
CXCL10 blockade protects mice from cyclophosphamide-induced cystitis
Upload
independentCategory
view
1download
0
Thrombopoietin Protects Against In Vitro and In VivoCardiotoxicity Induced by Doxorubicin
Karen Li, PhD; Rita Yn Tz Sung, MD; Wei Zhe Huang, MD; Mo Yang, MD, PhD;Nga Hin Pong, MD, MPhil; Shuk Man Lee, MPhil; Wood Yee Chan, PhD; Hailu Zhao, MD;
Man Yin To, BSc; Tai Fai Fok, MD; Chi Kong Li, MD; Yuek Oi Wong, PhD; Pak Cheung Ng, MD
Background—Doxorubicin (DOX) is an important antineoplastic agent. However, the associated cardiotoxicity, possiblymediated by the production of reactive oxygen species, has remained a significant and dose-limiting clinical problem.Our hypothesis is that the hematopoietic/megakaryocytopoietic growth factor thrombopoietin (TPO) protects againstDOX-induced cardiotoxicity and might involve antiapoptotic mechanism exerted on cardiomyocytes.
Methods and Results—In vitro investigations on H9C2 cell line and spontaneously beating cells of primary, neonatal ratventricle, as well as an in vivo study in a mouse model of DOX-induced acute cardiomyopathy, were performed. Ourresults showed that pretreatment with TPO significantly increased viability of DOX-injured H9C2 cells and beating ratesof neonatal myocytes, with effects similar to those of dexrazoxane, a clinically approved cardiac protective agent. TPOameliorated DOX-induced apoptosis of H9C2 cells as demonstrated by assays of annexin V, active caspase-3, andmitochondrial membrane potential. In the mouse model, administration of TPO (12.5 �g/kg IP for 3 alternate days)significantly reduced DOX-induced (20 mg/kg) cardiotoxicity, including low blood cell count, cardiomyocyte lesions(apoptosis, vacuolization, and myofibrillar loss), and animal mortality. Using Doppler echocardiography, we observedincreased heart rate, fractional shortening, and cardiac output in animals pretreated with TPO compared with thosereceiving DOX alone.
Conclusions—These data have provided the first evidence that TPO is a protective agent against DOX-induced cardiacinjury. We propose to further explore an integrated program, incorporating TPO with other protocols, for treatment ofDOX-induced cardiotoxicity and other forms of cardiomyopathy. (Circulation. 2006;113:2211-2220.)
Key Words: apoptosis � cardiomyopathy � doxorubicin � echocardiography � thrombopoietin
Doxorubicin (DOX), an anthracycline drug, is one of themost active antineoplastic agents developed to date for
the treatment of solid tumors and hematologic malignancies.However, its clinical use is limited by acute and chroniccardiotoxicities, which are dose related, cumulative, andessentially irreversible. It has been estimated that the normalheart can compensate for a lifetime dose of 400 to 550 mg/m2
of DOX.1,2 Beyond this level, severe injury, including car-diomyopathy and congestive heart failure, might occur. Heart
Clinical Perspective p 2220
failure in DOX-treated patients may take place many yearsafter treatment cessation, as reported by Steinherz et al3
(1991). Analyzing a group of 630 patients with breastcarcinoma and small-cell lung carcinoma, Swain et al4
reported that an estimated 26% of patients would experience
DOX-related congestive heart failure at a cumulative dose of550 mg/m2.
The pathogenesis of DOX-induced cardiotoxicity is notentirely clear. It has been suggested that the anticancer effectsand cardiotoxicity of DOX do not follow identical mecha-nisms.5,6 Available laboratory evidence shows that DOXinduces generation of reactive oxygen species. The increasein oxidative stress and depletion of endogenous antioxidantstrigger the intrinsic mitochondria-dependent apoptotic path-way in cardiomyocytes.5,7 Other outcomes include distur-bance of myocardial adrenergic function, intracellular cal-cium overload, and release of cardiotoxic cytokines.Numerous signal molecules, such as cytochrome c, superox-ide dismutase, creatine kinase, nitric oxide, Bcl-2, Bax, p53,and Fas, have been indicated in the reactive oxygen species–induced apoptotic pathways of cardiomyocytes.6–9
Received May 6, 2005; revision received October 3, 2005; second revision received January 26, 2006; third revision received March 8, 2006; acceptedMarch 9, 2006.
From the Departments of Pediatrics (K.L., R.Y.T.S., M.Y., N.H.P., S.M.L., M.Y.T., T.F.F., C.K.L., Y.O.W., P.C.N.), Anatomy (W.Y.C.), and Medicineand Therapeutics (H.Z.), The Chinese University of Hong Kong, Shatin, NT, Hong Kong; and Department of Cardiac Pulmonary Surgery, ShantouUniversity, Shantou, China (W.Z.H.).
The online-only Data Supplement can be found at http://circ.ahajournals.org/cgi/content/full/CIRCULATIONAHA.105.560250/DC1.Correspondence to Rita Yn Tz Sung, MD, Department of Pediatrics, The Chinese University of Hong Kong, 6th Floor, Clinical Sciences Block, The
Prince of Wales Hospital, Shatin, NT, Hong Kong. E-mail [email protected]© 2006 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.105.560250
2211 by guest on March 3, 2015http://circ.ahajournals.org/Downloaded from

Thrombopoietin (TPO) is an established cytokine forpromoting early hematopoietic progenitor cells, themegakaryocytic/platelet lineage, angiogenesis, and antiapo-ptosis.10,11 We hypothesize that TPO may protect againstcardiotoxicity induced by DOX. This is based on the rationalethat TPO possesses antiapoptotic functions mediated by theAkt prosurvival axis in hematopoietic stem cells andmegakaryocytes.11,12 The Akt pathway has been known toexert survival protections in cardiomyocytes.13 In addition,TPO and erythropoietin (EPO) have strong sequence homol-ogy, with 20% identity and 25% similarity in their receptor-binding regions.14 It was shown that TPO may exert itsmitogenic effects by binding to EPO receptors.15 EPO isreported to protect and promote cardiomyocytes.16,17 To testthe hypotheses, we performed 2 in vitro investigations as wellas 1 in vivo study in a mouse model of DOX-inducedcardiomyopathy.
MethodsDetails of methodology are described in the online-only DataSupplement.
In Vitro Model of Myocytes
Rat H9C2 Myoblast Cell LineThis embryonic line (American Type Tissue Collection, Manassas,Va; catalog No. CRL-1446) was maintained in Iscove’s modifiedDulbecco’s medium supplemented with 10% fetal calf serum andcultured in 5% CO2 at 37°C. All media and culture reagents wereproducts of Gibco (Grand Island, NY) unless specified otherwise.H9C2 cells were seeded at 2�104 cells per well (24 well plates) for24 hours, with or without preincubation with TPO (5, 10, 50, or 100ng/mL, overnight; Peprotech, Rocky Hill, NJ) or dexrazoxane[(�)-1,2-bis(dioxopiperazinyl-1-yl) propane] (50 �g/mL, 30 min-utes; Chiron, Amsterdam, the Netherlands). DOX (5 �g/mL; EbewePharma Ges, Austria) was added to the system for another 24 hours.Cell viability was measured by the MTT assay by adding 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (250 �g/mL;Sigma, St Louis, Mo) for 4 hours, and the optical density was readat 570 nm. Apoptotic cell death was analyzed by assays of annexinV, active caspase-3, and mitochondrial membrane potential (��m)with the use of flow cytometry.
Primary Neonatal Rat CardiomyocytesSpontaneously beating cells from heart ventricles of neonatal ratwere cultured18 and subjected to treatment with DOX (1 �mol/L or0.58 �g/mL), with and without pretreatment (1 hour before) withTPO (50 or 100 ng/mL) or dexrazoxane (5.8 �g/mL). Changes inbeating rates were captured by video camera.
