
Enzyme Replacement Therapy in Patients Who Have Mucopolysaccharidosis I and Are Younger Than 5...
12
DOI: 10.1542/peds.2006-2156 ; originally published online May 30, 2007; Pediatrics Yong Xue, Emil D. Kakkis and Nathalie Guffon J. Edmond Wraith, Michael Beck, Roderick Lane, Ans van der Ploeg, Elsa Shapiro, -L-Iduronidase (Laronidase) α Recombinant Human and Are Younger Than 5 Years: Results of a Multinational Study of Enzyme Replacement Therapy in Patients Who Have Mucopolysaccharidosis I http://pediatrics.aappublications.org/content/early/2007/05/30/peds.2006-2156.citation located on the World Wide Web at: The online version of this article, along with updated information and services, is of Pediatrics. All rights reserved. Print ISSN: 0031-4005. Online ISSN: 1098-4275. Boulevard, Elk Grove Village, Illinois, 60007. Copyright © 2007 by the American Academy published, and trademarked by the American Academy of Pediatrics, 141 Northwest Point publication, it has been published continuously since 1948. PEDIATRICS is owned, PEDIATRICS is the official journal of the American Academy of Pediatrics. A monthly by guest on June 9, 2013 pediatrics.aappublications.org Downloaded from
-
Upload
spanalumni -
Category
Documents
-
view
5 -
download
0
Transcript of Enzyme Replacement Therapy in Patients Who Have Mucopolysaccharidosis I and Are Younger Than 5...

DOI: 10.1542/peds.2006-2156; originally published online May 30, 2007;Pediatrics
Yong Xue, Emil D. Kakkis and Nathalie GuffonJ. Edmond Wraith, Michael Beck, Roderick Lane, Ans van der Ploeg, Elsa Shapiro,
-L-Iduronidase (Laronidase)αRecombinant Human and Are Younger Than 5 Years: Results of a Multinational Study of
Enzyme Replacement Therapy in Patients Who Have Mucopolysaccharidosis I
http://pediatrics.aappublications.org/content/early/2007/05/30/peds.2006-2156.citation
located on the World Wide Web at: The online version of this article, along with updated information and services, is
of Pediatrics. All rights reserved. Print ISSN: 0031-4005. Online ISSN: 1098-4275.Boulevard, Elk Grove Village, Illinois, 60007. Copyright © 2007 by the American Academy published, and trademarked by the American Academy of Pediatrics, 141 Northwest Pointpublication, it has been published continuously since 1948. PEDIATRICS is owned, PEDIATRICS is the official journal of the American Academy of Pediatrics. A monthly
by guest on June 9, 2013pediatrics.aappublications.orgDownloaded from

ARTICLE
Enzyme Replacement Therapy in Patients Who HaveMucopolysaccharidosis I and Are Younger Than 5Years: Results of a Multinational Study ofRecombinant Human �-L-Iduronidase (Laronidase)J. Edmond Wraith, MBChBa, Michael Beck, MDb, Roderick Lane, PhDc, Ans van der Ploeg, MD, PhDd, Elsa Shapiro, PhD, ABPPe,
Yong Xue, MD, PhDf, Emil D. Kakkis, MD, PhDg, Nathalie Guffon, MDh
aWillink Biochemical Genetics Unit, Royal Manchester Children’s Hospital, Manchester, United Kingdom; bDepartment of Pediatrics, Children’s Hospital, University ofMainz, Mainz, Germany; cSleep and Respiratory Services, Great Ormond Street Hospital, London, United Kingdom; dDepartment of Pediatrics, Sophia Children’s Hospital,Rotterdam, Netherlands; eDivision of Pediatric Clinical Neuroscience, University of Minnesota, Minneapolis, Minnesota; fGenzyme Europe BV, Naarden, Netherlands;gBioMarin Pharmaceutical Inc, Novato, California; hDepartment of Pediatrics, Centre de Reference des Maladies Hereditaires du Metabolisme, Hopital Edouard Herriot,Lyon, France
Financial Disclosure: Dr Wraith provided paid and unpaid consultancy services for Genzyme, received travel expenses to attend meetings, and participated in clinical trials sponsored by Genzyme; Dr Beck hasreceived unrestricted grants from BioMarin and Genzyme; Dr Lane received payment from Genzyme to analyze, interpret, and report the sleep studies that were performed in this study; Dr van der Ploeg hasperformed clinical trials for Genzyme and has received travel expenses and honoraria to attend and present at scientific meetings; Dr Shapiro is a consultant on neuropsychological methods and has receivedtravel expenses to attend and present at scientific meetings; Dr Xue is employed by Genzyme; Dr Kakkis is employed by BioMarin and has financial conflicts related to laronidase; and Dr Guffon has performedclinical trials for Genzyme and has received travel expenses and honoraria to attend and present at scientific meetings.
ABSTRACT
OBJECTIVE.Our objective was to evaluate the safety, pharmacokinetics, and efficacyof laronidase in young, severely affected children with mucopolysaccharidosis I.
METHODS. This was a prospective, open-label, multinational study of 20 patients whohad mucopolysaccharidosis I and were �5 years old (16 with Hurler syndrome, 4with Hurler-Scheie syndrome) and were scheduled to receive intravenous laroni-dase at 100 U/kg (0.58 mg/kg) weekly for 52 weeks. Four patients underwentdosage increases to 200 U/kg for the last 26 weeks because of elevated urinaryglycosaminoglycan levels at week 22.
RESULTS. Laronidase was well tolerated at both dosages. Investigators reported im-proved clinical status in 94% of patients at week 52. The mean urinary glycos-aminoglycan level declined by �50% at week 13 and was sustained thereafter. Amore robust decrease in urinary glycosaminoglycan was observed in patients withlow antibody levels and those who were receiving the 200 U/kg dosage. Onexamination, the liver edge was reduced by 69.5% in patients with a palpable liverat baseline and week 52 (n � 10). The proportion of patients with left ventricularhypertrophy decreased from 53% to 17%. Global assessment of sleep studiesshowed improvement or stabilization in 67% of patients, and the apnea/hypopneaindex decreased by 5.8 events per hour (�8.5%) in those with abnormal baselinevalues. The younger patients with Hurler syndrome (�2.5 years) and all 4 patientswith Hurler-Scheie syndrome showed normal mental development trajectoriesduring the 1-year treatment period.
www.pediatrics.org/cgi/doi/10.1542/peds.2006-2156
doi:10.1542/peds.2006-2156
KeyWordsenzyme replacement therapy, Hurlersyndrome, laronidase, MPS I
AbbreviationsMPS I—mucopolysaccharidosis IHSCT—hematopoietic stem celltransplantationERT—enzyme replacement therapyAE—adverse eventIgG—immunoglobulin GAHI—apnea/hypopnea indexIAR—infusion-associated reaction
Accepted for publication Dec 13, 2006
Address correspondence to J. Edmond Wraith,MBChB, Royal Manchester Children’s Hospital,Willink Biochemical Genetics Unit, HospitalRoad, Pendlebury, Manchester M27 1HA,United Kingdom. E-mail: [email protected]
PEDIATRICS (ISSN Numbers: Print, 0031-4005;Online, 1098-4275). Copyright © 2007 by theAmerican Academy of Pediatrics
PEDIATRICS Volume 120, Number 1, July 2007 e37 by guest on June 9, 2013pediatrics.aappublications.orgDownloaded from

CONCLUSIONS. Laronidase seems to be well tolerated and toprovide clinical benefit in patients who have severe mu-copolysaccharidosis I and are �5 years old. Enzymereplacement therapy is not curative and may not im-prove all affected organs and systems in individualswhen irreversible changes have developed. The long-term clinical outcome and effects of antibodies andlaronidase dosing on glycosaminoglycan reduction war-rant additional investigation.
