
Molecular genetic study of Egyptian patients with macular corneal dystrophy
Upload
independentCategory
view
1download
0
NIM811, a cyclophilin inhibitor withoutimmunosuppressive activity, is beneficial in collagenVI congenital muscular dystrophy models
Alessandra Zulian1,4,{, Erika Rizzo1,4,{, Marco Schiavone2,{, Elena Palma1, Francesca Tagliavini5,
Bert Blaauw1, Luciano Merlini5, Nadir Mario Maraldi5, Patrizia Sabatelli5,6, Paola Braghetta3,
Paolo Bonaldo3, Francesco Argenton2 and Paolo Bernardi1,4,∗
1Department of Biomedical Sciences and 2Department of Biology and 3Department of Molecular Medicine, University of
Padova, I-35131 Padova, Italy, 4Consiglio Nazionale Delle Ricerche Neuroscience Institute, I-35131 Padova, Italy,5Laboratory of Musculoskeletal Cell Biology, Istituto Ortopedico Rizzoli, I-40136 Bologna, Italy and 6Consiglio Nazionale
Delle Ricerche, Institute of Molecular Genetics, I-40136 Bologna, Italy
Received February 28, 2014; Revised and Accepted May 19, 2014
Ullrich congenital muscular dystrophy (UCMD) and Bethlem myopathy (BM) are inherited muscle diseases dueto mutations in the genes encoding the extracellular matrix protein collagen (Col) VI. Opening of the cyclosporinA-sensitive mitochondrial permeability transition pore (PTP) is a causative event in disease pathogenesis, and apotential target for therapy. Here, we have tested the effect of N-methyl-4-isoleucine-cyclosporin (NIM811), anon-immunosuppressive cyclophilin inhibitor, in a zebrafish model of ColVI myopathy obtained by deletionof the N-terminal region of the ColVIa1 triple helical domain, a common mutation of UCMD. Treatment with anti-sense morpholino sequences targeting col6a1 exon 9 at the 1–4 cell stage (within 1 h post fertilization, hpf)caused severe ultrastructural and motor abnormalities as assessed by electron and fluorescence microscopy,birefringence, spontaneous coiling events and touch-evoked responses measured at 24–48 hpf. Structural andfunctional abnormalities were largely prevented when NIM811—which proved significantly more effective thancyclosporin A—was administered at 21 hpf, while FK506 was ineffective. Beneficial effects of NIM811 were alsodetected (i) in primary muscle-derived cell cultures from UCMD and BM patients, where the typical mitochondrialalterations and depolarizing response to rotenone and oligomycin were significantly reduced; and (ii) in theCol6a12/2 myopathic mouse model, where apoptosis was prevented and muscle strength was increased.Since the PTP of zebrafish shares its key regulatory features with the mammalian pore, our results suggestthat early treatment with NIM811 should be tested as a potential therapy for UCMD and BM.
INTRODUCTION
Deficiency of collagen VI (ColVI) due to mutations of COL6genes gives rise to three main muscle disorders, Bethlem myop-athy (BM, MIM #158810), Ullrich congenital muscular dys-trophy (UCMD, MIM #254090) (1) and myosclerosismyopathy (MIM #255600) (2). BM is characterized by axialand proximal muscle weakness, and a hallmark of this diseaseis the presence of contractures of the interphalangeal joints of
the last four fingers (3,4). The clinical features are extremelyheterogenous and range from mild to severe myopathy with pro-gressive muscular dystrophy (5). UCMD is a severe musculardystrophy characterized by early onset, rapidly progressivemuscle wasting and weakness, proximal joint contractures anddistal joint hyperlaxity. Rapid progression usually leads toearly death due to respiratory failure (6,7). Myosclerosis myop-athy is characterized by progressive joint contractures, scoliosis,mild girdle and proximal limb weakness and moderate distal
†These Authors contributed equally to this work.
∗To whom correspondence should be addressed at: Department of Biomedical Sciences, University of Padova, Via Ugo Bassi 58/B, I-35121 Padova, Italy.Tel: +39 (0)498276365; Fax: +39 (0)49 8276049; Email: [email protected]
# The Author 2014. Published by Oxford University Press. All rights reserved.For Permissions, please email: [email protected]
Human Molecular Genetics, 2014, Vol. 23, No. 20 5353–5363doi:10.1093/hmg/ddu254Advance Access published on May 22, 2014
at UniversitÃ
degli Studi di Padova on September 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

weakness. Muscles become thin and sclerotic with woody con-sistency, causing severe motility restriction (2). About 70 differ-ent mutations of the COL6 genes have so far been described inColVI myopathies, and it is becoming apparent that these disor-ders represent a clinical continuum (1,8).
ColVI myopathies share a common pathogenesis downstreamthe lack of ColVI, i.e. (i) mitochondrial dysfunction due to de-regulation of the permeability transition pore (PTP), an innermembrane high conductance channel that forms from dimersof the mitochondrial F-ATP synthase (9) and is involved inmany degenerative diseases (10); and (ii) defective autophagyimpairing clearance of dysfunctional mitochondria (11–13).The mitochondrial defect was first identified in the Col6a12/2
mouse model (14) and then detected in cultures from UCMDand BM patients (15–17). Genetic inactivation of Ppif [thegene encoding for mitochondrial cyclophilin (CyP) D in themouse] in Col6a12/2 mice (18) or PTP desensitization withcyclosporin A (CsA) cured the mouse model of the myopathy(14) and gave promising results in a pilot trial on five patients(19,20). However, the immunosuppressive effect of CsAremains a major source of concern for the long-term treatmentof patients affected by a disease where respiratory insufficiency,which can be precipitated by pulmonary infections, is a promin-ent cause of death (21).
Myopathic Col6a12/2 mice have also been successfullycured with D-MeAla3-EtVal4-cyclosporin (Debio025), a non-immunosuppressive derivative of CsA that inhibits CyPs butnot calcineurin (22). This finding suggests that, in principle,ColVI myopathies could be cured without exposing the patientsto the hazard of immunosuppression. However, the Col6a12/2
mouse is affected by a very mild myopathy with virtually no fi-brosis in spite of the total lack of ColVI (23). This is at strikingvariance with UCMD patients, where fibroadipose substitutionis prominent and may not be reversible (21). In a recent im-portant development, the severe form of ColVI myopathy hasbeen modeled in zebrafish by injection of morpholino targetingexon 9 of col6a1, which caused an in-frame deletion ofthe N-terminal portion of the triple helical domain of ColVIa1 (24), a dominant mutation in UCMD (25,26). Treatmentresulted in severe myopathy with early-onset motor deficits,severe ultrastructural changes, mitochondrial abnormalitiesand increased apoptosis that could be improved by CsA (24).Since the features of the mammalian PTP are indistinguishablefrom those of zebrafish (27), the latter organism provides aunique opportunity to test whether CyP inhibitors devoid of im-munosuppressive activity are effective in the therapy of thesevere UCMD-like myopathy. In the present study, we haveinvestigated the effects of N-methyl-4-isoleucine-cyclosporin(NIM811), a CyP inhibitor that desensitizes the PTP to Ca2+
(28), in exon 9 morphant zebrafish, in myopathic Col6a12/2
mice and in cell cultures established from muscle biopsies ofUCMD and BM patients.
