
Characterization of Individuals with Muscular Dystrophy from ...
Upload
independentCategory
view
3download
0
ELSEVIER Brain & Development 1996; 18:316-322
Case report
Merosin-negative congenital muscular dystrophy, occipital epilepsy with periodic spasms and focal cortical dysplasia.
Report of three Italian cases in two families
Antonella Pini a,*, Luciano Merlini b Fernando M.S. Tom6 c Martine Chevallay c Giuseppe Gobbi a
a Servizio di Neuropsichiatria Infantile, Arcispedale S. Maria Nuova, Viale Risorgimento, 80, Reggio Emilia, Italy b Neuromuscular Laboratory, Rizzoli Orthopaedic Institute, Bologna, Italy
c INSERM U. 153, Paris, France
Received 6 October 1995; accepted 5 February 1996
W e repor t clinical, EEG and neuroimaging findings of three patients in two Italian families with merosin-negative congenital muscular dystrophy (CMD), drug-resistant occipital epilepsy, diffuse persistent cerebral white mat ter changes and focal cortical dysplasia. Clinical and epilepsy histories, EEG and neuroimaging findings were very similar in all patients. Seizures started in childhood and mainly consisted of periodic spasms, a part icular type of partial seizure characterized by clusters of epileptic spasms. The motor expression of the spasms was very mild so that they had been frequently missed or misinterpreted as non-convulsive generalized absence seizures. Interictal EEG showed occipital spike-waves and bilateral synchronous slow spike-wave discharges. Ictal EEG showed prolonged periodic sequences of slow waves with associated fast rhy thm complexes, characteristic of periodic spasms. Two patients had normal intelligence, one patient presented moderate mental retardation. Focal cortical dysplasia in the posterior areas of the brain, in addition to marked diffuse white mat ter alterations, was detected in the magnetic resonance images of all patients. Findings in these patients indicate that in merosin-negative CMD brain involvement can include cortical dysplasia, in addition to white mat ter changes. In such cases the brain damage can lead to a childhood-onset localization-related symptomatic occipital epilepsy. Epileptic seizures and cortical dysplasia can be, however, difficult to detect in CMD. The clinical semiology of epileptic seizures may in fact be modified because of muscular weakness. This implies that epilepsy may be misdiagnosed or even missed and EEG-polymyographic recordings may be necessary to identify it. Similarly, cortical dysplasia may be very localized and visible by neuroimaging only if it is carefully investigated on the basis of epileptological and EEG-polymyographic findings.
Keywords: Congenital muscular dystrophy; Merosin deficiency; Epileptic spasm; Cortical dysplasia
1. INTRODUCTION
