
Genetic determinants of normal variation in coagulation factor (F) IX levels: genome-wide scan and...
9
Click here to load reader
Transcript of Genetic determinants of normal variation in coagulation factor (F) IX levels: genome-wide scan and...

O R I G I N A L A R T I C L E
Genetic determinants of normal variation in coagulation factor(F) IX levels: genome-w ide scan and examination of the FIXstructural geneM . K H A C H I D Z E , * A . B U I L , K . R . V I E L , * S . P O R T E R , D . W A R R E N , § D . K . M A C H I A H , * J . M . S O R I A , J . C . S O U T O , A . A M E R I , – M . L A T H R O P , * * J . B L A N G E R O , § J . F O N T C U B E R T A , S . T . W A R R E N , L . A L M A S Y § and T . E . H O W A R D **Department of Pathology and Laboratory- M edicine, Emory University, Atlanta, G A , USA; Departament d Hematologia, Unitat d Hemostasia iTrombosi, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain; Geospiza Inc., Seattle, W A; §Department of Genetics, Southw est Foundation,San Antonio, TX; –Department of Pediatrics, M edical College of Georgia, Augusta, G A , USA; **C entre National de Genotypage, Evry, France;and Department of Human Genetics, Emory University, Atlanta, G A , USA
To cite this article: Khachidze M , Buil A , V iel KR, Porter S, W arren D , M achiah DK, Soria J M , Souto JC , Ameri A , Lathrop M , Blangero J,Fontcuberta J, W arren ST, A lmasy L, Ho w ard TE. Genetic determinants of normal variation in coagulation factor (F) IX levels: genome- w ide scanand examination of the FIX structural gene. J Thromb Haemost 2006; 4: 1537–45.
Summary. Background:High-normal and elevatedplasma FIXactivity (FIX:C) levels are associated with increased risk forvenous- and possibly arterial-thrombosis. Objective: Becausethe broad normal range for FIX:C involves a substantialunknown genetic component, we sought to identify quantita-tive-trait loci (QTLs) for this medically important hemostasistrait. Methods: We performed a genome-wide screen and aresequencing-based variation scan of the known functionalregions of every distinct FIX gene (F9) in the genetic analysis ofidiopathic thrombophilia project (GAIT), a collection of 398Spanish-Caucasians from 21 pedigrees. Results: We found noevidence for linkage (LOD scores <1.5) despite genotypingmore than 540 uniformly-spaced microsatellites. We identified27 candidateF9 polymorphisms, including three in cis-elementsresponsible for the increase in FIX:C that occurs with aging,but found no significant genotype-specific di erences inmean FIX:C levels (P-values ‡ 0.11) despite evaluatingevery polymorphism in GAIT by marginal multicovariatemeasured-genotype association analysis. Conclusions: Theheritable component of interindividual FIX:C variability likelyinvolves a collection of QTLs with modest e ects that mayreside in genes other than F9. Nevertheless, because the allelesof these 27 polymorphisms exhibited a low overall degree oflinkage disequilibrium, we are currently defining their haplo-types to interrogate several highly-conserved non-exonicsequences and other F9 segments not examined here.
Keywords: association, linkage, linkage disequilibrium, quan-titative-trait loci, single-nucleotide polymorphism, thrombosis.
Introduction
Thromboembolic disorders are a major cause of mortality andmorbidity worldwide. The ability to identify accurately individ-uals at risk would advance management of these conditionsthroughmore effective prophylactic use of antithrombotic agents.The pathogenesis of thrombosis is complex, involving multiplegenetic and environmental factors. While notable prothromboticgene variants have been discovered [1,2], family studies indicateadditional heritable factors remain unidentified [3,4].
Individuals with elevated levels of coagulation factor (F) IX,the plasma glycoprotein whose activity (FIX:C) is deficient inpatients with hemophilia B, have long been suspected of beingprothrombotic [5,6]. Recent studies have established that FIXlevels constitute a quantitative-trait which, across its greaterthan 3-fold normal-range [6–9], correlates with the risk ofvenous and possibly arterial thrombosis [7,10,11]. Based on astudy of 398 Caucasian-subjects from 21 extended Spanishpedigrees, known as the Genetic Analysis of IdiopathicThrombophilia Project (GAIT), Souto et al. [12] estimated39% heritability for FIX:C.
To identify the quantitative-trait loci (QTLs) underlyingFIX:Cvariability,weexamined the study-subjects inGAITviaacomprehensive strategy that includedbothgenome-wideandF9screens.We conducted the genome-wide screenfirstbecause thisapproach, which is based on linkage analysis, does not requirethat candidate genes be specified a priori. Next, we identifiedevery polymorphism in known functional regions of F9, byresequencing all potentially distinct alleles in GAIT, and subse-quently quantified their allelic contributions to FIX:C using
Correspondence: Tom E. Howard, Department of Pathology andLaboratory Medicine, Emory University, WMRB, Room 7109B, 101Woodru Circle, Atlanta, GA 30322, USA.Tel.: +1 404 712 0144; fax: +1 404 712 9625; e-mail:[email protected]
Received 22 December 2005, accepted 5 April 2006
Journal of Thrombosis and Haemostasis, 4: 1537–1545
2006 International Society on Thrombosis and Haemostasis
associationanalysis.Finally,weexaminedF9 in51healthyunre-lated individuals from three ethnic groups as afirst step towardsdetermining whether FIX QTLs may differ by ethnicity.
M aterials and methods
M aterials
All F9 amplicons were generated by polymerase chain reaction(PCR) using HotStarTaq-Master Mix (Qiagen, Valencia, CA,USA) and purified with AmpPure (Agencourt Biosciences;Beverly, MA, USA).Oligonucleotides forPCR, sequencing andamplicon-length polymorphism (ALP) analyses were made byInvitrogen (Carlsbad, CA, USA); all sequences were based onthe genomic-DNA in GenBank accession K02402 and areavailableupon request.BigDye-terminator sequencingkitswerepurchased from ABI (FosterCity, CA, USA). CleanSEQ wasobtained from Agencourt. SeqMan (v5.03)was purchased fromDNASTAR (Madison, WI, USA). Phred, a gift from theUniversity of Washington, was used within the Finch Sequen-cing System, purchased from Geospiza (Seattle, WA, USA).
