
Facioscapulohumeral muscular dystrophy (FSHD): an enigma unravelled?
Upload
a-hikkoshiCategory
view
1download
0
Journal of the Neurological Sciences 297 (2010) 19–28
Contents lists available at ScienceDirect
Journal of the Neurological Sciences
j ourna l homepage: www.e lsev ie r.com/ locate / jns
Elevated plasma levels of tissue inhibitors of metalloproteinase-1 and theiroverexpression in muscle in human and mouse muscular dystrophy
Guilian Sun a,b, Kazuhiro Haginoya a,c,⁎, Yoko Chiba a, Mitsugu Uematsu a, Naomi Hino-Fukuyo a,Soichiro Tanaka c, Akira Onuma c, Kazuie Iinuma d, Shigeru Tsuchiya a
a Department of Pediatrics, Tohoku University School of Medicine, Sendai, Japanb Department of Pediatrics, The First Affiliated Hospital, China Medical University, Shenyang 110001, Chinac Department of Pediatric Neurology, Takuto Rehabilitation Center for Children, Sendai, Japand Ishinomaki Red-Cross Hospital, Ishinomaki, Japan
⁎ Corresponding author. Department of Pediatrics,Medicine, 1-1 Seiryomachi, Aobaku, Sendai 980-8574,fax: +81 22 717 7290.
E-mail address: [email protected] (K. Hagino
0022-510X/$ – see front matter © 2010 Elsevier B.V. Adoi:10.1016/j.jns.2010.06.031
a b s t r a c t
a r t i c l e i n f oArticle history:Received 26 December 2009Received in revised form 21 June 2010Accepted 30 June 2010Available online 23 July 2010
Keywords:Muscular dystrophyTissue inhibitors of metalloproteinaseFibrosisTransforming growth factor-β1Neuromuscular junction
To investigate the role of tissue inhibitors of metalloproteinases (TIMPs) in muscular dystrophy, weexamined the expression of TIMP-1 using plasma and biopsied muscle from patients with various musculardystrophies by ELISA, immunohistochemistry, and Western blot analysis. TIMP-1 immunolocalization wasalso studied in mouse models of muscular dystrophy. Plasma TIMP-1 was elevated and correlated with TGF-β1 in Duchenne muscular dystrophy (DMD) and congenital muscular dystrophy (CMD), but not in Beckermuscular dystrophy. In dystrophic human muscles, TIMP-1 was immunopositive in the regenerating andnon-regenerating muscle fibers, and interstitial cells that consist of activated fibroblasts and macrophages.TIMP-1 immunoreactivity was also closely associated with TGF-β1. Western blot analysis showed elevatedTIMP-1 protein in muscles in DMD. The semiquantitative analysis of TIMP-1 staining intensity and tissuefibrosis showed that TIMP-1 immunoreactivity is closely associated with the extent of tissue fibrosis inhuman and mouse dystrophic muscles. In conclusion, the present study implied that the TGF-β1–TIMP-1pathway is activated in dystrophic muscles and the overexpression of TIMP-1 may result in increaseddeposition of extracellular matrix leading to tissue fibrosis.
Tohoku University School ofJapan. Tel.: +81 22 717 7287;
ya).
ll rights reserved.
© 2010 Elsevier B.V. All rights reserved.
1. Introduction
Recent investigations into the genetic background of varioushuman muscular dystrophies have offered insights into the mecha-nism that controls muscle fiber degeneration. However, manyunanswered questions remain as to why and how musculardystrophies progress, despite the potential for vigorous muscle fiberregeneration. Key factors in the pathogenesis of muscular dystrophyare reduced satellite cell performance andmuscle regeneration, whichresult in fibrotic tissue replacement. Transforming growth factor-beta1 (TGF-β1) appears to be the most important mediator of bothconnective tissue proliferation and the regenerative failure indystrophic muscle [1]. TGF-β1 is overexpressed in the muscles ofmusclar dystrophy [1] and affects skeletal muscle cells directly toinduce collagen and fibronectin synthesis in rat myoblasts [2]. Arecent study showed that an anti-TGF-β1 antibody ameliorated TGF-β1-induced failure in muscle regeneration in a mouse model of
muscular dystrophy [3], demonstrating a major pathophysiologicalcontribution of TGF-β1 in the progression of muscular dystrophy.
The tissue inhibitors of metalloproteinases (TIMPs) are a family ofsecreted proteins that are endogeneous inhibitors of matrix metallo-proteinases (MMPs), including collagenases, stromelysins, gelati-nases, and membrane-type MMPs [4–6]. The balance betweenMMPs and TIMPs regulates ECM turnover and remodeling duringnormal development and tissue repair [4,6]. Loss of control of theMMP/TIMP balance is now well established as a causative factor inmany diseases that involve pathological tissue destruction and fibroticprocesses. To date, four mammalian TIMPs have been identified(TIMP-1, -2, -3, and -4). TIMP-1, a 28-kDa glycosylated protein, is themost ubiquitous, and binds non-covalently in a 1:1 complex withMMPs. TIMP-1 is capable of inhibiting in vitro the activities of allknown MMPs, but with different binding affinities; TIMP-1 is knownto have a high affinity for MMP-9 [7]. TIMPs have been reported to beinvolved in the inhibition of cancer cell and inflammatory cellinvasion [4], arthritis, liver cirrhosis [4,8], pulmonary fibrosis [9],human renal allograft interstitial fibrosis [10], hepatic fibrosis [11],and valvular disease of the heart [12].
Several studies have demonstrated that TGF-β1 increases theexpression of TIMP-1 [13,14]. TGF-β1 was identified as an importantregulator of MMP and TIMP expression in hepatic stellate cells (HSCs)
20 G. Sun et al. / Journal of the Neurological Sciences 297 (2010) 19–28
in vitro and is known to be involved in hepatic tissue repair [15,16].The fibrotic effect of TGF-β1 in lung fibroblasts results in an increase inthe secretion and deposition of total ECM and collagens because of adecrease in MMP-1 secretion and an increase in TIMP-1expression [14].
TIMPs have also been shown to possess biological propertiesindependent of MMP-inhibitory activity, including stimulation of cellproliferation in a wide array of cell types, including normalkeratinocytes, fibroblasts, breast cancer cells, and osteosarcoma cells[6,17,18], inhibition of apoptosis [19], inhibition of angiogenesis [20],and induction of MMP expression in vitro [21]. TIMP-1 has beenshown to induce the proliferation of skin fibroblasts in an autocrinefashion, suggesting that TIMPs themselves possess fibrogenic activity[22].
Given these properties, TIMPs may play a pathogenic role inmuscular dystrophy, but the possible roles of MMPs and TIMPs in thepathogenesis of muscular dystrophy remain unclear. Recent studiesrevealed that MMP-2 and MMP-9 are involved in the process ofmuscular degeneration and regeneration in human inflammatorymyopathies and muscular dystrophy animal models [23–25]. MMP-9expression is related to the inflammatory response and likely to theactivation of satelllite cells, whereas MMP-2 activation occursconcomitantly with the regeneration of new myofibers [23–25].Increased levels of TIMP-1 mRNA have been reported in muscle tissueand cultured myotubes in Duchenne muscular dystrophy (DMD)[26,27]. However, to our knowledge, no detailed immunolocalizationstudy of TIMP-1 in dystrophic muscles has previously been reported.The plasma levels of TIMP-1 in various types of human musculardystrophy have also not been examined.
Thus, we examined the expression of TIMP-1 in plasma andmusclebiopsies obtained from patients with various kinds of musculardystrophy by ELISA, immunohistochemistry, and Western blotanalysis. In addition to the human muscular dystrophies, we alsoperformed an immunolocalization study of TIMP-1 in a murinemuscular dystrophy model.
Table 1Clinical summary of the patients.
