
Degradation of tert-butyl formate and its intermediates by an ozone/UV process
Upload
independentCategory
view
2download
0
B R A I N R E S E A R C H 1 3 0 6 ( 2 0 1 0 ) 1 1 6 – 1 3 0
ava i l ab l e a t www.sc i enced i r ec t . com
www.e l sev i e r . com/ loca te /b ra i n res
Research Report
Dopaminergic neurotoxicity of HIV-1 gp120: Reactive oxygenspecies as signaling intermediates
Lokesh Agrawala, Jean-Pierre Louboutina, Elena Marusicha, Beverley A.S. Reyesb,Elisabeth J. Van Bockstaeleb, David S. Strayera,⁎aDepartment of Pathology, Anatomy, and Cell Biology, Thomas Jefferson University, Philadelphia, PA, USAbDepartment of Neurosurgery, Farber Institute for Neurosciences, Thomas Jefferson University, Philadelphia, PA, USA
A R T I C L E I N F O
⁎ Corresponding author. Department of PathoFax: +1 215 503 1156.
E-mail address: [email protected]: BUGT, bilirubin–UDP–glucur
Dopaminergic neurons; EGF, epidermal growgenase; GDNF, glial-derived neurotropic factocognitive/motor dysfunction; NCAM, neuralParkinson's disease; RN, non-dopaminergicreactive oxygen species; SN, substantia nighydroxylase; VOCC, voltage-operated calcium
0006-8993/$ – see front matter © 2009 Publisdoi:10.1016/j.brainres.2009.09.113
A B S T R A C T
Article history:Accepted 29 September 2009Available online 6 October 2009
We examined the role of reactive oxygen species (ROS) in loss of dopaminergic neurons(DNs) from the substantia nigra (SN) in neuroAIDS. The frequency of Parkinson-likesymptomatology, and DN loss, in neuroAIDS is often attributed to nonspecific DN fragility tooxidative stress. Cultured DN are more sensitive to ROS than non-dopaminergic neurons(RN): DN underwent apoptosis at far lower H2O2 concentrations than RN. Gene delivery ofglutathione peroxidase (GPx1), which detoxifies H2O2, largely protected both neuron types.HIV-1 envelope, gp120, which elicits oxidative stress in neurons, caused apoptosis morereadily in DN than in RN. However, unlike apoptosis caused by H2O2, gp120-induced DNapoptosis was specific: DNs were specifically more sensitive than RN to receptor-mediated[Ca2+]i fluxes triggered by gp120. Gp120-induced Ca2+ signaling in both neuron types wasinhibited by GPx1 or Cu/Zn superoxide dismutase (SOD1), implicating superoxide andperoxide in ligand (gp120)-induced signaling upstream of Ca2+ release from intracellularstores. In vivo, rats given 10 ng of gp120 stereotaxically showed rapid DN loss within the SN,while loss of RN in the SN and caudate–putamen (CP) was slower and required ≥100 ng ofgp120. Furthermore, gp120 injected into the CP was transported axonally retrograde to theSN, causing delayed DN loss there. This, too, was prevented by SOD1 or GPx1. DNs aretherefore specifically hypersensitive to gp120-induced apoptosis, signaling for whichinvolves ROS intermediates. These findings may help explain why DN loss andParkinson's-like dysfunction predominate in neuroAIDS and may apply to otherneurodegenerative diseases involving the SN.
© 2009 Published by Elsevier B.V.
Keywords:AIDSAntioxidantApoptosisDopaminergic neuronGene delivery
logy, Jefferson Medical College, 1020 Locust Street, Room 251, Philadelphia, PA 19107, USA.
du (D.S. Strayer).onysyl transferase; CP, caudate–putamen; DA, dopamine; DAT, dopamine transporter; DN,th factor; FITC, fluorescein isothiocyanate; GAPDH, glyceraldehyde-3-phosphate dehydro-r; GPx1, glutathione peroxidase; HAART, highly active antiretroviral therapy; MCMD, minorcell adhesion molecule; NT, neurotrace (Nissl stain); PBS, phosphate-buffered saline; PD,neurons; RNS, reactive nitrogen species; rSV40, recombinant SV40-derived vector; ROS,ra; SOD1, Cu/Zn superoxide dismutase; SOD2, Mn superoxide dismutase; TH, tyrosinechannels
hed by Elsevier B.V.

117B R A I N R E S E A R C H 1 3 0 6 ( 2 0 1 0 ) 1 1 6 – 1 3 0
1. Introduction
Parkinson's disease (PD) is characterized by motor, cognitive,and psychiatric disturbances. These include apathy, slowmovement and thought, impaired gait, rigidity and others, andlargely reflect the consequences of loss of dopaminergicneurons (DNs) from the substantia nigra (SN). The causes ofmost cases of PD are unknown, but progressive loss of DNsoften involves continuing oxidant damage to DN proteins,accumulation of these abnormally modified proteins andconsequent disruption of neuronal function (Bernheimer etal., 1973; Hornykiewicz, 2001; Zhang and Kaufman, 2006;Gorman, 2008; Levy et al., 2009).
Ongoing oxidative neuronal injury is also characteristic ofsymptomatic HIV-1 infection in the CNS, i.e., neuroAIDS(Steiner et al., 2006). HIV-1 enters the CNS shortly afterentering the body and replicates in macrophages and micro-glia there (Gartner, 2000; Rausch et al., 1999). Although highlyactive antiretroviral therapy (HAART) generally does notpenetrate the CNS well andmay not greatly impair replicationof HIV-1 within the CNS (McArthur et al., 1999), HAARTexposure may decrease CNS virus load (Sinclair et al., 2008).The consequences of CNS HIV-1 infection reflect in a majorway the neurotoxicity of two HIV-1 protein products, envelopegp120 (Env) and Tat (Kruman et al., 1998; Shi et al., 1998;Corasaniti et al., 1998a; Rappaport et al., 1999; Nath et al.,2000a; Corasantini et al., 2001). Although frank HIV-1-associ-ated dementia is now rare in the US, minor cognitive andmotor dysfunction (MCMD) is common. As life expectanciesimprove for HIV-1-positive patients whose peripheral HIV-1loads are controlled with HAART, prevalence of MCMD isrising (Sacktor et al., 2002; McArthur et al., 2005).
The pathogenesis of neuronal injury leading to symptom-atic neuroAIDS is complex and involves neuronal loss (Naviaand Rostasy, 2005; Everall et al., 2005; Langford et al., 2003;Gonzalez-Scarano and Martin-Garcia, 2005; Kaul et al., 2005;Rumbaugh and Nath, 2006; Lawrence and Major, 2002) anddysfunction due to excitotoxic activation of synaptic gluta-mate receptors, proinflammatory cytokines, proapoptoticsignaling elicited by gp120 and Tat in neurons and otherfactors (Kruman et al., 1998; Shi et al., 1998; Corasaniti et al.,1998b; Rappaport et al., 1999; Nath et al., 2000a; Corasantini etal., 2001; Watanabe et al., 2003; Betz et al., 1998). Several ofthese mechanisms involve generation of reactive oxygen(ROS) and nitrogen (RNS), causing oxidative damage to manycellular macromolecules, especially lipids and proteins(Turchan et al., 2003; Bandaru et al., 2007; Mattson et al.,2005; Steiner et al., 2006). Gene delivery of Cu/Zn superoxidedismutase (SOD1) and/or glutathione peroxidase (GPx1) toneurons using recombinant SV40-derived vectors (rSV40s)can protect neurons from the toxicity of gp120 and Tat in vitroand in vivo (Agrawal et al., 2006, 2007; Louboutin et al., 2007a,2007b).
Interestingly, the symptomatology and pathology of neu-roAIDS both reflect disproportionate damage to the dopami-nergic systemof the SN. Thus, the bradykinesia, bradyphrenia,abnormalities of gait, rigidity, and other characteristics ofParkinson's disease also characterize neuroAIDS (Nath et al.,2000b; Berger and Arendt, 2000; Koutsilieri et al., 2002a, 2002b;
Chang et al., 2008; Khanlou et al., 2009). Disproportionate DNloss is common in neuroAIDS, as are decreased dopamine (DA)and DA metabolites (Reyes et al., 1991; Silvers et al., 2006).Tyrosine hydroxylase (TH), which is critical for DA synthesis,and DA transporter (DAT), which is important for DA reuptakeinto DN, are often decreased also (Itoh et al., 2000; Wang et al.,2004; Silvers et al., 2007).
Compared to non-dopaminergic neurons (RNs), DNs arereported to be nonspecifically more sensitive to oxidativedamage (Fahn and Cohen, 1992; Jenner, 2003), particularly viadamage to their mitochondria (Ben-Shachar et al., 1995; Gluckand Zeevalk, 2004). However, the basis for the predilection forDN injury and loss in neuroAIDS has been unclear. We reporthere that DNs are nonspecifically more susceptible than non-dopaminergic neurons to ROS-induced apoptosis but that, aswell, gp120 toxicity for DN is largely specific for gp120, reflectsgp120-initiated proapoptotic signaling, localizes mainly to thecytosol, and occurs in DN far more readily than in otherneuron types.
ROS are involved in HIV-1 gp120-induced neurotoxicity assignaling intermediates activated by gp120–DN interaction.They may also be direct effectors of oxidant damage. Gp120 istoxic to DN at levels that are innocuous to RN. In addition, it isspecifically transported from other areas of the brain to theSN, where it causes selective loss of DN. Antioxidant genedelivery blocks gp120-induced proapoptotic signaling in vitroand protects DN viability in vitro and in vivo.
2. Methods
2.1. Cell lines and chemicals
COS-7 cell line was obtained from American Type CultureCollection (ATCC) and maintained in Dulbecco's modifiedEagle's medium (DMEM) supplemented with 10% calf serum(Hyclone, Logan, UT, USA), 2 mm L-glutamine and containing1.5 g/l sodium bicarbonate, 4.5 g/l glucose, 1.0 mm sodiumpyruvate, penicillin (200 U/ml), and streptomycin (100 g/ml).H2O2 was purchased from Sigma Chemical Co. (St. Louis,MO). Growth factors used as media supplements, epidermalgrowth factor (EGF), fibroblast growth factor (FGF), IL-1β,glial-derived neurotrophic factor (GDNF) were purchasedfrom Chemicon. Poly-D-lysine was obtained from SigmaChemical Co.
