
Flow cytometric quantification of HIV1 Tat protein in tat-transfected Jurkat T cell lines
Upload
independentCategory
view
5download
0
HIV-1 and Cardiomyocyte Apoptosis 97
Cardiovascular Toxicology Humana Press Volume 4, 200497
*Author to whom allcorrespondence and reprintrequests should beaddressed: Milan Fiala, MD,UCLA CHS 63-090,10833 Le Conte Avenue,Los Angeles, CA 90095-1760. E-mail: [email protected].
Received: 12/6/2003
Revised: 2/6/2004
Accepted: 2/6/2004
Cardiovascular Toxicology,vol. 4, no. 2, 97–107, 2004
HIV-1 Induces Cardiomyopathyby Cardiomyocyte Invasion and gp120,Tat, and Cytokine Apoptotic SignalingMilan Fiala,*,1,2 Waldemar Popik,3 Jian-Hua Qiao,4
Albert S. Lossinsky,5 Timothy Alce,3 Kenix Tran,1
Wendy Yang,1 Kenneth P. Roos,2 and James Arthos6
1Department of Medicine, Greater Los Angeles VA Medical Center;2Cardiovascular Research Laboratory; 3Oncology Center, The JohnsHopkins University School of Medicine, Baltimore, MD; 4Departmentof Pathology, David Geffen School of Medicine at UCLA, Los Angeles, CA;5Huntington Medical Research Institutes, Pasadena, CA; and 6Laboratoryof Immunoregulation, National Institute of Allergy and Infectious Diseases,National Institutes of Health, Bethesda, MD
Cardiovascular Toxicology (2004) 04 97–107 $25.00 (http://www.cardiotox.com)© Copyright 2004 by Humana Press Inc. All rights of any nature whatsoever reserved. 1530-7905/01Humana Press
AbstractWe examined heart tissues of AIDS patients with or without HIV cardiomy-
opathy (HIVCM) by immunohistochemistry, in situ polymerase chain reac-tion, in situ riboprobe hybridization, and the TUNEL technique for apoptosis.In HIVCM tissues, only inflammatory cells, but not endothelial cells or car-diomyocytes, displayed HIV-1 DNA and RNA. However, macrophages, lym-phocytes, and—in a patchy fashion—cardiomyocytes and endothelial cellsexhibited virus envelope protein gp120. Macrophages infiltrated the myocar-dium in a perivascular fashion and expressed tumor necrosis factor familyligands; adjacent cardiomyocytes suffered apoptosis. In vitro HIV-1 stronglyinvaded neonatal rat ventricular myocytes (NRVMs) and coronary arteryendothelial cells (CAECs) and induced microvilli but did not replicate. HIV-1,gp120, or Tat induced Erk 1/2 phosphorylation, activation of caspase-3, andapoptosis of NRVMs and CAECs; all of these were inhibited by a MAPK/ERK-kinase (MEK) inhibitor U0126. The pathogenesis of HIVCM involves HIV-1replication in inflammatory cells and induction of cardiomyocyte apoptosisby (1) the extrinsic pathway through apoptotic ligands and (2) the intrinsicpathway through direct virus entry and gp120- and Tat-proapoptotic signaling.Key Words: HIV cardiomyopathy; cardiomyocyte apoptosis; HIV-1 envelopeprotein gp120; HIV-1 protein Tat; macrophage.
Introduction
Cardiovascular complications of HIV-1 infection involve a range of conditions
from contractile dysfunction, arrhythmias, and pulmonary hypertension to end-
stage dilated HIV cardiomyopathy (HIVCM). Although HIV-1 infection is consid-
ered to be a necessary cause of HIVCM in severely immunosuppressed patients,
opportunistic viral and nonviral infections are contributory. HIV-1 invades the
Fiala (04-119).p65 8/10/2004, 3:26 PM97

98 Fiala et al.
Cardiovascular Toxicology Humana Press Volume 4, 2004
heart by transcellular passage through coronary
artery endothelial cells (CAECs) (1) and by “Tro-
jan” transport in infiltrating monocyte/macrophages
(2). HIV-1 invades neonatal rat ventricular myocytes
(NRVMs) by macropinocytosis but does not repli-
cate in these cells (3). Previous studies of HIVCM
hearts identified HIV-1 RNA and DNA in heart tis-
sues but did not identify the infected cells (4–6). Some
hearts with HIVCM have shown a complete lack of
HIV-1 footprints. Enteroviruses are considered to
cause viral myocarditis by overt or undetected infec-
tion of cardiomyocytes (7), and their presence has
been detected in some AIDS hearts (8). Besides end-
stage damage, HIV-1 also exerts earlier pathophys-
iological effects on heart contractility (9), possibly
mediated by oxidative and nitration stress, as sug-
gested in rat cardiomyocytes (10) and a murine model
(11). Nucleoside reverse transcriptase inhibitors cause
mitochondrial toxicities (12), which could also lead
to heart failure (13). Consequently, the role of HIV-1
in HIVCM has not been completely delineated.
A critical mechanism of HIVCM is cardiomyocyte
apoptosis, which is found in over 50% of cardiomyo-
cytes in a patchy fashion adjacent to infiltrating HIV-
1-infected macrophages. These macrophages pro-
duce inflammatory cytokine tumor necrosis factor-�
(TNF-�) which induces cardiomyocyte apoptosis by
the extrinsic pathway and the viral protein gp120,
which causes intrinsic apoptosis. In a previous study,
gp120 expression was significantly associated with
HIVCM, whereas HIV-1 proteins Tat and Nef were
not significantly associated (3).