In Vivo Mouse Model of DOX-Induced CardiotoxicityMale Balb/c mice (Laboratory Animal Services Centre, The ChineseUniversity of Hong Kong, Hong Kong) at 9 to 10 weeks of age wererandomly divided into 4 groups. The control group was given 3 dosesof normal saline intraperitoneally (IP) on alternate days. At day 0, theDOX group received a single dose of DOX dissolved in 0.9% NaClat 20 mg/kg. The DOX�TPO group was given a single dose of DOX(20 mg/kg IP) and 3 doses of TPO (12.5 �g/kg, dissolved in normalsaline) 1 day before (day �1) and 1 and 3 days after DOX injection.The TPO group received 3 doses of TPO but no DOX treatment.Animal viability was recorded daily for 5 days. In an independentexperiment, the viability was recorded for 8 days in 4 groups ofanimals receiving the same treatments but not subjected to anysample collection or daily manipulation. All procedures were ap-proved by the Animal Research Ethics Committee, The ChineseUniversity of Hong Kong.
EchocardiographyTransthoracic echocardiography (Sonos 7500, Philips Ultrasound,Bothell, Wash) was performed with the use of a linear array 6- to15-MHz transducer at baseline (day �1) and day 5 on animalsmaintained at a conscious state.19 All echocardiographic imageswere transmitted to the Xcelera image management workstation(Philips Medical Systems, Nederland BV, the Netherlands) for lateroffline measurements.
Blood Cell Count and HistopathologyPeripheral red blood cells, white blood cells, and platelets werecounted at baseline and day 5. On day 5, after measurement of bodyweight and echocardiographic parameters, all mice were killed underanesthesia with ketamine and xylazine. The heart tissue was fixed in4% formaldehyde, and 5-�m-thick paraffin sections were stainedwith hematoxylin-eosin for histological examination. The frequencyand severity of DOX-induced myocardial damage were evaluated bya blinded investigator using semiquantitative light microscopicanalysis of the sections. The severity of the damage was scored from0 to 3 according to the percentage of vacuolization and myofibrillarloss in 8 randomly assigned areas of each section and 2 sections perheart.20
Terminal Deoxynucleotidyltransferase–Mediated Nick-EndLabeling AssayAn independent experiment was performed on 4 groups of mice(n�5) treated according to the same protocol except that the animalswere euthanized at day 3 after DOX treatment and had received 2doses of TPO (day �1 and day 1). The terminal deoxynucleotidyl-transferase–mediated nick-end labeling assay was used for micro-scopic detection of apoptosis.
Statistical AnalysisResults on in vitro studies were analyzed with the Kruskal-Wallisranking test or Friedman test, and post hoc comparisons wereperformed by Wilcoxon rank sum test or the Mann-Whitney test,respectively. The levels of significance were adjusted by Bonferroniadjustment of multiple comparisons (Figures 1 to 5; eg, P�0.017 for3 comparisons on DOX versus control, DOX�TPO versus DOX,and TPO only versus control). For in vivo studies, heart functionparameters and blood cell counts in same animals at day �1 and day5 were compared with the Wilcoxon signed rank test, and differencesbetween groups at day 5 were compared by the Mann-Whitney test(Table 1). The survival rates of mice were compared by the log ranktest. Effects of TPO on trends of daily heart rate were examined bymultilevel modeling, and the likelihood ratio test was used to assessthe significance of estimates at the 5% level. Cardiomyopathy scores
Figure 1. Viability of H9C2 cells by MTT assay. H9C2 cells werecultured in the presence of DOX (5 �g/mL) with or without pre-treatment with TPO (5, 10, 50, 100 ng/mL, overnight) or dexra-zoxane (DEX; 50 �g/mL, 30 minutes). Results are expressed aspercentage of control cells (without DOX) as mean�SD. The cellviability was increased with pretreatment of TPO at 50 ng/mLand 100 ng/mL (Mann-Whitney test; n�4; *P�0.010 and*P�0.016, respectively).
2212 Circulation May 9, 2006
by guest on March 3, 2015http://circ.ahajournals.org/Downloaded from
of experimental animals were analyzed by the Kruskal-Wallisranking test and Mann-Whitney test (Table 2).
The authors had full access to the data and take full responsibilityfor its integrity. All authors have read and agree to the manuscript aswritten.
ResultsTPO Increased Viability of DOX-Treated H9C2 CellsResults of MTT assay on the H9C2 cell line demonstratedthat DOX (5 �g/mL, 24 hours) significantly reduced cellviability by 26.2�1.1% (Figure 1; n�4; P�0.001). Thesecells responded to TPO at a dose-dependent manner, and cell
viability was recovered to 85.6�4.4% (100 ng/mL TPO;P�0.016). Dexrazoxane at 50 �g/mL increased the cellviability to 82.8�2.0%.
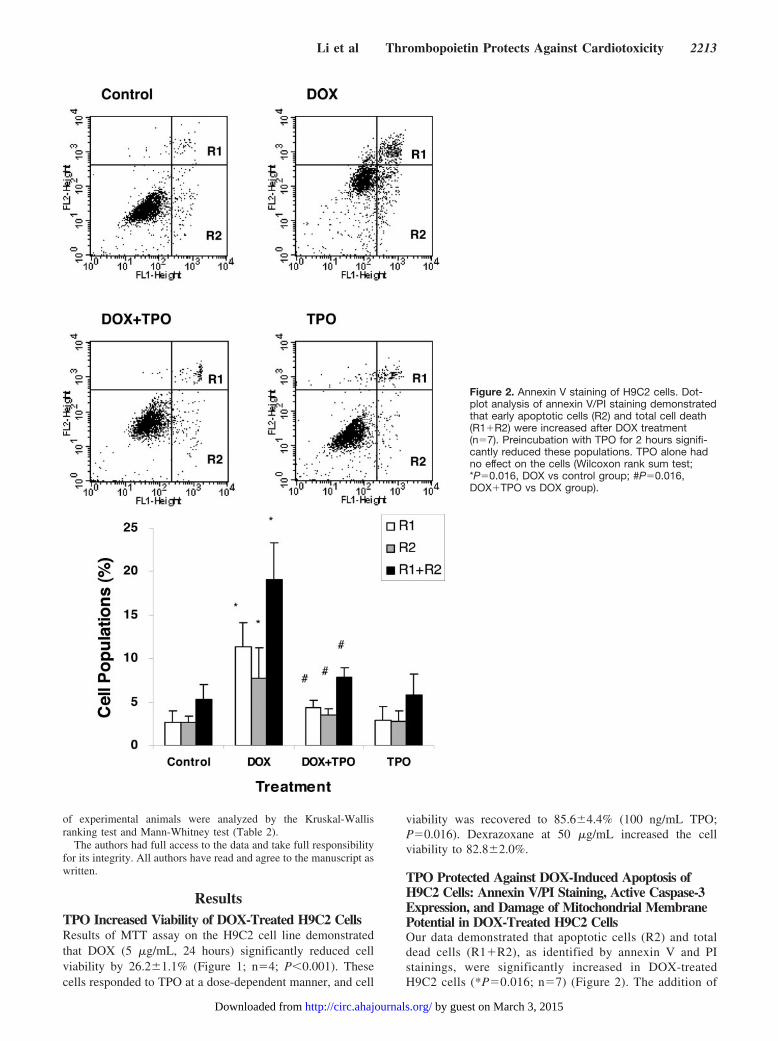
TPO Protected Against DOX-Induced Apoptosis ofH9C2 Cells: Annexin V/PI Staining, Active Caspase-3Expression, and Damage of Mitochondrial MembranePotential in DOX-Treated H9C2 CellsOur data demonstrated that apoptotic cells (R2) and totaldead cells (R1�R2), as identified by annexin V and PIstainings, were significantly increased in DOX-treatedH9C2 cells (*P�0.016; n�7) (Figure 2). The addition of
Figure 2. Annexin V staining of H9C2 cells. Dot-plot analysis of annexin V/PI staining demonstratedthat early apoptotic cells (R2) and total cell death(R1�R2) were increased after DOX treatment(n�7). Preincubation with TPO for 2 hours signifi-cantly reduced these populations. TPO alone hadno effect on the cells (Wilcoxon rank sum test;*P�0.016, DOX vs control group; #P�0.016,DOX�TPO vs DOX group).
