
Gastroenterology - BPG Management System
130
A Weekly Journal of Gastroenterology and Hepatology World Journal of Gastroenterology Indexed and Abstracted in: Current Contents ® /Clinical Medicine, Science Citation Index Expanded (also known as SciSearch ® ) and Journal Citation Reports/Science Edition, Index Medicus, MEDLINE and PubMed, Chemical Abstracts, EMBASE/Excerpta Medica, Abstracts Journals, Nature Clinical Practice Gastroenterology and Hepatology, CAB Abstracts and Global Health. ISI JCR 2003-2000 IF: 3.318, 2.532, 1.445 and 0.993. Volume 13 Number 13 April 7, 2007 World J Gastroenterol 2007 April 7; 13(13): 1893-2018 Online Submissions www.wjgnet.com/wjg/index.jsp www.wjgnet.com Printed on Acid-free Paper ISSN 1007-9327 CN 14-1219/R Local Post Offices Code No. 82-261 World Journal of Gastroenterology ® The WJG Press The WJG Press, Apartment 1066 Yishou Garden, 58 North Langxinzhuang Road, PO Box 2345, Beijing 100023, China Telephone: +86-10-85381892 Fax: +86-10-85381893 E-mail: [email protected] http: //www.wjgnet.com National Journal Award 2005 Volume 13 Number 13 April 7, 2007 World Journal of Gastroenterology www.wjgnet.com Volume 13 Number 13 Apr 07 2007 ISSN 1007-9327 CN 14-1219/R
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Gastroenterology - BPG Management System

A Weekly Journal of Gastroenterology and Hepatology
World Journal of Gastroenterology
Indexed and Abstracted in:Current Contents®/Clinical Medicine, Science Citation Index Expanded (also known as SciSearch®) and Journal Citation Reports/Science Edition, Index Medicus, MEDLINE and PubMed, Chemical Abstracts, EMBASE/Excerpta Medica, Abstracts Journals, Nature Clinical Practice Gastroenterology and Hepatology, CAB Abstracts and Global Health. ISI JCR 2003-2000 IF: 3.318, 2.532, 1.445 and 0.993.
Volume 13 Number 13April 7, 2007
World J Gastroenterol2007 April 7; 13(13): 1893-2018
Online Submissionswww.wjgnet.com/wjg/index.jsp
www.wjgnet.com Printed on Acid-free Paper
ISSN 1007-9327 CN 14-1219/R Local Post Offices Code No. 82-261
World Journal of Gastroenterology ®
The WJG Press
The WJG Press, Apartment 1066 Yishou Garden, 58 NorthLangxinzhuang Road, PO Box 2345, Beijing 100023, China
Telephone: +86-10-85381892Fax: +86-10-85381893
E-mail: [email protected]: //www.wjgnet.com
National Journal Award2005
Volume 13 Number 13April 7, 2007
World Journal of G
astroenterology ww
w.w
jgnet.com Volum
e 13 Num
ber 13 Apr 07 2007
ISSN 1007-9327CN 14-1219/R

