
Hydroxymethylglutaryl-coenzyme A reductase inhibition stimulates caspase-1 activity and Th1-cytokine...
11
Atherosclerosis 153 (2000) 303 – 313 Hydroxymethylglutaryl-coenzyme A reductase inhibition stimulates caspase-1 activity and Th1-cytokine release in peripheral blood mononuclear cells Marı ´a Teresa Montero a , Osvaldo Herna ´ndez a , Yajaira Sua ´rez a , Joaquı ´n Matilla a , Antonio J. Ferruelo a , Javier Martı ´nez-Botas a,1 , Diego Go ´ mez-Coronado a , Miguel A. Lasuncio ´n a,b, * a Ser6icio de Bioquı ´mica -In6estigacio ´n, Hospital Ramo ´n y Cajal, Ctra. Colmenar, km 9, 28034, Madrid, Spain b Departamento de Bioquı ´mica y Biologı ´a Molecular, Uni6ersidad de Alcala ´ , Alcala ´ de Henares, Spain Received 24 September 1999; received in revised form 24 January 2000; accepted 31 January 2000 Abstract T cells are prominent components of both early and late atherosclerotic lesions and the role of Th1/Th2 cells subsets in the evolution and rupture of the plaque is currently under investigation. Statins, which are inhibitors of 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) reductase, exert actions beyond that of simply lowering cholesterol levels, and some effects on immune function have been reported. We studied in vitro the effects of fluvastatin on Th1/Th2 cytokine release in relation to caspase-1 activation, in human peripheral-blood mononuclear cells (PBMC) stimulated or not with Mycobacterium tuberculosis. Fluvastatin treatment resulted in the activation of caspase-1 and in a small secretion of interleukin (IL)-1b, IL-18, and IFNg (Th1). In the presence of bacteria, the release of these cytokines was highly increased by the statin in a synergistic way. By contrast, production of IL-12, IL-10 and IL-4 were unaffected by the statin. Not only did mevalonate abolish the effects of the statin but it also prevented the caspase-1 activation induced by the bacteria, suggesting the involvement of isoprenoids in the response to M. tuberculosis. It is proposed that inhibition of HMG-CoA reductase may be immunoprotective by enhancing the Th1 response, which has therapeutical potential not only in atherosclerosis but also in infectious diseases. © 2000 Elsevier Science Ireland Ltd. All rights reserved. Keywords: Atherosclerosis; Infection; Interleukins; Caspase-1; Cholesterol www.elsevier.com/locate/atherosclerosis 1. Introduction Arteriosclerosis is a chronic inflammatory-fibrotic re- sponse to the accumulation of cholesterol in the artery wall [1,2], which has been regarded as a cell-mediated hypersensitivity reaction [3,4] that evoke the immune response akin to Mycobacterium tuberculosis [5,6]. Sev- eral reports demonstrated that IFN-g but not IL-4 is preferentially detected in atherosclerotic lesions, sug- gesting that T lymphocytes present in the plaque corre- spond predominantly to the Th1 phenotype [7,8]. In spite of that, a cross-regularory role of IL-12 and IL-10 also occurs in human atherosclerotic lesions, which may influence the differential development of T lymphocytes to the Th1 and Th2 phenotypes [7,9,10]. Interestingly, Zhou et al. reported that, in mouse, severe hypercholes- terolemia is associated with a switch from Th1 to Th2, resulting in the appearance of Th2 cytokines both in lymphoid organs and atherosclerotic lesions [11], which may have important consequences for the immune pro- cess that takes place in the plaque [7,12]. On the other hand, immunization of rabbits with M. tuberculosis heat shock protein 65 has been reported to result in the development of atherosclerotic lesions [13], which are aggravated when immunized animals are put on a hypercholesterolemic diet [14]. All this suggests a possi- * Corresponding author. Tel.: +34-1-3368077; fax: +34-1- 3369016. E-mail address: [email protected] (M.A. Lasuncio ´ n). 1 Present address: Department of Cell Biology, Baylor College of Medicine, Houston, TX 77030, USA. 0021-9150/00/$ - see front matter © 2000 Elsevier Science Ireland Ltd. All rights reserved. PII:S0021-9150(00)00417-2
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Hydroxymethylglutaryl-coenzyme A reductase inhibition stimulates caspase-1 activity and Th1-cytokine...

Atherosclerosis 153 (2000) 303–313
Hydroxymethylglutaryl-coenzyme A reductase inhibition stimulatescaspase-1 activity and Th1-cytokine release in peripheral blood
mononuclear cells
Marıa Teresa Montero a, Osvaldo Hernandez a, Yajaira Suarez a, Joaquın Matilla a,Antonio J. Ferruelo a, Javier Martınez-Botas a,1, Diego Gomez-Coronado a,
Miguel A. Lasuncion a,b,*a Ser6icio de Bioquımica-In6estigacion, Hospital Ramon y Cajal, Ctra. Colmenar, km 9, 28034, Madrid, Spain
b Departamento de Bioquımica y Biologıa Molecular, Uni6ersidad de Alcala, Alcala de Henares, Spain
Received 24 September 1999; received in revised form 24 January 2000; accepted 31 January 2000
Abstract
T cells are prominent components of both early and late atherosclerotic lesions and the role of Th1/Th2 cells subsets in theevolution and rupture of the plaque is currently under investigation. Statins, which are inhibitors of 3-hydroxy-3-methylglutarylCoA (HMG-CoA) reductase, exert actions beyond that of simply lowering cholesterol levels, and some effects on immune functionhave been reported. We studied in vitro the effects of fluvastatin on Th1/Th2 cytokine release in relation to caspase-1 activation,in human peripheral-blood mononuclear cells (PBMC) stimulated or not with Mycobacterium tuberculosis. Fluvastatin treatmentresulted in the activation of caspase-1 and in a small secretion of interleukin (IL)-1b, IL-18, and IFNg (Th1). In the presence ofbacteria, the release of these cytokines was highly increased by the statin in a synergistic way. By contrast, production of IL-12,IL-10 and IL-4 were unaffected by the statin. Not only did mevalonate abolish the effects of the statin but it also prevented thecaspase-1 activation induced by the bacteria, suggesting the involvement of isoprenoids in the response to M. tuberculosis. It isproposed that inhibition of HMG-CoA reductase may be immunoprotective by enhancing the Th1 response, which hastherapeutical potential not only in atherosclerosis but also in infectious diseases. © 2000 Elsevier Science Ireland Ltd. All rightsreserved.
