
Cytoskeleton actin-binding proteins in clinical behavior of ...
Upload
independentCategory
view
5download
0
IntroductionThe actin-based cytoskeleton is essential for cellular activitiesranging from maintenance of cell morphology to cellularlocomotion and also participates in regulation of secretion,endocytosis and transmembrane signaling (Mitchison andCramer, 1996; Schmidt and Hall, 1998). The assembly of thiscytoskeleton is regulated by multiple actin-binding proteinswhich allow for the diversity of actin filament forms andfunctions such as cytoskeletal remodeling, actin bundling andbranching (Ayscough, 1998). Although much attention hasbeen focused on the role of tyrosine kinases in cytoskeletalorganization, there is also substantial evidence that theelevation of cAMP and subsequent activation of cAMP-dependent protein kinase (PKA) alters the morphology ofepithelial cells and fibroblasts. Indeed, a cAMP-induced
alteration in the shape of epithelial cells was noted asearly as 1966 (Yasumura et al., 1966). Such changes inmorphology have been particularly well documented in theterminally differentiated gastric parietal cell in whichelevation of [cAMP]i induces dramatic changes in the actincytoskeleton which are correlated with the activation of HClsecretion (reviewed by Forte and Yao, 1996). In somecultured cells, elevation of [cAMP]i also leads to the loss ofstress fibers and focal adhesions (Schoenwaelder andBurridge, 1999).
Lasp-1 is a recently identified cAMP-dependent signalingprotein that may also be involved in tyrosine kinase signaling(Chew et al., 1998; Schreiber et al., 1998). It is widelyexpressed, but differentially distributed, in normal epithelialtissues and in brain (Chew et al., 2000; Chew et al., 1998;
4787
Lasp-1 has been identified as a signaling molecule that isphosphorylated upon elevation of [cAMP]i in pancreas,intestine and gastric mucosa and is selectively expressed incells within epithelial tissues. In the gastric parietal cell,cAMP-dependent phosphorylation induces the partialtranslocation of lasp-1 to the apically directed F-actin-richcanalicular membrane, which is the site of active HClsecretion. Lasp-1 is an unusual modular protein thatcontains an N-terminal LIM domain, a C-terminal SH3domain and two internal nebulin repeats. Domain-basedanalyses have recently categorized this protein as anepithelial representative of the nebulin family, which alsoincludes the actin binding, muscle-specific proteins,nebulin, nebulette and N-RAP.
In this study, we show that lasp-1 binds to non-musclefilamentous (F) actin in vitro in a phosphorylation-dependent manner. In addition, we provide evidence thatlasp-1 is concentrated within focal complexes as well as inthe leading edges of lamellipodia and the tips of filopodiain non-transformed gastric fibroblasts. In actin pull-downassays, the apparent Kd of bacterially expressed his-taggedlasp-1 binding to F-actin was 2 µM with a saturationstoichiometry of ~1:7. Phosphorylation of recombinantlasp-1 with recombinant PKA increased the Kd anddecreased the Bmax for lasp-1 binding to F-actin.
Microsequencing and site-directed mutagenesis localizedthe major in vivo and in vitro PKA-dependentphosphorylation sites in rabbit lasp-1 to S99 and S146.BLAST searches confirmed that both sites are conserved inhuman and chicken homologues. Transfection of lasp-1cDNA encoding for alanine substitutions at S99 and S146,into parietal cells appeared to suppress the cAMP-dependent translocation of lasp-1 to the intracellularcanalicular region. In gastric fibroblasts, exposure to theprotein kinase C activator, PMA, was correlated with thetranslocation of lasp-1 into newly formed F-actin-richlamellipodial extensions and nascent focal complexes. Sincelasp-1 does not appear to be phosphorylated by PKC, thesedata suggest that other mechanisms in addition to cAMP-dependent phosphorylation can mediate the translocationof lasp-1 to regions of dynamic actin turnover. Thelocalization of lasp-1 to these subcellular regions under arange of experimental conditions and the phosphorylation-dependent regulation of this protein in F-actin richepithelial cells suggests an integral and possibly cell-specificrole in modulating cytoskeletal/membrane-based cellularactivities.
Key words: Gastric parietal cell, Stomach, Rabbit, Proteinphosphorylation, Cytoskeleton, cAMP-dependent protein kinase
Summary
Lasp-1 binds to non-muscle F-actin in vitro and islocalized within multiple sites of dynamic actinassembly in vivoCatherine S. Chew*, Xunsheng Chen, John A. Parente, Jr ‡, Shannan Tarrer, Curtis Okamoto § and Hai-Yen QinInstitute of Molecular Medicine and Genetics, Medical College of Georgia, Augusta, GA 30912-3175, USA*Author for correspondence (e-mail: [email protected])‡Present address: Genzyme Corporation, One Mountain Road, Framingham, MA 01701, USA§Present address: Department of Pharmaceutical Sciences, University of Southern California, Los Angeles, CA 90089, USA
Accepted 15 September 2002Journal of Cell Science 115, 4787-4799 © 2002 The Company of Biologists Ltddoi:10.1242/jcs.00174
Research Article

4788
Schreiber et al., 1998). There is a particularly prominentexpression of this protein in the gastric parietal cell and inother F-actin rich ion-transporting cells including pancreaticand salivary duct cells as well as certain distal tubule andcollecting duct cells in the kidney (Chew et al., 2000). In theparietal cell, elevation of intracellular cAMP ([cAMP]i)induces a partial translocation of lasp-1 to the apicallydirected F-actin rich intracellular canaliculus, which is thesite of active HCl secretion. This stimulus-associatedphosphorylation and translocation of lasp-1 to the canalicularregion suggests that lasp-1 may play role in the regulation ofactin cytoskeleton plasticity and, possibly vesicle trafficking(Chew et al., 2000).
Lasp-1 was initially identified as pp40, a phosphoproteinthat migrated on SDS-PAGE gels with an apparent molecularmass of ~40 kDa (Chew and Brown, 1987). Phosphorylationof pp40 was increased in gastric parietal cells followingelevation of [cAMP]i and was correlated with histamine H2-receptor-activation of HCl secretion. Subsequently, pp40 wasisolated, sequenced and cloned (Chew et al., 1998) and shownto be identical to lasp-1 (LIM and SH3 domain-containingprotein), a product of the human gene, MLN 50, which isamplified in some cancers (Tomasetto et al., 1995). In additionto an N-terminal LIM domain and a carboxyl terminal SH3domain, lasp-1 contains two nebulin repeats. Although thespecific cellular functions of lasp-1 have not been defined, thepresence of several major protein-interacting motifs predictsmultiple binding partners. Sequence homology comparisons aswell as analyses of physical characteristics further suggest thatone or more these interacting proteins is likely to becytoskeletal (Chew et al., 1998; Schreiber et al., 1998). In thisregard, actin is a strong candidate because lasp-1 has beenlocalized to non-stress fiber, actin-rich subcellular regions(Chew et al., 2000; Schreiber et al., 1998) and also reportedlyassociates with actin on blot overlays and in GST pull downassays (Schreiber et al., 1998).
The initial goals of this study were to define the actin bindingproperties of lasp-1 and to determine if phosphorylation canmodulate the interaction. Our results demonstrate that lasp-1binds to filamentous (F) actin and that cAMP-dependentphosphorylation modifies this interaction in vitro. In the courseof these experiments, lasp-1 was found to be highly expressednot only in the gastric parietal cell but also to be present infocal adhesions and focal complexes as well in the extreme tipsof lamellipodia and filopodia in gastric mucosal fibroblasts.Since these subcellular regions are rich in F-actin and areassociated with a range of activities, including cell migrationand membrane trafficking, our results suggest that lasp-1 mayplay an important signaling-dependent role in the regulation ofone or more of these processes.
Materials and MethodsCellular modelsMixed gastric mucosal cells and parietal cells were isolated fromnembutal-anaesthetized, male New Zealand white rabbits and placedin primary culture as previously described (Chew et al., 1989). MadinDarby kidney (MDCK) cells, passage 20-25, were cultured usingstandard conditions. Transfection of various plasmids into gastric cellsand MDCK cells was accomplished using Effectene (Qiagen) aspreviously described (Parente et al., 1999).
DNA constructs and bacterial protein expressionPolyhistidine (his)-tagged lasp-1 protein was generated using thepET15b expression vector (Novagen, Madison, WI) by transformationinto BL21(DE3)pLysS bacteria (Promega, Madison, WI) andisopropylthio-(-D-galactoside) induction. His-tagged protein waspurified on a Hi Trap chelating column (Amersham-PharmaciaBiotech, Piscataway, NJ) followed by Mono Q purification aspreviously described (Chew et al., 1998). Immediately afterpurification, proteins were dialyzed, aliquoted and lyophilized.Glutathione-S-transferase (GST)-lasp-1 fusion protein was generatedby inserting cDNA encoding for the rabbit lasp-1 open reading framedownstream of the GST fragment in the pGEX4T-3 vector (PharmaciaBiotech, Piscataway, NJ) with BamHI and EcoRI restriction sites atthe 5′ and 3′ ends, respectively. Bacterial transformations andinductions were performed with lasp-1-pGEX4T-3 and emptypGEX4T-3 plasmid (to generate GST protein). A similar procedurewas used to generate mutated lasp-1 GST fusion proteins (below).GST-tagged proteins were purified using glutathione-sepharose 4Bbeads (Pharmacia Biotech).