MUCOPOLYSACCHARIDOSIS TYPE I (MPS I) is causedby a deficiency of the lysosomal enzyme �-L-
iduronidase, which leads to the progressive accumula-tion of the glycosaminoglycans dermatan and heparansulfate, ultimately interfering with cell functioning andcompromising virtually all tissues and organs.1 Histori-cally, patients with MPS I have been classified into 3clinical syndromes on the basis of differences in diseaseprogression: Hurler (severe), Hurler-Scheie (intermedi-ate), and Scheie (mild). Patients with Hurler syndromeexperience cognitive decline in early childhood, whereaspatients with Hurler-Scheie and Scheie syndromes haverelatively mild, if any, cognitive impairment. However,the 3 phenotypes are not always clearly delineated, be-cause there can be substantial overlap in symptom pre-sentation and considerable variability in both the sever-ity and the progression of the disease.1,2 The disease isbest described as a spectrum that ranges from severe(with cognitive decline) to attenuated (without cogni-tive decline).
Since 1980, hematopoietic stem cell transplantation(HSCT) has been used to treat patients with Hurler syn-drome, first using bone marrow and more recently usingumbilical cord blood.3 HSCT improves many of the so-matic features of MPS I and can preserve cognitive func-tion, but bone, heart valve, and eye disease seem to berecalcitrant to treatment. The procedure requires HLA-matched donor cells, is prone to engraftment failure, andis associated with considerable morbidity and mortality,which has limited its use to severely affected patientswho are early in the course of disease. Enzyme replace-ment therapy (ERT) with laronidase (recombinant hu-man �-L-iduronidase) has been shown to be a safe andeffective treatment in patients who have MPS I and areolder than 5 years.4–6 In the pivotal study to confirm thedrug’s safety and efficacy, the use of functional assess-ments as co-primary end points (forced vital capacityand the 6-minute walk test) precluded the enrollment ofyoung patients and those with severe cognitive impair-ment. To evaluate the safety, pharmacokinetics, and ef-ficacy of laronidase in a young and severely affectedpatient population, this study investigated the effects ofweekly laronidase infusions for 1 year in 20 patientswho had MPS I and were younger than 5 years.
METHODS
Study DesignThis was a 52-week, prospective, open-label, multi-center study of laronidase in 20 patients who had MPS Iand were enrolled at 4 sites in the United Kingdom,France, Germany, and the Netherlands. The primaryobjective of the study was to evaluate the safety oflaronidase in young and severely affected patients withMPS I. Secondary objectives were to evaluate the phar-macokinetics and efficacy of laronidase in this patientpopulation.
All patients were naive to laronidase therapy, had tobe younger than 5 years at initiation of treatment, andhad to have a diagnosis of MPS I confirmed by fibroblastor leukocyte �-L-iduronidase enzyme activity �10% ofnormal and by genotyping. Exclusion criteria includedhaving undergone or being under consideration forHSCT, acute hydrocephalus, clinically significant organicdisease not related to MPS I, administration of an inves-tigational drug within 30 days before study enrollment,or known hypersensitivity to components of the laroni-dase solution. All parents or legal guardians providedwritten informed consent to participate in the study. Theprotocol was approved by each site’s independent ethicscommittee. The study was designed and conducted incompliance with the principles of the International Con-ference on Harmonization of Technical Requirementsfor Registration of Pharmaceuticals for Human Useguidelines for Good Clinical Practice.
Laronidase TreatmentAll patients initially received weekly intravenous infu-sions of 100 U/kg (0.58 mg/kg) laronidase (recombinanthuman �-L-iduronidase (Aldurazyme; BioMarin Phar-maceutical, Inc, Novato, CA; and Genzyme Corp, Cam-bridge, MA). Laronidase was diluted in 100 mL or 250mL of 0.9% sodium chloride injection and infused over4 hours. Unlike in previous studies, laronidase was ad-ministered without 0.1% human serum albumin, in ac-cordance with the European summary of product char-acteristics. On the basis of the results of an interimanalysis that was performed on the first 13 patients, thestudy protocol was amended to allow the final 7 patientsto receive a dosage of 200 U/kg laronidase from week 26onward if their urinary glycosaminoglycan level at week22 was �200 �g/mg creatinine; 4 of these patients qual-ified and received the higher dosage for the second halfof the study. A Port-a-Cath was placed in 8 patients. Tominimize possible infusion-associated reactions (IARs),all patients received an antipyretic and an antihistaminebefore each infusion.
Evaluation of SafetySafety monitoring included adverse event (AE) report-ing, physical examination, clinical chemistries, hematol-
e38 WRAITH et al by guest on June 9, 2013pediatrics.aappublications.orgDownloaded from

ogy parameters, urinalysis, vital signs, and electrocardio-grams. AEs were characterized by severity and byrelationship to study drug. An infusion-associated reac-tion was defined as any AE that occurred from the startof an infusion to the end of the 30-minute postinfusionobservation period (longer if deemed necessary by theinvestigator) and that was considered to be drug relatedby the investigator.
Antibody TestingAntibody titers to laronidase (immunoglobulin G [IgG])were measured every 4 weeks with the use of a modifiedenzyme-linked immunosorbent assay and confirmed byimmunoprecipitation.7 IgE testing was to be performedafter moderate to severe IARs.
Pharmacokinetic AssessmentPharmacokinetic assessments were performed at weeks1, 13, 26, and 52. Plasma laronidase activity was deter-mined using 4-methylumbelliferyl iduronic acid as asubstrate.8 Pharmacokinetic parameters for laronidasewere calculated using standard noncompartmentalmethods.
Biochemical Evaluations and Efficacy OutcomeMeasuresBiochemical evaluations and clinical outcome measuresthat were relevant to this young severely affected MPS Ipatient population consisted of urinary glycosaminogly-can excretion, liver size, cardiac status, upper airwayobstruction during sleep, growth velocity, the investiga-tor’s global assessment, and mental development.
