
Thymoquinone Inhibits Murine Leukemia WEHI-3 Cells In Vivo and In Vitro

A
ocToftwsado(ppcbtv©
K
1
(dh
1d
Pharmacological Research 55 (2007) 318–328
In vitro and in vivo profiling of CHF5022 and CHF5074Two �-amyloid1–42 lowering agents
Bruno P. Imbimbo a,∗, Elda Del Giudice b, Valentina Cenacchi a, Roberta Volta a, Gino Villetti a,Fabrizio Facchinetti a, Benedetta Riccardi a, Paola Puccini a, Nadia Moretto c,
Francesca Grassi c, Simone Ottonello c, Alberta Leon b
a Research & Development Department, Chiesi Farmaceutici, Via Palermo 26/A, 43100 Parma, Italyb Research & Innovation, Via Svizzera 16, 35127 Padua, Italy
c Department of Biochemistry and Molecular Biology, University of Parma, Viale G.P. Usberti 23/A, 43100 Parma, Italy
Accepted 18 December 2006
bstract
Long-term use of non-steroidal anti-inflammatory drugs (NSAIDs) may delay or prevent the onset of Alzheimer’s disease (AD). A subsetf NSAIDs, including flurbiprofen, has been shown to selectively inhibit the production of �-amyloid1–42 (A�42), independently from theiryclooxygenase (COX) inhibiting activity. We evaluated the in vitro and in vivo profiles of CHF5022 and CHF5074, two flurbiprofen analogues.he in vitro A� inhibiting activity was evaluated in a human neuroglioma cell line (H4) carrying the double Swedish mutation (K595N/M596L)f the human amyloid precursor protein (APPsw). The in vitro anti-COX activity was evaluated using human recombinant enzymes isolatedrom transfected Sf-9 cells. The in vivo pharmacokinetic and pharmacodynamic profiles of the two compounds were evaluated in young APPswransgenic mice (Tg2576) after oral gavage (100 or 300 mg kg−1 day−1 for 4–5 days) and after medicated diet (375 ppm for 4 weeks). R-Flurbiprofenas used as comparator. In vitro, CHF5022 and CHF5074 were found to be 3- and 7-fold more potent than R-flurbiprofen in inhibiting A�42
ecretion (IC50s of 92, 40 and 268 �M, respectively). Differently from R-flurbiprofen, CHF5022 and CHF5074 did not affect COX-1 (at 100 �M)nd COX-2 (at 300 �M) activity. Similarly to R-flurbiprofen, no significant alteration in the expression profile of a subset of Notch intracellularomain-responsive genes was observed with either CHF5022 or CHF5074. In Tg2576 mice, CHF5022 was well tolerated when administered byral gavage (100 mg kg−1 day−1 for 5 days) or by medicated diet (56 mg kg−1 day−1 for 4 weeks). R-Flurbiprofen was poorly tolerated in the diet32 mg kg−1 day−1) with 55% of the animals dying during the first week of treatment. After 4–5 days of oral gavage, CHF5022 and CHF5074lasma and brain levels at 3 h were found to increase with the dose, leading to brain concentrations of about 10% and 5% of the correspondinglasma concentrations, respectively. In animals fed for 4 weeks with compound-supplemented diet, mean plasma (580 �M) and brain (20 �М)
oncentrations of CHF5022 were 8 and 15 times higher than those of R-flurbiprofen. Plasma A�42 concentration was dose-dependently decreasedy CHF5022 and CHF5074. Brain A� levels (formic acid-extractable) were not significantly affected by either compound, although A�42 levelsended to inversely correlate (P = 0.105) with CHF5022 concentration in the brain. CHF5022 and CHF5074 thus appear to have a promising initro and in vivo profile. This warrants further evaluation of their long-term effects on A� brain pathology.2007 Elsevier Ltd. All rights reserved.
a(g
eywords: �-Amyloid; �-Secretase modulators; Transgenic Tg2576 mouse
. Introduction
Long-term use of non-steroidal anti-inflammatory drugs
NSAIDs) may delay or prevent the onset of Alzheimer’sisease (AD). A subset of NSAIDs, including flurbiprofen,as been shown to selectively inhibit the production of �-∗ Corresponding author. Tel.: +39 0521 279278; fax: +39 0521 279579.E-mail address: [email protected] (B.P. Imbimbo).
[msAd�t
043-6618/$ – see front matter © 2007 Elsevier Ltd. All rights reserved.oi:10.1016/j.phrs.2006.12.010
myloid1–42 (A�42), independently from their cyclooxygenaseCOX) inhibiting activity [1–3] and to counteract the pro-ression of A� pathology in transgenic mouse models of AD4–8]. The proposed mechanism for this activity is an allostericodulation of presenilin-1, the major component of the �-
ecretase complex, that is responsible for the formation of
� [3,9–11]. These A�42 lowering NSAIDs differ from tra-itional �-secretase inhibitors because they do not inhibit the-secretase-mediated cleavages of either amyloid precursor pro-ein (APP) at the alternative � site, or Notch-1 at the S3

B.P. Imbimbo et al. / Pharmacologica
siadboCoerlcpCpna(SdhsiTio
2
2
HSCa
a3oatwcUoCsrToccC
idacbammfw3map(
2
mta(SCtBTwDmiCaa
Fig. 1. Structural formula of R-flurbiprofen, CHF5022 and CHF5074.
ite [1,12]. These �-secretase-mediated cleavages generate twontracellular domains with transcription factor activity knowns Notch intracellular domain (NICD) and APP intracellularomain (AICD), respectively. CHF5022 and CHF5074 are flur-iprofen analogues with A�42 lowering properties but devoidf anti-COX activity (Fig. 1) [13,14]. In rats, CHF5022 andHF5074 are well absorbed after oral administration (absoluteral bioavailability of 91% and 50%, respectively) and slowlyliminated from plasma (elimination half-lives of 33 and 21 h,espectively). Their high plasma protein binding (99%) regu-ates central nervous system penetration with concentrations inerebrospinal fluid around 2% of those in plasma [13]. In theresent study we compared the in vitro activity of CHF5022 andHF5074 with that of R-flurbiprofen, an A�42 lowering agentresently in Phase 3 clinical trials for AD [15]. The pharmacoki-etics and pharmacodynamics of the two new compounds werelso evaluated after multiple dosing in young transgenic miceTg2576) expressing the Swedish mutation of APP (APPsw).hort-term (4–5 days), dose–response (100 and 300 mg kg−1
ay−1) studies were initially carried out. Later, based on theigher brain penetration, clearer dose-proportionality and alightly better tolerability emerging from the short-term stud-es, CHF5022 was selected for a 4-week study in Tg2576 mice.he overall objective was the proper design of long-term stud-
es aimed at evaluating the effects of long-term administrationf the compound on brain A� pathology.
. Materials and methods
.1. Aβ42 and Aβ40 assay
A�42 and A�40 were measured in the culture medium of
4 cells, a human neuroglioma cell line expressing the doublewedish mutation (K595N/M596L) of human APP (APPsw).ells were seeded onto 24-well plates (2 × 105 cell well−1)nd allowed to grow to confluence for 24 h, in 5% CO2/95%fsfr
l Research 55 (2007) 318–328 319
ir in a humidified atmosphere. Increasing concentrations (fromto 300–400 �M) of the compounds were added to the cells
vernight in a final volume of 0.5 ml. R-Flurbiprofen was useds positive control (3–1000 �M). DMSO (1%) was used as nega-ive control. At the end of the incubation, 100 �l of supernatantsere removed and treated with a biotinylated mouse mono-
lonal antibody (4G8, Signet Laboratories Inc., Dedham, MA,SA), specifically recognizing the 17–24 amino acid regionf A� and two rabbit polyclonal antibodies (C-term 42 and-term 40, BioSource International, Camarillo, CA, USA),
pecifically recognizing the C-terminus of A�42 and A�40,espectively. Antigen–antibodies complexes were recognized byAG-donkey anti-rabbit IgG (Jackson ImmunoResearch Lab-ratories, Soham, UK). Streptavidin coated magnetic beadsaptured the complexes and the signals were read by an electro-hemiluminescence instrument (Origen M8 Analyzer, BioVerisorporation, Gaithersburg, MD, USA).
