
Genetic Variation in the Folate Receptor-α and ... - DiVA Portal
76
Genetic Variation in the Folate Receptor- α and Methylenetetrahydrofolate Reductase Genes as Determinants of Plasma Homocysteine Concentrations
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of Genetic Variation in the Folate Receptor-α and ... - DiVA Portal

Genetic Variation in the Folate Receptor-α and Methylenetetrahydrofolate Reductase Genes
as Determinants of Plasma Homocysteine Concentrations

To Edvin and Karl
Ja visst gör det ont när knoppar brister.Varför skulle annars våren tveka? Karin Boye

Örebro Studies in Medicine 25
Anna Böttiger
Genetic Variation in the Folate Receptor-α and Methylenetetrahydrofolate Reductase Genes
as Determinants of Plasma Homocysteine Concentrations

© Anna Böttiger, 2008
Title: Genetic Variation in the Folate Receptor-α and Methylenetetrahydrofolate Reductase Genes as Determinants
of Plasma Homocysteine Concentrations.
Publisher: Örebro University 2008 www.publications.oru.se
Editor: Maria Alsbjer [email protected]
Printer: Intellecta DocuSys, V Frölunda 11/2008
issn 1652-4063 isbn 978-91-7668-642-3

AbstractAbstractAbstractAbstract Anna Böttiger (2008): Genetic Variation in the Folate Receptor-α and Methylenetetrahy-drofolate Reductase Genes as Determinants of Plasma Homocysteine Concentrations, Örebro Studies in Medicine 25, 74 pp. Elevated total plasma homocysteine (tHcy) is a risk factor for cardiovascular disease and neurocognitive disease such as dementia. The B vitamins folate and B12 are the main de-terminants of tHcy. tHcy concentration can also be affected by mutations in genes coding for receptors, enzymes and transporters important in the metabolism of Hcy. This thesis focuses on mutations in the genes for folate receptor-α and methylenetetrahydrofolate reductase (MTHFR) and the effect they have on tHcy concentrations.
Six novel mutations in the gene for folate receptor-α were described in Paper I. Taken together they exist in a population with a prevalence of approximately 1% and thus are not unusual There may be an association of –69dupA and –18C>T to tHcy but for the 25-bp deletion, –856C>T, –921T>C and –1043G>A there is probably no associa-tion to tHcy. Mutation screening was continued and four additional mutations, 1314G>A, 1816delC, 1841G>A and 1928C>T, were described in Paper II. The preva-lences for the heterozygotes were between 0.5% and 13% in an elderly population. There was no significant difference in prevalence between the elderly subjects and pa-tients with dementia. The 1816(–)-allele and the 1841A-allele were in complete linkage and the haplotype 1816(–)-1841A may possibly have a tHcy raising effect. The 1314G>A and 1928C>T mutations had no association to tHcy.
The genotype prevalences and haplotype frequencies of the MTHFR 677C>T, 1298A>C and 1793G>A polymorphisms were determined in a population sample of Swedish children and adolescents (Paper III). The MTHFR 677T-allele was associated with increased tHcy concentrations in both children and adolescents. A small elevating effect of the 1298C-allele and a small lowering effect of the 1793A-allele could be shown. In an epidemiological sample of adults from the Canary Islands, Spain, data for serum folate and vitamin B12 were used for a broader study of the nutrigenetic impact on tHcy (Paper IV). The 677T-allele had a significant tHcy increasing effect in men but not in women. The 1298C-allele had a minor elevating effect on tHcy in men with the 677CT genotype. It was not possible to document any effect of the 1793A-allele on tHcy due to its low prevalence. A slightly superior explanatory power for the genetic impact was obtained using the MTHFR haplotypes in the analysis compared to the MTHFR 677C>T genotype-based approach in both the Swedish children and adolescents and in the Spanish adults. Therefore MTHFR haplotypes should be considered when analysing the impact of the MTHFR 677C>T, 1298A>C and 1793G>A polymorphisms on tHcy.
Notwithstanding the large geographical distance between our study populations the haplotype composition is quite similar. The MTHFR 677T-allele is slightly more preva-lent in Spain compared to Sweden but it has only an effect on tHcy in the Spanish men. Age, gender and factors linked to the ethnicity of the studied subjects, seem to be able to override the nutrigenetic impact of tHcy-raising genotypes or haplotypes in particular settings, such as in the Spanish women in our study. Gene-nutrient interactions on plasma tHcy levels thus may or may not exist in a certain population. The transferability of nutrigenetic findings may therefore be limited, and must be re-evaluated for each par-ticular setting of age-gender-ethnicity. Keywords: Folate receptor-α, folate, FOLR1, Haplotypes, Homocysteine, Methylenetet-rahydrofolate Reductase, MTHFR, Polymorphisms, Pyrosequencing®, SSCP. Anna Böttiger, Department of Clinical Chemistry, Örebro University Hospital, SE-701 85 Örebro, Sweden. E-mail: [email protected]


SammanfattningSammanfattningSammanfattningSammanfattning
Aminosyran homocystein (Hcy) uppkommer i kroppen genom metabolism av den livsnödvändiga aminosyran metionin. Förhöjda nivåer av total Hcy i plasma (tHcy) har visat sig i flera studier att vara en riskfaktor för hjärtkärlsjukdomar och neuro-kognitiva sjukdomar såsom demens. Det är i första hand B-vitaminerna folat och B12 som styr Hcy-nivåerna i blodet. Den individuella Hcy-koncentrationen kan också påverkas av mutationer i gener som kodar för receptorer, enzymer och transportörer som är viktiga i metabolismen av Hcy. Mutationer som påverkar individens tHcy-koncentration i relation till deras näringsintag är ett exempel på en forskningslinje som idag benämns Nutrigenetics, alltså samspelet nutrition-genetik-fenotyp. I den här avhandlingen har sambandet mellan mutationer i generna för folatreceptor-α och enzymet metylentetrahydrofolatreduktas och tHcy-koncentrationer studerats. I artikel I beskrivs sex nya mutationer i genen för folatreceptor-α. Dessa muta-tioner är inte helt ovanliga, sammantaget finns de i befolkningen i en prevalens av nästan 1 %. Ett samband med Hcy-koncentrationer kan vara möjligt för mutationer-na –69dupA och –18C>T men är mindre troligt för 25 bp-deletionen, –856C>T, –921T>C och –1043G>A. Fortsatt mutationsscreening (artikel II) från exon 1 i riktning nedströms ca 1 kb i genen visade fyra mutationer, varierande prevalenser för heterozygoterna mellan 0,5 % och 13 % i en äldre frisk befolkning. Det fanns ingen signifikant prevalensskillnad mellan dementa och en äldre frisk befolkning, men allelfrekvenserna för två av dem, g.1816delC och g.1841G>A, var tre gånger högre bland de dementa. De muterade allelerna 1816(–) och 1841A förekom alltid tillsammans hos de genotypade perso-nerna. Detta tyder på att de alltid ärvs tillsammans i en s.k. haplotyp. Den här ovan-liga haplotypen kan ha en liten Hcy-höjande effekt. De två vanligt förekommande mutationerna g.1314G>A och g.1928C>T verkade inte ha någon Hcy höjande effekt. I genen för metylentetrahydrofolatreduktas (MTHFR) finns flera mutationer beskrivna. I artikel III studerades sambandet mellan tre av dem (MTHFR 677C>T, 1298A>C och 1793G>A) och tHcy-nivåerna i 692 Mellansvenska barn och ungdo-mar. MTHFR 677C>T mutationen hade en tHcy-höjande effekt hos både barnen och ungdomarna. MTHFR 677C>T påverkar enzymets katalytiska egenskaper. MTHFR 1298A>C hade en viss tHcy-höjande effekt och en viss tHcy-sänkande effekt av 1793G>A mutationerna kunde beläggas. Genom utvidgning till haplotypbaserad ana-lys kunde en högre förklaringsgrad uppnås av variansen i tHcy-nivåer. Därför ska haplotyperna användas vid analys av MTHFR 677C>T, 1298A>C och 1793G>A mutationernas inflytande på tHcy. I artikel IV bestämdes genotypprevalenserna och haplotypprevalenserna av MTHFR mutationerna i 723 vuxna spanjorer bosatta på Kanarieöarna. I detta mate-rial fanns tillgång till personernas serum-folat och vitamin B12 nivåer och detta an-vändes för att göra en fördjupad analys av sambandet mellan genotyp-haplotyp-vitaminnivåer. I detta material kunde inte samma förklaringsgrad av tHcy-nivåer av MTHFR mutationerna uppnås som i artikel III. MTHFR 677C>T hade bara effekt på tHcy-koncentrationer hos män men inte hos kvinnor. MTHFR 1298A>C hade en viss tHcy-höjande effekt hos män med MTHFR 677CT-genotypen. Ingen effekt av MTHFR 1793G>A på Hcy-nivåer kunde påvisas vare sig hos kvinnor eller hos män. Detta visar att överförbarheten till andra länders förhållanden av slutsatser man dra-git av fynd rörandet sambandet mellan genotyp-fenotyp i ett visst land inte kan tas för given utan måste fastställas i varje aktuell befolkning med hänsynstagande till dess genetiska struktur och nutritionsmönster.


List of publicationsList of publicationsList of publicationsList of publications
I.
Börjel A.K, Yngve A, Sjöström M, Nilsson T.K. Novel mutations in the 5’UTR of
the FOLR1 gene.
Clinical Chemistry and Laboratory Medicine 2006;44(2): 161-167.
II.
Böttiger A.K, Hagnelius N.O, Nilsson T.K. Mutations in exons 2 and 3 of the
FOLR1 gene in demented and non-demented elderly subjects.
International Journal of Molecular Medicine 2007;20(5): 653-662.
III.
Böttiger A.K, Hurtig-Wennlöf A, Sjöström M, Yngve A, Nilsson T.K. Association
of total plasma homocysteine with methylenetetrahydrofolate reductase geno-
types 677C>T, 1298A>C, and 1793G>A and the corresponding haplotypes in
Swedish children and adolescents.
International Journal of Molecular Medicine 2007;19(4): 659-665.
IV.
Böttiger A.K, Nilsson T.K, Henriquez P, Serra Majem L. Plasma homocysteine
and MTHFR genotypes and haplotypes: gene-nutrient interactions in the Canary
Islands Nutrition Study (ENCA).
Manuscript (2008).

AbbreviationsAbbreviationsAbbreviationsAbbreviations
AdoHcy S-adenosylhomocysteine
APS Adenosine 5’phosphate
BHMT Betaine homocysteine methyltransferase
bp Base pair
CBS Cystathionine β-synthase
CTH Cystathionine �-lyase
CVD Cardiovascular disease
FOLR1 The gene for folate receptor-α
FR-αααα Folate receptor-α
Hcy Homocysteine
MAT Methionine adenosine transferase
Met Methionine
MTHFR 5,10-Methylenetetrahydrofolate reductase
MTR Methionine synthase
MTRR Methionine Synthase Reductase
NMDA N-methyl-D-aspartate
NTDs Neural tube defects
PCFT Proton-coupled folate transporter
PCR Polymerase Chain Reaction
PON1 Paraoxonase
PPi Pyrophosphate
RFC Reduced folate carrier
SAM S-adenosylmethionine
SNP Single Nucleotide Polymorphism
SSCP Single Strand Conformation Polymorphism
tHcy Total plasma homocysteine
THF Tetrahydrofolate
TCII Transcobalamin II
TF Transcription factor
UTR Untranslated region

ContentsContentsContentsContents
BACKGROUNDBACKGROUNDBACKGROUNDBACKGROUND _____________________________________________________ 13
HOMOCYSTEINE_____________________________________________________ 13 DETERMINANTS OF PLASMA HOMOCYSTEINE CONCENTRATIONS ________________ 14
INTRODUCTIONINTRODUCTIONINTRODUCTIONINTRODUCTION ____________________________________________________ 15
HOMOCYSTEINE METABOLISM__________________________________________ 15 FOLATE ___________________________________________________________ 16 FOLATE RECEPTOR-α_________________________________________________ 16 FOLATE RECEPTOR-α DURING DEVELOPMENT ______________________________ 18 METHYLENETETRAHYDROFOLATE REDUCTASE _____________________________ 18 MTHFR POLYMORPHISMS _____________________________________________ 19
MTHFR 677C>T (rs1801133) _______________________________________ 19 MTHFR 1298A>C (rs1801131) ______________________________________ 20 MTHFR 1793G>A (rs2274976) ______________________________________ 20 Other mutations in the MTHFR gene __________________________________ 21 MTHFR haplotypes _______________________________________________ 21 Composition of the MTHFR enzyme___________________________________ 22
OTHER GENETIC VARIATION IN THE HOMOCYSTEINE METABOLISM ______________ 23 HOMOCYSTEINE AS A RISK FACTOR FOR DISEASE____________________________ 25
How can homocysteine be pathogenic? ________________________________ 25 THE PRINCIPLE OF PYROSEQUENCING®___________________________________ 26
AIMS OF THIS THESISAIMS OF THIS THESISAIMS OF THIS THESISAIMS OF THIS THESIS ______________________________________________ 29
MATERIALS AND METHODMATERIALS AND METHODMATERIALS AND METHODMATERIALS AND METHODSSSS _________________________________________ 31
PATIENT SAMPLES INVESTIGATED FOR THCY OR MACROCYTOSIS _______________ 31 THE DEMENTIA, GENETICS AND MILIEU STUDY (DGM) ______________________ 31 ACTIVE SENIORS (AS)________________________________________________ 31 THE EUROPEAN YOUTH HEART STUDY (EYHS) ____________________________ 31 THE CANARY ISLANDS NUTRITION STUDY (ENCA) _________________________ 32 ETHICS____________________________________________________________ 32 MOLECULAR BIOLOGY TECHNIQUES _____________________________________ 32
DNA extraction from blood samples __________________________________ 32 Polymerase Chain Reaction (PCR) ___________________________________ 32 Mutation screening by Single Strand Conformation Polymorphism (SSCP)____ 33 DNA Sequencing__________________________________________________ 33 Analysis of polymorphisms by Pyrosequencing® ________________________ 34
MEASUREMENT OF THCY, FOLATE AND VITAMIN B12 ________________________ 34 STATISTICS ________________________________________________________ 34
RESULTSRESULTSRESULTSRESULTS __________________________________________________________ 35
NOVEL MUTATIONS IN THE PROMOTER REGION AND 5’-UTR OF THE FOLATE RECEPTOR-α GENE ___________________________________________________________ 35
Nomenclature of the mutations_______________________________________ 40 Transcription factors ______________________________________________ 41
PYROSEQUENCING® ASSAYS FOR MTHFR POLYMORPHISMS __________________ 42 PLASMA HOMOCYSTEINE LEVELS AND MTHFR POLYMORPHISMS ______________ 44

MTHFR polymorphisms ____________________________________________ 44 Association between MTHFR polymorphisms and tHcy concentrations _______ 48 MTHFR 677C>T _________________________________________________ 48 MTHFR 1298A>C ________________________________________________ 48 Haplotype analysis ________________________________________________ 49 MTHFR 1793G>A ________________________________________________ 51 Diplotypes_______________________________________________________ 52
DISCUSSION AND CONCLDISCUSSION AND CONCLDISCUSSION AND CONCLDISCUSSION AND CONCLUSIONSUSIONSUSIONSUSIONS ___________________________________ 55
NOVEL MUTATIONS IN THE PROMOTER REGION AND 5’-UTR OF THE FOLATE RECEPTOR-α GENE ___________________________________________________________ 55 MTHFR POLYMORPHISMS AND PLASMA HOMOCYSTEINE LEVELS_______________ 57
FUTURE PERSPECTIVESFUTURE PERSPECTIVESFUTURE PERSPECTIVESFUTURE PERSPECTIVES ____________________________________________ 59
TACKTACKTACKTACK ______________________________________________________________ 61
REFERENCESREFERENCESREFERENCESREFERENCES ______________________________________________________ 63

13
BackgroundBackgroundBackgroundBackground
An increased plasma concentration of homocysteine (Hcy) is an independent risk
factor for cardiovascular disease, for neural tube defects and other birth defects.5
Many studies have described a link between impaired Hcy metabolism and neu-
ropsychiatric disorders such as depression6 and cognitive impairment in the eld-
erly.5, 93 A severe mutation in the enzyme Cystathionine β-synthase (CBS) results
in the disease homocystinuria which has very high levels of plasma Hcy. The
clinical symptoms involve the eyes and the central nervous, skeletal and vascular
systems. Patients with this mutation are often mentally retarded, with only one
third having normal intelligence.2 Homocystinuria is associated with several other
mutations in enzymes involved in homocysteine metabolism. Common to all these
mutations is that they give rise to vascular pathology and mental disturbances. All
this suggests that high plasma concentrations of Hcy are probably detrimental to
the nervous system.
Folate receptor-α (FR- α) is a cell receptor that is one of the routes for the B-
vitamin folate to enter cells. Folate is one of the major determinants of plasma
Hcy concentrations, low folate intake leading to elevated plasma Hcy concentra-
tion.
Methylenetetrahydrofolate reductase (MTHFR) is an important enzyme in
Hcy metabolism, being a key enzyme in the process of transferring a methyl
group from folate to Hcy. To maintain normal concentrations of Hcy in plasma it
is important that both FR- α and MTHFR are functioning correctly. Mutations in
the genes for FR- α and MTHFR, depending on the location, can give rise to re-
ceptors or enzymes that are defective or are produced to a lesser extent and as a
consequence plasma Hcy concentrations will increase.
Homocysteine Homocysteine Homocysteine Homocysteine
Hcy is a sulphur-containing amino acid for which there is no genetic triplet. Hcy
is formed from methionine and was first described in 1932 by Butz and du Vi-
gneaud.12 They treated methionine with concentrated sulphuric acid and the
product they obtained was Hcy. Hcy was later (1962) identified in the urine of
some mentally retarded children and it was subsequently discovered that these
children had deficiency of the enzyme cystathionine β -synthase.5, 112

14
Figure 1. The homocysteine molecule.
Determinants of plasma homocysteine concentrationsDeterminants of plasma homocysteine concentrationsDeterminants of plasma homocysteine concentrationsDeterminants of plasma homocysteine concentrations
There are several factors which determine plasma levels of Hcy and vitamin defi-
ciency is one of the strongest. Folate, B12, B6, B2 and betaine are all important fac-
tors in Hcy metabolism. Furthermore, there are some common mutations in en-
zymes, transporters and receptors involved in Hcy metabolism that affect tHcy
levels. The liver and kidney are the most important organs for uptake and me-
tabolism of Hcy. Patients with renal diseases often have higher levels of tHcy
probably because of reduced renal clearance and reduced metabolism. 9, 34
Plasma tHcy concentration increases with age. The explanations are as fol-
lows: decreased kidney functions, general slowdown of metabolism, increased
intestinal malabsorption or insufficient nutritional supply. Men often have higher
tHcy than women. Before puberty girls and boys have similar tHcy levels (mean
values of about 6 μmol/L). Around the age of 40-42 years there is a difference of
about 2 μmol/L between men and women. One explanation is that men have rela-
tively more muscle mass and therefore produce more creatinine and when
creatinine is formed Hcy is also formed.5 Hormonal status is another explana-
tion.26
Life style factors are also important for tHcy levels. Smoking, high consump-
tion of alcohol and inadequate nutrition are factors that increase the tHcy levels.
High coffee consumption is also correlated with increased tHcy.74 In most cases a
combination of factors rather than a single factor lead to high levels of tHcy.5, 113
A decision limit for diagnosis of hyperhomocysteinaemia is set as 15 μmol/L in
our laboratory. Hyperhomocysteinaemia is classified in three levels: tHcy between
15 to 30 μmol/L is defined as moderate hyperhomocysteinaemia, tHcy between
31 and 100 μmol/L as intermediate hyperhomocysteinaemia and severe hyperho-
mocysteinaemia is defined as tHcy above 100 μmol/L.57

15
IntroductionIntroductionIntroductionIntroduction
Homocysteine metabolismHomocysteine metabolismHomocysteine metabolismHomocysteine metabolism
Hcy is formed by demethylation of methionine. Methionine is an essential amino
acid and is used both for protein synthesis and as a methyl group donor. Me-
thionine is activated to form S-adenosylmethionine (SAM), which is the universal
methyl group donor in several reactions e.g. methylation of DNA. When the
methyl group is removed from SAM through the enzyme methionine adenosine
transferase (MAT), S-adenosylhomocysteine (AdoHcy) is formed. AdoHcy is hy-
drolysed to generate Hcy and adenosine. Hcy metabolism has two pathways: re-
methylation back to methionine or transsulphuration to cystathionine (Figure 2).
Figure 2. The metabolism of homocysteine. Enzymes, receptors and transporters in the
metabolism of Hcy with some of the known polymorphisms are shown.71 Reproduced by
permission from the authors.