Congenital muscular dystrophies (CMD) constitute a group of syndromes which have in common muscle weakness at birth or within the first few months of life in association with a dys-
* Corresponding author. Fax: (39) (522) 296266.
trophic pattern on muscle biopsy [1]. The presence or absence of brain and eye involvement are additional findings which make it possible to differentiate classical CMD from the Fukuyama-type CMD (FCMD), muscle-eye-brain disease and Walker-Warburg syndrome. Nevertheless, following the description of non- Japanese cases with CMD, epileptic seizures and structural brain involvement similar but not identical to FCMD [2-9], nosological difficulties have often emerged. In such cases brain involvement mainly consists of diffuse white matter abnormalities, but cortical
0387-7604/96/$15.00 Copyright © 1996 Elsevier Science B.V. All rights reserved. PH S03 87-7604(96)00028-9
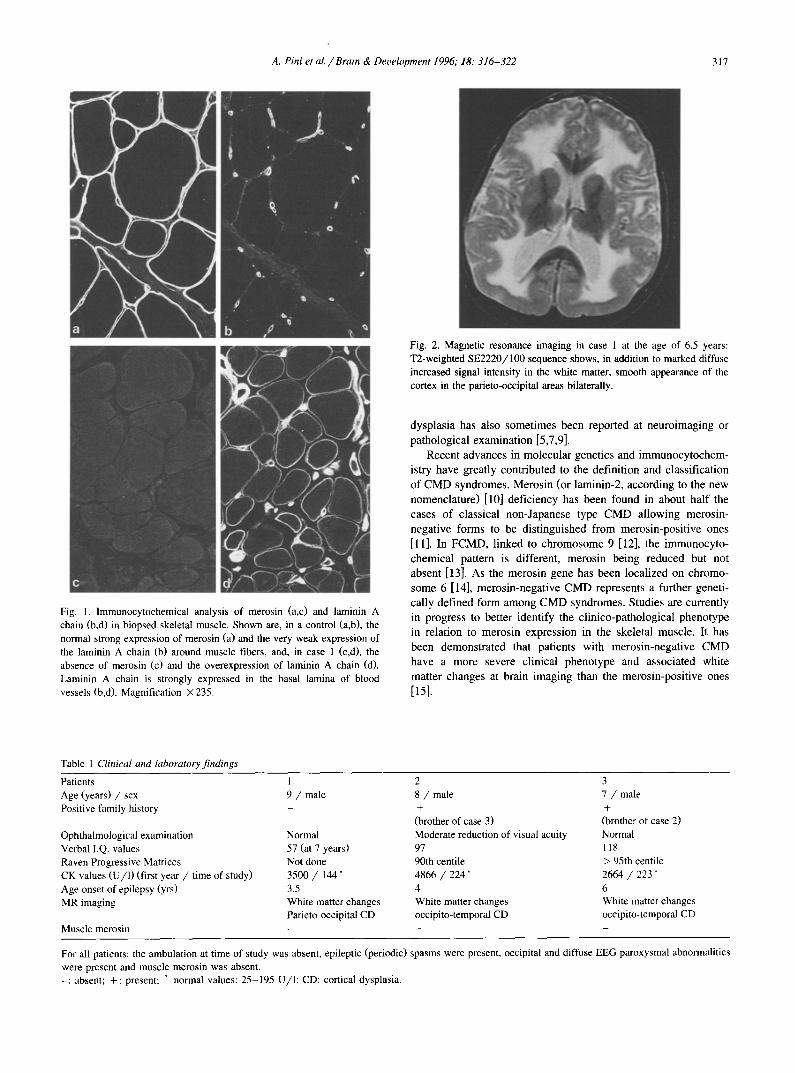
A. Pini et al./Brain & Development 1996; 18." 316-322 317
; ~t ~ r
Fig. 2. Magnetic resonance imaging in case 1 at the age of 6.5 years: T2-weighted SE2220/100 sequence shows, in addition to marked diffuse increased signal intensity in the white matter, smooth appearance of the cortex in the parieto-occipital areas bilaterally.
Fig. 1. Immunocytochemical analysis of merosin (a,c) and laminin A chain (b,d) in biopsed skeletal muscle. Shown are, in a control (a,b), the normal strong expression of merosin (a) and the very weak expression of the laminin A chain (b) around muscle fibers, and, in case 1 (c,d), the absence of merosin (c) and the overexpression of laminin A chain (d). Laminin A chain is strongly expressed in the basal lamina of blood vessels (b,d). Magnification × 235.
dysplasia has also sometimes been reported at neuroimaging or pathological examination [5,7,9].
Recent advances in molecular genetics and immunocytochem- istry have greatly contributed to the definition and classification of CMD syndromes. Merosin (or laminin-2, according to the new nomenclature) [10] deficiency has been found in about half the cases of classical non-Japanese type CMD allowing merosin- negative forms to be distinguished from merosin-positive ones [11]. In FCMD, linked to chromosome 9 [12], the immunocyto- chemical pattern is different, merosin being reduced but not absent [13]. As the merosin gene has been localized on chromo- some 6 [14], merosin-negative CMD represents a further geneti- cally defined form among CMD syndromes. Studies are currently in progress to better identify the clinico-pathological phenotype in relation to merosin expression in the skeletal muscle. It has been demonstrated that patients with merosin-negative CMD have a more severe clinical phenotype and associated white matter changes at brain imaging than the merosin-positive ones [15].