For expression in MDCK cells, constructs containing an N-terminalhemagglutinin (HA) tag upstream of the coding region for lasp-1 weresubcloned into the pcDNA3 vector (Invitrogen, Carlsbad, CA).Constructs were generated by PCR-amplification using anAdvantage -HF 2 PCR kit (Clontech, Palo Alto, CA). The pET15bplasmid containing lasp-1 cDNA served as the template for primersthat generated BamHI and EcoRI restriction sites at the 5′ and 3′ ends,respectively. Primers based on the rabbit lasp-1 cDNA sequence(GenBank accession # AF017438) were designed with OLIGO PrimerAnalysis software, version 6.6 for MacIntosh (National Biosciences,Plymouth, MN) and synthesized by Gibco BRL Life Sciences, PCRconditions were as follows: Sense primer: 5′ GCC GGA TCC ACCATG GGC TAC CCA TAC GAT GTT CCA GAT TAC GCT AACCCC AAC TGC GCC; anti-sense primer: 5′ GGC CGA ATT CTCAGA TGG CTT CCA CGT AGT T; Initial denaturation, 94°C, 30seconds followed by 35 cycles of 94°C, 30 seconds; 60°C, 30 seconds;72°C, 45 seconds and a final 10 minute extension at 72°C. PCRproducts were gel isolated and ligated into EcoRI and BamHI-digestedpcDNA3 vector. After transformation into Escherichia coliJM109bacteria, plasmids containing lasp-1 cDNA were isolated (QiagenMiniprep kit, Valencia, CA). Positive clones were identified by PCR.The sequences of all clones were confirmed prior to use (MedicalCollege of GA Core DNA Sequencing Facility; ABI Prism 377automated DNA sequencer; ABI Prism Cycle Sequencing DyeTerminator Ready Reaction kits). For transfections, plasmid DNA wasisolated and purified with Qiagen Maxiprep Endo-Free kits aspreviously described (Parente et al., 1999).
Site-directed mutagenesis was performed with the StratageneQuick-Change mutagenesis kit using pcDNA3 vector containingrabbit lasp-1 cDNA as a template. Primers for single serine to alaninesubstitutions were as follows: S146 (RRDA): sense, 5′ CGA GCGCCG GGA CGC CCA GGA CAG CAG C; antisense, 5′ GCT GCTGTC CTG GGC GTC CCG GCG CTC G; S99 (RGFA): sense, 5′GGG CAG AGG CTT CGC CGT GGT GGC AGA C; antisense, 5′GTC TGC CAC CAC GGC GAA GCC TCT GCC C. PCR reactionswere performed with Pfu taq using the following conditions: 95°C, 1minute then 16 cycles; 95°C, 30 seconds, 55°C, 1 minute, 68°C, 12minutes. After 16 cycles, 1 µl (10 U/µl) DPNI restriction enzyme wasadded to the reaction and, after a brief centrifugation, samplesincubated at 37°C for ~2 hours to digest supercoiled dsDNA. Vectorswere transformed into Epicurean Col:XL1 Blue Super Competentcells as per manufacturer’s instructions. Plasmid DNA was isolatedand sequences of all constructs confirmed after mutagenesis asdescribed above. For double S99/S146 (RRDA/RGFA) mutants, thesame strategy was employed using the RRDA mutant in pcDNA3vector as the starting material. For in vitro experiments with his-tagged lasp-1 mutants, pcDNA3 plasmids containing theappropriately mutated inserts were transferred to the pET15b plasmid
Journal of Cell Science 115 (24)

4789Lasp-1 binds to F-actin
using a PCR-based approach (Advantage -HF 2 PCR kit) asdescribed above with oligodeoxynucleotide primers containing NdeI(5′) and BamHI (3′) restriction sites respectively as follows: 5′ GGGAAT TCA TAT GAA CCC CAA CTGC GCC CGG TG and 5′ CCGGAT CCT TCA GAT GGC TTC CAC GTA GTT GGC A.
GST-based assaysGST ‘pull down’ assays were performed at 4°C using parietal cellextracts that were prepared by incubating freshly isolated cells for 15minutes in lysis buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 5 mMEDTA, 1 mM EGTA, 1% NP-40, 0.1% SDS, 10 mM NaF containingthe following inhibitors: 0.2 mM AEBSF, 5 mM benzamidine, 10µg/ml each of leupeptin, pepstatin). Cellular debris was removed bycentrifugation (10 minutes, 10,000 g) and supernatants (0.5-1 mgprotein in 0.5 ml) precleared by incubating with 150 µl of a PBS-washed, 50% GST-sepharose bead slurry for 1 hour on a nutator. Pre-cleared supernatants were collected by centrifugation (500 g, 5minutes) then incubated (90 minutes, nutator) with lasp-1-GST fusionprotein (30 µg) bound to washed glutathione-sepharose beads (7.5 µlbed volume) as per manufacturer’s instructions. All samples were runin duplicate with the following controls: GST protein + precleared celllysate + sepharose beads; precleared cell lysate + sepharose beads;GST lasp-1 + sepharose beads. Beads were harvested bycentrifugation (1500 g, 15 seconds), washed three times with cell lysisbuffer then solubilized with 40 µl of 2× SDS stop buffer. Supernatantswere analyzed on SDS-PAGE gels (8-12%), which were either silverstained (Investigator Silver stain kit, ESA, Chemsford, MA) or witha modified Coomassie Blue colloidal staining protocol (Chew et al.,1998), and by western blot with enhanced chemiluminescent (ECL)detection as previously described (Chew et al., 2000). Replicatewestern blots were probed for lasp-1 (anti-lasp-1 monoclonal antibody(mab, clone 3H8, diluted 1:1000) (Chew et al., 2000) and actin (anti-non-muscle actin mab, clone AC40, Sigma Aldrich, St Louis, MO,diluted 1:5000). ECL detection was performed with HRP-conjugatedsheep anti-mouse Ig (1:5000 dilution, Amersham Pharmacia,Piscataway, NJ) as the secondary antibody.
Actin interaction assaysActin co-sedimentation assays were performed with bacterially-expressed lasp-1. Monomeric (G)-actin was generated by incubatinghuman platelet actin (≥99% purity, 5:1 β/γ isoforms, Cytoskeleton,Denver, CO) at a concentration of 1 µg/ml in a buffer containing 5mM Tris-HCl, pH 8.0, 0.2 mM CaCl2, 0.2 mM ATP, 0.5 mM DTT for1-2 hours, 4°C. Lyophilized his-tagged or GST-tagged lasp-1 wasdissolved in the same buffer at a concentration of 1-2 µg/ml thencentrifuged (100,000 g, 1 hour, Airfuge (Beckman Instruments) toremove protein aggregates. Lasp-1 (0.5-16 µM) in the resultingsupernatants was incubated with G-actin (14-23 µM, 40 minutes, 4°C)then polymerized by addition actin polymerization buffer (2 mM Tris,pH 8, 50 mM KCl, 2 mM MgCl2, 1 mM ATP). After incubation for30 minutes, room temperature to allow actin polymerization to reacha steady state, samples were centrifuged (100,000 g, 1 hour) to pelletF-actin. Supernatants (which contained G-actin) and pellets weredissolved in SDS-PAGE buffer and resolved on 8% SDS-PAGE gels.Gels were stained with Coomassie Brilliant Blue R250. Images ofdestained gels were digitized with a Syngene Gene Genius system andbands quantitated with Gene Tools software (Synoptics, UK) usingBSA as a standard. BSA (Sigma) and α-actinin (Cytoskeleton) wereused respectively as negative and positive controls for F-actin co-sedimentation. In preliminary experiments in which actin waspolymerized prior to lasp-1 addition, similar amounts of lasp-1 werefound to co-sediment with F-actin as compared to experiments inwhich actin was polymerized after lasp-1 addition. This latterapproach was used in all subsequent experiments to avoid quantitationproblems associated with the transfer of small quantities of F-actin.
To assess the association of endogenous lasp-1 with F-actin, amodification of previously described methods was used (Weed et al.,2000). Parietal cells were temperature equilibrated then rapidlypelleted, rinsed in cold PBS then lysed by sonicating cells (3×10seconds, 4°C) in a lysis buffer containing 10 mM imidazole, pH 7.2,75 mM KCl, 5 mM MgCl2, 1 mM EDTA, 0.5 mM DTT plusproteolytic inhibitors (mini EDTA-free tablet, Roche Diagnostics,Mannheim, Germany). After centrifugation (30 minutes, 100,000 g,4°C), the resulting supernatants were preincubated with platelet-derived actin (5-8 µM). Samples were sedimented as for therecombinant protein following actin polymerization. Lasp-1associated with F-actin was detected by western blot using the 3H8monoclonal antibody and ECL detection as described above.Chemiluminescent signals on western blots were quantitated with aSyngene GeneGnome 16 bit CCD-based chemiluminescent detectionsystem and Gene Tools software (Synoptics).
