
Multimodal fundus imaging in Best vitelliform macular dystrophy

Ku70 regulates Bax-mediated pathogenesis inlaminin-a2-deficient human muscle cells andmouse models of congenital muscular dystrophy
Vivek K. Vishnudas1,2 and Jeffrey Boone Miller1,2,3,�
1Neuromuscular Biology & Disease Group and 2Paul D. Wellstone Muscular Dystrophy Cooperative Research Center,
Boston Biomedical Research Institute, 64 Grove Street, Watertown, MA 02478, USA and 3Department of Neurology,
Harvard Medical School, Boston, MA 02115, USA
Received July 17, 2009; Revised and Accepted August 17, 2009
The severely debilitating disease Congenital Muscular Dystrophy Type 1A (MDC1A) is caused by mutations inthe gene encoding laminin-a2. Bax-mediated muscle cell death is a significant contributor to the severe neu-romuscular pathology seen in the Lama2-null mouse model of MDC1A. To extend our understanding ofpathogenesis due to laminin-a2-deficiency, we have now analyzed molecular mechanisms of Bax regulationin normal and laminin-a2-deficient muscles and cells, including myogenic cells obtained from patients with aclinical diagnosis of MDC1A. In mouse myogenic cells, we found that, as in non-muscle cells, Bax co-immu-noprecipitated with the multifunctional protein Ku70. In addition, cell permeable pentapeptides designedfrom Ku70, termed Bax-inhibiting peptides (BIPs), inhibited staurosporine-induced Bax translocation andcell death in mouse myogenic cells. We also found that acetylation of Ku70, which can inhibit binding toBax and can be an indicator of increased susceptibility to cell death, was more abundant in Lama2-nullthan in normal mouse muscles. Furthermore, myotubes formed in culture from human laminin-a2-deficientpatient myoblasts produced high levels of activated caspase-3 when grown on poly-L-lysine, but not whengrown on a laminin-a2-containing substrate or when treated with BIPs. Finally, cytoplasmic Ku70 inhuman laminin-a2-deficient myotubes was both reduced in amount and more highly acetylated than innormal myotubes. Increased susceptibility to cell death thus appears to be an intrinsic property of humanlaminin-a2-deficient myotubes. These results identify Ku70 as a regulator of Bax-mediated pathogenesisand a therapeutic target in laminin-a2-deficiency.
INTRODUCTION
Aberrant activation of a mitochondrial cell death pathway is asignificant contributor to pathogenesis in mouse models ofseveral neuromuscular diseases including laminin-a2-deficiency (Congenital Muscular Dystrophy Type 1A;MDC1A), Oculopharyngeal Muscular Dystrophy (OPMD),Collagen VI-deficiency (Bethlem or Ullrich Congenital Mus-cular Dystrophy; BCMD, UCMD) and Spinal MuscularAtrophy (SMA) (1–8). In previous studies, for example, weshowed that inhibiting the mitochondrial cell death pathwaythrough genetic or pharmacological interventions decreasespathology and more than doubles lifespan inlaminin-a2-deficient (Lama22/2) mice which are a model
for human MDC1A (4,5,9). Because anti-cell death strategiesameliorate pathology in models of several skeletal muscle dis-eases, it may be that similar molecular mechanisms underlieexcessive activation of cell death pathways in pathogenesis.If so, a single therapeutic approach could be of benefit in mul-tiple diseases.
Further development of anti-cell death therapeutic strategiesfor neuromuscular diseases will benefit from additional under-standing of cell death pathways in diseased skeletal muscles.From previous studies which showed that inactivation of thepro-apoptotic protein Bax is sufficient to significantly decreasepathology in mouse models of MDC1A and SMA (4,8), itappeared that Bax is a key promoter of aberrant cell deathin at least these two disease models. The mechanisms that
�To whom correspondence should be addressed. Tel: þ1 6176587737; Fax: þ1 6179721761; Email: [email protected]
# The Author 2009. Published by Oxford University Press. All rights reserved.For Permissions, please email: [email protected]
Human Molecular Genetics, 2009, Vol. 18, No. 23 4467–4477doi:10.1093/hmg/ddp399Advance Access published on August 19, 2009
by guest on October 1, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

lead to Bax activation and cell death in neuromuscular dis-eases such as laminin-a2-deficiency remain, however, incom-pletely understood. In this work, therefore, we have examinedmolecular mechanisms that regulate Bax and cell death innormal and laminin-a2-deficient skeletal muscles and cells.
The most common form of MDC1A is caused by mutation ofthe human LAMA2 (mouse Lama2) gene that encodeslaminin-a2 (10–11). Loss of laminin-a2 function in this auto-somal recessive disease results in severe neuromuscular dys-function. Partial laminin-a2-deficiency is found in some otherdiseases, including Muscle–Eye–Brain disease, FukuyamaCongenital Muscular Dystrophy, and Walker-Warburg Syn-drome, though these diseases are caused by primary mutationsin genes other than LAMA2 (2). In skeletal muscles, laminin-a2assembles with laminin-b1 and -g1 to form laminin-211. Het-erotrimeric laminins that include laminin-a2 have beentermed merosins, and MDC1A has thus also been known asmerosin-deficient congenital muscular dystrophy. Laminin-a2has binding partners in both the extracellular matrix—entactin/nidogen, which in turn binds to collagen IV—and on the plasmamembrane—a-dystroglycan and a7-integrin (1).Laminin-a2-deficiency causes aberrant activation of Bax-mediated muscle cell death (4,5,12–13).
Recent work in non-skeletal muscle cells has shown thatinteraction of Bax with the Ku70 protein is an additionalmechanism to regulate cell death (14–17). Under normal con-ditions, Ku70 binds to Bax, and Bax is thereby retained in thecytoplasm in an inactive form. In response to a pro-deathsignal, however, acetylation of one or more critical lysineson Ku70 occurs and this modification inhibits Ku70 bindingand frees Bax for translocation to mitochondria (18). Ku70is acetylated by CBP and PCAF and deacetylated by SIRT1,SIRT3 and probably other deacetylases (14,18,19). Ku70 is amultifunctional protein that is found in both nuclei, where itparticipates in DNA break repair, and in the cytoplasm (15).Ku70 also appears to act as a Bax deubiquitinase (20). Todetermine if Ku70 regulates Bax-mediated pathogenesis inlaminin-a2-deficiency, we have now analyzed the Ku70/Baxpathway in normal and laminin-a2-deficient myogenic cellsand tissues. Taken together, our results identify Ku70 as a reg-ulator of Bax-mediated pathogenesis in laminin-a2-deficiencyand the Bax/Ku70 pathway as a therapeutic target inlaminin-a2-deficiency.
RESULTS
To analyze the role of Ku70 in normal and diseased skeletalmuscles, we studied systems with complementary experimen-tal advantages, including cells of the mouse C2C12 myogeniccell line; primary cells derived from muscles of wild-typeand genetically-modified mice; muscle tissues fromLama22/2 mice; and human myogenic cells obtained bothfrom healthy individuals and from three patients with a clini-cal diagnosis of MDC1A (Table 1 and Materials andMethods). Use of the mouse-derived materials circumventedthe experimental limits imposed by the limited mitotic capa-bility of the human cells and the lack of human musclebiopsy material. We first used immunoblots to demonstratethat Ku70 was expressed in both the cytoplasm and nuclei
of cultured primary mouse myoblasts (Fig. 1A). Less Ku70appeared to be in the cytoplasm in Bax-null than in wild-typecells, a difference which is consistent with the possibility thatthe cytoplasmic Ku70 was more rapidly degraded or translo-cated to the nucleus when binding to Bax was not possible.
We next used combined immunoprecipitation and immuno-blotting to demonstrate that Ku70 and Bax interact in mouseskeletal muscle tissue (Fig. 1B). Extracts of 6- or12-week-old mouse soleus muscles were immunoprecipitatedwith an antibody specific for Bax. Subsequently, the immuno-precipitated proteins were subjected to SDS-PAGE and immu-noblotting with antibodies specific for Ku70 or Bax. Ku70 wasfound with Bax in the precipitates, showing that Ku70 inter-acted with Bax either most likely by direct binding, as foundin other cell types, or indirectly as part of a multi-subunitcomplex.
To determine if laminin-a2-deficiency altered the interactionof Bax and Ku70, we compared the amounts of Ku70 thatco-immunoprecipitated with Bax from wild-type andLama22/2 mouse muscle extracts. We found a consistentdecrease in the amount of Ku70 that was co-immunoprecipitatedwith Bax from extracts of Lama2/2 soleus muscle comparedwith wild-type (Fig. 1B and C). This result suggests that asmaller percentage of Bax is in a complex with Ku70 inLama2/2 than in wild-type muscles. Because dissociation ofKu70 from Bax is associated with increased cell death, thisresult is consistent with the high levels of cell death previouslyfound in Lama2/2 mouse muscles (5).
