
Calcium and the damage pathways in muscular dystrophy
9
REVIEW / SYNTHE ` SE Calcium and the damage pathways in muscular dystrophy 1 David G. Allen, Othon L. Gervasio, Ella W. Yeung, and Nicholas P. Whitehead Abstract: Duchenne muscular dystrophy (DMD) is a severe muscle-wasting disease caused by the absence of the cytoske- letal protein dystrophin. Experiments on the mdx mouse, a model of DMD, have shown that mdx muscles are particularly susceptible to stretch-induced damage. In this review, we discuss evidence showing that a series of stretched contractions of mdx muscle fibres causes a prolonged increase in resting intracellular calcium concentration ([Ca 2+ ] i ). The rise in [Ca 2+ ] i is caused by Ca 2+ entry through a class of stretch-activated channels (SAC NSC ) for which one candidate gene is TRPC1. We review the evidence for activation of SAC NSC in muscle by reactive oxygen species (ROS) and suggest that stretch-induced ROS production is part of the pathway that triggers increased channel activity. When the TRPC1 gene was transfected into C2 myoblasts, expression occurred throughout the cell. Only when the TRPC1 gene was coexpressed with caveolin-3 did the TRPC1 protein express in the membrane. When TRPC1 was expressed in the membrane, it could be ac- tivated by ROS to produce Ca 2+ entry and this entry was inhibited by PP2, an inhibitor of src kinase. These results suggest that stretched contractions activate ROS production, which activates src kinase. Activity of this kinase causes opening of SAC NSC and allows Ca 2+ entry. This pathway appears to be a significant cause of muscle damage in DMD. Key words: Duchenne muscular dystrophy, mdx mouse, dystrophin, stretch-activated channels, TRPC1, intracellular cal- cium. Re ´sume ´: La dystrophie musculaire de Duchenne (DMD) est due a ` l’absence d’une prote ´ine cytosquelettique, la dystro- phine. Les e ´tudes sur la souris mdx, un mode `le de DMD, re ´ve `lent que les muscles de cette souris sont particulie `rement sensibles aux le ´sions cause ´es par l’e ´tirement. Dans cet article-synthe `se, nous de ´montrons qu’une se ´rie de contractions des fibres musculaires mdx e ´tire ´es suscite une augmentation prolonge ´e de la concentration intracellulaire de calcium au repos ([Ca 2+ ] i ). L’augmentation de [Ca 2+ ] i est cause ´e par l’entre ´e du calcium via un ensemble de canaux cationiques non se ´lectifs active ´s par l’e ´tirement (SAC NSC ) et qui sont code ´s par le ge `ne TRPC1. Nous de ´montrons que les SAC NSC dans le muscle peuvent e ˆtre active ´s par les espe `ces re ´actives de l’oxyge `ne (ROS), ce qui signifie que la production de ROS cause ´e par l’e ´tirement fait partie de la voie qui de ´clenche l’activite ´ des canaux. Apre `s la transfection du ge `ne TRPC1 dans les myo- blastes C2, on en observe l’expression dans toute la cellule. La prote ´ine TRPC1 est exprime ´e dans la membrane seulement quand le ge `ne codant la TRPC1 est coexprime ´ en pre ´sence de la cave ´oline-3. Quand TRPC1 est exprime ´ dans la mem- brane, il peut e ˆtre active ´ par les ROS de ´clenchant l’entre ´e de calcium qui peut e ˆtre contre ´e par PP2, un inhibiteur de la src kinase. Ces observations indiquent que les contractions de fibres musculaires e ´tire ´es activent la production des ROS les- quelles activent la src kinase. L’activite ´ des kinases de ´clenche l’ouverture des SAC NSC permettant ainsi l’entre ´e du Ca 2+ . Cette voie semble e ˆtre une cause importante de le ´sions musculaires dans la DMD. Mots-cle ´s : dystrophie musculaire de Duchenne, souris mdx, dystrophine, canaux active ´s par l’e ´tirement, TRPC1, calcium intracellulaire. [Traduit par la Re ´daction] Introduction Duchenne muscular dystrophy (DMD) is a severe muscle- wasting disease affecting about 1 in 3500 boys (for review see Emery and Muntoni 2003). Affected boys are apparently healthy at birth, and diagnosis is commonly made at 2– 4 years of age when the boys show the first signs of weak- ness and impaired mobility. Thereafter the disease pro- gresses relentlessly, with affected boys requiring a wheelchair by 8–12 years. Respiratory weakness develops slowly but becomes very severe, and in the past many boys died of respiratory failure and (or) infection in their late teens. However the development of nighttime ventilation Received 23 January 2009. Accepted 15 May 2009. Published on the NRC Research Press Web site at cjpp.nrc.ca on 11 February 2010. D.G. Allen, 2 O.L. Gervasio, and N.P. Whitehead. School of Medical Sciences and Bosch Institute, University of Sydney F13, NSW 2006, Australia. E.W. Yeung. Department of Rehabilitation Sciences, Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong. 1 This article is one of a selection of papers published in this special issue on Calcium Signaling. 2 Corresponding author (e-mail: [email protected]). 83 Can. J. Physiol. Pharmacol. 88: 83–91 (2010) doi:10.1139/Y09-058 Published by NRC Research Press Can. J. Physiol. Pharmacol. Downloaded from www.nrcresearchpress.com by 199.201.121.12 on 06/04/13 For personal use only.
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Calcium and the damage pathways in muscular dystrophy

REVIEW / SYNTHESE
Calcium and the damage pathways in musculardystrophy1
David G. Allen, Othon L. Gervasio, Ella W. Yeung, and Nicholas P. Whitehead
Abstract: Duchenne muscular dystrophy (DMD) is a severe muscle-wasting disease caused by the absence of the cytoske-letal protein dystrophin. Experiments on the mdx mouse, a model of DMD, have shown that mdx muscles are particularlysusceptible to stretch-induced damage. In this review, we discuss evidence showing that a series of stretched contractionsof mdx muscle fibres causes a prolonged increase in resting intracellular calcium concentration ([Ca2+]i). The rise in[Ca2+]i is caused by Ca2+ entry through a class of stretch-activated channels (SACNSC) for which one candidate gene isTRPC1. We review the evidence for activation of SACNSC in muscle by reactive oxygen species (ROS) and suggest thatstretch-induced ROS production is part of the pathway that triggers increased channel activity. When the TRPC1 gene wastransfected into C2 myoblasts, expression occurred throughout the cell. Only when the TRPC1 gene was coexpressed withcaveolin-3 did the TRPC1 protein express in the membrane. When TRPC1 was expressed in the membrane, it could be ac-tivated by ROS to produce Ca2+ entry and this entry was inhibited by PP2, an inhibitor of src kinase. These results suggestthat stretched contractions activate ROS production, which activates src kinase. Activity of this kinase causes opening ofSACNSC and allows Ca2+ entry. This pathway appears to be a significant cause of muscle damage in DMD.
Key words: Duchenne muscular dystrophy, mdx mouse, dystrophin, stretch-activated channels, TRPC1, intracellular cal-cium.
