
PHB2 Protects Sister-Chromatid Cohesion in Mitosis
10
Supplemental Data S1 PHB2 Protects Sister-Chromatid Cohesion in Mitosis Hideaki Takata, Sachihiro Matsunaga, Akihiro Morimoto, Nan Ma, Daisuke Kurihara, Rika Ono-Maniwa, Masatoshi Nakagawa, Takachika Azuma, Susumu Uchiyama, and Kiichi Fukui Supplemental Experimental Procedures Cells and Transfection HeLa cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO BRL) supplemented with 10% fetal-bovine serum (FBS; Equitech-Bio). For construction of HeLa cells transiently expressing GFP-PHB2 and Myc-PHB2, a cDNA of PHB2 (IMAGE: 3452360; Open Biosystems) was inserted into the pIC113 vector (kindly provided by Professor I.M. Cheeseman, Ludwig Institute for Cancer Research) pc DNA 3.1/myc-HisA (Invitrogen). We trans- fected HeLa cells with the GFP-PHB2-encoding plasmid by using the FuGene6 reagent (Roche). A GFP-tagged expression vector for H1.2 visualization was constructed as described previously [S1]. Antibodies For generating a rabbit PHB2 polyclonal antibody, a full-length PHB2 cDNA was inserted into the pDEST17 vector (Invitrogen). His-tagged PHB2 was expressed in Escherichia coli, purified with a fast-protein liquid chromatography system (GE Healthcare), and used to immunize a rabbit. The resulting antibodies raised were affinity purified with the antigen and used at a dilution of 1:1000. The other primary antibodies used in this study were as follows: anti-Scc1 rabbit polyclonal (kindly provided by Professor K. Yoko- mori, University of California), used at 1:100; anti-Sgo1 rabbit polyclonal (kindly provided by Prof. Y. Watanabe, The University of Tokyo), used at 1:1000; anti-B56a mouse monoclonal (BD Transduc- tion Laboratories), used at 1:100; anti-Bub1 mouse monoclonal (MBL), used at 1:20; anti-BubR1 mouse monoclonal (BD Transduc- tion Laboratories), used at 1:1000; anti-Mad2 rabbit polyclonal (Co- vance), used at 1:100; anti-CENP-A mouse monoclonal (Abcam), used at 1:50; anti-AIM1 (Aurora B) mouse monoclonal (BD Transduc- tion Laboratories), used at 1:200; anti-CENP-E mouse monoclonal (Abcam), used at 1:100; anti-CENP-F rabbit polyclonal (Novus Bio- logicals), used at 1:2000; anti-a-tubulin mouse monoclonal (Calbio- chem), used at 1:100; anti-g-tubulin rabbit (Sigma), used at 1:2000; anti-HEC1 mouse monoclonal (Affinity BioReagents), used at 1:1000; anti-Plk1 mouse monoclonal (Zymed Laboratories), used at 1:1000; anti-Cdc27 mouse monoclonal (Abcam), used at 1:2000; anti-securin mouse monoclonal (Abcam), used at 1:50; anti-Cyclin B1 rabbit (Sigma), used at 1:500; anti-phospho histone H3 (Thr3) rabbit monoclonal (Upstate Biotechnology), used at 1:500; anti- GFP rabbit polyclonal (Invitrogen), used at 1:300; and CREST (Cor- tex Biochem), used at 1:1000. RNAi and Rescue Experiments The siRNAs used in this study were as follows: PHB2 siRNA-1, 5 0 -GA AUCGUAUCUAUCUCACATT-3 0 ; PHB2 siRNA-2, 5 0 -CUGAACCCC UCU UGGAUUAAGTT-3 0 ; Aurora B siRNA [S2]; Mad2 siRNA [S3]; Plk1 siRNA [S4]; and HEC1 siRNA [S5]. The siRNAs were chemically synthesized, and HeLa cells were transfected with 120 nM of each siRNA duplex with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. For control transfection, a control siRNA duplex (5 0 -UUCUCCGAACGU GUCACGUTT-3 0 ; QIAGEN) was used. Cells were collected at 48 hr after transfection and sub- jected to further analyses. For siRNA rescue assay, three