
Electrochemical detection of harmful algae and other microbial contaminants in coastal waters using...
14
Electrochemical detection of harmful algae and other microbial contaminants in coastal waters using hand-held biosensors Michael J. LaGier a, * , Jack W. Fell b , Kelly D. Goodwin c a Cooperative Institute of Marine and Atmospheric Studies, Rosenstiel School of Marine and Atmospheric Science, University of Miami, 4600 Rickenbacker Causeway, Miami, FL 33149, USA b Marine Biology and Fisheries, Rosenstiel School of Marine and Atmospheric Science, University of Miami, 4600 Rickenbacker Causeway, Miami, FL 33149, USA c National Oceanographic and Atmospheric Administration, Atlantic Oceanographic and Meteorological Laboratories, Ocean Chemistry Division, 4301 Rickenbacker Causeway, Miami, FL 33149, USA Abstract Standard methods to identify microbial contaminants in the environment are slow, laborious, and can require specialized expertise. This study investigated electrochemical detection of microbial contaminants using commercially available, hand-held instruments. Elec- trochemical assays were developed for a red tide dinoflagellate (Karenia brevis), fecal-indicating bacteria (Enterococcus spp.), markers indicative of human sources of fecal pollution (human cluster Bacteroides and the esp gene of Enterococcus faecium), bacterial pathogens (Escherichia coli 0157:H7, Salmonella spp., Campylobacter jejuni, Staphylococcus aureus), and a viral pathogen (adenovirus). For K. bre- vis, two assay formats (Rapid PCR-Detect and Hybrid PCR-Detect) were tested and both provided detection limits of 10 genome equiv- alents for DNA isolated from K. brevis culture and amplified by PCR. Sensitivity with coastal water samples was sufficient to detect K. brevis that was ‘‘present’’ (61000 cells/l) without yielding false positive results and the electrochemical signal was significantly different than for samples containing cells at ‘‘medium’’ concentrations (100,000 to < 10 6 cells/l). Detection of K. brevis RNA was also shown. Multi-target capability was demonstrated with an 8-plex assay for bacterial and viral targets using isolated DNA, natural beach water spiked with human feces, and water and sediments collected from New Orleans, Louisiana following Hurricane Katrina. Furthermore, direct detection of dinoflagellate and bacterial DNA was achieved using lysed cells rather than extracted nucleic acids, allowing stream- lining of the process. The methods presented can be used to rapidly (3–5 h) screen environmental water samples for the presence of microbial contaminants and have the potential to be integrated into semi-automated detection platforms. Ó 2007 Elsevier Ltd. All rights reserved. Keywords: Electrochemical biosensor; Karenia brevis; Pathogen detection; Recreational water quality; Source tracking 1. Introduction Toxic algal blooms and microbial contaminants impact coastal water quality. As the nation’s coastal areas become more urbanized, poor water quality has increased eco- nomic, health, and environmental impacts. Harmful algal blooms (HABs) alone cost the United States 50 million dol- lars per year (Haugland et al., 2005), and sewage polluted waters adversely affect human health and the economy (Dwight et al., 2005; Leclerc et al., 2002). Environmental managers need rapid and accurate assessments of microbial water quality to restrict human access to contaminated waters and products. Monitoring techniques for HABs that rely on microscopy are time-consuming, labor intensive, error prone (Culverhouse et al., 2003) and require a signif- icant amount of taxonomic expertise (Millie et al., 1997). Standard culture techniques for bacterial indicators of fecal pollution are slow and do not return information on human pathogens or source tracking markers (Bower et al., 2005; Griffin et al., 2001; Scott et al., 2005). As a result, manage- ment decisions, ecological study and assessment of control 0025-326X/$ - see front matter Ó 2007 Elsevier Ltd. All rights reserved. doi:10.1016/j.marpolbul.2006.12.017 * Corresponding author. Tel.: +1 305 361 4316; fax: +1 305 361 4447. E-mail address: [email protected] (M.J. LaGier). www.elsevier.com/locate/marpolbul Marine Pollution Bulletin 54 (2007) 757–770
Transcript of Electrochemical detection of harmful algae and other microbial contaminants in coastal waters using...

www.elsevier.com/locate/marpolbul
Marine Pollution Bulletin 54 (2007) 757–770
Electrochemical detection of harmful algae and othermicrobial contaminants in coastal waters using hand-held biosensors
Michael J. LaGier a,*, Jack W. Fell b, Kelly D. Goodwin c
a Cooperative Institute of Marine and Atmospheric Studies, Rosenstiel School of Marine and Atmospheric Science, University of Miami,
4600 Rickenbacker Causeway, Miami, FL 33149, USAb Marine Biology and Fisheries, Rosenstiel School of Marine and Atmospheric Science, University of Miami, 4600 Rickenbacker Causeway,
Miami, FL 33149, USAc National Oceanographic and Atmospheric Administration, Atlantic Oceanographic and Meteorological Laboratories, Ocean Chemistry Division,
4301 Rickenbacker Causeway, Miami, FL 33149, USA
Abstract
Standard methods to identify microbial contaminants in the environment are slow, laborious, and can require specialized expertise.This study investigated electrochemical detection of microbial contaminants using commercially available, hand-held instruments. Elec-trochemical assays were developed for a red tide dinoflagellate (Karenia brevis), fecal-indicating bacteria (Enterococcus spp.), markersindicative of human sources of fecal pollution (human cluster Bacteroides and the esp gene of Enterococcus faecium), bacterial pathogens(Escherichia coli 0157:H7, Salmonella spp., Campylobacter jejuni, Staphylococcus aureus), and a viral pathogen (adenovirus). For K. bre-
vis, two assay formats (Rapid PCR-Detect and Hybrid PCR-Detect) were tested and both provided detection limits of 10 genome equiv-alents for DNA isolated from K. brevis culture and amplified by PCR. Sensitivity with coastal water samples was sufficient to detectK. brevis that was ‘‘present’’ (61000 cells/l) without yielding false positive results and the electrochemical signal was significantly differentthan for samples containing cells at ‘‘medium’’ concentrations (100,000 to < 106 cells/l). Detection of K. brevis RNA was also shown.Multi-target capability was demonstrated with an 8-plex assay for bacterial and viral targets using isolated DNA, natural beach waterspiked with human feces, and water and sediments collected from New Orleans, Louisiana following Hurricane Katrina. Furthermore,direct detection of dinoflagellate and bacterial DNA was achieved using lysed cells rather than extracted nucleic acids, allowing stream-lining of the process. The methods presented can be used to rapidly (3–5 h) screen environmental water samples for the presence ofmicrobial contaminants and have the potential to be integrated into semi-automated detection platforms.� 2007 Elsevier Ltd. All rights reserved.
Keywords: Electrochemical biosensor; Karenia brevis; Pathogen detection; Recreational water quality; Source tracking
1. Introduction
Toxic algal blooms and microbial contaminants impactcoastal water quality. As the nation’s coastal areas becomemore urbanized, poor water quality has increased eco-nomic, health, and environmental impacts. Harmful algalblooms (HABs) alone cost the United States 50 million dol-lars per year (Haugland et al., 2005), and sewage pollutedwaters adversely affect human health and the economy
0025-326X/$ - see front matter � 2007 Elsevier Ltd. All rights reserved.
doi:10.1016/j.marpolbul.2006.12.017
* Corresponding author. Tel.: +1 305 361 4316; fax: +1 305 361 4447.E-mail address: [email protected] (M.J. LaGier).
(Dwight et al., 2005; Leclerc et al., 2002). Environmentalmanagers need rapid and accurate assessments of microbialwater quality to restrict human access to contaminatedwaters and products. Monitoring techniques for HABs thatrely on microscopy are time-consuming, labor intensive,error prone (Culverhouse et al., 2003) and require a signif-icant amount of taxonomic expertise (Millie et al., 1997).Standard culture techniques for bacterial indicators of fecalpollution are slow and do not return information on humanpathogens or source tracking markers (Bower et al., 2005;Griffin et al., 2001; Scott et al., 2005). As a result, manage-ment decisions, ecological study and assessment of control

758 M.J. LaGier et al. / Marine Pollution Bulletin 54 (2007) 757–770
measures are difficult. Biosensors could improve monitor-ing by providing a means to achieve rapid, on-site identifi-cation of microbial contaminants. A robust sensor designis sought, in particular, a sensor that can simultaneouslydetect multiple targets such as harmful algae, fecal indica-tors, human pathogens, and source tracking markers.
The electrochemical detection of nucleic acids is quali-fied to meet the size, cost, and power requirements ofon-site water quality testing (Kerman et al., 2004; Wang,2000). The potential for electrochemical methods to beused as a tool for environmental monitoring depends, inpart, on the availability of reproducible biosensors. Wojcie-chowski and colleagues (Wojciechowski et al., 1999)described electrochemical biosensors that enable sensitivedetection of DNA or RNA after PCR or Reverse Trans-criptase polymerase chain reaction (Attatippaholkunet al., 2003), respectively. The system, marketed by Ald-eron Biosciences, Inc. (www.alderonbiosciences.com), usesa hand-held, battery-powered, electrochemical monitorand disposable carbon sensors. One method of detectionused with this system, termed ‘‘Rapid PCR-Detect,’’ iden-tifies and quantifies PCR products through biotin and fluo-rescein labeling during PCR. A second method, ‘‘HybridPCR-Detect,’’ provides added specificity by includinghybridization to a DNA probe.
Numerous approaches for detecting microbial nucleicacids electrochemically have been formulated and tested(Drummond et al., 2003; Kerman et al., 2004; LaGieret al., 2005; Metfies et al., 2005; Zhang et al., 2003). Previ-ous studies using the Alderon system detected single-basemutations in a human gene (Wojciechowski et al., 1999),enterotoxin genes from cultures of the bacterium Staphylo-
coccus aureus (Aitichou et al., 2004), and DNA isolatedfrom algal cultures (Pfiesteria piscicida and Cryptoperidi-
niopsoid spp.) (Litaker et al., 2001). All of these studiesused single-plex reactions to detect nucleic acids isolatedfrom pure culture. Environmental applications requiredetection in natural samples, with a preference for multi-target detection. Therefore, the use of electrochemical bio-sensors for environmental microbiology applicationsneeded further testing.
The feasibility of using electrochemical biosensors todetect microbes in environmental samples was tested usingKarenia brevis as a model. K. brevis is the causative agent ofrecurring red tide blooms in the Gulf of Mexico and occa-sional blooms off the southeastern coast of the UnitedStates (Steidinger et al., 1998; Tester and Steidinger,1997). The lipophilic toxin produced by K. brevis, breve-toxin, can result in massive fish and marine mammal kills(Landsberg, 2002). Aerosolized brevetoxin toxin can causerespiratory distress (Kirkpatrick et al., 2004) when levels ofK. brevisexceed 1000 cells/l The harvesting of shellfish isprohibited when cell concentrations reach 5000 K. brevis
cells per liter (http://www.floridaaquaculture.com).The potential to develop multi-target electrochemical
detection was tested here using an 8-plex assay of microbesimportant to microbial water quality monitoring that
included fecal-indicating bacteria (Enterococcus spp.), bac-terial markers of human fecal pollution (the human-specificHF8 cluster of Bacteroides and the esp gene of Enter-
ococcus faecium), bacterial pathogens (Escherichia coli
0157:H7, Salmonella spp., Campylobacter jejuni and S. aur-
eus), and a viral pathogen (human adenovirus). To addressshortfalls associated with the standard culture methodsused to monitor recreational water quality, the multi-targetassay was designed to return an indication of fecal contam-ination along with information on whether the contamina-tion was from a human source and if selected humanpathogens are present (Griffin et al., 2001).