We next used Bax-inhibiting peptides (BIPs) to examine therole of Ku70 in muscle cell death. BIPs are simple pentapep-tides with a sequence from the region of Ku70 that binds toBax. Previous work in non-muscle cells has shown that BIPscan bind to Bax and can inhibit apoptotic cell death by inhibit-ing Bax activation and translocation to mitochondria(15,16,21). We synthesized BIPs based on mouse and humanKu70 sequences either as the simple pentapeptide or with anovel modified structure that included fluorescein isothiocya-nate (FITC, to demonstrate cell permeability) linked bybeta-alanine to the BIP pentapeptide sequences. Thus, themouse-specific BIP (mBIP) was the simple pentapeptideVPTLK and the fluorescent version (fl-mBIP) wasFITC-ßA-VPTLK, whereas the human-specific BIP (hBIP)was PMLKE and the fluorescent version (fl-hBIP) wasFITC-ßA-PMLKE. A pentapeptide, IPMIK that does nothave Bax-inhibiting activity was similarly prepared with orwithout the FITC tag for use as a control. FITC-tagged BIPswere used for the short-term experiments described inFigures 2 and 3, whereas untagged BIPs were used for thelonger duration experiments described in Figures 6 and 7.
Our initial experiments showed that treatment with fl-mBIP,when compared with treatment with control peptide, signifi-cantly decreased the percentage of C2C12 myoblasts withabnormally-shaped nuclei (i.e. condensed, fragmented orwith an irregular or blebbed perimeter) that were induced bystaurosporine (Fig. 2). Staurosporine is a non-selectivekinase inhibitor that induces Bax-mediated apoptotic celldeath (cf. 22). We previously showed that induction by staur-osporine of such nuclear abnormalities correlates with theinduction of Annexin V staining and subsequent cell death(5). In our earlier study, we showed that myoblasts from
4468 Human Molecular Genetics, 2009, Vol. 18, No. 23
by guest on October 1, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

Bax-null mice were more resistant to staurosporine than myo-blasts from Baxþ/þ wild-type mice (22). Consistent withthat study, we found that nuclear abnormalities induced by
staurosporine in C2C12 myoblasts, which are Bax-positive,was likely Bax-dependent, because staurosporine treatmentinduced a significantly higher percentage of abnormal nucleiin C2C12 myoblasts than in Bax-null primary myoblasts(Fig. 2C). All cells in fl-mBIP-treated cultures showed cyto-plasmic FITC fluorescence, indicating that the peptidesentered cells (not shown). Treatment with fl-mBIP also inhib-ited staurosporine-induced abnormalities in C2C12 myotubes(not shown).
Figure 1. Ku70 expression in normal and diseased muscle cells and tissue.(A) In primary cultures of mouse myoblasts, Ku70 was found in bothnuclear and cytoplasmic fractions, with relatively less Ku70 in the cytoplasmof Bax-null than in wild-type cells. Primary cultures were established withmyoblasts prepared from lower hindlimb muscles of mice with normal Bax(Baxþ/þ) or mice that were Bax-null (Bax2/2). Ku70 expression was analyzedby immunoblot (IB) in cellular fractions enriched for nuclear (Nuc) or cyto-plasmic (Cyt) proteins. (B) In extracts of mouse hindlimb muscles, antibodiesspecific for Bax also co-immunoprecipitated Ku70, indicating that the Bax andKu70 interact. Extracts of mouse soleus muscles were prepared and immuno-precipitated (IP) with an antibody specific for Bax. The immunoprecipitateswere analyzed by SDS-PAGE and immunoblotting (IB) with antibodiesspecific to Ku70 or Bax as indicated. (C) Less Ku70 wasco-immunoprecipitated with Bax from extracts of Lama2/2 muscles thanfrom extracts of wild-type mouse muscles. Graph shows quantification ofimmunoprecipitations performed as in (B). Error bars ¼ range, n ¼ 2.
Figure 2. Mouse Bax-inhibiting peptide (mBIP) inhibitedstaurosporine-induced nuclear abnormalities in C2C12 cells. (A) After 6 htreatment with 0.1 mM staurosporine (STS), a large percentage of the nucleiin cultures of C2C12 myoblasts showed abnormal morphology indicative ofincipient cell death (5). (B) In contrast, when 200 mM mBIP was addedwith the staurosporine, a lower percentage of nuclei had abnormal mor-phology. (C) Quantitative analyses showed that the percentage of abnormalnuclei was high after 6 h of staurosporine treatment (þSTS), whereas thenumber of abnormal nuclei was lower when mBIP (þmBIP), but not200 mM control peptide (þcontP), was added with the staurosporine. Similarresults were obtained with differentiated C2C12 myotubes (not shown). Inaddition, primary cultures of mouse Bax-null (Bax2/2) myoblasts were resist-ant to staurosporine, showing that staurosporine-induced nuclear abnormalitieswere Bax-dependent. �, P , 0.05; ��, P , 0.01; n.s., not significant. Errorbars ¼ SE, n ¼ 3. Bar in A ¼ 10 mm.
Table 1. Clinical data on laminin-a2-deficient and normal human myoblast donors
Sample ID (Diagnosis) Laminin-a2a Gender (Age at biopsy) Serum Creatine Kinase (Units/l) (Normal , 175) Clinical observations
38/03 (MDC1A) Absent Male (4 months) 1000 HypotoniaNo respiratory distressNormal mental stage
96/04 (MDC1A) Absent Male (8 months) 1700 Hypotonia9/03 (MDC1A) Absent Female (12 months) 4000 Hypotonia
No respiratory distress363/07 (Normal) Normal Male (21 years) Normal Normal2/08 (Normal) Normal Male (36 years) Normal Normal
aLaminin-a2 expression determined by immunohistochemistry or immunoblotting.
Human Molecular Genetics, 2009, Vol. 18, No. 23 4469
by guest on October 1, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from
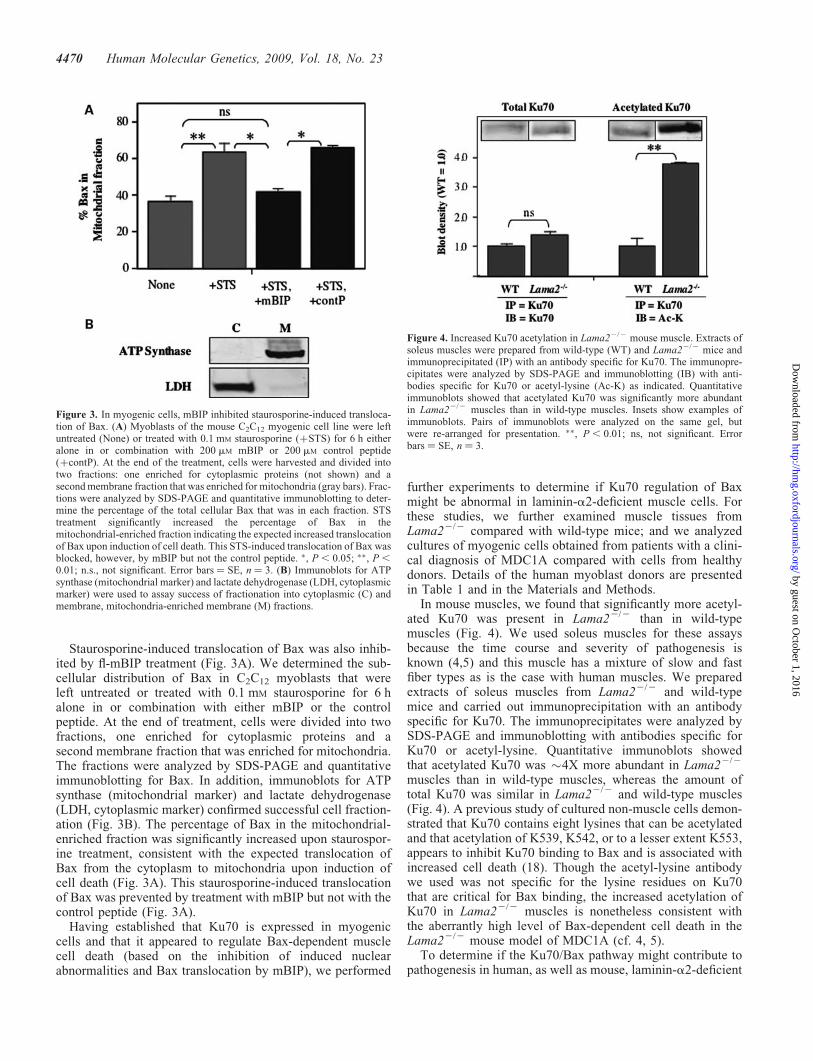
Staurosporine-induced translocation of Bax was also inhib-ited by fl-mBIP treatment (Fig. 3A). We determined the sub-cellular distribution of Bax in C2C12 myoblasts that wereleft untreated or treated with 0.1 mM staurosporine for 6 halone in or combination with either mBIP or the controlpeptide. At the end of treatment, cells were divided into twofractions, one enriched for cytoplasmic proteins and asecond membrane fraction that was enriched for mitochondria.The fractions were analyzed by SDS-PAGE and quantitativeimmunoblotting for Bax. In addition, immunoblots for ATPsynthase (mitochondrial marker) and lactate dehydrogenase(LDH, cytoplasmic marker) confirmed successful cell fraction-ation (Fig. 3B). The percentage of Bax in the mitochondrial-enriched fraction was significantly increased upon staurospor-ine treatment, consistent with the expected translocation ofBax from the cytoplasm to mitochondria upon induction ofcell death (Fig. 3A). This staurosporine-induced translocationof Bax was prevented by treatment with mBIP but not with thecontrol peptide (Fig. 3A).
