and/or EEG abnormalities present amongautistic individuals.
Elucidating the process underlying the abnormalCa2þ homeostasis described in this study is of utmostimportance. Unfortunately post-mortem studies donot allow us to determine whether the enhanced Ca2þ
levels documented in Figure 2a primarily reflectexcessive in vivo extracellular Ca2þ concentrations,intracellular Ca2þ storage, or both; these possibilitieswill have to be teased out using cell cultureparadigms. However, due to the dynamic nature ofCa2þ homeostasis, in either case cytosolic Ca2þ
spikes can be predicted to display increased ampli-tude and/or duration,46 resulting in enhanced AGCtransport activity. Although rare de novo mutationsraising intracellular Ca2þ levels can occasionallycause autism, a more likely scenario envisions eitherthe coexistence in the same individual of severalunfavorable common gene variants, or environmentaland stochastic contributions acting upon a vulnerablegenetic background. According to post-mortem andin vivo studies, an ongoing immune response couldprovide such a contribution by raising intracellular
Ca2þ levels through cytokines, such as tumor necrosisfactor-a and receptors, like CD38.28,47,48 Indeed, theexistence of an immune dysreactivity is also sup-ported by the association between macrocephaly andmacrosomy in autism with a history of immune/allergic disorders, either in the patient or in his/herfirst-degree relatives.9 Immune mechanisms may alsomediate the vulnerability to autism conferred byrecently described 16p11.2 microdeletions.49 Anotherpredicable pathophysiological consequence of anexcessive, immune-driven Ca2þ entry into cellswould be a progressive decrease in bone density,which has been described in at least two reports50,51
and may be frequently overlooked especially inyounger patients. Finally, an 8-year-old female control(case no. UMB1706) was excluded from this studyafter consistently showing an EGTA-sensitive, 50–75%increase in asp/glu exchange rates. Interestingly, thisgirl died of a heart transplant rejection, and it isvery intriguing that a massive immune responsewas followed in a nonautistic individual by anAGC activation reaching levels intermediate betweenthose recorded in autistic patients and in othercontrols. Clearly, an immune response can raiseCa2þ levels and boost asp/glu exchange rates, asreported in the present study. However, the oppositecould also occur: mutations or polymorphic variantsin genes encoding Ca2þ channels or glutamatereceptors could produce an increase in intracellularCa2þ and/or in oxidative stress (see below), able totrigger an immune response similar to the oneobserved in autistic brains. We are currently in theprocess of correlating AGC activity and levels ofoxidative stress with markers of immune activation,measured in the same tissue specimens assessed inthis study.
We found no evidence of genetic contributions toautism vulnerability by SLC25A12 gene variants inour sample. These negative findings are in accordancewith several prior reports.14,52,53 In addition toobvious genetic heterogeneity in complex disorders,other factors could have potentially contributed to therepeated nonreplication of the two initial positivereports:15,16 (1) prominent differences in LD patterns,previously well documented for example at thePRKCB1 locus27 between our sample and an Irishsample largely overlapping with the one assessed bySegurado et al.;16 (2) spurious positive findings basedon the two SNPs (rs2292813 and rs2056202) display-ing the lowest heterozygosity (HZ) in the region (seeobserved HZ in Supplementary Table S6); (3) possiblegenetic contributions to a clinical endophenotypespecifically characterized by prominent routines andrituals.54 In our sample, the presence of motorstereotypies at intake is not significantly associatedwith allele G at rs7586207 in haplotype block 1(w2 = 1.527, 2 d.f., P = 0.466), or allele T at rs6724337 inhaplotype block 2 (w2 = 2.488, 2 d.f., P = 0.288).Altogether, we do not exclude that rare polymorph-isms or functional variants located outside thegenomic regions sequenced here could conceivably
Altered calcium homeostasis in autismL Palmieri et al
49
Molecular Psychiatry

provide contributions to enhanced SLC25A12 geneexpression, although this seems rather unlikely giventhe tight LD pattern present in the region (Figure 4).Instead the present study clearly demonstrates thatthese genetic variants, if at all present, play a minorrole compared to the extent of AGC activationproduced by excessive Ca2þ levels in the neocortexof ASD patients.