RESULTS
NIM811 rescues structural and functional abnormalitiesof ColVI exon 9 morphant zebrafish
To obtain a zebrafish model of ColVI-related myopathies, weused a morpholino specifically directed against the col6a1
exon 9 splicing region, which results in an in-frame deletion ofthe N-terminal region of the ColVI a1 triple helical domainmatching the most frequent mutation of UCMD (24). In wild-type zebrafish embryos, ColVI was expressed at the myotendi-nous junction (MTJ), which can be identified byb-dystroglycanstaining (Fig. 1A and A′). In exon 9 morphants, ColVI expressionwas severely reduced (Fig. 1B), and the MTJ displayed an aber-rant profile (Fig. 1B′). Ninety-seven percent of exon 9 morphantembryos (out of 121 injected eggs) showed hypoplastic and bentbodies at both 24 and 48 hpf (examples are provided in Fig. 1, topright panel), and these morphological abnormalities matchedstructural myofiber alterations. Indeed, in wild type embryos,myofibers were aligned along the axis of contraction and mito-chondria were regularly distributed within the fibers (Fig. 1C–E), while after ColVI depletion the myofibrillar material was par-tially lost, myofibrils in adjacent myocytes were misaligned andthere was no obvious pattern that could be related to the axisof contraction (Fig. 1F) with several twisted myofibrils(Fig. 1F, arrowheads). A marked alteration of mitochondrial dis-tribution matched myofibrillar derangements, with clusters of
Figure 1. Effect of an exon 9 morpholino targeting col6a1 mRNA on ColVI ex-pression, morphology and muscle structure of zebrafish embryos. Top left, frozensections from control (A and A′) or exon 9 morphant zebrafishembryos (B and B′)were immunolabeled with anti-ColVI (A and B) or anti-b-dystroglycan (b-DG)antibodies (A′ and B′). Bar, 5 mm. Top right, microscopic analysis of control(CTL MO) and exon 9 morphant embryos (Ex9 MO) at 24 and 48 hpf. Bottom,confocal analysis of 48 hpf control (C–E) and exon 9 morphant embryos(F–H) stained with phalloidin (C and F), Tom20 (D and G). (E and H) Mergedimages of phalloidin and Tom20 staining. Bar, 30 mm.
5354 Human Molecular Genetics, 2014, Vol. 23, No. 20
at UniversitÃ
degli Studi di Padova on September 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

mitochondria corresponding to the areas of myofibrillar disor-ganization (Fig. 1G and H).
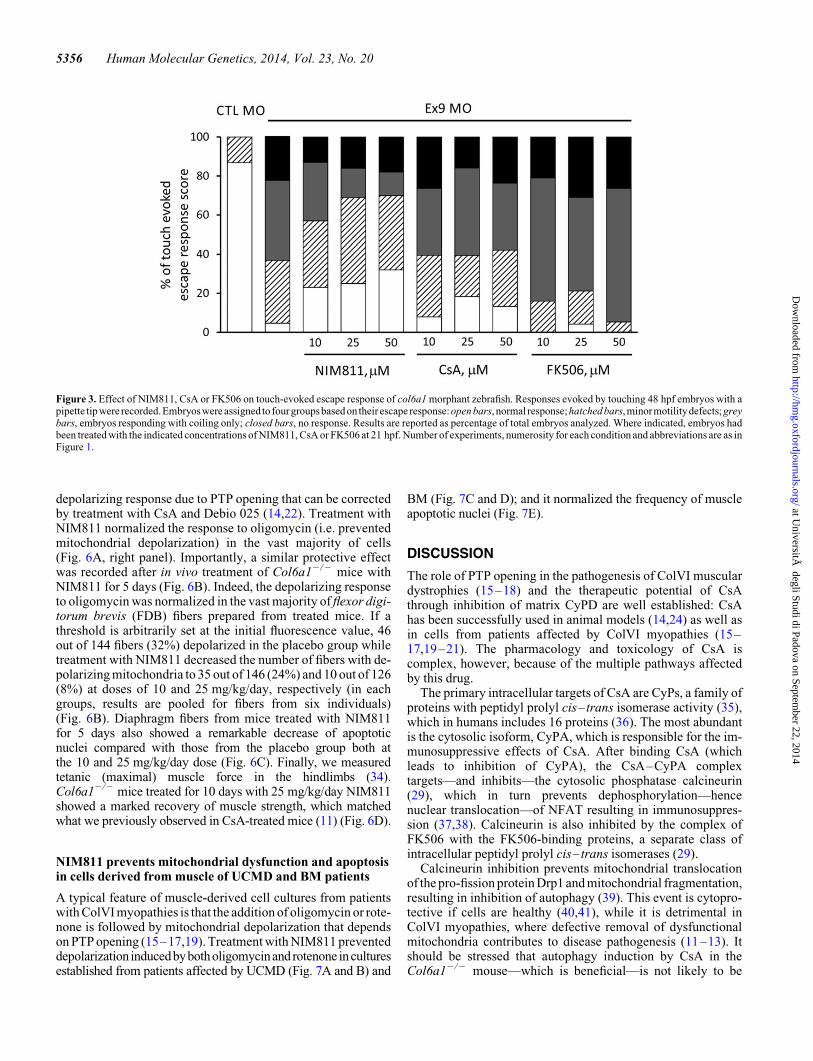
Morphant zebrafish had a severe motor impairment, as mea-sured both on spontaneous coiling events (Fig. 2) and touch-evoked responses (Fig. 3). Treatment with NIM811 fully restoredspontaneous coiling events at a concentration of 25 mM, whereasCsA gave a very limited rescue only at 50 mM and FK506, a calci-neurin inhibitor that acts through a CyP-independent mechanism(29), was totally ineffective (Fig. 2). Of note, as also reported byTelfer et al. (24), suppression of p53 expression by co-injectionof an appropriate morpholino did not improve spontaneouscoiling (data not shown), suggesting that the phenotypic effectwas not due to an off-site p53 knockdown (30). Touch-evokedresponses were also ameliorated by NIM811, marginally affectedby CsA and unaffected by FK506, while no treatment was able torescue the small subset of paralyzed embryos (Fig. 3).