Urinary glycosaminoglycan excretion was deter-mined in the first morning void using an automateddimethylmethylene blue dye–binding procedure in acentral laboratory (BioMarin Pharmaceutical Inc) andexpressed as micrograms per milligram of creatinine.4
Liver size was evaluated by the distance of the liveredge below the right costal margin at the midclavicularline at physical examination. Quantitative volumetricmeasurements by MRI and/or computed tomographywould have required anesthesia and were consideredtoo high a risk for these patients.
Two-dimensional echocardiography was performedand interpreted by local cardiologists according to a cen-tralized protocol. Left ventricular mass z scores werecalculated using normative data from Children’s Hospital(Boston, MA).9 In addition, local cardiologists assessedthe degree of left ventricular hypertrophy and valvularappearance.
Polysomnograms with resulting apnea/hypopnea in-dex (AHI) scores (the total number of episodes of apneaand hypopnea per hour of sleep) were interpreted cen-trally by a single, independent sleep study expert. Upperairway obstruction during sleep should be considered asa continuum that may not fully be represented by theAHI, especially in young children. Therefore, the sever-
ity of the upper airway obstruction during sleep was alsoassessed as mild, moderate, or severe by the same expertusing a nonlinear clinical global assessment scale basedon well-defined criteria that combined clinical observa-tion and sleep laboratory testing results.10,11
Height and weight were measured at baseline and atweeks 13, 26, and 52 and expressed as height-for-ageand weight-for-age z scores using the Centers for DiseaseControl and Prevention/National Center for Health Sta-tistics clinical growth charts.12,13 z scores were also calcu-lated for a historical control group that consisted ofuntreated patients with MPS I in the same age range(6–72 months) from the MPS I Registry (BioMarin andGenzyme, data on file).14
Investigators provided a global assessment of the pa-tient’s clinical status using a 7-point scale (marked, mod-erate, or slight decline; no change; mild, moderate, ormarked improvement) before and after 13, 26, and 52weeks of treatment with laronidase. Exploratory assess-ment on cognitive function was performed at baselineand weeks 26 and 52 using the Griffiths Mental Devel-opment Scales, which is validated for children from birthto 8 years and is widely used in Europe.15 Each patient’smental development trajectory was calculated by plot-ting the patient’s mental age equivalent versus the pa-tient’s chronological age for each study time point. Thenormal trajectory for mental development has the men-tal age corresponding to the chronological age.
StatisticsNo hypothesis testing was performed in this open-labelstudy. Continuous data were summarized by usingmeans, medians, and ranges. Categorical data were sum-marized by using frequencies and distributions. All anal-yses were performed with the use of SAS 8.0 software(SAS Institute, Cary, NC).
RESULTS
PatientsTwenty patients who received a diagnosis of MPS I at amean age of 1.3 years (ranging from prenatal diagnosisto 4.5 years) were enrolled in the study at a mean age of2.9 years (range: 0.5–5.1 years; Table 1). Patients 101 to109 were enrolled at the study site in the United King-dom, 201 to 207 in France, 301 and 302 in Germany,and 401 to 403 at the site in the Netherlands. As assessedby the principal investigators at the study sites, 16 (80%)patients had a diagnosis of severe Hurler syndrome and4 (20%) patients had the attenuated Hurler-Scheie syn-drome. Nearly all were white (90%) and there was aslight excess of boys (60%). The mean height/length-for-age z score was �1.56, and 40% of patients had shortstature (less than �2 z scores). The most common mu-tations identified were W402X (45%) and Q70X (20%).
PEDIATRICS Volume 120, Number 1, July 2007 e39 by guest on June 9, 2013pediatrics.aappublications.orgDownloaded from

Of the 20 enrolled patients, 18 (90%) completed the52-week study.
SafetyAll patients experienced at least 1 AE, the majority ofwhich were not related to laronidase but to underlyingdisease. The most commonly reported AEs (�50% ofpatients) were pyrexia, diarrhea, vomiting, cough, rhi-norrhea, rhinitis, and rash. Seven (35%) patients expe-rienced 33 IARs (any AE that occurred from the start ofthe infusion to the end of the 30-min postinfusion ob-servation period and that was considered to be drugrelated by the investigator), the most common of whichwere pyrexia (12 events in 6 patients) and chills (7events in 4 patients). Three of these were seriousand occurred in patient 403 during infusion 12 (mildincrease in blood pressure and heart rate, moderatedecrease in oxygen saturation). The infusion was tem-porarily stopped, and the patient was treated with para-cetamol (acetaminophen) and supplemental oxygen.The infusion was resumed 20 minutes later, and thepatient experienced no additional IARs during the trial.Of the 4 patients whose dosage was increased from 100to 200 U/kg laronidase after week 26, 1 had no IARs ateither dosage. IARs that were reported only with the 200U/kg dosage were mild tremor (week 33) and moderatecrepitations, wheezing, and respiratory distress (week39). IARs were easily managed by reduction of the in-fusion rate, temporary interruption of the infusion, oradministration of an antihistamine and/or antipyretic.
Two patients died during the study as a consequenceof events related to their underlying disease. A 13-month-old girl with Hurler syndrome died of cardiacfailure after 25 weeks of laronidase therapy, but noautopsy was performed. She had an episode of cardiacfailure before the study and became cyanotic 6 weeksbefore her death. A 3-year-old boy with Hurler syn-drome underwent surgery for bilateral hip dysplasia anddied of a postsurgical complication (accidental extuba-tion with unsuccessful reintubation and tracheostomy)at week 48.
There were no clinically meaningful changes for anyof the serum chemistry, hematologic, or urinary param-eters assessed, and changes in vital signs or physicalexamination parameters during the study were unre-markable.
All patients in this study developed IgG antibodies tolaronidase with a mean time to seroconversion of 25.8days. Antibody titers generally rose during the first 20weeks of the study; the highest measured titer duringthe study was 1:204 800. After 1 year of laronidasetreatment, 4 patients had titers �1:1600 (1 patient wasseronegative), and 12 patients had titers �1:1600. Therewas no apparent correlation between the time to sero-conversion or antibody titer and the incidence of IARs.In the 4 patients (marked with a ‘‘b’’ in Table 1) whoreceived the increased dosage of laronidase, the subse-quent antibody titers did not change appreciably. Twopatients tested negative for IgE antibodies after moderateIARs.