Our results point toward the possible existence of aprotective SLC25A12 gene variant in a sizable groupof unaffected siblings. This cannot be conclusivelydemonstrated with our sample size of 104 familiesincluding one or more unaffected sibling. Unfortu-nately, no other study has to this date reported onunaffected siblings14–16,52–54 and genotyping for theAutism Genetic Resource Exchange (AGRE) data set isonly available for the same two SNPs discussed above(rs2292813 and rs2056202), displaying low informa-tives and in the case of rs2292813 also located outsideof the haplotype blocks depicted in Figure 4. It will beimportant to see whether our results are confirmed inlarger samples, for at least two reasons: on one hand,they suggest that the status of ‘unaffected sibling’could define an individual possessing geneticallydetermined compensatory mechanisms not availableto autistic patients, rather than merely identifyingfamily members who have not inherited the full arrayof disease-producing alleles. Our recent identificationof another protective variant in the glyoxalase Igene,35 in conjunction with gain-of-function muta-tions in the CACNA1F gene consistently yieldingnight blindness while causing autism or epilepsy onlyin a subset of carriers,44 provide converging evidenceof genetic backgrounds effectively protecting fromautism. On the other hand, our results would providefurther support for key contributions of Ca2þ -trig-gered AGC1 activity to autism pathogenesis. In-creased asp/glu exchange rates provide morereducing equivalents (that is, NADH) to the respira-tory chain and could foster oxidative stress,24 whichwas previously found increased measuring peripheralmarkers in autism.55 Also overexpression of AGC1 incell culture has been recently found associated with abiphasic response, characterized initially by en-hanced neurite outgrowth, which subsequently slowsdown and ends in early cell death.56 This response isseemingly compatible with an initial overproductionof ATP paralleled by a progressive build up ofoxidative stress leading to cell damage. Oxidativestress, in addition to lipid and protein oxidation,55
can also produce genomic instability and stimulatecell cycle progression, pathophysiological eventslikely to be important in autism pathogenesis9,57,58 Inthis regard, the interindividual variability in oxida-tive damage reported in our study is not at allsurprising, as the balance between ROS productionand antioxidant agents leaves ample room for geneticand environmental influences.
The present results can potentially pave the path totargeted preventive and therapeutic strategies. Oneimportant example is represented by thimerosal, an
ethyl-mercury compound used as a preservative invaccines.59,60 Thimerosal has drawn attention follow-ing initial anecdotal reports by some parents linkingvaccinations to behavioral regression and to the onsetof autism in their child within a matter of days or fewweeks. Thimerosal is a Ca2þ -mobilizing agent, cap-able of releasing Ca2þ from intracellular stores andincreasing Ca2þ entry.61 Despite its short half-lifecompared to inorganic mercury, it undergoes prefer-ential accumulation in the CNS, affecting the micro-glia and producing strain-dependent neurotoxiceffects in rodents.62,63 This strain dependency, inconjunction with the present data, suggests thatthimerosal could contribute to produce an unba-lanced Ca2þ homeostasis in genetically vulnerableindividuals. Indeed, a postnatal exposure to thimer-osal is not reconcilable with the prenatal onset ofneurodevelopmental anomalies leading to autism.Also large retrospective epidemiological studies con-firm that thimerosal neither causes autism, norprovides large-scale contributions to its pathogen-esis.2,60 However, our results suggest that thimerosalcould conceivably precipitate an abrupt onset in asubset of children who would have otherwise devel-oped autistic symptoms more insidiously. At thesame time, we cannot exclude that thimerosal andother Ca2þ -mobilizing environmental factors couldalso push genetically vulnerable individuals alongthe autism-spectrum toward more severe forms of thedisease. On the basis of the present study, theelimination of thimerosal from vaccines, undertakenin the United States and Canada, is a well-justifiedsafety measure.