2. Materials and methods
2.1. Sources of DNA and environmental samples
Algae used in this study were obtained from the Prova-soli-Guillard National Center for Culture of Marine Phyto-plankton (CCMP) and maintained by the Toxic AlgalCulture Core of the Oceans and Human Health Center atthe University of Miami, unless otherwise noted (Table 1).Cultures were grown at 22 �C in a light:dark cycle asdescribed by Brand (1990). Cultures were pelleted or fil-tered onto 5 lm, 47 mm mixed esters of cellulose (MEC)membrane filters (Millipore Corp., Bedford, MA) andDNA was extracted using the FastDNA SPIN Kit forSoil (Qbiogene, Irvine, CA). The quality and amount ofextracted DNA was assessed by spectroscopy (Ausubelet al., 1999). Genome equivalents (cell numbers) were esti-mated from DNA concentrations based on data indicatingthat a K. brevis cell contains a single DNA genome weighingapproximately 100 pg (Lidie et al., 2005; Rizzo et al., 1982).
DNA from E. coli (ATCC #25922) and E. faecalis
(ATCC #29212) was isolated from cultures using theFastDNA SPIN Kit for Soil (Qbiogene, Irvine, CA).DNA from S. aureus was obtained from the ATCC(#700699D). DNA from E. coli 0157:H7, C. jejuni, andS. typhi was obtained from the New York State Depart-ment of Health courtesy of Dr. Nick Cirino. AdenovirusDNA consisted of a plasmid containing the PCR target(He and Jiang, 2005) and was provided by Dr. Sunny Jiangof the University of California at Irvine. A plasmid con-taining cloned DNA from the human-specific esp gene ofE. faecium (Scott et al., 2005) was provided by Dr. JoanRose of Michigan State University. The human-specificHF8 cluster of Bacteroides (Bernhard and Field, 2000)was PCR amplified from human fecal DNA and clonedinto the pCR2.1-TOPO vector (Invitrogen, Carlsbad, CA).
Coastal water samples for analysis of the toxic dinofla-gellate K. brevis were obtained from the Rookery BayNational Estuarine Research Reserve (NERR) in Naples,Florida from the sites Caxambas Pass, Goodland, Hender-son Creek, Marco Pass, and the 951 Launch Ramp asdescribed in Goodwin et al. (Goodwin et al., 2005). In sum-mary, paired water samples were collected in sterile whirl-packs and shipped overnight to the Florida Fish and
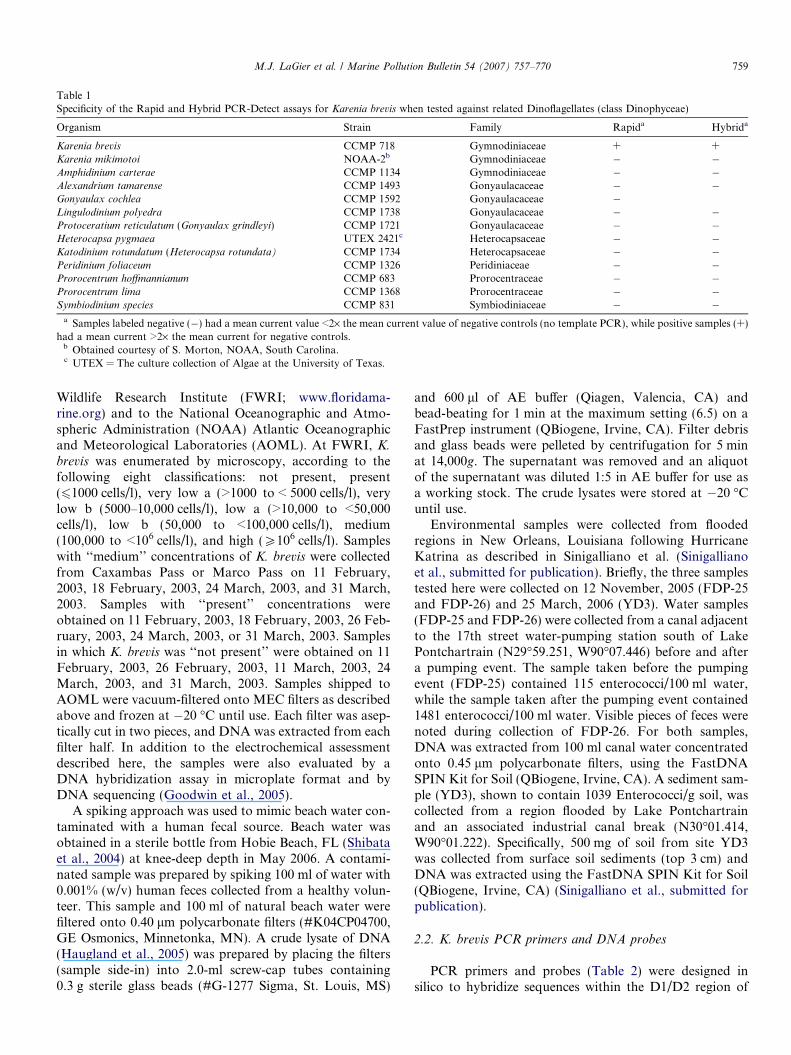
Table 1Specificity of the Rapid and Hybrid PCR-Detect assays for Karenia brevis when tested against related Dinoflagellates (class Dinophyceae)
Organism Strain Family Rapida Hybrida
Karenia brevis CCMP 718 Gymnodiniaceae + +Karenia mikimotoi NOAA-2b Gymnodiniaceae � �Amphidinium carterae CCMP 1134 Gymnodiniaceae � �Alexandrium tamarense CCMP 1493 Gonyaulacaceae � �Gonyaulax cochlea CCMP 1592 Gonyaulacaceae �Lingulodinium polyedra CCMP 1738 Gonyaulacaceae � �Protoceratium reticulatum (Gonyaulax grindleyi) CCMP 1721 Gonyaulacaceae � �Heterocapsa pygmaea UTEX 2421c Heterocapsaceae � �Katodinium rotundatum (Heterocapsa rotundata) CCMP 1734 Heterocapsaceae � �Peridinium foliaceum CCMP 1326 Peridiniaceae � �Prorocentrum hoffmannianum CCMP 683 Prorocentraceae � �Prorocentrum lima CCMP 1368 Prorocentraceae � �Symbiodinium species CCMP 831 Symbiodiniaceae � �
a Samples labeled negative (�) had a mean current value <2· the mean current value of negative controls (no template PCR), while positive samples (+)had a mean current >2· the mean current for negative controls.
b Obtained courtesy of S. Morton, NOAA, South Carolina.c UTEX = The culture collection of Algae at the University of Texas.
M.J. LaGier et al. / Marine Pollution Bulletin 54 (2007) 757–770 759
Wildlife Research Institute (FWRI; www.floridama-rine.org) and to the National Oceanographic and Atmo-spheric Administration (NOAA) Atlantic Oceanographicand Meteorological Laboratories (AOML). At FWRI, K.
brevis was enumerated by microscopy, according to thefollowing eight classifications: not present, present(61000 cells/l), very low a (>1000 to < 5000 cells/l), verylow b (5000–10,000 cells/l), low a (>10,000 to <50,000cells/l), low b (50,000 to <100,000 cells/l), medium(100,000 to <106 cells/l), and high (P106 cells/l). Sampleswith ‘‘medium’’ concentrations of K. brevis were collectedfrom Caxambas Pass or Marco Pass on 11 February,2003, 18 February, 2003, 24 March, 2003, and 31 March,2003. Samples with ‘‘present’’ concentrations wereobtained on 11 February, 2003, 18 February, 2003, 26 Feb-ruary, 2003, 24 March, 2003, or 31 March, 2003. Samplesin which K. brevis was ‘‘not present’’ were obtained on 11February, 2003, 26 February, 2003, 11 March, 2003, 24March, 2003, and 31 March, 2003. Samples shipped toAOML were vacuum-filtered onto MEC filters as describedabove and frozen at �20 �C until use. Each filter was asep-tically cut in two pieces, and DNA was extracted from eachfilter half. In addition to the electrochemical assessmentdescribed here, the samples were also evaluated by aDNA hybridization assay in microplate format and byDNA sequencing (Goodwin et al., 2005).
A spiking approach was used to mimic beach water con-taminated with a human fecal source. Beach water wasobtained in a sterile bottle from Hobie Beach, FL (Shibataet al., 2004) at knee-deep depth in May 2006. A contami-nated sample was prepared by spiking 100 ml of water with0.001% (w/v) human feces collected from a healthy volun-teer. This sample and 100 ml of natural beach water werefiltered onto 0.40 lm polycarbonate filters (#K04CP04700,GE Osmonics, Minnetonka, MN). A crude lysate of DNA(Haugland et al., 2005) was prepared by placing the filters(sample side-in) into 2.0-ml screw-cap tubes containing0.3 g sterile glass beads (#G-1277 Sigma, St. Louis, MS)
and 600 ll of AE buffer (Qiagen, Valencia, CA) andbead-beating for 1 min at the maximum setting (6.5) on aFastPrep instrument (QBiogene, Irvine, CA). Filter debrisand glass beads were pelleted by centrifugation for 5 minat 14,000g. The supernatant was removed and an aliquotof the supernatant was diluted 1:5 in AE buffer for use asa working stock. The crude lysates were stored at �20 �Cuntil use.
Environmental samples were collected from floodedregions in New Orleans, Louisiana following HurricaneKatrina as described in Sinigalliano et al. (Sinigallianoet al., submitted for publication). Briefly, the three samplestested here were collected on 12 November, 2005 (FDP-25and FDP-26) and 25 March, 2006 (YD3). Water samples(FDP-25 and FDP-26) were collected from a canal adjacentto the 17th street water-pumping station south of LakePontchartrain (N29�59.251, W90�07.446) before and aftera pumping event. The sample taken before the pumpingevent (FDP-25) contained 115 enterococci/100 ml water,while the sample taken after the pumping event contained1481 enterococci/100 ml water. Visible pieces of feces werenoted during collection of FDP-26. For both samples,DNA was extracted from 100 ml canal water concentratedonto 0.45 lm polycarbonate filters, using the FastDNASPIN Kit for Soil (QBiogene, Irvine, CA). A sediment sam-ple (YD3), shown to contain 1039 Enterococci/g soil, wascollected from a region flooded by Lake Pontchartrainand an associated industrial canal break (N30�01.414,W90�01.222). Specifically, 500 mg of soil from site YD3was collected from surface soil sediments (top 3 cm) andDNA was extracted using the FastDNA SPIN Kit for Soil(QBiogene, Irvine, CA) (Sinigalliano et al., submitted forpublication).