The cytotoxicity potential of test compounds was assessedn the same cells of the A� assay (H4) with the 3-[4,5-imethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT)ssay. MTT is a soluble pale yellow salt that is reduced by mito-hondrial succinate dehydrogenase to form an insoluble darklue formazan product to which the cell membrane is imperme-ble. The ability of cells to reduce MTT provides an indication ofitochondria integrity and activity and it may be interpreted as aeasure of viability and/or cell number. After medium removal
or A�42 and A�40 determination, cells were incubated for 3 hith 500 �l culture medium containing 0.5 mg ml−1 MTT, at7 ◦C, 5% CO2 and saturated humidity. After removal of theedium, 500 �l of 100% DMSO were added to each well. The
mount of formed formazan was determined reading the sam-les at 570 nm (background 630 nm) using a microplater readermodel 450, Bio-Rad, Hercules, CA, USA).
.2. COX-1 and COX-2 assay
The inhibition of the cyclooxygenase activity was estimatedeasuring PGE2 production from arachidonic acid according
o a modified version of the method described by Glaser etl. [16]. Recombinant human prostaglandin H2 synthase-1PGHS-1) and -2 (PGHS-2) were expressed in transfectedpodoptera frugiperda (Sf-9) cells (Invitrogen, San Diego,A, USA). The microsomal fractions were prepared from the
ransfected cells and used to assay the enzymatic activities.riefly, the enzymes (2 �g) reconstituted in a buffer (100 mMris–HCl, pH 8.0) containing 2 mM phenol, were preincubatedith vehicle (DMSO) or test compounds in DMSO (1%MSO in the final assay) for 20 min at 22 ◦C. The reactionixture was completed with 1 �M hematin. The reaction was
nitiated adding arachidonic acid (4 and 2 �M for COX-1 andOX-2, respectively) and the mixture was incubated for 5 mint 22 ◦C for COX-1 assay, or for 10 min at 25 ◦C for COX-2ssay. For control measurements, arachidonic acid was omitted
rom the reaction mixture. The reactions were stopped by theequential addition of 1 M HCl and 1 M Tris–HCl (pH 8.0),ollowed by cooling to 4 ◦C. The amount of PGE2 present in theeaction mixture was quantified using an enzyme-immunoassay
3 logica
dt
2g
AsOCHMSwwoFwDwnsT
R(dtpt5T(t5ofs(owSrwoT(ptaatvnlt
(r
2
i(sswcG
2
oCfwg3
2
oCf3b
2
vlmCdosw
2
ssOtcsew
20 B.P. Imbimbo et al. / Pharmaco
etection kit (Cayman Chemical, Ann Arbor, MI, USA) andhe measurements were made with a microplate reader.
.3. Expression profiling of AICD- and NICD-responsiveenes
The inhibition of �-secretase mediated cleavage at the � site ofPP and/or at the S3 site of Notch was estimated by gene expres-
ion profiling of a subset of AICD- and NICD-responsive genes.ligonucleotides (60mer) matching 8 AICD (KTN1,FST, TIEG,ASP7, CALB1, KAI1, FMR1, FMR2) and 20 NICD (HES1,ES7, HEY2, HEY1, HEYL, ASCL1, NEUROG1, GAA, CD4,YOG, CTNNB1, NFKBIA, GSK3B, UTG1A9, DF, GCG, CCK,
CT, SST, CDKN1A) responsive genes [17–19] were designedith the OligoArray 1.0 program [20], chemically synthesizedith a 5′-amino linker modification and spotted in duplicatento charged nylon membranes (Eurogentec, Liege, Belgium).our “housekeeping genes” (GAPD, HPRT1, ACTB, B2M)ere also spotted in duplicate, along with sonicated genomicNA from human placenta (20 spots). The latter, togetherith 20 empty spots, served as technical controls for dataormalization and analysis. A list of the genes and of the corre-ponding oligonucleotides utilised is provided in Supplementaryable 1.
H4 cells were incubated for 12 h in the presence of either-Flurbiprofen (250 �M), CHF5022 (100 �M) or CHF5074
100 �M). DMSO (1%, v/v) and DAPT (1 �M) (N-[N-(3,5-ifluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester), araditional �-secretase inhibitor, were used as negative andositive controls, respectively. Total RNA was extracted withhe Trizol reagent (Invitrogen), reverse-transcribed (60 min at0 ◦C) and radioactively labelled as previously described [21].he resulting cDNAs were purified on Bio-Spin 6 columns
Bio-Rad Laboratories, Segrate, Italy) and quantified by scin-illation counting. Filters were prehybridised (2 h at 42 ◦C) with.0 �g of denatured Cot-1 DNA, 5.0 �g of poly-dA and 4 mlf hybridisation solution (5× SSC, 0.5% SDS, 5× Denhardt’s),ollowed by the addition of the radiolabelled cDNAs and sub-equent hybridisation for 18 h at 42 ◦C. After washing at 55 ◦Ctwo washes with 2× SSC–0.05% SDS and 1× SSC–0.1% SDS;ne wash with 0.1× SSC–0.1% SDS; 20 min each), membranesere aligned on a phosphorimaging screen (Super ResolutionR, Perkin-Elmer, Branchburg, NJ) and exposed for 24–72 h atoom temperature. Target intensities, measured and visualizedith a Cyclone phosphorimager (Perkin-Elmer) at a resolutionf 42 �m per pixel, were digitized with the OptiQuant software.hree technical replicates were conducted for each treatment
“T”) and for the DMSO control (“C”) and three images of com-arable overall intensity for each condition, as determined withhe Quantity One program (Bio-Rad), were subjected to furthernalysis using ImaGene 5.0 (Biodiscovery, Marina Del Rey, CA)s described previously [20]. Signals from spots consistentlyagged as “flag 0” in the three replicates as well as within indi-
idual duplicates from the same membrane were averaged, andormalized signal intensity ratios (IT/IC and IC/IT) were calcu-ated. Genes with an IT/IC or an IC/IT value greater than or equalo 1.5 were judged as upregulated or downregulated in treatedtoi−
l Research 55 (2007) 318–328
“T”; NSAIDs plus DAPT) versus control (“C”; DMSO) cells,espectively.
.4. Studies in Tg2576 transgenic mice
Young male and female transgenic mice (Tg2576) express-ng the human APP gene with the Swedish double mutationK670N/M671L) under the transcriptional control of the ham-ter prion protein promoter [22] were used for the in vivotudies. Male animals were housed singly in individual cageshile female animals were placed in groups of 3–5 animals per
age. The experiments were performed in accordance with EECuidelines (86/609/ECC) for the use of laboratory animals.
.4.1. Study 1Twenty-one male mice of 4–5 months of age were given by
ral gavage vehicle (Kool-Aid 7.5 ml kg−1) or a suspension ofHF5022 (100 or 300 mg kg−1 day−1 in Kool-Aid) once daily
or 5 days. This vehicle was selected to replicate that reportedith flurbiprofen in similar studies [1,3]. On Day 5, mice wereiven a final dose of 100 or 300 mg kg−1 or vehicle and sacrificedh later, as described below.
.4.2. Study 2Seventeen female mice of 5–7 months of age were given by
ral gavage vehicle (Kool-Aid 7.5 ml kg−1) or a suspension ofHF5074 (100 or 300 mg kg−1 day−1 in Kool-Aid) once daily
or 4 days. On Day 4, mice were given a final dose of 100 or00 mg kg−1 or vehicle and sacrificed 3 h later, as describedelow.
.4.3. Study 3Thirty-three male and female mice of 4–5 months were given
ehicle or CHF5022 or R-flurbiprofen-supplemented chow adibitum for 4 weeks. There were 11 animals in each treat-
ent group. R-Flurbiprofen (Sigma, St. Louis, MO, USA) andHF5022 were formulated into standard, colour-coded, rodentiet by Charles River (Calco, Italy) at a final drug concentrationf 375 ppm. The concentration of the drugs in the diet was theame as that used for ibuprofen in previous studies [4,5,7]. Bodyeight and food consumption were monitored every 3–4 days.
.5. Plasma and brain Aβ measurements
Twenty-four hours before starting treatment, one bloodample was collected by means of retro-bulbar puncture for mea-urement of baseline plasma, A�40 and A�42 concentrations.n the last day of treatment, mice were sacrificed by decapita-
ion. Blood samples were collected in EDTA-coated tubes andentrifuged at 800 rpm for 20 min to separate plasma. Plasmaamples were divided into two aliquots of approximately 100 �lach and stored at −80 ◦C until A� and drug assay. The brainsere quickly removed and placed on an ice-cold plate. Cor-
ex and hippocampus were dissected and immediately frozenn dry ice and stored at −80 ◦C for A� assay. The remain-ng brain was immediately frozen on dry ice and stored at
80 ◦C for drug level measurements. Plasma was diluted 1:4

logica
ft(cw(MiwSptrrdrtut
2
pdaof1twotcpnata2ta4
2
Ce
wC
faOb
ipcAbAdrtCu
3
3
(i(C
siv2Abi(o(
acA1A(dNICD-responsive genes (Table 2).