Keywords: Atherosclerosis; Infection; Interleukins; Caspase-1; Cholesterol
www.elsevier.com/locate/atherosclerosis
1. Introduction
Arteriosclerosis is a chronic inflammatory-fibrotic re-sponse to the accumulation of cholesterol in the arterywall [1,2], which has been regarded as a cell-mediatedhypersensitivity reaction [3,4] that evoke the immuneresponse akin to Mycobacterium tuberculosis [5,6]. Sev-eral reports demonstrated that IFN-g but not IL-4 ispreferentially detected in atherosclerotic lesions, sug-gesting that T lymphocytes present in the plaque corre-
spond predominantly to the Th1 phenotype [7,8]. Inspite of that, a cross-regularory role of IL-12 and IL-10also occurs in human atherosclerotic lesions, which mayinfluence the differential development of T lymphocytesto the Th1 and Th2 phenotypes [7,9,10]. Interestingly,Zhou et al. reported that, in mouse, severe hypercholes-terolemia is associated with a switch from Th1 to Th2,resulting in the appearance of Th2 cytokines both inlymphoid organs and atherosclerotic lesions [11], whichmay have important consequences for the immune pro-cess that takes place in the plaque [7,12]. On the otherhand, immunization of rabbits with M. tuberculosisheat shock protein 65 has been reported to result in thedevelopment of atherosclerotic lesions [13], which areaggravated when immunized animals are put on ahypercholesterolemic diet [14]. All this suggests a possi-
* Corresponding author. Tel.: +34-1-3368077; fax: +34-1-3369016.
E-mail address: [email protected] (M.A. Lasuncion).1 Present address: Department of Cell Biology, Baylor College of
Medicine, Houston, TX 77030, USA.
0021-9150/00/$ - see front matter © 2000 Elsevier Science Ireland Ltd. All rights reserved.PII: S 0 0 2 1 -9150 (00 )00417 -2

M.T. Montero et al. / Atherosclerosis 153 (2000) 303–313304
ble link between cholesterol metabolism and the cell-mediated response pattern. Additional evidence forthis comes from the pharmacological effects ofstatins, which are competitive inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase,a key enzyme in cholesterol biosynthesis. These drugsare being widely used to lower cholesterol levelsaimed to reduce atherosclerosis, yet the benefit ofsuch treatment is greater than expected in terms ofthe ensuing reduction in LDL-cholesterol levels,which has led to the hypothesis that statins exert ac-tions beyond that of simply lowering cholesterol levels[15,16]. In keeping with this, some effects of statinson the inflammatory response have been reported[17–23].
Human Th1 cells induce IL-1b production uponcell–cell contact with monocytes [24], a cytokine thathas been found in abundance in atherosclerotic le-sions [3] and, on the other hand, has been referred asa mediator of the inflammatory process seen in tuber-culosis [25]. IL-1b is synthesized as a 31-kDa precur-sor devoid of a conventional signal sequence and,unlike most other peptide hormones, is not releasedvia the classic secretion pathway. This cytokine isprocessed to its proinflammatory 17-kDa form bycaspase-1, formerly IL-1b converting enzyme (ICE)[26], the founder member of a growing family of cys-teine proteases (caspases) with a substrate cleavagespecificity for Asp at P1 [27]. In the monocyte/macrophage, caspase-1 has been also shown to acti-vate the so-called interferon g (IFNg)-inducing factor(IGIF or IL-18) [28,29], which is a 18 kDa polypep-tide that is essential for the effective induction andactivation of Th1 (IFNg) cells in conjunction withIL-12 [30,31]. In turn, IFNg is a major activator ofmonocytes that increases their antigen-presenting ca-pacity and primes for the production of proinflamma-tory cytokines [32]. Therefore, caspase-1 appears toplay a central role in the coordinated function oflymphocytes and monocytes in the Th1 immune re-sponse, by triggering the secretion of both IL-1b andIL-18. Present study sought to explore the possibilitythat statins influence the Th1/Th2 cell-mediated re-sponse by evaluating the action of this drug on theproduction of cytokines by human peripheral bloodmononuclear cells (PBMC) stimulated with M.tuberculosis.
2. Methods
2.1. PBMC isolation and culture conditions
PBMC were isolated from buffy coats from normaldonors over a Ficoll–Hypaque gradient, according tothe method of Boyum [33], and cultured on 12 well
plates at 2×106 cells/ml in RPMI 1640 supplementedwith 10% heat inactivated fetal calf serum, L-glu-tamine, penicillin, streptomycin and gentamycin. Incu-bations were performed at 37°C in a humidifiedatmosphere containing 5% CO2 in air. The cultureswere or not supplemented with fluvastatin dissolvedin DMSO (final concentration in the media 0.04%),or mevalonate as indicated. After a 12 h incubationin the mentioned conditions, the cultures were supple-mented with heat inactivated M. tuberculosis H37 RA(10 mg/ml) or placebo, without any other change inthe medium, and the incubation was prolonged for anadditional 24 h. At the end of the incubation, themedia were collected, cells in suspension were re-moved by centrifugation, and the supernatants werefrozen until cytokine determinations.
2.2. Immunoblot analysis
For these analyses both cells adhered and not ad-hered were combined. First, cells in suspension weresedimented by centrifugation and the supernatantswere reserved. Then, adherent cells were scraped andcombined with the non-adherent cells. The whole cellswere washed with PBS, resuspended in lysis buffer(62.5 mM Tris ClH pH 6.8, 2% SDS, supplementedwith a suitable antiprotease cocktail) and sonicated at0°C. The cell supernatants were treated with affigelblue (Bio Rad, Hercules, California) to remove thealbumin, and then concentrated three times by usingUltrafree concentrators, cut-off 10 000 (Millipore,Bedford, MA). Proteins from both cell lysates andsupernatants were subjected to electrophoresis accord-ing to Laemmli [34], on a discontinuous 6–11% SDS-PAGE, using a mini-gel system (Hoeffer ScientificInstruments, CA) and then transferred to Immobilonmembranes (pore size 0.45 mm, Millipore), using aSemi-phor semi-dry transfer unit (Hoeffer ScientificInstruments). Buffers used for transfer were: Anode 1:300 mM Tris, 10% methanol, pH 10.4; Anode 2: 25mM Tris, 10% methanol, pH 10.4; Cathode: 25 mMTris, 40 mM 6-aminocaproic acid, 20% methanol, pH9.4. A constant current of 0.22 mA/cm2 was appliedfor 18 h. The blots were blocked overnight at 4°C in5% non-fat dry milk in 20 mM Tris, 500 mM NaCl,pH 7.5. Then the blots were probed for 6 h withanti-human IL-1b serum (Santa Cruz Biotechnology,Santa Cruz, CA) diluted 1/500 in 2% non-fat drymilk in TTBS (20 mM Tris, 150 mM NaCl, 0.05%Tween 20, pH 7.5). After four, 10 min washes inTTBS, the blots were incubated with anti-goat alka-line phosphatase-conjugated serum (Santa Cruz Bio-technology), diluted 1/1000 in 2% non-fat dry milk inTTBS for 45 min and finally washed and developedaccording to the supplier. Incubations were performedat room temperature.

M.T. Montero et al. / Atherosclerosis 153 (2000) 303–313 305
2.3. Cytokine determinations
At the end of incubations, cell supernatants werecollected by centrifugation as mentioned above andcytokines measured by ELISA techniques. Im-munoassay kits for IL-1b and IFNg were obtainedfrom R&D Systems (Minneapolis, MN), IL-4 was fromICN Pharmaceuticals (Costa Mesa, CA), IL-12 andIL-10 were purchased from Diaclone Research (Be-sancon, France) and IL-18 was from Medical & Biolog-ical Laboratories (Nagoya, Japan). Assays wereperformed according to the manufacturer’s instructions.