To elucidate which cells are the source and targets
of infectious HIV-1 and cytokines in the cardiovascu-
lar system, we examined virus nucleic acids and pro-
teins and cardiomyocyte apoptosis in the hearts of
infected patients. HIV-1 DNA was found only in in-
flammatory cells, whereas viral protein was detected
in inflammatory cells, endothelial cells, and cardio-
myocytes. We investigated the relationship between
cardiomyocyte apoptosis and infected macrophages
to detect a paracrine mechanism of apoptosis in heart
tissues. In HIVCM heart tissues, but not in other
AIDS heart tissues without HIVCM, cardiomyocytes
suffered an extremely high rate of apoptosis in a focal
fashion. Using cultured cardiomyocytes and endo-
thelial cells, we examined gp120- and Tat-induced
cell signaling and demonstrated the role of Erk phos-
phorylation in apoptosis.
Materials and Methods
Heart Tissues
This study was performed with formalin-fixed heart
tissues from the left ventricle of 21 patients with AIDS,
of which 10 were diagnosed as HIVCM and 11 had no
history or biochemical evidence of heart failure. The
tissues were provided by the Manhattan HIV Brain
Bank (R24MH59724) and Texas Repository for AIDS
Neuropathogenesis Research (R24MH 59656 and
R24NS 45491). The UCLA Office for Protection of
Research Subjects approved all work with human
tissues and blood cells. We previously described the
immunohistochemistry (IHC) of 17 hearts from this
group of patients (3).
HIV-1 and HIV-1 Proteins
HIV-1
HIV-1NL4-3, incorporating Vpr-GFP (named HIV-
1NL4-3 Vpr-GFP), was produced by cotransfection of
293 cells with pNL4-3 DNA and an expression vec-
tor coding for GFP-Vpr fusion protein (3).
gp120
Recombinant soluble gp120 from macrophage-
tropic strain JR-FL was produced in Chinese hamster
ovary (CHO) cells, essentially as described for simian
immunodeficiency virus (SIV) envelope (14). An HIV-
1 env was inserted into a mammalian expression vec-
tor and transfected into the CHO cell. A cell clone
expressing a high level of soluble gp120 was selected.
Concentrated culture supernatants of this clone were
purified on a lectin column, eluted with �-mannopyra-
noside, and then passed through an anti-�-macroglo-
bulin column.
Tat
HIV-1 Tat was expressed in Escherichia coli by
using wild-type pGEM2 Tat bacterial expression
vector and purified on an Aquapore RP-300 column
(Applied Biosystems, Foster City, CA) by reverse-
phase chromatography, as described (15).
Cell Culture
Primary CAECs were purchased from Clonetics/
BioWhittaker (Walkersville, MD) and cultured by
using an EGM®-2-MV Bullet Kit medium, as described
(16). NRVMs were prepared from neonatal rat heart
tissues with full approval of the UCLA Animal Re-
search Committee, as described (3). CD4- HeLa cells
Fiala (04-119).p65 8/10/2004, 3:26 PM98

HIV-1 and Cardiomyocyte Apoptosis 99
Cardiovascular Toxicology Humana Press Volume 4, 2004
were obtained from S. Chow, UCLA Department of
Pharmacology.
In Situ Riboprobe Hybridization (ISH)
The technique followed that of Bruggeman et al.
(17,18) with the following modifications relating to
the choice of probe derivation and labeling: Sense
and antisense nef mRNAs were transcribed from plas-
mid pGM93 (AIDS Research and Reference Reagent
Program, NIH). For fluorescein-labeled sense RNA,
pGM93 was digested with EcoRI and transcribed
with T7 RNA polymerase and fluorescein-dUTP by
using a kit from Roche (Mannheim, Germany). Fluo-
rescein antisense transcripts were synthesized from
HindIII-digested pGM93 by using SP6 RNA poly-
merase and fluorescein-dUTP (Roche). The transcrip-
tion products were then purified by using an RNeasy
kit (Qiagen, Valencia, CA). The 1.1-kb sense and anti-
sense transcripts were randomly fragmented by incu-
bation in 0.1M carbonate buffer (pH 10.2) at 60°C for
45 min and subsequent neutralization with 100 mM
sodium acetate (pH 6.0) and 0.5% (v/v) acetic acid.
Paraffin-embedded heart and lung sections were
de-paraffinized, subjected to Target Retrieval Solu-
tion (DAKO, Carpinteria, CA), treated with protein-
ase K (RTU), treated with 0.3% hydrogen peroxide in
methanol, prehybridized to a prehybridization solu-
tion (4XSSC, 0.45 formamide, sheared herring sperm
DNA [Sigma, St. Louis, MO] and heated transfer RNA
[Sigma]), and hybridized to the probe (30 ng RNA)
in situ hybridization solution (DAKO) overnight at
42°C in a humidified chamber. The sections were
washed in 2XSSC once at room temperature and once
at 42°C, and then in 0.2XSSC at 50°C. The brown
color of the probe was developed by using Genpoint
fluorescein-horseradish peroxidase (HRP) (DAKO),
as recommended by the manufacturer. In some expe-
riments, a blue color was developed by using an anti-
fluorescein isothiocyanate (FITC)-alkaline phospha-
tase (Sigma) and BCIP/nitroblue tetrazolium (NBT)
substrate (DAKO). The section was counterstained
by using Methyl Green.