Resume : La dystrophie musculaire de Duchenne (DMD) est due a l’absence d’une proteine cytosquelettique, la dystro-phine. Les etudes sur la souris mdx, un modele de DMD, revelent que les muscles de cette souris sont particulierementsensibles aux lesions causees par l’etirement. Dans cet article-synthese, nous demontrons qu’une serie de contractions desfibres musculaires mdx etirees suscite une augmentation prolongee de la concentration intracellulaire de calcium au repos([Ca2+]i). L’augmentation de [Ca2+]i est causee par l’entree du calcium via un ensemble de canaux cationiques non selectifsactives par l’etirement (SACNSC) et qui sont codes par le gene TRPC1. Nous demontrons que les SACNSC dans le musclepeuvent etre actives par les especes reactives de l’oxygene (ROS), ce qui signifie que la production de ROS causee parl’etirement fait partie de la voie qui declenche l’activite des canaux. Apres la transfection du gene TRPC1 dans les myo-blastes C2, on en observe l’expression dans toute la cellule. La proteine TRPC1 est exprimee dans la membrane seulementquand le gene codant la TRPC1 est coexprime en presence de la caveoline-3. Quand TRPC1 est exprime dans la mem-brane, il peut etre active par les ROS declenchant l’entree de calcium qui peut etre contree par PP2, un inhibiteur de la srckinase. Ces observations indiquent que les contractions de fibres musculaires etirees activent la production des ROS les-quelles activent la src kinase. L’activite des kinases declenche l’ouverture des SACNSC permettant ainsi l’entree du Ca2+.Cette voie semble etre une cause importante de lesions musculaires dans la DMD.
Mots-cles : dystrophie musculaire de Duchenne, souris mdx, dystrophine, canaux actives par l’etirement, TRPC1, calciumintracellulaire.
[Traduit par la Redaction]
IntroductionDuchenne muscular dystrophy (DMD) is a severe muscle-
wasting disease affecting about 1 in 3500 boys (for reviewsee Emery and Muntoni 2003). Affected boys are apparentlyhealthy at birth, and diagnosis is commonly made at 2–4 years of age when the boys show the first signs of weak-ness and impaired mobility. Thereafter the disease pro-gresses relentlessly, with affected boys requiring awheelchair by 8–12 years. Respiratory weakness developsslowly but becomes very severe, and in the past many boysdied of respiratory failure and (or) infection in their lateteens. However the development of nighttime ventilation
Received 23 January 2009. Accepted 15 May 2009. Publishedon the NRC Research Press Web site at cjpp.nrc.ca on11 February 2010.
D.G. Allen,2 O.L. Gervasio, and N.P. Whitehead. School ofMedical Sciences and Bosch Institute, University of Sydney F13,NSW 2006, Australia.E.W. Yeung. Department of Rehabilitation Sciences, HongKong Polytechnic University, Hung Hom, Kowloon, HongKong.
1This article is one of a selection of papers published in thisspecial issue on Calcium Signaling.
2Corresponding author (e-mail: [email protected]).
83
Can. J. Physiol. Pharmacol. 88: 83–91 (2010) doi:10.1139/Y09-058 Published by NRC Research Press
Can
. J. P
hysi
ol. P
harm
acol
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
199.
201.
121.
12 o
n 06
/04/
13Fo
r pe
rson
al u
se o
nly.
and improved small respirators has delayed the respiratorycomplications so that many affected boys currently surviveuntil their 20s or 30s. The heart is also affected, and cardiacfailure is an increasingly frequent complication as affectedindividuals live longer.
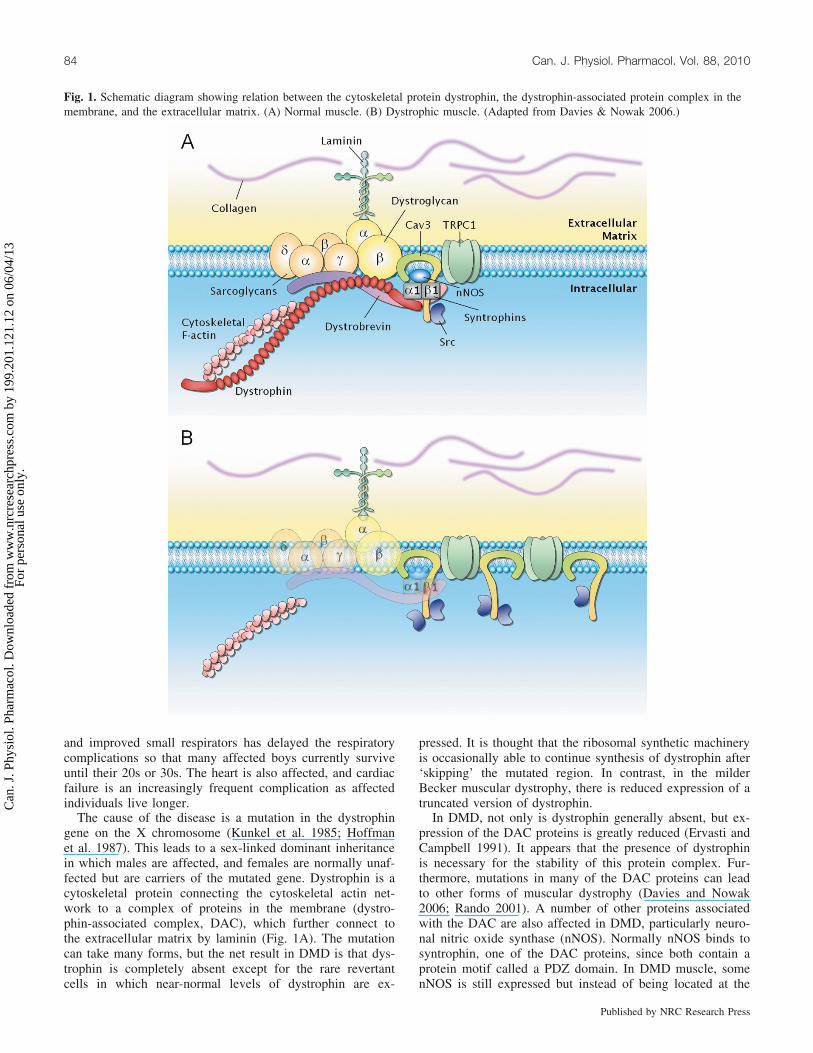
The cause of the disease is a mutation in the dystrophingene on the X chromosome (Kunkel et al. 1985; Hoffmanet al. 1987). This leads to a sex-linked dominant inheritancein which males are affected, and females are normally unaf-fected but are carriers of the mutated gene. Dystrophin is acytoskeletal protein connecting the cytoskeletal actin net-work to a complex of proteins in the membrane (dystro-phin-associated complex, DAC), which further connect tothe extracellular matrix by laminin (Fig. 1A). The mutationcan take many forms, but the net result in DMD is that dys-trophin is completely absent except for the rare revertantcells in which near-normal levels of dystrophin are ex-
pressed. It is thought that the ribosomal synthetic machineryis occasionally able to continue synthesis of dystrophin after‘skipping’ the mutated region. In contrast, in the milderBecker muscular dystrophy, there is reduced expression of atruncated version of dystrophin.
In DMD, not only is dystrophin generally absent, but ex-pression of the DAC proteins is greatly reduced (Ervasti andCampbell 1991). It appears that the presence of dystrophinis necessary for the stability of this protein complex. Fur-thermore, mutations in many of the DAC proteins can leadto other forms of muscular dystrophy (Davies and Nowak2006; Rando 2001). A number of other proteins associatedwith the DAC are also affected in DMD, particularly neuro-nal nitric oxide synthase (nNOS). Normally nNOS binds tosyntrophin, one of the DAC proteins, since both contain aprotein motif called a PDZ domain. In DMD muscle, somenNOS is still expressed but instead of being located at the
Fig. 1. Schematic diagram showing relation between the cytoskeletal protein dystrophin, the dystrophin-associated protein complex in themembrane, and the extracellular matrix. (A) Normal muscle. (B) Dystrophic muscle. (Adapted from Davies & Nowak 2006.)
