
Large scale analysis of the mutational landscape in β-glucuronidase: A major player of...
9
Research Paper Large scale analysis of the mutational landscape in β-glucuronidase: A major player of mucopolysaccharidosis type VII Faez Iqbal Khan a , Mohd. Shahbaaz a , Krishna Bisetty a , Abdul Waheed b , William S. Sly b , Faizan Ahmad c , Md. Imtaiyaz Hassan c, ⁎ a Department of Chemistry, Durban University of Technology, Durban 4000, South Africa b Edward A. Doisy Department of Biochemistry and Molecular Biology, Saint Louis University School of Medicine, St. Louis, MO 63104, USA c Center for Interdisciplinary Research in Basic Sciences, Jamia Millia Islamia, Jamia Nagar, New Delhi, 110025, India abstract article info Article history: Received 14 July 2015 Received in revised form 17 August 2015 Accepted 23 September 2015 Available online xxxx Keywords: Lysosomal storage diseases β-glucuronidase Mucopolysaccharidosis type VII Sly syndrome Mutation analysis MD simulation Enzyme deficiency The lysosomal storage disorders are a group of 50 unique inherited diseases characterized by unseemly lipid storage in lysosomes. These malfunctions arise due to genetic mutations that result in deficiency or reduced ac- tivities of the lysosomal enzymes, which are responsible for catabolism of biological macromolecules. Sly syn- drome or mucopolysaccharidosis type VII is a lysosomal storage disorder associated with the deficiency of β- glucuronidase (EC 3.2.1.31) that catalyzes the hydrolysis of β-D-glucuronic acid residues from the non- reducing terminal of glycosaminoglycan. The effects of the disease causing mutations on the framework of the sequences and structure of β-glucuronidase (GUSBp) were analyzed utilizing a variety of bioinformatic tools. These analyses showed that 211 mutations may result in alteration of the biological activity of GUSBp, including previously experimentally validated mutations. Finally, we refined 90 disease causing mutations, which presum- ably cause a significant impact on the structure, function, and stability of GUSBp. Stability analyses showed that mutations p.Phe208Pro, p.Phe539Gly, p.Leu622Gly, p.Ile499Gly and p.Ile586Gly caused the highest impact on GUSBp stability and function because of destabilization of the protein structure. Furthermore, structures of wild type and mutant GUSBp were subjected to molecular dynamics simulation to examine the relative structural behaviors in the explicit conditions of water. In a broader view, the use of in silico approaches provided a useful understanding of the effect of single point mutations on the structure-function relationship of GUSBp. © 2015 Elsevier B.V. All rights reserved. 1. Introduction The lysosomal membrane-bound systems are the primary intracel- lular apparatus for the degradation of endogenous and exogenous biologically occurring macromolecules and the successive recycling to their respective fundamental monomeric constituents (Parenti et al., 2015). It is also involved in the processing of several vital metabolites (Futerman and van Meer, 2004). A heritable imperfection in the proteins accountable for preserving the lysosomal system leads to the accumulation of moderately degraded biomolecules in the lysosome, which is the primary cause of lysosomal storage diseases (LSD) (Futerman and van Meer, 2004), a group of 50 genetically inherited dis- eases that occur due to imperfections in the post-translational modifica- tions, metabolic trafficking, and activity of lysosomal hydrolases (Shayman, 2014). All LSDs are inherited as autosomal recessive traits, except for Hunter syndrome, Fabry disease and Danon disease (Filocamo and Morrone, 2011). These are X-linked disorders. These disorders weaken the removal and intracellular turnover of a variety of substrates like sphingolipids, glycoproteins, glycosaminoglycans, etc. Accumulation of these substrates in the lysosome results in the disruption of biochemical and cellular proceedings and eventually causes cell and tissue damage in various organ systems (Ballabio and Gieselmann, 2009; Futerman and van Meer, 2004). These storage compounds intervene with several processes, such as modulation of re- ceptor response and activation by non-physiologic ligands, affecting intracellular effectors of signal transduction cascades (Ballabio and Gieselmann, 2009). LSDs are quite rare, generally occurring at a frequency of 1 in 8000 live births (Meikle et al., 1999) and causing se- vere physical and neurological manifestations in affected patients (Lachmann, 2011; Vitner et al., 2010). Mucopolysaccharidosis type VII (MPS VII), better known as Sly syndrome, is an autosomal recessive lysosomal storage disease charac- terized by the patient's inability to metabolize the glycosaminoglycan- containing glucuronic acid due to malfunction of GUSBp (Naz et al., 2013; Sly et al., 1973). This disorder is normally lethal, degenerative, Gene xxx (2015) xxx–xxx Abbreviations: LSD, lysosomal storage disease; MPSVII, mucopolysaccharidosis type VII; MD, molecular dynamics; GUSB, β-glucuronidase; WT, wild type; RMSD, root mean square deviations; RMSF, root mean square fluctuations; Rg, radius of gyration; ns, nano second. ⁎ Corresponding author. E-mail address: [email protected] (M.I. Hassan). GENE-40880; No. of pages: 9; 4C: http://dx.doi.org/10.1016/j.gene.2015.09.062 0378-1119/© 2015 Elsevier B.V. All rights reserved. Contents lists available at ScienceDirect Gene journal homepage: www.elsevier.com/locate/gene Please cite this article as: Khan, F.I., et al., Large scale analysis of the mutational landscape in β-glucuronidase: A major player of mucopolysaccharidosis type VII, Gene (2015), http://dx.doi.org/10.1016/j.gene.2015.09.062
Transcript of Large scale analysis of the mutational landscape in β-glucuronidase: A major player of...

Gene xxx (2015) xxx–xxx
GENE-40880; No. of pages: 9; 4C:
Contents lists available at ScienceDirect
Gene
j ourna l homepage: www.e lsev ie r .com/ locate /gene
Research Paper
Large scale analysis of the mutational landscape in β-glucuronidase: Amajor player of mucopolysaccharidosis type VII
Faez Iqbal Khan a, Mohd. Shahbaaz a, Krishna Bisetty a, Abdul Waheed b, William S. Sly b,Faizan Ahmad c, Md. Imtaiyaz Hassan c,⁎a Department of Chemistry, Durban University of Technology, Durban 4000, South Africab Edward A. Doisy Department of Biochemistry and Molecular Biology, Saint Louis University School of Medicine, St. Louis, MO 63104, USAc Center for Interdisciplinary Research in Basic Sciences, Jamia Millia Islamia, Jamia Nagar, New Delhi, 110025, India
Abbreviations: LSD, lysosomal storage disease; MPSVVII; MD, molecular dynamics; GUSB, β-glucuronidase; Wsquare deviations; RMSF, root mean square fluctuations;second.⁎ Corresponding author.
E-mail address: [email protected] (M.I. Hassan).
http://dx.doi.org/10.1016/j.gene.2015.09.0620378-1119/© 2015 Elsevier B.V. All rights reserved.
Please cite this article as: Khan, F.I., et amucopolysaccharidosis type VII, Gene (2015
a b s t r a c t
a r t i c l e i n f oArticle history:Received 14 July 2015Received in revised form 17 August 2015Accepted 23 September 2015Available online xxxx
Keywords:Lysosomal storage diseasesβ-glucuronidaseMucopolysaccharidosis type VIISly syndromeMutation analysisMD simulationEnzyme deficiency
The lysosomal storage disorders are a group of 50 unique inherited diseases characterized by unseemly lipidstorage in lysosomes. These malfunctions arise due to genetic mutations that result in deficiency or reduced ac-tivities of the lysosomal enzymes, which are responsible for catabolism of biological macromolecules. Sly syn-drome or mucopolysaccharidosis type VII is a lysosomal storage disorder associated with the deficiency of β-glucuronidase (EC 3.2.1.31) that catalyzes the hydrolysis of β-D-glucuronic acid residues from the non-reducing terminal of glycosaminoglycan. The effects of the disease causing mutations on the framework of thesequences and structure of β-glucuronidase (GUSBp) were analyzed utilizing a variety of bioinformatic tools.These analyses showed that 211 mutations may result in alteration of the biological activity of GUSBp, includingpreviously experimentally validatedmutations. Finally, we refined 90 disease causingmutations, which presum-ably cause a significant impact on the structure, function, and stability of GUSBp. Stability analyses showed thatmutations p.Phe208Pro, p.Phe539Gly, p.Leu622Gly, p.Ile499Gly and p.Ile586Gly caused the highest impact onGUSBp stability and function because of destabilization of the protein structure. Furthermore, structures ofwild type andmutant GUSBpwere subjected tomolecular dynamics simulation to examine the relative structuralbehaviors in the explicit conditions of water. In a broader view, the use of in silico approaches provided a usefulunderstanding of the effect of single point mutations on the structure-function relationship of GUSBp.
