
PEG-urokinase nanogels with enhanced stability and controllable bioactivity
Upload
independentCategory
view
1download
0
ORIGINAL ARTICLE
Generation of optimized and urokinase-targetedoncolytic Sendai virus vectors applicable for varioushuman malignancies
H Kinoh1, M Inoue2, A Komaru1,3, Y Ueda1, M Hasegawa2 and Y Yonemitsu1
1Department of Gene Therapy, Chiba University Graduate School of Medicine, Chiba, Japan; 2DNAVEC Corporation, Tsukuba, Ibaraki,Japan and 3Department of Urology, Chiba University Graduate School of Medicine, Chiba, Japan
We previously reported the development of a prototype‘oncolytic Sendai virus (SeV) vector’ formed by introducingtwo major genomic modifications to the original SeV, namelydeletion of the matrix (M) gene to avoid budding of secondaryviral particles and manipulation of the trypsin-dependentcleavage site of the fusion (F) gene to generate protease-specific sequences. As a result, the ‘oncolytic SeV’ thatwas susceptible to matrix metalloproteinases (MMPs) wasshown to selectively kill MMP-expressing tumors throughsyncytium formation in vitro and in vivo. However, its efficacyhas been relatively limited because of the requirementof higher expression of MMPs and smaller populations ofMMP-expressing tumors. To overcome these limitations, wehave designed an optimized and dramatically powerfuloncolytic SeV vector. Truncation of 14-amino acid residues
of the cytoplasmic domain of F protein resulted in dramaticenhancement of cell-killing activities of oncolytic SeV, and thecombination with replacement of the trypsin cleavage site withthe new urokinase type plasminogen activator (uPA)-sensitivesequence (SGRS) led a variety of human tumors, includingprostate (PC-3), renal (CAKI-I), pancreatic (BxPC3) and lung(PC14) cancers, to extensive death through massive cell-to-cell spreading without significant dissemination to the sur-rounding noncancerous tissue in vivo. These results indicate adramatic improvement of antitumor activity; therefore, exten-sive utility of the newly designed uPA-targeted oncolytic SeVhas significant potential for treating patients bearing uroki-nase-expressing cancers in clinical settings.Gene Therapy (2009) 16, 392–403; doi:10.1038/gt.2008.167;published online 27 November 2008
Keywords: uPA; MMP; oncolytic Sendai virus vector
Introduction
In recent decades, gene therapy has been anticipated as anew therapeutic strategy to treat patients with intractablemalignancies. A number of clinical studies of genetherapy for malignancies have been done worldwide,but the therapy’s efficacy was relatively limited. There-fore, improvement in some aspects of gene therapy,including the gene transfer efficiency without significantdamage to noncancerous tissue, has been much desired.To overcome the limited efficacy of gene transferring bycurrent replication-deficient viruses in vivo, tumor-selective and replication-competent oncolytic viruseshave been generated, and clinical studies on the use ofsuch viral vectors have been reported.1,2 These viruses,including an oncolytic adenovirus and a herpes virus,can replicate specifically in the cancer cells and spread insitu, exhibiting oncolytic activity through a directcytopathic effect during intracellular viral replicationand budding. Clinical studies on the use of oncolytic
viruses actually showed reduced tumor size in somepatients, suggesting proof of the strategy’s concept andits potential efficacy; however, cancer cells infected bythese viruses released active virus particles to theadjacent noncancerous tissue as well as to systemiccirculation, presenting a potential risk for adverse effectsthrough systemically disseminated virus infection, so-called ‘viremia.’ In fact, some patients who receivedoncolytic viruses available at present demonstrated flu-like symptoms, including fever and chills, probablythrough viremia-induced systemic innate immune re-sponses. Thus the development of a new mode ofoncolytic viruses that do not produce secondary virusparticles causing systemic viremia is highly desirablebecause of safety concerns.
Sendai virus (SeV) is an enveloped virus with a linear,nonsegmented negative-strand RNA genome of approxi-mately 15.4 kb, which belongs to the large virus familyParamyxoviridae, comprising a variety of mammalianpathogens. SeV displays a narrow spectrum of tissuetropism in susceptible hosts, actually growing in therespiratory tract of mice or in the allantoic cells ofembryonated chicken eggs with little appreciable spread-ing to other tissues in these host organisms, even thoughits receptor is sialic acid residues that are ubiquitousthroughout the body. This restricted tropism primarilydepends on the specific tissue proteases required for
Received 27 June 2008; revised 27 October 2008; accepted 28 October2008; published online 27 November 2008
Correspondence: Dr H Kinoh, Department of Gene Therapy, ChibaUniversity Graduate School of Medicine, 1-8-1 Inohana, Chuo-ku,Chiba 260-8670, Japan.E-mail: [email protected]
Gene Therapy (2009) 16, 392–403& 2009 Macmillan Publishers Limited All rights reserved 0969-7128/09 $32.00
www.nature.com/gt

cleavage activation of viral fusion (F) glycoprotein, andthus the proteases needed for infectivity of progeny (thecapacity to penetrate into and initiate infection in thenext cell) are available only on the surface of thoselimited types of tissue.3
Using sophisticated manipulating technology of thegenome of SeV, namely ‘Reverse Genetics,’ we recentlygenerated SeV vectors lacking envelope-related genes,including fusion (F) gene-deleted (SeV/DF),4 matrix (M)gene-deleted (SeV/DM),5 hemagglutinin/neuraminidase(HN) gene-deleted (SeV/DHN), both M and F genes-deleted (SeV/DMDF)6 and all of the envelope-relatedgenes-deleted (SeV/DMDFDHN).7 Among the proteinscontained in the viral lipid bilayer, M protein plays acentral role in secondary virus assembly and buddingfrom infected cells. Therefore, deletion of the M genefrom SeV almost completely abolished virus maturationduring formation of infectious particles in infectedcells and instead caused cell-to-cell vector spreadingthrough membrane fusion, forming large syncytiaand resulting in cell death by the active F proteinunder supplementation of trypsin.5 This suggested to usthat cell-to-cell spread of this vector could be manipu-lated by the presence of selected F protein-activatingproteases.
On the basis of these features of SeV vectors, wepreviously reported the development of a prototypeoncolytic M gene-deleted SeV vector by replacing thetrypsin-sensitive amino-acid residues of F protein at thematrix metalloproteinase (MMP)-selective cleavage site(-PLGMTS-).8 As expected, this new class of SeV vectorsspread widely from cell to cell through fusion of theplasma membranes, forming syncytia among the MMP-expressing tumor cells in vitro, and led the tumor toextensive death in vivo, without releasing secondaryvector particles.8 Our subsequent studies examining theutility of the vector among various human cancer celllines from different origins, however, showed that theefficacy of this prototype vector was relatively limitedbecause of (1) the requirement for a relatively highexpression level of MMPs for efficient cell death and (2)the limited numbers of MMP-expressing tumor cells, lessthan 20% (unpublished observation).
To overcome these problems, we have been focusingon the manipulation of the cytoplasmic domain of Fprotein and urokinase-type plasminogen activator (uPA)for the past 2 years. Recent studies showed thattruncation of the cytoplasmic domains, not only ofretrovirus and herpes virus glycoproteins,9 but alsothose of Paramyxoviridae (that is, measles virus andNewcastle disease virus) increases their fusion activity.10
Moreover, the plasminogen activator (PA) system con-sists of two PAs, urokinase (uPA) and tissue type (tPA),which activate plasminogen to the active plasmin. Thisserine protease can degrade extracellular matrix compo-nents directly, for example, fibronectin and proteo-glycans, or indirectly by activating other proteases,including MMPs. Importantly, uPA has been shown tobe expressed in various types of human malignanciesmore frequently (over 50%) than MMPs.
We here report the optimized design of protease-specific oncolytic recombinant SeV vectors that havebeen generated by truncation of the F protein cytoplas-mic domain and by the optimization of catalytic activityagainst MMPs and uPA. These new vectors showed
dramatically enhanced antitumor activities in vitro andin vivo.