World Journal ofGastroenterology®
Volume 13 Number 13April 7, 2007
Contents
www.wjgnet.com
1893 Innovative therapeutics for infl ammatory bowel disease Yamamoto-Furusho JK
1897 Treatment of hepatitis C virus infection Weigand K, Stremmel W, Encke J
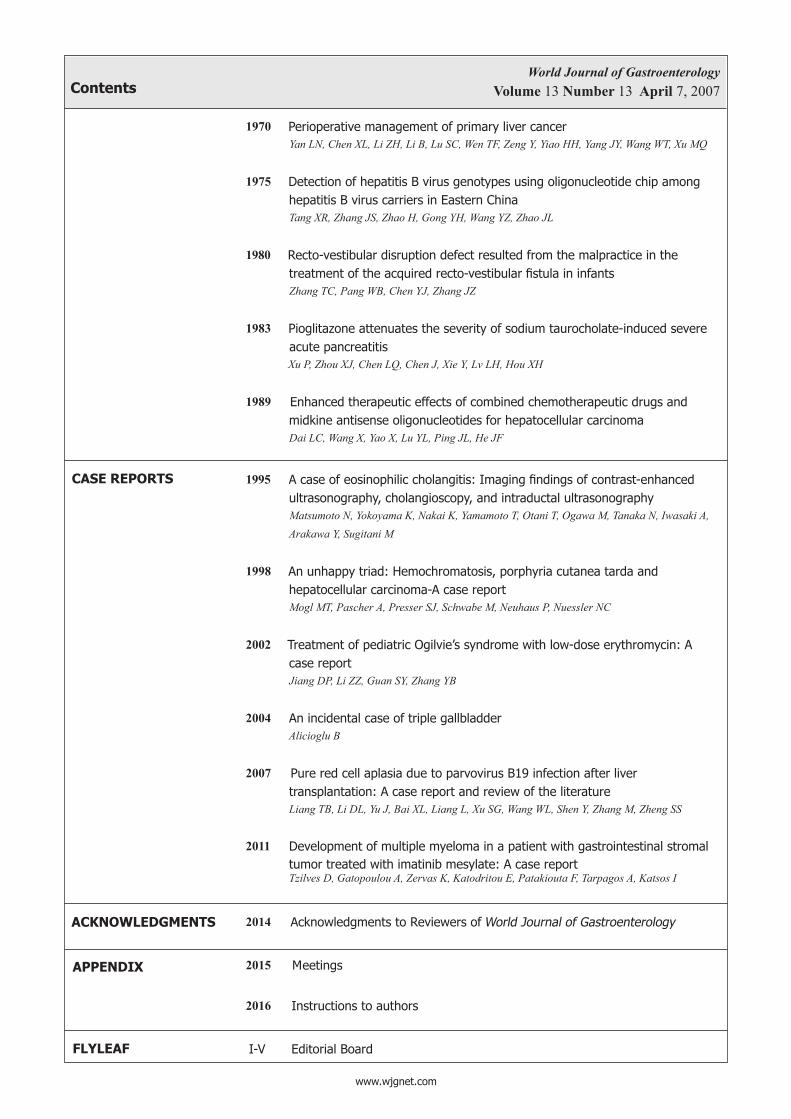
1906 Clinical characteristics of idiopathic portal hypertension Harmanci O, Bayraktar Y
1912 Hepatic venous outfl ow obstruction: Three similar syndromes Bayraktar UD, Seren S, Bayraktar Y
1928 Is portal vein cavernous transformation a component of congenital hepatic fi brosis?
Yonem O, Bayraktar Y
1930 Clinical characteristics of caroli’s disease Yonem O, Bayraktar Y
1934 Clinical characteristics of Caroli's syndrome Yonem O, Bayraktar Y
1938 Suppression of human colon tumor growth by adenoviral vector-mediated NK4 expression in an athymic mouse model
Jie JZ, Wang JW, Qu JG, Hung T
1947 Celecoxib attenuates 5-fl uorouracil-induced apoptosis in HCT-15 and HT-29 human colon cancer cells
Lim YJ, Rhee JC, Bae YM, Chun WJ
1953 Protective effect of curcumin against liver warm ischemia/reperfusion injury in rat model is associated with regulation of heat shock protein and antioxidant enzymes
Shen SQ, Zhang Y, Xiang JJ, Xiong CL
1962 Lactobacillus plantarum inhibits epithelial barrier dysfunction and interleukin-8 secretion induced by tumor necrosis factor-α
Ko JS, Yang HR, Chang JY, Seo JK
1966 Timing of mortality in severe acute pancreatitis: Experience from 643 patients
Fu CY, Yeh CN, Hsu JT, Jan YY, Hwang TL
RAPID COMMUNICATION
National Journal Award2005
REVIEW
The WJG Press
EDITORIAL
COLORECTAL CANCER
BASIC RESEARCH
TOPIC HIGHLIGHT
www.wjgnet.com
1970 Perioperative management of primary liver cancer Yan LN, Chen XL, Li ZH, Li B, Lu SC, Wen TF, Zeng Y, Yiao HH, Yang JY, Wang WT, Xu MQ
1975 Detection of hepatitis B virus genotypes using oligonucleotide chip among hepatitis B virus carriers in Eastern China
Tang XR, Zhang JS, Zhao H, Gong YH, Wang YZ, Zhao JL
1980 Recto-vestibular disruption defect resulted from the malpractice in the treatment of the acquired recto-vestibular fi stula in infants
Zhang TC, Pang WB, Chen YJ, Zhang JZ
1983 Pioglitazone attenuates the severity of sodium taurocholate-induced severe acute pancreatitis Xu P, Zhou XJ, Chen LQ, Chen J, Xie Y, Lv LH, Hou XH
1989 Enhanced therapeutic effects of combined chemotherapeutic drugs and midkine antisense oligonucleotides for hepatocellular carcinoma
Dai LC, Wang X, Yao X, Lu YL, Ping JL, He JF
1995 A case of eosinophilic cholangitis: Imaging fi ndings of contrast-enhanced ultrasonography, cholangioscopy, and intraductal ultrasonography
Matsumoto N, Yokoyama K, Nakai K, Yamamoto T, Otani T, Ogawa M, Tanaka N, Iwasaki A, Arakawa Y, Sugitani M
1998 An unhappy triad: Hemochromatosis, porphyria cutanea tarda and hepatocellular carcinoma-A case report
Mogl MT, Pascher A, Presser SJ, Schwabe M, Neuhaus P, Nuessler NC
2002 Treatment of pediatric Ogilvie’s syndrome with low-dose erythromycin: A case report
Jiang DP, Li ZZ, Guan SY, Zhang YB
2004 An incidental case of triple gallbladder Alicioglu B
2007 Pure red cell aplasia due to parvovirus B19 infection after liver transplantation: A case report and review of the literature
Liang TB, Li DL, Yu J, Bai XL, Liang L, Xu SG, Wang WL, Shen Y, Zhang M, Zheng SS
2011 Development of multiple myeloma in a patient with gastrointestinal stromal tumor treated with imatinib mesylate: A case report
Tzilves D, Gatopoulou A, Zervas K, Katodritou E, Patakiouta F, Tarpagos A, Katsos I
2014 Acknowledgments to Reviewers of World Journal of Gastroenterology
2015 Meetings
2016 Instructions to authors
I-V Editorial Board
CASE REPORTS
ContentsWorld Journal of Gastroenterology
Volume 13 Number 13 April 7, 2007
ACKNOWLEDGMENTS
APPENDIX
FLYLEAF

Online Submissions
International Subscription
www.wjgnet.com
ContentsWorld Journal of Gastroenterology
Volume 13 Number 13 April 7, 2007
HONORARY EDITORS-IN-CHIEFKe-Ji Chen, BeijingLi-Fang Chou, TaipeiZhi-Qiang Huang, BeijingShinn-Jang Hwang, TaipeiMin-Liang Kuo, TaipeiNicholas F LaRusso, RochesterJie-Shou Li, NanjingGeng-Tao Liu, BeijingLein-Ray Mo, TainanFa-Zu Qiu, WuhanEamonn M Quigley, CorkDavid S Rampton, LondonRudi Schmid, Kentfi eldNicholas J Talley, RochesterGuido NJ Tytgat, AmsterdamH-P Wang, TaipeiJaw-Ching Wu, TaipeiMeng-Chao Wu, ShanghaiMing-Shiang Wu, TaipeiJia-Yu Xu, ShanghaiTa-Sen Yeh, Taoyuan PRESIDENT AND EDITOR-IN-CHIEFLian-Sheng Ma, Beijing EDITOR-IN-CHIEFBo-Rong Pan, Xi'an ASSOCIATE EDITORS-IN-CHIEFGianfranco D Alpini, TempleBruno Annibale, RomaRoger William Chapman, OxfordChi-Hin Cho, Hong KongAlexander L Gerbes, MunichShou-Dong Lee, TaipeiWalter Edwin Longo, New HavenYou-Yong Lu, BeijingMasao Omata, TokyoHarry HX Xia, Hanover
SCIENCE EDITORSDirector: Jing Wang, BeijingDeputy Director: Jian-Zhong Zhang, Beijing
MEMBERSYe Liu, BeijingXing-Xia Yang, Beijing
LANGUAGE EDITORSDirector: Jing-Yun Ma, BeijingDeputy Director: Xian-Lin Wang, Beijing
MEMBERSGianfranco D Alpini, TempleBS Anand, HoustonRichard B Banati, LidcombeGiuseppe Chiarioni, ValeggioJohn Frank Di Mari, TexasShannon S Glaser, Temple Mario Guslandi, MilanoMartin Hennenberg, BonnAtif Iqbal, OmahaManoj Kumar, NepalPatricia F Lalor, BirminghamMing Li, New OrleansMargaret Lutze, ChicagoJing-Yun Ma, BeijingDaniel Markovich, BrisbaneSabine Mihm, GöttingenFrancesco Negro, GenèveBernardino Rampone, SienaRichard A Rippe, Chapel HillStephen E Roberts, Swansea Ross C Smith, SydneySeng-Lai Tan, SeattleXian-Lin Wang, BeijingEddie Wisse, KeerbergenDaniel Lindsay Worthley, BedfordLi-Hong Zhu, Beijing