Having established that Ku70 is expressed in myogeniccells and that it appeared to regulate Bax-dependent musclecell death (based on the inhibition of induced nuclearabnormalities and Bax translocation by mBIP), we performed
further experiments to determine if Ku70 regulation of Baxmight be abnormal in laminin-a2-deficient muscle cells. Forthese studies, we further examined muscle tissues fromLama22/2 compared with wild-type mice; and we analyzedcultures of myogenic cells obtained from patients with a clini-cal diagnosis of MDC1A compared with cells from healthydonors. Details of the human myoblast donors are presentedin Table 1 and in the Materials and Methods.
In mouse muscles, we found that significantly more acetyl-ated Ku70 was present in Lama22/2 than in wild-typemuscles (Fig. 4). We used soleus muscles for these assaysbecause the time course and severity of pathogenesis isknown (4,5) and this muscle has a mixture of slow and fastfiber types as is the case with human muscles. We preparedextracts of soleus muscles from Lama22/2 and wild-typemice and carried out immunoprecipitation with an antibodyspecific for Ku70. The immunoprecipitates were analyzed bySDS-PAGE and immunoblotting with antibodies specific forKu70 or acetyl-lysine. Quantitative immunoblots showedthat acetylated Ku70 was �4X more abundant in Lama22/2
muscles than in wild-type muscles, whereas the amount oftotal Ku70 was similar in Lama22/2 and wild-type muscles(Fig. 4). A previous study of cultured non-muscle cells demon-strated that Ku70 contains eight lysines that can be acetylatedand that acetylation of K539, K542, or to a lesser extent K553,appears to inhibit Ku70 binding to Bax and is associated withincreased cell death (18). Though the acetyl-lysine antibodywe used was not specific for the lysine residues on Ku70that are critical for Bax binding, the increased acetylation ofKu70 in Lama22/2 muscles is nonetheless consistent withthe aberrantly high level of Bax-dependent cell death in theLama22/2 mouse model of MDC1A (cf. 4, 5).
To determine if the Ku70/Bax pathway might contribute topathogenesis in human, as well as mouse, laminin-a2-deficient
Figure 3. In myogenic cells, mBIP inhibited staurosporine-induced transloca-tion of Bax. (A) Myoblasts of the mouse C2C12 myogenic cell line were leftuntreated (None) or treated with 0.1 mM staurosporine (þSTS) for 6 h eitheralone in or combination with 200 mM mBIP or 200 mM control peptide(þcontP). At the end of the treatment, cells were harvested and divided intotwo fractions: one enriched for cytoplasmic proteins (not shown) and asecond membrane fraction that was enriched for mitochondria (gray bars). Frac-tions were analyzed by SDS-PAGE and quantitative immunoblotting to deter-mine the percentage of the total cellular Bax that was in each fraction. STStreatment significantly increased the percentage of Bax in themitochondrial-enriched fraction indicating the expected increased translocationof Bax upon induction of cell death. This STS-induced translocation of Bax wasblocked, however, by mBIP but not the control peptide. �, P , 0.05; ��, P ,
0.01; n.s., not significant. Error bars ¼ SE, n ¼ 3. (B) Immunoblots for ATPsynthase (mitochondrial marker) and lactate dehydrogenase (LDH, cytoplasmicmarker) were used to assay success of fractionation into cytoplasmic (C) andmembrane, mitochondria-enriched membrane (M) fractions.
Figure 4. Increased Ku70 acetylation in Lama22/2 mouse muscle. Extracts ofsoleus muscles were prepared from wild-type (WT) and Lama22/2 mice andimmunoprecipitated (IP) with an antibody specific for Ku70. The immunopre-cipitates were analyzed by SDS-PAGE and immunoblotting (IB) with anti-bodies specific for Ku70 or acetyl-lysine (Ac-K) as indicated. Quantitativeimmunoblots showed that acetylated Ku70 was significantly more abundantin Lama22/2 muscles than in wild-type muscles. Insets show examples ofimmunoblots. Pairs of immunoblots were analyzed on the same gel, butwere re-arranged for presentation. ��, P , 0.01; ns, not significant. Errorbars ¼ SE, n ¼ 3.
4470 Human Molecular Genetics, 2009, Vol. 18, No. 23
by guest on October 1, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

muscle cells, we compared cultures of proliferating and differ-entiated myogenic cells that were obtained from three inde-pendent patients with a clinical diagnosis of MDC1A tosimilar cultures of myoblasts obtained from healthy humandonors (Materials and Methods; Table 1). Our initial charac-terization confirmed that the patient cells were myogenicand had the expected laminin-a2-deficient phenotype(Fig. 5). More than 90% of the cells in low density, proliferat-ing cultures of both the wild-type and laminin-a2-deficientcells were desmin-positive myoblasts (Fig. 5A and D).When grown on a poly-L-lysine substrate, both the normalhuman and the patients’ laminin-a2-deficient myoblastswere capable of forming multinucleate myotubes thatexpressed myosin heavy chain within 3–4 days after switchingto low serum differentiation medium (Fig. 5B and E). Finally,laminin-a2 was expressed by myotubes formed from wild-type human myoblasts, but, as would be expected for patientswith MDC1A, laminin-a2 was not detectable in myotubesformed from each of the three patient lines (Fig. 5C and Fand not shown). Though the clinical observations, includingearly onset and apparently complete lack of laminin-a2protein, support the MDC1A diagnosis, we currently havemutation information for only one of the three lines (seeMaterials and Methods) and causality still needs to be defini-tively established. Thus, we will refer to the human cells aslaminin-a2-deficient, though we expect that they are frombona fide MDC1A patients.
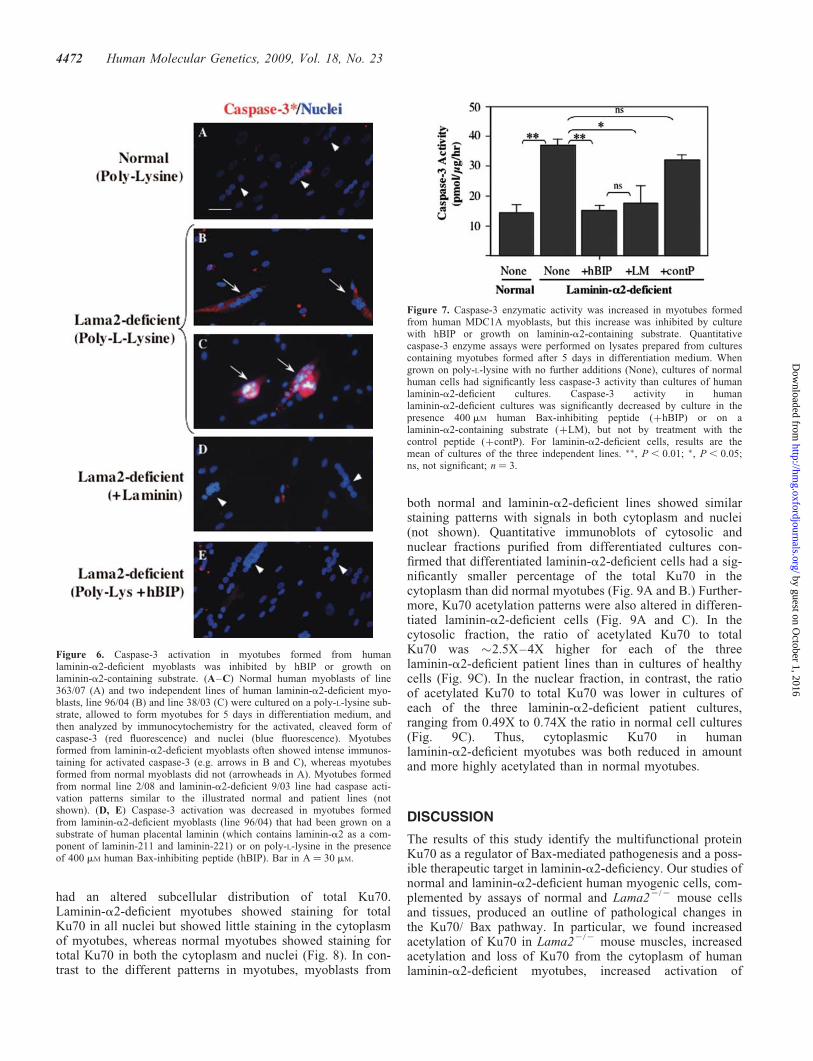
Though human laminin-a2-deficient myoblasts formedmyotubes when cultured on poly-L-lysine, these myotubesexhibited several abnormalities indicative of instability andincreased susceptibility to cell death. First, we noted thatmany of the laminin-a2-deficient myotubes, but not normalmyotubes, became rounded and began to detach from thedish after 4 or 5 days in differentiation medium. Second,immunoprecipitation and immunoblotting analyses of Ku70expression and acetylation similar to those in Figure 4showed that the ratio of acetylated Ku70 to total Ku70 was2.8+ 0.55 (Ave.+SE, n ¼ 3) times higher in cultures oflaminin-a2-deficient myotubes than in cultures of normalmyotubes. This increase in acetylated Ku70 in thelaminin-a2-deficient human myotubes was similar in magni-tude to that found in the Lama22/2 mouse muscles (Fig. 4)Third, Annexin V bound to many laminin-a2-deficient myo-tubes, but not to normal myotubes, which is consistent withongoing cell death of laminin-a2-deficient myotubes (notshown). Finally, laminin-a2-deficient myotubes, but notnormal myotubes, showed high levels of activated caspase-3(a marker for incipient cell death) at 4–5 days after theywere formed (Figs 6 and 7). After 5 days in differentiationmedium, for example, we found that many of the myotubesformed from human laminin-a2-deficient myoblasts showedintense immunostaining for activated caspase-3 (Fig. 6B andC), whereas myotubes formed from wild-type myoblasts didnot (Fig. 6A). Myotubes formed from each of the three avail-able lines of patient myoblasts (which were obtained fromthree individual donors) showed similar patterns of intenseimmunostaining for activated caspase-3 when grown onpoly-L-lysine (Fig. 6B and C, and not shown). Quantitativemeasurements of caspase-3 enzyme activity also demonstratedthat caspase-3 enzymatic activity was significantly higher indifferentiated laminin-a2-deficient than in normal cultures(Fig. 7). (N.B., because Lama22/2 mouse myoblasts did notadhere to poly-L-lysine, we could not carry out a similar exper-iment with primary cultures of mouse myogenic cells.)