2.2. Antibodies
Different primary antibodies were used: rabbit anti-humanSOD1 (1:100), mouse anti-GPX1 (1:100) (Stressgen, Victoria,British Columbia, Canada), mouse anti-TH (Immunostar,Hudson, WI), anti-GAPDH (Chemicon, International, Teme-cula, CA), and anti-gp120 (AIDS Reagent Program). Secondaryantibodies were used at a 1:100 dilution: fluorescein isothio-cyanate- (FITC) and tetramethylrhodamine isothiocyanate(TRITC)-conjugated goat anti-mouse, TRITC-conjugated goatanti-rabbit, FITC-conjugated sheep anti-rabbit (Sigma Chem-ical Co.), FITC-, and TRITC-conjugated donkey anti-mouseantibodies (Jackson ImmunoResearch Laboratories Inc., WestGrove, PA, USA).

118 B R A I N R E S E A R C H 1 3 0 6 ( 2 0 1 0 ) 1 1 6 – 1 3 0
2.3. Preparation of non-dopaminergic neurons (RNs) anddopaminergic neurons (DNs)
The preparation of RN has been described earlier (Agrawal etal., 2006). Briefly, fetal brain (obtained from the Human FetalTissue Bank, Albert Einstein College of Medicine, Bronx, NY,USA) was homogenized in Hank's balanced salt solution Ca2+-and Mg2+-free, containing 0.05% Trypsin and 100 U of DNase.Mixed brain cultures were passed through 170 μm nylonmesh, then plated on poly-D-lysine-coated 24-well plates or4-chamber slides. Nonadherent cells were removed bywashing with DMEM/F-12 (Invitrogen, Carlsbad, CA, USA).Adherent neuronal cultures were treated with cytosinearabinoside (ara C – 1 M) for 2 weeks. Immunostaining withMAP-2 antibody confirmed the preponderantly neuronalnature of these cell preparations (>98%, Agrawal et al.,2006). Dopaminergic cells were prepared from fetal brains(12–16 weeks) similar to procedures for differentiating NT2-derived progenitors into midbrain DN neurons (Ravindranand Rao, 2006). We first enriched the population of RN bysorting nestin-positive cells with magnetic beads conjugatedto antibody to neural cell adhesion molecule (NCAM) (MACS,Miltenyi Inc.) according to the manufacturer's instructions.Sorted cells were treated to drive differentiation to DN, firstusing epidermal growth factor (EGF, 50 ng/ml)+fibroblastgrowth factor (FGF; 50 ng/ml) for 1 week, then 2 weeks ofinterleukin 1-β (IL-1β; 1–2 μg/ml)+glial cell-derived neuro-tropic factor (GDNF; 1–5 μg/ml) on poly-L-lysine-coated platesor slides.
2.4. Animals
Female Sprague-Dawley rats (200–250 g) were purchased fromCharles River Laboratories (Wilmington, MA, USA). Protocolsfor injecting and euthanizing animals were approved byThomas Jefferson University IACUC and are consistent withAAALAC standards.
2.5. Vector production
The general principles for making recombinant, Tag-deleted,replication-defective SV40 viral vectors have been previouslyreported (Strayer, 1999). SOD1 and GPx1 transgenes weresubcloned into pT7[RSV-LTR], in which transgene expressionis controlled by the Rous sarcoma virus long-terminal repeat(RSV-LTR) as a promoter. Cloned rSV40 genomes were excisedfrom carrier plasmids, gel-purified, recircularized, then trans-fected into COS-7 cells. These cells supply the large T-antigen(Tag) and SV40 capsid proteins, needed to produce recombi-nant replication-defective SV40 viral vectors, in trans (Strayeret al., 1997). Crude virus stocks were prepared as cell lysates,band-purified by discontinuous sucrose density gradientultracentrifugation and titered by quantitative PCR (Q-PCR;Stratagene Inc.) (Strayer et al., 2001, 2006). SV(HBS), whichencodes hepatitis B surface antigen (HBsAg), was used asnegative control recombinant virus for in vitro experiments(Kondo et al., 1998). SV(HBS) cannot be used in vivo becauseimmunity to the expressed HBsAg may lead to elimination oftransduced cells in immunocompetent rats. SV(BUGT), whichcarries cDNA for human bilirubin–uridine 5′-diphosphate–
glucuronysyl transferase (BUGT), was used as negative controlvector for in vivo studies (Sauter et al., 2000).
2.6. Apoptosis and transduction and challenge studiesin vitro
To test gp120-induced apoptosis, both RNs and DNs weretreatedwith different doses of HIV-1BaL gp120 (0.1, 1.0, 10, and100 ng/culture) obtained from AIDS Reagent Program, NIH.Peroxide (H2O2) was used at 0, 0.1, 1.0, 10, and 100 μM in bothRN and DN cultures. Two days post-treatment, the cells wereprocessed for TUNEL assay, whichwas performed according tomanufacturer's instructions (Roche, Indianapolis, IN, USA). Allcells in eachwell of an 8-well chamber slidewere counted. Thecell density among the wells was uniform. For transductionand challenge studies, RNs or DNswere transducedwith SV40-derived viruses SV(SOD1) and SV(GPx1) on days 0, 3, and 5withMOI of 10, then 3 and again, 3, respectively. The cells weretested for transgene expression by Western blot analysis andimmunostaining. The cells were maintained for 5 days inDMEM supplemented with 2% fetal bovine serum. Thetransduced and mock-transduced cells showed the samedegree of viability (>95%). Recombinant HIV-1BaL-gp120 orH2O2 (80 μM for RNs, 3 μM for DNs) was then added to culturesbefore performing TUNEL assay.
2.7. Western blot analysis
Cells were grown on either 24-well plates or four-chamberslides treated with poly-D-lysine. At the indicated times post-transduction, the transduced cells were harvested by trypsi-nization and counted. The cells were lysed using a buffercontaining aprotonin and phenyl methyl sulfonyl fluoride(PMSF). Total protein was estimated using BCA protein assaykit (Pierce, Rockford, IL, USA). Total cell proteins (50 μg) wereloaded in each well, then transferred to Immobilon-P polyvinylidine difluoride (PVDF) membrane (Millipore Inc., USA).Equal lane loading was assessed using anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (1:5000). Blotswere executed according to manufacturer's instructions(Roche). Anti-SOD1 and anti-GPx1 were used at dilutions of1:500 and 1:100, respectively, and anti-mouse/anti-rabbithorseradish peroxidase (HRP) was used at a dilution of1:5000 before the addition of a chemiluminescence substrate(Roche).
2.8. Enzymatic activity assays for SOD1 and GPx1
Kinetic assays for both SOD1 and GPx1 have been describedearlier (Agrawal et al., 2006). SOD1 activity was measuredusing SOD1 kit (R&D Systems, Minneapolis, MN, USA). TheGPx1 assay was performed using a total GPx1 assay kit(Zeptometrix, Buffalo, NY, USA), following the manufacturer'sinstructions.
2.9. In vivo injection of gp120: transduction of antioxidantvectors (Louboutin et al., 2007a, 2007b)
Ratswere anesthetizedwith isofluraneUPS (Baxter HealthcareCorp., Deerfield, IL, USA) (1.0 U of isoflurane per 1.5 l of O2 per

119B R A I N R E S E A R C H 1 3 0 6 ( 2 0 1 0 ) 1 1 6 – 1 3 0
minute) and placed in a stereotaxic apparatus (Stoelting Corp.,Wood Dale, IL, USA) for cranial surgery. Body temperature wasmaintained at 37 °C by using a feedback-controlled heater(Harvard Apparatus, Boston, MA, USA). Glass micropipettes(1.2 mm outer diameter; World Precisions Instruments Inc.,Sarasota, FL, USA) with tip diameters of 15 μm were backfilledwith 5 μl of HIV BAL-gp120 containing 10, 25, 50, 100, 250, and500 ng of the protein. The gp120-filled micropipettes wereplaced in the CP or SN using coordinates obtained from the ratbrain atlas of Paxinos and Watson (1986). For transductionwith antioxidant vectors, glass micropipettes were backfilledwith 5 μl of SV(SOD1) or SV(GPx1) or SV(BUGT) viral vector,which contains approximately 107 particles. The vector-filledmicropipettes were placed in the CP (1 month) beforechallenge with gp120 (500 ng into the CP). Saline was used asthe negative control, as was the contralateral side of theunilaterally injected brains.
For injection into the CP, a burr hole was placed +0.48 mmanterior to bregma and −3.0 mm lateral to the sagittal suture.Once centered, the micropipette was placed 6.0 mm ventralfrom the top of the brain. The same coordinates were used forinjection of gp120 into the CP. For SN injection, a burr hole wasplaced 5.3 mm posterior to bregma and 2 mm lateral to thesagittal suture. Once centered, the micropipette was placed7.8mmventral fromthe topof thebrain.Thevectorwasgivenbya Picospritzer II (General Valve Corp., Fairfield, NJ, USA) pulse ofcompressed N2 of 10ms in duration at 20 psi until the fluid wascompletely ejected from the pipette. Following surgery, animalswere housed individually with free access to water and food.
Rats injected with gp120 into the CP (6 h, 24 h, 2 days,4 days, and 7 days) and SN (4 days and 7 days) wereanesthetized by intraperitoneal (i.p.) injection of sodiumpentobarbital (Abbott Laboratories, North Chicago, IL, USA) at60 mg kg−1 and perfused transcardially through the ascendingaorta with 10 ml or heparinized saline followed by ice-cold 4%paraformaldehyde (Electron Microscopy Sciences, FortWashington, PA, USA) in 0.1 M phosphate buffer (pH 7.4).Immediately following perfusion–fixation, the rat brains weredissected out, placed in 4% paraformaldehyde for 24 h, then ina 30% sucrose solution for 24 h and frozen in methyl butanecooled in liquid nitrogen. The samples were cut on a cryostat(10 mm sections).