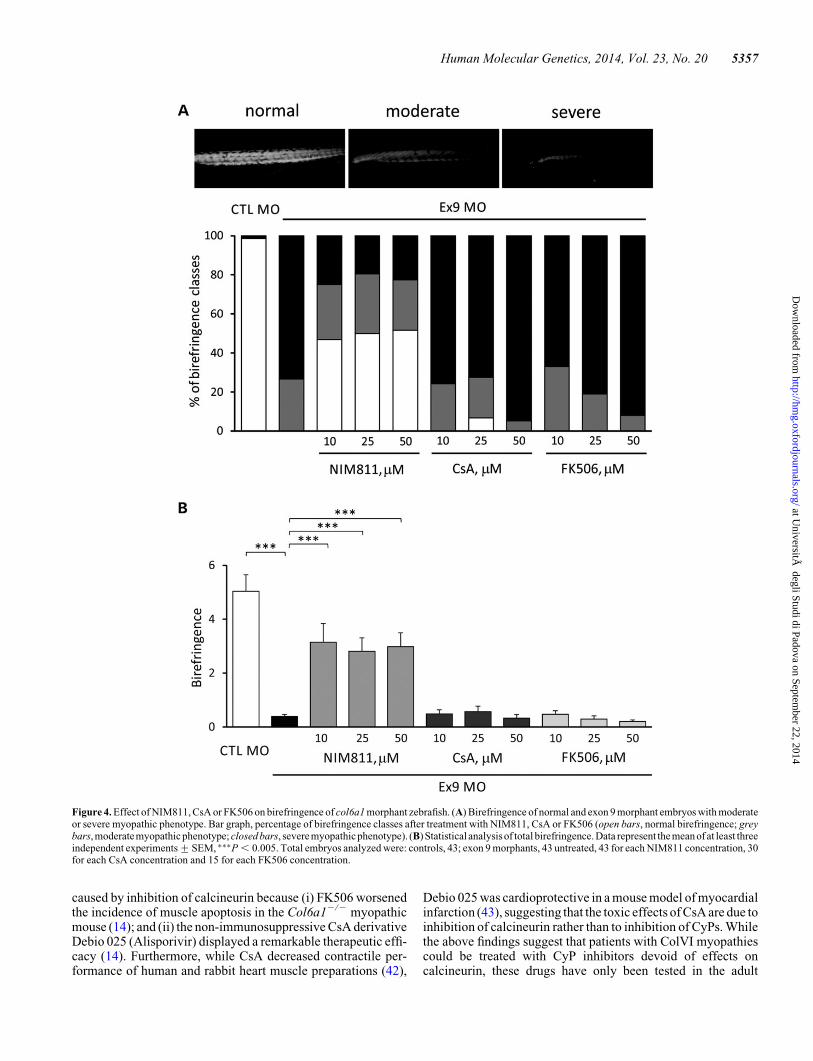
In order to obtain information on structural muscle organiza-tion in vivo, we measured birefringence with polarized light mi-croscopy (31), a technique that allows detection of musclestructural defects in several zebrafish models of muscular dys-trophy (32). The appearance of a normal zebrafish is illustratedin Figure 4A, which also provides examples of moderate andsevere phenotypes of exon 9 morphant individuals. Treatmentwith NIM811 allowed recovery of a normal birefringence in�50% of exon 9 morphants, while treatment with CsA led to amarginal rescue at 25 mM and to worsening of the severe pheno-type at 50 mM, no effect being observed with FK506 (Fig. 4A).Unbiased statistical analysis based on the total birefringencescore for all conditions indicates that only NIM811 allowed re-covery, the effect being seen at concentrations of 10 mM
(Fig. 4B).Exon 9 morphant embryos analyzed at 48 hpf displayed
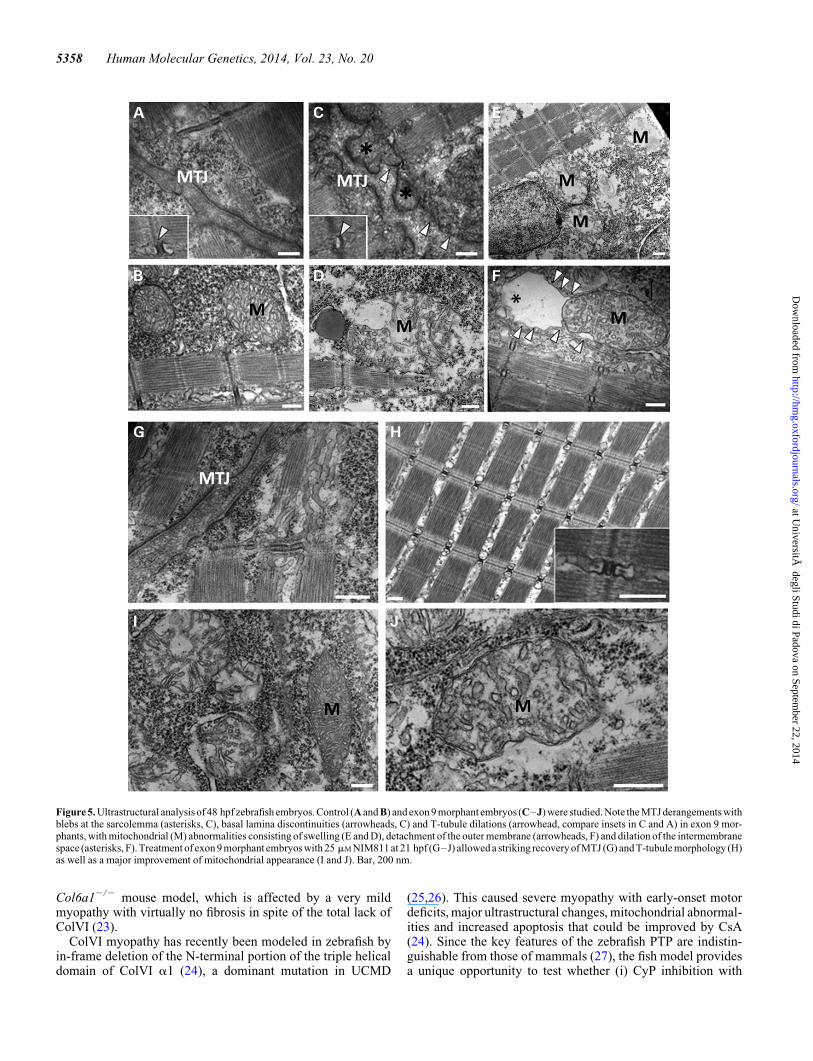
ultrastructural abnormalities, which correlated well with the
impaired motor function. The organization of muscle fibersshowed areas of myofibrillar disorganization and MTJ derange-ment (Fig. 5, compare panels A and C) with blebs of sarcolemma(Fig. 5, asterisks in panel C, compare with panel A), basal laminadiscontinuities (Fig. 5, arrowheads in panel C), and partial lossof myofibril attachment to the MTJ of sarcolemma in thealtered areas; in addition, exon 9 morphants displayed diffuseT-tubule dilations (Fig. 5, arrowheads, compare insets inpanels C and A), while the sarcoplasmic reticulum appearednormal. Most mitochondria (M, Fig. 5E, D and F, comparewith panel B) appeared enlarged and swollen with reducednumber of cristae, detachment of the outer membrane (Fig. 5F,arrowheads) and marked dilation of the intermembrane space(Fig. 5F, asterisk). Strikingly, in vivo treatment with 25 mM
NIM811 in fish water rescued the overall organization ofmuscle fibers including the MTJ derangement, T-tubule dila-tions and overall mitochondrial morphology as indicated bythe lack of intermembrane space dilations and swelling(Fig. 5G–J).