COPY EDITORSGianfranco D Alpini, Temple
Sujit Kumar Bhattacharya, KolkataFilip Braet, SydneyKirsteen N Browning, Baton RougeRadha K Dhiman, ChandigarhJohn Frank Di Mari, TexasShannon S Glaser, TempleMartin Hennenberg, BonnEberhard Hildt, BerlinPatricia F Lalor, BirminghamMing Li, New OrleansMargaret Lutze, ChicagoMI Torrs, JaénSri Prakash Misra, AllahabadGiovanni Monteleone, RomeGiovanni Musso, TorinoValerio Nobili, RomeOsman Cavit Ozdogan, IstanbulFrancesco Perri, San Giovanni RotondoThierry Piche, NiceBernardino Rampone, SienaRichard A Rippe, Chapel HillRoss C Smith, SydneyDaniel Lindsay Worthley, BedfordGeorge Y Wu, FarmingtonJian Wu, Sacramento
EDITORIAL ASSISTANTYan Jiang, Beijing
PUBLISHED BYThe WJG Press
PRINTED BYPrinted in Beijing on acid-free paper by Beijing Kexin Printing House
COPYRIGHT© 2007 Published by The WJG Press. All r ights reserved; no part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic,
mechanical, photocopying, recording, or otherwise without the prior permission of The WJG Press. Authors are required to grant WJG an exclusive licence to publish. Print ISSN 1007-9327 CN 14-1219/R
SPECIAL STATEMENT All articles published in this journal represent the viewpoints of the authors except where indicated otherwise.
EDITORIAL OFFICEWorld Journal of Gastroenterology, The WJG Press, Apartment 1066 Yishou Garden, 58 North Langxinzhuang Road, PO Box 2345, Beijing 100023, ChinaTelephone: +86-10-85381892Fax: +86-10-85381893E-mail: [email protected]://www.wjgnet.com
SUBSCRIPTION ANDAUTHOR REPRINTSJing Wang The WJG Press, Apartment 1066 Yishou Garden, 58 North Langxinzhuang Road,PO Box 2345, Beijing 100023, ChinaTelephone: +86-10-85381892Fax: +86-10-85381893E-mail: [email protected] http://www.wjgnet.com
SUBSCRIPTION INFORMATIONInstitutional Price 2007: USD 1500.00Personal Price 2007: USD 700.00
INSTRUCTIONS TO AUTHORSFull instructions are available online at http://www.wjgnet.com/wjg/help/instructions.jsp. If you do not have web access please contact the editorial offi ce.
World Journal of Gastroenterology ( World J Gastroenterol , WJG ), a leading international journal in gastroenterology and hepatology, has an established reputation for publishing fi rst class research on esophageal cancer, gastric cancer, liver cancer, viral hepatitis, colorectal cancer, and H pylori infection, providing a forum for both clinicians and scientists, and has been indexed and abstracted in Current Contents®/Clinical Medicine, Science Citation Index Expanded (also known as SciSearch®) and Journal Citation Reports/Science Edition, Index Medicus, MEDLINE and PubMed, Chemical Abstracts, EMBASE/Excerpta Medica, Abstracts Journals, Nature Clinical Practice Gastroenterology and Hepatology, CAB Abstracts and Global Health. ISI JCR 2003-2000 IF: 3.318, 2.532, 1.445 and 0.993. WJG is a weekly journal published by The WJG Press. The publication date is on 7th, 14th, 21st, and 28th every month. The WJG is supported by The National Natural Science Foundation of China, No. 30224801 and No.30424812, which was founded with a name of China National Journal of New Gastroenterology on October 1,1995, and renamed as WJG on January 25, 1998.
INSIDE BACK COVER
Responsible E-Editor for this issue: Wen-Hua Ma
COPY EDITOR FOR THIS ISSUE: Thierry Piche, MD, PhD
Responsible S-Editor for this issue: Jing Wang
INSIDE FRONT COVER

PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1893-1896www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
Innovative therapeutics for inflammatory bowel disease
Jesus K Yamamoto-Furusho
www.wjgnet.com
EDITORIAL
Jesus K Yamamoto-Furusho, Inflammatory Bowel Disease Clinic, Department of Gastroenterology, Instituto Nacional de Ciencias Médicas y Nutrición. Vasco de Quiroga 15, Colonia Sección XVI, Tlalpan, México CP 14000, DF MéxicoCorrespondence to: Jesus K Yamamoto-Furusho, MD, PhD, MSc, Head of Inflammatory Bowel Disease Clinic, Department of Gastroenterology, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán. Vasco de Quiroga 15, Col. Sección XVI, Tlalpan, México CP 14000, DF Mexico. [email protected]: +52-55-5533418 Fax: +52-55-56550942Received: 2007-02-27 Accepted: 2007-03-21
AbstractInf lammatory bowel diseases (IBD) are chronic inflammatory conditions of the gastrointestinal tract, which clinically present as one of two disorders, Crohn’s disease or ulcerative colitis. Mainstays of drug treatments for IBD include aminosalicylates, corticosteroids and immunosuppressants such as azathioprine, methotrexate and cyclosporin. Advances in basic research of the pathophysiological process in IBD have been applied to generate a variety of new therapeutics targeting at different levels of the inflammatory processes. New therapies are classified as: (1) Anti-TNFα antibodies; (2) Recombinant cytokines; (3) Selective adhesion blockade; (4) Growth factors; (5) Innate immunostimulation; (6) Nucleic acid based therapies; (7) Gene therapy; (8) Autologous bone-marrow transplantation; (9) Helminths and (10) Extracorporeal immunomodulat ion. Al l treatments have the potential to provide more effective and safe treatment for IBD.
© 2007 The WJG Press. All rights reserved.
Key words: Novel agents; Inflammatory bowel diseases; Biologic therapy; Future agents
Yamamoto-Furusho JK. Innovative therapeutics for inflammatory bowel disease. World J Gastroenterol 2007; 13(13): 1893-1896
http://www.wjgnet.com/1007-9327/13/1893.asp
INTRODUCTIONInflammatory bowel diseases (IBD) including ulcerative colitis (UC) and Crohn’s disease (CD) are chronic inflammatory conditions affecting the gastrointestinal tract.