In vitro and in vivo phosphorylation site analysesTo generate phosphorylated lasp-1 for actin co-sedimentation assays,lyophilized his- or GST-tagged lasp-1 (0.5-1 µg/µl) was dissolved in1× cAMP-dependent protein kinase buffer (50 mM Tris-HCl, pH 7.5,10 mM MgCl2). Following addition of recombinant cAMP-dependentprotein kinase (catalytic subunit, New England Biolabs, Beverly, MA;1-2 U/µg lasp-1), reactions were initiated with 1 mM ATP andcontinued for 10 minutes, 30°C. For controls, 1 µg synthetic rabbitprotein kinase inhibitor (PKI, Sigma Chemicals, St Louis, MO) wasincluded in the reaction mixture. 32P-labeling using [γ-32P]ATP as asubstrate (0.2 mM, specific activity 400 cpm/pmol) was used toconfirm that this concentration of PKI completely blocked thephosphorylation of lasp-1 by cAMP-dependent protein kinase. At theend of the incubation period, samples were immediately dialyzed (20×volume of 5 mM Tris-HCl, pH 8.0) and concentrated by centrifugation(2500 g, 4°C) in Centricons (Amicon, Beverly, MA). To prevent anyfurther phosphorylation in the ATP-containing actin polymerizationbuffer, PKI was added to samples in which it was not added initially.Phosphorylated lasp-1 was incubated with actin and polymerizationperformed as described above. Because recombinant protein kinasemigrates close to the phosphorylated form of his-tagged lasp-1 onSDS-PAGE gels, western blot analyses were also performed using thelasp-1 mab in conjunction with ECL detection as above. Quantitationwas performed using multiple film exposures to ensure film linearityas previously described (Chew et al., 2000) or with the SyngeneGeneGnome system as above.
To identify in vitro cAMP-dependent protein kinasephosphorylation sites, 20 µg of his-tagged lasp-1 was phosphorylatedwith 15 U recombinant cAMP-dependent protein kinase catalyticsubunit (New England Biolabs) for 5 minutes, 30°C in kinase buffer(25 mM HEPES, pH 7.4, 10 mM Mg (C2H3O2)2, 0.33 mM DTT)containing 0.1 mM [γ-32P]ATP (specific activity, 36,000 cpm/pmol).Phosphorylated protein was resolved on an 8% SDS-PAGE gel andsubjected to ‘in gel’ tryptic digestion as previously described (Chewet al., 1998; Parente et al., 1996). Peptides in digests were resolvedon aµRPC C2/C18 column (0-40% linear acetonitrile gradient, 100µl/minutes) using a Pharamacia SMART system then re-purified onthe same column at a flow rate of 50 µl/minutes. Peaks containingradiolabeled peptides were identified by Cerenkov counting of eachfraction (Parente et al., 1996). Sequencing by Edman degradation wasperformed at the Emory University Microchemical Core Facility,Atlanta, GA. This facility also performed mass spectrum analysescomparing signals in V8 digests of His-tagged lasp-1 before and afterphosphorylation with recombinant cAMP-dependent protein kinasecatalytic subunit.
Time course experiments were performed by adding [γ-32P]ATP(final concentration 0.2 mM, SA, 400 cpm/pmol) to temperature-equilibrated (5 minutes, 30°C) assay tubes containing 10 µg his-tagged lasp-1 plus recombinant cAMP-dependent protein kinase

4790
catalytic subunit (20 U) in kinase buffer. Aliquots (~1 µg lasp-1) werewithdrawn at different time points, placed in an equal volume of 2×SDS stop solution, boiled for 3 minutes and subjected to SDS-PAGE.Radiolabeled bands were located by autoradiography, excised andquantitated by Cerenkov counting (Parente et al., 1996).
In vivo phosphorylation site analyses were performed with MDCKcells. Exponentially growing cells were transfected with the pcDNA3vector containing HA-tagged wild-type lasp-1 cDNA and cDNA fromphosphorylation site mutants using Effectene (Qiagen) as previouslydescribed (Parente et al., 1999). Forty-eight hours later, cells wereincubated with forskolin (10 µM, 15 minutes) or an equal volume ofDMSO vehicle. For one dimensional (1D) Mr band shift analyses,cells were rinsed in cold PBS and immediately lysed in 1× SDS stopbuffer. Lysates were fractionated on SDS-PAGE gels and lasp-1detected by western blot with ECL detection as previously described(Chew et al., 2000). For two dimensional (2D) analyses, cells werelysed with hot 0.3% SDS-1% βME, 10 mM Tris, pH 7.4. Lysates wereprecipitated at room temperature with 4× volumes of acetone andredissolved in rehydration buffer (8 M urea, 2% CHAPS, 18 mMDTT, 0.5% IPG buffer, pH 3-10 (Amersham-Pharmacia), 0.001%bromphenol blue). First dimension IEF was performed on IPG strips(pH 3-10 NL or L) with an IPGPhor (Amersham-Pharmacia) asfollows: 12 hour rehydration; 500 V, 1 hour; 1000 V, 1 hour; 8000V→28,000 volt hours. For second dimension SDS-PAGE, strips wereincubated for 15 minutes, room temperature in SDS equilibrationbuffer (50 mM Tris, pH 8.8, 6 M urea, 30% glycerol, 2% SDS, 65mM DTT, 0.001% bromphenol blue). Resolved proteins weretransferred to nitrocellulose for western blot analyses of lasp-1 usingthe lasp-1 mab and ECL detection. Lasp-1 phosphorylation wasdefined as an acidic shift resulting from an addition of negativelycharged phosphate residue(s) to the protein (the predicted acidic shiftswere confirmed in metabolic 32P labeling experiments as previouslydescribed (Chew et al., 1998). Phosphorylation site analyses wereperformed with the Phosphobase program on the Center for BiologicalSequence Analysis (CBS) web site (Kreegipuu et al., 1999).
Indirect immunofluorescence microscopyEndogenous lasp-1 was localized by indirect immunofluorescence(primary antibody, anti-lasp-1 mab 3H8); secondary antibody, cyanine
(Cy)-5-labeled goat anti-mouse IgG (Jackson Immunoresearch Labs,Westgrove, PA) with primary and secondary antibody controls aspreviously described (Chew et al., 2000). In brief, gastric cells grownon glass coverslips were fixed with 4% paraformaldehyde,permeabilized with 0.2% Triton X-100, blocked with 5% non-fat milk(BioRad) in PBS and sequentially incubated with the lasp-1 antibody(diluted 1:50 in 1% milk/PBS) followed by the Cy-5-labeledsecondary antibody (diluted 1:100 in 0.1% milk/PBS). PBS rinses (3-6×5 minutes) were performed after each incubation step. In mostexperiments, cells were dual labeled for F-actin by adding OregonGreen phalloidin (1:400 dilution, Molecular Probes, Eugene, OR)simultaneously with the secondary antibody. Transfected lasp-1 andmutants were immunolocalized using a similar protocol withmonoclonal anti-HA antibody (BabCo/Covance, 1:1,000), Alexa 488chicken anti-mouse secondary antibody (Molecular Probes, 1:100).Dual labeling for F-actin was accomplished using Alexa 647-labeledphalloidin Molecular Probes, 1:400). Fluorescently labeled cells wereoptically sectioned using a Molecular Dynamics 2010 confocalmicroscope equipped with a krypton/argon laser (Chew et al., 2000).
Statistical analysesWhere appropriate, values are expressed as means±s.e.m. with nrepresenting the number of independent experiments. For pairedsamples, data was analyzed for statistical significance using theStudent’s t-test for paired comparisons. Analysis of variance andDunnett’s tests were used to analyze multiple comparisons (Chew andBrown, 1987).
ResultsLasp-1 binds to non-muscle F-actin in vitro Detailed domain-based analyses coupled with the inability todetect lasp-1 expression in skeletal, cardiac and non-vascularsmooth muscle have led us to propone this protein as anepithelial representative of the nebulin repeat family of proteins(Chew et al., 2002). The primary member, nebulin, is a large(~600-800 kDa) skeletal muscle protein that is thought to playa role in defining the length of thin filaments. Nebulin repeat
Journal of Cell Science 115 (24)
Fig. 1.Native and recombinant his-tagged lasp-1 co-sediment with purified non-muscle F-actin butGST-tagged lasp-1 does not co-sediment with G-actin in cell lysates. (A) Coomassie-blue-stainedSDS-PAGE gel showing typical actin co-sedimentation assay. Lasp-1 (8 µM, arrow) or α-actinin (2µM, star, positive control) were incubated with actin (23 µM, arrowhead). After actin polymerization,supernatants (lanes 1-5) and pellets (lanes 6-10) were prepared and resolved by electrophoresis. Lanes1,2,6,7: lasp-1 without F-actin. Lanes 3,4,8,9: lasp-1+F-actin. Lanes 5,10: α-actinin+F-actin. Std:BioRad precision Mr standard (100, 75, 50, 37, 25 kDa). Note that his-tagged lasp-1 migrates with an
apparent molecular mass of ~38 kDa. Previous analyses withless precise Mr standards reported an apparent molecular massof ~41 kDa for his-tagged lasp-1 and ~40 kDa for native lasp-1(Chew and Brown, 1987; Chew et al., 1998). (B) Western blotof actin co-sedimentation of duplicate samples of endogenouslasp-1 in parietal cell extracts. Lasp-1 was co-sedimented with7.5 µM F-actin as described in Materials and Methods. Similarresults were obtained in three independent experiments.(C) Quantitation of lasp-1 association with F-actin at differentconcentrations of lasp-1. The F-actin concentration was 14 µM.Corrections for nonspecific precipitation of his-tagged lasp-1were performed for each concentration. Values aremeans±s.e.m. for n=4 independent experiments. (D) Westernblot of GST pull-down assay fractions using an actin antibodyshowing that similar amounts of actin in samples with GST-sepharose vs GST-tagged lasp-1. As expected, no signal wasdetected in the absence of parietal cell lysate. Similar resultswere obtained in four independent experiments. See Materialsand Methods for details.