silent mutations were introduced into Myc- or GFP-PHB2 vector, changing nucleotide sequences at 814–822 position of PHB2 to 5 0 -TACCTGACC-3 0 (italic nucleotides indicate silent mutations). The RNAi-refractory constructs were transfected into HeLa cells with FuGENE6 (Rosh) at 4 hr before transfection of siRNA. Immunofluorescence Microscopy HeLa cells grown on coverslips were fixed with 4% paraformalde- hyde (PFA) in phosphate-buffered saline (PBS [pH 7.4]) for 15 min at 37 C. Alternatively, for Mad2, CENP-F, and B56a staining, cells were fixed with 100% ice-cold methanol for 10 min at 220 C. For ki- netochores-fiber analysis, cells were incubated in a calcium- containing buffer (100 mM PIPES [pH 6.8], 1 mM MgCl 2 , 0.1 mM CaCl 2 , and 0.1% Triton X-100) for 2 min at 37 C to depolymerize the nonkinetochore microtubules; this was followed by fixation in the same buffer supplemented with 4% PFA. For depolymerization of unstable kinetochore microtubules, cells were incubated in ice- cold medium for 10 min prior to incubation in the calcium-containing buffer. Fixed cells were permeabilized with 0.2% Triton X-100 in PBS for 10 min at room temperature and blocked with 1% BSA-PBS for 15 min. Metaphase-chromosome spreads were prepared as de- scribed previously [S6]. Next, the cells were incubated with primary antibodies for 1 hr at room temperature, and this was followed by incubation with Alexa 488- or TRITC-conjugated secondary anti- bodies (1:200 dilution) for 1 hr. After washing, the cells were stained with 1 mg/ml of 4 0 ,6-diamidino-2-phenylindole (DAPI) in Vectorshield mounting medium (Vector Laboratories) and observed under an Axioplan II imaging fluorescence microscope (Carl Zeiss) equipped with a cooled CCD camera (MicroMax; Roper Scientific) driven by the IP Lab software. We captured deconvoluted images with a cooled CCD camera (CH 350/L; Roper Scientific) by using an IX- 70 microscope (Olympus) and analyzed them by using the Delta vision software (Applied Precision). Live-Cell Imaging HeLa cells stably expressing GFP-histone H1.2 were cultured in 35 mm glass-bottom dishes (Matsunami) and transfected with siRNAs with Lipofectamine 2000 (Invitrogen). The medium was changed to a CO 2 -independent medium (GIBCO BRL) supple- mented with 10% FBS, 0.1 mg/ml of penicillin/streptomycin, 20 mM glutamine, and 100 mM HEPES at 1 hr before imaging. The dishes were placed on the inverted platform of a fluorescence micro- scope (IX-81; Olympus) equipped with a CO 2 chamber set at 37 C. Fluorescence images were acquired every 3 min with a 403 objec- tive controlled with the MetaMorph software (Universal Imaging Corporation). Stacks of images were assembled and processed with the MetaMorph software. Fluorescent In Situ Hybridization RP11-113F1 BAC clone in the tff1 locus on chromosome 21 was obtained from BACPAC Resources in the Children’s Hospital and Research Center (Oakland, CA). FISH probes were labeled with a nick-translation kit (Roche) and digoxigenin-11-dUTP (Roche). Human cot-1 DNA (Invitrogen) and salmon sperm DNA (Wako) were added, and probes were precipitated and resuspended in hybridization buffer (50% formamide, 10% dextrane sulfate, and 23 SSC). HeLa cells were synchronized by double-thymidine treat- ment, and interphase nuclei were obtained according to the method by Schmitz et al. [S7]. After transfection of control