Results
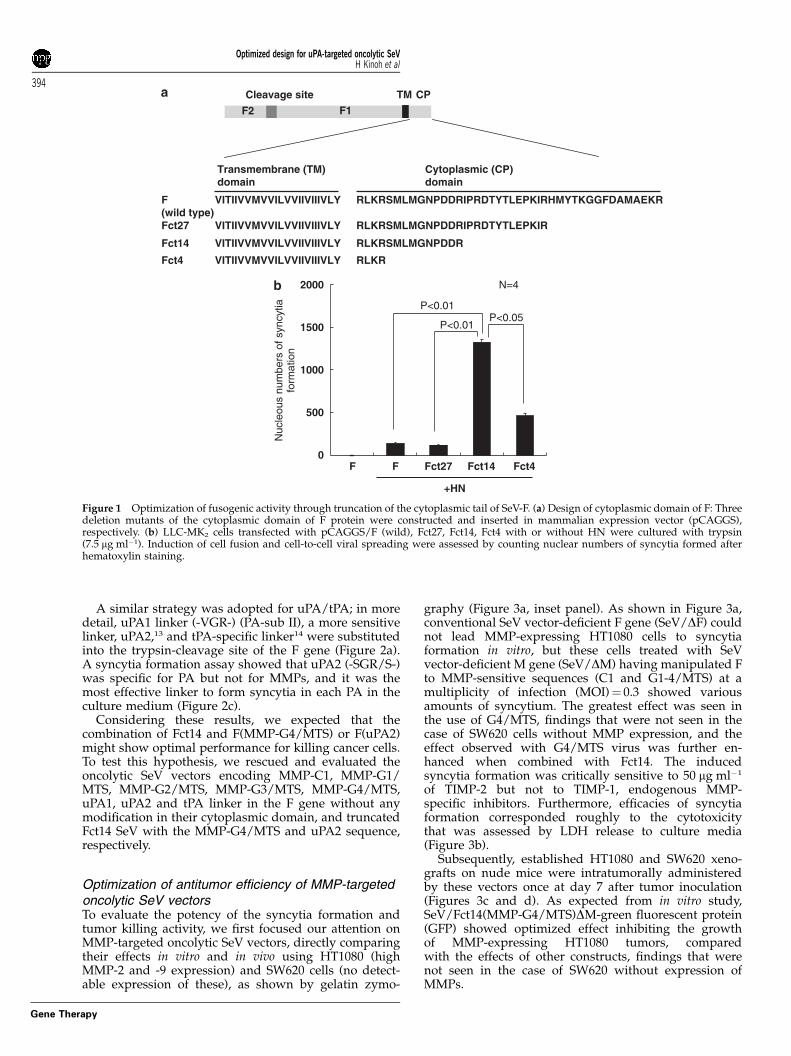
Optimization of truncation for the cytoplasmic domainof the F gene of SeV to enhance its fusogenic activityAs an initial step, we optimized the truncation of thecytoplasmic tail of wild-type F gene transfected bypCAGGS to LLC-MK2 cells cultivated with trypsin. Weexamined three deleting mutants (Fct27, Fct14, Fct4), asshown in Figure 1a, and the fusion efficiency wasquantified by counting the nuclei per formed syncy-tium.8 As shown in Figure 1b, syncytia formation wasobserved only in cells with cotransfection of pairedmembrane glycoproteins (F and HN). The highest ratioof fusion activity, six times higher than that seen by wild-type F gene cotransfected (Po0.01), was seen in the useof a truncated clone (Fct14); therefore, we concludedthat Fct14 should be used in our new design of anoncolytic SeV.
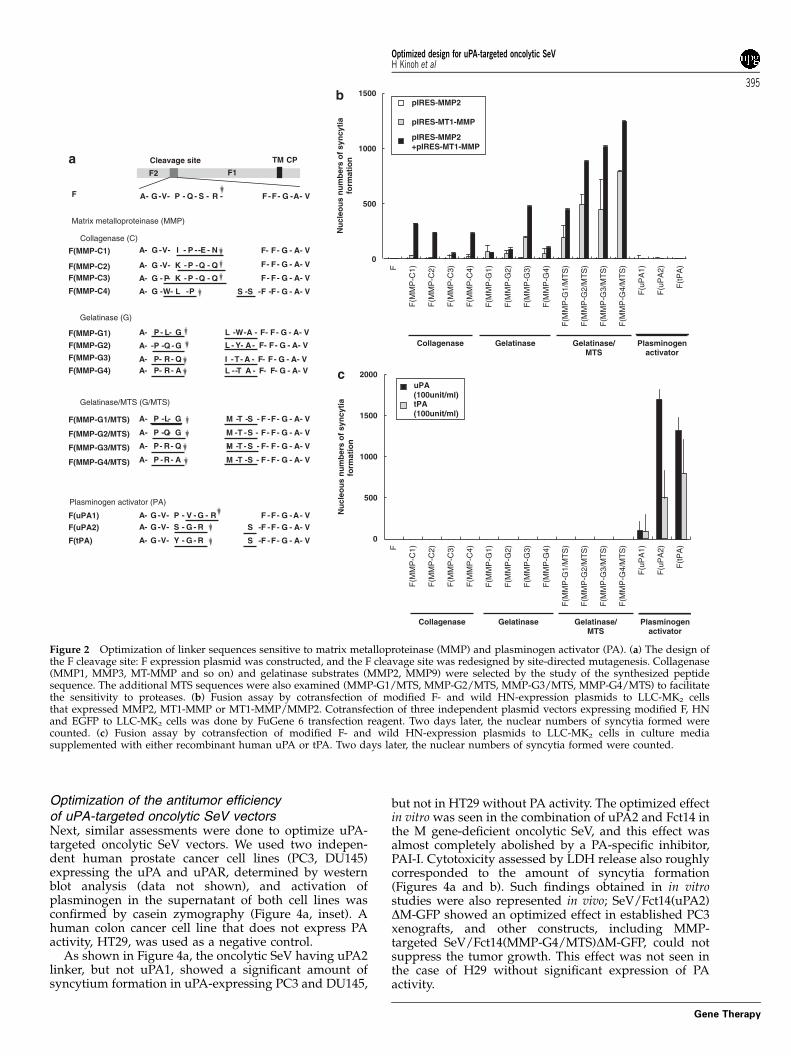
Optimization of the sensitive linker sequencein a protease-specific processing site of the F geneAs a next step, we optimized the sensitive linkersequence in the processing site of F gene against theMT1-MMP and MMP2, urokinase-type (uPA) or tissue-type (tPA) plasminogen activators, that is, proteases thatwere shown to be expressed by various human cancers.The candidate sequences were based on the informationdetermined previously by phage display.11 Each plasmidsubcloned with corresponding mutant F genes shown inFigure 2a was cotransfected with HN gene expressionvector to LLC-MK2 cells, and fusion activity wasdetermined as described above. In cases of collagenaseand gelatinase, cDNA of MT1-MMP was also cotrans-fected, because MT1-MMP is required for efficientactivation of MMP-2. Expression of active MMP-2 inthis experiment was confirmed by zymography (data notshown). Replacement of the trypsin cleavage site (-QSR-)in the collagenase, gelatinase or uPA/tPA cleavable sitewould add amino acids upstream of the fusion peptide.Active fusion peptides of Paramyxovirus highly conservethe hydrophobic sequence, consisting of phenylalanineat the N terminus of F1. Previously, we revealed that theaddition of the sequence –MTS-, but not –LGL- or –LWA-, at the N terminus of activated fusion peptide (F1) of SeVwas susceptible to replacement with an efficient mem-brane fusion; therefore, –MTS- was also examined todetermine whether it improved the original gelatinasesequence.
Syncytia formation of active MMP2 (MT1-MMP/MMP2)-expressing cells was examined by the additionof each plasmid vector expressing MMP-C1-4, -G1-4,-G1-4/MTS linker. As shown in Figure 2b, MMP-C1, -C2,-C4 linker formed active MMP-2-specific syncytia for-mation, whereas PA-sensitive linkers did not. MMP-G4/MTS showed the highest efficiency in forming syncytiaunder cotransfection of both MT1-MMP and MMP2. Ithas been reported that MMP-G2 (-PQG/LYA-) is anefficient sequence in MMP-expressing HT-1080 cell, inthe use of recombinant measles virus,12 but this was notthe case with SeV under similar experimental conditionsusing HT-1080 cells (data not shown).
Optimized design for uPA-targeted oncolytic SeVH Kinoh et al
393
Gene Therapy
A similar strategy was adopted for uPA/tPA; in moredetail, uPA1 linker (-VGR-) (PA-sub II), a more sensitivelinker, uPA2,13 and tPA-specific linker14 were substitutedinto the trypsin-cleavage site of the F gene (Figure 2a).A syncytia formation assay showed that uPA2 (-SGR/S-)was specific for PA but not for MMPs, and it was themost effective linker to form syncytia in each PA in theculture medium (Figure 2c).
Considering these results, we expected that thecombination of Fct14 and F(MMP-G4/MTS) or F(uPA2)might show optimal performance for killing cancer cells.To test this hypothesis, we rescued and evaluated theoncolytic SeV vectors encoding MMP-C1, MMP-G1/MTS, MMP-G2/MTS, MMP-G3/MTS, MMP-G4/MTS,uPA1, uPA2 and tPA linker in the F gene without anymodification in their cytoplasmic domain, and truncatedFct14 SeV with the MMP-G4/MTS and uPA2 sequence,respectively.
