
Gold nanoparticle-based electrochemical biosensors
19
Electrochimica Acta 53 (2008) 5848–5866 Contents lists available at ScienceDirect Electrochimica Acta journal homepage: www.elsevier.com/locate/electacta Gold nanoparticle-based electrochemical biosensors Jos ´ e M. Pingarr ´ on ∗ , Paloma Y ´ a˜ nez-Sede ˜ no, Araceli Gonz ´ alez-Cort ´ es Department of Analytical Chemistry, Faculty of Chemistry, University Complutense of Madrid, 28040 Madrid, Spain article info Article history: Received 25 January 2008 Received in revised form 26 February 2008 Accepted 1 March 2008 Available online 18 March 2008 Keywords: Electrochemical biosensors Gold nanoparticles Enzyme electrodes Immunosensors DNA sensors abstract The unique properties of gold nanoparticles to provide a suitable microenvironment for biomolecules immobilization retaining their biological activity, and to facilitate electron transfer between the immobi- lized proteins and electrode surfaces, have led to an intensive use of this nanomaterial for the construction of electrochemical biosensors with enhanced analytical performance with respect to other biosensor designs. Recent advances in this field are reviewed in this article. The advantageous operational character- istics of the biosensing devices designed making use of gold nanoparticles are highlighted with respect to non-nanostructured biosensors and some illustrative examples are commented. Electrochemical enzyme biosensors including those using hybrid materials with carbon nanotubes and polymers, sol–gel matrices, and layer-by-layer architectures are considered. Moreover, electrochemical immunosensors in which gold nanoparticles play a crucial role in the electrode transduction enhancement of the affinity reaction as well as in the efficiency of immunoreagents immobilization in a stable mode are reviewed. Similarly, recent advances in the development of DNA biosensors using gold nanoparticles to improve DNA immobilization on electrode surfaces and as suitable labels to improve detection of hybridization events are considered. Finally, other biosensors designed with gold nanoparticles oriented to electrically contact redox enzymes to electrodes by a reconstitution process and to the study of direct electron transfer between redox proteins and electrode surfaces have also been treated. © 2008 Elsevier Ltd. All rights reserved. 1. Introduction In last years an intensive research effort has been performed in the field of analytical electrochemistry seeking for designs of electrochemical biosensors capable to provide better analytical characteristics in terms of sensitivity, selectivity, reliability, ease of fabrication and use and low cost. Nowadays it is well established that the performance of biosensors depends greatly on the influ- ence imposed on biomolecules by immobilization. In this context, the use of nanomaterials for the construction of biosensing devices constitutes one of the most exciting approaches. The extremely promising prospects of these devices accrue from the unique prop- erties of nanomaterials [1]. Although different nanomaterials are employed, this article is devoted to the use of gold nanoparticles for the construction of electrochemical biosensors that allow the achievement of enhanced analytical performance with respect to other designs. The ability of gold nanoparticles to provide a stable immo- bilization of biomolecules retaining their bioactivity is a major advantage for the preparation of biosensors. Furthermore, gold nanoparticles permit direct electron transfer between redox pro- ∗ Corresponding author. Tel.: +34 91 3944315; fax: +34 91 3944329. E-mail address: [email protected] (J.M. Pingarr ´ on). teins and bulk electrode materials, thus allowing electrochemical sensing to be performed with no need for electron transfer mediators. Characteristics of gold nanoparticles such as high surface-to-volume ratio, high surface energy, ability to decrease proteins–metal particles distance, and the functioning as electron- conducting pathways between prosthetic groups and the electrode surface, have been claimed as reasons to facilitate electron transfer between redox proteins and electrode surfaces [2,3]. Gold nanopar- ticles have also demonstrate to constitute useful interfaces for the electrocatalysis of redox processes of molecules such as H 2 O 2 , O 2 or NADH involved in many significant biochemical reactions [4]. In this article, the recent advances in the construction of gold nanoparticles-based electrochemical biosensors are reviewed and some illustrative examples commented. Enzyme biosensors, immunosensors and DNA sensors are considered separately. Due to the broad range of application of gold nanoparticles, many synthetic procedures can be found in the literature in order to control the size, monodisperse, morphology and surface chemistry of gold nanoparticles. A nice recent review from Guo and Wang, [5] reports novel nanoparticle preparation methods using differ- ent surface protectors (peptides, lipids, polysaccharides, polymers, dendrimers, etc.). Moreover, Luo et al. have reported a minireview addressing recent advances in nanoparticle-based electrochemical sensors and biosensors [6]. 0013-4686/$ – see front matter © 2008 Elsevier Ltd. All rights reserved. doi:10.1016/j.electacta.2008.03.005
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Gold nanoparticle-based electrochemical biosensors

Electrochimica Acta 53 (2008) 5848–5866
Contents lists available at ScienceDirect
Electrochimica Acta
journa l homepage: www.e lsev ier .com/ locate /e lec tac ta
Gold nanoparticle-based electrochemical biosensors
Jose M. Pingarron ∗, Paloma Yanez-Sedeno, Araceli Gonzalez-CortesDepartment of Analytical Chemistry, Faculty of Chemistry, University Complutense of Madrid, 28040 Madrid, Spain
goldtheire sursorsn thisvicesnsors
e usincturel roleuno
ent oas suesignution
ve als
a r t i c l e i n f o
Article history:Received 25 January 2008Received in revised form 26 February 2008Accepted 1 March 2008Available online 18 March 2008
Keywords:Electrochemical biosensorsGold nanoparticlesEnzyme electrodesImmunosensorsDNA sensors
a b s t r a c t
The unique properties ofimmobilization retaininglized proteins and electrodof electrochemical biosendesigns. Recent advances iistics of the biosensing denon-nanostructured biosebiosensors including thosand layer-by-layer architenanoparticles play a cruciaas in the efficiency of immadvances in the developmon electrode surfaces andFinally, other biosensors dto electrodes by a reconstitand electrode surfaces ha
1. Introduction
In last years an intensive research effort has been performedin the field of analytical electrochemistry seeking for designs ofelectrochemical biosensors capable to provide better analyticalcharacteristics in terms of sensitivity, selectivity, reliability, easeof fabrication and use and low cost. Nowadays it is well establishedthat the performance of biosensors depends greatly on the influ-ence imposed on biomolecules by immobilization. In this context,the use of nanomaterials for the construction of biosensing devicesconstitutes one of the most exciting approaches. The extremelypromising prospects of these devices accrue from the unique prop-erties of nanomaterials [1]. Although different nanomaterials areemployed, this article is devoted to the use of gold nanoparticlesfor the construction of electrochemical biosensors that allow theachievement of enhanced analytical performance with respect toother designs.
The ability of gold nanoparticles to provide a stable immo-bilization of biomolecules retaining their bioactivity is a majoradvantage for the preparation of biosensors. Furthermore, goldnanoparticles permit direct electron transfer between redox pro-
∗ Corresponding author. Tel.: +34 91 3944315; fax: +34 91 3944329.E-mail address: [email protected] (J.M. Pingarron).
0013-4686/$ – see front matter © 2008 Elsevier Ltd. All rights reserved.doi:10.1016/j.electacta.2008.03.005
nanoparticles to provide a suitable microenvironment for biomoleculesbiological activity, and to facilitate electron transfer between the immobi-faces, have led to an intensive use of this nanomaterial for the constructionwith enhanced analytical performance with respect to other biosensorfield are reviewed in this article. The advantageous operational character-
designed making use of gold nanoparticles are highlighted with respect toand some illustrative examples are commented. Electrochemical enzyme
g hybrid materials with carbon nanotubes and polymers, sol–gel matrices,s are considered. Moreover, electrochemical immunosensors in which goldin the electrode transduction enhancement of the affinity reaction as well
reagents immobilization in a stable mode are reviewed. Similarly, recentf DNA biosensors using gold nanoparticles to improve DNA immobilizationitable labels to improve detection of hybridization events are considered.ed with gold nanoparticles oriented to electrically contact redox enzymesprocess and to the study of direct electron transfer between redox proteins
o been treated.© 2008 Elsevier Ltd. All rights reserved.
teins and bulk electrode materials, thus allowing electrochemicalsensing to be performed with no need for electron transfermediators. Characteristics of gold nanoparticles such as highsurface-to-volume ratio, high surface energy, ability to decrease
proteins–metal particles distance, and the functioning as electron-conducting pathways between prosthetic groups and the electrodesurface, have been claimed as reasons to facilitate electron transferbetween redox proteins and electrode surfaces [2,3]. Gold nanopar-ticles have also demonstrate to constitute useful interfaces forthe electrocatalysis of redox processes of molecules such as H2O2,O2 or NADH involved in many significant biochemical reactions[4].In this article, the recent advances in the construction ofgold nanoparticles-based electrochemical biosensors are reviewedand some illustrative examples commented. Enzyme biosensors,immunosensors and DNA sensors are considered separately. Dueto the broad range of application of gold nanoparticles, manysynthetic procedures can be found in the literature in order tocontrol the size, monodisperse, morphology and surface chemistryof gold nanoparticles. A nice recent review from Guo and Wang,[5] reports novel nanoparticle preparation methods using differ-ent surface protectors (peptides, lipids, polysaccharides, polymers,dendrimers, etc.). Moreover, Luo et al. have reported a minireviewaddressing recent advances in nanoparticle-based electrochemicalsensors and biosensors [6].

himic
J.M. Pingarron et al. / Electroc2. Gold nanoparticle-based electrochemical enzymebiosensors
Much of the research on biosensors involving gold nanopar-ticles has been devoted to enzyme electrodes (Table 1). Amongthe various strategies followed, a useful and simple way con-sists on the direct deposition of nanoparticles onto the electrodesurface. For example, tyrosinase immobilization by cross-linkingonto a GCE modified with electrodeposited gold nanoparticleswas used to prepare a biosensor which showed a high activitytowards various phenolic compounds [7]. Also, a glucose biosensorwas prepared by covalent attachment of GOx to a gold nanoparti-cle monolayer-modified Au electrode [8]. Another example is theconstruction of a xanthine oxidase biosensor for the determina-tion of hypoxanthine. This biosensor makes use of a carbon pasteelectrode modified with electrodeposited gold nanoparticles, ontowhich the enzyme was cross-linked with glutaraldehyde and BSA[9]. The XOD–nAu–CPE configuration allowed Hx detection to becarried out at 0.00 V, with the subsequent minimization of inter-ferences such as ascorbic acid. The detection limit achieved for Hx,2.2 × 10−7 M, was similar to the best reported values with biosensordesigns without gold nanoparticles working at much more extremepotentials. Electrodeposition of gold nanoparticles onto a planarAuE was also used to create a favorable surface for the attach-ment of acetylcholinesterase. The enzyme-modified electrode wasemployed to detect nM concentrations of pesticide carbofuran[10].
Modification of electrode surfaces with self-assembled mono-layers (SAMs) of thiols provides a simple way to design tailoredmaterials that can be further used as functionalized sites to immo-bilize gold nanoparticles and enzymes [11]. A comparison of theanalytical performance of different GOx biosensor designs basedon several SAM-modified electrodes showed that a configura-tion involving colloidal gold bound to cysteamine monolayersself-assembled on a gold disk electrode exhibited a high sensitiv-ity and a long biosensor lifetime in comparison with other GOxbiosensors [12]. Schemes displaying the different strategies for GOxbiosensors preparation using tailored gold nanoparticle-modifiedelectrodes are shown in Fig. 1.
Multilayer films of GOx/gold nanoparticles on the Au electrodesurface using cysteamine as a covalent attachment cross-linkerwere prepared by layer-by-layer (LBL) technique, which hasattracted much attention because of its simplicity and wide choiceof methods. The bioelectrocatalytic response was directly corre-
lated to the number of deposited bilayers, that is, to the amount ofactive enzyme immobilized on the Au electrode surface [13].Peroxidase biosensors based on SAM-modified electrodes havealso been reported. For example, Au colloids onto a cysteaminemonolayer on the gold electrode surface were used to study thedirect electron transfer of immobilized HRP. A pair of redox peaksattributed to the direct redox reaction of the enzyme was observed.The biosensor exhibited an excellent electrocatalytic responseto the reduction of H2O2 without need for an electron media-tor [14]. Recently, a disposable pseudomediatorless amperometricbiosensor was fabricated for H2O2 determination by modificationof an indium–tin oxide (ITO) electrode with (3-mercaptopropyl)trimethoxysilane. The stable nano-Au-SH monolayer was preparedthrough covalent linking of gold nanoparticles and thiol groupson the ITO surface. HRP and tetramethyl benzidine, as the elec-tron transfer mediator, were finally coentrapped by colloidal goldnanoparticles. The results showed that the AuS monolayer not onlycould steadily immobilize HRP but also efficiently retain HRP bioac-tivity [15]. Gold nanoparticles have also been self-assembled tohollow porous thiol-functionalized poly(divinylbenzene-co-acrylicacid) nanospheres. An HRP biosensor was prepared by chemisorb-
a Acta 53 (2008) 5848–5866 5849
ing gold nanoparticles onto the thiol groups of nanospheresand enzyme immobilization on the gold nanoparticles surface[16].
The incorporation of nanomaterials into composite electrodematrices constitutes another useful strategy for the preparation ofenzyme biosensors with improved analytical performance. Thesedevices exhibit the characteristics of the involved nanomateri-als and also the advantages of composite electrodes, such as lowbackground currents, a great versatility because it is possible toincorporate different substances into the bulk of the electrodematrix, and an easy surface regeneration. In this context, vari-ous research activities have been directed towards combining theadvantageous features of colloidal gold and carbon paste electrodes.A reagentless glucose biosensor based on direct electron trans-fer of GOx [17], an HRP biosensor [18], and a Tyr biosensor forphenol detection [19] have been constructed by immobilizing thecorresponding enzymes onto electrodes prepared by mixing col-loidal gold with the carbon paste components. Based on a similarmethodology, a composite electrode was also recently preparedby modifying glassy carbon microparticles with gold nanoparti-cles and xanthine oxidase for xanthine and hypoxanthine detection[20].
Enzyme biosensors using composite graphite-Teflon electrodesmodified with gold nanoparticles have been prepared by our group.The biosensors design is based on a graphite–Teflon compositematrix in which the enzyme(s) and colloidal gold nanoparti-cles are physically incorporated. Based on this methodology,a tyrosinase biosensor with improved stability and sensitiv-ity with respect to other configurations was fabricated. TheTyr–Aucoll–graphite–Teflon biosensor exhibited suitable ampero-metric responses at −0.10 V for different alkyl- and chlorophenols.The detection limits obtained were 3 nM for catechol and approx-imately 20 nM for other phenolic compounds. The presence ofcolloidal gold into the composite matrix gave rise to enhancedkinetics of both the enzyme reaction and the electrochemicalreduction of the corresponding o-quinones at the electrode, thusproviding a high sensitivity. The biosensor exhibited an excellentrenewability by simple polishing with a lifetime of at least 39 dayswithout apparent loss of enzyme activity [21].
Other useful strategies include, for example, the use of theLBL technique with which poly(amidoamine) dendrimers withcobalt hexacyanoferrate-modified gold nanoparticles were alter-nated with poly(vinylsulfonic acid) layers on ITO electrodes. Thisfilm was used as substrate for immobilization of GOx in the pres-
ence of bovine serum albumin and glutaraldehyde as cross-linker.The modified electrode was successfully applied as a biosensor forthe amperometric measurement of glucose at 0.0 V vs SCE [22].Furthermore, controlled multilayer films of sulfonate-capped goldnanoparticles/thionine were prepared on a gold electrode throughelectrostatic and covalent interactions. Such superstructures canthus provide an ideal matrix for the construction of bienzymaticsensors, where thionine molecules acted as a mediator for elec-tron transfer. Periodate-oxidized glucose oxidase and HRP werecovalently attached to the multilayer precursor film. The resultingbiosensor exhibited good electrocatalytic response toward glucoseand the response increased with the number of thionine layers [23].2.1. Enzyme biosensors based on hybrid materials with carbonnanotubes
Conjugation of gold nanoparticles with other nanomaterials andbiomolecules is an attractive research area within nanobiotechnol-ogy [24]. In this context, carbon nanotubes (CNTs) have attractedmuch interest in fundamental and applied research due to theirunique properties. Electrode modification with CNTs gives elec-
aracelig
Resaltado