2.2. K. brevis PCR primers and DNA probes
PCR primers and probes (Table 2) were designed insilico to hybridize sequences within the D1/D2 region of

760 M.J. LaGier et al. / Marine Pollution Bulletin 54 (2007) 757–770
the ribosomal RNA large subunit (LSU) of K. brevis
(Goodwin et al., 2005). Forward primers were labeled withbiotin and reverse primers were labeled with fluoroscein(FITC). Both assays used the same forward primer(Brevis-1-PCR-F). However, the Rapid PCR-Detect assayused a reverse primer specific for K. brevis (Karenia-1-PCR-1); whereas, the Hybrid PCR-Detect used a generaleukaryotic reverse primer (Goodwin et al., 2005; Kieslinget al., 2002) labeled with free phosphate at the 5 0 end(R635-1-PCR-R). The Hybrid PCR-Detect assay employedthe Karenia-1-PCR-1 sequence as a probe. All primers andprobes were synthesized by Sigma-Genosys (www.sigma-genosys.com). Probe and primer sequences were alignedwith Karenia LSU rRNA sequences available from Gen-Bank (http://www.nlm.nih.gov) using the default settingsof the AlignX module of Vector NTI software (Invitrogen,Calrsbad, CA).
2.3. PCR amplification of algal DNA
DNA was amplified from algal cultures and coastalwater samples by standard PCR on an Eppendorf Master-cycler using K. brevis primers as summarized in Table 2.For the Rapid PCR-Detect assay, the PCR contained5 ll of 10· DyNAzyme II buffer (contains 1.5 mM MgCl2),0.2 mM dNTP, 10 pmol forward primer, 10 pmol reverseprimer, 1 ll DNA, 1 U DyNAzyme II DNA polymerase(Finnzymes, www.finnzymes.fi), and nuclease-free waterfor a total reaction volume of 50 ll. PCRs were similarfor the Hybrid PCR-Detect assay except that 50 pmol ofeach primer was used.
B2 Thermal cycling for the Rapid PCR-Detect assayconsisted of one cycle at 94 �C for 10 min, 30 cycles of94 �C for 30 s, 58 �C for 30 s, and 72 �C for 30 s; followedby a final extension step at 70 �C for 8 min. Thermalcycling for the Hybrid PCR-Detect assay consisted of onecycle at 94 �C for 10 min, 30 cycles of 94 �C for 1 min,55 �C for 1 min, and 72 �C for 1 min; followed by one finalextension step at 70 �C for 8 min. Amplicons used for elec-trochemical analysis were confirmed by agarose gel electro-phoresis (Ausubel et al., 1999). The amplicon sizes for theRapid and Hybrid PCR-Detect assays were 138 and 322base pairs, respectively.
2.4. Reverse transcriptase PCR of K. brevis
Total RNA was isolated from K. brevis using theRNeasy kit (Qiagen, Valencia, CA) according to the man-
Table 2PCR primers and probes used in the Rapid and Hybrid PCR-Detect assays d
Primer or probea Nucleotide sequence (50 ! 3 0)
Brevis-1-PCR-F Biotin-TGTTGTCTAAGGTGATAGCTTGCKarenia-1-PCR-R FITC-GAAGCAAATTACCATGTCCCTAGR635-1-PCR-R Phosphate-GGTCCGTGTTTCAAGACGG
a Sequences previously published in Goodwin et al., 2005; FITC = fluoresce
ufacturer’s instructions. RNA quality and quantity wasassessed by agarose gel electrophoresis and spectroscopyprior to using RT-PCR to detect the LSU rRNA of K. bre-
vis. Initially, total isolated RNA was synthesized into com-plementary DNA using the protocol included with theAccess RT-PCR System (Promega, Madison, WI). Each25 ll reaction contained 1 ll (10 ng) K. brevis total RNA,12.5 ll of AccessQuick 2 · Master Mix, 0.5 ll of AMVReverse Transcriptase, 10 pmol of each primer (Table 2),and nuclease-free water. For both the Rapid and HybridPCR-Detect assays, the first-strand cDNA synthesis wascarried out at 45 �C for 45 min in an Eppendorf Mastercy-cler prior to PCR. PCR thermal cycling for the RapidPCR-Detect assay consisted of one cycle at 94 �C for10 min, 30 cycles of 94 �C for 30 s, 58 �C for 30 s, and72 �C for 30 s; followed by a final extension step at 70 �Cfor 8 min. Thermal cycling for the Hybrid PCR-Detectassay consisted of one cycle at 94 �C for 10 min, 30 cyclesof 94 �C for 1 min, 55 �C for 1 min, and 72 �C for 1 min;followed by one final extension step at 70 �C for 8 min.Successful amplification was confirmed by agarose gelelectrophoresis.
2.5. Filter PCR of K. brevis
K. brevis cells grown in culture were counted, seriallydiluted into 100 ml of natural seawater culture media(Andersen et al., 1997), and concentrated onto replicate25 mm membrane filters (0.45 lm Supor-450; Pall Corp.,East Hills, NY). For PCR amplification, a 5 mm diametercircular section of each filter was removed from the centerof each membrane using a standard paper hole puncherand placed into a 500 ll PCR tube using sterile forceps(Kirchman et al., 2001). To prevent contamination betweenPCRs, the hole puncher was treated with DNA AWAY
(Molecular BioProducts, San Diego, CA), 100% ethanoland flamed between samples. The PCR and cycling condi-tions were identical to the Rapid PCR-Detect protocolexcept a final reaction volume of 100 ll was used and15 lg of bovine serum albumin (10 mg/ml stock; A7030;Sigma, St. Louis, MS) was added to each reaction. Electro-chemical detection of PCR products was carried out usingthe standard Rapid PCR-Detect protocol.
2.6. Amplification of bacterial and viral targets
Monoplex PCR amplification for bacterial and viraltargets was carried out as described above for the Rapid
eveloped for Karenia brevis
Tm (�C) Use
62.1 Forward primer for Rapid and Hybrid62.4 Reverse primer for Rapid and probe for Hybrid65.5 Reverse primer for Hybrid
in.

M.J. LaGier et al. / Marine Pollution Bulletin 54 (2007) 757–770 761
PCR-Detect protocol for algal DNA except that 5 ll (1 ng)of DNA was used and 15 lg (1.5 ll) of 10 mg/ml stockbovine serum albumin (#A7030; Sigma, St. Louis, MS).For detection of Salmonella, each 50 ll PCR also con-tained 2% formamide and an additional 1 U of DNA poly-merase. Amplification of beach water spiked with humanfeces was carried out by using 5 ll of crude lysate diluted1:5 as a template in each 50 ll reaction. PCR products wereanalyzed using gel electrophoresis. Table 3 summarizes theprimers, references, and thermal cycling conditions usedfor the multi-target assays.
2.7. Electrochemical detection of K. brevis
A single-target format was used for electrochemical detec-tion of K. brevis. Disposable carbon sensors coated withNeutrAvidinTM were obtained from Alderon Biosciences
Table 3PCR primers and conditions used in the multi-target electrochemical assay fo
Target Gene Primer label-name-sequence(lM per PCR)
Human-specificEnterococcus faeciumb
esp Biotin-espF-TATGAAAGC(0.3)FITC-espR-ACGTCGAAAGTTCGATT
Human-specificBacteroidesb
16S, HF8 genecluster
Biotin-HF183F-ATCATGAGTTCACATGTFITC-Bac708R-CAATCGGAGTTCTTCGT
Campylobacter jejuni c HipO Biotin-CjF1-TGCTAGTGACAAAAGAATT (0.5)FITC-CjR1-TCATTTCGCAAAAAAAT
Salmonella spp.c IpaB Biotin-IpaBF-GGACTTTTTAAAAGCGGFITC-IpaBR-GCCTCTCCCAGAGCCGT
E. coli 0157:H7c rfb Biotin-0157PF8-CGTGATGATGTTGAGTTFITC-0157PR8-AGATTGGTTGGCATTAC
Human Adenovirusc Hexon Biotin-AD2F-CCCTGGTAKCCRATRTTFITC-AD3R-GACTCYTCWGTSAGYG
Enterococcus spp.d 23S rRNA Biotin-ECST748F-AGAAATTCCAAACGAAFITC-ENC854R-CAGTGGTCTACCTCCAT
Staphylococcus aureusc clfA Biotin-clfAF-GCAAAATCCACAGGAAACGA (0.1)FITC-clfAR-CTTGATCTCCAGCCATAATTGGTGG
a For all reactions and targets, the PCR thermal cycling was preceded byextension step (70 �C 8 min).
b Human fecal marker.c Waterborne pathogen.d Fecal indicator; FITC = fluorescein.
(Durham, NC). The screen printed sensors incorporate aconventional three-electrode configuration comprised of acircular (5 mm diameter) working electrode, a counter elec-trode, and a silver (Ag/AgCl) reference electrode, all printedon a polycarbonate substrate (4.5 cm · 1.5 cm). Both work-ing and counter electrodes were made of heat-cured carboncomposite inks. A ring-shaped layer printed around the threeelectrodes comprised the reservoir of the electrochemical cell,which can accommodate a volume of up to 50 ll.
During PCR, amplicons were labeled on one end withbiotin to allow capture onto the NeutrAvidinTM-coated elec-trodes (i.e., biosensors). Amplicons were also labeled withfluorescein during PCR (Rapid PCR-Detect) or duringhybridization to a probe post k-exonuclease digestion(Hybrid PCR-Detect). The amount of DNA bound to thesensor was proportional to the electrochemical current gen-erated via horseradish peroxidase (HRP) chemistry and
r bacterial and viral targets
, 5 0 ! 30 Cyclinga Reference
ACAAGTT 94 �C 1 min; 58 �C 1 min; 72 �C1 min; 40 cycles
Scott et al. (2005)
TCC (0.3)
CCG (0.4)94 �C 30 s; 59 �C 30 s; 72 �C 30 s;40 cycles
Bernhard and Field(2000)
G (0.4)
GGTTG 94 �C 30 s; 60 �C 30 s; 72 �C 30 s;40 cycles
LaGier et al. (2004)
CCAAA (0.5)
CGG (0.3)94 �C 1 min; 62 �C 1 min; 72 �C1 min; 35 cycles
Kong et al. (2002)
CTGG (0.3)
G (1.0)94 �C 30 s; 55 �C 30 s; 72 �C 30 s;40 cycles
Maurer et al.(1999)
TG (1.0)
GTA (0.3)94 �C 30 s; 60 �C 30 s; 72 �C 30 s;40 cycles
He and Jiang(2005)
GCC (0.3)
CTTG (0.9)94 �C 30 s; 60 �C 30 s; 72 �C 30 s;30 cycles
Haugland et al.(2005)
CATT (0.3)
AGCACA 94 �C 1 min; 55 �C 1 min; 72 �C1 min; 40 cycles
Mason et al. (2001)
(0.1)
an initial heat denaturation step (94 �C 10 min) and followed by a final

762 M.J. LaGier et al. / Marine Pollution Bulletin 54 (2007) 757–770
intermittent pulse amperometry (IPA). The HRP was con-jugated to the amplicon via fluorescein, oxidized by hydro-gen peroxide (Eq. (1)), and regenerated upon donation ofan electron to 3,3 0,5,5 0-tetramethylbenzidine (TMB) (Eq.(2)). The sensor reader generated a charged electrode sur-face by applying millisecond pulses of �100 mV potentialand the TMB was regenerated (Eq. (3)) upon transferringan electron to the carbon sensor surface, thus completingthe catalytic cycle.