2.4. Caspase-1 acti6ity
At the end of the incubation, both the adhered andnot adhered cells were collected, washed with PBS andlysed in a lysis buffer (0.5% Nonidet P-40, 0.5 mM
EDTA, 150 mM NaCl, 50 mM Tris pH 7.5, 1 mMphenylmethylsulfonyl fluoride, 1 mg/ml aprotinin, 50mg/ml antipain) [35]. The lysates were centrifuged at10 000×g for 10 min. at 4°C, and the supernatantswere collected. A volume of 200 ml aliquots of theextracts were incubated with the fluorogenic peptideDABCYL-YVADAPV-EDANS (Bachem AG, Buben-dorf, Switzerland), a specific substrate for caspase-1, ata final concentration of 10 mM in 2.5 ml of reactionbuffer (100 mM HEPES, pH 7.5, 10% (w/v) sucrose,0.1% (w/v) 3-[(3-cholamido-propyl) dimethylammonio]-1-propane sulfonate (CHAPS), 10 mM dithio-threitol(DTT), 0.1 mg/ml ovalbumin) for 1 hour at 37°C, andthe released EDANS was measured on a fluorescencespectrophotometer (Hitachi, Model 244-A) using anexcitation wavelength of 340 nm and emission of 490nm [36]. Activity was calculated as the change influorescence, and expressed as arbitrary units of fluores-cence (a.u.f) per mg of protein. Specificity of the reac-tion was assessed by adding equimolar amounts of thespecific inhibitor Ac-YVAD-CHO (Bachem AG,Bubendorf, Switzerland).
2.5. Analysis of apoptosis
Apoptosis was assessed by using the Apoptosis De-tection System kit (Promega Corporation, Madison,WI) following the manufacturer instructions, and it wasanalyzed by flow cytometry.
2.6. Statistical analysis
Values are means9SEM. The statistical significanceof changes was assessed at the 5% level using one-wayrepeated measures analysis of variance (parametric) orthe Friedman repeated measures ANOVA method onranks (non-parametric), where appropriate, and pair-wise multiple comparisons were run using the Student–Newman–Keuls method, by using the Sigma StatStatistical Analysis System package (Jandel Scientific).
3. Results
3.1. HMG-CoA reductase inhibition promotes IL-1b
and IFNg release by PBMC
PBMC were used instead of isolated leukocyte popu-lations to preserve the regulatory interactions betweenlymphocytes and monocytes/macrophages. Treatmentof PBMC with fluvastatin was observed to slightlyinduce the release of IL-1b to the medium in a dose-de-pendent manner (Fig. 1A, control conditions). M. tu-berculosis induced the release of IL-1b, yet in culturespre-treated with the statin, this stimulation was syner-gistic, reaching values up to ten times higher than those
Fig. 1. Dose-response effect of fluvastatin on IL-1b (A) and IFNg (B)release by human peripheral blood mononuclear cells (PBMC)treated or not with M. tuberculosis. PBMC were incubated in thepresence of increasing concentrations of fluvastatin for 12 h and thenfor an additional 24 h, after supplementing the medium with heat-in-activated M. tuberculosis H37 RA (10 mg/ml) () or placebo – –control-("). At the end of the 36-h incubation, both IL-1b and IFNgwere determined in the supernatants by specific ELISA. Data corre-spond to the means9S.E.M. of four independent experiments. Theeffect of fluvastatin on IL-1b and IFNg release, both in control andM. tuberculosis-stimulated conditions, was statistically significant asdetermined by ANOVA.
M.T. Montero et al. / Atherosclerosis 153 (2000) 303–313306
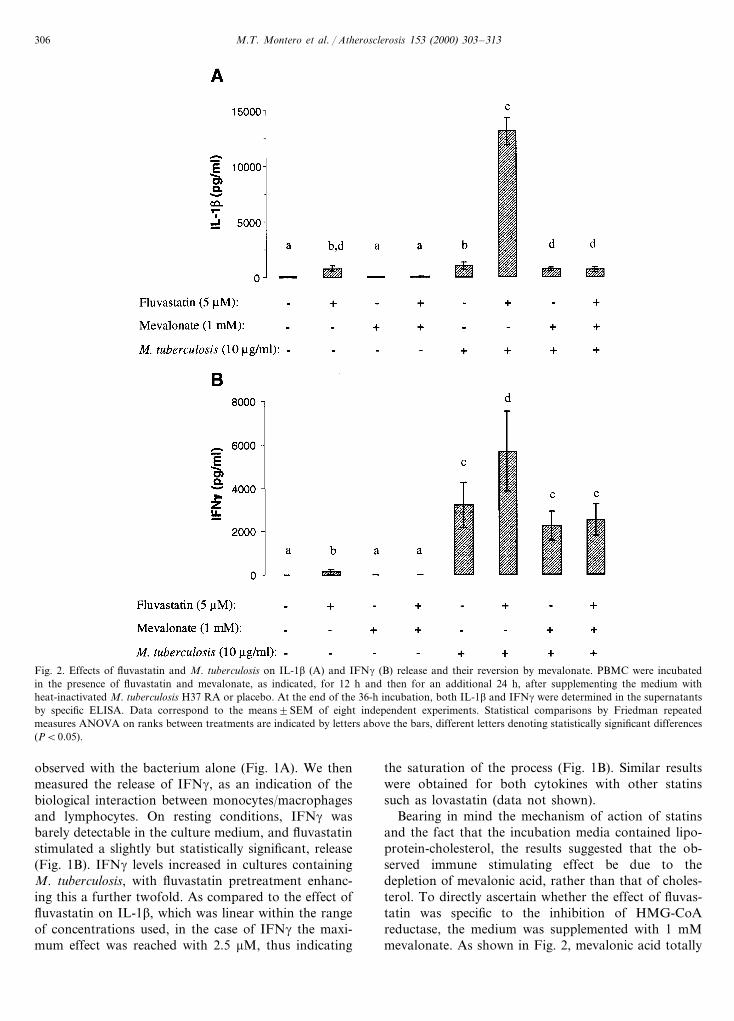
Fig. 2. Effects of fluvastatin and M. tuberculosis on IL-1b (A) and IFNg (B) release and their reversion by mevalonate. PBMC were incubatedin the presence of fluvastatin and mevalonate, as indicated, for 12 h and then for an additional 24 h, after supplementing the medium withheat-inactivated M. tuberculosis H37 RA or placebo. At the end of the 36-h incubation, both IL-1b and IFNg were determined in the supernatantsby specific ELISA. Data correspond to the means9SEM of eight independent experiments. Statistical comparisons by Friedman repeatedmeasures ANOVA on ranks between treatments are indicated by letters above the bars, different letters denoting statistically significant differences(PB0.05).
observed with the bacterium alone (Fig. 1A). We thenmeasured the release of IFNg, as an indication of thebiological interaction between monocytes/macrophagesand lymphocytes. On resting conditions, IFNg wasbarely detectable in the culture medium, and fluvastatinstimulated a slightly but statistically significant, release(Fig. 1B). IFNg levels increased in cultures containingM. tuberculosis, with fluvastatin pretreatment enhanc-ing this a further twofold. As compared to the effect offluvastatin on IL-1b, which was linear within the rangeof concentrations used, in the case of IFNg the maxi-mum effect was reached with 2.5 mM, thus indicating
the saturation of the process (Fig. 1B). Similar resultswere obtained for both cytokines with other statinssuch as lovastatin (data not shown).