5850J.M
.Pingarronet
al./Electrochimica
Acta
53(2008)
5848–5866
Table 1Gold nanoparticle-based electrochemical enzyme biosensors
Enzyme(s)/electrode Immobilization mode Detection Performance Analyte/sample Analytical characteristics Ref.
GOx/gold Covalent attachment of GOx to a nAumonolayer-modified Au E
Amperom. (E = 0.3 V vsSCE)
KappM = 4.3 mM Glucose Linear range: 2.0 × 10−5 to
5.7 × 10−3 M; slope:8.8 �A mM−1 cm−2; LOD:8.2 �M
[8]
GOx/GCE GOx and the redox mediator TTF coimmobilized bycross-linking with glutaraldehyde on gold-modifiedelectrodes with either Cyst or MPA monolayers
Amperom. (E = 0.2 V) Useful lifetime: 28 days0.05 M PBS, pH 7.4
Glucose Linear range: 0.01–10 mM;LOD: 0.7 × 10−5 M; slope:1.02 ± 0.06 mA M−1
[12]
GOx/CPE GOx adsorbed on a colloidal gold modified CPE CV Electron transfer rateconstant: (38.9 ± 5.3) s−1
PBS 0.1 M pH 5
Glucose/serum sample Linear range:0.04–0.28 mM; LOD:0.01 mM; slope:8.4 �A mM−1
[17]
GOx/CNT–Teflon compositeelectrode
GOx and colloidal gold incorporated into compositeelectrode
Amperom. (E = +0.5 V) KappM = 14.9 mM, lifetime: 3
monthsGlucose/sport beverages Linear range: 0.05–1 mM;
LOD: 17 �M; slope:2.6 mA M−1
[25]
GOx/GCE Nanocomposite of nAu and conducting polyanilinenanofibers used to immobilize GOx
Amperom. (E = +0.6 V vsSCE)
Repeatability: R.S.D. = 4.8%(n = 7)
Glucose/blood serumsamples
Linear range: 1.0 × 10−6 to8.0 × 10−4 M; LOD:5.0 × 10−7 M
[30]
GOx/gold Electrochemical deposition of chitosan–GOx–goldnanoparticles biocomposite
Amperom. (E = 0.7 V) 0.1 M phosphate buffer, pH7.4
Glucose/serum sample Linear range:0.005–2.4 mM; LOD:2.7 �M
[32]
GOx/platinum Layer-by-layer assembled chitosan/nAu/GOxmultilayer films
Amperom. (E = 600 mV) KappM = 10.5 mM, 0.1 M PBS,
pH 6.8Glucose/human serum Linear range: 0.5–32 mM [34]
GOx/gold Layer-by-layer covalent attachment of IO4−-oxidized
GOx and nAu using cysteamine as cross-linkerAmperom. (E = 0.25 V) 10 mL 0.1 M PBS, pH 6.8
containing 0.25 mMferrocenemethane
Glucose Linear range:1.0 × 10−5–1.3 × 10−2 M;slope: 5.72 �A mM−1 cm−2
[13]
GOx/ITO PAMAM dendrimers with cobalthexacyano-ferrate-modified nAu alternated with PVSlayers. Cobalt hexacyanoferrate as redox mediator GOximmob. with Glu as cross-linker
Amperom. (E = 0.0 V vs SCE) KappM = 2.03 mM Glucose Linear range: up to 1.5 mM;
LOD: 17 �M; slope:33.6 ± 0.2 nA mM−1 cm−2
[22]
GOx/gold Immob. GOx on nAu, which were self-assembled on Auelectrode modified with thiol-containingthree-dimensional network of silica gel
Amperom. (E = 0.3 V vs SCE) R.S.D. = 1.9% (n = 8) Glucose Slope: 8.3 �A mM−1 cm−2;LOD: 23 �M
[23]
GOx/gold Multilayer nAu/MWNT/GOx membrane prepared byelectrostatic assembly using positively chargedpoly(dimethyldiallylammonium) Cl− to connect themlayer-by-layer
Amperom. (E = −0.2 V) KappM = 10.6 mM Glucose Linear range: up to 9.9 mM;
LOD: 128 �M[95]
GOx/gold Multilayer film of (PDDA/GNPs)n/PDDA constructed onthe gold electrode by electrostatic layer-by-layerself-assembly of PDDA and nAu. GOx was then sorbedonto this multilayer film
Amperom. (E = 0.3 V) 0.1 M PBS, pH 7.0 Glucose Au/(PDDA/GNPs)3/PDDA/GOx,slope: 5.2 �A mM−1 cm−2;Au/(PDDA/GNPs)6/PDDA/GOx,slope: 13.6 �A mM−1 cm−2;Au/(PDDA/GNPs)9/PDDA/GOx,slope: 22.6 �A mM−1 cm−2
[96]
GOx/microporous goldelectrode
Co-immob. GOx and nAu on thedl-thiorphan/1,8-octanedithiol mixed layer modifiedelectrode
Amperom. (E = +0.45 mV) 0.05 M PBS, pH 7 Glucose Linear range: up to22.0 mM
[97]
GOx/GCE Incorporating GOx within the electrodepositedchitosan–gold nanoparticle hybrid film on a PrussianBlue modified electrode
Amperom. (E = −0.05 V) Repeatability: R.S.D. = 8.3% Glucose/human serum Linear range:1.0 × 10−6–1.6 × 10−3 M;LOD: 6.9 × 10−7 M; slope:69.26 �A mM−1 cm−2
[98]
GOx/HRP/gold Covalently attachment of periodate-oxidized glucoseoxidase and HRP on multilayer films ofsulfonate-capped gold nanoparticles/thionine
Amperom. (E = −18 mV) KappM = 1.21 mM Glucose and H2O2 Linear range:
6.0 × 10−6–1.1 × 10−3 M;LOD: 3.5 × 10−5 M; slope:3.8 �A mM−1 cm−2
[99]
HRP/gold Au colloids associated with a cysteamine monolayer ongold electrode surface. HRP immob. on colloidal gold
Amperom. (E = −0.3 V) KappM = 2.3 mM, R.S.D. = 3%
(n = 10), 0.1 M PBS pH 7H2O2 Linear range:
0.0014–2.8 mM; LOD:0.58 �M
[14]

J.M.Pingarron
etal./Electrochim
icaA
cta53
(2008)5848–5866
5851Table 1 (Continued )
Enzyme(s)/electrode Immobilization mode Detection Performance Analyte/sample Analytical characteristics Ref.
HRP/ITO Covalent linking nAu-SH monolayer on the surface ofthe ITO; HRP and TMB (mediator) coentrapped by thecolloidal gold
Amperom. (E = −700 mV) KappM = 2.2 mM,
repeteability: R.S.D. = 2.7%(n = 8)
H2O2 Linear range:0.005–1.5 mM; LOD: 1 �M
[15]
HRP/gold Gold electrode immersed in hollow porousthiol-functionalized poly(DVB-co-AA) nanospherelatex to assemble the nanospheres; nAu chemisorbedonto thiol groups of the nanospheres
Amperom. (E = −150 mV) Repeatability: R.S.D. = 1.8%(n = 9), 0.1 M PBS, pH 7.0
H2O2 Linear range: 1.0 �M to8.0 mM; LOD: 0.5 �M
[16]
HRP/CPE HRP–CP–colloidal gold Amperom. (E = −0.4 V vsSCE)
Electron transfer rateconstant: 6.04 ± 0.18 s−1;Kapp
M = 3.7 ± 0.7 mM,lifetime: 40 days (storageperiod); 0.1 M PBS, pH 7.4
H2O2 Linear range: 0.48–50 �M;LOD: 0.21 �M
[18]
HRP/GCE Covalent incorp. of CNTs and nAu onto poly(thionine)film deposited by electropolymerization on GCE. HRPimmob. by incubation in 15 mg mL−1 HRP solutionovernight at 4 ◦C
Amperom. (E = 0.4 V vs SCE) 0.1 M PBS, pH 7.0 H2O2 Linear range:5.0 × 10−6–7.0 × 10−3 M;LOD: 3.0 × 10−6 M
[28]
HRP/GCE Electropolymer p-ABSA on the GCE by CV. Thionineadsorbed to the film and nAu layer assembled for HRPimmob.
Amperom. (E = −0.45 V) pH 7 acetate buffersolutions
H2O2 Linear range:2.6 × 10−6–8.8 × 10−3 M;LOD: 6.4 × 10−7 M
[31]
HRP/gold Chitosan film electrochem. deposited on Au electrode,self-assembling gold nanoparticles on chitosan, andincubating in HRP solution
Amperom. (E = −250 mV) Mediator: methylene blue;lifetime: four weeks
H2O2 Linear range:0.008–15 mM; LOD: 2.4 �M
[33]
HRP/ITO HRP entrapped in colloidal gold modified chitosanmembrane (Au–chitosan)
Amperom. (E = −510 mV) Repeatability: R.S.D. = 1%(n = 34), lifetime: 7 daysstorage
H2O2 Linear range: 0.01–0.5 mM;LOD: 5 �M
[35]
HRP/screen-printed carbonelectrode (SPCE)
HRP bound with nAu in the matrix of chitosan on SPCE Amperom. (E = −400 mV) KappM = 1.3 mM,
R.S.D. = 4.9% (n = 30), 0.1 Mcitrate buffer, pH 6.5
H2O2 Linear range:0.01–11.3 mM; LOD:0.65 �M
[36]
HRP/gold Self-assembling nAu to a thiol-containing sol–gelnetwork. HRP adsorbed onto the surface of the nAu
Amperom. (E = −0.25 V) R.S.D.: 2.1% (n = 9), lifetime:120 days
H2O2 Linear range: 5.0 �M to10.0 mM; LOD: 2.0 �M
[38]
HRP/platinum Polythionine (mediator) embedded in Nafion film onthe surface of platinum electrode. HRP bound by goldnanoparticles and gelatin coated on theNafion-modified electrode
Chronoamp R.S.D.: 2.2% (n = 5) H2O2 Linear range:0.05–30.6 mM; LOD:0.02 mM
[100]
HRP/gold Colloidal gold supported by thiol-tailed groups ofcysteamine monolayer. HRP immobilized onnanometer-sized colloidal gold
Amperom. (E = −0.1 V vsSCE)
KappM decreases with
diameter of colloidal gold;0.1 M PBS, pH 6.9
H2O2 Linear range: 0.39 �M to0.33 mM; LOD: 0.15 �M
[101]
HRP/gold Precursor film formed on the Au electrode surface byself-assembly of l-cysteine and the adsorption of nAu.Toluidine blue and nAu were assembled onto filmthrough layer-by-layer assembly technique
H2O2 Linear range:1.5 × 10−7–8.6 × 10−3 M;LOD: 7.0 × 10−8 M
[102]
HRP/ITO Modified with APTMS. colloidal gold particleschemisorbed onto the amine groups of the APTMS.HRP adsorbed onto the surface of the colloidal gold
Amperom. (E = −0.25 V) Repeatability: R.S.D. = 2.4%(n = 8), 0.05 M PBS, pH 7
H2O2 Linear range: 0.020–8 mM;LOD: 8.0 �M
[103]
HRP/CPE l-Cysteine–gold particle nanocomposite immobilizedin a Nafion membrane on a GC electrode. HRP cov.cross-linked on the GC/NCGN surface
CV Repeatability.: R.S.D. = 4.7%(n = 5), 0.1 M PBS, pH 7
H2O2 Linear range:1.60 × 10−5–1.10 × 10−3 M;LOD: 5.50 × 10−6 M
[104]
XOD/CPE XOD immob. by cross-linking with glutaraldehyde andBSA on CPE modified with electrodeposited nAu
Amperom. (E = +600;0.00 mV vs Ag/AgCl)
KappM = 10 × 10−6 M Hypoxanthine/sardines
chickenLinear range: 0.5–10.0 �M;LOD: 2.2 × 10−7 M; slope:(2.29 ± 0.03) × 104 �A M−1
[9]
XOD/GCPE–nAu compositeelectrode
XOD-based GCPE prepared by hand mixing of 80:20 (%,w/w)
Chronoamp. (E = +700 mV) R.S.D. = 6.31%, 5 �M X(n = 5), R.S.D. = 3.57% 80 �MHx (n = 5)
Xanthine (X) hypoxanthine(Hx)/canned tuna fish
Linear range:5.0 × 10−7–1.0 × 10−5 M(X);5.0 × 10−6–1.5 × 10−4 M(Hx); slope = 0.24 �A �M−1
(X); 0.14 �A �M−1 (Hx)
[20]