4791Lasp-1 binds to F-actin
fragments bind to actin (Wang, 1996) and also promote actinpolymerization and bundling (Chen et al., 1993; Gonsior et al.,1998). Although lasp-1 contains only two nebulin repeats anddoes not appear to modulate either actin polymerization orbundling (Chew et al., 2002), it has been reported to bind toactin on blot overlays and in GST pull down assays (Schreiberet al., 1998).
To characterize further the interactions between lasp-1 andactin, actin co-sedimentation assays were performed usinghighly purified, platelet-derived non-muscle actin in order tomimic epithelial cell physiology as closely as possible. Ininitial experiments we confirmed that both endogenous andbacterially expressed lasp-1 cosediment with F-actin (Fig.1A,B). The well-characterized actin binding protein, α-actinin,also co-sedimented with F-actin in these assays but BSA,which does not bind actin, did not (Fig. 1A). Cumulativedata from several independent experiments demonstratedreproducible and saturable lasp-1-F-actin co-sedimentationthat was significantly greater than controls (Fig. 1C).
To determine if lasp-1 also binds to monomeric (G)-actin, arange of assay conditions were tested including GST pull downassays with GST-lasp vs GST alone, blot overlay assays with32P-labeled lasp-1, and far western blots using his-tagged lasp-1 in conjunction with the anti-lasp-1 monoclonal antibody. Noactin binding was detected in blot overlays or far westerns (notshown). Although actin was present in GST pull downs, therewas no significant difference between samples containingGST-lasp vs GST alone (Fig. 1D). Thus, a variety ofapproaches indicated that lasp-1 does not bind to monomericactin; however, additional studies are requiredto establish this point unequivocally.
Identification of major cAMP-dependentphosphorylation sites in vitro and in vivoby microsequencing, mass spectrometryand site-directed mutagenesisBefore testing the effects of phosphorylationon the interaction between recombinant lasp-1 and F-actin, it was essential to define thepattern of in vitro phosphorylation and todetermine if the phosphorylation sites targetedby PKA in vitro were the same as thoseregulated by cAMP in vivo. In intact cells,lasp-1 is phosphorylated on serine residuesfollowing elevation of [cAMP]i (Chew et al.,1998). This could be the result or either adirect or an indirect involvement of cAMP-dependent protein kinase (PKA). Previouswork predicted that lasp-1 is a direct substratefor PKA because the recombinant his-taggedprotein was strongly phosphorylated byrecombinant cAMP-dependent protein kinasecatalytic subunit, but not by several otherserine/threonine kinases including proteinkinase C (Chew et al., 1998). As shown in Fig.2A, the kinetics of lasp-1 phosphorylationwith PKA (recombinant catalytic subunit)is also rapid and monophasic, reaching amaximum within ~30 minutes.
Two cAMP-dependent protein kinase
phosphorylation consensus sites, both of which contain serineresidues, have been identified in the rabbit lasp-1 protein (Chewet al., 1998; Chew et al., 2002). Blast searches of the GenBankhave confirmed that these sites are conserved in the human(NM_006148) and chicken (#BI394039) homologues but onlythe K/RGFS99 site is conserved in rat (NM_032613) and mouse(NM_010688) (C.S.C., unpublished). As shown in the diagramin Fig. 2A, the first consensus phosphorylation site falls betweenthe two nebulin repeats (RGFS99) and the second is immediatelydownstream of this region (RRDS146). To confirm that these sitesare, indeed, targeted by PKA, his-tagged lasp-1 was incubatedwith recombinant cAMP-dependent protein kinase catalyticsubunit (using [γ-32P]ATP as a substrate) then subjected totryptic digestion followed by micro-HPLC purification andmicrosequencing (as described in Materials and Methods). BothPKA consensus sites were tentatively identified within the threemajor peaks containing phosphorylated peptides (Fig. 2B). Massspectrum analyses were also performed on the same preparationof his-tagged lasp-1 before and after phosphorylation withrecombinant PKA. Nanocolumn eluates were analyzed using ananospray device and positive, negative and phosphate ion scans.Positive and negative scans revealed that proteins were digestedand fragments produced. Analysis of his-tagged lasp-1 revealedonly background signal in the phosphate ion scans. Significantphosphate ion peaks were identified in the 50% methanoleluate of phosphorylated lasp-1. Based on the predictedproteolytic map, two proteolytic fragments were identifiedwith a high degree of certainty (GFS99VADTPELQR) and(MGPSGGEGAEPERRDS146QDSSNYR).
Fig. 2.Time course of phosphorylation of lasp-1 by cAMP-dependent protein kinase andtentative identification of the major in vitro phosphorylation sites. (A) Top panel:diagram showing location of the two known cAMP-dependent protein kinase consensussites in nebulin repeat region. Lower panel: time course of phosphorylation of his-taggedlasp-1 by a recombinant subunit of cAMP-dependent protein kinase (see Materials andMethods for details). (B) Chromatogram of tryptic digest of lasp-1. 20 µg of his-taggedlasp-1 was phosphorylated with recombinant catalytic subunit of cAMP-dependentprotein kinase using [γ-32P]ATP as a substrate. Radiolabeled protein was isolated on anSDS-PAGE gel, subjected to in-gel tryptic digest and labeled peptides resolved on aSMART system with an acetonitrile gradient (flow rate, 100 µl/minute). Peaks shown inthe figure were further resolved using a slower flow rate (50 µl/minute) thenmicrosequenced. Parentheses in the sequences locate the predicted position of arginineand other residues present in the deduced lasp-1 sequence. Since trypsin normallycleaves at arginine residues, these amino acids were not expected, nor were theyidentified in sequencing analyses. However, because it is known that trypsin does notreadily cleave R-X-Ser(P), it is likely that the tryptic fragment in the second peakcontained the RRDS sequence in lower abundance compared with theMGPSGGEGAEPE fragment.

4792
Because other serine residues were present in the sequencedpeptides and in one of the two mass spectrum-based phosphateanalysis products, neither sequencing nor mass spectrumanalyses unequivocally identified Ser99 and Ser146 as specificphosphorylation sites. Therefore, additional analyses wereperformed with mutated his-tagged lasp-1 proteins in whichone or both of these putative cAMP-dependent protein kinasephosphorylation sites were mutated to alanines. As shownin Fig. 3A, cAMP-dependent protein kinase catalyzed 32Pincorporation into both RRDA146 and RGFA99 mutants (65%and 26% of total 32P incorporation into wild-type lasp-1,respectively). There was also a lesser degree of 32Pincorporation into the double (R/R) mutant. The combined datasupported the conclusion that the most significant in vitrophosphorylation catalyzed by cAMP-dependent protein kinaseoccurs at the Ser99 and Ser146 residues with Ser99 being themost prominent site.
Mutation of Ser146, but not Ser99, to alanine blocked thephosphorylation-induced Mr band shift (Fig. 3A). Thus, thisband shift, which has been observed both in vivo and in vitro(Chew et al., 2000; Chew et al., 1998), appeared to result fromthe phosphorylation of Ser146. To test this hypothesis moredirectly, MDCK cells were transiently transfected withpcDNA3 plasmids containing HA-tagged wild-type lasp-1cDNA or lasp-1 cDNA containing S→A substitutions forSer99 and Ser146respectively. Transfected cells were incubatedwith forskolin (10 µM, 15 minutes) to elevate endogenouscAMP and to activate endogenous cAMP-dependent proteinkinase. Cell lysates were analyzed for lasp-1 phosphorylationby western blot. [The lasp-1 mab (clone 3H8) was used fordirect analysis of the expressed rabbit lasp-1 protein becausethis antibody does not recognize the endogenous canineprotein (see mock transfection lanes in Fig. 3B).] In theseexperiments, S→A mutation of the RRDS146, but not theRGFS99 site, did indeed block the Mr band shift of lasp-1 (Fig.3B). This experimental approach confirmed that Ser146 is anin vivo target for cAMP-dependent protein kinase and thatphosphorylation of this site induces a Mr band shift.
To determine if the RGFS99 site (Fig. 3A) is also targeted bycAMP-dependent protein kinase in vivo, pcDNA3 plasmidscontaining either wild-type lasp-1, the RRDA146 construct, orthe double mutant (R/R) construct were transfected into MDCKcells. Cells were stimulated with forskolin (as above), lysed andsubjected to 2D gel electrophoresis followed by western blotanalysis with the anti-lasp-1 mab. Fig. 3C demonstrates thatforskolin stimulation of cells transfected with wild-type lasp-1led to the expected shift in apparent Mr which was accompaniedby an acidic shift (presumably reflecting the increased negativecharge of the phosphate groups). With the RRDA146 mutant,there was an acidic shift, but no shift in apparent Mr. Both theacidic and Mr shifts were abolished in the double mutant. Thecombined results from microsequencing, mass spectrum andmutational analysis studies, therefore, demonstrate that Ser99
and Ser146 are the major in vitro targets of cAMP-dependentprotein kinase and are consistent with direct PKAphosphorylation of these sites in vivo.
