
Influenza A Virus Infection Inhibits the Efficient Recruitment of Th2 Cells into the Airways and the...
12
of July 14, 2015. This information is current as Eosinophilia Airways and the Development of Airway Efficient Recruitment of Th2 Cells into the Influenza A Virus Infection Inhibits the Moll and Klaus J. Erb Heidrun Hambrecht, Udo Herz, Harald Renz, Edgar Schmitt, Gisela Wohlleben, Justus Müller, Ursula Tatsch, Christine http://www.jimmunol.org/content/170/9/4601 doi: 10.4049/jimmunol.170.9.4601 2003; 170:4601-4611; ; J Immunol References http://www.jimmunol.org/content/170/9/4601.full#ref-list-1 , 21 of which you can access for free at: cites 51 articles This article Subscriptions http://jimmunol.org/subscriptions is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/ji/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/cgi/alerts/etoc Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 2003 by The American Association of 9650 Rockville Pike, Bethesda, MD 20814-3994. The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on July 14, 2015 http://www.jimmunol.org/ Downloaded from by guest on July 14, 2015 http://www.jimmunol.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
5 -
download
0
Transcript of Influenza A Virus Infection Inhibits the Efficient Recruitment of Th2 Cells into the Airways and the...

of July 14, 2015.This information is current as
EosinophiliaAirways and the Development of AirwayEfficient Recruitment of Th2 Cells into the Influenza A Virus Infection Inhibits the
Moll and Klaus J. ErbHeidrunHambrecht, Udo Herz, Harald Renz, Edgar Schmitt,
Gisela Wohlleben, Justus Müller, Ursula Tatsch, Christine
http://www.jimmunol.org/content/170/9/4601doi: 10.4049/jimmunol.170.9.4601
2003; 170:4601-4611; ;J Immunol
Referenceshttp://www.jimmunol.org/content/170/9/4601.full#ref-list-1
, 21 of which you can access for free at: cites 51 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2003 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on July 14, 2015http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on July 14, 2015
http://ww
w.jim
munol.org/
Dow
nloaded from

Influenza A Virus Infection Inhibits the EfficientRecruitment of Th2 Cells into the Airways and theDevelopment of Airway Eosinophilia1
Gisela Wohlleben,* Justus Muller,† Ursula Tatsch,* Christine Hambrecht,* Udo Herz,‡
Harald Renz,‡ Edgar Schmitt,§ Heidrun Moll,* and Klaus J. Erb2*
Most infections with respiratory viruses induce Th1 responses characterized by the generation of Th1 and CD8� T cells secretingIFN-�, which in turn have been shown to inhibit the development of Th2 cells. Therefore, it could be expected that respiratoryviral infections mediate protection against asthma. However, the opposite seems to be true, because viral infections are oftenassociated with the exacerbation of asthma. For this reason, we investigated what effect an influenza A (flu) virus infection has onthe development of asthma. We found that flu infection 1, 3, 6, or 9 wk before allergen airway challenge resulted in a strongsuppression of allergen-induced airway eosinophilia. This effect was associated with strongly reduced numbers of Th2 cells in theairways and was not observed in IFN-�- or IL-12 p35-deficient mice. Mice infected with flu virus and immunized with OVAshowed decreased IL-5 and increased IFN-�, eotaxin/CC chemokine ligand (CCL)11, RANTES/CCL5, and monocyte chemoat-tractant protein-1/CCL2 levels in the bronchoalveolar lavage fluid, and increased airway hyperreactivity compared with OVA-immunized mice. These results suggest that the flu virus infection reduced airway eosinophilia by inducing Th1 responses, whichlead to the inefficient recruitment of Th2 cells into the airways. However, OVA-specific IgE and IgG1 serum levels, blood eosin-ophilia, and goblet cell metaplasia in the lung were not reduced by the flu infection. Flu virus infection also directly induced AHRand goblet cell metaplasia. Taken together, our results show that flu virus infections can induce, exacerbate, and suppress featuresof asthmatic disease in mice. The Journal of Immunology, 2003, 170: 4601–4611.
A sthma is an allergic disorder characterized by chronicairway inflammation and airway hyperreactivity (AHR)3
induced by allergen-specific Th2 cells secreting IL-4,IL-5, and IL-13 (1). The secretion of these cytokines leads to theproduction of allergen-specific IgE by B cells, the development ofairway eosinophilia, the induction of airway smooth muscle con-traction, and mucus production, respectively. Recent studies alsoindicate that chemokines such as eotaxin/CC chemokine ligand(CCL)11, RANTES/CCL5, monocyte chemoattractant protein(MCP)-1/CCL2, and MCP-4/CCL13 are also critically involved inthe development of asthma (2, 3).
Although relatively much is known about the immunologicalmechanisms responsible for the development of asthma, its inci-dence, severity, and mortality rate have steadily increased over the
past decades. The reason for this alarming development remainsunknown. However, it appears that environmental factors may beresponsible for this development, because the genetic predisposi-tion of the affected population has not changed. One environmentalfactor believed to possibly have a strong impact on the develop-ment of asthma is the lack of exposure to infectious diseases earlyduring infancy (4, 5). The rationale behind this hypothesis (alsoreferred to as the hygiene hypothesis) is that children infected withviruses or bacteria early in life do not develop asthma becausethese infections establish a Th1-biased immunity leading to theinhibition of allergen-specific Th2 cell priming or Th2 effectorfunction. It is believed that this effect is mediated by IFN-� se-creted by Th1 cells, because it has been demonstrated that IFN-�can suppress the development of Th2 cells both in vitro and in vivo(6, 7). Supporting this view are numerous studies showing that theexposure of the lung to bacteria or bacterial products inhibits thedevelopment of asthma in animal models with the effect beingassociated with increased Th1 immune responses (8–10).
Infections with respiratory viruses such as rhinovirus, respira-tory syncytial virus (RSV), and influenza A (flu) virus also induceTh1 responses. It is conceivable that infections with these virusescould also suppress the development of asthma. However, the op-posite seems to be true, because most epidemiological studiesshow a clear association between infections with respiratory vi-ruses and the exacerbation of asthma in humans (11–14). Animalexperiments addressing this issue have yielded somewhat conflict-ing results with some reports indicating that respiratory viral in-fections increase allergen-induced Th2 responses in the lung andothers reporting no effect or a suppressive effect (15–26). For thisreason, we investigated how an intranasal (i.n.) infection with fluvirus influenced the development of allergen-induced Th2 re-sponses in the lung of mice. In contrast to most previously pub-lished reports addressing similar questions (15, 22, 25, 26), we
*Center for Infectious Diseases and †Department of Pathology, University of Wurz-burg, Wurzburg, Germany; ‡Department of Clinical Chemistry and Molecular Diag-nostics, Central Laboratory, Hospital of the Philipps University, Marburg, Germany;and §Department of Immunology, University of Mainz, Mainz, Germany
Received for publication December 9, 2002. Accepted for publication February20, 2003.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by the Bundesministerium fur Bildung und Forschung andthe Bayerisches Staatsministerium fur Wissenschaft, Forschung und Kunst.2 Address correspondence and reprint requests to Dr. Klaus J. Erb, Center for Infec-tious Diseases, University of Wurzburg, Rontgenring 11, 97070 Wurzburg, Germany.E-mail address: [email protected] Abbreviations used in this paper: AHR, airway hyperreactivity; CCL, CC chemo-kine ligand; MCP, monocyte chemoattractant protein; RSV, respiratory syncytial vi-rus; flu, influenza A; i.n., intranasal; EID50, 50% egg infectious dose; BAL, bron-choalveolar lavage; MLN, mediastinal lymph node; RT, room temperature; MCh50,concentration of methacholine that causes a 50% reduction in expiratory airflow;MESLN, mesenteric lymph node; BCG, Mycobacterium bovis bacillus Calmette-Guérin; MIP, macrophage-inflammatory protein.
The Journal of Immunology
Copyright © 2003 by The American Association of Immunologists, Inc. 0022-1767/03/$02.00
by guest on July 14, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

used an OVA immunization protocol where the initial allergen-specific Th2 response does not develop in the lung. The allergen-specific Th2 response is induced by an OVA/alum protocol, lead-ing to the recruitment of Th2 cells into the airways and resulting inairway eosinophilia, mucus production, and AHR. Using this ex-perimental protocol, we have previously reported that the applica-tion of mycobacteria into the lung of mice leads to an almost totallack of eosinophils and Th2 cells in the airways after i.n. allergenchallenge (27). In this study, we report that, similar to the myco-bacterial infection, flu virus infection suppressed the developmentof allergen-induced airway eosinophilia and the efficient recruit-ment of Th2 cells into the airways. However, in contrast to my-cobacterial infection, allergen-induced AHR and goblet cell meta-plasia were not inhibited by the flu virus infection.
Materials and MethodsMice
The experiments were performed with C57BL/6 mice or IFN-�- and IL-12p35-deficient mice (both on C57BL/6 background). In the experimentsusing Th2 cells from mice transgenic for the OVA323–339-specific TCR-��(BALB/c genetic background), BALB/c mice were used as the recipients ofthe T cells. C57BL/6 and BALB/c mice were purchased from CharlesRiver (Sulzfeld, Germany), and IFN-�-deficient mice were purchased fromThe Jackson Laboratory (Bar Harbor, ME). IL-12 p35-deficient mice weregenerously provided by M. Kopf (Swiss Federal Institute of Technology,Zurich, Switzerland). Mice transgenic for the OVA323–339-specificTCR-�� were provided by E. Schmitt (University of Mainz, Mainz, Ger-many). All animals used for the experiments were between 5 and 7 wk ofage and housed in a conventional animal facility.