The increased level of activated caspase-3 seen inlaminin-a2-deficient myotubes was not found, however,when the patients’ myogenic cells were either grown on a sub-strate that contained laminin-a2 or were treated with thehuman Bax-inhibiting peptide (hBIP) (Figs 6D, E and 7). Todetermine if provision of laminin-a2 could preventcaspase-3 activation, we cultured patient cells on a substrateof human placental laminin, which contains laminin-a2 as asubunit of laminin-211 and laminin-221. Both immunostainingand enzyme assays showed that differentiated laminin-a2-deficient cultures grown on the human placental lamininsubstrate had approximately the same caspase-3 levels as wild-type cells, which was significantly less than in thelaminin-a2-deficient cultures grown on poly-L-lysine(Figs 6D and 7). Similarly, differentiated laminin-a2-deficientcultures grown on poly-L-lysine in the absence laminin-a2 butin the presence of hBIP had about the same caspase-3 levels aswild-type cells, which was significantly less than inlaminin-a2-deficient cultures grown similarly in the absenceof hBIP (Figs 6E and 7).
When we examined Ku70 expression by immunostaining,we found that, when compared with normal myotubes, myo-tubes formed from the laminin-a2-deficient patient myoblasts
Figure 5. Myotubes formed from myoblasts obtained from donors with a diag-nosis of MDC1A did not express laminin-a2. Myoblasts obtained fromhumans with normal laminin-a2 expression and from patients with a clinicaldiagnosis of MDC1A were cultured and analyzed at different stages of differ-entiation for muscle markers. (A, D) At low density in proliferation medium,.90% of both normal and MDC1A diagnosis cells expressed desmin asexpected for myoblasts. (B, E) After 4 days in differentiation medium on apoly-L-lysine substrate, both normal and MDC1A diagnosis myoblastsformed multinucleate myotubes that expressed myosin heavy chain(MyHC). (C, F) Laminin-a2 was expressed by myotubes formed fromnormal myoblasts (examples indicated by arrows in C) but not by myotubesformed from MDC1A diagnosis myoblasts (examples indicated by arrowheadsin F). Bar in F ¼ 25 mM.
Human Molecular Genetics, 2009, Vol. 18, No. 23 4471
by guest on October 1, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from
had an altered subcellular distribution of total Ku70.Laminin-a2-deficient myotubes showed staining for totalKu70 in all nuclei but showed little staining in the cytoplasmof myotubes, whereas normal myotubes showed staining fortotal Ku70 in both the cytoplasm and nuclei (Fig. 8). In con-trast to the different patterns in myotubes, myoblasts from
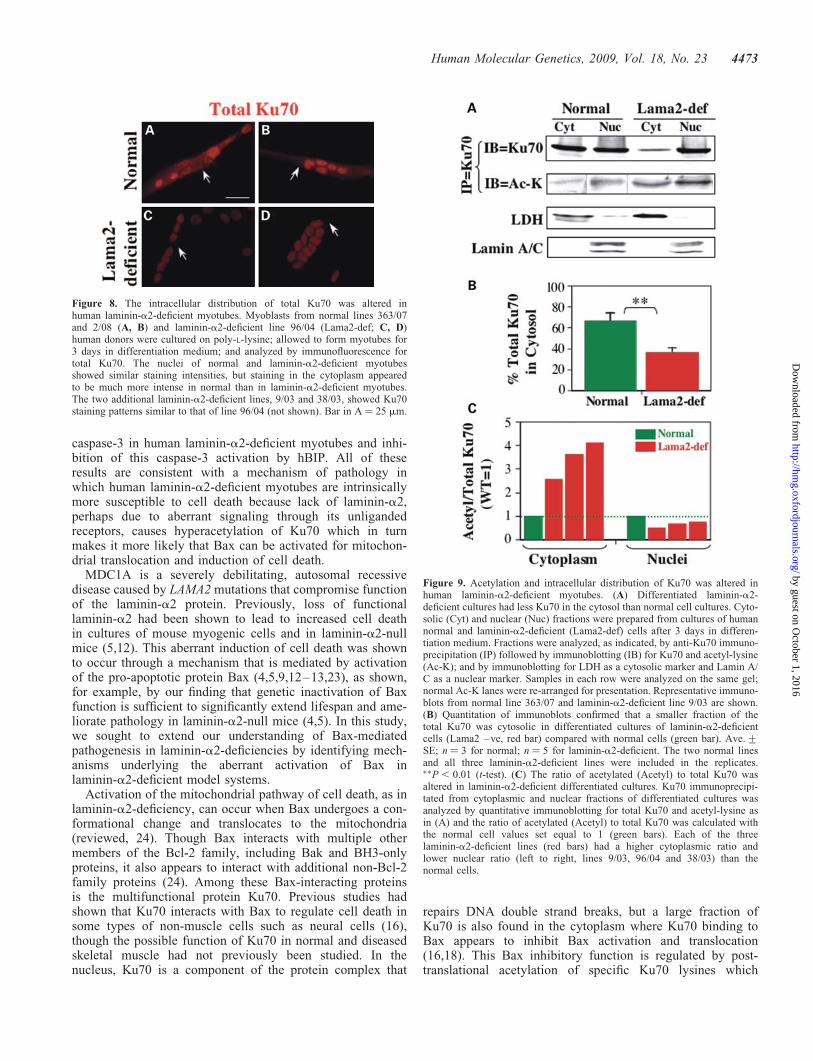
both normal and laminin-a2-deficient lines showed similarstaining patterns with signals in both cytoplasm and nuclei(not shown). Quantitative immunoblots of cytosolic andnuclear fractions purified from differentiated cultures con-firmed that differentiated laminin-a2-deficient cells had a sig-nificantly smaller percentage of the total Ku70 in thecytoplasm than did normal myotubes (Fig. 9A and B.) Further-more, Ku70 acetylation patterns were also altered in differen-tiated laminin-a2-deficient cells (Fig. 9A and C). In thecytosolic fraction, the ratio of acetylated Ku70 to totalKu70 was �2.5X–4X higher for each of the threelaminin-a2-deficient patient lines than in cultures of healthycells (Fig. 9C). In the nuclear fraction, in contrast, the ratioof acetylated Ku70 to total Ku70 was lower in cultures ofeach of the three laminin-a2-deficient patient cultures,ranging from 0.49X to 0.74X the ratio in normal cell cultures(Fig. 9C). Thus, cytoplasmic Ku70 in humanlaminin-a2-deficient myotubes was both reduced in amountand more highly acetylated than in normal myotubes.