Subjects and phenotype
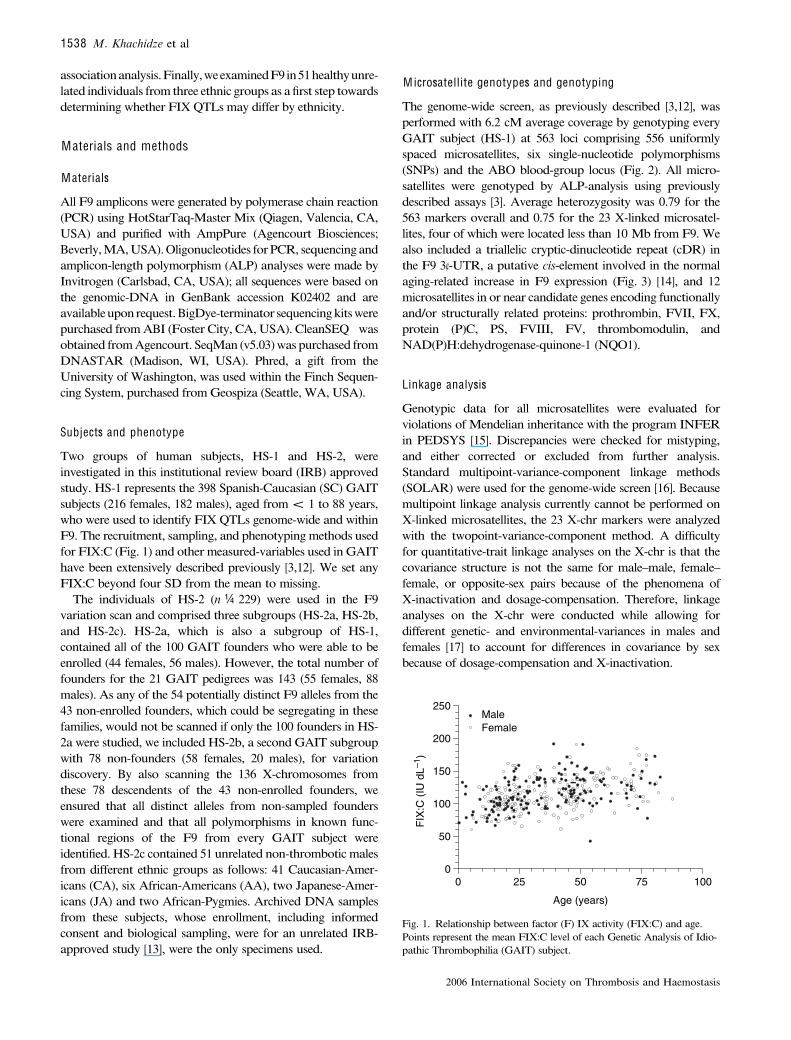
Two groups of human subjects, HS-1 and HS-2, wereinvestigated in this institutional review board (IRB) approvedstudy. HS-1 represents the 398 Spanish-Caucasian (SC) GAITsubjects (216 females, 182 males), aged from < 1 to 88 years,who were used to identify FIX QTLs genome-wide and withinF9. The recruitment, sampling, and phenotypingmethods usedfor FIX:C (Fig. 1) and othermeasured-variables used in GAIThave been extensively described previously [3,12]. We set anyFIX:C beyond four SD from the mean to missing.
The individuals of HS-2 (n ¼ 229) were used in the F9variation scan and comprised three subgroups (HS-2a, HS-2b,and HS-2c). HS-2a, which is also a subgroup of HS-1,contained all of the 100 GAIT founders who were able to beenrolled (44 females, 56 males). However, the total number offounders for the 21 GAIT pedigrees was 143 (55 females, 88males). As any of the 54 potentially distinct F9 alleles from the43 non-enrolled founders, which could be segregating in thesefamilies, would not be scanned if only the 100 founders in HS-2a were studied, we included HS-2b, a second GAIT subgroupwith 78 non-founders (58 females, 20 males), for variationdiscovery. By also scanning the 136 X-chromosomes fromthese 78 descendents of the 43 non-enrolled founders, weensured that all distinct alleles from non-sampled founderswere examined and that all polymorphisms in known func-tional regions of the F9 from every GAIT subject wereidentified. HS-2c contained 51 unrelated non-thromboticmalesfrom different ethnic groups as follows: 41 Caucasian-Amer-icans (CA), six African-Americans (AA), two Japanese-Amer-icans (JA) and two African-Pygmies. Archived DNA samplesfrom these subjects, whose enrollment, including informedconsent and biological sampling, were for an unrelated IRB-approved study [13], were the only specimens used.
M icrosatellite genotypes and genotyping
The genome-wide screen, as previously described [3,12], wasperformed with 6.2 cM average coverage by genotyping everyGAIT subject (HS-1) at 563 loci comprising 556 uniformlyspaced microsatellites, six single-nucleotide polymorphisms(SNPs) and the ABO blood-group locus (Fig. 2). All micro-satellites were genotyped by ALP-analysis using previouslydescribed assays [3]. Average heterozygosity was 0.79 for the563 markers overall and 0.75 for the 23 X-linked microsatel-lites, four of which were located less than 10 Mb from F9. Wealso included a triallelic cryptic-dinucleotide repeat (cDR) inthe F9 3¢-UTR, a putative cis-element involved in the normalaging-related increase in F9 expression (Fig. 3) [14], and 12microsatellites in or near candidate genes encoding functionallyand/or structurally related proteins: prothrombin, FVII, FX,protein (P)C, PS, FVIII, FV, thrombomodulin, andNAD(P)H:dehydrogenase-quinone-1 (NQO1).
Linkage analysis
Genotypic data for all microsatellites were evaluated forviolations of Mendelian inheritance with the program INFERin PEDSYS [15]. Discrepancies were checked for mistyping,and either corrected or excluded from further analysis.Standard multipoint-variance-component linkage methods(SOLAR) were used for the genome-wide screen [16]. Becausemultipoint linkage analysis currently cannot be performed onX-linked microsatellites, the 23 X-chr markers were analyzedwith the twopoint-variance-component method. A difficultyfor quantitative-trait linkage analyses on the X-chr is that thecovariance structure is not the same for male–male, female–female, or opposite-sex pairs because of the phenomena ofX-inactivation and dosage-compensation. Therefore, linkageanalyses on the X-chr were conducted while allowing fordifferent genetic- and environmental-variances in males andfemales [17] to account for differences in covariance by sexbecause of dosage-compensation and X-inactivation.
0 25 50 75 1000
50
100
150
200
250
Age (years)
FIX
:C (
IU d
L–1)
MaleFemale
Fig. 1. Relationship between factor (F) IX activity (FIX:C) and age.Points represent the mean FIX:C level of each Genetic Analysis of Idio-pathic Thrombophilia (GAIT) subject.
1538 M. Khachidze et al
2006 International Society on Thrombosis and Haemostasis

Variance-component methods are vulnerable to deviationsfrom normality in trait distribution, particularly to high levelsof kurtosis [18]. The FIX:C distribution in GAIT exhibited akurtosis of 0.03. Statistical-genetic theory demonstrates thatthis level will not affect the distribution of LOD scores and thatthe standard nominal P-values for LOD scores are appropriatefor the FIX:C linkage screen [19]. Allele frequencies wereestimated in GAIT, and the marker maps for multipointanalyses were obtained from ABI and the Marshfield Clinic(Marshfield, WI, USA). Because 12 GAIT families wereascertained through thrombophilic probands, all analysesincluded an ascertainment correction achieved by conditioningthe likelihood of these pedigrees on the likelihoods of theirrespective probands [20].