and PHB2 siRNAs for 48 hr, cells were treated with trypsin and harvested with centrifu- gation at 310 g. Cells were then fixed in methanol/acetic (3:1, vol/vol) and dropped onto the glass slides. The glass slide was incubated at 37 C for 16 hr, denatured in 70% formamide/23 SSC at 78 C for 1.5 min, and dehydrated by passage through an ethanol series (70%, 90%, and 100%). The labeled probes were denatured at 92 C for 10 min, cooled on ice, and then applied to the slides. The slides with the probe were denatured at 80 C for 2 min and incu- bated for 12 hr at 37 C in a humid chamber. The hybridized probe
Transcript of PHB2 Protects Sister-Chromatid Cohesion in Mitosis

Supplemental Data S1
PHB2 Protects Sister-ChromatidCohesion in Mitosis
Hideaki Takata, Sachihiro Matsunaga,Akihiro Morimoto, Nan Ma, Daisuke Kurihara,
Rika Ono-Maniwa, Masatoshi Nakagawa,Takachika Azuma, Susumu Uchiyama,
and Kiichi Fukui
Supplemental Experimental Procedures
Cells and Transfection
HeLa cells were grown in Dulbecco’s modified Eagle’s medium
(DMEM; GIBCO BRL) supplemented with 10% fetal-bovine serum
(FBS; Equitech-Bio). For construction of HeLa cells transiently
expressing GFP-PHB2 and Myc-PHB2, a cDNA of PHB2 (IMAGE:
3452360; Open Biosystems) was inserted into the pIC113 vector
(kindly provided by Professor I.M. Cheeseman, Ludwig Institute for
Cancer Research) pc DNA 3.1/myc-HisA (Invitrogen). We trans-
fected HeLa cells with the GFP-PHB2-encoding plasmid by using
the FuGene6 reagent (Roche). A GFP-tagged expression vector for
H1.2 visualization was constructed as described previously [S1].
Antibodies
For generating a rabbit PHB2 polyclonal antibody, a full-length
PHB2 cDNA was inserted into the pDEST17 vector (Invitrogen).
His-tagged PHB2 was expressed in Escherichia coli, purified with
a fast-protein liquid chromatography system (GE Healthcare), and
used to immunize a rabbit. The resulting antibodies raised were
affinity purified with the antigen and used at a dilution of 1:1000.
The other primary antibodies used in this study were as follows:
anti-Scc1 rabbit polyclonal (kindly provided by Professor K. Yoko-
mori, University of California), used at 1:100; anti-Sgo1 rabbit
polyclonal (kindly provided by Prof. Y. Watanabe, The University of
Tokyo), used at 1:1000; anti-B56a mouse monoclonal (BD Transduc-
tion Laboratories), used at 1:100; anti-Bub1 mouse monoclonal
(MBL), used at 1:20; anti-BubR1 mouse monoclonal (BD Transduc-
tion Laboratories), used at 1:1000; anti-Mad2 rabbit polyclonal (Co-
vance), used at 1:100; anti-CENP-A mouse monoclonal (Abcam),
used at 1:50; anti-AIM1 (Aurora B) mouse monoclonal (BD Transduc-
tion Laboratories), used at 1:200; anti-CENP-E mouse monoclonal
(Abcam), used at 1:100; anti-CENP-F rabbit polyclonal (Novus Bio-
logicals), used at 1:2000; anti-a-tubulin mouse monoclonal (Calbio-
chem), used at 1:100; anti-g-tubulin rabbit (Sigma), used at 1:2000;
anti-HEC1 mouse monoclonal (Affinity BioReagents), used at
1:1000; anti-Plk1 mouse monoclonal (Zymed Laboratories), used
at 1:1000; anti-Cdc27 mouse monoclonal (Abcam), used at 1:2000;
anti-securin mouse monoclonal (Abcam), used at 1:50; anti-Cyclin
B1 rabbit (Sigma), used at 1:500; anti-phospho histone H3 (Thr3)
rabbit monoclonal (Upstate Biotechnology), used at 1:500; anti-
GFP rabbit polyclonal (Invitrogen), used at 1:300; and CREST (Cor-
tex Biochem), used at 1:1000.