CHF5022 was less potent than CHF5074 in inhibiting A�42secretion (IC50 = 92 �M, Fig. 2) and retained a modest activ-ity on A�40 at non-cytotoxic concentrations (−22.2 ± 4.8% at
Table 1Effects of R-flurbiprofen, CHF5022 and CHF5074 on human recombinant COX-1 and COX-2 activity from Sf-9 insect cells
Compound COX-1 (% of control) COX2 (% of control)
100 �Ma 300 �Ma 300 �Ma
R-Flurbiprofen 50.0 ± 5.0 12.0 ± 2.6 54.0 ± 5.5CHF5022 99.3 ± 0.3 97.0 ± 10.8 129.4 ± 9.1
B.P. Imbimbo et al. / Pharmaco
or A�42 and 1:20 for A�40. For measurement of A�, brainissue samples were homogenized in 70% formic acid at 1:10w/v). Homogenates were agitated at 4◦ C for 3 h and thenentrifuged at 15,000 × g for 25 min at 4 ◦C. The supernatantsere collected and neutralized with 1 M Tris, pH 11 at 1:20
w/v) dilution with 3× protease inhibitor mixture (Boehringerannheim, Mannheim, Germany). Levels of A�40 and A�42
n plasma and in brain homogenate supernatants were measuredith commercial ELISA kits (The Genetics Company, Zurich,witzerland). The micro-titre plates were coated with capturingurified monoclonal antibodies specifically recognizing the C-erminus of human A�40 (clone G2-10, reactive to aminoacidesidues 31–40, isotype IgG2b, kappa) or A�42 (clone G2-13,eactive to aminoacid residues 33–42, isotype IgG1, kappa). Asetection antibody, a monoclonal biotin conjugated antibodyecognizing the N-terminus of human A� (clone W0-2, reac-ive to aminoacid residues 4–10, isotype IgG2a, kappa) wassed. The assay was linear in the range 25–500 pg ml−1 andhe detection limit was 25 pg ml−1.
.6. Plasma and brain drug measurements
CHF5022 and CHF5074 levels in plasma and in brain sam-les were measured by liquid chromatography as previouslyescribed [12]. Briefly, samples were prepared by adding 300 �lcetonitrile and 40 �l phosphoric acid 40% to 100 �l plasmar brain homogenate and placing the mixture in a vortexor 5 s. Plasma and brain samples were then centrifuged at4,000 rpm for 5 min and the supernatants (15 and 50 �l, respec-ively) were injected into the HPLC system. Equipment systemsith fluorescence (Waters 474, Waters, Guyancourt, France)r mass spectrometry (API 2000, Applied Biosystems, Fos-er City, CA, USA) detectors were used. The chromatographiconditions were adapted to each compound to obtain goodeak separation and detection sensitivity. A mixture of ammo-ium formate (20 mM) buffer–acetonitrile–methanol was useds mobile phase for the fluorescence detector. For CHF5022,he assay was linear in the range 20–4000 ng g−1 in the brainnd 5–1000 ng ml−1 in plasma with limits of quantitation of0 ng g−1 in the brain and 5 ng ml−1 in plasma. For CHF5074,he assay was linear between 400 and 20,000 ng g−1 in the brainnd 100–8500 ng ml−1 in plasma with limits of quantitation of00 ng g−1 in the brain and 100 ng ml−1 in plasma.
.7. Statistical analysis
The effects of test compounds on in vitro A� secretion andOX activity were expressed as percent of controls. IC50s werestimated by logistic regression analysis.
Expression profilings of NICD- and AICD-responsive genesere analysed with the Bayesian t-test implemented in theyber-T software (http://visitor.ics.uci.edu/genex/cybert/).
In the in vivo studies, two-way analysis of variance (ANOVA)
or repeated measures was used to detect significant treatmentnd time differences in plasma A�40 and A�42 concentrations.ne-way ANOVA was used to test treatment differences inrain A� levels. After a significant factor effect by ANOVA,C
Ep
l Research 55 (2007) 318–328 321
ndividual group differences were analysed using Holm-Sidak’srocedure for multiple comparisons versus control group (vehi-le). In Study 3, using male and female animals, a two-wayNOVA was used to test treatment and gender differences inrain A� levels. When appropriate, plasma and brain A�40 and�42 levels were transformed (log or square root) to normalizeata and to obtain homoscedasticity. Exact probabilities (P) wereeported for multiple comparisons. Calculations were done withhe statistical software SigmaStatTM 2.0 for WindowsTM (SPSS,hicago, IL, USA). Data are presented either as individual val-es or as mean ± S.E.M. (standard error of mean) in the text.
. Results
.1. In vitro studies
In H4 cells, cytotoxicity of the tested compounds was modest<15%) at concentrations up to 100 �M. Significant toxic-ty (30–40%) was observed at 1000 �M with R-flurbiprofen40.3 ± 3.0%) and at 300 �M with CHF5074 (29.4 ± 5.9%) andHF5022 (37.7 ± 1.3%).
R-Flurbiprofen concentration-dependently inhibited A�42ecretion with an IC50 of 268 �M (Fig. 2). A�40 was not inhib-ted at concentrations up to 300 �M (103.5 ± 1.7% of the controlalues). At 500–1000 �M, A�40 secretion was inhibited by0–40%, but these concentrations were also cytotoxic (Fig. 2).t 100 and 300 �M, R-flurbiprofen inhibited COX-1 activityy 50 ± 5% and 88 ± 3%, respectively (Table 1). At 300 �M,t also displayed a significant inhibitory activity on COX-2−46 ± 6%). At 250 �M, R-flurbiprofen did not influence anyf the tested AICD- and NICD-responsive genes while DAPT1 �M) downregulated the expression of five genes (Table 2).
CHF5074 showed a clear concentration-dependent inhibitoryctivity on A�42 secretion (IC50 = 40 �M, Fig. 2). At non-ytotoxic concentrations, no inhibition of the production of�40 was observed (100.4 ± 3.8% of the control value at00 �M, Fig. 2). COX-1 activity was not inhibited at 100 �M.t 300 �M, an inhibitory effect was detected on COX-1
−40 ± 13%) but not on COX-2 (Table 1). At 100 �M, CHF5074id not alter the expression levels of any of the tested AICD- or
HF5074 99.5 ± 2.5 59.9 ± 12.5 110.7 ± 9.6
nzyme activity is expressed as percent of control values (mean ± S.E.M., n = 3er experiment).a Compound concentration.
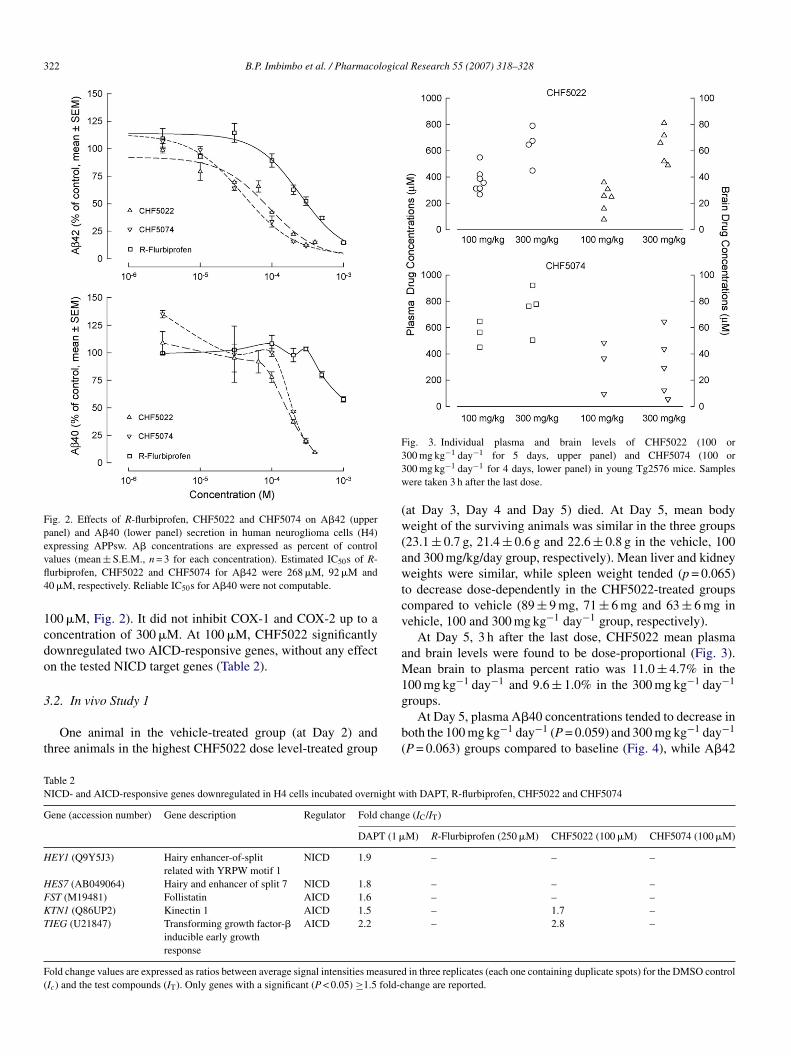
322 B.P. Imbimbo et al. / Pharmacological Research 55 (2007) 318–328
Fig. 2. Effects of R-flurbiprofen, CHF5022 and CHF5074 on A�42 (upperpanel) and A�40 (lower panel) secretion in human neuroglioma cells (H4)expressing APPsw. A� concentrations are expressed as percent of controlvfl4
1cdo
3
t
F33w
(w(awtcv
aM1
TN
G
H
HFKT
F(
alues (mean ± S.E.M., n = 3 for each concentration). Estimated IC50s of R-urbiprofen, CHF5022 and CHF5074 for A�42 were 268 �M, 92 �M and0 �M, respectively. Reliable IC50s for A�40 were not computable.