TABLE 1 Baseline Patient Characteristics
Patient Age atDiagnosis, y
Age atEnrollment, y
Gender Genotype Phenotypea
101 0.9 4.3 M W402X/Q70X Hurler102 4.5 4.8 F P533R/P533R Hurler-Scheie104 2.1 2.4 M Q70X/W402X Hurler105 1.0 3.1 F Q70X/W402X Hurler106 0.1 4.8 F A36E/Q70X Hurler-Scheie107 2.3 2.5 M Q380R/T388R Hurler-Scheie108b �0.3 3.9 M W402X/W402X Hurler109b �0.6 3.0 F W402X/W402X Hurler201 0.9 1.3 M W402X/W402X Hurler202 0.7 1.3 M W402X/c206delA Hurler203 2.1 3.3 M W402X/Q70X Hurler204 2.7 4.0 F W402X/P533R Hurler-Scheie205 0.6 1.3 M c.35_4del/c.1524� 1G3A Hurler206 2.1 2.4 M W402X/A327P Hurler207 0.5 0.8 M W402X/W402X Hurler301 0.6 3.6 M W402X/W402X Hurler302 0.3 0.5 F Y202X/Y202X Hurler401b 1.5 5.1c F Q70X/W402X Hurler402 2.4 4.7 M A327P/L218P Hurler403b 1.0 1.2 F Q70X/Q70X HurlerMean 1.3 2.9 12 M/8 F 4 Hurler-Scheie/16 Hurler
For all patient numbers, the first digit of the number is site specific. M indicates male; F, female.a As assessed clinically by individual investigators.b Patients who received 200 U/kg after week 26.c Patient 401 was 5 years 1 month of age but was enrolled because of severe disease status.
e40 WRAITH et al by guest on June 9, 2013pediatrics.aappublications.orgDownloaded from

PharmacokineticsThe range of individual patient values, although vari-able, was reasonably consistent across 52 weeks of treat-ment. The mean area under the curve increased from0.58 � 0.59 hours/U per mL at the first infusion to 0.94� 0.97 hours/U per mL at week 52, but the change wasnot significant. There was no apparent relationship be-tween area under the curve and antibody titer over the52 week period. The mean half-life ranged from 0.55hours to 1.55 hours with large variability in individualvalues. The mean volume of distribution corrected forbody weight decreased from 0.753 � 0.497 L/kg to 0.246� 0.210 L/kg during treatment. There was a trend to-ward a lower volume of distribution as the antibodylevel increased, suggesting that the binding of antibodiesto laronidase keeps more laronidase in the plasma, awayfrom the renal epithelial cells that are believed to be thesource of urinary glycosaminoglycans, resulting in asmaller distribution volume. Pharmacokinetic analyseswere hampered by the relatively small sample size andlarge intragroup variations that did not allow for anymeaningful correlation analyses.
Urinary Glycosaminoglycans and Efficacy OutcomeMeasuresAfter initiation of treatment with laronidase, urinaryglycosaminoglycan levels showed a sharp decline withinthe first 13 weeks followed by a plateau. The meanreduction in urinary glycosaminoglycan level after 52weeks of treatment was 61.3% for all patients, 59.1% forpatients who were treated with 100 U/kg throughoutthe study, and 67.7% for the 4 patients who received200 U/kg after week 26 (Table 2).
At week 26, the mean percentage decrease was com-parable between the 2 dosage groups. Whereas patientswho were treated with 100 U/kg showed no additionalchange in the mean urinary glycosaminoglycan levelbetween weeks 26 and 52, the level was further reduced
by a mean of 8.6% in the 4 patients who were treatedwith 200 U/kg. Urinary glycosaminoglycan levels overtime in individual study patients are presented in Fig 1.Urinary glycosaminoglycan levels from healthy childrenwho were of the same age and assessed in a separatestudy are provided for comparison purposes.
All 20 patients had a palpable liver edge at baseline;however, at week 52, the liver edge was palpable in only10 patients. The mean distance of the liver edge belowthe right costal margin in the 10 patients with a palpableliver during the entire study decreased from 6.0 cm to1.7 cm (mean decrease: 69.5%; Table 2). Liver volumeswere also assessed by the investigators as being normalor abnormal (ie, hepatomegaly). At the start of study,the liver size was abnormal in all patients, whereas after52 weeks of treatment, the liver size had normalized in50% (9 of 18) of assessable patients.
The number of patients who were categorized as hav-ing mild left ventricular hypertrophy by echocardiogra-phy decreased from 52.6% (10 of 19) of assessable pa-tients at the start of the study to 16.7% (3 of 18) ofassessable patients at the final study visit. At the start ofstudy, the mean left ventricular mass was abnormallyhigh as demonstrated by a mean z score of 3.8 (range:0.6–8.4). At the end of study, the mean left ventricularmass z score had decreased by 0.9 (�11.3%) for the 17patients with available data (Table 2). For the 14 patientswith left ventricular mass z scores �2 at baseline, themean reduction in z score was 1.3 (�28.7%), demon-strating that the largest decreases in left ventricular masswere observed in the patients with hypertrophy at base-line.
Six patients had normal AHI values at baseline (AHI�10 events per hour); in 4 of these patients, the AHIremained normal for the rest of the study, and in 2patients, the AHI increased to 11.6 and 14.7 events perhour. AHI changes were assessed in the subgroup of
TABLE 2 Key Efficacy Outcomes
100 U/kg(n � 12)
100–200 U/kg(n � 4)
AvailableResults
Urinary glycosaminoglycans level, �g/mg creatinineBaseline 526.5� 175.7 670.7� 183.9Week 26 224.8� 109.0 284.6� 84.9Week 52 223.1� 163.3 201.1� 71.9Change, baseline to week 26, % �59.7� 13.0 �56.1� 13.7Change, baseline to week 52, % �59.1� 21.4 �67.7� 14.2
Liver edge BRCM (n � 10), cma
Baseline 6.0� 2.13Week 52 1.7� 0.98Change, baseline to week 52, % �69.5� 15.34
Left ventricular mass z score (n � 17)Baseline 3.8� 2.23Week 52 2.9� 1.60Change, baseline to week 52, % �0.9� 1.58
Data are shown as mean � SD. BRCM indicates below the right costal margin.a Calculated only for the patients who had a palpable liver during the whole study.
PEDIATRICS Volume 120, Number 1, July 2007 e41 by guest on June 9, 2013pediatrics.aappublications.orgDownloaded from
patients (n � 9) who had clinically significant abnormalbaseline values (AHI �10) and available results at theend of study. In this subgroup, the mean AHI decreasedfrom 45.3 to 39.6 events per hour, which corresponds toan 8.5% reduction. Predefined clinical significance (ie, a25% reduction in events per hour) was reached by 5patients (33% of the patients with available data). Of the9 patients with abnormal baseline AHI values, 5 patientshad tonsillectomies and/or adenoidectomies performedbetween baseline and week 11 that might have con-founded the results. However, most of the AHI improve-ments occurred between weeks 26 and 52, suggestingthat the benefit was attributable to laronidase ratherthan surgery.
In the 15 patients with available sleep study results atboth baseline and week 52, global assessment of thesleep study data by the central expert revealed that 5(33%) patients showed improvement (1 mild, 3 moder-ate, and 1 marked), 5 (33%) patients showed no change,and 5 (33%) patients showed worsening (1 marked, 1moderate, and 3 slight).