DISCUSSION
The results of this study identify the multifunctional proteinKu70 as a regulator of Bax-mediated pathogenesis and a poss-ible therapeutic target in laminin-a2-deficiency. Our studies ofnormal and laminin-a2-deficient human myogenic cells, com-plemented by assays of normal and Lama22/2 mouse cellsand tissues, produced an outline of pathological changes inthe Ku70/ Bax pathway. In particular, we found increasedacetylation of Ku70 in Lama22/2 mouse muscles, increasedacetylation and loss of Ku70 from the cytoplasm of humanlaminin-a2-deficient myotubes, increased activation of
Figure 6. Caspase-3 activation in myotubes formed from humanlaminin-a2-deficient myoblasts was inhibited by hBIP or growth onlaminin-a2-containing substrate. (A–C) Normal human myoblasts of line363/07 (A) and two independent lines of human laminin-a2-deficient myo-blasts, line 96/04 (B) and line 38/03 (C) were cultured on a poly-L-lysine sub-strate, allowed to form myotubes for 5 days in differentiation medium, andthen analyzed by immunocytochemistry for the activated, cleaved form ofcaspase-3 (red fluorescence) and nuclei (blue fluorescence). Myotubesformed from laminin-a2-deficient myoblasts often showed intense immunos-taining for activated caspase-3 (e.g. arrows in B and C), whereas myotubesformed from normal myoblasts did not (arrowheads in A). Myotubes formedfrom normal line 2/08 and laminin-a2-deficient 9/03 line had caspase acti-vation patterns similar to the illustrated normal and patient lines (notshown). (D, E) Caspase-3 activation was decreased in myotubes formedfrom laminin-a2-deficient myoblasts (line 96/04) that had been grown on asubstrate of human placental laminin (which contains laminin-a2 as a com-ponent of laminin-211 and laminin-221) or on poly-L-lysine in the presenceof 400 mM human Bax-inhibiting peptide (hBIP). Bar in A ¼ 30 mM.
Figure 7. Caspase-3 enzymatic activity was increased in myotubes formedfrom human MDC1A myoblasts, but this increase was inhibited by culturewith hBIP or growth on laminin-a2-containing substrate. Quantitativecaspase-3 enzyme assays were performed on lysates prepared from culturescontaining myotubes formed after 5 days in differentiation medium. Whengrown on poly-L-lysine with no further additions (None), cultures of normalhuman cells had significantly less caspase-3 activity than cultures of humanlaminin-a2-deficient cultures. Caspase-3 activity in humanlaminin-a2-deficient cultures was significantly decreased by culture in thepresence 400 mM human Bax-inhibiting peptide (þhBIP) or on alaminin-a2-containing substrate (þLM), but not by treatment with thecontrol peptide (þcontP). For laminin-a2-deficient cells, results are themean of cultures of the three independent lines. ��, P , 0.01; �, P , 0.05;ns, not significant; n ¼ 3.
4472 Human Molecular Genetics, 2009, Vol. 18, No. 23
by guest on October 1, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from
caspase-3 in human laminin-a2-deficient myotubes and inhi-bition of this caspase-3 activation by hBIP. All of theseresults are consistent with a mechanism of pathology inwhich human laminin-a2-deficient myotubes are intrinsicallymore susceptible to cell death because lack of laminin-a2,perhaps due to aberrant signaling through its unligandedreceptors, causes hyperacetylation of Ku70 which in turnmakes it more likely that Bax can be activated for mitochon-drial translocation and induction of cell death.
MDC1A is a severely debilitating, autosomal recessivedisease caused by LAMA2 mutations that compromise functionof the laminin-a2 protein. Previously, loss of functionallaminin-a2 had been shown to lead to increased cell deathin cultures of mouse myogenic cells and in laminin-a2-nullmice (5,12). This aberrant induction of cell death was shownto occur through a mechanism that is mediated by activationof the pro-apoptotic protein Bax (4,5,9,12–13,23), as shown,for example, by our finding that genetic inactivation of Baxfunction is sufficient to significantly extend lifespan and ame-liorate pathology in laminin-a2-null mice (4,5). In this study,we sought to extend our understanding of Bax-mediatedpathogenesis in laminin-a2-deficiencies by identifying mech-anisms underlying the aberrant activation of Bax inlaminin-a2-deficient model systems.
Activation of the mitochondrial pathway of cell death, as inlaminin-a2-deficiency, can occur when Bax undergoes a con-formational change and translocates to the mitochondria(reviewed, 24). Though Bax interacts with multiple othermembers of the Bcl-2 family, including Bak and BH3-onlyproteins, it also appears to interact with additional non-Bcl-2family proteins (24). Among these Bax-interacting proteinsis the multifunctional protein Ku70. Previous studies hadshown that Ku70 interacts with Bax to regulate cell death insome types of non-muscle cells such as neural cells (16),though the possible function of Ku70 in normal and diseasedskeletal muscle had not previously been studied. In thenucleus, Ku70 is a component of the protein complex that
repairs DNA double strand breaks, but a large fraction ofKu70 is also found in the cytoplasm where Ku70 binding toBax appears to inhibit Bax activation and translocation(16,18). This Bax inhibitory function is regulated by post-translational acetylation of specific Ku70 lysines which
Figure 9. Acetylation and intracellular distribution of Ku70 was altered inhuman laminin-a2-deficient myotubes. (A) Differentiated laminin-a2-deficient cultures had less Ku70 in the cytosol than normal cell cultures. Cyto-solic (Cyt) and nuclear (Nuc) fractions were prepared from cultures of humannormal and laminin-a2-deficient (Lama2-def) cells after 3 days in differen-tiation medium. Fractions were analyzed, as indicated, by anti-Ku70 immuno-precipitation (IP) followed by immunoblotting (IB) for Ku70 and acetyl-lysine(Ac-K); and by immunoblotting for LDH as a cytosolic marker and Lamin A/C as a nuclear marker. Samples in each row were analyzed on the same gel;normal Ac-K lanes were re-arranged for presentation. Representative immuno-blots from normal line 363/07 and laminin-a2-deficient line 9/03 are shown.(B) Quantitation of immunoblots confirmed that a smaller fraction of thetotal Ku70 was cytosolic in differentiated cultures of laminin-a2-deficientcells (Lama2 –ve, red bar) compared with normal cells (green bar). Ave.+SE; n ¼ 3 for normal; n ¼ 5 for laminin-a2-deficient. The two normal linesand all three laminin-a2-deficient lines were included in the replicates.��P , 0.01 (t-test). (C) The ratio of acetylated (Acetyl) to total Ku70 wasaltered in laminin-a2-deficient differentiated cultures. Ku70 immunoprecipi-tated from cytoplasmic and nuclear fractions of differentiated cultures wasanalyzed by quantitative immunoblotting for total Ku70 and acetyl-lysine asin (A) and the ratio of acetylated (Acetyl) to total Ku70 was calculated withthe normal cell values set equal to 1 (green bars). Each of the threelaminin-a2-deficient lines (red bars) had a higher cytoplasmic ratio andlower nuclear ratio (left to right, lines 9/03, 96/04 and 38/03) than thenormal cells.
Figure 8. The intracellular distribution of total Ku70 was altered inhuman laminin-a2-deficient myotubes. Myoblasts from normal lines 363/07and 2/08 (A, B) and laminin-a2-deficient line 96/04 (Lama2-def; C, D)human donors were cultured on poly-L-lysine; allowed to form myotubes for3 days in differentiation medium; and analyzed by immunofluorescence fortotal Ku70. The nuclei of normal and laminin-a2-deficient myotubesshowed similar staining intensities, but staining in the cytoplasm appearedto be much more intense in normal than in laminin-a2-deficient myotubes.The two additional laminin-a2-deficient lines, 9/03 and 38/03, showed Ku70staining patterns similar to that of line 96/04 (not shown). Bar in A ¼ 25 mm.
Human Molecular Genetics, 2009, Vol. 18, No. 23 4473
by guest on October 1, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

prevent binding of Ku70 to Bax (18), thereby freeing Bax toundergo the activating conformational change and transloca-tion to mitochondria in response to pro-apoptotic signals.Because Bax-mediated cell death is a significant contributorto pathology in laminin-a2-deficiency, these previous findingsin non-muscle cells prompted us to examine the role of Ku70in normal and diseased skeletal muscle.
Our finding that BIPs can inhibit signs of induced and spon-taneous cell death provides strong evidence that Ku70 inter-action with Bax regulates cell death in normal and inlaminin-a2-deficient skeletal muscle cells. The BIPs weredesigned to bind to and prevent activation of Bax evenunder pro-cell death conditions when binding of Ku70 toBax is inhibited by acetylation (15,16,21). Here we foundthat appropriate species-specific BIPs inhibited both thestaurosporine-induced cell death in normal mouse myogeniccells and the aberrant activation of caspase-3 that occurs inhuman myotubes formed from laminin-a2-deficient patientmyoblasts in culture.