00 �M, Fig. 2). It did not inhibit COX-1 and COX-2 up to aoncentration of 300 �M. At 100 �M, CHF5022 significantlyownregulated two AICD-responsive genes, without any effectn the tested NICD target genes (Table 2).
.2. In vivo Study 1
One animal in the vehicle-treated group (at Day 2) andhree animals in the highest CHF5022 dose level-treated group
g
b(
able 2ICD- and AICD-responsive genes downregulated in H4 cells incubated overnight w
ene (accession number) Gene description Regulator Fold chang
DAPT (1 �
EY1 (Q9Y5J3) Hairy enhancer-of-splitrelated with YRPW motif 1
NICD 1.9
ES7 (AB049064) Hairy and enhancer of split 7 NICD 1.8ST (M19481) Follistatin AICD 1.6TN1 (Q86UP2) Kinectin 1 AICD 1.5IEG (U21847) Transforming growth factor-�
inducible early growthresponse
AICD 2.2
old change values are expressed as ratios between average signal intensities measuredIc) and the test compounds (IT). Only genes with a significant (P < 0.05) ≥1.5 fold-c
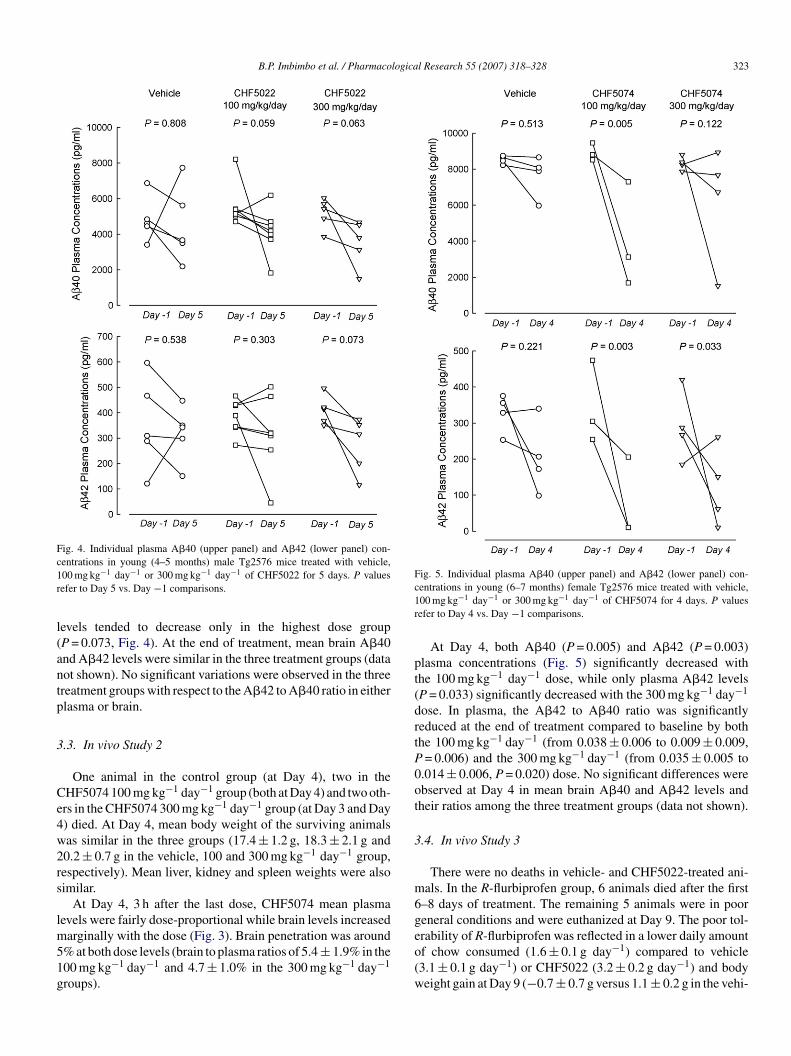
ig. 3. Individual plasma and brain levels of CHF5022 (100 or00 mg kg−1 day−1 for 5 days, upper panel) and CHF5074 (100 or00 mg kg−1 day−1 for 4 days, lower panel) in young Tg2576 mice. Samplesere taken 3 h after the last dose.
at Day 3, Day 4 and Day 5) died. At Day 5, mean bodyeight of the surviving animals was similar in the three groups
23.1 ± 0.7 g, 21.4 ± 0.6 g and 22.6 ± 0.8 g in the vehicle, 100nd 300 mg/kg/day group, respectively). Mean liver and kidneyeights were similar, while spleen weight tended (p = 0.065)
o decrease dose-dependently in the CHF5022-treated groupsompared to vehicle (89 ± 9 mg, 71 ± 6 mg and 63 ± 6 mg inehicle, 100 and 300 mg kg−1 day−1 group, respectively).
At Day 5, 3 h after the last dose, CHF5022 mean plasmand brain levels were found to be dose-proportional (Fig. 3).ean brain to plasma percent ratio was 11.0 ± 4.7% in the
00 mg kg−1 day−1 and 9.6 ± 1.0% in the 300 mg kg−1 day−1
roups.At Day 5, plasma A�40 concentrations tended to decrease in
oth the 100 mg kg−1 day−1 (P = 0.059) and 300 mg kg−1 day−1
P = 0.063) groups compared to baseline (Fig. 4), while A�42
ith DAPT, R-flurbiprofen, CHF5022 and CHF5074
e (IC/IT)
M) R-Flurbiprofen (250 �M) CHF5022 (100 �M) CHF5074 (100 �M)
– – –
– – –– – –– 1.7 –– 2.8 –
in three replicates (each one containing duplicate spots) for the DMSO controlhange are reported.
B.P. Imbimbo et al. / Pharmacological Research 55 (2007) 318–328 323
Fig. 4. Individual plasma A�40 (upper panel) and A�42 (lower panel) con-c1r
l(antp
3
Ce4w2rs
lm51g
Fig. 5. Individual plasma A�40 (upper panel) and A�42 (lower panel) con-c1r
pt(drtP0ot
3
m6g
entrations in young (4–5 months) male Tg2576 mice treated with vehicle,00 mg kg−1 day−1 or 300 mg kg−1 day−1 of CHF5022 for 5 days. P valuesefer to Day 5 vs. Day −1 comparisons.
evels tended to decrease only in the highest dose groupP = 0.073, Fig. 4). At the end of treatment, mean brain A�40nd A�42 levels were similar in the three treatment groups (dataot shown). No significant variations were observed in the threereatment groups with respect to the A�42 to A�40 ratio in eitherlasma or brain.
.3. In vivo Study 2
One animal in the control group (at Day 4), two in theHF5074 100 mg kg−1 day−1 group (both at Day 4) and two oth-rs in the CHF5074 300 mg kg−1 day−1 group (at Day 3 and Day) died. At Day 4, mean body weight of the surviving animalsas similar in the three groups (17.4 ± 1.2 g, 18.3 ± 2.1 g and0.2 ± 0.7 g in the vehicle, 100 and 300 mg kg−1 day−1 group,espectively). Mean liver, kidney and spleen weights were alsoimilar.
At Day 4, 3 h after the last dose, CHF5074 mean plasmaevels were fairly dose-proportional while brain levels increased
arginally with the dose (Fig. 3). Brain penetration was around% at both dose levels (brain to plasma ratios of 5.4 ± 1.9% in the00 mg kg−1 day−1 and 4.7 ± 1.0% in the 300 mg kg−1 day−1
roups).
eo(w
entrations in young (6–7 months) female Tg2576 mice treated with vehicle,00 mg kg−1 day−1 or 300 mg kg−1 day−1 of CHF5074 for 4 days. P valuesefer to Day 4 vs. Day −1 comparisons.