In Situ Polymerase Chain Reaction
(IS-PCR) for HIV-1 DNA
The IS-PCR technique is based on incorporation
of FITC-labeled dUTP into PCR-amplified DNA by
using HIV-1 gag GE1 (5'GAAGGAGCCACCCCACA
AGATT) and GE2 (5'TAGGTGGATTATTTGTCAT
CCA) primers, as previously published (19), with some
modifications. Tissues were deparaffinized in xylene
and alcohol and digested with proteinase K (1:200
for 15 min at room temperature; Promega,Madison,
WI). The PCR reaction was performed in a volume of
50 µL by using a PCR Core Kit (Roche) with FITC-
dUTP (1:8), Ampli Taq-Gold polymerase (5 units;
Applied Biosystems) (20), and forward (100 pM) and
reverse (100 pM) Gag primers. Ampli Taq-Gold poly-
merase was added at 70°C. The PCR reaction was per-
formed in a PCR machine (PTC-100, MJ Research,
Waltham, MA) at 94°C for 2 min 30 s, at 55°C for
1 min 30 s, and then for 20 cycles of 94°C for 55 s
and 55°C for 45 s. The reaction mix was enclosed by
using a coverslip sealed with a nail polish and min-
eral oil overlay. After the reaction, the coverslip was
removed and the slide was washed once with xylene
for 10 min, once with 100% ethanol for 5 min, and
twice with 0.5XSSC for 3 min. The tissue was then
blocked with 0.6% hydrogen peroxide in methanol
for 5 min, rinsed with water, and stained by using
a DAKO GenPoint Fluorescein method (DAKO),
which detected incorporated FITC-dUTP by tyra-
mide signal amplification or an anti-FITC-alkaline
phosphatase/BCIP/NBT technique.
Immunohistochemistry
Formalin-fixed tissues were immunostained by
using an Envision Double Stain System (DAKO) after
antigen retrieval, as described (2). The cardiomyocyte
diameter was measured at a magnification of �40
after staining with wheat germ agglutinin (Sigma).
Apoptosis and Caspase-3 Assays
To induce apoptosis, CAECs or NRVMS were
treated with recombinant gp120JRFL (5–500 ng/mL)
or Tat (50 ng/mL) for 4 h or overnight. Apoptotic
cells were detected by (1) the terminal deoxynucleo-
tidyl transferase-mediated dUTP nick-end labeling
(TUNEL) technique (Apoptosis Detection Kit with
dUTP, Promega) by using the FITC-conjugated nucle-
otide mix and simultaneous staining with DAPI or
Hoechst 33342 (Molecular Probes, Eugene, OR), or
by (2) the FAM FLICA DEVD-FMK caspase-3/7
assay kit (Immunochemistry Technologies, Bloom-
ington, MN). Active caspase-3 was assayed by using
the Caspase-3 Colorimetric Protease kit (BioSource,
Camarillo, CA) and one 60-mm culture dish of CAECs
or one 100-mm dish of NRVMs per experimental
Fiala (04-119).p65 8/10/2004, 3:26 PM99

100 Fiala et al.
Cardiovascular Toxicology Humana Press Volume 4, 2004
sample (50–200 µg of protein). The reaction was
developed by using a Reaction Buffer® and DEVD-
pNA substrate at 37°C for 2 h and read at 405 nm in
a spectrophotometer.
Western Blotting
After reaching confluence, approx 1 million cells
in a 60-mm culture dish were maintained overnight
in an EBM-2 basal medium (Clonetics/BioWhittaker)
without serum. The cells were stimulated by using
100 µL of prewarmed viral protein for 1–20 min and
harvested into 250 µL of ice-cold RIPA buffer (50
mM HEPES at pH 7.4, 10 mM EDTA, 150 mM NaCl,
1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate,
complete protease inhibitor [Roche], and phospha-
tase inhibitor cocktail [Sigma]). The lysate was incu-
bated for 5 min on ice, centrifuged at maximum speed
in a microfuge, and frozen until the assay was per-
formed by Western blotting, as described (1).
Transmission Electron Microscopy (TEM)
and Scanning Electron Microscopy (SEM)
TEM and SEM were performed with cells grown
in Transwell inserts, as described (1). For TEM, the
inserts were gently washed with PBS and fixed with
3% glutaraldehyde buffered with 0.1M sodium phos-
phate or sodium cacodylate salts (pH 7.4) for 1 h at
25°C, stored in a 0.1M sodium cacodylate buffer con-
taining 0.2M sucrose, postfixed in 1% osmium tetrox-
ide in a 0.1M sodium cacodylate buffer for 1 h at
4°C, dehydrated in a graded series of ethanol, and
embedded in Epon. For SEM, after osmication and
dehydration, the inserts were dried with hexamethyl-
disalisane (21), attached to aluminum SEM stubs by
using double-sticky carbon tape, and then coated with
gold and palladium and scanned on an FEI ESEM
XL30 SEM.
Results
HIV Cardiomyopathy Is Hypertrophic and
Associated with Epi-, Peri-, and Endocarditis
In our material comprising the heart tissues of
21 patients with AIDS, 10 hearts showed HIVCM, 1
HIVCM heart also showed epi- and pericarditis and
vasculitis, and 2 hearts had endocarditis. The hearts
with HIVCM were hypertrophic (the average weight
of the hearts with HIVCM was 580 g, as compared
with the 337-g average weight of the hearts without
HIVCM). The average cardiomyocyte diameter in
the hearts with HIVCM was increased (41 µ vs 23 µ).
HIV-1 DNA and RNA
Are Restricted to Inflammatory Cells,
but Envelope Protein gp120 Is Detected
in Inflammatory Cells and Cardiomyocytes
We initially tested IS-PCR, ISH, and IHC by using
infected and uninfected CD4 HeLa cells. Positive
staining was detected only in infected cells using
HIV-1 primers, antisense RNA probe, or anti-gp120
antibody, respectively (Fig. 1). The results with un-
infected cells or when the correct primers, probe, or
antibody were omitted were negative with the excep-
tion of a weak staining of multinucleated giant cells
by both sense and antisense riboprobes. To detect
HIV-1 infection in heart tissues from patients with
and without HIVCM, we performed IS-PCR, ISH,
and IHC. These techniques detected virus footprints
only in HIVCM hearts. Virus DNA was detected by IS-
PCR in inflammatory cells, macrophages (some mul-
tinucleated), and T cells, whereas endothelial cells
and cardiomyocytes were negative (Fig. 1b,c). ISH
showed HIV-1 RNA in inflammatory cells with a halo
of positive reactivity surrounding these cells (Fig. 1i).