Pharmacological treatments able to modulate extra-cellular Ca2þ entry, intracellular Ca2þ release fromthe endoplasmic reticulum or putative upstreamimmune mechanisms affecting either or both thepathways are, at least in principle, already available.However, caution should be exercised in translatingthe present findings into therapeutic interventionsprior to at least one replication in an independentcohort of brain samples and to assessments of Ca2þ
homeostasis in vivo. In particular, our findings in noway support the use of Ca2þ chelation as a therapeu-tic approach in autism. Ca2þ chelation has not onlybeen purported of benefit in few anecdotal reportsand small-sized open trials, but also carries asubstantial risk to produce hypocalcemia, resultingin recent deaths of autistic children.64,65 Pharmacolo-gically reducing Ca2þ entry into cells or bluntingthe oxidative damage produced by AGC activationseemingly represent more amenable and less danger-ous therapeutic strategies. It is nonetheless difficult topredict the actual efficacy of treatments initiatedduring childhood on pathogenetic mechanisms activesince early prenatal development. In this regard, theidentification and functional characterization of pro-tective SLC25A12 gene variants, if existent, couldprovide additional critical information on the con-tribution of AGC activation and oxidative stress toautism pathogenesis.
Altered calcium homeostasis in autismL Palmieri et al
50
Molecular Psychiatry

Acknowledgments
We gratefully acknowledge all the patients and familieswho generously contributed to these studies, theAutism Tissue Program, Harvard Brain Tissue ResourceCenter and NICHD Brain and Tissue Bank for provid-ing the brain tissue samples, and Roberto Rigardettoand Franco Nardocci for contributing to patientrecruitment. This work was supported by MIUR PRIN2005052128 and 2006058195 (LP, PS and AMP), MIURFIRB (LP) and the Fondation Jerome Lejeune (AMP).
References
1 American Psychiatric Association. Diagnostic and StatisticalManual of Mental Disorders, 4th edn. American PsychiatricAssociation: Washington, DC, 1994.
2 Rutter M. Incidence of autism spectrum disorders: changes overtime and their meaning. Acta Paediatr 2005; 94: 2–15.
3 Persico AM, Bourgeron T. Searching for ways out of the autismmaze: genetic, epigenetic and environmental clues. TrendsNeurosci 2006; 29: 349–358.
4 Piven J, Palmer P, Jacobi D, Childress D, Arndt S. Broader autismphenotype: evidence from a family history study of multiple-incidence autism families. Am J Psychiatry 1997; 154: 185–190.
5 Berney TP. Autism, an evolving concept. Br J Psychiatry 2000; 176:20–25.
6 Miller MT, Stromland K, Ventura L, Johansson M, Bandim JM,Gillberg C. Autism associated with conditions characterized bydevelopmental errors in early embryogenesis: a mini review. Int JDev Neurosci 2005; 23: 201–219.
7 Bauman ML, Kemper TL. Neuroanatomic observations of the brainin autism: a review and future directions. Int J Dev Neurosci 2005;23: 183–187.
8 Teitelbaum O, Benton T, Shah PK, Prince A, Kelly JL, Teitelbaum P.Eshkol-Wachman movement notation in diagnosis: the earlydetection of Asperger’s syndrome. Proc Natl Acad Sci USA 2004;101: 11909–11914.
9 Sacco R, Militerni R, Frolli A, Bravaccio C, Gritti A, Elia M et al.Clinical, morphological, and biochemical correlates of headcircumference in autism. Biol Psychiatry 2007; 62: 1038–1047.
10 Wakefield AJ, Ashwood P, Limb K, Anthony A. The significance ofileo-colonic lymphoid nodular hyperplasia in children with autisticspectrum disorder. Eur J Gastroenterol Hepatol 2005; 17: 827–836.
11 Jyonouchi H, Geng L, Ruby A, Zimmerman-Bier B. Dysregulatedinnate immune responses in young children with autism spectrumdisorders: their relationship to gastrointestinal symptoms anddietary intervention. Neuropsychobiology 2005; 51: 77–85.