H2O2 þ 2Hþ þHRPred ! HRPox þ 2H2O ð1Þ2TMBþHRPox ! 2TMBþ þHRPred ð2Þ2TMBþ þ 2e� ! 2TMB ð3Þ
With the Rapid PCR-Detect assay, amplicons werelabeled with biotin on one end and fluorescein on the otherduring PCR (Table 2). Reagents supplied with the RapidPCR-Detect Reagent Kit (Alderon Biosciences, Durham,NC) were used. Sensors were pre-washed with 100 ll of1· wash solution for 10 min at room temperature. The buf-fer was removed just prior to addition of sample to ensurethat the sensors were not dry. Following PCR, 1 ll ofamplicon was diluted 1:100 in 100 ll of nuclease-free water.A drop (40 ll) of diluted amplicon was placed on a pre-washed sensor and allowed to bind for 5 min at room tem-perature. Using a squirt bottle, sensors were washed twicewith 1· wash solution to remove unbound constituents.The wash solution was removed and the sensor was incu-bated for 5 min with 50 ll of horseradish peroxidase(HRP)-conjugated anti-fluorescein antibody (0.75 U/ll) atroom temperature. Sensors were washed twice with 1·wash solution and inserted into an AndCare 100 Sensorreader (Alderon Biosciences, Durham, NC). The electro-chemical reaction was initiated by adding 50 ll of HRPsubstrate (0.4 g/ll of TMB containing 0.02% hydrogen per-oxidase). A reading was obtained in approximately 30 s.The sensor reader was set to perform IPA analysis accord-ing to the following parameters: 15 s delay, �100 mV pulsepotential (versus Ag/AgCl reference electrode), 10 ms pulsetime, 10 s measurement time, 10 lA current range, and5 Hz frequency. A total of 50 current measurements werecollected from each sensor during the IPA analysis. Eachreported current value was the mean of the last five mea-surements collected. The mean value from each sensor wellwas displayed on the instrument following IPA.
With Hybrid PCR-Detect, PCR amplicons were labeledwith biotin and a free phosphate group (Table 2). Additionof k-exonuclease was then used to convert the doublestranded PCR product into single-stranded DNA mole-cules labeled at the 5 0 end with biotin. Reagents suppliedwith the Hybrid PCR-Detect Reagent Kit (Alderon Biosci-ences, Durham, NC) were used. After PCR, 3 ll of ampli-con was combined with 0.5 ll of k-exonuclease (1 U/ll),1.5 ll of 10 · k-exonuclease buffer and 10 ll of nuclease-free water (final volume of 15 ll). This reaction wasincubated for 10 min at room temperature to allow thek-exonuclease to degrade the DNA strand containing the
5 0 phosphate group. The resulting single-stranded DNAwas hybridized to a fluorescein-labeled probe (Table 2,Karenia-1-PCR-R) by combining the following to form a50 ll reaction: 1 lM probe, 10 ll of 5· hybridization buf-fer, 23 ll of nuclease-free H2O and 15 ll of the k-exonucle-ase digested PCR product. The mixture was heated for5 min at 80 �C and incubated for an additional 5 min atroom temperature. A drop (40 ll) of hybridized PCR prod-uct was placed on a pre-washed sensor and analysis pro-ceeded as described for the Rapid PCR-Detect assayexcept the final wash step was performed three times priorto placing the sensor into the AndCare 100 Sensor reader.
2.8. Multi-target electrochemical detection of bacteria, virus,
and source tracking markers
A multi-target format was used for detection of bacte-rial and viral targets. Specifically, the assay targeted: (1)Enterococcus spp., which are used to indicate the presenceof fecal pollution in environmental waters (Cabelli et al.,1979; Durfour, 1984; EPA, 2003); (2) the human-specificHF8 cluster of Bacteroides (Bernhard and Field, 2000);(3) the esp gene from E. faecium, which is used as a proxyfor human fecal pollution (Scott et al., 2005); the water-borne bacterial pathogens; (4) E. coli 0157:H7, (5) Salmo-
nella spp., (6) C. jejuni and (7) S. aureus (Percival et al.,2004); and (8) adenoviruses known to cause disease inhumans via a waterborne route of transmission (Fongand Lipp, 2005).
Sample analysis was as described for the Rapid PCR-Detect assay except that 8-well carbon sensor strips andthe AndCare 800 Sensor reader (Alderon Biosciences, Dur-ham, NC) were used. That is, the method was identical tothat described for Rapid PCR-Detect of K. brevis exceptthat each surface of an eight-well sensor array (rather thana single well sensor) was exposed to a different microbialtarget.
2.9. Data analysis and controls
Student’s t-test and regression analysis were used toevaluate statistical significance and the electrochemicalresponse versus DNA concentration (GraphPad Prism,San Diego, CA). Duplicate negative controls were run foreach electrochemical assay and a positive response was cal-culated as an electrochemical signal greater than 2· themean current signal of the negative controls. The negativecontrols contained PCR and electrochemical reagentsexcept template DNA. Positive controls were included ineach assay, consisting of all PCR and electrochemicalreagents plus a known amount of target DNA. TriplicatePCR and triplicate electrodes were used to relate electro-chemical signals to amount of template DNA. All environ-mental samples tested (Section 2.1) were previously shownnot to contain PCR inhibitors (Goodwin et al., 2005;Sinigalliano et al., submitted for publication).
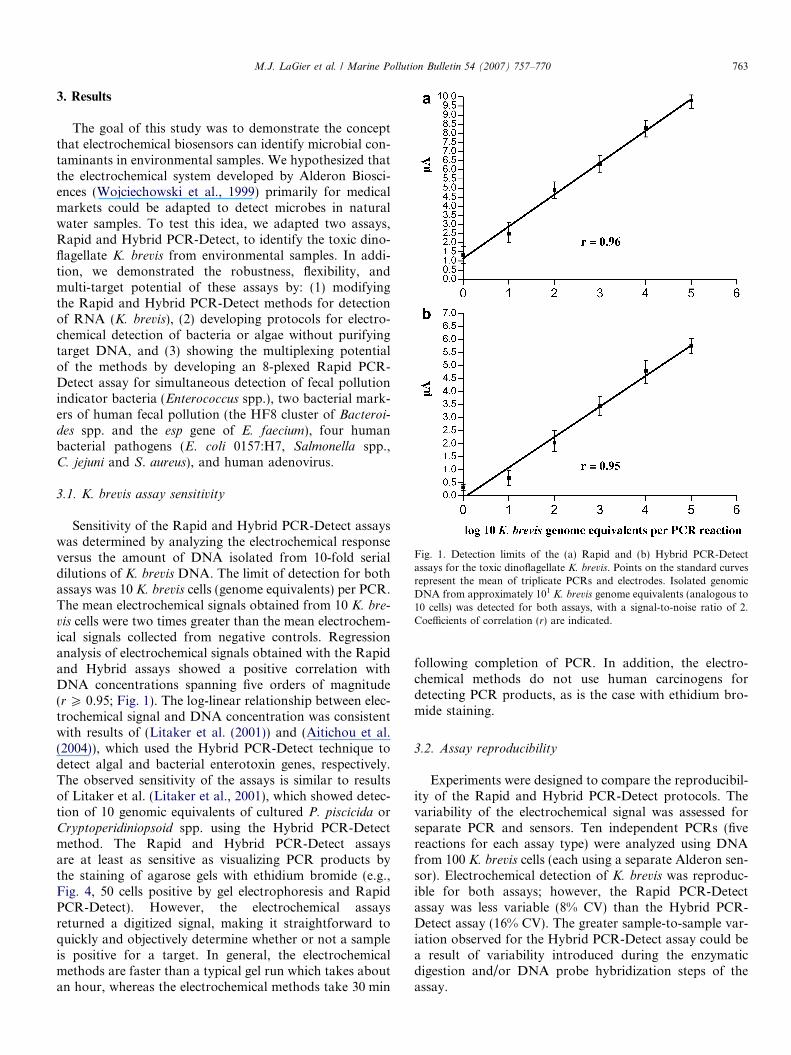
Fig. 1. Detection limits of the (a) Rapid and (b) Hybrid PCR-Detectassays for the toxic dinoflagellate K. brevis. Points on the standard curvesrepresent the mean of triplicate PCRs and electrodes. Isolated genomicDNA from approximately 101 K. brevis genome equivalents (analogous to10 cells) was detected for both assays, with a signal-to-noise ratio of 2.Coefficients of correlation (r) are indicated.
M.J. LaGier et al. / Marine Pollution Bulletin 54 (2007) 757–770 763
3. Results
The goal of this study was to demonstrate the conceptthat electrochemical biosensors can identify microbial con-taminants in environmental samples. We hypothesized thatthe electrochemical system developed by Alderon Biosci-ences (Wojciechowski et al., 1999) primarily for medicalmarkets could be adapted to detect microbes in naturalwater samples. To test this idea, we adapted two assays,Rapid and Hybrid PCR-Detect, to identify the toxic dino-flagellate K. brevis from environmental samples. In addi-tion, we demonstrated the robustness, flexibility, andmulti-target potential of these assays by: (1) modifyingthe Rapid and Hybrid PCR-Detect methods for detectionof RNA (K. brevis), (2) developing protocols for electro-chemical detection of bacteria or algae without purifyingtarget DNA, and (3) showing the multiplexing potentialof the methods by developing an 8-plexed Rapid PCR-Detect assay for simultaneous detection of fecal pollutionindicator bacteria (Enterococcus spp.), two bacterial mark-ers of human fecal pollution (the HF8 cluster of Bacteroi-
des spp. and the esp gene of E. faecium), four humanbacterial pathogens (E. coli 0157:H7, Salmonella spp.,C. jejuni and S. aureus), and human adenovirus.
3.1. K. brevis assay sensitivity
Sensitivity of the Rapid and Hybrid PCR-Detect assayswas determined by analyzing the electrochemical responseversus the amount of DNA isolated from 10-fold serialdilutions of K. brevis DNA. The limit of detection for bothassays was 10 K. brevis cells (genome equivalents) per PCR.The mean electrochemical signals obtained from 10 K. bre-
vis cells were two times greater than the mean electrochem-ical signals collected from negative controls. Regressionanalysis of electrochemical signals obtained with the Rapidand Hybrid assays showed a positive correlation withDNA concentrations spanning five orders of magnitude(r P 0.95; Fig. 1). The log-linear relationship between elec-trochemical signal and DNA concentration was consistentwith results of (Litaker et al. (2001)) and (Aitichou et al.(2004)), which used the Hybrid PCR-Detect technique todetect algal and bacterial enterotoxin genes, respectively.The observed sensitivity of the assays is similar to resultsof Litaker et al. (Litaker et al., 2001), which showed detec-tion of 10 genomic equivalents of cultured P. piscicida orCryptoperidiniopsoid spp. using the Hybrid PCR-Detectmethod. The Rapid and Hybrid PCR-Detect assaysare at least as sensitive as visualizing PCR products bythe staining of agarose gels with ethidium bromide (e.g.,Fig. 4, 50 cells positive by gel electrophoresis and RapidPCR-Detect). However, the electrochemical assaysreturned a digitized signal, making it straightforward toquickly and objectively determine whether or not a sampleis positive for a target. In general, the electrochemicalmethods are faster than a typical gel run which takes aboutan hour, whereas the electrochemical methods take 30 min
following completion of PCR. In addition, the electro-chemical methods do not use human carcinogens fordetecting PCR products, as is the case with ethidium bro-mide staining.