RNAi and Rescue Experiments
The siRNAs used in this study were as follows: PHB2 siRNA-1, 50-GA
AUCGUAUCUAUCUCACATT-30; PHB2 siRNA-2, 50-CUGAACCCC
UCU UGGAUUAAGTT-30; Aurora B siRNA [S2]; Mad2 siRNA [S3];
Plk1 siRNA [S4]; and HEC1 siRNA [S5]. The siRNAs were chemically
synthesized, and HeLa cells were transfected with 120 nM of each
siRNA duplex with Lipofectamine 2000 (Invitrogen) according to
the manufacturer’s instructions. For control transfection, a control
siRNA duplex (50-UUCUCCGAACGU GUCACGUTT-30; QIAGEN)
was used. Cells were collected at 48 hr after transfection and sub-
jected to further analyses.
For siRNA rescue assay, three silent mutations were introduced
into Myc- or GFP-PHB2 vector, changing nucleotide sequences at
814–822 position of PHB2 to 50-TACCTGACC-30 (italic nucleotides
indicate silent mutations). The RNAi-refractory constructs were
transfected into HeLa cells with FuGENE6 (Rosh) at 4 hr before
transfection of siRNA.
Immunofluorescence Microscopy
HeLa cells grown on coverslips were fixed with 4% paraformalde-
hyde (PFA) in phosphate-buffered saline (PBS [pH 7.4]) for 15 min
at 37�C. Alternatively, for Mad2, CENP-F, and B56a staining, cells
were fixed with 100% ice-cold methanol for 10 min at 220�C. For ki-
netochores-fiber analysis, cells were incubated in a calcium-
containing buffer (100 mM PIPES [pH 6.8], 1 mM MgCl2, 0.1 mM
CaCl2, and 0.1% Triton X-100) for 2 min at 37�C to depolymerize
the nonkinetochore microtubules; this was followed by fixation in
the same buffer supplemented with 4% PFA. For depolymerization
of unstable kinetochore microtubules, cells were incubated in ice-
cold medium for 10 min prior to incubation in the calcium-containing
buffer. Fixed cells were permeabilized with 0.2% Triton X-100 in PBS
for 10 min at room temperature and blocked with 1% BSA-PBS for
15 min. Metaphase-chromosome spreads were prepared as de-
scribed previously [S6]. Next, the cells were incubated with primary
antibodies for 1 hr at room temperature, and this was followed by
incubation with Alexa 488- or TRITC-conjugated secondary anti-
bodies (1:200 dilution) for 1 hr. After washing, the cells were stained
with 1 mg/ml of 40,6-diamidino-2-phenylindole (DAPI) in Vectorshield
mounting medium (Vector Laboratories) and observed under an
Axioplan II imaging fluorescence microscope (Carl Zeiss) equipped
with a cooled CCD camera (MicroMax; Roper Scientific) driven by
the IP Lab software. We captured deconvoluted images with
a cooled CCD camera (CH 350/L; Roper Scientific) by using an IX-
70 microscope (Olympus) and analyzed them by using the Delta
vision software (Applied Precision).
Live-Cell Imaging
HeLa cells stably expressing GFP-histone H1.2 were cultured in
35 mm glass-bottom dishes (Matsunami) and transfected with
siRNAs with Lipofectamine 2000 (Invitrogen). The medium was
changed to a CO2-independent medium (GIBCO BRL) supple-
mented with 10% FBS, 0.1 mg/ml of penicillin/streptomycin,
20 mM glutamine, and 100 mM HEPES at 1 hr before imaging. The
dishes were placed on the inverted platform of a fluorescence micro-
scope (IX-81; Olympus) equipped with a CO2 chamber set at 37�C.
Fluorescence images were acquired every 3 min with a 403 objec-
tive controlled with the MetaMorph software (Universal Imaging
Corporation). Stacks of images were assembled and processed
with the MetaMorph software.
Fluorescent In Situ Hybridization
RP11-113F1 BAC clone in the tff1 locus on chromosome 21 was
obtained from BACPAC Resources in the Children’s Hospital and
Research Center (Oakland, CA). FISH probes were labeled with
a nick-translation kit (Roche) and digoxigenin-11-dUTP (Roche).