16
Re-methylation: Hcy can be re-methylated back to methionine by the en-
zyme methionine synthase (MTR) which requires folate, in the form of 5-
methyltetrahydrofolate (5-methyl-THF), as methyl donor and vitamin B12 in the
form of methylcobalamin as cofactor. The enzyme MTHFR catalyses the reduc-
tion of 5,10-methylenetetrahydrofolate to 5-methyl-THF. In the liver where me-
thionine metabolism is very active, the enzyme betaine homocysteine methyltrans-
ferase (BHMT) participates in the re-methylation of Hcy. The methyl group
comes, in this reaction, from betaine or trimethylglycine.5, 10, 91, 101
Transsulphuration: In this pathway Hcy condenses with the amino acid ser-
ine to form cystathionine, catalysed by the enzyme cystathionine β -synthase,
which requires pyridoxyl 5’phosphate (the active form of vitamin B6) for its activ-
ity. Cystathionine is further metabolized to cysteine and α-ketobutyrate. Excess
cysteine can be excreted in the urine.5, 10, 91, 101 The transsulphuration pathway is
activated only if there is an excess of dietary methionine 101 and the pathway is
limited to the liver, kidney, pancreas and small intestine.10
FolateFolateFolateFolate
Folates are one-carbon donors required in Hcy metabolism, for methylation reac-
tions and also for purine and thymidylate biosynthesis. Following absorption in
the intestines by the proton-coupled folate transporter (PCFT) folates are deliv-
ered via the hepatic portal system to the liver where they are stored as polygluta-
mate derivatives. Folate, mostly in the form of 5-methyl-THF, is released from the
liver into the blood stream and transported into most body cells by either folate
receptors (FR) or by the reduced folate carrier (RFC).121 Folate is of great impor-
tance in foetal development and optimal folate status has been shown to protect
against neural tube defects (NTDs).1, 31, 80 Studies have also shown that folate is of
importance for cognitive functions in adults. Durga et al reported that deficient
levels of erythrocyte folate were linked to poor performance in tests probing age-
related cognitive decline 29 and in a more recent study the same group showed
that 3-year folic acid supplementation improved performance in tests that meas-
ure information-processing speed and memory in older adults with raised tHcy
concentrations.30 In the Nun study it was demonstrated that low folate concentra-
tions were related to atrophy of the neocortex in persons with a significant num-
ber of Alzheimer disease lesions.96
Folate receptorFolate receptorFolate receptorFolate receptor----αααα
Folate is a water-soluble B vitamin and it needs to be actively transported into the
cells. Folate receptor-α (FR-α), a membrane receptor, has 100-200 times greater

17
affinity for plasma folate than the ubiquitous reduced folate carrier (RFC).56 FR-α
is attached to the cell surface by a glycosyl phosphatidylinositol (GPI) anchor
which recycles between the extracellular and endocytic compartments.31 FR-α has
a limited tissue distribution, being present in placenta, kidney and choroid plexus
(a network of small blood vessels in the ventricles of the brain which produce
cerebrospinal fluid) and it is possible that the primary role of the receptor is to
concentrate and/or conserve folate in selected compartments, such as in the foetus
and the central nervous system.68 Tissues that lack FR-α have their need for folate
uptake met by RFC.31 The gene for FR-α, named FOLR1, consists of seven exons
which spans over 6.70 kbp32 and is located on chromosome 11q13.7 The gene has
two TATA-less promoters (P1 and P4) located upstream of exon 1 and 4. P1 gen-
erates several transcripts whereas P4 only give rise to a single transcript. The
transcription of the FR-α gene in the kidney, testis and brain appear to originate
from P1. In the lung, placenta, salivary gland, uterus, breast and stomach the
transcription appears to be primarily driven by P4.31 Exon 4 to 7 encodes the FR-
α protein.32 Two studies attempted to identify mutations in exons 3 to 7 without
success.4, 45 Heil et al suggested that FR-α plays such an important part in the de-
velopment of the embryo that abnormalities in the genes coding for this receptor
are lethal.45 However, other studies have reported that there are some mutations
in the highly conserved exons. De Marco et al reported insertions of pseudogene-
specific mutations in exon 7 and 3’-UTR of the FOLR1. They also reported the
finding of a subject with a gene conversion event in the coding region.22 A re-
cently published Chinese study has screened the exons in a gastric cancer cell line
and in gastric tumours for mutations. They report the finding of two polymor-
phisms A1314G and C1816delC in exons 2 and 3.120
Figure 3. Gene structure of the folate receptor-α gene. The figures and letters indicate
exons and introns, respectively. The two promoters P1 and P4 are marked. The figures
indicate the number of base pairs in the corresponding exon or intron.31 Reproduced by
permission from the authors.

18
Folate receptorFolate receptorFolate receptorFolate receptor----αααα during development during development during development during development
Folate is essential for normal foetal development and deficiency during pregnancy
of this vitamin has been shown to result in NTDs.1, 31, 80 More support for the im-
portant role of folate during pregnancy was obtained from transgenic mouse
models. Mouse embryos which were homozygous for a deletion in Folbp1 (the
murine homolog to FR-α) that knocked out the gene, died in the uterus, exhibit-
ing failure of neural closure.79 Mice heterozygous for the Folb1 deletion showed
normal development despite having approximately 30% lower circulating folate
compared to wild type mice.79
Methylenetetrahydrofolate reductaseMethylenetetrahydrofolate reductaseMethylenetetrahydrofolate reductaseMethylenetetrahydrofolate reductase
Methylenetetrahydrofolate reductase (MTHFR) is a flavoprotein that is involved
in the methylation of Hcy. In an NADPH linked process it catalyses the reduction
of 5’10-methylenetetrahydrofolate to 5-methyl-THF which is the main compo-
nent of plasma folate. 5-methyl-THF acts as the major methyl donor in the con-
version of Hcy to methionine.41, 103, 116 Human MTHFR is a dimer consisting of
two monomers of approximately 70 kDa each and each subunit contains nonco-
valently bound FAD. Each monomer contains a catalytical domain that binds the
FAD cofactor and folate, and a regulatory domain that binds SAM.103, 116 Studies
on porcine MTHFR have shown that the activity of MTHFR is allosterically in-
hibited by SAM in response to the level of methionine in the cell.98, 116 Studies on
human recombinant MTHFR have shown that the human enzyme has properties
that are generally similar to the porcine MTHFR.116 Goyette et al mapped the
gene for MTHFR to chromosomal region 1p36.3 (Figure 4).41 The gene contains
11 exons ranging in size from 102 bp to 432 bp.40
Figure 4. The gene for MTHFR is located at the end of the short arm of chromosome 1.
Reproduced with permission from NCBI (http://ghr.nlm.nih.gov/gene=mthfr).

19
There is no TATA-box in the promoter region but it does contain CpG-islands.37
There are multiple transcriptional and translational start sites in the gene for
MTHFR and there are several alternative splicing sites in exon 1. Homberger et al
described three different transcripts of the gene which differed in the first exons49,
which leads to several isoforms of MTHFR.102 The alternative splicing sites in
exon 1 generate several 5’UTRs. The length of the 5’UTR has been shown to in-
fluence translational efficiency, typically the shorter and less GC-rich 5’UTR the
more efficient translation.102, 106 In humans a MTHFR polypeptide of about 77
kDa has been found and a polypeptide of about 70 kDa has been found solely in
the human liver.36 MTHFR is a key enzyme in Hcy-metabolism and to retain Hcy
levels within the normal range the enzyme must be functional. Mutations in the
gene, which are not uncommon, can affect the activity of the enzyme and thus the
tHcy concentrations.
MTHFR polymorphismsMTHFR polymorphismsMTHFR polymorphismsMTHFR polymorphisms
MTHFR 677C>T (rs1801133)
In 1988 Kang et al detected a variant of MTHFR that was associated with de-
creased enzyme activity and thermolability.59 Some years later the same group
found that this variant was associated with increased tHcy concentrations and
they also demonstrated that it was associated with cardiovascular disease.58 In
1995 Frosst et al identified the mutation in the MTHFR gene that caused the de-
creased enzyme activity and the thermolability. The mutation was a C to T substi-
tution at nucleotide 677 (exon 4) and it converted an alanine residue to a valine
residue at codon 222.36, 101 Frosst et al found that the three genotypes (CC (wild
type), CT and TT) were all significantly different with respect to enzyme thermo-
lability. They also demonstrated that individuals with the TT-genotype had sig-
nificantly elevated tHcy concentrations.36 The 677C>T polymorphism leads to
altered binding of FAD and thus reduced activity.42, 116
The 677C>T polymorphism exists in most populations but the distribution
of the allele shows marked ethnic and geographical variation. The TT genotype is
most common in northern China (20%), southern Italy (26%) and Mexico
(32%). There are also geographical gradients in Europe (north to south increase)
and in China (north to south decrease). The frequency of TT is low among new-
borns of African ancestry, intermediate among newborns of European ancestry
and high among newborns of American Hispanic origin.111
The reported frequencies in the SNP database (rs1801133) for the MTHFR
677T-allele range from 0.237 to 0.250 in Europeans, 0.333 and 0.511 in Asian
populations and 0.108 to 0.110 in African populations.21 Numerous studies have

20
shown that the TT genotype is associated with increased plasma Hcy concentra-
tions and in particular when folate status is low43, 44, 54 but the polymorphism has
no effect on tHcy when folate status is high.39, 54
The negative effect of the 677C>T polymorphism has been well investigated.
However, the survival of the 677C>T polymorphism probably reflects benefits of
the T-allele in some circumstances. Studies have shown that it may have a protec-
tive effect against colon cancer when folate status is adequate66 and also against
leukemias.95, 110 A proposed mechanism for the protection could be that, due to
lower activity of the enzyme, there is an increased availability of 5,10-
methylenetetrahydrofolate for thymidine synthesis. This provides nucleotide pools
for DNA synthesis and repair and may reduce uracil mis-incorporation into
DNA. Uracil incorporation can result in chromosomal strand breaks during exci-
sion repair.102
Durga et al reported that subjects with the MTHFR 677TT genotype per-
formed better on cognitive tests and this association was strongest among those
with high folate intake. In addition the MTHFR 677TT genotype offered protec-
tion against hearing loss when folate status was above the population median.28, 29
MTHFR 1298A>C (rs1801131)
The MTHFR 1298 A>C polymorphism was described in 1998 in two studies.105,
107 This SNP, an A>C transversion at nucleotide 1298 (exon 7), leads to a gluta-
mate to alanine substitution at codon 429101. The 1298A>C polymorphism has an
impact on the activity of the enzyme but less than that of the 677C>T SNP105, 108
and in contrast to the 677C>T SNP it does not give a thermolabile enzyme.108 The
SNP is found in the C-terminal regulatory domain.108 The effect of the MTHFR
1298A>C polymorphism on tHcy is not clear. Some studies report no effect on
tHcy 35, 108 while others report that the 1298C-allele is associated with increased
tHcy.103 However, several studies do report an association to increased tHcy
when present together with the 677C>T SNP.105, 108
The reported frequencies of the MTHFR 1298C-allele, in dbSNP, range
from 0.292 to 0.358 in Europeans, from 0.146 to 0.202 in Asian populations and
from 0.102 to 0.108 African populations south of the Sahara.19 The MTHFR
1298C-allele is uncommon in black populations. 89
MTHFR 1793G>A (rs2274976)
Rady et al described, in 2001, a novel polymorphism in the MTHFR gene, the
1793G>A mutation.81 The mutation is a G to A change in exon 11, resulting in an
amino acid substitution of arginine to glutamine at codon 594.81 The MTHFR
1793G>A has a lower prevalence compared to the 677C>T and 1298A>C poly-

21
morphisms and it is much less studied. The association of the 1793G>A poly-
morphism to tHcy is not clear.
The reported frequencies of the MTHFR 1793A-allele are between 0.043
and 0.058 in Europeans, between 0.089 and 0.100 for Asians and 0.033 in Afri-
can populations south of the Sahara. The polymorphism is most common in
Asian populations and the MTHFR 1793AA prevalence can be as high as 4.5%.20
Other mutations in the MTHFR gene
Several other mutations have been found in the MTHFR gene. Some rare muta-
tions, most of them being located in the 5’region encoding the catalytical domain,
have been reported in homocystinuric patients. Most of these mutations are mis-
sense mutations but some are nonsense and splice site mutations and one deletion
has been described. These mutations are rare but lead to severe deficiency of the
enzyme with residual enzyme activity between 0 – 20% activity of the wildtype
enzyme. The mutations are associated with homocystinuria as well as severe neu-
rological and vascular abnormalities.90, 102
MTHFR haplotypes
The effect of the simultaneous occurrence of the MTHFR 677C>T and 1298A>C
polymorphisms upon tHcy is equivocal.15, 24, 63, 99, 103 The haplotype structure is
debatable: some subjects with two mutations on the same DNA strand (haplotype
677T-1298C) have even been reported.13, 53, 64, 103 The physical distance between
the MTHFR 677C>T and the 1298A>C polymorphisms is only 2.1 kb and it is
likely that this is too short for recombination to occur.97 The 677T variant may
have arisen later than the 1298C variant on a chromosome harbouring 1298A.86,
103 The haplotype might exist but can only be demonstrated in very large popula-
tion studies such as the study by Ulvik et al where they have genotyped more than
10 000 persons for the MTHFR 677C>T and 1298A>C polymorphisms.103
Wakutani et al presented a haplotype analysis of the three MTHFR muta-
tions 677 C>T, 1298 A>C and 1793 G>A. They found that the MTHFR gene had
four major haplotypes (Figure 5) and that one of them was protective against the
development of late-onset Alzheimer’s disease.104

22
Figure 5. A schematic presentation of the estimated regional haplotypes of the MTHFR
gene.104 Wakutani et al found four haplotypes (A, B, C, and D) for the three-locus sys-
tem, MTHFR 677C>T, 1298A>C, and 1793G>A.
Composition of the MTHFR enzyme
The MTHFR enzyme is a dimer consisting of two monomers that associate “head
to tail”. Each polypeptide has a catalytical and a regulatory domain. In Figure 6
the translation of the common haplotypes CA, CC and TA into polypeptides and
the six dimers is illustrated. Depending on the genotype, 1 or 3 of these configu-
rations is present for any given individual.103 The different configurations have
different stability. In vitro studies of the enzyme by Yamada et al showed that the
enzyme dimer dissociates into monomers upon dilution or heating but that folate
stabilises the enzyme.116 The dimer configuration E (see Figure 6) is the less stable
configuration because it arise from two copies of the 677T-allele. Configuration F
represents an enzyme that has the genotype 677 CT and 1298 AC, the compound
heterozygote. This enzyme is also unstable. However, these figures are based on
the assumption that all dimers are equally stable. Instability of configuration E
could shift for example the equilibrium for the CTAA genotype towards more
enzymes in the stable D configuration at the expense of A and E.103

23
Figure 6. A schematic figure over the composition of the MTHFR enzyme dimer variants
that occur due to the 677C>T and 1298A>C polymorphisms.103 On top, the three com-
mon alleles based on the 677 and 1298 positions and in the middle the associated poly-
peptides. Bottom, the polypeptides combine “head to tail” in six different configurations.
Reproduced with permission from the authors.
Other genetic variation in the homocysteine metabolismOther genetic variation in the homocysteine metabolismOther genetic variation in the homocysteine metabolismOther genetic variation in the homocysteine metabolism
Besides the MTHFR polymorphisms there are several other mutations in trans-
porters and enzymes involved in Hcy metabolism (Table I). For most of the muta-
tions it is uncertain if they have effects on tHcy. In the enzyme cystathionine β-
synthase there are some mutations that give total deficiency of the enzyme. Total
deficiency of the cystathionine β -synthase is one cause of homocystinuria. 17 In
addition to cystathionine β -synthase deficiency there can be deficiency of other
enzymes e.g. transcobalamin II (TCII) that cause homocystinuria. TC II is respon-
sible for the influx of vitamin B12 into the cells. B12 is needed for the conversion of
5-methyl-THF and Hcy to tetrahydrofolate and methionine.91 In the gene for
transcobalamin II there is a common mutation, 776C>G, which results in a sub-
stitution of proline for arginine at codon 259. Studies have shown that the mu-
tated genotype have lower levels of holo-TC (transcobalamin bound to transco-
balamin II)70 and that it influences the risk of spontaneous abortions in hu-
mans.119 Some enzymes and transporters in the metabolism of Hcy that contain
polymorphisms55 are listed in Table I (courtesy of Dr. Johan Hultdin, Umeå).