Discovery and genotyping of F9 polymorphisms
F9 sequence variations were discovered and genotyped bydirectly sequencing nine amplicons, designated 1–9, thatcontained all known functional regions of this locus (Fig. 3B).These amplicons, located between sense-strand nucleotides1,965–35,850 in GenBank sequence K02402 [21], were gener-ated using genomic-DNA from individuals in HS-2 and theremaining GAIT subjects in HS-1. In order to define preciselythe F9 regions investigated (Table 1), the 5¢- and 3¢-mostnucleotide of each amplicon are designated with the numberof the corresponding base in K02402 (Fig. 3B). All amplifi-cations were performed using reaction conditions and ther-mal-cycling parameters that are available upon request. Awater negative-control was included for each amplicon, inevery run, and all were validated by 1%-agarose-gel electro-phoresis. All amplicons were purified using AmpPure , asdescribed by the manufacturer, and subjected to cycle-sequencing using reaction conditions and thermal-cyclingparameters that are available upon request. All sequencing-products were purified using CleanSEQ , as described by themanufacturer, and electrophoresed on either an ABI Prism377 or 3100 instrument. AB1-files were uploaded into theFinch server and assessed for overall quality; any sequencewith an average Phred quality score (Q) less than 30 [22,23]was repeated. The sequence of each amplicon was determinedfrom both strands for variation discovery and one strand forpolymorphism genotyping.
All F9 variants (Fig. 3A) were identified by manuallyreviewing trace files in SeqMan multiple-sequence alignments.To minimize false-positives, all poor quality data (averageQ < 30) were first removed. To be considered a true sequencevariation, the allele(s) had to be consistent in both the forward-and reverse-strand reactions. Taq-induced errors are unlikelyunder the conditions used as the amount of genomic-DNA ineach reaction provided several thousand haploid genomeequivalents as template and amplicons were subjected to directsequencing. However, before a previously unknown F9variation was designated as naturally occurring, the minor-allele had to be present in at least two subjects in HS-2 or beconfirmed by directly-sequencing a second preparation of thisamplicon derived from an independent PCR that used the samegenomic-DNA as template. To confirm the sequencing-basedgenotypes of 5:32197:6/4 in the cDR (Fig. 3), fluorescently-labeled amplicons were generated, electrophoresed on an ABIPrism 3100 instrument, and evaluated by ALP-analysis usingGeneScan (v3.7).
Linkage disequilibrium (LD)
We determined the pairwise LD between alleles of the 23 F9polymorphisms that were informative in at least the SC and/orCA subjects (Fig. 4). Allele frequencies for these polymor-phisms were estimated in 148 unrelated Caucasian individualsincluding the 100 SC subjects in HS-2a, seven additional GAITmales whose genotypes were inferred from family information,and the 41 CA males in HS-2c (Table 2). Weighted D and r2
were calculated for all consecutive SNP pairs using maximum-likelihood haplotype frequencies estimated by the expectation-maximization method implemented in GENECOUNTING[24]. The small number of JA and individuals of African-descent (AD) precluded accurate frequency estimates for F9haplotypes found only in the non-Caucasian populationssampled, that is, those defined by SNPs monomorphic inCaucasians (G13516A, G30249A, A30555G, and G31093A).Thus we could neither investigate the possibility for ethnicdifferences in LD patterns across F9 nor determine thedegree of LD within polymorphism pairs involving these fourSNPs.
M arginal measured-genotype association analysis
Blinded genotypic data from the entire GAIT cohort for the 17F9 polymorphisms that were informative in SC subjects(Table 2) were analyzed with INFER for violations ofMendelian inheritance. All disparities were checked formistyping and either corrected or excluded from furtheranalysis. Marginal association analysis was performed, usingthe measured-genotype approach [20], to test for genotype-specific differences in mean FIX:C while allowing for non-independence among family members. As males have just oneallele, we used a dosage-compensation model in whichhemizygous males had a gene effect equivalent to femaleshomozygous for the same allele.
Table 1 F9 regions resequenced
Genic region bp
Coding 1386Non-coding 8815
Promoter 10015¢-UTR 29Intronic* 62693¢-UTR 13893¢-Genomic 127
Total 10201
*Approximately 4 kb of intronic sequence studied in HS-1.
Comprehensive search for FIX QTLs 1539
2006 International Society on Thrombosis and Haemostasis

Endogenous, genetic and environmental covariates
GAITdata forage, sex, smoking status, oral contraceptive (OC)use, ABO-genotype, body mass index (BMI), diabetes-mellitusstatus, and plasma levels of total cholesterol (TC), high-densitylipoprotein (HDL), low-density lipoprotein (LDL), very low-density lipoprotein (VLDL), triglycerides (TG), lipoprotein(a),von Willebrand factor antigen (VWF:Ag) and FVIII:C weresubjected to both univariate and multivariate analyses withSOLAR [16]. We used indicator variables to represent ABO-genotypes, with theOO-genotype being the reference level, andmodeled separate mean FVIII:C levels for each.
Results
FIX:C is correlated w ith age in univariate analysis
Because FIX:C was one of more than 25 hemostasis traitsassayed in GAIT, duplicate measures were available for only342 subjects; genomic-DNA from one was not available forstudy. In the remaining 341 individuals, FIX:C ranged from 42to 237 IU dL )1 (mean ¼ 115 IU dL )1; SD ¼ 26 IU dL )1).Data from one male, whose FIX:C (237 IU dL )1) was greaterthan 4 SD above the mean (219 IU dL )1) was excluded fromfurther analysis. FIX:C in these 340 subjects (185 females, 155males), which ranged from 42 to 191 IU dL )1 and demonstra-
ted the greater than threefold concentration range reported forother non-hemophilic populations [6–9], are presented in Fig. 1as a function of age. When evaluated independently from othermeasured covariates, we observed significant relationshipsbetween FIX:C and both age (P ¼ 0.0000021) and age2 (P ¼0.016), but not for age3 (P ¼ 0.582) or sex (P ¼ 0.237),confirming the widely reported observation that FIX:Cincreases with age in univariate analysis [8,9,14].
No major autosomal FIX determinants identified
We genotyped all GAIT subjects for 540 autosomal polymor-phisms, including 506 uniformly spaced microsatellites and 34random markers located in or near obvious candidate genes,and applied multipoint-variance-component methods to iden-tify regions on chromosomes 1–22 in linkage with FIX:C(Fig. 2). As no LOD scores greater than 1.5 were observed, noregions met criteria suggestive for linkage. This findingindicates there may be no major autosomal determinants ofFIX variability, at least in the SCpopulation sampled in GAIT.However, despite the use of a high-density map providing6.2 cM average genome coverage, two chr6 regions were notadequately screened. Referred to as 20 cM gaps, these regionsspan the 7.6 Mb chr6p21.2–12.3 segment flanked by D6S1610and D6S452, and the 29Mb chr6q15–22.31 segment flanked byD6S462 and D6S287, which contain 96 and 104 genes,respectively (available upon request). Because of the smallnumber of subjects, the power to detect QTLs with moderateeffects is only modest.