12 Reichelt WH, Knivsberg AM, Nodland M, Stensrud M, Reichelt KL.Urinary peptide levels and patterns in autistic children from sevencountries, and the effect of dietary intervention after 4 years. DevBrain Dysfunct 1997; 10: 44–55.
13 Chugani DC, Sundram BS, Behen M, Lee ML, Moore GJ. Evidenceof altered energy metabolism in autistic children. Prog Neuropsy-chopharmacol Biol Psychiatry 1999; 23: 635–641.
14 Correia C, Coutinho AM, Diogo L, Grazina M, Marques C, Miguel Tet al. Brief report: high frequency of biochemical markers formitochondrial dysfunction in autism: no association with themitochondrial aspartate/glutamate carrier SLC25A12 gene.J Autism Dev Disord 2006; 36: 1137–1140.
15 Ramoz N, Reichert JG, Smith CJ, Silverman JM, Bespalova IN,Davis KL et al. Linkage and association of the mitochondrialaspartate/glutamate carrier SLC25A12 gene with autism. Am JPsychiatry 2004; 161: 662–669.
16 Segurado R, Conroy J, Meally E, Fitzgerald M, Gill M, Gallagher L.Confirmation of association between autism and the mitochon-drial aspartate/glutamate carrier SLC25A12 gene on chromosome2q31. Am J Psychiatry 2005; 162: 2182–2184.
17 Palmieri F. The mitochondrial transporter family (SLC25): physio-logical and pathological implications. Pflugers Arch 2004; 447:689–709.
18 Palmieri L, Pardo B, Lasorsa FM, del Arco A, Kobayashi K, Iijima Met al. Citrin and aralar1 are Ca2þ -stimulated aspartate/glutamatetransporters in mitochondria. EMBO J 2001; 20: 5060–5069.
19 Satrustegui J, Pardo B, del Arco A. Mitochondrial transporters asnovel targets for intracellular calcium signaling. Physiol Rev 2007;87: 29–67.
20 Ramos M, del Arco A, Pardo B, Martinez-Serrano A, Martinez-Morales JR, Kobayashi K et al. Developmental changes in theCa2þ -regulated mitochondrial aspartate–glutamate carrier aralar1in brain and prominent expression in the spinal cord. Brain ResDev Brain Res 2003; 143: 33–46.
21 del Arco A, Morcillo J, Martinez-Morales JR, Galian C, Martos V,Bovolenta P et al. Expression of the aspartate/glutamate mitochon-drial carriers aralar1 and citrin during development and in adultrat tissues. Eur J Biochem 2002; 269: 3313–3320.
22 Tufty RM, Kretsinger RH. Troponin and parvalbumin calciumbinding regions predicted in myosin light chain and T4 lysozyme.Science 1975; 187: 167–169.
23 Lasorsa FM, Pinton P, Palmieri L, Fiermonte G, Rizzuto R, Palmieri F.Recombinant expression of the Ca2þ -sensitive aspartate/glutamatecarrier increases mitochondrial ATP production in agonist-stimu-lated CHO cells. J Biol Chem 2003; 278: 38686–38692.
24 Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium,ATP, and ROS: a mitochondrial love–hate triangle. Am J PhysiolCell Physiol 2004; 287: C817–C833.
25 Zilbovicius M, Meresse I, Chabane N, Brunelle F, Samson Y,Boddaert N. Autism, the superior temporal sulcus and socialperception. Trends Neurosci 2006; 29: 359–366.
26 Campbell DB, D’Oronzio R, Garbett K, Ebert PJ, Mirnics K, Levitt Pet al. Disruption of cerebral cortex MET signaling in autismspectrum disorder. Ann Neurol 2007; 62: 243–250.
27 Lintas C, Sacco R, Garbett K, Mirnics K, Militerni R, Bravaccio Cet al. Involvement of the PRKCB1 gene in autistic disorder:significant genetic association and reduced neocortical geneexpression. Mol Psychiatry 2008; [e-pub ahead of print].
28 Garbett K, Ebert PJ, Mitchell A, Lintas C, Manzi B, Mirnics Ket al. Immune transcriptome alterations in the temporal cortex ofsubjects with autism. Neurobiol Dis 2008; 30: 303–311.