24
Table I. Common polymorphisms in enzymes and transporters involved in Hcy metabo-
lism.
Gene, polymorphism
Function
Frequency
Allele frequency
Methylenetetrahydrofolate Re-ductase (MTHFR 677C>T and 1298A>C)
Formation of 5-methyl tetrahydro-folate.
App 10 % homozy-gotes for both 677C-T and 1298A-C.
677C: 0.71 677T: 0.29 1298A: 0.68 1298C: 0.32
Methionine synthase (MTR 2756A>G)
Mediates the re-methylation of Hcy to methionine
AA: 61 % AG: 29 % GG: 10 %
A: 0.76 G: 0.24
Methionine synthase reductase (MTRR 66A>G)
Reactivation of methionine synthase
AA: 28 % AG: 49 % GG: 23 %
A: 0.52 G: 0.48
Betaine-homocysteine methyl-transferase (BHMT 742G>A)
Remethylation of Hcy to methionine
GG: 49 % GA: 39 % AA: 12 %
G: 0.68 A:0.32
Cystathion beta-synthase (CBS 844ins68)
Converts Hcy to methionine and has effects on methyla-tion capacity
No insertion: 90 % Heterozygotes: 10 %
No insertion: 0.95 Insertion: 0.05
Paraoxonase (PON1- LEU55MET/108C-T and GLN192ARG)
Functions as a thio-lactonase of Hcy and may lead to the formation of homo-cysteine thiolactone which may induce protein alteration.
LEU55MET LEU/LEU: 34 % LEU/MET: 53 % MET/MET: 13 % GLN192ARG GLN/GLN: 48 % GLN/ARG: 30 % ARG/ARG 22 %
LEU:0.60 MET:0.40 GLN:0.63 ARG:0.37
Transcobalamin II (TCII P259R, 776C>G)
Mediates the uptake of B12 in the cells.
775 GG: 30 % 775 GC: 50 % 775CC: 20 %
G: 0.55 C: 0.45
Glutamate carboxypeptidase II (GCP II 1571C-T)
Zinc dependent enzyme, which de-grades polygluta-mates to monoglu-tamates in the intes-tine.
CC: 89.6 %
CT: 10.1 %
TT: 0.3 %
C: 0.95
T: 0.05

25
Homocysteine as a risk factor for diseaseHomocysteine as a risk factor for diseaseHomocysteine as a risk factor for diseaseHomocysteine as a risk factor for disease
Hcy is a risk factor for several diseases. Neural tube defects (NTDs) arise early in
embryogenesis following a failure of the neural tube to close. Several studies have
reported that pregnant women who are folate deficient have a greatly increased
risk of having babies with NTDs.82, 83, 118 Elevated tHcy as been observed in NTD
mothers.82, 83, 122 Vitamin B12 deficiency during pregnancy results in elevated Hcy
values in the foetus and increases the risk of NTDs. Studies have also shown that
there is an increased risk of early spontaneous abortion among women with low
plasma folate levels.118 Folate supplementation decreases the occurrence and re-
occurrence of NTDs82, 118, 122 by up to 70-100%.45, 83
Elevated tHcy is a well-known risk factor for cardiovascular disease
(CVD).60, 109 Specifically, the participation of Hcy in thrombosis has been docu-
mented in many studies.8, 77 Many studies have also shown that elevated plasma
Hcy is a risk factor for neuropsychiatric disorders such as depression6 and cogni-
tive impairment such as dementia and Alzheimer’s disease in the elderly.5, 93
How can homocysteine be pathogenic?
There are several hypotheses to explain the pathogenicity of Hcy. The most
commonly suggested toxic mechanisms are oxidative injury by different mecha-
nisms: impaired methylation, impaired DNA synthesis/repair and excitotoxic ef-
fects (damage to nerve cells) mediated by N-methyl-D-aspartate (NMDA) gluta-
mate subreceptors. Other suggested mechanisms are interaction with inflamma-
tory mechanisms, and protein homocysteinylation.14, 27, 48, 62, 67, 78
As regards cardiovascular disease elevated Hcy levels are associated amongst
other thing with reduced vasodilatation. Hcy can for instance induce direct dam-
age to endothelial cells and also increase platelet activity. Hyperhomocysteinae-
mia inhibits nitric oxide synthase which leads to a decreased bioavailability of
nitric oxide. At normal levels of tHcy nitric oxide detoxifies Hcy by forming S-
nitroso-homocysteine which is a vasodilator.101 When Hcy is in excess it is not
totally detoxified by nitric oxide, the remainder being auto-oxidized with another
Hcy molecule and free radicals are generated that are toxic to endothelial cells.101,
117 Normally free radicals are neutralized by glutathione but excess Hcy decreases
glutathione perioxidase activity.101 Another mechanism of endothelial injury by
Hcy is by the reduced catabolism of asymmetric dimethylarginine which is a
strong inhibitor of nitric oxide synthase.101 When Hcy is in excess it may be con-
verted to the thioester homocysteine-thiolactone. In association with low-density
lipoprotein it produces atherogenic oxidative damage to the endothelium.101 Hcy

26
may activate platelets, increase platelet aggregation and adhesion. In homocys-
teinaemia platelet thromboxane A2 biosynthesis is increased and this may be a
contributor to the risk of thrombosis.101
Hcy has been found to be a risk factor for neurocognitive disease and the
link may be due to cerebrovascular as well as to direct toxic effects.75 Elevated
Hcy increases oxidative stress and neurons are very sensitive to attacks by free
radicals.5, 112 In neuron cell culture, folate deficiency and Hcy have been shown to
damage and kill the cells.69 The neuron damaging effect may also be caused by
Hcy acting as an agonist at the neurotransmitter glutamate binding site of the
NMDA receptor.65 Another hypothesis worthy of mention is that Hcy accelerates
dementia by stimulating amyloid beta deposition in the brain.75
Another overall explanation as to why Hcy is associated with disease is to
view it as a marker of disturbed intracellular metabolism of methyl groups. Hcy
induces DNA breakage possibly through impaired DNA transmethylation since
SAM levels are reduced as a consequence of folate and vitamin B12 deficiencies.
Damage to DNA means that the cells ATP reserves are depleted in attempting to
repair the DNA. Cellular depletion of ATP is thought to be an important factor in
neurodegeneration, for instance, in Alzheimer’s disease.69
The principle of PThe principle of PThe principle of PThe principle of Pyrosequencing®yrosequencing®yrosequencing®yrosequencing®
Pyrosequencing® technology is a sequencing by synthesis technique for quantita-
tive analysis of DNA sequences. The sequencing by synthesis technique offers the
advantage of real-time detection84 and the Pyrosequencing® technology was de-
scribed in the late 1990’s by a Swedish research group.84, 85 The method is based
on indirect luminometric quantification of pyrophosphate (PPi) that is released as
a result of nucleotide incorporation onto an amplified template.
PCR is performed using one of the primers in the PCR reaction modified
with biotin at the 5’-end leading to amplicons with a biotin molecule at the 5’-
end. The biotin binds to Sepharose beads and can therefore be transferred in the
different washing steps needed before sequencing, see Paper II. After the washing
steps, a sequencing primer is added and the DNA template is made single
stranded by heating to 80°C. The sequencing primer, which is complementary to
the DNA sequence just before the SNP or site of interest, is hybridized to the sin-
gle stranded DNA at some stage when the mixture is cooled down to room tem-
perature. The DNA fragment (with the hybridized sequencing primer) is incu-
bated with the enzymes: DNA polymerase, ATP sulphurylase, firefly luciferase,
and apyrase (a nucleotide-degrading enzyme), and the substrates: adenosine

27
5’phosphosulfate (APS) and luciferin. Repeated cycles of nucleotide addition are
performed. A nucleotide will only be incorporated into the growing DNA strand
if it is complementary to the base in the DNA template. Each incorporation event
is accompanied by release of PPi. The amount released PPi is proportional to the
number of incorporated nucleotides. ATP sulphurylase converts PPi to ATP in the
presence of APS. This ATP drives the conversion of luciferin to oxyluciferin that
generates light. The light is detected and seen as a peak in the pyrogram. The
height of each peak is proportional to the number of nucleotides incorporated.
Unincorporated nucleotides and the produced ATP are degraded between each
cycle by the apyrase.85 The complementary DNA strand is built up and the nu-
cleotide sequence is determined from the signal peaks in the pyrogram. Pyrose-
quencing® can be used in a broad range of applications such as SNP genotyping,
de novo mutation detection, gene identification and microbial genotyping 3 and
also for detecting DNA Methylation pattern in genes.94
Figure 7. The principle of pyrosequencing. When the nucleotide is complementary to the
DNA template, light is produced by the firefly enzyme, luciferase, and the light is de-
tected as a peak in the pyrogram (www.biotage.se). Reproduced by permission from Bio-
tage.


29
Aims of this ThesisAims of this ThesisAims of this ThesisAims of this Thesis
The aims of these studies were to investigate mutations in the genes for folate re-
ceptor-α and methylenetetrahydrofolate reductase and how they affect tHcy con-
centrations.
I. To search for novel mutations in the promoter region and the 5’-UTR in
the folate receptor-α gene.
II. To investigate a possible association between the discovered mutations
and tHcy.
III. To analyse possible associations between FOLR1-mutations and demen-
tia.
IV. To determine the prevalence of the MTHFR polymorphisms 677C>T,
1298A>C and 1793G>A in a population of Swedish children and adoles-
cents and also in healthy Spanish adults. To construct haplotypes from
the MTHFR polymorphisms and to compare the prevalences of the geno-
types and haplotypes between the two populations.
V. To investigate the association between MTHFR genotypes and haplo-
types and tHcy.


31
Materials and methodsMaterials and methodsMaterials and methodsMaterials and methods
Patient samples investigated for tHcy or macrocytosisPatient samples investigated for tHcy or macrocytosisPatient samples investigated for tHcy or macrocytosisPatient samples investigated for tHcy or macrocytosis
Four hundred and forty five (445) patient samples which were sent to our labora-
tory for analysis of tHcy were studied. In addition patient samples sent to our
laboratory for analysis of complete blood count (performed on a Celldyn 4000
instrument, Abbott Laboratories, IL, USA) were reviewed to identify cases of
non-anaemic or mildly anaemic macrocytosis with the following characteristics: a
mean corpuscular volume of >100 fl and a Hb of >105 g/l. Samples with these
characteristics, n = 333, represent a patient group with an increased prevalence of
high levels of tHcy.
The Dementia, Genetics and Milieu study (DGM)The Dementia, Genetics and Milieu study (DGM)The Dementia, Genetics and Milieu study (DGM)The Dementia, Genetics and Milieu study (DGM)
The Dementia, Genetics and Milieu study (DGM) is a case control study at the
University Hospital, Örebro, Sweden, with a study population of 202 consecutive
patients (106 women and 96 men) who were referred, mainly by general practi-
tioners, to the memory care unit at the Department of Geriatrics for diagnostic
assessment and treatment. Every patient in the study group underwent a thorough
clinical investigation, including medical history, treatment and drug history,
physical as well as neurological and psychiatric examinations. All were screened
with Mini Mental State Examination (MMSE) and Clock Drawing Test (CDT).
Computed tomography (CT) or magnetic resonance imaging (MRI) of the brain
was performed on all but nine subjects (96%). Routine analyses of the CSF bio-
markers: total tau protein, phosphorylated tau protein and β-amyloid protein,
were performed at the Department of Psychiatry and Neurochemistry, Institute of
Clinical Neuroscience, Sahlgrenska University Hospital, Mölndal, Sweden. The
ICD-10 criteria were used to divide patients into different diagnostic categories.114
Active Seniors (AS)Active Seniors (AS)Active Seniors (AS)Active Seniors (AS)
Active Seniors (AS) is a sample of 389 senior citizens, 262 women and 127 men.
Which were used as a control group to the DGM subjects. The subjects, from
central Sweden, were all retired and lived independently in their own homes. All
were Caucasians and most of them were born in the 1920s and 1930s, mean age
at sampling being 74 years.
The European Youth Heart Study (EYHS)The European Youth Heart Study (EYHS)The European Youth Heart Study (EYHS)The European Youth Heart Study (EYHS)
The European Youth Heart Study (EYHS) is a cross-sectional school based popu-
lation study on risk factors for future cardiovascular disease in children. In this

32
study 692 children and adolescents (336 girls and 356 boys) in school grade 3 or
9 (aged 9-10 to 15-16 years) from schools in the central Sweden, participated.51, 52
The Canary Islands Nutrition Study (ENCA)The Canary Islands Nutrition Study (ENCA)The Canary Islands Nutrition Study (ENCA)The Canary Islands Nutrition Study (ENCA)
The Canary Islands Nutrition Study (ENCA) is a cross-sectional study that was
carried out to survey the nutritional status and selected metabolic and genetic
variables in a population from the Canary Islands, Spain. Sampling procedure
and participation rates have been described previously.47, 92 Blood samples for
DNA analysis were obtained from 723 subjects (395 women and 328 men) and
serum samples were obtained from 523 subjects (297 women and 226 men).
EthicsEthicsEthicsEthics
The studies were approved by the local Research committees. Studies from Öre-
bro University Hospital were approved by the committee of Örebro County
Council until 2003. From 2004 and onwards the committee in Uppsala approved
the studies from Örebro. All participants received written information about the
studies, and gave a specific and written informed consent to the studies.
Molecular biology techniquesMolecular biology techniquesMolecular biology techniquesMolecular biology techniques
DNA extraction from blood samples
Human genomic DNA was extracted and purified from 200 μL whole blood anti-
coagulated with EDTA using the QIAamp DNA Blood Mini Kit by the spin pro-
cedure, according to the manufacturer’s instructions (QIAGEN Inc., Valencia,
CA, USA). DNA was eluted in 200 μL elution buffer (Buffer AE). The concentra-
tions of the DNA measured by absorbance at 260 nm were usually between 20-
50 ng/μL. The purity was determined by calculating the ratio of the absorbance at
260 nm to the absorbance at 280 nm. Pure DNA has a ratio of 1.7-1.9 and the
typical ratio of our DNA was 1.5-1.9. The purified DNA was stored at -20°C.
Polymerase Chain Reaction (PCR)
All amplicons were amplified by PCR. The Primers were ordered from SGS
(Köping, Sweden) or from biomers.net (Ulm, Germany). The PCR was performed
with the HotStarTaqDNA Polymerase Kit (Qiagen Inc.). The reaction mixture
(50 μL total volume) contained 0.4 μmol/L of each of the primers, 1.25 units of
Taq polymerase, 1.5 mmol/L MgCl2, and 0.2 mmol/L each of dGTP, dATP, dTTP
and dCTP; 15 –30 ng of DNA was added as template. Optimization of the PCR
was performed using an Eppendorf Mastercycler Gradient (Eppendorf Nordic
Aps, Horsholm, Denmark). A PCR amplification consists of an activation step of

33
the polymerase, needed when using the HotStar Taq DNA Polymerase, at 95°C
for 15 min, followed by 35-45 cycles of denaturation of the genomic DNA at
94°C for 1 min, annealing of the primers at a temperature between 55-65°C for 1
min, and extension at 72°C for 1 min. After the repeated cycles there is a final
extension at 72°C for 7 min.
Mutation screening by Single Strand Conformation Polymorphism (SSCP)
SSCP is a technique for detecting mutations. The principle of SSCP is based on
the fact that electrophoretic mobility of DNA in a gel is sensitive to both size and
shape and that single stranded DNA form secondary structures which depends on
the base composition. A mutation or even a SNP will cause a different secondary
structure and therefore also a different mobility pattern in the gel.
The amplicon to be screened for mutations was amplified with PCR. 4 to 10
μL PCR product (depending on the DNA concentration) and 10-16 μL SSCP
loading solution containing 95% (v/v) de-ionized formamide and 5% bromophe-
nol blue (bromophenol blue 25 g/L and glycerol 630 g/L) were mixed to a total
volume of 20 μL. Polyacrylamide gels (Amersham Pharmacia Biotech AB, Upp-
sala, Sweden) were hydrated with buffer. To denature the DNA, the mixture was
heated at 95°C for 7 min. Immediately after heating the mixture, it was put on ice
before being loaded into the gel. Electrophoresis was carried out using Genephor
(Amersham Pharmacia Biotech AB). Optimization was performed using the
GeneGel SSCP Starter Kit, and gels were stained using the PlusOne DNA Silver
Staining Kit, both from Amersham Pharmacia Biotech AB.
DNA Sequencing
DNA sequence analysis was performed on the amplicons using the ABI Prism Big
Dye Terminator Cycle Sequencing Ready Reaction Kit v2.0 (PE Biosystems, Fos-
ter City, CA, USA). This special PCR-step incorporates stop nucleotides which
are each labelled with a different coloured fluorescent dye at every base in the
amplicon. At the end of the PCR there are PCR fragments with different lengths
but all are terminated with a stop nucleotide. A detector reads the colour of the
fluorescent label and a computer puts together the nucleotide sequence. Sequenc-
ing was performed by capillary electrophoresis on an ABI Prism 310 Genetic Ana-
lyzer.

34
Analysis of polymorphisms by Pyrosequencing®
The MTHFR polymorphisms were analysed with pyrosequencing. MTHFR
677C>T was amplified according to the Pyrosequencing® Assay Protocol “Geno-
typing of the C677T variant in the human methylene tetrahydrofolate reductase
(MTHFR) gene”, version 1 (Biotage AB, Uppsala, Sweden). For the MTHFR
1298A>C and 1793G>A polymorphisms we developed our own genotyping pro-
tocols, as specified in Paper II. For good results a relatively large amount of DNA
template (PCR product) is required for the pyrosequencing. In our own protocols
for the MTHFR 1298A>C and the 1793G>A analyses, sufficient DNA was ob-
tained from 38 PCR cycles. Both of the assays for the polymorphism at nt 1298
and the polymorphism at nt 1793 are reversed assays. In reversed assays the se-
quencing primer is complementary to the forward DNA strand and as a result the
complementary DNA strand is built as the reverse strand. Therefore, the change
is detected as a genotype of opposite nucleotides. The reason why some assays are
made in this reverse way is to avoid artefacts because the biotinylated primer may
develop primer-dimer (if the primer is complementary to itself or if two primers
can bind to each other) or hairpins (the primer structure can be complementary
within itself and can stick together).
Measurement of tHcy, folate and vitamin BMeasurement of tHcy, folate and vitamin BMeasurement of tHcy, folate and vitamin BMeasurement of tHcy, folate and vitamin B12121212
Total Hcy levels were measured in our laboratory upon an IMx® instrument
(Abbott Laboratories, IL, USA). All coefficients of variation were <7.5%. In pa-
per IV tHcy was analysed at the University of Barcelona’s Clinical Hospital upon
an AXSYM instrument (Abbot). All coefficients of variation were <6.3%. Serum
folate and vitamin B12 were measured at the Haematology Unit of the University
Hospital of Gran Canaria using an AXSYM. We used serum folate (S-folate) in-
stead of erythrocyte folate (Ery-folate) because it better reflects the actual intra-
cellular folate status. Ery-folate reflects the folate status when the erythrocyte was
formed and does not reflect the actual folate intake. It takes 4-5 months before a
decrease in folate intake affects the levels of Ery-folate.50
StatisticsStatisticsStatisticsStatistics
Data was analysed with SPSS 11.5 for Windows (SPSS Inc., Chicago, IL, USA)
and Statistix8 (Analytical Software, Tallahassee, FL, USA).