No X-linked FIX determinants identified
Seventeen of the 23 X-linked microsatellites genotyped inGAIT were uniformly distributed across the chr; six othermarkers (DXS1227, DXS8043, DXS52, DXS1073,DXS1253E, and cDR) were selected for their locations in ornear either F9 or the gene encoding FVIII (F8), the pro-cofactor for catalytically active FIX. Analogous to the auto-somes, we found no X-chr regions that met genome-widecriteria suggestive for linkage; LOD scores for these 23markerswere all less than 1.0, including 0.35 for the cDR and 0.76, thehighest, for DXS986. As FIX levels are reported to increasewith age in humans [8,9,14], we modeled age as a covariate.However, the linkage signals remained unchanged after inclu-ding age, age2 and age · sex. Despite using equally informativeX-linked- and autosomal-microsatellites, X-linked loci inclu-ding F9may contain determinants of FIX variance because X-chr markers can currently only be evaluated with less powerfultwopoint linkage methods. Considered together, these resultssuggest that a collection of QTLs with modest effects is mostlikely responsible for the heritable variance in FIX:C.
M ultiple novel F9 polymorphisms
Because association analysis is statistically superior to linkageanalysis for detecting QTLs within candidate genes, we identi-
cM0 50 100
150
cM0 50 100
150
200
250
300
chr1
LOD
01
2
chr2
chr3
chr4
chr5
chr6
chr7
chr8
chr12
chr13
chr14
chr15
chr16
chr17
chr18
chr19
chr9
chr10
chr11
chr20
chr21
chr22
Fig. 2. Autosomal linkage results from the genome-wide screen fordeterminants of factor (F)IX activity (FIX:C) levels in GAIT.
1540 M. Khachidze et al
2006 International Society on Thrombosis and Haemostasis
fied all polymorphisms in known functional regions of F9 bysequencing two subgroupsofGAIT subjects, HS-2a andHS-2b.Although HS-2a contained all enrolled founders, 43 GAITfounders could not be enrolled for reasons previously described[3,12]. Consequently, any distinct F9 allele passed-down bythese non-enrolled founders would be missed if the variationscan were limited to HS-2a. By including the descendents ofthese 43 non-enrolled founders (HS-2b), we ensured that allnaturally occurring polymorphisms in the known functionalregionsof every foundingF9allele segregated inGAITwouldbeidentified. By doing so, we maximized the probability ofdiscovering FIX QTLs in the structural locus by associationanalysis, as these sequence variations would likely contain thefunctional subset of GAIT F9 polymorphisms.
We resequenced the known functional F9 regions includingall exons, 1 kb of the contiguous promoter region, 50 bp ofeach intronic junction, and 127 bp of 3¢-genomic-DNA(Table 1) [5,6,14,25]. We directly sequenced nine 500–3000 bp F9 amplicons generated from each person in HS-2a
and HS-2b (Fig. 3). Using RepeatMasker (http://repeatmas-ker.org) to screen the 38059 bp of K02402 for repeats and lowcomplexity sequences [26] in Repbase Update [27], we foundthat repetitive elements comprise 33% (12665 bp) of thestructural locus and that 40.2% of all non-repetitive F9sequences were covered in our resequencing scan. As the firststep towards determining whether heritable FIX determinantsin the SC population are conserved across ethnic groups, weresequenced the same F9 regions in a group containing 51healthy unrelated male subjects (HS-2c) of different ethnicity,including: 41 CA, 6 AA, 2 JA, and 2 AP. The eight people ofAD, including 6 AA and 2 AP, comprised the second largestethnic group examined in this study.
We identified 27 polymorphisms distributed throughout F9including seven in the promoter, two in coding sequences, 12 inintrons, two in the 3¢-UTR, and three in contiguous 3¢-genomic-DNA (Table 2). Eleven of these polymorphisms, including fourin the promoter (G )816A, G )653C, A )634G, and T )609C),five in introns (C10118G, C13162T, G13516A, G30249A, and
Table 2 Characteristics, status, and allelic associations of F9 polymorphisms with FIX:C
F9 polymorphisms* MAF F9 variation databases FIX:C association–
TU GenBank SC CA AD JA T HBMD VDR dbSNP P-value
G-816A 46565745 1.4 NP NP NP NP NF NF NF 0.193G-793A 48397547 33.6 49.0 13.0 NP 47.0 + F9-000716 rs411017 0.242C-698T 48397548 33.6 49.0 13.0 NP 47.0 + F9-000811 rs378815 0.229G-653C 46532015 0.7 2.0 NP NP 2.0 NF NF NF 0.695A-634G 46532016 0.7 NP NP NP NP NF NF NF 0.174T-609C 46532017 0.7 NP NP NP NP NF NF NF 0.305T-561G 48397549 8.4 15 50.0 NP 20.0 + F9-000948 rs4149657 0.113A192G 48397550 0.7 NP NP NP NP + NF rs3817939 0.425C10118G 46532020 NP 29.0 25.0 50.0 29.0 NF NF NF NAA10365G 48397558 NP 2.0 NP NP 2.0 NF F9-011902 rs3134809 NAA10512G 48397559 NP 5.0 NP NP 4.0 + F9-012049 rs6049 NAA11113G 48397551 28.0 32.0 13.0 NP 27.0 NF F9-012651 rs398101 0.920T11269A 48397552 5.6 7.0 NP NP 6.0 NF F9-012806 rs4149755 0.215C13162T 46532021 NP 2.0 NP NP 2.0 NF NF NF NAG13516A 46532022 NP NP NP 50.0 2.0 NF NF NF NAA20002C 48397553 28.0 NP NT NT NT + F9-021554 rs422187 0.575A20422G 48397554 27.3 32.0 25.0 NP 29.0 +§ F9-021975 rs6048 0.630G30249A 46532023 NP NP 13.0 NP 2.0 NF NF NF NAC30294T 46532024 NP 2.0 NP NP 2.0 NF NF NF NAA30555G 48397560 NP NP 13.0 NP 2.0 NF F9-032113 rs4149750 NAG31093A 48397561 NP NP 13.0 NP 2.0 + NF NF NAG32056A 48397555 17.5 24.0 25.0 NP 24.0 NF F9-033614 rs440051 0.538A32168T 46532018 2.8 2.0 NP NP 2.0 NF NF NF 0.5425:32197:6/4 48397557 25.0/10.0 25.0/10.0 13.0/0.0 NP 22.0/8.0 NF NF NF >0.450T32770C 48397562 NP 2.0 NP NP 2.0 + NF rs3788871 NAA32781G 46532019 0.7 2.0 NP NP 2.0 NF NF NF 0.394C32847T 48397556 1.4 2.0 NP NP 2.0 NF NF rs428199 0.759
FIX, factor IX; F9, FIX gene; FIX:C, FIX activity; MAF, minor-allele frequencies; TU, transcription unit; SC, Spanish-Caucasian; CA,Caucasian- American; AD, African-descent; JA, Japanese-American; T, total; HBMD, hemophilia-B mutation database; VDR, variationdiscovery resources (UW-FHCRC); dbSNP, database of single-nucleotide polymorphisms (NCBI); NP, not polymorphic; NT, not tested; NF,not found; NA, not applicable.*The alleles, position in TU (start-site: nucleotide 1), and GenBank number (dbSNP) are listed for each polymorphism. MAF estimated in unrelated subjects: HS-2a (SC); HS-2c: either its ethnic subgroups (CA, AD, and JA) or overall (T). Designates databases with F9 polymorphisms: HBMD (+ designates known polymorphisms;§denotes that A20422G is listed as 20421G>A and 147Ala>Thr); VDR (polymorphisms designated by gene and unique identifier); and dbSNP(polymorphisms designated by unique identifier).–P-values from marginal measured-genotype association analysis.