3.2. Assay reproducibility
Experiments were designed to compare the reproducibil-ity of the Rapid and Hybrid PCR-Detect protocols. Thevariability of the electrochemical signal was assessed forseparate PCR and sensors. Ten independent PCRs (fivereactions for each assay type) were analyzed using DNAfrom 100 K. brevis cells (each using a separate Alderon sen-sor). Electrochemical detection of K. brevis was reproduc-ible for both assays; however, the Rapid PCR-Detectassay was less variable (8% CV) than the Hybrid PCR-Detect assay (16% CV). The greater sample-to-sample var-iation observed for the Hybrid PCR-Detect assay could bea result of variability introduced during the enzymaticdigestion and/or DNA probe hybridization steps of theassay.
764 M.J. LaGier et al. / Marine Pollution Bulletin 54 (2007) 757–770
3.3. K. brevis assay specificity
The probes and PCR primers used in both assay for-mats were specific for K. brevis when tested againstDNA isolated from 12 different toxic dinoflagellates,including DNA from a close relative of K. brevis, Kareniamikimotoi (Table 1). K. mikimotoi differs from K. brevis byfour nucleotides within the region targeted by the PCRprimers and DNA probe. As predicted by our sequenceanalysis efforts, only PCRs containing K. brevis DNA gaverise to positive electrochemical signals (Table 1). A fewrecently described Karenia species differ from K. brevis
by only one (K. asterichroma and K. bidigitata) or two(K. papilionacea) nucleotides within the region of thePCR primers/probes. Although published reports existdescribing K. asterichroma, K. bidigitata and K. papiliona-
cea (De Salas et al., 2004; Haywood et al., 2004), ourattempts to obtain samples of these species were notsuccessful.
3.4. Electrochemical detection of K. brevis in coastal water
samples
The Rapid and Hybrid PCR-Detect assays developedfor K. brevis successfully detected this toxic dinoflagellate
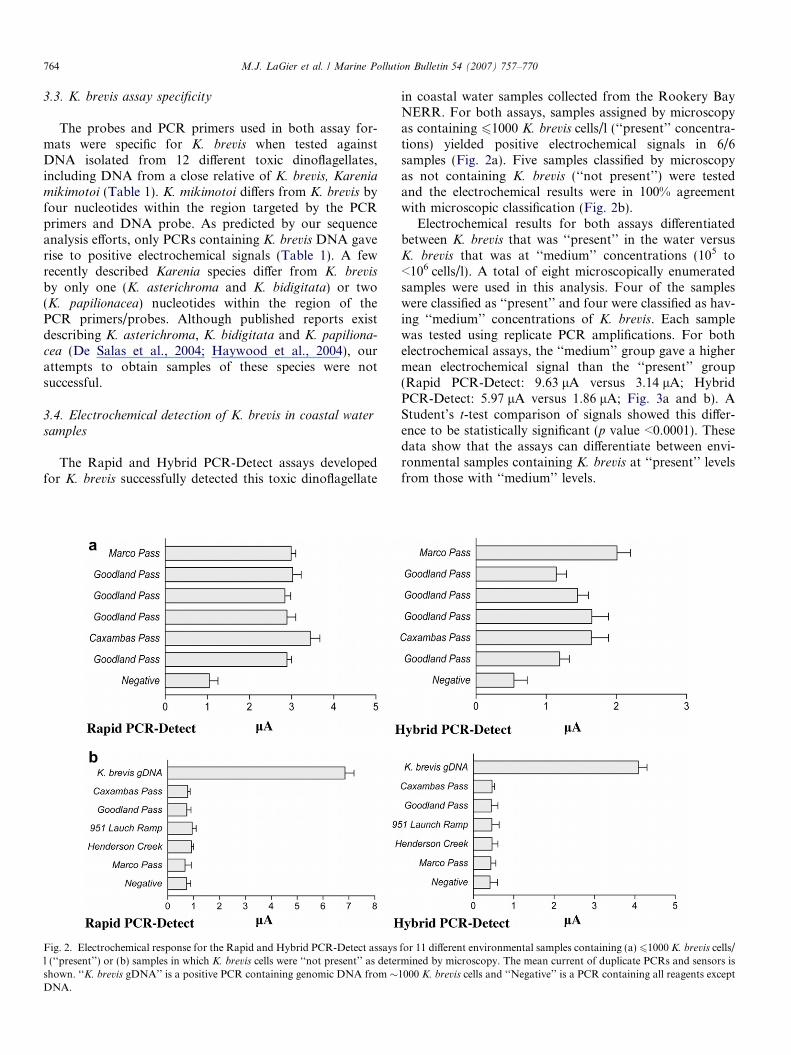
Fig. 2. Electrochemical response for the Rapid and Hybrid PCR-Detect assaysl (‘‘present’’) or (b) samples in which K. brevis cells were ‘‘not present’’ as detershown. ‘‘K. brevis gDNA’’ is a positive PCR containing genomic DNA from �DNA.
in coastal water samples collected from the Rookery BayNERR. For both assays, samples assigned by microscopyas containing 61000 K. brevis cells/l (‘‘present’’ concentra-tions) yielded positive electrochemical signals in 6/6samples (Fig. 2a). Five samples classified by microscopyas not containing K. brevis (‘‘not present’’) were testedand the electrochemical results were in 100% agreementwith microscopic classification (Fig. 2b).
Electrochemical results for both assays differentiatedbetween K. brevis that was ‘‘present’’ in the water versusK. brevis that was at ‘‘medium’’ concentrations (105 to<106 cells/l). A total of eight microscopically enumeratedsamples were used in this analysis. Four of the sampleswere classified as ‘‘present’’ and four were classified as hav-ing ‘‘medium’’ concentrations of K. brevis. Each samplewas tested using replicate PCR amplifications. For bothelectrochemical assays, the ‘‘medium’’ group gave a highermean electrochemical signal than the ‘‘present’’ group(Rapid PCR-Detect: 9.63 lA versus 3.14 lA; HybridPCR-Detect: 5.97 lA versus 1.86 lA; Fig. 3a and b). AStudent’s t-test comparison of signals showed this differ-ence to be statistically significant (p value <0.0001). Thesedata show that the assays can differentiate between envi-ronmental samples containing K. brevis at ‘‘present’’ levelsfrom those with ‘‘medium’’ levels.
for 11 different environmental samples containing (a) 61000 K. brevis cells/mined by microscopy. The mean current of duplicate PCRs and sensors is
1000 K. brevis cells and ‘‘Negative’’ is a PCR containing all reagents except
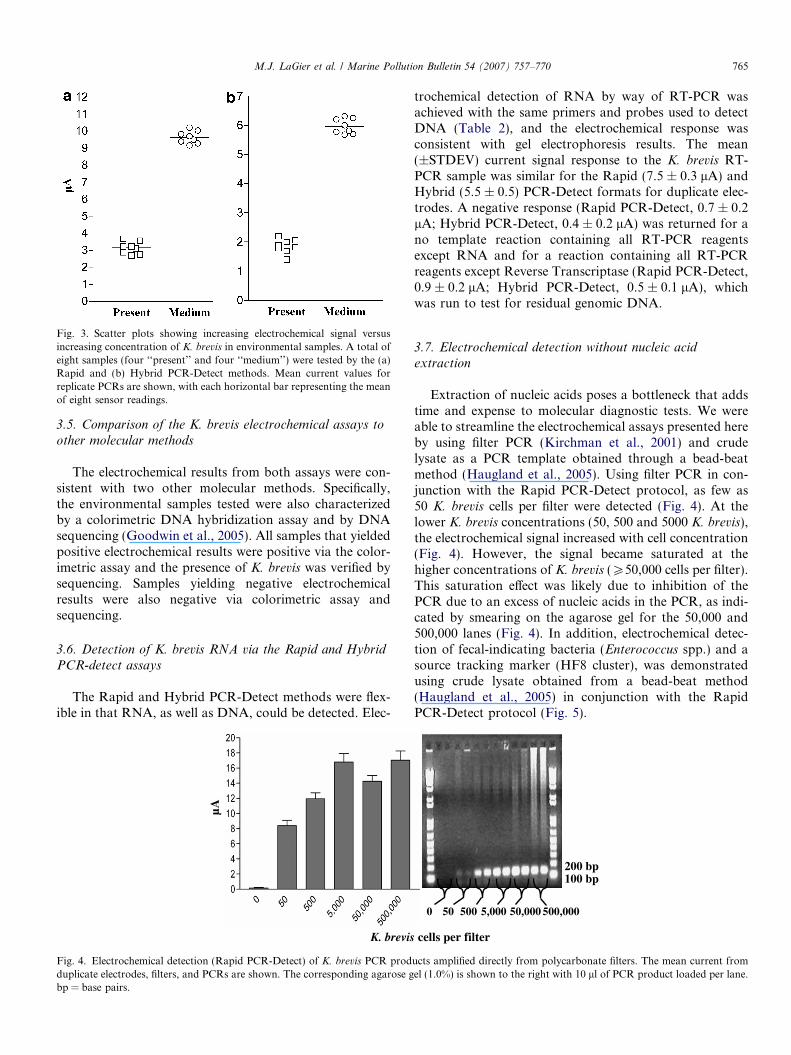
Fig. 3. Scatter plots showing increasing electrochemical signal versusincreasing concentration of K. brevis in environmental samples. A total ofeight samples (four ‘‘present’’ and four ‘‘medium’’) were tested by the (a)Rapid and (b) Hybrid PCR-Detect methods. Mean current values forreplicate PCRs are shown, with each horizontal bar representing the meanof eight sensor readings.
M.J. LaGier et al. / Marine Pollution Bulletin 54 (2007) 757–770 765
3.5. Comparison of the K. brevis electrochemical assays to
other molecular methods
The electrochemical results from both assays were con-sistent with two other molecular methods. Specifically,the environmental samples tested were also characterizedby a colorimetric DNA hybridization assay and by DNAsequencing (Goodwin et al., 2005). All samples that yieldedpositive electrochemical results were positive via the color-imetric assay and the presence of K. brevis was verified bysequencing. Samples yielding negative electrochemicalresults were also negative via colorimetric assay andsequencing.
3.6. Detection of K. brevis RNA via the Rapid and Hybrid
PCR-detect assays
The Rapid and Hybrid PCR-Detect methods were flex-ible in that RNA, as well as DNA, could be detected. Elec-
µA
K. brevis
Fig. 4. Electrochemical detection (Rapid PCR-Detect) of K. brevis PCR prodduplicate electrodes, filters, and PCRs are shown. The corresponding agarose gbp = base pairs.
trochemical detection of RNA by way of RT-PCR wasachieved with the same primers and probes used to detectDNA (Table 2), and the electrochemical response wasconsistent with gel electrophoresis results. The mean(±STDEV) current signal response to the K. brevis RT-PCR sample was similar for the Rapid (7.5 ± 0.3 lA) andHybrid (5.5 ± 0.5) PCR-Detect formats for duplicate elec-trodes. A negative response (Rapid PCR-Detect, 0.7 ± 0.2lA; Hybrid PCR-Detect, 0.4 ± 0.2 lA) was returned for ano template reaction containing all RT-PCR reagentsexcept RNA and for a reaction containing all RT-PCRreagents except Reverse Transcriptase (Rapid PCR-Detect,0.9 ± 0.2 lA; Hybrid PCR-Detect, 0.5 ± 0.1 lA), whichwas run to test for residual genomic DNA.