84 Can. J. Physiol. Pharmacol. Vol. 88, 2010
Published by NRC Research Press
Can
. J. P
hysi
ol. P
harm
acol
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
199.
201.
121.
12 o
n 06
/04/
13Fo
r pe
rson
al u
se o
nly.

DAC, it is distributed more widely in the cell, and NO pro-duction is reduced to about 25% of normal (Froehner 2002).Figure 1B illustrates the situation in DMD with dystrophinabsent, the DAC proteins reduced in expression, and otherproteins, to be discussed below, upregulated.
The primary cause of the disease is the mutation in thedystrophin gene leading to absence of dystrophin and associ-ated loss of the DAC. The consequences of loss of dystro-phin and the DAC are uncertain. It is generally thought thatdystrophin has a role in stabilizing the cell membrane, andthat in its absence, membrane damage is more frequent
(Petrof et al. 1993; Miyake and McNeil 2003). The basisfor this hypothesis is the appearance of localized cell dam-age in association with localized membrane damage (Mokriand Engel 1975) and the fact that soluble proteins, such ascreatine kinase, leak from the cell into the plasma. In addi-tion, damaged muscle cells have a very high total Ca2+ con-tent that is consistent with Ca2+ leaking into the cell throughdamaged membranes (Bodensteiner and Engel 1978). Ofcourse, other pathways of Ca2+ entry may also be enhancedin the disease, as discussed below.
An alternative view is that dystrophin and the DAC havea protein scaffolding role and are involved in the localiza-tion and regulation of a series of proteins so that, in theirabsence, the abnormal function of these proteins triggers thedamage pathways. Obviously these two hypotheses are notnecessarily mutually exclusive, but in this article we reviewevidence from our own laboratory and others that identifiesa surface membrane channel Ca2+ whose activity is in-creased in DMD and appears to make a major contributionto the disease.
Stretch-induced Ca2+ entryMuscles can contract isometrically, can shorten, or can be
stretched by a force greater than the muscle produces. Eachtype of contraction occurs normally under some circumstan-ces, but contraction in which stretch occurs are recognizedas more likely to provoke muscle damage. For example,under normal conditions, walking down a mountain, inwhich the quadriceps group are repeatedly stretched andcontribute to braking, is liable to cause immediate weaknessfollowed on subsequent days by pain, swelling, and inflam-mation (delayed onset muscle soreness, DOMS). The mech-anism of DOMS has attracted considerable interest, andexcessive Ca2+ entry appears to be one contributing factor(Morgan and Allen 1999; Warren et al. 2001). The link be-tween excessive Ca2+ entry and muscle weakness is uncer-tain, but a popular hypothesis is that activation of calpainsmay be partly responsible (Allen 2001; Verburg et al.2006). This type of mild muscle damage is self-limiting andthere is usually complete recovery of function after 1–2 weeks. When the damage is severe, however, fibre dam-age can occur, and then activated satellite cells divide andproduce myoblasts that fuse and replace the damaged regionof the fibre (Charge and Rudnicki 2004).
An early observation in the mdx mouse, which also lacksdystrophin and is a model of DMD, is that this stretch-induceddamage is much more severe (Head et al. 1992; Petrof et al.1993). Thus it is widely thought that excessive stretch-inducedmuscle damage, possibly complicated by a failure in the repairprocess, is an important early component of the damage thatcharacterizes DMD. This view is further supported by the evi-dence that expression of a shortened form of dystrophin inmdx muscle reduces the propensity to stretch-induced muscledamage (Deconinck et al. 1996).
We became interested in the cellular mechanisms ofstretch-induced muscle damage and developed an isolatedmodel of this condition (Balnave and Allen 1995). Single fi-bres are dissected from the mouse flexor digitorum brevisand connected between a force transducer and a motor. Thefibres can be stimulated to produce tetani, and the motor can
Fig. 2. Resting [Ca2+]i in single fibres of wild-type and mdx mousemuscle before and after 10 stretched contractions. *, Significantdifference at p < 0.05 between wild-type and mdx; #, significantlydifferent at p < 0.05 from control period before stretched contrac-tions. (Based on Yeung et al. 2005.)
Fig. 3. Resting [Ca2+]i in single fibres of mdx muscle after 10 iso-metric tetani and 10 stretched tetani. Solid data points show the in-crease in [Ca2+]i after stretched contractions. Open points show thedata when streptomycin (200 mmol/L) was applied for 10 min afterthe stretched contractions. Fluorescence intensity is from intracellu-lar fluo-3 and represents [Ca2+]i. *, Significantly different at p <0.05 from data points in the absence of streptomycin. (Modifiedfrom Yeung et al. 2005.)
Allen et al. 85
Published by NRC Research Press
Can
. J. P
hysi
ol. P
harm
acol
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
199.
201.
121.
12 o
n 06
/04/
13Fo
r pe
rson
al u
se o
nly.
impose a stretch during the tetanus. In addition, the intracel-lular Ca2+ concentration ([Ca2+]i) can be monitored. In ouroriginal study we measured the reduction in isometric ten-sion caused by a series of stretched tetani and showed thatthere was a reduction in the accompanying tetanic [Ca2+]i.If the reduction in tetanic [Ca2+]i was reversed with caffeine,one component of the reduction in force was eliminated,demonstrating unequivocally that failure of sarcoplasmic re-ticulum (SR) Ca2+ release was a contributor to the stretch-induced force reduction. We also noted a very small in-
crease in resting [Ca2+]i occurring over 30 min after thestretched contractions; more recent data confirming thispoint are shown in Fig. 2.