Bearing in mind the mechanism of action of statinsand the fact that the incubation media contained lipo-protein-cholesterol, the results suggested that the ob-served immune stimulating effect be due to thedepletion of mevalonic acid, rather than that of choles-terol. To directly ascertain whether the effect of fluvas-tatin was specific to the inhibition of HMG-CoAreductase, the medium was supplemented with 1 mMmevalonate. As shown in Fig. 2, mevalonic acid totally

M.T. Montero et al. / Atherosclerosis 153 (2000) 303–313 307
prevented fluvastatin-induced stimulation of IL-1b andIFNg secretion, both in the presence and in the absenceof M. tuberculosis. Surprisingly, mevalonate supplemen-tation also reduced the release of IL-1b promoted bythe bacteria alone, thus suggesting that this immuno-logical response to M. tuberculosis is dependent onmevalonic acid availability. In order to elucidate themechanism underlying this phenomenon, the effect ofinhibitors of prenyl:protein transferases on IL-1b re-lease was determined. Both manumycin A and R(+ )-limonene, separately, at a concentration of 0.1 mM,enhanced the release of IL-1b in PBMC stimulated withM. tuberculosis, to 186930% (PB0.05, n=3) and to191930% (PB0.05, n=3), respectively, suggestingthat inhibition of protein prenylation is involved in thestimulation of IL-1b release.
3.2. HMG-CoA reductase inhibition acti6ates caspase-1enzyme
To determine whether statin-induced IL-1b releaseinvolved normal processing of the cytokine IL-1b,forms appearing in cell lysates and culture supernatantswere examined by gel electrophoresis and immunoblot-ting (Fig. 3). As a general observation, intracellularIL-1b corresponded to the 34 kDa pro-form, while thatin the culture supernatants corresponded to the 17 kDaactive form. Under control conditions, without im-munological stimulation, pro-forms were scarcely de-tected within the cells, with a slight increase in thosetreated with mevalonate (Fig. 3a), whereas negligibleamounts of mature IL-1b were detected in the superna-tants by this method regardless of the study conditions(Fig. 3c). In contrast, M. tuberculosis induced a signifi-
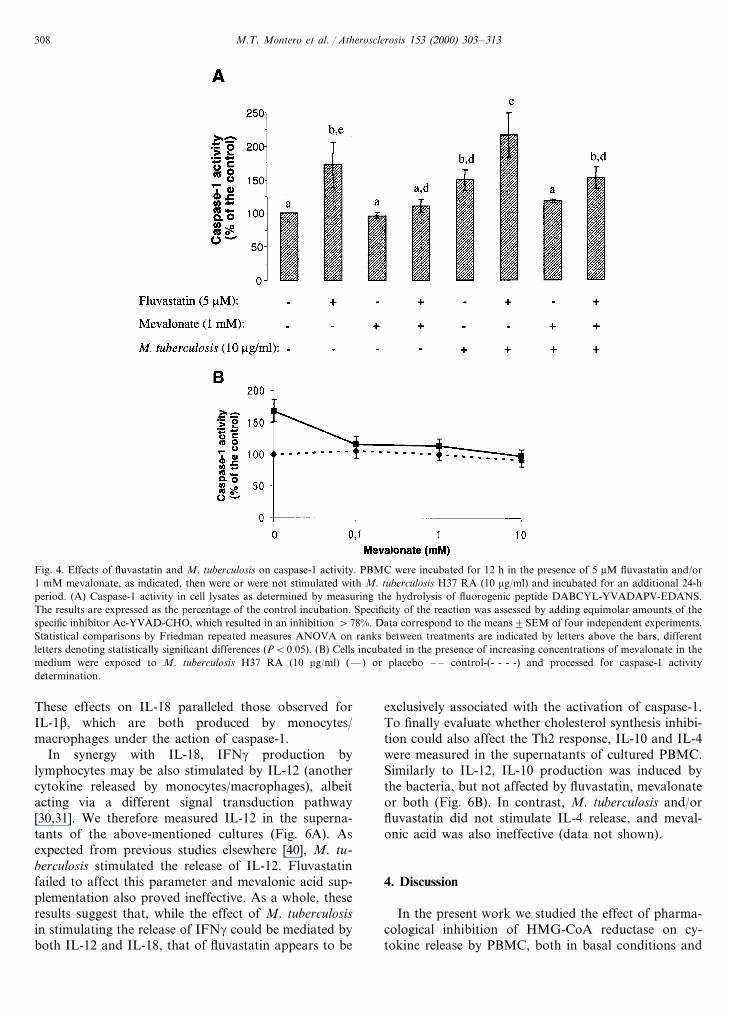
cant increase in intracellular pro-IL-1b (Fig. 3b), andthe mature 17-kDa form was also detected in thesupernatants (Fig. 3d). In cultures exposed to the bacte-ria, fluvastatin pretreatment yielded a marked decreasein the content of intracellular pro-IL-1b, along with aconcurrent increase in the mature form in cell superna-tants. This effect of fluvastatin proved to be specific,since it was totally prevented by mevalonic acid supple-mentation. The result reflects that inhibition of meval-onate biosynthesis affects the processing and release ofIL-1b. Moreover, in cultures stimulated with M. tuber-culosis alone, provision of exogenous mevalonate de-creased the amount of mature IL-1b in the supernatantand simultaneously increased the cellular content of thepro-form, thus firmly suggesting that mevalonic acid orsome isoprene derivative modulates the post-transla-tional processing of IL-1b. We therefore proceeded todetermine caspase-1 activity in cell lysates, after incu-bating PBMC under the above conditions. In Fig. 4Awe show that both fluvastatin and M. tuberculosis,separately, increased caspase-1 activity significantly,and their effects were additive although not signifi-cantly different from those of fluvastatin alone (Fig.4A). Mevalonic acid circumvented the effect of thestatin and, most interestingly, also reduced M. tubercu-losis-induced activation, an effect that was detectedeven at lower concentrations of mevalonic acid (Fig.4B). These results are in line with those on IL1-brelease and maturation described above, and indicatethat mevalonic acid deprivation results in caspase-1activation. Based on the involvement of caspase en-zymes on programmed cell death [37], we analyzed theinduction of apoptosis in our conditions. Neitherfluvastatin treatment nor the exposure to M. tuberculo-sis stimulated DNA fragmentation, as assessed byTUNEL assay, or increased the proportion of cells withDNA content lower than 2n (subG1), as measured bylabeling with propidium iodide and flow cytometryanalysis (data not shown). These results indicate thatnone of the treatments induced apoptosis in PBMC atthe time studied. Therefore, the changes in cytokineproduction mentioned above could not be ascribed toapoptosis induction.
3.3. Effect of HMG-CoA reductase inhibition on theproduction of IL-18, IL-12 and IL-4
Caspase-1 also participates in the maturation andrelease of IL-18 [8,9], a potent co-stimulatory factor forT lymphocyte production of IFNg [38,39]. As shown inFig. 5, both fluvastatin and M. tuberculosis, separately,stimulated the release of IL-18 and, when used incombination, their effect was greater than additive. Onthe other side, mevalonic acid supplementation abro-gated the effect of fluvastatin, both in stimulated and inunstimulated cells, indicating it was specific (Fig. 5).