Li et al Thrombopoietin Protects Against Cardiotoxicity 2213
by guest on March 3, 2015http://circ.ahajournals.org/Downloaded from
TPO reduced the proportion of these populations(#P�0.016; n�7) to near control levels. TPO alone had nonoticeable effect on the apoptosis of H9C2 cells.
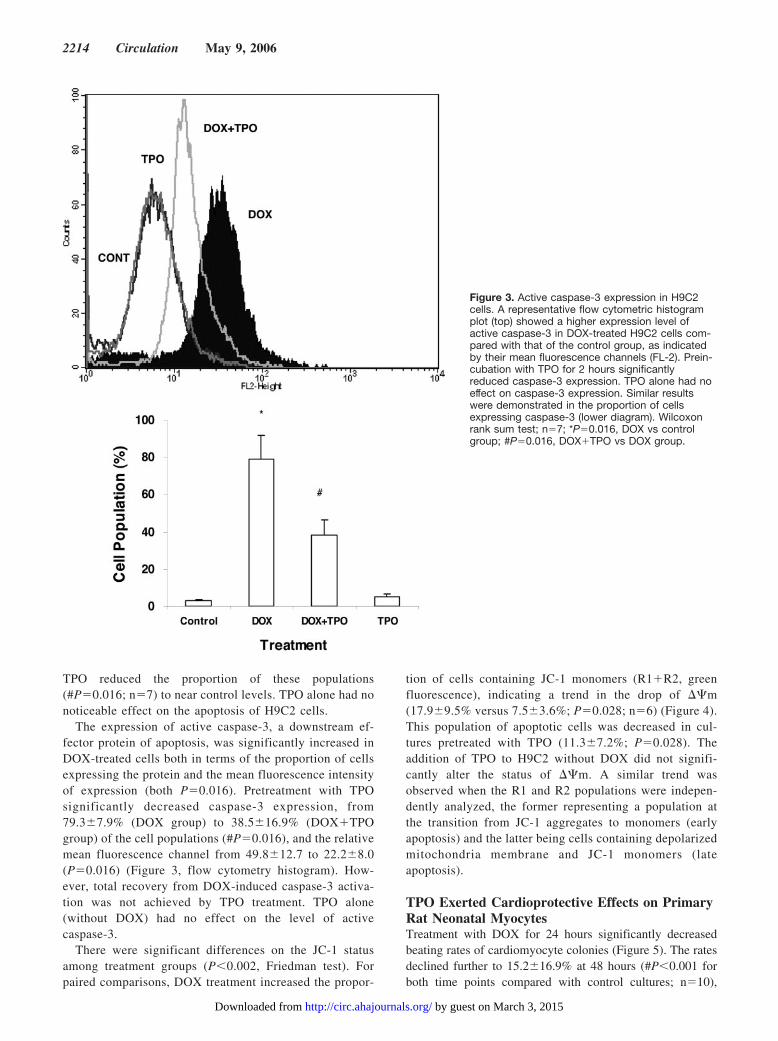
The expression of active caspase-3, a downstream ef-fector protein of apoptosis, was significantly increased inDOX-treated cells both in terms of the proportion of cellsexpressing the protein and the mean fluorescence intensityof expression (both P�0.016). Pretreatment with TPOsignificantly decreased caspase-3 expression, from79.3�7.9% (DOX group) to 38.5�16.9% (DOX�TPOgroup) of the cell populations (#P�0.016), and the relativemean fluorescence channel from 49.8�12.7 to 22.2�8.0(P�0.016) (Figure 3, flow cytometry histogram). How-ever, total recovery from DOX-induced caspase-3 activa-tion was not achieved by TPO treatment. TPO alone(without DOX) had no effect on the level of activecaspase-3.
There were significant differences on the JC-1 statusamong treatment groups (P�0.002, Friedman test). Forpaired comparisons, DOX treatment increased the propor-
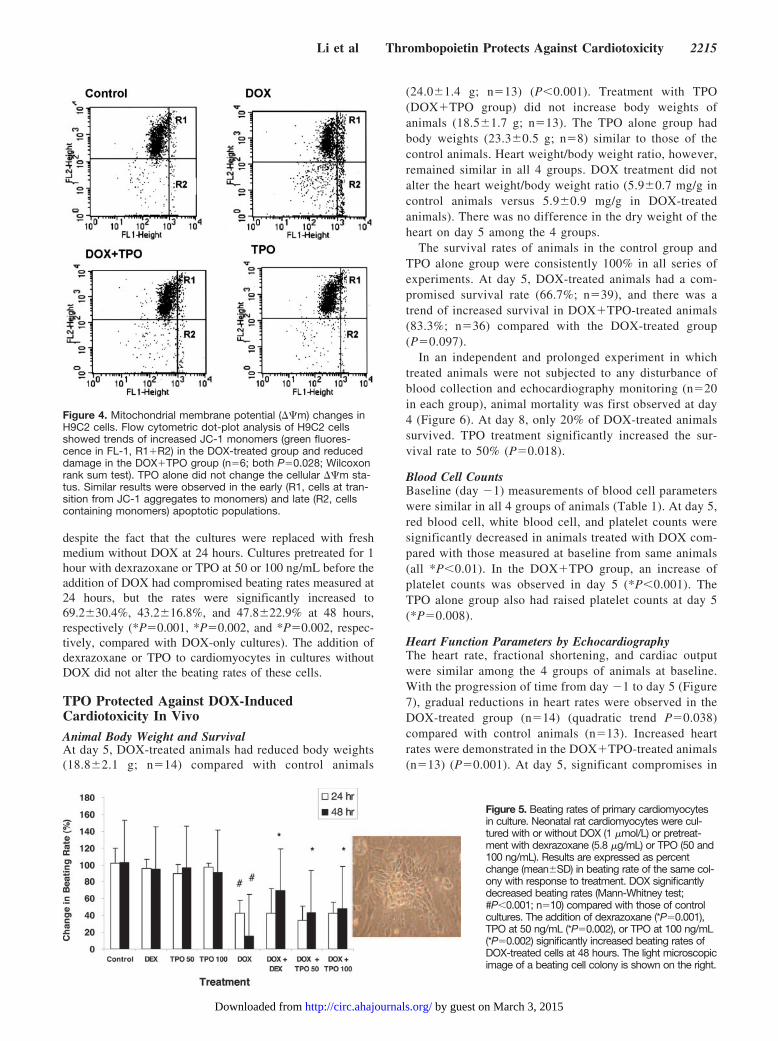
tion of cells containing JC-1 monomers (R1�R2, greenfluorescence), indicating a trend in the drop of ��m(17.9�9.5% versus 7.5�3.6%; P�0.028; n�6) (Figure 4).This population of apoptotic cells was decreased in cul-tures pretreated with TPO (11.3�7.2%; P�0.028). Theaddition of TPO to H9C2 without DOX did not signifi-cantly alter the status of ��m. A similar trend wasobserved when the R1 and R2 populations were indepen-dently analyzed, the former representing a population atthe transition from JC-1 aggregates to monomers (earlyapoptosis) and the latter being cells containing depolarizedmitochondria membrane and JC-1 monomers (lateapoptosis).
TPO Exerted Cardioprotective Effects on PrimaryRat Neonatal MyocytesTreatment with DOX for 24 hours significantly decreasedbeating rates of cardiomyocyte colonies (Figure 5). The ratesdeclined further to 15.2�16.9% at 48 hours (#P�0.001 forboth time points compared with control cultures; n�10),
Figure 3. Active caspase-3 expression in H9C2cells. A representative flow cytometric histogramplot (top) showed a higher expression level ofactive caspase-3 in DOX-treated H9C2 cells com-pared with that of the control group, as indicatedby their mean fluorescence channels (FL-2). Prein-cubation with TPO for 2 hours significantlyreduced caspase-3 expression. TPO alone had noeffect on caspase-3 expression. Similar resultswere demonstrated in the proportion of cellsexpressing caspase-3 (lower diagram). Wilcoxonrank sum test; n�7; *P�0.016, DOX vs controlgroup; #P�0.016, DOX�TPO vs DOX group.