Patient Diagnosis Sex Age ofonset (y)
Age atbiopsy (y)
Clinicalsymptomsat biopsy
Serum CK(IU/L)
1 DMD M No symptom 2 HyperCKemia 16,3402 DMD M 4 8 Abnormal gait 13,3603 DMD M 4 5 Frequent falling
down14,550
4 DMD M 2 3.1 Hypotonia 13,5965 DMD M 0.8 2.7 unsteady gait 18,6006 DMD M 3 8 Abnormal gait 13,6007 DMD M 1.5 3.5 Abnormal gait 16,1308 DMD M 0 0.9 Hypotonia 23,3009 BMD M 5 7 Muscle pain 384410 BMD M No symptom 5 HyperCKemia 184711 BMD M No symptom 3.9 HyperCKemia 500012 CMDa F 1.5 2.2 Gait disturbance 96513 CMDa M 0 0.1 Floppy infant 246314 CMDa M 0 0.4 Floppy infant 863015 CMDa M 0.3 0.7 Hypotonia 609216 CMDb M 0.5 0.8 Motor delay 631217 CMDb F 0.4 18 Motor delay 30018 CMDb F 0.2 0.6 Hypotonia 360019 CMDb M 0.5 2.7 Motor delay 184720 CMDb M 0.3 7 Motor delay 900021 CMDb M 0.4 7 Hypotonia 393022 CMDb F 0.1 0.7 Floppy infant 272623 SMA M 1.2 1.8 Motor delay b20024 SMA M 0.2 0.4 Floppy infant b20025 CCD F 0.9 9 Abnormal gait 20626 MTM M 0 0.8 Floppy infant 67
DMD: Duchenne muscular dystrophy, BMD: Becker muscular dystrophy, CMDa:merosin-positve congenital muscular dystrophy, CMDb: Fukuyama-type congenitalmuscular dystrophy; SMA: spinal muscular atrophy, CCD: central core disease, MTM:myotubular myopathy, and CK: creatine kinase.
2. Materials and methods
2.1. ELISA to determine of plasma TIMP-1 and TGF-β1
Blood samples were collected in tubes containing EDTA-Na2 usingstandard venipuncture techniques, after obtaining informed consentfrom the patients' families. Cellular elements and platelets wereremoved by centrifugation (3000 rpm, 10 min, 4 °C). All sampleswerestored at or below −20 °C until measurements were made.
Concentrations of TIMP-1 were determined with commercialsandwich-type ELISA kits (Fuji Chemical Industries, Takaoka, Japan).Plasma TGF-β1 was determined using the Predicta TGF-β1 ELISA kitpurchased from Genzyme (Cambridge, MA, USA) as describedpreviously [28]. These assays were carried out according to thesuppliers' instructions. The detection limits for TIMP-1 and TGF-β1were 5 ng/mL and 0.5 ng/mL, respectively.
We studied plasma samples from 26 patients with DMD (1–15 years old), 14 with congenital muscular dystrophy (CMD)including 8 with Fukuyama-type CMD (FCMD; 1–13.8 years old)and 6 with merosin-positive CMD (0.5–12.7 years old), and 9 withBecker muscular dystrophy (BMD; 4–15 years old). The controlsample included 41 patients with non-muscle disorders, including33 with epilepsy (0.5–16 years old), four with mental retardation(2.5–15.8 years old), and one each with tic, cranial nerve palsy,headache, and focal scleroderma.
The results are expressed as the means±SEM and were comparedusing the Mann–Whitney U-test. Values of pb0.05 were deemed to bestatistically significant. Statistical analyses were performed usingStatView (J5.0; SAS Inc., Cary, NC, USA).
2.2. TIMP-1 immunohistochemistry
We studied diagnostic muscle biopsies from 27 patients aged 0.1–18 years. We obtained written informed consent from each patient'sfamily to use these specimens for research as well as diagnosis. Theinstitutional ethics committee at Tohoku University School ofMedicine (Japan) approved this study. Among the biopsies collected,we identified eight patients with DMD, seven with FCMD, four withmerosin-positive CMD, three with BMD, two with spinal muscularatrophy, one with central core disease, and one with myotubularmyopathy; five patients were normal. Diagnosis was based on clinical,laboratory, muscle biopsy histochemistry, dystrophin and merosinimmunohistochemistry, multiplex PCR for the dystrophin gene, andmutational analysis of the Fukutin gene. A clinical summary of thepatients is presented in Table 1.
We also obtained muscle samples from three different strains ofmice: a control strain (B-10), dy/dy (a model of merosin-deficientcongenital muscular dystrophy), and mdx (a model of DMD). Theprotocol for the use of the mice was approved by the AnimalExperimentation Committee of Tohoku University School of Medicine.We used samples of hamstring muscle that were obtained from two16-week-old mice of each strain after the animals had been killed bycervical dislocation. At this age, mdx mice show almost normal dailyactivity with hypertrophied skeletal muscles, whereas dy/dy miceshow profound limb weakness with difficulty in crawling withseverely atrophied skeletal muscles.
All muscle samples were frozen immediately in isopentane andstored at −80 °C until used for immunohistochemistry. Transversesections (10 μm) of fresh-frozen muscle tissue were used forimmunohistochemistry. For immunodetection of TIMP-1, we useda specific rabbit anti-human TIMP-1 antibody (1:1000 dilution;Chemicon, Temecula, CA, USA) as the primary antibody. Sectionsof muscle tissue were stained using indirect immunoperoxidase
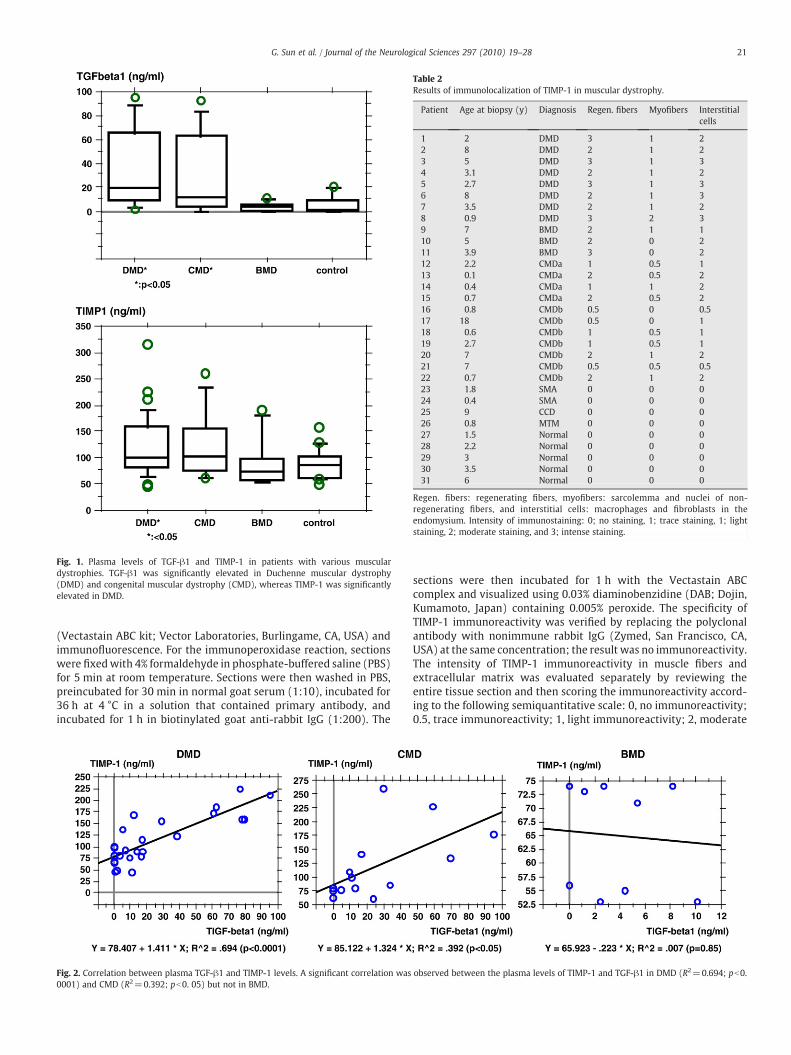
Fig. 1. Plasma levels of TGF-β1 and TIMP-1 in patients with various musculardystrophies. TGF-β1 was significantly elevated in Duchenne muscular dystrophy(DMD) and congenital muscular dystrophy (CMD), whereas TIMP-1 was significantlyelevated in DMD.
Table 2Results of immunolocalization of TIMP-1 in muscular dystrophy.