Table 1 Clinical and laboratory findings
Patients Age (years) / sex Positive family history
Ophthalmological examination Verbal I.Q. values Raven Progressive Matrices CK values (U/l) (first year / time of study) Age onset of epilepsy (yrs) MR imaging
Muscle merosin
1 2 3
9 / male 8 / male 7 / male - + +
(brother of case 3) (brother of case 2) Normal Moderate reduction of visual acuity Normal 57 (at 7 years) 97 118 Not done 90th centile > 95th centile 3500 / 144" 4866 / 224* 2664 / 223 * 3.5 4 6 White matter changes White matter changes White matter changes Parieto-occipital CD occipito-temporal CD occipito-temporal CD
For all patients: the ambulation at time of study was absent, epileptic (periodic) spasms were present, occipital and diffuse EEG paroxysmal abnormalities were present and muscle merosin was absent. - : absent; + : present; * normal values: 25-195 U/ l ; CD: cortical dysplasia.
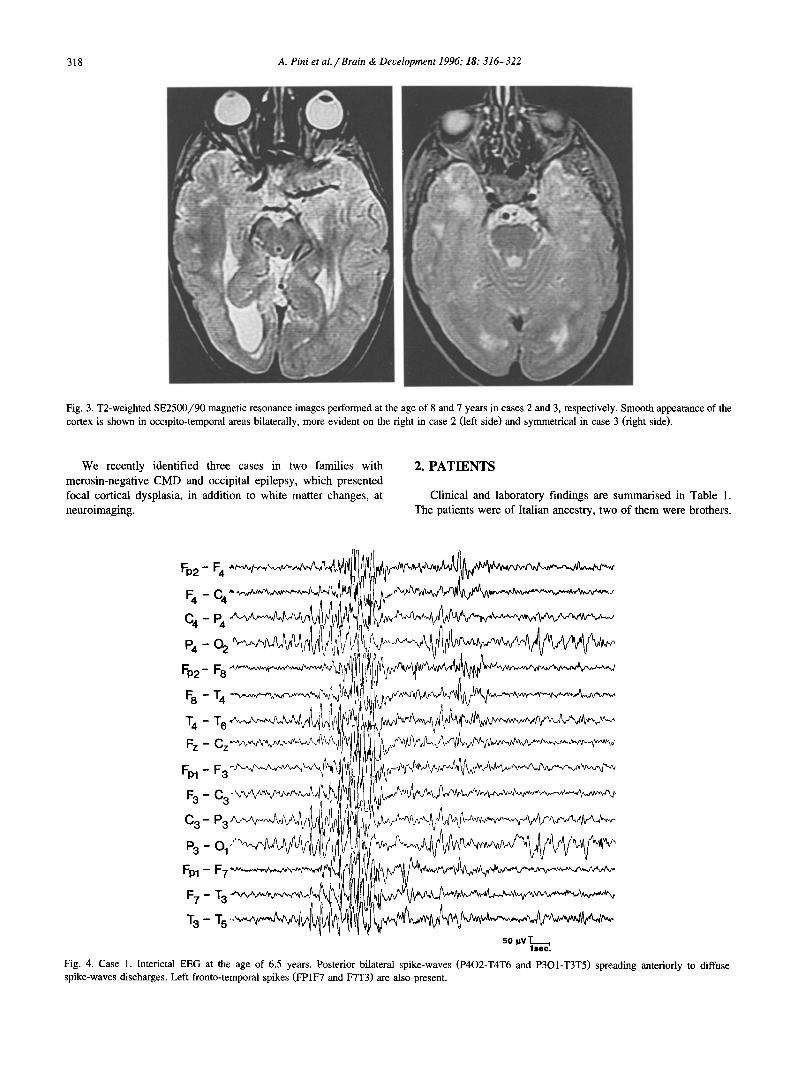
318 A. Pini et aL / Brain & Deuelopment 1996; 18:316-322
Fig. 3. T2-weighted SE2500/90 magnetic resonance images performed at the age of 8 and 7 years in cases 2 and 3, respectively. Smooth appearance of the cortex is shown in occipito-temporal areas bilaterally, more evident on the right in case 2 (left side) and symmetrical in case 3 (right side).
We recently identified three cases in two families with merosin-negative CMD and occipital epilepsy, which presented focal cortical dysplasia, in addition to white matter changes, at neuroimaging.
2 . P A T I E N T S
Clinical and laboratory findings are summarised in Table 1. The patients were of Italian ancestry, two of them were brothers.