© 2015 Elsevier B.V. All rights reserved.
1. Introduction
The lysosomal membrane-bound systems are the primary intracel-lular apparatus for the degradation of endogenous and exogenousbiologically occurring macromolecules and the successive recycling totheir respective fundamental monomeric constituents (Parenti et al.,2015). It is also involved in the processing of several vital metabolites(Futerman and van Meer, 2004). A heritable imperfection in theproteins accountable for preserving the lysosomal system leads to theaccumulation of moderately degraded biomolecules in the lysosome,which is the primary cause of lysosomal storage diseases (LSD)(Futerman and vanMeer, 2004), a group of 50 genetically inherited dis-eases that occur due to imperfections in the post-translationalmodifica-tions, metabolic trafficking, and activity of lysosomal hydrolases
II, mucopolysaccharidosis typeT, wild type; RMSD, root meanRg, radius of gyration; ns, nano
l., Large scale analysis of t), http://dx.doi.org/10.1016/j
(Shayman, 2014). All LSDs are inherited as autosomal recessive traits,except for Hunter syndrome, Fabry disease and Danon disease(Filocamo and Morrone, 2011). These are X-linked disorders. Thesedisorders weaken the removal and intracellular turnover of a varietyof substrates like sphingolipids, glycoproteins, glycosaminoglycans,etc. Accumulation of these substrates in the lysosome results in thedisruption of biochemical and cellular proceedings and eventuallycauses cell and tissue damage in various organ systems (Ballabio andGieselmann, 2009; Futerman and van Meer, 2004). These storagecompounds intervene with several processes, such asmodulation of re-ceptor response and activation by non-physiologic ligands, affectingintracellular effectors of signal transduction cascades (Ballabio andGieselmann, 2009). LSDs are quite rare, generally occurring at afrequency of 1 in 8000 live births (Meikle et al., 1999) and causing se-vere physical and neurological manifestations in affected patients(Lachmann, 2011; Vitner et al., 2010).
Mucopolysaccharidosis type VII (MPS VII), better known as Slysyndrome, is an autosomal recessive lysosomal storage disease charac-terized by the patient's inability to metabolize the glycosaminoglycan-containing glucuronic acid due to malfunction of GUSBp (Naz et al.,2013; Sly et al., 1973). This disorder is normally lethal, degenerative,
he mutational landscape in β-glucuronidase: A major player of.gene.2015.09.062

2 F.I. Khan et al. / Gene xxx (2015) xxx–xxx
and progressive in nature and results in extensive deviation in theexpression of many phenotypes (Shipley et al., 1993). Various manifes-tations are associated with GUSBp deficiency including skeletal anoma-lies, hepatosplenomegaly, dysostosis multiplex, coarse features, andvarying levels of mental retardation (Metcalf et al., 2009; Shipleyet al., 1993). In addition, non-immune hydrops fetalis conditions havebeen reported before birth with MPS VII-affected fetuses (Liebherret al., 2014; Molyneux et al., 1997). Therefore, in order to understandthe origin of these abnormalities, a proper insight into GUSBp structureis essential.
The GUSB gene is located on the long (q) arm of chromosome 7 atposition 21.11 (7q21.11). More precisely, the GUSB gene is locatedfrom base pair 65,960,683 to base pair 65,982,313 on chromosome 7.GUSBmRNA (1953-bp in dimension) encodes a 651-amino acid residueprecursor polypeptide. After cleavage of a 22-amino acid N-terminalsignal peptide and glycosylation, a 78-kDa monomer is transported tolysosomes and becomes the mature active enzyme (Brot et al., 1978;Oshima et al., 1987). The overall structure of GUSB contains threedomains, a jelly roll-like domain containing the lysosomal targeting res-idues, a TIM barrel domain (active site), and an immunoglobulin regionconstant domain (Hassan et al., 2013; Jain et al., 1996).
There are a total of 55mutations reported in GUSB protein accordingto the Human GeneMutation Database (HGMD). There were 49 uniquemutations observed in the patients suffering from Sly disease (illustrat-ed in Fig. 1), of which 36 were missense mutations, two splice sitealterations, five deletions, and six nonsense mutations (Tomatsu et al.,2009). The percentage of each form of mutation in a whole set of 103mutant alleles was 78.6% for missense mutations, 2.9% for splice-sitemutations, 5.8% for deletions, and 12.6% for nonsense mutations(Tomatsu et al., 2009). Therefore,missensemutations like p.Leu176Phe,p.Pro415Leu, p.Pro408Ser, p.Ala619Val, p.Arg216Trp, p.Arg382Cys, andp.Arg477Trp (Tomatsu et al., 1990, 1991) were highly frequent,whilep.Leu176Phe was the most prevalent in the patients (Vervoort et al.,1996; Wu et al., 1994). In this paper, we have analyzed the amino acidsequence and structure of GUSBp in order to predict the impact of
Fig. 1. Cartoon model showing the structure of GUSBp w
Please cite this article as: Khan, F.I., et al., Large scale analysis of tmucopolysaccharidosis type VII, Gene (2015), http://dx.doi.org/10.1016/j
mutations on structure, function and stability. We further aimed togain a deeper insight into understanding the mechanism of Sly diseaseat molecular level. In this paper we describe the emerging understand-ing of the interplay between these essential metabolic systems, Sly dis-ease, and the GUSBpmutation, with a particular focus on their potentialas a therapeutic target for MPS VII.
2. Methods
2.1. Sequence-based mutation analysis
The primary phase included the creation of sequence based pointmutations at every constituent amino acid position of GUSBp (primaryaccession no. P08236). This analysis resulted in the classification of dis-ease causing mutations, which were further validated by observingtheir effect on the structure of this enzyme (PDB ID — 3HN3). By utiliz-ing the information obtained from these analyses, we identified 90disease-causing mutations in GUSBp. Among these, eight alterationswere selected for the molecular dynamics (MD) simulations becauseof the availability of the experimental data in the literature (Storchet al., 2003).
We used bioinformatic platforms to perform mutational analysis onthe sequence of GUSBp. The Sorting Intolerant from Tolerant (SIFT)(http://sift.jcvi.org/) (Sim et al., 2012) and PMUT (http://mmb2.pcb.ub.es:8080/PMut/) (Ferrer-Costa et al., 2005) servers were used foridentification and classification of pathological mutations from neutralmutations. The SIFT algorithm calculates the outcome of variations incoding on protein functions (Sim et al., 2012). It uses sequence homol-ogy to predict the probability of whether an amino acid substitutionmay result in an unfavorable effect on the function of the protein (Simet al., 2012). The fundamental postulation is that evolutionarilyconservedmotifs or regions have a tendency to be less tolerant tomuta-tion, and hence, replacement of amino acid in these fractions is morelikely to influence the protein structure and function (Sim et al.,2012). Similarly, PMUT also allows a rapid and precise calculation of
ith 49 experimentally validated mutations indicated.
he mutational landscape in β-glucuronidase: A major player of.gene.2015.09.062

3F.I. Khan et al. / Gene xxx (2015) xxx–xxx
the pathological nature of single point mutations using the neuralnetwork algorithms (Ferrer-Costa et al., 2005). The fast scanning ofmutational hot spots was performed using three protocols, first, alaninescanning, second, massive mutation and third, genetically accessiblemutations (Ferrer-Costa et al., 2005). These algorithms identified theputative disease causing mutational sites in GUSBp, which were usedas the candidate sites for further analyses.