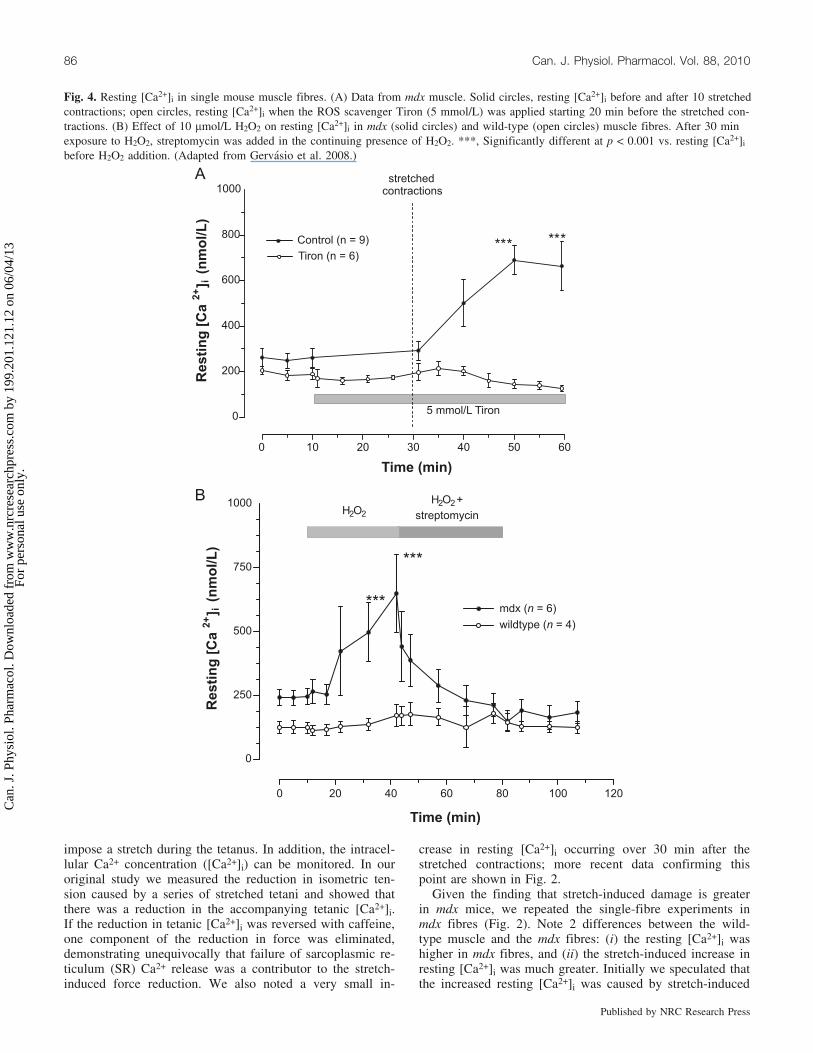
Given the finding that stretch-induced damage is greaterin mdx mice, we repeated the single-fibre experiments inmdx fibres (Fig. 2). Note 2 differences between the wild-type muscle and the mdx fibres: (i) the resting [Ca2+]i washigher in mdx fibres, and (ii) the stretch-induced increase inresting [Ca2+]i was much greater. Initially we speculated thatthe increased resting [Ca2+]i was caused by stretch-induced
Fig. 4. Resting [Ca2+]i in single mouse muscle fibres. (A) Data from mdx muscle. Solid circles, resting [Ca2+]i before and after 10 stretchedcontractions; open circles, resting [Ca2+]i when the ROS scavenger Tiron (5 mmol/L) was applied starting 20 min before the stretched con-tractions. (B) Effect of 10 mmol/L H2O2 on resting [Ca2+]i in mdx (solid circles) and wild-type (open circles) muscle fibres. After 30 minexposure to H2O2, streptomycin was added in the continuing presence of H2O2. ***, Significantly different at p < 0.001 vs. resting [Ca2+]i
before H2O2 addition. (Adapted from Gervasio et al. 2008.)
86 Can. J. Physiol. Pharmacol. Vol. 88, 2010
Published by NRC Research Press
Can
. J. P
hysi
ol. P
harm
acol
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
199.
201.
121.
12 o
n 06
/04/
13Fo
r pe
rson
al u
se o
nly.

tears in the membrane, but examination of images of theelevated [Ca2+]i or [Na+]i (which also increases afterstretched contractions) did not reveal any near-membrane lo-calized increases in [Ca2+]i or [Na+]i consistent with tears(Yeung et al. 2003; Yeung et al. 2005). For this reason weconsidered whether the increased resting [Ca2+]i occurs byCa2+ entry through a channel. One possibility is the stretch-activated channels first described by Guharay and Sachs(1984). These channels are permeable to most cations andallow the entry of Na+ and Ca2+ under normal conditions;this class of stretch-activated channels (SAC) is thereforepermeable to nonspecific cations and is designated SACNSC.Of particular interest is that these channels have been shownto be more active in mdx muscle (Franco-Obregon and Lans-man 2002; Ducret et al. 2006).
We therefore examined whether blockers of the SACNSC(for review see Hamill and McBride 1996) modulate theresting [Ca2+]i in mdx fibres (Fig. 3). The results were dra-matic and showed that streptomycin (100 mmol/L) com-pletely prevented the rise of [Ca2+]i and caused the resting[Ca2+]i to fall below that of the control (pre-drug applica-tion) period. Two other chemically dissimilar blockers ofSACNSC, gadolinium (Gd3+) and the spider venom toxinGsMTx-4 (Suchyna et al. 2000), produced similar effects.This suggests that the stretch-induced Ca2+ influx is throughSACNSC and is the cause of the elevated resting [Ca2+]i.
These results showed that stretch-induced Ca2+ entry oc-curred in the mdx muscle through a channel whose pharma-cology is that of SACNSC. In other experiments we haveshown SACNSC blockers minimized the reduction of isomet-ric force caused by stretch-induced muscle damage andminimized the muscle damage caused by downhill running
in intact mdx mice (Yeung et al. 2005). In addition, SACNSCblockers reduced the increase in membrane permeabilitycaused by stretched contractions, suggesting that the in-creased permeability is a secondary consequence of Ca2+ en-try rather than a primary consequence of membrane tears(Whitehead et al. 2006). These data all support the idea thatCa2+ entry through SACNSC makes an important contributionto the muscle damage in mdx mice.
Candidate genes for SACNSC
Maroto et al. (2005) purified SACNSC from frog oocytesand showed that these channels could be reconstituted in lip-osomes, had a molecular weight of 80 kDa, and bound atransient receptor potential canonical 1 (TRPC1) antibody.They also expressed human TRPC1 in both oocytes andChinese hamster ovary (CHO) cells and showed increasedexpression of the SACNSC channels. Finally, they showedthat antisense RNA to human TRPC1 reduced theexpression of both endogenous oocyte SACNSC and humanTRPC1-expressed channels. On the basis of these findings,they proposed that TRPC1 formed the SACNSC inmammalian tissues. Subsequently, however, many of thesame authors (Gottlieb et al. 2008) showed that expressionof human TRPC1 in green monkey kidney (COS cell line)or CHO cells did not cause a significant increase in theendogenous level of SACNSC activity. In the CHO cells, theexpressed TRPC1 occurred in punctate regions distributedthroughout the intracellular space, and the protein did not in-sert in the membrane. It is a frequent observation that exo-genously expressed TRPC1 does not insert in the membrane(Beech et al. 2003), and several studies have identifiedfactors that improve functionality of expressed TRPC1. Forinstance, coexpression with TRPC3 (Lintschinger et al.2000) and the presence of cholesterol also appear criticalfor channel function (Bergdahl et al. 2003). Our studyshowed that coexpression with caveolin-3 increased the frac-tion of TRPC1 expressed in the membrane, which allowedus to measure the Ca2+ influx produced by activation ofTRPC1 (Gervasio et al. 2008). Studies of the store-operatedCa2+ entry in mdx muscle found that this pathway was alsoupregulated (Tutdibi et al. 1999), and this pathway was re-duced by antisense downregulation of TRPC1 and TRPC4(Vandebrouck et al. 2002). Later these authors found thatthe same channel seemed to be both store-operated andstretch-activated and was blocked by several SACNSC block-ers (Ducret et al. 2006). Our conclusion is that TRPC1 isstill a viable candidate gene for SACNSC.