Patient Age at biopsy (y) Diagnosis Regen. fibers Myofibers Interstitialcells
1 2 DMD 3 1 22 8 DMD 2 1 23 5 DMD 3 1 34 3.1 DMD 2 1 25 2.7 DMD 3 1 36 8 DMD 2 1 37 3.5 DMD 2 1 28 0.9 DMD 3 2 39 7 BMD 2 1 110 5 BMD 2 0 211 3.9 BMD 3 0 212 2.2 CMDa 1 0.5 113 0.1 CMDa 2 0.5 214 0.4 CMDa 1 1 215 0.7 CMDa 2 0.5 216 0.8 CMDb 0.5 0 0.517 18 CMDb 0.5 0 118 0.6 CMDb 1 0.5 119 2.7 CMDb 1 0.5 120 7 CMDb 2 1 221 7 CMDb 0.5 0.5 0.522 0.7 CMDb 2 1 223 1.8 SMA 0 0 024 0.4 SMA 0 0 025 9 CCD 0 0 026 0.8 MTM 0 0 027 1.5 Normal 0 0 028 2.2 Normal 0 0 029 3 Normal 0 0 030 3.5 Normal 0 0 031 6 Normal 0 0 0
Regen. fibers: regenerating fibers, myofibers: sarcolemma and nuclei of non-regenerating fibers, and interstitial cells: macrophages and fibroblasts in theendomysium. Intensity of immunostaining: 0; no staining, 1; trace staining, 1; lightstaining, 2; moderate staining, and 3; intense staining.
21G. Sun et al. / Journal of the Neurological Sciences 297 (2010) 19–28
(Vectastain ABC kit; Vector Laboratories, Burlingame, CA, USA) andimmunofluorescence. For the immunoperoxidase reaction, sectionswere fixedwith 4% formaldehyde in phosphate-buffered saline (PBS)for 5 min at room temperature. Sections were then washed in PBS,preincubated for 30 min in normal goat serum (1:10), incubated for36 h at 4 °C in a solution that contained primary antibody, andincubated for 1 h in biotinylated goat anti-rabbit IgG (1:200). The
Fig. 2. Correlation between plasma TGF-β1 and TIMP-1 levels. A significant correlation was0001) and CMD (R2=0.392; pb0. 05) but not in BMD.
sections were then incubated for 1 h with the Vectastain ABCcomplex and visualized using 0.03% diaminobenzidine (DAB; Dojin,Kumamoto, Japan) containing 0.005% peroxide. The specificity ofTIMP-1 immunoreactivity was verified by replacing the polyclonalantibody with nonimmune rabbit IgG (Zymed, San Francisco, CA,USA) at the same concentration; the result was no immunoreactivity.The intensity of TIMP-1 immunoreactivity in muscle fibers andextracellular matrix was evaluated separately by reviewing theentire tissue section and then scoring the immunoreactivity accord-ing to the following semiquantitative scale: 0, no immunoreactivity;0.5, trace immunoreactivity; 1, light immunoreactivity; 2, moderate
observed between the plasma levels of TIMP-1 and TGF-β1 in DMD (R2=0.694; pb0.
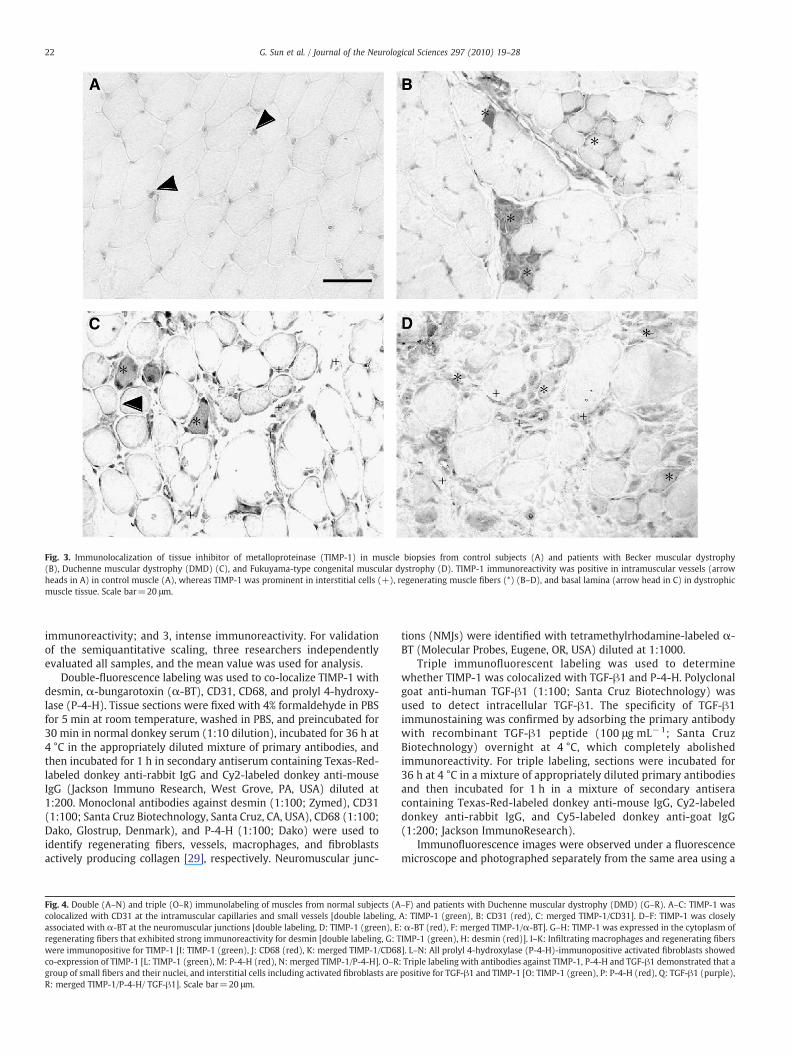
Fig. 3. Immunolocalization of tissue inhibitor of metalloproteinase (TIMP-1) in muscle biopsies from control subjects (A) and patients with Becker muscular dystrophy(B), Duchenne muscular dystrophy (DMD) (C), and Fukuyama-type congenital muscular dystrophy (D). TIMP-1 immunoreactivity was positive in intramuscular vessels (arrowheads in A) in control muscle (A), whereas TIMP-1 was prominent in interstitial cells (+), regenerating muscle fibers (*) (B–D), and basal lamina (arrow head in C) in dystrophicmuscle tissue. Scale bar=20 μm.
22 G. Sun et al. / Journal of the Neurological Sciences 297 (2010) 19–28
immunoreactivity; and 3, intense immunoreactivity. For validationof the semiquantitative scaling, three researchers independentlyevaluated all samples, and the mean value was used for analysis.
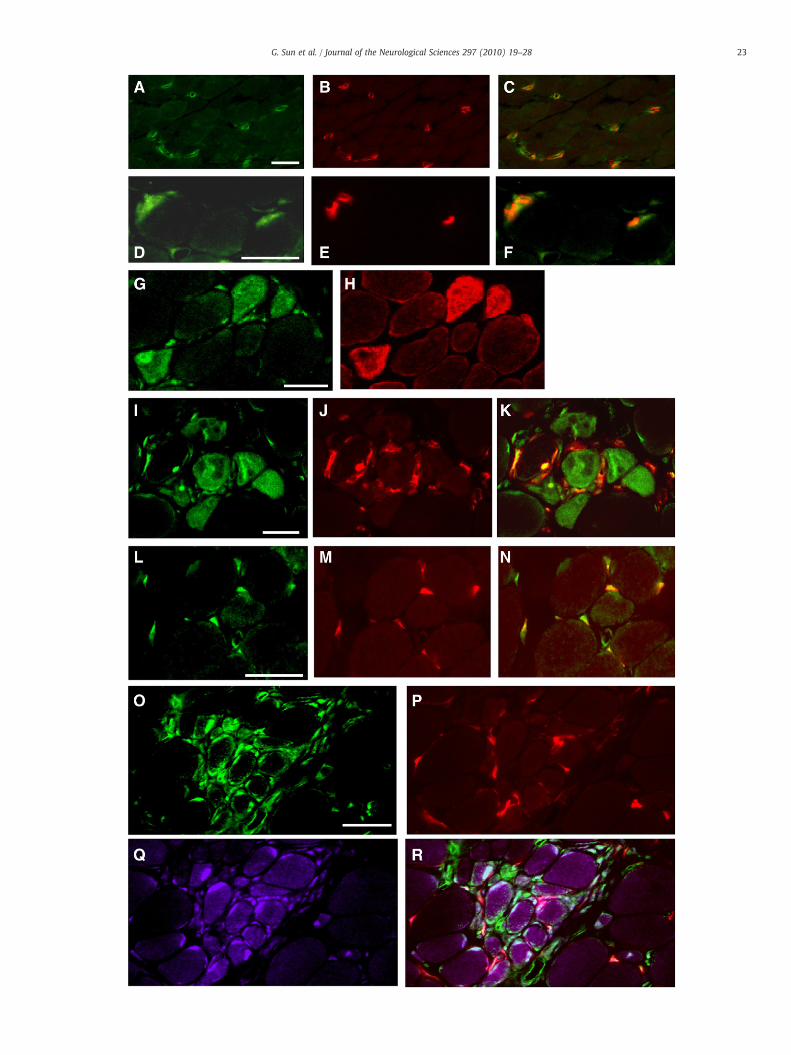
Double-fluorescence labeling was used to co-localize TIMP-1 withdesmin, α-bungarotoxin (α-BT), CD31, CD68, and prolyl 4-hydroxy-lase (P-4-H). Tissue sections were fixed with 4% formaldehyde in PBSfor 5 min at room temperature, washed in PBS, and preincubated for30 min in normal donkey serum (1:10 dilution), incubated for 36 h at4 °C in the appropriately diluted mixture of primary antibodies, andthen incubated for 1 h in secondary antiserum containing Texas-Red-labeled donkey anti-rabbit IgG and Cy2-labeled donkey anti-mouseIgG (Jackson Immuno Research, West Grove, PA, USA) diluted at1:200. Monoclonal antibodies against desmin (1:100; Zymed), CD31(1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA), CD68 (1:100;Dako, Glostrup, Denmark), and P-4-H (1:100; Dako) were used toidentify regenerating fibers, vessels, macrophages, and fibroblastsactively producing collagen [29], respectively. Neuromuscular junc-
Fig. 4. Double (A–N) and triple (O–R) immunolabeling of muscles from normal subjects (Acolocalized with CD31 at the intramuscular capillaries and small vessels [double labeling,associated with α-BT at the neuromuscular junctions [double labeling, D: TIMP-1 (green), Eregenerating fibers that exhibited strong immunoreactivity for desmin [double labeling, G: Twere immunopositive for TIMP-1 [I: TIMP-1 (green), J: CD68 (red), K: merged TIMP-1/CD68co-expression of TIMP-1 [L: TIMP-1 (green), M: P-4-H (red), N: merged TIMP-1/P-4-H]. O–Rgroup of small fibers and their nuclei, and interstitial cells including activated fibroblasts areR: merged TIMP-1/P-4-H/ TGF-β1]. Scale bar=20 μm.