Phosphorylation of lasp-1 inhibits cosedimentation withF-actin in vitroTo determine if phosphorylation of Ser99 and Ser146 by
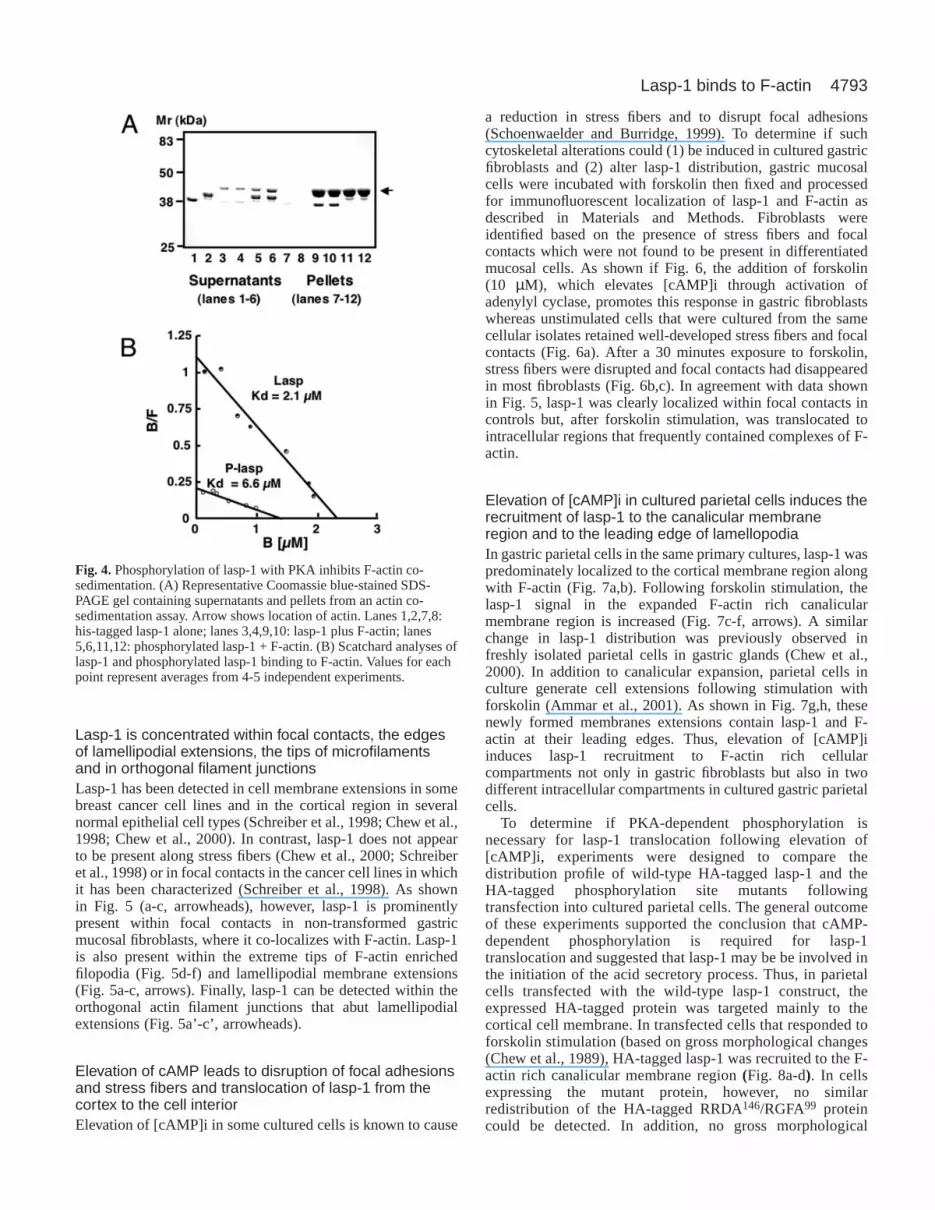
cAMP-dependent protein kinase modifies the interactionbetween lasp-1 and F-actin, his-tagged lasp-1 wasphosphorylated with recombinant PKA prior to actincosedimentation assay. As shown in Fig. 4, phosphorylationsuppressed the interaction (as detected by both Coomassieblue staining (Fig. 4A) and western blot (not shown)). Inmore detailed kinetic analyses, his-tagged lasp-1 was foundto bind to F-actin in a saturable, concentration-dependentmanner with a Kd of 2.2±0.3 µM (n=4) and stoichiometry of1 mol lasp-1:7 mol actin (Fig. 4B). PKA-dependentphosphorylation of his-tagged lasp-1 increased the Kd bymore than three times (Fig. 4B).
Journal of Cell Science 115 (24)
Fig. 3.Serine to alanine mutations of the predicted cAMP-dependentprotein kinase consensus phosphorylation sites in lasp-1 inhibits thephosphorylation of lasp-1 by cAMP-dependent protein kinase in vivoand in vitro. (A) For in vitro analyses, his-tagged wild-type andmutated lasp-1 were phosphorylated and resolved on an SDS-PAGEgel as described in Fig. 2. Inset shows autoradiographic data. Bandswere excised from the gel and radiolabel incorporation quantitatedby Cerenkov counting (graph). Mutations were as follows: RRDA,Ser99; RGFA, Ser146; R/R, both sites mutated. Values are expressedas a percentage of total counts present in wild-type lasp-1.(B) Western blot analysis of expressed wild-type (WT) and mutated(RGFA146, RRDA99) lasp-1 constructs following transfection ofpcDNA3 vectors into MDCK cells. Transfected and mock-transfected (Mock, empty vector) cells were incubated with DMSOvehicle or forskolin (10 µM, 15 minutes) and lysed; extracts wereanalyzed using the lasp-1 mab as described in Materials andMethods. Mr band shifts are known to correlate with increasedphosphorylation in vivo and in vitro (Chew et al., 1998; Chew et al.,2000). (C) Two dimensional western blot analyses of extractsprepared from transfected MDCK cells as described in panel B andMaterials and Methods. Note the acidic shift in the RRDA but not thedouble (R/R) mutant following forskolin stimulation. These datasupport the conclusion that both Ser99 and Ser146 are in vivophosphorylation sites.
4793Lasp-1 binds to F-actin
Lasp-1 is concentrated within focal contacts, the edgesof lamellipodial extensions, the tips of microfilamentsand in orthogonal filament junctionsLasp-1 has been detected in cell membrane extensions in somebreast cancer cell lines and in the cortical region in severalnormal epithelial cell types (Schreiber et al., 1998; Chew et al.,1998; Chew et al., 2000). In contrast, lasp-1 does not appearto be present along stress fibers (Chew et al., 2000; Schreiberet al., 1998) or in focal contacts in the cancer cell lines in whichit has been characterized (Schreiber et al., 1998). As shownin Fig. 5 (a-c, arrowheads), however, lasp-1 is prominentlypresent within focal contacts in non-transformed gastricmucosal fibroblasts, where it co-localizes with F-actin. Lasp-1is also present within the extreme tips of F-actin enrichedfilopodia (Fig. 5d-f) and lamellipodial membrane extensions(Fig. 5a-c, arrows). Finally, lasp-1 can be detected within theorthogonal actin filament junctions that abut lamellipodialextensions (Fig. 5a’-c’, arrowheads).
Elevation of cAMP leads to disruption of focal adhesionsand stress fibers and translocation of lasp-1 from thecortex to the cell interiorElevation of [cAMP]i in some cultured cells is known to cause
a reduction in stress fibers and to disrupt focal adhesions(Schoenwaelder and Burridge, 1999). To determine if suchcytoskeletal alterations could (1) be induced in cultured gastricfibroblasts and (2) alter lasp-1 distribution, gastric mucosalcells were incubated with forskolin then fixed and processedfor immunofluorescent localization of lasp-1 and F-actin asdescribed in Materials and Methods. Fibroblasts wereidentified based on the presence of stress fibers and focalcontacts which were not found to be present in differentiatedmucosal cells. As shown if Fig. 6, the addition of forskolin(10 µM), which elevates [cAMP]i through activation ofadenylyl cyclase, promotes this response in gastric fibroblastswhereas unstimulated cells that were cultured from the samecellular isolates retained well-developed stress fibers and focalcontacts (Fig. 6a). After a 30 minutes exposure to forskolin,stress fibers were disrupted and focal contacts had disappearedin most fibroblasts (Fig. 6b,c). In agreement with data shownin Fig. 5, lasp-1 was clearly localized within focal contacts incontrols but, after forskolin stimulation, was translocated tointracellular regions that frequently contained complexes of F-actin.
Elevation of [cAMP]i in cultured parietal cells induces therecruitment of lasp-1 to the canalicular membraneregion and to the leading edge of lamellopodiaIn gastric parietal cells in the same primary cultures, lasp-1 waspredominately localized to the cortical membrane region alongwith F-actin (Fig. 7a,b). Following forskolin stimulation, thelasp-1 signal in the expanded F-actin rich canalicularmembrane region is increased (Fig. 7c-f, arrows). A similarchange in lasp-1 distribution was previously observed infreshly isolated parietal cells in gastric glands (Chew et al.,2000). In addition to canalicular expansion, parietal cells inculture generate cell extensions following stimulation withforskolin (Ammar et al., 2001). As shown in Fig. 7g,h, thesenewly formed membranes extensions contain lasp-1 and F-actin at their leading edges. Thus, elevation of [cAMP]iinduces lasp-1 recruitment to F-actin rich cellularcompartments not only in gastric fibroblasts but also in twodifferent intracellular compartments in cultured gastric parietalcells.