The etiopathogenesis has not been fully elucidated but is currently presumed to result from a complex interplay among genetic, environmental, microbial and immune factors[1].
The current state of therapy for patients with IBD is not very satisfactory. In patients with active CD, an adequate course of corticosteroids induces remissions in only 60% to 70% and in this group with response, many patients relapse during tapering of the steroids dose[2]. About 33% to 50% of patients with fulminant ulcerative colitis who are admitted to the hospital have their colon removed during the same admission because of failure of conventional medical therapy[3].
Patients with IBD frequently experience relapse and traditional medical treatments are not potent enough to keep in remission for long-term periods.
Biological approach to IBD therapy has developed in recent years as a result of understanding of the mucosal immunology processes in intestinal inflammation. The first effective biological therapy approved for commercial use is infliximab, a chimeric monoclonal antibody directed against tumour necrosis factor α (TNFα) , for the treatment of CD in 1998 and UC in 2006 by the FDA. Infliximab is indicated for induction and maintenance of remission in patients with active luminal inflammatory CD refractory to conventional therapy, those with draining enterocutaneous and perianal fistulas and recently, for patients with moderate to severe active UC despite treatment with concurrent medications. However, a meta-analysis of 34 studies (896 patients with UC) showed that long-term remission (9 mo) was 39%[4].
Conventional treatment including infliximab can provide clinical benefit, reduce signs and symptoms of disease, and improve quality of life, however, most do not significantly alter the long-term course of the disease or the underlying immunopathology.
New therapies, which are targeted at specific disease mechanisms, have the potential to provide more effective and safe treatments for human diseases as shown in Figure 1.
THERAPEUTIC APPROACHESThere are several categories of treatments that are relevant to IBD, including (1) New anti-TNFα antibodies; (2) Recombinant cytokines; (3) Selective adhesion blockade; (4) Growth factors; (5) Immunostimulation; (6) Nucleic acid based therapies; (7) Gene therapy; (8) Autologous bone-marrow transplantation; (9) Helminths, (10) Extracorporeal immunomodulation. A number of new anti-TNFα agents are currently being investigated in phase Ⅲ studies in CD,

including a pegylated humanized anti-TNFα fragment, CDP870 (certolizumab) and a fully human anti-TNFα antibody (Adalimumab) that binds with a high affinity and specificity to human soluble TNFα.
An initial randomized controlled trial (RCT) of certolizumab in active CD demonstrated a significant benefit in achieving remission, through not response[5]. Another trial demonstrated a benefit in a subgroup of CD patients with elevated CRP (10 mg per litre) and found a statistically significant response in the highest dose tested[6].
Recently, CHARM and CLASSIC Ⅱ trials demon-strated the efficacy and safety of adalimumab in the induction and maintenance of clinical remission in moderate to severe CD for up to 56 wk[7,8]. An important aspect of anti-TNFα agents is the development of anti-idiotype antibodies directed towards the murine portion of the chimeric antibody resulting in hypersensitivity and decreased responsiveness. On the other hand, adalimumab i s a human ized monoc lona l ag ent to wh ich the sensitization and development of anti-idiotype antibodies are much less than infliximab.
A second approach has been to block steps in T-cell development by targeting cytokines involved in T-cell differentiation and activation such as IL-12. A recent RCT of 79 patients with CD receiving 1 mg or 3 mg of anti-IL-12 monoclonal antibody versus placebo demonstrated a response in 75% of CD patients compared with 25% in the placebo group[9]. Other antibodies have been generated against T-cell subsets including CD3+ cells (visilizumab) and CD25+ cells (daclizumab and basiliximab) for UC. Pilot studies have shown promising results in steroid-resistant UC patients[10].
IL-6 participates in a variety of critical functions, including T cell growth and differentiation, as well as B-cell proliferation. A pilot study showed in patients with active CD treated with an antibody directed against the IL-6 receptor (Atlizumab), 80% responded at the full dose compared with 31% in the placebo group[11].
RDP58 is an oral decapeptide that decreases the activity of TNFα, IL-2, IL-12 and IFN-γ. Phase Ⅱ studies showed high remission rates and minimal toxicity in UC patients[12]. Most of the strategies have targeted to interrupt cytokine activity through inhibition of pathways
promoting cytokine production, or by directly blocking cytokine action.
A different approach aims to block leukocyte migration to sites of inflammation by interfering with cell adhesion molecules. There are three targeting therapies in clinical trials to date including natalizumab (integrin α4 subunit) and MLN-02 (integrin α4β7). An RCT in 248 patients with moderate to severe CD randomly assigned to receive natalizumab or placebo, demonstrated high rates of clinical remission and response, and improved quality of life and C-reactive protein levels[13]. Studies of MLN-02 have demonstrated equivocal results in CD[14] but promising effects in UC, with responses and remission rates of 66% and 33% compared with 33% and 15%, respectively, in those receiving placebo[15].
Augmenting intestinal repair could be another approach for patients with IBD. Topical administration of epidermal growth factor (EGF) seemed to be effective in promoting both initial response and long-term maintenance in 20 patients with left-sided UC[16].
Growth hormone and a high-protein diet resulted in a significant decrease in Crohn´s disease activity index (CDAI) in the active treatment group as compared with placebo group[17]. This effect is attributed to growth hormone because it might enhance the intestinal barrier function, including decreasing intestinal permeability and increasing intestinal protein turnover as well as has stimulatory effects on neutrophil function.
Recent evidence has shown the importance of innate immune mechanisms in sustaining homeostasis in the gastrointestinal mucosa and some defects in these intestinal innate immune responses in patients with CD[18]. Some reports suggest that agents that act to enhance innate immune defenses can indeed confer therapeutic benefit. Two agents (G-CSF or filgastrim and GM-CSF sargramostin) have been evaluated in patients with CD. The endogenous growth factor granulocyte-macrophage colony-stimulating factor (GM-CSF) performs important funct ions in both the phag ocyt ic and ep i the l i a l components of intestinal early innate immune defense. GM-CSF is expressed by both CD4+ T cells and Paneth cells in the intestine, and its receptors have been recently demonstrated to be present on intestinal epithelial cells which proliferate in response to GM-CSF in vitro[19,20]. Both pilot studies suggested a benefit[21,22], though GM-CSF appeared more effective. A recent RCT of 124 patients found a significant benefit in response at 100-point decrease in CDAI and in remission. The response was sustained for a mean of 8-10 wk after discontinuation of therapy[23].