Endothelial cells adjacent to adherent mononuclear
cells were positive in a patchy fashion (Fig. 1h). HIV-
1 gp120 was detected in perivascular macrophages
(Fig. 1e), in a sarcomeric pattern in cardiomyocytes
(Fig. 1f), and in some endothelial cells. Six of the
10 hearts with HIVCM but none of the 4 without
HIVCM were positive for gp120 on infiltrating macro-
phages and lymphocytes. As noted previously, some
inflammatory cells expressed Nef and Tat, but these
viral proteins were not significantly associated with
HIVCM (3).
Apoptotic Cardiomyocytes
Are Found Adjacent to HIV-1-Infected
Perivascular Macrophages
Inflammatory cells infiltrated HIVCM hearts in a
patchy and perivascular distribution, and some were
diffusely scattered among cardiomyocytes. Apoptotic
cardiomyocytes were detected by TUNEL technique
adjacent to gp120-positive perivascular macrophages
(Fig. 2). We previously showed that TNF-� family
ligands Fas and TNF-��caspases-3 and -9, as shown in
Fig. 2 (3), and caspase-8 (22,3) were strongly expressed
in heart tissues with HIVCM.
Fiala (04-119).p65 8/10/2004, 3:26 PM100

HIV-1 and Cardiomyocyte Apoptosis 101
Cardiovascular Toxicology Humana Press Volume 4, 2004
Fig. 1. Heart tissues of patients with HIVCM show HIV-1 DNA and RNA in inflammatory cells and gp120 in inflam-
matory cells, cardiomyocytes, and endothelial cells. (A) IS-PCR with Gag primers (Genpoint-FITC-HRP). a, HIV-1-
infected HeLa-CD4 (inset shows uninfected cells). b and c, Heart tissue from a patient with HIVCM shows positive staining
of inflammatory perivascular cells but no staining of cardiomyocytes (inset shows negative staining in control heart
tissue). (B) IHC for gp120. a, HIV-1-infected HeLa-CD4 (inset shows uninfected HeLa-CD4; HRP-diaminobenzidine).
b, Perivascular CD68-positive macrophages (alkaline phosphatase-BCIP) are positive for gp120. c, Cardiomyocytes in
patchy distribution display positive staining for gp120 in sarcomeric pattern. (C) ISH with HIV-1 riboprobe (Genpoint-
FITC-HRP). a, HIV-1-infected HeLa-CD4 (inset shows uninfected HeLa-CD4). b, Positive staining of two mononuclear
cells and adjacent endothelial cells. c, Positive staining of inflammatory cells surrounded by a halo of positive staining
(inset shows negative staining of control heart tissue). Color image available for viewing at www.humanapress.com.
In Vitro Effects of HIV-1
Proteins on NRVMs and CAECs
To analyze the role of HIV-1 proteins in cardio-
myocyte apoptosis, we induced apoptosis with recom-
binant gp120 or Tat in cultured cells.
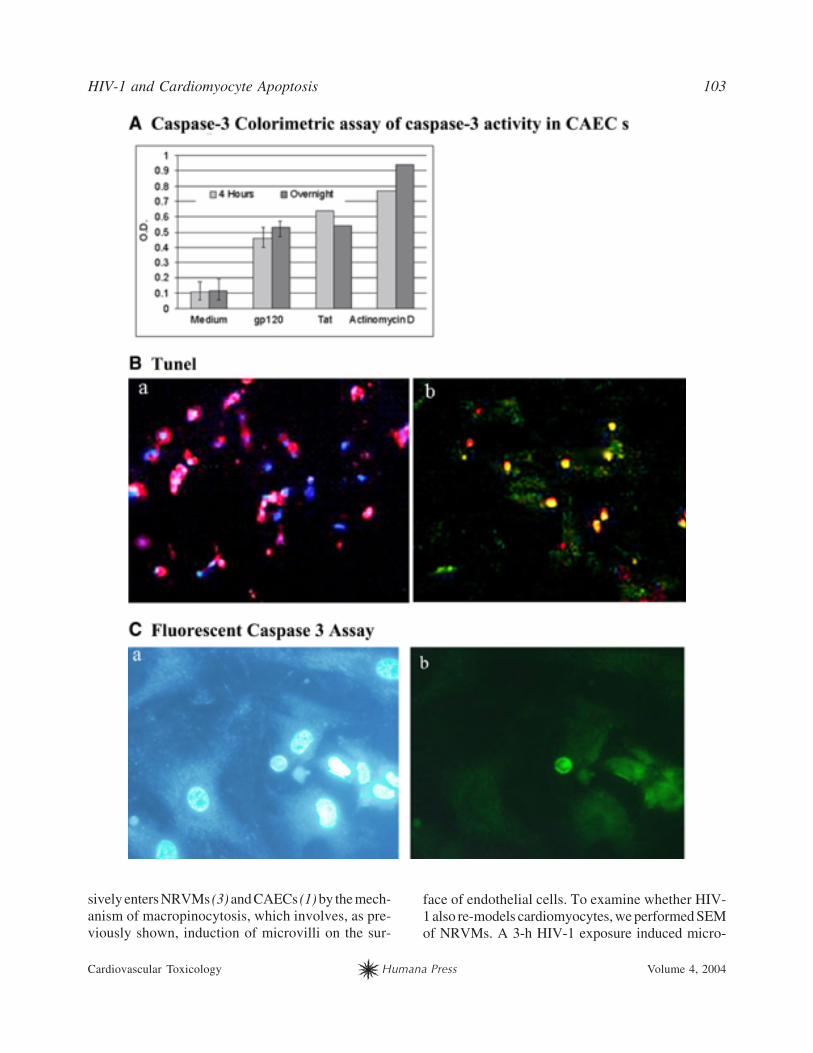
gp120 Activates Caspase-3in CAECs and NRVMsand Induces Apoptosis
gp120 induced apoptosis of NRVMs, as shown by
TUNEL assay (Fig. 3B), and CAECs, as shown by cas-
Fiala (04-119).p65 8/10/2004, 3:26 PM101