Human cot-1 DNA (Invitrogen) and salmon sperm DNA (Wako)
were added, and probes were precipitated and resuspended in
hybridization buffer (50% formamide, 10% dextrane sulfate, and
23 SSC). HeLa cells were synchronized by double-thymidine treat-
ment, and interphase nuclei were obtained according to the method
by Schmitz et al. [S7]. After transfection of control and PHB2 siRNAs
for 48 hr, cells were treated with trypsin and harvested with centrifu-
gation at 310 g. Cells were then fixed in methanol/acetic (3:1, vol/vol)
and dropped onto the glass slides. The glass slide was incubated at
37�C for 16 hr, denatured in 70% formamide/23 SSC at 78�C for
1.5 min, and dehydrated by passage through an ethanol series
(70%, 90%, and 100%). The labeled probes were denatured at
92�C for 10 min, cooled on ice, and then applied to the slides. The
slides with the probe were denatured at 80�C for 2 min and incu-
bated for 12 hr at 37�C in a humid chamber. The hybridized probe

was detected with the fluorescent antibody enhancer set for DIG
detection kit (Roche). DNA was 1 mg/ml of DAPI in Vectorshield
mounting medium (Vector Laboratories). Acquisition of deconvo-
luted images was performed as described above.
Supplemental References
S1. Gambe, A.E., Maniwa-Ono, R., Matsunaga, S., Kutsuna, N.,
Higaki, T., Higashi, T., Hasezawa, S., Uchiyama, S., and Fukui,
K. (2007). Development of a multistage classifier for a monitoring
system of cell activity based on imaging of chromosomal
dynamics. Cytometry 71A, 286–296.
S2. Ditchfield, C., Johnson, V.L., Tighe, A., Ellston, R., Haworth, C.,
Johnson, T., Mortlock, A., Keen, N., and Taylor, S.S. (2003).
Aurora B couples chromosome alignment with anaphase by
targeting BubR1, Mad2, and Cenp-E to kinetochores. J. Cell
Biol. 161, 267–280.
S3. Tang, Z., Sun, Y., Harley, S.E., Zou, H., and Yu, H. (2004). Human
Bub1 protects centromeric sister-chromatid cohesion through
Shugoshin during mitosis. Proc. Natl. Acad. Sci. USA 101,
18012–18017.
S4. Tang, Z., Shu, H., Qi, W., Mahmood, N.A., Mumby, M.C., and Yu,
H. (2006). PP2A is required for centromeric localization of Sgo1
and proper chromosome segregation. Dev. Cell 10, 575–585.
S5. Joseph, J., Liu, S.T., Jablonski, S.A., Yen, T.J., and Dasso, M.
(2004). The RanGAP1-RanBP2 complex is essential for microtu-
bule-kinetochore interactions in vivo. Curr. Biol. 14, 611–617.
S6. Ma, N., Matsunaga, S., Takata, H., Maniwa-Ono, R., Uchiyama,
S., and Fukui, K. (2007). Nucleolin functions in nucleolus forma-
tion and chromosome congression. J. Cell Sci. 120, 2091–2105.
S7. Schmitz, J., Watrin, E., Lenart, P., Mechtler, K., and Peters, J.M.
(2007). Sororin is required for stable binding of cohesin to chro-
matin and for sister chromatid cohesion in interphase. Curr. Biol.
17, 630–636.
S8. Kasashima, K., Ohta, E., Kagawa, Y., and Endo, H. (2006). Mito-
chondrial functions and estrogen receptor-dependent nuclear
translocation of pleiotropic human prohibitin 2. J. Biol. Chem.
281, 36401–36410.
Figure S1. Localization Analysis of PHB2 during Mitosis
(A) HeLa cells expressing GFP-PHB2 (green) were fixed with 4% PFA, and centromeres were detected by CREST (red). DNA was stained with
DAPI (blue). The exogenous GFP-PHB2 is localized in the cytoplasm and the chromosomes of prophase and prometaphase cells, whereas it is
mainly localized in the cytoplasm and spindles of metaphase and anaphase cells. The scale bar represents 5 mm.