Nucleic acid based therapies have focused on the use of antisense phosphorothioate oligonucleotide to the p65 subunit of NF-κB and ICAM-1 antisense oligonucleotide (Alicaforsen, ISIS-2302). In a pilot study 11 steroid-refractory or resistant IBD patients were given a single dose of rectal antisense NF-κB p65 oligonucleotide. An improvement in clinical, endoscopic and histologic scores was seen at d 7 in 71% of the treatment group compared with 25% of the placebo group[24].
ISIS-2302 is a 20-base pair complementary nucleotide
Immune stimulation
Growth factors
Probiotics
Inflammatorycascade
Abnormal T cell function
Inhibiting celladhesion
Sargramostim
MLN02
α4β7
Alicaforsen
ICAM1 IL-12 IL-6TNFα
InfliximabAdalimumabCertolizumab
Anti-IL-12 Atlizumab
α4 Natalizumab
IL-2
Daclizumab
Th1Visilizumab
Th2
IFN IL 10
Fontolizumab
Figure 1 Current and potential therapeutic agents target at different pathways in inflammatory bowel disease.
1894 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

chain which hybridizes with ICAM-1 mRNA and is thus degraded by RNAse-H and the message and expression of ICAM-1 is therefore decreased. In patients treated with doses between 300-350 mg infused 3 times per week for 4 wk, it seemed to have higher benefit in those with active CD[25].
Gene therapy strategies using plasmid IL-10 vectors or an adenovirus IL-10 construct seem to be a potent approach for the treatment of IBD. Barbara et al [26] reported that gene transfer was achieved by intraperitoneal injection of non-replicating human adenovirus bearing IL-10 gene, either 24 h before or 1 h after intrarectal administration of trinitrobenzenesulfonic acid (TNBS) in rats. IL-10 gene transfer prior to colitis improved colitis macroscopically and histologically.
Allogenic bone-marrow transplants in CD patients were noted to induce prolonged disease remission, providing evidence of the role of bone-marrow T cells in this disease.
The goal of autologous haematopoietic stem-cell transplantation (HSCT) is resetting T cell responses by eliminating all circulating T cells. A phase I study in 12 patients with refractory CD showed that 11 patients entered a sustained remission (CDAI < 150). After a median follow-up of 18.5 mo, only one patient has developed a recurrence of active CD, which occurred 15 mo after HSCT[27].
Another strategy for resetting the T-cell repertoire has been proposed based on the helminths. The immune system has evolved with the presence of these helminthes, which function to expand the T regulatory cell population, enhancing IL-10 production and shifting a TH1 type process more towards TH2[28]. An RCT of using 2500 Trichiuris suis ova, a pig worm, administered orally once every two weeks, in UC has shown a response of 44% of the treatment group versus 14% of receiving placebo[29]. An open label study of 29 patients with active CD identified high rates of response (79.3%) and remission (72.4%)[30].
Early reports on the benefit of lymphocyte apheresis to treat CD described selective extracorporeal T cell depletion by ultracentrifugation in patients with active CD, the majority of whom went into long-lasting remission and decreased steroid use to zero. Two strategies that decrease specific lymphocyte subpopulations were developed in Japan. One technique is a leukocyte removal filter column-CellsorbaTM. All leukocyte subpopulations, 99% of granulocytes and macrophages and 40%-60% of lymphocytes as well as platelets, are trapped within the filter made of polyester fibers. In patients with CD an intensive induction protocol-leukocytopheresis every week for 5 wk induced remission in 50% of the patients. An alternative leukocytopheresis method, Adacolumn, uses a column adsorbing granulocytes and monocytes/macrophages to 2 mm in diameter of cellulose acetate beads, without significantly adsorbing lymphocytes. Several trials demonstrated a positive effect of the column in active UC[31,32].
Immunostimulatory DNA sequences, ISS-DNA, also known as CpG DNA, are unmethylated CpG dinucleotides within consensus sequences present in bacterial and viral genomes. ISS-DNA and their synthetic analogues activate
innate immunity via Toll-like receptor 9. Liposomal immunostimulatory DNA sequence (ISS-ODN) was shown to prevent and ameliorate the severity of colitis in animal models[33] and therefore may be effective also in the treatment of human IBD. Clinical trials to test their efficacies are underway.
CONCLUSIONCurrently, numerous innovative agents for IBD have been available for the treatment of patients with IBD refractory to conventional therapy including infliximab. In the near future, the enticing concept of combining different therapeutic approaches targets at different components of the inflammatory process. Intensive basic research continues in order to generate new targets or novel approaches that intervene at other steps in the pathophysiological processes.