2.10. Immunocytochemistry
For immunofluorescence, coronal cryostat sections (10 μmthick) were processed for indirect immunofluorescence.Blocking was performed by incubating for 60 min with 10%goat serum, or 10% donkey serum, depending on the source ofthe secondary antibody to be used, in phosphate-bufferedsaline (PBS) (pH 7.4). Then, sections were incubated withantibodies diluted according to manufacturer's recommenda-tions: 1 h with primary antibody, then 1 h with secondaryantibody diluted at 1:100, all at room temperature. Doubleimmunofluorescence was performed as described previously(Rouger et al., 2001). All incubations were followed byextensive washing with PBS. To stain nuclei, we usedmounting medium containing 4′,6-diamidino-2-phenylindole(Vector Laboratories, Burlingame, CA, USA). Specimens werefinally examined under a Leica DMRBE microscope (Leica
Microsystems, Wetzlar, Germany). Negative controls con-sisted of preincubation with PBS and 0.1% bovine serumalbumin, substitution of non-immune isotype-matched con-trol antibody for primary antibody, and/or omission of theprimary antibody.
2.11. Staining of neurons using neurotrace (NT)
After rehydration in 0.1 M PBS (pH 7.2) sections were treatedwith PBS plus 0.1% Triton X-100 for 10 min, washed twice for5 min in PBS, and then stained by NT (Molecular Probes Inc.,Eugene, OR, USA) (1:100), a fluorescent Nissl stain, for 20min atroom temperature. Sections were washed in PBS plus 0.1%Triton X-100, then with PBS, and were left undisturbed for 2 hat room temperature in PBS before being counterstained with4′,6-diamidino-2-phenylindole. Combination stainingwith NT+ antibodywas done by primary and secondary immunostain-ing first (see above), followed by staining with NT fluorescentNissl stain. Experiments were repeated three times and werecarried out the same day for the different sections considered.
2.12. General morphology
Microscopic morphology of the brain was assessed by neutralred (NR) staining of cryostat sections.
2.13. Statistical analysis
Comparisons of 2 groups used the Mann–Whitney test (2-tailed P value). Comparisons of more than 2 groups used theKruskal–Wallis test. Differences between groups were consid-ered significant if P<0.05.
3. Results
3.1. Cultured DNs express tyrosine hydroxylase asa marker for DNs and are more sensitive than RNsto oxidative stress
Fully differentiated, DNs were prepared as described inMethods, from 12 to 16-week-old fetal brain, then comparedto RNs for expression of the DN marker tyrosine hydroxylase(TH) by immunohistology (Fig. 1a) and Western blotting (Fig.1b). DNs were positive for TH by both assays, compared tocontrol cultures. On Western analysis, TH was visualized as aprominent band around 65 kDa. These cells were also positivefor protein markers such as nestin, NCAM (neuronal celladhesion molecule), and MAP-2 (not shown). Morphologicallyfully differentiated cells developed rounded cell bodies withneuronal morphological characteristics, forming bipolar andmultipolar extensions that formed networks (Fig. 1a).
We then tested these cultured DN for sensitivity tooxidative stress, compared to RN by exposing them to 0 to100 μM H2O2. Apoptotic cells were enumerated in all cultures(Fig. 1c). Treatment with H2O2 at 10 μM and 100 μM causedsignificantly more apoptosis (p<0.05) in DNs than in RNs.Therefore, DNs are more sensitive than RNs to oxidant stress:they underwent apoptosis at H2O2 concentrations that werefar less toxic for RNs.

120 B R A I N R E S E A R C H 1 3 0 6 ( 2 0 1 0 ) 1 1 6 – 1 3 0
3.2. Protecting RNs and DNs from gp120-inducedapoptosis using antioxidant gene delivery
The ability of H2O2 to cause apoptosis in DNs and in RNsshould be prevented by detoxification of the cytosolic H2O2.We then tested whether antioxidant gene delivery couldprotect DNs and RNs from apoptosis caused by H2O2. Wetransduced DNs and RNs with SV(GPx1), which detoxifiesH2O2, SV(SOD1), which detoxifies O2
−, or (control) SV(HBS) andmock transduction. Transgene expression in DNs was ascer-tained by enzyme activity assays (Table 1) and byWestern blotanalysis (Fig. 2a). Neurons were then challenged by exposingthem to 3 μM H2O2 (DNs) or 80 μM H2O2 (RNs), and apoptoticneurons were enumerated. Prior transduction with SV(GPx1)largely protected both DNs and RNs from the respectiveconcentrations of H2O2. The level of protection was significantcompared to control-transduced cultures (Fig. 2b). SV(SOD1)and the control rSV40, which would not be expected to protectfrom H2O2, did not. Thus, compared to RNs, DNs are more
fragile in a setting of oxidant stress and undergo apoptosisthat can prevent gene delivery of an enzyme, GPx1, capable ofcatalyzing the conversion of H2O2 to water.
3.3. DNs are more sensitive than non-dopaminergicneurons (RNs) to gp120-induced apoptosis
HIV-1 neurotoxicity involves generation of ROS, leading to celldeath by apoptosis. The documented heightened susceptibil-ity of SN neurons to HIV-1 neurotoxicity may reflect enhancedsensitivity to the HIV-1 envelope glycoprotein, gp120. Wetherefore compared dose–response relationships in RNs andDNs for gp120-induced apoptosis in vitro. Cultured RNs andDNswere incubatedwith 0–100 ng/ml of gp120 for 6–72 h, afterwhich TUNEL assays were used to enumerate apoptotic cells.DN were more sensitive, and more rapidly sensitive, to gp120(Figs. 3a and b), although the kinetics of gp120-inducedapoptosis may vary with the concentration of gp120. Theyalso demonstrated maximal apoptosis at gp120 concentra-tions 1:10 as high as was observed in RN/DN achievedmaximal apoptosis at ≈1–10 ng/ml of gp120, while RNs showedmaximal apoptosis at 10–100 ng/ml. Longer incubation timesand higher gp120 concentrations did not increase TUNELpositivity. Thus, gp120 causes apoptosis of DNs much morerapidly and at much more lower doses of gp120 than areneeded to achieve same effect in RN.
The centrality of ROS to HIV-1 neurotoxicity, and the abilityof antioxidant gene delivery to protect non-dopaminergicneurons from apoptosis caused by HIV-1 proteins, raises thequestion of to what extent ROS may participate in theheightened sensitivity of DNs to gp120. We tested thisquestion by transducing DN with SV(SOD1) or SV(GPx1) (or,as controls, SV(HBS), and mock). We then challenged themwith HIV-1 gp120 and measured apoptosis by TUNEL. Prior
Fig. 1 – Characterization of dopaminergic neurons andtheir sensitivity to H2O2. Fully differentiated, dopaminergicneurons (DN) were prepared from 12 to 16-week-oldcultured fetal brain as described in Methods, thenassayed, compared to non-dopaminergic neurons (RN) forexpression of the DN marker tyrosine hydroxylase (TH).(a) Immunocytochemistry. Cultured neurons wereimmunostained with anti-TH antibody and visualizedby immunofluorescence, with phase-contrastphotomicrographs shown for comparison. (b) Westernblotting. Protein preparations from DN and RN wereelectrophoresed on SDS–PAGE and blotted to PVDFmembranes, which were then probed using anti-THantibody. Filters were stripped and reprobed for GAPDH asa loading control. This is representative of two independentblots. (c) Sensitivity of DNs to H2O2-induced oxidant stress,compared to RN. Cultured DN or RN were exposed tovarious concentrations of H2O2 (0.1 μM to 80 μM), afterwhich TUNEL+ cells were enumerated as a measure ofH2O2-induced apoptosis. Levels of apoptosis for all doses lessthan 0.1 μM H2O2, including 0 μM H2O2, were virtuallyidentical, and are represented by the <0.01 μM data point.Differences between DN and RN were statistically significant(P at least <0.05) for all H2O2 concentrations ≥1 μM.

Table 1 – Antioxidant enzyme activity followingtransduction.
Treatment SOD1 activity (U/L) GPx1 activity (U/L)
SV(HBS) 442±45 123±19SV(GPx1) 257±65 775±41SV(SOD1) 4758±552 198±28
The enzymatic activities of SOD1 and GPx1 were quantitated asdescribed in Methods in cultures enriched for dopaminergicneurons. Data represent the means of at least 3 independentdeterminations. Differences in SOD1 activity between SV(SOD1)-transduced DN cultures and either SV(HBS)- or SV(GPx1)-transduced cultures were significantly different (P<0.01), as weredifferences in GPx1 activity between SV(GPx1)-transduced DNcultures and either SV(HBS)- or SV(SOD1)-transduced cultures.
Fig. 2 – Antioxidant gene delivery and protection of culturedDNs from H2O2-induced apoptosis. Dopaminergic (DNs) andnon-dopaminergic neurons (RNs) were transduced with SV(GPx1), carrying the cDNA for human glutathione peroxidase(GPx1), SV(SOD1), carrying the cDNA for human Cu/Znsuperoxide dismutase (SOD1) or, as a control, SV(HBS),carrying the cDNA for hepatitis B surface antigen. (a)Transgene expression was confirmed by Western blotting.Electrophoresed proteins were blotted, then probed withanti-SOD1 antibody. Filters were then stripped and reprobedwith anti-GPx1. Finally, filters were stripped again, andprobed with antibody to GAPDH as a loading control.Molecular size marker positions are indicated on the left. (b)Antioxidant protection from H2O2-induced apoptosis. DNcultures were treated with 3 mM H2O2, while RN cultureswere treated with 80 μM H2O2. One day later, apoptotic cellswere enumerated by TUNEL. These data represent theaverages of at least 4 independent determinations.Differences between SV(GPx1) treatment and all othertreatments are significant (*P at least <0.05 for allcomparisons, for both DNs and RNs).
121B R A I N R E S E A R C H 1 3 0 6 ( 2 0 1 0 ) 1 1 6 – 1 3 0
gene delivery of the antioxidant enzymes protected DNsvirtually completely from gp120-induced apoptosis (Fig. 3c).