Comprehensive search for FIX QTLs 1541
2006 International Society on Thrombosis and Haemostasis
C30294T), one each in the 3¢-UTR (A32168T) and 3¢-genomic-DNA (A32781G), were absent from public databases (http://www.kcl.ac.uk/ip/petergreen/haemBdatabase.html;http://pga.gs.washington.edu; http://www.ncbi.nlm.nih.gov/books/bookres.fcgi/handbook/ch5d1.pdf). Of two coding sequence polymor-phisms (A20422G, G31093A), both previously known, oneencodes a non-synonymous substitution (Thr148Ala) [28];G31093A encodes Gln324Gln (Fig. 3C).
All sequence variations identifiedwere biallelic SNPs, exceptfor the cDR, in the 3¢-UTR, whichwas comprised of a triallelicGT-repeat (GTn,where n ¼ 4, 5, or 6),whose 5¢-most base is atpre-mRNA position 32197 (nucleotide 35162; K02402), and apreviously unknown SNP A32168T (Fig. 3). As GT5 repre-sents the major-allele and GT6 is the more common minor-allele, we designated this polymorphism 5:32197:6/4. To obtainallele frequency estimates accurately reflecting the populationsampled in GAIT, values in Table 2 are based only on the 100unrelated people in HS-2a. When considered independently
from ethnicity, the minor-allele frequencies (MAFs) of thesevariations ranged from 0.7–47% (Table 2).
Some M A Fs vary w ith ethnicity
While MAF for a few polymorphisms, e.g. C10118G, weresimilar across the ethnicities studied, several were not (Table 2).For example, while G13516A was not polymorphic in GAITand demonstrated an overall MAF of 2% in HS-2c, theminor-allele was observed in one of two JA subjects. Eleven otherSNPs were defined analogously by a single individual from oneethnic group, including eight restricted to CA and three toindividuals of AD. Furthermore, four SNPs that were variablein SC subjects were monomorphic in CA. Likewise, while theMAF ofC10118G was 29% in CA, as well as 25% and 50% inindividuals of AD and JA, respectively, the minor-allele wasabsent in GAIT. However, the small numbers of individualsfrom other ethnic groups are insufficient for accurate frequencycomparisons.
Patchy LD across F9
Because of their location in F9, each of the 27 polymorphismsmay be functional and represent potentialQTLs. To determineif these polymorphisms should be evaluated separately forassociation with FIX:C, we analyzed their pairwise LD.Because only ten non-Caucasian X-chromosomes were exam-ined, AD (n ¼ 8) and JA (n ¼ 2), our estimated allelefrequencies were based only on the 148 unrelated Caucasians
0 5000 10 000 15 000 20 000 25 000 30 000 35 000 40 000
G-816AG-793AC-698TG-653CA-634GT-609CT-561GA192G
C10118GA10365GA10512GA11113GT11269AC13162TA20002CA20422GC30294TG32056AA32168T
5:32197:6/4T32770CA32781GC32847T
F9 polymorphisms: physical spacing (bp)
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.0
0.1
F9
poly
mor
phis
ms:
alle
les
and
geni
c po
sitio
n in
the
TU
LD
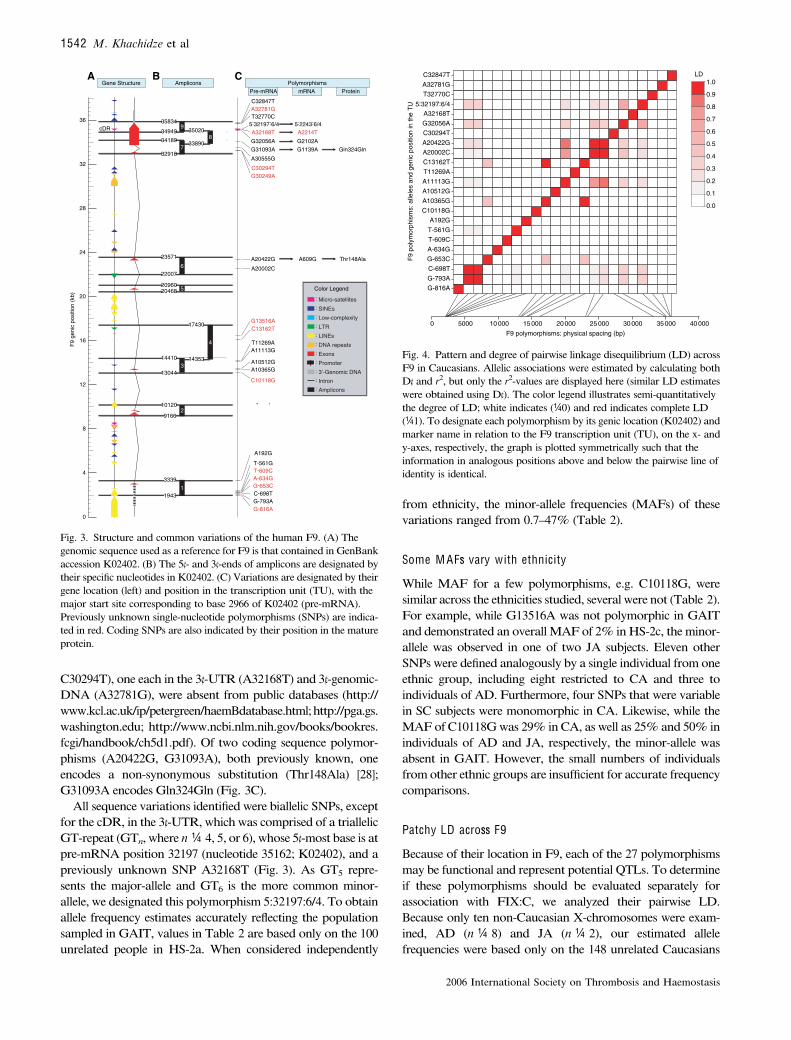
Fig. 4. Pattern and degree of pairwise linkage disequilibrium (LD) acrossF9 in Caucasians. Allelic associations were estimated by calculating bothD¢ and r2, but only the r2-values are displayed here (similar LD estimateswere obtained using D¢). The color legend illustrates semi-quantitativelythe degree of LD; white indicates (¼0) and red indicates complete LD(¼1). To designate each polymorphism by its genic location (K02402) andmarker name in relation to the F9 transcription unit (TU), on the x- andy-axes, respectively, the graph is plotted symmetrically such that theinformation in analogous positions above and below the pairwise line ofidentity is identical.