tions (NMJs) were identified with tetramethylrhodamine-labeled α-BT (Molecular Probes, Eugene, OR, USA) diluted at 1:1000.
Triple immunofluorescent labeling was used to determinewhether TIMP-1 was colocalized with TGF-β1 and P-4-H. Polyclonalgoat anti-human TGF-β1 (1:100; Santa Cruz Biotechnology) wasused to detect intracellular TGF-β1. The specificity of TGF-β1immunostaining was confirmed by adsorbing the primary antibodywith recombinant TGF-β1 peptide (100 μg mL− 1; Santa CruzBiotechnology) overnight at 4 °C, which completely abolishedimmunoreactivity. For triple labeling, sections were incubated for36 h at 4 °C in a mixture of appropriately diluted primary antibodiesand then incubated for 1 h in a mixture of secondary antiseracontaining Texas-Red-labeled donkey anti-mouse IgG, Cy2-labeleddonkey anti-rabbit IgG, and Cy5-labeled donkey anti-goat IgG(1:200; Jackson ImmunoResearch).
Immunofluorescence images were observed under a fluorescencemicroscope and photographed separately from the same area using a
–F) and patients with Duchenne muscular dystrophy (DMD) (G–R). A–C: TIMP-1 wasA: TIMP-1 (green), B: CD31 (red), C: merged TIMP-1/CD31]. D–F: TIMP-1 was closely: α-BT (red), F: merged TIMP-1/α-BT]. G–H: TIMP-1 was expressed in the cytoplasm ofIMP-1 (green), H: desmin (red)]. I–K: Infiltrating macrophages and regenerating fibers]. L–N: All prolyl 4-hydroxylase (P-4-H)-immunopositive activated fibroblasts showed: Triple labeling with antibodies against TIMP-1, P-4-H and TGF-β1 demonstrated that apositive for TGF-β1 and TIMP-1 [O: TIMP-1 (green), P: P-4-H (red), Q: TGF-β1 (purple),
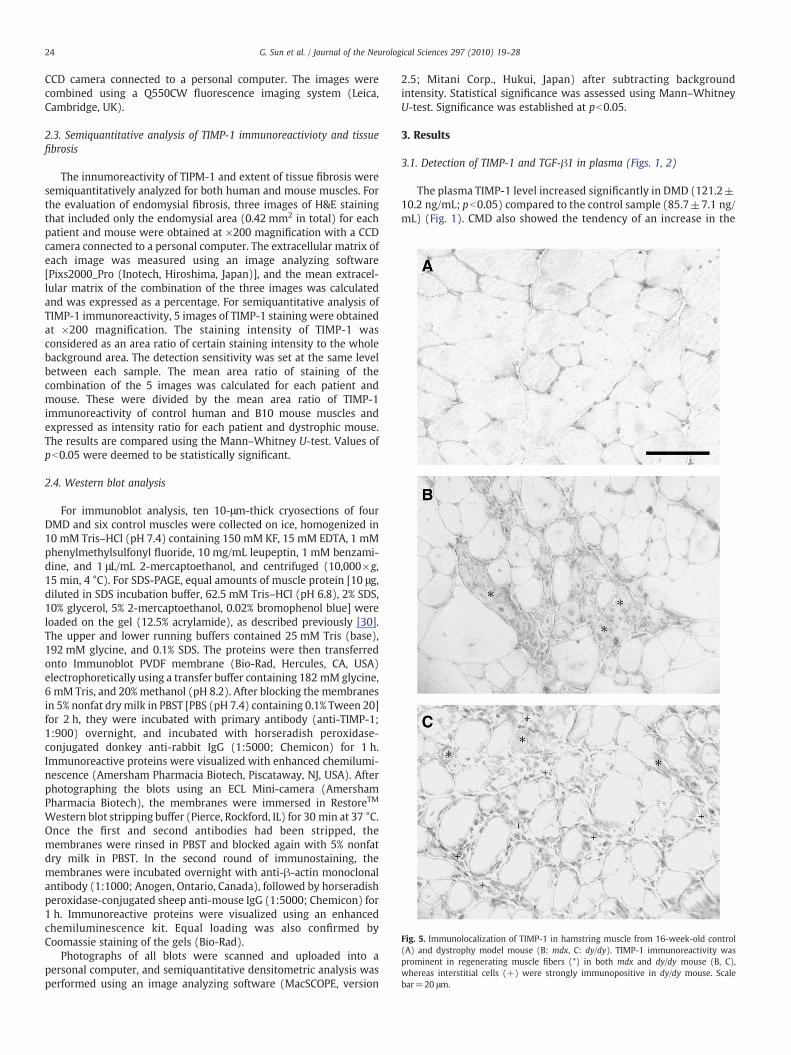
Fig. 5. Immunolocalization of TIMP-1 in hamstring muscle from 16-week-old control(A) and dystrophy model mouse (B: mdx, C: dy/dy). TIMP-1 immunoreactivity wasprominent in regenerating muscle fibers (*) in both mdx and dy/dy mouse (B, C),whereas interstitial cells (+) were strongly immunopositive in dy/dy mouse. Scalebar=20 μm.
24 G. Sun et al. / Journal of the Neurological Sciences 297 (2010) 19–28
CCD camera connected to a personal computer. The images werecombined using a Q550CW fluorescence imaging system (Leica,Cambridge, UK).
2.3. Semiquantitative analysis of TIMP-1 immunoreactivioty and tissuefibrosis
The innumoreactivity of TIPM-1 and extent of tissue fibrosis weresemiquantitatively analyzed for both human and mouse muscles. Forthe evaluation of endomysial fibrosis, three images of H&E stainingthat included only the endomysial area (0.42 mm2 in total) for eachpatient and mouse were obtained at ×200 magnification with a CCDcamera connected to a personal computer. The extracellular matrix ofeach image was measured using an image analyzing software[Pixs2000_Pro (Inotech, Hiroshima, Japan)], and the mean extracel-lular matrix of the combination of the three images was calculatedand was expressed as a percentage. For semiquantitative analysis ofTIMP-1 immunoreactivity, 5 images of TIMP-1 staining were obtainedat ×200 magnification. The staining intensity of TIMP-1 wasconsidered as an area ratio of certain staining intensity to the wholebackground area. The detection sensitivity was set at the same levelbetween each sample. The mean area ratio of staining of thecombination of the 5 images was calculated for each patient andmouse. These were divided by the mean area ratio of TIMP-1immunoreactivity of control human and B10 mouse muscles andexpressed as intensity ratio for each patient and dystrophic mouse.The results are compared using the Mann–Whitney U-test. Values ofpb0.05 were deemed to be statistically significant.