2214 Circulation May 9, 2006
by guest on March 3, 2015http://circ.ahajournals.org/Downloaded from
despite the fact that the cultures were replaced with freshmedium without DOX at 24 hours. Cultures pretreated for 1hour with dexrazoxane or TPO at 50 or 100 ng/mL before theaddition of DOX had compromised beating rates measured at24 hours, but the rates were significantly increased to69.2�30.4%, 43.2�16.8%, and 47.8�22.9% at 48 hours,respectively (*P�0.001, *P�0.002, and *P�0.002, respec-tively, compared with DOX-only cultures). The addition ofdexrazoxane or TPO to cardiomyocytes in cultures withoutDOX did not alter the beating rates of these cells.
TPO Protected Against DOX-InducedCardiotoxicity In Vivo
Animal Body Weight and SurvivalAt day 5, DOX-treated animals had reduced body weights(18.8�2.1 g; n�14) compared with control animals
(24.0�1.4 g; n�13) (P�0.001). Treatment with TPO(DOX�TPO group) did not increase body weights ofanimals (18.5�1.7 g; n�13). The TPO alone group hadbody weights (23.3�0.5 g; n�8) similar to those of thecontrol animals. Heart weight/body weight ratio, however,remained similar in all 4 groups. DOX treatment did notalter the heart weight/body weight ratio (5.9�0.7 mg/g incontrol animals versus 5.9�0.9 mg/g in DOX-treatedanimals). There was no difference in the dry weight of theheart on day 5 among the 4 groups.
The survival rates of animals in the control group andTPO alone group were consistently 100% in all series ofexperiments. At day 5, DOX-treated animals had a com-promised survival rate (66.7%; n�39), and there was atrend of increased survival in DOX�TPO-treated animals(83.3%; n�36) compared with the DOX-treated group(P�0.097).
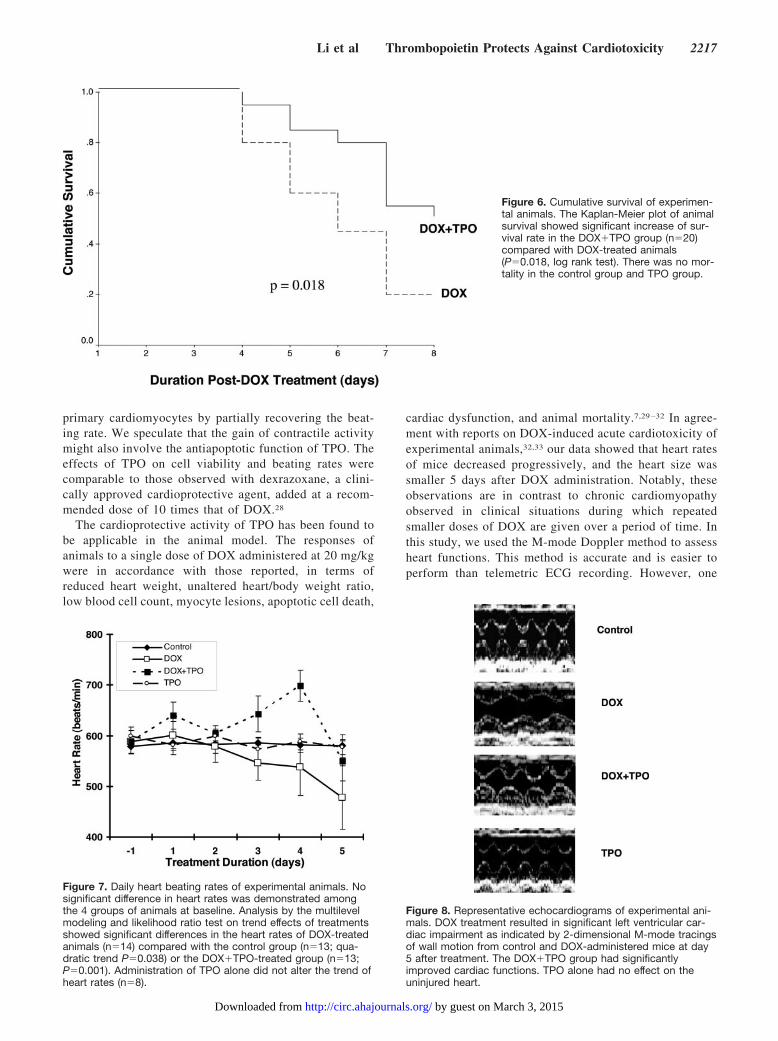
In an independent and prolonged experiment in whichtreated animals were not subjected to any disturbance ofblood collection and echocardiography monitoring (n�20in each group), animal mortality was first observed at day4 (Figure 6). At day 8, only 20% of DOX-treated animalssurvived. TPO treatment significantly increased the sur-vival rate to 50% (P�0.018).
Blood Cell CountsBaseline (day �1) measurements of blood cell parameterswere similar in all 4 groups of animals (Table 1). At day 5,red blood cell, white blood cell, and platelet counts weresignificantly decreased in animals treated with DOX com-pared with those measured at baseline from same animals(all *P�0.01). In the DOX�TPO group, an increase ofplatelet counts was observed in day 5 (*P�0.001). TheTPO alone group also had raised platelet counts at day 5(*P�0.008).
Heart Function Parameters by EchocardiographyThe heart rate, fractional shortening, and cardiac outputwere similar among the 4 groups of animals at baseline.With the progression of time from day �1 to day 5 (Figure7), gradual reductions in heart rates were observed in theDOX-treated group (n�14) (quadratic trend P�0.038)compared with control animals (n�13). Increased heartrates were demonstrated in the DOX�TPO-treated animals(n�13) (P�0.001). At day 5, significant compromises in
Figure 4. Mitochondrial membrane potential (��m) changes inH9C2 cells. Flow cytometric dot-plot analysis of H9C2 cellsshowed trends of increased JC-1 monomers (green fluores-cence in FL-1, R1�R2) in the DOX-treated group and reduceddamage in the DOX�TPO group (n�6; both P�0.028; Wilcoxonrank sum test). TPO alone did not change the cellular ��m sta-tus. Similar results were observed in the early (R1, cells at tran-sition from JC-1 aggregates to monomers) and late (R2, cellscontaining monomers) apoptotic populations.
Figure 5. Beating rates of primary cardiomyocytesin culture. Neonatal rat cardiomyocytes were cul-tured with or without DOX (1 �mol/L) or pretreat-ment with dexrazoxane (5.8 �g/mL) or TPO (50 and100 ng/mL). Results are expressed as percentchange (mean�SD) in beating rate of the same col-ony with response to treatment. DOX significantlydecreased beating rates (Mann-Whitney test;#P�0.001; n�10) compared with those of controlcultures. The addition of dexrazoxane (*P�0.001),TPO at 50 ng/mL (*P�0.002), or TPO at 100 ng/mL(*P�0.002) significantly increased beating rates ofDOX-treated cells at 48 hours. The light microscopicimage of a beating cell colony is shown on the right.