5852J.M
.Pingarronet
al./Electrochimica
Acta
53(2008)
5848–5866
Table 1 (Continued )
Enzyme(s)/electrode Immobilization mode Detection Performance Analyte/sample Analytical characteristics Ref.
XOD/gold Electron mediator, Prussian Blue deposited on theelectrode; XOD entrapped onto pPy film byelectrooxidation of XOD and pyrrole; colloidal goldadsorbed onto enzyme electrode
Amperom. (E = −0.15 V) KappM = 43.2 �M Xanthine Linear range: 1–20 �M [105]
ADH/gold Colloidal gold self-assembled through the thiol groupsof an 1,6-hexanedithiol monolayer and immobilizationof ADH on colloidal gold
EIS KappM = 6.03 × 10−4 M, 0.1 M
PBS, pH 7.4 + 0.1 M NaClEthanol Linear range:
1.0 × 10−5–1.3 × 10−2 M;LOD: 8.0 × 10−6 M
[106]
LDH/gold ADH/gold nAu self-assembled on a thiol-terminated, MPTSsol–gel-derived, 3-D, silicate network and enlarged byhydroxylamine seeding. Adsorption of MPTSsol-enzyme biocomposite on AuE gold
Amperom. (E = −5 mV) KappM (lactate) = 0.91 mM,
KappM (ethanol) = 2 mM 0.1 M
PBS, pH 7.2
Lactate, alcohol Slope: 446.8 nA mM−1
(lactate); 47 nA mM−1
(ethanol); LOD: 0.1 �M(lactate); 20 �M (ethanol)
[39]
AChE/gold Electrodeposition of colloidal gold nanoparticles ontoAuE; AChE adsorption
Amperom. (E = 680 mV) KappM = 0.18 ± 0.02 mM, 0.1 M
PBS, pH 8Carbofuran Linear range: 10–135 nM;
LOD: 33 nM[10]
AChE/GCE Silica sol–gel assembling nAu composite coated on aGCE. Then coated with AChE solutions
CV R.S.D. = 1.7–2.3%, lifetime:30 days
monocrotophos, methylparathion, carbaryl
[37]
Tyr/CPE Tyr immob. on a colloidal gold modified CPE Amperom. (E = −150 mV) KappM
= 54 ± 3 �M, 0.1 MPBS, pH 7
Phenol Linear range: 4–48 �M;LOD: 6.1 nM;slope = 12.3 �A cm−2 �M−1
[19]
Tyr/GCE Immob. by cross-linking with glutaraldehyde of theenzyme onto a GCE modified with electrodepositednAu
Amperom. (E = −0.1 V) 0.1 M PBS, pH 7.4, usefullifetime: >18 days
Phenoliccompounds/wines
Linear range:(0.005–0.5) × 10−4 M(catechol); LOD: 0.15 �M;slope: 0.107 A M−1
[7]
Tyr/graphite–Tefloncomposite electrode
Tyrosinase and colloidal gold nanoparticlesincorporated by simple physical inclusion
Amperom. (E = −0.10 V) KappM : 8.9 �M (catechol);
6.6 �M (phenol), lifetime:39 days
Phenols/spring and wastewaters
Linear range: 0.010–8.0 �M(catechol); 0.025–4.0 �M(phenol);slope = 746 mA M−1
(catechol); LOD: 3 nM(catechol)
[21]
GOx/CGE Tyr/GCE Immobilization of GOx or tyrosinase in chemicallysynthesized gold polypyrrole nanocomposite
Amperom. (E = 0.70 V),GOx; (E = −0.15 V), Tyr
KappM (GOx) = 43 �M, Kapp
M(Tyr) = 48 �A
Phenol (Ph), glucose (Glu) Linear range: 1.0–20 �M(Ph); 0.05–0.6 mM (Glu);slope: 497.1 mA M−1 (Ph);1.089 mA M−1 (Glu); LOD:3 × 10−8 M (Ph); 2 × 10−6 M(Glu)
[29]
Abbreviations: AChE: acetylcholinesterase, ADH: alcohol dehydrogenase, Amperom.: amperometry; APTMS: (3-aminopropyl)trimethoxysilane, ASV: anodic stripping voltammetry, AuE: gold electrode; chronoamp.: chronoam-perometry, CPE: carbon paste electrode, CNT: carbon nanotubes, cov.: covalently, CPE: carbon paste electrode, CV: cyclic voltammetry, DPV: differential pulse voltammetry, EIS: electrochemical impedance spectroscopy,Electrochem.: electrochemically, GC: glassy carbon, GCE: glassy carbon electrode, GCPE: glassy carbon paste electrode, GOx: glucose oxidase, Glu: glutaraldehyde, HRP: peroxidase, immob.: immobilization, incorp.: incorpo-ration; ITO: indium tin oxide, Kapp
M : apparent Michaelis–Menten constant, LOD: limit of detection, MP-11: microperoxidase 11, MPTS: (3-mercaptopropyl)trimethoxysilane, MWNT: multiwalled carbon nanotube, nAu: goldnanoparticles, p-ABSA: p-aminobenzene sulfonic acid, PBS, phosphate buffer solution; PDDA: poly(diallyldimethylammonium chloride), PVB: polivinil-butiral, PVS: poly(vinylsulfonic acid), SCE: saturated calomel electrode,TMB: tetramethyl benzidine, TTF: tetrathiafulvalene; Tyr: tyrosinase, XOD: xanthine oxidase.

J.M. Pingarron et al. / Electrochimica Acta 53 (2008) 5848–5866 5853
fferenOx/cy
Fig. 1. Schemes of different GOx biosensors constructed by means of digold–cysteamine–AuE; (B) GOx/colloidal gold–cysteamine/cysteamine–AuE; (C) Gacid (MPA)–electrodeposited gold nanoparticles–GCE (adapted from Ref. [12]).
trocatalytic ability towards the electrooxidation of NADH or H2O2.Hybrid nanoparticles/nanotubes materials have shown to possessinteresting properties, which can be profited for the development ofelectrochemical biosensors. A colloidal gold–CNT composite elec-trode using Teflon as the non-conducting binding material showedsignificantly improved responses to H2O2 when compared withother carbon composite electrodes, including those based on CNTs.The incorporation of GOx into the new composite matrix allowed
the preparation of a mediatorless glucose biosensor with a remark-ably higher sensitivity than that from other GOx–CNT bioelectrodes[25].Hybrid composites can be also prepared by selective attachmentof gold nanoparticles to carbon nanotubes surfaces. This requiresprevious CNTs functionalization to immobilize gold nanoparticles.So, cationic polyethyleneamine or anionic citrate used as disper-sants can change the surface properties of CNTs yielding acidic orbasic surfaces [26]. Then, CNTs could be successfully coated withgold nanoparticles by electrostatic interaction. A nanohybrid filmconsisted of MWCNTs with attached gold nanoparticles was usedto immobilize microperoxidase (MP-11) by covalent binding [27].The MP-11 modified film was used to coat a GCE at which theenzyme exhibited direct electron transfer retaining its bioelectro-catalytic activity for H2O2 reduction. In other configuration, a directelectrochemical biosensing platform was fabricated by covalentincorporation of CNTs and gold nanoparticles onto a GCE modi-fied with electropolymerized poly(thionine). The synergic effectsof the composite nanomaterials together with the excellent medi-ating ability of the redox polymer, allowed faster electron transferand higher enzyme immobilization efficiency than the designed
t tailored gold nanoparticle-modified electrode surfaces: (A) GOx/colloidalsteamine–electrodeposited gold nanoparticles–GCE or GOx/3-mercaptopropionic
based on CNTs or gold nanoparticles, as it was demonstrated usingHRP as a test enzyme model [28].
2.2. Enzyme biosensors based on hybrid materials with polymers
Electropolymerization is widely accepted as an appropriatemethodology for the preparation of suitable immobilization matri-ces for biomolecules. This is because polymer characteristics such
as film thickness, permeation and charge transport properties canbe easily controlled by adjusting the electrochemical parame-ters of polymer grown on the electrode surface. Immobilizationof enzymes into/onto various conductive polymers has receivedgreat attention because of their unique characteristics. The mainlimitation is the high amount of biomolecule and monomer nec-essary for the immobilization. The presence of gold nanoparticlesin the conductive polymer matrix provides enhanced electrochemi-cal activity/conductivity and avoids enzyme leaking while allowingrapid diffusion of substrate and product using small amount ofenzyme. Furthermore, when gold nanoparticles are incorporatedto the electrode, a chemical polymerization method instead ofthe electrodeposition process, that can affect the enzyme, can beapplied. So, gold polypyrrole (Au–PPy) nanocomposites were syn-thetized and deposited onto a GCE surface by polymerization ofPy in the presence of AuCl4− onto the electrode, giving PPy andAu0 followed by the enzyme immobilization [29]. Polyaniline hasbeen successfully used to design nanocomposite bioelectrodes. Aglucose biosensor based on a mixture of gold nanoparticles andconductive polyaniline with GOx and Nafion on the surface ofnanocomposite was developed [30]. A H2O2 biosensor was fabri-
himic
5854 J.M. Pingarron et al. / Electroccated by electropolymerization of p-aminobenzene sulfonic acidusing cyclic voltammetry. Then, thionine was adsorbed to the filmto form a composite membrane which yielded an interface contain-ing amine groups to assemble gold nanoparticles. HRP was finallyadsorbed and the biosensor responded to H2O2 with a detectionlimit of 6.4 × 10−7 M [31].
Chitosan is a natural polymer with one amino group and twohydroxyl groups in the repeating hexosamide residue. Its excel-lent film forming and adhesion ability, together with non-toxicity,biocompatibility, cheapness and susceptibility to chemical mod-ification have led to use it for immobilization of biomolecules,especially enzymes. Adsorption of colloidal gold nanoparticles ona chitosan membrane provides an assembly of gold nanoparti-cle multilayers and a suitable microenvironment similar to thenative environment of biomolecules. Using this approach, amper-ometric GOx [32], and HRP [33] biosensors were prepared byself-assembling gold nanoparticles on chitosan hydrogel modifiedAu electrodes. Chitosan membranes modified with colloidal goldand enzymes have also been used with other electrode materials.So, an amperometric glucose biosensor based on LBL assembly ofmultilayer films composed of chitosan, gold nanoparticles and GOxwas prepared on Pt electrodes. The results showed an excellentcatalytic ability to glucose, and demonstrated that gold nanoparti-cles efficiently improved the electron transfer between analyte andelectrode surface [34]. Moreover, an HRP biosensor was preparedby entrapping the enzyme in the gold nanoparticle-modified chi-tosan membrane onto an ITO electrode [35]. This design profits thespecial characteristics of such electrodes to fabricate a disposablebiosensor combined with flow injection analysis for the rapid deter-mination of H2O2. An equivalent design, based on a screen-printedcarbon electrode was also developed with the inherent advantagesof screen-printing technology [36].
2.3. Enzyme biosensors based on sol–gel matrices
Sol–gel technology provides unique means to preparethree-dimensional networks suited for the encapsulation ofbiomolecules. Sol–gel hybrid materials prepared by physicallyencapsulating gold nanoparticles into porous sol–gel networkshave been used for the fabrication of biosensors. An example isthe construction of an acetylcholinesterase biosensor in which thesol–gel-derived silicate network assembling gold nanoparticlesprovided a biocompatible microenvironment that stabilized theenzyme bioactivity and prevented its leaking out from the inter-
face. Typical pesticides such as monocrotophos, methyl parathionand carbaryl were determined with high sensitivity, accuracy, lowcost and simplified procedures [37].The opportunity to combine sol–gel and self-assembly tech-nologies to prepare biosensors has emerged using thiolated siliconalkoxides. Gold nanoparticles can be assembled both inside thenetwork and on the sol–gel surface. This technology exhibits thebenefits of coupling self-assembly, nanoparticles, and the increasedsurface area of three-dimensional electrodes. Gold nanoparticlesimmobilized by silica gel three-dimensional network can act astiny conducting centers and facilitate the electron transfer [38]. Ahighly sensitive nanostructured electrochemical biosensor basedon the integrated assembly of dehydrogenase enzymes and goldnanoparticles was prepared using this approach. The Au nanopar-ticles were self-assembled on a thiol-terminated, sol–gel-derived,3-D, silicate network and enlarged by hydroxylamine seeding. Goldnanoparticles efficiently catalyzed the oxidation of NADH with anoverpotential decrease of 915 mV in the absence of redox media-tor. The nanostructured electrode showed high sensitivity towardNADH with an amperometric detection limit of 5 nM, and it was suc-cessfully applied for the determination of lactate and ethanol [39].
a Acta 53 (2008) 5848–5866
3. Gold nanoparticle-based electrochemicalimmunosensors
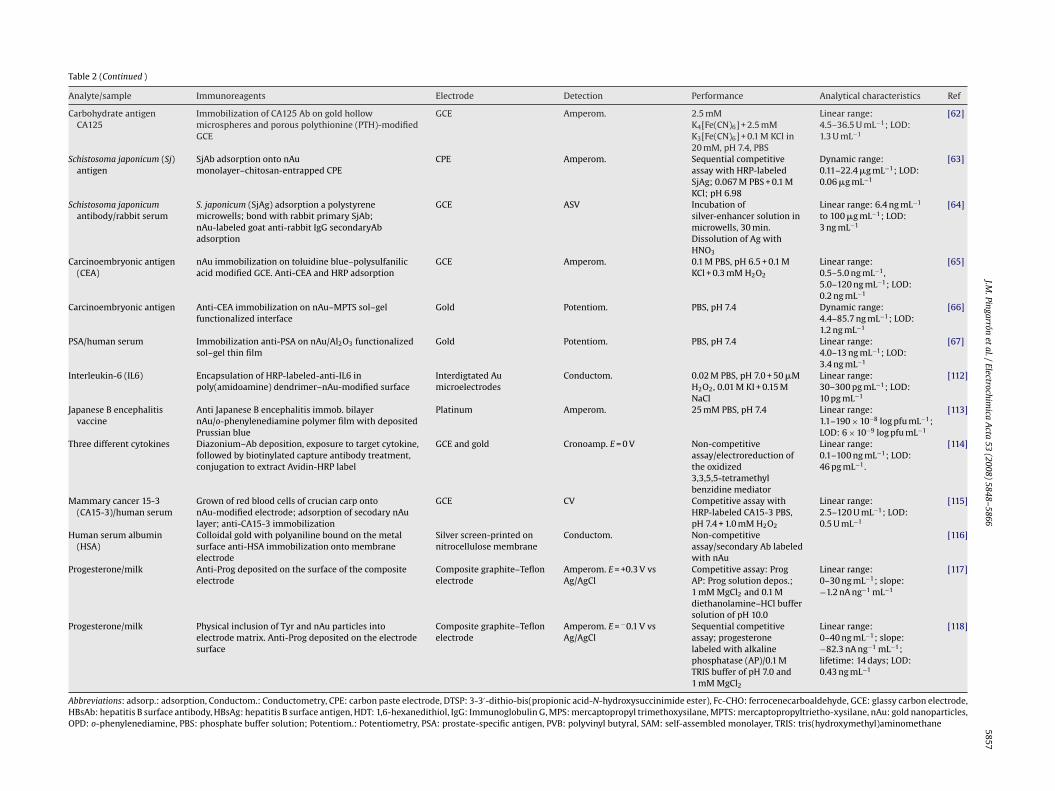
Table 2 summarizes recent approaches in the construction ofelectrochemical immunosensors in which gold nanoparticles playa crucial role both in the enhancement of the electrochemicalsignal transducing the binding reaction of antigens at antibody-immobilized surfaces, and in the ability of increasing the amountof immobilized immunoreagents in a stable mode.
Hepatitis B virus surface antigen was detected using electro-chemical impedance spectroscopy (EIS) through immobilization ofthe antibody onto gold nanoparticles-modified 4-aminothiophenolself-assembled monolayers [40] or on the surface of a PtE modi-fied with colloidal gold and polyvinyl butyral [41]. Potentiometricand amperometric immunosensors for HBsAg detection were alsoconstructed by electrostatic adsorption of the antibody onto goldnanoparticles/tris(2,2-bipyridyl)cobalt (III) multilayer films [42]or by self-assembling gold nanoparticles to a thiol-containingsol–gel network and assembling hepatitis B surface antibody ontothe surface of the gold nanoparticles [43]. A gold nanoparticle-based biomolecular immobilization method based on Nafion andgelatin was also described [44]. The detection of HBsAg was per-formed by measuring changes in the electric potential beforeand after the antigen–antibody reaction. Furthermore, a differentstrategy was used to fabricate an amperometric immunosen-sor based on immobilization of hepatitis B antibody on a goldelectrode modified with gold nanoparticles and horseradish per-oxidase [45]. In this immunosensor, HRP was incorporated insteadof BSA to block the possible remaining active sites of the goldnanoparticles monolayer, in order to avoid non-specific adsorptionand also amplify the response of the antigen–antibody reaction.Fig. 2 shows the steps involved in the immunosensor prepara-tion.
The self-assembling technique and the opposite-chargedadsorption methodology were combined for the immobilizationof diphtheria antibody, and applied to prepare an immunosensorfor detecting diphtheria antigen. Anti-Diph was immobilized onnanometer-sized colloidal gold particles associated with polyvinylbutyral on a PtE [46]. Furthermore, a potentiometric immunosen-sor for direct and rapid detection of diphtherotoxin was developedby self-assembling of monoclonal diphtheria antibody onto a PtEbased on the use of a gold nanoparticles–silica nanoparticles mix-ture and polyvinyl butyral as matrices. Anti-Diph was adsorbedonto the surface of the nanoparticles mixture, and then they
were entrapped into polyvinyl butyral sol–gel network on a PtE.The immobilized DiphAb exhibited direct potentiometric responsetoward DiphAg. The immunosensor using the nanoparticles mix-ture exhibited much higher sensitivity, better reproducibility, andlong-term stability than those constructed with gold nanoparticlesor silica nanoparticles alone [47].�-Fetoprotein (AFP), an oncofetal glycoprotein, is a widelyused tumor marker for the diagnosis of patients with germ celltumors and hepatocellular carcinoma. An immunosensor for AFPwas prepared by entrapping thionine into Nafion to form a com-posite Thi/Nf membrane, which yields an interface containingamine groups to assemble gold nanoparticles layer for immobi-lization of �-1-fetoprotein antibody. After the immunosensor wasincubated with AFP, the CVs current decreased linearly in con-centration ranges of AFP [48]. More recently, an amperometricenzyme immunosensor with amplified sensitivity, based on layer-by-layer assembly of gold nanoparticles and thionine immobilizedon a Nafion-modified electrode surface by electrostatic adsorption,has been reported for the detection of AFP. As in the configura-tion cited above, HRP was employed not only to block the possibleremaining active sites of the nano-Au monolayer but also to amplify