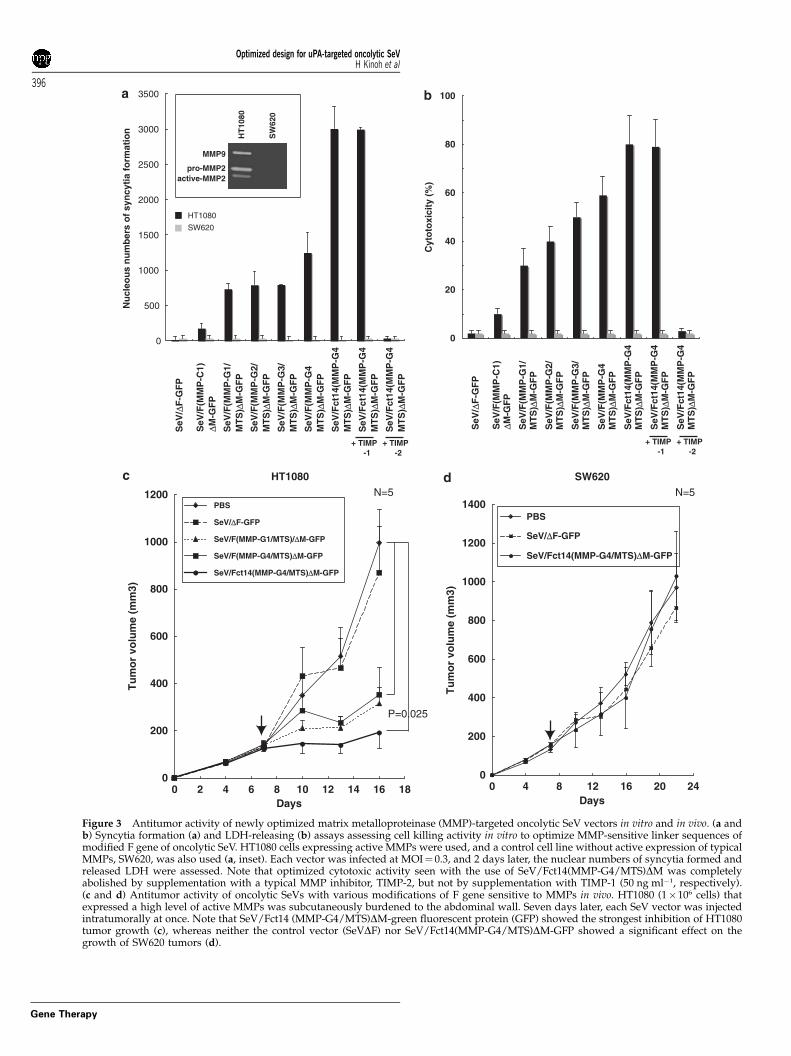
Optimization of antitumor efficiency of MMP-targetedoncolytic SeV vectorsTo evaluate the potency of the syncytia formation andtumor killing activity, we first focused our attention onMMP-targeted oncolytic SeV vectors, directly comparingtheir effects in vitro and in vivo using HT1080 (highMMP-2 and -9 expression) and SW620 cells (no detect-able expression of these), as shown by gelatin zymo-
graphy (Figure 3a, inset panel). As shown in Figure 3a,conventional SeV vector-deficient F gene (SeV/DF) couldnot lead MMP-expressing HT1080 cells to syncytiaformation in vitro, but these cells treated with SeVvector-deficient M gene (SeV/DM) having manipulated Fto MMP-sensitive sequences (C1 and G1-4/MTS) at amultiplicity of infection (MOI)¼ 0.3 showed variousamounts of syncytium. The greatest effect was seen inthe use of G4/MTS, findings that were not seen in thecase of SW620 cells without MMP expression, and theeffect observed with G4/MTS virus was further en-hanced when combined with Fct14. The inducedsyncytia formation was critically sensitive to 50 mg ml�1
of TIMP-2 but not to TIMP-1, endogenous MMP-specific inhibitors. Furthermore, efficacies of syncytiaformation corresponded roughly to the cytotoxicitythat was assessed by LDH release to culture media(Figure 3b).
Subsequently, established HT1080 and SW620 xeno-grafts on nude mice were intratumorally administeredby these vectors once at day 7 after tumor inoculation(Figures 3c and d). As expected from in vitro study,SeV/Fct14(MMP-G4/MTS)DM-green fluorescent protein(GFP) showed optimized effect inhibiting the growthof MMP-expressing HT1080 tumors, comparedwith the effects of other constructs, findings that werenot seen in the case of SW620 without expression ofMMPs.
RLKRSMLMGNPDDRIPRDTYTLEPKIRHMYTKGGFDAMAEKRF(wild type)
VITIIVVMVVILVVIIVIIIVLY
RLKRSMLMGNPDDRIPRDTYTLEPKIRFct27 VITIIVVMVVILVVIIVIIIVLY
RLKRSMLMGNPDDRFct14 VITIIVVMVVILVVIIVIIIVLY
RLKRFct4 VITIIVVMVVILVVIIVIIIVLY
Cytoplasmic (CP) domain
Transmembrane (TM)domain
Cleavage site
F2 F1
TM CP
+HN
Nuc
leou
s nu
mbe
rs o
f syn
cytia
form
atio
n
0
500
1000
1500
2000
F F Fct27 Fct14 Fct4
P<0.01P<0.05
P<0.01
N=4
Figure 1 Optimization of fusogenic activity through truncation of the cytoplasmic tail of SeV-F. (a) Design of cytoplasmic domain of F: Threedeletion mutants of the cytoplasmic domain of F protein were constructed and inserted in mammalian expression vector (pCAGGS),respectively. (b) LLC-MK2 cells transfected with pCAGGS/F (wild), Fct27, Fct14, Fct4 with or without HN were cultured with trypsin(7.5 mg ml�1). Induction of cell fusion and cell-to-cell viral spreading were assessed by counting nuclear numbers of syncytia formed afterhematoxylin staining.
Optimized design for uPA-targeted oncolytic SeVH Kinoh et al
394
Gene Therapy
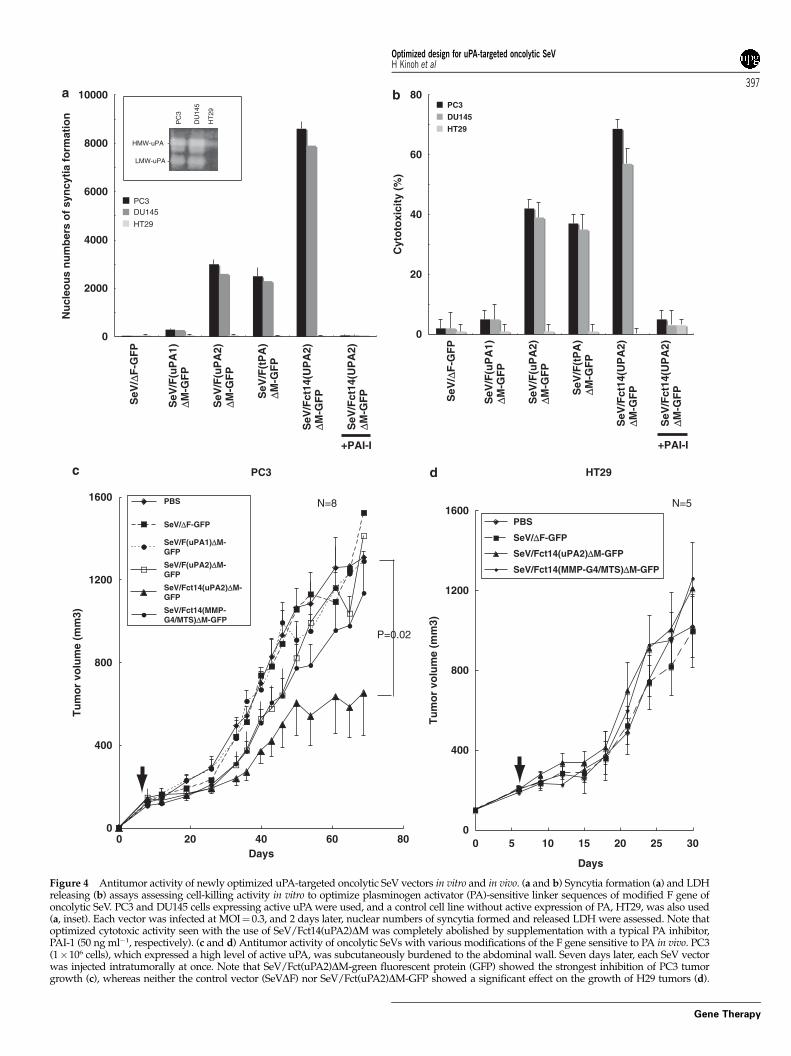
Optimization of the antitumor efficiencyof uPA-targeted oncolytic SeV vectorsNext, similar assessments were done to optimize uPA-targeted oncolytic SeV vectors. We used two indepen-dent human prostate cancer cell lines (PC3, DU145)expressing the uPA and uPAR, determined by westernblot analysis (data not shown), and activation ofplasminogen in the supernatant of both cell lines wasconfirmed by casein zymography (Figure 4a, inset). Ahuman colon cancer cell line that does not express PAactivity, HT29, was used as a negative control.
As shown in Figure 4a, the oncolytic SeV having uPA2linker, but not uPA1, showed a significant amount ofsyncytium formation in uPA-expressing PC3 and DU145,
but not in HT29 without PA activity. The optimized effectin vitro was seen in the combination of uPA2 and Fct14 inthe M gene-deficient oncolytic SeV, and this effect wasalmost completely abolished by a PA-specific inhibitor,PAI-I. Cytotoxicity assessed by LDH release also roughlycorresponded to the amount of syncytia formation(Figures 4a and b). Such findings obtained in in vitrostudies were also represented in vivo; SeV/Fct14(uPA2)DM-GFP showed an optimized effect in established PC3xenografts, and other constructs, including MMP-targeted SeV/Fct14(MMP-G4/MTS)DM-GFP, could notsuppress the tumor growth. This effect was not seen inthe case of H29 without significant expression of PAactivity.