REFERENCES1 Podolsky DK. Inflammatory bowel disease. N Engl J Med
2002; 347: 417-4292 Munkholm P, Langholz E, Davidsen M, Binder V. Frequency
of glucocorticoid resistance and dependency in Crohn's disease. Gut 1994; 35: 360-362
3 Travis SP, Farrant JM, Ricketts C, Nolan DJ, Mortensen NM, Kettlewell MG, Jewell DP. Predicting outcome in severe ulcerative colitis. Gut 1996; 38: 905-910
4 Gisbert JP, González-Lama Y, Maté J. Systematic review: Infliximab therapy in ulcerative colitis. Aliment Pharmacol Ther 2007; 25: 19-37
5 Winter TA, Wright J, Ghosh S, Jahnsen J, Innes A, Round P. Intravenous CDP870, a PEGylated Fab' fragment of a humanized antitumour necrosis factor antibody, in patients with moderate-to-severe Crohn's disease: an exploratory study. Aliment Pharmacol Ther 2004; 20: 1337-1346
6 Schreiber S, Rutgeerts P, Fedorak RN, Khaliq-Kareemi M, Kamm MA, Boivin M, Bernstein CN, Staun M, Thomsen OØ, Innes A. A randomized, placebo-controlled trial of certolizumab pegol (CDP870) for treatment of Crohn's disease. Gastroenterology 2005; 129: 807-818
7 Colombel JF, Sandborn WJ, Rutgeerts P, Enns R, Hanauer SB, Panaccione R, Schreiber S, Byczkowski D, Li J, Kent JD, Pollack PF. Adalimumab for maintenance of clinical response and remission in patients with Crohn's disease: the CHARM trial. Gastroenterology 2007; 132: 52-65
8 Sandborn WJ, Hanauer SB, Rutgeerts P, Fedorak RN, Lukas M, MacIntosh DG, Panaccione R, Wolf D, Kent JD, Bittle B, Li J, Pollack PF. Adalimumab for maintenance treatment of Crohn's disease: results of the CLASSIC II trial. Gut 2007; 56:1232-1239
9 Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, Dolin B, Goodman N, Groden C, Hornung RL, Quezado M, Yang Z, Neurath MF, Salfeld J, Veldman GM, Schwertschlag U, Strober W. Anti-interleukin-12 antibody for active Crohn's disease. N Engl J Med 2004; 351: 2069-2079
10 Creed TJ, Probert CS, Norman MN, Moorghen M, Shepherd NA, Hearing SD, Dayan CM. Basiliximab for the treatment of steroid-resistant ulcerative colitis: further experience in moderate and severe disease. Aliment Pharmacol Ther 2006; 23: 1435-1442
11 Ito H, Takazoe M, Fukuda Y, Hibi T, Kusugami K, Andoh A, Matsumoto T, Yamamura T, Azuma J, Nishimoto N, Yoshizaki K, Shimoyama T, Kishimoto T. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn's disease. Gastroenterology 2004; 126: 989-996; discussion 947
12 Travis S, Yap LM, Hawkey C, Warren B, Lazarov M, Fong T,
Yamamoto-Furusho JK. Novel agents in IBD 1895
www.wjgnet.com

Tesi RJ. RDP58 is a novel and potentially effective oral therapy for ulcerative colitis. Inflamm Bowel Dis 2005; 11: 713-719
13 Ghosh S, Goldin E, Gordon FH, Malchow HA, Rask-Madsen J, Rutgeerts P, Vyhnálek P, Zádorová Z, Palmer T, Donoghue S. Natalizumab for active Crohn's disease. N Engl J Med 2003; 348: 24-32
14 Feagan B, Greenberg G, Wild G. Efficacy and safety of a humanized alpha4beta7 antibody in active Crohn’s disease. Gastroenterology 2003; 124: A25
15 Feagan BG, Greenberg GR, Wild G, Fedorak RN, Paré P, McDonald JW, Dubé R, Cohen A, Steinhart AH, Landau S, Aguzzi RA, Fox IH, Vandervoort MK. Treatment of ulcerative colitis with a humanized antibody to the alpha4beta7 integrin. N Engl J Med 2005; 352: 2499-2507
16 Sinha A, Nightingale J, West KP, Berlanga-Acosta J, Playford RJ. Epidermal growth factor enemas with oral mesalamine for mild-to-moderate left-sided ulcerative colitis or proctitis. N Engl J Med 2003; 349: 350-357
17 Slonim AE, Bulone L, Damore MB, Goldberg T, Wingertzahn MA, McKinley MJ. A preliminary study of growth hormone therapy for Crohn's disease. N Engl J Med 2000; 342: 1633-1637
18 Yamamoto-Furusho JK, Korzenik JR. Crohn's disease: innate immunodeficiency? World J Gastroenterol 2006; 12: 6751-6755
19 Fukuzawa H, Sawada M, Kayahara T, Morita-Fujisawa Y, Suzuki K, Seno H, Takaishi S, Chiba T. Identification of GM-CSF in Paneth cells using single-cell RT-PCR. Biochem Biophys Res Commun 2003; 312: 897-902
20 Ramsay RG, Micallef SJ, Williams B, Lightowler S, Vincan E, Heath JK, Mantamadiotis T, Bertoncello I. Colony-stimulating factor-1 promotes clonogenic growth of normal murine colonic crypt epithelial cells in vitro. J Interferon Cytokine Res 2004; 24: 416-427
21 Dieckgraefe BK, Korzenik JR. Treatment of active Crohn's disease with recombinant human granulocyte-macrophage colony-stimulating factor. Lancet 2002; 360: 1478-1480
22 Korzenik JR, Dieckgraefe BK. An open-labelled study of granulocyte colony-stimulating factor in the treatment of active Crohn's disease. Aliment Pharmacol Ther 2005; 21:
391-40023 Korzenik JR, Dieckgraefe BK, Valentine JF, Hausman DF,
Gilbert MJ. Sargramostim for active Crohn's disease. N Engl J Med 2005; 352: 2193-2201
24 Loftberg R, Neurath M, Ost A, Petterson S. Topical NF-κB p65 antisense oligonucleotides in patients with active distal colonic IBD. A randomized, controlled pilot trial. Gastroenterology 2002; 122: A60
25 Barish CF. Alicaforsen therapy in inflammatory bowel disease. Expert Opin Biol Ther 2005; 5: 1387-1391
26 Barbara G, Xing Z, Hogaboam CM, Gauldie J, Collins SM. Interleukin 10 gene transfer prevents experimental colitis in rats. Gut 2000; 46: 344-349
27 Oyama Y, Craig RM, Traynor AE, Quigley K, Statkute L, Halverson A, Brush M, Verda L, Kowalska B, Krosnjar N, Kletzel M, Whitington PF, Burt RK. Autologous hematopoietic stem cell transplantation in patients with refractory Crohn's disease. Gastroenterology 2005; 128: 552-563
28 Doetze A, Satoguina J, Burchard G, Rau T, Löliger C, Fleischer B, Hoerauf A. Antigen-specific cellular hyporesponsiveness in a chronic human helminth infection is mediated by T(h)3/T(r)1-type cytokines IL-10 and transforming growth factor-beta but not by a T(h)1 to T(h)2 shift. Int Immunol 2000; 12: 623-630
29 Summers RW, Elliott DE, Urban JF, Thompson RA, Weinstock JV. Trichuris suis therapy for active ulcerative colitis: a randomized controlled trial. Gastroenterology 2005; 128: 825-832
30 Summers RW, Elliott DE, Urban JF, Thompson R, Weinstock JV. Trichuris suis therapy in Crohn's disease. Gut 2005; 54: 87-90
31 Kohgo Y, Ashida T, Maemoto A, Ayabe T. Leukocytapheresis for treatment of IBD. J Gastroenterol 2003; 38 Suppl 15: 51-54
32 Hanai H. Positions of selective leukocytapheresis in the medical therapy of ulcerative colitis. World J Gastroenterol 2006; 12: 7568-7577
33 Rachmilewitz D, Katakura K, Karmeli F, Hayashi T, Reinus C, Rudensky B, Akira S, Takeda K, Lee J, Takabayashi K, Raz E. Toll-like receptor 9 signaling mediates the anti-inflammatory effects of probiotics in murine experimental colitis. Gastroenterology 2004; 126: 520-528
S- Editor Zhu LH L- Editor Zhu LH E- Editor Zhou T
1896 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1897-1905www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
Treatment of hepatitis C virus infection
Kilian Weigand, Wolfgang Stremmel, Jens Encke
www.wjgnet.com
REVIEW
Kilian Weigand, Wolfgang Stremmel, Jens Encke, University of Heidelberg, Department of Gastroenterology and Hepatology, Im Neuenheimer Feld 410, Heidelberg 69120, Germany Correspondence to: Professor J Encke, University of Heidelberg, Department of Gastroenterology, Im Neuenheimer Feld 410, Hei-delberg D-69120, Germany. [email protected]: +49-6221-5638825 Fax: +49-6221-566858Received: 2007-01-11 Accepted: 2007-03-14
AbstractAcute and chronic hepatitis C virus (HCV) infection remains a serious health problem worldwide, however, there has been advancement in the treatment of HCV infection due to standard treatment using pegylated interferon and ribavirin. The literature indicates that therapy for HCV is becoming more individualized. In addition to considering genotype and viral RNA levels before treatment, achievement of an early virologic response (EVR) and a rapid virologic response (RVR) is now possible during therapy. Moreover, problem patients, such as non-responders, relapsers, HIV or HBV co-infected patients, patients with liver cirrhosis, and pre- or post-liver transplantation patients are an increasing fraction of the patients requiring treatment. This article reviews the literature regarding standard treatments and problem patients with acute and chronic HCV infection. It also includes discussion on contraindications and side effects of treatment with interferon and ribavirin, as well as new drug development.