0
4
8
12
16
20
24
28
32
36cDR
10120
9
7
8
6
5
2
1
G-793AC-698TG-653C
T-561G
G-816A
A-634GT-609C
A192G
C10118G
A10365GA10512G
A11113GT11269A
C13162TG13516A
A20422G
A20002C
G30249AC30294T
A30555G
G31093AG32056A
T32770CA32781GC32847T
A32168T
Thr148Ala
Gln324Gln
A B C
G1139AG2102A
A2214T
9166
3339
1943
14410
17430
2046820960
22007
23571
32918
34189 33890
3502034949
35834
314353
4
A609G
: Intron
: 3’-Genomic DNA
: Exons
: DNA repeats
: LINEs
: LTR
: Low-complexity
: Micro-satellites: SINEs
: Amplicons
: Promoter
Color Legend
5:32197:6/4
Pre-mRNA mRNA ProteinPolymorphisms
5:2243:6/4
AmpliconsGene Structure
13044
F9
geni
c po
sitio
n (k
b)
Fig. 3. Structure and common variations of the human F9. (A) Thegenomic sequence used as a reference for F9 is that contained in GenBankaccession K02402. (B) The 5¢- and 3¢-ends of amplicons are designated bytheir specific nucleotides in K02402. (C) Variations are designated by theirgene location (left) and position in the transcription unit (TU), with themajor start site corresponding to base 2966 of K02402 (pre-mRNA).Previously unknown single-nucleotide polymorphisms (SNPs) are indica-ted in red. Coding SNPs are also indicated by their position in the matureprotein.
1542 M. Khachidze et al
2006 International Society on Thrombosis and Haemostasis

in HS-2a, HS-2c, and the seven additional GAIT subjectsdecribed in Methods. Using these estimates, we found lowoverall LD across F9 in Caucasian populations (Fig. 2). Infact, of the 23 sites polymorphic in either SC or CA, 12 werenot in LD with any other (D¢ + r2 < 0.2). Nevertheless, thealleles in two pairs of polymorphisms (G-793A|C-698T,A10365G|C13162T) exhibited complete LD (D¢ ¼ r2 ¼ 1) inthe analyzed sample for both D¢ and r2; alleles in four otherpairs (A20002C|A20422G, A11113G|A20422G, A11113G|A20002C, and G32056A|5:32197:6/4) demonstrated moderate(0.4 £ LD < 0.6) to high (0.6 £ LD < 1) LD.
No major FIX determinants in F9
Because of the weak overall pattern of LD across F9, wegenotyped every GAIT subject for the subset of poly-morphisms variable in the SC population, including 17 (16SNPs and 5:32197:6/4) whose minor-alleles were found in atleast one person in HS-2a or HS-2b (Table 2). We thenevaluated the relationship between the genotypes of each F9polymorphism and FIX:C independently by marginal associ-ation analysis using the measured-genotype approach [29].After allowing for non-independence among family members,we found no significant genotype-specific differences in meanFIX:C (P-values ‡ 0.11) (Table 2). These results suggest thatnaturally occurring variations in known functional F9 regionsare not likely to be determinants ofFIX:C variability, at least inthe SC population.
FIX:C is not associated w ith age in multivariate analysis
The identification of common variants in both age-related, cis-acting regulatory elements, without a clear association withFIX:C, led us to reexamine the relationship of FIX with age inthe context of other endogenous, genetic and environmental
variables, for which data was available (Table 3). Afterseparately evaluating the relationship of FIX:C with eachcovariate independently, we found the following to have atleast suggestive associations (P £ 0.10): age, smoking status,BMI, ABO-genotype, and TC, LDL, HDL, VLDL, TG,VWF:Ag, and FVIII:C levels. Because the correlation coeffi-cients between TC and LDL, and TG and VLDL wereboth greater than 0.90 (P < 0.05), we evaluated FIX:C bymultivariate analysis using a model that included only TC andTG, together with age, sex, smoking status, BMI, ABO-genotype, and levels of HDL, VWF:Ag and FVIII:C. Despitenot finding an association between sex and FIX:C, we chooseto include sex in the model for final analysis (Table 3). Whilethree endogenous parameters (TG, FVIII:C, BMI) and oneenvironmental factor (smoking) were correlated significantlywith FIX:C (P-values < 0.005), age was not.
D iscussion
FIX:C, a quantitative-trait exhibiting a broad normal range innon-hemophilic populations, represents a risk factor for venousthromboembolism [7,10] and possibly arterial thrombosis [11].Despite mounting evidence that a substantial heritable com-ponent contributes to interindividual variance in FIX [4,12]and pleiotropically influences thrombosis risk [3], the underly-ing QTLs have not been identified. We used genome-wide andcandidate-gene strategies to identify determinants of FIXvariability. The genome-wide linkage screen was performedinitially because FIX is a complex trait, involving multiplegenetic and environmental factors, and this approach does notrequire candidate genes to be specified a priori. This strategy’susefulness has been demonstrated by numerous studies thathave identified heritable determinants of continuous traits inseveral physiologic systems and their correlated complexdiseases [30–34].
We conducted the genome-wide screen in GAIT because, inaddition to exhibiting a high FIX:C heritability, these familiesrepresented the general SC population sampled. This is the firstreported use of a genome-wide approach to identify FIXQTLs. No autosomal regions met criteria for linkage withFIX:C (all LOD scores < 1.5), despite the use of multipoint-variance-component methods and 563 markers that provided6.2 cM genome coverage (Fig. 2). These results suggest that the39% heritability found in GAIT [12] is likely because of acollection of QTLs with modest effects. Although none of the204 genes contained in the two 20 cM gaps on chr6 are obviouscandidates, genotyping additional microsatellites in theseregions could help to determine if they contain determinantsof FIX variance. Similarly, no single region of the X-chr metgenome-wide criteria for linkage. However, male hemizygosityprecludes the use of multipoint linkage analysis for X-linkedmarkers, including those that were near or in F9. Therefore, itis not possible to exclude formally the possibility of an X-chrlocus with a modest effect on FIX:C.
Given markers that are themselves functional or in high LDwith an un-typed functional site, association analysis may be
Table 3 Multicovariate analysis of FIX:C levels in the Genetic Analysis ofIdiopathic Thrombophilia Project (GAIT)
Covariate b-coe cient P-value
Age (years) )8.963 · 10 ) 2 0.3728Sex 0.295 0.9193Age · Sex 9.764 · 10 ) 2 0.4169Age2 )5.671 · 10 ) 4 0.8914Age2 · Sex )2.566 · 10 ) 3 0.6303Smoking (yes/no) 6.972 0.0021AA )3.110 0.4271AO 0.361 0.8953AB 1.109 0.8864BO )5.394 0.1955TC (mM) 1.008 0.4519HDL (mM) 5.792 0.1146TG (mM) 15.212 <0.0001BMI (kg m ) 2) 1.752 <0.0001FVIII:C (IU dL ) 1) 0.137 <0.0001VWF:Ag (%) 4.695 · 10 ) 2 0.2639
TC, total cholesterol; HDL, high-density lipoprotein; TG, triglycer-ides; BMI, body mass index; FVIII:C, factor VIII activity; VWF:Ag,von Willebrand factor antigen.