Nu
cleo
us
nu
mb
ers
of
syn
cyti
afo
rmat
ion
F
F(u
PA
1)
F(u
PA
2)
F(t
PA
)
F(M
MP
-C1)
F(M
MP
-C2)
F(M
MP
-C3)
F(M
MP
-C4)
F(M
MP
-G1)
F(M
MP
-G2)
F(M
MP
-G3)
F(M
MP
-G4)
F(M
MP
-G1/
MT
S)
F(M
MP
-G2/
MT
S)
F(M
MP
-G3/
MT
S)
F(M
MP
-G4/
MT
S)
Plasminogenactivator
Collagenase Gelatinase Gelatinase/MTS
0
500
1000
1500pIRES-MMP2
pIRES-MT1-MMP
pIRES-MMP2+pIRES-MT1-MMP
Nu
cleo
us
nu
mb
ers
of
syn
cyti
afo
rmat
ion
F
F(u
PA
1)
F(u
PA
2)
F(t
PA
)
F(M
MP
-C1)
F(M
MP
-C2)
F(M
MP
-C3)
F(M
MP
-C4)
F(M
MP
-G1)
F(M
MP
-G2)
F(M
MP
-G3)
F(M
MP
-G4)
F(M
MP
-G1/
MT
S)
F(M
MP
-G2/
MT
S)
F(M
MP
-G3/
MT
S)
F(M
MP
-G4/
MT
S)
Plasminogenactivator
Collagenase Gelatinase Gelatinase/MTS
0
500
1000
1500
2000uPA(100unit/ml)tPA(100unit/ml)
Cleavage site
F2 F1
F G V P Q S R F F G A V-- - - - - - - - -A-
TM CP
F(MMP-G1/MTS)
F(MMP-G2/MTS)
F(MMP-G4)
F(MMP-C1)
F(MMP-C2)F(MMP-C3)
F(MMP-C4)
F(MMP-G3/MTS)
F(MMP-G4/MTS)
F(MMP-G3)
M T S F F G A V- - - - - - --A - -P GL
F(MMP-G2) -A --P GQ F F G A V- - - -L A- - -Y
F F G A V- - - --A - -P GQ M T S- - -
F(MMP-G1) G-A - -P L F F G A V- - - -L W A- - -
F F G A V- - - - -- - -A V- -G P E NI
F F G A V- - - - -- - -A V- -G Q QPK
F F G A V- - - --A - -G P - - -Q QPK
F F G A V- - - - - -- - -A G -W L P S S
F F G A V- - - -R Q- - -A M T S-- -P
R Q- - -A I A F F G A V- - - - - - -TP
M T S F F G A V- - - - - - -R A- - -A P
L T A F F G A V-- - - - - -R A- - -A P
-
-
-
Gelatinase/MTS (G/MTS)
Gelatinase (G)
Matrix metalloproteinase (MMP)
F(uPA1)F(uPA2)
F(tPA)
G V P V R F F G A V- - - - - - - - -A- GGG V F F G A V- - - - - - - -A- -R SS
G V F F G A V- - - - - - -A- - -RG SY
Plasminogen activator (PA)
Collagenase (C)
Figure 2 Optimization of linker sequences sensitive to matrix metalloproteinase (MMP) and plasminogen activator (PA). (a) The design ofthe F cleavage site: F expression plasmid was constructed, and the F cleavage site was redesigned by site-directed mutagenesis. Collagenase(MMP1, MMP3, MT-MMP and so on) and gelatinase substrates (MMP2, MMP9) were selected by the study of the synthesized peptidesequence. The additional MTS sequences were also examined (MMP-G1/MTS, MMP-G2/MTS, MMP-G3/MTS, MMP-G4/MTS) to facilitatethe sensitivity to proteases. (b) Fusion assay by cotransfection of modified F- and wild HN-expression plasmids to LLC-MK2 cellsthat expressed MMP2, MT1-MMP or MT1-MMP/MMP2. Cotransfection of three independent plasmid vectors expressing modified F, HNand EGFP to LLC-MK2 cells was done by FuGene 6 transfection reagent. Two days later, the nuclear numbers of syncytia formed werecounted. (c) Fusion assay by cotransfection of modified F- and wild HN-expression plasmids to LLC-MK2 cells in culture mediasupplemented with either recombinant human uPA or tPA. Two days later, the nuclear numbers of syncytia formed were counted.
Optimized design for uPA-targeted oncolytic SeVH Kinoh et al
395
Gene Therapy
Days
Tu
mo
r vo
lum
e (m
m3)
SW620
Nu
cleo
us
nu
mb
ers
of
syn
cyti
a fo
rmat
ion
HT1080
Tu
mo
r vo
lum
e (m
m3)
MMP9
pro-MMP2active-MMP2
HT
1080
SW
620
0
500
1000
1500
2000
2500
3000
3500
HT1080
SW620
SeV
/ ∆F
-GF
P
SeV
/F(M
MP
-C1)
∆M-G
FP
SeV
/F(M
MP
-G1/
MT
S) ∆
M-G
FP
SeV
/F(M
MP
-G2/
MT
S) ∆
M-G
FP
SeV
/F(M
MP
-G3/
MT
S) ∆
M-G
FP
SeV
/F(M
MP
-G4
MT
S) ∆
M-G
FP
SeV
/Fct
14(M
MP
-G4
MT
S) ∆
M-G
FP
SeV
/Fct
14(M
MP
-G4
MT
S) ∆
M-G
FP
SeV
/Fct
14(M
MP
-G4
MT
S) ∆
M-G
FP
+ TIMP-1
+ TIMP-2
+ TIMP-1
+ TIMP-2
Cyt
oto
xici
ty (
%)
100
80
60
40
20
0
N=5 N=5
P=0.025
SeV
/ ∆F
-GF
P
SeV
/F(M
MP
-C1)
∆M-G
FP
SeV
/F(M
MP
-G1/
MT
S) ∆
M-G
FP
SeV
/F(M
MP
-G2/
MT
S) ∆
M-G
FP
SeV
/F(M
MP
-G3/
MT
S) ∆
M-G
FP
SeV
/F(M
MP
-G4
MT
S) ∆
M-G
FP
SeV
/Fct
14(M
MP
-G4
MT
S) ∆
M-G
FP
SeV
/Fct
14(M
MP
-G4
MT
S) ∆
M-G
FP
SeV
/Fct
14(M
MP
-G4
MT
S) ∆
M-G
FP
0
200
400
600
800
1000
1200
1400
0 4 8 12 16 20 24
PBS
SeV/∆F-GFP
SeV/Fct14(MMP-G4/MTS)∆M-GFP
Days
0
200
400
600
800
1000
1200
0 2 4 6 8 10 12 14 16 18
PBS
SeV/∆F-GFP
SeV/F(MMP-G1/MTS)/∆M-GFP
SeV/F(MMP-G4/MTS)∆M-GFP
SeV/Fct14(MMP-G4/MTS)∆M-GFP
Figure 3 Antitumor activity of newly optimized matrix metalloproteinase (MMP)-targeted oncolytic SeV vectors in vitro and in vivo. (a andb) Syncytia formation (a) and LDH-releasing (b) assays assessing cell killing activity in vitro to optimize MMP-sensitive linker sequences ofmodified F gene of oncolytic SeV. HT1080 cells expressing active MMPs were used, and a control cell line without active expression of typicalMMPs, SW620, was also used (a, inset). Each vector was infected at MOI¼ 0.3, and 2 days later, the nuclear numbers of syncytia formed andreleased LDH were assessed. Note that optimized cytotoxic activity seen with the use of SeV/Fct14(MMP-G4/MTS)DM was completelyabolished by supplementation with a typical MMP inhibitor, TIMP-2, but not by supplementation with TIMP-1 (50 ng ml�1, respectively).(c and d) Antitumor activity of oncolytic SeVs with various modifications of F gene sensitive to MMPs in vivo. HT1080 (1�106 cells) thatexpressed a high level of active MMPs was subcutaneously burdened to the abdominal wall. Seven days later, each SeV vector was injectedintratumorally at once. Note that SeV/Fct14 (MMP-G4/MTS)DM-green fluorescent protein (GFP) showed the strongest inhibition of HT1080tumor growth (c), whereas neither the control vector (SeVDF) nor SeV/Fct14(MMP-G4/MTS)DM-GFP showed a significant effect on thegrowth of SW620 tumors (d).
Optimized design for uPA-targeted oncolytic SeVH Kinoh et al
396
Gene Therapy
0
400
800
1200
1600
0 20 40 60 80
PBS
SeV/∆F-GFP
SeV/F(uPA1)∆M-GFP
SeV/F(uPA2)∆M-GFP
SeV/Fct14(uPA2)∆M-GFP
SeV/Fct14(MMP-G4/MTS)∆M-GFP
+PAI-I
PC3
Nu
cleo
us
nu
mb
ers
of
syn
cyti
a fo
rmat
ion
HT29
Days
Tu
mo
r vo
lum
e (m
m3)
Days
Tu
mo
r vo
lum
e (m
m3)
HMW-uPA -
LMW-uPA -
PC
3
DU
145
HT
29PC3DU145
HT29
0
2000
4000
6000
8000
10000PC3
DU145
HT29
80
60
40
20
0
Cyt
oto
xici
ty (
%)
N=8 N=5
P=0.02
0
400
800
1200
1600
0 5 10 15 20 25 30
PBS
SeV/∆F-GFP
SeV/Fct14(uPA2)∆M-GFP
SeV/Fct14(MMP-G4/MTS)∆M-GFP
SeV
/ ∆F
-GF
P
SeV
/F(u
PA
1)∆M
-GF
P
SeV
/F(u
PA
2)∆M
-GF
P
SeV
/F(t
PA
)∆M
-GF
P
SeV
/Fct
14(U
PA
2)∆M
-GF
P
SeV
/Fct
14(U
PA
2)∆M
-GF
P
+PAI-IS
eV/ ∆
F-G
FP
SeV
/F(u
PA
1)∆M
-GF
P
SeV
/F(u
PA
2)∆M
-GF
P
SeV
/F(t
PA
)∆M
-GF
P
SeV
/Fct
14(U
PA
2)∆M
-GF
P
SeV
/Fct
14(U
PA
2)∆M
-GF
P
Figure 4 Antitumor activity of newly optimized uPA-targeted oncolytic SeV vectors in vitro and in vivo. (a and b) Syncytia formation (a) and LDHreleasing (b) assays assessing cell-killing activity in vitro to optimize plasminogen activator (PA)-sensitive linker sequences of modified F gene ofoncolytic SeV. PC3 and DU145 cells expressing active uPA were used, and a control cell line without active expression of PA, HT29, was also used(a, inset). Each vector was infected at MOI¼ 0.3, and 2 days later, nuclear numbers of syncytia formed and released LDH were assessed. Note thatoptimized cytotoxic activity seen with the use of SeV/Fct14(uPA2)DM was completely abolished by supplementation with a typical PA inhibitor,PAI-1 (50 ng ml�1, respectively). (c and d) Antitumor activity of oncolytic SeVs with various modifications of the F gene sensitive to PA in vivo. PC3(1�106 cells), which expressed a high level of active uPA, was subcutaneously burdened to the abdominal wall. Seven days later, each SeV vectorwas injected intratumorally at once. Note that SeV/Fct(uPA2)DM-green fluorescent protein (GFP) showed the strongest inhibition of PC3 tumorgrowth (c), whereas neither the control vector (SeVDF) nor SeV/Fct(uPA2)DM-GFP showed a significant effect on the growth of H29 tumors (d).