2.4. Western blot analysis
For immunoblot analysis, ten 10-μm-thick cryosections of fourDMD and six control muscles were collected on ice, homogenized in10 mM Tris–HCl (pH 7.4) containing 150 mM KF, 15 mM EDTA, 1 mMphenylmethylsulfonyl fluoride, 10 mg/mL leupeptin, 1 mM benzami-dine, and 1 μL/mL 2-mercaptoethanol, and centrifuged (10,000×g,15 min, 4 °C). For SDS-PAGE, equal amounts of muscle protein [10 μg,diluted in SDS incubation buffer, 62.5 mM Tris–HCl (pH 6.8), 2% SDS,10% glycerol, 5% 2-mercaptoethanol, 0.02% bromophenol blue] wereloaded on the gel (12.5% acrylamide), as described previously [30].The upper and lower running buffers contained 25 mM Tris (base),192 mM glycine, and 0.1% SDS. The proteins were then transferredonto Immunoblot PVDF membrane (Bio-Rad, Hercules, CA, USA)electrophoretically using a transfer buffer containing 182 mM glycine,6 mM Tris, and 20% methanol (pH 8.2). After blocking the membranesin 5% nonfat drymilk in PBST [PBS (pH 7.4) containing 0.1% Tween 20]for 2 h, they were incubated with primary antibody (anti-TIMP-1;1:900) overnight, and incubated with horseradish peroxidase-conjugated donkey anti-rabbit IgG (1:5000; Chemicon) for 1 h.Immunoreactive proteins were visualized with enhanced chemilumi-nescence (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Afterphotographing the blots using an ECL Mini-camera (AmershamPharmacia Biotech), the membranes were immersed in RestoreTM
Western blot stripping buffer (Pierce, Rockford, IL) for 30 min at 37 °C.Once the first and second antibodies had been stripped, themembranes were rinsed in PBST and blocked again with 5% nonfatdry milk in PBST. In the second round of immunostaining, themembranes were incubated overnight with anti-β-actin monoclonalantibody (1:1000; Anogen, Ontario, Canada), followed by horseradishperoxidase-conjugated sheep anti-mouse IgG (1:5000; Chemicon) for1 h. Immunoreactive proteins were visualized using an enhancedchemiluminescence kit. Equal loading was also confirmed byCoomassie staining of the gels (Bio-Rad).
Photographs of all blots were scanned and uploaded into apersonal computer, and semiquantitative densitometric analysis wasperformed using an image analyzing software (MacSCOPE, version
2.5; Mitani Corp., Hukui, Japan) after subtracting backgroundintensity. Statistical significance was assessed using Mann–WhitneyU-test. Significance was established at pb0.05.
3. Results
3.1. Detection of TIMP-1 and TGF-β1 in plasma (Figs. 1, 2)
The plasma TIMP-1 level increased significantly in DMD (121.2±10.2 ng/mL; pb0.05) compared to the control sample (85.7±7.1 ng/mL) (Fig. 1). CMD also showed the tendency of an increase in the
25G. Sun et al. / Journal of the Neurological Sciences 297 (2010) 19–28
plasma levels of TIMP-1 (122.5±17.8 ng/mL; p=0.17). However,the plasma TIMP-1 levels were not significantly elevated in BMD(90.0±16.9 ng/mL; p=0.39). The plasma TGF-β1 level increasedsignificantly in DMD (21.4±5.0 ng/mL; pb0.05) and CMD (29.2±9.3 ng/mL; pb0.05) compared to the controls (6.1±1.1 ng/mL) butnot in BMD (3.3±1.3 ng/mL). A significant correlation was observedbetween the plasma levels of TIMP-1 and TGF-β1 in DMD(R2=0.694; pb0. 0001) and CMD (R2=0.392; pb0. 05) but not inBMD (Fig. 2).
3.2. Immunolocalization of TIMP-1 in human control and dystrophicmuscles (Table 2; Figs. 3, 4)
In control muscle biopsies, TIMP-1 immunoreactivity was ob-served in intramuscular vessels as confirmed by co-localization withCD31 (Fig. 4; A–C) and in intramuscular nerve twigs (data not shown)and NMJs as confirmed by co-localization with α-BT (Fig. 4; D–F).These findings suggest physiological roles for TIMP-1 at these sites. Inall dystrophic muscles, cytoplasm and nuclei of regenerating muscle
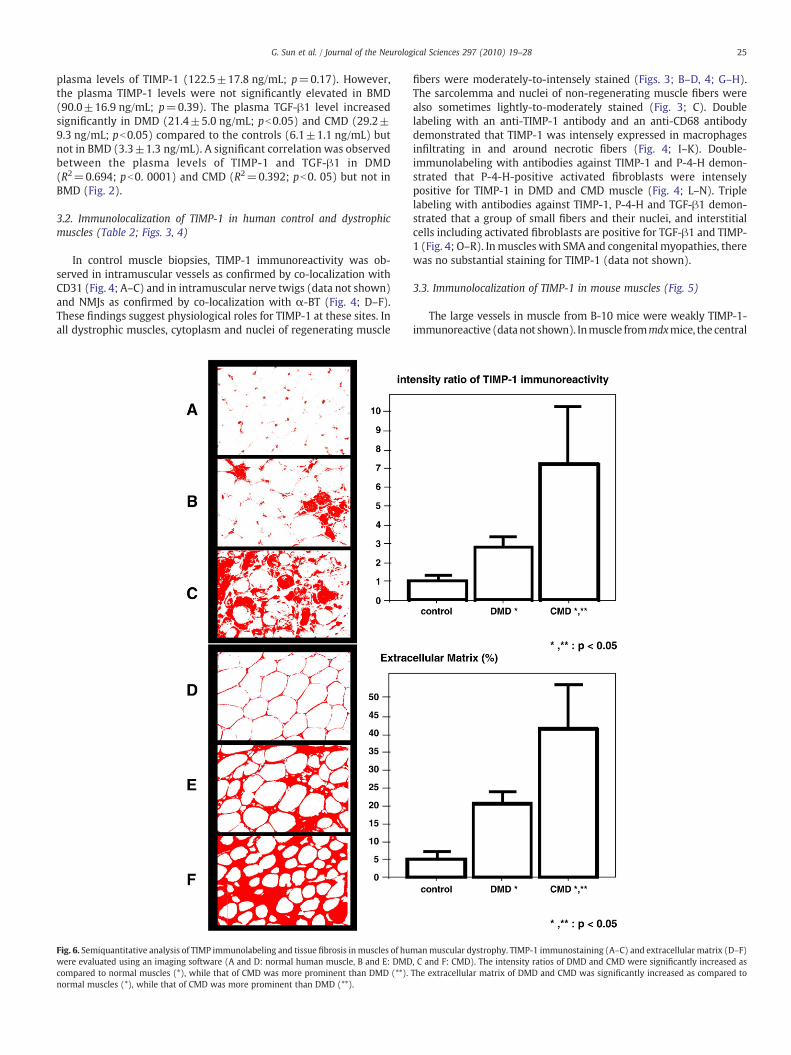
Fig. 6. Semiquantitative analysis of TIMP immunolabeling and tissue fibrosis inmuscles of huwere evaluated using an imaging software (A and D: normal human muscle, B and E: DMDcompared to normal muscles (*), while that of CMD was more prominent than DMD (**).normal muscles (*), while that of CMD was more prominent than DMD (**).
fibers were moderately-to-intensely stained (Figs. 3; B–D, 4; G–H).The sarcolemma and nuclei of non-regenerating muscle fibers werealso sometimes lightly-to-moderately stained (Fig. 3; C). Doublelabeling with an anti-TIMP-1 antibody and an anti-CD68 antibodydemonstrated that TIMP-1 was intensely expressed in macrophagesinfiltrating in and around necrotic fibers (Fig. 4; I–K). Double-immunolabeling with antibodies against TIMP-1 and P-4-H demon-strated that P-4-H-positive activated fibroblasts were intenselypositive for TIMP-1 in DMD and CMD muscle (Fig. 4; L–N). Triplelabeling with antibodies against TIMP-1, P-4-H and TGF-β1 demon-strated that a group of small fibers and their nuclei, and interstitialcells including activated fibroblasts are positive for TGF-β1 and TIMP-1 (Fig. 4; O–R). Inmuscles with SMA and congenital myopathies, therewas no substantial staining for TIMP-1 (data not shown).