Fig. 3. Fluvastatin stimulates the processing of pro-IL-1b. PBMCwere incubated for 12 h in the presence of 5 mM fluvastatin and/or 1mM mevalonate, as indicated, then were or were not stimulated withM. tuberculosis H37 RA (10 mg/ml) and incubated for an additional24-h period. Detection of pro-IL-1b (34 kDa) and mature IL-1b (17kDa) forms in cell extracts (a, b) and supernatants (c, d) by SDS-PAGE and immunoblot analysis using anti-human IL-1b antibody.Lane 1, control; lane 2, fluvastatin (5 mM); lane 3, mevalonate (1mM); lane 4, fluvastatin (5 mM) plus mevalonate (1 mM).
M.T. Montero et al. / Atherosclerosis 153 (2000) 303–313308
Fig. 4. Effects of fluvastatin and M. tuberculosis on caspase-1 activity. PBMC were incubated for 12 h in the presence of 5 mM fluvastatin and/or1 mM mevalonate, as indicated, then were or were not stimulated with M. tuberculosis H37 RA (10 mg/ml) and incubated for an additional 24-hperiod. (A) Caspase-1 activity in cell lysates as determined by measuring the hydrolysis of fluorogenic peptide DABCYL-YVADAPV-EDANS.The results are expressed as the percentage of the control incubation. Specificity of the reaction was assessed by adding equimolar amounts of thespecific inhibitor Ac-YVAD-CHO, which resulted in an inhibition \78%. Data correspond to the means9SEM of four independent experiments.Statistical comparisons by Friedman repeated measures ANOVA on ranks between treatments are indicated by letters above the bars, differentletters denoting statistically significant differences (PB0.05). (B) Cells incubated in the presence of increasing concentrations of mevalonate in themedium were exposed to M. tuberculosis H37 RA (10 mg/ml) (—) or placebo – – control-(- - - -) and processed for caspase-1 activitydetermination.
These effects on IL-18 paralleled those observed forIL-1b, which are both produced by monocytes/macrophages under the action of caspase-1.
In synergy with IL-18, IFNg production bylymphocytes may be also stimulated by IL-12 (anothercytokine released by monocytes/macrophages), albeitacting via a different signal transduction pathway[30,31]. We therefore measured IL-12 in the superna-tants of the above-mentioned cultures (Fig. 6A). Asexpected from previous studies elsewhere [40], M. tu-berculosis stimulated the release of IL-12. Fluvastatinfailed to affect this parameter and mevalonic acid sup-plementation also proved ineffective. As a whole, theseresults suggest that, while the effect of M. tuberculosisin stimulating the release of IFNg could be mediated byboth IL-12 and IL-18, that of fluvastatin appears to be
exclusively associated with the activation of caspase-1.To finally evaluate whether cholesterol synthesis inhibi-tion could also affect the Th2 response, IL-10 and IL-4were measured in the supernatants of cultured PBMC.Similarly to IL-12, IL-10 production was induced bythe bacteria, but not affected by fluvastatin, mevalonateor both (Fig. 6B). In contrast, M. tuberculosis and/orfluvastatin did not stimulate IL-4 release, and meval-onic acid was also ineffective (data not shown).
4. Discussion
In the present work we studied the effect of pharma-cological inhibition of HMG-CoA reductase on cy-tokine release by PBMC, both in basal conditions and

M.T. Montero et al. / Atherosclerosis 153 (2000) 303–313 309
under the stimulation with M. tuberculosis, with the aimto evaluate whether treatment with statins could affectthe Th1 immune response.
In principle, Th1 response is characterized by releaseof IFNg and IL-2 by T lymphocytes, which is usuallyaccompanied by IL-1b secretion from monocyte/macrophages [24], all defining a proinflammatory re-sponse. Release of mature IL-1b requires two signals:the primary stimulus to promote transcription andtranslation of the pro-form, the second to activatecaspase-1 [41,42]. This second stimulus provides anadditional checkpoint whereby cells regulate the biosyn-thesis of this interleukin. It is worth mentioning thatseveral pathogens act upon caspase-1, either activatingor inhibiting it [43–45], which highlights the impor-tance of this enzyme in the inflammatory response toinfection. In our conditions we observed that the re-sponse to M. tuberculosis comprised both the inductionof pro-IL-1b synthesis and caspase-1 activation. How-ever, enzyme activation induced by the bacteria was ofa relatively small magnitude and, as in the case oflipopolysaccharide (LPS) [46], a significant proportionof pro-IL-1b is accumulated within the cells. Pretreat-ment with fluvastatin further increased caspase-1 activ-ity when cells were exposed to the bacterium and theamount of IL-1b released was greatly enhanced, the cellbecoming depleted of pro-forms. The synergistic natureof this effect could thus be attributed to action atdifferent levels, namely, the pathogen preferentially in-ducing pro-IL-1b expression and the statin activatingcaspase-1. Indeed, fluvastatin treatment would not seemto induce pro-IL-1b expression, as indicated by thescant detection of the pro-form in lysates from non-
stimulated cells (Fig. 2A), which resulted in limitedsecretion of mature IL-1b despite the fact that caspase-1 was activated. In keeping with this interpretation,when fluvastatin-pretreated cells were exposed to M.tuberculosis, release of IL-1b was very high, likely be-cause pro-forms were induced and proteolytic process-ing was maximally stimulated. Accordingly, theseresults suggest that inhibition of mevalonate synthesisconstitutes a second signal that triggers IL-1b process-ing and release. This response appeared not to beassociated with apoptosis induction, since in our condi-tions programmed cell death was not stimulated byeither fluvastatin or the bacteria. Apparently, this lastresult is in contrast with previous ones by us [18] andothers [47,48], who reported the induction of apoptosisin established cell lines by effect of statins. It should benoted, however, that this effect of HMG-CoA reduc-tase inhibitors is detectable in proliferating but not inquiescent cells [48], thus, our results in unstimulatedPBMC are in line with the latter observations.
Similarly to caspase-3 [48], caspase-1 activation byeffect of fluvastatin herein shown appears to be related,at least in part, to the inhibition of protein prenylationsecondary to mevalonate deficiency, since direct in-hibitors of protein prenylation stimulated IL-1b secre-tion, as shown in the present work. Interestingly,bisphosphonates, which are antiresorptive drugs thatco-laterally affect protein prenylation [49], have beenshown to activate caspase-3 [50] and to enhance theIL-1b release induced by LPS in macrophages [51,52].
One of the most intriguing observations was theinteraction of mevalonic acid with the response to M.tuberculosis. Supplementation with mevalonate slightly,
Fig. 5. Effects of fluvastatin and M. tuberculosis on IL-18 release. PBMC were incubated in the presence of fluvastatin and mevalonate, asindicated, for 12 h and then for an additional 24 h, after supplementing the medium with heat-inactivated M. tuberculosis H37 RA or placebo.At the end of the 36-h incubation, IL-18 was determined in the supernatants by specific ELISA. Data correspond to the means9SEM of eightindependent experiments. Statistical comparisons by Friedman repeated measures ANOVA on ranks between treatments are indicated by lettersabove the bars, different letters denoting statistically significant differences (PB0.05).