3.4. DN hypersensitivity to gp120-induced apoptosisreflects, at least in part, specific DN receptivity to gp120
Having determined that DNs undergo apoptosis when chal-lenged with concentrations of gp120 that do not elicitsignificant apoptosis in RNs and that this response involvedROS, we asked whether these observations might involvegp120-specific responses or, on the other hand, nonspecific
DN hypersensitivity to ROS (as in Fig. 2). This was tested byexamining [Ca2+]i fluxes in RNs and DNs as a function of gp120concentration.
Cells were loaded with Fura-2AM and then treated withHIV-1BaL gp120. [Ca2+]i was measured using a split beamfluorescence photometer. In parallel, cultures of RNs or DNswere preloaded with BAPTA-AM, a Ca2+ chelator, to blockcytosol Ca2+ signaling.
DNs were specifically more responsive to gp120 than wereRNs. That is, gp120-induced changes in [Ca2+]i followed adifferent dose–response curve in DNs from that seen in RNs(Figs. 4a and b). RNs did not initiate [Ca2+]i fluxes in response to10 ng/ml gp120. Rather, gp120-initiated [Ca2+]i fluxes wereobserved in RNs at 100 ng/ml (Fig. 4a). In contrast, DNs didgenerate [Ca2+]i fluxes upon exposure to 10 ng/ml of HIV-1BaLgp120 (Fig. 4b). The observed [Ca2+]i fluxes elicited by gp120 inRNs and DNswere blocked by preincubating cells with BAPTA-AM, suggesting that release of Ca2+ from intracellular storeswas a very early step in the gp120-initiated Ca2+ signaling. Thelong shoulder in each case probably represents influx fromextracellular fluid. If cells were cultured in Ca2+-free medium,this second phase did not occur, although the initial Ca2+
release peak remained intact (not shown). Thus, in addition toshowing heightened susceptibility to apoptosis on oxidantexposure, dopaminergic neurons were also specifically highlyresponsive to apoptosis signaling triggered by gp120.
3.5. In DNs and in RNs, ROS act as signalingintermediates in gp120-induced apoptosis
The effectiveness of antioxidant gene delivery in protectingDNs and RNs from gp120-induced apoptosis raises thequestion of how gp120-elicited ROS participate in the injuryof DNs and RNs. Using the same system described above, wemeasured [Ca2+]i signaling in DNs and RNs transducedwith SV(SOD1) or SV(GPx1). Prior transduction with either SV(SOD1) orSV(GPx1) altered gp120-induced [Ca2+]i transients in both RNs(Fig. 4c) and DNs (Fig. 4d). In RNs, prior transduction witheither antioxidant vector individually prevented gp120-in-duced [Ca2+]i fluxes (Fig. 4b). In DNs, transduction with SV(SOD1) and SV(GPx1) largely blocked gp120-mediatedincreases in intracellular [Ca2+]i (Fig. 4b). Thus, HIV-1 envelopegp120 elicits [Ca2+]i fluxes as part of proapoptotic signalingprocesses in both RNs and DNs, and ROS participate as early
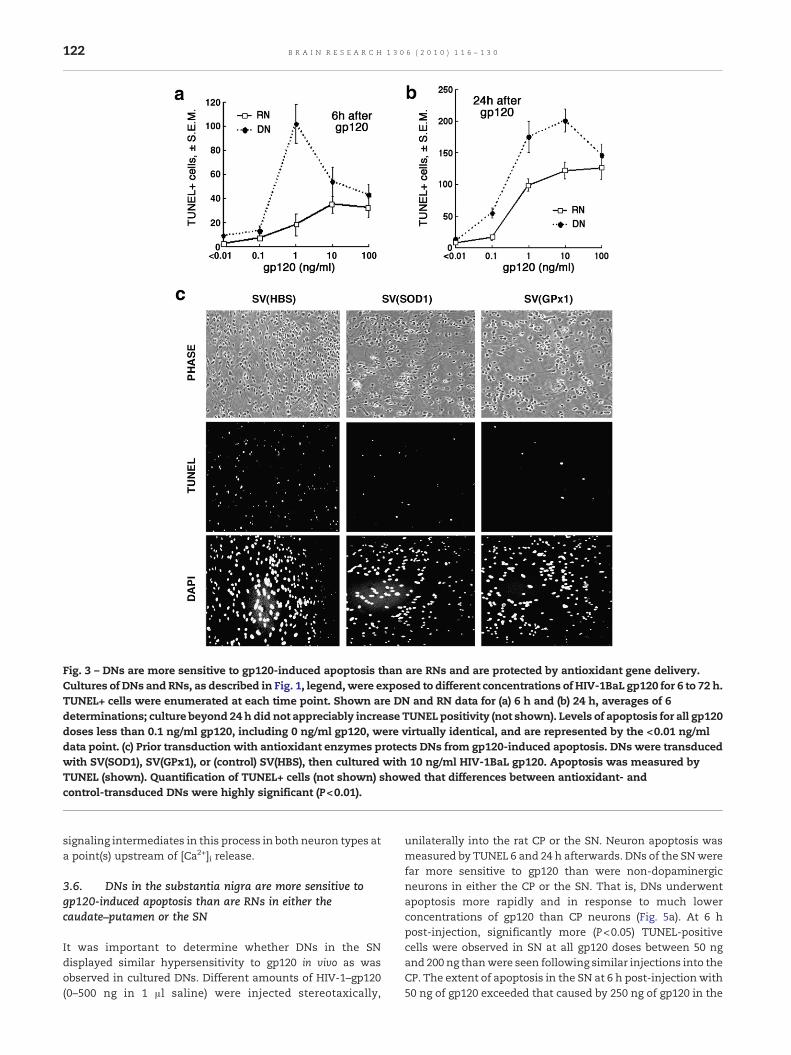
Fig. 3 – DNs are more sensitive to gp120-induced apoptosis than are RNs and are protected by antioxidant gene delivery.Cultures of DNs and RNs, as described in Fig. 1, legend,were exposed to different concentrations of HIV-1BaL gp120 for 6 to 72 h.TUNEL+ cells were enumerated at each time point. Shown are DN and RN data for (a) 6 h and (b) 24 h, averages of 6determinations; culture beyond 24 h did not appreciably increase TUNEL positivity (not shown). Levels of apoptosis for all gp120doses less than 0.1 ng/ml gp120, including 0 ng/ml gp120, were virtually identical, and are represented by the <0.01 ng/mldata point. (c) Prior transduction with antioxidant enzymes protects DNs from gp120-induced apoptosis. DNs were transducedwith SV(SOD1), SV(GPx1), or (control) SV(HBS), then cultured with 10 ng/ml HIV-1BaL gp120. Apoptosis was measured byTUNEL (shown). Quantification of TUNEL+ cells (not shown) showed that differences between antioxidant- andcontrol-transduced DNs were highly significant (P<0.01).
122 B R A I N R E S E A R C H 1 3 0 6 ( 2 0 1 0 ) 1 1 6 – 1 3 0
signaling intermediates in this process in both neuron types ata point(s) upstream of [Ca2+]i release.
3.6. DNs in the substantia nigra are more sensitive togp120-induced apoptosis than are RNs in either thecaudate–putamen or the SN
It was important to determine whether DNs in the SNdisplayed similar hypersensitivity to gp120 in vivo as wasobserved in cultured DNs. Different amounts of HIV-1–gp120(0–500 ng in 1 μl saline) were injected stereotaxically,
unilaterally into the rat CP or the SN. Neuron apoptosis wasmeasured by TUNEL 6 and 24 h afterwards. DNs of the SNwerefar more sensitive to gp120 than were non-dopaminergicneurons in either the CP or the SN. That is, DNs underwentapoptosis more rapidly and in response to much lowerconcentrations of gp120 than CP neurons (Fig. 5a). At 6 hpost-injection, significantly more (P<0.05) TUNEL-positivecells were observed in SN at all gp120 doses between 50 ngand 200 ng thanwere seen following similar injections into theCP. The extent of apoptosis in the SN at 6 h post-injection with50 ng of gp120 exceeded that caused by 250 ng of gp120 in the

Fig. 4 – DN apoptosis due to gp120 reflects specific recognition of gp120 with ROS acting as key signaling intermediates. (a, b)Differential sensitivity of DN and RN to gp120-induced calcium signaling. Non-dopaminergic neurons (a) or dopaminergicneurons (b) were loaded with Fura-2AM, then treated with either 10 ng/ml or 100 ng/ml HIV-1BaL gp120. [Ca2+]i was measuredusing a split beam fluorescence photometer, as described in Methods. In parallel, cultures of RNs or DNs were preloadedwith BAPTA-AM, a Ca2+ chelator, to bind cytosol Ca2+. [Ca2+]i was measured by comparing differential absorbance of Fura-2 at340 and 380 nm. Measurements were done for 300 s with a total of 100 frames (1 frame=3 s). Gp120 was added at 1 min (arrow,frame 20). Data represent average of three independent experiments. (c, d) Gp120-induced calcium release is blocked whenperoxide and superoxide are removed by transduction with SV(SOD1) or SV(GPx1). RNs (c) and DNs (d) were transduced withSV(HBS), (SV(SOD1)), or SV(GPx1), then loaded with Fura-2AM and challenged with HIV-1BaL gp120 (100 ng/ml for RNs,10 ng/ml for DNs). [Ca2+]i was measured in a split beam fluorescence photometer as described above. Data are representativeaverages of ≥10 individual cells in each of three independent experiments.