3.7. Electrochemical detection without nucleic acid
extraction
Extraction of nucleic acids poses a bottleneck that addstime and expense to molecular diagnostic tests. We wereable to streamline the electrochemical assays presented hereby using filter PCR (Kirchman et al., 2001) and crudelysate as a PCR template obtained through a bead-beatmethod (Haugland et al., 2005). Using filter PCR in con-junction with the Rapid PCR-Detect protocol, as few as50 K. brevis cells per filter were detected (Fig. 4). At thelower K. brevis concentrations (50, 500 and 5000 K. brevis),the electrochemical signal increased with cell concentration(Fig. 4). However, the signal became saturated at thehigher concentrations of K. brevis (P50,000 cells per filter).This saturation effect was likely due to inhibition of thePCR due to an excess of nucleic acids in the PCR, as indi-cated by smearing on the agarose gel for the 50,000 and500,000 lanes (Fig. 4). In addition, electrochemical detec-tion of fecal-indicating bacteria (Enterococcus spp.) and asource tracking marker (HF8 cluster), was demonstratedusing crude lysate obtained from a bead-beat method(Haugland et al., 2005) in conjunction with the RapidPCR-Detect protocol (Fig. 5).
cells per filter
100 bp200 bp
0 50 500 5,000 50,000 500,000
ucts amplified directly from polycarbonate filters. The mean current fromel (1.0%) is shown to the right with 10 ll of PCR product loaded per lane.
0
1
2
3
4
5
6
Entero
cocc
us faec
ium
(esp
)
Bacte
roid
esHF8 c
lust
er
Campylo
bacte
r jeju
ni
Escher
ichia
coli (
0157
:H7)
Human
Aden
ovirus
Salmonell
a spec
ies
Staphylo
cocc
us aure
us
Entero
cocc
us spec
ies
No template
Target DNA
Beach water
Contaminated beach water
µA
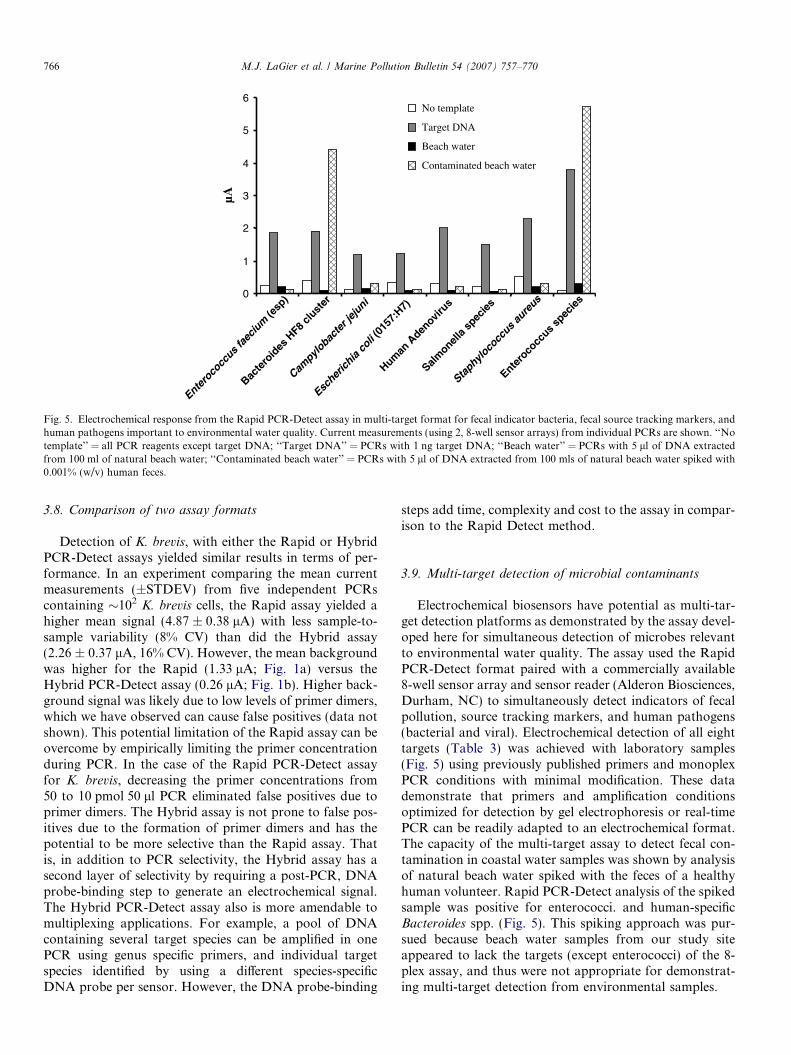
Fig. 5. Electrochemical response from the Rapid PCR-Detect assay in multi-target format for fecal indicator bacteria, fecal source tracking markers, andhuman pathogens important to environmental water quality. Current measurements (using 2, 8-well sensor arrays) from individual PCRs are shown. ‘‘Notemplate’’ = all PCR reagents except target DNA; ‘‘Target DNA’’ = PCRs with 1 ng target DNA; ‘‘Beach water’’ = PCRs with 5 ll of DNA extractedfrom 100 ml of natural beach water; ‘‘Contaminated beach water’’ = PCRs with 5 ll of DNA extracted from 100 mls of natural beach water spiked with0.001% (w/v) human feces.
766 M.J. LaGier et al. / Marine Pollution Bulletin 54 (2007) 757–770
3.8. Comparison of two assay formats
Detection of K. brevis, with either the Rapid or HybridPCR-Detect assays yielded similar results in terms of per-formance. In an experiment comparing the mean currentmeasurements (±STDEV) from five independent PCRscontaining �102 K. brevis cells, the Rapid assay yielded ahigher mean signal (4.87 ± 0.38 lA) with less sample-to-sample variability (8% CV) than did the Hybrid assay(2.26 ± 0.37 lA, 16% CV). However, the mean backgroundwas higher for the Rapid (1.33 lA; Fig. 1a) versus theHybrid PCR-Detect assay (0.26 lA; Fig. 1b). Higher back-ground signal was likely due to low levels of primer dimers,which we have observed can cause false positives (data notshown). This potential limitation of the Rapid assay can beovercome by empirically limiting the primer concentrationduring PCR. In the case of the Rapid PCR-Detect assayfor K. brevis, decreasing the primer concentrations from50 to 10 pmol 50 ll PCR eliminated false positives due toprimer dimers. The Hybrid assay is not prone to false pos-itives due to the formation of primer dimers and has thepotential to be more selective than the Rapid assay. Thatis, in addition to PCR selectivity, the Hybrid assay has asecond layer of selectivity by requiring a post-PCR, DNAprobe-binding step to generate an electrochemical signal.The Hybrid PCR-Detect assay also is more amendable tomultiplexing applications. For example, a pool of DNAcontaining several target species can be amplified in onePCR using genus specific primers, and individual targetspecies identified by using a different species-specificDNA probe per sensor. However, the DNA probe-binding
steps add time, complexity and cost to the assay in compar-ison to the Rapid Detect method.
3.9. Multi-target detection of microbial contaminants
Electrochemical biosensors have potential as multi-tar-get detection platforms as demonstrated by the assay devel-oped here for simultaneous detection of microbes relevantto environmental water quality. The assay used the RapidPCR-Detect format paired with a commercially available8-well sensor array and sensor reader (Alderon Biosciences,Durham, NC) to simultaneously detect indicators of fecalpollution, source tracking markers, and human pathogens(bacterial and viral). Electrochemical detection of all eighttargets (Table 3) was achieved with laboratory samples(Fig. 5) using previously published primers and monoplexPCR conditions with minimal modification. These datademonstrate that primers and amplification conditionsoptimized for detection by gel electrophoresis or real-timePCR can be readily adapted to an electrochemical format.The capacity of the multi-target assay to detect fecal con-tamination in coastal water samples was shown by analysisof natural beach water spiked with the feces of a healthyhuman volunteer. Rapid PCR-Detect analysis of the spikedsample was positive for enterococci. and human-specificBacteroides spp. (Fig. 5). This spiking approach was pur-sued because beach water samples from our study siteappeared to lack the targets (except enterococci) of the 8-plex assay, and thus were not appropriate for demonstrat-ing multi-target detection from environmental samples.

µA
YD3
FDP-26
FDP-25
No template
*
**
Fig. 6. Multi-target detection (Rapid PCR-Detect) of fecal indicatorbacteria (Enterococcus spp.) and human-specific Bacteroides (HF8 cluster)amplified from sediment (YD3) and water (FDP-25 and FDP-26) samplescollected from New Orleans, LA following Hurricane Katrina. Meancurrent measurements from duplicate sensors and PCRs are shown. ‘‘Notemplate’’ = all PCR reagents except target DNA. Samples marked withan asterisk (*) are electrochemical positive, based on having a meancurrent >2· the mean current of no template PCR controls.
M.J. LaGier et al. / Marine Pollution Bulletin 54 (2007) 757–770 767
Environmental samples (sediment and water) collectedin New Orleans, Louisiana following Hurricanes Katrinaand Rita were also tested with the multi-target assay. Lim-ited DNA sample availability restricted testing to Entero-cocci and Bacteroides spp. The two water samples testedwere collected from a canal before and after a water-pump-ing event. The sample taken before the pumping event(FDP-25) was reported to contain 115 enterococci/100 mlwater, and the sample taken after the pumping event(FDP-26) was reported to contain 1481 enterococci/100 ml water (Sinigalliano et al., submitted for publica-tion). By electrochemical analysis, FDP-25 was negativefor enterococci and the human-marker of Bacteroidesspp.; whereas, FDP-26 was positive for enterococci andthe human-marker of Bacteroides spp. (Fig. 6). Thesediment sample (YD3), reported to contain 1039 entero-cocci/g soil (Sinigalliano et al., submitted for publication),was positive for enterococci and negative for human-mar-ker Bacteroides in this study (Fig. 6). These results indicatethat the multi-target assay can be used to detect microbialcontaminants from environmental sediment and watersamples while returning information regarding the sourcesof fecal contamination.
4. Discussion
The utility of electrochemical methods for water qualityapplications depends in part on availability, ease of use,and versatility. The reagents, biosensors, and sensor read-ers used in this study were all commercially available.The electrochemical formats, particularly the RapidPCR-Detect, were relatively simple to use. For example,the multi-target assay design was based on previously pub-lished primers, and thus requires little modification toenable the 8-target sensor array to work ‘‘out of thebox’’. The methods described here were highly versatile
as demonstrated by detection of both DNA and RNA ofa toxic dinoflagellate and detection of a variety of bacterialand viral targets. The demonstrated ability to detect RNAcould be useful for the differentiation of live versus deadmicrobes in the environment, as RNA is a better indicatorof cellular viability (Keer and Birch, 2003). The successfuldetection of RNA also allows for the identification ofmicrobes with an RNA genome. Furthermore, the devel-oped assays were sensitive (Figs. 1, 2 and 4), rapid (canbe completed in 3–5 h), used hand-held sensor readers thatare relatively affordable (www.alderonbiosciences.com),returned a digitized signal which allowed rapid and objec-tive determination of a positive result, and did not requirethe isolation of nucleic acids prior to detection (Figs. 4 and5).