M.T. Montero et al. / Atherosclerosis 153 (2000) 303–313310
Fig. 6. Effects of fluvastatin and M. tuberculosis on IL-12 (A) and IL-10 (B) release. PBMC were incubated in the presence of fluvastatin andmevalonate, as indicated, for 12 h and then for an additional 24 h, after supplementing the medium with heat-inactivated M. tuberculosis H37 RAor placebo. At the end of the 36-h incubation, both IL-12 p70 and IL-10 were determined in the supernatants by specific ELISA. Data correspondto the means9SEM of eight (IL-12) or four (IL-10) independent experiments. Statistical comparisons by Friedman repeated measures ANOVAon ranks between treatments are indicated by letters above the bars, different letters denoting statistically significant differences (PB0.05).
yet consistently, prevented caspase-1 activation and re-duced the secretion of IL-1b induced by the pathogen.This further indicates that caspase-1 activation is re-sponsive to the availability of mevalonic acid. It is notknown yet whether M. tuberculosis inhibits the meval-onate pathway to induce caspase-1 activation in thehost, a possibility which will have to be expresslyevaluated by further studies. In this regard, a reductionin HMG-CoA reductase activity has been reported inmurine macrophages primed in vivo with Corynebac-terium parvum [53].
IL-18 is a cytokine that acts as a costimulatory factorfor the production of IFNg [28–31,38,39], one of the
most important cytokine that seems to be involved inthe development of the atherosclerotic lesion [8,11,54].As for IL-1b, cleavage by caspase-1 of pro-IL-18 isrequired before the mature form is secreted. We ob-served that treatment with fluvastatin gave rise to anincrease of IL-18 secretion in non-stimulated cells and,more intensely, in M. tuberculosis-stimulated cells,likely due to caspase-1 activation. It is to note that,IL-18 release in response to fluvastatin was as high asto M. tuberculosis when added separately to the cul-tures. This relatively high response to fluvastatin maybe attributed to constitutive gene expression of IL-18,as recently reported in unstimulated freshly isolated

M.T. Montero et al. / Atherosclerosis 153 (2000) 303–313 311
human PBMC [55]. Despite that IL-18 levels weresimilar, the release of IFNg was higher in response toM. tuberculosis than to the statin, likely because thebacteria also stimulated the release of IL-12 (Fig. 6),and both interleukins cooperate together for the stimu-lation of IFNg release, as recently reported by others[30]. Given that in our cell system, release of IL-12/IL-10 was not affected by the statin, the increased produc-tion of IFNg induced by the drug in the presence ofbacteria must be mainly ascribed to the increment ofIL-18 and/or IL-1b rather than to changes in IL-12 orIL-10. This is the first evidence that statins couldenhance Th1 cytokine production.
IFNg has been implicated in the development ofatherosclerotic, since mice lacking IFNg receptors aremore resistant to atherosclerosis [54]. This together withthe frequent detection of this cytokine in humanatherosclerotic plaques has led to the notion that firststeps in lesion formation are governed by a Th1-cellresponse [7,8,10,54]. However, once the lesion has beenestablished, IFNg may be beneficial since this cytokinereduces smooth muscle cell and macrophage prolifera-tion [56,57] and collagen synthesis [58], prevents metal-loproteinase activation in the plaque [59], anddownregulates scavenger receptor expression [60]. Be-sides, since IFNg plays a crucial role as activator ofdiverse macrophage effector functions, it could protectthe lesion against intracellular infectious agents thatwould complicate the lesions, as is the case of Chlamy-dia pneumonie. Thus, the influence of local secretion ofIFNg on atherosclerotic plaque disruption is notevident.
In advanced lesions, the presence of type Th2 (IL-10and IL-4) in conjunction with type Th1 cytokines havebeen recently reported [7,9,10]. Since Th1 and Th2 cellsreciprocally inhibit each other, it has been hypothesizedthat IL-10 locally produced may protect the atheroscle-rotic lesion from excessive inflammatory response, bydownregulating Th1-cell activation [9,10]. However,Th2 cytokines induce a humoral response, implying achange in IgG isotype, IgE production, activation ofcomplement components that mediate cytolysis [61] andactivation of different effector cells, such as basophils,eosinophils and mast cells, which release powerful va-soactive substances and potent proteolytic enzymes[62,63]. Interestingly, mast cells were recently shown toaccumulate in coronary plaques at the site of rupture[64] and an increase of degranulated mast cells in theadventicia backing ruptured plaques has been reported[65]. Moreover, there is evidence from other systems,that inflammatory lesions mediated by mixed Th1+Th2 (or Th0) T cell activity are susceptible to necrosis,and disease progression is associated to a worse prog-nostic [66,67]. We, therefore, hypothesize that a typeTh1-driven inflammatory lesions, while promoting le-sion growth, ensues the stabilization of the plaque,
whereas the combination of cytokines type Th1-Th2correlates with an increase in tissue damage, resulting innecrosis and plaque rupture. In the light of presentresults, we suggest that statins could be immunoprotec-tive by preserving and/or reestablishing a Th1 cytokineprofile in the atheroma, which helps to stabilize theplaque.
Systemic administration of cytokines for the restora-tion of a protective cytokine profile is currently re-garded as a therapeutic strategy to resolve criticaldiseases [68,69]. Results presented herein, besides illus-trating a link between cholesterol metabolism and theinflammatory response, provide a tool for modulatingthe cytokine secretion pattern Th1, which has a poten-tial to be used for therapeutic purposes. Since thismodulation is synergistic with an immune stimulus,cytokine regulation could be expected to be principallyaffected at the lesion site, hence side effects due tosystemic action would be avoided. Accordingly, phar-macological inhibition of mevalonate synthesis could bean alternative adjuvant to boost the immune Th1-typeresponse to M. tuberculosis, but until more direct clini-cal assays are performed, the benefit of such a pharma-cological intervention must remain speculative.
Acknowledgements
This study was supported by grants from the Fondode Investigacion Sanitaria (FIS 97/0389 and 99/0286)and Comision Interministerial de Ciencia y Tecnologıa(SAF 96/0011), Spain. We thank Miguel Martın for hisexcellent technical assistance and Novartis for provid-ing us with fluvastatin. We are deeply indebted to allDonor Unit personnel, at the Service of Hematology,Hospital La Paz, Madrid, Spain, for supplying thebuffy coats, and to Dr. Enrique Gomez Mampaso, atthe Service of Microbiology, Hospital Ramon y Cajal,Madrid, for providing the heat-inactivated M. tubercu-losis H37 RA.
References
[1] Ross R. The pathogenesis of atherosclerosis: a perspective forthe 1990s. Nature 1993;362:801–8.
[2] Hansson GK. Cell-mediated immunity in atherosclerosis. CurrOpin Lipidol 1997;8:301–11.
[3] Kishikawa H, Shimokama T, Watanabe T. Localization of Tlymphocytes and macrophages expressing IL-1, IL-2 receptor,IL-6 and TNF in human aortic intima. Role of cell-mediatedimmunity in human atherogenesis. Virchows Archiv A PatholAnat 1993;423:433–42.