J.M.Pingarron
etal./Electrochim
icaA
cta53
(2008)5848–5866
5855Table 2Gold nanoparticle-based electrochemical immunosensors
Analyte/sample Immunoreagents Electrode Detection Performance Analytical characteristics Ref
HBsAg HBsAb immobilized ontonAu–4-aminothiophenol-modified electrode
Gold EIS 2.5 mM Fe(CN)64− + 2.5 mM
Fe(CN)63− in 0.1 M
KCl + 20 mM PBS, pH 7.4
Linear range:0.5–200 �g L−1; LOD:50 ng L−1
[40]
HBsAg/human serum HBsAb immobilized on colloidalgold/polyvinylbutyral-modified electrode
Platinum EIS [Fe(CN)63−]/[Fe(CN)6
4−]1:1 as a redox probe inPBS + 0.1 M KCl, pH 7.4
Linear range:20–160 ng mL−1; LOD:7.8 ng mL−1; lifetime: 1month
[41]
HBsAg/human serum Co(bpy)33+ and nAu assembled onto Nafion-modified
Pt electrode by layer-by-layer technique. HBsAbelectrostatically adsorbed onto nAu
Platinum Potentiom., Amperom.(E = −0.35 V)
PBS, pH 7.0, lifetime: 1month
Linear range:0.05–4.5 �g mL−1; LOD:0.005 �g mL−1 (amp.);0.015 �g mL−1 (pot.)
[42]
HBsAg/human serum Electrode immersion in MPS sol–gel solution; nAuadsorption onto thiol groups of sol–gel network;HBsAb assembled onto nAu
Gold Potentiom. Amperom. PBS, pH 7.4, lifetime: 1month
Linear range:4–960 ng mL−1; LOD:1.9 ng mL−1
[43]
HBsAg/human serum HBsAb immobilized on disk electrode with nAu,Nafion, and gelatine
Platinum Potentiom. 0.1 M PBS, pH 7.4 Linear range:4–800 ng mL−1; LOD:1.3 ng mL−1
[44]
HBsAg/clinical samples HBsAb immobilization ontonAu/thionine/Nafion-HRP-modified electrode
Gold Amperom. 0.1 M HAc–NaAc buffersolution + 0.27 mM H2O2;pH 6.5
Linear range:2.6–563 ng mL−1; LOD:0.85 ng mL−1
[45]
Diphtheria antigen Anti-Diph immob. onto colloidal gold/polyvinylbutyral-modified electrode
Platinum Potentiom. pH 7.0, PBS + KCl 0.1 M Linear range:24–1280 ng mL−1; LOD:7.8 ng mL−1; R.S.D. = 2.3%(n = 13)
[46]
Diphthero toxin DiphAb adsorp. onto nAu–SiO2 mixture; entrappedinto PVB sol–gel network
Platinum Potentiom. PBS, pH 7.0 Linear range:5.0 × 10−3–1.2 �g mL−1;LOD: 1.1 × 10−3 �g mL−1
[47]
Diphtheria antigen/serum Anti-Diph immobilized onto nAu/polyvinylbutyral-modified electrode
Platinum Potenciom. pH 7.0, PBS + KCl 0.1 M,lifetime: 4 months (n = 40)
Linear range:4.4–960 ng mL−1; LOD:2.4 ng mL−1
[107]
�-1-Fetoprotein(AFP)/human serum
Anti-AFP immobilization ontonanoparticles/thionine/Nafion-membrane-modifiedelectrode
Gold CV, Amperom. 0.1 M HAc–NaAc buffersolution + 0.1 M KCl, pH 5.5
Linear range:5.0–200 ng mL−1; LOD:2.4 ng mL−1; repeatability:R.S.D. = 3.8% (n = 50)
[48]
�-1-Fetoprotein(AFP)/human serum
HRP immobilization ontoanti-AFP/nAu/thionine/Nafion-modified electrode
Gold Amperom. 0.1 M HAc–NaAc buffersolution + 0.1 M KCl of pH5.5; lifetime: 3 months
Linear range:1–250 ng mL−1; LOD:0.56 ng mL−1
[49]
�-Fetoprotein/serum Anti-AFP immobilization onto1,6-hexanedithiol/cysteamine
Miniaturized gold Amperom. E = −0.4 V Non-competitive assaywith HRP-labeledsecondary antibody. PBS,pH 7.4; lifetime: 90 days
Linear range:15–350 ng mL−1; LOD:5 ng mL−1
[50]
Human IgG/human serum IgG immobilization into nAu/MPTS sol–gel electrode Gold EIS 10 mM PBS + 0.1 M KCl, pH7.4 + 5 mM Fe(CN)63−/Fe(CN)6
4−
Linear range:8.3–2128 ng mL−1; LOD:3.3 ng mL−1
[51]
hIgG Anti-IgG immobilization on microtiter plates wells. IgGincubation for 1 h at 37 ◦C
CPE ASV Non-competitive assay.Biotin-modified magneticparticles–avidin nAu reactwith dethiobiotinylated Ab.Dissolution of nAu withHBr/Br2
Dynamic range:0.2–5 ng mL−1; LOD:0.1 ng mL−1
[52]
hIgG/human serum Ab adsorption on CPE; bond antigen and colloidalgold-labeled Ab
CPE ASV Non-competitiveassay/0.05 M PBS, pH 7.4
Linear range:10–500 ng mL−1; LOD:4.0 ng mL−1
[53]

5856J.M
.Pingarronet
al./Electrochimica
Acta
53(2008)
5848–5866Table 2 (Continued )
Analyte/sample Immunoreagents Electrode Detection Performance Analytical characteristics Ref
IgG Antibody immobilization on colloidalgold/HDT-modified electrode
Gold EIS Non-competitive assay;amplification with colloidalgold-labeled goat anti-hIgGand colloidal gold-labeledrabbit anti-goat IgG Ab.20 mM PBS + 0.1 M KCl,5 mM Fe(CN)6
3−/Fe(CN)64− ,
pH 7.4
Linear range:15.3–328.3 ng L−1; LOD:4.1 ng L−1
[54]
IgG/human serum Incubation in polystyrene microwells: anti-IgG,overnight; IgG and nAu-labeled anti-IgG, 1 h
GCE ASV Incubation ofsilver-enhancer solution inmicrowells, 30 min.Dissolution of Ag withHNO3
Linear range: 2 ng mL−1 to27 �g mL−1; LOD:1 ng mL−1
[55]
IgG/serum Immobilization of anti-IgG on colloidalgold-mercaptoethylamine-modified gold electrode
Gold Potentiom. PBS, pH 7 Linear range:12–800 ng mL−1; LOD:3.4 ng mL−1; repeatability:R.S.D. = 5.9%
[108]
IgG/human serum Anti-IgG immob. in poly(amido-amine)film and coreshell SiO2/nAu. Anti-IgG labeled with Fc-CHO, as amediator, adsorbed onto nAu. GOx back-filled intoanti-IgG-modified surface
Platinum CV 10 mM phosphate buffersolution, pH 7.0
Linear range:5.0–9600 �g mL−1; LOD:0.8 ng mL−1
[109]
IgG Anti-IgG adsorption on a porous gold film producedelectrochemically (E = −0.5 V vs Ag/AgCl for 50 s) onGCE
GCE CV; EIS Non-competitive assaywith HRP-labeledsecondary Ab. PBS + 0.1 MKCl + 10 mMFe(CN)6
3−/Fe(CN)64− , pH
7.4
Linear range:0.011–11 ng mL−1; LOD:0.009 ng mL−1
[110]
IgG Physical inclusion of Tyr and nAu on the electrodemixture. Incubation in protein A and anti-IgGimmobilization
Composite graphite–Teflonelectrode
EIS; Amperom. E = −0.1 V vsAg/AgCl
Non-competitive assaywith alkaline phosphatase(AP)-labeled anti-IgG/0.1 MTRIS buffer + 1 mM MgCl2,pH 7.0
Linear range:5–100 ng mL−1; LOD:2.6 ng mL−1
[111]
Aflatoxin B1 (AFB1) HRP and anti-AFB1 immob. ontonAu/2-amino-ethanethiol microelectrode
Interdigitated goldmicroelectrodes
Conductim. 0.02 M PBS, pH 7.0 + 0.05 MKI, 80 �M H2O2 + 0.15 MNaCl
Linear range:0.5–10 ng mL−1; LOD:0.1 ng mL−1
[56]
Carcinoma antigen 125(CA125)/human serum
Immobilization of anti-CA125 on nAu and thionine(Thi)-modified CPE
CPE Amperom. pH 7.0, HAc–NaAc buffer Dynamic range:10–30 U mL−1; LOD:1.8 mL−1
[57]
Carcinomaantigen-125/humanserum
CA125 antigen immobilization on gold nanoparticlesstabilized with cellulose acetate membrane
GCE DPV Competitive immunoassaywith HRP labeled CA125Ab, OPD and H2O2 asenzyme substrates/0.1 MPBS pH 5.5
Linear range: 0–30 U mL−1;LOD: 1.73 U mL−1
[58]
Human chorionicgonatrophin/serum
Encapsulation of HRP-labeled hCG antibody in colloidalgold nanoparticle/titania sol–gel composite membrane
GCE EIS 0.1 M PBS, pH 7.1 Linear range:0.5–5.0 mIU mL−1,5.0–30 mIU mL−1; LOD:0.3 mIU mL−1
[59]
Dust mite allergen Allergen Der f2 immobilization onto electrodepositedgold-modified electrode
GCE EIS 2.5 mM Fe(CN)64− + 2.5 mM
Fe(CN)63− + 0.1 M KCl in
20 mM, pH 7.4, PBS
[60]
Carbohydrate antigen 19-9(CA19-9)/human serum
HRP-labeled-CA19-9 Ab immobilization ontonAu-modified electrode
CPE CV; EIS; DPV PBS, pH 7.0 Linear range: 2–30 U mL−1;LOD: 1.37 U mL−1
[61]
J.M.Pingarron
etal./Electrochim
icaA
cta53
(2008)5848–5866
5857Table 2 (Continued )
Analyte/sample Immunoreagents Electrode Detection Performance Analytical characteristics Ref
Carbohydrate antigenCA125
Immobilization of CA125 Ab on gold hollowmicrospheres and porous polythionine (PTH)-modifiedGCE
GCE Amperom. 2.5 mMK4[Fe(CN)6] + 2.5 mMK3[Fe(CN)6] + 0.1 M KCl in20 mM, pH 7.4, PBS
Linear range:4.5–36.5 U mL−1; LOD:1.3 U mL−1
[62]
Schistosoma japonicum (Sj)antigen
SjAb adsorption onto nAumonolayer–chitosan-entrapped CPE
CPE Amperom. Sequential competitiveassay with HRP-labeledSjAg; 0.067 M PBS + 0.1 MKCl; pH 6.98
Dynamic range:0.11–22.4 �g mL−1; LOD:0.06 �g mL−1
[63]
Schistosoma japonicumantibody/rabbit serum
S. japonicum (SjAg) adsorption a polystyrenemicrowells; bond with rabbit primary SjAb;nAu-labeled goat anti-rabbit IgG secondaryAbadsorption
GCE ASV Incubation ofsilver-enhancer solution inmicrowells, 30 min.Dissolution of Ag withHNO3
Linear range: 6.4 ng mL−1
to 100 �g mL−1; LOD:3 ng mL−1
[64]
Carcinoembryonic antigen(CEA)
nAu immobilization on toluidine blue–polysulfanilicacid modified GCE. Anti-CEA and HRP adsorption
GCE Amperom. 0.1 M PBS, pH 6.5 + 0.1 MKCl + 0.3 mM H2O2
Linear range:0.5–5.0 ng mL−1,5.0–120 ng mL−1; LOD:0.2 ng mL−1
[65]
Carcinoembryonic antigen Anti-CEA immobilization on nAu–MPTS sol–gelfunctionalized interface
Gold Potentiom. PBS, pH 7.4 Dynamic range:4.4–85.7 ng mL−1; LOD:1.2 ng mL−1
[66]
PSA/human serum Immobilization anti-PSA on nAu/Al2O3 functionalizedsol–gel thin film
Gold Potentiom. PBS, pH 7.4 Linear range:4.0–13 ng mL−1; LOD:3.4 ng mL−1
[67]
Interleukin-6 (IL6) Encapsulation of HRP-labeled-anti-IL6 inpoly(amidoamine) dendrimer–nAu-modified surface
Interdigtated Aumicroelectrodes
Conductom. 0.02 M PBS, pH 7.0 + 50 �MH2O2, 0.01 M KI + 0.15 MNaCl
Linear range:30–300 pg mL−1; LOD:10 pg mL−1
[112]
Japanese B encephalitisvaccine
Anti Japanese B encephalitis immob. bilayernAu/o-phenylenediamine polymer film with depositedPrussian blue
Platinum Amperom. 25 mM PBS, pH 7.4 Linear range:1.1–190 × 10−8 log pfu mL−1;LOD: 6 × 10−9 log pfu mL−1
[113]
Three different cytokines Diazonium–Ab deposition, exposure to target cytokine,followed by biotinylated capture antibody treatment,conjugation to extract Avidin-HRP label
GCE and gold Cronoamp. E = 0 V Non-competitiveassay/electroreduction ofthe oxidized3,3,5,5-tetramethylbenzidine mediator
Linear range:0.1–100 ng mL−1; LOD:46 pg mL−1.
[114]
Mammary cancer 15-3(CA15-3)/human serum
Grown of red blood cells of crucian carp ontonAu-modified electrode; adsorption of secodary nAulayer; anti-CA15-3 immobilization
GCE CV Competitive assay withHRP-labeled CA15-3 PBS,pH 7.4 + 1.0 mM H2O2
Linear range:2.5–120 U mL−1; LOD:0.5 U mL−1
[115]
Human serum albumin(HSA)
Colloidal gold with polyaniline bound on the metalsurface anti-HSA immobilization onto membraneelectrode
Silver screen-printed onnitrocellulose membrane
Conductom. Non-competitiveassay/secondary Ab labeledwith nAu
[116]
Progesterone/milk Anti-Prog deposited on the surface of the compositeelectrode
Composite graphite–Teflonelectrode
Amperom. E = +0.3 V vsAg/AgCl
Competitive assay: ProgAP: Prog solution depos.;1 mM MgCl2 and 0.1 Mdiethanolamine–HCl buffersolution of pH 10.0
Linear range:0–30 ng mL−1; slope:−1.2 nA ng−1 mL−1
[117]
Progesterone/milk Physical inclusion of Tyr and nAu particles intoelectrode matrix. Anti-Prog deposited on the electrodesurface
Composite graphite–Teflonelectrode
Amperom. E = −0.1 V vsAg/AgCl
Sequential competitiveassay; progesteronelabeled with alkalinephosphatase (AP)/0.1 MTRIS buffer of pH 7.0 and1 mM MgCl2
Linear range:0–40 ng mL−1; slope:−82.3 nA ng−1 mL−1;lifetime: 14 days; LOD:0.43 ng mL−1
[118]
Abbreviations: adsorp.: adsorption, Conductom.: Conductometry, CPE: carbon paste electrode, DTSP: 3-3′-dithio-bis(propionic acid-N-hydroxysuccinimide ester), Fc-CHO: ferrocenecarboaldehyde, GCE: glassy carbon electrode,HBsAb: hepatitis B surface antibody, HBsAg: hepatitis B surface antigen, HDT: 1,6-hexanedithiol, IgG: Immunoglobulin G, MPS: mercaptopropyl trimethoxysilane, MPTS: mercaptopropyltrietho-xysilane, nAu: gold nanoparticles,OPD: o-phenylenediamine, PBS: phosphate buffer solution; Potentiom.: Potentiometry, PSA: prostate-specific antigen, PVB: polyvinyl butyral, SAM: self-assembled monolayer, TRIS: tris(hydroxymethyl)aminomethane