To determine if PKA-dependent phosphorylation isnecessary for lasp-1 translocation following elevation of[cAMP]i, experiments were designed to compare thedistribution profile of wild-type HA-tagged lasp-1 and theHA-tagged phosphorylation site mutants followingtransfection into cultured parietal cells. The general outcomeof these experiments supported the conclusion that cAMP-dependent phosphorylation is required for lasp-1translocation and suggested that lasp-1 may be be involved inthe initiation of the acid secretory process. Thus, in parietalcells transfected with the wild-type lasp-1 construct, theexpressed HA-tagged protein was targeted mainly to thecortical cell membrane. In transfected cells that responded toforskolin stimulation (based on gross morphological changes(Chew et al., 1989), HA-tagged lasp-1 was recruited to the F-actin rich canalicular membrane region (Fig. 8a-d). In cellsexpressing the mutant protein, however, no similarredistribution of the HA-tagged RRDA146/RGFA99 proteincould be detected. In addition, no gross morphological
Fig. 4.Phosphorylation of lasp-1 with PKA inhibits F-actin co-sedimentation. (A) Representative Coomassie blue-stained SDS-PAGE gel containing supernatants and pellets from an actin co-sedimentation assay. Arrow shows location of actin. Lanes 1,2,7,8:his-tagged lasp-1 alone; lanes 3,4,9,10: lasp-1 plus F-actin; lanes5,6,11,12: phosphorylated lasp-1 + F-actin. (B) Scatchard analyses oflasp-1 and phosphorylated lasp-1 binding to F-actin. Values for eachpoint represent averages from 4-5 independent experiments.

4794
changes in cells expressing the HA-taggedRRDA146/RGFA99 protein were identified(Fig. 8e-h).
Lasp-1 is recruited into some regions ofdynamic actin assembly independent of[cAMP]i elevationSince other signaling pathways may also beinvolved in regulating lasp-1 interactionswith the actin cytoskeleton, we soughtto determine if cAMP-independentmanipulations that induce the formation oflamellipodial extensions would alter thedistribution of lasp-1. A well recognizedstrategy for inducing the formation of cellularextensions is to expose fibroblasts deprived ofgrowth factors to the phorbol ester, PMA.Growth factor deprivation reduces theformation of lamellipodial extensions and,under these conditions, actin incorporationoccurs only at the sites of protrusive activity(Chan et al., 1998). The addition of PMAinduces lamellipodial formation and actinassembly (Downey et al., 1992; Schliwa et al.,1984). Thus, sites of protrusive activity can beidentified under growth factor-free conditionsand newly formed membrane extensions canbe identified following the addition of PMA(Kellie et al., 1985). Data depicted in Fig. 9support this prediction. Twenty-four hoursafter the removal of growth factors, lasp-1was prominently localized within focalcontacts and microspikes present at the tips ofplasma membrane extensions in gastricfibroblasts (Fig. 9a-c). Within two minutes
Journal of Cell Science 115 (24)
Fig. 5.Subcellular distribution patterns of lasp-1 in gastric fibroblasts. (a,a’,d) Lasp-1 immunoreactivity detected with Cy5-labelled secondaryantibody; (b,b’,e) F-actin staining with Oregon green phalloidin; (c,c’,f) merged images (lasp-1 red; F-actin green). Bars, 10 µM (a,c); 5 µM (f).
Fig. 6.Exposure of gastric fibroblasts to the adenylyl cyclase activator, forskolin,disrupts stress fibers and focal contacts and induces the translocation of lasp-1 fromfocal contacts to the cell interior. Control, a-c; forskolin, d-i. (a,d.g) lasp-1; (b,e,h) F-actin; (c-f) merged images (lasp-1, red; F-actin, green). Data are representative of fiveindependent experiments. Bars, 10 µM.

4795Lasp-1 binds to F-actin
after addition of PMA (100 nM), lamellipodial extensionsbegan to form and weak lasp-1 and F-actin signals could bedetected within these newly generated structures (Fig. 9d-f).After 5 minutes, an uninterrupted band of co-localized lasp-1and F-actin became obvious at the membrane-actin interfaceof growing extensions (Fig. 9g-h). At 10 minutes there wassome broadening of this band and focal contacts in most cellsappeared as punctate regions in which lasp-1 and F-actinsignals persisted (Fig. 9j-l). Taken together, these results
demonstrate the recruitment of lasp-1 to F-actin rich regionsthat are associated with active membrane protrusion in gastricfibroblasts. Since lasp-1 does not appear to be a direct targetof PKC (Chew et al., 1998), the recruitment mechanism isprobably not dependent on direct phosphorylation by PKC butmay be initiated by the phosphorylation of associatedprotein(s).
Fig. 7. In cultured gastric parietal cells, forskolin stimulation inducesthe recruitment of lasp-1 to the F-actin rich canalicular membraneregion as well as to newly formed membrane extensions. Parietalcells in primary culture can be identified based on several differentcriteria including cell size and general morphology, autofluorescencewhen excited at lower wavelengths, lack of stress fibers or focalcontacts, and the expansion of the canaliculi following secretagoguestimulation (Chew et al., 1989) (C.S.C., unpublished). (a,c,e,g) lasp-1; (b,d,f,h) F-actin. The cell in the top panel was treated with DMSOvehicle. Cells in the lower panels were stimulated with forskolin (10µM, 30 minutes). Panels e-h contain images of the same cellacquired from the mid-section (e,f) and near the base (g,h). Arrowsindicate position of expanded intracellular canaliculi. Arrowheads,newly formed cell extensions. Data representative of six independentexperiments. Bars, 10 µM.
Fig. 8. In transfected parietal cells, expression HA-tagged lasp-1,forskolin stimulation also induces the recruitment of this protein tothe canalicular region. However, mutation of the two major cAMP-dependent phosphorylation sites, S99 and S146, to alanines appears toblock this cAMP-dependent process and may also inhibit the acidsecretory response. Panels a,c,e,g show signals derived from HA-tagged lasp-1 construct (a,c) and for HA-tagged double S99/S146
(RRDA/RGFA) mutants (e,g). Corresponding F-actin signals in thesame cells are shown in panels on the right. Arrows indicate locationof expended intracellular canaliculi; arrowheads indicate intracellularcanaliculi in non-transfected cells. Note non-transfected cell in panelh (actin signal only)is clearly stimulated by forskolin addition but thetransfected cell is not. Data representative of six independentexperiments. Bars, 10 µM.

4796
DiscussionPresent results are the first to identify lasp-1 as an F-actinbinding protein and to confirm and extend earlier observations(Chew et al., 2000; Schreiber et al., 1998) that the subcellulardistribution of lasp-1 is at least partially correlated with that ofF-actin under a range of conditions, including those whichdrastically modify the actin cytoskeletal dynamics andstructure. The F-actin binding studies presented here suggestthat this association involves a direct interaction betweenlasp-1 and F-actin. The cellular observations further suggestthat this interaction may be regulated in vivo by bothphosphorylation-dependent and -independent mechanisms.The Kd for lasp-1 binding to F-actin in vitro falls within the
range previously reported for several other actinbinding proteins (Pollard, 1999) as does thesaturation stoichometry (Ishikawa et al., 1994).This low saturation stoichiometry is consistent withbinding along filaments rather than a monomericassociation (Pollard, 1999).
Two major serine phosphorylation sites havebeen identified in lasp-1 and several lines ofevidence indicate that this protein is a directdownstream substrate for PKA in vivo. Based onthe parietal cell transfection studies it appears thatcAMP-dependent phosphorylation of lasp-1 isrequired for the recruitment of this protein to theintracellular canalicular region of parietal cells.Although these findings were consistent, it shouldbe noted that there are several potential problemsin their interpretation. First, the mutated proteinsdisplayed reduced affinity for lasp-1 in vitro. Thus,it is possible that vivo functions were similarlycompromised. Also, the transfection efficiencywith parietal cells is low and not all parietal cellsin primary culture respond to acid secretoryagonists with obvious changes in cytoskeletalmorphology (Parente et al., 1999). Clearly, newexperimental approaches will be required toconfirm or discount a direct role for cAMP-dependent phosphorylation in lasp-1 translocationand to define the specific cellular activities that aremodulated during this translocation process.
In gastric fibroblasts, it appears that lasp-1 canalso recruited to regions of dynamic actin/membrane turnover by cAMP-independentmechanisms. In addition, lasp-1 appears to retainits association with F-actin in forskolin-stimulatedfibroblasts in which stress fibers are disrupted.Thus, this novel signaling protein may also beregulated either directly or indirectly by severalmechanisms, with the predominant mechanismbeing dependent on the specific cell type. Tyrosinephosphorylation might also play a regulatory rolein some cell types. Although we have not been ableto detect tyrosine phosphorylation of lasp-1 in theterminally differentiated parietal cell (C.S.C., X.C.and H.-Y.Q., unpublished), there are two conservedtyrosine phosphorylation consensus sites in lasp-1and this protein can be phosphorylated on tyrosineresidues in cell lines overexpressing Src kinase(Schreiber et al., 1998). The finding that a
cytoskeletal-associated protein is differentially phosphorylatedin normal epithelial cells has precedence in that cytoskeletal-membrane linking protein, ezrin, is phosphorylated on tyrosineresidues in A431 cells (Bretscher, 1989) but on serine/threonine residues in the parietal cell (Urushidani et al., 1989).These differences in phosphorylation patterns may reflectdifferences in cellular protein kinase expression profiles and/oralterations in the coupling of signaling pathways to thecytoskeleton.
The observation that PKA-dependent phosphorylationreduces the affinity of lasp-1 for F-actin in vitro raisesinteresting questions regarding in vivo mechanisms. If PKA-dependent phosphorylation also reduces the affinity of lasp-1
Journal of Cell Science 115 (24)
Fig. 9. In gastric mucosal fibroblasts, lasp-1 is recruited to regions of activemembrane-cytoskeletal rearrangements following the activation of cAMP-independent signaling pathways. Gastric fibroblasts in mixed gastric mucosalprimary cultures were growth-factor deprived for 24 hours, treated with 100 nMPMA (0 minutes, a-c; 2 minutes, d-f; 5 minutes, g-i; 10 minutes, j-l) and then fixedand stained for lasp-1 and F-actin as described in Materials and Methods and Fig.5. Left panels, lasp-1; center panels, F-actin; right panels, merged images (lasp-1,red; F-actin, green). Arrows indicate locations of microspikes and nascent focalcomplexes. Data representative of three independent experiments. Bars, 5 µM.