123B R A I N R E S E A R C H 1 3 0 6 ( 2 0 1 0 ) 1 1 6 – 1 3 0
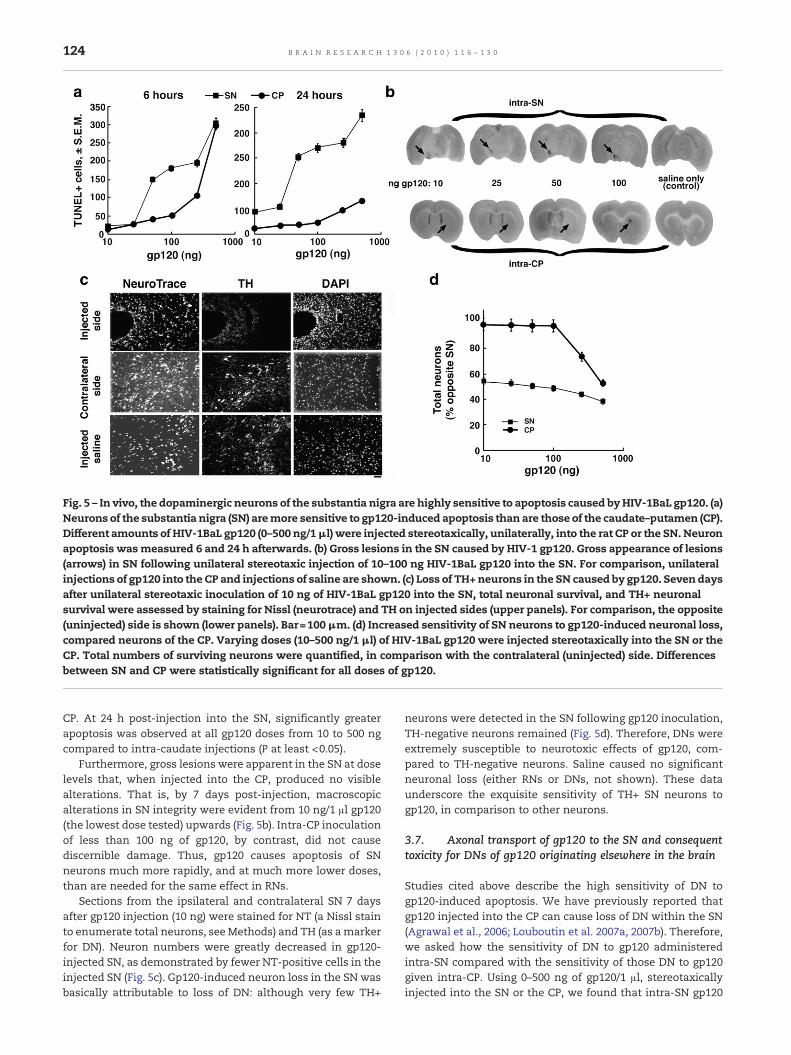
Fig. 5 – In vivo, the dopaminergic neurons of the substantia nigra are highly sensitive to apoptosis caused byHIV-1BaL gp120. (a)Neurons of the substantia nigra (SN) aremore sensitive to gp120-induced apoptosis than are those of the caudate–putamen (CP).Different amounts of HIV-1BaL gp120 (0–500 ng/1μl) were injected stereotaxically, unilaterally, into the rat CP or the SN. Neuronapoptosis wasmeasured 6 and 24 h afterwards. (b) Gross lesions in the SN caused by HIV-1 gp120. Gross appearance of lesions(arrows) in SN following unilateral stereotaxic injection of 10–100 ng HIV-1BaL gp120 into the SN. For comparison, unilateralinjections of gp120 into the CP and injections of saline are shown. (c) Loss of TH+neurons in the SN caused by gp120. Seven daysafter unilateral stereotaxic inoculation of 10 ng of HIV-1BaL gp120 into the SN, total neuronal survival, and TH+ neuronalsurvival were assessed by staining for Nissl (neurotrace) and TH on injected sides (upper panels). For comparison, the opposite(uninjected) side is shown (lower panels). Bar=100μm. (d) Increased sensitivity of SN neurons to gp120-induced neuronal loss,compared neurons of the CP. Varying doses (10–500 ng/1 μl) of HIV-1BaL gp120 were injected stereotaxically into the SN or theCP. Total numbers of surviving neurons were quantified, in comparison with the contralateral (uninjected) side. Differencesbetween SN and CP were statistically significant for all doses of gp120.
124 B R A I N R E S E A R C H 1 3 0 6 ( 2 0 1 0 ) 1 1 6 – 1 3 0
CP. At 24 h post-injection into the SN, significantly greaterapoptosis was observed at all gp120 doses from 10 to 500 ngcompared to intra-caudate injections (P at least <0.05).
Furthermore, gross lesions were apparent in the SN at doselevels that, when injected into the CP, produced no visiblealterations. That is, by 7 days post-injection, macroscopicalterations in SN integrity were evident from 10 ng/1 μl gp120(the lowest dose tested) upwards (Fig. 5b). Intra-CP inoculationof less than 100 ng of gp120, by contrast, did not causediscernible damage. Thus, gp120 causes apoptosis of SNneurons much more rapidly, and at much more lower doses,than are needed for the same effect in RNs.
Sections from the ipsilateral and contralateral SN 7 daysafter gp120 injection (10 ng) were stained for NT (a Nissl stainto enumerate total neurons, see Methods) and TH (as amarkerfor DN). Neuron numbers were greatly decreased in gp120-injected SN, as demonstrated by fewer NT-positive cells in theinjected SN (Fig. 5c). Gp120-induced neuron loss in the SN wasbasically attributable to loss of DN: although very few TH+
neurons were detected in the SN following gp120 inoculation,TH-negative neurons remained (Fig. 5d). Therefore, DNs wereextremely susceptible to neurotoxic effects of gp120, com-pared to TH-negative neurons. Saline caused no significantneuronal loss (either RNs or DNs, not shown). These dataunderscore the exquisite sensitivity of TH+ SN neurons togp120, in comparison to other neurons.
3.7. Axonal transport of gp120 to the SN and consequenttoxicity for DNs of gp120 originating elsewhere in the brain
Studies cited above describe the high sensitivity of DN togp120-induced apoptosis. We have previously reported thatgp120 injected into the CP can cause loss of DN within the SN(Agrawal et al., 2006; Louboutin et al. 2007a, 2007b). Therefore,we asked how the sensitivity of DN to gp120 administeredintra-SN compared with the sensitivity of those DN to gp120given intra-CP. Using 0–500 ng of gp120/1 μl, stereotaxicallyinjected into the SN or the CP, we found that intra-SN gp120

125B R A I N R E S E A R C H 1 3 0 6 ( 2 0 1 0 ) 1 1 6 – 1 3 0
administration was more toxic to SN neurons than was intra-CP administration (P at least <0.05 at all dose levels).Nonetheless, the extent of SN neuron loss after intra-CPinoculation of gp120 was considerable.
We therefore tested whether this observation mightreflect retrograde transport of gp120 to the SN, e.g., via thenigrostriatal tract. Antegrade transport, e.g., via the GABAergic
Fig. 6 – Gp120 localizes to axons in vitro and in vivo. (a) Localizatprimary human neurons were treated with HIV-1BaL gp120 andthe HIV-1-gp120. Gp120 immunostaining is shown overlaid on ppositivity within neuron cell bodies is highlighted by arrows andw(DAPI) in lower micrographs (representative axonal gp120 localiznoted above). The intensity of the DAPI signal was muted to facil(b) In vivo localization of gp120 in axons in the SN following intrainto the CP was assessed in the SN 4 days later. Nigrostriatal bucryostat sections using the atlas of Paxinos and Watson (1986). Ihighlighted by arrowheads. Cer P: cerebral peduncle; Aq: aquedupars reticulata. Upper row: bar=100 μm; bar, insert=20 μm. Low
striatonigral pathway, might also be responsible, althoughantegrade transport is much slower than retrograde trans-port. We thus asked whether neurons exposed to gp120localize the HIV-1 envelope protein in axons in vitro and invivo. Cultured human neurons (RNs) were treated with gp120and studied at various times for localization of the HIV-1 Envprotein (Fig. 6a). Immunostaining for gp120 was performed
ion of gp120 in axons of cultured human neurons. Culturedstudied at various times thereafter for immunolocalization ofhase-contrast micrograph (upper frames) (representativeithin axons by arrowheads) and overlaid on nuclear staining
ation highlighted by arrows and in axons by arrowheads, asitate visualization of the gp120 immunostaining. Bar=20 μm.-CP inoculation. Localization of gp120 that had been injectedndle (NS) and SN pars compacta (SNC) were determined onmmunostaining demonstrating gp120 in axons of the SN isct of sylvius; Am N: amygdaloid nuclei; SNR: substantia nigraer row: bar=100 μm; bar, insert=15 μm.

126 B R A I N R E S E A R C H 1 3 0 6 ( 2 0 1 0 ) 1 1 6 – 1 3 0
from 0 min to 24 h after exposure to gp120. Gp120 wasdetected in neuronal cell bodies and in axons as early as10 min after gp120 exposure. By 1–2 h post-treatment withgp120, the majority of the gp120 was localized within axons,withminimal staining in cell bodies (Fig. 6a). By 2 h, gp120 wasseen only in axons. Thus, gp120 was internalized by neuronsand persisted within axons long after it became undetectablein perikaryons.
We confirmed these in vitro data in studies done in vivo usinggp120-injected rat brains (Fig. 6b). Gp120 was injected into theCP. We analyzed the SN 4 days later. By immunostaining, gp120was detected in the SN pars compacta (SNpc), both in axons(upper frames) andwithincell bodies (lower frames) on the sameside as was injected (ipsilateral). Overlays show higher magni-fication, with arrows highlighting gp120-containing axons. Forcomparison, the contralateral SNpc was negative (rightmostframes). Thus, following inoculation into the CP, gp120 wastransported retrograde into the SN, where it led to loss of DN.