Fp2- F 4
F 4 - C 4
c4-P4
P4-02 Fp2- F8
%- - r 4
r4 -T 6 Fz - C~
Fpl - F a
%-% C 3 - Pa
Fpl - F 7
F 7 - T 3
T3-Ts 50 pV ls-L.s~ee,
Fig. 4. Case 1. Interictal EEG at the age of 6.5 years. Posterior bilateral spike-waves (P402-T4T6 and P301-T3T5) spreading anteriorly to diffuse spike-waves discharges. Left fronto-temporal spikes (FP1F7 and F7T3) are also present.

A. Pini et al./Brain & Deuelopment 1996; 18:316-322 319
Clinical signs of CMD were present since birth in all cases, with floppiness, muscle weakness and multiple contractures. Motor skills were delayed. After surgical release of contractures, pa- tients were able to stand with calipers, though this ability was subsequently lost. Weakness also involved facial muscles result- ing in mandibular ptosis and, with time, severe open bite maloc- clusion. Cranial circumference was normal. Serum creatine ki- nase levels were markedly increased in the first year of life in all patients. Ophthalmological examination showed a moderate re- duction of visual acuity in patient 2. Ocular funduscopy and visual evoked potentials were normal in all patients.
Muscle biopsy showed typical dystrophic changes with exten- sive connective tissue and fat proliferation. Indirect immunofluo- rescence microscopy was peformed on 7-p,m thick cryosections from the muscle biopsy specimens using monoclonal anti-M (a 2) at l: 1000 (Chemicon), and anti-A (ct 1) at 1:500, anti-B 1 (131) at 1:500 and anti-B2 ('¢1) at 1:200 (Gibco BRL) laminin chain antibodies. Goat anti-mouse fluorescein conjugated secondary antibody (Amersham) was used at 1:200. Incubations with all antibodies were performed at room temperature during 1 h. All dilutions and washings were in 0.2 M phosphate-buffered saline. The immunocytochemical study showed a complete deficiency in merosin (absence of chain M and overexpression of chain A in laminin surrounding the muscle fibres) in all patients (Fig. 1).
Intellectual development was normal in patients 2 and 3, their verbal IQ at the age of 8 and 7 being 97 and 118, respectively. Raven Progressive Matrices scores were 90th and > 95th centile,
respectively. Moderate mental retardation was present in patient l, whose verbal IQ value was 57 at the age of 7. CT scan of the brain showed marked diffuse white matter hypodensity, un- changed on repeated examinations at different ages in each patient (first examination: 4, 5 and 4 years, respectively; subse- quent examination: 6, 8 and 7 years, respectively). Mild ventricu- lar dilatation was also present, more pronounced on the right side in patient 2. Brain magnetic resonance imaging (MRI) confirmed the involvement of the periventricular and of the centrum semio- vale white matter; in addition smooth appearance of the parieto- occipital areas in patient 1 (Fig. 2) and of the occipito-temporal areas in the two brothers (Fig. 3) was present, indicating focal cortical dysplasia.
2.1. Ep i l epsy h is tor ies
Epileptic seizures started after the age of 3 years (age range: 3.5-6 years). In all patients the main type of seizure was a cluster, lasting several minutes, composed of repeated 'absence- like' attacks with a duration of approximately 1 second and recurring every 10-15 seconds. During the cluster the children seemed to be inattentive and their vigilance levels progressively lowered. Each 'absence-like' attack was characterized by fixed gaze with a raising of eyebrows; in patient 1 a very slight lowering of the head also occurred. This seizure type occurred every day, mainly on awakening.
In addition, patients 1 and 2 presented, both during sleep and at awaking state, occasional short-lasting convulsive seizures
c,
F 8 - T , ' ! / , ~ ~/V~,N,.,,~. ' ,;
T -
Fp -F ,"' 'I' ~ ' - "
F-T~ , , 7 3 ~
R . D e l I . . ~ , . ~ . . . . . . ~ : . ;
L.Delt . . . . . . . . - . . . . . . . . . . . = . . . . . . . . . . . . . . . . . . . I . . . . ~-~ . . . . . . . . .
E KG ~ ~ l & , ~ t , ~ k k t ~ . t Q £ l ~ J
Thor .Resp. ~ - ~ _ . . - - _ ~ _ ~ N f _ ~
Fig. 5. Case 1. Ictal EEG 2 min and 30 s after seizure onset: periodically repetitive diffuse slow-wave complexes associated with posterior dominant fast activity. Each EEG complex is concomitant to an increase of muscular activity on deltoid muscle surface electrodes and to an interruption of respiratory rhythm.