Although attention paid to electrochemical methods asmicrobial detection tools has grown (Drummond et al.,2003; Kerman et al., 2004; LaGier et al., 2005; Metfieset al., 2005), there is a lack of published investigations test-ing whether electrochemical methods can detect microbialcontaminants in environmental samples. Without this keydata, the usefulness of electrochemical sensors for environ-mental microbiology applications cannot be fully judged.In this study, electrochemical detection of the toxic dinofla-gellate K. brevis was successfully demonstrated usingcoastal water samples. The assay was sensitive enough todetect K. brevis that was ‘‘present’’ (61000 cells) in sampleswithout returning false positive results for samples in whichK. brevis was ‘‘not present’’ (Fig. 2). Furthermore, the elec-trochemical response was significantly different for samplescontaining K. brevis at ‘‘present’’ versus ‘‘medium’’ concen-trations (100,000 to <106 cells/l cells) (Fig. 3). Hence, theRapid and Hybrid PCR-Detect assays could be usefulresearch tools to screen for the presence or absence ofK. brevis with the potential to differentiate between broadclasses of abundance.
The multi-target assay was designed to return an indica-tion of fecal contamination along with information onwhether contamination was from a human source and ifselected pathogens were present. Such an tool would beuseful for managers of recreational water quality (Boweret al., 2005) because even though high levels of fecal-indi-cating bacteria appear to pose a public health risk (Cabelliet al., 1979; Dufour, 1984; EPA, 2003; Wade et al., 2003),the presence of indicators is not always correlated with thepresence of pathogens (Lipp et al., 2001; Noble and Furh-man, 2001), and standard methods (EPA, 2003) do notidentify the source of the fecal contamination (Scottet al., 2005).
The multi-target assay was tested against laboratorysamples and beach water spiked with human feces. RapidPCR-Detect analysis successfully detected all eight targetsin laboratory samples, and the beach water sample contam-inated with human feces was positive for enterococci andhuman-specific Bacteroides (Fig. 5). Both Enterococcus
spp. and Bacteroides spp. are commonly found in humanfeces (Tortora et al., 2001), and the human-specific

768 M.J. LaGier et al. / Marine Pollution Bulletin 54 (2007) 757–770
Bacteroides marker (HF8) is estimated to be present at5 · 1010 copies/g of feces (Bernhard and Field, 2000). Inaddition, the reaction for detecting the human-specific Bac-
teroides marker has been shown to be sensitive, with a posi-tive visualization of PCR products from sewage samplesdiluted 1:150,000 (Bower et al., 2005). Lack of detectionfor the other targets may have been due to lack of presencein the human fecal sample, but was not due to PCR inhibi-tion (data not shown). The sample was from a healthy vol-unteer, thus the pathogens Salmonella spp., C. jejuni,E. coli 0157:H7, and human adenovirus were not expectedin the sample. The esp gene is not found in all humans(Scott et al., 2005), and only 30–50% of humans carry S.
aureus (Youmans et al., 1985). The lack of an esp signalalso might be due the relatively low sensitivity of thePCR used to amplify the esp gene, which in our handswas 10·less sensitive then the PCR used to amplify Bacte-
roides belonging the HF8 cluster (10 plasmid copies/PCRfor esp versus 1 plasmid/PCR for HF8).
In addition, the multi-target assay was tested againstwater and sediment samples collected from flooded siteswithin New Orleans following Hurricane Katrina. Thewater samples were collected before (FDP-25) and after(FDP-26) a pumping event. Visual observation confirmedthe infusion of fecal waste after pumping (Sinigallianoet al., submitted for publication). The electrochemical datashowed that the enterococci detected after pumping wereassociated with feces of human origin (Fig. 6) and suggestthe usefulness of the electrochemical assay as a sourcetracking tool. The lack of an enterococci signal in sampleFDP-25 is likely due to the constraint of DNA availability.The filter for water sample FDP-25 should have contained115 enterococci prior to DNA extraction. Even assumingno loss of target DNA during the extraction procedure,each PCR (containing 1 ll of genomic DNA) run onFDP-25 would have contained �2 genome equivalents ofenterococci, which is less than the reported sensitivity ofthe enterococci primers (five genome equivalents perPCR) (He and Jiang, 2005).
The sediment sample (YD3) was positive for enterococciand negative for human-marker Bacteroides (Fig. 6), indi-cating that the enterococci signal was not the result ofrecent human contamination. Nonetheless, this samplewas positive by PCR for Bfidobacteria adolescentis (Kinget al., 2007; Sinigalliano et al., submitted for publication),a species with a human-dominated host range (Bonjochet al., 2004). The lack of a HF8 signal might be a resultof PCR-detectable Bacteroides not persisting in aerobicenvironments for more than 1–2 days, based on resultsfor Bacteroides distasonis (Kreader, 1998). Alternatively,perhaps B. adolescentis is more prevalent in human fecesin comparison to the HF8 marker, and thus more likelyto be detected.
Although the multi-target assay requires more field-test-ing, the data (Figs. 5 and 6) reinforce single-plex results forK. brevis (Figs. 2 and 3) demonstrating the capacity of elec-trochemical methods to detect microbial contaminants
from environmental samples. The multi-target data alsosuggest that electrochemical methods could be used tosimultaneously return information on fecal indicators,source tracking markers, and perhaps pathogen presence,particularly if adequate sample concentration and efficientnucleic acid recovery can be achieved. A limitation of usingthe multi-target assay for analyzing large numbers of fieldsamples is the use of monoplex PCR prior to electrochem-ical detection (i.e. simultaneous electrochemical detectionusing sensor arrays). Multiplex PCR coupled with theHybrid PCR-Detect method may be utilized to more effi-ciently analyze large sets of field samples. In addition, theintegration of the multi-target electrochemical assays intoautomated devices should minimize limitations associatedwith monoplex PCR (Farmer et al., 2006).
Efficient upstream processing of environmental samplesis critical to the success of downstream molecular detectionmethods, but sample concentration and nucleic acid extrac-tion pose major obstacles to the development of rapid andpractical methods for water quality monitoring (Noble andWeisberg, 2005). The sensitivity achieved with isolatedDNA (Fig. 1) typically is not observed with filtered envi-ronmental water samples (Figs. 2 and 6, sample FDP-25)in part because of poor DNA recovery during the extrac-tion and isolation of nucleic acids. For instance, averageextraction efficiencies associated with commercial kits canrange from 2–30% for environmental soil samples (Mumyand Findlay, 2004). Thus, in addition to streamlining theupstream processing of environmental samples for molecu-lar analysis, eliminating the nucleic acid isolation step(Figs. 4 and 5) may minimize losses in sensitivity due topoor extraction efficiencies.
The Rapid and Hybrid PCR-Detect assays use hand-held instruments, compact sensors, and produce digitizedresults and thus are amendable to field detection. However,the PCR and electrochemical steps of the assays should beintegrated into a single device to be more practical for fieldapplications. Ongoing efforts include building electrochem-ical instruments for multi-target microbial detection(Farmer et al., 2006).
Acknowledgements
We thank Dr. Marek Wojciechowski and the AlderonBiosciences staff for providing AndCare Sensor readersduring the course of this study. We thank G. Scorzetti,L. Brand, and S. Morton for dinoflagellate samples; J.Rose, T. Scott, S. Jiang, and N. Cirino for bacterial andviral samples; and the Rookery Bay NERR for environ-mental samples. We are grateful to H. Solo-Gabriele andC. Sinigialliano of the University of Miami Center ofExcellence for Oceans and Human Health (NSF#0CE0432368/ NIEHS #P50 ES12736) for post-Katrinasamples. Financial support is gratefully acknowledgedfrom the National Science Foundation (OCE-332918),the NIEHS Marine and Freshwater Biomedical SciencesCenter Grant (ES 05705) to the University of Miami, the

M.J. LaGier et al. / Marine Pollution Bulletin 54 (2007) 757–770 769
Cooperative Institute of Estuarine and EnvironmentalTechnology (CICEET), and the NOAA Oceans and Hu-man Health Initiative Capacity Building Grant. Thisresearch was carried out in part under the auspices of theCooperative Institute for Marine and Atmospheric Studies(CIMAS), a joint institute of the University of Miami andthe National Oceanic and Atmospheric Administration,cooperative agreement #NA17RJ1226.
References
Aitichou, M., Henkens, R., Sultana, A.M., Ulrich, R.G., Sofi Ibrahim,M., 2004. Detection of Staphylococcus aureus enterotoxin a and bgenes with PCR-EIA and a hand-held electrochemical sensor. Molec-ular and Cellular Probes 18, 373–377.
Andersen, R.A., Morton, S.L., Sexton, J.P., 1997. Provasoli-Guillardnational center for culture of marine phytoplankton 1997 list ofstrains. Journal of Phycology 33 (suppl.), 1–75.
Attatippaholkun, W.H., Attatippaholkum, M.K., Nisalak, A., Vaughn,D.W., Innis, B.L., 2003. A novel method for the preparation of largecDNA fragments from dengue-3 RNA genome by long RT-PCRamplification. Southeast Asian Journal of Tropical Medicine andPublic Health 31, 126–133.
Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G.,Smith, J.A., Struhl, K., 1999. Short Protocols in Molecular Biology,4th ed. John Wiley and Sons, New York.
Bernhard, A.E., Field, K.G., 2000. A PCR assay to discriminate humanand ruminant feces on the basis of host differences in Bacteroides-
Prevotella genes encoding 16S rRNA. Applied and EnvironmentalMicrobiology 66, 4571–4574.
Bonjoch, X., Balleste, E., Blanch, A.R., 2004. Multiplex PCR with 16SrRNA Gene-Targeted Primers of Bifidobacterium spp. To IdentifySources of Fecal Pollution. Applied and Environmental Microbiology70, 3171–3175.
Bower, P.A., Scopel, C.O., Jensen, E.T., Depas, M.M., McLellan, S.L.,2005. Detection of genetic markers of fecal indicator bacteria in LakeMichigan and determination of their relationship to Escherichia coli
densities using standard microbiological methods. Applied and Envi-ronmental Microbiology 71, 8305–8313.
Brand, L.E., 1990. The isolation and culture of microalgae for biotech-nological applications. In: Labeda, D.P. (Ed.), Isolation of Biotech-nological Organisms from Nature. McGraw-Hill, New York, pp. 81–115.
Cabelli, V.J., Dufour, A.P., Levin, M.A., McCabe, J., Haberman, P.W.,1979. Relationship of microbial indicators to health effects at bathingbeaches. American Journal of Public Health 69, 690–696.
Culverhouse, P.F., Williams, R., Reguera, B., Herry, V., Gonzalez-Gil, S.,2003. Do experts make mistakes? A comparison of human andmachine identification of dinoflagellates. Marine Ecology ProgressSeries 247, 17–25.