35
ResultsResultsResultsResults
Novel mutations in the promoter region and 5’Novel mutations in the promoter region and 5’Novel mutations in the promoter region and 5’Novel mutations in the promoter region and 5’----UTR UTR UTR UTR
of the Folate receof the Folate receof the Folate receof the Folate recepppptortortortor----αααα gene gene gene gene
In an initial study (not included in this thesis) we searched for mutations in the
promoter region and 5’-UTR of the folate receptor-α gene. The studied region
was between nt –188 and nt +272 and the transcription start site was according
to Elwood et al.32 Seven hundred and seventy eight (778) patient samples (445
samples sent to our laboratory for analysis of Hcy and 333 samples sent to our
laboratory for complete blood count) were analysed with single strand conforma-
tion polymorphism (SSCP) and DNA sequencing. Two different novel mutations
were discovered. Three patients had a 25-bp deletion and three patients had a
duplication of an A (–69dupA).73
In Paper I we extended our search by screening the 692 EYHS subjects be-
tween the same nucleotides as the previously screened patients (nt –188 and nt
+272). None of the children or adolescents in the EYHS group had the –69dupA
mutation but we discovered a novel –18C>T mutation which we did not find
among the patients (see Figure 8). After this initial study we continued with fur-
ther mutation screening upstream of transcription start site, this time between nt
–1110 to nt –425. In this study we selected the 92 patients with the highest levels
of tHcy from the 445 patients, which were used in our previous study. We dis-
covered 3 novel mutations, one subject had two mutations very close to each
other; –856C>T and –921T>C, and two subjects had the same mutation, –
1043G>A. Among the 92 patients with hyperhomocysteinaemia 6 patients had a
mutation in the studied regions (nt –188 to nt +272 and nt –1110 to nt –425)
which is 5.4 %. Eight of 692 (1.2 %) of the children had a mutation between nt –
188 and nt +272.

36
Figure 8. DNA Sequencing chromatogram which shows the –18C>T mutation found in
three children. Upper chromatogram shows the forward sequence and lower chroma-
togram the reverse sequence. The arrows point at the mutation which shows as two
peaks instead of one.
To find out whether or not these mutations had an impact on the tHcy levels we
genotyped both the EYHS subjects and the patients for the MTHFR 677C>T
polymorphism, because subjects with the MTHFR 677TT genotype have signifi-
cantly higher tHcy levels than others (see Paper III). None of the patients or the
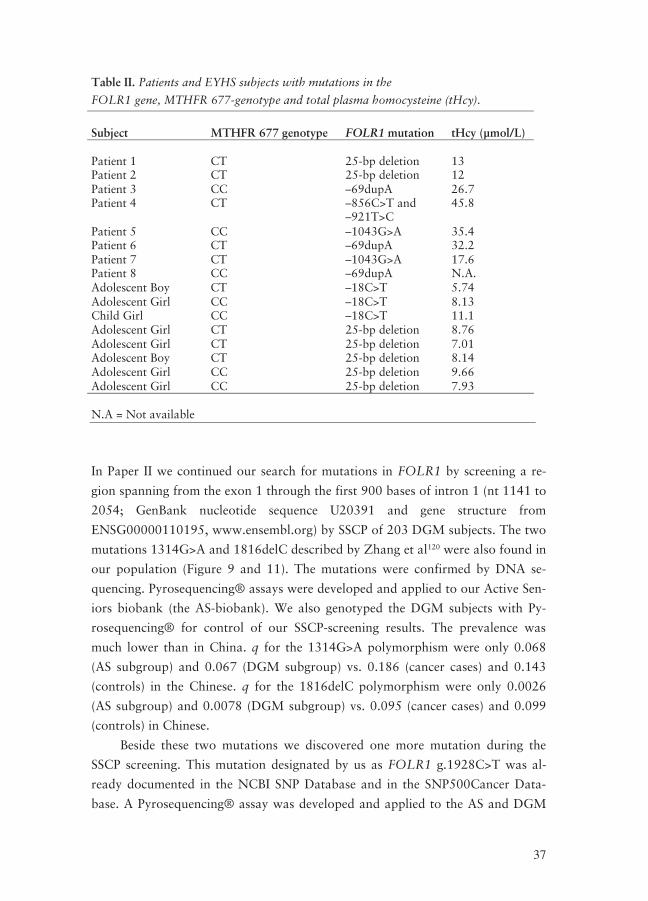
the EYHS subjects had the MTHFR 677 TT genotype (Table II), implying that
the high values of tHcy in some of these subjects can be due to the FOLR1 muta-
tions.
37
Table II. Patients and EYHS subjects with mutations in the
FOLR1 gene, MTHFR 677-genotype and total plasma homocysteine (tHcy).
Subject
MTHFR 677 genotype
FOLR1 mutation
tHcy (μmol/L)
Patient 1
CT
25-bp deletion
13
Patient 2 CT 25-bp deletion 12 Patient 3 CC –69dupA 26.7 Patient 4 CT –856C>T and
–921T>C 45.8
Patient 5 CC –1043G>A 35.4 Patient 6 CT –69dupA 32.2 Patient 7 CT –1043G>A 17.6 Patient 8 CC –69dupA N.A. Adolescent Boy CT –18C>T 5.74 Adolescent Girl CC –18C>T 8.13 Child Girl CC –18C>T 11.1 Adolescent Girl CT 25-bp deletion 8.76 Adolescent Girl CT 25-bp deletion 7.01 Adolescent Boy CT 25-bp deletion 8.14 Adolescent Girl CC 25-bp deletion 9.66 Adolescent Girl CC 25-bp deletion 7.93 N.A = Not available
In Paper II we continued our search for mutations in FOLR1 by screening a re-
gion spanning from the exon 1 through the first 900 bases of intron 1 (nt 1141 to
2054; GenBank nucleotide sequence U20391 and gene structure from
ENSG00000110195, www.ensembl.org) by SSCP of 203 DGM subjects. The two
mutations 1314G>A and 1816delC described by Zhang et al120 were also found in
our population (Figure 9 and 11). The mutations were confirmed by DNA se-
quencing. Pyrosequencing® assays were developed and applied to our Active Sen-
iors biobank (the AS-biobank). We also genotyped the DGM subjects with Py-
rosequencing® for control of our SSCP-screening results. The prevalence was
much lower than in China. q for the 1314G>A polymorphism were only 0.068
(AS subgroup) and 0.067 (DGM subgroup) vs. 0.186 (cancer cases) and 0.143
(controls) in the Chinese. q for the 1816delC polymorphism were only 0.0026
(AS subgroup) and 0.0078 (DGM subgroup) vs. 0.095 (cancer cases) and 0.099
(controls) in Chinese.
Beside these two mutations we discovered one more mutation during the
SSCP screening. This mutation designated by us as FOLR1 g.1928C>T was al-
ready documented in the NCBI SNP Database and in the SNP500Cancer Data-
base. A Pyrosequencing® assay was developed and applied to the AS and DGM

38
biobanks. We found 4.9% heterozygotes in the AS subgroup (q=0.024) and 4.7%
heterozygotes in the DGM subgroup (q=0.023) for this SNP. The reported preva-
lence of heterozygotes in the NCBI SNP Database is 10.7% in Caucasians.
We discovered an additional mutation, a G>A substitution, on sequencing
the subjects with an indeterminant appearance on the SSCP gels. We designated
this mutation FOLR1 g. 1841G>A and it had already been reported to the NCBI
SNP and the SNP500Cancer Databases.76 A Pyrosequencing® assay was devel-
oped for this mutation and applied to the AS and DGM biobanks. A main finding
was that all subjects carrying the g.1816delC (3 DGM subjects and 2 AS subjects)
were also heterozygotes for the g.1841G>A polymorphism. This explains why we
could not identify the mutation on the SSCP gels. For the amplicon, where the
g.1816delC, g.1841G>A and g.1928C>T mutations were found, only three dif-
ferent patterns appeared on the SSCP gel: wildtype pattern, 1816delC–1841G>A
pattern and finally a pattern for the 1928C>T mutation (Figure 10 and 11). We
were not able to identify the separate occurrence of the g.1816delC and
g.1841G>A mutations and conclude that they are in linkage.
Figure 9. SSCP gel showing screening of nt 1141 to 1493 in the FOLR1 gene. The ar-
rows point to patients heterozygote for the FOLR1 g.1314G>A mutation.

39
Figure 10. SSCP gel showing screening of nt 1723 to 2054 in the FOLR1 gene. The ar-
row points to a patient heterozygote for the FOLR1 g.1928C>T mutation.
Figure 11. SSCP gel from the mutation screening of nt 1723 to 2054 in the FOLR1 gene.
The arrow points to a patient heterozygote for the FOLR1 g.1816delC and also het-
erozygote for the FOLR1 g.1841G>A mutation.

40
Nomenclature of the mutations
The naming of the discovered mutations in the FOLR1 gene has been confusing
and has changed over time. Recommendations for the description of changes in
DNA sequences has been published by den Dunnen et al 25 and the Human Ge-
nome Variation Society have a homepage where they update nomenclature rec-
ommendations (http://www.hgvs.org/mutnomen/recs-DNA.html).
In Paper I we named the discovered mutations after the transcription start
site. Transcription start site is defined as nucleotide 1 and the first nucleotide up-
stream of the transcription start site is –1. However, on the Human Genome
Variation Society homepage (in their updated version, autumn 2008) they dis-
courage the use of the transcription start site as nucleotide 1 because there may
exist several transcripts of the same gene. Therefore, in Paper II, we named the
mutations after a sequence, U20391, which we designated as the reference se-
quence. The first nucleotide in that sequence is nucleotide 1 and no + or – signs
are used.
Another way of naming changes in DNA sequences is by using a cDNA se-
quence. Nucleotide 1 is the first nucleotide of the translated exons which means
that the nucleotide 5’ of the ATG-translation initiation codon is –1. Mutations in
introns are designated on the basis of the exons, for example if there is a change
in the intron, 3 nucleotides 5’ of exon 3, then that mutation would be named Ex3
–3X>Y.
The additional four mutations that we discovered in Paper II were already
present in the NCBI SNP Database and in the SNP500Cancer Database.
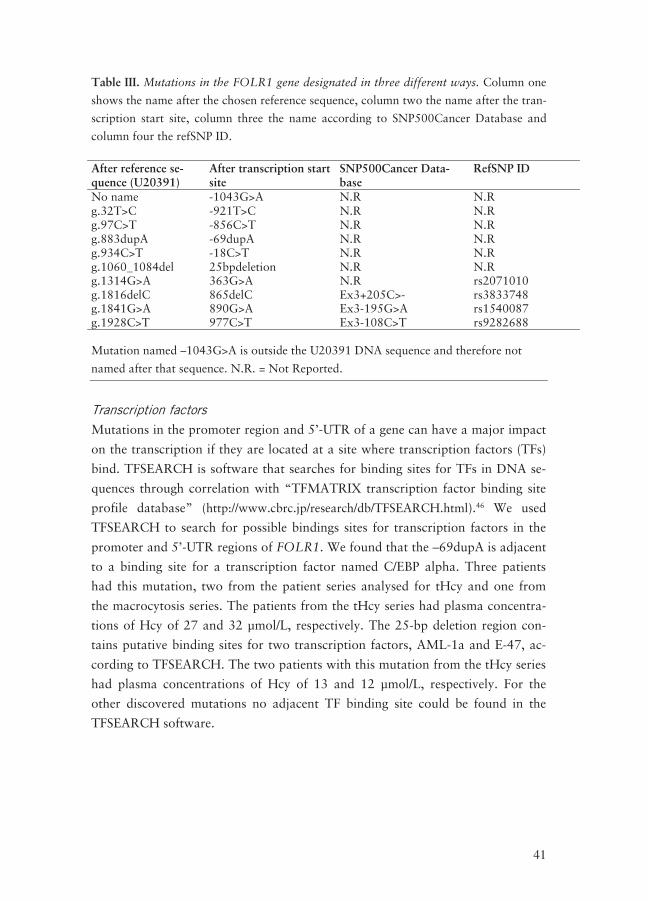
In Table III some different variants of names for the FOLR1 mutations are
listed.
41
Table III. Mutations in the FOLR1 gene designated in three different ways. Column one
shows the name after the chosen reference sequence, column two the name after the tran-
scription start site, column three the name according to SNP500Cancer Database and
column four the refSNP ID.
After reference se-quence (U20391)
After transcription start site
SNP500Cancer Data-base
RefSNP ID
No name -1043G>A N.R N.R g.32T>C -921T>C N.R N.R g.97C>T -856C>T N.R N.R g.883dupA -69dupA N.R N.R g.934C>T -18C>T N.R N.R g.1060_1084del 25bpdeletion N.R N.R g.1314G>A 363G>A N.R rs2071010 g.1816delC 865delC Ex3+205C>- rs3833748 g.1841G>A 890G>A Ex3-195G>A rs1540087 g.1928C>T 977C>T Ex3-108C>T rs9282688 Mutation named –1043G>A is outside the U20391 DNA sequence and therefore not
named after that sequence. N.R. = Not Reported.
Transcription factors
Mutations in the promoter region and 5’-UTR of a gene can have a major impact
on the transcription if they are located at a site where transcription factors (TFs)
bind. TFSEARCH is software that searches for binding sites for TFs in DNA se-
quences through correlation with “TFMATRIX transcription factor binding site
profile database” (http://www.cbrc.jp/research/db/TFSEARCH.html).46 We used
TFSEARCH to search for possible bindings sites for transcription factors in the
promoter and 5’-UTR regions of FOLR1. We found that the –69dupA is adjacent
to a binding site for a transcription factor named C/EBP alpha. Three patients
had this mutation, two from the patient series analysed for tHcy and one from
the macrocytosis series. The patients from the tHcy series had plasma concentra-
tions of Hcy of 27 and 32 μmol/L, respectively. The 25-bp deletion region con-
tains putative binding sites for two transcription factors, AML-1a and E-47, ac-
cording to TFSEARCH. The two patients with this mutation from the tHcy series
had plasma concentrations of Hcy of 13 and 12 μmol/L, respectively. For the
other discovered mutations no adjacent TF binding site could be found in the
TFSEARCH software.

42
Pyrosequencing® assays for MTHFR polymorphismsPyrosequencing® assays for MTHFR polymorphismsPyrosequencing® assays for MTHFR polymorphismsPyrosequencing® assays for MTHFR polymorphisms
We used Pyrosequencing® to genotype the MTHFR 677C>T, 1298A>C and
1793G>A polymorphisms. We consider that pyrosequencing is a more reliable
method than Restriction Fragment Length Polymorphism (RFLP) which is still a
common method of genotype analysis. The MTHFR 677C>T polymorphism was
analysed according to the Pyrosequencing Assay Protocol for the MTHFR
677C>T polymorphism. Pyrograms for the three genotypes are shown in Figure
12.
C/C
���
���
���
���
� � � � � �
� �
�
C/T
���
���
���
� � � � � �
�
� T/T
���
���
���
���
���
���
���
���
���
���
���
���
���
� � � � � �
�
Figure 12. Three different subjects with different genotypes for the MTHFR 677C>T.
Upper panel left, a subject with the CC genotype (wild type), upper panel right, a het-
erozygote subject with the CT genotype and lower panel, a homozygote subject (TT
genotype).
For the other polymorphisms (MTHFR 1298A>C and 1793G>A) we developed
our own assays. The Genbank sequence NT_021937 was used for development
of the assay for the MTHFR 1298A>C polymorphism. Primers for the PCR were
constructed with the primer3 software.88 The amplicon should be as short as pos-
sible. Pyrosequencing® recommends amplicons shorter than 300 bp and opti-
mally about 100 bp. Two different primers are shown in Figures 13 and 14, one
that gives a forward assay and one that gives a reverse assay. The biotinylated
primer is the most important primer in the analysis because it is used as the tem-
plate for the sequencing. For a forward assay a reverse biotinylated primer should
be used and for a reverse assay the forward primer should be biotinylated. In Fig-
ure 13 the best primer for the forward assay is shown. This primer pair can build
primer-dimers and also a hairpin structure. In Figure 14 the best biotinylated
primer for the reverse assay is shown. This primer can also form primer-dimer
and hairpin structures, but the difference between the primers is that the last
primer has a free 3’-end. In the first primer the DNA polymerase can start to in-

43
corporate nucleotides in this position. Usually, a forward assay is chosen if possi-
ble, but in this case, because of the problems with the biotinylated primer for the
forward assay, we chose a reverse assay for the analysis of MTHFR 1298A>C.
Figure 13. Suggested biotin primer for the forward assay for the MTHFR 1298A>C. The
primer can form a primer-dimer and also a hairpin structure. In the hairpin structure, the
3’-end is complementary and therefore the DNA polymerase can start to incorporate
nucleotides at that position.
Figure 14. Selected biotin primer for the MTHFR 1298A>C assay. This primer gives a
reverse assay.
The sequence primer was designed using the SNP Primer Design 1.01 software
(Biotage). The sequence primer should be complementary to the DNA template
and as close to the polymorphism as possible. Selected primers and sequence are
shown in Figure 15.