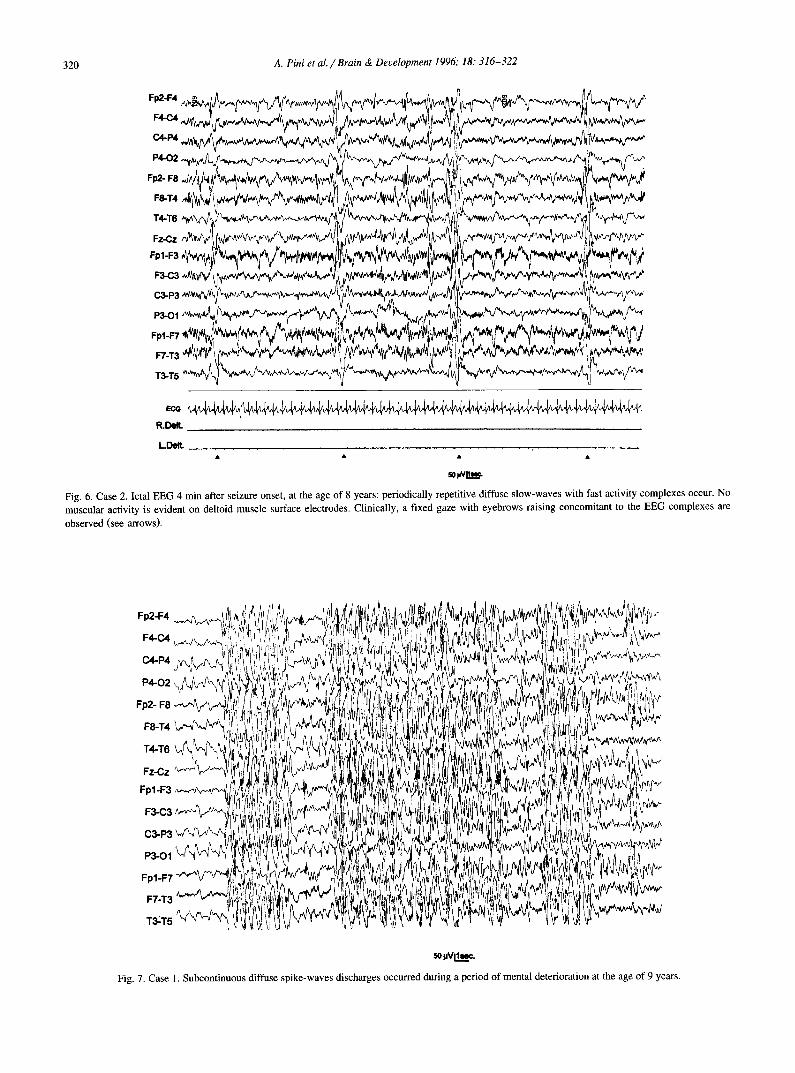
320 A. Pini et al . / Brain & Development 1996; 18." 316-322
R.I~IIt.
L.I~I.
n 0 ~ v ~
Fig. 6. Case 2. Ictal EEG 4 min after seizure onset, at the age of 8 years: periodically repetitive diffuse slow-waves with fast activity complexes occur. No muscular activity is evident on deltoid muscle surface electrodes. Clinically, a fixed gaze with eyebrows raising concomitant to the EEG complexes are
observed (see arrows).
~.v~.
Fig. 7. Case 1. Subcontinuous diffuse spike-waves discharges occurred during a period of mental deterioration at the age of 9 years.

A. Pini et al./Brain & Development 1996; 18:316-322 321
with apnea, cyanosis, minimal increase in muscle tone, hand tremor and impaired consciousness.
~-Vinyl-GABA controlled the clusters of 'absence-like' at- tacks for a short while in each patient. Nevertheless, because of the recurrence of seizures, dose increments were required in two patients (cases 1 and 2) to keep them under control. Other seizures were resistant to multiple anticonvulsants.
Patient 1 presented, at the age of 9, apparent mental deteriora- tion with fluctuating reduction of vigilance and attention levels. In that period, true absence seizures with fixed gaze and impair- ment of consciousness occurred many times during the day, in addition to the convulsive seizures and cluster-seizures described above. Ethosuximide was added to the therapy with rapid im- provement in psychological performances and disappearance of absence seizures.