Li et al Thrombopoietin Protects Against Cardiotoxicity 2215
by guest on March 3, 2015http://circ.ahajournals.org/Downloaded from
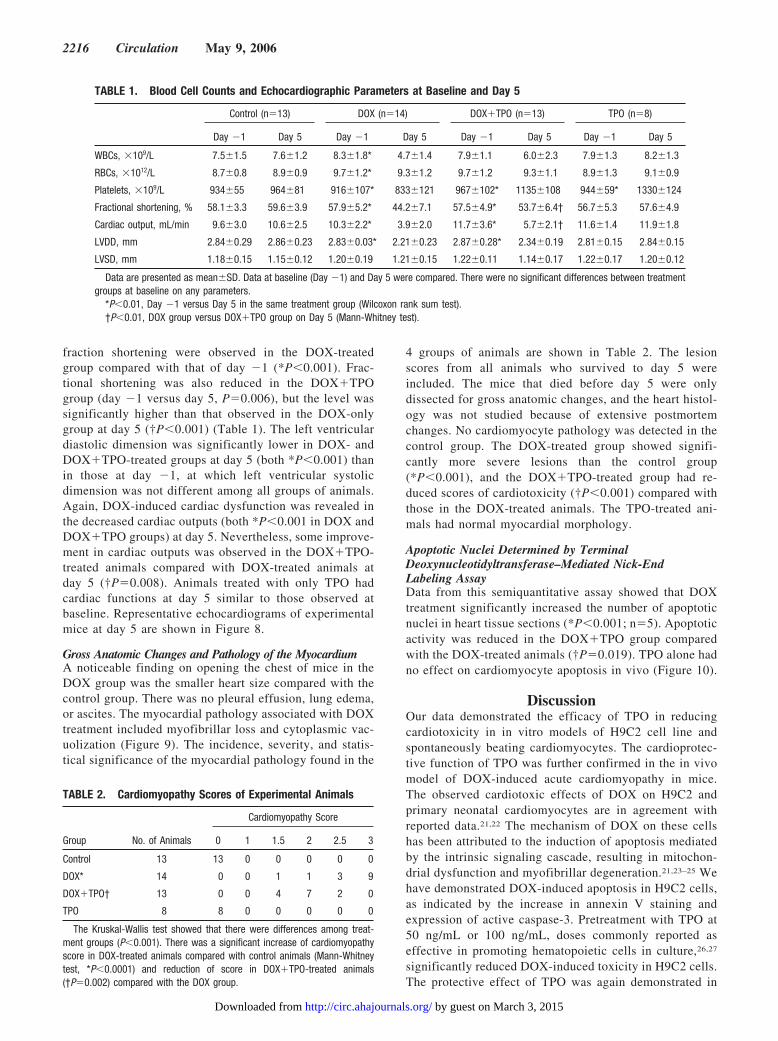
fraction shortening were observed in the DOX-treatedgroup compared with that of day �1 (*P�0.001). Frac-tional shortening was also reduced in the DOX�TPOgroup (day �1 versus day 5, P�0.006), but the level wassignificantly higher than that observed in the DOX-onlygroup at day 5 (†P�0.001) (Table 1). The left ventriculardiastolic dimension was significantly lower in DOX- andDOX�TPO-treated groups at day 5 (both *P�0.001) thanin those at day �1, at which left ventricular systolicdimension was not different among all groups of animals.Again, DOX-induced cardiac dysfunction was revealed inthe decreased cardiac outputs (both *P�0.001 in DOX andDOX�TPO groups) at day 5. Nevertheless, some improve-ment in cardiac outputs was observed in the DOX�TPO-treated animals compared with DOX-treated animals atday 5 (†P�0.008). Animals treated with only TPO hadcardiac functions at day 5 similar to those observed atbaseline. Representative echocardiograms of experimentalmice at day 5 are shown in Figure 8.
Gross Anatomic Changes and Pathology of the MyocardiumA noticeable finding on opening the chest of mice in theDOX group was the smaller heart size compared with thecontrol group. There was no pleural effusion, lung edema,or ascites. The myocardial pathology associated with DOXtreatment included myofibrillar loss and cytoplasmic vac-uolization (Figure 9). The incidence, severity, and statis-tical significance of the myocardial pathology found in the
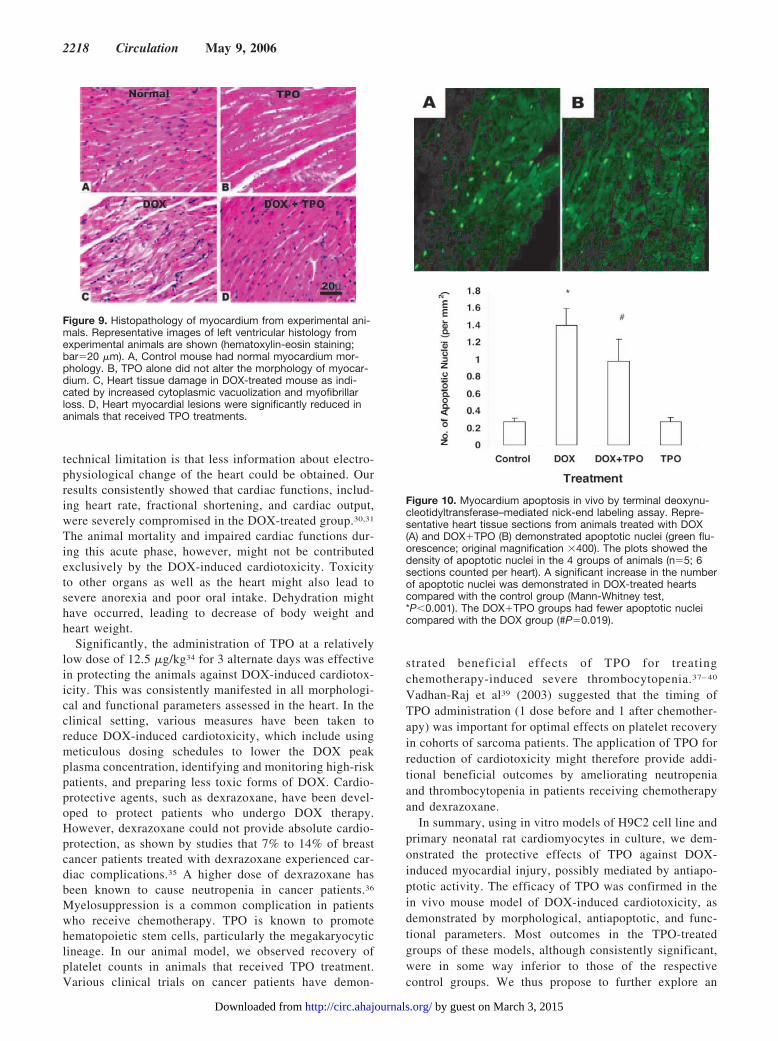
4 groups of animals are shown in Table 2. The lesionscores from all animals who survived to day 5 wereincluded. The mice that died before day 5 were onlydissected for gross anatomic changes, and the heart histol-ogy was not studied because of extensive postmortemchanges. No cardiomyocyte pathology was detected in thecontrol group. The DOX-treated group showed signifi-cantly more severe lesions than the control group(*P�0.001), and the DOX�TPO-treated group had re-duced scores of cardiotoxicity (†P�0.001) compared withthose in the DOX-treated animals. The TPO-treated ani-mals had normal myocardial morphology.
Apoptotic Nuclei Determined by TerminalDeoxynucleotidyltransferase–Mediated Nick-EndLabeling AssayData from this semiquantitative assay showed that DOXtreatment significantly increased the number of apoptoticnuclei in heart tissue sections (*P�0.001; n�5). Apoptoticactivity was reduced in the DOX�TPO group comparedwith the DOX-treated animals (†P�0.019). TPO alone hadno effect on cardiomyocyte apoptosis in vivo (Figure 10).