OVA immunization protocol
Mice were injected i.p. with 2 �g of OVA (Sigma-Aldrich, St. Louis, MO)in 200 �l of alum adjuvant (Serva, Heidelberg, Germany) on day 0 andboosted again i.p. with 2 �g of OVA/alum on day 14. Ten days after thesecond i.p. immunization, mice were anesthetized by an i.p. injection of amixture of ketamine and xylazine (Sigma-Aldrich) and treated i.n. with 50�l of PBS containing 100 �g of OVA.
Infection of mice with flu virus
The HKx31 (H3N2) flu virus was grown in the allantoic fluid of 10-day-oldembryonated eggs. Mice were anesthetized by i.p. inoculation with Avertin(2,2,2-tribromoethanol) and infected i.n. with 30 �l of PBS containing 2 �105 50% egg infectious dose (EID50) of the HKx31 flu virus at the indi-cated time points before the i.n. application of OVA. For some experi-ments, mice were also infected with 1 � 106, 1 � 105, 1 � 104, or 1 � 103
EID50 of the HKx31 flu virus.
Infection of mice with Nippostrongylus brasiliensis
Naive mice and mice infected with flu virus were infected with 1000 L3larvae of N. brasiliensis i.p. as described previously (8).
Bronchoalveolar lavage (BAL)
Six days after the i.n. OVA challenge, the mice of the different groups weresacrificed, the trachea were cannulated, and a BAL was performed byflushing lung and airways five times with 1 ml of PBS. BAL cells werecounted and spun onto glass slides using a cytospin (Shandon SouthernProducts, Asmoor, U.K.) and afterward stained with Diff-Quik according tothe manufacturer’s instructions (Roche Diagnostics, Mannheim, Germany).Percentages of macrophages, lymphocytes, neutrophils, and eosinophils weredetermined microscopically using standard histological criteria.
Culture conditions of cells
Single-cell suspensions from the mediastinal lymph nodes (MLN; 2 � 106
cells/ml) of the different groups of mice were prepared and cultured inRPMI 1640 medium (Sigma-Aldrich) supplemented with sodium bicar-bonate (3.024 g/l), 10 �g/ml streptomycin, 10 U/ml penicillin, 50 �M2-ME, and 10% FCS. The cell preparations were stimulated with a mAb toCD3� (145-2C11; 25 �g/ml) together with 200 U/ml recombinant humanIL-2 (Novartis, Basel, Switzerland), 40 �g/ml OVA (Sigma-Aldrich), ormedium. After 48 h, the culture supernatants were harvested and tested forthe presence of cytokines by ELISA.
Cytokine and chemokine ELISA
For the detection of the different cytokines and chemokines, sandwichELISAs were performed using the following Abs: biotinylated rat anti-mouse IL-4 (BVD4-1D11) and unconjugated rat anti-mouse IL-4 (BVD6-24GL); biotinylated rat anti-mouse IL-5 (TRFK4) and unconjugated ratanti-mouse IL-5 (TRFK5); biotinylated polyclonal goat anti-mouse IL-13and unconjugated rat anti-mouse IL-13 (38213.11); biotinylated anti-mouse IFN-� (AN-18.17.24) and unconjugated rat anti-mouse IFN-� (R4-6A2); biotinylated polyclonal goat anti-mouse eotaxin/CCL11 and uncon-jugated polyclonal goat anti-mouse eotaxin/CCL11; and biotinylatedpolyclonal goat anti-mouse RANTES/CCL5 and unconjugated mAb ratanti-mouse RANTES/CCL5 (53433). For the detection of MCP-1/CCL2,the OptEIA set from BD PharMingen (San Diego, CA) was used. The Absfor the detection of IL-4, IL-5, and IFN-� were purchased from BD Phar-Mingen, and the Abs for the detection of IL-13, eotaxin/CCL11, andRANTES/CCL5 were purchased from R&D Systems (Wiesbaden, Germa-ny). The ELISA were performed in polyvinyl chloride microtiter plates(Dynatech, Denkendorf, Germany) according to the instructions of themanufacturer of the Abs. The binding reactions were visualized with aconjugate of peroxidase-labeled streptavidin (DAKO, Glostrup, Denmark)and the substrate ABTS (Sigma-Aldrich). The amounts of cytokines andchemokines present in the samples were determined by including serialdilutions of the different murine cytokines or murine chemokines (recom-binant cytokines and chemokines were purchased from BD PharMingen).The BAL fluid was concentrated 3-fold on ultrafiltration membranes pur-chased from Sigma-Aldrich (Ultrafree-15 centrifuge filter units with Bi-omax-5K membrane; Millipore, Bedford, MA). Briefly, for 3-fold concen-tration, BAL fluid was centrifuged for 15 min in the indicated centrifugefilter units (2000 rpm) at 4°C.
OVA-specific IgE and IgG1
OVA-specific Ig levels were determined by coating microtiter plates withOVA (10 �g/ml) or anti-IgE mAb (1 �g/ml) and then blocking with 10%BSA for 60 min at room temperature (RT). Two-fold dilutions of serumwere added and incubated for 2 h at RT. Appropriate dilutions of biotin-ylated rat anti-mouse IgG1 or OVA-labeled biotin (for the detection ofOVA-specific IgE) were added for 2 h at RT. The binding reactions werevisualized as described in Cytokine and chemokine ELISA. As a reference,the serum of untreated control mice was also included in the ELISA. OVA-specific IgG1 and IgE titers were defined as the serum dilution at which theOD was at least 2-fold higher than the OD measured when using a 1/10dilution of serum from untreated control mice. All Abs were purchasedfrom BD PharMingen.
Flow cytometric analysis
CD4� and CD8� T cells from the BAL producing IL-4 or IFN-� weredetected by using two-color FACS analysis performed with a FACScan(BD Biosciences, Mountain View, CA). For this purpose, BAL cells fromthe different groups of mice were spun down and then resuspended inRPMI 1640 medium containing 10% FCS. The cells were then stimulatedwith phorbol ester (5 �g/ml) and calcium ionophore (0.5 �M) for 6 h (bothreagents from Sigma-Aldrich). Brefeldin A (2 �g/ml; Sigma-Aldrich) wasadded for the last 2 h of the in vitro culture period. The stainings wereperformed according to the instructions from BD PharMingen. Briefly,after the 6 h stimulation, BAL cells were washed and stained with anti-CD4- or anti-CD8-FITC-labeled mAb. The cells were then fixed with 4%formalin in PBS for 20 min and later incubated with anti-CD16/CD32 mAb(2.4G2; Fc Block; 5 �g/ml). After 30 min, PE-labeled anti-IL-4 mAb(11B11) or PE-labeled anti-IFN-� was added. All Abs were purchasedfrom BD PharMingen. No CD8� T cells producing IL-4 above backgroundstaining (using isotype-matched mAb) could be detected in the BAL of anyof the mice analyzed (data not shown).
Body plethysmography
AHR was assessed by head-out body plethysmography as described pre-viously (28). Briefly, mice were placed in four body plethysmographs at-tached to a exposure chamber (Crown Glass, Somerville, NJ). Airflow wasmeasured with a PTM 378/1.2 pneumotachograph (Hugo Sachs Electron-ics, March-Hugstetten, Germany) and a 8-T2 differential pressure trans-ducer (Gaeltec, Dunvegan, U.K.). Airflow in response to various concen-trations of methacholine (25, 50, 75, 100, and 150 mg/ml; 1 min) wasdelivered by a jet nebulizer (Pari-Boy; Pari-Werke, Starnberg, Germany).The concentration of methacholine that caused a 50% reduction in expi-ratory airflow (MCh50) was determined.
4602 FLU VIRUS INFECTION INHIBITS ALLERGEN-INDUCED Th2 RESPONSES
by guest on July 14, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

Generation of OVA-specific Th2 cells and adoptive transfers ofMLN cells
CD4�MEL-14high T cells were isolated from spleen cells of mice trans-genic for the OVA323–339-specific TCR�� (DO11.10) on a BALB/c geneticbackground by positive selection using high-gradient magnetic cell sepa-ration in combination with MultiSort beads (MACS; Miltenyi Biotec, Ber-gisch-Gladbach, Germany) according to the manufacturer’s instructions.The CD4 sort, as well as the MEL-14 sort, was performed twice. MEL-14highCD4� T cells were enriched �99% and showed no proliferative re-sponse in the presence of Con A or soluble anti-��TCR mAb, which in-dicates negligible contamination with accessory cells. Primary stimulationwas conducted by incubating 1 � 106 CD4� T cells on anti-� TCR clono-type-specific mAb (KJ1-26; 5 �g/ml)-coated 24-well plates in a total vol-ume of 1.0 ml of culture medium with the addition of IL-4 (1000 U/ml) andanti-IFN-� mAb (XMG1.2; 10 �g/ml). After 96 h, the developing T cellswere transferred to uncoated 24-well culture dishes, and 0.5 ml of IL-2-containing (human rIL-2; 1000 U/ml) culture medium was added. After anadditional 48 h, the T cells were collected and transferred into BALB/cmice as indicated in Fig. 3. Simultaneously, aliquots of the generated Th2cells were restimulated by immobilized anti-��TCR mAb (5 �g/ml) todetermine their cytokine profile. After an additional 18 to 24 h, superna-tants were collected and assayed for cytokines. Th2 cells secreted only IL-4and IL-5 and no or very little IFN- � (data not shown). Flow cytometryrevealed that all T cell populations used for restimulation consisted of�99% CD4� Th cells. For the adoptive transfer experiments, BALB/cmice immunized with OVA were treated i.v. with 9 � 106 flu virus-im-mune MLN cells (n � 8) (from mice infected 2 wk with 2 � 105 EID50 fluvirus) or 9 � 106 mesenteric lymph node (MESLN) cells from naiveBALB/c mice as controls (n � 7) 1 wk before the i.n. application of OVA.Numbers of eosinophils were detected in the BAL fluid of the differentgroups of mice (6 days after the i.n. challenge with OVA) as described inBronchoalveolar lavage.