5858 J.M. Pingarron et al. / Electrochimica Acta 53 (2008) 5848–5866
ion of
Fig. 2. Scheme showing the stepwise HBsAg immunosensor preparation: (a) formatmonolayer; (d) binding of HBsAg; (e) blocking with HRP (adapted from Ref. [45]).the response of the antigen–antibody reaction [49]. Furthermore,mixed self-assembled monolayers, gold nanoparticles and enzymeamplification were used together with microelectronic technology,to develop an immunosensor for AFP based on microelectrodesand microwells systems constructed by SU-8 photoresist on siliconwafer [50].
Many examples on the determination of human IgG can be foundin the literature. For example, a capacitive sensing method usinggold nanoparticles self-assembled to a sol–gel-modified electrodewas developed for the direct detection of the human IgG in serum.Since the formed mercaptopropyltriethoxysilane film was ultra-thin, the immobilization density of antibodies was high becauseof the high surface–volume ratio of the assembled gold nanoparti-cles. The capacitive immunosensor provided a high sensitivity andno cross-reactivity was observed with other proteins [51].
Another method for the determination of human IgG employedgold nanoparticle accumulation using magnetic particles [52]. Goatanti-human IgG was immobilized on the wells of microtiter plates.The human IgG analyte was first captured by the primary antibodyand then sandwiched by secondary antibody labeled with dethio-
biotin. The conjugate of biotin-modified magnetic particles withcaptured avidin functionalized gold nanoparticles, was reactedwith dethiobiotinylated antibody followed by addition of biotinsolution to wash it down, which caused the dissociation of theconjugates. The release of gold nanoparticles was then followedby anodic striping voltammetry. This example shows that nanopar-ticles can be used not only as medium to retain biomolecules with ahigh stability, but also to provide versatile labels for the amplifica-tion of biosensing events. The secondary dissolution of the capturednanoparticles enables the amplified detection of the respective ana-lyte by the release of many ions/molecules as a result of a singlerecognition event. This methodology was applied in a sensitiveimmunosensor for human IgG based on a sandwich-type assayusing colloidal gold as electrochemical label [53]. As it is displayedin Fig. 3, the capture protein was immobilized on a carbon pasteelectrode through passive adsorption to bind quantitatively withthe corresponding antigen and colloidal gold-labeled antibody. Inorder to detect the amount of colloidal gold captured on the elec-trode surface, it was oxidized electrochemically to produce AuCl4−ions which were strongly adsorbed on the electrode surface anddetermined by adsorptive voltammetry.
a Nafion monolayer; (b) adsorption of thionine; (c) formation of gold nanoparticles
A highly sensitive electrochemical impedance immunosensorwas also developed using an amplification procedure with a col-loidal gold-labeled antibody as the primary amplifying probe, and amultistep amplification sequence. Rabbit anti-human IgG antibodywas immobilized through a self-assembled colloidal gold layer ona gold electrode. The analyte, human IgG, was detected through theimpedance measurements with the sensing interface modified bythe rabbit anti-human IgG antibody. In the primary amplification,the colloidal gold-labeled goat anti-human IgG antibody was usedto amplify the electron transfer resistance resulting from the anti-gen binding to the immunosensor surface. A further amplificationwas performed through sequential binding of the colloidal gold-labeled rabbit anti-goat IgG and the colloidal gold-labeled goatanti-human IgG antibodies [54].
Other configurations for IgG made use of silver precipitationon colloidal gold labels. After silver metal dissolution in an acidicsolution, the antigen was indirectly determined by anodic strippingvoltammetry at a GCE [55].
Aflatoxin B1 (AFB1) was determined using an electrode fab-ricated by self-assembling HRP and AFB1 antibody onto gold
nanoparticles. These provided a suitable microenvironment forthe immobilized biomolecules and decreased the electron transferimpedance. The formation of the antibody–antigen complex by asimple one-step immunoreaction introduced a barrier for the directelectrical communication between the immobilized HRP and theelectrode surface. Therefore, local conductivity variations could bedetected giving a good conductometric response relative to AFB1[56].Determination of the carcinoma antigen 125 was carried outby using carbon electrodes modified with gold nanoparticles. Anelectrochemical immunosensor was designed by immobilizinganti-CA125 on a thionine and gold nanoparticles-modified carbonpaste interface [57]. The same antibody was also immobilized oncolloidal gold nanoparticles to form a bioconjugate stabilized with acellulose acetate membrane on the surface of a GCE. This configura-tion was proposed to develop a competitive immunoassay format todetect CA125 antigen with HRP-labeled CA125 antibody as tracer,and o-phenylenediamine and hydrogen peroxide as enzyme sub-strates [58].
An electrochemical immunoassay for human chorionicgonadotrophin (hCG) was proposed by using a conductive

himic
ich i
J.M. Pingarron et al. / Electroc
Fig. 3. Scheme displaying the construction and the transduction principle of a sandwfrom Ref. [53]).
colloidal gold nanoparticle/titania sol–gel composite membranedeposited on a GCE via a vapor deposition method. Horseradishperoxidase-labeled hCG antibody (HRP-anti-hCG) was encapsu-lated into the composite architecture. Similarly to that commentedabove, the formation of the immunoconjugate between hCG andthe immobilized HRP-anti-hCG introduced a barrier for the directelectrical communication between the immobilized enzyme andthe electrode surface that can be monitorized by EIS [59].
Impedance sensing was also used to detect allergen–antibodyinteraction on a GCE modified by gold electrodeposition [60].In this application, allergen Der f2 was immobilized throughgold nanocrystals deposited on the electrode surface, the charge-transfer kinetics of the [Fe(CN)6]3−/4− redox pair being monitored.The allergen–antibody interactions that occurred on the electrodesurface altered the interfacial electron transfer resistance, RCT, bypreventing the redox species approaching the electrode. The resultsshowed that RCT increased with increasing concentration of mon-oclonal antibodies.
Carbohydrate antigen 19-9 (CA19-9) is one of the most impor-tant carbohydrate tumor markers, expressed in many malignanciesas pancreatic, colorectal, gastric and hepatic carcinomas. Animmunosensor for the rapid determination of CA19-9 in humanserum has been developed by immobilization of the antibodyin colloidal gold nanoparticle-modified carbon paste electrodesand monitoring of the direct electrochemistry of HRP labeledto a CA19-9 antibody. The formation of the antigen–antibodycomplex blocked the electron transfer of HRP toward the elec-
trode substrate which resulted in a significant current decrease[61]. Another carbohydrate antigen, CA125, was detected usinganti-CA125 gold hollow microspheres and porous polythionine-modified GCEs. The gold hollow microspheres greatly amplifiedthe coverage of anti-CA125 molecules on the electrode surface.Electrochemical detection was accomplished by the amperometricchanges occurring before and after the antigen–antibody interac-tion [62].A nanogold monolayer modified chitosan-entrapped car-bon paste electrode was used to construct an amperometricimmunosensor for Schistosoma japonicum (Sj) antigen. A sequentialcompetitive immunoassay configuration was employed by loadingof SjAb on nAu monolayer, then blocking in BSA solution, fol-lowed by a competitive incubation in the buffer containing the SjAganalyte and SjAg labeled with HRP. Amperometric detection wasmade using hydroquinone as the enzyme substrate [63]. A silver-enhanced colloidal gold metalloimmunoassay was also proposedfor the determination of Schistosoma japonicum antibody in rabbitserum. The adult worm antigen SjAg was adsorbed on the walls ofa polystyrene microwell and then reacted with the desired SjAb.Colloidal gold-labeled goat anti-rabbit IgG secondary antibody was
a Acta 53 (2008) 5848–5866 5859
mmunosensor for human IgG using colloidal gold as electrochemical label (adapted
adsorbed on the walls of the polystyrene microwells through thereaction with SjAb, followed by the silver enhancement process,dissolution of silver metal atoms in an acidic solution, and deter-mination of dissolved silver ions by anodic stripping voltammetryat a GCE [64].
Carcinoembryonic antigen (CEA) is a well-known marker asso-ciated with progression of colorectal tumors. A current amplifiedimmunosensor for its determination was fabricated by coat-ing negatively charged polysulfanilic acid modified GCEs withpositively charged toluidine blue. This approach provided an inter-face containing amine groups to assemble gold nanoparticlesfor immobilization of the carcinoembryonic antibody and HRP[65]. A dual-amplification strategy was proposed via backfillinggold nanoparticles on (3-mercaptopropyl) trimethoxysilane sol–gel(MPTS) functionalized interface. MPTS acted as a building block forthe electrode surface modification as well as a matrix for ligandfunctionalization with first amplification. The second signal ampli-fication strategy was based on the backfilling immobilization ofnanogold particles to the immunosensor surface. Using the non-competitive design, membrane potential change occurred beforeand after the antigen–antibody interaction [66].
Electrochemical detection of prostate-specific antigen has beencarried out by using a colloidal gold/alumina-derived sol–gel film[67]. An anti-PSA antibody-functionalized colloidal gold/aluminasol–gel film was prepared and PSA detection was accomplished onthe basis of the potential change occurring before and after theantigen–antibody interaction.
4. Gold nanoparticle-based electrochemical DNAbiosensors
Electrochemical DNA biosensors constitute useful analyticaltools for sequence-specific DNA diagnosis and detection due totheir inherent advantages of low cost, sensitivity and rapidity ofresponse [68]. Similarly to other electrochemical biosensors, goldnanoparticles can play an important role both in DNA immobi-lization on electrode surfaces and as suitable labels to improvedetection of hybridization events. Table 3 summarizes some of therecent approaches in this field.
Electrode surface-immobilization techniques for the achieve-ment of stable and highly dense single-stranded DNA (ssDNA)monolayers are a key aspect in the development of DNA biosensors.In this context, gold nanoparticle films provide a suitable means forthe ssDNA immobilization. For example, self-assembly of colloidalAu onto a gold electrode resulted in an easier attachment of anoligonucleotide with a mercaptohexyl group at the 5′-phosphateend and, therefore, an increased capacity for nucleic acid detec-tion. The ssDNA surface density on the colloidal Au-modified gold