A number of other genes have been proposed to encodemechanosensitive channels that might be SACNSC. A strongcontender is TRPV2, which was originally identified as aCa2+-permeable channel, activated by insulin-like growthfactor 1 (IGF-1) (Kanzaki et al. 1997), that was shown tobe stretch-activated in d-sarcoglycan-deficient hamster(Nakamura et al. 2001). Subsequently it was renamed thegrowth factor-induced channel and was said to show homol-ogy with TRPV1 (Iwata et al. 2003). Most recently thechannel has been identified as TRPV2 and shown to be amajor pathway for Ca2+ entry in muscles of both mdx miceand the d-sarcoglycan-deficient hamster. Of particularinterest is the demonstration that downregulation of TRPV2
Fig. 5. Fluorescence images of C2 myoblast cells after transfectionwith caveolin-3 and yellow fluorescent protein (Cav-3-YFP) and(or) TRPC1 and cyan fluorescent protein (TRPC1-CFP). Note thatboth caveolin-3 and TRPC1 show additional targeting to the surfacemembrane when cotransfected (arrows). Scale bar represents10 mm. (Adapted from Gervasio et al. 2008.)
Allen et al. 87
Published by NRC Research Press
Can
. J. P
hysi
ol. P
harm
acol
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
199.
201.
121.
12 o
n 06
/04/
13Fo
r pe
rson
al u
se o
nly.
in the mdx mouse caused reduced Ca2+ entry and amelio-rated a number of features of the dystrophic phenotype(Iwata et al. 2009). Thus TRPV2 is an additional candidatefor the Ca2+ entry pathway of muscular dystrophy.
In adult cardiac ventricular myocytes, SACNSC have notbeen identified by patch clamping, despite repeated attempts(Zeng et al. 2000). Nevertheless, when whole ventricularmyocytes were stretched, stretch-activated currents werereadily discernable and these were blocked by Gd3+
(Kamkin et al. 2003) and GsMTx-4 (Dyachenko et al.2009a). Dyachenko et al. also showed that TRPC6 was ex-pressed in the T-tubules of ventricular cells and that an anti-body to the intracellular pore site on TRPC6 blocked thechannel when applied to the intracellular site. Thus TRPC6appears to be a strong contender for the SACNSC in mouseventricular cells, and the location of TRPC6 in the T-tubularmembrane probably explains the failure to identify it bypatch clamping.
Pathway for activation of SACNSC
Although Ca2+ entry after stretched contractions appearsto arise through SACNSC, this does not necessarily indicatethat the channel is being directly activated by stretch. Oneissue that points to this conclusion is the time course. Patchclamp studies have shown that SACNSC are activated in afew hundred milliseconds after an abrupt application of neg-ative pressure to the patch and are turned off with a compa-rable time course (Yeung et al. 2005; Suchyna and Sachs2007). This suggests that since the stretch imposed on amuscle during a tetanus lasts several hundred milliseconds,the influx of Ca2+ ought to occur during, or immediatelyafter, the tetanus. In fact no Ca2+ influx is detectable at thatstage (Balnave and Allen 1995), and the observed rise inresting [Ca2+]i occurs over 10–20 min after the stretchedcontractions when the muscle is back at the control length.Furthermore blockers of SACNSC are effective when appliedimmediately after the stretches, at which time the musclelength, and presumably the degree of channel stretch, are
back to the control state (Yeung et al. 2005). These observa-tions lead to the conclusion that the SACNSC are not beingdirectly activated by stretch.
Several of the TRP channels are activated by reactiveoxygen species (ROS) (Groschner et al. 2004; Poteser et al.2006), and there is evidence that ROS production is in-creased in DMD and the mdx mouse (Rando 2002). Thisled us to test whether ROS are involved in the opening ofSACNSC (Gervasio et al. 2008). We first used Tiron, a ROSscavenger, which when applied to the isolated musclefibres before the stretched contractions, prevented thestretch-induced rise of [Ca2+]i (Fig. 4A). As an additionalapproach, we used exogenous application of a ROS, specifi-cally H2O2 (10 mmol/L), which caused a very large increasein resting [Ca2+]i in mdx fibres but not in wild-type fibres.Importantly the increase in [Ca2+]i caused by H2O2 was in-hibited by streptomycin, showing that the influx of Ca2+
was through SACNSC (Fig. 4B).These data suggest that the opening of SACNSC in mdx
muscle may arise through some stretch-dependent produc-tion of ROS. It is puzzling that H2O2 had such a large effecton SACNSC in mdx muscle while having no obvious effecton wild-type fibres (Fig. 4B). This suggests that a ROS tar-get is greatly sensitized in mdx muscle; we have suggestedthat src kinase is the target of ROS and have shown that itis overexpressed in mdx muscle (Gervasio et al. 2008). It ispossible that the greatly increased sensitivity arises from in-creased levels or locations of src kinase.
Expression of TRPC1 in cell linesGiven the uncertainty about the gene which encodes
SACNSC, we decided to express TRPC1 in a mammaliancell line and examine its properties directly. The plasmidfor TRPC1 with attached green fluorescent protein wastransfected into C2 cells, a myoblast cell line, and expres-sion was observed mainly in the cytoplasm and with verylittle in the membrane. To examine the interaction with cav-eolin-3, the muscle-specific scaffolding protein that is essen-
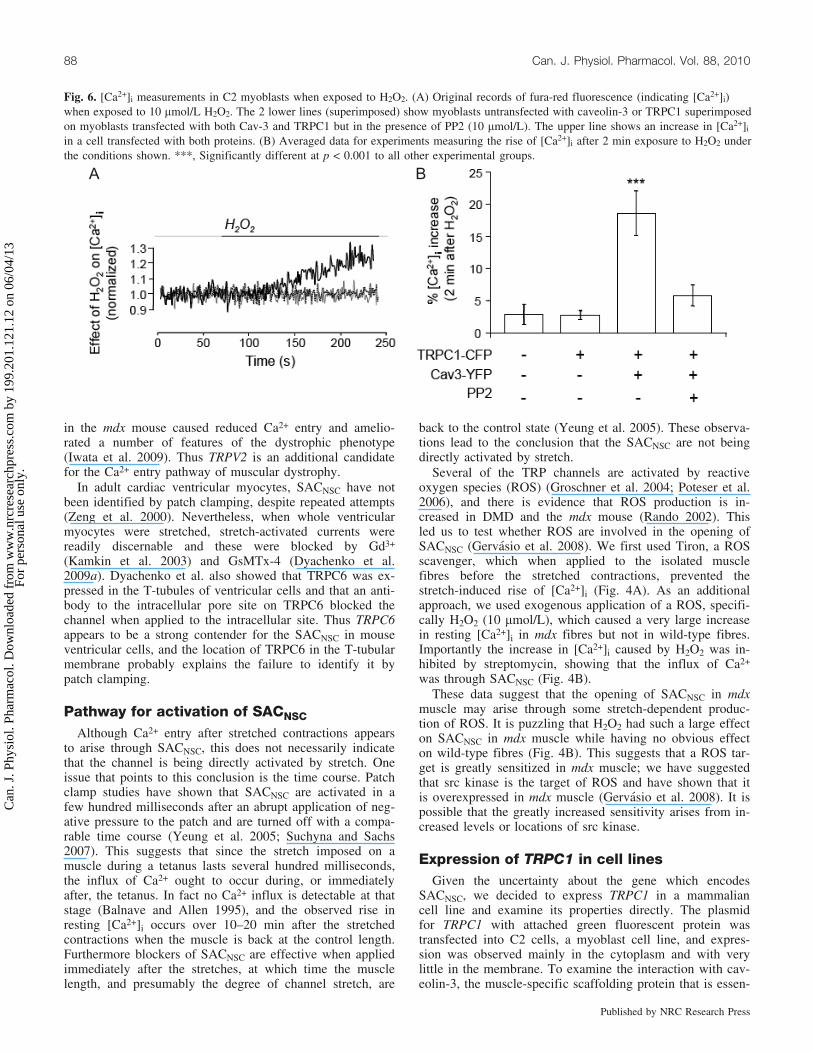
Fig. 6. [Ca2+]i measurements in C2 myoblasts when exposed to H2O2. (A) Original records of fura-red fluorescence (indicating [Ca2+]i)when exposed to 10 mmol/L H2O2. The 2 lower lines (superimposed) show myoblasts untransfected with caveolin-3 or TRPC1 superimposedon myoblasts transfected with both Cav-3 and TRPC1 but in the presence of PP2 (10 mmol/L). The upper line shows an increase in [Ca2+]i
in a cell transfected with both proteins. (B) Averaged data for experiments measuring the rise of [Ca2+]i after 2 min exposure to H2O2 underthe conditions shown. ***, Significantly different at p < 0.001 to all other experimental groups.