Furthermore, the I-Mutant 2.0 (http://folding.biofold.org/i-mutant/i-mutant2.0.html) (Capriotti et al., 2005) and MuStab (http://bioinfo.ggc.org/mustab/) (Teng et al., 2010) servers were used for predictingthe effect of mutations on the stability of the protein. I-Mutant 2.0 isbased on Support Vector Machine (SVM) protocol that estimates theprotein stability change in terms of free energy change (i.e., Gibb's freeenergy (ΔΔG) values) upon single point mutation. It can also be usedas a regression predictor for estimating the related ΔΔG values and aclassifier for predicting the sign of the protein stability change uponmutation with an accuracy of 77%. The MuStab server also performs asimilar analysis with an accuracy of 84.59%..
2.2. Structure-based mutation analysis
Numerousmethodswere used for estimating the effect ofmutationson the structure of GUSBp. GUSBp structure was mutated using variousbioinformatic tools, such as Site DirectedMutator (SDM) server (http://mordred.bioc.cam.ac.uk/~sdm/sdm.php) (Worth et al., 2011), FoldX(Schymkowitz et al., 2005), Cologne University Protein Stability Analy-sis Tool (CUPSAT) (http://cupsat.tu-bs.de/) (Parthiban et al., 2006)and Swiss-PDB Viewer (Deep View) (Kaplan and Littlejohn, 2001), togenerate the structural mutants. The stability of wild type (WT) andmutant GUSBp were compared. The SDM server is based on a statisticalpotential energy function that utilizes the nature-specific amino-acidreplacement frequencies within homologs to calculate a stability scorein terms of change in ΔΔG. FoldX is an empirical force-field tool devel-oped for a quick assessment of the consequence of mutations on thefolding, stability and dynamics of proteins. Similarly, CUPSAT utilizesthe structural environment specific torsion angle and atom potentialsto predict stability changes (i.e., ΔΔG) upon point mutations. TheDeep view was also used for analyzing the mutation pattern in GUSB,by first performing the mutation and the energy minimization usingthe steepest descent algorithm executed in GROMOS from Deepview(Kaplan and Littlejohn, 2001). The pattern of mutations is understoodin terms of contact maps and hydrogen bonding (HB) plots generatedusing the CCP4 suite (Bronstein et al., 1994) and Discovery Studio 3.5(Accelrys, 2013).
2.3. Molecular dynamics simulations
Many remarkable biological functions in protein and their profounddynamicmechanism can be revealed by studying their internalmotions(Khan et al., 2015). Molecular dynamics (MD) simulation providesconformational differences between the native and the mutated pro-teins to identify the effect of mutation on stability and flexibility(Anwer et al., 2015; Stephens et al., 2014). The MD simulations onselected disease associated mutationswere performed using GROMACS4.6.5 molecular mechanics package (Van Der Spoel et al., 2005). Theresulting mutants were soaked in a cubic box of water molecules witha dimension of 10 Å i.e., setting the box edge 10 Å from the moleculeperiphery. The boundary of the system was created using the editconfmodule while solvation was performed using the genbox. The editconfmodule of GROMACS is used to manipulate the coordinate file to definea box, apply rotations and translations, and allow us to specify a centerfor our system, while the genbox generates solvent box, solvates theprotein and adds extra molecules on random position. The charges onthe protein were neutralized using the verlet cut-off scheme by the ad-dition of Na+ and Cl− ions to maintain neutrality. The systemwas thenminimized in order to combat inappropriate geometry and structure
Please cite this article as: Khan, F.I., et al., Large scale analysis of tmucopolysaccharidosis type VII, Gene (2015), http://dx.doi.org/10.1016/j
distortion using 1500 steps of steepest descent. This process wasexecuted until a maximum force (b1000 kJ/mol/nm) was reached.Equilibration was achieved using the NVT (constant number of parti-cles, volume, and temperature) and NPT ensembles (constant numberof particles, pressure, and temperature). After the equilibration phase,the particle-mesh Ewald summation method (Horng et al., 2005) wasapplied, and then a 10 ns production phase was performed to increasethe probability of the system. The production stage of MD is used tosample the structural characteristics and dynamics of GUSBp. Theresulting trajectories were analyzed using utilities present in theGROMACS. The outputs were analyzed using the GRACE (GRaphingAdvanced Computation and Exploration of data) plotting tool. Allgraphic presentations of the 3D model were prepared using Discoverystudio 4.0 and Visual Molecular Dynamics (Humphrey et al., 1996).
2.4. Hydrogen bond analysis
Different types of non-covalent interactions in the protein structurewere calculated at the mutated positions using the CONTACT programof the CCP4 suite, which analyses contacts between subsets of atomsin a PDB file. The mutations were created using the program BuildMutants and the hydrogen bonds in the protein were visualized. The ef-fects of amino acid mutations on the macromolecule properties werepredicted using Discovery Studio.
3. Results and discussion
The output generated from the PMut, SIFT, I-MUTANT 2.0 (sequenceand structure based), MuStab, CUPSAT and SDM server suggested thatthere are 211 lethal mutations present in the structure of GUSBp(Table S1). These mutations were further grouped into a class of 90mutations according to the disease causing nature (Table 1). These mu-tations were classified into seven categories based on the nature of theamino acid mutations in order to understand their characteristicchange. Furthermore, the contact map analyses were carried out onthe experimentally determined mutations to understand the extent ofthe changes in terms of the interaction of different residues. We haveclassified all disease causing mutations into seven categories as de-scribed here in detail.
3.1. Neutral by neutral
There were 122 mutations observed in the framework of GUSBp(Table S1). These mutations cause either excess stabilization or destabi-lization of the enzyme, which may lead to the development of diseaselike symptoms. There were 18 mutations that resulted in additionalstabilization of GUSBp and that may have been responsible for MPSVIIdisease. The substitution p.Ser71Leu was the most stabilizing type ofmutation with a ΔΔG score of 4.70 and a tolerance index of 0.00. Thesolvent accessibility of the wild type was 21.4% i.e., partially accessibleand that of the mutant type was 13.9%. The total energy of thenative and mutant p.Ser71Leu structure was −28,954.71 kJ/mol and−27,648.186 kJ/mol, respectively. The substitution of the serine residueoften leads to a change in the preference of the enzyme (Arima et al.,2014) andmay be important for understanding the development of bet-ter therapeutic agents against diseases like tuberculosis (TB) (Purohitet al., 2011). Therefore, thesemutationswere considered important tar-gets for investigational studies. Several other types of experimentallyvalidated mutations were p.Pro30Cys, p.Ser52Phe, p.Tyr320Cys,p.Tyr495Cys, and p.Tyr508Cys. Based on the generated contact mapsof mutants and wild types, they may be responsible for this geneticdisorder (Tomatsu et al., 2009)..
Furthermore, 34 highly destabilizing mutations of the GUSBp struc-turewere also predicted thatmay affect the structure and causeMPS VIIdisease. The p.Phe208Pro, p.Phe539Gly and p.Leu622Gly were thoughtto be the most deleterious mutations with ΔΔG scores of −7.15,
he mutational landscape in β-glucuronidase: A major player of.gene.2015.09.062

Table 1List of amino acid substitutions causing malfunction or associated with disease in proteinGUSBp. The mutations selected for MD simulations are indicated in red.