102 Fiala et al.
Cardiovascular Toxicology Humana Press Volume 4, 2004
Fig. 2. Apoptotic cardiomyocytes are adjacent to infected perivascular macrophages. (A–C) Small-vessel vasculitisin the myocardium (hematoxylin-eosin). (D) The vasculitis involves infiltration by CD68-positive macrophages (alka-line phosphatase-BCIP). (E) The macrophages are gp120-positive (alkaline phosphatase-BCIP). (F) The cardiomyocytesadjacent to perivascular macrophages are TUNEL-positive (HRP-diaminobenzidine). Color image available for viewing
at www.humanapress.com.
Fig. 3. (Opposite page) gp120 and Tat activate caspase-3 and induce apoptosis of cardiomyocytes and CAECs. (A)gp120, Tat, or actinomycin D cleaves DEVD-p-nitroanilide (OD at 400 nm; colorimetric apoptosis assay [Biosource]in CAECs treated with gp120 [50 ng/mL] for 18 h). (B) gp120 induces NRVM apoptosis (fluorescent TUNEL assay incultured NRVMs treated with gp120 [50 ng/mL] for 48 h). a, Primary NRVM cultures comprise mostly cardiomyocytes(note most nuclei [DAPI-positive] colocalize with the cytoplasm stained red by antisarcomeric antibody MF-20/TR).b, Most cardiomyocytes colocalize with TUNEL staining and are apoptotic. (C) gp120 induces CAEC apoptosis (fluo-rescent caspase-3 assay in CAECs treated with gp120 [50 ng/mL] for 18 h). a, Hoechst 33342 staining of apoptotic nuclei(note shrunken [bright]) nuclei in the right lower corner). b, Fluorescent caspase-3 staining of cells with apoptotic nuclei(FLICA assay). Color image available for viewing at www.humanapress.com.
pase-3 colorimetric assay and fluorescent caspase-
3/Hoechst 33342 staining (Fig. 3C). A MEK inhib-
itor U0126 inhibited caspase-3 induction by viral
proteins. In CAECs and NRVMs, gp120 and Tat acti-
vated caspase-3 to a similar extent after 4 h or over-
night stimulation. The average ratio of caspase-3
optical density (OD) of CAECs treated with gp120
(50 ng/mL) compared with OD of CAECs treated
with medium was 2.18 (four experiments), and the
average ratio of OD of NRVMs treated with gp120
(50 ng/mL) compared with OD of NRVMs treated
with medium was 1.6 (two experiments). The stimu-
lation of caspase-3 by Tat (50 ng/mL) was 2.5-fold
in CAECs and 1.7-fold in NRVMs (Fig. 3A). U0126
decreased caspase-3 induction to baseline or below
the baseline and, as shown previously, reduced apop-
tosis by 80% (3).
gp120 Induces Phosphorylation of Erk1/2
To determine the initial virus interaction with car-
diomyocytes and CAECs, we examined mitogen-
activated protein kinase (MAPK) cell signaling by
gp120 and Tat. gp120 induced Erk 1/2 phosphoryla-
tion more rapidly (2 min) in NRVMs (Fig. 4A) than
in CAECs (10 min; Fig. 4B). Tat also induced signal-
ing in CAECs with slower kinetics (10 min; Fig. 4C).
The MEK inhibitor U0126 abolished Erk 1/2 phos-
phorylation in NRVMs by gp120 (Fig. 4A). No c-Jun
N-terminal kinase (JNK) signaling by gp120 was
found.
HIV-1 Enters and Remodels
CAECs and NRVMs
As previously shown by confocal microscopy,
within 3 h after exposure, HIV-1NL4-3 Vpr-GFP mas-
Fiala (04-119).p65 8/10/2004, 3:26 PM102
HIV-1 and Cardiomyocyte Apoptosis 103
Cardiovascular Toxicology Humana Press Volume 4, 2004
sively enters NRVMs (3) and CAECs (1) by the mech-
anism of macropinocytosis, which involves, as pre-
viously shown, induction of microvilli on the sur-
face of endothelial cells. To examine whether HIV-
1 also re-models cardiomyocytes, we performed SEM
of NRVMs. A 3-h HIV-1 exposure induced micro-
Fiala (04-119).p65 8/10/2004, 3:26 PM103

104 Fiala et al.
Cardiovascular Toxicology Humana Press Volume 4, 2004
Fig. 4. gp120 and Tat induce Erk 1/2 phosphorylation
in cardiomyocytes and CAECs. (A) gp120 treatment of
NRVMs induced Erk1/2 phosphorylation with a maximum
at 2 min and a second maximum at 15 min postexposure.
U0126 blocked Erk 1/2 phosphorylation. (B) gp120 treat-
ment of CAECs induced Erk1/2 phosphorylation with a
maximum at 10 min and a second maximum at 30 min.
(C) Tat treatment of CAECs induced Erk1/2 phosphory-
lation with a maximum at 10 min.