Histological analysis
Tissues from flu virus-infected and/or OVA-immunized mice were fixed in10% phosphate-buffered formalin for 24 h and embedded in paraffin wax.Sections (2–3 �m) were cut and stained using standard histological pro-tocols with H&E and periodic acid-Schiff reaction. The stained sectionswere visualized by light microscopy.
Cutaneous anaphylaxis
Active cutaneous anaphylaxis was tested in flu virus/OVA-treatedC57BL/6 mice (flu virus infection 3 wk before OVA airway challenge),OVA only-treated C57BL/6 mice, or C57BL/6 control mice. Ten days afterOVA airway challenge, mice were injected i.v. with 200 �l of 0.5% Evansblue in PBS (Sigma-Aldrich). Subsequently, the skin of the belly wasshaved, and 50 �l of PBS, 50 �l of PBS containing OVA (50 �g/ml), orcompound 48/80 (5 mg/ml; Sigma-Aldrich) was injected intradermally intotwo premarked sites on the skin. After 15 min, mice were sacrificed. Pos-itive reactions to OVA resulted in mast cell degranulation and fluid ex-travasation, which led to the formation of a blue patch around the injectionsite. Mast cell reactivity was considered positive when the wheals were �3mm. Control mice (n � 6) not subjected to OVA immunization did notshow any mast cell reactivity after the application of OVA. In contrast, allthe mice immunized with OVA (n � 5), and almost all the mice infectedwith flu virus and immunized with OVA (five of six mice) showed cuta-neous mast cell reactivity after the application of OVA. None of the micehad positive skin reactivity after the application of PBS, and all the micereacted to compound 48/80.
Statistical analysis
Statistical significance was analyzed by Student’s t test.
ResultsInfection with flu virus suppresses the development ofallergen-induced airway eosinophilia
To address the question of whether an infection with flu virusmodulates the development of allergen-induced Th2 responses,C57BL/6 mice were infected once with the virus at different timepoints and subjected to an OVA immunization protocol (Fig. 1A).Fig. 1B shows that C57BL/6 mice infected with flu virus 9, 6, 3, or1 wk before OVA airway challenge showed a strong decrease ineosinophil numbers in the airways in comparison to OVA only-
immunized C57BL/6 mice. The strongest inhibitory effect wasseen when the infection was performed 1 wk before OVA airwaychallenge. Furthermore, we also found that using 20- or 200-foldless flu virus for the i.n. infection 1 wk before i.n. OVA challengealso suppressed allergen-induced airway eosinophilia by 90 � 3 or77 � 22%, respectively, in comparison to OVA only-immunizedC57BL/6 mice (mean of six mice per group with SD). Histological
FIGURE 1. Intranasal infection with flu virus strongly inhibits the de-velopment of airway eosinophilia and reduces the amount of IL-5 secretedby the T cells from the lymph node of the lung. A, Experimental designused to investigate the influence of a flu infection on OVA-induced eosin-ophilia in the lung. B, C57BL/6 mice were subjected to the OVA immu-nization scheme and infected i.n. with 2 � 105 EID50 of flu virus 1, 3, 6,or 9 wk before i.n. OVA challenge. Six days following OVA airway chal-lenge, BALs were performed, and the cells were counted and stained withH&E, and the different cell types were identified microscopically. Shownare the mean numbers of eosinophils � SD present in the BALs of thedifferent groups of mice (8–20 mice/group from one to three separate ex-periments). C, In parallel to the BALs, single-cell suspensions (2 � 105
cells/well) from MLN of the different groups of mice were prepared andstimulated in vitro for 48 h with anti-CD3/IL-2. Shown is the mean amountof IL-5 � SD present in the MLN cultures from the different groupsof mice. The experiments were repeated at least once with similar results.�, p � 0.05; ��, p � 0.01; ���, p � 0.001, compared to OVA only-treatedmice.
4603The Journal of Immunology
by guest on July 14, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

examinations of lung tissue from C57BL/6 mice immunized withOVA and previously infected with flu virus 1, 3, or 6 wk before thei.n. application of OVA confirmed the strong reduction in lungeosinophil numbers in the airways (data not shown). Moreover, thequality of the inflammatory response also differed in the individualgroups of C57BL/6 mice. OVA-sensitized C57BL/6 mice showedmostly eosinophil and lymphocyte infiltrations in the lung,whereas neutrophils, macrophages, and lymphocytes dominated inthe flu virus-infected and flu virus-infected/OVA-immunizedC57BL/6 mice. Similar results were obtained when the numbers ofthe different cell types were determined in the BAL fluid of theC57BL/6 mice analyzed (data not shown).
The observed reduction in eosinophil numbers correlated withsignificantly reduced amounts of IL-5 present in cultures of MLNcells stimulated in vitro from the groups of C57BL/6 mice infected1, 3, or 6 wk, but not 9 wk before OVA i.n. challenge (Fig. 1C).No IL-4 could be detected in any of the cultures. MLN cells fromthe different groups of C57BL/6 mice incubated with mediumalone and MLN cells from untreated C57BL/6 control mice stim-ulated in vitro produced no detectable IL-5 (data not shown).
Numbers of Th2 cells present in the airways after allergenairway challenge are strongly reduced by an infection withflu virus
The flu virus infection suppressed the development of airway eo-sinophilia more strongly than the amount of IL-5 secreted by Tcells from the MLN. Furthermore, significant inhibition of airwayeosinophilia did not correlate with reduced amounts of IL-5 se-creted by MLN cells in vitro in C57BL/6 mice infected with fluvirus 9 wk before OVA i.n. challenge. For this reason, we analyzedwhether the strong suppression of airway eosinophilia in the fluvirus-infected C57BL/6 mice could be explained by a strongerreduction of Th2 cell numbers in the airways. Table I shows thatthe amount of CD4� Th2 cells present in the airways of flu virus-infected/OVA-treated C57BL/6 mice was strongly reduced incomparison to the amount of Th2 cells detected in the BAL fluidof C57BL/6 mice that were only immunized with OVA. Interest-ingly, suppression of allergen-induced eosinophilia and the reduc-tion of Th2 cells present in the BAL fluid correlated with a sig-nificant increase in CD8� but not CD4� T cells producing IFN-�(Table I). Recently, it was reported that Th2 cells show impairedhoming into Th1 cell-mediated inflamed sites (29–31). Therefore,it is possible that the flu virus infection reduced the numbers ofTh2 cells in the airways by interfering with the efficient recruit-ment of Th2 cells. However, it is also possible that Th2 cell ex-pansion in the airways may be inhibited by the flu virus infection.Figure 2 shows that CD4� Th2 cells producing IL-4 could already
be detected 1 day after OVA airway challenge in the BAL ofOVA-sensitized C57BL/6 mice. The percentage of CD4� T cellsproducing IL-4 gradually increased over the next 5 days (startingat 1.27% at day 1 and reaching 7.75% at day 6 after allergenchallenge). No IL-4 producers were detectable in non-OVA-im-munized C57BL/6 mice or C57BL/6 mice immunized i.p. withOVA/alum and i.n. challenged with PBS instead of OVA (data notshown). In contrast, the percentage of CD4� T cells secreting IL-4remained very low during the entire 6 days after OVA challenge inthe flu virus-infected C57BL/6 mice (0.3–0.62% with an isotypebackground staining of 0.1–0.2%). The numbers of total CD4� Tcells did not significantly differ in the two groups of C57BL/6 mice(data not shown). These results suggest that the flu virus infectioninterfered with the efficient recruitment of Th2 cells into the air-ways. However, although likely, it is not clear whether the Th2cells detected in the BAL fluid were OVA specific. For this reason,we generated OVA-specific CD4� Th2 cells in vitro (derived fromDO11.10 ��TCR transgenic mice on BALB/c background) andinjected the Th2 cells i.v. into BALB/c control mice or BALB/cmice that had been infected with flu virus 1 wk previously. Themice were then treated three times with OVA i.n. to induce therecruitment of the Th2 cells into the airways (Fig. 3A). Figure 3, Band C, shows that the percentage and the total numbers of CD4�
T cells expressing the transgenic TCR were similar in the MLN ofOVA-treated and flu virus-infected/OVA-treated BALB/c mice.However, flu virus-infected/OVA-treated BALB/c mice showed a
Table I. Flu virus infection leads to a strong reduction in the amountof CD4� Th2 cells present in the airways of OVA-treated micea
Treatment CD4�/IL-4� CD4�/IFN-�� CD8�/IFN-��
PBS �1 �1 �1OVA 31.2 � 21 12 � 8.4 3.3 � 2.61 wk flu/OVA 5 � 5** 8.3 � 3.6 26.7 � 18.2*3 wk flu/OVA 5 � 4** 8.2 � 2.9 20.2 � 6.5***
a C57BL/6 mice were subjected to the OVA immunization protocol and infectedi.n. with flu virus 1 or 3 wk prior to OVA airway challenge as described in Fig. 1.BALs were performed, and the BAL cells were stimulated and stained for expressionof CD4 and IL-4, CD4 and IFN-�, or CD8 and IFN-�, as described in Materials andMethods. As controls, PBS- or OVA only-treated mice were used. Shown are cellnumbers (�103) per milliliter of BAL fluid. Shown are means of eight mice pergroup � SD.