© 2007 The WJG Press. All rights reserved.
Key words: Hepatitis C virus; Acute and chronic HCV infection; Treatment; Pegylated interferon; Ribavirin; Sustained virologic response; Non-responders; Relapsers
Weigand K, Stremmel W, Encke J. Treatment of hepatitis C virus infection. World J Gastroenterol 2007; 13(13): 1897-1905
http://www.wjgnet.com/1007-9327/13/1897.asp
INTRODUCTIONInfection with hepatitis C virus (HCV) remains a severe life-threatening medical and public health problem worldwide. Every year there are an estimated 3 to 4 million new cases of infection due to transfusion contamination, contaminated injection needles, and parenteral exposure[1-5].
There is a lower rate of infection for sexual transmission[6]. About 55%-85% of individuals with acute HCV infection become chronically infected and are at risk for developing hepatocellular injury, liver cirrhosis, hepatocellular carcinoma or liver failure[7-9].
In total, More than 170 million individuals (> 2% of the world’s population) are infected with HCV[10]. While prevention of primary infection is possible, vertical transmission of HCV remains a significant problem especially in developing countries.
The transition from acute to chronic infection is only partly understood. However, early treatment with pegylated interferon (PEG-IFN) alpha to prevent chronic infection is effective in up to 95% of patients with acute hepatitis[11-13]. Determining the optimal treatment for chronically infected individuals is a remaining question. To date, standard treatment for chronically infected patients is the combination of PEG-IFN alpha with ribavirin[14]. Recent studies have demonstrated that a relatively high number of patients acquire sustained virologic response (SVR), defined as non-detectable serum virus RNA levels by qualitative PCR 6 mo after end of treatment[15,16], and this is the primary goal of therapy. However, a large number of patients remain viraemic and chronically infected. In addition, many patients suffer from severe side effects while receiving this combination therapy[14-17]. These are the reasons for attempts to find medications with higher SVRs, better tolerability and shorter treatment regimens[18-21]. Moreover, alternative therapeutic regimens, such as an effective therapeutic or prophylactic vaccine for HCV infection, are being sought after and developed[22-26].
TREATMENT OF ACUTE HEPATITIS CDiagnosis of acute HCV infection is a rare event since acutely infected individuals are mostly asymptomatic[27,28]. Also, social problems within high risk groups (especially injection drug users) keep these individuals from seeing physicians.
An optimal treatment for acute HCV infections has not been established. There are several studies showing excellent responses using IFNa[11]. The best results, with a SVR in over 95% of the patients, were achieved by using 5 million international units (MIU) of IFN daily for 4 wk, followed by 5 MIU three times weekly for another 20 wk. This treatment was well tolerated in most cases. Another recent study achieved a SVR in 87% of patients, using 6 MIU of IFN injected intramuscularly daily for 4 wk[29]. In acute HCV, genotype and RNA serum levels seem to have no influence on treatment outcomes[11,30]. While undergoing treatment, patients need to be monitored at

least every four weeks for transaminases, HCV antibodies and serum RNA levels.
Since spontaneous viral clearance is documented in up to 50% of acutely infected individuals[13,30-32], some authors believe treatment should be initiated after three to four months of observation. The data to date show a worse outcome following this policy[11,13], but patients avoid the potential severe side effects of IFN therapy[33]. Also, this scheme gives an opportunity for patients with contraindications to IFN therapy; i.e. pregnancy, acute alcohol or i.v. drug abuse, and psychiatric diseases such as severe depression, to resolve these problems before starting therapy[14,17,34]. Nevertheless, delaying therapy for acute HCV infection beyond three months after onset of the disease cannot be recommended since one study showed this resulted in a dramatic drop of SVR rates (from 87% to 53%)[29]. Even if IFN monotherapy is sufficient for the therapy of acute HCV infection[35], preliminary data suggest PEG-IFN to be as effective as the IFN regime used in the reported German trial[35]. Recently, Wiegand et al[12] published a trial using PEG-IFN alpha-2b 1.5 μg per kg body weight with patients that had acute HCV infection. In patients adherent to therapy, a SVR of 94% after therapy and 89% after a follow up of 24 wk was achieved. In non-adherent patients (less than 80% of PEG-IFN application in 80% of scheduled treatment duration), the rates dropped to 82% and 71%, respectively. Furthermore, Kamal et al[36] presented a study demonstrating a combination therapy of PEG-IFN and ribavirin to be more effective than PEG-IFN monotherapy (increase of SVR from 80% with monotherapy to 85% with combination therapy). In addition, it was shown that the time point to start treatment after onset of disease is very important. A recent trial demonstrated that overall SVR dropped from 95% to 92% and then to 76% when treatment was started 8, 12 or 20 wk after onset of disease[37], respectively. Considering these data, we suggest treating acute HCV infection for 24 wk, starting immediately or at three months after onset of the disease, using a combination of PEG-INF and ribavirin in a dosage recommended for treatment of chronic HCV infection (see below).