S. no Amino acid substitution Tolerance indexa
Degree of riskb
Validation
(E–experimental,T–theoretical)
1. Gly25Cys 0.00 High T2. Gly26Ile 0.00 High T3. Pro30Cys 0.00 High E4. Glu37Phe 0.01 High T5. Cys38Glu 0.08 High E6. Ser52Phe 0.04 High E7. Ser71Leu 0.00 High T8. Pro73Cys 0.01 High T9. Pro80Cys 0.00 High T10. Phe83Gln 0.01 High T11. Val99Lys 0.05 High T12. Trp100Pro 0.00 High T13. Trp111Pro 0.01 High T14. His142Trp 0.01 High T15. Ser154Phe 0.00 High T16. Leu176Phe 0.08 High E17. Phe200Lys 0.00 High T18. Phe208Pro 0.00 High T19. Arg216Trp 0.03 High E20. Val218Ser 0.00 High T21. Leu220Pro 0.00 High T22. Pro224Cys 0.02 High T23. Val237Ser 0.04 High T24. Leu243Pro 0.15 High E25. Val282Trp 0.00 High T26. Tyr299Pro 0.00 High T27. Val305Pro 0.00 High T28. Gly314Gln 0.00 High T29. Tyr320Cys 0.01 High E30. Gly325Cys 0.00 High T31. Ile338Pro 0.00 High T32. Asn339Ser 0.00 High E33. Phe345Pro 0.01 High T34. Asn349Leu 0.01 High T35. Glu352Val 0.01 High T36. Phe361Asn 0.02 High T37. Leu366Pro 0.02 High T38. Asp369Cys 0.00 High T39. Arg382Cys 0.00 High E40. Pro387Lys 0.00 High T41. Cys396Lys 0.01 High T42. Ile401Gly 0.01 High T43. Val403Gln 0.00 High T44. Pro408Ser 0.26 High E45. Pro415Leu 0.69 High E46. Met430Asp 0.01 High T47. Arg435Pro 0.25 High E
48. Arg436Val 0.00 High T49. Lys438Cys 0.00 High T50. His440Gly 0.00 High T51. Pro441Cys 0.00 High T52. Ala442Trp 0.01 High T53. Val443Gln 0.00 High T54. Met445Gln 0.01 High T55. Val448Asn 0.01 High T56. Asn450Cys 0.00 High T57. Tyr462Lys 0.01 High T58. Val466Arg 0.01 High T59. Leu473Glu 0.00 Medium high T60. Arg477Trp 0.00 High E61. Phe481Ser 0.06 High T62. Tyr495Cys 0.13 High E63. Val496Ser 0.03 High T64. Ile499Gly 0.01 High T65. Cys500Asn 0.04 High T66. Asn502Cys 0.00 High T67. Tyr508Cys 0.01 High E68. Phe525Pro 0.02 High T69. Trp528Val 0.02 High T70. Lys534Leu 0.01 High T71. Ile536His 0.03 High T72. Ile537His 0.00 High T73. Tyr541Gly 0.00 High T74. His550Cys 0.00 High T75. His570Cys 0.00 High T76. Leu573Glu 0.01 High T77. Asp574Trp 0.00 High T78. Val581His 0.04 High T79. Ile586Gly 0.06 High T80. Phe592Gly 0.00 High T81. Asn604Cys 0.00 High T82. Lys605Phe 0.01 High T83. Ile608Gly 0.00 High T84. Ala618Trp 0.00 High T85. Ala619Val 0.00 High E86. Leu621Asp 0.00 High T87. Leu622Gly 0.01 High T88. Arg623Trp 0.00 High T89. Arg625Trp 0.00 High T90. Asn631Trp 0.00 High T
aTolerance index (TI) is the normalized probability of amino acid substitution at a partic-ular position. The mutation with TI value (≤0.05) are assumed to be deleterious and vice-versa.bDegree of risk is calculated using the consensus of the outcomesof all software used in theadopted pipeline.
4 F.I. Khan et al. / Gene xxx (2015) xxx–xxx
−6.42 and −6.80, respectively. In addition, the p.Ile499Gly andp.Ile586Gly substitutions have ΔΔG scores of−6.21. The solvent acces-sibility of wild type residues was 1.1%, 2.8%, 2.2%, 1.5% and 0%. For mu-tant type residues, solvent accessibility was 19.6%, 42.3%, 20.6%, 34.3%
Please cite this article as: Khan, F.I., et al., Large scale analysis of tmucopolysaccharidosis type VII, Gene (2015), http://dx.doi.org/10.1016/j
and 25.2% for positions 208, 499, 586, 592 and 622, respectively. Theminimized total energy as calculated by Deep View of the five mutantsreferenced above was −27,861.256 kJ/mol, −28,882.879 kJ/mol,−28,872.096 kJ/mol, −28,802.490 kJ/mol and −28,859.408 kJ/mol,respectively.We also performedHB plot analysis and contact map anal-ysis on the p.Leu243Pro and p.Asn339Ser mutations since these wereobserved to be associated with disease (Tomatsu et al., 2009). Thesubstitution, p.Phe208Prowas assumed to be significant since phenylal-anine mutations may produce resistance to various drugs (Loo andClarke, 1993)..
3.2. Neutral by basic
There were 17 mutations found in the structure of GUSBp that weregroupedunder this category (Table S1). Therewas no stabilizing diseaseassociated with mutations observed in this category, while ninemutations are predicted that cause destabilization of the enzyme struc-ture. These substitutions were p.Val99Lys, p.Phe200Lys, p.Pro387Lys,p.Cys396Lys, p.Tyr462Lys, p.Val466Arg, p.Ile536His, p.Ile537His, andp.Val581His. Among these mutations, p.Val99Lys, p.Pro387Lys,and p.Cys396Lys showed the highest deleterious effects with ΔΔGscores of−5.15,−5.09, and−5.37, respectively. p.Gly136Arg was pre-dicted to be a disease-associated mutation with a destabilizing effect.Due to its ΔΔG score of −0.98, this experimentally verified mutationmay be responsible for Sly disease in humans (Tomatsu et al., 2009).The calculated total energy of the three deleteriousmutants, p.Val99Lys,p.Phe200Lys, and p.Cys396Lys were−20,860.982 kJ/mol,−28,048.387kJ/mol, and −28,115.736 kJ/mol, respectively. Further, the solvent ac-cessibility of these mutants was 9.5%, 24%, and 0.5%, respectively. Themutation p.Cys396Lys showed the highest deleterious effect and wasimportant for these studies because the cysteine mutation affects thestability of the protein (Vivian et al., 2003)..
3.3. Neutral by acidic
Cysteine mutations likely cause many diseases, such as vonWillebrand disease and Sly disease (Ahmad et al., 2013; Hassan et al.,2012; Schooten et al., 2005; Tomatsu et al., 2009). Therefore, p.Cys38Glumay be considered as a disease-causing mutation in the group of ninesuch substitutions. There were five mutations of this type that causehigh destabilization of GUSBp and may have resulted in disease likesymptoms (Table S1). Furthermore, p.Met430Asp showed the mostdeleterious effect, with a ΔΔG score of −4.90 and total energy of−28,521.766kJ/mol, which indicated its effect on the stability of the en-zyme. The solvent accessibility of p.Met430Aspwas observed to be 5.1%as compared to that of the native structure, which was calculated to be0.3%. These analyses will be useful in curing the genetic disease causedby the substitution of the amino acids (Xiong et al., 2007)..
3.4. Basic by basic
The only observedmutation of this category in the structure of GUSBwas p.Arg382His, an experimentally validated substitution associatedwith Sly disease (Tomatsu et al., 2009). The total energy afterminimiza-tion of the mutant structure was −27,189.594 kJ/mol and the solventaccessibility was 3.1%, with a ΔΔG score of 0.18. This mutation showeda consistent decrease in stability of the protein molecule in the predict-ed results of the I-MUTANT, MuStab, and CUPSAT servers (Table S1)..