[4] Nagornev VA. Kinetics of vascular wall cells and atherogenesis.Arkh Pathol 1998;60:39–43.
[5] Berliner JA, Navab M, Fogelman AM, et al. Atherosclerosis:basic mechanisms. Oxidation, inflammation, and genetics. Circu-lation 1995;91:2488–96.

M.T. Montero et al. / Atherosclerosis 153 (2000) 303–313312
[6] Seely S. Is atherosclerosis an overreaction of the immune system?Int J Cardiol 1998;66:323–4.
[7] Uyemura K, Demer LL, Castle SC, et al. Cross-regulatory rolesof interleukin(IL)-12 and IL-10 in atherosclerosis. J Clin Invest1996;97:2130–8.
[8] Frostegard J, Ulfgren A, Nyberg P, et al. Cytokine expression inadvanced human atherosclerotic plaques: dominance of pro-infl-ammatory (Th1) and macrophage-stimulating cytokines.Atherosclerosis 1999;145:33–43.
[9] Mallat Z, Heymes C, Ohan J, Faggin E, Leseche G, Tedgui A.Expression of interleukin-10 in advanced human atheroscleroticplaques: relation to inducible nitric oxide synthase expressionand cell death. Arterioscler Thromb Vasc Biol 1999;19:611–6.
[10] Lee TS, Yen HC, Pan CC, Chau LY. The role of interleukin 12in the development of atherosclerosis in ApoE-deficient mice.Arterioscler Thromb Vasc Biol 1999;19:734–42.
[11] Zhou X, Paulsson G, Stemme S, Hansson G. Hypercholes-terolemia is associated with a T helper (Th)1/Th2 switch of theautoimmune response in atherosclerotic apo E-knockout mice. JClin Invest 1998;101:1717–25.
[12] Folcik VA, Aamir R, Cathcart MK. Cytokine modulation ofLDL oxidation by activated human monocytes. ArteriosclerThromb Vasc Biol 1997;17:1954–61.
[13] Wick G, Schett G, Amberger A, Kleindienst R, Xu Q. Isatherosclerosis an immunologically mediated disease? ImmunolToday 1995;16:27–33.
[14] Xu Q, Kleindienst R, Schett G, et al. Regression of arterioscle-rotic lesions induced by immunization with heat shock protein65-containing material in normocholesterolemic, but not hyperc-holesterolemic, rabbits. Atherosclerosis 1996;123:146–55.
[15] Massy ZA, Keane WF, Kasiske BL. Inhibition of the meval-onate pathway: benefits beyond cholesterol reduction? Lancet1996;347:102–3.
[16] Rossen RD. HMG-CoA reductase inhibitors: a new class ofanti-inflammatory drugs? J Am Coll Cardiol 1997;30:218–9.
[17] Chakrabarti R, Engleman EG. Interrelationships between meval-onate metabolism and the mitogenic signaling pathway in Tlymphocyte proliferation. J Biol Chem 1991;266:12216–22.
[18] Carrero P, Ortega H, Martınez-Botas J, Gomez-Coronado D,Lasuncion MA. Flavonoid-induced ability of minimally modifiedlow-density lipoproteins to support lymphocyte proliferation.Biochem Pharmacol 1998;55:1125–9.
[19] Weber C, Erl W, Weber KS, Weber PC. HMG-CoA reductaseinhibitors decrease CD11b expression and CD11b-dependentadhesion of monocytes to endothelium and reduce increasedadhesiveness of monocytes isolated from patients with hyperc-holesterolemia. J Am Coll Cardiol 1997;30:1212–7.
[20] Han DC, Kim JH, Cha MK, et al. Effect of HMG-CoAreductase inhibition on TGF-b1 mRNA expression in diabeticrat glomeruli. Kidney Int 1995;48:S61–5.
[21] Terkeltaub R, Solan J, Barry M, Santoro D, Bockoch GM. Roleof mevalonate pathway of isoprenoid synthesis in IL-8 genera-tion by activated monocytic cells. J Leuk Biol 1994;55:749–55.
[22] Colli S, Eligini S, Lalli M, Camera M, Paoletti R, Tremoli E.Vastatins inhibit tissue factor in cultured human macrophages. Anovel mechanism of protection against atherotrombosis. Arte-rioscler Thromb Vasc Biol 1997;17:265–71.
[23] Pahan K, Sheikh FG, Namboodiri AM, Singh I. Lovastatin andphenylacetate inhibit the induction of nitric oxide synthase andcytokines in rat primary astrocytes, microglia and macrophages.J Clin Invest 1997;100:2671–9.
[24] Chizzolini C, Chicheportiche R, Burger D, Dayer JM. HumanTh1 cells preferentially induce interleukin (IL)-1beta while Th2cells induce IL-1 receptor antagonist production upon cell/cellcontact with monocytes. Eur J Immunol 1997;27:171–7.
[25] Mendez-Samperio P, Hernandez-Garay M, Badillo-Flores A,Nunez-Vazquez A. Down-modulation of mycobacterial-induced
IL-1b production in human mononuclear cells by IL-4. Clin ExpImmunol 1996;104:374–9.
[26] Thornberry NA, Bull HG, Calaycay JR, et al. A novel het-erodimeric cysteine protease is required for interleukin-1b pro-cessing in monocytes. Nature 1992;356:768–74.
[27] Howard AD, Kostura MJ, Thornberry NA, et al. IL-1-convert-ing enzyme requires aspartic acid residues for processing of theIL-1b precursor at two distinct sites and does not cleave 31-kDaIL-1a. J Immunol 1991;147:2964–9.
[28] Gu Y, Kuida K, Tsutsui H, et al. Activation of interferon-ginducing factor mediated by interleukin-1b converting enzyme.Science 1997;275:206–9.
[29] Ghayur T, Banerjee S, Hugunin M, et al. Caspase-1 processesIFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature 1997;286:619–23.
[30] Okamura H, Kashiwamura S, Tsutsui H, Yoshimoto T, Nakan-ishi K. Regulation of interferon-gamma production by IL-12 andIL-18. Curr Opin Immunol 1998;10:259–64.
[31] Garcia VE, Uyemura K, Sieling PA, et al. IL-18 promotes Type1 cytokine production from NK cells and T cells in humanintracellular infection. J Immunol 1999;162:6114–21.
[32] Billiau A. Interferon-g: biology and role in pathogenesis. AdvImmunol 1996;62:61–130.
[33] Boyum A. Separation of leucocytes from blood and bone mar-row. Scand J Clin Invest 1968;21:77–89.
[34] Laemmli UK. Cleavage of structural proteins during the assem-bly of the head of bacteriophage T4. Nature 1970;227:680–5.
[35] Sekine C, Yagita H, Kobata T, Hasunuma T, Nishioka K,Okumura K. Fas-mediated stimulation induces IL-8 secretion byrheumatoid artritis synoviocytes independently of CPP32-medi-ated apoptosis. Biochem Biophys Res Commun 1996;228:14–20.
[36] Thornberry NA. Interleukin-1b converting enzyme. Meth Enzy-mol 1994;244:615–31.