(B) Deconvoluted images of the prophase cell expressing GFP-PHB2. HeLa cells expressing GFP-PHB2 were pre-extracted by 0.01% Triton
X-100 before 4% PFA fixation and immunostained with GFP antibody (green) and CREST (red). DNA was stained with DAPI (blue). The scale
bar represents 5 mm.
S2

Figure S2. RNAi of PHB2 Using siRNAs with Different Sequences
For the control siRNA, we used a random RNA sequence. For the PHB2 siRNA, two siRNAs (PHB2 siRNA-1 and PHB2 siRNA-2) with different
sequences were designed.
(A) Immunoblotting analysis revealed that the two siRNAs cause 70% and 90% reductions in the expression of PHB2, respectively.
(B) At 48 hr after transfection with each siRNA, HeLa cells were fixed with 4% PFA and stained with DAPI so that the chromosome morphologies
could be observed. Depletion of PHB2 with either siRNA causes similar aberrant chromosomes. However, when we used PHB2 siRNA-2, the
significant reduction in PHB2 decreased the cell viability, as previously reported [S8]. In fact, a further reduction in PHB2 after a long-term
incubation with PHB2 siRNA-1 also caused cell death. Therefore, we used PHB2 siRNA-1 for our experiments. The scale bar represents 10 mm.
(C) The percentage of defects in sister-chromatid cohesion. The defects in sister-chromatid cohesion observed in PHB2 RNAi cells expressing
Myc or GFP (w45%) are rescued by the expression of RNAi-refractory Myc- or GFP-PHB2 (w15%). n = 5, >100 cells were counted in each
experiment.
(D) The expression levels of exogenous PHB2 were confirmed by immunoblotting. By the transfection of PHB2 siRNA-1, the expressions of
endogenous PHB2 and exogenous Myc-PHB2 (WT; non-RNAi-resistant form) were significantly decreased. Similarly, RNAi-resistant GFP-
PHB2 also showed a significant decrease of its expression by PHB2 RNAi (data not shown). By contrast, the expression of exogenous RNAi-
resistant Myc-PHB2 did not show any significant decrease of its expression.
S3

Figure S3. Defects in Chromosome Dynamics in PHB2-Depleted Cells
(A) PHB2 was depleted in HeLa cells stably expressing GFP-H1.2. The number in each panel shows the time point (min) after nuclear-envelope
breakdown (NEBD). In PHB2-depleted cells, four types of aberrant chromosome behaviors (I–IV) are observed: I, chromosomes never form a dis-
tinct metaphase plate and are scattered throughout the cytoplasm; II, chromosomes are aligned at the metaphase plate but begin to scatter
throughout the cytoplasm after a long period of arrest at metaphase; III, chromosomes are segregated without alignment at the metaphase plate;
and IV, chromosomes are aligned at the metaphase plate, but the cells are arrested at metaphase for a long time. These aberrations are not
observed in control cells. The scale bar represents 5 mm.
(B) The time intervals between NEBD and the onset of anaphase were measured in movies of control and PHB2 RNAi cells. PHB2-depleted cells
show a significant delay in the onset of anaphase. Note that PHB2-depleted cells can be identified by their aberrant chromosome movement.
(C) Frequencies of the aberrations of chromosome dynamics in PHB2 RNAi cells. Categories I–IV correspond to the numerals indicated in (A).