3.3. Immunolocalization of TIMP-1 in mouse muscles (Fig. 5)
The large vessels in muscle from B-10 mice were weakly TIMP-1-immunoreactive (datanot shown). Inmuscle frommdxmice, the central
manmuscular dystrophy. TIMP-1 immunostaining (A–C) and extracellular matrix (D–F), C and F: CMD). The intensity ratios of DMD and CMD were significantly increased asThe extracellular matrix of DMD and CMD was significantly increased as compared to
26 G. Sun et al. / Journal of the Neurological Sciences 297 (2010) 19–28
nuclei and cytoplasm of regenerating fibers were lightly-to-moderatelyimmunoreactive (Fig. 5B). Inmuscle from dy/dymouse, the anti-TIMP-1antibody stained the central nuclei and cytoplasmof regeneratingfibers.However, in addition, endomysial interstitial cells were moderately-to-intensely immunopositive (Fig. 5C) in dy/dy mouse. The intenseimmunostaining of interstitial cells in dy/dy mice compared to mdxmice was the major difference between these animal strains.
3.4. Semiquantitative analysis of TIMP-1 immunoreactivioty and tissuefibrosis (Figs. 6 and 7)
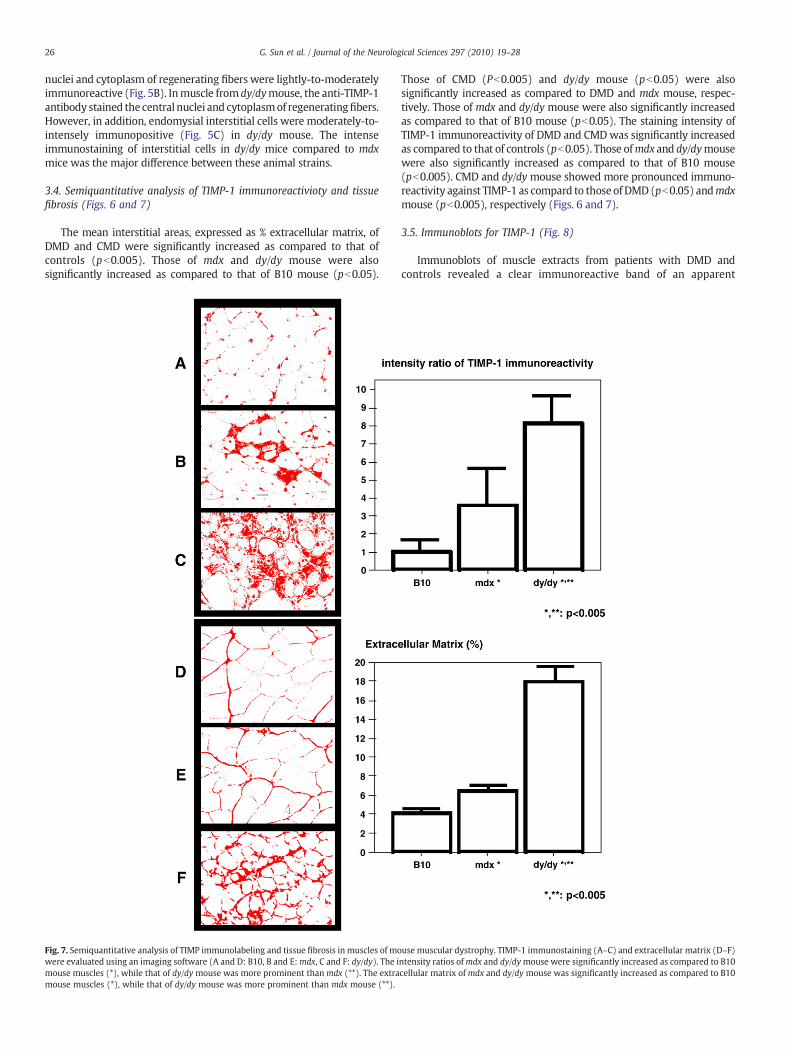
The mean interstitial areas, expressed as % extracellular matrix, ofDMD and CMD were significantly increased as compared to that ofcontrols (pb0.005). Those of mdx and dy/dy mouse were alsosignificantly increased as compared to that of B10 mouse (pb0.05).
Fig. 7. Semiquantitative analysis of TIMP immunolabeling and tissue fibrosis in muscles of mowere evaluated using an imaging software (A and D: B10, B and E:mdx, C and F: dy/dy). The imouse muscles (*), while that of dy/dy mouse was more prominent thanmdx (**). The extramouse muscles (*), while that of dy/dy mouse was more prominent than mdx mouse (**).
Those of CMD (Pb0.005) and dy/dy mouse (pb0.05) were alsosignificantly increased as compared to DMD and mdx mouse, respec-tively. Those of mdx and dy/dy mouse were also significantly increasedas compared to that of B10 mouse (pb0.05). The staining intensity ofTIMP-1 immunoreactivity of DMD and CMDwas significantly increasedas compared to that of controls (pb0.05). Those ofmdx and dy/dymousewere also significantly increased as compared to that of B10 mouse(pb0.005). CMD and dy/dy mouse showed more pronounced immuno-reactivity against TIMP-1 as compard to thoseof DMD(pb0.05) andmdxmouse (pb0.005), respectively (Figs. 6 and 7).
3.5. Immunoblots for TIMP-1 (Fig. 8)
Immunoblots of muscle extracts from patients with DMD andcontrols revealed a clear immunoreactive band of an apparent
use muscular dystrophy. TIMP-1 immunostaining (A–C) and extracellular matrix (D–F)ntensity ratios ofmdx and dy/dymouse were significantly increased as compared to B10cellular matrix of mdx and dy/dy mouse was significantly increased as compared to B10
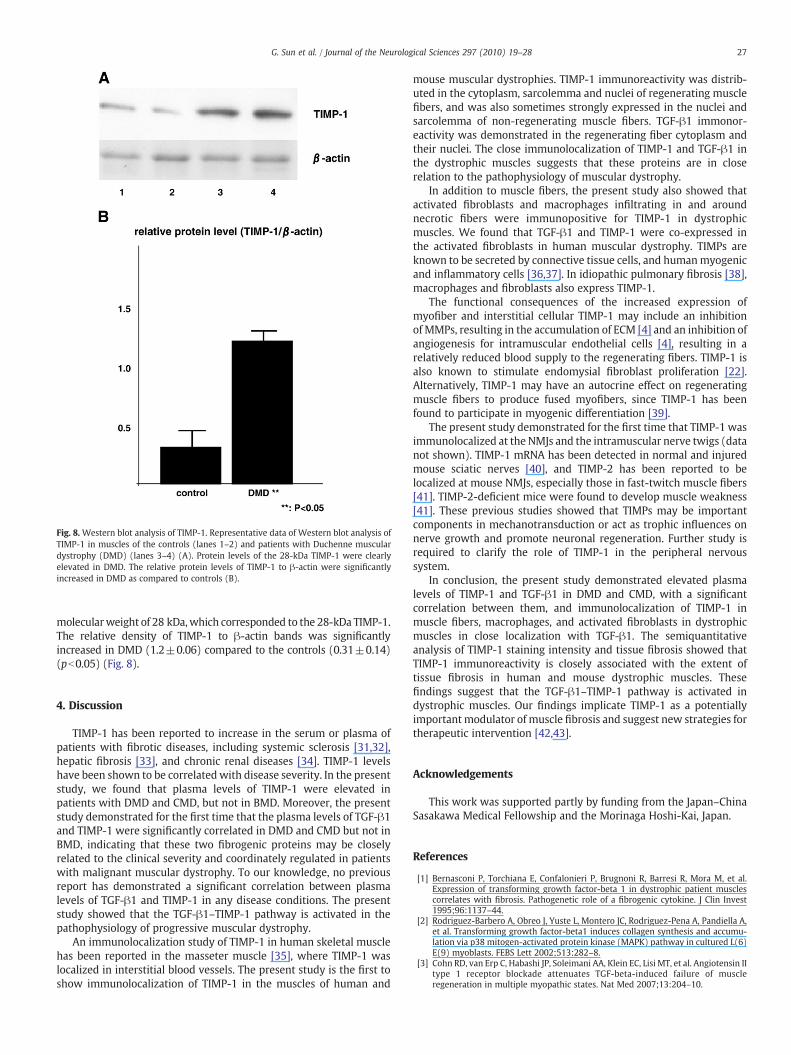
Fig. 8. Western blot analysis of TIMP-1. Representative data of Western blot analysis ofTIMP-1 in muscles of the controls (lanes 1–2) and patients with Duchenne musculardystrophy (DMD) (lanes 3–4) (A). Protein levels of the 28-kDa TIMP-1 were clearlyelevated in DMD. The relative protein levels of TIMP-1 to β-actin were significantlyincreased in DMD as compared to controls (B).