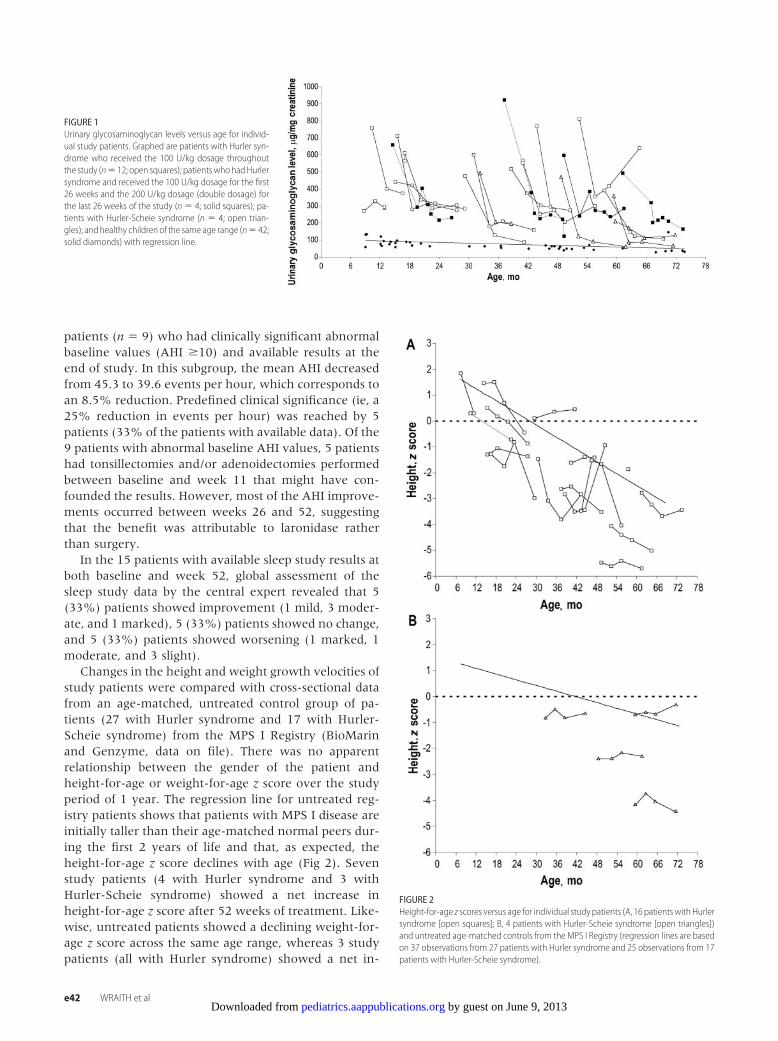
Changes in the height and weight growth velocities ofstudy patients were compared with cross-sectional datafrom an age-matched, untreated control group of pa-tients (27 with Hurler syndrome and 17 with Hurler-Scheie syndrome) from the MPS I Registry (BioMarinand Genzyme, data on file). There was no apparentrelationship between the gender of the patient andheight-for-age or weight-for-age z score over the studyperiod of 1 year. The regression line for untreated reg-istry patients shows that patients with MPS I disease areinitially taller than their age-matched normal peers dur-ing the first 2 years of life and that, as expected, theheight-for-age z score declines with age (Fig 2). Sevenstudy patients (4 with Hurler syndrome and 3 withHurler-Scheie syndrome) showed a net increase inheight-for-age z score after 52 weeks of treatment. Like-wise, untreated patients showed a declining weight-for-age z score across the same age range, whereas 3 studypatients (all with Hurler syndrome) showed a net in-
FIGURE 2Height-for-age z scores versus age for individual study patients (A, 16 patientswithHurlersyndrome [open squares]; B, 4 patients with Hurler-Scheie syndrome [open triangles])and untreated age-matched controls from the MPS I Registry (regression lines are basedon 37 observations from 27 patients with Hurler syndrome and 25 observations from 17patients with Hurler-Scheie syndrome).
FIGURE 1Urinary glycosaminoglycan levels versus age for individ-ual study patients. Graphed are patients with Hurler syn-drome who received the 100 U/kg dosage throughoutthe study (n� 12; open squares); patientswhohadHurlersyndrome and received the 100 U/kg dosage for the first26 weeks and the 200 U/kg dosage (double dosage) forthe last 26 weeks of the study (n � 4; solid squares); pa-tients with Hurler-Scheie syndrome (n � 4; open trian-gles); and healthy children of the same age range (n� 42;solid diamonds) with regression line.
e42 WRAITH et al by guest on June 9, 2013pediatrics.aappublications.orgDownloaded from
crease in weight-for-age z score during the 1-year studyduration (data not shown).
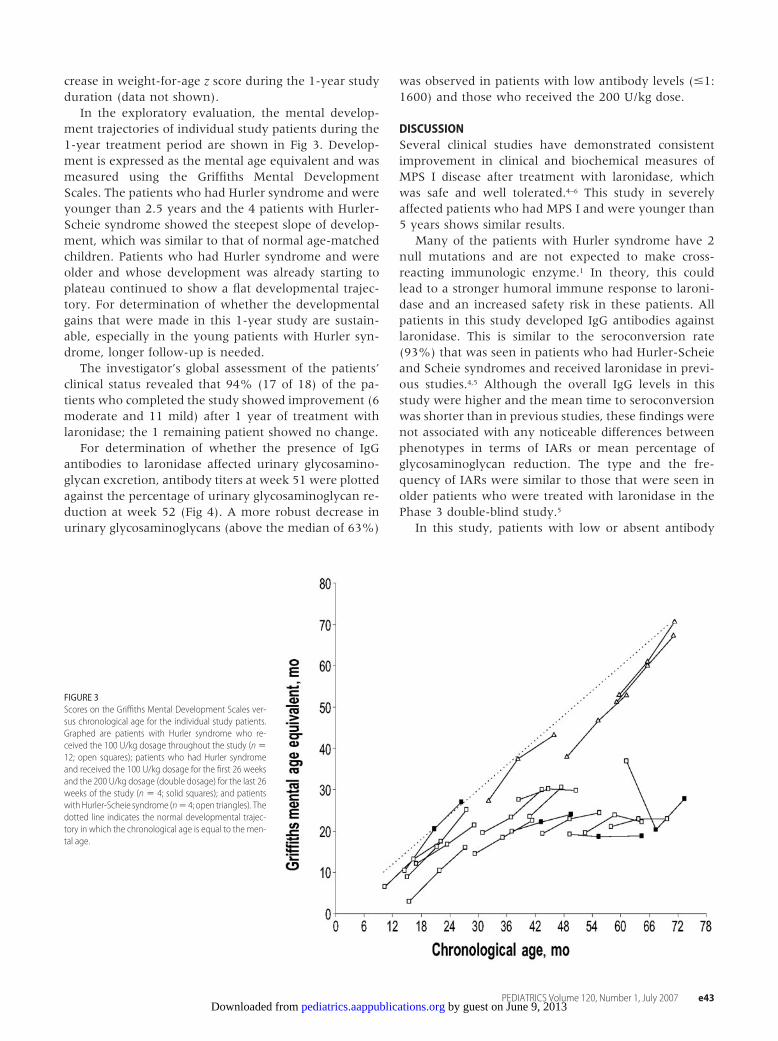
In the exploratory evaluation, the mental develop-ment trajectories of individual study patients during the1-year treatment period are shown in Fig 3. Develop-ment is expressed as the mental age equivalent and wasmeasured using the Griffiths Mental DevelopmentScales. The patients who had Hurler syndrome and wereyounger than 2.5 years and the 4 patients with Hurler-Scheie syndrome showed the steepest slope of develop-ment, which was similar to that of normal age-matchedchildren. Patients who had Hurler syndrome and wereolder and whose development was already starting toplateau continued to show a flat developmental trajec-tory. For determination of whether the developmentalgains that were made in this 1-year study are sustain-able, especially in the young patients with Hurler syn-drome, longer follow-up is needed.