S4
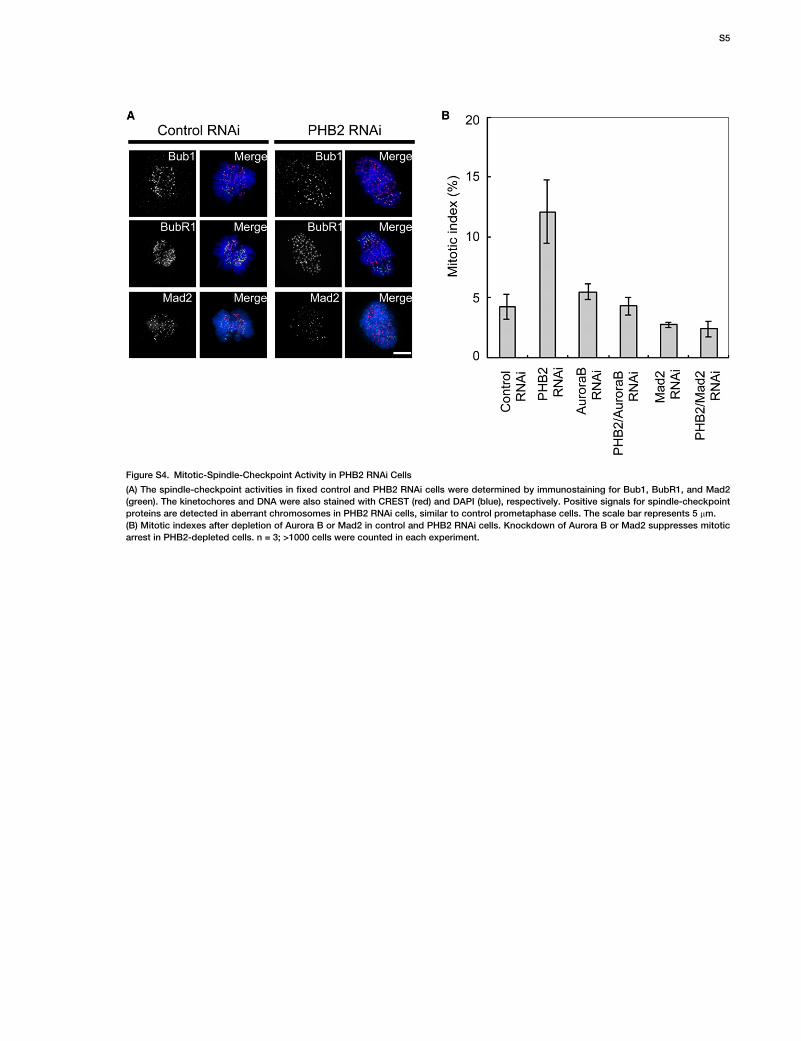
Figure S4. Mitotic-Spindle-Checkpoint Activity in PHB2 RNAi Cells
(A) The spindle-checkpoint activities in fixed control and PHB2 RNAi cells were determined by immunostaining for Bub1, BubR1, and Mad2
(green). The kinetochores and DNA were also stained with CREST (red) and DAPI (blue), respectively. Positive signals for spindle-checkpoint
proteins are detected in aberrant chromosomes in PHB2 RNAi cells, similar to control prometaphase cells. The scale bar represents 5 mm.
(B) Mitotic indexes after depletion of Aurora B or Mad2 in control and PHB2 RNAi cells. Knockdown of Aurora B or Mad2 suppresses mitotic
arrest in PHB2-depleted cells. n = 3; >1000 cells were counted in each experiment.
S5

Figure S5. Mitotic-Spindle Morphologies in PHB2 RNAi Cells
(A) Mitotic spindles were visualized by immunostaining for a-tubulin (green) and g-tubulin (red). DNA was stained with DAPI (blue). The scale bar
represents 5 mm.
(B) Three types of aberrant spindle morphologies, namely multipolar, elongated, and disorganized, are observed in PHB2-depleted cells.
(C) Frequencies of the spindle-aberration phenotypes shown in (B). Elongated mitotic spindles are frequently observed in PHB2-depleted cells.
n = 5; >1000 cells were counted in each experiment.
(D) Intercentrosome distances in control metaphase cells and PHB2-depleted mitotic cells. We determined the intercentrosome distance by
measuring the distance between the two g-tubulin signals in a cell; n = 10.
(E) Kinetochore microtubule stability in RNAi-treated cells. Control, PHB2, and Hec1 siRNA-transfected cells were cold-treated, processed for
immunostaining, and stained with an a-tubulin antibody (green) and DAPI (blue). The Hec1 siRNA was used as a control for destabilizing kinet-
ochores-microtubule formation. The fluorescence intensity of the kinetochore microtubules is decreased in PHB2-depleted cells, similar to that
of kinetochores microtubules in Hec1-depleted cells. The scale bar represents 5 mm.