2.2. Interictal and ictal EEG findings
Interictal EEG during wakefulness revealed normal back- ground alpha activity in all cases. Posterior spike-wave dis- charges occurred; they sometimes spreaded to anteriorly predomi- nant diffuse spike-waves discharges, to constitute bilaterally syn- chronous slow spike-wave activity (Fig. 4).
The polygraphic-EEG recording of a cluster-seizure in patient 1 at the age of 6.5 years showed that the periodically repetitive 'absence like' attacks corresponded to spasms. Each spasm was concomitant with a diffuse EEG complex consisting of slow-wave burst followed by occipito-temporal fast activity (Fig. 5). The seizure stopped after 8.5 min. The same ictal EEG pattern was recorded in the two brothers during a prolonged cluster-seizure (Fig. 6). During the period of mental deterioration associated with absence seizures in patient 1, EEG showed almost subcon- tinuous diffuse spike-wave discharges, as for non-convulsive epileptic status (Fig. 7).
3. DISCUSSION
We have described three Italian patients in two families showing a very similar clinical, EEG and neuroimaging picture. They presented merosin-negative CMD, drug resistant occipital epilepsy and posterior focal cortical dysplasia. Occipital and diffuse paroxysmal discharges with frontal predominance as in secondary bilateral synchrony [16] were the main interictal EEG characteristics.
The most frequent seizure type consisted of periodic spasms (PS). As the motor expression of the spasms was very mild in patient 1 and limited to facial musculature in the other two patients the seizure had been clinically interpreted as repeated absence seizures. The ictal polygraphic-EEG recording allowed us to correctly diagnose this type of seizure as PS in each patient. PS is a recently described complicated type of epileptic seizure composed of several clinical and EEG manifestations: be- havioural change is associated with a prolonged cluster of epilep- tic spasms, each of them related to a slow-wave with superim- posed fast rhythm EEG complex [17,18]. Because of the associa- tion of focal and generalized clinico-electroencephalographic signs, PS is considered as a partial seizure with a particular type of secondary generalization [19].
Epileptic spasms, including PS, are a frequent feature of cortical dysplasia [17,19-22]. While in diffuse cortical dysplasia,
such as lissencephaly, epilepsy usually starts in the first months of life, in focal cortical dysplasia seizures can also start later, in childhood or even in adult life [20,22].
Because of that we suspected the presence of cortical malfor- mation in our patients. MRI allowed us to visualize, in addition to marked diffuse white matter changes, the smooth appearance of the cerebral cortex in the posterior areas indicative of focal cortical dysplasia. It was not possible to better identify the type of cortical dysplasia; smooth surface can in fact correspond to pachygyria or to polymicrogyria [20,23].
To our knowledge this clinico-electroencephalographic and neuroimaging picture has never been described in CMD, includ- ing the merosin-negative type. In FCMD, febrile or non-febrile seizures have been observed in about half of the cases, generally with onset before the age of three [24,25]. In most of the cases convulsions were generalized; infantile spasms have only rarely been reported [25]. The major cerebral pathological change is micropolygyria-pachygyria involving several lobes [25,26]; mod- erate to severe mental retardation is present in all typical cases. Our patients presented with symptoms similar to FCMD, though differences were also evident. Cortical dysplasia appeared limited to posterior areas only and no patient showed severe mental retardation (verbal IQ was normal in two cases, and 57 in one case). Moreover, epilepsy started later and consisted of localiza- tion-related occipital epilepsy with different types of seizures, including PS.
Epileptic seizures and cortical dysplasia (associated or not) have been also reported in some western CMD cases with normal intelligence and white matter abnormalities at neuroimaging or pathological examination [2-9]. When in such cases epilepsy occurred, onset was in childhood, as was the case in our patients, but the reported seizures were mainly generalized and epileptic spasms were never reported. Nevertheless, details on the type of epilepsy and polymyographic-EEG recordings have frequently been lacking, data being consequently difficult to compare. The general impression is that when epileptic seizures were well controlled by monotherapy, brain involvement did not include cortical dysplasia. On the contrary, as in our cases, drug-resistant epileptic seizures occurred in the presence of cortical malforma- tion.