3.8. Intra-CP delivery of antioxidant enzymes protects theTH+ neurons in the SN from gp120 injected into the CP
Having established that DN cells can be transduced andprotected from gp120-induced apoptosis using antioxidant
Fig. 7 – Intra-CP gene delivery of antioxidant enzymes protects THinjected stereotaxically into the CPwith SV(GPx1), SV(SOD1), or (c500 ng of gp120 1 week afterwards. (a) Quantification of protectiogene delivery. The SN was analyzed for neuron populations remaare represented as percentages of opposite sides. (b) IllustrationRepresentative immunostained sections for TH and for NT in SV(Bsimilar to SV(GPx1) recipients (not shown). Bar=50 μm. Slight difvariations in the positioning of the sections between the ipsilate
enzymes delivered by SV(SOD1) and SV(GPx1) in vitro, wetested whether delivery of these antioxidant enzymes to theCP would similarly protect DN in the SN from gp120 injectedinto the CP. Rats injected stereotaxically into the CP with SV(GPx1), SV(SOD1), or, as a control, SV(BUGT) were challengedintra-CP 4 weeks later with 500 ng of gp120 in 1 μl of saline.Neuronal survival was assayed 1 week after challenge. SNsections were analyzed for total neurons by quantitating NT-positive and for DNs by enumerating TH+ cells. The latter werecompared with the contralateral SN, both in terms of viabletotal neurons (NT+ cells) and viable DN (TH+) cells. SN neuronsimmunopositive for TH were greatly reduced in rats givenintra-CP gp120, compared to those receiving the control vectorSV(BUGT) (Figs. 7a and b). By contrast, when the CP waspreviously injected either with SV(GPx1) or with SV(SOD1),numerous TH+ cells in the SN were largely protected from theretrograde toxicity of gp120 injected into the CP. Antioxidantprotection of TH+ neurons in the SN was highly significant(p<0.01), compared to control vector (SV(BUGT)) recipients(Figs. 7a and b). It should be emphasized that the retrogradetransport of gp120 into the SN after intra-CP gp120 led topredominant loss of dopaminergic neurons (TH+, NT+) in theSN. Other SN neurons (TH−, NT+) were generally spared.Furthermore, transduction of the CP using SV(SOD1) or SV
+ neurons in the SN fromgp120 injected into the CP. Ratswereontrol) SV(BUGT), then challengedwith intra-CP inoculation ofn from gp120 provided to SN neurons by intra-CP antioxidantining. Neurotrace-+ (NT) and TH+ cells were enumerated andof antioxidant protection of SN neurons from gp120.UGT) and SV(GPx1) transduced rats. SV(SOD1) recipientswereferences in neuron density are seen, most likely due to slightral and contralateral SNs, relative to collections of neurons.

127B R A I N R E S E A R C H 1 3 0 6 ( 2 0 1 0 ) 1 1 6 – 1 3 0
(GPx1) protected DN from the toxicity of gp120 delivered to theCP. Therefore, TH+ neurons of the SN are susceptible to thetoxicity of gp120 that may be transported in axons from otherparts of the brain, and these DNs can be protected byantioxidant gene delivery to places from which the gp120 isdelivered to the SN.
4. Discussion
Gp120 binds neuron cell membrane coreceptors (CCR3, CCR5,CXCR4) and elicits apoptosis (Garden et al., 2004) via G protein-coupled pathways (Kruman, 1998; Kaul and Lipton, 1999).Soluble gp120 also increases glial cell release of arachidonate,which impairs astrocyte and neuron reuptake of glutamate(Lipton et al., 1993) leading to prolonged activation of N-methyl-D-aspartate (NMDA) receptor with consequent disrup-tion of cellular Ca2+ homeostasis (Kaul and Lipton, 2006). Asecond kind of calcium-permeable membrane receptor affect-ed by gp120 is voltage-operated calcium channels (VOCCs)(Lipton, 1991). At least in part, activation of VOCCs by gp120appears to be due to membrane depolarization, which occursas a result of glutamate receptor-mediated ion flux and bymechanisms involving amiloride-sensitive Na+/H+ exchangeand K+ efflux in astrocytes (Benos et al., 1994).
Here, we examined the basis for the frequency of Parkin-son's-like symptomatology and pathology in neuroAIDS. We,therefore, tested the relationship between HIV-1-relatedoxidative stress and loss of DN in the substantia nigra. In sodoing, we documented both the heightened nonspecificsensitivity of DN to apoptosis caused by peroxide and thefact that DNs are also specifically more sensitive to apoptosiselicited by HIV-1 envelope glycoprotein, gp120.
To perform these studies, we generated DNs from primaryhuman fetal brain tissue. The method we used is similar to oneusedbyRavindranandRao (2006) todifferentiateNT-2 cells overtime with a cocktail of growth factors, without retinoic acid. Byimmunostaining, resulting neuron cultures were overwhelm-ingly TH+. Using these cells and applying H2O2 as a nonspecificoxidant stress, DNs underwent apoptosis at much lower H2O2
concentrations than non-dopaminergic neurons (RNs). Bothtypes of neurons were protected by SV(GPx1) genedelivery, butnotbySV(SOD1).This isasanticipated, sinceGPx1convertsH2O2
to H2O, while SOD1 does not detoxify H2O2.The nonspecific hypersensitivity of DN to exogenous ROS
does not explain the heightened sensitivity of DN to apoptosiselicited by HIV-1 envelope glycoprotein, gp120, however,because gp120 is able to elicit apoptosis-inducing Ca2+
signaling at far lower concentrations of gp120 in DNs thanare required for RN. DN apoptosis caused by gp120 is thereforea signaling process initiated by a specific interaction betweengp120 and the neuron, and DN are more responsive than areRN to such signal initiation or propagation.
The effectiveness of gene delivery of both antioxidantenzymes, SOD1 and GPx1, in protecting DNs from gp120-induced apoptosis indicates that superoxide (O2
−, converted toH2O2 by SOD1) and peroxide (H2O2, converted to H2O by GPx1)are required. Thus, fact that gp120-initiated [Ca2+] fluxes occuralmost immediately upon exposure to gp120 indicates that[Ca2+] release is central to gp120-induced signaling. Further-
more, the parallel abilities of SOD1 and GPx1 both to blockgp120-initiated [Ca2+]i fluxes and to protect from gp120-induced apoptosis strongly suggests that O2
− and H2O2 arerequired for HIV-1 envelope-induced apoptosis in neurons andthat they participate in proapoptotic pathways at a point(s)upstreamof Ca2+ release in the proapoptotic signaling process.The slightly reduced inhibition of [Ca2+]i fluxes by SOD1,compared to GPx1 and compared to SOD1 inhibition of gp120-induced apoptosis, may reflect alternate signaling pathwaysin gp120-induced Ca2+ release that do not involve O2
− and donot signal apoptosis.
The basis for DN hypersensitivity to gp120-induced ROSsignaling is not yet known. It may reflect greater binding ofgp120 by DNs than by RNs, differences in coreceptor density ordistribution, generation of more ROS signaling species at thecell membrane, more effective signaling by ROS and/or othermechanisms.
In addition to their roles in mediating oxidative damage tocellular proteins and lipids, ROS are important components ofproapoptotic signaling that is activated by HIV-1 gp120 in DNsand in RNs. As such, their involvement supports the conclu-sion that the exquisite sensitivity of DNs to HIV-1 neurotox-icity reflects a specific response to gp120, in addition to—orinstead of—nonspecific DN sensitivity to ROS-mediatedcellular damage. Similar data have been obtained using theother major HIV-1-encoded neurotoxin, Tat (L. Agrawal et al.,in preparation). The prevalence of Parkinson-like DN loss andclinical presentation in neuroAIDS thus represents, at least inpart, specific DN sensitivity to these HIV-1 proteins.
It is now clear that ROS and RNS are important signalingintermediates in many receptor driven and other processes(Matsuzawa and Ichijo, 2005; Stone and Yang, 2006). Produc-tion of superoxide, peroxide and other oxygen radicals occursat the cell membrane and is activated by receptor-triggeredenzymes, particularly NADPH oxidase (NOX) (Finkel, 2003).NOXes, are multimeric enzymes activated by some cellmembrane receptors. These activated receptors bind sulfhy-dryl groups at the enzymes' active sites, activating them toproduce O2
− or H2O2. The specificity of the interaction and thenature of the result depend in part on the magnitude and theduration of the increase in ROS. Transient, relatively low-levelexposure to O2
− or to H2O2 may activate cell proliferation, oftenmediated by the ERK family of MAP kinases (Matsuzawa andIchijo, 2005). Longer and greater exposures may have theopposite effect, leading to apoptosis, by a number of potentialmechanisms. Proapoptotic signaling may be mediated by p38and JNK groups of MAP kinases, particularly apoptosissignaling kinase-1 (ASK-1) (Benhar et al., 2001). Alternatively,ER Ca2+ release may affect the balance of calcium withinnearby mitochondria and lead to apoptosis via mechanismsrelating to release of mitochondrial constituents like cyto-chrome C and Apaf-1 into the cytosol (Gorman, 2008; Pinton etal., 2008).
The direct toxic effects of HIV-1 gp120 on DN are likelymagnified by the retrograde axonal transport of gp120 fromsites distant from the SN, with loss of DN as a result. AlthoughDN in the SN were less sensitive to gp120 administered intra-CP than to gp120 given directly intra-SN, the transport of gp120from the CP to the SN likely provides an additional means bywhich gp120 exerts its toxicity for dopaminergic neurons. By

128 B R A I N R E S E A R C H 1 3 0 6 ( 2 0 1 0 ) 1 1 6 – 1 3 0
1 week after intra-CP inoculation of HIV-1 Env, TH+ neuronswere all but absent from the SN. Only TH-negative neuronsremained. It is of note that similar DN loss via axonal transportis also seen with HIV-1 Tat (L. Agrawal, in preparation).
Our findings have potential therapeutic implications. First,DNs are protected by antioxidant enzyme gene delivery.Antioxidant compounds such as the glutathione precursor, N-acetylcysteine, have shownneuroprotective activity in vitro, andare being studied in early clinical trials for treatment ofneuroAIDS (reviewed, Steiner et al., 2006), although antioxidantneuroprotection remains a future goal. It is unclear to whatextent gp120 neurotoxicity in vivo is a direct effect of the HIV-1Env glycoprotein, or perhaps an indirect consequence of gp120actions on other cell types. In either event, our data suggest thatuse of antioxidant genedeliverywithapermanentgenedeliveryvehicle like SV40 to target a particular area of the brain, such asthe SN, offers the potential to protect from a relatively commonconsequence of HIV-1 encephalopathy. Furthermore, we havedemonstrated effective gene delivery throughout the CNS.Intra-CSF administration of gene transfer vectors providesantioxidant protection from local gp120 challenge comparableto that observed with local gene delivery at the site ofsubsequent challenge (Louboutin et al., 2007a, 2007b).