At Day 4, both A�40 (P = 0.005) and A�42 (P = 0.003)lasma concentrations (Fig. 5) significantly decreased withhe 100 mg kg−1 day−1 dose, while only plasma A�42 levelsP = 0.033) significantly decreased with the 300 mg kg−1 day−1
ose. In plasma, the A�42 to A�40 ratio was significantlyeduced at the end of treatment compared to baseline by bothhe 100 mg kg−1 day−1 (from 0.038 ± 0.006 to 0.009 ± 0.009,= 0.006) and the 300 mg kg−1 day−1 (from 0.035 ± 0.005 to
.014 ± 0.006, P = 0.020) dose. No significant differences werebserved at Day 4 in mean brain A�40 and A�42 levels andheir ratios among the three treatment groups (data not shown).
.4. In vivo Study 3
There were no deaths in vehicle- and CHF5022-treated ani-als. In the R-flurbiprofen group, 6 animals died after the first
–8 days of treatment. The remaining 5 animals were in pooreneral conditions and were euthanized at Day 9. The poor tol-
rability of R-flurbiprofen was reflected in a lower daily amountf chow consumed (1.6 ± 0.1 g day−1) compared to vehicle3.1 ± 0.1 g day−1) or CHF5022 (3.2 ± 0.2 g day−1) and bodyeight gain at Day 9 (−0.7 ± 0.7 g versus 1.1 ± 0.2 g in the vehi-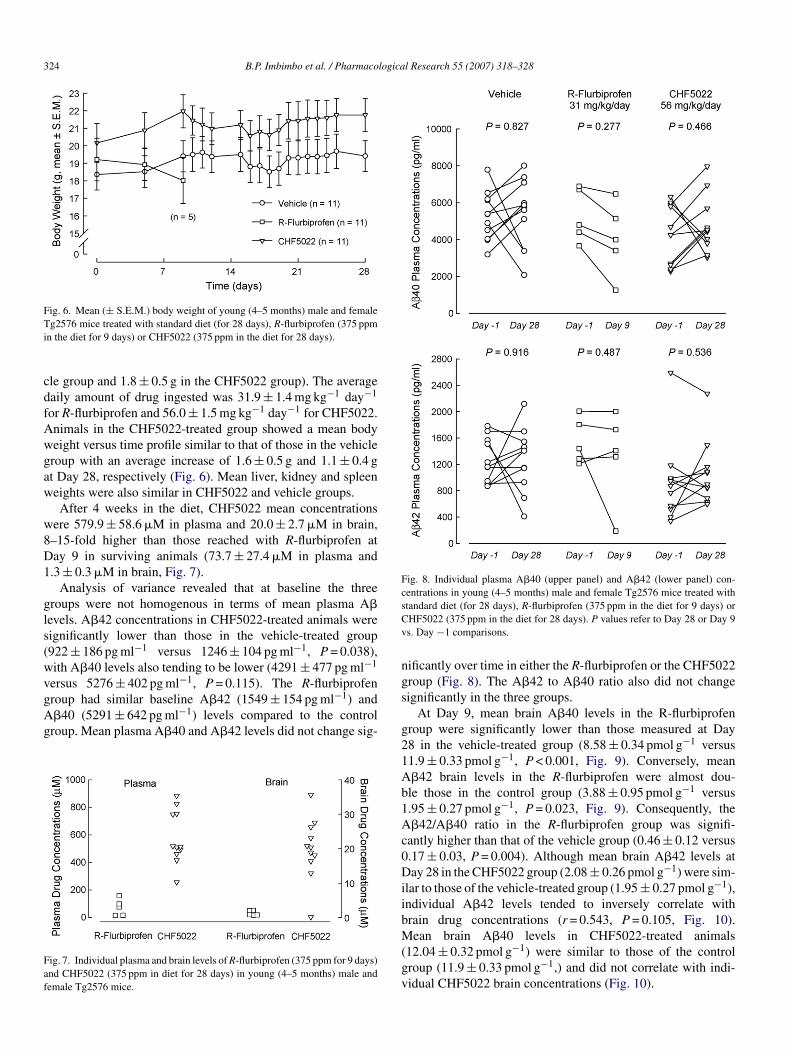
324 B.P. Imbimbo et al. / Pharmacological Research 55 (2007) 318–328
Fig. 6. Mean (± S.E.M.) body weight of young (4–5 months) male and femaleTi
cdfAwgaw
w8D1
gls(wvgAg
Faf
Fig. 8. Individual plasma A�40 (upper panel) and A�42 (lower panel) con-centrations in young (4–5 months) male and female Tg2576 mice treated withsCv
ng
g2576 mice treated with standard diet (for 28 days), R-flurbiprofen (375 ppmn the diet for 9 days) or CHF5022 (375 ppm in the diet for 28 days).
le group and 1.8 ± 0.5 g in the CHF5022 group). The averageaily amount of drug ingested was 31.9 ± 1.4 mg kg−1 day−1
or R-flurbiprofen and 56.0 ± 1.5 mg kg−1 day−1 for CHF5022.nimals in the CHF5022-treated group showed a mean bodyeight versus time profile similar to that of those in the vehicleroup with an average increase of 1.6 ± 0.5 g and 1.1 ± 0.4 gt Day 28, respectively (Fig. 6). Mean liver, kidney and spleeneights were also similar in CHF5022 and vehicle groups.After 4 weeks in the diet, CHF5022 mean concentrations
ere 579.9 ± 58.6 �M in plasma and 20.0 ± 2.7 �M in brain,–15-fold higher than those reached with R-flurbiprofen atay 9 in surviving animals (73.7 ± 27.4 �M in plasma and.3 ± 0.3 �M in brain, Fig. 7).
Analysis of variance revealed that at baseline the threeroups were not homogenous in terms of mean plasma A�evels. A�42 concentrations in CHF5022-treated animals wereignificantly lower than those in the vehicle-treated group922 ± 186 pg ml−1 versus 1246 ± 104 pg ml−1, P = 0.038),ith A�40 levels also tending to be lower (4291 ± 477 pg ml−1
ersus 5276 ± 402 pg ml−1, P = 0.115). The R-flurbiprofen
roup had similar baseline A�42 (1549 ± 154 pg ml−1) and�40 (5291 ± 642 pg ml−1) levels compared to the controlroup. Mean plasma A�40 and A�42 levels did not change sig-ig. 7. Individual plasma and brain levels of R-flurbiprofen (375 ppm for 9 days)nd CHF5022 (375 ppm in diet for 28 days) in young (4–5 months) male andemale Tg2576 mice.
s
g21Ab1Ac0DiibM(gv
tandard diet (for 28 days), R-flurbiprofen (375 ppm in the diet for 9 days) orHF5022 (375 ppm in the diet for 28 days). P values refer to Day 28 or Day 9s. Day −1 comparisons.
ificantly over time in either the R-flurbiprofen or the CHF5022roup (Fig. 8). The A�42 to A�40 ratio also did not changeignificantly in the three groups.