NIM811 prevents mitochondrial dysfunction and apoptosis,and rescues muscle force in Col6a12/2 mice
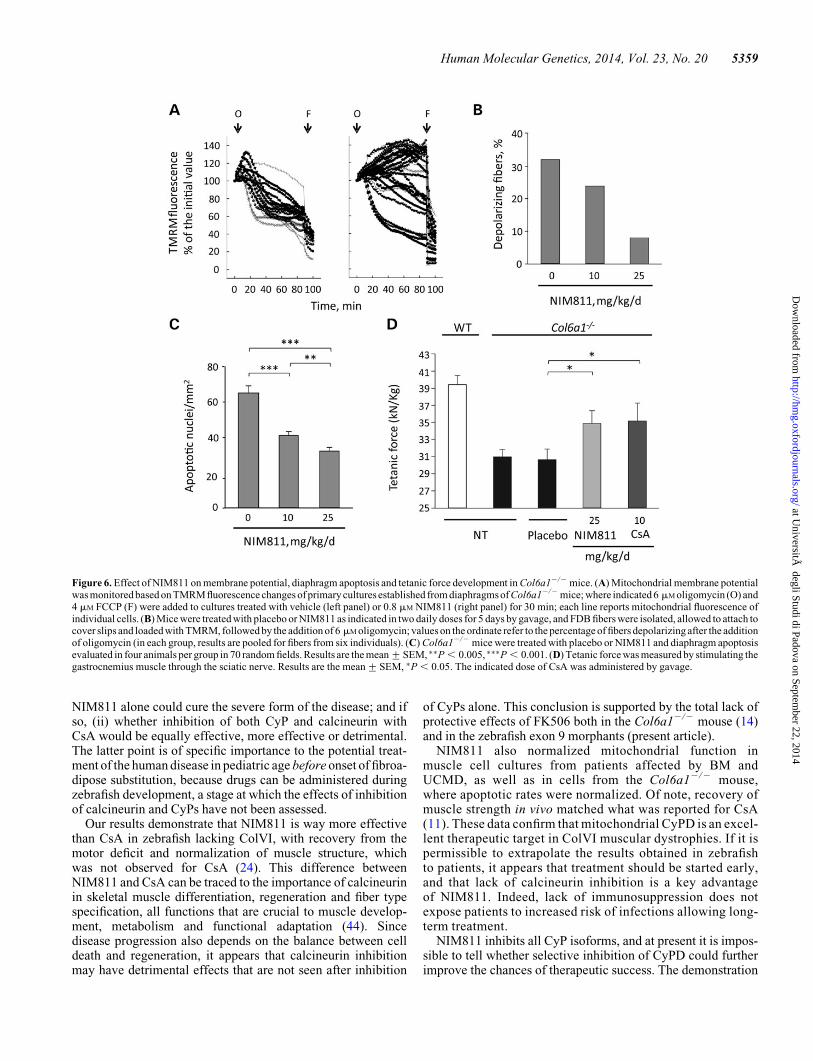
In order to assess the efficacy of NIM811 also in the well-characterized Col6a12/2 mouse model (14,23), we studied themitochondrial transmembrane potential in situ based on themitochondrial fluorescence of tetramethylrhodamine methylester (TMRM), a probe that accumulates in polarized mitochon-dria and is released when the transmembrane potential decreases(33). Addition of oligomycin (inhibitor of the mitochondrialF-ATP synthase) to primary cultures derived from diaphragmof Col6a12/2 mice caused the expected decrease of TMRMmitochondrial fluorescence (Fig. 6A, left panel), an anomalous
Figure 2. Effect of NIM811, CsA or FK506 on spontaneous coiling events of col6a1 morphant zebrafish. Spontaneous coiling events were recorded in 24 hpf controland exon 9 morphant embryos in the presence of the indicated concentrations of NIM811, CsA or FK506 added at 21 hpf. Data represent the mean of at least threeindependent experiments+SEM. ∗∗∗P , 0.005. Total embryos analyzed were: controls, 160; exon 9 morphants, 177 untreated, 100 for each NIM811 concentration,50 for each CsA concentration and 20, 18 and 16 for concentrations of FK506 of 10, 25 and 50 mM, respectively. Abbreviations are as in Figure 1.
Human Molecular Genetics, 2014, Vol. 23, No. 20 5355
at UniversitÃ
degli Studi di Padova on September 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
depolarizing response due to PTP opening that can be correctedby treatment with CsA and Debio 025 (14,22). Treatment withNIM811 normalized the response to oligomycin (i.e. preventedmitochondrial depolarization) in the vast majority of cells(Fig. 6A, right panel). Importantly, a similar protective effectwas recorded after in vivo treatment of Col6a12/2 mice withNIM811 for 5 days (Fig. 6B). Indeed, the depolarizing responseto oligomycin was normalized in the vast majority of flexor digi-torum brevis (FDB) fibers prepared from treated mice. If athreshold is arbitrarily set at the initial fluorescence value, 46out of 144 fibers (32%) depolarized in the placebo group whiletreatment with NIM811 decreased the number of fibers with de-polarizing mitochondria to 35 out of 146 (24%) and 10 out of 126(8%) at doses of 10 and 25 mg/kg/day, respectively (in eachgroups, results are pooled for fibers from six individuals)(Fig. 6B). Diaphragm fibers from mice treated with NIM811for 5 days also showed a remarkable decrease of apoptoticnuclei compared with those from the placebo group both atthe 10 and 25 mg/kg/day dose (Fig. 6C). Finally, we measuredtetanic (maximal) muscle force in the hindlimbs (34).Col6a12/2 mice treated for 10 days with 25 mg/kg/day NIM811showed a marked recovery of muscle strength, which matchedwhat we previously observed in CsA-treated mice (11) (Fig. 6D).
NIM811 prevents mitochondrial dysfunction and apoptosisin cells derived from muscle of UCMD and BM patients
A typical feature of muscle-derived cell cultures from patientswith ColVI myopathies is that the addition of oligomycin or rote-none is followed by mitochondrial depolarization that dependson PTP opening (15–17,19). Treatment with NIM811 preventeddepolarization inducedbybotholigomycinand rotenone inculturesestablished from patients affected by UCMD (Fig. 7A and B) and
BM (Fig. 7C and D); and it normalized the frequency of muscleapoptotic nuclei (Fig. 7E).
DISCUSSION
The role of PTP opening in the pathogenesis of ColVI musculardystrophies (15–18) and the therapeutic potential of CsAthrough inhibition of matrix CyPD are well established: CsAhas been successfully used in animal models (14,24) as well asin cells from patients affected by ColVI myopathies (15–17,19–21). The pharmacology and toxicology of CsA iscomplex, however, because of the multiple pathways affectedby this drug.
The primary intracellular targets of CsA are CyPs, a family ofproteins with peptidyl prolyl cis–trans isomerase activity (35),which in humans includes 16 proteins (36). The most abundantis the cytosolic isoform, CyPA, which is responsible for the im-munosuppressive effects of CsA. After binding CsA (whichleads to inhibition of CyPA), the CsA–CyPA complextargets—and inhibits—the cytosolic phosphatase calcineurin(29), which in turn prevents dephosphorylation—hencenuclear translocation—of NFAT resulting in immunosuppres-sion (37,38). Calcineurin is also inhibited by the complex ofFK506 with the FK506-binding proteins, a separate class ofintracellular peptidyl prolyl cis–trans isomerases (29).
Calcineurin inhibition prevents mitochondrial translocationof the pro-fission protein Drp1 and mitochondrial fragmentation,resulting in inhibition of autophagy (39). This event is cytopro-tective if cells are healthy (40,41), while it is detrimental inColVI myopathies, where defective removal of dysfunctionalmitochondria contributes to disease pathogenesis (11–13). Itshould be stressed that autophagy induction by CsA in theCol6a12/2 mouse—which is beneficial—is not likely to be
Figure 3. Effect of NIM811, CsA or FK506 on touch-evoked escape response of col6a1 morphant zebrafish. Responses evoked by touching 48 hpf embryos with apipette tip were recorded. Embryos were assigned to four groupsbasedon their escape response:open bars, normal response;hatched bars, minormotilitydefects;greybars, embryos responding with coiling only; closed bars, no response. Results are reported as percentage of total embryos analyzed. Where indicated, embryos hadbeen treated with the indicated concentrations of NIM811, CsA or FK506 at 21 hpf. Number of experiments, numerosity for each condition and abbreviations are as inFigure 1.