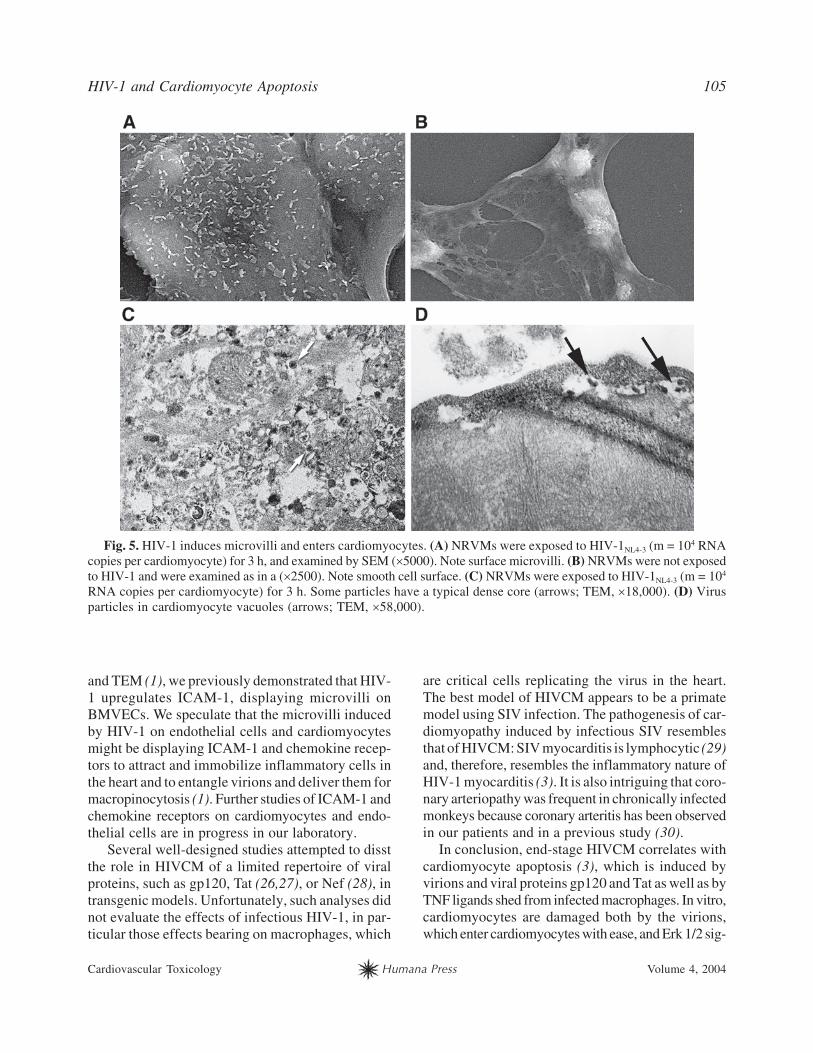
villi, as shown by SEM (Fig. 5, cf. A,B). HIV-1NL4-3
Vpr-GFP strongly enters NRVMs, as observed by
confocal microscopy (3). TEM of NRVMs exposed
to HIV-1 for 3 h showed virion-sized (100-mµ diam-
eter) particles in vacuoles (Fig. 5D) and scattered in
the cytoplasm. Some of these particles had a typical
HIV nucleocapsid core (Fig. 5C).
Discussion
This study by nucleic acid hybridization tech-
niques complements previous studies by IHC (3) and
underlines the critical role of macrophage infiltra-
tion and production of viral and cytokine products
inducing cardiomyocyte apoptosis. The hearts with
HIVCM display multifaceted pathology with focal
myocarditis, complicated in some cases by epicardi-
tis, vasculitis in small intramyocardial vessels, myo-
cardial infarction, and endocarditis. The root of large
vessels is infiltrated by HIV-1-infected macrophages.
Coronary vessels were not available for examination
in this study. The hearts with HIVCM have cardio-
megaly (22,3) and hypertrophic cardiomyocytes. IHC
of HIVCM shows inflammatory pathology with infil-
tration by macrophages and T cells in a patchy peri-
vascular pattern. However, because of its focal nature,
unless many sections are examined, the inflamma-
tion may remain undetected.
As shown by nucleic acid hybridization, HIV-1
was found to productively infect inflammatory cells,
macrophages, and lymphocytes. We did not find evi-
dence by hybridization techniques of HIV-1 infec-
tion of cardiomyocytes, which had been previously
suspected (4,6,23). Although no viral DNA was found
in cardiomyocytes or endothelial cells, these cells
displayed gp120, indicating that the virus may invade
them without replication. HIV-1 proteins that are re-
leased when virions are degraded in lysosomes (as
observed in brain microvascular endothelial cells
[BMVECs; 1]), in particular gp120, could cause mito-
chondrion-controlled apoptosis by binding to cardio-
myocyte mitochondria. gp120 induced cytochrome
c release from NRVM mitochondria (3). Therefore,
cardiomyocyte apoptosis seems to be produced in a
paracrine fashion by death receptor family ligands
and viral products from infected macrophages.
The in vitro effects of HIV-1 observed in BMVECs
(1), CAECs (16), and NRVMs (this study) explain
the mechanisms of HIV-1 entry and apoptosis of tar-
get cells in the heart and probably the brain. First,
HIV-1 easily enters CAECs and BMVECs by macro-
pinocytosis (1) and traverses these cells into target
organs by transcellular and pericellular processes
(24,25). Second, in target organs, HIV-1 infects and
is replicated by perivascular macrophages, and the
extent of macrophage infiltration appears to deter-
mine the risk of HIVCM (2). Third, HIV-1 and gp120
signal in CAECs and NRVMs through the MAPK
pathway by Erk1/2 phosphorylation, and this signal-
ing is necessary for HIV-1- or gp120-induced apop-
tosis. Finally, HIV-1 remodels cardiomyocytes by
inducing microvilli on the cell surface. Using IEM
Fiala (04-119).p65 8/10/2004, 3:26 PM104
HIV-1 and Cardiomyocyte Apoptosis 105
Cardiovascular Toxicology Humana Press Volume 4, 2004
Fig. 5. HIV-1 induces microvilli and enters cardiomyocytes. (A) NRVMs were exposed to HIV-1NL4-3 (m = 104 RNA
copies per cardiomyocyte) for 3 h, and examined by SEM (�5000). Note surface microvilli. (B) NRVMs were not exposed
to HIV-1 and were examined as in a (�2500). Note smooth cell surface. (C) NRVMs were exposed to HIV-1NL4-3 (m = 104
RNA copies per cardiomyocyte) for 3 h. Some particles have a typical dense core (arrows; TEM, �18,000). (D) Virus
particles in cardiomyocyte vacuoles (arrows; TEM, �58,000).