�, p � 0.05; ��, p � 0.01; and ���, p � 0.001 compared to OVA only-treatedmice.
FIGURE 2. Flu virus infection inhibited the expansion or recruitment ofTh2 cells in the airways of OVA-immunized mice after allergen airwaychallenge. OVA-immunized C57BL/6 mice were either infected i.n. with2 � 105 EID50 of flu virus 1 wk before OVA airway challenge or leftuninfected as an OVA control (Fig. 1A). A BAL was performed on bothgroups of mice 1, 2, 3, and 6 days following the OVA airway challenge.BAL cells were then stimulated, fixed, and stained for the expression ofCD4 and IL-4 as described in Materials and Methods. Specificity of Abbinding was controlled by staining with irrelevant isotype-matched controlAbs (data not shown). Shown are FACS stainings gated on CD4� T cellsrepresentative of six mice per group. The percentage of CD4� T cellsproducing IL-4 is indicated.
4604 FLU VIRUS INFECTION INHIBITS ALLERGEN-INDUCED Th2 RESPONSES
by guest on July 14, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

strong decrease in the numbers of ��TCR transgenic CD4� T cellsin the BAL fluid in comparison with BALB/c mice treated onlywith OVA. Furthermore, airway eosinophilia induced by the trans-ferred Th2 cells was also strongly reduced in the flu virus-infectedBALB/c mice (Fig. 3C). The application of OVA alone without thetransfer of Th2 cells did not lead to an increase in eosinophils in
the airways (data not shown). Taken together, these data suggestthat an infection with flu virus interferes with the efficient recruit-ment of Th2 cells into the airways after allergen challenge.
Flu virus-induced suppression of airway eosinophilia isassociated with reduced amounts of IL-5 and increased amountsof IFN-�, eotaxin/CCL11, RANTES/CCL5, and MCP-1/CCL2present in the airways
The results in the previous section suggest that the flu virus infec-tion may suppress the development of airway eosinophilia by re-ducing the amount of Th2 cells secreting IL-5 in the lung, becauseIL-5 is necessary for the development and recruitment of eosino-phils (32, 33). Supporting this view was the finding that the IL-5levels in the BAL of flu virus-infected/OVA-treated C57BL/6mice were significantly lower in comparison to the levels detectedin the BAL fluid of OVA only-immunized C57BL/6 mice (Fig.4A). However, a recent publication has shown that the productionof eotaxin/CCL11 is also needed for the efficient recruitment ofeosinophils into the airways (33). Therefore, it may be possiblethat the flu virus infection reduced the amounts of eotaxin/CCL11present in the airways after allergen challenge, thereby suppressingthe development of airway eosinophilia. Surprisingly, we foundthat the amount of eotaxin/CCL11 was increased in the BAL fluidof flu virus-infected/OVA-treated C57BL/6 mice in comparison toOVA only-immunized C57BL/6 mice (Fig. 4C). Interestingly,RANTES/CCL5, MCP-1/CCL2, and IFN-� were also strongly el-evated in the BAL fluid of flu virus-infected/OVA-treatedC57BL/6 mice (Fig. 4, B, D, and E). In a separate experiment, wefound that C57BL/6 mice infected with flu virus for 2 wk alsoproduced detectable levels of IFN-� (960 � 420 pg/ml), eotaxin/CCL11 (160 � 40 ng/ml), RANTES/CCL5 (320 � 100 ng/ml),and MCP-1/CCL2 (87 � 25 ng/ml) but no IL-5 (�0.2 U/ml) in theairways (mean values of eight mice � SD). These results suggestthat flu virus-induced suppression of airway eosinophilia may bedue to increased IFN-� and decreased IL-5 levels but not eotaxin/CCL11, RANTES/CCL5, or MCP-1/CCL2 levels in the airways.
IFN-�-deficient mice do not show flu virus-induced suppressionof airway eosinophilia
Table I and Figure 4 show that flu virus-induced suppression ofairway eosinophilia correlated with increased amounts of IFN-�and CD8� T cells secreting IFN-� in the BAL fluid of flu virus-infected/OVA-treated C57BL/6 mice in comparison to OVA only-immunized C57BL/6 mice. These findings suggest that Th1 immuneresponses may be responsible for the inhibition of allergen-inducedTh2 responses in the airways of flu virus-infected C57BL/6 mice.Supporting this view are our results showing that flu virus infection-mediated suppression of airway eosinophilia was not detected in IFN-�-deficient mice (C57BL/6 background) (Fig. 5A). In addition, wefound that the T cells from the MLN of IFN-� deficient mice infectedwith flu virus and immunized with OVA or only immunized withOVA secreted strongly elevated levels of IL-5 after in vitro stimula-tion in comparison with those of C57BL/6 control mice (Fig. 5B).Interestingly, Figure 5C shows that IFN-�-deficient mice (C57BL/6background) infected with flu virus alone, but not C57BL/6 controlmice, also developed airway eosinophilia 1 and 2 wk after infection.MLN cells from IFN-�-deficient mice infected with flu virus alsoshowed increased secretion of IL-5 after in vitro stimulation withanti-CD3 (Fig. 5D) and flu virus Ag (data not shown) in comparisonto MLN cells from C57BL/6 control mice infected with flu virus.These results suggest that IFN-� is not only responsible for flu virus-induced suppression of allergic Th2 responses but also for the weakTh2 responses normally observed in mice infected with flu virus.IL-12 p35 (C57BL/6 background)-deficient mice infected with flu
FIGURE 3. Intranasal infection with flu virus inhibits the recruitment ofin vitro-generated OVA-specific CD4� Th2 cells into the airways and re-duces airway eosinophilia. A, OVA-specific Th2 cells derived fromDO11.10 TCR transgenic mice (BALB/c background) were generated asdescribed in Materials and Methods. OVA-transgenic T cells (5 � 106)were then i.v. injected into BALB/c control mice or BALB/c mice that hadbeen infected with 2 � 105 EID50 of flu virus 1 wk before. One and 3 daysafter the i.v. application of the Th2 cells, the BALB/c mice were treatedwith OVA i.n. B, Three days after the last OVA airway challenge, a BALwas performed and the MLN cells were isolated. BAL and MLN cells werestained with Abs against CD4 and an Ab against the transgenic TCR.Shown are the FACS stainings of the BAL and MLN cells from one controland one flu-infected mouse that are representative for eight mice per group.The percentage of CD4� T cells expressing the transgenic TCR is indi-cated. C, The total amount of CD4� T cells expressing the transgenic TCRpresent in the BAL and MLN together with the total eosinophil numbers ofindividual mice are also shown.
4605The Journal of Immunology
by guest on July 14, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

virus 1 wk before OVA i.n. challenge also showed no flu virus-induced suppression of airway eosinophilia 6 days after OVA airwaychallenge in comparison with IL-12 p35-deficient mice only immu-nized with OVA (43.5 � 7.9 vs 45.6 � 19.5 eosinophils/ml BALfluid � 104 (n � 4–6/group � SD)). Taken together, these resultsclearly indicate that an infection with flu virus inhibits the develop-ment of airway eosinophilia by inducing Th1 immune responses.