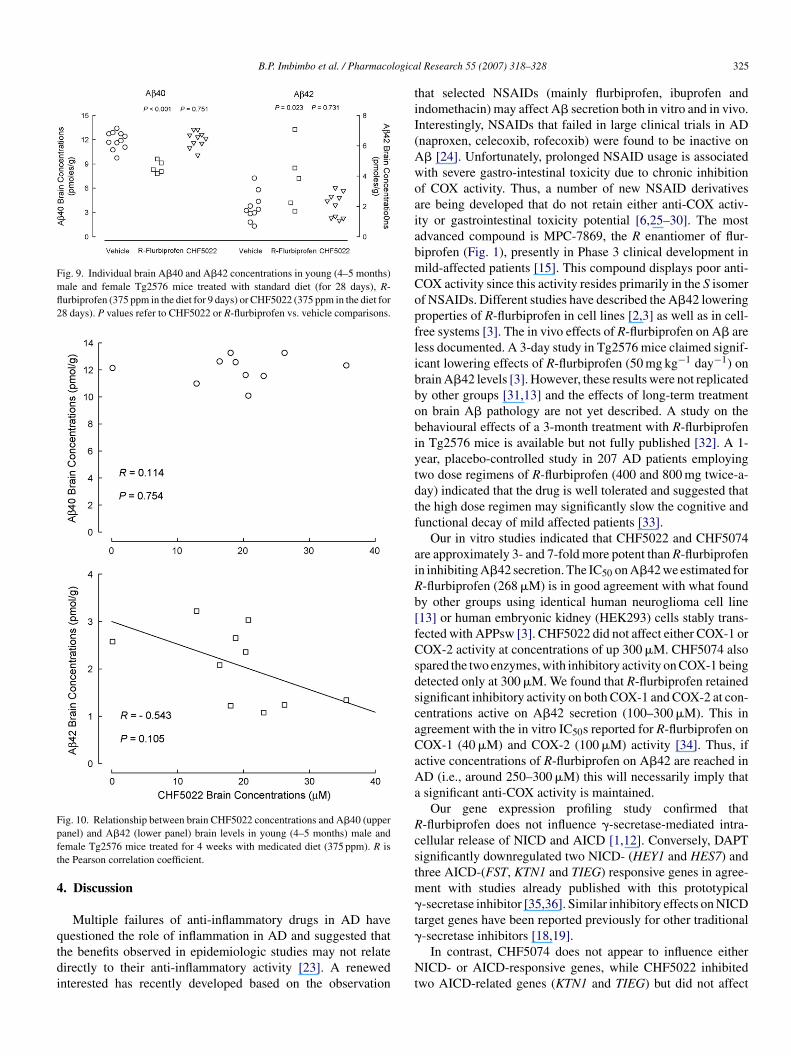
At Day 9, mean brain A�40 levels in the R-flurbiprofenroup were significantly lower than those measured at Day8 in the vehicle-treated group (8.58 ± 0.34 pmol g−1 versus1.9 ± 0.33 pmol g−1, P < 0.001, Fig. 9). Conversely, mean�42 brain levels in the R-flurbiprofen were almost dou-le those in the control group (3.88 ± 0.95 pmol g−1 versus.95 ± 0.27 pmol g−1, P = 0.023, Fig. 9). Consequently, the�42/A�40 ratio in the R-flurbiprofen group was signifi-
antly higher than that of the vehicle group (0.46 ± 0.12 versus.17 ± 0.03, P = 0.004). Although mean brain A�42 levels atay 28 in the CHF5022 group (2.08 ± 0.26 pmol g−1) were sim-
lar to those of the vehicle-treated group (1.95 ± 0.27 pmol g−1),ndividual A�42 levels tended to inversely correlate withrain drug concentrations (r = 0.543, P = 0.105, Fig. 10).
ean brain A�40 levels in CHF5022-treated animals12.04 ± 0.32 pmol g−1) were similar to those of the controlroup (11.9 ± 0.33 pmol g−1,) and did not correlate with indi-idual CHF5022 brain concentrations (Fig. 10).
B.P. Imbimbo et al. / Pharmacologica
Fig. 9. Individual brain A�40 and A�42 concentrations in young (4–5 months)male and female Tg2576 mice treated with standard diet (for 28 days), R-flurbiprofen (375 ppm in the diet for 9 days) or CHF5022 (375 ppm in the diet for28 days). P values refer to CHF5022 or R-flurbiprofen vs. vehicle comparisons.
Fpft
4
qtdi
tiI(AwoaiabmCopflibbobiytdtf
aiRb[fCsdscaCaAa
Rcstm�t
ig. 10. Relationship between brain CHF5022 concentrations and A�40 (upperanel) and A�42 (lower panel) brain levels in young (4–5 months) male andemale Tg2576 mice treated for 4 weeks with medicated diet (375 ppm). R ishe Pearson correlation coefficient.
. Discussion
Multiple failures of anti-inflammatory drugs in AD have
uestioned the role of inflammation in AD and suggested thathe benefits observed in epidemiologic studies may not relateirectly to their anti-inflammatory activity [23]. A renewednterested has recently developed based on the observation�
Nt
l Research 55 (2007) 318–328 325
hat selected NSAIDs (mainly flurbiprofen, ibuprofen andndomethacin) may affect A� secretion both in vitro and in vivo.nterestingly, NSAIDs that failed in large clinical trials in ADnaproxen, celecoxib, rofecoxib) were found to be inactive on� [24]. Unfortunately, prolonged NSAID usage is associatedith severe gastro-intestinal toxicity due to chronic inhibitionf COX activity. Thus, a number of new NSAID derivativesre being developed that do not retain either anti-COX activ-ty or gastrointestinal toxicity potential [6,25–30]. The mostdvanced compound is MPC-7869, the R enantiomer of flur-iprofen (Fig. 1), presently in Phase 3 clinical development inild-affected patients [15]. This compound displays poor anti-OX activity since this activity resides primarily in the S isomerf NSAIDs. Different studies have described the A�42 loweringroperties of R-flurbiprofen in cell lines [2,3] as well as in cell-ree systems [3]. The in vivo effects of R-flurbiprofen on A� areess documented. A 3-day study in Tg2576 mice claimed signif-cant lowering effects of R-flurbiprofen (50 mg kg−1 day−1) onrain A�42 levels [3]. However, these results were not replicatedy other groups [31,13] and the effects of long-term treatmentn brain A� pathology are not yet described. A study on theehavioural effects of a 3-month treatment with R-flurbiprofenn Tg2576 mice is available but not fully published [32]. A 1-ear, placebo-controlled study in 207 AD patients employingwo dose regimens of R-flurbiprofen (400 and 800 mg twice-a-ay) indicated that the drug is well tolerated and suggested thathe high dose regimen may significantly slow the cognitive andunctional decay of mild affected patients [33].
Our in vitro studies indicated that CHF5022 and CHF5074re approximately 3- and 7-fold more potent than R-flurbiprofenn inhibiting A�42 secretion. The IC50 on A�42 we estimated for-flurbiprofen (268 �M) is in good agreement with what foundy other groups using identical human neuroglioma cell line13] or human embryonic kidney (HEK293) cells stably trans-ected with APPsw [3]. CHF5022 did not affect either COX-1 orOX-2 activity at concentrations of up 300 �M. CHF5074 also
pared the two enzymes, with inhibitory activity on COX-1 beingetected only at 300 �M. We found that R-flurbiprofen retainedignificant inhibitory activity on both COX-1 and COX-2 at con-entrations active on A�42 secretion (100–300 �M). This ingreement with the in vitro IC50s reported for R-flurbiprofen onOX-1 (40 �M) and COX-2 (100 �M) activity [34]. Thus, ifctive concentrations of R-flurbiprofen on A�42 are reached inD (i.e., around 250–300 �M) this will necessarily imply thatsignificant anti-COX activity is maintained.
Our gene expression profiling study confirmed that-flurbiprofen does not influence �-secretase-mediated intra-ellular release of NICD and AICD [1,12]. Conversely, DAPTignificantly downregulated two NICD- (HEY1 and HES7) andhree AICD-(FST, KTN1 and TIEG) responsive genes in agree-ent with studies already published with this prototypical-secretase inhibitor [35,36]. Similar inhibitory effects on NICD
arget genes have been reported previously for other traditional
-secretase inhibitors [18,19].In contrast, CHF5074 does not appear to influence eitherICD- or AICD-responsive genes, while CHF5022 inhibited
wo AICD-related genes (KTN1 and TIEG) but did not affect

3 logica
tfiCth
Coti(a[ratiotbfC553tpaw(rap[mRp
watoAtwmoilf(aisesC
dibo(
Cbsstc
CitAop
cbi(loibbmAbArISasiavr
o(fliiepmps
26 B.P. Imbimbo et al. / Pharmaco
ranscriptional activities mediated by Notch processing. If con-rmed in vivo, the lack of effects on Notch nuclear signalling byHF5074 and CHF5022 may have important consequences in
erms of safety, since the interference with Notch is a potentiallyarmful effect of traditional �-secretase inhibitors [37,18].
Our studies indicated that after multiple oral administration,HF5022 is absorbed dose-dependently, with a fair penetrationf the brain (≈10%) of young Tg2575 mice. Oral administra-ion of CHF5074 appears to be less efficient than CHF5022 bothn terms of dose-proportionality and ability to reach the brain≈5%). Studies in rats have indicated a higher oral bioavail-bility for CHF5022 compared to CHF5074 (91% versus 50%)13]. The studies in non-transgenic rats have also indicated aather scarce central nervous system penetration of CHF5022nd CHF5074, the cerebrospinal fluid level concentrations ofhe two compounds being 1.7% and 1.3% of the correspond-ng plasma concentrations [13]. The higher brain penetrationbserved for CHF5022 and CHF5074 in Tg2576 mice comparedo non-transgenic rats may be due to the well known increasedlood–brain barrier permeability of organic molecules observedor this transgenic AD mice model [38,39]. After 4 weeks ofHF5022 administration with the diet, at an estimated dose of6 mg kg−1 day−1, mean plasma levels were higher than after a-day course of a 100 mg kg−1 day−1 dose (580 ± 59 �M versus72 ± 35 �M). This is likely due to the long plasma elimina-ion half-life (32 h in rats) [13] that leads to accumulation inlasma when the frequency of administration is high like indiet regimen. Brain CHF5022 levels after the diet regimenere lower than those after oral gavage of 100 mg kg−1 day−1
20 ± 3 �M versus 37 ± 14 �M) equalling only 4% of the cor-esponding plasma levels. This may be due to saturation of thective transport of the CHF5022 fraction unbound to plasmarotein across the blood-brain barrier as occurring for ibuprofen40]. Nevertheless, CHF5022 administration in the diet achieveduch higher central exposure (15-fold) compared to that of-flurbiprofen, confirming its better oral availability and brainenetration in non-transgenic rats [13].