Despite many parallels in clinical presentation and strikingpredilection for DN, it is not clear to what extent our data forneuroAIDSmayhelpunderstand thepathogenesisof Parkinson'sdisease. Themechanisms bywhich PD develops are obscure andmay involve any number of factors not known to be related toneuroAIDS: purine recycling (Lewers et al., 2008), increasedsensitivity to PGE (Carrasco et al., 2008) and to proteinmisfolding(McNaught et al., 2006; Rideout et al., 2005; Xiong et al., 2009),dysfunction of mitochondrial electron transport complex I(Ahmadi et al., 2008), etc. The ability of cytosolic SOD (SOD1) toprotect from gp120-induced injury contrasts both with theimportance of mitochondrial SOD (SOD2) in limiting the mito-chondrial dysfunction in PD and with the inability of SOD2 genedelivery to mitigate gp120-induced apoptosis (L. Agrawal, pre-liminary studies). However, DN sensitivity to oxidative stress, therole of glutathione inprotectingDNs fromROS—and its depletionin PD—and the common factor of the added oxidative stresscausedbyDNmetabolitesareall parallel in theDNloss seen inPDand neuroAIDS.
Therefore, the sensitivity of dopaminergic neurons to HIV-1-related neurotoxicity reflects a specific sensitivity to HIV-1gp120. This sensitivity is mediated by ROS that serve assignaling intermediates, over and above possible effects of ROSin damaging cellular macromolecules. These data suggest thatROS play evenmore important mechanistic roles in neuroAIDSthan has previously been realized. They also suggest thattargeting ROS, whether by gene delivery or other techniques,may be worth considering as an approach to treating thisincreasingly more prevalent complication of HIV-1 infection.
Acknowledgments
This work was supported by NIH grant MH70287. We areindebted to the Human Fetal Tissue Bank at Albert EinsteinCollege of Medicine for the primary tissue from which neuroncultures were prepared.
R E F E R E N C E S
Agrawal, L., Louboutin, J.P., Reyes, B.A.S., Van Bockstaele, E.J.,Strayer, D.S., 2006. Antioxidant enzyme gene delivery toprotect from HIV-1 gp120-induced neuronal apoptosis. GeneTher. 13, 1645–1656.
Agrawal, L., Louboutin, J.P., Strayer, D.S., 2007. Preventing HIV-1Tat-induced apoptosis using antioxidant enzymes:mechanistic and therapeutic implications. Virology 363,462–472.
Ahmadi, F.A., Grammatopoulos, T.N., Poczobutt, A.M., Jones, S.M.,Snell, L.D., Das, M., Zawada, W.M., 2008. Dopamine selectivelysensitizes dopaminergic neurons to rotenone-inducedapoptosis. Neurochem. Res. 33, 886–901.
Bandaru, V.V.R., McArthur, J.C., Sacktor, N., Cutler, R.G., Knapp,E.L., Mattson, M.P., Haughey, N.J., 2007. Associative andpredictive biomarkers of dementia in HIV-1-infected patients.Neurology 68, 1481–1487.
Ben-Shachar, D., Zuk, R., Glinka, Y., 1995. Dopamine neurotoxicity:inhibition of mitochondrial respiration. J. Neurochem. 64,718–723.
Benhar, M., Dalyot, I., Engelberg, D., Levitzki, A., 2001. EnhancedROS production in oncogenically transformed cells potentiatesc-Jun N-terminal kinase and p38 mitogen-activated proteinkinase activation and sensitization to genotoxic stress. Mol.Cell. Biol. 21, 6913–6926.
Benos, D.J., Hahn, B.H., Bubien, J.K., Ghosh, S.K., Mashburn, N.A.,Chaikin, M.A., Shaw, G.M., Benveniste, E.N., 1994. Envelopeglycoprotein gp120 of human immunodeficiency virus type 1alters ion transport in astrocytes: implications for AIDSdementia complex. Proc. Natl. Acad. Sci. U. S. A. 91,494–498.
Berger, J.R., Arendt, G., 2000. HIV dementia: the role of the basalganglia and dopaminergic systems. J. Psychopharm. 14,214–221.
Bernheimer, H., Birkmayer, W., Hornykiewicz, O., Jellinger, K.,Seitelberger, F., 1973. Brain dopamine and the syndromes ofParkinson and Huntington. Clinical, morphological andneurochemical correlations. J. Neurol. Sci. 20, 415–455.
Betz, A., Shakui, L.P., Davidson, B.L., 1998. Gene transfer to rodentbrain with recombinant adenoviral vectors: effects of infusionparameters, infectious titer, and virus concentration ontransduction volume. Exp. Neurol. 150, 136–142.
Carrasco, E., Werner, P., Casper, D., 2008. Prostaglandin receptorEP2 protects dopaminergic neurons against 6-OHDA-mediatedlow oxidative stress. Neurosci. Lett. 441, 44–49.
Chang, L., Wang, G.J., Volkow, N.D., Ernst, T., Telang, F., Logan, J.,Fowler, J.S., 2008. Decreased brain dopamine transporters arerelated to cognitive deficits in HIV patients with and withoutcocaine abuse. NeuroImage 42, 869–878.
Corasaniti, M.T., Bagetta, G., Rotiroti, D., Nistico, G., 1998a. The HIVenvelope protein gp120 in the nervous system. Biochem.Pharmacol. 56, 153–156.
Corasaniti, M.T., Bagetta, G., Rotiroti, D., Nistico, G., 1998b. The HIVenvelope protein gp120 in the nervous system: interactionswith nitric oxide, interleukin-1beta and nerve growth factorsignaling, with pathological implications in vivo and in vitro.Biochem. Pharmacol. 56, 153–156.
Corasantini, M.T., Piccirilli, S., Paoletti, A., Nistico, R., Stringaro, A.,Malorni, W., Finazzi-Agro, A., Bagetta, G., 2001. Evidence thatthe HIV-1 coat protein gp120 causes neuronal apoptosis in theneocortex of rat via a mechanism involving CXCR4 chemokinereceptor. Neurosci. Lett. 312, 67–70.
Everall, I.P., Hansen, L.A., Masliah, E., 2005. The shifting patterns ofHIV encephalitis neuropathology. Neurotox. Res. 8, 51–61.
Fahn, S., Cohen, G., 1992. The oxidant stress hypothesis inParkinson's disease: evidence supporting it. Ann. Neurol. 32,804–812.

129B R A I N R E S E A R C H 1 3 0 6 ( 2 0 1 0 ) 1 1 6 – 1 3 0
Finkel, T., 2003. Oxidant signals and oxidative stress. Curr. Opin.Cell Biol. 15, 247–254.
Garden, G.A., Guo, W., Jayadev, S., Tun, C., Balcaitis, S., Choi, J.,Montine, T.J., Moller, T., Morrison, R.S., 2004. HIV associatedneurodegeneration requires p53 in neurons and microglia.FASEB J. 1141–1143.
Gartner, S., 2000. HIV infection and dementia. Science 287,602–604.
Gluck, M.R., Zeevalk, G.D., 2004. Inhibition of brain mitochondrialrespiration by dopamine and its metabolites: implications forParkinson's disease and catecholamine-associated diseases.J. Neurochem. 91, 788–795.
Gonzalez-Scarano, F., Martin-Garcia, J., 2005. Theneuropathogenesis of AIDS. Nat. Rev. Immunol. 5, 69–81.
Gorman, A.M., 2008. Neuronal cell death in neurodegenerativediseases: recurring themes around protein handling. J. Cell.Mol. Med. 12, 2263–2280.
Hornykiewicz, O., 2001. Chemical neuroanatomy of the basalganglia—normal and in Parkinson's disease. J. Chem.Neuroanat. 22, 3–12.
Itoh, K., Mehraein, P., Weis, S., 2000. Neuronal damage of thesubstantia nigra in HIV-1 infected brains. Acta Neuropathol. 99,376–384.
Jenner, P., 2003. Oxidative stress in Parkinson's disease. Ann.Neurol. 53 (Suppl 3), S36–S38.
Kaul, M., Lipton, S.A., 1999. Chemokines and activatedmacrophages in HIV gp120-induced neuronal apoptosis.Proc. Natl. Acad. Sci. U. S. A. 96, 8212–8216.
Kaul, M., Lipton, S.A., 2006. Mechanisms of neuronal injury anddeath in HIV-1 associated dementia. Curr. HIV Res. 4, 307–318.
Kaul, M., Zheng, J., Okamoto, S., Gendelman, H.E., Lipton, S.A.,2005. HIV-1 infection and AIDS: consequences for the centralnervous system. Cell Death Differ. 1, 878–892.
Khanlou, N., Moore, D.J., Chana, G., Cherner, M., Lazzaretto, D.,Dawes, S., Grant, I., Masliah, E., Everall, I.P., HNRC Group, 2009.Increased frequency of α-synuclein in the substantia nigra inhuman immunodeficiency virus infection. J. Neurovirol. 15,131–138.
Kondo, R., Feitelson, M.A., Strayer, D.S., 1998. Use of SV40 toimmunize against hepatitis B surface antigen: implications forthe use of SV40 for gene transduction and its use as animmunizing agent. Gene Ther. 5, 575–582.
Koutsilieri, E., Sopper, S., Scheller, C., ter Meulen, V., Riederer, P.,2002a. Parkinsonism in HIV dementia. J. Neural. Transm. 109,767–775.
Koutsilieri, E., Sopper, S., Scheller, C., ter Meulen, V., Riederer, P.,2002b. Involvement of dopamine in the progression of the AIDSdementia complex. J. Neural. Transm. 109, 399–410.
Kruman, I.I., Nath, A., Mattson, M.P., 1998. HIV-1 protein Tatinduces apoptosis of hippocampal neurons by a mechanisminvolving caspase activation, calcium overload, and oxidativestress. Exp. Neurol. 154, 276–288.
Langford, T.D., Letendre, S.L., Larrea, G.J., Maslia, E., 2003.Changing patterns in the neuropathologenesis of HIV duringthe HAART era. Brain Pathol. 13, 195–210.
Lawrence, D.M., Major, E.O., 2002. HIV-1 and the brain: connectionsbetween HIV-1-associated dementia, neuropathology, andneuroimmunology. Microbes Infect. 4, 301–308.