S6

Figure S6. Localizations of Outer Kinetochore Proteins in PHB2-Depleted Cells
(A) CENP-E, CENP-F, and Hec1 (green) were visualized by immunostaining in control and PHB2 RNAi cells. The centromeres and DNA were
stained with CREST (red) and DAPI (blue), respectively. Scale bars represent 5 mm.
(B) Fluorescence intensities of the outer kinetochore proteins. After immunostaining for CENP-E, CENP-F, and Hec1, the fluorescence intensities
of the proteins in cells were measured with the MetaMorph software. After PHB2 depletion, the intensity of each outer kinetochore protein is
decreased to <50% of the level in control cells; n = 5.
S7

Figure S7. Sister-Chromatid Cohesion in
RNAi Cells during S/G2 Phase
(A) HeLa cells were synchronized by double-
thymidine treatment. Cells were harvested
5 hr after release from the second arrest.
FISH analysis with a probe specific for the
trisomic tff1 locus on chromosome 21 could
successfully detect three pairs of centromere
signals (green) both in control and PHB2
RNAi cells. DNA was counterstained with
DAPI (blue). The paired signals indicated by
white arrows were magnified (6003) in the
lower-left inset. When HeLa cells were har-
vested at 9 hr after release from the second
arrest, PHB2-depleted mitotic cells showed
chromosome aberrations. These results
indicated that PHB2 were depleted in most
of G2-phase nuclei observed in the FISH
experiment and showed defects in chromo-
some congression when they entered mito-
sis. The scale bar represents 5 mm.
(B) The distance between paired FISH signals
in RNAi cells. There was no significant differ-
ence in the interchromatid distance between
control (0.40 6 0.12 mm) and PHB2 RNAi cells
(0.36 6 0.16 mm); n > 20 per condition.
Figure S8. Percentages of Aberrant Chromosomes in the Presence
or Absence of MG132 in RNAi Cells
At 45 hr after siRNA transfection, HeLa cells were incubated with
20 mM MG132 for 3 hr to inhibit the anaphase transition. The percent-
ages of aberrant chromosomes in mitotic cells among either control
or PHB2 RNAi cells are not affected by the MG132 treatment. This
indicates that the aberrant chromosomes in PHB2-depleted cells
are generated before the onset of anaphase and that prolonged
metaphase cannot rescue the chromosome congression defects
in PHB2-depleted cells. n = 3; >1000 cells were counted in each
experiment.
S8

Figure S9. Immunostaining of Scc1 in Inter-
phase and Anaphase
(A) HeLa cells were pre-extracted by 0.5%
Triton X-100 and fixed with 4% PFA. Scc1
was detected by Scc1 antibody (green), and
centromeres were visualized by CREST.
There was no difference in Scc1 localization
of interphase nuclei between control and
PHB2 RNAi cells. This indicated that loading
of PHB2 into interphase nuclei occurred nor-
mally in PHB2-depleted cells. The scale bar
represents 5 mm.
(B) In control cells, Scc1 signals were ob-
served at the centromeric region in prometa-
phase. In anaphase, Scc1 signals disap-
peared. The scale bar represents 5 mm.
S9

Figure S10. Immunoblotting to Confirm the
Protein Expression Levels in RNAi Cells
(A) The efficiencies of double knockdown of
PHB2/Aurora B, PHB2/Mad2, and PHB2/
Plk1 were investigated by immunoblotting.
(B) The expression levels of the cell cohesin
subunit Scc1 and cycle-related proteins
CDC27, CDC20, and Plk1 do not change after
PHB2 RNAi. There are no differences in the
expression levels of any of the cell-cycle-
related proteins between control and PHB2
RNAi cells. These results indicate that the
cell-cycle arrest observed in PHB2-depleted
cells is caused by spindle-checkpoint activa-
tion rather than failure of metaphase exit.
S10




