The development of systemic allergen-specific Th2 responses,AHR, and goblet cell metaplasia are not suppressed by infectionwith flu virus
Our results thus far clearly show that the flu virus infection sup-pressed the development of allergen-specific Th2 responses in the
lung. Because Th2 responses, in particular airway eosinophilia, areknown to induce airway bronchoconstriction, we analyzed whetherthe flu virus infection inhibited the development of AHR in OVA-immunized C57BL/6 mice. Figure 6D shows that the flu virusinfection 1 or 3 wk before OVA challenge exacerbated AHR inC57BL/6 mice immunized with OVA. In addition, we found thatC57BL/6 mice infected with flu virus for 2 wk, but not 4 or 7 wk,also developed AHR (MCh50 � SD: control mice, 91.1 � 31.1(n � 6); 2 wk flu-infected mice, 40.9 � 25.6 (n � 7; p � 0.05,comparison between control and 2 wk-infected mice); 4 wk fluvirus-infected mice, 71.6 � 18.7 (n � 8); and 7 wk flu-infectedmice, 91.2 � 55.2 (n � 7)). The T cells from the MLN of C57BL/6mice infected with flu virus for 2 wk secreted increased amounts of
FIGURE 5. The suppression of allergen-inducedeosinophilia after flu virus infection is dependent onIFN-�. OVA-immunized C57BL/6 control and IFN-�-deficient (C57BL/6 background) mice were eitherinfected i.n. with flu virus 1 wk before OVA airwaychallenge or left uninfected as an OVA control (Fig.1A). Six days following OVA airway challenge,BALs were performed, and the MLN cells weretreated as described in Fig. 1. Shown are the meannumbers of eosinophils present in the BALs (A) andthe amount of IL-5 detected in the MLN cell culturesstimulated in vitro for 48 h with anti-CD3/IL-2 (B)from the different groups of mice (mean from 8–10mice/group with SD). In a separate experiment, IFN-�-deficient and C57BL/6 control mice were infectedi.n. with 2 � 105 EID50 of flu virus. One and 2 wklater, BALs were performed, and the MLN cells weretreated as described in Fig. 1. Shown are the meanamounts of eosinophils present in the BAL fluid (C)and the mean amount of IL-5 detected in the cell cul-tures stimulated with anti-CD3/IL-2 (D) of C57BL/6control and IFN-�-deficient mice (mean from sixmice per group with SD). The experiments shown inC and D were repeated once with similar results. �,p � 0.05 compared to OVA-Wt (A and B) or Flu-Wt(C and D).
FIGURE 4. The production of IL-5, IFN-�, eotaxin/CCL11, RANTES/CCL5, and MCP-1/CCL2 after allergen challenge in the lung is not suppressedby infection with flu virus. C57BL/6 mice were treated as described in Fig. 3. One day after OVA airway challenge, BALs were performed, and the BALfluid was concentrated 3-fold. Shown are the amounts of IL-5, IFN-�, eotaxin/CCL11, RANTES/CCL5, and MCP-1/CCL2 in the BALs of six to sevenC57BL/6 mice per group with SD, as determined by ELISA. �, p � 0.05; ��, p � 0.01; ���, p � 0.001, compared to OVA control mice.
4606 FLU VIRUS INFECTION INHIBITS ALLERGEN-INDUCED Th2 RESPONSES
by guest on July 14, 2015http://w
ww
.jimm
unol.org/D
ownloaded from
IL-13 after in vitro stimulation with anti-CD3 (560 � 250 pg/ml)(mean of 7 mice per group with SD; the experiment was repeatedonce with similar results). The amount of IL-13 in the BAL fluidof infected C57BL/6 mice and in the cultures from MLN ofnoninfected C57BL/6 mice stimulated with anti-CD3 was belowthe detection level of 100 pg/ml of the IL-13 ELISA used.
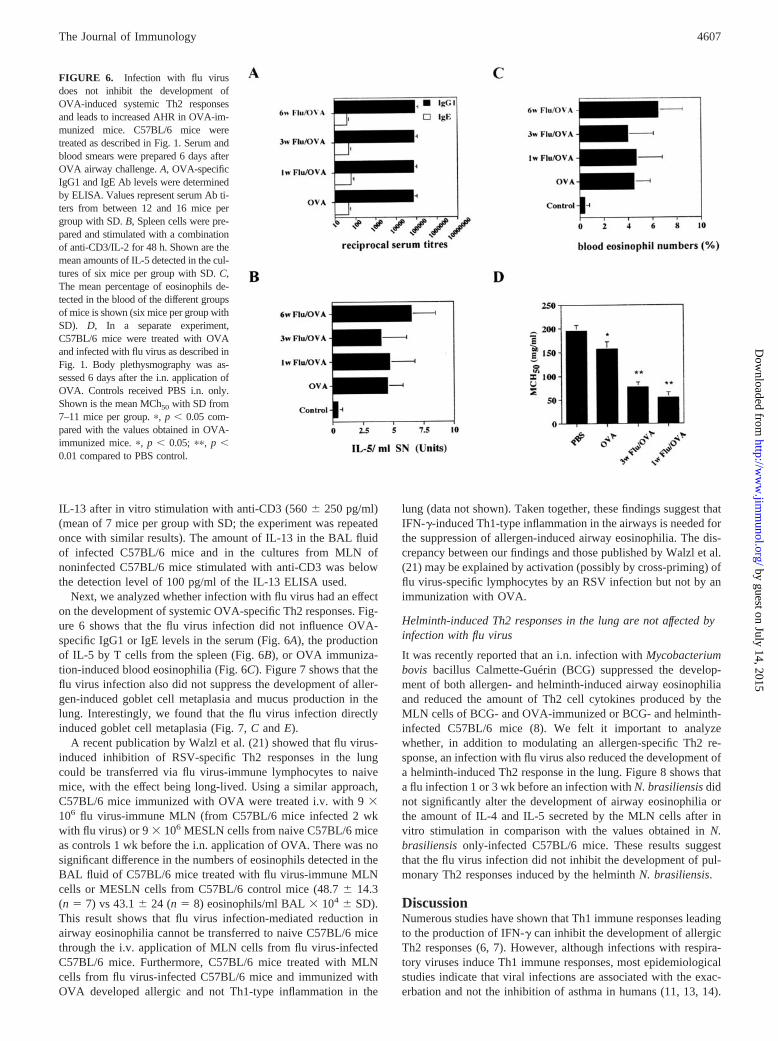
Next, we analyzed whether infection with flu virus had an effecton the development of systemic OVA-specific Th2 responses. Fig-ure 6 shows that the flu virus infection did not influence OVA-specific IgG1 or IgE levels in the serum (Fig. 6A), the productionof IL-5 by T cells from the spleen (Fig. 6B), or OVA immuniza-tion-induced blood eosinophilia (Fig. 6C). Figure 7 shows that theflu virus infection also did not suppress the development of aller-gen-induced goblet cell metaplasia and mucus production in thelung. Interestingly, we found that the flu virus infection directlyinduced goblet cell metaplasia (Fig. 7, C and E).
A recent publication by Walzl et al. (21) showed that flu virus-induced inhibition of RSV-specific Th2 responses in the lungcould be transferred via flu virus-immune lymphocytes to naivemice, with the effect being long-lived. Using a similar approach,C57BL/6 mice immunized with OVA were treated i.v. with 9 �106 flu virus-immune MLN (from C57BL/6 mice infected 2 wkwith flu virus) or 9 � 106 MESLN cells from naive C57BL/6 miceas controls 1 wk before the i.n. application of OVA. There was nosignificant difference in the numbers of eosinophils detected in theBAL fluid of C57BL/6 mice treated with flu virus-immune MLNcells or MESLN cells from C57BL/6 control mice (48.7 � 14.3(n � 7) vs 43.1 � 24 (n � 8) eosinophils/ml BAL � 104 � SD).This result shows that flu virus infection-mediated reduction inairway eosinophilia cannot be transferred to naive C57BL/6 micethrough the i.v. application of MLN cells from flu virus-infectedC57BL/6 mice. Furthermore, C57BL/6 mice treated with MLNcells from flu virus-infected C57BL/6 mice and immunized withOVA developed allergic and not Th1-type inflammation in the
lung (data not shown). Taken together, these findings suggest thatIFN-�-induced Th1-type inflammation in the airways is needed forthe suppression of allergen-induced airway eosinophilia. The dis-crepancy between our findings and those published by Walzl et al.(21) may be explained by activation (possibly by cross-priming) offlu virus-specific lymphocytes by an RSV infection but not by animmunization with OVA.
Helminth-induced Th2 responses in the lung are not affected byinfection with flu virus
It was recently reported that an i.n. infection with Mycobacteriumbovis bacillus Calmette-Guérin (BCG) suppressed the develop-ment of both allergen- and helminth-induced airway eosinophiliaand reduced the amount of Th2 cell cytokines produced by theMLN cells of BCG- and OVA-immunized or BCG- and helminth-infected C57BL/6 mice (8). We felt it important to analyzewhether, in addition to modulating an allergen-specific Th2 re-sponse, an infection with flu virus also reduced the development ofa helminth-induced Th2 response in the lung. Figure 8 shows thata flu infection 1 or 3 wk before an infection with N. brasiliensis didnot significantly alter the development of airway eosinophilia orthe amount of IL-4 and IL-5 secreted by the MLN cells after invitro stimulation in comparison with the values obtained in N.brasiliensis only-infected C57BL/6 mice. These results suggestthat the flu virus infection did not inhibit the development of pul-monary Th2 responses induced by the helminth N. brasiliensis.
DiscussionNumerous studies have shown that Th1 immune responses leadingto the production of IFN-� can inhibit the development of allergicTh2 responses (6, 7). However, although infections with respira-tory viruses induce Th1 immune responses, most epidemiologicalstudies indicate that viral infections are associated with the exac-erbation and not the inhibition of asthma in humans (11, 13, 14).