3.5. Basic by neutral
This group contained 28 disease-causing mutations dominated byeleven stability increasing substitutions in the structure of GUSBp. Thelysine mutation has been shown to be important for the regulation ofp53 (Nakamura et al., 2000). Therefore, p.Lys534Leu may be associatedwith Sly disease. The SIFT calculated the tolerance index for this
he mutational landscape in β-glucuronidase: A major player of.gene.2015.09.062
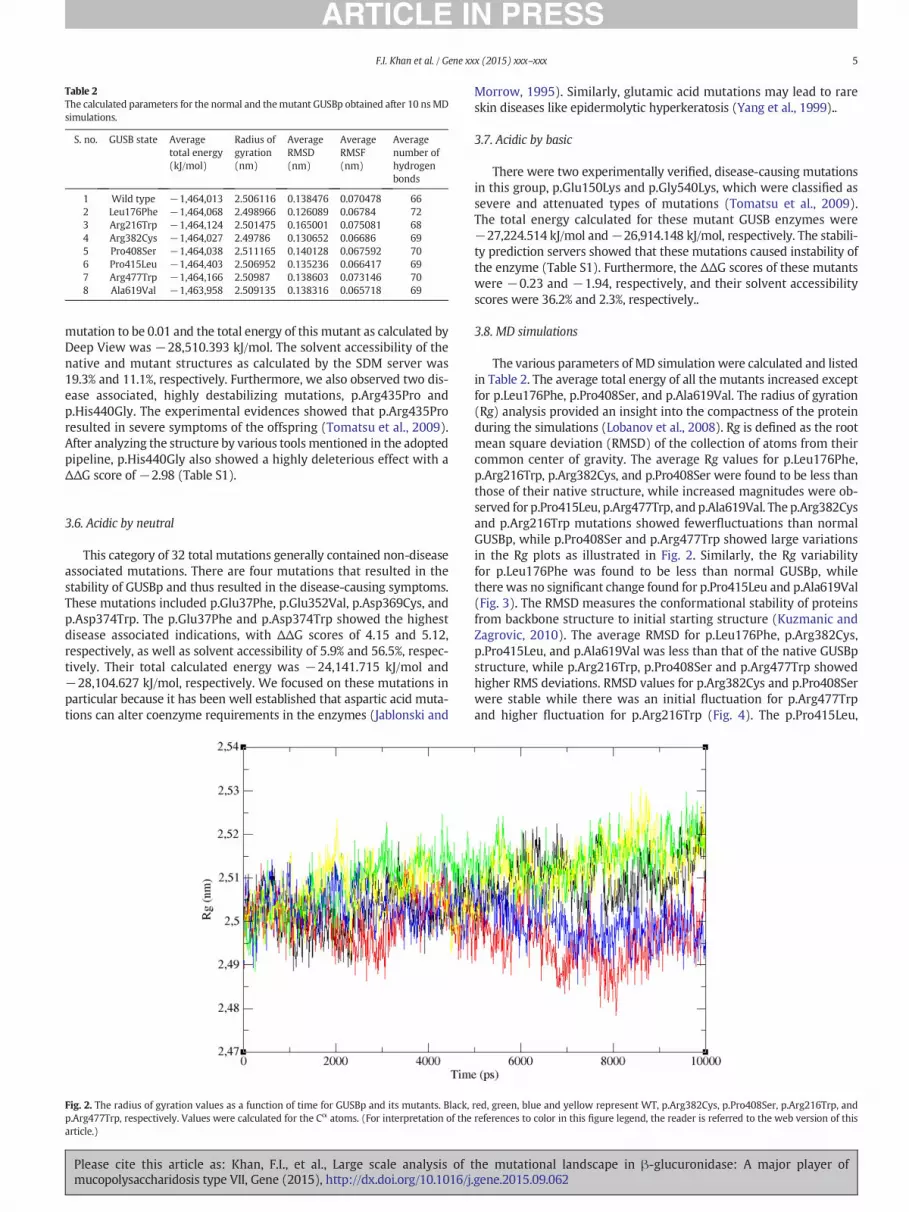
Table 2The calculated parameters for the normal and themutant GUSBp obtained after 10 nsMDsimulations.
S. no. GUSB state Averagetotal energy(kJ/mol)
Radius ofgyration(nm)
AverageRMSD(nm)
AverageRMSF(nm)
Averagenumber ofhydrogenbonds
1 Wild type −1,464,013 2.506116 0.138476 0.070478 662 Leu176Phe −1,464,068 2.498966 0.126089 0.06784 723 Arg216Trp −1,464,124 2.501475 0.165001 0.075081 684 Arg382Cys −1,464,027 2.49786 0.130652 0.06686 695 Pro408Ser −1,464,038 2.511165 0.140128 0.067592 706 Pro415Leu −1,464,403 2.506952 0.135236 0.066417 697 Arg477Trp −1,464,166 2.50987 0.138603 0.073146 708 Ala619Val −1,463,958 2.509135 0.138316 0.065718 69
5F.I. Khan et al. / Gene xxx (2015) xxx–xxx
mutation to be 0.01 and the total energy of this mutant as calculated byDeep View was −28,510.393 kJ/mol. The solvent accessibility of thenative and mutant structures as calculated by the SDM server was19.3% and 11.1%, respectively. Furthermore, we also observed two dis-ease associated, highly destabilizing mutations, p.Arg435Pro andp.His440Gly. The experimental evidences showed that p.Arg435Proresulted in severe symptoms of the offspring (Tomatsu et al., 2009).After analyzing the structure by various tools mentioned in the adoptedpipeline, p.His440Gly also showed a highly deleterious effect with aΔΔG score of −2.98 (Table S1).
3.6. Acidic by neutral
This category of 32 total mutations generally contained non-diseaseassociated mutations. There are four mutations that resulted in thestability of GUSBp and thus resulted in the disease-causing symptoms.These mutations included p.Glu37Phe, p.Glu352Val, p.Asp369Cys, andp.Asp374Trp. The p.Glu37Phe and p.Asp374Trp showed the highestdisease associated indications, with ΔΔG scores of 4.15 and 5.12,respectively, as well as solvent accessibility of 5.9% and 56.5%, respec-tively. Their total calculated energy was −24,141.715 kJ/mol and−28,104.627 kJ/mol, respectively. We focused on these mutations inparticular because it has been well established that aspartic acid muta-tions can alter coenzyme requirements in the enzymes (Jablonski and
Fig. 2. The radius of gyration values as a function of time for GUSBp and its mutants. Black,p.Arg477Trp, respectively. Values were calculated for the Cα atoms. (For interpretation of thearticle.)
Please cite this article as: Khan, F.I., et al., Large scale analysis of tmucopolysaccharidosis type VII, Gene (2015), http://dx.doi.org/10.1016/j
Morrow, 1995). Similarly, glutamic acid mutations may lead to rareskin diseases like epidermolytic hyperkeratosis (Yang et al., 1999)..
3.7. Acidic by basic
There were two experimentally verified, disease-causing mutationsin this group, p.Glu150Lys and p.Gly540Lys, which were classified assevere and attenuated types of mutations (Tomatsu et al., 2009).The total energy calculated for these mutant GUSB enzymes were−27,224.514 kJ/mol and−26,914.148 kJ/mol, respectively. The stabili-ty prediction servers showed that these mutations caused instability ofthe enzyme (Table S1). Furthermore, the ΔΔG scores of these mutantswere −0.23 and −1.94, respectively, and their solvent accessibilityscores were 36.2% and 2.3%, respectively..