Levy, O.A., Malagelada, C., Greene, L.A., 2009. Cell death pathwaysin Parkinson's disease: proximal triggers, distal effectors, andfinal steps. Apoptosis 14, 478–500.
Lewers, J.C., Ceballos-Picot, I., Shirley, T.L., Mockel, L., Egami, K.,Jinnah, H.A., 2008. Consequences of impaired purine recyclingin dopaminergic neurons. Neuroscience 152, 761–772.
Lipton, S.A., 1991. Calcium channel antagonists and humanimmunodeficiency virus coat protein-mediated neuronalinjury. Ann. Neurol. 30, 110–114.
Lipton, S.A., Choi, Y.B., Pan, Z.H., Lei, S.Z., Chen, H.S., Sucher, N.J.,Loscalzo, J., Singel, D.J., Stamler, J.S., 1993. A redox-based
mechanism for the neuroprotective and neurodestructiveeffects of nitric oxide and related nitroso-compounds. Nature364, 626–632.
Louboutin, J.P., Agrawal, L., Reyes, B.A.S., van Bockstaele, E.J.,Strayer, D.S., 2007a. Protecting neurons from HIV-1gp120-induced oxidant stress using both localizedintracerebral and generalized intraventricular administrationof antioxidant enzymes delivered by SV40-derived vectors.Gene Ther. 14, 1650–1661.
Louboutin, J.P., Reyes, B., Agrawal, L., Van Bockstaele, E., Strayer,D.S., 2007b. Strategies for CNS-directed gene delivery: in vivogene transfer to the brain using SV40-derived vectors. GeneTher. 14, 939–949.
Matsuzawa, A., Ichijo, H., 2005. Stress-responsive protein kinasesin redox-regulated apoptosis signaling. Antioxid. Redox Signal.7, 472–481.
Mattson, M.P., Haughey, N.J., Nath, A., 2005. Cell death in HIVdementia. Cell Death Differ. 893–904.
McArthur, J.C., Sacktor, M.D., Selnes, O., 1999. Humanimmunodeficiency virus-associated dementia. Sem. Neurol.19, 129–150.
McArthur, J.C., Brew, B.J., Nath, A., 2005. Neurologicalcomplications of HIV infection. Lancet Neurol. 4, 543–555.
McNaught, K.S., Jackson, T., Jnobaptiste, R., Kapustin, A., Olanow,C.W., 2006. Proteasomal dysfunction in sporadic Parkinson'sdisease. Neurology 23 (Suppl 4), S37–S49.
Nath, A., Haughey, N.J., Jones, M., Anderson, C., Bell, J.E., Geiger,J.D., 2000a. Synergistic neurotoxicity by humanimmunodeficiency virus proteins Tat and gp120: protectionby memantine. Ann. Neurol. 47, 186–194.
Nath, A., Anderson, C., Jones, M., Maragos,W., Booze, R., Mactutus,C., Bell, J., Hauser, K.F., Mattson, M., 2000b. Neurotoxicity anddysfunction of dopaminergic systems associated with AIDSdementia. J. Psychopharm. 14, 222–227.
Navia, B.A., Rostasy, K., 2005. The AIDS demential complex:clinical and basic neuroscience and implications for novelmolecular therapies. Neurotox. Res. 8, 3–24.
Paxinos, G., Watson, C., 1986. The Rat Brain in StereotaxicCoordinates. Academic Press, New York.
Pinton, P., Giorgi, C., Siviero, R., Zecchini, E., Rizzuto, R., 2008.Calcium and apoptosis: ER–mitochondria Ca2+ transfer in thecontrol of apoptosis. Oncogene 27, 6407–6418.
Rappaport, J., Joseph, J., Croul, S., Alexander, G., Del Valle, L.,Amini, S., Khalili, K., 1999. Molecular pathway involved inHIV-1-induced CNS pathology: role of viral regulatory proteinTat. J. Leukoc. Biol. 65, 458–465.
Rausch, D.M., Murray, E.A., Eiden, L.E., 1999. The SIV-infectedrhesus monkey model for HIV-associated dementia andimplications for neurological diseases. J. Leukoc. Biol. 65,466–474.
Ravindran, G., Rao, H.S., 2006. Enriched NCAM-positive cells formfunctional dopaminergic neurons in the rat model ofParkinson's disease. Stem Cells Dev. 15, 575–582.
Reyes, M.G., Faraldi, F., Senseng, C.S., Flowers, C., Fariello, R., 1991.Nigral degeneration in acquired immune deficiency syndrome(AIDS). Acta Neuropathol. 82, 39–44.
Rideout, H.J., Lang-Rollin, I.C.J., Savalle, M., Stefanis, L., 2005.Dopaminergic neurons in rat ventral midbrain culturesundergo selective apoptosis and form inclusions, but do notup-regulate iHSP70, following proteasome inhibition.J. Neurochem. 93, 1304.
Rouger, K., Louboutin, J.P., Villanova, M., Cherel, Y., Fardeau, M.,2001. X-linked vacuolated myopathy: TNF-alpha andIFN-gamma expression in muscle fibers with MHC class I onsarcolemma. Am. J. Pathol. 158, 355–359.
Rumbaugh, J.A., Nath, A., 2006. Developments in HIVneuropathogenesis. Curr. Pharm. Des. 12, 1023–1044.
Sacktor, N., McDermott, M.P., Marder, K., Schifitto, G., Selnes, O.A.,McArthur, J.C., Stern, Y., Albert, S., Palumbo, D., Kieburtz, K., De

130 B R A I N R E S E A R C H 1 3 0 6 ( 2 0 1 0 ) 1 1 6 – 1 3 0
Marcaida, J.A., Cohen, B., Epstein, L., 2002. HIV associatedcognitive impairment before and after the advent ofcombination therapy. J. Neurovirol. 8, 136–142.
Sauter, B.V., Parashar, B., Chowdhury, N.R., Kadakol, A., Ilan, Y.,Singh, H., Milano, J., Strayer, D.S., Chowdhury, J.R., 2000. Genetransfer to the liver using a replication-deficient recombinantSV40 vector results in long-term amelioration of jaundice inGunn rats. Gastroenterol 119, 1348–1357.
Shi, B., Raina, J., Lorenzo, A., Busciglio, J., Gabzuda, D., 1998.Neuronal apoptosis induced by HIV-1 Tat protein and TNF-α:potentiation of neurotoxicity mediated by oxidative stress andimplications for HIV-1 dementia. J. Neurovirol. 4, 281–290.
Silvers, J.M., Aksenov, M.Y., Aksenova, M.V., Beckley, J., Olton, P.,Mactutus, C.F., Booze, R.M., 2006. Dopaminergic markerproteins in the substantia nigra of human immunodeficiencyvirus type 1-infected brains. J. Neurovirol. 12, 140–145.
Silvers, J.M., Aksenova, M.V., Aksenov, M.Y., Mactutus, C.F., Booze,R.M., 2007. Neurotoxicity of HIV-1 Tat protein: involvement ofD1 dopamine receptor. Neurotoxicology 28, 1184–1190.
Sinclair, E., Ronquillo, R., Lollo, N., Deeks, S.G., Hunt, P., Yian-noutsos, C.T., Spudich, S., Price, R.W., 2008. Antiretroviraltreatment effect on immune activation reduces cerebrospinalfluid HIV-1 infection. JAIDS 47, 544–552.
Steiner, J., Haughey, N., Li, W., Venkatesan, A., Anderson, C., Reid,R., Malpica, T., Pochernich, C., Butterfield, D.A., Nath, A., 2006.Oxidative stress and therapeutic applications in HIV dementia.Antiox. Redox Signal. 8, 2089–2100.
Stone, J.R., Yang, S., 2006. Hydrogen peroxide: a signalingmessenger. Antioxid. Redox Signal. 8, 243–270.
Strayer, D.S., 1999. Gene therapy using SV40-derived vectors: whatdoes the future hold? J. Cell. Physiol. 181, 375–384.
Strayer, D.S., Kondo, R., Milano, J., Duan, L.X., 1997. Use ofSV40-based vectors to transduce foreign genes to normalhuman peripheral blood mononuclear cells. Gene Ther. 4,219–225.
Strayer, D.S., Lamothe, M., Wei, D., Milano, J., Kondo, R., 2001.Generation of recombinant SV40 vectors for gene transfer.Methods Mol. Biol. 65, 87–102.
Strayer, D.S., Mitchell, C., Maier, D.A., Nichols, C.N., 2006.Papovaviruses: SV40. In: Friedmann, T., Rossi, J. (Eds.), GeneTransfer: Delivery and Expression of DNA and RNA, ALaboratory Manual, Ch. 24. Cold Spring Harbor Press,pp. 273–287.
Turchan, J., Pocernich, C.B., Gairola, C., Chauhan, A., Schifitto, G.,Butterfield, D.A., et al., 2003. Oxidative stress in HIV dementedpatients and protection ex vivo with novel antioxidants.Neurology 60, 307–314.
Wang, G.J., Chang, L., Volkow, N.D., Telang, F., Logan, J.,Ernst, T., Fowler, J.S., 2004. Decreased brain dopaminergictransporters in HIV-associated dementia patients. Brain 127,2452–2458.
Watanabe, Y., Chu, Y., Andresen, J.J., Nakane, H., Faraci, F.M.,Heistad, D.D., 2003. Gene transfer of extracellular superoxidedismutase reduces cerebral vasospasm after subarachnoidhemorrhage. Stroke 34, 434–440.
Xiong, H., Wang, D., Chen, L., Choo, Y.S., Ma, H., Tang, C., Xia, K.,Jiang, W., Ronai, Z., Zhuang, X., Zhang, Z., 2009. Parkin, PINK1,and DJ-1 form a ubiquitin E3 ligase complex promotingunfolded protein degradation. J. Clin. Invest. 119, 650–666.
Zhang, K., Kaufman, R.J., 2006. The unfolded protein response: astress signaling pathway critical for health and disease.Neurology 66, S102–S109.
Copyright © 2022 FDOKUMEN




