[37] Enari M, Talanian RV, Wong WW, Nagata S. Sequential activa-tion of ICE-like and CPP32-like proteases during Fas-mediatedapoptosis. Nature 1996;380:723–6.
[38] Kohno K, Kataoka J, Ohtsuki T, et al. IFN-gamma-inducingfactor (IGF) is a costimulatory factor on the activation of Th1but not Th2 cells and exerts its effect independently of IL-12. JImmunol 1997;158:1541–50.
[39] Bohn E, Sing A, Zumbihl R, et al. IL-18 (IFN-gamma-inducingfactor) regulates early cytokine production in, and promotesresolution of, bacterial infection in mice. J Immunol1998;160:299–307.
[40] Ladel CH, Szalay G, Riedel D, Kaufmann SH. Interleukin-12secretion by Mycobacterium tuberculosis-infected macrophages.Infect Immun 1997;65:1936–8.
[41] Chin J, Kostura MJ. Dissociation of IL-1b synthesis and secre-tion in human blood monocytes stimulated with bacterial cellwall products. J Immunol 1993;151:5574–85.
[42] Miossec C, Decoen MC, Durand L, Fassy F, Diu-Hercend A.Use of monoclonal antibodies to study interleukin-1 beta-con-verting enzyme expression: only precursor forms are detected ininterleukin-1 beta-secreting cells. Eur J Immunol 1996;26:1032–42.
[43] Ray CA, Black RA, Kronheim SR, et al. Viral inhibition ofinflammation: cowpox virus encodes an inhibitor of the inter-leukin-1 beta converting enzyme. Cell 1992;69:597–604.
[44] Hilbi H, Chen Y, Thirumalai K, Zychlinsky K. The interleukin1 beta-converting enzyme, caspase-1, is activated during Shigellaflexneri induced apoptosis in human monocyte derivedmacrophages. Infect Immun 1997;65:5165–70.
[45] Barsig J, Kaufmann SH. The mechanism of cell death in Listeriamonocytogenes-infected murine macrophages is distinct fromapoptosis. Infect Immun 1997;65:4075–81.
[46] Schumann RR, Belka C, Reuter D, et al. Lipopolysaccharideactivates caspase-1 (interleukin-1-converting enzyme) in culturedmonocytic and endotelial cells. Blood 1998;91:577–84.

M.T. Montero et al. / Atherosclerosis 153 (2000) 303–313 313
[47] Marcelli M, Cunningham GR, Haidacher SJ, et al. Caspase-7 isactivated during lovastatin-induced apoptosis of the prostatecancer cell line LNCaP. Cancer Res 1998;58:76–83.
[48] Vitale M, Di Matola T, Rossi G, Laezza C, Fenzi G, Bifulco M.Prenyltransferase inhibitors induce apoptosis in proliferatingthyroid cells through a p53-independent, CrmA-sensitive, andcaspase-3-like protease-dependent mechanism. Endocrinology1999;140:698–704.
[49] Luckman SP, Hughes DE, Coxon FP, Graham R, Russell G,Rogers MJ. Nitrogen-containing bisphosphonates inhibit themevalonate pathway and prevent post-translational prenylationof GTP-binding proteins, including Ras. J Bone Miner Res1998;13:581–9.
[50] Coxon FP, Benford HL, Russell RG, Rogers MJ. Protein syn-thesis is required for caspase activation and induction of apopto-sis by bisphosphonate drugs. Mol Pharmacol 1998;54:631–8.
[51] Monkkonen J, Simila J, Rogers MJ. Effects of tiludronate andibandronate on the secretion of proinflammatory cytokines andnitric oxide from macrophages in vitro. Life Sci 1998;62:95–102.
[52] Makkonen N, Salminen A, Rogers MJ, et al. Contrasting effectsof alendronate and clodronate on RAW 264 macrophages: therole of a bisphophonate metabolite. Eur J Pharm Sci1999;8:109–18.
[53] Kraemer FB, Tavangar K, Gandjei RK, Kirlew K, Behr SR.Effects of activation on lipid and lipoprotein metabolism inmurine macrophages. Arteriosclerosis 1990;10:8–16.
[54] Gupta S, Pablo AM, Jiang X, Wang N, Tall AR, Schindler C.IFN-g potentiates atherosclerosis in ApoE knockout mice. J ClinInvest 1997;99:2752–61.
[55] Puren AJ, Fantuzzi G, Dinarello CA. Gene expression, synthe-sis, and secretion of interleukin 18 and interleukin 1 beta aredifferentially regulated in human blood mononuclear cells andmouse spleen cells. Proc Natl Acad Sci USA 1999;96:2256–61.
[56] Hansson GK, Holm J, Holm S, Fotev Z, Hedrich HJ, FingerleJ. T lymphocytes inhibit the vascular response to injury. ProcNatl Acad Sci USA 1991;88:10530–4.
[57] Dey A, Kim L, Li W. Gamma interferon induces expression ofMad1 gene in macrophage, which inhibits colony-stimulatingfactor-1-dependent mitogenesis. J Cell Biochem 1999;72:232–41.
[58] Amento EP, Ehsani N, Palmer H, Libby P. Cytokines andgrowth factors positively and negatively regulate interstitial col-lagen gene expression in human vascular smooth muscle cells.Arterioscler Thromb 1991;11:1223–30.
[59] George SJ. Tissue inhibitors of metalloproteinases and metallo-proteinases in atherosclerosis. Curr Opin Lipidol 1998;9:413–23.
[60] Geng YJ, Hansson GK. Interferon-gamma inhibits scavengerreceptor expression and foam cell formation in human mono-cyte-derived macrophages. J Clin Invest 1992;89:1322–30.
[61] Niculescu F, Rus H. Atherosclerosis and the immune system[letter]. Arch Intern Med 1999;159:315.
[62] Metzler B, Xu Q. The role of mast cells in atherosclerosis. IntArch Allergy Immunol 1997;114:10–4.
[63] Galli SJ. New concepts about the mast cell. N Engl J Med1993;328:257–65.
[64] Kovanen PT, Kaartinen M, Paavonen T. Infiltrates of activatedmast cells at the site of coronary atheromatous erosion orrupture in myocardial infarction. Circulation 1995;92:1084–8.
[65] Laine P, Kaartinen M, Penttila A, Panula P, Paavonen T,Kovanen PT. Association between myocardial infarction and themast cells in the adventitia of the infarct-related coronary artery.Circulation 1999;99:361–9.
[66] Rook GAW, Hernandez-Pando R. The pathogenesis of tubercu-losis. Ann Rev Microbiol 1996;50:259–84.
[67] Barnes FP, Modlin RL. Human cellular immune responses toMycobacterium tuberculosis. Curr Top Microbiol Immunol1996;215:197–219.
[68] Docke WD, Randow F, Syrbe U, et al. Monocyte deactivationin septic patients: restoration by IFN-g treatment. Nature Med1997;3:678–81.
[69] Maini A, Morse PD, Wang CY, Jones RP, Haas GP. Newdevelopments in the use of cytokines for cancer therapy. Anti-cancer Res 1997;17:3803–8.
.




