Recently the involvement of laminin M chain (merosin) has been discovered in a percentage of patients with classical non- Fukuyama type CMD [11]. The lack of merosin, which is proba- bly the cause of muscle tissue lesions, does identify a particular form of CMD. As a consequence, efforts are currently directed toward the phenotypic characterization of non-Fukuyama CMD in relation to the merosin status. The study of Philpot et al. [15] demonstrated that children with merosin-deficient CMD have a more severe clinical phenotype and more significant white matter changes at brain MRI than the merosin-positive cases. In accor- dance with these results, none of our patients has achieved independent ambulation and all presented with marked diffuse white matter changes at neuroimaging. Nevertheless, brain in- volvement in our patients was complicated by the presence of posterior cortical dysplasia and occipital drug-resistant partial epilepsy with epileptic spasms. Similar findings have recently been reported in an Italian patient with merosin-positive CMD, marked intellectual deficit, childhood-onset epilepsy requiring trials with multiple anticonvulsants, white matter abnormalities and posterior cortical dysplasia at neuroimaging [27]. However, epileptic spasms were not reported, white matter abnormalities

322 A. Pini et al. / Brain & Development 1996; 18:316-322
improved with time and the muscle immunocytochemical pattern was different.
In conclusion, results in our patients indicate that focal corti- cal dysplasia may occur in patients with merosin-negative CMD and marked persistent diffuse white matter changes. The brain damage may lead, in these patients, to a childhood-onset localiza- tion-related symptomatic occipital epilepsy with PS, whose clini- cal course may include transient mental deterioration concomitant with non-convulsive epileptic status.
Further studies are necessary to confirm whether this clinical, EEG and neuroimaging association is related to merosin-negative CMD or not.
It is our opinion that for a correct phenotypic definition of CMD syndromes it is important to take into account that the semiology of epileptic seizures may sometimes be only minimal, because of the muscle weakness. This implies that epilepsy may be misdiagnosed or even missed and EEG-polymyographic recordings may be necessary to identify it. Similarly, cortical dysplasia may be very localized and visible by MRI only if it is carefully investigated on the basis of epileptological and EEG- polymyographic findings.
ACKNOWLEDGEMENTS We are grateful to Mr. V. Mezzacani, Mr. M. Bonazzi and Mr. C. Basile for technical assistance. The authors wish to thank Dr. G. Crisi and Dr. P. Ambrosetto for discussion of neuroim- ages. The authors also thank Mr. S. Jewkes for revising the English text. Dr. L. Merlini was supported by Italian Telethon (No. 430).
R E F E R E N C E S
1. Dubowitz V. Congenital muscular dystrophy. In: Emery AEH, ed. Diagnostic criteria for neuromuscular disorders. Baarn, The Nether- lands: European Neuromuscular Centre, 1994: pp. 32-4.
2. Bernier JP, Brooke MH, Naidich TP, Carrol JE. Myoencephalopathy: cerebral hypomyelination revealed by CT scan of the head in a muscle disease. Ann Neurol 1979; 6: 165.
3. Echenne B, Astruc J, Brunel D, Pages M, Baldet P, Martinazzo G. Congenital muscular dystrophy and rigid spine syndrome. Neurope- diatrics 1983; 14: 97-101.
4. Echenne B, Pages M, Marty-Double C. Congenital muscular dystro- phy with cerebral white matter spongiosis. Brain Dev (Tokyo) 1984; 6: 491-5.
5. Egger J, Kendall BE, Erdohazi M, Lake BD, Wilson J, Brett EM. Involvement of the central nervous system in congenital muscular dystrophies. Dev Med Child Neurol 1983; 25: 32-42.
6. Martinelli P, Gabellini AS, Ciucci G, Govoni E, Vitali S, Gulli MR. Congenital muscular dystrophy with central nervous system involve- ment: Case report. Eur Neurol 1987; 26: 17-22.
7. Stern LM, Albertyn L, Manson JI. Fukuyama congenital muscular dystrophy in two Australian female siblings. Dev Med Child Neurol 1990; 32: 808-13.
8. Streib EW, Lucking CH. Congenital muscular dystrophy with leukoencephalopathy. Eur Neurol 1989; 29:211-5.
9. Trevisan CP, Carollo C, Segalla P, Angelini C, Drigo P, Giordano R. Congenital muscular dystrophy: brain alterations in an unselected
series of Western patients. J Neurol Neurosurg Psychiatry 1991; 54: 330-4.