In addition to the BIP studies, our work provided additionalevidence that Ku70 regulates Bax-mediated cell death in dis-eased skeletal muscle. In particular, we found thatLama22/2 muscles and human laminin-a2-deficientmyotube cultures contained higher levels of acetylatedKu70, particularly in the cytoplasm, than did the correspond-ing normal muscles or myotube cultures. Furthermore, wefound that Ku70 co-immunoprecipitated with Bax fromextracts of mouse skeletal muscles, but that less Ku70co-immunoprecipitated with Bax from Lama22/2 thannormal muscles. The combination of a high level of Ku70acetylation with less binding of Ku70 to Bax would beexpected for tissues undergoing high levels of Bax-mediatedcell death, which is the case in Lama22/2 mouse muscles(4,5). Thus, the differences that we found between normaland Lama22/2 muscles in Ku70 acetylation and Baxbinding are consistent with a role for Ku70 in regulation ofBax-mediated cell death in skeletal muscles.
Our studies of human laminin-a2-deficient myogenic cellsin culture are the first to show that these cells recapitulatean important disease phenotype. Previous important studieswith laminin-a2-deficient variants of the mouse C2C12 myo-genic cell line showed that myotubes were unstable in theabsence of laminin-a2 and showed high levels of cell deaththrough the mitochondrial pathway (12–13,23). Thosestudies also identified kinase signaling pathways involved incell death induced by laminin-a2-deficiency and showed thatthe high levels of cell death could be inhibited by overexpres-sion of Bcl-2 (which inhibits Bax function) or by differen-tiation in the presence of laminin-a2. Here we showed that,when grown in the absence of laminin-a2, the humanlaminin-a2-deficient myoblasts similarly formed unstablemyotubes and that these myotubes developed high levels ofactivated caspase-3 after 4–5 days in differentiationmedium. The laminin-a2-deficient myotubes also showedincreased levels of acetylated Ku70 and Annexin V stainingcompared with normal myotubes in culture. Though lowlevels of activated caspase-3 have been reported to be requiredfor myogenic cell differentiation (25), the high levels of acti-vated caspase-3 in laminin-a2-deficient myotubes were clearlyabnormal. A complete absence of laminin-a2, as in the cells
from patients with a MDC1A diagnosis that we studied, maybe necessary to produce the abnormal myotube phenotypeswe observed. A previous study did not find abnormalities inmyotubes that were formed from myoblasts of a patient witha different form of congenital muscular dystrophy (not dueto LAMA2 mutation) in which laminin-a2 processing wasaltered but expression continued (26). In addition, the differen-tiation state of cells may affect outcome, as a study of mito-chondria in one line of MDC1A myoblasts did not findabnormalities (27), though myotubes were not examined.The abnormal myotube phenotypes that we identified wereconsistent over three independent lines of laminin-a2-deficientmyoblasts and could be prevented either by growth on alaminin-a2-containing substrate or by treatment with hBIP.Thus, the human laminin-a2-deficient myogenic cell culturesappear to provide a relevant system in which to identify patho-logical mechanisms and test therapeutic approaches.
Our findings identify Ku70 regulation of Bax as potentialtherapeutic target for MDC1A and perhaps additional neuro-muscular diseases. Genetic and pharmaceutical approachesthat inhibit disease-induced mitochondrial abnormalitieshave been shown to improve outcomes in mouse models ofmultiple neuromuscular diseases including MDC1A, OPMD,SMA and UCMD (1–8). Developing therapies based on theKu70/Bax pathway could include testing BIP function invivo. One study, for example, has shown that BIPs deliveredinto the eye can inhibit experimentally-induced retinalganglion cell death (28), but whether administration throughthe circulation is possible is not known. In addition, becauseacetylation of Ku70 is central to regulation of Bax activation,an understanding of the specific acetyltransferases and deace-tylases that act on Ku70 in diseased skeletal muscles couldprovide additional targets for therapeutic development. In non-muscle cells, Ku70 is acetylated by CBP and PCAF and dea-cetylated by SIRT1, SIRT3 and probably other deacetylases(14,16,18,29). In normal and diseased skeletal muscles,however, it remains to be determined which specific deacety-lases and acetyltransferases regulate Ku70 functions andwhere within the cell they function. Therapeutic strategiesthat preserve normal mitochondrial function by inhibitingaberrantly activated cell death pathways could have significanttherapeutic benefit.
MATERIALS AND METHODS
Mice
Heterozygous Lama2dy-W/þ mice, which carry the targeteddy-W mutation in the Lama2 gene (30), have been maintainedin our laboratory for .5 years by breeding with C57BL/6Jmice (4,5,9). Mice obtained from crosses of Lama2þ/2 hetero-zygotes were genotyped at weaning by PCR (4). Breedingpairs of heterozygous B6.129X1-Baxtm1Sjk mice (31) wereobtained from the Jackson Laboratory and mated in our lab-oratory. Progeny were genotyped (4), and muscle tissue orcells for primary cultures were obtained from tissues of theresulting wild-type or Bax-null littermates at 4–6 weeks ofage.
4474 Human Molecular Genetics, 2009, Vol. 18, No. 23
by guest on October 1, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

Cells and culture
Myoblasts derived from biopsies obtained from three donorswith a clinical diagnosis of MDC1A and from two normalindividuals were provided by the Muscle Tissue Culture Col-lection at the University of Munich. Because of the young ageof the MDC1A donors, it was not possible to obtain age-matched normal donors. All experiments with primaryhuman cells were performed with cells at no later than25 population doublings. All available clinical observations onthe donors are shown in Table 1. For MDC1A line 96/04,direct sequencing of LAMA2 gene exons and short stretchesof intron flanking sequences was performed (Prevention Gen-etics, Marshfield, WI, USA). Fifteen polymorphisms weredetected including two previously undocumented variants:c.8845-8 T.G and c.1789þ10 C.T (nomenclature describedin Ref. 32). The c.8845-8 T.G variant at the junction ofintron 62 and exon 63 creates a new splice site and is likelycausative, whereas the c.1789þ10 C.T variant currentlyhas an unknown functional effect. Additional investigationsof mRNA and genomic sequences are required to determineif these mutations are causal and to search further for del-etions, which can be missed by exon sequencing and areoften found in MDC1A (33). We added the 96/04 sequenceinformation to the Leiden Open Variation LAMA2 poly-morphism database which was accessed at http://www.dmd.nl/nmdb2/home.php?select_db=LAMA2. Sequences of theLAMA2 genes of the 9/03 and 38/03 lines remain to be inves-tigated.
Human myoblasts from normal individuals and MDC1Apatients were grown in skeletal muscle cell growth mediumsupplemented with 15% fetal bovine serum, 50 mg/ml fetuin,1 ng/ml basic fibroblast growth factor, 10 ng/ml epidermalgrowth factor, 10 mg/ml insulin and 0.4 mg/ml dexamethasone(Promocell, Heidelberg, Germany). Differentiation wasinduced by replacing the fetal bovine serum with 2% horseserum. Cells of the mouse C2C12 myogenic cell line andprimary cultures of mouse myoblasts derived from lower hind-limb muscles were grown and induced to differentiate as pre-viously described (5,22,34,35). Culture dishes for C2C12 cellswere coated with gelatin; for Baxþ/þ or Bax2/2 primary myo-blasts with ECL matrix (Entactin–Collagen–Laminin,Upstate Biotechnology, Lake Placid, NY, USA); and fornormal and MDC1A human myoblasts with eitherpoly-L-lysine (Sigma-Aldrich, St. Louis, MO, USA) or 1 mg/cm2 human placental laminin (L6274, Sigma-Aldrich).
Antibodies, reagents and assays
Antibodies for activated caspase-3 and mouse acetyl-lysinewere from Cell Signaling Technology (Beverly, MA, USA).Goat anti-LDH, anti-Bax mAb 6A7 and goat anti-Ku70 werefrom Santa Cruz Biotechnology (Santa Cruz, CA, USA).Rabbit anti-Bax, mouse anti-Ku70 and mouse anti-beta-ATPsynthase were from BD Biosciences (San Jose, CA, USA).Anti-desmin mAb was from Sigma-Aldrich (St. Louis, MO,USA). The anti-myosin heavy chain mAb F59 was as pre-viously described (33,36,37). Dr Lydia Sorokin (Universityof Munster) generously provided the rat anti-laminin-a2mAb 4H8-2 which reacts with an epitope located within the
L4b (previously IVa) globular domain encoded by exons23–27 in the N-terminal portion of the laminin-a2 molecule(38,39). Staurosporine was from Sigma-Aldrich. Caspase-3enzyme activity was measured with the CaspACE system(Promega, Madison, WI, USA).