44
cctcttctacctgaagagcaagtcccccaaggaggagctgctgaagatgtggggggaggagctgaccagtgaagc/aaag
tgtctttgaagtctttgttctttacctctcgggagaacca
Figure 15. MTHFR 1298A>C polymorphism and surrounding sequence with forward,
reverse and sequence primers. Forward and reverse primers are shaded with grey and the
sequence primer is underlined. The polymorphism (c/a) is also shaded with grey.
For the MTHFR 1793G>A, the same GenBank sequence as for the 1298A>C was
used and a reverse assay was chosen. Primers for the MTHFR polymorphisms are
shown in Paper I’s Table 2.
Plasma homocysteine levels and MTHFR polymorphismsPlasma homocysteine levels and MTHFR polymorphismsPlasma homocysteine levels and MTHFR polymorphismsPlasma homocysteine levels and MTHFR polymorphisms
The aim of Papers III and IV was to study the three common and well-known
polymorphisms in the gene for MTHFR (677C>T, 1298A>C and 1793G>A) in
relation to tHcy concentrations in two different populations. In Paper III Swedish
children and adolescents (the EYHS study) were investigated and in Paper IV the
subjects were Spanish adults (the ENCA study). In Paper IV we extended the
genotype and haplotype analysis of the MTHFR 677, 1298, and 1793 polymor-
phisms and their relationship to tHcy to include the nutritional biomarkers serum
folate and vitamin B12.
MTHFR polymorphisms
We genotyped 692 Swedish children and adolescents and 723 Spanish subjects for
the three MTHFR polymorphisms, 677C>T, 1298A>C and 1793G>A. The preva-
lences for the polymorphisms are shown in Figure 16.
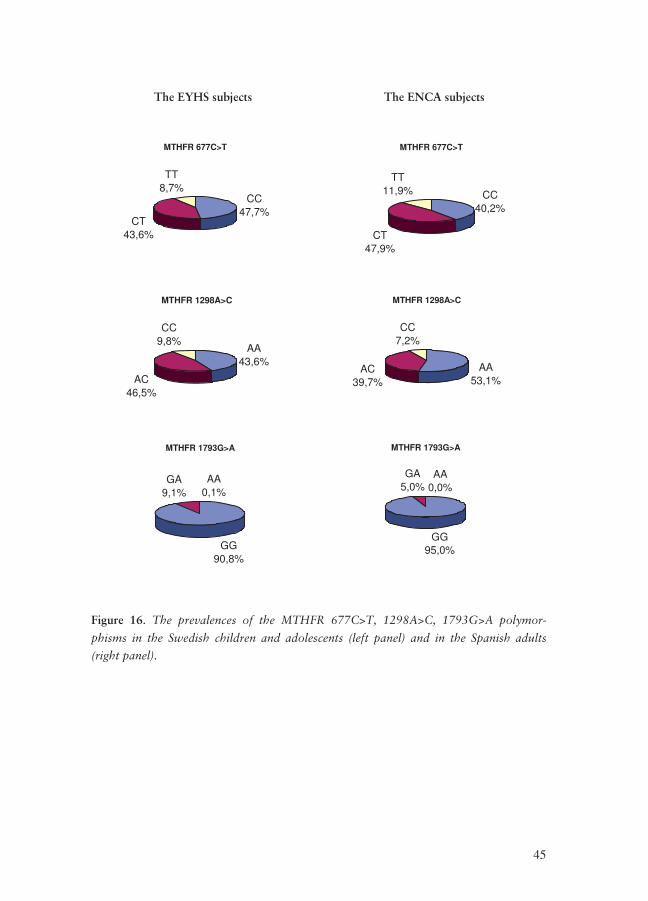
45
The EYHS subjects The ENCA subjects
MTHFR 677C>T
CC47,7%
CT43,6%
TT8,7%
MTHFR 677C>T
CC40,2%
CT47,9%
TT11,9%
MTHFR 1298A>C
AA43,6%
AC46,5%
CC9,8%
MTHFR 1298A>C
AA53,1%
AC39,7%
CC7,2%
MTHFR 1793G>A
GG90,8%
GA9,1%
AA0,1%
MTHFR 1793G>A
GG95,0%
GA5,0%
AA0,0%
Figure 16. The prevalences of the MTHFR 677C>T, 1298A>C, 1793G>A polymor-
phisms in the Swedish children and adolescents (left panel) and in the Spanish adults
(right panel).
46
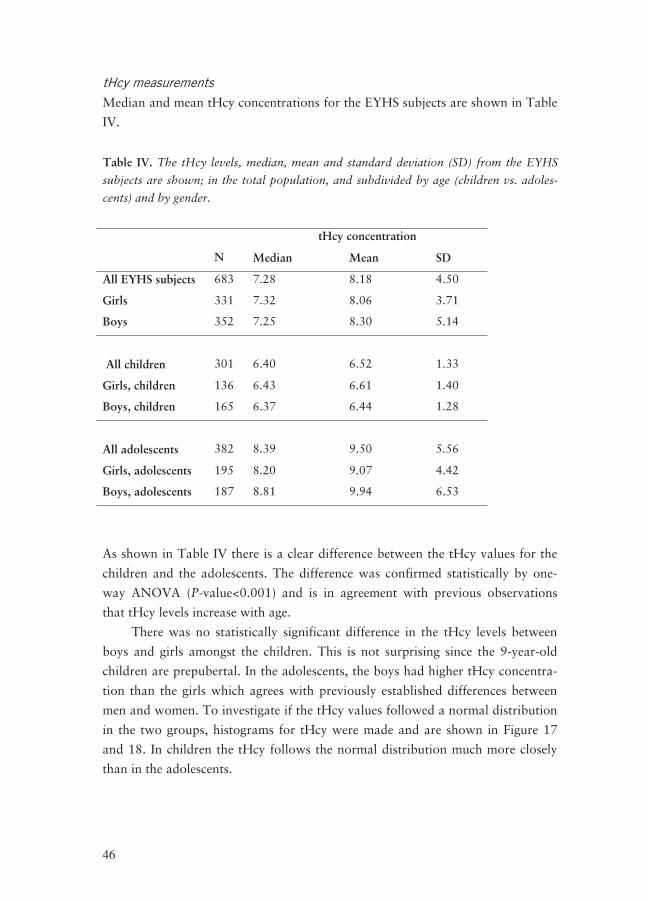
tHcy measurements
Median and mean tHcy concentrations for the EYHS subjects are shown in Table
IV.
Table IV. The tHcy levels, median, mean and standard deviation (SD) from the EYHS
subjects are shown; in the total population, and subdivided by age (children vs. adoles-
cents) and by gender.
tHcy concentration
N Median Mean SD
All EYHS subjects 683 7.28 8.18 4.50
Girls 331 7.32 8.06 3.71
Boys 352 7.25 8.30 5.14
All children
301
6.40
6.52
1.33
Girls, children 136 6.43 6.61 1.40
Boys, children 165 6.37 6.44 1.28
All adolescents
382
8.39
9.50
5.56
Girls, adolescents 195 8.20 9.07 4.42
Boys, adolescents 187 8.81 9.94 6.53
As shown in Table IV there is a clear difference between the tHcy values for the
children and the adolescents. The difference was confirmed statistically by one-
way ANOVA (P-value<0.001) and is in agreement with previous observations
that tHcy levels increase with age.
There was no statistically significant difference in the tHcy levels between
boys and girls amongst the children. This is not surprising since the 9-year-old
children are prepubertal. In the adolescents, the boys had higher tHcy concentra-
tion than the girls which agrees with previously established differences between
men and women. To investigate if the tHcy values followed a normal distribution
in the two groups, histograms for tHcy were made and are shown in Figure 17
and 18. In children the tHcy follows the normal distribution much more closely
than in the adolescents.
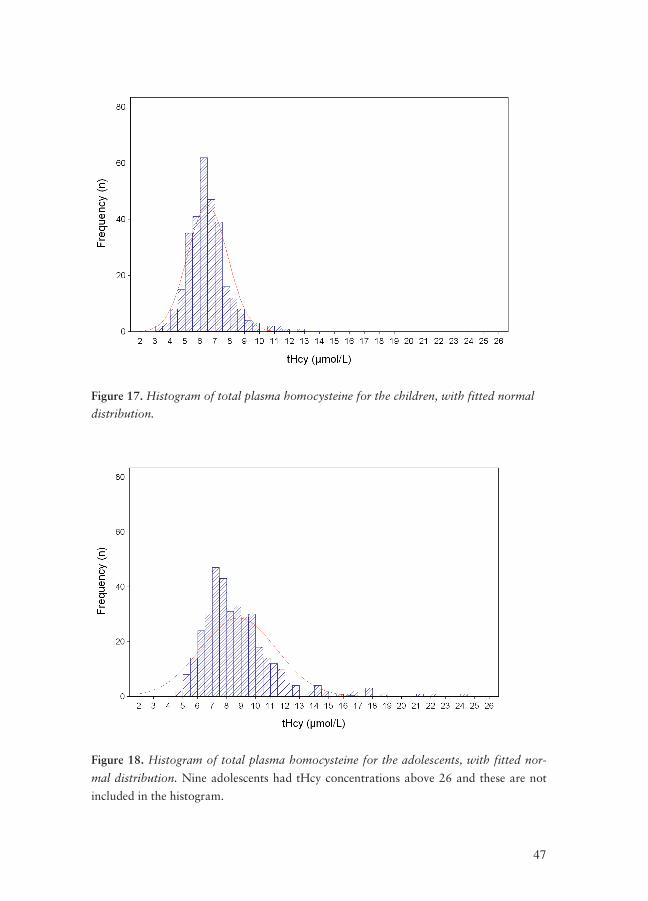
47
Figure 17. Histogram of total plasma homocysteine for the children, with fitted normal
distribution.
Figure 18. Histogram of total plasma homocysteine for the adolescents, with fitted nor-
mal distribution. Nine adolescents had tHcy concentrations above 26 and these are not
included in the histogram.

48
In the Spanish subjects men had, as expected, higher tHcy levels than women.
Association between MTHFR polymorphisms and tHcy concentrations
The correlation between the MTHFR polymorphism and tHcy levels were inves-
tigated with the statistical models ANOVA and ANCOVA.
For the Swedish EYHS subjects the correlation was analysed by a three-way
ANOVA with the factors: age (children and adolescents), gender (boys and girls)
and MTHFR genotype in three levels (CC,CT and TT for 677C>T; AA, AC and
CC for 1298A>C; and GG, GA and AA for 1793G>A).
For the Spanish ENCA subjects the correlation was analysed with ANCOVA
where all values were adjusted for age, serum folate and vitamin B12.
The MTHFR 677 TT genotype has such a major impact on tHcy concentra-
tions that smaller effects from the other polymorphisms can be hidden behind this
large effect. It was therefore necessary to analyse the effects of 1298A>C and
1793G>A isolated from the MTHFR 677 T-allele. Likewise, to obtain the effect
of the 677T-allele upon tHcy the effects of the 1298C-allele and 1793A-allele
were excluded by only including subjects that had the wild type genotype for
1298A>C and 1793G>A.
MTHFR 677C>T
A three-way ANOVA showed that the MTHFR 677C>T was associated with
tHcy in both children and adolescents (Table II, Paper III). The effect of the
MTHFR 677C>T polymorphism on tHcy was also analysed in subjects that were
non-mutated for the 1298A>C and 1793G>A polymorphism and the result
showed that the 677T allele was associated with significantly increased tHcy in
both children and adolescents (Table III, Paper III).
In the ENCA study the MTHFR 677C>T polymorphism had a significant effect
on tHcy in men but not in women (Table III, Paper IV).
MTHFR 1298A>C
The effect of the 1298 C-allele on tHcy concentrations was analysed in subjects
with the 677 CC-genotype and also in subjects with the 677 CT-genotype. To
rule out the effect of the MTHFR 1793G>A polymorphism, subjects with the
1793GA or AA genotype were excluded from the analysis.
In the EYHS study the 1298A>C polymorphism was significantly associated
with increased tHcy levels in children with the MTHFR 677CC genotype and in
adolescents with the MTHFR 677CT genotype (Table IV, Paper III). The com-

49
pound heterozygotes had 1.1 μmol/L higher mean tHcy levels compared to sub-
jects with the 677CT-1298AA genotype.
In the ENCA study there was a significant effect of the MTHFR 1298C-
allele in men with the MTHFR 677CT genotype (Table IV, Paper IV). The com-
pound heterozygotes (677CT-1298AC) had 1.7 μmol/L higher mean tHcy con-
centration compared with the subjects that had the 677CT-1298AA genotype.
Haplotype analysis
Haplotype analysis for the three MTHFR polymorphisms was also performed.
MTHFR haplotype prevalences were calculated based on the genotype preva-
lences of the individual polymorphisms, using the Arlequin software for the
EYHS study and manually for the ENCA study. From the MTHFR 677C>T and
MTHFR 1298A>C loci three haplotypes were obtained; CA, CC and TA. This is
consistent with our finding that no subject was homozygote for a mutation at one
locus and heterozygote at the other locus. When we did haplotype analysis with
the three locus system, 677C>T, 1298A>C, and 1793G>A only one more haplo-
type appeared and this is because of the strong linkage between the 1298 locus
and the 1793 locus. The prevalent haplotypes were CAG, CCG, CCA and TAG.
To analyse the gene dose effect of the haplotypes on tHcy levels, the EYHS
subjects were divided into gender and age groups, and one-way ANOVA analysis
was performed. The subjects could have the values zero, one or two for each hap-
lotype. The CA haplotype is the haplotype that is non-mutated on both loci, the
TA haplotype is mutated on nt 677 and the CC haplotype is mutated on nt 1298.
As can be seen in Table V, the TA haplotype has a positive impact on Hcy levels
in all groups, the CA haplotype is negatively associated with tHcy in all groups
except for girls in the childrens group where it is not significant. The CC haplo-
type is negatively associated with tHcy in adolescents but not in children.
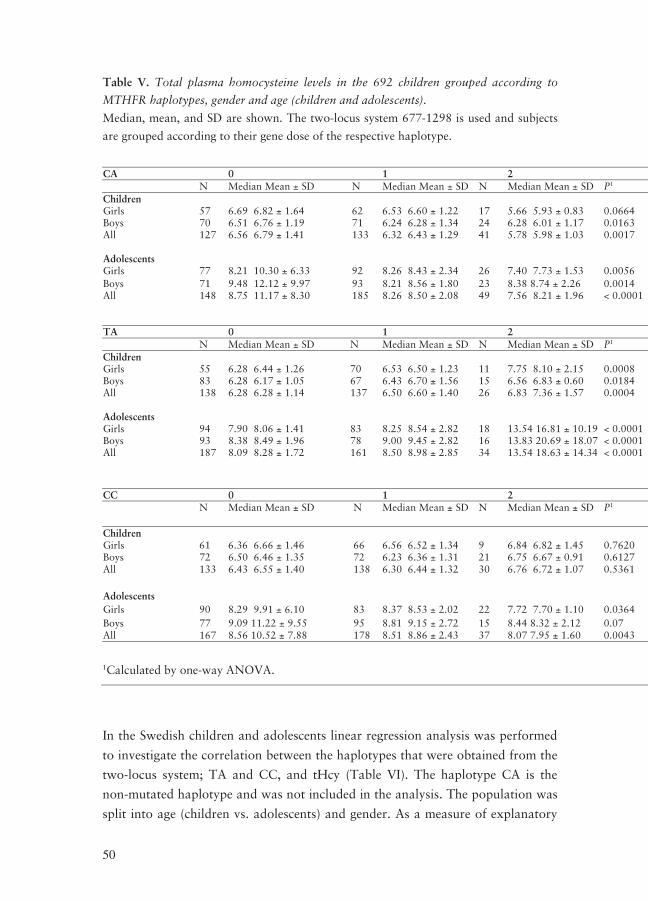
50
Table V. Total plasma homocysteine levels in the 692 children grouped according to
MTHFR haplotypes, gender and age (children and adolescents).
Median, mean, and SD are shown. The two-locus system 677-1298 is used and subjects
are grouped according to their gene dose of the respective haplotype.
CA 0 1 2 N Median Mean ± SD N Median Mean ± SD N Median Mean ± SD P1 Children Girls 57 6.69 6.82 ± 1.64 62 6.53 6.60 ± 1.22 17 5.66 5.93 ± 0.83 0.0664 Boys 70 6.51 6.76 ± 1.19 71 6.24 6.28 ± 1.34 24 6.28 6.01 ± 1.17 0.0163 All 127 6.56 6.79 ± 1.41 133 6.32 6.43 ± 1.29 41 5.78 5.98 ± 1.03 0.0017 Adolescents Girls 77 8.21 10.30 ± 6.33 92 8.26 8.43 ± 2.34 26 7.40 7.73 ± 1.53 0.0056 Boys 71 9.48 12.12 ± 9.97 93 8.21 8.56 ± 1.80 23 8.38 8.74 ± 2.26 0.0014 All 148 8.75 11.17 ± 8.30 185 8.26 8.50 ± 2.08 49 7.56 8.21 ± 1.96 < 0.0001 TA 0 1 2 N Median Mean ± SD N Median Mean ± SD N Median Mean ± SD P1 Children Girls 55 6.28 6.44 ± 1.26 70 6.53 6.50 ± 1.23 11 7.75 8.10 ± 2.15 0.0008 Boys 83 6.28 6.17 ± 1.05 67 6.43 6.70 ± 1.56 15 6.56 6.83 ± 0.60 0.0184 All 138 6.28 6.28 ± 1.14 137 6.50 6.60 ± 1.40 26 6.83 7.36 ± 1.57 0.0004 Adolescents Girls 94 7.90 8.06 ± 1.41 83 8.25 8.54 ± 2.82 18 13.54 16.81 ± 10.19 < 0.0001 Boys 93 8.38 8.49 ± 1.96 78 9.00 9.45 ± 2.82 16 13.83 20.69 ± 18.07 < 0.0001 All 187 8.09 8.28 ± 1.72 161 8.50 8.98 ± 2.85 34 13.54 18.63 ± 14.34 < 0.0001
CC 0 1 2 N Median Mean ± SD N Median Mean ± SD N Median Mean ± SD P1
Children Girls 61 6.36 6.66 ± 1.46 66 6.56 6.52 ± 1.34 9 6.84 6.82 ± 1.45 0.7620 Boys 72 6.50 6.46 ± 1.35 72 6.23 6.36 ± 1.31 21 6.75 6.67 ± 0.91 0.6127 All 133 6.43 6.55 ± 1.40 138 6.30 6.44 ± 1.32 30 6.76 6.72 ± 1.07 0.5361
Adolescents Girls 90 8.29 9.91 ± 6.10 83 8.37 8.53 ± 2.02 22 7.72 7.70 ± 1.10 0.0364
Boys 77 9.09 11.22 ± 9.55 95 8.81 9.15 ± 2.72 15 8.44 8.32 ± 2.12 0.07 All 167 8.56 10.52 ± 7.88 178 8.51 8.86 ± 2.43 37 8.07 7.95 ± 1.60 0.0043
1Calculated by one-way ANOVA.
In the Swedish children and adolescents linear regression analysis was performed
to investigate the correlation between the haplotypes that were obtained from the
two-locus system; TA and CC, and tHcy (Table VI). The haplotype CA is the
non-mutated haplotype and was not included in the analysis. The population was
split into age (children vs. adolescents) and gender. As a measure of explanatory
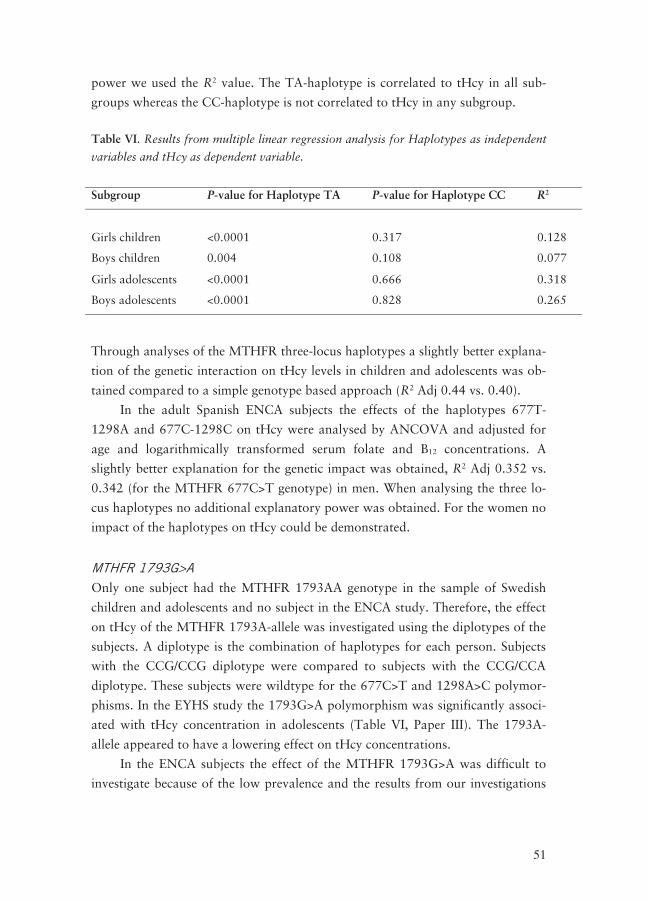
51
power we used the R2 value. The TA-haplotype is correlated to tHcy in all sub-
groups whereas the CC-haplotype is not correlated to tHcy in any subgroup.
Table VI. Results from multiple linear regression analysis for Haplotypes as independent
variables and tHcy as dependent variable.
Subgroup P-value for Haplotype TA P-value for Haplotype CC R2
Girls children
<0.0001
0.317
0.128
Boys children 0.004 0.108 0.077
Girls adolescents <0.0001 0.666 0.318
Boys adolescents <0.0001 0.828 0.265
Through analyses of the MTHFR three-locus haplotypes a slightly better explana-
tion of the genetic interaction on tHcy levels in children and adolescents was ob-
tained compared to a simple genotype based approach (R2 Adj 0.44 vs. 0.40).
In the adult Spanish ENCA subjects the effects of the haplotypes 677T-
1298A and 677C-1298C on tHcy were analysed by ANCOVA and adjusted for
age and logarithmically transformed serum folate and B12 concentrations. A
slightly better explanation for the genetic impact was obtained, R2 Adj 0.352 vs.
0.342 (for the MTHFR 677C>T genotype) in men. When analysing the three lo-
cus haplotypes no additional explanatory power was obtained. For the women no
impact of the haplotypes on tHcy could be demonstrated.
MTHFR 1793G>A
Only one subject had the MTHFR 1793AA genotype in the sample of Swedish
children and adolescents and no subject in the ENCA study. Therefore, the effect
on tHcy of the MTHFR 1793A-allele was investigated using the diplotypes of the
subjects. A diplotype is the combination of haplotypes for each person. Subjects
with the CCG/CCG diplotype were compared to subjects with the CCG/CCA
diplotype. These subjects were wildtype for the 677C>T and 1298A>C polymor-
phisms. In the EYHS study the 1793G>A polymorphism was significantly associ-
ated with tHcy concentration in adolescents (Table VI, Paper III). The 1793A-
allele appeared to have a lowering effect on tHcy concentrations.
In the ENCA subjects the effect of the MTHFR 1793G>A was difficult to
investigate because of the low prevalence and the results from our investigations