5860J.M
.Pingarronet
al./Electrochimica
Acta
53(2008)
5848–5866
Table 3Gold nanoparticle-based electrochemical DNA biosensors
Electrode material Immobilization technique Hybridization Detection Target Analytical characteristics Ref.
Gold Colloidal Au-cysteamine-modified electrode; ssDNA(oligonucleotide with a mercaptohexyl group at the5′-phosphate end) immobilized onto nAu
ssDNA-containing goldelectrode exposed toferrocene-carbo-xaldehyde-labeledcomplementary ssDNAsolution
DPV (E = 0–0.80 V vsAg/AgCl)
ssDNA (20-mer) Linear range:1.0 × 10−9–5.0 × 10−7 M;LOD: 5.0 × 10−10 M
[69]
Gold Colloidal Au-cysteamine-modified electrode dippedinto ssDNA solution 120 min
Hybridized with thenAg–DNA probe
EIS; oxidized silverdissolution; ASV of AgI at acarbon fibermicroelectrode
ssDNA (30-mer) Linear range: 10–800 pM;LOD: 5 pM
[70]
Gold Self-assembling of bilayer two-dimensional3-mercaptopropyltrimethoxy-silane and nAuchemisorption onto the thiol groups of the secondsilane layer. DNA self-assembling onto nAu
DNA probe incubation intarget DNA solution 30 min
EIS cDNA Linear range:1.0 × 10−8–1.0 × 10−6 M;LOD: 5.0 × 10−9 M
[119]
Gold Electrodeposition of nAu (at −0.2 V). Immersion intoPBS (pH 6.83, 50 mmol L−1 NaCl) with 0.01 mmol L−1
ssDNA probe for 5 h
ssDNA probe modifiedelectrodes immersed intophosphate buffer solutionwith target DNA 4 h atroom temperature
CV; methylene blue aselectroactive indicator
16-mer oligo-nucleotide55-mer oligo-nucleotide
LOD: 10−9 and 10−11 M,respectively
[120]
Gold Electrochemical deposition of nAu; immobilization ofsynthesized thiol-linked DNA probes
Interaction of DNA-specificsequence binding drugswith duplex DNAmonolayers immobilizedon a gold surface
EIS Nogalamycin,mythramycin, netropsin
LOD: 5, 10, 40 nM,respectively
[80]
GCE Target ssDNA immobilization into chitosan Hybridization to thenAu-labeledoligonucleotides DNAprobe
Silver deposition ontoDNA/nAu electrode; DPV
DNA Linearity range: 0.1–5 nM;LOD: 50 pM
[73]
GCE Accumulation of DNA for 30 min at +1.5 V on GCE;electrodeposition of nAu at −0.2 V for 5 min
– DPV (lifetime: 15 days) Norepinephrine inpresence of ascorbic acid
Linear range:5 × 10−7–8 × 10−5 M(containing 1 mM ascorbicacid); LOD: 5 nM
[121]
GCE 2,6-Pyridinedicarboxylic acid (PDC) electropolymer;nAu immob. on PDC by electrodeposition andimmersing adsorption. Immobilization of DNA probe
Hybridization reaction byelectrode immersion into astirred 50 �M target DNAsolution
EIS Sequence-specific PAT genefragment
Linear range:1.0 × 10−10–1.0 × 10−5 M;LOD: 2.4 × 10−11 M
[76]
GCE nAu–DNA solution dropping onto GCE surface;immersion of modified electrode in SH–ssDNA solution14 h
Immersing MB markedssDNA/nAu–DNA/GCE inTRIS–HCl buffer solution(pH 8.0) containing 21-merssDNA; incubating at 37 ◦Cfor 1 h
DPV; methylene blue aselectroactive indicator
cDNA (21-mer) Linear range:1.52 × 10−10–4.05 × 10−8 M;LOD: 10−10 M
[71]
CPE Chitosan layer preparation onto SAM-modified nAu.Monobases attachment onto the chitosan-coated nAuthrough 5′-phosphate group
Hybridization betweenmu-tant site andmonobase-modified nAu inpresence of DNApolymerase I (Klenowfragment)
SWV Single-nucleotidepolymorphisms (SNPs)
[78]
Carbon SPE Biotinylated probe immobilized onto magneticstreptavidin-coated beads
Hybridization of targetoligo-nucleotide tomagnetic bead-linkedoligo-nucleotide probes,followed by bindingstreptavidin-coated metalnanoparticles to capturedDNA
PSA; dissolution of goldtag; amplification bycatalytic precipitation ofgold (>80-fold)
DNA R.S.D. = 12% (n = 8) LOD: 10 ng mL−1 (1.5 nM) [72]

J.M. Pingarron et al. / ElectrochimicTa
ble
3(C
onti
nued
)
Elec
trod
em
ater
ial
Imm
obil
izat
ion
tech
niq
ue
Hyb
rid
izat
ion
Det
ecti
onTa
rget
An
alyt
ical
char
acte
rist
ics
Ref
.
Car
bon
SPE
Un
iqu
ebi
nd
ing
even
tbe
twee
nE.
coli
sin
gle-
stra
nd
edD
NA
bin
din
gp
rote
inan
dsi
ngl
e-st
ran
ded
olig
o-n
ucl
eoti
des
con
juga
ted
ton
Au
SSB
atta
chm
ent
onto
aSA
Mof
sin
gle-
stra
nd
edol
igon
ucl
eoti
de
mod
ified
nA
uan
dth
ere
sult
ing
Au
-tag
ged
SSB
SWV
DN
Am
=0.
23n
An
M−1
;LO
D:
2.17
pM
targ
etD
NA
[75]
Pen
cilg
rap
hit
eel
ectr
ode
(PG
E)C
oval
ent
imm
obil
izat
ion
ofta
rget
DN
Aon
PGE;
Inos
ine-
subs
titu
ted
pro
bes
cova
len
tly
atta
ched
onto
l-cy
stei
ne
SAM
pre
form
edon
nA
u
Targ
et-i
mm
obil
ized
PGEs
dip
ped
into
the
nA
un
anop
arti
cle
tagg
edca
ptu
rep
robe
solu
tion
for
1h
DPV
(oxi
d.s
ign
alof
coll
oid
algo
ld)
Fact
orV
Leid
enm
uta
tion
LOD
:0.
78fm
ol[7
7]
ITO
Imm
obil
izat
ion
ofD
NA
onn
Au
-(3-
amin
opro
pyl)
trim
eth
oxys
ilan
e-m
odifi
edIT
OD
PV,p
H7.
0,PB
SM
ifep
rist
one
Lin
ear
ran
ge:
4×
10−7
–×
10−6
M;
LOD
:2
×10
−7m
olL−1
.
[79]
ITO
ITO
elec
trod
esm
odifi
edby
the
pol
ymer
scop
olet
inem
bed
ded
wit
hst
rept
avid
in;
imm
obil
izat
ion
of5′ -
biot
inyl
ated
olig
onu
cleo
tid
ep
robe
s(S
tach
ybot
rys
char
taru
man
dE.
coli)
via
the
avid
in–b
ioti
nco
up
lin
g
Incu
bati
onin
targ
etp
ath
ogen
PCR
pro
du
cts
orsy
nth
etic
olig
o-n
ucl
eoti
des
;n
Au
labe
lbou
nd
toth
ehy
brid
ized
targ
etby
exp
osin
gth
eel
ectr
ode
surf
ace
tost
rept
avid
in–g
old
solu
tion
chro
noa
mp
erom
etri
csi
lver
elec
trod
epos
itio
non
ton
Au
labe
lan
dPS
Aof
silv
er
DN
ALi
nea
rra
nge
:5
pM
to10
nM
;LO
D:
2.0
×10
−12
M[7
4]
Abb
revi
atio
ns:
ASV
:an
odic
stri
pp
ing
volt
amm
etry
,CPE
:ca
rbon
pas
teel
ectr
ode,
DPV
:d
ifer
enti
alp
uls
evo
ltam
met
ry,E
IS:
elec
troc
hem
ical
imp
edan
cesp
ectr
osco
py,M
B:
met
hyle
ne
blu
e,n
Au
:G
old
nan
opar
ticl
e,PA
T:p
hos
-p
hin
oth
rici
nac
etyl
tran
sfer
ase,
PSA
:p
oten
tiom
etri
cst
rip
pin
gan
alys
is,S
PE:
scre
en-p
rin
ted
elec
trod
e,SS
B:
sin
gle-
stra
nd
brea
ks,S
WV
:sq
uar
ew
ave
volt
amm
etry
.
a Acta 53 (2008) 5848–5866 5861
electrode was 1.0 × 1014 molecules cm−2, ca. 10 times larger thanon a bare gold electrode [69].
DNA was also immobilized successfully on colloidal goldnanoparticles associated with a cysteamine monolayer on a goldelectrode surface. Self-assembly of colloidal gold onto the modi-fied electrode enlarged the electrode surface area and enhancedgreatly the amount of immobilized ssDNA. The single-stranded tar-get DNA immobilized on the gold electrode hybridized with silvernanoparticle-oligonucleotide DNA probe, which was followed bythe release of the silver metal atoms anchored on the hybrids byoxidative metal dissolution, and the indirect determination of thereleased solubilized Ag+ ions by anodic stripping voltammetry at acarbon fiber microelectrode. The method allowed the detection oftarget nucleotides at a level as low as 5 pM [70].
DNA hybridization was also investigated at a goldnanoparticles–DNA-modified GCE. Thiol-modified-probe oligonu-cleotides were assembled on the modified electrode surface.The electrochemical response of the probe immobilization andhybridization with target DNA was monitored by DPV using methy-lene blue as electroactive indicator. The Au nano DNA-modifiedGCE increased greatly the active sites and enhanced the responsesignal during immobilization and hybridization [71].
In order to achieve adequate sensitivity, the development ofnovel amplification methodologies for quantitative DNA sensingevents is essential. Gold nanoparticles can play a crucial role to thisrespect. So, DNA hybridization detection was accomplished by elec-trochemical stripping of the colloidal gold tag. In this protocol, thehybridization of a target oligonucleotide to magnetic bead-linkedoligonucleotide probes was followed by binding of streptavidin-coated metal nanoparticles to the captured DNA, dissolution of thenanometer-sized gold tag, and potentiometric stripping measure-ments of the dissolved metal tag at single-use thick-film carbonelectrodes. An advanced magnetic processing technique was usedto isolate the DNA duplex and to provide low-volume mixing. Fur-ther signal amplification, and lowering of the detection limits tothe nanomolar and picomolar domains, was achieved by precipi-tating gold or silver, respectively, onto the colloidal gold label. Thiselectrochemical stripping metallogenomagnetic approach coupledthe inherent signal amplification of stripping metal analysis withdiscrimination against non-hybridized DNA, the use of microlitersample volumes, and disposable transducers [72]. A scheme of thesensor preparation and functioning is displayed in Fig. 4.
The use of gold nanoparticle–DNA probes and subsequent sig-nal amplification by silver enhancement constitutes a somewhatsimilar approach. The assay relied on the electrostatic adsorption
of target oligonucleotides onto the sensing surface of a GCE andits hybridization to the gold nanoparticle-labeled oligonucleotidesDNA probe. After silver deposition onto gold nanoparticles, bind-ing events between probe and target were monitored by the DPVsignal of the large number of silver atoms anchored on the hybridsat the electrode surface. The signal intensity difference permittedto distinguish between the match of two perfectly matched DNAstrands and the near-perfect match where just one base pair waswrong. Due to this ‘nanoparticle-promoted’ silver signal amplifica-tion, a detection limit of 50 pM of complementary oligonucleotidescould be obtained [73].Pathogens, bacteria and viruses, can be detected via theirunique, respective nucleic acid sequences. An electrochemicalmethodology that enabled the rapid identification of differ-ent DNA sequences on microfabricated electrodes was basedon an electropolymerization process on a patterned ITO-coatedglass electrode, followed by a selective immobilization ofbiotin-tagged probes on individually addressable spots via thebiotin–streptavidin linkage. An exemplary target mixture con-taining Escherichia coli and Stachybotrys chartarum, an airborne