DiscussionOur data demonstrated the efficacy of TPO in reducingcardiotoxicity in in vitro models of H9C2 cell line andspontaneously beating cardiomyocytes. The cardioprotec-tive function of TPO was further confirmed in the in vivomodel of DOX-induced acute cardiomyopathy in mice.The observed cardiotoxic effects of DOX on H9C2 andprimary neonatal cardiomyocytes are in agreement withreported data.21,22 The mechanism of DOX on these cellshas been attributed to the induction of apoptosis mediatedby the intrinsic signaling cascade, resulting in mitochon-drial dysfunction and myofibrillar degeneration.21,23–25 Wehave demonstrated DOX-induced apoptosis in H9C2 cells,as indicated by the increase in annexin V staining andexpression of active caspase-3. Pretreatment with TPO at50 ng/mL or 100 ng/mL, doses commonly reported aseffective in promoting hematopoietic cells in culture,26,27
significantly reduced DOX-induced toxicity in H9C2 cells.The protective effect of TPO was again demonstrated in
TABLE 1. Blood Cell Counts and Echocardiographic Parameters at Baseline and Day 5
Control (n�13) DOX (n�14) DOX�TPO (n�13) TPO (n�8)
Day �1 Day 5 Day �1 Day 5 Day �1 Day 5 Day �1 Day 5
WBCs, �109/L 7.5�1.5 7.6�1.2 8.3�1.8* 4.7�1.4 7.9�1.1 6.0�2.3 7.9�1.3 8.2�1.3
RBCs, �1012/L 8.7�0.8 8.9�0.9 9.7�1.2* 9.3�1.2 9.7�1.2 9.3�1.1 8.9�1.3 9.1�0.9
Platelets, �109/L 934�55 964�81 916�107* 833�121 967�102* 1135�108 944�59* 1330�124
Fractional shortening, % 58.1�3.3 59.6�3.9 57.9�5.2* 44.2�7.1 57.5�4.9* 53.7�6.4† 56.7�5.3 57.6�4.9
Cardiac output, mL/min 9.6�3.0 10.6�2.5 10.3�2.2* 3.9�2.0 11.7�3.6* 5.7�2.1† 11.6�1.4 11.9�1.8
LVDD, mm 2.84�0.29 2.86�0.23 2.83�0.03* 2.21�0.23 2.87�0.28* 2.34�0.19 2.81�0.15 2.84�0.15
LVSD, mm 1.18�0.15 1.15�0.12 1.20�0.19 1.21�0.15 1.22�0.11 1.14�0.17 1.22�0.17 1.20�0.12
Data are presented as mean�SD. Data at baseline (Day �1) and Day 5 were compared. There were no significant differences between treatmentgroups at baseline on any parameters.
*P�0.01, Day �1 versus Day 5 in the same treatment group (Wilcoxon rank sum test).†P�0.01, DOX group versus DOX�TPO group on Day 5 (Mann-Whitney test).
TABLE 2. Cardiomyopathy Scores of Experimental Animals
Cardiomyopathy Score
Group No. of Animals 0 1 1.5 2 2.5 3
Control 13 13 0 0 0 0 0
DOX* 14 0 0 1 1 3 9
DOX�TPO† 13 0 0 4 7 2 0
TPO 8 8 0 0 0 0 0
The Kruskal-Wallis test showed that there were differences among treat-ment groups (P�0.001). There was a significant increase of cardiomyopathyscore in DOX-treated animals compared with control animals (Mann-Whitneytest, *P�0.0001) and reduction of score in DOX�TPO-treated animals(†P�0.002) compared with the DOX group.
2216 Circulation May 9, 2006
by guest on March 3, 2015http://circ.ahajournals.org/Downloaded from
primary cardiomyocytes by partially recovering the beat-ing rate. We speculate that the gain of contractile activitymight also involve the antiapoptotic function of TPO. Theeffects of TPO on cell viability and beating rates werecomparable to those observed with dexrazoxane, a clini-cally approved cardioprotective agent, added at a recom-mended dose of 10 times that of DOX.28
The cardioprotective activity of TPO has been found tobe applicable in the animal model. The responses ofanimals to a single dose of DOX administered at 20 mg/kgwere in accordance with those reported, in terms ofreduced heart weight, unaltered heart/body weight ratio,low blood cell count, myocyte lesions, apoptotic cell death,
cardiac dysfunction, and animal mortality.7,29 –32 In agree-ment with reports on DOX-induced acute cardiotoxicity ofexperimental animals,32,33 our data showed that heart ratesof mice decreased progressively, and the heart size wassmaller 5 days after DOX administration. Notably, theseobservations are in contrast to chronic cardiomyopathyobserved in clinical situations during which repeatedsmaller doses of DOX are given over a period of time. Inthis study, we used the M-mode Doppler method to assessheart functions. This method is accurate and is easier toperform than telemetric ECG recording. However, one
Figure 6. Cumulative survival of experimen-tal animals. The Kaplan-Meier plot of animalsurvival showed significant increase of sur-vival rate in the DOX�TPO group (n�20)compared with DOX-treated animals(P�0.018, log rank test). There was no mor-tality in the control group and TPO group.
Figure 7. Daily heart beating rates of experimental animals. Nosignificant difference in heart rates was demonstrated amongthe 4 groups of animals at baseline. Analysis by the multilevelmodeling and likelihood ratio test on trend effects of treatmentsshowed significant differences in the heart rates of DOX-treatedanimals (n�14) compared with the control group (n�13; qua-dratic trend P�0.038) or the DOX�TPO-treated group (n�13;P�0.001). Administration of TPO alone did not alter the trend ofheart rates (n�8).
Figure 8. Representative echocardiograms of experimental ani-mals. DOX treatment resulted in significant left ventricular car-diac impairment as indicated by 2-dimensional M-mode tracingsof wall motion from control and DOX-administered mice at day5 after treatment. The DOX�TPO group had significantlyimproved cardiac functions. TPO alone had no effect on theuninjured heart.
Li et al Thrombopoietin Protects Against Cardiotoxicity 2217
by guest on March 3, 2015http://circ.ahajournals.org/Downloaded from
technical limitation is that less information about electro-physiological change of the heart could be obtained. Ourresults consistently showed that cardiac functions, includ-ing heart rate, fractional shortening, and cardiac output,were severely compromised in the DOX-treated group.30,31
The animal mortality and impaired cardiac functions dur-ing this acute phase, however, might not be contributedexclusively by the DOX-induced cardiotoxicity. Toxicityto other organs as well as the heart might also lead tosevere anorexia and poor oral intake. Dehydration mighthave occurred, leading to decrease of body weight andheart weight.
Significantly, the administration of TPO at a relativelylow dose of 12.5 �g/kg34 for 3 alternate days was effectivein protecting the animals against DOX-induced cardiotox-icity. This was consistently manifested in all morphologi-cal and functional parameters assessed in the heart. In theclinical setting, various measures have been taken toreduce DOX-induced cardiotoxicity, which include usingmeticulous dosing schedules to lower the DOX peakplasma concentration, identifying and monitoring high-riskpatients, and preparing less toxic forms of DOX. Cardio-protective agents, such as dexrazoxane, have been devel-oped to protect patients who undergo DOX therapy.However, dexrazoxane could not provide absolute cardio-protection, as shown by studies that 7% to 14% of breastcancer patients treated with dexrazoxane experienced car-diac complications.35 A higher dose of dexrazoxane hasbeen known to cause neutropenia in cancer patients.36
Myelosuppression is a common complication in patientswho receive chemotherapy. TPO is known to promotehematopoietic stem cells, particularly the megakaryocyticlineage. In our animal model, we observed recovery ofplatelet counts in animals that received TPO treatment.Various clinical trials on cancer patients have demon-
strated beneficial effects of TPO for treatingchemotherapy-induced severe thrombocytopenia.37– 40
Vadhan-Raj et al39 (2003) suggested that the timing ofTPO administration (1 dose before and 1 after chemother-apy) was important for optimal effects on platelet recoveryin cohorts of sarcoma patients. The application of TPO forreduction of cardiotoxicity might therefore provide addi-tional beneficial outcomes by ameliorating neutropeniaand thrombocytopenia in patients receiving chemotherapyand dexrazoxane.