CHF5022 was very well tolerated in young Tg2576 micehen administered in the diet at 375 ppm for 4 weeks (estimated
ssumed dose of 56 mg kg−1 day−1). No deaths occurred duringhe 4-week treatment period. Body weight and body weight gainver time was very similar to that of the vehicle-treated group.t week 4, mean liver, kidney and spleen weights were similar to
hose of the control group. R-Flurbiprofen was poorly tolerated,ith 5 out of 11 animals dying during the first week of treat-ent. The surviving animals were in poor condition with loss
f body weight. Although no formal histopathological exam-nations were performed, the deaths were attributed to gastriciability of this drug. Although the R enantiomer of flurbipro-en is a poor COX inhibitor, in mouse it may be significantly15–24%) converted to the pharmacologically active S-form [41]nd this may explain the gastric liability observed for this drugn the Tg2576 mice. On the other hand, CHF5022 does not show
ignificant anti-COX activity and this may explain its good tol-rability in vivo. The observation that in the short-term studiesome animals died in both the vehicle and the CHF5022 andHF5074 treated groups (100 and 300 mg kg−1 day−1) may beestb
l Research 55 (2007) 318–328
ue to the oral gavage procedure rather than to intrinsic toxic-ty of the drugs. In a 2-week formal toxicity study in mice, noehavioural, laboratory or histopathological toxic effects werebserved with CHF5022 at doses of 30 or 100 mg kg−1 day−1
Villetti, personal communication).Oral administration of doses of 100 or 300 mg kg−1 day−1 of
HF5022 or CHF5074 may lower A�40 and A�42 in plasmaut a dose of 56 mg kg−1 day−1 of CHF5022 in the diet failed tohow a significant effect on plasma A�. The latter observationhould be taken cautiously since baseline A� plasma levels inhe CHF5022 group were significantly lower than those in theontrol group.
Neither CHF5022 nor CHF5074 given by gavage orHF5022 given with the diet were able to significantly mod-
fy formic acid-extractable A� in the brain. R-Flurbiprofen inhe diet significantly lowered brain A�40 levels and increased�42 concentration, compared to vehicle. These contradictorybservations are unclear and could be spurious since group com-arisons were made at different time points (Day 9 and Day 28).
The type of solvent used to extract brain A� (formic acid)ould have influenced the outcome of the present study. It haseen shown that in Tg2576 mice ibuprofen has a marked lower-ng effect on brain A�42 extracted with sodium dodecyl sulphateSDS) but not extracted with formic acid [7]. A significantowering effect of indomethacin and of its derivative DP-155n brain SDS-extractable A�42 has been recently describedn young Tg2576 mice [27]. A lack of central effects of flur-iprofen in Tg2576 mice has been observed when guanidineuffer was used to extract brain A� [31,13]. Thus, NSAIDsay have differential effects on “soluble” (SDS-extractable�) and “insoluble” (formic acid- or guanidine-extractable)rain A� pools. Soluble A� could represent newly formed� from the APP metabolism while insoluble A� could rep-
esent the A� pools in equilibrium with fibrils or plaques.ndeed, further analyses of brain homogenates derived fromtudy 2, which showed a significant inhibition of plasma A�fter CHF5074 (100 mg kg−1 day−1), revealed lower, though notignificant mean A� levels following extraction with 2% SDSn drug-treated animals compared to controls (A�40 = 8.5 ± 0.4nd 9.2 ± 0.7 pmoles g−1 and A�42 = 0.19 ± 0.014 pmoles g−1
ersus 0.21 ± 0.02 pmoles g−1 after CHF5074 and vehicle,espectively).
Another factor potentially influencing the effect of NSAIDsn brain A� is the length of the treatment. Long-term treatments3–8 months) with ibuprofen [4,5,7] indomethacin [42,43] orurbiprofen derivatives [6,8] have shown a significant reduction
n brain A� pathology in transgenic mice. Short-term admin-stration (3–8 days) of NSAIDs has failed to show significantffect on brain A� levels [31,13,26]. It could be that, afterrolonged treatment, NSAIDs accumulate within the neuronalembrane where �-secretase is localized. Alternatively, it is
ossible that NSAIDs do not act centrally but rather progres-ively reduce peripheral A�42 levels, which results in enhanced
fflux of A�42 from the brain [24]. However, doubts still per-ist on the low potency of these A�42 lowering agents andheir low brain penetration. This could be due to insufficientrain concentrations reached by these compounds, which are
logica
nci3ct
ai2metbec
D
Vch
A
Iae
A
i
R
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
B.P. Imbimbo et al. / Pharmaco
ot compatible with their micromolar potency. The highest brainoncentrations of CHF5022 and CHF5074 reached in our stud-es were 64 and 37 �M, respectively (after a dose regimen of00 mg kg−1 day−1 for 4–5 days). These values approach theorresponding in vitro IC50s on A�42 (92 and 40 �M, respec-ively).
In conclusion, our studies indicated that in vitro CHF5022nd CHF5074 are 3- and 7-fold more potent than R-flurbiprofenn inhibiting A�42 secretion and did not affect COX-1 and COX-
activity at A� inhibiting concentrations. In young Tg2576ice, the two compounds appear to be orally absorbed and to
nter the central nervous system. CHF5022 is better toleratedhan R-flurbiprofen and displayed higher systemic exposure andrain penetration. Long-term studies in transgenic mice mod-ls of AD are warranted to evaluate the effects of these newompounds on brain A� pathology.
isclosure statement
Bruno P. Imbimbo, Valentina Cenacchi, Roberta Volta, Ginoilletti, Fabrizio Facchinetti, Benedetta Riccardi and Paola Puc-ini are employee at Chiesi Farmaceutici. They own no stock norold any options to purchase stock in the company.
cknowledgements
The study was sponsored by Chiesi Farmaceutici, Parma,taly. We thank Roberta Ruotolo (Department of Biochemistrynd Molecular Biology, University of Parma) for help with genexpression data analysis.
ppendix A. Supplementary data
Supplementary data associated with this article can be found,n the online version, at doi:10.1016/j.phrs.2006.12.010.
eferences
[1] Weggen S, Eriksen JL, Das P, Sagi S, Wang R, Pietrzik CU, et al. A subsetof NSAIDs lower amyloidogenic A�42 independently of cyclooxygenaseactivity. Nature 2001;414:212–6.
[2] Morihara T, Chu T, Ubeda O, Beech W, Cole GM. Selective inhi-bition of A�42 production by NSAID R-enantiomers. J Neurochem2002;83:1009–12.
[3] Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, et al.NSAIDs and enantiomers of flurbiprofen target �-secretase and lower A�42in vivo. J Clin Invest 2003;112:440–9.
[4] Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, et al. Ibuprofensuppresses plaque pathology and inflammation in a mouse model forAlzheimer’s disease. J Neurosci 2000;20:5709–14.
[5] Lim GP, Yang F, Chu T, Gahtan E, Ubeda O, Beech W, et al. Ibuprofeneffects on Alzheimer pathology and open field activity in APPsw transgenicmice. Neurobiol Aging 2001;22:983–91.
[6] Jantzen PT, Condor KE, Di Carlo G, Wenk GL, Wallace JL, Rojiani AM, etal. Microglial activation and �-amyloid deposit reduction caused by a nitric-
oxide-releasing non-steroidal anti-inflammatory drug in amyloid precursorprotein plus presenilin-1 transgenic mice. J Neurosci 2002;22:2246–54.[7] Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, et al. Anti-inflammatory drug therapy alters �-amyloid processing and deposition inan animal model of Alzheimer’s disease. J Neurosci 2003;23:7504–9.
[
l Research 55 (2007) 318–328 327
[8] van Groen T, Kadish I. Transgenic AD model mice, effects of potentialanti-AD treatments on inflammation and pathology. Brain Res Brain ResRev 2005;48:370–8.