and TEM (1), we previously demonstrated that HIV-
1 upregulates ICAM-1, displaying microvilli on
BMVECs. We speculate that the microvilli induced
by HIV-1 on endothelial cells and cardiomyocytes
might be displaying ICAM-1 and chemokine recep-
tors to attract and immobilize inflammatory cells in
the heart and to entangle virions and deliver them for
macropinocytosis (1). Further studies of ICAM-1 and
chemokine receptors on cardiomyocytes and endo-
thelial cells are in progress in our laboratory.
Several well-designed studies attempted to disst
the role in HIVCM of a limited repertoire of viral
proteins, such as gp120, Tat (26,27), or Nef (28), in
transgenic models. Unfortunately, such analyses did
not evaluate the effects of infectious HIV-1, in par-
ticular those effects bearing on macrophages, which
are critical cells replicating the virus in the heart.
The best model of HIVCM appears to be a primate
model using SIV infection. The pathogenesis of car-
diomyopathy induced by infectious SIV resembles
that of HIVCM: SIV myocarditis is lymphocytic (29)
and, therefore, resembles the inflammatory nature of
HIV-1 myocarditis (3). It is also intriguing that coro-
nary arteriopathy was frequent in chronically infected
monkeys because coronary arteritis has been observed
in our patients and in a previous study (30).
In conclusion, end-stage HIVCM correlates with
cardiomyocyte apoptosis (3), which is induced by
virions and viral proteins gp120 and Tat as well as by
TNF ligands shed from infected macrophages. In vitro,
cardiomyocytes are damaged both by the virions,
which enter cardiomyocytes with ease, and Erk 1/2 sig-
Fiala (04-119).p65 8/10/2004, 3:26 PM105

106 Fiala et al.
Cardiovascular Toxicology Humana Press Volume 4, 2004
naling through viral proteins gp120 and Tat. Inhibition
of cell signaling by gp120 inhibits cardiomyocyte
apoptosis. Inhibitors of MAPK signaling might be
useful in the therapy of HIVCM.
Acknowledgments
We thank Sergei Nekhai for the supply of recom-
binant Tat; Sam Chow for CD4-HeLa; M. Ross, L.
Bruggeman, and P. Klotman for advice regarding
the IS-PCR and ISH techniques; and Mithulan Jega-
pragasan for cardiomyocyte measurements. The AIDS
Research and Reference Reagent Program, NIAID,
NIH provided the listed reagents.
References
1. Liu, N.Q., Lossinsky, A.S., Popik, W., Li, X., Gujuluva,
C., Kriederman, B., et al. (2002). Human immunodefi-
ciency virus type 1 enters brain microvascular endothelia
by macropinocytosis dependent on lipid rafts and the mito-
gen-activated protein kinase signaling pathway. J. Virol. 76:
6689–6700.
2. Liu, Q.N., Reddy, S., Sayre, J.W., Pop, V., Graves, M.C.,
and Fiala, M. (2001). Essential role of HIV type 1-infected
and cyclooxygenase 2-activated macrophages and T cells
in HIV type 1 myocarditis. AIDS Res. Hum. Retroviruses
17:1423–1433.
3. Twu, C., Liu, N.Q., Popik, W., Bukrinsky, M., Sayre, J.,
Roberts, J., et al. (2002). Cardiomyocytes undergo apop-
tosis in human immunodeficiency virus cardiomyopathy
through mitochondrion- and death receptor-controlled path-
ways. Proc. Natl. Acad. Sci. USA 99:14386–14391.
4. Grody, W.L.C. and Lewis, W. (1990). Infection of the
human heart by the human immunodeficiency virus. Am.
J. Cardiol. 66:203–206.
5. Lipshultz, S.E., Fox, C.H., Perez-Atayde, A.R., Sanders,
S.P., Colan, S.D., McIntosh, K., et al. (1990). Identifica-
tion of human immunodeficiency virus-1 RNA and DNA
in the heart of a child with cardiovascular abnormalities and
congenital acquired immune deficiency syndrome. Am. J.
Cardiol. 66:246–250.
6. Rodriguez, E.R., Nasim, S., Hsia, J., Sandin, R.L., Ferreira,
A., Hilliard, B.A., et al. (1991). Cardiac myocytes and den-
dritic cells harbor human immunodeficiency virus in infected
patients with and without cardiac dysfunction: detection
by multiplex, nested, polymerase chain reaction in individ-
ually microdissted cells from right ventricular endomyocar-
dial biopsy tissue. Am. J. Cardiol. 68:1511–1520.
7. Kandolf, R., Ameis, D., Kirschner, P., Canu, A., and Hof-
schneider, P.H. (1987). In situ detection of enteroviral
genomes in myocardial cells by nucleic acid hybridization:
an approach to the diagnosis of viral heart disease. Proc.
Natl. Acad. Sci. USA 84:6272–6276.
8. Cioc, A.M. and Nuovo, G.J. (2002). Histologic and in situ
viral findings in the myocardium in cases of sudden, unex-
pected death. Mod. Pathol. 15:914–922.
9. Chen, F., Shannon, K., Ding, S., Silva, M.E., Wetzel, G.T.,
Klitzner, T.S., et al. (2002). HIV type 1 glycoprotein 120
inhibits cardiac myocyte contraction. AIDS Res. Hum. Retro-
viruses 18:777–784.