Optimized design for uPA-targeted oncolytic SeVH Kinoh et al
397
Gene Therapy

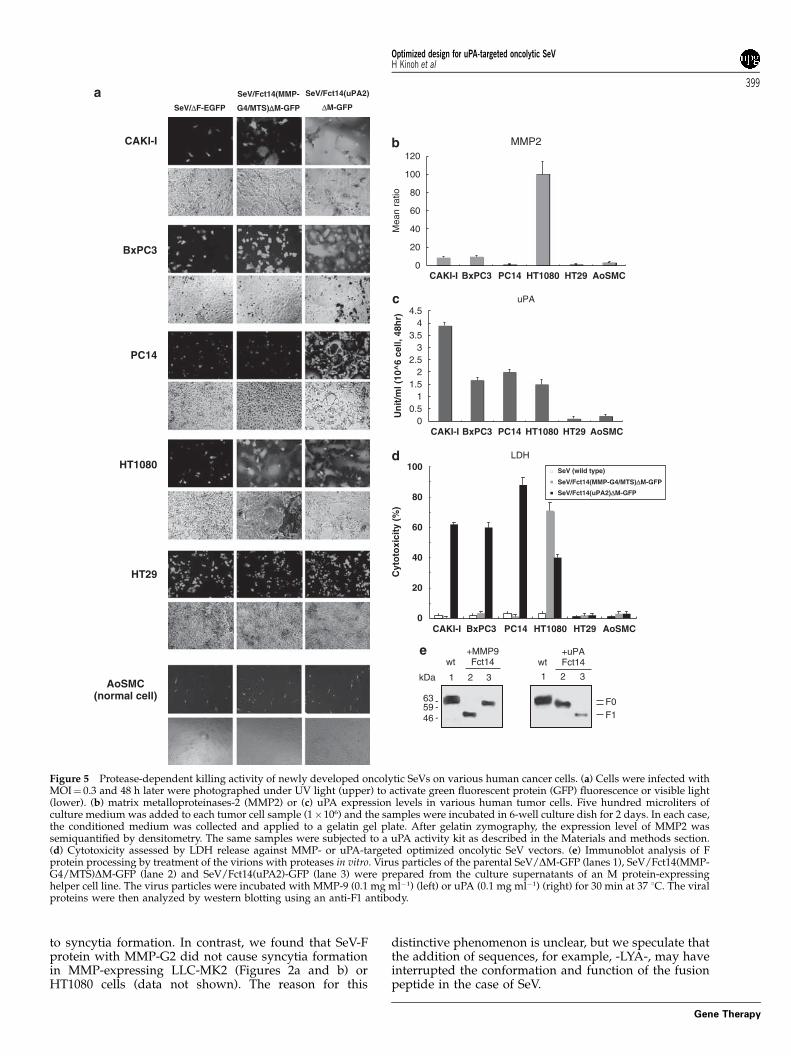
New oncolytic SeV vectors selectively inducedprotease-dependent syncytia formation and inhibitedtumor growth through extensive killing of MMP- oruPA-expressing cells
Protease-dependent syncytia formation was examinedin vitro, using these optimized MMP-targeted(SeV/Fct14(MMP-G4/MTS)DM-GFP) and uPA-targetedvectors (SeV/Fct14(uPA2)DM-GFP). Figures 5b andc shows the enzymatic activity of MMP2 and uPA inseveral human tumor cell lines and normal human cell(human aortic smooth muscle cells). HT1080 showed thehighest MMP2 expression, and CAKI-I and BxPC3expressed a low level, whereas HT29 and PC14 ex-pressed MMP2 at undetectable levels (Figure 5b). Most ofthe tested tumor cell lines (CAKI-I, BxPC3, PC14,HT1080) expressed both uPA protein and uPAR proteinby western blot analysis (data not shown) and exhibitedenzymatic activity (Figure 5c), whereas HT29 expressedonly uPAR protein but not urokinase activity.
Infection with the MMP-targeted oncolytic SeV vectorinduced extensive syncytia in HT1080 and small syncytiain CAKI-I and BxPC3, but not in HT29 and aortic smoothmuscle cells (Figure 5a), reflecting their MMP2 expres-sion levels. Infection with a uPA-targeted vector formedlarge syncytia in all tumor cell lines tested except forHT29 and aortic smooth muscle cells (Figure 5a). Theformation of syncytia nearly corresponded to the level ofcytotoxicity (Figure 5d), at least in vitro.
To demonstrate the protease-mediated cleavage of theengineered F protein, we collected the MMP-targetedand uPA-targeted vector particles without any proteaseand digested them by recombinant MMP9 or uPA in vitro(Figure 5e). The virus induced fusion and virus spread-ing in a highly specific protease-dependent mannerstrictly paralleled the cleavability of the F0 precursoron the virions into F1 and F2 by the respective proteases;specific cleavage of F0 by MMP9 and uPA for MMP-targeted (SeV/Fct14(MMP-G4/MTS)DM-GFP) (Figure5e, lane 2) and uPA-targeted SeV vector (SeV/Fct14(u-PA2)DM-GFP) (Figure 5e, lane 3), respectively. ControlSeV/DM-GFP, which have wild type F (Figure 5e, lane 1),were not processed by both protease.
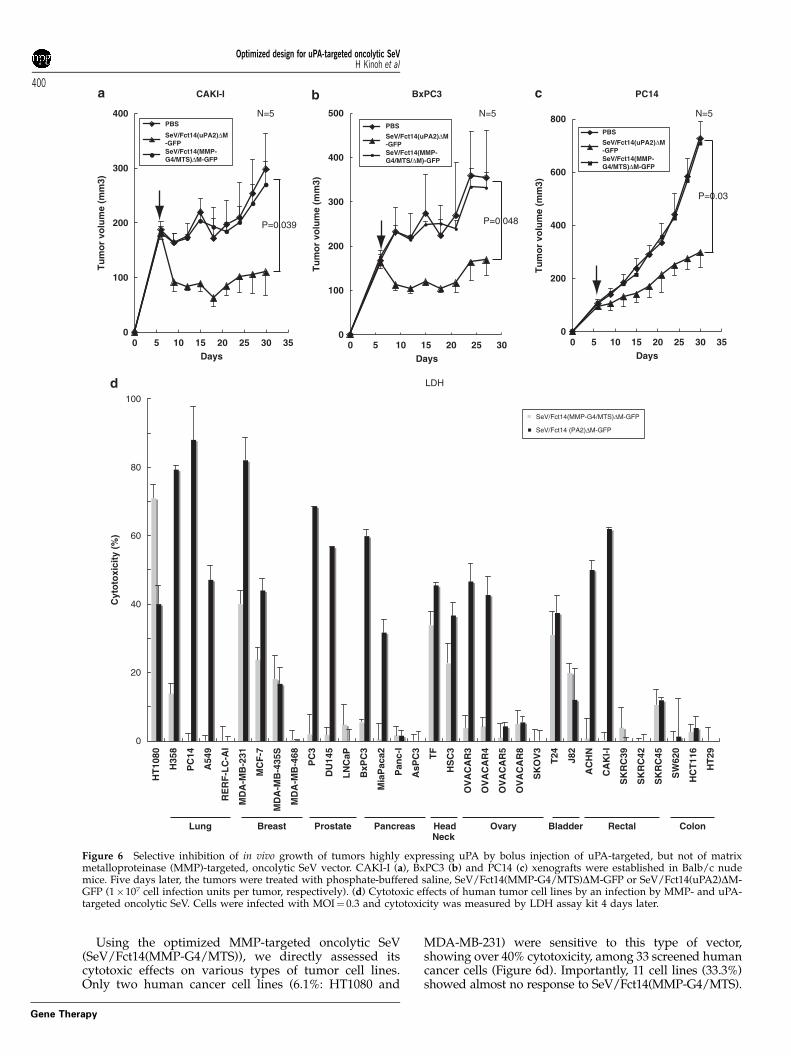
Subsequently, we assessed whether these findingsobtained in vitro would occur in vivo. The growth ofHT29 tumors established in the flanks of athymic nudemice, tumors that expressed almost no MMP or uPA, as anegative control, was rarely affected by the bolusintratumor injection of either MMP-targeted vector(SeV/Fct14(MMP-G4/MTS)DM-GFP) or uPA-targetedvector (SeV/Fct14(uPA2)DM-GFP) on day 6, as shownin Figure 5d. In contrast, the growth of dermal tumors ofCAKI-I, BxPC3 and PC14 were significantly suppressedby intratumor injection of uPA-targeted vector (CAKI-I:P¼ 0.039, BxPC3: P¼ 0.048 and PC14: P¼ 0.03) but notby MMP-targeted vector (Figures 6a–c), reflecting thecytotoxic activity seen in vitro (Figure 5d).