52
of the correlation between the polymorphism and tHcy levels did not show any
significant association (Table V, Paper IV).
Diplotypes
Diplotypes were constructed from the haplotypes for the EYHS and ENCA sub-
jects and six diplotypes were obtained from the two-locus system, 677C>T-
1298A>C, and ten were obtained from the three locus system, 677C>T-
1298A>C-1793G>A. The diplotype of each subject was decided for both the two
locus and the three locus systems. The prevalences for the diplotypes are shown in
Tables VII and VIII. The prevalence of the non-mutated diplotype CA/CA is al-
most the same in Sweden and Spain (13.2% vs. 13.4%). The 677T-allele is more
common in Spain than in Sweden but the compound heterozygotes TA/CC are
more common in Sweden than in Spain. The prevalences in the Swedish subjects
and the Spanish subjects are fairly similar despite the large geographical distance.
Table VII. Diplotypes from the two-locus
system MTHFR 677C>T and 1298A>C in
the EYHS and ENCA subjects.
Prevalence (%)
Diplotype 677-1298 EYHS ENCA
CA/CA
13.2
13.4
CA/TA 21.8 27.8
TA/TA 8.7 11.7
CA/CC 24.7 19.7
CC/CC 9.8 7.6
TA/CC 21.8 19.8
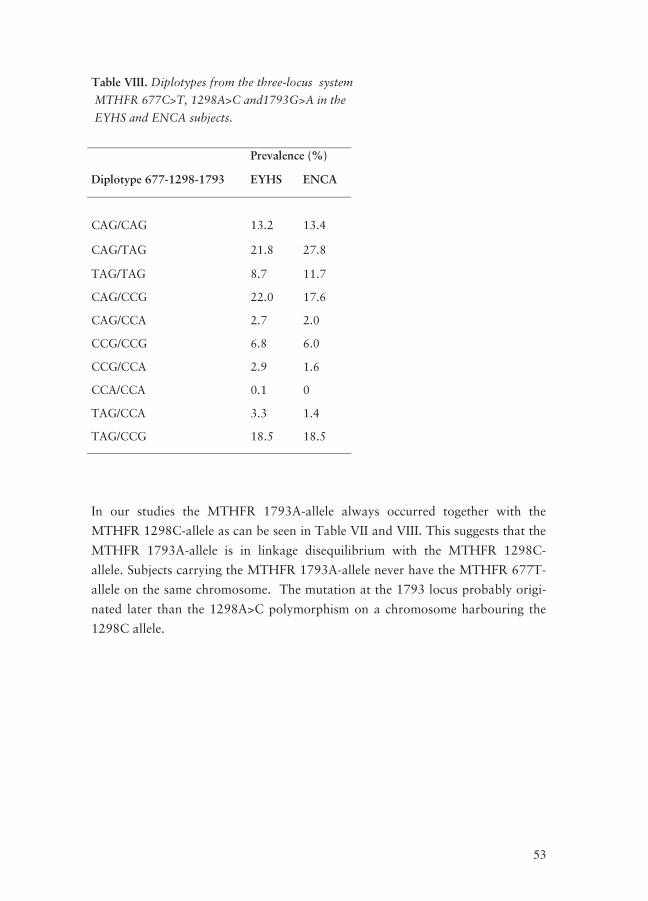
53
Table VIII. Diplotypes from the three-locus system
MTHFR 677C>T, 1298A>C and1793G>A in the
EYHS and ENCA subjects.
Prevalence (%)
Diplotype 677-1298-1793 EYHS ENCA
CAG/CAG
13.2
13.4
CAG/TAG 21.8 27.8
TAG/TAG 8.7 11.7
CAG/CCG 22.0 17.6
CAG/CCA 2.7 2.0
CCG/CCG 6.8 6.0
CCG/CCA 2.9 1.6
CCA/CCA 0.1 0
TAG/CCA 3.3 1.4
TAG/CCG 18.5 18.5
In our studies the MTHFR 1793A-allele always occurred together with the
MTHFR 1298C-allele as can be seen in Table VII and VIII. This suggests that the
MTHFR 1793A-allele is in linkage disequilibrium with the MTHFR 1298C-
allele. Subjects carrying the MTHFR 1793A-allele never have the MTHFR 677T-
allele on the same chromosome. The mutation at the 1793 locus probably origi-
nated later than the 1298A>C polymorphism on a chromosome harbouring the
1298C allele.


55
DiscussDiscussDiscussDiscussion and Conclusionsion and Conclusionsion and Conclusionsion and Conclusions
Novel mutations in the promoter region and 5’Novel mutations in the promoter region and 5’Novel mutations in the promoter region and 5’Novel mutations in the promoter region and 5’----UTR UTR UTR UTR
of the Folate receptorof the Folate receptorof the Folate receptorof the Folate receptor----αααα gene gene gene gene
Mutations in the promoter region and 5’-UTR of a gene can have great impact on
the transcription of the gene if they are situated in a binding site for transcription
factors. Folate receptor-α is a cell membrane receptor with a high affinity for
folate and is one of the main routes for folate to enter the cells. There have been
few published reports describing mutations in the exons of the FOLR1 gene. We
felt motivated to search for mutations in the 5’region upstream transcription start
and the 5’-UTR of FOLR1 because FR-α is so important for folate utilisation.
Although mutations may not give rise to dysfunctional receptors they may affect
the relative amount of receptors present on the cell surface. Our results showed
that this region of the FR-α gene does have mutations. In Paper I we described six
novel mutations in the FOLR1 gene: the 25-bp deletion, −69dupA, −18C>T,
−856C>T, −921T>C and −1043G>A.
The region of the FOLR1 gene which we screened for mutations was ana-
lysed with TFSEARCH software, which searches for putative TF binding sites in
DNA sequences. We found that in the 25-bp region, which was deleted in three of
the patients and in five of the EYHS group, there were putative binding sites for
two TFs. For two of the three patients tHcy concentrations were available, which
were only mildly elevated (13 and 12 μmol/L). Three of the five EYHS subjects
with the 25-bp deletion had slightly elevated tHcy concentrations and this could
only be explained by low folate intake in one of them. The 25-bp deletion had a
similar prevalence in both the EYHS subjects and elevated tHcy patients which
suggests that transcription factors that bind to the 25-bp region in FOLR1 are
not involved in the transcription of the gene.
Three patients (one from the macrocytosis series and two from the tHcy se-
ries) had the −69dupA mutation. Analysis with TFSEARCH revealed that there is
a binding site for the TF C/EBP- alpha adjacent to the duplication. The two sub-
jects from the tHcy patient series had plasma concentrations of Hcy of 27 and 32
μmol/L, which are clearly elevated. Not one of the three subjects had the MTHFR
677TT genotype, which means that the high tHcy levels may be due to the
−69dupA mutation. We did not find the −69dupA mutation in the EYHS group
that was screened at the same region as the patients. Possibly the −69dupA has an
impact on tHcy levels which explains why it was only present in the patient sam-
ples.

56
The −18C>T mutation was not present in patients with elevated levels of
tHcy which is an argument against a role in explaining elevated tHcy concentra-
tions. However, in two of the EYHS subjects their elevated tHcy concentration
could not be explained by folate intake or the MTHFR 677C>T polymorphism.
This raises the possibility that the –18C>T mutation influences tHcy levels.
The −856C>T, −921T>C and −1043G>A mutations were discovered in the
tHcy patient series. We know that the MTHFR 677TT genotype is not the cause
of their high tHcy concentrations but we lack additional information that could
explain the elevated tHcy levels. More studies of these mutations are needed to
clarify their impact on tHcy.
Zhang et al reported finding two mutations in the exons 2 and 3 in the
FOLR1 gene in Chinese patients.120 In Paper II we screened the DGM biobank for
mutations in the same part of the FOLR1 gene as the described Chinese muta-
tions. We found the previously described Chinese mutations; 1314G>A and
1816delC and also two other mutations; 1841G>A and 1928C>T. We developed
Pyrosequencing assays for these mutations and genotyped our biobanks of elderly
Swedes (AS and DGM).
One of the hypotheses in Paper II was that there might be an increased
prevalence of the FOLR1 mutations in the patients diagnosed for dementia com-
pared to the healthy elderly subjects. The 1816delC and the 1841G>A mutations
were more prevalent in the DGM patients but the difference was not statistically
significant. In our subjects the 1816delC allele always occurred concurrently with
the 1841A-allele and we propose that these mutations are in complete linkage
with one other. The distance between the mutations is only 25 bp and therefore it
is unlikely that recombination could occur between them. It would be interesting
to investigate the prevalence of this haplotype in other populations.
The 1314G>A and 1928C>T mutations had no statistically significant asso-
ciation to tHcy but the double mutated haplotype may possibly have a small ef-
fect upon raising tHcy. More studies upon the effect of this haplotype on tHcy
are needed, especially in populations with a higher prevalence.
Some of the mutations that we discovered were already present in the NCBI
SNP Database and also in the SNP500Cancer Database. However the allele fre-
quencies reported for European populations in the databases were not in accor-
dance with our frequencies. This shows that there are regional differences and
that allele frequencies need to be determined in each geographical area.

57
MTHFR polymorphisms and plasma homocysteine levelsMTHFR polymorphisms and plasma homocysteine levelsMTHFR polymorphisms and plasma homocysteine levelsMTHFR polymorphisms and plasma homocysteine levels
In Paper III and IV we studied the association between the MTHFR 677C>T,
1298A>C and 1793G>A polymorphisms to tHcy. In the EYHS study there was
no difference in tHcy concentrations between boys and girls in the younger age
group (children) but in the adolescent group, boys had higher tHcy concentra-
tions than girls. In the ENCA study men had higher tHcy concentration than
women. In the EYHS study we found a significant association between the
MTHFR 677C>T and tHcy concentrations in both children and adolescents
whereas in the ENCA study we found an association between MTHFR 677C>T
and tHcy in men but not in women. This gender difference in the effect of the
MTHFR 677 T-allele on tHcy has been reported previously.11, 16, 61 This suggests
that in females with the MTHFR 677TT genotype the reduced level of 5’-methyl-
THF does not affect tHcy levels. Estrogen has been suggested as a possible expla-
nation to the gender difference in the effect of the MTHFR 677T-allele on tHcy
since estrogen has been shown to lower tHcy concentration.26 In our EYHS study,
the effect of the 677T-allele on tHcy was significant in girls although it was of a
smaller magnitude than that in boys of the same age. Possibly the girls in our
study were too young; estrogen levels were not sufficient to lower the tHcy.
The effect of the MTHFR 1298A>C polymorphism on tHcy has been de-
bated. In the EYHS study we found that the 1298C-allele was associated with
elevated tHcy in children with the MTHFR 677CC genotype. In both Swedish
adolescents and in Spanish men the compound heterozygotes (677CT-1298AC)
had approximately 13% higher tHcy concentrations compared to subjects with
the 677CT-1298AA genotype. In the Spanish women no effect of the MTHFR
1298C-allele on tHcy was found.
The MTHFR 1793G>A polymorphism is much less studied than the
MTHFR 677C>T and 1298A>C. The 1793G>A polymorphism has a much lower
prevalence (only one adolescent had the AA genotype in the EYHS study and no
subject in the ENCA study) compared to the other two MTHFR polymorphisms
and therefore its effect on tHcy is difficult to investigate. In the EYHS study, the
A-allele had a minor tHcy-lowering effect in the adolescents. The prevalence of
the MTHFR 1793A-allele was very low in the Spanish subjects and no effect of
the 1793A-allele on tHcy could be discerned.
We constructed haplotypes from the MTHFR 677C>T, 1298A>C and
1793G>A polymorphisms. The effect of the MTHFR haplotypes on tHcy was
analysed. A slightly superior explanatory power for the genetic impact was ob-
tained using the MTHFR haplotypes in the analysis compared to the MTHFR
677C>T genotype-based approach in both the Swedish children and adolescents

58
and in the Spanish adults. Therefore MTHFR haplotypes should be considered
when analysing the impact of the MTHFR 677C>T, 1298A>C and 1793G>A
polymorphisms on tHcy.
In our studies we did not find any subject with three or four mutations at
the MTHFR 677C>T and 1298A>C loci. We used Pyrosequencing® for the geno-
typing, which is a real-time sequencing method. It is possible that the MTHFR
677T-1298C haplotype exists but in such a low prevalence that it is only found in
very large population studies such as the study by Ulvik et al.103 However, the
finding of the haplotype may be due to genotyping errors. In close proximity to
the MTHFR 1298A>C polymorphism, there is another mutation, 1317T>C,
which creates a cleavage site for the MboII restriction enzyme. When using this
enzyme in RFLP genotyping, errors may occur because the enzyme will not dis-
criminate between the two polymorphisms.87, 107
Regardless of the large geographical distance between our study populations
the haplotype composition is quite similar. The MTHFR 677T-allele is slightly
more prevalent in Spain compared to Sweden but it has only an effect on tHcy in
the Spanish men. Age, gender and factors linked to the ethnicity of the studied
subjects, seem to be able to override the nutrigenetic impact of tHcy-raising geno-
types or haplotypes in particular settings, such as in the Spanish women in our
study. Gene-nutrient interactions on tHcy levels may thus differ between popula-
tions. The transferability of nutrigenetic findings may therefore be limited, and
must probably be re-evaluated for each particular setting of age-gender-ethnicity.

59
Future perspectivesFuture perspectivesFuture perspectivesFuture perspectives
The search for mutations in the regulatory region of the FOLR1 gene is in its
early stages. Mutations that affect the expression of a gene can be situated thou-
sands of bp upstream of the transcription start. For instance, the SNP that gives
rise to lactose tolerance is at position –13910 bp from the transcription start.33 So
far we have surveyed up to approximately 1000 bp upstream from the transcrip-
tion start in the FOLR1 gene, and we have already found six novel mutations.
The finding of these mutations motivates continuing screening for additional mu-
tations in this region.
A Pyrosequencing® method was not a good choice for genotyping the
–69dupA mutation because of the adjacent sequence (wildtype 6A and mutant
7A), since the method cannot distinguish between 6 As or 7 As in sequence. An-
other genotyping method, Denaturating High Performance Liquid Chromatogra-
phy (DHPLC), is therefore to be set up for this mutation. DHPLC has been suc-
cessfully applied to genotyping and mutation screening.38, 115 Using the DHPLC
method more subjects from different populations will be genotyped for the –
69dupA mutation and hopefully more persons with this unusual mutation will be
found, making it possible to ascertain the association to tHcy.
Proton-Coupled Folate Transporter (PCFT) has been recently described.
PCFT is a low-pH transporter and it represents the mechanism by which folates
are absorbed in the small intestine.100, 121 The gene for PCFT is less studied but
mutations in this gene have already been described, for instance in a woman with
hereditary folate malabsorption.72 In our research group, mutation screening of
the promoter part of the PCFT gene has commenced.
Epigenetics describe changes of the phenotype, which do not involve changes
in the DNA sequence. DNA methylation is an important and common form of
the epigenetic control of genes.18 Enzymes, transporters and receptors in Hcy me-
tabolism are of interest in studies upon DNA methylation because the methyl
groups are derived from S-adenosylmethionine (SAM), which is derived from me-
thionine. Our hypothesis is that subjects with elevated tHcy levels may have a
different methylation pattern in the FOLR1 gene. Investigation of the methyla-
tion pattern of the promoter part and exon 1 in the FOLR1 gene have started and
future studies are planned to determine the methylation pattern in RFC and PCFT
genes.
Depending on nutrient intake (gene-nutrient interaction) and possibly on
other genes (gene-gene interactions), the MTHFR 677C>T, 1298A>C and
1793G>A polymorphisms affect tHcy concentrations differently in different

60
populations. Therefore, studies from one country cannot necessarily be applied to
another country. Several countries are now fortifying food with folate. The rate
of neural tube defects (NTDs) has decreased in Canada after the start of such
folate fortification.23 It will be interesting to follow the impact of fortification will
upon other diseases linked to raised tHcy concentrations. However, as noted
above the result from one nutrigenetic study from one geographical region is dif-
ficult to apply to another population from a different geographical region.