10. Burgeson RE, Chiquet M, Deutzmann R, et al. A new nomenclature for the laminins. Matrix Biol 1994, 14: 209-11.
11. Tome FMS, Evangelista T, Leclerc A, et al. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci Paris Life Sci 1994; 317: 351-7.
12. Toda T, Segawa M, Nomura Y, et al. Localization of a gene for Fukuyama type congenital muscular dystrophy to chromosome 9q31-33. Nat Genet 1993; 5: 283-6.
13. Arahata K, Hayashi Y, Mizuno Y, Yoshida M, Ozawa E. Dystrophin-associated glycoprotein and dystrophin co-localisation at sarcolemma in Fukuyama congenital muscular dystrophy. Lancet 1993; 342: 623-4.
14. Hillaire D, Leclerc A, Faure S, et al. Localization of merosin-nega- tive congenital muscular dystrophy to chromosome 6q2 by homozy- gosity mapping. Hum Molec Genet 1994; 3: 1657-61.
15. Philpot J, Sewry C, Pennock J, Dubowitz V. Clinical phenotype in congenital muscular dystrophy: correlation with expression of merosin in skeletal muscle. Neuromusc Disord 1995; 5: 301-5.
16. Blume WT, Pillay N. Electrographic and clinical correlates of sec- ondary bilateral synchrony. Epilepsia 1985, 26: 636-41.
17. Gobbi G, Bruno L, Pini A, Rossi Giovanardi P, Tassinari CA. Periodic spasms: an unclassified type of epileptic seizure in child- hood. Dev Med Child Neurol 1987; 29: 766-75.
18. Roger J, Dulac O. West syndrome: history and nosology. In: Dulac O, Chugani HT, Dalla Bernardina, eds. Infantile spasms and West syndrome. Philadelphia: W.B. Saunders, 1994: pp. 6-11.
19. Gobbi G, Parmeggiani A, Pini A, Tassinari CA. Periodic spasm. A particular type of infantile spasms. In: Ohtahara S, Roger J, eds. New trends in pediatric epileptology. Okayama: Okayama University Medical School, 1991: pp. 111-8.
20. Aicardi J. The agyria-pachygyria complex: a spectrum of cortical malformations. Brain Dev (Tokyo) 1991; 13: 1-8.
21. DaUa Bernardina B, Watanabe K. Interictal EEG: variation and pitfalls. In: Dulac O, Chugani HT, Dalla Bemardina, eds. Infantile spasms and West syndrome. Philadelphia: W B Saunders, 1994: pp. 63-81.
22. Vigevano F, Fusco L, Ricci S, Granata T, Viani F. Dysplasias. In: Dulac O, Chugani HT, Dalla Bemardina B, eds. Infantile spasms and West syndrome. Philadelphia: W B Saunders, 1994: pp. 178-91.
23. Barkovich AJ, Koch TK, Carrol CL. The spectrum of lissencephaly: report of ten patients analyzed by magnetic resonance imaging. Ann Neurol 1991; 30: 139-46.
24. Segawa M, Nomura Y, Hachimori K, Shinoyama N, Hosaka A, Mizuno Y. Fukuyama type congenital muscular dystrophy as a natural model of childhood epilepsy. Brain Dev (Tokyo) 1979; 2: 113-9.
25. Fukuyama Y, Osawa M, Suzuki H. Congenital progressive muscular dystrophy of the Fukuyama type - - clinical, genetic and pathological considerations. Brain Dev (Tokyo) 1981; 3: 1-29.
26. Takashima S, Becker LE, Chart F, Takada K. A Golgi study of the cerebral cortex in Fukuyama-type congenital muscular dystrophy, Walker-type 'lissencephaly', and classical lissencephaly. Brain Dev (Tokyo) 1987; 9: 621-6.
27. Trevisan CP, Martinello F, Ferruzza E, Angelini C. Divergence of central nervous system involvement in 2 Italian sisters with congeni- tal muscular dystrophy: a clinical and neuroradiological follow-up. Eur Neurol 1995; 35: 230-5.
Copyright © 2022 FDOKUMEN




