Finally, we screened the oncolytic activity of theseoptimized vectors using 33 established human cancercell lines from various origins. As shown in Figure 6d,extensive oncolytic/cytotoxic activity (440%) of uPA-type vectors was illustrated on 42.4% of these cancerlines (14/33 cell lines), whereas only 2 cancers(6.1%) were sensitive to the MMP-targeted vector. As anumber of publications show that the uPA system
appears to be active in the majority of various cancersin situ,15,16 these results suggest the more extensiveutility of uPA-targeted oncolytic SeV than that of MMP-targeting SeV.
Discussion
In a previous study, we developed a novel prototype ofoncolytic recombinant SeV by introducing two genomicmodifications to its genome, resulting in cell-to-cellspread through fusion of plasma membranes formingsyncytia only among the MMP-expressing tumor cells,and successful inhibition of the tumor growth in vivo,without releasing secondary vector particles.8 On thebasis of this concept, in this study, we developeddramatically improved vectors by modifying the F genesensitive to uPA and MMP associated with truncation ofits cytoplasmic tail. These newly optimized vectorsshowed markedly enhanced cancer killing activity in aprotease-dependent manner in vitro and in vivo, and,importantly, modification of amino-acid residues of the Fgene for targeting uPA dramatically expanded the cell-killing spectrum of oncolytic SeV. These results suggestthe potential utility of SeV/Fct14(PA2)DM in clinicalgene therapy for cancer patients.
Some recent studies showed that artificial truncationof the cytoplasmic domain of glycoprotein of retrovirus,9
herpes virus17 as well as paramyxovirus10 enhanced theirfusogenic activity. Considering this information, wefound that the modification of the F gene of SeV(truncation of 14 C-terminal amino acids) resulted in adramatic increase of fusogenic activity, namely a sixfoldincrease in the number of syncytia formed comparedwith that seen by the original F protein (Figure 1). Similarfindings were also seen in the case of measles viruses,another member of Paramyxoviridae; modification of the Fgene (truncation of 17 C-terminal amino acids) enhancedits fusogenic activity.10 The precise mechanism related toenhancement of fusogenic activity caused by the cyto-plasmic tail of Paramyxoviridae, however, has been largelyunknown. A possible explanation is the loss of interac-tion among H, F and M, because the direct interaction ofthe cytoplasmic tails of H/F and M were shown todownregulate viral fusion activity.10,18 However, thismay not be the case, because the M gene was deleted inthe oncolytic SeV vector used in this study. Furtherstudies will be needed to clarify the possible molecularmechanism underlying fusogenic activity and the cyto-plasmic domain of F protein.
Subsequently, we further investigated the optimalamino-acid sequence of the cleavage site against MMPsand uPA/tPA to enhance the catalytic potency ofoncolytic SeV. We found more sensitive linker sequences(MMP-G4/MTS: -PRAMTS-; uPA2: -SGRS-) than that ofthe prototype vectors (MMP-G1/MTS: -PLGMTS- anduPA1: -VGR-) (Figure 2); however, the sequence-specificsensitivity may depend on the type of virus. Forexample, MMP-activated recombinant retroviruses couldbe generated by the introduction of -PLGLYA- hexamer,and a similar sequence (MMP-G2: -PQGLYA-) could beadapted to the oncolytic measles virus vector, asdemonstrated by Springfeld et al.12 Measles virus-Fprotein containing MMP-G2 (-PQGLYA-) sequence,called ‘MMP-A1’ in their study, could lead HT1080 cells
Optimized design for uPA-targeted oncolytic SeVH Kinoh et al
398
Gene Therapy
to syncytia formation. In contrast, we found that SeV-Fprotein with MMP-G2 did not cause syncytia formationin MMP-expressing LLC-MK2 (Figures 2a and b) orHT1080 cells (data not shown). The reason for this
distinctive phenomenon is unclear, but we speculate thatthe addition of sequences, for example, -LYA-, may haveinterrupted the conformation and function of the fusionpeptide in the case of SeV.
BxPC3
PC14
CAKI-I
SeV/Fct14(MMP-
G4/MTS)∆∆M-GFP
HT1080
SeV/Fct14(uPA2)
∆M-GFP
HT29
SeV/∆F-EGFP
MMP2
Mea
n ra
tioU
nit
/ml (
10^6
cel
l, 48
hr)
uPA
Cyt
oto
xici
ty (
%)
100
80
60
40
20
0
LDH
AoSMC(normal cell)
0
20
40
60
80
100
120
CAKI-I BxPC3 HT1080PC14 HT29 AoSMC
00.5
11.5
22.5
33.5
44.5
CAKI-I BxPC3 HT1080PC14 HT29 AoSMC
CAKI-I BxPC3 HT1080PC14 HT29 AoSMC
1 2 3 1 2 3
F0F1
+MMP9Fct14
+uPAFct14
SeV (wild type)
SeV/Fct14(MMP-G4/MTS)∆M-GFP
SeV/Fct14(uPA2)∆M-GFP
wt wt
kDa
635946 -
--
Figure 5 Protease-dependent killing activity of newly developed oncolytic SeVs on various human cancer cells. (a) Cells were infected withMOI¼ 0.3 and 48 h later were photographed under UV light (upper) to activate green fluorescent protein (GFP) fluorescence or visible light(lower). (b) matrix metalloproteinases-2 (MMP2) or (c) uPA expression levels in various human tumor cells. Five hundred microliters ofculture medium was added to each tumor cell sample (1�106) and the samples were incubated in 6-well culture dish for 2 days. In each case,the conditioned medium was collected and applied to a gelatin gel plate. After gelatin zymography, the expression level of MMP2 wassemiquantified by densitometry. The same samples were subjected to a uPA activity kit as described in the Materials and methods section.(d) Cytotoxicity assessed by LDH release against MMP- or uPA-targeted optimized oncolytic SeV vectors. (e) Immunoblot analysis of Fprotein processing by treatment of the virions with proteases in vitro. Virus particles of the parental SeV/DM-GFP (lanes 1), SeV/Fct14(MMP-G4/MTS)DM-GFP (lane 2) and SeV/Fct14(uPA2)-GFP (lane 3) were prepared from the culture supernatants of an M protein-expressinghelper cell line. The virus particles were incubated with MMP-9 (0.1 mg ml�1) (left) or uPA (0.1 mg ml�1) (right) for 30 min at 37 1C. The viralproteins were then analyzed by western blotting using an anti-F1 antibody.
Optimized design for uPA-targeted oncolytic SeVH Kinoh et al
399
Gene Therapy
Using the optimized MMP-targeted oncolytic SeV(SeV/Fct14(MMP-G4/MTS)), we directly assessed itscytotoxic effects on various types of tumor cell lines.Only two human cancer cell lines (6.1%: HT1080 and
MDA-MB-231) were sensitive to this type of vector,showing over 40% cytotoxicity, among 33 screened humancancer cells (Figure 6d). Importantly, 11 cell lines (33.3%)showed almost no response to SeV/Fct14(MMP-G4/MTS).
CAKI-I
0
100
200
300
400
0 5 10 15 20 25 30 35
PBS
SeV/Fct14(uPA2)∆M-GFPSeV/Fct14(MMP-G4/MTS)∆M-GFP
Days
Tu
mo
r vo
lum
e (m
m3)
P=0.039
N=5
BxPC3
0
100
200
300
400
500
0 5 10 15 20 25 30
PBS
SeV/Fct14(uPA2)∆M-GFPSeV/Fct14(MMP-G4/MTS/∆M)-GFP
Days
Tu
mo
r vo
lum
e (m
m3)
N=5
P=0.048
PC14
Days
Tu
mo
r vo
lum
e (m
m3)
0
200
400
600
800
0 5 10 15 20 25 30 35
PBS
SeV/Fct14(uPA2)∆M-GFPSeV/Fct14(MMP-G4/MTS)∆M-GFP
N=5
P=0.03
Cyt
oto
xici
ty (
%)
Lung Breast Prostate
HT
1080
H35
8
PC
14
A54
9
RE
RF
-LC
-AI
MD
A-M
B-2
31
MD
A-M
B-4
68
MC
F-7
MD
A-M
B-4
35S
PC
3
DU
145
LN
CaP
BxP
C3
Mia
Pac
a2
Pan
c-I
AsP
C3
TF
HS
C3
OV
AC
AR
3
OV
AC
AR
4
OV
AC
AR
5
OV
AC
AR
8
SK
OV
3
T24 J82
AC
HN
CA
KI-
I
SK
RC
39
SK
RC
42
SK
RC
45
SW
620
HC
T11
6
HT
29
Pancreas HeadNeck
Ovary Bladder Rectal Colon
0
20
40
60
80
100
SeV/Fct14 (PA2)∆M-GFP
SeV/Fct14(MMP-G4/MTS)∆M-GFP
LDH
Figure 6 Selective inhibition of in vivo growth of tumors highly expressing uPA by bolus injection of uPA-targeted, but not of matrixmetalloproteinase (MMP)-targeted, oncolytic SeV vector. CAKI-I (a), BxPC3 (b) and PC14 (c) xenografts were established in Balb/c nudemice. Five days later, the tumors were treated with phosphate-buffered saline, SeV/Fct14(MMP-G4/MTS)DM-GFP or SeV/Fct14(uPA2)DM-GFP (1�107 cell infection units per tumor, respectively). (d) Cytotoxic effects of human tumor cell lines by an infection by MMP- and uPA-targeted oncolytic SeV. Cells were infected with MOI¼ 0.3 and cytotoxicity was measured by LDH assay kit 4 days later.