FIGURE 6. Infection with flu virusdoes not inhibit the development ofOVA-induced systemic Th2 responsesand leads to increased AHR in OVA-im-munized mice. C57BL/6 mice weretreated as described in Fig. 1. Serum andblood smears were prepared 6 days afterOVA airway challenge. A, OVA-specificIgG1 and IgE Ab levels were determinedby ELISA. Values represent serum Ab ti-ters from between 12 and 16 mice pergroup with SD. B, Spleen cells were pre-pared and stimulated with a combinationof anti-CD3/IL-2 for 48 h. Shown are themean amounts of IL-5 detected in the cul-tures of six mice per group with SD. C,The mean percentage of eosinophils de-tected in the blood of the different groupsof mice is shown (six mice per group withSD). D, In a separate experiment,C57BL/6 mice were treated with OVAand infected with flu virus as described inFig. 1. Body plethysmography was as-sessed 6 days after the i.n. application ofOVA. Controls received PBS i.n. only.Shown is the mean MCh50 with SD from7–11 mice per group. �, p � 0.05 com-pared with the values obtained in OVA-immunized mice. �, p � 0.05; ��, p �0.01 compared to PBS control.
4607The Journal of Immunology
by guest on July 14, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

Furthermore, animal experiments addressing this issue haveyielded somewhat conflicting results with some reports indicatingthat viral infections can both increase or suppress allergen-inducedTh2 responses in the lung, depending upon the virus used and thetime point of infection (15–26). The aim of our study was to an-alyze whether an infection with flu virus interfered with the de-velopment of Th2 responses in the lung induced by the recruitmentof Th2 cells into the airways after the i.n. application of allergen.We found that infection with flu virus 1, 3, 6, or 9 wk before i.n.allergen challenge strongly inhibited the development of airwayeosinophilia. The finding that blood eosinophil levels were notreduced by the flu virus infection suggests that the recruitment ofeosinophils into the airways may have been impaired by the fluvirus infection. One possibility is that the flu virus infection in-hibited the allergen-induced production of MCP-1/CCL2, macro-phage inflammatory protein (MIP)-1�/CCL3, RANTES/CCL5,MCP-3/CCL7, eotaxin/CCL11, or macrophage-derived chemo-kine/CCL22 in the lung, because these chemokines are associatedwith the recruitment of eosinophils into the airways (2, 34). How-ever, our results clearly show that the production of MCP-1/CCL2,RANTES/CCL5, and eotaxin/CCL11 were not impaired inC57BL/6 mice immunized with OVA and infected with the fluvirus. Interestingly, we found that the flu virus infection directlyinduced the production of these chemokines in the airways. Fur-thermore, recent publications have shown that infection of epithe-lial cells or macrophages with flu virus induced the production ofRANTES/CCL5 and MCP-1/CCL2, or RANTES/CCL5, MIP-1�/CCL3, MCP-1/CCL2 and MCP-3/CCL7 by these cells, respec-tively (35, 36). Taken together, these results suggest that the in-hibition of allergen-induced airway eosinophilia by infection withflu virus was not due to a reduction in the production of RANTES/
CCL5, MIP-1�/CCL3, MCP-1/CCL2, MCP-3/CCL7, or eotaxin/CCL11 in the airways.
Recent publications indicate that, in addition to chemokines,adhesion molecules such as VCAM-1 or ICAM-1 expressed byendothelial cells may also be involved in the efficient accumulationof eosinophils into the airways (34). Therefore, it is possible thatthe flu virus infection reduced the expression of these or otheradhesion molecules on the endothelial cells, thereby interferingwith the homing of eosinophils into the airways. Supporting thisview are the recently published results of Yang et al. (37). Theyshowed that BCG infection-induced inhibition of established al-lergic inflammatory responses was associated with decreasedVCAM-1 expression in the lung.
The suppression of allergen-induced airway eosinophilia ob-served in the flu-infected mice correlated with a strong reduction inthe numbers of Th2 cells present in the airways. A possible ex-planation for this finding may be that the flu virus infection re-duced the amount of total Th2 cells generated by the two i.p.applications of OVA/alum. However, we found that OVA-specificIgG1 and IgE serum levels, blood eosinophil levels, and IL-5 pro-duction by T cells in the spleen were not reduced by infection withflu virus. This result indicates that numbers of OVA-specific Th2cells present in the periphery of OVA-treated C57BL/6 mice didnot differ from the amount of Th2 cells present in the periphery offlu virus-infected/OVA-treated C57BL/6 mice. Furthermore, wealso found that a flu virus infection strongly reduced the numbersof in vitro-generated and i.v.-injected OVA-specific Th2 cellsdetected in the airways of BALB/c mice challenged with OVAi.n. The eosinophilia induced by the OVA-specific Th2 cells wasalso strongly reduced in the flu virus-infected BALB/c mice incomparison to BALB/c control mice. Taken together, these results
FIGURE 7. OVA-induced goblet cell metaplasia inthe lung was not inhibited by infection with flu virus.C57BL/6 mice were treated as described in Fig. 1.Sections of the lung were subjected to a periodic acid-Schiff reaction staining. Shown are untreated C57BL/6mice (A), OVA-immunized C57BL/6 mice (B),C57BL/6 mice infected with flu virus for 2 wk (C),C57BL/6 mice infected with flu virus 1 wk beforeOVA airway challenge (D), C57BL/6 mice infectedwith flu virus for 4 wk (E), and C57BL/6 mice in-fected with flu virus 3 wk before OVA airway chal-lenge (F). Representative examples of five mice pergroup. Goblet cells are indicated by arrows (�200magnification).
4608 FLU VIRUS INFECTION INHIBITS ALLERGEN-INDUCED Th2 RESPONSES
by guest on July 14, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

suggest that a flu virus infection interfered with the efficient re-cruitment of Th2 cells into the airways.
Reduced production of IL-5 in the airways due to the inhibitionof Th2 cell recruitment to this site may also be the most likelyexplanation for the suppression of airway eosinophilia, because ithas been shown that IL-5 is necessary for the efficient recruitmentand survival of eosinophils in the airways (32, 33, 38). However,we cannot completely rule out the possibility that Th2 cells effec-tively home into the airways of flu-infected mice but can then nolonger proliferate. Supporting this possibility is the finding of Leeet al. (39) showing that clonal expansion of T cells in the lung wasimpaired by Th1 cell-mediated inflammation. In contrast, Harris etal. (40) recently reported that activated T cells undergo clonal ex-pansion in the LN but not in the lung after migrating to this site.Therefore, in our opinion, the most likely explanation for thestrongly reduced numbers of Th2 cells in the airways of flu-in-fected mice is the impaired recruitment and not impaired clonalexpansion of Th2 cells. This is further supported by our findingthat 3-fold fewer numbers of Th2 cells were already detected 24 hafter the i.n. application of OVA in flu virus-infected/OVA-treatedC57BL/6 mice in comparison to OVA only-treated mice. How-ever, this effect may be limited to the airways, because the num-bers of adoptively transferred OVA-specific transgenic Th2 cells
detected in the MLN were not affected by the flu virus infection.Furthermore, the amount of IL-5 secreted by the T cells from theMLN of flu virus-infected/OVA-treated C57BL/6 mice was alsonot strongly reduced in comparison to the amount of IL-5 secretedby the MLN of OVA-immunized mice after in vitro stimulation.
The mechanism of how a flu virus infection interferes with therecruitment of Th2 cells into the airways is not known. However,impaired Th2 cell recruitment may be due to a reduction in theexpression of chemokines or adhesion molecules in the lungneeded for the efficient recruitment of Th2 cells into the airways.Prospective candidates are the chemokines macrophage-derivedchemokine/CCL22, T cell activation-3/CCL1, and thymus and ac-tivation-regulated chemokine/CCL17, and the adhesion moleculesVCAM-1, fibronectin CS1, and ICAM-1, -2, and -3 (2, 41). Wealso cannot rule out the possibility that the flu virus infection alsoleads to a change in chemokine receptor expression or integrins onthe surface of Th2 cells needed for homing into the airways.
Although it is not clear how an infection with flu virus inhibitsthe development of allergen-induced Th2 responses in the lung,our experiments using IFN-�- and IL-12 p35-deficient mice sug-gest that it is due to Th1 immune responses. Furthermore, thesuppression of allergen-induced Th2 responses by the flu virusinfection correlated with a significant increase in IFN-� levels and
FIGURE 8. Flu virus infection did not inhibit thedevelopment of airway eosinophilia in mice infectedwith the helminth N. brasiliensis. C57BL/6 mice werecoinfected with flu virus 1 (A) or 3 (B) wk before aninfection with N. brasiliensis as described in Materialsand Methods. As controls, C57BL/6 mice were alsoonly infected with N. brasiliensis. Ten days after in-fection with the helminths, BALs were performed, andthe cells were counted and stained with H&E, and thedifferent cell types were identified microscopically. Inparallel to the BALs, single-cell suspensions (2 �105cells/well) from MLN of the different groups ofmice were prepared and stimulated in vitro for 48 h onanti-CD3-bound plates in the presence of IL-2. Shownare the mean numbers of eosinophils present in theBALs and the mean amounts of IL-4 and IL-5 presentin the MLN cultures from six to eight mice (A) or fivemice (B) per group with SD. The experiment shown inA was repeated once with similar results.