29 Darley-Usmar VM, Rickwood D, Wilson MT (eds). Mitochondria,A Practical Approach. IRL Press: Washington, DC, 1987.
30 Palmieri F, Indiveri C, Bisaccia F, Iacobazzi V. Mitochondrialmetabolite carrier proteins: purification, reconstitution, andtransport studies. Methods Enzymol 1995; 260: 349–369.
31 Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2þ
indicators with greatly improved fluorescence properties. J BiolChem 1985; 260: 3440–3450.
32 Conciatori M, Stodgell CJ, Hyman SL, O’Bara M, Militerni R,Bravaccio C et al. Morphogenetic effect of the HOXA1 A218Gpolymorphism on head circumference in patients with autism.Biol Psychiatry 2004; 55: 413–419.
33 Lord C, Rutter M, DiLavore PC, Risi S. ADOS, Autism DiagnosticObservation Schedule. Western Psychological Services: LosAngeles, 2002 (Italian version ed. by Tancredi R, Saccani M,Persico AM, Parrini B, Igliozzi R and Faggioli R. OrganizzazioniSpeciali: Florence, 2005).
34 Rutter M, Le Couter A, Lord C. ADI-R, Autism Diagnostic Interview—Revised. Western Psychological Services: Los Angeles, 2003 (Italianversion ed. by Faggioli R, Saccani M, Persico AM, Tancredi R, ParriniB and Igliozzi R. Organizzazioni Speciali: Florence, 2005).
35 Sacco R, Papaleo V, Hager J, Rousseau F, Moessner R, Militerni Ret al. Case–control and family-based association studies ofcandidate genes in autistic disorder and its endophenotypes:TPH2 and GLO1. BMC Med Genet 2007; 8: 11.
36 Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualiz-ation of LD and haplotype maps. Bioinformatics 2005; 21: 263–265.
37 Horvath S, Xu X, Lake SL, Silverman EK, Weiss ST, Laird NM.Family-based tests for associating haplotypes with generalphenotype data: application to asthma genetics. Genet Epidemiol2004; 26: 61–69.
38 Spielman RS, Ewens WJ. The TDT and other family-based tests forlinkage disequilibrium and association. Am J Hum Genet 1996; 59:983–989.
39 Dudbridge F. Pedigree disequilibrium tests for multilocus haplo-types. Genet Epidemiol 2003; 25: 115–121.
Altered calcium homeostasis in autismL Palmieri et al
51
Molecular Psychiatry

40 Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel Bet al. The structure of haplotype blocks in the human genome.Science 2002; 296: 2225–2229.
41 Pritchard JK, Stephens M, Donnelly P. Inference of populationstructure using multilocus genotype data. Genetics 2000; 155:945–959.
42 Krey JF, Dolmetsch RE. Molecular mechanisms of autism: a possiblerole for Ca2þ signaling. Curr Opin Neurobiol 2007; 17: 112–119.
43 Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P,Bloise R et al. Ca(V)1.2 calcium channel dysfunction causes amultisystem disorder including arrhythmia and autism. Cell 2004;119: 19–31.
44 Hope CI, Sharp DM, Hemara-Wahanui A, Sissingh JI, Lundon P,Mitchell EA et al. Clinical manifestations of a unique X-linkedretinal disorder in a large New Zealand family with a novelmutation in CACNA1F, the gene responsible for CSNB2. ClinExperiment Ophthalmol 2005; 33: 129–136.
45 Laumonnier F, Roger S, Guerin P, Molinari F, M’rad R, Cahard Det al. Association of a functional deficit of the BKCa channel, asynaptic regulator of neuronal excitability, with autism and mentalretardation. Am J Psychiatry 2006; 163: 1622–1629.
46 Rizzuto R, Bernardi P, Pozzan T. Mitochondria as all-round playersof the calcium game. J Physiol 2000; 529: 37–47.
47 Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, PardoCA. Neuroglial activation and neuroinflammation in the brain ofpatients with autism. Ann Neurol 2005; 57: 67–81.