5356 Human Molecular Genetics, 2014, Vol. 23, No. 20
at UniversitÃ
degli Studi di Padova on September 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
caused by inhibition of calcineurin because (i) FK506 worsenedthe incidence of muscle apoptosis in the Col6a12/2 myopathicmouse (14); and (ii) the non-immunosuppressive CsA derivativeDebio 025 (Alisporivir) displayed a remarkable therapeutic effi-cacy (14). Furthermore, while CsA decreased contractile per-formance of human and rabbit heart muscle preparations (42),
Debio 025 was cardioprotective in a mouse model of myocardialinfarction (43), suggesting that the toxic effects of CsA are due toinhibition of calcineurin rather than to inhibition of CyPs. Whilethe above findings suggest that patients with ColVI myopathiescould be treated with CyP inhibitors devoid of effects oncalcineurin, these drugs have only been tested in the adult
Figure 4. Effect of NIM811, CsA or FK506 on birefringence of col6a1 morphant zebrafish. (A) Birefringence of normal and exon 9 morphant embryos with moderateor severe myopathic phenotype. Bar graph, percentage of birefringence classes after treatment with NIM811, CsA or FK506 (open bars, normal birefringence; greybars, moderatemyopathic phenotype; closed bars, severe myopathicphenotype). (B) Statistical analysis of total birefringence. Data represent the mean of at least threeindependent experiments+SEM, ∗∗∗P , 0.005. Total embryos analyzed were: controls, 43; exon 9 morphants, 43 untreated, 43 for each NIM811 concentration, 30for each CsA concentration and 15 for each FK506 concentration.
Human Molecular Genetics, 2014, Vol. 23, No. 20 5357
at UniversitÃ
degli Studi di Padova on September 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
Col6a12/2 mouse model, which is affected by a very mildmyopathy with virtually no fibrosis in spite of the total lack ofColVI (23).
ColVI myopathy has recently been modeled in zebrafish byin-frame deletion of the N-terminal portion of the triple helicaldomain of ColVI a1 (24), a dominant mutation in UCMD
(25,26). This caused severe myopathy with early-onset motordeficits, major ultrastructural changes, mitochondrial abnormal-ities and increased apoptosis that could be improved by CsA(24). Since the key features of the zebrafish PTP are indistin-guishable from those of mammals (27), the fish model providesa unique opportunity to test whether (i) CyP inhibition with
Figure 5. Ultrastructural analysis of 48 hpf zebrafish embryos. Control (A and B) and exon 9 morphant embryos (C–J) were studied. Note the MTJ derangements withblebs at the sarcolemma (asterisks, C), basal lamina discontinuities (arrowheads, C) and T-tubule dilations (arrowhead, compare insets in C and A) in exon 9 mor-phants, with mitochondrial (M) abnormalities consisting of swelling (E and D), detachment of the outer membrane (arrowheads, F) and dilation of the intermembranespace (asterisks, F). Treatment of exon 9 morphant embryos with 25 mM NIM811 at 21 hpf (G–J) allowed a striking recovery of MTJ (G) and T-tubule morphology (H)as well as a major improvement of mitochondrial appearance (I and J). Bar, 200 nm.
5358 Human Molecular Genetics, 2014, Vol. 23, No. 20
at UniversitÃ
degli Studi di Padova on September 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
NIM811 alone could cure the severe form of the disease; and ifso, (ii) whether inhibition of both CyP and calcineurin withCsA would be equally effective, more effective or detrimental.The latter point is of specific importance to the potential treat-ment of the human disease in pediatric age before onset of fibroa-dipose substitution, because drugs can be administered duringzebrafish development, a stage at which the effects of inhibitionof calcineurin and CyPs have not been assessed.
Our results demonstrate that NIM811 is way more effectivethan CsA in zebrafish lacking ColVI, with recovery from themotor deficit and normalization of muscle structure, whichwas not observed for CsA (24). This difference betweenNIM811 and CsA can be traced to the importance of calcineurinin skeletal muscle differentiation, regeneration and fiber typespecification, all functions that are crucial to muscle develop-ment, metabolism and functional adaptation (44). Sincedisease progression also depends on the balance between celldeath and regeneration, it appears that calcineurin inhibitionmay have detrimental effects that are not seen after inhibition
of CyPs alone. This conclusion is supported by the total lack ofprotective effects of FK506 both in the Col6a12/2 mouse (14)and in the zebrafish exon 9 morphants (present article).
NIM811 also normalized mitochondrial function inmuscle cell cultures from patients affected by BM andUCMD, as well as in cells from the Col6a12/2 mouse,where apoptotic rates were normalized. Of note, recovery ofmuscle strength in vivo matched what was reported for CsA(11). These data confirm that mitochondrial CyPD is an excel-lent therapeutic target in ColVI muscular dystrophies. If it ispermissible to extrapolate the results obtained in zebrafishto patients, it appears that treatment should be started early,and that lack of calcineurin inhibition is a key advantageof NIM811. Indeed, lack of immunosuppression does notexpose patients to increased risk of infections allowing long-term treatment.
NIM811 inhibits all CyP isoforms, and at present it is impos-sible to tell whether selective inhibition of CyPD could furtherimprove the chances of therapeutic success. The demonstration
Figure 6. Effect of NIM811 on membrane potential, diaphragm apoptosis and tetanic force development in Col6a12/2 mice. (A) Mitochondrial membrane potentialwas monitored based on TMRM fluorescence changes of primary cultures established from diaphragms of Col6a12/2 mice; where indicated 6 mM oligomycin (O) and4 mM FCCP (F) were added to cultures treated with vehicle (left panel) or 0.8 mM NIM811 (right panel) for 30 min; each line reports mitochondrial fluorescence ofindividual cells. (B) Mice were treated with placebo or NIM811 as indicated in two daily doses for 5 days by gavage, and FDB fibers were isolated, allowed to attach tocover slips and loaded with TMRM, followed by the addition of 6 mM oligomycin; values on the ordinate refer to the percentage of fibers depolarizing after the additionof oligomycin (in each group, results are pooled for fibers from six individuals). (C) Col6a12/2 mice were treated with placebo or NIM811 and diaphragm apoptosisevaluated in four animals per group in 70 random fields. Results are the mean+SEM, ∗∗P , 0.005, ∗∗∗P , 0.001. (D) Tetanic force was measured by stimulating thegastrocnemius muscle through the sciatic nerve. Results are the mean+SEM, ∗P , 0.05. The indicated dose of CsA was administered by gavage.
Human Molecular Genetics, 2014, Vol. 23, No. 20 5359
at UniversitÃ
degli Studi di Padova on September 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

that isoform-specific inhibitors of CyPs can be synthesized (45)opens new perspectives to the development of CyPD-selectiveinhibitors, and holds further promise for treatment of diseaseswhere the PTP is involved.
MATERIALS AND METHODS
Zebrafish and embryo maintenance
Adult zebrafish were maintained in the facility of University ofPadova containing aerated, 28.58C-conditioned saline wateraccording to standard protocols. Fish were kept under a 14 hlight–10 h dark cycle. For mating, males and females were
separated in the late afternoon and the next morning werefreed to start courtship, which ended with eggs deposition and fe-cundation. Eggs were collected, washed with fish water (0.5 mM
NaH2PO4, 0.5 mM NaHPO4, 0.2 mg/l methylene blue, 3 mg/linstant ocean) and maintained at 28.58C in fish water supplemen-ted with an antibiotic-antimycotic cocktail (50 mg/ml ampicil-lin, 100 units/ml penicillin and 0.1 mg/ml streptomycin,Biochrom, 3.3 mg/ml amphotericin B, Bristol-Myers-Squibb).
Treatment with antisense morpholino
In order to affect ColVI synthesis and secretion, we used a pub-lished exon 9 morpholino (24), which targets zebrafish col6a1
Figure 7. Effect of NIM811 on oligomycin- and rotenone-induced mitochondrial depolarization and apoptosis in cell cultures from UCMD and BM patients. (A–D)Mitochondrial membrane potential was monitored based on TMRM fluorescence changes of primary cultures established from muscle biopsies of UCMD (A and B)or BM patients (C and D). Where indicated by arrows, 6 mM oligomycin (O), 2 mM rotenone (R) and 4 mM FCCP (F) were added in the absence (open symbols) or presence(closedsymbols) of 0.8 mM NIM811added30 minbefore thebeginningof the recordings (time0). Data represent the meanofat least six independentexperiments+SEM,and the line marked by one asterisk indicates the time points with P , 0.05 for no treatment compared with NIM811. (E) Primary myoblast cultures from one UCMDpatient and one BM patient were plated and scored for the presence of apoptotic nuclei by the TUNEL reaction. Where indicated, cells had been incubated withNIM811 for 2 h. Data are mean of three independent experiments+SEM, ∗P , 0.05 and ∗∗P , 0.01 for NIM811-treated versus non-treated cells.