27G. Sun et al. / Journal of the Neurological Sciences 297 (2010) 19–28
molecularweight of 28 kDa,which corresponded to the 28-kDa TIMP-1.The relative density of TIMP-1 to β-actin bands was significantlyincreased in DMD (1.2±0.06) compared to the controls (0.31±0.14)(pb0.05) (Fig. 8).
4. Discussion
TIMP-1 has been reported to increase in the serum or plasma ofpatients with fibrotic diseases, including systemic sclerosis [31,32],hepatic fibrosis [33], and chronic renal diseases [34]. TIMP-1 levelshave been shown to be correlatedwith disease severity. In the presentstudy, we found that plasma levels of TIMP-1 were elevated inpatients with DMD and CMD, but not in BMD. Moreover, the presentstudy demonstrated for the first time that the plasma levels of TGF-β1and TIMP-1 were significantly correlated in DMD and CMD but not inBMD, indicating that these two fibrogenic proteins may be closelyrelated to the clinical severity and coordinately regulated in patientswith malignant muscular dystrophy. To our knowledge, no previousreport has demonstrated a significant correlation between plasmalevels of TGF-β1 and TIMP-1 in any disease conditions. The presentstudy showed that the TGF-β1–TIMP-1 pathway is activated in thepathophysiology of progressive muscular dystrophy.
An immunolocalization study of TIMP-1 in human skeletal musclehas been reported in the masseter muscle [35], where TIMP-1 waslocalized in interstitial blood vessels. The present study is the first toshow immunolocalization of TIMP-1 in the muscles of human and
mouse muscular dystrophies. TIMP-1 immunoreactivity was distrib-uted in the cytoplasm, sarcolemma and nuclei of regenerating musclefibers, and was also sometimes strongly expressed in the nuclei andsarcolemma of non-regenerating muscle fibers. TGF-β1 immonor-eactivity was demonstrated in the regenerating fiber cytoplasm andtheir nuclei. The close immunolocalization of TIMP-1 and TGF-β1 inthe dystrophic muscles suggests that these proteins are in closerelation to the pathophysiology of muscular dystrophy.
In addition to muscle fibers, the present study also showed thatactivated fibroblasts and macrophages infiltrating in and aroundnecrotic fibers were immunopositive for TIMP-1 in dystrophicmuscles. We found that TGF-β1 and TIMP-1 were co-expressed inthe activated fibroblasts in human muscular dystrophy. TIMPs areknown to be secreted by connective tissue cells, and humanmyogenicand inflammatory cells [36,37]. In idiopathic pulmonary fibrosis [38],macrophages and fibroblasts also express TIMP-1.
The functional consequences of the increased expression ofmyofiber and interstitial cellular TIMP-1 may include an inhibitionof MMPs, resulting in the accumulation of ECM [4] and an inhibition ofangiogenesis for intramuscular endothelial cells [4], resulting in arelatively reduced blood supply to the regenerating fibers. TIMP-1 isalso known to stimulate endomysial fibroblast proliferation [22].Alternatively, TIMP-1 may have an autocrine effect on regeneratingmuscle fibers to produce fused myofibers, since TIMP-1 has beenfound to participate in myogenic differentiation [39].
The present study demonstrated for the first time that TIMP-1 wasimmunolocalized at the NMJs and the intramuscular nerve twigs (datanot shown). TIMP-1 mRNA has been detected in normal and injuredmouse sciatic nerves [40], and TIMP-2 has been reported to belocalized at mouse NMJs, especially those in fast-twitch muscle fibers[41]. TIMP-2-deficient mice were found to develop muscle weakness[41]. These previous studies showed that TIMPs may be importantcomponents in mechanotransduction or act as trophic influences onnerve growth and promote neuronal regeneration. Further study isrequired to clarify the role of TIMP-1 in the peripheral nervoussystem.
In conclusion, the present study demonstrated elevated plasmalevels of TIMP-1 and TGF-β1 in DMD and CMD, with a significantcorrelation between them, and immunolocalization of TIMP-1 inmuscle fibers, macrophages, and activated fibroblasts in dystrophicmuscles in close localization with TGF-β1. The semiquantitativeanalysis of TIMP-1 staining intensity and tissue fibrosis showed thatTIMP-1 immunoreactivity is closely associated with the extent oftissue fibrosis in human and mouse dystrophic muscles. Thesefindings suggest that the TGF-β1–TIMP-1 pathway is activated indystrophic muscles. Our findings implicate TIMP-1 as a potentiallyimportant modulator of muscle fibrosis and suggest new strategies fortherapeutic intervention [42,43].
Acknowledgements
This work was supported partly by funding from the Japan–ChinaSasakawa Medical Fellowship and the Morinaga Hoshi-Kai, Japan.
References
[1] Bernasconi P, Torchiana E, Confalonieri P, Brugnoni R, Barresi R, Mora M, et al.Expression of transforming growth factor-beta 1 in dystrophic patient musclescorrelates with fibrosis. Pathogenetic role of a fibrogenic cytokine. J Clin Invest1995;96:1137–44.
[2] Rodriguez-Barbero A, Obreo J, Yuste L, Montero JC, Rodriguez-Pena A, Pandiella A,et al. Transforming growth factor-beta1 induces collagen synthesis and accumu-lation via p38 mitogen-activated protein kinase (MAPK) pathway in cultured L(6)E(9) myoblasts. FEBS Lett 2002;513:282–8.
[3] Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, et al. Angiotensin IItype 1 receptor blockade attenuates TGF-beta-induced failure of muscleregeneration in multiple myopathic states. Nat Med 2007;13:204–10.

28 G. Sun et al. / Journal of the Neurological Sciences 297 (2010) 19–28
[4] Gomez DE, Alonso DF, Yoshiji H, Thorgeirsson UP. Tissue inhibitors ofmetalloproteinases: structure, regulation and biological functions. Eur J Cell Biol1997;74:111–22.
[5] Yong VW, Krekoski CA, Forsyth PA, Bell R, Edwards DR. Matrix metalloproteinasesand diseases of the CNS. Trends Neurosci 1998;21:75–80.
[6] Chirco R, Liu XW, Jung KK, Kim HR. Novel functions of TIMPs in cell signaling.Cancer Metastasis Rev 2006;25:99–113.
[7] Goldberg GI, Strongin A, Collier IE, Genrich LT, Marmer BL. Interaction of 92-kDatype IV collagenase with the tissue inhibitor of metalloproteinases preventsdimerization, complex formation with interstitial collagenase, and activation ofthe proenzyme with stromelysin. J Biol Chem 1992;267:4583–91.
[8] Chambers AF, Matrisian LM. Changing views of the role of matrix metalloprotei-nases in metastasis. J Natl Cancer Inst 1997;89:1260–70.
[9] Madtes DK, Elston AL, Kaback LA, Clark JG. Selective induction of tissue inhibitor ofmetalloproteinase-1 in bleomycin-induced pulmonary fibrosis. Am J Respir CellMol Biol 2001;24:599–607.
[10] Nicholson ML, Waller JR, Bicknell GR. Renal transplant fibrosis correlates withintragraft expression of tissue inhibitor of metalloproteinase messenger RNA. Br JSurg 2002;89:933–7.
[11] Nie QH, Cheng YQ, Xie YM, Zhou YX, Bai XG, Cao YZ. Methodologic research onTIMP-1, TIMP-2 detection as a new diagnostic index for hepatic fibrosis and itssignificance. World J Gastroenterol 2002;8:282–7.
[12] Soini Y, Satta J, Maatta M, Autio-Harmainen H. Expression of MMP2, MMP9, MT1-MMP, TIMP1, and TIMP2 mRNA in valvular lesions of the heart. J Pathol 2001;194:225–31.