48 Jin D, Liu HX, Hirai H, Torashima T, Nagai T, Lopatina O et al.CD38 is critical for social behaviour by regulating oxytocinsecretion. Nature 2007; 446: 41–45.
49 Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, BadnerJA et al. Recurrent 16p11.2 microdeletions in autism. Hum MolGenet 2008; 17: 628–638.
50 Lohiya GS, Tan-Figueroa L, Iannucci A. Identification of low bonemass in a developmental center: finger bone mineral densitymeasurement in 562 residents. J Am Med Dir Assoc 2004; 5: 371–376.
51 Jaffe JS, Timell AM, Gulanski BI. Prevalence of low bone density inwomen with developmental disabilities. J Clin Densitom 2001; 4:25–29.
52 Blasi F, Bacchelli E, Carone S, Toma C, Monaco AP, Bailey AJ et al.SLC25A12 and CMYA3 gene variants are not associated withautism in the IMGSAC multiplex family sample. Eur J Hum Genet2006; 14: 123–126.
53 Rabionet R, McCauley JL, Jaworski JM, Ashley-Koch AE,Martin ER, Sutcliffe JS et al. Lack of association between autismand SLC25A12. Am J Psychiatry 2006; 163: 929–931.
54 Silverman JM, Buxbaum JD, Ramoz N, Schmeidler J, ReichenbergA, Hollander E et al. Autism-related routines and ritualsassociated with a mitochondrial aspartate/glutamate carrierSLC25A12 polymorphism. Am J Med Genet B NeuropsychiatrGenet 2007; 147B: 408–410.
55 Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysio-logy 2006; 13: 171–181.
56 Lepagnol-Bestel AM, Maussion G, Boda B, Cardona A, Iwayama Y,Delezoide AL et al. SLC25A12 expression is associated withneurite outgrowth and is upregulated in the prefrontal cortex ofautistic subjects. Mol Psychiatry 2008; 13: 385–397.
57 Jacquemont ML, Sanlaville D, Redon R, Raoul O, Cormier-Daire V,Lyonnet S et al. Array-based comparative genomic hybridisationidentifies high frequency of cryptic chromosomal rearrangementsin patients with syndromic autism spectrum disorders. J MedGenet 2006; 43: 843–849.
58 Autism Genome Project Consortium, Szatmari P, Paterson AD,Zwaigenbaum L, Roberts W, Brian J, Liu XQ et al. Mapping autismrisk loci using genetic linkage and chromosomal rearrangements.Nat Genet 2007; 39: 319–328.
59 Bernard S, Enayati A, Roger H, Binstock T, Redwood L. The role ofmercury in the pathogenesis of autism. Mol Psychiatry 2002; 7:S42–S43.
60 Nelson KB, Bauman ML. Thimerosal and autism? Pediatrics 2003;111: 674–679.
61 Elferink JG. Thimerosal: a versatile sulfhydryl reagent, calciummobilizer, and cell function-modulating agent. Gen Pharmacol1999; 33: 1–6.
62 Burbacher TM, Shen DD, Liberato N, Grant KS, Cernichiari E,Clarkson T. Comparison of blood and brain mercury levels ininfant monkeys exposed to methylmercury or vaccines containingthimerosal. Environ Health Perspect 2005; 113: 1015–1021.
63 Hornig M, Chian D, Lipkin WI. Neurotoxic effects of postnatal thi-merosal are mouse strain dependent. Mol Psychiatry 2004; 9: 833–845.
64 Brown MJ, Willis T, Omalu B, Leiker R. Deaths resultingfrom hypocalcemia after administration of edetate disodium:2003–2005. Pediatrics 2006; 118: e534–e536.
65 Sinha Y, Silove N, Williams K. Chelation therapy and autism. BMJ333: 756.
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
Altered calcium homeostasis in autismL Palmieri et al
52
Molecular Psychiatry
Copyright © 2022 FDOKUMEN




