5360 Human Molecular Genetics, 2014, Vol. 23, No. 20
at UniversitÃ
degli Studi di Padova on September 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

mRNA leading to translation of a truncated, dominant negative,version of the ColVI a1 chain. Embryos from Zebrafish wildtype incrosses were injected with morpholino at 1–4 cell stageusing a WPI pneumatic PicoPump PV820 injector. Morpholinowas injected at a concentration of 0.1 mM, corresponding toabout 4 ng per embryo.
Ultrastructural analysisWild type and exon 9 morphant zebrafish embryos (48 hpf) werefixed with Karnovsky fixative (2.5% glutaraldehyde and 2% par-aformaldheyde in 0.1 M cacodylate buffer) for 3 h at 48C, washedwith 0.1 M cacodilate buffer, post-fixed with osmium tetroxidefor 2 h and embedded in EPON 812 as previously described(24). Ultrathin sections were stained with uranyl acetate andlead citrate and observed at Philips EM400 operating at 100 kV.
NIM811, CsA and FK506 treatment
Morphant embryos were dechorionated at 20 hpf and then incu-bated with drugs from 21 hpf until the time of observation (24 or48 hpf as specified). NIM811 and CsA (Novartis Pharma AG) orFK506 (Sigma, St Louis, MO, USA) were used at 10, 25 and50 mM and dissolved in 0.01, 0.03 and 0.06% DMSO in fishwater. Vehicle control treatment consisted of 0.06% DMSO infish water.
Immunofluorescence analysis of zebrafish cryosections
Zebrafish embryos (48 hpf) were frozen in isopentane pre-chilledin liquid nitrogen and stored at 2808C. Seven-millimeter-thickfrozen sections were immunolabeled with anti-ColVI (Abcam)and anti-b-dystroglycan antibodies (Novocastra) and revealedwith TRITC- and FITC-conjugated secondary antibodies, re-spectively (Dako). Samples were mounted with anti-fading (Mo-lecular Probes) and observed with an epifluorescence NikonE600 microscope. For confocal analysis (Fig. 1C–H), embryoswere fixed with 4% paraformaldehyde in phosphate buffersaline (PBS), washed with PBS and permeabilized with 0.15%Triton X-100. Immunolabeling was performed with a polyclonalanti-Tom20 antibody (Santa Cruz) revealed with a FITC-conjugated anti-rabbit secondary antibody (Dako), and actinwas stained with rhodamine-conjugated phalloidin (Sigma).Imaging was performed with an A1-R confocal laser scanningmicroscope (Nikon, Melville, NY, USA) equipped with 488and 561 nm laser lines to elicit FITC and TRITC fluorescence, re-spectively. Each confocal image was obtained by maximum in-tensity projection of 10 optical sections scanned in the centralregion of the sample (recorded at z-step size of 300 nm).
Motor activity
Spontaneous coiling rate was recorded as the number of eventsobserved in 15 s for individual embryos at 24 hpf using light mi-croscopy. For the touch escape response assay, we observed theability to escape after touching embryos with a small tip at48 hpf. We subdivided embryos into four groups according totheir ability to escape: ‘normal’ means embryos with normal mo-tility to swim; ‘motor impairments’ means embryos with minormotility disruptions; ‘only coiling events’ means embryos circ-ling without the ability to escape; ‘paralyzed’ means embryoswith no motility.
Birefringence assay
Muscle birefringence was analyzed by taking advantage ofmuscle fibers anisotropy. Briefly, we placed anesthetizedembryos on a glass and analyzed muscle light refraction byusing two polarizing filters. The first filter produces the polarizedlight to illuminate the sample and the second polarizing filter,called the analyzer, restricts detected light to refracted lightcoming from muscle fibers. In particular, the top polarizingfilter was twisted of a 908 angle until the light refractingthrough the muscle was visible. To perform these experiments,we used a Leica M165FC stereomicroscope. We calculated inte-grated area of birefringence by using ImageJ software, asdescribed (46). Birefringence values ≥3 × 106 (typical of wildtype individuals) were rated as normal, values between 2.9 and1 × 106 were considered as an indication of mild disease andvalues ≤1 × 106 were rated as an indication of severe myopathy.
Mice
Col6a12/2 mice (23) had free access to a standard diet and werekept under controlled conditions of temperature and humidity ona 12 h light/12 h dark cycle. Mice were treated with placebo,10 mg or 25 mg NIM811/kg/day by gavage in two separatedoses for 5 and 10 days. Mice were observed every day untilthe end of the treatment period in order to detect mortality, mor-bidity or any abnormal clinical event. No abnormal behavioral ordigestive signs were found during treatment, nor other signs ofpoor clinical condition. Body weight measurements at days 1,3 and 5 displayed small variations that were not significantly dif-ferent for the various treatment groups. All in vivo experimentswere approved by the competent Authority of the Universityof Padova and authorized by the Italian Ministry of Health.
Muscle isolation
Fibers from FDB muscles were isolated exactly as described pre-viously (33). Fibers were then plated onto glass cover slipscoated with laminin (3 mg/cm2) and cultured in Dulbecco’smodified Eagle medium (DMEM) containing 10% fetal bovineserum. During the experiment, FDB fibers were placed in 1 mlTyrode’s buffer and loaded with 20 nM of TMRM as describedpreviously (14).
In vivo muscle mechanics
In vivo determination of force and contraction kinetics of gastro-cnemius muscle were carried out as previously described (47).
Terminal deoxynucleotidyl transferase-mediateddUTP nick end labelling (TUNEL) assay
Seven-micrometer-thick sections of muscle diaphragm wereprepared after 4% paraformaldehyde fixation and paraffinembedding. TUNEL was done using the ApopTag in situ apop-tosis detection kit. Samples were stained with peroxidase-diaminobenzidine to detect TUNEL-positive nuclei andcounterstained with Hoechst 33258 to identify all nuclei, asdescribed previously (14). Total and TUNEL-positive nucleiwere determined in randomly selected fibers using a Zeiss
Human Molecular Genetics, 2014, Vol. 23, No. 20 5361
at UniversitÃ
degli Studi di Padova on September 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

Axioplan microscope equipped with a Leica DC 500 camera. Inthe muscle cultures, TUNEL assays were performed with theDead End Fluorometric in situ apoptosis detection system(Promega Italia, Milan, Italy). Muscle cultures were permeabi-lized in methanol–acetone 50:50 at 2208C for 10 min. Afterdrying, slides were washed in PBS, incubated with equilibrationbuffer for 10 min and treated with buffer containing fluorescentnucleotides, rTdT enzyme and Hoechst 33258 (Sigma) for 1 h at378C. SSC solution was used to block the activity of rTdTenzyme, before washing and preparing slides for microscopyanalysis.