Immunoblots and fractionation
Proteins were transferred to nitrocellulose membranes andsignals were detected with Alexa-680-conjugated secondaryantibodies with appropriate species specificity. Immunoblotswere quantified using the quantification tool in the Odyssey2.0 software that accompanies the LI-COR Odyssey infraredsystem (LI-COR Biosciences, Lincoln, NE, USA). Subcellularfractionations were performed by incubating cells in isotonic0.05% digitonin buffer for 10 min at 48C followed by centrifu-gation to separate the cytosolic supernatant from the membrane/mitochondrial/organellar/nuclear pellet as described (40). Thisfractionation has been validated for use in Bax translocationstudies (40). To further purify nuclei, the pellet was resuspendedin 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM
dithiothreitol, 0.05% NP-40, pH 7.9 for 10 min at 48Cfollowed by centrifugation at 3000g for 10 min as described(http://www.abcam.com/index.html?pageconfig=resource&rid=11408). This second pellet was then resuspended in5 mM HEPES, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM dithio-threitol, 26% glycerol (v/v), pH 7.9; and then adjusted to300 mM NaCl, homogenized on ice in a glass homogenizer,and centrifuged at 24 000g for 20 min at 48C. The resultingpellet, containing purified nuclei, was used for immunoprecipi-tation or immunoblotting. When using immunoblots to compareproteins in different cell fractions, we analyzed an equal percen-tage of the total fractionated material, e.g. 10% of the nuclearand 10% of the cytoplasmic preparation, in each lane.
Immunoprecipitation
The buffer used for cell lysis and Ku70 immunoprecipitationconsisted of 20 mM HEPES, pH 7.4, 1% Triton X-100,200 mM NaCl, 1 mM EDTA, 5 mM sodium pyrophosphate,20 mM beta-glycerophosphate, 50 mM NaF, 1 mM sodiumorthovandate and 1X protease inhibitor cocktail (Calbiochem,San Diego, CA, USA). Bax immunoprecipitation was carriedout as previously described (41). Frozen soleus tissue was pul-verized under liquid nitrogen in a mortar and pestle and thenhomogenized using a Polytron in appropriate lysis bufferand sonicated on ice. Insoluble material was removed by cen-trifugation at 12 000g for 10 min at 48C. Protein concentrationwas determined by BCA assay (Bio-Rad Laboratories, Her-cules, CA, USA).
Synthesis of BIPs
Sequences of the control peptide, and the mouse and humanBIP peptide sequences were adopted from previous studies(15,16). The fl-hBIP and fl-mBIP peptides were labeled atthe N-terminus with FITC using beta-alanine as a linkerbetween the BIP peptide and the FITC label. Peptides wereprepared by solid state peptide synthesis using an automatedpeptide synthesizer (ABI Model 433A) and Fmoc (fluorenyl-
Human Molecular Genetics, 2009, Vol. 18, No. 23 4475
by guest on October 1, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

methoxycarbonyl) to block a-amino groups. Coupling ofside-chain protected amino acids to a nascent peptide wasaccomplished by converting the a-carboxyl groups toactive benzotriazole esters using the coupling reagent2-(1H-Benzotriazole-1-yl)-1,1,3,3-teteramethyluronium hexa-fluorophosphate. The completed peptide was cleaved fromthe resin and deprotected using a 95% trifluoroacetic acidcocktail containing scavenger molecules (phenol, thioanisoleand ethanedithiol). Final purification employed preparativereversed phase HPLC on C18 columns. The quality andidentity of the purified peptide was ascertained by massspectrometry and analytical rpHPLC. Lyophilized peptideswere resuspended at 200 mM in dimethylsulfoxide anddilutions from this stock solution were made in culturemedium. Initial experiments tested final concentrations ofpeptides of 200—1 mM for efficacy.
ACKNOWLEDGEMENTS
We thank Dr Paul Leavis (Boston Biomedical Research Insti-tute) for peptide synthesis; Dr Sachiko Homma (Boston Bio-medical Research Institute) for much helpful advice; MaryLou Beermann and Maggie Ardelt for technical assistance;Dr Lydia Sorokin (University of Munster) for the laminin-a2mAb; and Dr Rosario Santos (Centro de Genetica MedicaJ. Magalhaes, INSA Porto, Portugal) and Dr Tom Winder(Prevention Genetics Inc.) for analysis and discussion of line96/04 polymorphisms. In addition, we thank the MuscleTissue Culture Collection (MTCC) at the University ofMunich, particularly Dr Peter Schneiderat and Ira Kaus, forarranging to provide the myoblasts obtained from MDC1Apatients. The MTCC is part of the German network on muscu-lar dystrophies (MD-NET, service structure S1, 01GM0601)funded by the German Ministry of Education and Research(BMBF, Bonn, Germany). The MTCC is a partner ofEurobiobank (www.eurobiobank.org) and TREAT-NMD(www.treat-nmd.eu).
Conflict of Interest statement. None declared.
FUNDING
This work was supported by the National Institutes of Health(5R01HL064641, 1U54HD060848); the National ResearchInitiative Competitive Grant Program of the United StatesDepartment of Agriculture (2006-35206-16622); and the Mus-cular Dystrophy Association of the United States (114981).
REFERENCES
1. Angelin, A., Tiepolo, T., Sabatelli, P., Grumati, P., Bergamin, N., Golfieri,C., Mattioli, E., Gualandi, F., Ferlini, A., Merlini, L. et al. (2007)Mitochondrial dysfunction in the pathogenesis of Ullrich congenitalmuscular dystrophy and prospective therapy with cyclosporins. Proc.Natl. Acad. Sci. USA, 104, 991–996.
2. Davies, J.E., Wang, L., Garcia-Oroz, L., Cook, L.J., Vacher, C.,O’Donovan, D.G. and Rubinsztein, D.C. (2005) Doxycycline attenuatesand delays toxicity of the oculopharyngeal muscular dystrophy mutationin transgenic mice. Nat. Med., 11, 672–677.
3. Irwin, W.A., Bergamin, N., Sabatelli, P., Reggiani, C., Megighian, A.,Merlini, L., Braghetta, P., Columbaro, M., Volpin, D., Bressan, G.M.,
Bernardi, P. and Bonaldo, P. (2003) Mitochondrial dysfunction andapoptosis in myopathic mice with collagen VI deficiency. Nat. Genet., 35,367–371.
4. Girgenrath, M., Dominov, J.A., Kostek, C.A. and Miller, J.B. (2004)Inhibition of apoptosis improves outcome in a model of congenitalmuscular dystrophy. J. Clin. Invest., 114, 1635–1639.
5. Girgenrath, M., Beermann, M.L., Vishnudas, V.K., Homma, S. andMiller, J.B. (2008) Pathology is alleviated by doxycycline in alaminin-alpha2-null model of congenital muscular dystrophy. Ann.
Neurol., 65, 47–56. First published on Dec. 11, 2008 10.1002/ana.21523.6. Millay, D.P., Sargent, M.A., Osinska, H., Baines, C.P., Barton, E.R.,
Vuagniaux, G., Sweeney, H.L., Robbins, J. and Molkentin, J.D. (2008)Genetic and pharmacologic inhibition of mitochondrial-dependentnecrosis attenuates muscular dystrophy. Nat Med., 14, 442–447.
7. Miller, J.B. and Girgenrath, M. (2006) Role of apoptosis in neuromusculardiseases and potential usefulness of anti-apoptosis therapy. Trends Mol.
Med., 12, 279–286.8. Tsai, M.S., Chiu, Y.T., Wang, S.H., Hsieh-Li, H.M., Lian, W.C. and Li,
H. (2006) Abolishing Bax-dependent apoptosis shows beneficial effectson spinal muscular atrophy model mice. Mol. Ther., 13, 1149–1155.
9. Dominov, J.A., Kravetz, A.J., Ardelt, M., Kostek, C.A., Beermann, M.L.and Miller, J.B. (2005) Muscle-specific BCL2 expression amelioratesmuscle disease in laminin-alpha2-deficient, but not dystrophin-deficient,mice. Hum. Mol. Genet, 14, 1029–1040.
10. Allamand, V. and Guicheney, P. (2002) Merosin-deficient congenitalmuscular dystrophy, autosomal recessive (MDC1A, MIM#156225,LAMA2 gene coding for alpha2 chain of laminin). Eur. J. Hum. Genet.,10, 91–94.
11. Miyagoe-Suzuki, Y., Nakagawa, M. and Takeda, I. (2000) Merosin andcongenital muscular dystrophy. Microsc. Res. Tech., 48, 181–191.
12. Vachon, P.H., Loechel, F., Xu, H., Wewer, U.M. and Engvall, E. (1996)Merosin and laminin in myogenesis; specific requirement for merosin inmyotube stability and survival. J. Cell Biol., 134, 1483–1497.
13. Laprise, P., Vallee, K., Demers, M.J., Bouchard, V., Poirier, E.M., Vezina,A., Reed, J.C., Rivard, N. and Vachon, P.H. (2003) Merosin (laminin-2/4)-driven survival signaling: complex modulations of Bcl-2 homologs.J. Cell Biochem., 89, 1115–1125.