3.8. MD simulations
The various parameters of MD simulation were calculated and listedin Table 2. The average total energy of all the mutants increased exceptfor p.Leu176Phe, p.Pro408Ser, and p.Ala619Val. The radius of gyration(Rg) analysis provided an insight into the compactness of the proteinduring the simulations (Lobanov et al., 2008). Rg is defined as the rootmean square deviation (RMSD) of the collection of atoms from theircommon center of gravity. The average Rg values for p.Leu176Phe,p.Arg216Trp, p.Arg382Cys, and p.Pro408Ser were found to be less thanthose of their native structure, while increased magnitudes were ob-served for p.Pro415Leu, p.Arg477Trp, and p.Ala619Val. The p.Arg382Cysand p.Arg216Trp mutations showed fewerfluctuations than normalGUSBp, while p.Pro408Ser and p.Arg477Trp showed large variationsin the Rg plots as illustrated in Fig. 2. Similarly, the Rg variabilityfor p.Leu176Phe was found to be less than normal GUSBp, whilethere was no significant change found for p.Pro415Leu and p.Ala619Val(Fig. 3). The RMSD measures the conformational stability of proteinsfrom backbone structure to initial starting structure (Kuzmanic andZagrovic, 2010). The average RMSD for p.Leu176Phe, p.Arg382Cys,p.Pro415Leu, and p.Ala619Val was less than that of the native GUSBpstructure, while p.Arg216Trp, p.Pro408Ser and p.Arg477Trp showedhigher RMS deviations. RMSD values for p.Arg382Cys and p.Pro408Serwere stable while there was an initial fluctuation for p.Arg477Trpand higher fluctuation for p.Arg216Trp (Fig. 4). The p.Pro415Leu,
red, green, blue and yellow represent WT, p.Arg382Cys, p.Pro408Ser, p.Arg216Trp, andreferences to color in this figure legend, the reader is referred to the web version of this
he mutational landscape in β-glucuronidase: A major player of.gene.2015.09.062
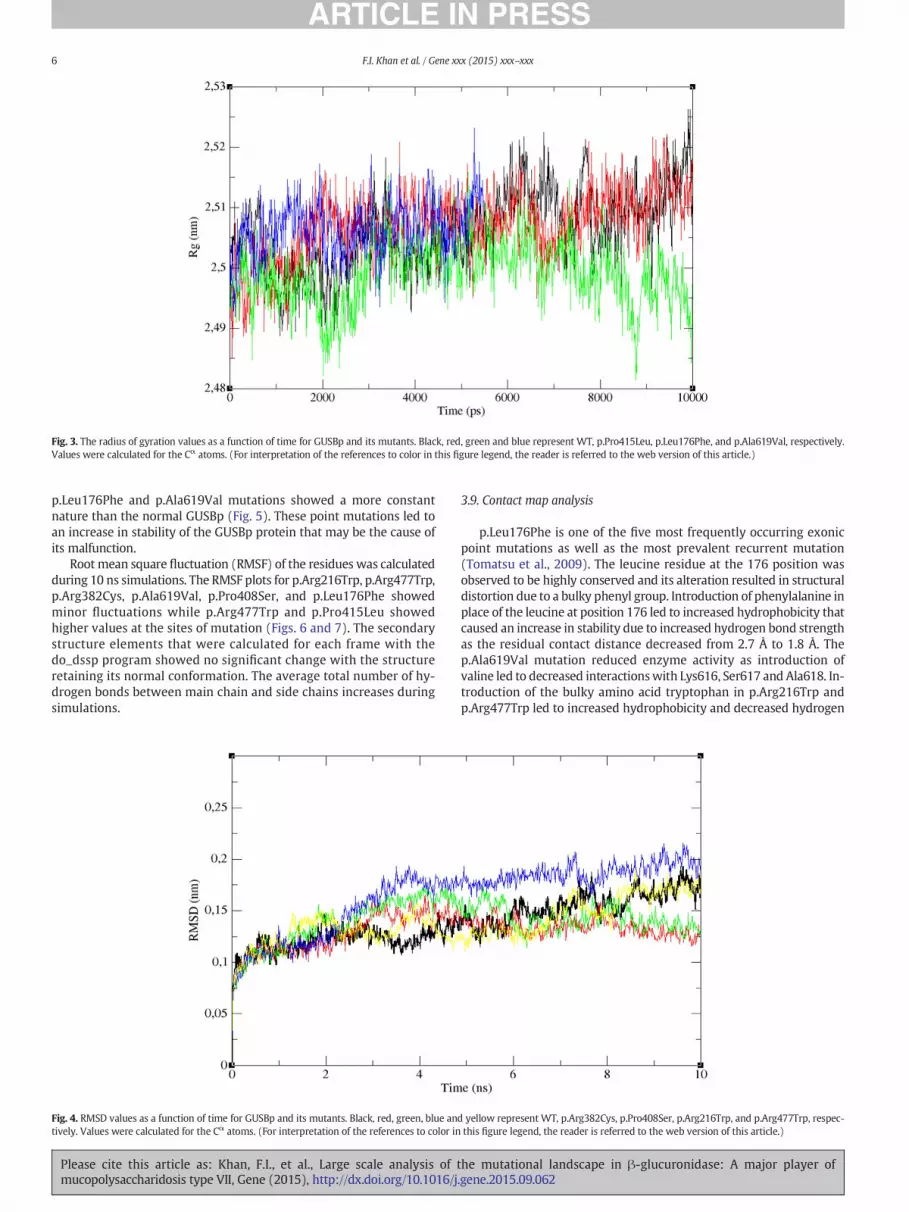
Fig. 3. The radius of gyration values as a function of time for GUSBp and its mutants. Black, red, green and blue represent WT, p.Pro415Leu, p.Leu176Phe, and p.Ala619Val, respectively.Values were calculated for the Cα atoms. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
6 F.I. Khan et al. / Gene xxx (2015) xxx–xxx
p.Leu176Phe and p.Ala619Val mutations showed a more constantnature than the normal GUSBp (Fig. 5). These point mutations led toan increase in stability of the GUSBp protein that may be the cause ofits malfunction.
Root mean square fluctuation (RMSF) of the residues was calculatedduring 10 ns simulations. The RMSF plots for p.Arg216Trp, p.Arg477Trp,p.Arg382Cys, p.Ala619Val, p.Pro408Ser, and p.Leu176Phe showedminor fluctuations while p.Arg477Trp and p.Pro415Leu showedhigher values at the sites of mutation (Figs. 6 and 7). The secondarystructure elements that were calculated for each frame with thedo_dssp program showed no significant change with the structureretaining its normal conformation. The average total number of hy-drogen bonds between main chain and side chains increases duringsimulations.
Fig. 4. RMSD values as a function of time for GUSBp and its mutants. Black, red, green, blue antively. Values were calculated for the Cα atoms. (For interpretation of the references to color in
Please cite this article as: Khan, F.I., et al., Large scale analysis of tmucopolysaccharidosis type VII, Gene (2015), http://dx.doi.org/10.1016/j
3.9. Contact map analysis
p.Leu176Phe is one of the five most frequently occurring exonicpoint mutations as well as the most prevalent recurrent mutation(Tomatsu et al., 2009). The leucine residue at the 176 position wasobserved to be highly conserved and its alteration resulted in structuraldistortion due to a bulky phenyl group. Introduction of phenylalanine inplace of the leucine at position 176 led to increased hydrophobicity thatcaused an increase in stability due to increased hydrogen bond strengthas the residual contact distance decreased from 2.7 Å to 1.8 Å. Thep.Ala619Val mutation reduced enzyme activity as introduction ofvaline led to decreased interactionswith Lys616, Ser617 and Ala618. In-troduction of the bulky amino acid tryptophan in p.Arg216Trp andp.Arg477Trp led to increased hydrophobicity and decreased hydrogen
d yellow represent WT, p.Arg382Cys, p.Pro408Ser, p.Arg216Trp, and p.Arg477Trp, respec-this figure legend, the reader is referred to the web version of this article.)
he mutational landscape in β-glucuronidase: A major player of.gene.2015.09.062

Fig. 5.RMSdeviation values as a function of time forGUSBp and itsmutants. Black, red, green andblue representWT, p.Pro415Leu, p.Leu176Phe, andp.Ala619Val, respectively. Valueswerecalculated for the Cα atoms. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
7F.I. Khan et al. / Gene xxx (2015) xxx–xxx
bonds with neighboring amino acids. Though cysteine is uncommon inproteins, replacement of cysteine with arginine at position 382 resultedin decreased side chainflexibility, while introduction of serine increasedthe side chain hydrogen bonding in Pro408Ser.