61
TTTTackackackack
Studierna i den här avhandlingen genomfördes på Kliniskt kemiska laboratoriet
på Universitetssjukhuset i Örebro.
Ett stort tack till Institutionen för vård och omsorg, Örebro universitet, Nyckel-
fonden, Örebro och Forskningskommittén, Örebro läns landsting för det ekono-
miska stöd jag erhållit och som gjorde mina forskarstudier möjliga.
Under de år som mina doktorandstudier pågått är det många som har stöttat mig
och gjort det möjligt att genomföra den här avhandlingen.
Jag vill rikta ett speciellt tack till min handledare Torbjörn Nilsson som gjorde
det möjligt för mig att påbörja och genomföra mina doktorandstudier. Din entu-
siasm för forskning har inspirerat mig och genom åren har du alltid tagit dig tid
att svara på frågor och diskutera resultat. Jag har alltid känt att jag haft ditt stöd.
Jag vill tacka mina medförfattare Nils-Olof Hagnelius, Anita Hurtig-Wennlöf,
Agneta Yngve och Michael Sjöström för ett bra och trevligt samarbete. Nisse tack
också för att jag fick möjlighet att delta i en demensutredning. Anita, tack för allt
du lärt mig om SPSS och för alla oändliga timmar vi spenderat med statistik. Ag-
neta och Michael, jag är tacksam för att jag fått möjlighet att arbeta med EYHS.
Professor Lluís Serra-Majem and co-worker Patricia Henriquez, thank you for
giving us the opportunity to work with the ENCA study.
Ian Jones, för allt stöd under avhandlingsarbetet framförallt för utmärkt kor-
rekturläsning och språkgranskning. Jag kan inte tacka nog för din uppbackning
ända in i det sista!
Ett tack också till mina nuvarande doktorandkollegor Lovisa Olsson och Helena
Isaksson samt Victoria Hahn-Strömberg med vilka jag har delat det vardagliga
arbetet såväl i labbet som framför datorn med artiklar och avhandling och inte
minst fikapauser. Lovisa jag är tacksam för all hjälp jag fått med mitt labbarbete
och för allt du lärt mig inom detta område. Helena ett speciellt tack till dig för
alla goda skratt och för att du spelar så bra banjomusik när man minst anar det
samt för all hjälp med allt möjligt och omöjligt och tack Victoria, för all upp-
muntran och peppning.

62
Jag tänker också på Anita Koskela, Margareta Karlsson, Zarah Löf-Öhlin, Jose-
fin Yngve, Gabriella Risberg, Ravi Vumma och Margit Laanpere. Ni har alla på
ett eller annat sätt bidragit till den här avhandlingen.
Ett stort tack till vill jag ge till Lars Breimer för din språkgranskning av kappan
och till Margareta Landin för snabb och utomordentligt bra hjälp med EndNote
och referenserna.
Jag vill också tacka Lena Björkman för all hjälp med ansökningar om forsknings-
medel och forskarveckor samt för administrationen av alla tilldelade medel. Ewa-
Lena Sjöberg, tacka för ditt stöd och engagemang under alla år.
Medarbetarna på Klin kem lab vill jag tacka för ert bidrag i form av en mycket
trivsam arbetsplats. Ett speciellt tack till mina kollegor på molekylärlab: Kenneth,
Mats, Britt-Marie, Bella, Gisela, ni har gjort alla timmar jag spenderat i labbet
väldigt trevliga!
Jag vill också tacka min familj och mina vänner: Karl och Edvin, mina älskade
barn, vad vore livet utan er? Ni gör det omöjligt att tänka på forskning när man
är med er. Min man Björn, för ditt enorma stöd under alla år och för att du alltid
tror på mig! Tack också för ditt tålamod speciellt under sista tiden. Mina föräld-
rar Sinikka och Agne, er vill jag tacka för ert eviga stöd. Tack också för att ni
sitter barnvakt när som helst och var som helst. Min bror Per och min svägerska
Maria som alltid uppmuntrat mig, jag hoppas vi kan ses oftare nu. Min framlidna
mormor Maire, jag vet att du, kanske mer än någon annan, ville komma till min
disputation. Kanske är du med mig ändå? Min moster Hillevi med familj, för alla
veckor jag har bott i ert meditationsrum under mina kurser i Stockholm. Min
svärmor Maj, vill jag tacka för din uppmuntran. Alla vänner, några nämnda men
inga är glömda: Maria F för alla våra diskussioner om hur det är att vara dokto-
rand, och Maria B, Emma, Johanna och Carola för att ni är sådana goda vänner
samt Thomas & Madde med familj för att vi kan dela allt familjekaos.

63
ReferencesReferencesReferencesReferences
1. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. MRC Vitamin Study Research Group. Lancet 1991; 338(8760): 131-7.
2. Abbott MH, Folstein SE, Abbey H, Pyeritz RE. Psychiatric manifestations of homocystinuria due to cystathionine beta-synthase deficiency: preva-lence, natural history, and relationship to neurologic impairment and vi-tamin B6-responsiveness. Am J Med Genet 1987; 26(4): 959-69.
3. Ahmadian A, Ehn M, Hober S. Pyrosequencing: history, biochemistry and future. Clin Chim Acta 2006; 363(1-2): 83-94.
4. Barber RC, Shaw GM, Lammer EJ, Greer KA, Biela TA, Lacey SW, Was-serman CR, Finnell RH. Lack of association between mutations in the folate receptor-alpha gene and spina bifida. Am J Med Genet 1998; 76(4): 310-7.
5. Bolander-Gouaille C, Bottiglieri T. Homocysteine Related Vitamins and Neuropsychiatric Disorders. Paris: Springer; 2003.
6. Bottiglieri T, Laundy M, Crellin R, Toone BK, Carney MW, Reynolds EH. Homocysteine, folate, methylation, and monoamine metabolism in depres-sion. J Neurol Neurosurg Psychiatry 2000; 69(2): 228-32.
7. Bowcock AM, Lacey S, Saltman D, Mohandas TK, Kamen B, Taggart RT. Localization of the folate receptor gene to chromosome 11q13. Cytoge-netic Cell Genetics 1991; 58.
8. Brattström L, Wilcken DE. Homocysteine and cardiovascular disease: cause or effect? Am J Clin Nutr 2000; 72(2): 315-23.
9. Brosnan JT. Homocysteine in the kidney. In: Ralph C, Jacobsen DW, edi-tors. Homocysteine in health and Disease. Cambridge: Cambridge Univer-sity Press; 2001.
10. Brosnan JT, Jacobs RL, Stead LM, Brosnan ME. Methylation demand: a key determinant of homocysteine metabolism. Acta Biochim Pol 2004; 51(2): 405-13.
11. Brown KS, Kluijtmans LA, Young IS, Murray L, McMaster D, Woodside JV, Yarnell JW, Boreham CA, McNulty H, Strain JJ, McPartlin J, Scott JM, Mitchell LE, Whitehead AS. The 5,10-methylenetetrahydrofolate re-ductase C677T polymorphism interacts with smoking to increase homo-cysteine. Atherosclerosis 2004; 174(2): 315-22.
12. Butz LW, du Vigneaud V. The formation of a homologue of cystine by the decomposition of methionine with sulfuric acid. 1932; 99(1): 135-42.

64
13. Callejón G, Mayor-Olea A, Jiménez AJ, Gaitán MJ, Palomares AR, Martínez F, Ruiz M, Reyes-Engel A. Genotypes of the C677T and A1298C polymorphisms of the MTHFR gene as a cause of human sponta-neous embryo loss. Hum Reprod 2007; 22(12): 3249-54.
14. Campise M, Bamonti F, Novembrino C, Ippolito S, Tarantino A, Cornelli U, Lonati S, Cesana BM, Ponticelli C. Oxidative stress in kidney transplant patients. Transplantation 2003; 76(10): 1474-8.
15. Chango A, Boisson F, Barbe F, Quilliot D, Droesch S, Pfister M, Fillon-Emery N, Lambert D, Fremont S, Rosenblatt DS, Nicolas JP. The effect of 677C-->T and 1298A-->C mutations on plasma homocysteine and 5,10-methylenetetrahydrofolate reductase activity in healthy subjects. Br J Nutr 2000; 83(6): 593-6.
16. Chango A, Potier De Courcy G, Boisson F, Guilland JC, Barbe F, Perrin MO, Christides JP, Rabhi K, Pfister M, Galan P, Hercberg S, Nicolas JP. 5,10-methylenetetrahydrofolate reductase common mutations, folate sta-tus and plasma homocysteine in healthy French adults of the Supplementa-tion en Vitamines et Mineraux Antioxydants (SU.VI.MAX) cohort. Br J Nutr 2000; 84(6): 891-6.
17. Cortese C, Motti C. MTHFR gene polymorphism, homocysteine and car-diovascular disease. Public Health Nutrition. 2001; 4(2B): 493-7.
18. Das PM, Singal R. DNA methylation and cancer. J Clin Oncol 2004; 22(22): 4632-42.
19. Database of Single Nucleotide Polymorphisms (dbSNP [rs1801131]). Be-thesda (MD): National Center for Biotechnology Information, National Library of Medicine. [cited 2008 Oct 9] http://www.ncbi.nlm.nih.gov/SNP/
20. Database of Single Nucleotide Polymorphisms (dbSNP [rs2274976]). Be-thesda (MD): National Center for Biotechnology Information, National Library of Medicine. [cited 2008 Oct 9] http://www.ncbi.nlm.nih.gov/SNP/
21. Database of Single Nucleotide Polymorphisms (dbSNP [rs 1801133]). Be-thesda (MD): National Center for Biotechnology Information, National Library of Medicine [cited 2008 Sept 10] http://www.ncbi.nlm.nih.gov/SNP/
22. De Marco P, Moroni A, Merello E, de Franchis R, Andreussi L, Finnell RH, Barber RC, Cama A, Capra V. Folate pathway gene alterations in pa-tients with neural tube defects. Am J Med Genet 2000; 95(3): 216-23.
23. De Wals P, Tairou F, Van Allen MI, Uh SH, Lowry RB, Sibbald B, Evans JA, Van den Hof MC, Zimmer P, Crowley M, Fernandez B, Lee NS, Ni-yonsenga T. Reduction in neural-tube defects after folic acid fortification in Canada. N Engl J Med 2007; 357(2): 135-42.

65
24. Dekou V, Whincup P, Papacosta O, Ebrahim S, Lennon L, Ueland PM, Refsum H, Humphries SE, Gudnason V. The effect of the C677T and A1298C polymorphisms in the methylenetetrahydrofolate reductase gene on homocysteine levels in elderly men and women from the British re-gional heart study. Atherosclerosis 2001; 154(3): 659-66.
25. den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutation: a discussion. Hum Mutat 2000; 15: 7-12.
26. Dimitrova KR, DeGroot K, Myers AK, Kim YD. Estrogen and homocys-teine. Cardiovasc Res 2002; 53(3): 577-88.
27. Doshi S, McDowell I, Moat S, Lewis M, Goodfellow J. Folate improves endothelial function in patients with coronary heart disease. Clin Chem Lab Med 2003; 41(11): 1505-12.
28. Durga J, Anteunis LJ, Schouten EG, Bots ML, Kok FJ, Verhoef P. Associa-tion of folate with hearing is dependent on the 5,10-methylenetetrahdyrofolate reductase 677C-->T mutation. Neurobiol Aging 2006; 27(3): 482-9.
29. Durga J, van Boxtel MP, Schouten EG, Bots ML, Kok FJ, Verhoef P. Folate and the methylenetetrahydrofolate reductase 677C-->T mutation correlate with cognitive performance. Neurobiol Aging 2006; 27(2): 334-43.
30. Durga J, van Boxtel MP, Schouten EG, Kok FJ, Jolles J, Katan MB, Ver-hoef P. Effect of 3-year folic acid supplementation on cognitive function in older adults in the FACIT trial: a randomised, double blind, controlled trial. Lancet 2007; 369(9557): 208-16.
31. Elnakat H, Ratnam M. Role of folate receptor genes in reproduction and related cancers. Front Biosci 2006; 11: 506-19.
32. Elwood PC, Nachmanoff K, Saikawa Y, Page ST, Pacheco P, Roberts S, Chung KN. The divergent 5' termini of the alpha human folate receptor (hFR) mRNAs originate from two tissue-specific promoters and alternative splicing: characterization of the alpha hFR gene structure. Biochemistry 1997; 36(6): 1467-78.
33. Enattah NS, Sahi T, Savilahti E, Terwilliger JD, Peltonen L, Järvelä I. Iden-tification of a variant associated with adult-type hypolactasia. Nat Genet 2002; 30(2): 233-7.
34. Friedman AN, Bostom AG, Selhub J, Levey AS, Rosenberg IH. The kidney and homocysteine metabolism. J Am Soc Nephrol 2001; 12(10): 2181-9.

66
35. Friso S, Girelli D, Trabetti E, Stranieri C, Olivieri O, Tinazzi E, Martinelli N, Faccini G, Pignatti PF, Corrocher R. A1298C methylenetetrahydro-folate reductase mutation and coronary artery disease: relationships with C677T polymorphism and homocysteine/folate metabolism. Clin Exp Med 2002; 2(1): 7-12.
36. Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Matthews RG, Bo-ers GJ, den Heijer M, Kluijtmans LA, van den Heuvel LP, et al. A candi-date genetic risk factor for vascular disease: a common mutation in me-thylenetetrahydrofolate reductase. Nat Genet 1995; 10(1): 111-3.
37. Gaughan DJ, Barbaux S, Kluijtmans LA, Whitehead AS. The human and mouse methylenetetrahydrofolate reductase (MTHFR) genes: genomic or-ganization, mRNA structure and linkage to the CLCN6 gene. Gene 2000; 257(2): 279-89.
38. Gerhardus A, Schleberger H, Schlegelberger B, Gadzicki D. Diagnostic accuracy of methods for the detection of BRCA1 and BRCA2 mutations: a systematic review. Eur J Hum Genet 2007; 15(6): 619-27.
39. Girelli D, Friso S, Trabetti E, Olivieri O, Russo C, Pessotto R, Faccini G, Pignatti PF, Mazzucco A, Corrocher R. Methylenetetrahydrofolate reduc-tase C677T mutation, plasma homocysteine, and folate in subjects from northern Italy with or without angiographically documented severe coro-nary atherosclerotic disease: evidence for an important genetic-environmental interaction. Blood 1998; 91(11): 4158-63.
40. Goyette P, Pai A, Milos R, Frosst P, Tran P, Chen Z, Chan M, Rozen R. Gene structure of human and mouse methylenetetrahydrofolate reductase (MTHFR). Mamm Genome 1998; 9(8): 652-6.
41. Goyette P, Sumner JS, Milos R, Duncan AM, Rosenblatt DS, Matthews RG, Rozen R. Human methylenetetrahydrofolate reductase: isolation of cDNA mapping and mutation identification. Nat Genet 1994; 7(4): 551.
42. Guenther BD, Sheppard CA, Tran P, Rozen R, Matthews RG, Ludwig ML. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomo-cysteinemia. Nat Struct Biol 1999; 6(4): 359-65.
43. Guttormsen AB, Ueland PM, Nesthus I, Nygard O, Schneede J, Vollset SE, Refsum H. Determinants and vitamin responsiveness of intermediate hy-perhomocysteinemia (> or = 40 micromol/liter). The Hordaland Homocys-teine Study. J Clin Invest 1996; 98(9): 2174-83.
44. Harmon DL, Woodside JV, Yarnell JW, McMaster D, Young IS, McCrum EE, Gey KF, Whitehead AS, Evans AE. The common 'thermolabile' variant of methylene tetrahydrofolate reductase is a major determinant of mild hyperhomocysteinaemia. QJM 1996; 89(8): 571-7.

67
45. Heil SG, van der Put NM, Trijbels FJ, Gabreels FJ, Blom HJ. Molecular genetic analysis of human folate receptors in neural tube defects. Eur J Hum Genet 1999; 7(3): 393-6.
46. Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Ignatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, Podkolodny NL, Kolchanov NA. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res 1998; 26(1): 362-7.
47. Henríquez P, Doreste J, Deulofeu R, Fiuza MD, Serra-Majem L. Nutri-tional determinants of plasma total homocysteine distribution in the Ca-nary Islands. Eur J Clin Nutr 2007; 61(1): 111-8.
48. Hofmann MA, Lalla E, Lu Y, Gleason MR, Wolf BM, Tanji N, Ferran LJ Jr, Kohl B, Rao V, Kisiel W, Stern DM, Schmidt AM. Hyperhomocys-teinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J Clin Invest 2001; 107(6): 675-83.
49. Homberger A, Linnebank M, Winter C, Willenbring H, Marquardt T, Harms E, Koch HG. Genomic structure and transcript variants of the hu-man methylenetetrahydrofolate reductase gene. Eur J Hum Genet 2000; 8(9): 725-9.
50. Hultberg B, Isaksson A, Nilsson K, Gustafson L. [Optimal use of markers for cobalamin/folate status. Results from a survey in a group of psycho-geriatric patients]. Läkartidningen 2003; 100(40): 3136-40.
51. Hurtig Wennlöf A, Yngve A, Nilsson TK, Sjöström M. Serum lipids, glu-cose and insulin levels in healthy schoolchildren aged 9 and 15 years from Central Sweden: reference values in relation to biological, social and life-style factors. Scand J Clin Lab Invest 2005; 65(1): 65-76.
52. Hurtig Wennlöf A, Yngve A, Sjöström M. Sampling procedure, participa-tion rates and representativeness in the Swedish part of the European Youth Heart Study (EYHS). Public Health Nutr 2003; 6(3): 291-9.
53. Isotalo PA, Wells GA, Donnelly JG. Neonatal and fetal methylenetetrahy-drofolate reductase genetic polymorphisms: an examination of C677T and A1298C mutations. Am J Hum Genet 2000; 67(4): 986-90.
54. Jacques PF, Bostom AG, Williams RR, Ellison RC, Eckfeldt JH, Rosenberg IH, Selhub J, Rozen R. Relation between folate status, a common mutati-on in methylenetetrahydrofolate reductase, and plasma homocysteine con-centrations. Circulation 1996; 93(1): 7-9.
55. Janosikova B, Zavadakova P, Kozich V. Single-nucleotide polymorphisms in genes relating to homocysteine metabolism: how applicable are public SNP databases to a typical European population? Eur J Hum Genet 2005; 13(1): 86-95.