88 Can. J. Physiol. Pharmacol. Vol. 88, 2010
Published by NRC Research Press
Can
. J. P
hysi
ol. P
harm
acol
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
199.
201.
121.
12 o
n 06
/04/
13Fo
r pe
rson
al u
se o
nly.

tial for caveolae, we coexpressed caveolin-3 and TRPC1 andfound improved membrane localization of TRPC1 underthese circumstances (Fig. 5). We also demonstrated fluores-cence resonance energy transfer (FRET) between caveolin-3and TRPC1, showing that the proteins are closely colocal-ized and presumably binding partners. It is already knownthat caveolin-3 binds to b-dystroglycan (Sotgia et al. 2000);therefore, on the basis of the evidence that TRPC1 binds tocaveolin-3, we show in Fig. 1B caveolin-3 and TRPC1 aspart of the proteins associated with the DAC. Caveolin-3 isupregulated in DMD and the mdx mouse (Repetto et al.1999; Vaghy et al. 1998; Gervasio et al. 2008); TRPC1 isalso upregulated (Williams and Allen 2007; Gervasio et al.2008). Thus in Fig. 1B we show these 2 proteins to be upre-gulated in mdx muscle, whereas the proteins of the DACsare downregulated.
Above, we have presented findings that suggest the phys-iological stimulus to SACNSC is increased levels of ROS,and in particular, we have shown that H2O2 is capable ofcausing channel opening in mdx but not wild-type muscle.For this reason we tested whether H2O2 is capable of open-ing TRPC1 channels when expressed in C2 cells (Gervasioet al. 2008). We did this by measuring [Ca2+]i in the cellsafter 2 min exposure to 10 mmol/L H2O2, and the main re-sults are shown in Fig. 6. Note that exposure to H2O2 hadno effect on the intracellular calcium in cells transfectedwith either caveolin-3 or TRPC1 alone. Only when bothwere transfected together and there was good expression ofthe 2 proteins in the cell membrane did we observe a robustincrease in [Ca2+]i in response to H2O2. Because the mecha-nism by which H2O2 affects TRP channels is unclear, wetested whether src kinase is involved, since this kinase is ac-tivated by ROS (Chen et al. 2001). We first showed that srckinase was present in the cell line and activated by H2O2, asjudged by phosphorylation of the Y418 residue. Wethen used the specific src kinase inhibitor PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazole[3,4-d]pyrimidine) andshowed that in the presence of 10 mmol/L PP2, the H2O2-generated Ca2+ influx was inhibited. As a control, weshowed that the inactive analogue PP3 had no such effect.
Thus these experiments demonstrate that when TRPC1 iscotransfected with caveolin-3, it expresses in the membraneand can act as a Ca2+-permeable channel activated by srckinase when this has been activated by ROS. These resultsare consistent with SACNSC in the mdx muscle beingTRPC1, although they do not prove this to be the case.These experiments do not address the possible source of theROS, but we speculate that this is NADPH oxidase, as sug-gested by experiments on other SACs (Browe and Baum-garten 2004; Dyachenko et al. 2009b). NADPH oxidase isthought to be expressed in lipid rafts that provide the mem-brane for caveolae, so it is closely located to the other rele-vant proteins. As src kinase also binds to caveolin-3(Kawasaki et al. 2006), it appears that a number of proteinscritical to the pathogenic Ca2+ influx of DMD are colocatedin the caveolae (Fig. 1B). These observations all point to ab-normal expression of proteins within the signaling complexof the caveolae as making a critical contribution to the earlypathology of DMD.
ConclusionsThe role of excessive Ca2+ entry as a contributor to
stretch-induced muscle damage is well established. Weargue that this Ca2+ entry is through a channel with thepharmacology of a stretch-activated channel. The molecularidentity of this channel is uncertain and TRPC1, TRPC6,and TRPV2 are all candidates. The mechanism of activationof this channel is also uncertain, and we review our evi-dence that ROS, probably produced by NADPH oxidase,are an important precursor and that the ROS may well oper-ate by activation of src kinase, which directly activates thechannel. It has already been established that either blockingstretch-activated channels or using ROS scavengers amelio-rates the consequences of dystrophy in the mdx mouse. Thepathways of activation of the stretch-activated channel de-scribed here provide several new therapeutic targets that de-serve exploration.
AcknowledgementsWe are grateful to the National Health and Medical Re-
search Council of Australia for funds to support this work.E.W. acknowledges support from the Research Grants Coun-cil of the Hong Kong Special Administrative Region, China.
ReferencesAllen, D.G. 2001. Eccentric muscle damage: mechanisms of early
reduction of force. Acta Physiol. Scand. 171(3): 311–319.doi:10.1046/j.1365-201x.2001.00833.x. PMID:11412143.
Balnave, C.D., and Allen, D.G. 1995. Intracellular calcium andforce in single mouse muscle fibres following repeated contrac-tions with stretch. J. Physiol. 488(Pt 1): 25–36. PMID:8568662.
Beech, D.J., Xu, S.Z., McHugh, D., and Flemming, R. 2003.TRPC1 store-operated cationic channel subunit. Cell Calcium,33(5-6): 433–440. doi:10.1016/S0143-4160(03)00054-X. PMID:12765688.
Bergdahl, A., Gomez, M.F., Dreja, K., Xu, S.Z., Adner, M., Beech,D.J., et al. 2003. Cholesterol depletion impairs vascular reactiv-ity to endothelin-1 by reducing store-operated Ca2+ entry depen-dent on TRPC1. Circ. Res. 93(9): 839–847. doi:10.1161/01.RES.0000100367.45446.A3. PMID:14551243.
Bodensteiner, J.B., and Engel, A.G. 1978. Intracellular calcium ac-cumulation in Duchenne dystrophy and other myopathies: astudy of 567,000 muscle fibers in 114 biopsies. Neurology,28(5): 439–446. PMID:76996.
Browe, D.M., and Baumgarten, C.M. 2004. Angiotensin II (AT1)receptors and NADPH oxidase regulate Cl– current elicited byb1 integrin stretch in rabbit ventricular myocytes. J. Gen. Phy-siol. 124(3): 273–287. doi:10.1085/jgp.200409040. PMID:15337822.
Charge, S.B., and Rudnicki, M.A. 2004. Cellular and molecularregulation of muscle regeneration. Physiol. Rev. 84(1): 209–238. doi:10.1152/physrev.00019.2003. PMID:14715915.