The investigator’s global assessment of the patients’clinical status revealed that 94% (17 of 18) of the pa-tients who completed the study showed improvement (6moderate and 11 mild) after 1 year of treatment withlaronidase; the 1 remaining patient showed no change.
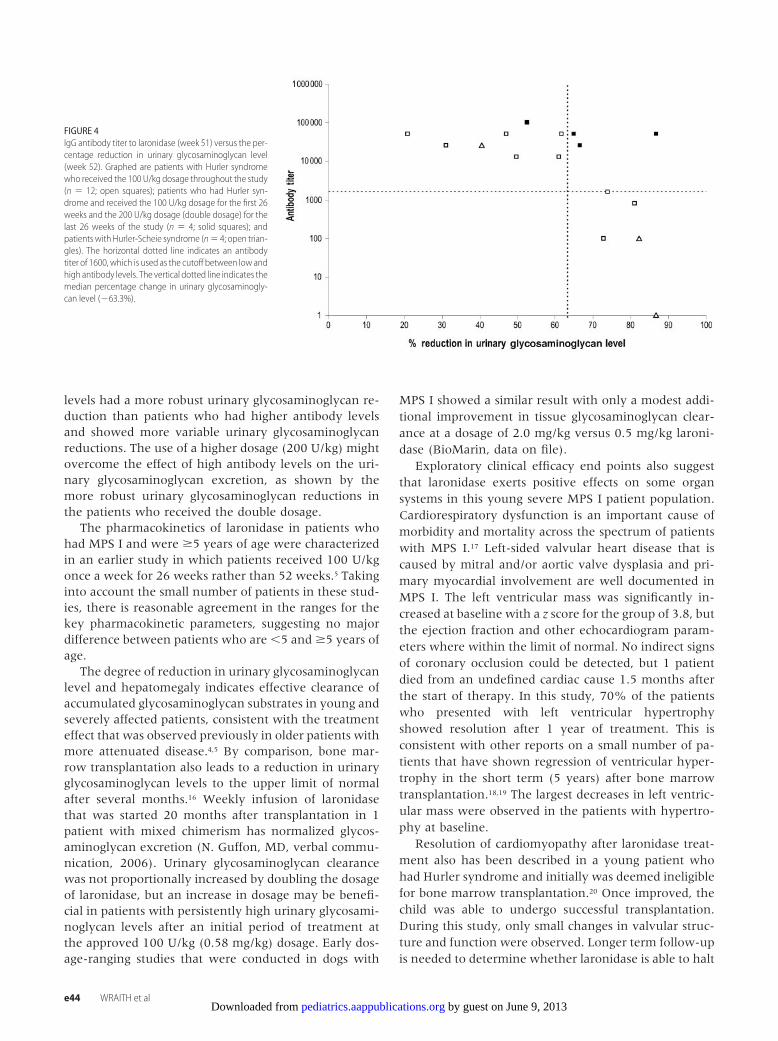
For determination of whether the presence of IgGantibodies to laronidase affected urinary glycosamino-glycan excretion, antibody titers at week 51 were plottedagainst the percentage of urinary glycosaminoglycan re-duction at week 52 (Fig 4). A more robust decrease inurinary glycosaminoglycans (above the median of 63%)
was observed in patients with low antibody levels (�1:1600) and those who received the 200 U/kg dose.
DISCUSSIONSeveral clinical studies have demonstrated consistentimprovement in clinical and biochemical measures ofMPS I disease after treatment with laronidase, whichwas safe and well tolerated.4–6 This study in severelyaffected patients who had MPS I and were younger than5 years shows similar results.
Many of the patients with Hurler syndrome have 2null mutations and are not expected to make cross-reacting immunologic enzyme.1 In theory, this couldlead to a stronger humoral immune response to laroni-dase and an increased safety risk in these patients. Allpatients in this study developed IgG antibodies againstlaronidase. This is similar to the seroconversion rate(93%) that was seen in patients who had Hurler-Scheieand Scheie syndromes and received laronidase in previ-ous studies.4,5 Although the overall IgG levels in thisstudy were higher and the mean time to seroconversionwas shorter than in previous studies, these findings werenot associated with any noticeable differences betweenphenotypes in terms of IARs or mean percentage ofglycosaminoglycan reduction. The type and the fre-quency of IARs were similar to those that were seen inolder patients who were treated with laronidase in thePhase 3 double-blind study.5
In this study, patients with low or absent antibody
FIGURE 3Scores on the Griffiths Mental Development Scales ver-sus chronological age for the individual study patients.Graphed are patients with Hurler syndrome who re-ceived the 100 U/kg dosage throughout the study (n �12; open squares); patients who had Hurler syndromeand received the 100 U/kg dosage for the first 26 weeksand the 200 U/kg dosage (double dosage) for the last 26weeks of the study (n � 4; solid squares); and patientswithHurler-Scheie syndrome (n� 4; open triangles). Thedotted line indicates the normal developmental trajec-tory in which the chronological age is equal to the men-tal age.
PEDIATRICS Volume 120, Number 1, July 2007 e43 by guest on June 9, 2013pediatrics.aappublications.orgDownloaded from
levels had a more robust urinary glycosaminoglycan re-duction than patients who had higher antibody levelsand showed more variable urinary glycosaminoglycanreductions. The use of a higher dosage (200 U/kg) mightovercome the effect of high antibody levels on the uri-nary glycosaminoglycan excretion, as shown by themore robust urinary glycosaminoglycan reductions inthe patients who received the double dosage.
The pharmacokinetics of laronidase in patients whohad MPS I and were �5 years of age were characterizedin an earlier study in which patients received 100 U/kgonce a week for 26 weeks rather than 52 weeks.5 Takinginto account the small number of patients in these stud-ies, there is reasonable agreement in the ranges for thekey pharmacokinetic parameters, suggesting no majordifference between patients who are �5 and �5 years ofage.