68
56. Kamen BA, Smith AK. A review of folate receptor alpha cycling and 5-methyltetrahydrofolate accumulation with an emphasis on cell models in vitro. Adv Drug Deliv Rev 2004; 56(8): 1085-97.
57. Kang SS, Wong PW, Malinow MR. Hyperhomocyst(e)inemia as a risk fac-tor for occlusive vascular disease. Annu Rev Nutr 1992; 12: 279-98.
58. Kang SS, Wong PW, Susmano A, Sora J, Norusis M, Ruggie N. Thermola-bile methylenetetrahydrofolate reductase: an inherited risk factor for coro-nary artery disease. Am J Hum Genet 1991; 48(3): 536-45.
59. Kang SS, Zhou J, Wong PW, Kowalisyn J, Strokosch G. Intermediate ho-mocysteinemia: a thermolabile variant of methylenetetrahydrofolate reduc-tase. Am J Hum Genet 1988; 43(4): 414-21.
60. Klerk M, Verhoef P, Clarke R, Blom HJ, Kok FJ, Schouten EG. MTHFR 677C-->T polymorphism and risk of coronary heart disease: a meta-analysis. JAMA 2002; 288(16): 2023-31.
61. Kluijtmans LA, Young IS, Boreham CA, Murray L, McMaster D, McNulty H, Strain JJ, McPartlin J, Scott JM, Whitehead AS. Genetic and nutritional factors contributing to hyperhomocysteinemia in young adults. Blood 2003; 101(7): 2483-8.
62. Knott L, Hartridge T, Brown NL, Mansell JP, Sandy JR. Homocysteine oxidation and apoptosis: a potential cause of cleft palate. In Vitro Cell Dev Biol Anim 2003; 39(1-2): 98-105.
63. Lievers KJ, Boers GH, Verhoef P, den Heijer M, Kluijtmans LA, van der Put NM, Trijbels FJ, Blom HJ. A second common variant in the methyl-enetetrahydrofolate reductase (MTHFR) gene and its relationship to MTHFR enzyme activity, homocysteine, and cardiovascular disease risk. J Mol Med 2001; 79(9): 522-8.
64. Linnebank M, Homberger A, Nowak-Göttl U, Marquardt T, Harms E, Koch HG. Linkage disequilibrium of the common mutations 677C > T and 1298A > C of the human methylenetetrahydrofolate reductase gene as proven by the novel polymorphisms 129C > T, 1068C > T. Eur J Pediatr 2000; 159(6): 472-3.
65. Lipton SA, Kim WK, Choi YB, Kumar S, D'Emilia DM, Rayudu PV, Ar-nelle DR, Stamler JS. Neurotoxicity associated with dual actions of homo-cysteine at the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A 1997; 94(11): 5923-8.
66. Ma J, Stampfer MJ, Giovannucci E, Artigas C, Hunter DJ, Fuchs C, Willett WC, Selhub J, Hennekens CH, Rozen R. Methylenetetrahydro-folate reductase polymorphism, dietary interactions, and risk of colorectal cancer. Cancer Res 1997; 57(6): 1098-102.

69
67. Mansoor MA, Guttormsen AB, Fiskerstrand T, Refsum H, Ueland PM, Svardal AM. Redox status and protein binding of plasma aminothiols dur-ing the transient hyperhomocysteinemia that follows homocysteine ad-ministration. Clin Chem 1993; 39(6): 980-5.
68. Massaro EJ, Rogers JM. Folate and Human Development. Oxford: Hu-mana Press; 2002.
69. Mattson MP, Shea TB. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci 2003; 26(3): 137-46.
70. McCaddon A, Blennow K, Hudson P, Hughes A, Barber J, Gray R, Davies G, Williams JH, Duguid J, Lloyd A, Tandy S, Everall M, Cattell H, Ellis D, Palmer M, Bogdanovic N, Gottfries CG, Zetterberg H, Rymo L, Regland B. Transcobalamin polymorphism and serum holo-transcobalamin in rela-tion to Alzheimer's disease. Dement Geriatr Cogn Disord 2004; 17(3): 215-21.
71. Meyer K, Fredriksen A, Ueland PM. High-level multiplex genotyping of polymorphisms involved in folate or homocysteine metabolism by matrix-assisted laser desorption/ionization mass spectrometry. Clin Chem 2004; 50(2): 391-402.
72. Min SH, Oh SY, Karp GI, Poncz M, Zhao R, Goldman ID. The clinical course and genetic defect in the PCFT gene in a 27-year-old woman with hereditary folate malabsorption. J Pediatr 2008; 153(3): 435-7.
73. Nilsson TK, Börjel AK. Novel insertion and deletion mutations in the 5'-UTR of the folate receptor-alpha gene: an additional contributor to hyper-homocysteinemia? Clin Biochem 2004; 37(3): 224-9.
74. Nurk E, Tell GS, Vollset SE, Nygård O, Refsum H, Nilsen RM, Ueland PM. Changes in lifestyle and plasma total homocysteine: the Hordaland Homocysteine Study. Am J Clin Nutr 2004; 79(5): 812-9.
75. Obeid R, Herrmann W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett 2006; 580(13): 2994-3005.
76. Packer BR, Yeager M, Burdett L, Welch R, Beerman M, Qi L, Sicotte H, Staats B, Acharya M, Crenshaw A, Eckert A, Puri V, Gerhard DS, Chanock SJ. SNP500Cancer: a public resource for sequence validation, as-say development, and frequency analysis for genetic variation in candidate genes. Nucleic Acids Res 2006; 34(Database issue): D617-21.
77. Panagiotakos DB, Pitsavos C, Zeimbekis A, Chrysohoou C, Stefanadis C. The association between lifestyle-related factors and plasma homocysteine levels in healthy individuals from the "ATTICA" Study. Int J Cardiol 2005; 98(3): 471-7.

70
78. Parnetti L, Bottiglieri T, Lowenthal D. Role of homocysteine in age-related vascular and non-vascular diseases. Aging (Milano) 1997; 9(4): 241-57.
79. Piedrahita JA, Oetama B, Bennett GD, van Waes J, Kamen BA, Richard-son J, Lacey SW, Anderson RG, Finnell RH. Mice lacking the folic acid-binding protein Folbp1 are defective in early embryonic development. Nat Genet 1999; 23(2): 228-32.
80. Pitkin RM. Folate and neural tube defects. Am J Clin Nutr 2007; 85(1): 285S-8S.
81. Rady PL, Szucs S, Grady J, Hudnall SD, Kellner LH, Nitowsky H, Tyring SK, Matalon RK. Genetic polymorphisms of methylenetetrahydrofolate reductase (MTHFR) and methionine synthase reductase (MTRR) in ethnic populations in Texas; a report of a novel MTHFR polymorphic site, G1793A. Am J Med Genet 2002; 107(2): 162-8.
82. Relton CL, Wilding CS, Laffling AJ, Jonas PA, Burgess T, Binks K, Tawn EJ, Burn J. Low erythrocyte folate status and polymorphic variation in folate-related genes are associated with risk of neural tube defect preg-nancy. Mol Genet Metab 2004; 81(4): 273-81.
83. Relton CL, Wilding CS, Pearce MS, Laffling AJ, Jonas PA, Lynch SA, Tawn EJ, Burn J. Gene-gene interaction in folate-related genes and risk of neural tube defects in a UK population. J Med Genet 2004; 41(4): 256-60.
84. Ronaghi M, Karamohamed S, Pettersson B, Uhlen M, Nyren P. Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem 1996; 242(1): 84-9.
85. Ronaghi M, Uhlen M, Nyren P. A sequencing method based on real-time pyrophosphate. Science 1998; 281(5375): 363, 5.
86. Rosenberg N, Murata M, Ikeda Y, Opare-Sem O, Zivelin A, Geffen E, Seligsohn U. The frequent 5,10-methylenetetrahydrofolate reductase C677T polymorphism is associated with a common haplotype in whites, Japanese, and Africans. Am J Hum Genet 2002; 70(3): 758-62.
87. Rozen R. Genetic modulation of homocysteinemia. Semin Thromb He-most 2000; 26(3): 255-61.
88. Rozen S, Skaletsky H. Primer3 on the WWW for general users and for bi-ologist programmers. Methods Mol Biol 2000; 132: 365-86.
89. Scholtz CL, Odendaal HJ, Thiart R, Loubser L, Hillermann R, Delport R, Vermaak WJ, Kotze MJ. Analysis of two mutations in the MTHFR gene associated with mild hyperhomocysteinaemia--heterogeneous distribution in the South African population. S Afr Med J 2002; 92(6): 464-7.

71
90. Schwahn B, Rozen R. Polymorphisms in the methylenetetrahydrofolate reductase gene: clinical consequences. Am J Pharmacogenomics 2001; 1(3): 189-201.
91. Selhub J. Homocysteine metabolism. Annu Rev Nutr 1999; 19: 217-46.
92. Serra Majem L, Ribas Barba L, Armas Navarro A, Alvarez León E, Sierra A. [Energy and nutrient intake and risk of inadequate intakes in Canary Is-lands (1997-98)]. Arch Latinoam Nutr 2000; 50(1 Suppl 1): 7-22.
93. Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D'Agostino RB, Wilson PW, Wolf PA. Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. N Engl J Med 2002; 346(7): 476-83.
94. Shames DS, Minna JD, Gazdar AF. Methods for detecting DNA methyla-tion in tumors: from bench to bedside. Cancer Lett 2007; 251(2): 187-98.
95. Skibola CF, Smith MT, Kane E, Roman E, Rollinson S, Cartwright RA, Morgan G. Polymorphisms in the methylenetetrahydrofolate reductase gene are associated with susceptibility to acute leukemia in adults. Proc Natl Acad Sci U S A 1999; 96(22): 12810-5.
96. Snowdon DA, Tully CL, Smith CD, Riley KP, Markesbery WR. Serum folate and the severity of atrophy of the neocortex in Alzheimer disease: findings from the Nun study. Am J Clin Nutr 2000; 71(4): 993-8.
97. Stegmann K, Ziegler A, Ngo ET, Kohlschmidt N, Schröter B, Ermert A, Koch MC. Linkage disequilibrium of MTHFR genotypes 677C/T-1298A/C in the German population and association studies in probands with neural tube defects(NTD). Am J Med Genet 1999; 87(1): 23-9.
98. Sumner J, Jencks DA, Khani S, Matthews RG. Photoaffinity labeling of methylenetetrahydrofolate reductase with 8-azido-S-adenosylmethionine. J Biol Chem 1986; 261(17): 7697-700.
99. Szczeklik A, Sanak M, Jankowski M, Dropi ski J, Czachór R, Musial J, Axenti I, Twardowska M, Brzostek T, Tendera M. Mutation A1298C of methylenetetrahydrofolate reductase: risk for early coronary disease not associated with hyperhomocysteinemia. Am J Med Genet 2001; 101(1): 36-9.
100. Thwaites DT, Anderson CM. H+-coupled nutrient, micronutrient and drug transporters in the mammalian small intestine. Exp Physiol 2007; 92(4): 603-19.
101. Trabetti E. Homocysteine, MTHFR gene polymorphisms, and cardio-cerebrovascular risk. J Appl Genet 2008; 49(3): 267-82.

72
102. Tran P, Leclerc D, Chan M, Pai A, Hiou-Tim F, Wu Q, Goyette P, Artigas C, Milos R, Rozen R. Multiple transcription start sites and alternative splicing in the methylenetetrahydrofolate reductase gene result in two en-zyme isoforms. Mamm Genome 2002; 13(9): 483-92.
103. Ulvik A, Ueland PM, Fredriksen A, Meyer K, Vollset SE, Hoff G, Schneede J. Functional inference of the methylenetetrahydrofolate reductase 677C > T and 1298A > C polymorphisms from a large-scale epidemiological study. Hum Genet 2007; 121(1): 57-64.
104. Wakutani Y, Kowa H, Kusumi M, Nakaso K, Yasui K, Isoe-Wada K, Yano H, Urakami K, Takeshima T, Nakashima K. A haplotype of the me-thylenetetrahydrofolate reductase gene is protective against late-onset Alz-heimer's disease. Neurobiol Aging 2004; 25(3): 291-4.
105. van der Put NM, Gabreels F, Stevens EM, Smeitink JA, Trijbels FJ, Eskes TK, van den Heuvel LP, Blom HJ. A second common mutation in the me-thylenetetrahydrofolate reductase gene: an additional risk factor for neu-ral-tube defects? Am J Hum Genet 1998; 62(5): 1044-51.
106. van der Velden AW, Thomas AA. The role of the 5' untranslated region of an mRNA in translation regulation during development. Int J Biochem Cell Biol 1999; 31(1): 87-106.
107. Weisberg I, Tran P, Christensen B, Sibani S, Rozen R. A second genetic polymorphism in methylenetetrahydrofolate reductase (MTHFR) associ-ated with decreased enzyme activity. Mol Genet Metab 1998; 64(3): 169-72.
108. Weisberg IS, Jacques PF, Selhub J, Bostom AG, Chen Z, Curtis Ellison R, Eckfeldt JH, Rozen R. The 1298A-->C polymorphism in methylenetetra-hydrofolate reductase (MTHFR): in vitro expression and association with homocysteine. Atherosclerosis 2001; 156(2): 409-15.
109. Verhoef P, Stampfer M. Epidemiology of Vascular and Thrombotic Asso-ciations. In: Carmel R, Jacobsen DW, editors. Homocysteine in Health and Disease. Cambridge: Cambridge University Press; 2001.
110. Wiemels JL, Smith RN, Taylor GM, Eden OB, Alexander FE, Greaves MF. Methylenetetrahydrofolate reductase (MTHFR) polymorphisms and risk of molecularly defined subtypes of childhood acute leukemia. Proc Natl Acad Sci U S A 2001; 98(7): 4004-9.

73
111. Wilcken B, Bamforth F, Li Z, Zhu H, Ritvanen A, Renlund M, Stoll C, Alembik Y, Dott B, Czeizel AE, Gelman-Kohan Z, Scarano G, Bianca S, Ettore G, Tenconi R, Bellato S, Scala I, Mutchinick OM, Lopez MA, de Walle H, Hofstra R, Joutchenko L, Kavteladze L, Bermejo E, Martínez-Frias ML, Gallagher M, Erickson JD, Vollset SE, Mastroiacovo P, Andria G, Botto LD, Redlund M. Geographical and ethnic variation of the 677C>T allele of 5,10 methylenetetrahydrofolate reductase (MTHFR): findings from over 7000 newborns from 16 areas world wide. J Med Ge-net 2003; 40(8): 619-25.
112. Wilcken DEL, Wilcken B. Historical Overview and Recent Perspectives. In: Carmel R, Jacobsen DW, editors. Homocysteine in Health and Disease. Cambridge: Cambridge University Press; 2001.
113. Vollset SE, Refsum H, Nygård O, Ueland PM. Lifestyle Factors Associated with Hyperhomocysteinemia. In: Carmel R, Jacobsen DW, editors. Homo-cysteine in Health and Disease. Cambridge: Cambridge University Press; 2001.
114. World Health Organization. The ICD-10 Classification of Mental and Behavioural Disorders. Geneva: WHO; 1992.
115. Xiao W, Oefner PJ. Denaturing high-performance liquid chromatography: A review. Hum Mutat 2001; 17(6): 439-74.
116. Yamada K, Chen Z, Rozen R, Matthews RG. Effects of common poly-morphisms on the properties of recombinant human methylenetetrahydro-folate reductase. Proc Natl Acad Sci U S A 2001; 98(26): 14853-8.
117. Yang F, Tan HM, Wang H. Hyperhomocysteinemia and atherosclerosis. Sheng Li Xue Bao 2005; 57(2): 103-14.
118. Zetterberg H. Methylenetetrahydrofolate reductase and transcobalamin genetic polymorphisms in human spontaneous abortion: biological and clinical implications. Reprod Biol Endocrinol 2004; 2(1): 7.
119. Zetterberg H, Regland B, Palmer M, Rymo L, Zafiropoulos A, Arvanitis DA, Spandidos DA, Blennow K. The transcobalamin codon 259 polymor-phism influences the risk of human spontaneous abortion. Hum Reprod 2002; 17(12): 3033-6.
120. Zhang G, Zhang QY, Miao XP, Lin DX, Lu YY. Polymorphisms and mu-tations of the folate receptor-alpha gene and risk of gastric cancer in a Chinese population. Int J Mol Med 2005; 15(4): 627-32.
121. Zhao R, Goldman ID. The molecular identity and characterization of a Proton-coupled Folate Transporter--PCFT; biological ramifications and impact on the activity of pemetrexed. Cancer Metastasis Rev 2007; 26(1): 129-39.

74
122. Zhu H, Wicker NJ, Shaw GM, Lammer EJ, Hendricks K, Suarez L, Canfi-eld M, Finnell RH. Homocysteine remethylation enzyme polymorphisms and increased risks for neural tube defects. Mol Genet Metab 2003; 78(3): 216-21.

Publikationer i serien Örebro Studies in Medicine
1. Bergemalm, Per-Olof (2004). Audiologic and cognitive long-term sequelae from closed head injury.
2. Jansson, Kjell (2004). Intraperitoneal Microdialysis. Technique and Results.
3. Windahl, Torgny (2004). Clinical aspects of laser treatment of lichen sclerosus and squamous cell carcinoma of the penis.
4. Carlsson, Per-Inge (2004). Hearing impairment and deafness. Genetic and environmental factors – interactions – consequences. A clinical audiological approach.
5. Wågsäter, Dick (2005). CXCL16 and CD137 in Atherosclerosis.
6. Jatta, Ken (2006). Inflammation in Atherosclerosis.
7. Dreifaldt, Ann Charlotte (2006). Epidemiological Aspects on Malignant Diseases in Childhood.
8. Jurstrand, Margaretha (2006). Detection of Chlamydia trachomatis and Mycoplasma genitalium by genetic and serological methods.
9. Norén, Torbjörn (2006). Clostridium difficile, epidemiology and antibiotic resistance.
10. Anderzén Carlsson, Agneta (2007). Children with Cancer – Focusing on their Fear and on how their Fear is Handled.
11. Ocaya, Pauline (2007). Retinoid metabolism and signalling in vascular smooth muscle cells.
12. Nilsson, Andreas (2008). Physical activity assessed by accelerometry in children.
13. Eliasson, Henrik (2008). Tularemia – epidemiological, clinical and diagnostic aspects.
14. Walldén, Jakob (2008). The influence of opioids on gastric function: experimental and clinical studies.
15. Andrén, Ove (2008). Natural history and prognostic factors in localized prostate cancer.
16. Svantesson, Mia (2008). Postpone death? Nurse-physician perspectives and ethics rounds.

17. Björk, Tabita (2008). Measuring Eating Disorder Outcome – Definitions, dropouts and patients’ perspectives.
18. Ahlsson, Anders (2008). Atrial Fibrillation in Cardiac Surgery.
19. Parihar, Vishal Singh (2008). Human Listeriosis – Sources and Routes.
20. Berglund, Carolina (2008). Molecular Epidemiology of Methicillin-Resistant Staphylococcus aureus. Epidemiological aspects of MRSA and the dissemination in the community and in hospitals.
21. Nilsagård, Ylva (2008). Walking ability, balance and accidental falls in persons with Multiple Sclerosis.
22. Johansson, Ann-Christin (2008). Psychosocial factors in patients with lumbar disc herniation: Enhancing postoperative outcome by the identification of predictive factors and optimised physiotherapy.
23. Larsson, Matz (2008). Secondary exposure to inhaled tobacco products.
24. Hahn-Strömberg, Victoria (2008). Cell adhesion proteins in dif-ferent invasive patterns of colon carcinoma: A morphometric and molecular genetic study.
25. Böttiger, Anna (2008). Genetic Variation in the Folate Receptor-α and Methylenetetrahydrofolate Reductase Genes as Determinants of Plasma Homocysteine Concentrations.




