Chen, K., Vita, J.A., Berk, B.C., and Keaney, J.F., Jr. 2001. c-JunN-terminal kinase activation by hydrogen peroxide in endothe-lial cells involves SRC-dependent epidermal growth factor re-ceptor transactivation. J. Biol. Chem. 276(19): 16045–16050.doi:10.1074/jbc.M011766200. PMID:11278982.
Davies, K.E., and Nowak, K.J. 2006. Molecular mechanisms ofmuscular dystrophies: old and new players. Nat. Rev. Mol. CellBiol. 7(10): 762–773. doi:10.1038/nrm2024. PMID:16971897.
Deconinck, N., Ragot, T., Marechal, G., Perricaudet, M., and Gillis,J.M. 1996. Functional protection of dystrophic mouse (mdx)
Allen et al. 89
Published by NRC Research Press
Can
. J. P
hysi
ol. P
harm
acol
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
199.
201.
121.
12 o
n 06
/04/
13Fo
r pe
rson
al u
se o
nly.

muscles after adenovirus-mediated transfer of a dystrophin mini-gene. Proc. Natl. Acad. Sci. U.S.A. 93(8): 3570–3574. doi:10.1073/pnas.93.8.3570. PMID:8622977.
Ducret, T., Vandebrouck, C., Cao, M.L., Lebacq, J., and Gailly, P.2006. Functional role of store-operated and stretch-activatedchannels in murine adult skeletal muscle fibres. J. Physiol.575(Pt 3): 913–924. doi:10.1113/jphysiol.2006.115154. PMID:16825296.
Dyachenko, V., Husse, B., Rueckschloss, U., and Isenberg, G.2009a. Mechanical deformation of ventricular myocytes modu-lates both TRPC6 and Kir2.3 channels. Cell Calcium, 45(1):38–54. doi:10.1016/j.ceca.2008.06.003. PMID:18635261.
Dyachenko, V., Rueckschloss, U., and Isenberg, G. 2009b. Modula-tion of cardiac mechanosensitive ion channels involves superox-ide, nitric oxide and peroxynitrite. Cell Calcium, 45(1): 55–64.doi:10.1016/j.ceca.2008.06.002. PMID:18639930.
Emery, A.E., and Muntoni, F. 2003. Duchenne muscular dystrophy.3rd ed. Oxford University Press.
Ervasti, J.M., and Campbell, K.P. 1991. Membrane organization ofthe dystrophin-glycoprotein complex. Cell, 66(6): 1121–1131.doi:10.1016/0092-8674(91)90035-W. PMID:1913804.
Franco-Obregon, A., and Lansman, J.B. 2002. Changes in mechan-osensitive channel gating following mechanical stimulation inskeletal muscle myotubes from the mdx mouse. J. Physiol.539(Pt 2): 391–407. doi:10.1113/jphysiol.2001.013043. PMID:11882673.
Froehner, S.C. 2002. Just say NO to muscle degeneration? TrendsMol. Med. 8(2): 51–53. doi:10.1016/S1471-4914(02)02275-X.PMID:11815264.
Gervasio, O.L., Whitehead, N.P., Yeung, E.W., Phillips, W.D., andAllen, D.G. 2008. TRPC1 binds to caveolin-3 and is regulatedby Src kinase: role in Duchenne muscular dystrophy. J. CellSci. 121(Pt 13): 2246–2255. doi:10.1242/jcs.032003. PMID:18544631.
Gottlieb, P., Folgering, J., Maroto, R., Raso, A., Wood, T.G., Kur-osky, A., et al. 2008. Revisiting TRPC1 and TRPC6 mechano-sensitivity. Pflugers Arch. 455(6): 1097–1103. doi:10.1007/s00424-007-0359-3. PMID:17957383.
Groschner, K., Rosker, C., and Lukas, M. 2004. Role of TRP chan-nels in oxidative stress. Novartis Found. Symp. 258: 222–230,discussion 231–235, 263–266. doi:10.1002/0470862580.ch16.PMID:15104185.
Guharay, F., and Sachs, F. 1984. Stretch-activated single ion chan-nel currents in tissue-cultured embryonic chick skeletal muscle.J. Physiol. 352: 685–701. PMID:6086918.
Hamill, O.P., and McBride, D.W., Jr. 1996. The pharmacology ofmechanogated membrane ion channels. Pharmacol. Rev. 48(2):231–252. PMID:8804105.
Head, S.I., Williams, D.A., and Stephenson, D.G. 1992. Abnormal-ities in structure and function of limb skeletal muscle fibres ofdystrophic mdx mice. Proc Biol Sci, 248(1322): 163–169.doi:10.1098/rspb.1992.0058. PMID:1352891.
Hoffman, E.P., Brown, R.H., Jr., and Kunkel, L.M. 1987. Dystro-phin: the protein product of the Duchenne muscular dystrophylocus. Cell, 51(6): 919–928. doi:10.1016/0092-8674(87)90579-4.PMID:3319190.
Iwata, Y., Katanosaka, Y., Arai, Y., Komamura, K., Miyatake, K.,and Shigekawa, M. 2003. A novel mechanism of myocyte de-generation involving the Ca2+-permeable growth factor-regulatedchannel. J. Cell Biol. 161(5): 957–967. doi:10.1083/jcb.200301101. PMID:12796481.
Iwata, Y., Katanosaka, Y., Arai, Y., Shigekawa, M., and Waka-bayashi, S. 2009. Dominant-negative inhibition of Ca2+ influx
via TRPV2 ameliorates muscular dystrophy in animal models.Hum. Mol. Genet. 18(5): 824–834. PMID:19050039.
Kamkin, A., Kiseleva, I., and Isenberg, G. 2003. Ion selectivity ofstretch-activated cation currents in mouse ventricular myocytes.Pflugers Arch. 446(2): 220–231. PMID:12739160.
Kanzaki, M., Nie, L., Shibata, H., and Kojima, I. 1997. Activationof a calcium-permeable cation channel CD20 expressed in Balb/c 3T3 cells by insulin-like growth factor-I. J. Biol. Chem.272(8): 4964–4969. doi:10.1074/jbc.272.8.4964. PMID:9030557.
Kawasaki, B.T., Liao, Y., and Birnbaumer, L. 2006. Role of Src inC3 transient receptor potential channel function and evidence fora heterogeneous makeup of receptor- and store-operated Ca2+
entry channels. Proc. Natl. Acad. Sci. U.S.A. 103(2): 335–340.doi:10.1073/pnas.0508030102. PMID:16407161.
Kunkel, L.M., Monaco, A.P., Middlesworth, W., Ochs, H.D., andLatt, S.A. 1985. Specific cloning of DNA fragments absentfrom the DNA of a male patient with an X chromosome dele-tion. Proc. Natl. Acad. Sci. U.S.A. 82(14): 4778–4782. doi:10.1073/pnas.82.14.4778. PMID:2991893.
Lintschinger, B., Balzer-Geldsetzer, M., Baskaran, T., Graier, W.F.,Romanin, C., Zhu, M.X., and Groschner, K. 2000. Coassemblyof Trp1 and Trp3 proteins generates diacylglycerol- and Ca2+-sensitive cation channels. J. Biol. Chem. 275(36): 27799–27805.PMID:10882720.