5862 J.M. Pingarron et al. / Electrochimic
gold nanoparticles.
Fig. 4. Schematic representation of the DNA sensor preparation and functioning(adapted from Ref. [72]).
pathogen, was then introduced. Recognition of the DNA hybridiza-tion event of the immobilized probes with the target pathogenPCR products or synthetic oligonucleotides was achieved by anelectrochemistry-based technique utilizing the preferential cat-alytic silver electrodeposition process on the DNA-linked nanogoldshells. The gold nanoparticle label was specifically bound to theformed hybrid, followed by a silverstaining step, in which silver-enhancing solutions were used to improve the signal of the goldparticles by forming shells of silver surrounding the gold core.The silver deposits were dissolved by an acid treatment and mea-sured by potentiometric stripping analysis [74]. Another designuses the unique binding event between E. coli single-stranded DNAbinding protein (SSB) and single-stranded oligonucleotides conju-gated to gold (Au) nanoparticles. SSB was attached onto a SAM ofsingle-stranded oligonucleotide-modified Au nanoparticle, and theresulting Au-tagged SSB was used as the hybridization label. Theamplified oxidation signal of Au nanoparticles provided a detection
limit of 2.17 pM target DNA [75].The security of the food made from transgenic plants is aworldwide matter of concern. An important associated aspectis the accurate, sensitive, and rapid detection of the trans-genic plants. DNA electrochemical sensors are most likelyto become an analytical tool for the transgenic plant prod-ucts. Recently, a DNA electrochemical biosensor was describedfor electrochemical impedance spectroscopy detection of thesequence-specific DNA related to PAT transgene in the transgenicplants. Poly-2,6-pyridinedicarboxylic acid film (PDC) was fabri-cated by electropolymerizing 2,6-pyridinedicarboxylic acid on aGCE. Gold nanoparticles were attached on the PDC/GCE, and thenprobe DNA was immobilized by the interaction of gold nanopar-ticles with DNA. The electron transfer resistance of the electrodesurface in [Fe(CN)6]3−/4− solution increased after the immobiliza-tion of the DNA probe on the nAu/PDC/GCE. The hybridization ofthe DNA probe with complementary DNA made electron trans-fer resistance increase further. The presence of gold nanoparticleson the PDC dramatically enhanced the amount of the immobilizedprobe DNA and greatly improved the sensitivity of DNA detection.
a Acta 53 (2008) 5848–5866
The difference between the electron transfer resistance value at thessDNA/nAu/PDC/GCE and at the dsDNA/nAu/PDC/GCE was used asthe signal for detecting the PAT gene fragment with a detectionlimit of 2.4 × 10−11 M [76].
A genosensor based on the modification of a pencil graphiteelectrode with target DNA and further hybridization with comple-mentary probes conjugated to Au nanoparticles was developed forthe detection of the Factor V Leiden mutation from PCR ampli-fied real samples. Specific probes were immobilized onto theAu nanoparticles in two different modes: (a) inosine-substitutedprobes were covalently attached from their amino groups at the5′-end using N-(3-dimethylamino)propyl)-N-ethylcarbodiimidehydrochloride and N-hydroxysulfosuccinimide onto a carboxylate-terminated l-cysteine self-assembled monolayer preformed onthe Au nanoparticles; (b) probes with a hexanethiol group attheir 5′-phosphate end formed a SAM on Au nanoparticles. Theappearance of the Au oxidation signal at +1.20 V was employed todetect hybridization. Discrimination between the homozygous andheterozygous mutations was established by comparing the peakcurrents of the Au signals. The detection limit for the PCR ampliconswas found to be as low as 0.78 fmol [77].
Electrochemical genosensors offer a promising alternative tocarry out applications directed to gene analysis, detection of geneticdisorders, tissue matching, and forensic applications due to theirhigh sensitivity, small dimensions, low cost and compatibility withmicro-fabrication technology. Furthermore, increasing informationabout the human genome requires fast and simple methods for thedetection of single-nucleotide polymorphisms (SNPs). SNPs can bealso coded by monitoring the changes in the electrochemical signalof the monobase-modified colloidal gold nanoparticles. A chitosanlayer formed on the alkanethiol SAM-modified Au nanoparticle canbe used to attach the monobases through their 5′-phosphate groupvia the formation of a phosphoramidate bond with the free aminogroups of chitosan. If there is a SNP in DNA and the mismatchedbases are complementary to the monobase, Au nanoparticles accu-mulate on the electrode surface in the presence of DNA polymeraseI (Klenow fragment), thus resulting in a significant change in the Auoxide wave. Monobase-modified Au nanoparticles showed not onlythe presence of a SNP, but also identified which bases were involvedwithin the pair. A model study was performed by using a synthetic21-base DNA probe related to tumor necrosis factor (TNF-r) alongwith its all possible mutant combinations [78]. Fig. 5 shows as thebases involved in a SNP can be identified by comparing the voltam-metric signals obtained from the four different monobase-modified
Another interesting research area is the preparation of DNA-modified electrodes to study the interaction of DNA with othermolecules. For example, a simple, stable and repeatable approachbased on the preparation of a DNA-modified ITO electrode was usedfor the determination of mifepristone, which is a kind of proges-terone receptor compound mainly used in anti-pregnancy that canbe intercalated into a DNA double helix. The electrode consistedof (3-aminopropyl)trimethoxysilane, gold nanoparticles and DNAmolecules self-assembled to the ITO electrode surface [79]. Fur-thermore, the analysis of the interfacial interaction between DNAbinding drugs, mostly used as antibiotics and in cancer therapy,with immobilized DNA probes has a particular role in the ratio-nal design of new DNA binding drugs and drug screening. Theseinterfacial interactions can be investigated by impedance spec-troscopy. Using gold nanoparticle-deposited substrates, impedancespectroscopy resulted in a 20-40-fold increase in the detectionlimit. Arrays of deposited gold nanoparticles on gold electrodesoffered a convenient tool to subtly control probe immobilizationto ensure suitably adsorbed DNA orientation and accessibility ofother binding molecules [80].

J.M. Pingarron et al. / Electrochimica Acta 53 (2008) 5848–5866 5863
d in a
Fig. 5. Scheme displaying voltammetric identification of the bases involve5. Other biosensors designed with gold nanoparticles
Gold nanoparticles have also been used for direct electrical con-tacting of redox proteins to electrode supports. The study on thedirect electrochemistry of proteins is of high significance both fromtheoretical and practical points of view because understandingof these reactions can provide insight into physiological electrontransfer process. An interesting approach in this context is proposedby Willner and co-workers involving the orientation of the enzymeson electrodes by a reconstitution process [24,81–82]. The precisepositioning of relay units between the protein redox center andthe electrode results in effective electron transfer. Gold nanopar-ticles have been employed for the reconstitution of apo-glucoseoxidase on a 1.4-nanometer gold nanocrystal functionalized withthe cofactor FAD [83]. The electron transfer turnover rate of the
reconstituted bioelectrocatalytic system is approximately 5000 persecond, noticeably higher than that the rate at which molecularoxygen accepts electrons. The gold nanoparticle acts as an elec-trical nanoplug for the alignment of the enzyme on the electrodesupport and for the electrical wiring of its redox center. This con-cept was extended to pyrroloquinoline quinone (PQQ)-dependentenzymes by the reconstitution of apo-glucose dehydrogenaseon PQQ-functionalized gold nanoparticles assembled on a goldelectrode surface [84]. The electrical contacting of GDH in thissystem was improved approximately 25-fold as compared to asystem where polyaniline acted as the matrix for the electricalcontacting between the reconstituted enzyme and the electrodesupport.Some other examples of approaches to study direct electrontransfer between other redox proteins and electrode surfaces aresummarized in Table 4. Hemoglobin (Hb) possesses functionalgroups that can be readily oxidized or reduced by chemical redoxagents, but it does not easily undergo facile redox reactions at bareelectrodes due to the very slow electron transfer process. This slowelectron exchange may be caused by the unfavorable orientationof Hb molecules on electrode surface, which increases the distance
SNP using monobase-modified Au nanoparticles (adapted from Ref. [78]).
between its heme center and electrode surface. Gold nanoparticles,acting as an electron transfer bridge, can greatly promote the directelectron transfer between Hb and a modified GCE without theaid of any electron mediator [85]. Furthermore, direct electrontransfer between hemoglobin and a gold electrode was alsoobserved by immobilization of the protein on gold nanoparticlesassociated with a 1,4-benzenedimethanethiol monolayer on theelectrode surface [86]. These designs were also applied for theelectrocatalytic detection of hydrogen peroxide. Another approachfor the construction of an amperometric third-generation H2O2biosensor made use of Hb immobilized on MWCNTs and col-loidal gold nanoparticles [87]. Also, an amperometric sensor forhydrogen peroxide was developed based on the immobilization ofmyoglobin and colloidal gold nanoparticles on a GCE by a Nafionfilm. The immobilized Mb displayed a pair of well-defined and
nearly reversible cyclic voltammetric peaks with a formal potentialof −0.373 V, and exhibited excellent electrocatalytic response tothe reduction of H2O2 [88]. Recently, the direct electrochemistryof Mb in films composed of gold nanoparticles encapsulated insidepolyamidoamine dendrimers assembled on pyrolytic graphiteelectrodes was used to demonstrate the conductive effect of goldnanoparticles on bridging electron transfer between the proteinand electrodes [89]. The alternate adsorption of negatively chargedgold nanoparticles and positively charged Mb on pyrolytic graphiteelectrodes was used to assemble gold–Mb layer-by-layer films. Thedirect electrochemistry of Mb at these modified electrodes wasstudied and was used to electrocatalyze the reduction of oxygenand hydrogen peroxide [90].Cytochrome P450scc electron transfer at rhodium–graphiteelectrodes modified with gold nanoparticles was studied [91]. Inthis case, the electrodes were modified by gold nanoparticles dropcasting onto the rhodium–graphite surface, and the mediating roleof nanoparticles in the cytochrome electron transfer was verifiedby ac impedance spectroscopy. An application of this design wasmade for the detection of low concentrations of cholesterol. Theelectrocatalytic activity of cytochrome c immobilized on a col-

5864
J.M.Pingarron
etal./Electrochim
icaA
cta53
(2008)5848–5866
Table 4Other biosensors designed with gold nanoparticles
Protein/electrode material Immobilization Detection Performance Analyte Analytical characteristics REF.
Hb/GCE Hb immob. onto nAu-sulfhydryl-monolayer modifiedelectrode
Amperom. Repeatability: R.S.D. = 3.8%(n = 11)
H2O2 Linear range:2.0 × 10−6–2.4 × 10−4 M;LOD: 9.1 × 10−7 M
[85]
Hb/GCE Hb/nAu multilayer films assembled onto Hb/MWNTfilm through layer-by-layer assembly technique
Amperom. KappM = 0.26 mM H2O2 Linear range:
2.1 × 10−7–3.0 × 10−3 M;LOD: 8.0 × 10−8 M
[87]
Hb/AuE Hb immobilization on nAu/1,4-benzenedimethanethiolmonolayer modified electrode
Amperom. (E = −200 mV) H2O2 Linear range: 2–18 �M [86]
Mb/GCE Immobilization of myoglobin (Mb) and colloidal goldnanoparticles on GCE by a Nafion film
Amperom. (E = −0.45 V vsSCE)
R.S.D. = 5.8% (n = 6);lifetime: 30 days
H2O2 Linear range:1.5 × 10−6–9.0 × 10−5 M;LOD: 5.0 × 10−7 M
[88]
Mb/basal plane pyrolyticgraphite
nAu encapsulated in the cavities of the PAMAMmolecules. The PAMAM-Au nanocomposites wereassembled layer-by-layer with myoglobin (Mb) into{PAMAM-Au/Mb}n films
Amperom. (E = −0.1 V) H2O2 Slope of the calibrationcurve of the{PAMAM-Au/Mb}n filmlarger than that of the{PAMAM/Mb}n films
[89]
Mb/plane pyrolyticgraphite
The oppositely charged myoglobin and different sizedgold nanoparticles were assembled into {Au/Mb}nlayer-by-layer films by electrostatic interaction
Amperom. (E = −0.1 V) H2O2 [90]
Cyt c/screen-printedrhodium graphiteelectrodes
Cytochrome P450scc and nAu immobilized onrhodium–graphite electrode by deposition
Amperom. (E = −400 mV) Cholesterol Linear range: 10–70 �M;slope: 0–13 �A �M−1
[91]
Cyt c/CPE Cytochrome c immobilized on colloidal gold CV 0.1 M PBS; pH 7 H2O2 Linear range: 10 �M to1 mM
[92]
Living pancreaticadenocarcinomacells/composite CPE
Physical inclusion of nAu onto CP; immobilizationliving pancreatic adenocarcinoma cells derived fromascites
CV Decreasing of irreversiblevoltammetric response ofguanine in presence ofadriamycin
[93]
Leukemia K562 cells/GCE Leukemia K562 cells immobilized in a microporouscellulose membrane modified with nAu
CV Decreasing of irreversiblevoltammetric response inpresence of methotrexate
[94]
Abbreviations: CPE: carbon paste electrode, CV, cyclic voltammetry; Cyt c, cytochrome c; Hb, hemoglobine; Mb, mioglobine; nAu: gold nanoparticle, PBS: phosphate buffer solution