In summary, using in vitro models of H9C2 cell line andprimary neonatal rat cardiomyocytes in culture, we dem-onstrated the protective effects of TPO against DOX-induced myocardial injury, possibly mediated by antiapo-ptotic activity. The efficacy of TPO was confirmed in thein vivo mouse model of DOX-induced cardiotoxicity, asdemonstrated by morphological, antiapoptotic, and func-tional parameters. Most outcomes in the TPO-treatedgroups of these models, although consistently significant,were in some way inferior to those of the respectivecontrol groups. We thus propose to further explore an
Figure 9. Histopathology of myocardium from experimental ani-mals. Representative images of left ventricular histology fromexperimental animals are shown (hematoxylin-eosin staining;bar�20 �m). A, Control mouse had normal myocardium mor-phology. B, TPO alone did not alter the morphology of myocar-dium. C, Heart tissue damage in DOX-treated mouse as indi-cated by increased cytoplasmic vacuolization and myofibrillarloss. D, Heart myocardial lesions were significantly reduced inanimals that received TPO treatments.
Figure 10. Myocardium apoptosis in vivo by terminal deoxynu-cleotidyltransferase–mediated nick-end labeling assay. Repre-sentative heart tissue sections from animals treated with DOX(A) and DOX�TPO (B) demonstrated apoptotic nuclei (green flu-orescence; original magnification �400). The plots showed thedensity of apoptotic nuclei in the 4 groups of animals (n�5; 6sections counted per heart). A significant increase in the numberof apoptotic nuclei was demonstrated in DOX-treated heartscompared with the control group (Mann-Whitney test,*P�0.001). The DOX�TPO groups had fewer apoptotic nucleicompared with the DOX group (#P�0.019).
2218 Circulation May 9, 2006
by guest on March 3, 2015http://circ.ahajournals.org/Downloaded from

integrated program, incorporating TPO with other protec-tive reagents/protocols (eg, dexrazoxane and EPO) againstDOX-induced cardiac dysfunction. Our data have providedthe first evidence on the potential of TPO as a protectiveagent against DOX-induced apoptosis and cardiotoxicity.The clinical applications of TPO in this category ofpatients and other forms of cardiomyopathy deserve fur-ther investigation.
AcknowledgmentsThis work was supported by strategic grant SRP2/02 and direct grantCUHK4521/05M from The Chinese University of Hong Kong.
DisclosuresNone.
References1. Hequet O, Le QH, Moullet I, Pauli E, Salles G, Espinouse D, Dumontet
C, Thieblemont C, Arnaud P, Antal D, Bouafia F, Coiffier B. Subclinicallate cardiomyopathy after doxorubicin therapy for lymphoma in adults.J Clin Oncol. 2004;22:1864–1871.
2. Lipshultz SE, Lipsitz SR, Sallan SE, Dalton VM, Mone SM, GelberRD, Colan SD. Chronic progressive cardiac dysfunction years afterdoxorubicin therapy for childhood acute lymphoblastic leukaemia.J Clin Oncol. 2005;23:2629 –2636.
3. Steinherz LJ, Steinherz PG, Tan CT, Heller G, Murphy ML. Cardiactoxicity 4 to 20 years after completing anthracycline therapy. JAMA.1991;266:1672–1677.
4. Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patientstreated with doxorubicin. Cancer. 2003;97:2869 –2879.
5. Kluza J, Marchetti P, Gallego M-A, Lancel S, Fournier C, Loyens A,Beauvillain J-C, Bailly C. Mitochondrial proliferation during apopto-sis induced by anticancer agents: effects of doxorubicin and mitox-antrone on cancer and cardiac cells. Oncogene. 2004;23:7018 –7030.
6. Wang S, Konorev EA, Kotamraju S, Joseph J, Kalivendi S, Kalyanaraman B.Doxorubicin induces apoptosis in normal and tumor cells via distinctlydifferent mechanisms. J Biol Chem. 2004;279:25535–25543.
7. Childs AC, Phaneuf SL, Dirks AJ, Phillips T, Leeuwenburgh C.Doxorubicin treatment in vitro causes cytochrome c release and car-diomyocyte apoptosis, as well as increased mitochondrial efficiency,superoxide dismutase activity, and Bcl-2: Bax ratio. Cancer Res.2002;62:4592– 4598.
8. Nakamura T, Ueda Y, Juan Y, Katsuda S, Takahashi H, Koh E.Fas-mediated apoptosis in Adriamycin-induced cardiomyopathy inrats in vivo study. Circulation. 2000;102:572–578.
9. Abd El-Gawad HM, El-Sawalhi MM. Nitric oxide and oxidative stressin brain and heart of normal rats treated with doxorubicin: role ofaminoguanidine. J Biochem Mol Toxicol. 2004;18:69 –77.
10. Kuter DJ, Begley CG. Recombinant human thrombopoietin: basicbiology and evaluation of clinical studies. Blood. 2002;100:3457–3469.
11. Majka M, Ratajczak J, Villaire G, Kubiczek K, Marquez LA,Janowska-Wieczorek A, Ratajczak MZ. Thrombopoietin, but not cy-tokines binding to gp130 protein– coupled receptors, activatesMAPKp42/44, AKT, and STAT proteins in normal human CD34�
cells, megakaryocytes, and platelets. Exp Hematol. 2002;30:751–760.12. Sigurjonsson OE, Gudmundsson KO, Haraldsdottir V, Rafnar T,
Agnarsson BA, Gudmundsson S. Flt3/Flk-2 ligand in combinationwith thrombopoietin decreases apoptosis in megakaryocyte devel-opment. Stem Cells Dev. 2004;13:183–191.
13. Latronico MV, Costinean S, Lavitrano ML, Peschle C, Condorelli G.Regulation of cell size and contractile function by Akt in cardiomyo-cytes. Ann N Y Acad Sci. 2004;1015:250 –260.
14. De Sauvage EJ, Hass PE, Spencer SD, Malloy BE, Gurney AL,Spencer SA, Darbonne WC, Henzel WJ, Wong SC, Kuang WJ, OlesKJ, Hultgren B, Solberg LA, Goeddel DV, Eaton DL. Stimulation ofmegakaryocytopoiesis and thrombopoiesis by the c-mpl ligand.Nature. 1994;369:533–538.
15. Rouleau C, Cui K, Feldman L. A functional erythropoietin receptor isnecessary for the action of thrombopoietin on erythroid cells lackingc-mpl. Exp Hematol. 2004;32:140 –148.
16. Tramontano AF, Muniyappa R, Black AD, Blendea MC, Cohen I,Deng L, Sowers JR, Cutaia MV, El-Sherif N. Erythropoietin protectscardiac myocytes from hypoxia-induced apoptosis through an Akt-dependent pathway. Biochem Biophys Res Commun. 2003;308:990 –994.
17. Parsa CJ, Matsumoto A, Kim J, Riel RU, Pascal LS, Walton GB,Thompson RB, Petrofski JA, Annex BH, Stamler JS, Koch W. A novelprotective effect of erythropoietin in the infarcted heart. J Clin Invest.2003;112:999 –1007.
18. Reinecke H, Zhang M, Bartosek T, Murry CE. Survival, integration,and differentiation of cardiomyocyte grafts: a study in normal andinjured rat hearts. Circulation. 1999;100:193–202.
19. Georgakopoulos D, Kass DA. Minimal force-frequency modulation ofinotropy and relaxation of in situ murine heart. J Physiol. 2001;534:535–545.
20. Herman EH, Zhang J, Ferrans VJ. Comparison of the protectiveeffects of desferrioxamine and ICRF-187 against doxorubicin-inducedtoxicity in spontaneously hypertensive rat. Cancer ChemotherPharmacol. 1994;35:93–100.
21. Green PS, Leeuwenburgh C. Mitochondrial dysfunction is an earlyindicator of doxorubicin-induced apoptosis. Biochim Biophys Acta.2002;1588:94 –101.
22. Abou-El-Hassan MA, Rabelink MJ, van der Vijgh WJ, Bast A,Hoeben RC. A comparative study between catalase gene therapy andthe cardioprotector monohydroxyethylrutoside (Mono HER) in pro-tecting against doxorubicin-induced cardiotoxicity in vitro. Br JCancer. 2003;89:2140 –2146.
23. Sussman MA, Hamm-Alvarez SF, Vilalta PM, Welch S, Kedes L.Involvement of phosphorylation in doxorubicin-mediated myofibrildegeneration, an immunofluorescence microscopy analysis. Circ Res.1997;80:52– 61.