The degree of reduction in urinary glycosaminoglycanlevel and hepatomegaly indicates effective clearance ofaccumulated glycosaminoglycan substrates in young andseverely affected patients, consistent with the treatmenteffect that was observed previously in older patients withmore attenuated disease.4,5 By comparison, bone mar-row transplantation also leads to a reduction in urinaryglycosaminoglycan levels to the upper limit of normalafter several months.16 Weekly infusion of laronidasethat was started 20 months after transplantation in 1patient with mixed chimerism has normalized glycos-aminoglycan excretion (N. Guffon, MD, verbal commu-nication, 2006). Urinary glycosaminoglycan clearancewas not proportionally increased by doubling the dosageof laronidase, but an increase in dosage may be benefi-cial in patients with persistently high urinary glycosami-noglycan levels after an initial period of treatment atthe approved 100 U/kg (0.58 mg/kg) dosage. Early dos-age-ranging studies that were conducted in dogs with
MPS I showed a similar result with only a modest addi-tional improvement in tissue glycosaminoglycan clear-ance at a dosage of 2.0 mg/kg versus 0.5 mg/kg laroni-dase (BioMarin, data on file).
Exploratory clinical efficacy end points also suggestthat laronidase exerts positive effects on some organsystems in this young severe MPS I patient population.Cardiorespiratory dysfunction is an important cause ofmorbidity and mortality across the spectrum of patientswith MPS I.17 Left-sided valvular heart disease that iscaused by mitral and/or aortic valve dysplasia and pri-mary myocardial involvement are well documented inMPS I. The left ventricular mass was significantly in-creased at baseline with a z score for the group of 3.8, butthe ejection fraction and other echocardiogram param-eters where within the limit of normal. No indirect signsof coronary occlusion could be detected, but 1 patientdied from an undefined cardiac cause 1.5 months afterthe start of therapy. In this study, 70% of the patientswho presented with left ventricular hypertrophyshowed resolution after 1 year of treatment. This isconsistent with other reports on a small number of pa-tients that have shown regression of ventricular hyper-trophy in the short term (5 years) after bone marrowtransplantation.18,19 The largest decreases in left ventric-ular mass were observed in the patients with hypertro-phy at baseline.
Resolution of cardiomyopathy after laronidase treat-ment also has been described in a young patient whohad Hurler syndrome and initially was deemed ineligiblefor bone marrow transplantation.20 Once improved, thechild was able to undergo successful transplantation.During this study, only small changes in valvular struc-ture and function were observed. Longer term follow-upis needed to determine whether laronidase is able to halt
FIGURE 4IgG antibody titer to laronidase (week 51) versus the per-centage reduction in urinary glycosaminoglycan level(week 52). Graphed are patients with Hurler syndromewho received the 100 U/kg dosage throughout the study(n � 12; open squares); patients who had Hurler syn-drome and received the 100 U/kg dosage for the first 26weeks and the 200 U/kg dosage (double dosage) for thelast 26 weeks of the study (n � 4; solid squares); andpatients with Hurler-Scheie syndrome (n� 4; open trian-gles). The horizontal dotted line indicates an antibodytiter of 1600, which is used as the cutoff between low andhigh antibody levels. The vertical dotted line indicates themedian percentage change in urinary glycosaminogly-can level (�63.3%).
e44 WRAITH et al by guest on June 9, 2013pediatrics.aappublications.orgDownloaded from

or slow the progression of valvular disease and avoid theneed for valve replacement surgery.
Exploratory mental development testing indicatedthat the patients with Hurler-Scheie syndrome had anormal to above-normal rate of cognitive growth duringthe 1-year study. Similarly, the younger (�2.5 years ofage) patients with Hurler syndrome showed an increasein cognitive function at a rate similar to that of healthychildren. In contrast, the older patients with Hurler syn-drome did not show any significant gains or loss incognition.
Because the intravenously infused enzyme is not ex-pected to cross the blood-brain barrier in appreciableamounts at the administered dosage levels,21 it is possiblethat some of the developmental gains that were ob-served were indirectly related to improvement in overallhealth status. Long-term follow-up is needed to distin-guish direct from indirect effects. The children werereported to benefit from the treatment as assessed by theinvestigator global assessment.
There are some limitations in the design and themethods that were used in this study. First, several fac-tors precluded performing a randomized, controlledstudy, including the low incidence of MPS I, the rapidlyprogressive and fatal course of Hurler disease, the fam-ilies’ unwillingness to receive HSCT as an alternativetherapy, and ethical concerns about withholding a med-ication that was anticipated to receive regulatory ap-proval during the study. Historical comparison waslargely impossible because of the paucity of data fromuntreated patients with Hurler syndrome. The relativelyshort follow-up period hampers definitive conclusion onthe impact of laronidase on the long-term disease pro-gression. Comparisons from the University of Minnesotadatabase suggest little difference early in cognitive de-velopment from HSCT (E. Shapiro, PhD, written com-munication, 2006). A group of 15 children who had MPSI and underwent HSCT and were the same age and hadthe same gender distribution had the same median de-velopmental quotient of 65 as observed in our studygroup after 1 year of follow-up. Both groups were betterthan untreated children from the same database whosemedian developmental quotient was 50 at a median ageof 3.3 years. Longer term follow-up, particularly of theyoung patients with Hurler syndrome in this study, willclarify the cognitive development trajectory of patientswho have Hurler syndrome and are treated with ERT.
The age range of the patients commanded choice ofsome nonquantitative explorative efficacy measures.Most, if not all, patients would not have been able tocooperate with sophisticated testing methods (eg, forcedvital capacity, 6-minute walk test) as used in previoustrials. In addition, the high risks that are related toanesthesia precluded the use of MRI. Because of thesmall sample size and variability, it was not possible toestablish correlations between urinary glycosaminogly-
can reductions and improvements in clinical end pointsin this trial.
CONCLUSIONSLaronidase seems to be well tolerated and clinically ben-eficial in severely affected patients who have MPS I andare younger than 5 years. ERT is not curative and maynot improve all affected organs and systems in individ-uals when irreversible changes have developed. Evi-dence of clinical benefit is provided by improvements inhepatomegaly, sleep disorder, left ventricular mass,growth velocity, mental development, and the investi-gator’s global assessment after 1 year of laronidase ther-apy. The impact on serious morbidity and mortality willrequire the long-term observation of a larger number ofpatients. Such data are best collected through the MPS IRegistry, which is gathering data on all patients acrossthe spectrum regardless of treatment status to under-stand better the natural history of the disease and todetermine the long-term impact of ERT.
The age at diagnosis, the severity of existing symp-toms, the expected disease progression, and the avail-ability of a donor in the case of transplantation arefactors to take in account when discussing treatmentoptions with parents. In this trial, all parents and guard-ians were fully informed of the risk/benefit profiles ofHSCT and laronidase. A variety of reasons led parents tochoose ERT for their children who were younger than 5years: delayed diagnosis (after 2 years of age), significantdevelopmental delay, lack of a compatible HSCT donor,non-Hurler phenotype (no expected neurocognitive re-gression), and concern over the safety risk of transplan-tation. MPS I is a rapidly progressive disorder; as forother lysosomal disorders, initiation of treatment asearly as possible in the disease course is required toprevent and/or minimize irreversible damage.