4. Conclusions
In this study, we have successfully analyzed the impact of pointmutations in the structure, function, and stability of GUSBp, using insilico methods to understand the molecular basis of MPS VII disease.
Fig. 6. Average RMSD values as a function of amino acid sequence numbers for GUSBp and ip.Arg216Trp, and p.Arg477Trp, respectively. Values were calculated for the Cα atoms. (For inweb version of this article.)
Please cite this article as: Khan, F.I., et al., Large scale analysis of tmucopolysaccharidosis type VII, Gene (2015), http://dx.doi.org/10.1016/j
Out of a total of 211 characterized deleterious mutations, we identified90 that are predicted to be disease-causingmutations. Stability analysespredicted mutations that had maximum impact on the structure andstability of the GUSBp, namely p.Phe208Pro, p.Phe539Gly, p.Leu622Gly,p.Ile499Gly, and p.Ile586Gly. These notable mutations resulted inthe destabilization of the protein structure, causing malfunction of theprotein and, presumably MPS VII disease. On the other hand,p.Glu37Phe, p.Ser71Leu, p.Lys534Leu, and p.Asp574Trp mutationswere predicted to increase the protein stability. However, these muta-tions were found to be associated with disease, presumably due to
ts mutants. Black, red, green, blue and yellow represent WT, p.Arg382Cys, p.Pro408Ser,terpretation of the references to color in this figure legend, the reader is referred to the
he mutational landscape in β-glucuronidase: A major player of.gene.2015.09.062

Fig. 7. Average RMSD values as a function of amino acid sequence numbers for GUSBp and its mutants. Black, red, green and blue represent WT, p.Pro415Leu, p.Leu176Phe, andp.Ala619Val, respectively. Values were calculated for the Cα atoms.
8 F.I. Khan et al. / Gene xxx (2015) xxx–xxx
reduction in accessibility of the substrate to the protein. These outcomescan be further utilized for other experimental studies in order to under-stand the mechanisms of Sly syndrome in detail.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.gene.2015.09.062.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgments
The authors would like to express sincere gratitude to the Centre forHigh Performance Computing, in South Africa for providing the compu-tational infrastructure. We thank Tracey L. Baird for editorial assistancein preparing this manuscript. FA and MIH are thankful to the Depart-ment of Science and Technology, India and Indian Council of MedicalResearch, India for financial support.
References
Accelrys, 2013. Discovery studio modeling environment, Release 3.5. Accelrys SoftwareInc., San Diego.
Ahmad, F., Jan, R., Kannan, M., Obser, T., Hassan, M.I., Oyen, F., Budde, U., Saxena, R.,Schneppenheim, R., 2013. Characterisation of mutations and molecular studies oftype 2 von Willebrand disease. Thromb. Haemost. 109, 39–46.
Anwer, K., Sonani, R., Madamwar, D., Singh, P., Khan, F., Bisetty, K., Ahmad, F., Hassan, M.I.,2015. Role of N-terminal residues on folding and stability of C-phycoerythrin:simulation and urea-induced denaturation studies. J. Biomol. Struct. Dyn. 33,121–133.
Arima, J., Tanaka, A., Morimoto, M., Mori, N., 2014. Mutation of active site serine residuewith cysteine displays change in acyl-acceptor preference of beta-peptidylaminopeptidase from Pseudomonas aeruginosa PAO1. Appl. Microbiol. Biotechnol.98, 1631–1640.
Ballabio, A., Gieselmann, V., 2009. Lysosomal disorders: from storage to cellular damage.Biochim. Biophys. Acta 1793, 684–696.
Bronstein, I., Fortin, J., Stanley, P.E., Stewart, G.S., Kricka, L.J., 1994. Chemiluminescent andbioluminescent reporter gene assays. Anal. Biochem. 219, 169–181.
Brot, F.E., Bell Jr., C.E., Sly, W.S., 1978. Purification and properties of beta-glucuronidasefrom human placenta. Biochemistry 17, 385–391.
Capriotti, E., Fariselli, P., Casadio, R., 2005. I-Mutant2.0: predicting stability changes uponmutation from the protein sequence or structure. Nucleic Acids Res. 33,W306–W310.
Please cite this article as: Khan, F.I., et al., Large scale analysis of tmucopolysaccharidosis type VII, Gene (2015), http://dx.doi.org/10.1016/j
Ferrer-Costa, C., Gelpi, J.L., Zamakola, L., Parraga, I., de la Cruz, X., Orozco, M., 2005. PMUT:a web-based tool for the annotation of pathological mutations on proteins.Bioinformatics 21, 3176–3178.
Filocamo, M., Morrone, A., 2011. Lysosomal storage disorders: molecular basis andlaboratory testing. Hum. Genomics 5, 156–169.
Futerman, A.H., van Meer, G., 2004. The cell biology of lysosomal storage disorders. Nat.Rev. Mol. Cell Biol. 5, 554–565.
Hassan, M.I., Saxena, A., Ahmad, F., 2012. Structure and function of von Willebrand factor.Blood Coagul. Fibrinolysis 23, 11–22.
Hassan, M.I., Waheed, A., Grubb, J.H., Klei, H.E., Korolev, S., Sly, W.S., 2013. High resolutioncrystal structure of human beta-glucuronidase reveals structural basis of lysosometargeting. PLoS One 8, e79687.
Horng, J.C., Tracz, S.M., Lumb, K.J., Raleigh, D.P., 2005. Slow folding of a three-helix proteinvia a compact intermediate. Biochemistry 44, 627–634.
Humphrey, W., Dalke, A., Schulten, K., 1996. VMD: visual molecular dynamics. J. Mol.Graphs. 14 (33–38), 27–38.
Jablonski, S.A., Morrow, C.D., 1995. Mutation of the aspartic acid residues of the GDD se-quence motif of poliovirus RNA-dependent RNA polymerase results in enzymes withaltered metal ion requirements for activity. J. Virol. 69, 1532–1539.
Jain, S., Drendel, W.B., Chen, Z.W., Mathews, F.S., Sly, W.S., Grubb, J.H., 1996. Structure ofhuman beta-glucuronidase reveals candidate lysosomal targeting and active-sitemotifs. Nat. Struct. Biol. 3, 375–381.
Kaplan, W., Littlejohn, T.G., 2001. Swiss-PDB viewer (deep view). Brief. Bioinform. 2,195–197.
Khan, F.I., Govender, A., Permaul, K., Singh, S., Bisetty, K., 2015. Thermostable chitinase IIfrom Thermomyces lanuginosus SSBP: cloning, structure prediction and molecular dy-namics simulations. J. Theor. Biol. 374, 107–114.
Kuzmanic, A., Zagrovic, B., 2010. Determination of ensemble-average pairwise rootmean-square deviation from experimental B-factors. Biophys. J. 98, 861–871.
Lachmann, R.H., 2011. Enzyme replacement therapy for lysosomal storage diseases. Curr.Opin. Pediatr. 23, 588–593.
Liebherr, R.B., Renner, M., Gorris, H.H., 2014. A single molecule perspective on thefunctional diversity of in vitro evolved beta-glucuronidase. J. Am. Chem. Soc. 136,5949–5955.
Lobanov, M., Bogatyreva, N.S., Galzitskaia, O.V., 2008. Radius of gyration is indicator ofcompactness of protein structure. Mol. Biol. 42, 701–706.
Loo, T.W., Clarke, D.M., 1993. Functional consequences of phenylalanine mutations in thepredicted transmembrane domain of P-glycoprotein. J. Biol. Chem. 268, 19965–19972.