De Salas, M.F., Bolch, C.J.S., Hallegraeff, G.M., 2004. Karenia asteri-
chroma spp. nov. (Gymnodiniales, Dinophyceae), a new dinoflagellatespecies associated with finfish aquaculture mortality in Tasmania,Australia. Phycologia 43, 624–631.
Drummond, T.G., Hill, M.G., Barton, J.K., 2003. Electrochemical DNAsensors. Nature Biotechnology 21, 1192–1199.
Dufour, A.P., 1984. Health effects criteria for fresh recreational waters.EPA-600/1-84/004. Office of Research and Development, USEPA.
Durfour, A.P., 1984. Bacterial indicators of recreational water quality.Can. J. Public Health 75, 49–56.
Dwight, R.H., Fernandez, L.M., Baker, D.B., Semenza, J.C., Olson, B.H.,2005. Estimating the economic burden from illnesses associated withrecreational coastal water pollution–a case study in Orange County,California. Journal of Environmental Management 76, 95–103.
EPA, 2003. Guidelines establishing test procedures for the analysis ofpollutants; Analytical methods for biological pollutants in ambient
water; Final Rule. Federal Register V68, No. 139 40 CFR Part 136,43272–43283.
Farmer, A.S., LaGier, M.J., Goodwin, K.D., Ivanov, S., Steimle, G.,Fries, D., 2006. Portable sensor development towards PCR-basedelectrochemical detection. Florida Marine Biotechnology Summit V,18.
Fong, T.T., Lipp, E.K., 2005. Enteric viruses of humans and animals inaquatic environments: health risks, detection, and potential waterquality assessment tools. Microbiol. Mol. Biol. Rev. 69, 357–371.
Goodwin, K.D., Cotton, S.A., Scorzetti, G., Fell, J.W., 2005. A DNAhybridization assay to identify toxic dinoflagellates in coastal waters:detection of Karenia brevis in the Rookery Bay National EstuarineResearch Reserve. Harmful Algae 4, 411–422.
Griffin, D.W., Lipp, E.K., McLaughlin, M.R., Rose, J.B., 2001. Marinerecreation and public health microbiology: quest for the idealindicator. BioScience 51, 817–825.
Haugland, R.A., Siefring, S.C., Wymer, L.J., Brenner, K.P., Dufour, A.P.,2005. Comparison of Enterococcus measurements in freshwater at tworecreational beaches by quantitative polymerase chain reaction andmembrane filter culture analysis. Water Research 39, 559–568.
Haywood, A.J., Steidinger, K.A., Truby, E.W., Bergquist, P.R., Berg-quist, P.L., Adamson, J., Mackenzie, L., 2004. Comparative morphol-ogy and molecular phylogenetic analysis of three new species of thegenus Karenia (Dinophyceae) from New Zealand. Journal of Phycol-ogy 40, 165–179.
He, J.W., Jiang, S., 2005. Quantification of Enterococci and Humanadenoviruses in environmental samples by real-Time PCR. Appliedand Environmental Microbiology 71, 2250–2255.
Keer, J.T., Birch, L., 2003. Molecular methods for the assessment ofbacterial viability. Journal of Microbiological Methods 53, 175–183.
Kerman, K., Kobayashi, M., Tamiya, E., 2004. Recent trends inelectrochemical DNA biosensing technology. Measurement ScienceTechnology 15, R1–R11.
Kiesling, T.L., Wilkinson, E., Rabalais, J., Ortner, P.B., McCabe, M.M.,Fell, J.W., 2002. Rapid identification of adult and macular stages ofcopepods using DNA hybridization methodology. Marine Biotech-nology 4, 30–39.
King, E.L., Bachoon, D.S., Gates, K.W., 2007. Rapid detection of humanfecal contamination in estuarine environments by PCR targeting ofBifidobacteria adolescentis. Journal of Microbiological Methods 68,76–81.
Kirchman, D.L., Yu, L., Fuchs, B.M., Amaan, R., 2001. Structure ofcommunities in aquatic systems as revealed by filter PCR. AquaticMicrobial Ecology 26, 13–22.
Kirkpatrick, B., Fleming, L.E., Squicciarini, D., Backer, L.C., Clark, R.,Abraham, W., Benson, J., Cheng, Y.S., Johnson, D., Pierce, R., 2004.Literature review of Florida red tide: implications for human healtheffects. Harmful Algae 3, 99–115.
Kong, R.Y.C., Lee, S.K.Y., Lee, T.F.W., Law, S.H.W., Wu, R.S.S., 2002.Rapid detection of six types of bacterial pathogens in marine waters bymultiplex PCR. Water Research 36, 2802–2812.
Kreader, C.A., 1998. Persistence of PCR-detectable bacteroides distasonisfrom human feces in river water. Applied and Environment Microbi-ology 64, 4103–4105.
LaGier, M.J., Scholin, C.A., Fell, J.W., Wang, J., Goodwin, K.D., 2005.An electrochemical RNA hybridization assay for detection of the fecalindicator bacterium Escherichia coli. Marine Pollution Bulletin 50,1251–1261.
LaGier, M.J., Joseph, L.A., Passaretti, T.V., Musser, K.A., Cirino, N.M.,2004. A real-time multiplexed PCR assay for rapid detection anddifferentiation of Campylobacter jejuni and Campylobacter coli.Molecular and Cellular Probes 18, 275–282.
Landsberg, J.H., 2002. The effects of harmful algal blooms on aquaticorganisms. Reviews in Fisheries Science 10, 113–390.
Leclerc, H., Schwarzbrod, L., Dei-Cas, E., 2002. Microbial agentsassociated with waterborne diseases. Critical Reviews in Microbiology28, 371–409.

770 M.J. LaGier et al. / Marine Pollution Bulletin 54 (2007) 757–770
Lidie, K.B., Ryan, J.C., Barbier, M., Van Dolah, F.M., 2005. Geneexpression in Florida red tide dinoflagellate Karenia brevis: analysis ofan expressed sequence tag library and development of DNA micro-array. Marine Biotechnology 7, 481–493.
Lipp, E.K., Farrah, S.A., Rose, J.B., 2001. Assessment and impact ofmicrobial fecal pollution and human enteric pathogens in a coastalcommunity. Marine Pollution Bulletin 42, 286–293.
Litaker, W., Sundseth, R., Wojciechowski, M., Bonaventura, C., Hen-kens, R., Tester, P. 2001. Electrochemical detection of DNA or RNAfrom harmful algal bloom species. in: Harmful Algal Blooms 2000,Paris, pp. 242–245.
Mason, W.J., Blevins, J.S., Beenken, K., Wibowo, N., Ojha, N., Smeltzer,M.S., 2001. Multiplex PCR protocol for the diagnosis of Staphylo-coccal infection. Journal of Clinical Microbiology 39, 3332–3338.
Maurer, J.J., Schmidt, D., Petrosko, P., Sanchez, S., Bolton, L., Lee,M.D., 1999. Development of primers to O-antigen biosynthesis genesfor specific detection of Escherichia coli O157 by PCR. Applied andEnvironmental Microbiology 65, 2954–2960.
Metfies, K., Huljic, S., Lange, M., Medlin, L.K., 2005. Electrochemicaldetection of the toxic dinoflagellate Alexandrium ostenfeldi with aDNA-biosensor. Biosensors and Bioelectronics 20, 1349–1357.
Millie, D.F., Schofield, O.M., Vinyard, B.T., 1997. Detection of harmfulalgal blooms using photopigments and absorption signatures: a casestudy of the Florida red tide dinoflagellate, Gymnodinium breve.Limnology and oceanography 42, 1240–1251.
Mumy, K.L., Findlay, R.H., 2004. Convenient determination of DNAextraction efficiency using an external DNA recovery standard andquantitative-competitive PCR. Journal of Microbiological Methods57, 259–268.
Noble, R.T., Furhman, J.A., 2001. Enteroviruses detected by reversetranscriptase polymerase chain reaction from the coastal waters ofSanta Monica Bay, California: low correlation to bacterial indicatorlevels. Hydrobiologia 460, 175–184.
Noble, R.T., Weisberg, S.B., 2005. A review of technologies beingdeveloped for rapid detection of bacteria in recreational waters.Journal of Water and Health 3, 381–392.
Percival, S.L., Chalmers, R.M., Embrey, M., Hunter, P.R., Sellwood, J.,Wyn-Jones, P., 2004. Microbiology of Waterborne Diseases. ElsevierAcademic Press, pp. 21–209.
Rizzo, P.J., Jones, M., Ray, S.M., 1982. Isolation and properties ofisolated nuclei from the Florida red tide dinoflagellate Gymnodinium
breve. Journal of Phycology 29, 217–222.Scott, T.M., Jenkins, T.M., Lukasik, J., Rose, J.B., 2005. Potential use of
a host associated molecular marker in Enterococcus faecium as anindex of human fecal pollution. Environmental Science and Technol-ogy 39, 283–287.
Shibata, T., Solo-Gabriele, H.M., Fleming, L.E., Elmir, S., 2004.Monitoring marine recreational water quality using multiple microbialindicators in an urban tropical environment. Water Research 38, 3119–3131.
Sinigalliano, C.D., Gidley, M.L., Shibata, T., Dixon, T.H., Whitman, D.,Bachoon, D., Brand, L., Amaral-Zettler, L., Gast, R., Nigro, O.D.,Steward, G.F., Hou, A., Mathews, J., Laws, E., Fujioka, R., Solo-Gabriele, H.M., Fleming, L.E., submitted for publication. Microbialmeasurements in water and sediments of the New Orleans area in theaftermath of hurricanes Katrina and Rita.
Steidinger, K.A., Landsberg, J.H., Truby, E.W., Roberts, B.S., 1998. Firstreport of Gymnodinium pulchellum (Dinophyceae) in North Americaand associated fish kills in the Indian River, Florida. Journal ofPhycology 34, 431–437.
Tester, P.A., Steidinger, K.A., 1997. Gymnodinium breve red tide blooms:initiation, transport, and consequences of surface circulation. Limnol-ogy and oceanography 42, 1039.
Tortora, G.J., Funke, B.R., Case, C.L., 2001. Microbiology, An Intro-duction, 7th ed. Addison Wesley Longman, Inc., San Francisco.
Wade, T.J., Pai, N., Eisenberg, J.N.S., Colford Jr., J.M., 2003. Do U.S.Environmental Protection Agency water quality guidelines for recre-ational waters prevent gastrointestinal illness? A systematic review andmeta-analysis. Environmental Health Perspectives 111, 1102–1109.
Wang, J., 2000. Analytical Electrochemistry, 2nd ed. John Wiley and SonsPress, New York.
Wojciechowski, M., Sundseth, R., Moreno, M., Henkens, R., 1999.Multichannel electrochemical detection system for quantitative mon-itoring of PCR amplification. Clinical Chemistry 45, 1690–1693.
Youmans, G.P., Paterson, P.Y., Sommers, H.M., 1985. The Biologicaland Clinical Basis of Infectious Diseases, 3rd ed. The W.B. SaundersCompany, Philadelphia.
Zhang, Y., Kim, H.H., Heller, A., 2003. Enzyme-amplified amperometricdetection of 3000 copies of DNA in a 10-ll droplet at 0.5 fMconcentration. Analytical Chemistry 85, 3267–3269.




