4797Lasp-1 binds to F-actin
for F-actin in vivo, why does lasp-1 translocate to the F-actinrich canalicular membrane region in the parietal cell? There area several possible explanations. In the gastric parietal cell, thereare at least two distinct pools of actin. At the cell cortex,γ-actin predominates whereas β-actin predominates in thecanalicular region (Yao et al., 1995) The cortical actin pool isexquisitely sensitive to latrunculin B, which binds monomericactin, whereas the formation of microvillar filaments appear tobe highly resistant to this inhibitor as well as to cytochalasinD, which severs actin filaments (Ammar et al., 2001). Thus, atthe cell cortex, a reduced affinity for actin could assist in therapid remodeling of the actin cytoskeleton (Pollard, 1999). Thesource of lasp-1 that translocates to the canalicular region isnot yet established. However, both detergent-soluble and-insoluble pools of lasp-1 have been identified (Chew et al.,2002; Chew et al., 1998) so it is likely that lasp-1 is distributedwithin several subcellular compartments, possibly withstronger and weaker linkages to the actin cytoskeleton. Sincelasp-1 contains both LIM and SH3 protein associationdomains, it is also likely that other interacting proteins play adirect and/or indirect role in regulating the affinity of lasp-1for F-actin in vivo. The phosphorylation of lasp-1 by otherprotein kinase(s) may also regulate this interaction. Finally, thebehavior of actin itself is extensively modified by cellmembranes as well as by cytosolic extracts (Jahraus et al.,2001). Thus, the local intracellular environment may alsodifferentially affect the association between lasp-1 and F-actindepending on the specific pool of actin that is present.
The presence of lasp-1 in both classical and nascent focalcontacts or adhesions in gastric fibroblasts is intriguing and itwill be important to determine if this association is regulated byvesicular trafficking and/or phosphorylation-dependentmechanisms and to determine whether F-actin and/or otherproteins within these complexes interact directly with lasp-1.Although classical focal contacts are viewed as relatively stablestructures, nascent focal contacts and the cortical actincytoskeleton appear to contain critical sites for the initiation ofactin polymerization (Beningo et al., 2001; Small et al., 1998;Wang, 1985). To date, more than 50 proteins have been detectedin focal contacts and it is becoming increasing evident that thereis molecular diversity as well as complex signaling activity andvesicular trafficking of proteins into and out of these regions(Sastry and Burridge, 2000). Since no systematic orcomprehensive search for focal contact proteins has beenconducted, the extent of cell-type specific distribution has notyet been established (Zamir and Geiger, 2001). However,because lasp-1 does not appear to be present in focal contacts ofthe human BT-474 breast cancer cell line (Schreiber et al., 1998),the distribution of this signaling protein may indeed be cell-typespecific. Defining the distribution pattern and functions of lasp-1 in a range transformed and non-transformed cell lines as wellas normal epithelial cells may provide useful insights into thefunctions of this recently identified signaling molecule.
The specific localization of lasp-1 to proximal junctionalbranch points within the cortical actin filament network as wellas in lamellipodial tips, where the Arp 2/3 complex proteinshave also been localized (Svitkina and Borisy, 1999), suggeststhe possibility that lasp-1 might interact either directly orindirectly with this important actin-nucleating complex. Theactin-binding protein, cortactin (Schuuring et al., 1993), whichis also known as amplaxin and oncogene EMS-1 gene, has
recently been found to bind to the Arp 2/3 complex therebyregulating actin polymerization (Uruno et al., 2001; Weaver etal., 2001). Lasp-1 shares a relatively high degree of homologywith cortactin in the SH3 domain and proline-rich regions aswell as in their actin binding regions (Sparks et al., 1996)(C.S.C., unpublished). As with lasp-1, in vitro phosphorylationof cortactin suppresses F-actin binding (Huang et al., 1997) andcortactin undergoes phosphorylation-dependent translocationin intact cells (Ozawa et al., 1995). Cortactin also has no directhomologue in yeast and is primarily localized within inperipheral cell regions associated with dynamic actin assemblyin cultured cells (Wu and Parsons, 1993). Most earlier studiesof cortactin focused on pp60 src kinase-dependent tyrosinephosphorylation. However, epidermal growth factor inducesthe phosphorylation of cortactin on serine and threonineresidues, and it is the phosphorylation of these residues whichinduces a mobility shift on western blots (Campbell et al.,1999). In addition, it has recently been proposed that cortactintranslocation is mediated by serine/threonine phosphorylationrather than by tyrosine phosphorylation (Lopez et al., 2001).Thus, cortactin, like ezrin and possibly lasp-1 may bedifferentially regulated by serine/threonine kinases andtyrosine kinase dependent on the developmental stage and/orcell type involved.
Because lasp-1 lacks the conserved Arp 2/3 interaction site(the acidic ‘A’ domain (DD/EW), which is present in cortactinas well as WASP-family members and several other Arp 2/3binding proteins (Olazabal and Machesky, 2001), it appearsunlikely that there is a direct interaction between lasp-1 andthe Arp 2/3 complex. However, lasp-1 could modulate thiscomplex indirectly. Several SH3 domain-containing proteinshave been found to bind to proline-rich sequences in WASPand N-WASP (Higgs and Pollard, 2001). In addition, the largeGTPase, dynamin, which has been implicated in endocytotictrafficking (Kessels et al., 2000), has been shown to complexwith WASP indirectly through an interaction with the WASP-binding protein, syndapin I (Qualmann et al., 1999). Recentwork has shown that lasp-1 associates with dynamin II in vitro(Okamoto, 2001). Thus, it will be of interest to determinewhether or not lasp-1 has the ability to modulate the Arp 2/3complex via direct connections with WASP family membersand/or indirect associations via dynamin, for example. Theanswer to this question is particularly critical given the currentlack of knowledge of the molecular mechanisms involved indirecting membrane trafficking to sites of actin polymerizationat the leading edge of migrating cells and in the formation ofmicrovillar extensions in ion-transporting cells such as thegastric parietal cell. Another important goal for future researchwill be to identify additional protein binding partners for lasp-1 and to determine whether it is an indirect modulator of actinassembly and/or membrane trafficking and cell adhesion andmotility.
We are grateful to Carlos Isales and Wendy Bolag for sharingequipment and expertise and to James Goldenring and Jennifer Grinerfor supplying MDCK cells. We also thank Carolyn Leitner in theMCG Molecular Biology Core Facility for DNA sequencing, Jon Pohlat the Emory University Microsequencing Facility for phosphopeptidesequencing, and Boris Babakov and Steve Vogel in the MCG ImagingCore for valuable assistance with sequencing and image production,respectively. This work was supported by National Institutes of Healthgrants R01 DK31900 and F32 DK09447.

4798
ReferencesAmmar, D. A., Nguyen, P. N. and Forte, J. G. (2001). Functionally distinct
pools of actin in secretory cells. Am. J. Physiol. Cell Physiol.281, C407-C417.
Ayscough, K. R. (1998). In vivo functions of actin-binding proteins. Curr.Opin. Cell Biol.10, 102-111.
Beningo, K. A., Dembo, M., Kaverina, I., Small, J. V. and Wang, Y. L.(2001). Nascent focal adhesions are responsible for the generation of strongpropulsive forces in migrating fibroblasts. J. Cell Biol.153, 881-888.
Bretscher, A. (1989). Rapid phosphorylation and reorganization of ezrin andspectrin accompany morphological changes induced in A-431 cells byepidermal growth factor. J. Cell Biol.108, 921-930.
Campbell, D. H., Sutherland, R. L. and Daly, R. J. (1999). Signalingpathways and structural domains required for phosphorylation ofEMS1/cortactin. Cancer Res.59, 5376-5385.
Chan, A. Y., Raft, S., Bailly, M., Wyckoff, J. B., Segall, J. E. and Condeelis,J. S. (1998). EGF stimulates an increase in actin nucleation and filamentnumber at the leading edge of the lamellipod in mammary adenocarcinomacells. J. Cell Sci.111, 199-211.
Chen, M. J., Shih, C. L. and Wang, K. (1993). Nebulin as an actin zipper.A two-module nebulin fragment promotes actin nucleation and stabilizesactin filaments. J. Biol. Chem.268, 20327-20334.
Chew, C. S. and Brown, M. R. (1987). Histamine increases phosphorylationof 27- and 40-kDa parietal cell proteins. Am. J. Physiol.253, G823-G829.
Chew, C. S., Ljungstrom, M., Smolka, A. and Brown, M. R. (1989).Primary culture of secretagogue-responsive parietal cells from rabbit gastricmucosa. Am. J. Physiol.256, G254-G263.
Chew, C. S., Parente, J. A., Jr, Zhou, C., Baranco, E. and Chen, X. (1998).Lasp-1 is a regulated phosphoprotein within the cAMP signaling pathwayin the gastric parietal cell. Am. J. Physiol.275, C56-C67.
Chew, C. S., Parente, J. A., Jr, Chen, X., Chaponnier, C. and Cameron,R. S. (2000). The LIM and SH3 domain-containing protein, lasp-1, may linkthe cAMP signaling pathway with dynamic membrane restructuringactivities in ion transporting epithelia. J. Cell Sci.113, 2035-2045.