24. Wang L, Ma W, Markovich R, Chen J-W, Wang PH. Regulation ofcardiomyocyte apoptotic signaling by insulin-like growth factor I.Circ Res. 1998;83:516 –522.
25. Spallarossa P, Garibaldi S, Altieri P, Fabbi P, Manca V, Nasti S,Rossettin P, Ghigliotti G, Ballestrero A, Patrone F, Barsotti A,Brunelli C. Carvedilol prevents doxorubicin-induced free radicalrelease and apoptosis in cardiomyocytes in vitro. J Mol Cell Cardiol.2004;37:837– 846.
26. Liu J, Li K, Yuen PMP, Fok TF, Yau FW, Yang M, Li CK. Ex vivoexpansion of enriched CD34� cells from neonatal blood in thepresence of thrombopoietin, a comparison with cord blood and bonemarrow. Bone Marrow Transplant. 1999;24:247–252.
27. Chen W, Antonenko S, Sederstrom JM, Liang X, Chan ASH, KanzlerH, Blom B, Blazar BR, Liu Y-J. Thrombopoietin cooperates withFLT-3-ligand in the generation of plasmacytoid dendritic cell pre-cursors from human hematopoietic progenitors. Blood. 2004;103:2547–2553.
28. Imondi AR, Torre PD, Mazuè G, Sullivan TM, Robbins TL,Hangerman LM, Podestà A, Pinciroli G. Dose-response relationshipof dexrazoxane for prevention of doxorubicin-induced cardiotoxicityin mice, rats and dogs. Cancer Res. 1996;56:4200 – 4204.
29. Torre PD, Imondi AR, Bernardi C, Podestà A, Moneta D, RiflettutoM, Mazuè G. Cardioprotection by dexrazoxane in rats treated withdoxorubicin and paclitaxel. Cancer Chemother Pharmacol. 1999;44:138 –142.
30. Nozaki N, Shishido T, Takeishi Y, Kubota I. Modulation ofdoxorubicin-induced cardiac dysfunction in toll-like receptor-2– knockout mice. Circulation. 2004;110:2869 –2874.
31. Olson LE, Bedja D, Alvey SJ, Cardounel AJ, Gabrielson KL, ReevesRH. Protection from doxorubicin-induced cardiac toxicity in micewith a null allele of carbonyl reductase 1. Cancer Res. 2003;63:6602– 6606.
32. Liu X, Chen Z, Chua CC, Ma Y, Youngberg GA, Hamdy R, Chua BH.Melatonin as an effective protector against doxorubicin-induced car-diotoxicity. Am J Physiol. 2002;283:H254 –H263.
33. Matoba S, Hwang PM, Nguyen T, Shizukuda Y. Evaluation of pulsedDoppler tissue velocity imaging for assessing systolic function ofmurine global heart failure. J Am Soc Echocariogr. 2005;18:148 –154.
34. Shibuya K, Akahori H, Takahashi K, Tahara E, Kato T, Miyazaki H.Multilineage hematopoietic recovery by a single injection ofpegylated recombinant human megakaryocyte growth and devel-opment factor in myelosuppressed mice. Blood. 1998;91:37– 45.
Li et al Thrombopoietin Protects Against Cardiotoxicity 2219
by guest on March 3, 2015http://circ.ahajournals.org/Downloaded from

35. Swain SM, Vici P. The current and future role of dexrazoxane as acardioprotectant in anthracycline treatment: expert panel review. JCancer Res Clin Oncol. 2004;130:1–7.
36. Hochster H, Liebes L, Wadler S, Oratz R, Wernz JC, Meyers M,Green M, Blum RH, Speyer JL. Pharmacokinetics of the cardiopro-tector ADR-529 (ICRF-187) in escalating doses combined withfixed-dose doxorubicin. J Natl Cancer Inst. 1992;84:1725–1730.
37. Fanucchi M, Glaspy J, Crawford J, Garst J, Figlin R, Sheridan W,Menchaca D, Tomita D, Ozer H, Harker L. Effects of polyethyleneglycol– conjugated recombinant human megakaryocyte growth anddevelopment factor on platelet counts after chemotherapy for lungcancer. N Engl J Med. 1997;336:404 – 409.
38. Vadhan-Raj S, Kavanagh JJ, Freedman RS, Folloder J, Currie LM,Bueso-Ramos C, Verschraegen CF, Narvios AB, Connor J, Hoots WK,Broemeling LD, Lichtiger B. Safety and efficacy of transfusions of
autologous cryopreserved platelets derived from recombinant humanthrombopoietin to support chemotherapy-associated severe thrombocy-topenia: a randomised cross-over study. Lancet. 2002;359:2145–2152.
39. Vadhan-Raj S, Patel S, Bueso-Ramos C, Folloder J, Papadopolous N,Burgess A, Broemeling LD, Broxmeyer HE, Benjamin RS.Importance of predosing of recombinant human thrombopoietin toreduce chemotherapy-induced early thrombocytopenia. J Clin Oncol.2003;21:3158 –3167.
40. Angiolillo AL, Davenport V, Bonilla MA, van de Ven C, Ayello J,Militano O, Miller LL, Krailo M, Reaman G, Cairo MS, for theChildren’s Oncology Group. A phase I clinical, pharmacologic, andbiologic study of thrombopoietin and granulocyte colony-stimulatingfactor in children receiving ifosfamide, carboplatin, and etoposidechemotherapy for recurrent or refractory solid tumors: a Children’sOncology Group experience. Clin Cancer Res. 2005;11:2644 –2650.
CLINICAL PERSPECTIVEAnthracycline drugs are highly effective antineoplastic agents for the treatment of solid tumors and hematologicmalignancies. However, their clinical use is limited by delayed cardiotoxicity, which is dose related, cumulative, andessentially irreversible. In the clinical setting, various measures have been taken to reduce anthracycline-inducedcardiotoxicity, including dosing schedules to lower peak plasma concentrations, identifying and monitoring high-riskpatients, and preparing less toxic forms of the drugs. Cardioprotective agents, such as dexrazoxane, have been developedto protect patients who undergo anthracycline therapy. However, dexrazoxane treatment could not provide absolutecardioprotection, and a high dose might result in neutropenia. The present study has presented the first evidence thatthrombopoietin (TPO), a known hematopoietic growth factor, provided protection against doxorubicin-induced cardiotox-icity in in vitro and in vivo models. The mechanism was possibly associated with antiapoptotic activities. The resultsindicate the potential of using TPO as a protective agent against doxorubicin-induced cardiac injury. The administrationof TPO to patients may provide additional benefits by ameliorating neutropenia and thrombocytopenia resulting fromchemotherapy and dexrazoxane treatment. We thus propose to further explore an integrated program, incorporating TPOwith other protective reagents/protocols (eg, dexrazoxane and erythropoietin) against doxorubicin-induced cardiacdysfunction in cancer patients.
2220 Circulation May 9, 2006
by guest on March 3, 2015http://circ.ahajournals.org/Downloaded from

NgCheungYee Chan, Hailu Zhao, Man Yin To, Tai Fai Fok, Chi Kong Li, Yuek Oi Wong and Pak
Karen Li, Rita Yn Tz Sung, Wei Zhe Huang, Mo Yang, Nga Hin Pong, Shuk Man Lee, WoodDoxorubicin
Thrombopoietin Protects Against In Vitro and In Vivo Cardiotoxicity Induced by
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2006 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation doi: 10.1161/CIRCULATIONAHA.105.560250
2006;113:2211-2220; originally published online May 1, 2006;Circulation.
http://circ.ahajournals.org/content/113/18/2211World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org/content/suppl/2006/05/02/CIRCULATIONAHA.105.560250.DC1.htmlData Supplement (unedited) at:
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialCirculationin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on March 3, 2015http://circ.ahajournals.org/Downloaded from
Copyright © 2022 FDOKUMEN




