[13] Verrecchia F, Chu ML, Mauviel A. Identification of novel TGF-beta/Smad genetargets in dermal fibroblasts using a combined cDNA microarray/promotertransactivation approach. J Biol Chem 2001;276:17058–62.
[14] Eickelberg O, Kohler E, Reichenberger F, Bertschin S, Woodtli T, Erne P, et al.Extracellular matrix deposition by primary human lung fibroblasts in response toTGF-beta1 and TGF-beta3. Am J Physiol 1999;276:L814–24.
[15] Knittel T, MehdeM, Kobold D, Saile B, Dinter C, Ramadori G. Expression patterns ofmatrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells of rat liver: regulation by TNF-alpha and TGF-beta1. J Hepatol1999;30:48–60.
[16] Jia JD, Bauer M, Cho JJ, Ruehl M, Milani S, Boigk G, et al. Antifibrotic effect ofsilymarin in rat secondary biliary fibrosis is mediated by downregulation ofprocollagen alpha1(I) and TIMP-1. J Hepatol 2001;35:392–8.
[17] Nemeth JA, Rafe A, Steiner M, Goolsby CL. TIMP-2 growth-stimulatory activity: aconcentration- and cell type-specific response in the presence of insulin. Exp CellRes 1996;224:110–5.
[18] Hayakawa T, Yamashita K, Tanzawa K, Uchijima E, Iwata K. Growth-promotingactivity of tissue inhibitor of metalloproteinases-1 (TIMP-1) for a wide range ofcells. A possible new growth factor in serum. FEBS Lett 1992;298:29–32.
[19] Preaux AM, D'Ortho P, Bralet MP, Laperche Y, Mavier P. Apoptosis of humanhepatic myofibroblasts promotes activation of matrix metalloproteinase-2.Hepatology 2002;36:615–22.
[20] Guedez L, McMarlin AJ, Kingma DW, Bennett TA, Stetler-Stevenson M, Stetler-Stevenson WG. Tissue inhibitor of metalloproteinase-1 alters the tumorigenicityof Burkitt's lymphoma via divergent effects on tumor growth and angiogenesis.Am J Pathol 2001;158:1207–15.
[21] Clark IM, Powell LK, Cawston TE. Tissue inhibitor of metalloproteinases (TIMP-1)stimulates the secretion of collagenase from human skin fibroblasts. BiochemBiophys Res Commun 1994;203:874–80.
[22] Kikuchi K, Kadono T, Furue M, Tamaki K. Tissue inhibitor of metalloproteinase 1(TIMP-1) may be an autocrine growth factor in scleroderma fibroblasts. J InvestDermatol 1997;108:281–4.
[23] Kherif S, Lafuma C, Dehaupas M, Lachkar S, Fournier JG, Verdiere-Sahuque M,et al. Expression of matrix metalloproteinases 2 and 9 in regenerating skeletalmuscle: a study in experimentally injured and mdx muscles. Dev Biol 1999;205:158–70.
[24] Kieseier BC, Schneider C, Clements JM, Gearing AJ, Gold R, Toyka KV, et al.Expression of specific matrix metalloproteinases in inflammatory myopathies.Brain 2001;124:341–51.
[25] Fukushima K, Nakamura A, Ueda H, Yuasa K, Yoshida K, Takeda S, et al. Activationand localization of matrix metalloproteinase-2 and -9 in the skeletal muscle of themuscular dystrophy dog (CXMDJ). BMC Musculoskelet Disord 2007;8:54.
[26] vonMoers A, Zwirner A, Reinhold A, Bruckmann O, van Landeghem F, Stoltenburg-Didinger G, et al. Increased mRNA expression of tissue inhibitors of metallopro-teinase-1 and -2 in Duchenne muscular dystrophy. Acta Neuropathol 2005;109:285–93.
[27] Zanotti S, Saredi S, Ruggieri A, Fabbri M, Blasevich F, Romaggi S, et al. Alteredextracellular matrix transcript expression and protein modulation in primaryDuchenne muscular dystrophy myotubes. Matrix Biol 2007;26:615–24.
[28] Ishitobi M, Haginoya K, Zhao Y, Ohnuma A, Minato J, Yanagisawa T, et al. Elevatedplasma levels of transforming growth factor beta1 in patients with musculardystrophy. NeuroReport 2000;11:4033–5.
[29] Sundberg C, IvarssonM, Gerdin B, Rubin K. Pericytes as collagen-producing cells inexcessive dermal scarring. Lab Invest 1996;74:452–66.
[30] Zhao Y, Haginoya K, Sun G, Dai H, Onuma A, Iinuma K. Platelet-derived growthfactor and its receptors are related to the progression of human musculardystrophy: an immunohistochemical study. J Pathol 2003;201:149–59.
[31] Young-Min SA, Beeton C, Laughton R, Plumpton T, Bartram S, Murphy G, et al.Serum TIMP-1, TIMP-2, and MMP-1 in patients with systemic sclerosis, primaryRaynaud's phenomenon, and in normal controls. Ann RheumDis 2001;60:846–51.
[32] Kim WU, Min SY, Cho ML, Hong KH, Shin YJ, Park SH, et al. Elevated matrixmetalloproteinase-9 in patients with systemic sclerosis. Arthritis Res Ther 2005;7:R71–9.
[33] Zhang BB, Cai WM, Weng HL, Hu ZR, Lu J, Zheng M, et al. Diagnostic value ofplatelet derived growth factor-BB, transforming growth factor-beta1, matrixmetalloproteinase-1, and tissue inhibitor of matrix metalloproteinase-1 in serumand peripheral blood mononuclear cells for hepatic fibrosis. World J Gastroenterol2003;9:2490–6.
[34] Horstrup JH, Gehrmann M, Schneider B, Ploger A, Froese P, Schirop T, et al.Elevation of serum and urine levels of TIMP-1 and tenascin in patients with renaldisease. Nephrol Dial Transplant 2002;17:1005–13.
[35] Singh A, Nelson-Moon ZL, Thomas GJ, Hunt NP, Lewis MP. Identification of matrixmetalloproteinases and their tissue inhibitors type 1 and 2 in human massetermuscle. Arch Oral Biol 2000;45:431–40.
[36] Guerin CW, Holland PC. Synthesis and secretion of matrix-degrading metallopro-teases by human skeletal muscle satellite cells. Dev Dyn 1995;202:91–9.
[37] Koskinen SO, Wang W, Ahtikoski AM, Kjaer M, Han XY, Komulainen J, et al. Acuteexercise induced changes in rat skeletal muscle mRNAs and proteins regulatingtype IV collagen content. Am J Physiol Regul Integr Comp Physiol 2001;280:R1292–300.
[38] Selman M, Ruiz V, Cabrera S, Segura L, Ramirez R, Barrios R, et al. TIMP-1, -2, -3,and -4 in idiopathic pulmonary fibrosis. A prevailing nondegradative lungmicroenvironment? Am J Physiol Lung Cell Mol Physiol 2000;279:L562–74.
[39] Lewis MP, Tippett HL, Sinanan AC, Morgan MJ, Hunt NP. Gelatinase-B (matrixmetalloproteinase-9; MMP-9) secretion is involved in the migratory phase ofhuman and murine muscle cell cultures. J Muscle Res Cell Motil 2000;21:223–33.
[40] Platt CI, Krekoski CA, Ward RV, Edwards DR, Gavrilovic J. Extracellular matrix andmatrix metalloproteinases in sciatic nerve. J Neurosci Res 2003;74:417–29.
[41] Lluri G, Langlois GD, McClellan B, Soloway PD, Jaworski DM. Tissue inhibitor ofmetalloproteinase-2 (TIMP-2) regulates neuromuscular junction development viaa beta1 integrin-mediated mechanism. J Neurobiol 2006;66:1365–77.
[42] Parsons CJ, Bradford BU, Pan CQ, Cheung E, Schauer M, Knorr A, et al. Antifibroticeffects of a tissue inhibitor of metalloproteinase-1 antibody on established liverfibrosis in rats. Hepatology 2004;40:1106–15.
[43] Hu YB, Li DG, Lu HM. Modified synthetic siRNA targeting tissue inhibitor ofmetalloproteinase-2 inhibits hepatic fibrogenesis in rats. J Gene Med 2007;9:217–29.
Copyright © 2022 FDOKUMEN




