Optimized design for uPA-targeted oncolytic SeVH Kinoh et al
400
Gene Therapy

Recently some researchers and we reported on thegeneration of plasmid DNA, adenovirus vector carryingviral envelope glycoproteins, measles vector and Sendaivirus vector activatable by MMPs.8,12,19,20 Their datashow that those vectors can target the cytotoxicity of thefusogenic membrane glycoprotein in MMP-overexpres-sing tumor lines and xenografts, and maintain significantantitumor activity both in vitro and in vivo. MMP-targeted vectors, irrespective of the vectors’ backbone,may be confronted with a similar difficulty in clinicalsettings.
To overcome such a potential issue, we here furthergenerated a new group of vectors that could be usefulagainst a broad range of human tumors, namely theoptimized uPA-targeted oncolytic SeVs. We here as-sessed uPA-sensitive peptide substrates, F(uPA1) (-VGR-)and F(uPA2) (-SGRS-)13 as well as tPA-sensitive residueF(tPA) (-YGRS-).21 Cell fusion activity was optimal withthe use of F(uPA2), and suboptimal activity was seen inthe case of F(tPA) (Figure 2c). As western blot analysesrevealed significant expression of tPA in a few cancer celllines among 33 cell lines assessed (as listed in Figure 6d(data not shown)), the uPA2 substrate was utilized forsubsequent studies. The result was that greater numbersof human tumor cells (42.4 and 51.5% for 440 and 430%cytotoxicity, respectively) were highly sensitive to newand optimized uPA-targeted oncolytic SeVs. Thesefindings suggest that the targeting uPA should extendthe potential utility of oncolytic SeVs in clinical settings.
In addition, there are several theoretical advantages ofM gene-deleted paramyxovirus-based vectors over thecurrent oncolytic viruses. First, abolishment of secondaryviral particles through deletion of the M gene mayreduce the risk of their systemic spread. Second, weshould encounter the direct and indirect antitumor effectof the fusogenic membrane glycoprotein of envelopedviral vectors for gene therapy, including measles virusF/H proteins, gibbon ape leukemia virus envelopeprotein, vesicular stomatitis virus G protein and HN/Fproteins of our oncolytic SeVs. Virus-mediated fusogenicmembrane glycoprotein expression in target tumor cellshas shown direct cytotoxicity, associated with a ‘bystan-der-like effect,’ against human tumor xenografts in vivowith the use of plasmid DNA, adenoviral or lentiviralvectors.22,23 In addition to the direct cytotoxic effect,fusogenic membrane glycoprotein delivery can initiatesystemic antitumor immunity, thus indirectly suppres-sing the growth of distant tumors.24 We also observedthat treatment of solid tumors by uPA-targeted oncolyticSeVs used in this study resulted in the establishment oftumor-specific antitumor immunity to prevent the recur-rence of tumors (Kinoh et al., unpublished data). Thesemultiple mechanisms for antitumor activity of uPA-targeted oncolytic SeVs show promise for the develop-ment of treatments for patients with intractable and faradvanced malignancies in the clinical setting.
In summary, we successfully developed new andoptimized oncolytic recombinant SeVs that target uPAand MMP for extensive killing of cancer cells in vitro andin vivo. We believe that this novel type of oncolytic virusthat works without spreading secondary particles mayopen the way to developing an oncolytic strategy forcancer patients through not only possible improvementof the treatment’s efficacy, but also its safety in clinicalsettings.
Materials and methods
Cell lines and reagentsTumor cell lines used in this study were obtained fromthe American Type Culture Collection (ATCC, Rockville,MD, USA) or the European collection of animal cellcultures (Wiltshire, UK) or RIKEN (Tsukuba, Japan).Each cell line was maintained in directed mediumsupplemented with 10% fetal bovine serum. The follow-ing cell lines were used: PC3, DU145, CAKI-I, HT1080,SW620, HT29, LLCMK2, PC14, BxPC3. Normal humanaortic smooth muscle cells were purchased from TakaraBiotech (Otsu, Japan). Recombinant human PAI-I, humanTIMP-1 and human TIMP-2 (Calbiochem, Darmstadt,Germany) at final concentrations of 50 mg ml�1 were usedas protease-specific inhibitors.
Plasmid construction of F gene and site-specificmutagenesisModification of the F protein cleavage site and deletionof the cytoplasmic domain of the F gene were done aftersubcloning to the expression vector pCAGGS25 with theQuick-Change Mutagenesis Kit (Stratagene, La Jolla, CA,USA) according to the manufacturer’s instructions. Theintegrity of mutant constructs was confirmed by directDNA sequencing.
Construction of MMP2 or MT1-MMP-expressingplasmid vectorsFull-length human MT1-MMP (MMP14) and humanMMP2 cDNAs were cloned from total RNA of HT1080cells by reverse transcription-PCR, and amplified frag-ments were inserted in a pIRES vector (Takara Biotech,Otsu, Japan). The full sequence of cDNAs were deter-mined by direct sequencing. The enzymatic activity ofthese genes was checked by gelatin zymography, as willbe described later.
Construction and recovery of improved oncolytic SeVvectorA scheme of the designs of SeV construction is shown inSupplementary Figure 1. The parent plasmid pSeV18+/DM-GFP, in which the GFP had been substituted for thedeleted M gene, was constructed as described before byInoue et al.5 The parent plasmid was digested with SalIand NheI, and the F gene fragment (9634 bp) wassubcloned into LITMUS 38 (New England Biolabs,Beverly, MA, USA). Site-directed mutagenesis wasperformed using the Quick-Change Mutagenesis Kit(Stratagene), and mutated F gene was returned to thepSeV18+/DF-GFP backbone.4 Recovery and amplifi-cation of the SeV vector were generated essentially asdescribed before;4,5 briefly, LLC-MK2 cells were trans-fected at room temperature with a plasmid mixturecontaining each plasmid [pSeV+18/F(MMP-C1 MMP-G1/MTS, G2/MTS, G3/MTS, G4/MTS, uPA1, uPA2 andtPA)DM-GFP], pSeV+18/Fct14(MMP-G4/MTS, uPA2)DM-GFP, pGEM-NP, pGEM-P and pGEM-L in 110 ml ofSuperfect transfection reagent (Qiagen, Tokyo, Japan).The transfected cells were maintained for 3 h in 3 ml ofOpti-MEM (Gibco-BRL) plus 3% fetal calf serum, washedthree times with modified Eagles’ medium (MEM) andincubated for 60 h in MEM containing araC (40 mg ml�1).The transfected cells were collected by centrifugation
Optimized design for uPA-targeted oncolytic SeVH Kinoh et al
401
Gene Therapy

at 1000 g for 5 min, resuspended in Opti-MEM and lysedby three cycles of freezing and thawing. The lysatesolution was incubated on the F/M-expressingLLC-MK2 cells in a 24-well plate.5 Twenty-four hourslater, cells were washed three times with MEM andincubated for 3–6 days in MEM containing araC and5 U ml�1 collagenase type IV, Clostridium histolyticum (ICNBiochemical Inc., Costa Mesa, CA, USA) or 7.5 mg ml�1
trypsin plus 10 ng ml�1 urokinase (Cosmobio, Tokyo,Japan). Virus yield is expressed in cell infection units, asdescribed previously.4
Fusion assay and cytotoxicity assayCells were plated in 96-well dishes at a density of5� 103 cells per well and infected as described abovewith each SeV vector at an MOI of 0.3. On day 4, cellswere fixed with 4% paraformaldehyde. Cells were thenstained with hematoxylin, rinsed and covered with100 ml phosphate-buffered saline. The number (N) ofsyncytia per well (0.3 cm2) was counted in triplicatewells, and the average nucleus number was calculated.All experiments were repeated at least four times. Toevaluate the fusogenic activity against MMP-expressingcells, Sendai envelope expression plasmid (pCAGGS/Fand pCAGGS/HN) and MMP expression plasmid weretransfected into LLCMK2 in 96-well plates using trans ITreagent. The number of nuclei in the fusion cells wascounted as described above. Cytotoxicity was deter-mined by LDH assay using the Cytotoxicity DetectionKit (Roche Diagnostics KK, Tokyo, Japan) according tothe manufacturer’s instructions.
Casein and gelatin zymographyGelatin and casein zymography was performed usingthe method outlined by Heussen and Dowdle.26 Briefly,serum-free supernatants from each cell culture wereanalyzed under nonreducing conditions on 7.5% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (Sigma,St Louis, MO, USA) containing 0.1% casein plus 4 mg ofplasminogen or 0.1% gelatin. The gel was then incubatedat room temperature in a 2.5% Triton X-100 solutioncontaining 0.05% sodium azide and 0.05 M Tris (pH 7.5;Sigma) followed by incubation at 37 1C in 0.1 M Tris-HCl(pH 8.3)/0.5 M sodium chloride solution for 16 h. The gelwas then stained with a 0.25% Coomassie Blue/1.1%acetic acid/45.5% methanol solution for 40 min, followedby destaining in a 25% methanol/10% acetic acidmixture. Enzyme activity was visualized as clear zoneson a blue background. Semiquantification of MMP2 wasdone by the method of Togawa et al.27 Densitometry wasdone using NIH Image.