14. Sundaresan, N.R., Samant, S.A., Pillai, V.B., Rajamohan, S.B. and Gupta,M.P. (2008) SIRT3 is a stress-responsive deacetylase in cardiomyocytesthat protects cells from stress-mediated cell death by deacetylation ofKu70. Mol. Cell. Biol., 28, 6384–6401.
15. Gomez, J.A., Gama, V., Yoshida, T., Sun, W., Hayes, P., Leskov, K.,Boothman, D. and Matsuyama, S. (2007) Bax-inhibiting peptides derivedfrom Ku70 and cell-penetrating pentapeptides. Biochem. Soc. Trans., 35,797–801.
16. Li, Y., Yokota, T., Gama, V., Yoshida, T., Gomez, J.A., Ishikawa, K.,Sasaguri, H., Cohen, H.Y., Sinclair, D.A., Mizusawa, H. and Matsuyama,S. (2007) Bax-inhibiting peptide protects cells from polyglutaminetoxicity caused by Ku70 acetylation. Cell Death Differ., 14, 2058–2067.
17. Mazumder, S., Plesca, D., Kinter, M. and Almasan, A. (2007) Interactionof a cyclin E fragment with Ku70 regulates Bax-mediated apoptosis. Mol.
Cell. Biol., 27, 3511–3520.18. Cohen, H.Y., Lavu, S., Bitterman, K.J., Hekking, B., Imahiyerobo, T.A.,
Miller, C., Frye, R., Ploegh, H., Kessler, B.M. and Sinclair, D.A. (2004)Acetylation of the C terminus of Ku70 by CBP and PCAF controlsBax-mediated apoptosis. Mol. Cell., 13, 627–638.
19. Cohen, H.Y., Miller, C., Bitterman, K.J., Wall, N.R., Hekking, B.,Kessler, B., Howitz, K.T., Gorospe, M., de Cabo, R. and Sinclair, D.A.(2004) Calorie restriction promotes mammalian cell survival by inducingthe SIRT1 deacetylase. Science, 305, 390–392.
20. Amsel, A.D., Rathaus, M., Kronman, N. and Cohen, H.Y. (2008)Regulation of the proapoptotic factor Bax by Ku70-dependentdeubiquitylation. Proc. Natl. Acad. Sci. USA, 105, 5117–5122.
21. Iriyama, T., Kamei, Y., Kozuma, S. and Taketani, Y. (2009)Bax-inhibiting peptide protects glutamate-induced cerebellar granule celldeath by blocking Bax translocation. Neurosci. Lett., 451, 11–15.
22. Nowak, J.A., Malowitz, J., Girgenrath, M., Kostek, C.A., Kravetz, A.J.,Dominov, J.A. and Miller, J.B. (2004) Immortalization of myogenic cellsdoes not require loss of p16INK4a, p19ARF, or p53 and is accelerated byinactivation of Bax. BMC Cell Biol., 5, 1.
23. Laprise, P., Poirier, E.M., Vezina, A., Rivard, N. and Vachon, P.H. (2002)Merosin–integrin promotion of skeletal myofiber cell survival:
4476 Human Molecular Genetics, 2009, Vol. 18, No. 23
by guest on October 1, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

differentiation state-distinct involvement of p60Fyn tyrosine kinase andp38alpha stress-activated MAP kinase. J. Cell Physiol., 191, 69–81.
24. Er, E., Oliver, L., Cartron, P.F., Juin, P., Manon, S. and Vallette, F.M.(2006) Mitochondria as the target of the pro-apoptotic protein Bax.Biochim. Biophys. Acta., 1757, 1301–1311.
25. Fernando, P., Kelly, J.F., Balazsi, K., Slack, R.S. and Megeney, L.A.(2002) Caspase 3 activity is required for skeletal muscle differentiation.Proc. Natl. Acad. Sci. USA, 99, 11025–11030.
26. Lattanzi, G., Muntoni, F., Sabatelli, P., Squarzoni, S., Maraldi, N.M.,Cenni, V., Villanova, M., Columbaro, M., Merlini, L. and Marmiroli, S.(2000) Unusual laminin alpha2 processing in myoblasts from a patientwith a novel variant of congenital muscular dystrophy. Biochem. Biophys.Res. Commun., 277, 639–642.
27. Hicks, D., Lampe, A.K., Laval, S.H., Allamand, V., Jimenez-Mallebrera,C., Walter, M.C., Muntoni, F., Quijano-Roy, S., Richard, P., Straub, V.,Lochmuller, H. and Bushby, K.M. (2009) Cyclosporine A treatment forUllrich congenital muscular dystrophy: a cellular study of mitochondrialdysfunction and its rescue. Brain, 132, 147–155.
28. Qin, Q., Patil, K. and Sharma, S.C. (2004) The role of Bax-inhibitingpeptide in retinal ganglion cell apoptosis after optic nerve transection.Neurosci. Lett., 372, 17–21.
29. Jeong, J., Juhn, K., Lee, H., Kim, S.H., Min, B.H., Lee, K.M., Cho, M.H.,Park, G.H. and Lee, K.H. (2007) SIRT1 promotes DNA repair activity anddeacetylation of Ku70. Exp. Mol. Med., 39, 8–13.
30. Kuang, W., Xu, H., Vachon, P.H., Liu, L., Loechel, F., Wewer, U.M. andEngvall, E. (1999) Merosin-deficient congenital muscular dystrophy:partial genetic correction in two mouse models. J. Clin. Invest., 102,844–852.
31. Knudson, C.M., Tung, K.S., Tourtellotte, W.G., Brown, G.A. andKorsmeyer, S.J. (1995) Bax-deficient mice with lymphoid hyperplasia andmale germ cell death. Science, 270, 96–99.
32. den Dunnen, J.T. and Antonarakis, S.E. (2000) Mutation nomenclatureextensions and suggestions to describe complex mutations: A discussion.Hum. Mutat., 15, 7–12.
33. Oliveira, J., Santos, R., Soares-Silva, I., Jorge, P., Vieira, E., Oliveira,M.E., Moreira, A., Coelho, T., Ferreira, J.C., Fonseca, M.J. et al. (2008)LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophypatients. Clin. Genet., 74, 502–512.
34. Block, N.E. and Miller, J.B. (1992) Expression of MRF4, a myogenichelix-loop-helix protein, produces multiple changes in the myogenicprogram of BC3H-1 cells. Mol. Cell. Biol., 12, 2484–2492.
35. Dominov, J.A., Dunn, J.J. and Miller, J.B. (1998) Bcl-2 expressionidentifies an early stage of myogenesis and promotes clonal expansion ofmuscle cells. J. Cell Biol., 142, 537–544.
36. Miller, J.B., Teal, S.B. and Stockdale, F.E. (1989) Evolutionarilyconserved sequences of striated muscle myosin heavy chain isoforms:Epitope mapping by cDNA expression. J. Biol. Chem., 264, 13122–13130.
37. Smith, T.H., Block, N.E., Rhodes, S.J., Konieczny, S.F. and Miller, J.B.(1993) A unique pattern of expression of the four muscle regulatoryfactors distinguishes somitic from embryonic, fetal, and newborn mousemyogenic cells. Development, 117, 1125–1133.
38. Allamand, V., Sunada, Y., Salih, M.A., Straub, V., Ozo, C.O., Al-Turaiki,M.H., Akbar, M., Kolo, T., Colognato, H., Zhang, X. et al. (1997) Mildcongenital muscular dystrophy in two patients with an internally deletedlaminin alpha2-chain. Hum. Mol. Genet., 6, 747–752.
39. Allamand, V., Bidou, L., Arakawa, M., Floquet, C., Shiozuka, M.,Paturneau-Jouas, M., Gartioux, C., Butler-Browne, G.S., Mouly, V.,Rousset, J.P. et al. (2008) Drug-induced readthrough of premature stopcodons leads to the stabilization of laminin alpha2 chain mRNA in CMDmyotubes. J. Gene Med., 10, 217–224.
40. Wang, J., Wei, Q., Wang, C.Y., Hill, W.D., Hess, D.C. and Dong, Z.(2004) Minocycline up-regulates Bcl-2 and protects against cell death inmitochondria. J. Biol. Chem., 279, 19948–19954.
41. Yamaguchi, H., Paranawithana, S.R., Lee, M.W., Huang, Z., Bhalla, K.N.and Wang, H.G. (2002) Epothilone B analogue (BMS-247550)-mediatedcytotoxicity through induction of Bax conformational change in humanbreast cancer cells. Cancer Res., 62, 466–471.
Human Molecular Genetics, 2009, Vol. 18, No. 23 4477
by guest on October 1, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from
Copyright © 2022 FDOKUMEN




