Comprehensive search for FIX QTLs 1543
2006 International Society on Thrombosis and Haemostasis

able to detect loci with smaller effects. This is supported byfindings from a similar study to identify variance determinantsof FVIII:C [35]. Although FVIII:C represents one of the mostheritable coagulation phenotypes in GAIT, we found noevidence for linkage with microsatellites in or near F8. Usingassociation analysis, however, we found that D1241E, aconservative non-hemophilic B-domain substitution, representsa FVIII QTL. After identifying all naturally occurring F9polymorphisms in the known functional regions (Table 1) ofevery distinct allele segregated in GAIT, we used associationanalysis to evaluate the relationshipbetween the structural locusand FIX:C. Because the pattern of LD across F9 was weakoverall (Fig. 4), we examined each polymorphism (Fig. 3)separately for potential contributions to FIX:C variability bymarginalmeasured-genotype association analysis. However,nosignificant differences in mean FIX:C levels were found (P-values ‡ 0.11). While these findings suggest that the structurallocus is not a determinant of FIX:C variance, our study wasconfined toknown functional regions that compriseonly 40%of all non-repetitive F9 sequences (Fig. 3). Using PhastCons(http://www.genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid¼41178675&c¼chrX&g¼phastConsElements) [36–38], we iden-tified17non-exonicF9segments thatarehighly conservedacrossvertebrates (not shown). These segments, ranging from 58–449 bp and containing 15 SNPs from dbSNP, displayedconservation scores greater than the mean score for all exonicsequence. None of these candidate cis-acting regulatory ele-ments,whichmay containdeterminantsofFIX:Cvariance,wereexamined here. To interrogate these segments by associationanalysis, we are currently defining naturally occurring alleliccombinations of the F9 polymorphisms identified here. Bytakingadvantageof thegreateroveralldegreeofLDprovidedbyhaplotypes, this approach should increase the power of theGAIT sample to detect functional variants in these regions.
Limiting factors of the present genome-wide and candi-date-gene studies may be considered in the followingcategories: sample size, phenotype (FIX:C), and ethnicity.To overcome limitations related to the current sample size, alarger family study of the SC population (GAIT-II) isunderway. FIX:C is measured under conditions which maynot recapitulate precisely those in vivo and requires interac-tions between additional complex hemostasis traits, all ofwhich, except FIX, are derived from numerous unrelatedhealthy donors. As FIX:Ag measurements require a singleinteraction between FIX and reagent antibodies, this com-plimentary phenotype is being used, along with FIX:C, inGAIT-II. This will allow use of FIX:C and FIX:Agindependently and/or in combination, by dividing FIX:Cby FIX:Ag, as a parameter of specific activity. While ourfindings demonstrate that no single gene, including F9, islikely to be a major FIX variance determinant in the SCpopulation sampled by GAIT, it is not clear whether thisapplies to non-Caucasian populations or even non-Spanishsubgroups of Caucasians, such as CA. To determine whetherthere are ethnic differences in the types and/or frequencies ofF9 polymorphisms, we examined the same gene regions in a
group of 51 unrelated healthy male subjects (HS-2c) inclu-ding 41 CA, two JA, and eight of AD (Table 2). Although14 polymorphisms were restricted to Caucasians, such MAFdeviations are likely because of the small number of non-Caucasian F9 alleles sampled, that is, eight of AD and twofrom JA. While the MAF of some polymorphisms weresimilar across ethnic groups, there were several notableexceptions. Of the 27 variations identified, ten SNPs were notvariable in SC subjects, including C10118G whose MAFranged from 0.25 to 0.5% in HS-2c. Moreover, while theMAF of G13516A was 50% in the two JA individuals, itwas not polymorphic in other ethnic groups. Furthermore,G30249A, A30555G, and G31093A were restricted toindividuals of AD. As the results from this study may applyonly to the SC subjects sampled in GAIT, similar familystudies should be conducted in other ethnic populations.
Conclusions
Our findings suggest that no single gene, including F9, is likelyto be a major determinant of FIX:C. We hypothesize that acollection of QTLs with modest effect sizes and distinctgenomic locations underlie the variance in this phenotype. Weobserved a significant univariate correlation between FIX:Cand age, consistent with previous studies. However, as thiscorrelation canceled out in multivariate analysis, furtherinvestigation will be required to elucidate the relationshipbetween age and F9 expression. Finally, in vitro functionalinvestigations may be necessary to overcome the limitations ofthe current study.
Author contributions
Study design: J. M. Soria, J. C. Souto, J. Blangero, J.Fontcuberta, L. Almasy, and T. E. Howard; subject enroll-ment: J. C. Souto, J. Fontcuberta, S. T. Warren; dataacquisition: M. Khachidze, S. Porter, D. Machiah, J. M.Soria, M. Lathrop, T. E. Howard; data analysis and interpret-ation: M. Khachidze, A. Buil, K. R.Viel, S. Porter, D. Warren,L. Almasy, T. E. Howard;manuscript draft: M. Khachidze, A.Buil, S. Porter, S. T. Warren, L. Almasy, T. E. Howard;manuscript revision: M. Khachidze, A. Buil, K. R. Viel, S.Porter, A. Ameri, L. Almasy, T. E. Howard.
Ackno w ledgements
We thank all patients and other individuals for their partici-pation in this study. We also thank Pete Lollar for mentorshipand early support, Ming Shen for technical help, Bruce Evattand Connie Miller for helpful discussions regarding themanuscript, Russ Howard and Peter Hay for editing themanuscript, and Mary Howard for her enthusiastic support.This work was supported by: NIH-R01-HL-70751 (L. A.),NIH-K08-HL-71130 and -R01-HL-68016 (T. E. H.), FIS-02/0375, SAF2002-03449, FIS-RECAVA-C03/01, FIS-99/3048and -01/A046 (J. M. S. and A. B.).
1544 M. Khachidze et al
2006 International Society on Thrombosis and Haemostasis

References
1 Bertina RM, Koeleman BP, Koster T, Rosendaal FR, Dirven RJ, deRonde H, van der Velden PA, Reitsma PH. Mutation in bloodcoagulation factor V associated with resistance to activated protein C.Nature 1994; 369: 64–7.
2 Poort SR, Rosendaal FR, Reitsma PH, Bertina RM. A commongenetic variation in the 3¢-untranslated region of the prothrombin geneis associated with elevated plasma prothrombin levels and an increasein venous thrombosis. Blood 1996; 88: 3698–703.
3 Souto JC, Almasy L, Borrell M, Blanco-Vaca F, Mateo J, Soria JM,Coll I, Felices R, Stone W, Fontcuberta J, Blangero J. Genetic sus-ceptibility to thrombosis and its relationship to physiological riskfactors: the GAIT study. Genetic Analysis of Idiopathic Thrombo-philia. Am J Hum Genet 2000; 67: 1452–9.
4 Vossen CY, Hasstedt SJ, Rosendaal FR, Callas PW, Bauer KA, BrozeGJ, Hoogendoorn H, Long GL, Scott BT, Bovill EG. Heritability ofplasma concentrations of clotting factors and measures of a pre-thrombotic state in a protein C-deficient family. J Thromb Haemost2004; 2: 242–7.
5 Reiner AP, Davie EW. The physiology and biochemistry of factor IX.In: Bloom AL, Forbes CD, Thomas DP, Tuddenham EGD, eds.Haemostasis and Thrombosis, 3rd edn. Edniburgh, UK: ChurchillLivingstone, 1994: 309–32.