Maroto, R., Raso, A., Wood, T.G., Kurosky, A., Martinac, B., andHamill, O.P. 2005. TRPC1 forms the stretch-activated cationchannel in vertebrate cells. Nat. Cell Biol. 7(2): 179–185.doi:10.1038/ncb1218. PMID:15665854.
Miyake, K., and McNeil, P.L. 2003. Mechanical injury and repairof cells. Crit. Care Med. 31(8 Suppl): S496–S501. doi:10.1097/01.CCM.0000081432.72812.16. PMID:12907878.
Mokri, B., and Engel, A.G. 1975. Duchenne dystrophy: electronmicroscopic findings pointing to a basic or early abnormality inthe plasma membrane of the muscle fiber. Neurology, 25(12):1111–1120. PMID:1105232.
Morgan, D.L., and Allen, D.G. 1999. Early events in stretch-in-duced muscle damage. J. Appl. Physiol. 87(6): 2007–2015.PMID:10601142.
Nakamura, T.Y., Iwata, Y., Sampaolesi, M., Hanada, H., Saito, N.,Artman, M., et al. 2001. Stretch-activated cation channels inskeletal muscle myotubes from sarcoglycan-deficient hamsters.Am. J. Physiol. Cell Physiol. 281(2): C690–C699. PMID:11443068.
Petrof, B.J., Shrager, J.B., Stedman, H.H., Kelly, A.M., and Swee-ney, H.L. 1993. Dystrophin protects the sarcolemma from stres-ses developed during muscle contraction. Proc. Natl. Acad. Sci.U.S.A. 90(8): 3710–3714. doi:10.1073/pnas.90.8.3710. PMID:8475120.
Poteser, M., Graziani, A., Rosker, C., Eder, P., Derler, I., Kahr, H.,et al. 2006. TRPC3 and TRPC4 associate to form a redox-sensi-tive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J. Biol.Chem. 281(19): 13588–13595. doi:10.1074/jbc.M512205200.PMID:16537542.
Rando, T.A. 2001. The dystrophin-glycoprotein complex, cellularsignaling, and the regulation of cell survival in the musculardystrophies. Muscle Nerve, 24(12): 1575–1594. doi:10.1002/mus.1192. PMID:11745966.
Rando, T.A. 2002. Oxidative stress and the pathogenesis of muscu-lar dystrophies. Am. J. Phys. Med. Rehabil. 81(11 Suppl): S175–S186. doi:10.1097/00002060-200211001-00018. PMID:12409822.
Repetto, S., Bado, M., Broda, P., Lucania, G., Masetti, E., Sotgia,F., et al. 1999. Increased number of caveolae and caveolin-3
90 Can. J. Physiol. Pharmacol. Vol. 88, 2010
Published by NRC Research Press
Can
. J. P
hysi
ol. P
harm
acol
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
199.
201.
121.
12 o
n 06
/04/
13Fo
r pe
rson
al u
se o
nly.

overexpression in Duchenne muscular dystrophy. Biochem. Bio-phys. Res. Commun. 261(3): 547–550. doi:10.1006/bbrc.1999.1055. PMID:10441463.
Sotgia, F., Lee, J.K., Das, K., Bedford, M., Petrucci, T.C., Ma-cioce, P., et al. 2000. Caveolin-3 directly interacts with the C-terminal tail of b-dystroglycan. Identification of a central WW-like domain within caveolin family members. J. Biol. Chem.275(48): 38048–38058. doi:10.1074/jbc.M005321200. PMID:10988290.
Suchyna, T.M., and Sachs, F. 2007. Mechanosensitive channelproperties and membrane mechanics in mouse dystrophic myo-tubes. J. Physiol. 581(Pt 1): 369–387. doi:10.1113/jphysiol.2006.125021. PMID:17255168.
Suchyna, T.M., Johnson, J.H., Hamer, K., Leykam, J.F., Gage,D.A., Clemo, H.F., et al. 2000. Identification of a peptide toxinfrom Grammostola spatulata spider venom that blocks cation-selective stretch-activated channels. J. Gen. Physiol. 115(5):583–598. doi:10.1085/jgp.115.5.583. PMID:10779316.
Tutdibi, O., Brinkmeier, H., Rudel, R., and Fohr, K.J. 1999. In-creased calcium entry into dystrophin-deficient muscle fibres ofMDX and ADR-MDX mice is reduced by ion channel blockers.J. Physiol. 515(Pt 3): 859–868. doi:10.1111/j.1469-7793.1999.859ab.x. PMID:10066910.
Vaghy, P.L., Fang, J., Wu, W., and Vaghy, L.P. 1998. Increasedcaveolin-3 levels in mdx mouse muscles. FEBS Lett. 431(1):125–127. doi:10.1016/S0014-5793(98)00738-8. PMID:9684879.
Vandebrouck, C., Martin, D., Colson-Van Schoor, M., Debaix, H.,and Gailly, P. 2002. Involvement of TRPC in the abnormal cal-cium influx observed in dystrophic (mdx) mouse skeletal musclefibers. J. Cell Biol. 158(6): 1089–1096. doi:10.1083/jcb.200203091. PMID:12235126.
Verburg, E., Dutka, T.L., and Lamb, G.D. 2006. Long-lasting mus-cle fatigue: partial disruption of excitation-contraction couplingby elevated cytosolic Ca2+ concentration during contractions.Am. J. Physiol. Cell Physiol. 290(4): C1199–C1208. doi:10.1152/ajpcell.00469.2005. PMID:16306125.
Warren, G.L., Ingalls, C.P., Lowe, D.A., and Armstrong, R.B.2001. Excitation-contraction uncoupling: major role in contrac-tion-induced muscle injury. Exerc. Sport Sci. Rev. 29(2): 82–87. doi:10.1097/00003677-200104000-00008. PMID:11337828.
Whitehead, N.P., Streamer, M., Lusambili, L.I., Sachs, F., and Al-len, D.G. 2006. Streptomycin reduces stretch-induced membranepermeability in muscles from mdx mice. Neuromuscul. Disord.16(12): 845–854. doi:10.1016/j.nmd.2006.07.024. PMID:17005404.
Williams, I.A., and Allen, D.G. 2007. Intracellular calcium hand-ling in ventricular myocytes from mdx mice. Am. J. Physiol.Heart Circ. Physiol. 292(2): H846–H855. doi:10.1152/ajpheart.00688.2006. PMID:17012353.
Yeung, E.W., Ballard, H.J., Bourreau, J.P., and Allen, D.G. 2003.Intracellular sodium in mammalian muscle fibers after eccentriccontractions. J. Appl. Physiol. 94(6): 2475–2482. PMID:12588791.
Yeung, E.W., Whitehead, N.P., Suchyna, T.M., Gottlieb, P.A.,Sachs, F., and Allen, D.G. 2005. Effects of stretch-activatedchannel blockers on [Ca2+]i and muscle damage in the mdxmouse. J. Physiol. 562(Pt 2): 367–380. doi:10.1113/jphysiol.2004.075275. PMID:15528244.
Zeng, T., Bett, G.C., and Sachs, F. 2000. Stretch-activated wholecell currents in adult rat cardiac myocytes. Am. J. Physiol. HeartCirc. Physiol. 278(2): H548–H557. PMID:10666087.
Allen et al. 91
Published by NRC Research Press
Can
. J. P
hysi
ol. P
harm
acol
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
199.
201.
121.
12 o
n 06
/04/
13Fo
r pe
rson
al u
se o
nly.




