Urokinase activity assayCells (1�106) were seeded in six-well dishes, and 0.5 mlof supernatant was collected after 48 h of incubationat 37 1C. The urokinase activity was measured witha Urokinase Activity Kit (Chemicon International,Temecula, CA, USA).
Western blot analysisThree Sendai virus vectors (SeV/DM-GFP, SeV/Fct14(MMP-G4/MTS)DM-GFP, SeV/Fct14(uPA2)DM-GFP)were prepared from the culture supernatants of an Mprotein-expressing helper cell line after infection of anMOI of 3 (cell infection units) and subsequent incubation
for 2 days in the absence of any protease. Three SeVs weredigested by human recombinant active MMP9(0.1 mg ml�1) (Calbiochem) or urokinase (Calbiochem) for30 min at 37 1C. The viral proteins were then analyzed bywestern blotting using an anti-F1 rabbit antiserum.Immunoblot analysis was carried out using a rabbitpeptide antiserum against the F1 (dilution, 1:1000),8 ananti-rabbit-horseradish peroxidase conjugate and theenhanced chemiluminescence detection system (Amer-sham Biosciences, Piscataway, NJ, USA).
In vivo xenograft modelFemale 7- to 8-week-old Balb/c nu/nu mice wereobtained from Charles River Co. (Shizuoka, Japan).Human tumor cell lines (HT1080, PC3, PC14, CAKI-I,BxPC3, SW620, HT29) (1�107 cells) were propagatedand implanted in Matrigel (Beckton Dickinson, Bedford,MA, USA). The cells were injected subcutaneously intothe right flank of mice. When the subcutaneous tumorsgrew to 4–8 mm in diameter (usually after 7 days), themice were divided into groups (n¼ 5–8 per group), andeach SeV vector (1�107 cell infection units) in a 100 mlvolume was injected intratumorally using a 26-gaugeneedle. Tumor volume was calculated with the formulaVolume¼ a2b/2, where a is the shortest diameter and b isthe longest diameter. Tumor sizes were expressed asmean volumes±s.e.
Statistical analysisStatistical significance was assessed with the Mann–Whitney U-test as appropriate.
Acknowledgements
This work was supported in part by Research Grantsfrom the 21st Century Center of Excellence Program,Chiba University Graduate School of Medicine, and byGrants-in-Aid (to YY) from the Japanese Ministry ofEducation, Culture, Sports, Science and Technology. Wethank B Moss, D Kolakofsky, I Saito and H Iba forsupplying experimental materials essential for thisstudy; K Ishida, T Kanaya, T Yamamoto, M Yoshizaki,A Tagawa, E Suzuki and N Kohno for their excellenttechnical assistance; and T Hironaka, T Zhu and A Iidafor helpful discussions.
References
1 Aghi M, Martuza RL. Oncolytic viral therapies—the clinicalexperience. Oncogene 2005; 24: 7802–7816.
2 Bell J. Replicating oncolytic virus therapeutics—Third Interna-tional Meeting. IDrugs 2005; 8: 360–363.
3 Tashiro M, McQueen NL, Seto JT. Determinants of organ tropismof sendai virus. Front Biosci 1999; 4: D642–D645.
4 Li HO, Zhu YF, Asakawa M, Kuma H, Hirata T, Ueda Y et al.A cytoplasmic RNA vector derived from nontransmissibleSendai virus with efficient gene transfer and expression. J Virol2000; 74: 6564–6569.
5 Inoue M, Tokusumi Y, Ban H, Kanaya T, Shirakura M,Tokusumi T et al. A new Sendai virus vector deficient in thematrix gene does not form virus particles and shows extensivecell-to-cell spreading. J Virol 2003; 77: 6419–6429.
6 Inoue M, Tokusumi Y, Ban H, Shirakura M, Kanaya T,Yoshizaki M et al. Recombinant Sendai virus vectors deleted in
Optimized design for uPA-targeted oncolytic SeVH Kinoh et al
402
Gene Therapy

both the matrix and the fusion genes: efficient gene transfer withpreferable properties. J Gene Med 2004; 6: 1069–1081.
7 Yoshizaki M, Hironaka T, Iwasaki H, Ban H, Tokusumi Y, Iida Aet al. Naked Sendai virus vector lacking all of the envelope-related genes: reduced cytopathogenicity and immunogenicity.J Gene Med 2006; 8: 1151–1159.
8 Kinoh H, Inoue M, Washizawa K, Yamamoto T, Fujikawa S,Tokusumi Y et al. Generation of a recombinant Sendai virus thatis selectively activated and lyses human tumor cells expressingmatrix metalloproteinases. Gene Therapy 2004; 11: 1137–1145.
9 Rein A, Mirro J, Haynes JG, Ernst SM, Nagashima K. Function ofthe cytoplasmic domain of a retroviral transmembrane protein:p15E-p2E cleavage activates the membrane fusion capabilityof the murine leukemia virus Env protein. J Virol 1994; 68:1773–1781.
10 Cathomen T, Naim HY, Cattaneo R. Measles viruses with alteredenvelope protein cytoplasmic tails gain cell fusion competence.J Virol 1998; 72: 1224–1234.
11 Ke SH, Coombs GS, Tachias K, Corey DR, Madison EL. Optimalsubsite occupancy and design of a selective inhibitor ofurokinase. J Biol Chem 1997; 272: 20456–20462.
12 Springfeld C, von Messling V, Frenzke M, Ungerechts G,Buchholz CJ, Cattaneo R. Oncolytic efficacy and enhanced safetyof measles virus activated by tumor-secreted matrix metallopro-teinases. Cancer Res 2006; 66: 7694–7700.
13 Ke SH, Coombs GS, Tachias K, Corey DR, Madison EL. Optimalsubsite occupancy and design of a selective inhibitor ofurokinase. J Biol Chem 1997; 272: 20456–20462.
14 Ding L, Coombs GS, Strandberg L, Navre M, Corey DR,Madison EL. Origins of the specificity of tissue-type plasmino-gen activator. Proc Natl Acad Sci USA 1995; 92: 7627–7631.
15 Egeblad M, Werb Z. New functions for the matrix metallopro-teinases in cancer progression. Nat Rev Cancer 2002; 2: 161–174.
16 Dano K, Behrendt N, Hoyer-Hansen G, Johnsen M, Lund LR,Ploug M et al. Plasminogen activation and cancer. ThrombHaemost 2005; 93: 676–681.
17 Gage PJ, Levine M, Glorioso JC. Syncytium-inducing mutationslocalize to two discrete regions within the cytoplasmic domainof herpes simplex virus type 1 glycoprotein B. J Virol 1993; 67:2191–2201.
18 Tahara M, Takeda M, Yanagi Y. Altered interaction of the matrixprotein with the cytoplasmic tail of hemagglutinin modulatesmeasles virus growth by affecting virus assembly and cell-cellfusion. J Virol 2007; 81: 6827–6836.
19 Johnson KJ, Peng KW, Allen C, Russell SJ, Galanis E. Targetingthe cytotoxicity of fusogenic membrane glycoproteins in gliomasthrough protease-substrate interaction. Gene Therapy 2003; 10:725–732.
20 Allen C, McDonald C, Giannini C, Peng KW, Rosales G,Russell SJ et al. Adenoviral vectors expressing fusogenicmembrane glycoproteins activated via matrix metalloproteinasecleavable linkers have significant antitumor potential in the genetherapy of gliomas. J Gene Med 2004; 6: 1216–1227.
21 Harris JL, Backes BJ, Leonetti F, Mahrus S, Ellman JA, Craik CS.Rapid and general profiling of protease specificity by usingcombinatorial fluorogenic substrate libraries. Proc Natl Acad SciUSA 2000; 97: 7754–7759.
22 Bateman AR, Harrington KJ, Kottke T, Ahmed A, Melcher AA,Gough MJ et al. Viral fusogenic membrane glycoproteins killsolid tumor cells by nonapoptotic mechanisms that promotecross presentation of tumor antigens by dendritic cells. CancerRes 2002; 62: 6566–6578.
23 Diaz RM, Bateman A, Emiliusen L, Fielding A, Trono D, RussellSJ et al. A lentiviral vector expressing a fusogenic glycoproteinfor cancer gene therapy. Gene Therapy 2000; 7: 1656–1663.
24 Linardakis E, Bateman A, Phan V, Ahmed A, Gough M, Olivier Ket al. Enhancing the efficacy of a weak allogeneic melanomavaccine by viral fusogenic membrane glycoprotein-mediatedtumor cell–tumor cell fusion. Cancer Res 2002; 62: 5495–5504.
25 Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene1991; 108: 193–199.
26 Heussen C, Dowdle EB. Electrophoretic analysis of plasminogenactivators in polyacrylamide gels containing sodium dodecylsulfate and copolymerized substrates. Anal Biochem 1980; 102:196–202.
27 Togawa D, Koshino T, Saito T, Takagi T, Machida J. Highlyactivated matrix metalloproteinase-2 secreted from clones ofmetastatic lung nodules of nude mice injected with humanfibrosarcoma HT1080. Cancer Lett 1999; 146: 25–33.
Supplementary Information accompanies the paper on Gene Therapy website (http://www.nature.com/gt)
Optimized design for uPA-targeted oncolytic SeVH Kinoh et al
403
Gene Therapy
Copyright © 2022 FDOKUMEN




