Patient and mouse muscle-derived cell cultures
BM and UCMD were diagnosed according to the criteria of theEuropean Neuromuscular Center (8). All patients were exam-ined and all underwent a muscle biopsy. All participants pro-vided written informed consent, and approval was obtainedfrom the Ethics Committee of the Rizzoli Orthopedic Institute(Bologna, Italy). Cell cultures were established using enzymaticand mechanical treatment and plating in DMEM plus 20%fetal calf serum (FCS) and antibiotics (penicillin, streptomycinand amphotericin B; Sigma) and then stored in liquid nitrogen.Mouse cell cultures were obtained from diaphragms aspreviously described (18).
Measurements of mitochondrial membrane potential
Mitochondrial membrane potential was measured by epifluores-cence microscopy based on the accumulation of TMRM asdescribed (15). Muscle-derived cell cultures (30 000–35 000cells) were seeded onto 24 mm diameter round glass coverslips, grown for 2 days in DMEM supplemented with 20%FCS and studied as described after loading with 10 nM TMRMin 1 ml of serum-free DMEM.
Statistical analysis
Data were analyzed using the unpaired Student’s t-test exceptfor Figures 2 and 4B, where ANOVA test was used. Valueswith P , 0.05 were considered significant and data are reportedas mean+SEM.
ACKNOWLEDGMENTS
NIM811and CsA were supplied by Novartis Pharma AG (Basel,Switzerland).
Conflict of Interest statement. None declared.
FUNDING
The study was supported in part by Research Grants fromNovartis Pharma AG (Basel) and Telethon, Italy (GGP11082).
REFERENCES
1. Lampe, A.K. and Bushby, K.M. (2005) Collagen VI related muscledisorders. J. Med. Genet., 42, 673–685.
2. Merlini, L., Martoni, E., Grumati, P., Sabatelli, P., Squarzoni, S., Urciuolo,A., Ferlini, A., Gualandi, F. and Bonaldo, P. (2008) Autosomal recessivemyosclerosis myopathy is a collagen VI disorder. Neurology, 71,1245–1253.
3. Bethlem, J. and Wijngaarden, G.K. (1976) Benign myopathy, withautosomal dominant inheritance. A report on three pedigrees. Brain, 99,91–100.
4. Merlini, L., Morandi, L., Granata, C. and Ballestrazzi, A. (1994) Bethlemmyopathy: early-onset benign autosomal dominant myopathy withcontractures. Description of two new families. Neuromuscul. Disord., 4,503–511.
5. Jobsis, G.J., Boers, J.M., Barth, P.G. and de Visser, M. (1999) Bethlemmyopathy: a slowly progressive congenital muscular dystrophy withcontractures. Brain, 122(Pt 4), 649–655.
6. Camacho Vanegas, O., Bertini, E., Zhang, R.Z., Petrini, S., Minosse, C.,Sabatelli, P., Giusti, B., Chu, M.L. and Pepe, G. (2001) Ullrich scleroatonicmuscular dystrophy is caused by recessive mutations in collagen type VI.Proc. Natl Acad. Sci. USA, 98, 7516–7521.
7. Demir, E., Sabatelli, P., Allamand, V., Ferreiro, A., Moghadaszadeh, B.,Makrelouf, M., Topaloglu, H., Echenne, B., Merlini, L. and Guicheney, P.(2002) Mutations in COL6A3 cause severe and mild phenotypes of Ullrichcongenital muscular dystrophy. Am. J. Hum. Genet., 70, 1446–1458.
8. Pepe, G., Bertini, E., Bonaldo, P., Bushby, K., Giusti, B., de Visser, M.,Guicheney, P., Lattanzi, G., Merlini, L., Muntoni, F. et al. (2002) Bethlemmyopathy (BETHLEM) and Ullrich scleroatonic muscular dystrophy: 100thENMC international workshop, 23–24 November 2001, Naarden, TheNetherlands. Neuromuscul. Disord., 12, 984–993.
9. Giorgio, V., von Stockum, S., Antoniel, M., Fabbro, A., Fogolari, F., Forte,M., Glick, G.D., Petronilli, V., Zoratti, M., Szabo, I. et al. (2013) Dimers ofmitochondrial ATP synthase form the permeability transition pore. Proc.
Natl Acad. Sci. USA, 110, 5887–5892.10. Bernardi, P. (2013) The mitochondrial permeability transition pore: A
mystery solved? Front. Physiol., 4, 95.
11. Grumati, P., Coletto,L., Sabatelli, P., Cescon, M., Angelin, A., Bertaggia, E.,Blaauw, B., Urciuolo, A., Tiepolo, T., Merlini, L. et al. (2010) Autophagy isdefective in collagen VI muscular dystrophies, and its reactivation rescuesmyofiber degeneration. Nat. Med., 16, 1313–1320.
12. Grumati, P., Coletto, L., Sandri, M. and Bonaldo, P. (2011) Autophagyinduction rescues muscular dystrophy. Autophagy, 7, 426–428.
13. Grumati, P., Coletto, L., Schiavinato, A., Castagnaro, S., Bertaggia, E.,Sandri, M. and Bonaldo, P. (2011) Physical exercise stimulates autophagy innormal skeletal muscles but is detrimental for collagen VI-deficient muscles.Autophagy, 7, 1415–1423.
14. Irwin, W.A., Bergamin, N., Sabatelli, P., Reggiani, C., Megighian, A.,Merlini, L., Braghetta, P., Columbaro, M., Volpin, D., Bressan, G.M. et al.
(2003) Mitochondrial dysfunction and apoptosis in myopathic mice withcollagen VI deficiency. Nat. Genet., 35, 267–271.
15. Angelin, A., Tiepolo, T., Sabatelli, P., Grumati, P., Bergamin, N., Golfieri,C., Mattioli, E., Gualandi, F., Ferlini, A., Merlini, L. et al. (2007)Mitochondrial dysfunction in the pathogenesis of Ullrich congenitalmuscular dystrophy and prospective therapy with cyclosporins. Proc. Natl
Acad. Sci. USA, 104, 991–996.16. Angelin, A., Bonaldo, P. and Bernardi, P. (2008) Altered threshold of the
mitochondrial permeability transition pore in Ullrich congenital musculardystrophy. Biochim. Biophys. Acta, 1777, 893–896.
17. Sabatelli, P., Palma, E., Angelin, A., Squarzoni, S., Urciuolo, A., Pellegrini,C., Tiepolo, T., Bonaldo, P., Gualandi, F., Merlini, L. et al. (2012) Criticalevaluation of the use of cell cultures for inclusion in clinical trials of patientsaffected by collagen VI myopathies. J. Cell. Physiol., 227, 2927–2935.
18. Palma, E., Tiepolo, T., Angelin, A., Sabatelli, P., Maraldi, N.M., Basso, E.,Forte, M.A., Bernardi, P. and Bonaldo, P. (2009) Genetic ablation ofcyclophilin D rescues mitochondrial defects and prevents muscle apoptosisin collagen VI myopathic mice. Hum. Mol. Genet., 18, 2024–2031.
19. Merlini, L., Angelin, A., Tiepolo, T., Braghetta, P., Sabatelli, P., Zamparelli,A., Ferlini, A., Maraldi, N.M., Bonaldo, P. and Bernardi, P. (2008)Cyclosporin A corrects mitochondrial dysfunction and muscle apoptosis inpatients with collagen VI myopathies. Proc. Natl Acad. Sci. USA, 105,5225–5229.
20. Merlini, L., Sabatelli, P., Armaroli, A., Gnudi, S., Angelin, A., Grumati, P.,Michelini, M.E., Franchella, A., Gualandi, F., Bertini, E. et al. (2011)Cyclosporine A in Ullrich congenital muscular dystrophy: long-term results.Oxid. Med. Cell Longev., 2011, 139194.