4609The Journal of Immunology
by guest on July 14, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

numbers of CD8� but not CD4� T cells producing IFN-�, sug-gesting that CD8� T cells secreting IFN-� were responsible for thesuppression of the allergen-induced Th2 responses. Previous stud-ies have also shown that flu virus-induced suppression of Th2 re-sponses was also due to the presence of IFN-� (25). However, incontrast to our finding, the inhibition of allergen-specific Th2 re-sponses could be explained by the suppression of Th2 cell develop-ment by IFN-�. In our system, the development of allergen-specificTh2 cells was not inhibited by the flu virus infection. Our results pointat an additional mechanism of how the presence of IFN-� may inter-fere with the development of allergic Th2 responses, namely, that theflu virus infection induces a strong IFN-�-mediated inflammatory re-sponse in the lung that results in the impaired recruitment of allergen-specific Th2 cells into the airways. Supporting this finding are resultsshowing a similar effect during mycobacterial infection and showingthat Th2 cells could not home efficiently into Th1 cell-mediated in-flamed sites (29, 30, 42). However, this may not be a general phe-nomenon, because it was also reported that Th1 and Th2 responsescan coexist at the same site (43). Furthermore, we also found thatinfection with flu virus did not decrease the development of Th2 re-sponses in the lung after infection with the helminth N. brasiliensis. Incontrast, previous studies have shown that an infection with BCG canreduce the development of N. brasiliensis-induced Th2 responses.The discrepancy between this finding and our results suggests thatinfections with mycobacteria are more efficient in inhibiting the de-velopment of Th2 responses than are infections with flu virus. Thisview is supported by the observation that mycobacterial infection wasmore efficient than flu virus infection in inhibiting the development ofallergen-induced airway eosinophilia (8, 27).
An unexpected finding was that infection with flu virus not onlyexacerbated but also directly induced AHR in the absence of air-way eosinophilia, because eosinophils have been shown to be nec-essary for the induction of AHR in some experimental systems(18). How can this occur? First, the flu virus infection may directlyinduce AHR by increasing airway smooth muscle contractionthrough the production of IL-1�. Rhinovirus-induced IL-1�-de-pendent increase in airway smooth-muscle contraction has beenshown to occur in vitro (44), and it has been shown that flu virusinfection leads to the production of IL-1� in the lung (45). Asecond possibility is that the flu virus infection induced the pro-duction of IL-13 in the airways. IL-13 has also been shown toinduce AHR independently of eosinophils (46). This would alsoexplain why allergen-induced mucus production was not inhibitedby the flu virus infection, because goblet cell metaplasia is alsoinduced by the production of IL-13 (46). Supporting this view wasour finding that T cells from the MLN of C57BL/6 mice infectedwith flu virus, but not from uninfected C57BL/6 control mice,produced IL-13 after in vitro stimulation with anti-CD3 and IL-2.A further source of IL-13 possibly inducing AHR are mast cells,because they can also produce and release IL-13 (47). In unisonwith this hypothesis is the report from Grunewald et al. (48), whoshowed that the application of flu virus Ag induced the degranu-lation of mast cells in the skin of mice previously infected with fluvirus. Furthermore, a dissociation between airway eosinophilia andAHR has also been demonstrated in other recent reports. Gerholdet al. (49) reported that the application of LPS before allergensensitization strongly reduced the development of OVA-inducedairway eosinophilia but not AHR, with the effect being dependenton IL-12. In addition, Hansen et al. (50) also reported that theapplication of allergen-specific Th1 cells could suppress the de-velopment of Th2 cell-induced airway eosinophilia but not AHR.Our observation together with these findings suggest that Th1 im-mune responses may be more efficient in inhibiting the develop-ment of Th2 cell-induced eosinophilia than AHR. However, Coyle
et al. (51) reported that IL-5-deficient mice infected with N. bra-siliensis also developed AHR in the absence of airway eosino-philia, suggesting that the dissociation between airway eosino-philia and AHR may not only be associated with the induction ofTh1 responses.
In conclusion, we found that a flu virus infection 1, 3, 6, or 9 wkbefore allergen airway challenge resulted in a strong reduction ofallergen-induced airway eosinophilia. This effect correlated with astrong reduction in the recruitment of allergen-specific Th2 cellsinto the airways but not MLN of the lungs. The detailed mecha-nism of how a flu virus infection mediates these effects remains tobe determined. However, our results suggest that it is dependentupon the induction of Th1 responses. Our experiments support theview that, generally, infections that induce strong Th1 responsesleading to the production of IFN-� have the potential to suppressthe development of local allergic Th2 cell-mediated responses inthe lung of mice and possibly also humans. The mechanism mayinvolve the inhibition of efficient Th2 cell homing into the airways.However, our results also indicate that a flu virus infection exac-erbates allergen-induced AHR and does not inhibit the develop-ment of goblet cell metaplasia. Our findings may also have impli-cations for human disease. Children infected with flu virus maydirectly develop AHR and thus virus-induced asthma. Further-more, previous studies indicate that a flu virus infection duringallergen sensitization may inhibit the development of tolerance andexacerbate allergic Th2 responses (15, 25). However, in the longterm, a flu virus infection may contribute to the protection againstasthma by reducing the development of eosinophilic inflammationin the airways.
References1. Romagnani, S. 2000. The role of lymphocytes in allergic disease. J. Allergy Clin.
Immunol. 105:399.2. Lukacs, N. W. 2001. Role of chemokines in the pathogenesis of asthma. Nat. Rev.
Immunol. 1:108.3. Gutierrez-Ramos, J. C., C. Lloyd, M. L. Kapsenberg, J. A. Gonzalo, and
A. J. Coyle. 2000. Non-redundant functional groups of chemokines operate in acoordinate manner during the inflammatory response in the lung. Immunol. Rev.177:31.
4. Erb, K., and G. Wohlleben. 2002. Novel vaccines protecting against the devel-opment of allergic disorders: a double-edged sword? Curr. Opin. Immunol. 14:633.
5. Herz, U., P. Lacy, H. Renz, and K. Erb. 2000. The influence of infections on thedevelopment and severity of allergic disorders. Curr. Opin. Immunol. 12:632.
6. Gajewski, T. F., and F. W. Fitch. 1988. Anti-proliferative effect of IFN-� inimmune regulation. I. IFN-� inhibits the proliferation of Th2 but not Th1 murinehelper T lymphocyte clones. J. Immunol. 140:4245.
7. O’Garra, A. 1998. Cytokines induce the development of functionally heteroge-neous T helper cell subsets. Immunity 8:275.
8. Erb, K. J., J. W. Holloway, A. Sobeck, H. Moll, and G. Le Gros. 1998. Infectionof mice with Mycobacterium bovis-bacillus Calmette-Guerin (BCG) suppressesallergen-induced airway eosinophilia. J. Exp. Med. 187:561.
9. Hansen, G., V. P. Yeung, G. Berry, D. T. Umetsu, and R. H. DeKruyff. 2000.Vaccination with heat-killed Listeria as adjuvant reverses established allergen-induced airway hyperreactivity and inflammation: role of CD8� T cells and IL-18. J. Immunol. 164:223.
10. Sur, S., J. S. Wild, B. K. Choudhury, N. Sur, R. Alam, and D. M. Klinman. 1999.Long term prevention of allergic lung inflammation in a mouse model of asthmaby CpG oligodeoxynucleotides. J. Immunol. 162:6284.
11. Sigurs, N., R. Bjarnason, F. Sigurbergsson, B. Kjellman, and B. Bjorksten. 1995.Asthma and immunoglobulin E antibodies after respiratory syncytial virus bron-chiolitis: a prospective cohort study with matched controls. Pediatrics 95:500.
12. Sigurs, N., R. Bjarnason, F. Sigurbergsson, and B. Kjellman. 2000. Respiratorysyncytial virus bronchiolitis in infancy is an important risk factor for asthma andallergy at age 7. Am. J. Respir. Crit. Care Med. 161:1501.
13. Eriksson, M., R. Bennet, and A. Nilsson. 2000. Wheezing following lower re-spiratory tract infections with respiratory syncytial virus and influenza A in in-fancy. Pediatr. Allergy Immunol. 11:193.
14. Neuzil, K. M., P. F. Wright, E. F. J. Mitchel, and M. R. Griffin. 2000. The burdenof influenza illness in children with asthma and other chronic medical conditions.J. Pediatr. 137:856.
15. Suzuki, S., Y. Suzuki, N. Yamamoto, Y. Matsumoto, A. Shirai, and T. Okubo.1998. Influenza A virus infection increases IgE production and airway respon-siveness in aerosolized antigen-exposed mice. J. Allergy Clin. Immunol. 102:732.
16. O’Donnell, D. R., and P. J. Openshaw. 1998. Anaphylactic sensitization toaeroantigen during respiratory virus infection. Clin. Exp. Allergy 28:1501.
4610 FLU VIRUS INFECTION INHIBITS ALLERGEN-INDUCED Th2 RESPONSES
by guest on July 14, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

17. Schwarze, J., E. Hamelmann, K. L. Bradley, K. Takeda, and E. W. Gelfand. 1997.Respiratory syncytial virus infection results in airway hyperresponsiveness andenhanced airway sensitization to allergen. J. Clin. Invest. 100:226.
18. Schwarze, J., G. Cieslewicz, E. Hamelmann, A. Joetham, L. D. Shultz,M. C. Lamers, and E. W. Gelfand. 1999. IL-5 and eosinophils are essential for thedevelopment of airway hyperresponsiveness following acute respiratory syncytialvirus infection. J. Immunol. 162:2997.