Meikle, P.J., Hopwood, J.J., Clague, A.E., Carey, W.F., 1999. Prevalence of lysosomal storagedisorders. JAMA 281, 249–254.
Metcalf, J.A., Zhang, Y., Hilton, M.J., Long, F., Ponder, K.P., 2009. Mechanism of shortenedbones in mucopolysaccharidosis VII. Mol. Genet. Metab. 97, 202–211.
Molyneux, A.J., Blair, E., Coleman, N., Daish, P., 1997. Mucopolysaccharidosis type VII asso-ciated with hydrops fetalis: histopathological and ultrastructural features with genet-ic implications. J. Clin. Pathol. 50, 252–254.
Nakamura, S., Roth, J.A., Mukhopadhyay, T., 2000. Multiple lysine mutations in the C-terminal domain of p53 interfere with MDM2-dependent protein degradation andubiquitination. Mol. Cell. Biol. 20, 9391–9398.
Naz, H., Islam, A., Waheed, A., Sly, W.S., Ahmad, F., Hassan, M.I., 2013. Human β-glucuron-idase: structure, function, and application in enzyme replacement therapy. Rejuvena-tion Res. 16, 352–363.
he mutational landscape in β-glucuronidase: A major player of.gene.2015.09.062

9F.I. Khan et al. / Gene xxx (2015) xxx–xxx
Oshima, A., Kyle, J.W., Miller, R.D., Hoffmann, J.W., Powell, P.P., Grubb, J.H., Sly, W.S.,Tropak, M., Guise, K.S., Gravel, R.A., 1987. Cloning, sequencing, and expression ofcDNA for human beta-glucuronidase. Proc. Natl. Acad. Sci. U. S. A. 84, 685–689.
Parenti, G., Andria, G., Ballabio, A., 2015. Lysosomal storage diseases: from pathophysiol-ogy to therapy. Annu. Rev. Med. 66, 471–486.
Parthiban, V., Gromiha, M.M., Schomburg, D., 2006. CUPSAT: prediction of proteinstability upon point mutations. Nucleic Acids Res. 34, W239–W242.
Purohit, R., Rajendran, V., Sethumadhavan, R., 2011. Relationship between mutation ofserine residue at 315th position in M. tuberculosis catalase-peroxidase enzyme andisoniazid susceptibility: an in silico analysis. J. Mol. Model. 17, 869–877.
Schooten, C.J., Tjernberg, P., Westein, E., Terraube, V., Castaman, G., Mourik, J.A.,Hollestelle, M.J., Vos, H.L., Bertina, R.M., Berg, H.M., et al., 2005. Cysteine-mutationsin von Willebrand factor associated with increased clearance. J. Thromb. Haemost.3, 2228–2237.
Schymkowitz, J., Borg, J., Stricher, F., Nys, R., Rousseau, F., Serrano, L., 2005. The FoldXwebserver: an online force field. Nucleic Acids Res. 33, W382–W388.
Shayman, J.A., 2014. Thematic review series: recent advances in the treatment oflysosomal storage diseases. J. Lipid Res. 55, 993–994.
Shipley, J.M., Klinkenberg, M., Wu, B.M., Bachinsky, D.R., Grubb, J.H., Sly, W.S., 1993.Mutational analysis of a patient with mucopolysaccharidosis type VII, and identifica-tion of pseudogenes. Am. J. Hum. Genet. 52, 517–526.
Sim, N.L., Kumar, P., Hu, J., Henikoff, S., Schneider, G., Ng, P.C., 2012. SIFT web server:predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40,W452–W457.
Sly, W.S., Quinton, B.A., McAlister, W.H., Rimoin, D.L., 1973. Beta glucuronidase de-ficiency: report of clinical, radiologic, and biochemical features of a newmucopolysaccharidosis. J. Pediatr. 82, 249–257.
Stephens, D.E., Khan, F.I., Singh, P., Bisetty, K., Singh, S., Permaul, K., 2014. Creation ofthermostable and alkaline stable xylanase variants by DNA shuffling. J. Biotechnol.187C, 139–146.
Storch, S., Wittenstein, B., Islam, R., Ullrich, K., Sly, W.S., Braulke, T., 2003. Mutationalanalysis in longest known survivor of mucopolysaccharidosis type VII. Hum. Genet.112, 190–194.
Teng, S., Srivastava, A.K., Wang, L., 2010. Sequence feature-based prediction of proteinstability changes upon amino acid substitutions. BMC Genomics 11 (Suppl. 2), S5.
Please cite this article as: Khan, F.I., et al., Large scale analysis of tmucopolysaccharidosis type VII, Gene (2015), http://dx.doi.org/10.1016/j
Tomatsu, S., Sukegawa, K., Ikedo, Y., Fukuda, S., Yamada, Y., Sasaki, T., Okamoto, H.,Kuwabara, T., Orii, T., 1990. Molecular basis of mucopolysaccharidosis type VII:replacement of Ala619 in beta-glucuronidase with Val. Gene 89, 283–287.
Tomatsu, S., Fukuda, S., Sukegawa, K., Ikedo, Y., Yamada, S., Yamada, Y., Sasaki, T.,Okamoto, H., Kuwahara, T., Yamaguchi, S., et al., 1991. Mucopolysaccharidosis typeVII: characterization of mutations and molecular heterogeneity. Am. J. Hum. Genet.48, 89–96.
Tomatsu, S., Montano, A.M., Dung, V.C., Grubb, J.H., Sly, W.S., 2009. Mutations and poly-morphisms in GUSB gene in mucopolysaccharidosis VII (Sly syndrome). Hum.Mutat. 30, 511–519.
Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A.E., Berendsen, H.J., 2005.GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718.
Vervoort, R., Islam, M.R., Sly, W.S., Zabot, M.T., Kleijer, W.J., Chabas, A., Fensom, A.,Young, E.P., Liebaers, I., Lissens, W., 1996. Molecular analysis of patients withbeta-glucuronidase deficiency presenting as hydrops fetalis or as earlymucopolysaccharidosis VII. Am. J. Hum. Genet. 58, 457–471.
Vitner, E.B., Platt, F.M., Futerman, A.H., 2010. Common and uncommon pathogeniccascades in lysosomal storage diseases. J. Biol. Chem. 285, 20423–20427.
Vivian, J.P., Hastings, A.F., Duggin, I.G., Wake, R.G., Wilce, M.C., Wilce, J.A., 2003. Theimpact of single cysteine residue mutations on the replication terminator protein.Biochem. Biophys. Res. Commun. 310, 1096–1103.
Worth, C.L., Preissner, R., Blundell, T.L., 2011. SDM—a server for predicting effects ofmutations on protein stability and malfunction. Nucleic Acids Res. 39, W215–W222.
Wu, B.M., Tomatsu, S., Fukuda, S., Sukegawa, K., Orii, T., Sly, W.S., 1994. Overexpressionrescues the mutant phenotype of L176F mutation causing beta-glucuronidase defi-ciency mucopolysaccharidosis in two Mennonite siblings. J. Biol. Chem. 269,23681–23688.
Xiong, A.S., Peng, R.H., Cheng, Z.M., Li, Y., Liu, J.G., Zhuang, J., Gao, F., Xu, F., Qiao, Y.S.,Zhang, Z., et al., 2007. Concurrent mutations in six amino acids in beta-glucuronidase improve its thermostability. Protein Eng. Des. Sel. 20, 319–325.
Yang, J.M., Nam, K., Kim, H.C., Lee, J.H., Park, J.K., Wu, K., Lee, E.S., Steinert, P.M., 1999. Anovel glutamic acid to aspartic acid mutation near the end of the 2B rod domain inthe keratin 1 chain in epidermolytic hyperkeratosis. J. Investig. Dermatol. 112,376–379.
he mutational landscape in β-glucuronidase: A major player of.gene.2015.09.062




