5362 Human Molecular Genetics, 2014, Vol. 23, No. 20
at UniversitÃ
degli Studi di Padova on September 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

21. Merlini, L. and Bernardi, P. (2008) Therapy of collagen VI-relatedmyopathies (Bethlem and Ullrich). Neurotherapeutics, 5, 613–618.
22. Tiepolo, T., Angelin, A., Palma, E., Sabatelli, P., Merlini, L., Nicolosi, L.,Finetti, F., Braghetta, P., Vuagniaux, G., Dumont, J.M. et al. (2009) Thecyclophilin inhibitor Debio 025 normalizes mitochondrial function, muscleapoptosis and ultrastructural defects in Col6a12/2 myopathic mice.Br. J. Pharmacol., 157, 1045–1052.
23. Bonaldo, P., Braghetta, P., Zanetti, M., Piccolo, S., Volpin, D. and Bressan,G.M. (1998) Collagen VI deficiency induces early onset myopathy in themouse: an animal model for Bethlem myopathy. Hum. Mol. Genet., 7,2135–2140.
24. Telfer, W.R., Busta, A.S., Bonnemann, C.G., Feldman, E.L. and Dowling,J.J. (2010) Zebrafish models of collagen VI-related myopathies. Hum. Mol.
Genet, 19, 2433–2444.
25. Pepe, G., Lucarini, L., Zhang, R.Z., Pan, T.C., Giusti, B., Quijano-Roy, S.,Gartioux, C., Bushby, K.M., Guicheney, P. and Chu, M.L. (2006) COL6A1genomic deletions in Bethlem myopathy and Ullrich muscular dystrophy.Ann. Neurol., 59, 190–195.
26. Lampe, A.K., Zou, Y., Sudano, D., O’Brien, K.K., Hicks, D., Laval, S.H.,Charlton, R., Jimenez-Mallebrera, C., Zhang, R.Z., Finkel, R.S. et al. (2008)Exon skipping mutations in collagen VI are common and are predictive forseverity and inheritance. Hum. Mutat., 29, 809–822.
27. Azzolin, L., Basso, E., Argenton, F. and Bernardi, P. (2010) MitochondrialCa2+ transport and permeability transition in zebrafish (Danio rerio).Biochim. Biophys. Acta, 1797, 1775–1779.
28. Waldmeier, P.C., Feldtrauer, J.J., Qian, T. and Lemasters, J.J. (2002)Inhibition of the mitochondrial permeability transition by thenonimmunosuppressive cyclosporin derivative NIM811. Mol. Pharmacol.,62, 22–29.
29. Liu, J., Farmer, J.D.J., Lane, W.S., Friedman, J., Weissman, I. and Schreiber,S.L. (1991) Calcineurin is a common target of cyclophilin-cyclosporinA andFKBP-FK506 complexes. Cell, 66, 807–815.
30. Robu, M.E., Larson, J.D., Nasevicius, A., Beiraghi, S., Brenner, C., Farber,S.A. and Ekker, S.C. (2007) p53 activation by knockdown technologies.PLoS Genet., 3, e78.
31. Bessarab, D.A., Chong, S.W., Srinivas, B.P. and Korzh, V. (2008) Six1a isrequired for the onset of fast muscle differentiation in zebrafish. Dev. Biol.,323, 216–228.
32. Bassett, D.I. and Currie, P.D. (2003) The zebrafish as a model for musculardystrophy and congenital myopathy. Hum. Mol. Genet., 12(Spec No. 2),R265–R270.
33. Irwin, W., Fontaine, E., Agnolucci, L., Penzo, D., Betto, R., Bortolotto, S.,Reggiani, C., Salviati, G. and Bernardi, P. (2002) Bupivacaine myotoxicityis mediated by mitochondria. J. Biol. Chem., 277, 12221–12227.
34. Rossi, R., Bottinelli, R., Sorrentino, V. and Reggiani, C. (2001) Response tocaffeine and ryanodine receptor isoforms in mouse skeletal muscles.Am. J. Physiol. Cell Physiol., 281, C585–C594.
35. Fischer, G., Wittmann-Liebold, B., Lang, K., Kiefhaber,T. and Schmid, F.X.(1989) Cyclophilin and peptidyl-prolyl cis-trans isomerase are probablyidentical proteins. Nature, 337, 476–478.
36. Wang, P. and Heitman, J. (2005) The cyclophilins. Genome Biol., 6,226.1–226.6.
37. Clipstone, N.A. and Crabtree, G.R. (1992) Identification of calcineurin as akey signalling enzyme in T-lymphocyte activation. Nature, 357, 695–697.
38. Walsh, C.T., Zydowsky, L.D. and McKeon, F.D. (1992) Cyclosporin A, thecyclophilin class of peptidylprolyl isomerases, and blockade of T cell signaltransduction. J. Biol. Chem., 267, 13115–13118.
39. Cereghetti, G.M., Stangherlin, A., Martins de Brito, O., Chang, C.R.,Blackstone, C., Bernardi, P. and Scorrano, L. (2008) Dephosphorylation bycalcineurin regulates translocation of Drp1 to mitochondria. Proc. NatlAcad. Sci. USA, 105, 15803–15808.
40. Cereghetti, G.M., Costa, V. and Scorrano, L. (2010) Inhibition ofDrp1-dependent mitochondrial fragmentation and apoptosis by apolypeptide antagonist of calcineurin. Cell Death Differ., 17, 1785–1794.
41. Gomes, L.C., Di Benedetto, G. and Scorrano, L. (2011) During autophagymitochondria elongate, are spared from degradation and sustain cellviability. Nat. Cell Biol., 13, 589–598.
42. Janssen, P.M., Zeitz, O., Keweloh, B., Siegel, U., Maier, L.S., Barckhausen,P., Pieske, B., Prestle, J., Lehnart, S.E. and Hasenfuss, G. (2000) Influence ofcyclosporine A on contractile function, calcium handling, and energetics inisolated human and rabbit myocardium. Cardiovasc. Res., 47, 99–107.
43. Gomez, L., Thibault, H., Gharib, A., Dumont, J.-M., Vuagniaux, G.,Scalfaro, P., Derumeaux, G. and Ovize, M. (2007) Inhibition ofmitochondrial permeability transition improves functional recovery andreduces mortality following acute myocardial infarction in mice.Am. J. Physiol. Heart Circ. Physiol., 293, H1654–H1661.
44. Sakuma, K. and Yamaguchi, A. (2010) The functional role of calcineurin inhypertrophy, regeneration, and disorders of skeletal muscle. J. Biomed.Biotechnol., 2010, 721219.
45. Daum, S., Schumann, M., Mathea, S., Aumuller, T., Balsley, M.A., Constant,S.L., de Lacroix, B.F., Kruska, F., Braun, M. and Schiene-Fischer, C. (2009)Isoform-specific inhibition of cyclophilins. Biochemistry, 48, 6268–6277.
46. Berger, J., Sztal, T. and Currie, P.D. (2012) Quantification of birefringencereadily measures the level of muscle damage in zebrafish. Biochem. Biophys.Res. Commun., 423, 785–788.
47. Blaauw, B., Canato, M., Agatea, L., Toniolo, L., Mammucari, C., Masiero,E., Abraham, R., Sandri,M., Schiaffino,S. and Reggiani,C. (2009) Inducibleactivation of Akt increases skeletal muscle mass and force without satellitecell activation. FASEB J., 23, 3896–3905.
Human Molecular Genetics, 2014, Vol. 23, No. 20 5363
at UniversitÃ
degli Studi di Padova on September 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
Copyright © 2022 FDOKUMEN




