[9] Takahashi Y, Hayashi I, Tominari Y, Rikimaru K, Morohashi Y, Kan T, et al.Sulindac sulfide is a noncompetitive �-secretase inhibitor that preferentiallyreduces A�42 generation. J Biol Chem 2003;278:18664–70.
10] Beher D, Clarke EE, Wrigley JD, Martin AC, Nadin A, Churcher I, et al.Selected non-steroidal anti-inflammatory drugs and their derivatives target�-secretase at a novel site. Evidence for an allosteric mechanism. J BiolChem 2004;279:43419–26.
11] Lleo A, Berezovska O, Herl L, Raju S, Deng A, Bacskai BJ, et al. Non-steroidal anti-inflammatory drugs lower A�42 and change presenilin 1conformation. Nat Med 2004;10:1065–6.
12] Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Golde TE, Koo EH. A�42-lowering nonsteroidal anti-inflammatory drugs preserve intramembranecleavage of the amyloid precursor protein (APP) and ErbB-4 recep-tor and signaling through the APP intracellular domain. J Biol Chem2003;278:30748–54.
13] Peretto I, Radaelli S, Parini C, Zandi M, Raveglia LF, Dondio G,et al. Synthesis and biological activity of flurbiprofen analogues asselective inhibitors of �-amyloid1–42 secretion. J Med Chem 2005;48:5705–20.
14] Raveglia L, Peretto I, Radaelli, S, Imbimbo BP, Rizzi A, Villetti G.1-Phenylalkanecarboxylic acid derivatives for the treatment of neurode-generative diseases, 2004, PCT WO 2004/074232 A1.
15] Wilcock G. Disease-modifying treatments for Alzheimer’s disease: a per-spective based on experience with R-flurbiprofen. Alzheimer Dementia2006;2:150–2.
16] Glaser K, Sung Mei-Li, O’Neill K, Belfast M, Hartman D, Carlson R, et al.Etodolac selectively inhibits human prostaglandin G/H 2 (PGHS-2) versushuman PGHS-1. Eur J Pharmacol 1995;281:107–11.
17] Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectorsof the Notch signaling pathway. J Cell Physiol 2003;194:237–55.
18] Milano J, McKay J, Dagenais C, Foster-Brown L, Pognan F, Gadient R, etal. Modulation of notch processing by �-secretase inhibitors causes intesti-nal goblet cell metaplasia and induction of genes known to specify gutsecretory lineage differentiation. Toxicol Sci 2004;82:341–58.
19] Searfoss GH, Jordan WH, Calligaro DO, Galbreath EJ, Schirtzinger LM,Berridge BR, et al. Adipsin, a biomarker of gastrointestinal toxicity medi-ated by a functional �-secretase inhibitor. J Biol Chem 2003;278:46107–16.
20] Rouillard JM, Herbert CJ, Zuker M. OligoArray: genome-scale oligonu-cleotide design for microarrays. Bioinformatics 2002;18:486–7.
21] Ruotolo R, Grassi F, Percudani R, Rivetti C, Martorana D, Maraini G,et al. Gene expression profiling in human age-related cataract. Mol Vis2003;9:538–48.
22] Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, etal. Correlative memory deficits, A� elevation, and amyloid plaques intransgenic mice. Science 1996;274:99–102.
23] Launer LJ. Nonsteroidal anti-inflammatory drugs and Alzheimer disease:what’s next? JAMA 2003;289:2865–7.
24] Imbimbo BP. The potential role of non-steroidal anti-inflammatorydrugs in treating Alzheimer’s disease. Expert Opin Investig Drugs2004;13:1469–81.
25] van Groen T, Kadish I. Transgenic AD model mice, effects of potentialanti-AD treatments on inflammation and pathology. Brain Res Brain ResRev 2005;48:370–8.
26] Stock N, Munoz B, Wrigley JD, Shearman MS, Beher D, Peachey J, et al.The geminal dimethyl analogue of flurbiprofen as a novel A�42 inhibitorand potential Alzheimer’s disease modifying agent. Bioorg Med Chem Lett2006;16:2219–23.
27] Dvir E, Friedman JE, Lee JY, Koh JY, Younis F, Raz S, et al. A novelphospholipid derivative of indomethacin, DP-155 [mixture of 1-steroyland 1-palmitoyl-2-{4-[1-(p-chlorobenzoyl)-5-methoxy-2-methyl-3-indol-
yl acetamido]butanoyl}-sn-glycero-3-phosophatidyl choline], shows supe-rior safety and similar efficacy in reducing brain amyloid beta in anAlzheimer’s disease model. J Pharmacol Exp Ther 2006;318:1248–56.28] Hobden A. Pharmaceutical composition and method for treating neurode-generative disorders, 2006, WO/2006/020850.

3 logica
[
[
[
[
[
[
[
[
[
[
[
[
[
[
28 B.P. Imbimbo et al. / Pharmaco
29] Hobden A. Pharmaceutical composition and method for treating neurode-generative disorders, 2006, WO/2006/020852.
30] Hobden A. Pharmaceutical composition and method for treating neurode-generative disorders, 2006, WO/2006/020853.
31] Lanz TA, Fici GJ, Merchant KM. Lack of specific amyloid-�1–42 sup-pression by nonsteroidal anti-inflammatory drugs in young, plaque-freeTg2576 mice and in guinea pig neuronal cultures. J Pharmacol Exp Ther2005;312:399–406.
32] Nicolle MM, Prescott S, Murphy MP, Kukar T, Eriksen JL, Koo EH, et al.Long-term but not short-term, R-flurbiprofen treatment ameliorates cogni-tive deficits in transgenic APP mice. In: 34th annual meeting of Society ofNeuroscience. 2004 [Program No. 674.6].
33] Wilcock G, Black S, Haworth J, Laughlin M, Hendrix S, Binger MH, etal. A placebo-controlled, double-blind trial of the selective A�42 lower-ing agent, Flurizan (MPC-7869, (R)-flurbiprofen) in patients with mild tomoderate Alzheimer’s diseases. In: Alzheimer’s Association InternationalConference on Prevention of Dementia. 2005 [Abstract O2-01-05].
34] Geisslinger G, Muth-Selbach U, Coste O, Vetter G, Schrodter A, SchaibleHG, et al. Inhibition of noxious stimulus-induced spinal prostaglandin E2release by flurbiprofen enantiomers: a microdialysis study. J Neurochem2000;74:2094–100.
35] Geling A, Steiner H, Willem M, Bally-Cuif L, Haass C. A gamma-secretaseinhibitor blocks Notch signaling in vivo and causes a severe neurogenicphenotype in zebrafish. EMBO Rep 2002;3:688–94.
36] Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, etal. Presenilin-dependent gamma-secretase processing of beta-amyloid pre-
[
l Research 55 (2007) 318–328
cursor protein at a site corresponding to the S3 cleavage of Notch. EMBORep 2001;2:835–41.
37] Hadland BK, Manley NR, Su D, Longmore GD, Moore CL, Wolfe MS, etal. �-Secretase inhibitors repress thymocyte development. Proc Natl AcadSci USA 2001;98:7487–91.
38] Ujiie M, Dickstein DL, Carlow DA, Jefferies WA. Blood-brain barrier per-meability precedes senile plaque formation in an Alzheimer disease model.Microcirculation 2003;10:463–70.
39] LaRue B, Hogg E, Sagare A, Jovanovic S, Maness L, Maurer C, etal. Method for measurement of the blood-brain barrier permeability inthe perfused mouse brain: application to amyloid-� peptide in wildtype and Alzheimer’s Tg2576 mice. J Neurosci Methods 2004;138:233–42.
40] Parepally JM, Mandula H, Smith QR. Brain uptake of nonsteroidal anti-inflammatory drugs: Ibuprofen, flurbiprofen, and indomethacin. Pharm Res2006;23:873–81.
41] Leipold DD, Kantoci D, Murray Jr ED, Quiggle DD, Wechter WJ. Bioin-version of R-flurbiprofen to S-flurbiprofen at various dose levels in rat,mouse, and monkey. Chirality 2004;16:379–87.
42] Quinn J, Montine T, Morrow J, Woodward WR, Kulhanek D, EckensteinF. Inflammation and cerebral amyloidosis are disconnected in an animal
model of Alzheimer’s disease. J Neuroimmunol 2003;137:32–41.43] Sung S, Yang H, Uryu K, Lee EB, Zhao L, Shineman D, et al. Mod-ulation of NF-�B activity by Indomethacin influences A� levels butnot APP metabolism in a model of Alzheimer disease. Am J Pathol2004;165:2197–206.
Copyright © 2022 FDOKUMEN




