19. Scheerens, J., G. Folkerts, H. Van Der Linde, P. J. Sterk, D. M. Conroy,T. J. Williams, and F. P. Nijkamp. 1999. Eotaxin levels and eosinophils in guineapig broncho-alveolar lavage fluid are increased at the onset of a viral respiratoryinfection. Clin. Exp. Allergy 29:74.
20. Dakhama, A., A. M. Bramley, N. G. Chan, K. O. McKay, R. R. Schellenberg, andR. G. Hegele. 1999. Effect of respiratory syncytial virus on subsequent allergicsensitization to ovalbumin in guinea-pigs. Eur. Respir. J. 13:976.
21. Walzl, G., S. Tafuro, P. Moss, P. J. Openshaw, and T. Hussell. 2000. Influenzavirus lung infection protects from respiratory syncytial virus-induced immuno-pathology. J. Exp. Med. 192:1317.
22. Yamamoto, N., S. Suzuki, Y. Suzuki, A. Shirai, M. Nakazawa, M. Suzuki,T. Takamasu, Y. Nagashima, M. Minami, and Y. Ishigatsubo. 2001. Immuneresponse induced by airway sensitization after influenza A virus infection de-pends on timing of antigen exposure in mice. J. Virol. 75:499.
23. Suzuki, M., S. Suzuki, N. Yamamoto, S. Komatsu, S. Inoue, T. Hashiba,M. Nishikawa, and Y. Ishigatsubo. 2000. Immune responses against replication-deficient adenovirus inhibit ovalbumin-specific allergic reactions in mice. Hum.Gene Ther. 11:827.
24. Adamko, D. J., B. L. Yost, G. J. Gleich, A. D. Fryer, and D. B. Jacoby. 1999.Ovalbumin sensitization changes the inflammatory response to subsequent para-influenza infection: eosinophils mediate airway hyperresponsiveness, m2 musca-rinic receptor dysfunction, and antiviral effects. J. Exp. Med. 190:1465.
25. Tsitoura, D. C., S. Kim, K. Dabbagh, G. Berry, D. B. Lewis, and D. T. Umetsu.2000. Respiratory infection with influenza A virus interferes with the induction oftolerance to aeroallergens. J. Immunol. 165:3484.
26. Hurst, S. D., B. W. Seymour, T. Muchamuel, V. P. Kurup, and R. L. Coffman.2001. Modulation of inhaled antigen-induced IgE tolerance by ongoing Th2 re-sponses in the lung. J. Immunol. 166:4922.
27. Major, T., G. Wohlleben, B. Reibetanz, and K. J. Erb. 2002. Application of heatkilled Mycobacterium bovis-BCG into the lung inhibits the development of al-lergen-induced Th2 responses. Vaccine 20:1532.
28. Neuhaus-Steinmetz, U., T. Glaab, A. Daser, A. Braun, M. Lommatzsch, U. Herz,J. Kips, Y. Alarie, and H. Renz. 2000. Sequential development of airway hyper-responsiveness and acute airway obstruction in a mouse model of allergic in-flammation. Int. Arch. Allergy Immunol. 121:57.
29. Austrup, F., D. Vestweber, E. Borges, M. Lohning, R. Brauer, U. Herz, H. Renz,R. Hallmann, A. Scheffold, A. Radbruch, and A. Hamann. 1997. P- and E-selectinmediate recruitment of T-helper-1 but not T-helper-2 cells into inflamed tissues.Nature 385:81.
30. Lukacs, N. W. 2000. Migration of helper T-lymphocyte subsets into inflamedtissues. J. Allergy Clin. Immunol. 106:264.
31. Iezzi, G., D. Scheidegger, and A. Lanzavecchia. 2001. Migration and function ofantigen-primed nonpolarized T lymphocytes in vivo. J. Exp. Med. 193:987.
32. Mould, A. W., A. J. Ramsay, K. I. Matthaei, I. G. Young, M. E. Rothenberg, andP. S. Foster. 2000. The effect of IL-5 and eotaxin expression in the lung oneosinophil trafficking and degranulation and the induction of bronchial hyperre-activity. J. Immunol. 164:2142.
33. Rothenberg, M. E., J. A. MacLean, E. Pearlman, A. D. Luster, and P. Leder.1997. Targeted disruption of the chemokine eotaxin partially reduces antigen-induced tissue eosinophilia. J. Exp. Med. 185:785.
34. Tachimoto, H., M. Ebisawa, and B. S. Bochner. 2002. Cross-talk between inte-grins and chemokines that influences eosinophil adhesion and migration. Int.Arch. Allergy Immunol. 128(Suppl. 1):18.
35. Matikainen, S., J. Pirhonen, M. Miettinen, A. Lehtonen, C. Govenius-Vintola,T. Sareneva, and I. Julkunen. 2000. Influenza A and Sendai viruses induce dif-ferential chemokine gene expression and transcription factor activation in humanmacrophages. Virology 276:138.
36. Julkunen, I., K. Melen, M. Nyqvist, J. Pirhonen, T. Sareneva, and S. Matikainen.2000. Inflammatory responses in influenza A virus infection. Vaccine 19(Suppl.1):S32.
37. Yang, X., Y. Fan, S. Wang, X. Han, J. Yang, L. Bilenki, and L. Chen. 2002.Mycobacterial infection inhibits established allergic inflammatory responses viaalteration of cytokine production and vascular cell adhesion molecule-1 expres-sion. Immunology 105:336.
38. Dent, L. A., M. Strath, A. L. Mellor, and C. J. Sanderson. 1990. Eosinophilia intransgenic mice expressing interleukin 5. J. Exp. Med. 172:1425.
39. Lee, S. C., Z. H. Jaffar, K. S. Wan, S. T. Holgate, and K. Roberts. 1999. Reg-ulation of pulmonary T cell responses to inhaled antigen: role in Th1- and Th2-mediated inflammation. J. Immunol. 162:6867.
40. Harris, N. L., V. Watt, F. Ronchese, and G. Le Gros. 2002. Differential T cellfunction and fate in lymph node and nonlymphoid tissues. J. Exp. Med. 195:317.
41. Yusuf-Makagiansar, H., M. E. Anderson, T. V. Yakovleva, J. S. Murray, andT. J. Siahaan. 2002. Inhibition of LFA-1/ICAM-1 and VLA-4/VCAM-1 as atherapeutic approach to inflammation and autoimmune diseases. Med. Res. Rev.22:146.
42. Erb, K. J., C. Trujillo, M. Fugate, and H. Moll. 2002. Infection with the helminthNippostrongylus brasiliensis does not interfere with efficient elimination of My-cobacterium bovis BCG from the lungs of mice. Clin. Diagn. Lab. Immunol.9:727.
43. Li, L., Y. Xia, A. Nguyen, L. Feng, and D. Lo. 1998. Th2-induced eotaxinexpression and eosinophilia coexist with Th1 responses at the effector stage oflung inflammation. J. Immunol. 161:3128.
44. Hakonarson, H., C. Carter, N. Maskeri, R. Hodinka, and M. M. Grunstein. 1999.Rhinovirus-mediated changes in airway smooth muscle responsiveness: inducedautocrine role of interleukin-1�. Am. J. Physiol. 277:13.
45. Van Reeth, K., H. Nauwynck, and M. Pensaert. 1998. Bronchoalveolar interfer-on-�, tumor necrosis factor-�, interleukin-1, and inflammation during acute in-fluenza in pigs: a possible model for humans? J. Infect. Dis. 177:1076.
46. Kuperman, D. A., X. Huang, L. L. Koth, G. H. Chang, G. M. Dolganov, Z. Zhu,J. A. Elias, D. Sheppard, and D. J. Erle. 2002. Direct effects of interleukin-13 onepithelial cells cause airway hyperreactivity and mucus overproduction inasthma. Nat. Med. 8:885.
47. Kobayashi, H., Y. Okayama, T. Ishizuka, R. Pawankar, C. Ra, and M. Mori.1998. Production of IL-13 by human lung mast cells in response to Fc� receptorcross-linkage. Clin. Exp. Allergy 28:1219.
48. Grunewald, S. M., C. Hahn, G. Wohlleben, M. Teufel, T. Major, H. Moll,E. B. Brocker, and K. J. Erb. 2002. Infection with influenza A virus leads to fluantigen-induced cutaneous anaphylaxis in mice. J. Invest. Dermatol. 118:645.
49. Gerhold, K., K. Blumchen, A. Bock, C. Seib, P. Stock, T. Kallinich, M. Lohning,U. Wahn, and E. Hamelmann. 2002. Endotoxins prevent murine IgE production,TH2 immune responses, and development of airway eosinophilia but not airwayhyperreactivity. J. Allergy Clin. Immunol. 110:110.
50. Hansen, G., G. Berry, R. H. DeKruyff, and D. T. Umets. 1999. Allergen-specificTh1 cells fail to counterbalance Th2 cell-induced airway hyperreactivity butcause severe airway inflammation. J. Clin. Invest. 103:175.
51. Coyle, A. J., G. Kohler, S. Tsuyuki, F. Brombacher, and M. Kopf. 1998. Eosin-ophils are not required to induce airway hyperresponsiveness after nematodeinfection. Eur. J. Immunol. 28:2640.
4611The Journal of Immunology
by guest on July 14, 2015http://w
ww
.jimm
unol.org/D
ownloaded from




