6 Thompson AR. Molecular Biology of Factor IX. In: Colman RW,Hirsh J, Marder VJ, Clowes AW, George JN, eds. Hemostasis andThrombosis: Basic Principles & Clinical Practice, 4th edn. Philadelphia,PA, USA: Lippincott Williams & Wilkins, 2001: 123–34.
7 van Hylckama Vlieg A, van der Linden IK, Bertina RM, RosendaalFR. High levels of factor IX increase the risk of venous thrombosis.Blood 2000; 95: 3678–82.
8 Sweeney JD, Hoernig LA. Age-dependent e ect on the level of factorIX. Am J Clin Pathol 1993; 99: 687–8.
9 Lowe GD, Rumley A, Woodward M, Morrison CE, Philippou H,Lane DA, Tunstall-Pedoe H. Epidemiology of coagulation factors,inhibitors and activation markers: the Third Glasgow MONICASurvey. I. Illustrative reference ranges by age, sex and hormone use. BrJ Haematol 1997; 97: 775–84.
10 Lowe GD. Factor IX and thrombosis. Br J Haematol 2001; 115: 507–13.
11 Ameri A, Kurachi S, SueishiK, Kuwahara M, KurachiK. Myocardialfibrosis inmice with overexpression of human blood coagulation factorIX. Blood 2003; 101: 1871–3.
12 Souto JC, Almasy L, Borrell M, Gari M, Martinez E, Mateo J, StoneWH, Blangero J, Fontcuberta J. Genetic determinants of hemostasisphenotypes in Spanish families. Circulation 2000; 101: 1546–51.
13 Zerylnick C, Torroni A, Sherman SL, Warren ST. Normal variation atthe myotonic dystrophy locus in global human populations. Am JHum Genet 1995; 56: 123–30.
14 Kurachi S, Deyashiki Y, Takeshita J, Kurachi K. Geneticmechanismsof age regulation of human blood coagulation factor IX. Science 1999;285: 739–43.
15 Dyke B. PEDSYS: A Pedigree Data Management System (User’sManual). San Antonio, TX, USA: Population Laboratory, Depart-ment of Genetics, Southwest Foundation for Biomedical Research,1995.
16 Williams JT, Van Eerdewegh P, Almasy L, Blangero J. Joint multi-point linkage analysis of multivariate qualitative and quantitativetraits. I. Likelihood formulation and simulation results. Am J HumGenet 1999; 65: 1134–47.
17 Towne B, Siervogel RM, Blangero J. E ects of genotype-by-sexinteraction on quantitative trait linkage analysis.Genet Epidemiol 1997;14: 1053–8.
18 Allison DB, Neale MC, Zannolli R, Schork NJ, Amos CI, Blangero J.Testing the robustness of the likelihood-ratio test in a variance-com-
ponent quantitative-trait loci-mapping procedure. Am J Hum Genet1999; 65: 531–44.
19 Blangero J, Williams JT, Almasy L. Variance component methods fordetecting complex trait loci. Adv Genet 2001; 42: 151–81.
20 Boehnke M, Lange K. Ascertainment and goodness of fit of variancecomponent models for pedigree data. Prog Clin Biol Res 1984; 147:173–92.
21 Yoshitake S, Schach BG, Foster DC, Davie EW, Kurachi K. Nuc-leotide sequence of the gene for human factor IX (antihemophilicfactor B). Biochemistry 1985; 24: 3736–50.
22 Ewing B, Green P. Base-calling of automated sequencer traces usingphred. II. Error probabilities. Genome Res 1998; 8: 186–94.
23 Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automatedsequencer traces using phred. I. Accuracy assessment. Genome Res1998; 8: 175–85.
24 Zhao JH, Sham PC. Faster haplotype frequency estimation usingunrelated subjects. Hum Hered 2002; 53: 36–41.
25 Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence(RefSeq): a curated non-redundant sequence database of genomes,transcripts and proteins. Nucleic Acids Res 2005; 33: D501–4.
26 Smit AF. The origin of interspersed repeats in the human genome.Curr Opin Genet Dev 1996; 6: 743–8.
27 Jurka J. Repbase update: a database and an electronic journal ofrepetitive elements. Trends Genet 2000; 16: 418–20.
28 Graham JB, Kunkel GR, Tennyson GS, Lord ST, Fowlkes DM. TheMalmo polymorphism of factor IX: establishing the genotypes byrapid analysis of DNA. Blood 1989; 73: 2104–7.
29 Hopper JL, Mathews JD. Extensions to multivariate normal modelsfor pedigree analysis. Ann Hum Genet 1982; 46: 373–83.
30 Almasy L, Soria JM, Souto JC, Coll I, Bacq D, Faure A, Mateo J,Borrell M, Munoz X, Sala N, Stone WH, Lathrop M, Fontcuberta J,Blangero J. A quantitative trait locus influencing free plasma protein Slevels on human chromosome 1q: results from the Genetic Analysis ofIdiopathic Thrombophilia (GAIT) project. Arterioscler Thromb VascBiol 2003; 23: 508–11.
31 Buil A, Soria JM, Souto JC, Almasy L, Lathrop M, Blangero J,Fontcuberta J. Protein C levels are regulated by a quantitative traitlocus on chromosome 16: results from the Genetic Analysis of Idio-pathic Thrombophilia (GAIT) Project. Arterioscler Thromb Vasc Biol2004; 24: 1321–5.
32 Comuzzie AG, Mitchell BD, Cole S, Martin LJ, Hsueh WC, Rain-water DL, Almasy L, Stern MP, Hixson J, MacCluer JW, Blangero J.The genetics of obesity in Mexican Americans: the evidence fromgenome scanning e orts in the San Antonio family heart study. HumBiol 2003; 75: 635–46.
33 Duggirala R, Blangero J, Almasy L, Dyer TD, Williams KL, LeachRJ, O’Connell P, Stern MP. Linkage of type 2 diabetes mellitus and ofage at onset to a genetic location on chromosome 10q in MexicanAmericans. Am J Hum Genet 1999; 64: 1127–40.
34 Soria JM, Almasy L, Souto JC, Bacq D, Buil A, Faure A, Martinez-Marchan E, Mateo J, Borrell M, Stone W, Lathrop M, Fontcuberta J,Blangero J. A quantitative-trait locus in the human factor XII geneinfluences both plasma factor XII levels and susceptibility tothrombotic disease. Am J Hum Genet 2002; 70: 567–74.
35 Machiah DK, Viel K, Almasy L, Souto JC, Soria JM, Blangero J,Fontcuberta J, Howard TE. A common SNP in the factorVIII (fVIII)gene encodes a conservative aspartate to glutamate substitution (As-p1241Glu) in the B-domain that influences fVIII activity levels. Blood2003; 102: 55a.
36 Siepel A, Haussler D. Combining phylogenetic and hidden Markovmodels in biosequence analysis. J Comput Biol 2004; 11: 413–28.
37 Chiaromonte F, Yap VB, Miller W. Scoring pairwise genomicsequence alignments. Pac Symp Biocomput 2002; 115–26.
38 Yang Z. A space-time process model for the evolution of DNAsequences. Genetics 1995; 139: 993–1005.
Comprehensive search for FIX QTLs 1545
2006 International Society on Thrombosis and Haemostasis




