10. Kan, H., Xie, Z., and Finkel, M.S. (2000). HIV gp120
enhances NO production by cardiac myocytes through p38
MAP kinase-mediated NF-kappaB activation. Am. J. Phys-
iol. Heart Circ. Physiol. 279:H3138–H3143.
11. Chaves A.A., Mihm M.J., Schanbacher B., Basuray, A.,
Liu, C.Y., Ayers, L.W., et al. (2003). Cardiomyopathy in
a murine model of AIDS: evidence of reactive nitrogen
species and corroboration in human HIV/AIDS cardiac tis-
sues. Cardiovasc. Res. 60:108–118.
12. Carr, A. and Cooper, D.A. (2000). Adverse effects of anti-
retroviral therapy. Lancet 356:1423–1430.
13. Frerichs, F.C., Dingemans, K.P., and Brinkman, K. (2002).
Cardiomyopathy with mitochondrial damage associated
with nucleoside reverse-transcriptase inhibitors. N. Engl.
J. Med. 347:1895–1896.
14. Mossman, S.P., Bex, F., Berglund, P., Arthos, J., O’Neil,
S.P., Riley, D., et al. (1996). Protection against lethal sim-
ian immunodeficiency virus SIVsmmPBj14 disease by a
recombinant Semliki Forest virus gp160 vaccine and by a
gp120 subunit vaccine. J. Virol. 70:1953–1960.
15. Deng, L., Ammosova, T., Pumfery, A., Kashanchi, F., and
Nekhai, S. (2002). HIV-1 tat interaction with RNA polymer-
ase II C-terminal domain (CTD) and a dynamic association
with CDK2 induce CTD phosphorylation and transcription
from HIV-1 promoter. J. Biol. Chem. 277:33922–33929.
16. Gujuluva, C., Burns, A.R., Pushkarsky, T., Popik, W.,
Berger, O., Bukrinsky, M., et al. (2001). HIV-1 penetrates
coronary artery endothelial cells by transcytosis. Mol. Med.
7:169–176.
17. Bruggeman, L.A., Dikman, S., Meng, C., Quaggin, S.E.,
Coffman, T.M., and Klotman, P.E. (1997). Nephropathy
in human immunodeficiency virus-1 transgenic mice is due
to renal transgene expression. J. Clin. Invest. 100:84–92.
18. Bruggeman, L.A., Ross, M.D., Tanji, N., Cara, A., Dikman,
S., Gordon, R.E., et al. (2000). Renal epithelium is a pre-
viously unrecognized site of HIV-1 infection. J. Am. Soc.
Nephrol. 11:2079–2087.
19. Strappe, P.M., Wang, T.H., McKenzie, C.A., Lowrie, S.,
Simmonds, P., and Bell, J.E. (1998). In situ polymerase
chain reaction amplification of HIV-1 DNA in brain tis-
sue. J. Virol. Methods 70:119–127.
20. Nuovo, G. (1997). PCR in Situ Hybridization: Protocols and
Amplifications, 3rd ed. New York, NY: Lippincott-Raven.
21. Lossinsky, A.S. and Shivers, R.R. (2003). Studies of cere-
bral endothelium by scanning and high-voltage electron
microscopy. In: Nag, S. (ed.). The Blood-Brain Barrier:
Biology and Research Protocols. Totowa, NJ: Humana
Press, pp 67–82.
22. October 2002. Available at: http://www.pnas.org. Accessed
February 27, 2004.
23. Barbaro, G. and Lipshultz, S.E. (2001). Pathogenesis of HIV-
associated cardiomyopathy. Ann. NY Acad. Sci. 946:57–81.
24. Fiala, M., Looney, D.J., Stins, M., Way, D.D., Zhang, L.,
Gan, X., et al. (1997). TNF-alpha opens a paracellular
route for HIV-1 invasion across the blood-brain barrier.
Mol. Med. 3:553–564.
Fiala (04-119).p65 8/10/2004, 3:26 PM106

HIV-1 and Cardiomyocyte Apoptosis 107
Cardiovascular Toxicology Humana Press Volume 4, 2004
25. Zhang, L., Looney, D., Taub, D., Chang, S.L., Way, D.,
Witte, et al. (1998). Cocaine opens the blood-brain barrier
to HIV-1 invasion. J. Neuro. Virol. 4:619–626.
26. Reid, W., Sadowska, M., Denaro, F., Rao, S., Foulke J. Jr.,
Hayes, N., et al. (2001). An HIV-1 transgenic rat that
develops HIV-related pathology and immunologic dysfunc-
tion. Proc. Natl. Acad. Sci. USA 98:9271–9276.
27. Bruggeman, L.A., Thomson, M.M., Nelson, P.J., Kopp,
J.B., Rappaport, J., Klotman, P.E., et al. (1994). Patterns
of HIV-1 mRNA expression in transgenic mice are tissue-
dependent. Virology 202:940–948.
28. Kay, D.G., Yue, P., Hanna, Z., Jothy, S., Tremblay, E.,
and Jolicoeur, P. (2002). Cardiac disease in transgenic
mice expressing human immunodeficiency virus-1 nef in
cells of the immune system. Am. J. Pathol. 161:321–335.
29. Shannon, R.P., Simon, M.A., Mathier, M.A., Geng, Y.J.,
Mankad, S., and Lackner, A.A. (2000). Dilated cardiomy-
opathy associated with simian AIDS in nonhuman pri-
mates. Circulation 101:185–193.
30. Barbaro, G., Barbarini, G., and Pellicelli, A.M. (2001).
HIV-associated coronary arteritis in a patient with fatal myo-
cardial infarction. N. Engl. J. Med. 344:1799–1800.
Fiala (04-119).p65 8/10/2004, 3:26 PM107
Copyright © 2022 FDOKUMEN




