himic
J.M. Pingarron et al. / Electrocloidal gold-modified carbon paste electrode was also profited forthe detection of hydrogen peroxide [92].
Electrochemical methods have been used in studying intactliving cells, i.e. by impedance monitoring or voltammetric mea-surements. The electrochemical signals from cells attached to theelectrode have been used to test the effect of drugs and other exoge-nous factors on cell viability. Gold nanoparticles have demonstratedto be efficient to immobilize cells by means of mercapto or primaryamine groups on the cell membrane as well as for preserving theactivity of immobilized living entities and preventing their leak-age from the electrode surface. For example, a composite electrodebased on colloidal gold nanoparticles and carbon paste has beenused to immobilize living pancreatic adenocarcinoma cells derivedfrom ascites and investigate the cytotoxic effect of the anti-tumordrug adriamycin. The method is based on the measurement of anirreversible voltammetric response from cells related to the oxida-tion of guanine, whose peak current decreases in the presence of thedrug [93]. Leukemia K562 cells were immobilized in a microporouscellulose membrane modified with colloidal gold nanoparticles.The electrochemical response from cells was used to evaluate theeffectiveness of anti-tumor drug methotrexate [94].
6. Conclusions
The intensive work made recently on the development ofgold nanopaticle-based biosensors shows clearly the potentialitiesand advantageous features of this approach to construct biosen-sors exhibiting enhanced performances with respect to previousdesigns. The unique properties of gold nanoparticles concerningimmobilization of biomolecules retaining their biological activityand as efficient conducting interfaces with electrocatalytic abilitymakes them a powerful tool to modify electrode materials and toconstruct robust and sensitive biosensors which can find applica-tion in many fields of interest.
Acknowledgement
Financial support from the Spanish Ministerio de Educacion yCiencia (Projects CTQ2006-02905 and CTQ2006-02743) is grate-fully acknowledged.
References
[1] E. Katz, I. Willner, J. Wang, Electroanalysis 16 (2004) 19.[2] P. Yanez-Sedeno, J.M. Pingarron, Anal. Bioanal. Chem. 382 (2005) 884.[3] S. Liu, D. Leech, H. Ju, Anal. Lett. 36 (2003) 1.[4] M.H. Rashid, R.R. Bhattacharjee, A. Kota, T.K. Mandal, Langmuir 22 (2006) 7141.[5] S. Guo, E. Wang, Anal. Chim. Acta 598 (2007) 181.[6] X. Luo, A. Morrin, A.J. Killard, Snyth. MR 18 (2006) 319.[7] C. Carralero, M.L. Mena, A. Gonzalez-Cortes, P. Yanez-Sedeno, J.M. Pingarron,
Anal. Chim. Acta 528 (2005) 1.[8] S. Zhang, N. Wang, H. Yu, Y. Niu, C. Sun, Bioelectrochemistry 67 (2005) 15.[9] L. Aguı, J. Manso, P. Yanez-Sedeno, J.M. Pingarron, Sens. Actuators B Chem. 113
(2006) 272.[10] O. Shulga, J.R. Kirchhoff, Electrochem. Commun. 9 (2007) 935.[11] J.J. Gooding, D.B. Hibbert, Trac-Trend Anal. Chem. 18 (1999) 525.[12] M.L. Mena, P. Yanez-Sedeno, J.M. Pingarron, Anal. Biochem. 336 (2005) 20.[13] W. Yang, J. Wang, S. Zhao, Y. Sun, C. Sun, Electrochem. Commun. 8 (2006) 665.[14] X. Yi, J. Huang-Xian, C. Hong-Yuan, Anal. Biochem. 278 (2000) 22.[15] J. Lin, W. Qu, S. Zhang, Anal. Biochem. 360 (2007) 288.[16] S. Xu, B. Peng, X. Han, Biosens. Bioelectron. 22 (2007) 1807.[17] S.Q. Liu, H.X. Ju, Biosens. Bioelectron. 19 (2003) 177.[18] S.Q. Liu, H.X. Ju, Anal. Biochem. 307 (2002) 110.[19] S. Liu, J. Yu, H. Ju, J. Electroanal. Chem. 540 (2003) 61.[20] M. Cubukcu, S. Timur, U. Anik, Talanta 74 (2007) 434.[21] C. Carralero, M.L. Mena, A. Gonzalez-Cortes, P. Yanez-Sedeno, J.M. Pingarron,
Biosens. Bioelectron. 22 (2006) 730.[22] F.N. Crespilho, M.E. Ghica, M. Florescu, F.C. Nart, O.N. Oliveira Jr., C.M.A. Brett,
Electrochem. Commun. 8 (2006) 1665.[23] Y. Sun, Y. Bai, W. Yang, C. Sun, Electrochim. Acta 52 (2007) 7352.[24] E. Katz, I. Willner, Angew. Chem. Int. Ed. 43 (2004) 6042.
a Acta 53 (2008) 5848–5866 5865
[25] J. Manso, M.L. Mena, P. Yanez-Sedeno, J.M. Pingarron, J. Electroanal. Chem. 603(2007) 1.
[26] L. Jiang, L. Gao, Carbon 41 (2003) 2923.[27] Y. Liug, M. Wang, F. Zhao, Z. Guo, H. Chen, S. Dong, J. Electroanal. Chem. 581
(2005) 1.[28] H. Feng, H. Wang, Y. Zhang, B. Yan, G. Shen, R. Yu, Anal. Sci. 23 (2007) 235.[29] J. Njagi, S. Andreescu, Biosens. Bioelectron. 23 (2007) 168.[30] Y. Xian, Y. Hu, F. Liu, Y. Xian, H. Wang, L. Jin, Biosens. Bioelectron. 21 (2003)
1996.[31] F. Gao, R. Yuan, Y. Chai, S. Chen, S. Cao, T. Tang, J. Biochem. Biopharm. Meth.
70 (2007) 407.[32] X.L. Luo, J.J. Xu, Y. Du, H.Y. Chen, Anal. Biochem. 334 (2004) 284.[33] X.L. Luo, J.J. Xu, Q. Zhang, G.J. Yang, H.Y. Chen, Biosens. Bioelectron. 21 (2005)
190.[34] B.Y. Wu, S.H. Hou, F. Yin, J. Li, Z.X. Zhao, Biosens. Bioelectron. 22 (2007) 838.[35] J. Lin, L. Zhang, S. Zhang, Anal. Biochem. 370 (2007) 180.[36] T. Tangkuaram, C. Ponchio, T. Kangkasomboon, P. Katikawong, W. Veerasai,
Biosens. Bioelectron. 22 (2007) 2071.[37] D. Du, S. Chen, J. Cai, A. Zhang, Talanta 74 (2008) 766.[38] J. Jia, B. Wang, A. Wu, G. Cheng, Z. Li, S. Dong, Anal. Chem. 74 (2002) 2217.[39] B.K. Jena, C.R. Raj, Anal. Chem. 78 (2006) 6332.[40] M. Wang, L. Wang, G. Wang, X. Ji, Y. Bai, T. Li, S. Gong, J. Li, Biosens. Bioelectron.
19 (2004) 575.[41] D. Tang, R. Yuan, Y. Chai, J. Dai, X. Zhong, Y. Liu, Bioelectrochemistry 65 (2004)
15.[42] D. Tang, T.R. Yuan, Y. Chai, Y. Fu, J. Dai, Y. Liu, X. Zhong, Biosens. Bioelectron.
21 (2005) 539.[43] D. Tang, R. Yuan, Y. Chai, X. Zhong, Y. Liu, J. Dai, Clin. Biochem. 39 (2006) 309.[44] D. Tang, R. Yuan, Y.Q. Chai, X. Zhong, Y. Liu, J Dai, L.Y. Zhang, Anal. Biochem.
333 (2004) 345.[45] Y. Zhuo, R. Yuan, Y. Chai, Y. Zhang, N. Wang, X. Li, Q. Zhu, N. Wang, Anal. Chim.
Acta 548 (2005) 205.[46] D. Tang, R. Yuan, Y. Chai, L. Zhang, X. Zhong, Y. Liu, J. Dai, Sens. Actuators B
Chem. 104 (2005) 199.[47] D. Tang, J.J. Ren, Electroanalysis 17 (2005) 2208.[48] Y. Zhuo, R. Yuan, Y. Chai, D. Tang, Y. Zhang, N. Wang, W. Li, Q. Zhu, Electrochem.
Commun. 7 (2005) 355.[49] Y. Zhuo, R. Yuan, Y. Chai, Y. Zhang, X. Li, N. Wang, O. Zhu, Sens. Actuators B
Chem. 114 (2006) 631.[50] Y.Y. Xu, C. Bian, S. Chen, S. Xia, Anal. Chim. Acta 561 (2006) 48.[51] Z.S. Wu, J.S. Li, M.H. Luo, G.L. Shen, R.Q. Yu, Anal. Chim. Acta 528 (2005) 235.[52] X. Mao, J. Jiang, Y. Huang, G. Shen, R. Yu, Sens. Actuators B Chem. 123 (2007)
198.[53] Z. Chen, Z. Peng, P. Zhang, X. Jin, J. Jiang, X. Zhang, G. Shen, R. Yu, Talanta 72
(2007) 1800.[54] H. Chen, J.-H. Jiang, Y. Huang, T. Deng, J.S. Li, G. Li, R.Q. Yu, Sens. Actuators B
Chem. 117 (2006) 211.[55] X. Chu, X. Fu, K. Chen, G.L. Shen, R.Q. Yu, Biosens. Bioelectron. 20 (2005) 1805.[56] Y. Liu, Z. Qin, X. Wu, H. Jiang, Biochem. Eng. J. 32 (2006) 211.[57] D. Tang, R. Yuan, Y. Chai, Anal. Chim. Acta 654 (2006) 158.[58] L. Wu, J. Chen, D. Du, H. Ju, Electrochim. Acta 51 (2006) 1208.[59] J. Chen, J. Tang, F. Yan, H Ju, Biomaterials 27 (2006) 2313.[60] H. Huang, P. Ran, Z. Liu, Bioelectrochemistry 70 (2007) 257.[61] D. Dan, X. Xiaoxing, W. Shengfu, Z. Aidong, Talanta 71 (2007) 1257.[62] X.H. Fu, Electroanalysis 19 (2007) 1831.[63] C.X. Lei, F.C. Gong, G.L. Shen, R.Q. Yu, Sens. Actuators B Chem. 96 (2003) 582.[64] X. Chu, Z.F. Xiang, X. Fu, S.P. Wang, G.L. Shen, R.Q. Yu, J. Immunol. Methods 301
(2005) 77.[65] X. Li, R. Yuan, Y. Chai, L. Zhang, Y. Zhuo, Y. Zhang, J. Biotechnol. 123 (2006) 356.[66] H.Z. An, R.T. Yuan, D.P. Tang, Y. Chai, N. Li, Electroanalysis 19 (2007) 479.[67] Y. Liu, Thin Solid Film 516 (2008) 1803.[68] K.J. Odenthal, J.J. Gooding, Analyst 132 (2007) 603.[69] H. Cai, C. Xu, P. He, Y. Fang, J. Electroanal. Chem. 510 (2001) 78.[70] M. Wang, C. Sun, L. Wang, X. Ji, Y. Bai, T. Li, J. Li, J. Pharm. Biomed. 33 (2003)
1117.[71] J. Kang, X. Li, G. Wu, Z. Wang, X. Lu, Anal. Biochem. 364 (2007) 165.[72] J. Wang, D. Xu, A.N. Kawde, R. Polsky, Anal. Chem. 73 (2001) 5576.[73] H. Cai, Y. Wang, P. He, Y. Fang, Anal. Chim. Acta 469 (2002) 165.[74] H. Cai, C. Shang, I.M. Hsing, Anal. Chim. Acta 523 (2004) 61.[75] K. Kerman, Y. Morita, Y. Takamura, M. Ozsoz, E. Tamiya, Anal. Chim. Acta 510
(2004) 169.[76] J. Yang, T. Yang, Y. Feng, K. Jiao, Anal. Biochem. 365 (2007) 24.[77] M. Ozsoz, A. Erdem, K Kerman, D. Ozkan, B. Tugrul, N. Topcuoglu, H. Ekren, M.
Taylan, Anal. Chem. 75 (2003) 2181.[78] K. Kerman, M. Saito, Y. Morita, Y. Takamura, M. Ozsoz, E. Tamiya, Anal. Chem.
76 (2004) 1877.[79] J. Xu, J.J. Zhu, Y. Zhu, K. Gu, H.Y. Chen, Anal. Lett. 34 (2001) 503.[80] C.Z. Li, Y. Liu, J.H.T. Luong, Anal. Chem. 77 (2005) 478.[81] B. Willner, E. Katz, I. Willner, Curr. Opin. Biotechnol. 17 (2006) 589.[82] I. Willner, B. Willner, E. Katz, Bioelectrochemistry 70 (2007) 2.[83] Y. Xiao, F. Patolsky, E. Katz, J.F. Hainfeld, I. Willner, Science 299 (2003) 1877.[84] M. Zayats, E. Katz, R. Baron, I. Willner, J. Am. Chem. Soc. 127 (2005) 12400.[85] L. Zhang, X. Jiang, E. Wang, S. Dong, Biosens. Bioelectron. 21 (2005) 337.[86] B. Fang, G. Wang, X. Yang, Q. Zha, W. Zhang, X. Kan, Anal. Lett. 37 (2004)
2911.

5866 J.M. Pingarron et al. / Electrochimic
[87] S. Chen, R. Yuan, Y. Chai, L. Zhang, N. Wang, X. Li, Biosens. Bioelectron. 22(2007) 1268.
[88] W. Yang, Y. Li, Y. Bai, C. Sun, Sens. Actuators B Chem. 115 (2006) 42.[89] H. Zhang, N. Hu, J. Phys. Chem. 111 (2007) 10583.[90] H. Zhang, H. Lu, N. Hu, J. Phys. Chem. 110 (2006) 2171.[91] V.V. Shumyantseva, S. Carrara, V. Bavastrello, D.J. Riley, T.V. Bulkoa, K.G.
Skryabinf, A.I. Archakova, C. Nicolinic, Biosens. Bioelectron. 21 (2005) 217.[92] H.X. Ju, S. Liu, B. Ge, F. Lisdat, F.W. Seller, Electroanalysis 14 (2002) 141.[93] D. Du, S. Liu, J. Chen, H. Ju, H. Lian, J. Li, Biomaterials 26 (2005) 6487.[94] F. Yan, J. Chen, H. Ju, Electrochem. Commun. 9 (2007) 293.[95] Y. Liu, S. Wu, H. Ju, L. Xu, Electroanalysis 19 (2007) 986.[96] S. Zhang, W. Yang, Y. Niu, Y. Li, M. Zhang, C. Sun, Anal. Bioanal. Chem. 384
(2006) 736.[97] F.H. Zhang, S.S. Cho, S.H. Yang, S.S. Seo, G.S. Cha, H. Nam, Electroanalysis 18
(2006) 217.[98] M.H. Xue, Q. Xu, M. Zhou, J.J. Zhu, Electrochem. Commun. 8 (2006) 1468.[99] S. Zhang, N. Wang, Y. Niu, C. Sun, Sens. Actuators B Chem. 109 (2005) 367.
[100] Q. Zhu, R. Yuan, Y. Chai, Y. Zhuo, Y. Zhang, X. Li, N. Wang, Anal. Lett. 39 (2006)483.
[101] Y. Xiao, H.X. Ju, H.Y. Chen, Anal. Chim. Acta 391 (1999) 73.[102] S. Chen, R. Yuan, Y. Chai, L. Xu, N.L. Wang, X.L. Zhang, Electroanalysis 18 (2006)
471.[103] L. Wang, E. Wang, Electrochem. Commun. 6 (2004) 225.[104] X. Li, J. Wu, N. Gao, G. Shen, R. Yu, Sens. Actuators B Chem. 117 (2006) 35.[105] Y. Liu, L. Nie, W. Tao, S. Yao, Electroanalysis 16 (2004) 1271.
a Acta 53 (2008) 5848–5866
[106] Y. Liu, F. Yin, Y. Long, Z. Zhang, S. Yao, J. Colloids Interf. Sci. 258 (2003) 75.[107] D. Tang, R. Yua, Y. Chai, X. Zhong, Y. Liu, J. Dai, Biochem. Eng. J. 22 (2004)
43.[108] Y. Fu, R. Yuan, D. Tang, Y. Chai, L. Xu, Colloid Surf. B 40 (2005) 61.[109] D. Tang, R. Yuan, Y. Chai, Bionsens Bioelectron (2005), doi:10.1016/
j.bios.2005.10.026.[110] Z. Chen, J. Jiang, G. Shen, R. Yu, Anal. Chim. Acta 553 (2005) 190.[111] V. Carralero, A. Gonzalez-Cortes, P. Yanez-Sedeno, J.M. Pingarron, Anal. Lett.
41 (2008) 244.[112] K.Z. Liang, W.J. Mu, M.Y. Huang, Y. Zhongxue, L. Qingke, Electroanalysis 18
(2006) 1505.[113] R. Yuan, L. Zhang, Q. Li, Y. Chai, S. Cao, Anal. Chim. Acta 531 (2005) 1.[114] R. Polsky, J.C. Harper, D.R. Wheeler, S.M. Dirk, D.C. Arango, S.M. Brozik, Biosens.
Bioelectron. 23 (2008) 757.[115] D. Tang, R. Yuan, Y. Chai, Biosens. Bioelectron. 22 (2007) 1116.[116] J.H. Kim, J.H. Cho, G.S. Cha, C.W. Lee, H.B. Kim, S.H. Paek, Biosens. Bioelectron.
14 (2000) 907.[117] V. Carralero, A. Gonzalez-Cortes, P. Yanez-Sedeno, J.M. Pingarron, Electroanal-
ysis 19 (2007) 853.[118] V. Carralero, A. Gonzalez-Cortes, P. Yanez-Sedeno, J.M. Pingarron, Anal. Chim.
Acta 596 (2007) 86.[119] Y. Fu, R. Yuan, L. Xu, Y. Chai, X. Zhong, D. Tang, Biochem. Eng. J. 23 (2005) 37.[120] S.F. Liu, Y.F. Li, J.R. Li, Biosens. Bioelectron. 21 (2005) 789.[121] L.P. Lu, S.Q. Wang, X.Q. Lin, Anal. Chim. Acta 519 (2004) 161.




