ACKNOWLEDGMENTSThis study was sponsored by BioMarin PharmaceuticalInc and Genzyme Corp.
We acknowledge the participation of study patientsand their families and the expert assistance of all study-site coordinators and personnel.
REFERENCES1. Neufeld E, Muenzer J. The mucopolysaccharidoses. In: Scriver
C, Beaudet A, Sly W, Valle D, eds. The Metabolic & MolecularBases of Inherited Disease. New York, NY: McGraw-Hill; 2001:3421–3452
2. Roubicek M, Gehler J, Spranger J. The clinical spectrum ofalpha-L-iduronidase deficiency. Am J Med Genet. 1985;20:471–481
3. Muenzer J, Fisher A. Advances in the treatment of mucopoly-saccharidosis type I. N Engl J Med. 2004;350:1932–1934
4. Kakkis ED, Muenzer J, Tiller GE, et al. Enzyme-replacementtherapy in mucopolysaccharidosis I. N Engl J Med. 2001;344:182–188
5. Wraith JE, Clarke LA, Beck M, et al. Enzyme replacement
PEDIATRICS Volume 120, Number 1, July 2007 e45 by guest on June 9, 2013pediatrics.aappublications.orgDownloaded from

therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombi-nant human alpha-L-iduronidase (laronidase). J Pediatr. 2004;144:581–588
6. Wraith JE. The first 5 years of clinical experience with laroni-dase enzyme replacement therapy for mucopolysaccharidosis I.Expert Opin Pharmacother. 2005;6:489–506
7. Richards SM, Olson TA, McPherson JM. Antibody response inpatients with Gaucher disease after repeated infusion withmacrophage-targeted glucocerebrosidase. Blood. 1993;82:1402–1409
8. Hopwood JJ, Muller V. Diagnostic enzymology of alpha-L-iduronidase with special reference to a sulphated disaccharidederived from heparin. Clin Sci (Lond). 1982;62:193–201
9. Colan SD, Parness IA, Spevak PJ, Sanders SP. Developmentalmodulation of myocardial mechanics: age- and growth-relatedalterations in afterload and contractility. J Am Coll Cardiol.1992;19:619–629
10. Leighton SE, Papsin B, Vellodi A, Dinwiddie R, Lane R. Disor-dered breathing during sleep in patients with mucopolysaccha-ridoses. Int J Pediatr Otorhinolaryngol. 2001;58:127–138
11. Gonsalez S, Hayward R, Jones B, Lane R. Upper airway ob-struction and raised intracranial pressure in children with cra-niosynostosis. Eur Respir J. 1997;10:367–375
12. Ogden CL, Kuczmarski RJ, Flegal KM, et al. Centers for DiseaseControl and Prevention 2000 growth charts for the UnitedStates: improvements to the 1977 National Center for HealthStatistics version. Pediatrics. 2002;109:45–60
13. Kuczmarski RJ, Ogden CL, Guo SS, et al. CDC growth charts
for the United States: methods and development. Vital HealthStat 11. 2002;(246):1–190
14. Pastores G. The MPS I Registry: centralized data collection todelineate the natural history and treatment of MPS I [abstract].Presented at: American Society of Human Genetics 54th An-nual Meeting; October 26–30, 2004; Toronto, Ontario, Canada
15. Griffiths R. The Abilities of Young Children. London, England:Child Development Research Centre; 1970
16. Souillet G, Guffon N, Maire I, et al. Outcome of 27 patientswith Hurler’s syndrome transplanted from either related orunrelated haematopoietic stem cell sources. Bone MarrowTransplant. 2003;31:1105–1117
17. Mohan UR, Hay AA, Cleary MA, Wraith JE, Patel RG. Cardio-vascular changes in children with mucopolysaccharide disor-ders. Acta Paediatr. 2002;91:799–804
18. Vinallonga X, Sanz N, Balaguer A, Miro L, Ortega JJ, CasaldaligaJ. Hypertrophic cardiomyopathy in mucopolysaccharidoses: re-gression after bone marrow transplantation. Pediatr Cardiol.1992;13:107–109
19. Gatzoulis MA, Vellodi A, Redington AN. Cardiac involvementin mucopolysaccharidoses: effects of allogeneic bone marrowtransplantation. Arch Dis Child. 1995;73:259–260
20. Grewal SS, Wynn R, Abdenur JE, et al. Safety and efficacy ofenzyme replacement therapy in combination with hematopoi-etic stem cell transplantation in Hurler syndrome. Genet Med.2005;7:143–146
21. Vogler C, Levy B, Grubb JH, et al. Overcoming the blood-brainbarrier with high-dose enzyme replacement therapy in murinemucopolysaccharidosis VII. Proc Natl Acad Sci U S A. 2005;102:14777–14782
e46 WRAITH et al by guest on June 9, 2013pediatrics.aappublications.orgDownloaded from

DOI: 10.1542/peds.2006-2156; originally published online May 30, 2007;Pediatrics
Yong Xue, Emil D. Kakkis and Nathalie GuffonJ. Edmond Wraith, Michael Beck, Roderick Lane, Ans van der Ploeg, Elsa Shapiro,
-L-Iduronidase (Laronidase)αRecombinant Human and Are Younger Than 5 Years: Results of a Multinational Study of
Enzyme Replacement Therapy in Patients Who Have Mucopolysaccharidosis I
ServicesUpdated Information &
/peds.2006-2156.citationhttp://pediatrics.aappublications.org/content/early/2007/05/30including high resolution figures, can be found at:
References
/peds.2006-2156.citation#ref-list-1http://pediatrics.aappublications.org/content/early/2007/05/30at:This article cites 18 articles, 5 of which can be accessed free
Citations
/peds.2006-2156.citation#related-urlshttp://pediatrics.aappublications.org/content/early/2007/05/30This article has been cited by 7 HighWire-hosted articles:
Permissions & Licensing
mlhttp://pediatrics.aappublications.org/site/misc/Permissions.xhttables) or in its entirety can be found online at: Information about reproducing this article in parts (figures,
Reprints http://pediatrics.aappublications.org/site/misc/reprints.xhtml
Information about ordering reprints can be found online:
rights reserved. Print ISSN: 0031-4005. Online ISSN: 1098-4275.Grove Village, Illinois, 60007. Copyright © 2007 by the American Academy of Pediatrics. All and trademarked by the American Academy of Pediatrics, 141 Northwest Point Boulevard, Elkpublication, it has been published continuously since 1948. PEDIATRICS is owned, published, PEDIATRICS is the official journal of the American Academy of Pediatrics. A monthly
by guest on June 9, 2013pediatrics.aappublications.orgDownloaded from




