Chew, C. S., Chen, X., Qin, H.-Y. and Stoming, T. (2002). New insights intosecond messenger regulation of parietal cell function by novel downstreamsignaling proteins. In Mechanisms and Consequences of Proton Transport(ed. T. Urushidani, J. G. Forte and G. Sachs), pp. 185-195. Boston: KluwerAcademic Publishers.
Downey, G. P., Chan, C. K., Lea, P., Takai, A. and Grinstein, S. (1992).Phorbol ester-induced actin assembly in neutrophils: role of protein kinaseC. J. Cell Biol.116, 695-706.
Forte, J. G. and Yao, X. (1996). The membrane-recruitment and recyclinghypothesis of gastric HCl secretion. Trends Cell Biol.6, 45-48.
Gonsior, S. M., Gautel, M. and Hinssen, H. (1998). A six-module humannebulin fragment bundles actin filaments and induces actin polymerization.J. Muscle Res. Cell Motil.19, 225-235.
Higgs, H. N. and Pollard, T. D. (2001). Regulation of actin filament networkformation through ARP2/3 complex: activation by a diverse array ofproteins. Annu. Rev. Biochem.70, 649-676.
Huang, C., Ni, Y., Wang, T., Gao, Y., Haudenschild, C. C. and Zhan, X.(1997). Down-regulation of the filamentous actin cross-linking activity ofcortactin by Src-mediated tyrosine phosphorylation. J. Biol. Chem.272,13911-13915.
Ishikawa, R., Hayashi, K., Shirao, T., Xue, Y., Takagi, T., Sasaki, Y. andKohama, K. (1994). Drebrin, a development-associated brain protein fromrat embryo, causes the dissociation of tropomyosin from actin filaments. J.Biol. Chem.269, 29928-29933.
Jahraus, A., Egeberg, M., Hinner, B., Habermann, A., Sackman, E.,Pralle, A., Faulstich, H., Rybin, V., Defacque, H. and Griffiths, G.(2001). ATP-dependent membrane assembly of F-actin facilitates membranefusion. Mol. Biol. Cell12, 155-170.
Kellie, S., Holme, T. C. and Bissell, M. J. (1985). Interaction of tumourpromoters with epithelial cells in culture. An immunofluorescence study.Exp. Cell Res.160, 259-274.
Kessels, M. M., Engqvist-Goldstein, A. E. and Drubin, D. G. (2000).Association of mouse actin-binding protein 1 (mAbp1/SH3P7), an Srckinase target, with dynamic regions of the cortical actin cytoskeleton inresponse to Rac1 activation. Mol. Biol. Cell11, 393-412.
Kreegipuu, A., Blom, N. and Brunak, S. (1999). PhosphoBase, adatabase of phosphorylation sites: release 2.0. Nucleic Acids Res27, 237-239.
Lopez, I., Duprez, V., Melle, J., Dreyfus, F., Levy-Toledano, S. andFontenay-Roupie, M. (2001). Thrombopoietin stimulates cortactin
translocation to the cytoskeleton independently of tyrosine phosphorylation.Biochem. J.356, 875-881.
Mitchison, T. J. and Cramer, L. P. (1996). Actin-based cell motility and celllocomotion. Cell 84, 371-379.
Okamoto, C. T., Chew, C. S. and Li, R. (2001). Characterization of lasp-1-dynamin II interactions in vitro and in vivo. FASEB J. 15, A499.
Olazabal, I. M. and Machesky, L. M. (2001). Abp1p and cortactin, new‘hand-holds’ for actin. J. Cell Biol.154, 679-682.
Ozawa, K., Kashiwada, K., Takahashi, M. and Sobue, K. (1995).Translocation of cortactin (p80/85) to the actin-based cytoskeleton duringthrombin receptor-mediated platelet activation. Exp. Cell Res.221, 197-204.
Parente, J. A., Goldenring, J. R., Petropoulos, A. C., Hellman, U. andChew, C. S. (1996). Purification, cloning, and expression of a novel,endogenous, calcium- sensitive, 28-kDa phosphoprotein. J. Biol. Chem.271,20096-20101.
Parente, J. A., Jr, Chen, X., Zhou, C., Petropoulos, A. C. and Chew, C. S.(1999). Isolation, cloning, and characterization of a new mammalian coroninfamily member, coroninse, which is regulated within the protein kinase Csignaling pathway. J. Biol. Chem.274, 3017-3025.
Pollard, T. (1999). Guidebook to the Cytoskeletal and Motor Proteins. NewYork: Oxford University.
Qualmann, B., Roos, J., DiGregorio, P. J. and Kelly, R. B. (1999). SyndapinI, a synaptic dynamin-binding protein that associates with the neuralWiskott-Aldrich syndrome protein. Mol. Biol. Cell10, 501-513.
Sastry, S. K. and Burridge, K. (2000). Focal adhesions: a nexus forintracellular signaling and cytoskeletal dynamics. Exp. Cell Res.261, 25-36.
Schliwa, M., Nakamura, T., Porter, K. R. and Euteneuer, U. (1984). Atumor promoter induces rapid and coordinated reorganization of actin andvinculin in cultured cells. J. Cell Biol.99, 1045-1059.
Schmidt, A. and Hall, M. N. (1998). Signaling to the actin cytoskeleton.Annu. Rev. Cell Dev. Biol.14, 305-338.
Schoenwaelder, S. M. and Burridge, K. (1999). Bidirectional signalingbetween the cytoskeleton and integrins. Curr. Opin. Cell Biol.11, 274-286.
Schreiber, V., Moog-Lutz, C., Regnier, C. H., Chenard, M. P., Boeuf, H.,Vonesch, J. L., Tomasetto, C. and Rio, M. C. (1998). Lasp-1, a novel typeof actin-binding protein accumulating in cell membrane extensions. Mol.Med.4, 675-687.
Schuuring, E., Verhoeven, E., Litvinov, S. and Michalides, R. J. (1993).The product of the EMS1 gene, amplified and overexpressed in humancarcinomas, is homologous to a v-src substrate and is located in cell-substratum contact sites. Mol Cell Biol 13, 2891-2898.
Small, J. V., Rottner, K., Kaverina, I. and Anderson, K. I. (1998).Assembling an actin cytoskeleton for cell attachment and movement.Biochim. Biophys. Acta1404, 271-281.
Sparks, A. B., Hoffman, N. G., McConnell, S. J., Fowlkes, D. M. and Kay,B. K. (1996). Cloning of ligand targets: systematic isolation of SH3 domain-containing proteins. Nat. Biotechnol.14, 741-744.
Svitkina, T. M. and Borisy, G. G. (1999). Arp2/3 complex and actindepolymerizing factor/cofilin in dendritic organization and treadmilling ofactin filament array in lamellipodia. J. Cell Biol.145, 1009-1026.
Tomasetto, C., Moog-Lutz, C., Regnier, C. H., Schreiber, V., Basset, P. andRio, M. C. (1995). Lasp-1 (MLN 50) defines a new LIM protein subfamilycharacterized by the association of LIM and SH3 domains. FEBS Lett.373,245-249.
Uruno, T., Liu, J., Zhang, P., Fan, Y., Egile, C., Li, R., Mueller, S. C. andZhan, X. (2001). Activation of Arp2/3 complex-mediated actinpolymerization by cortactin. Nat. Cell Biol.3, 259-266.
Urushidani, T., Hanzel, D. K. and Forte, J. G. (1989). Characterization ofan 80-kDa phosphoprotein involved in parietal cell stimulation. Am. J.Physiol.256, G1070-G1081.
Wang, K. (1996). Titin/connectin and nebulin: giant protein rulers of musclestructure and function. Adv. Biophys.33, 123-134.
Wang, Y. L. (1985). Exchange of actin subunits at the leading edge of livingfibroblasts: possible role of treadmilling. J. Cell Biol.101, 597-602.
Weaver, A. M., Karginov, A. V., Kinley, A. W., Weed, S. A., Li,Y., Parsons, J. T. and Cooper, J. A. (2001). Cortactin promotes andstabilizes Arp2/3-induced actin filament network formation. Curr. Biol.11, 370-374.
Weed, S. A., Karginov, A. V., Schafer, D. A., Weaver, A. M., Kinley, A. W.,Cooper, J. A. and Parsons, J. T. (2000). Cortactin localization to sites ofactin assembly in lamellipodia requires interactions with F-actin and theArp2/3 complex. J. Cell Biol.151, 29-40.
Journal of Cell Science 115 (24)

4799Lasp-1 binds to F-actin
Wu, H. and Parsons, J. T. (1993). Cortactin, an 80/85-kilodalton pp60srcsubstrate, is a filamentous actin-binding protein enriched in the cell cortex.J. Cell Biol.120, 1417-1426.
Yao, X., Chaponnier, C., Gabbiani, G. and Forte, J. G. (1995). Polarizeddistribution of actin isoforms in gastric parietal cells. Mol. Biol. Cell6, 541-557.
Yasumura, Y., Buonassisi, V. and Sato, G. (1966). Clonal analysis ofdifferentiated function in animal cell cultures. I. Possible correlatedmaintenance of differentiated function and the diploid karyotype. CancerRes.26, 529-535.
Zamir, E. and Geiger, B. (2001). Molecular complexity and dynamics of cell-matrix adhesions. J. Cell Sci.114, 3583-3590.
Copyright © 2022 FDOKUMEN




