TREATMENT OF CHRONIC HEPATITIS C The primary treatment goal for chronic HCV infection is, as mentioned previously, sustained virologic response (SVR)[38,39]. With the recommended treatment, SVR can be achieved in about 55% of patients who are chronically infected with genotype 1 of HCV, while with genotype 2 and 3 the efficacy is 80% or greater[15,16,40]. The standard therapy is PEG-IFN alpha-2a or PEG-IFN alpha-2b subcutaneously in combination with twice daily oral doses of ribavirin[15,16,41]. The combination has proven to be more efficient than monotherapy alone, even though the antiviral mechanism of ribavirin is not fully understood[42]. Ribavirin monotherapy has no therapeutic effect in HCV infected patients[43,44].
There are two widely accepted regimens that can be followed, with both showing comparable SVR rates. Published in 2001 by Manns et al[15], PEG-IFN alpha-2b in a dose of 1.5 μg per kg body weight once a week
subcutaneously plus oral ribavirin 800 mg daily led to a virus clearance of 54%. Higher ribavirin doses resulted in a lower efficacy. While genotype 1 must be treated for 48 wk to achieve the best results, treatment longer than 24 wk for genotype 2 and 3 did not raise SVR rates beyond 82%. A trial by Fried et al[16] using PEG-IFN alpha-2a demonstrated comparable results. Using PEG-IFN alpha-2a in a dose of 180 μg once a week subcutaneously plus 1000-1200 mg (depending on body weight, cut-off 75 kg) of oral ribavirin daily resulted in 56% of SVR in genotype 1 carriers, and 80% in genotype 2 and 3 carriers. Patients with genotype 1 were treated for 48 wk. In patients infected with genotype 2 and 3, a treatment period of 24 wk seemed to be sufficient. Recently, two studies have shown that treatment can be abbreviated in patients with low baseline levels of HCV-RNA (< 600 000 IU/mL) who become HCV-RNA negative. Treatment of genotype 2 and 3 for only 12 or 16 wk may be sufficient for a special population[45,46]. Therefore, therapy for HCV infection is becoming an individualized therapy.
In summary, for patients who are chronically infected with HCV, the recommendation is to use PEG-IFN alpha-2b 1.5 μg/kg per week or PEG-IFN alpha-2a 180 μg/wk plus ribavirin 1000-1200 mg/d (body weight dependent) for 48 wk for patients with genotype 1 or 24 wk for patients with genotype 2 or 3. For genotypes 4, 5 and 6, the data are not sufficient for the development of a guideline, but it has been suggested to treat patients with these genotypes in a similar way as for patients with genotype 1[47,48]. Recently, Hadziyannis et al[40] presented a study on 36 genotype 4 carriers. While patients in the short-term group (24 wk) had a SVR of 63%-67%, treatment for 48 wk resulted in a SVR of 82.
Induction therapy does not result in higher SVR rates, therefore a recommendation regarding induction therapy cannot be given[49,50], but there are ongoing individual trials. While it is recommended to treat patients with relapse of HCV or non-responders after IFN alpha monotherapy with the described combination of PEG-IFN and ribavirin[51-54], the treatment indication for non-responders or patients with relapse after treatment with PEG-IFN alpha and ribavirin is controversial. Recent studies do not offer a general recommendation. Controlled trials to answer this question are ongoing.
During therapy, monthly monitoring of side effects, blood count, transaminases, creatinine, urea and glucose should be made. For the first 2 mo of therapy, blood counts should be made every 2 wk. Thyroid function should be considered in 12 wk intervals by measuring thyroid stimulating hormone (TSH). To monitor treatment efficacy, HCV-RNA should be determined quantitatively before and 12 wk after the start of therapy. If the RNA level drops less than 2 logs-known as early virologic response (EVR)-or remains detectable at wk 24, a successful treatment is extremely unlikely and therapy should be stopped[15,16,55-58]. The same is true if after 24 wk HCV-RNA is still measurable[59].
As mentioned above, successful treatment is defined as SVR and undetectable HCV-RNA 6 mo after the end of therapy. A recently described predictive factor is the rapid virologic response (RVR), defined as undetectable
1898 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

HCV RNA levels at wk 4 after the start of treatment. For patients who have genotype 1 with a low baseline viral load (< 600 000 IU/mL) and who have achieved RVR under therapy with continuous undetectable HCV-RNA afterwards, a treatment course of 24 wk may be required to match comparable results to the standard treatment of 48 wk[60]. For patients with genotype 2 or 3 and a RVR followed by undetectable RNA levels, therapy for 16 or even 12 wk may be sufficient[46]. However, reducing treatment duration in these patient populations remains controversial[61]. Therapy to increase SVR and reduce side-effects for chronic HCV infected patients is becoming more often designed for the individual, with genotype, EVR, RVR and the HCV-RNA level before start of treatment as predictors for achieving a SVR.
TREATMENT OF HEPATITIS C INFECTION IN CHILDRENChildren suffering from chronic HCV infection generally show no symptoms. While biochemistry and histology are comparable to adults with HCV, the progression of hepatitis C seems to be slower compared to adults[62-64]. It has been shown that, in general, children tolerate IFN therapy relatively well. Side effects are usually mild or moderate. One study of 41 children receiving standard combination therapy showed an overall SVR of 61% one year after treatment[65]. Altogether response rates in children to INF monotherapy and combination therapy with INF and ribavirin seem to be equivalent to adults[14,64,66,67], PEG-IFN is not yet approved for use in children. Therefore, the present regime is 15 mg ribavirin per kg body weight per day plus 3 MIU/m2 body surface interferon alpha-2b three times per week[68]. This treatment appears to be reasonably safe and effective in children with hepatitis C. Prospective controlled trials evaluating combination therapy with PEG-INF are being developed.
SIDE-EFFECTS AND CONTRAINDICATIONS FOR TREATMENT OF HEPATITIS CINFECTION Should everybody with chronic HCV infection be treated? Patients with signs of hepatitis, such as elevated transaminases (serum alanine aminotransferase level, ALT), and beginning fibrosis in liver biopsy are thought to be the most appropriate group to undergo therapy[14,69]. Nevertheless, patients with normal transaminases have the same outcome as individuals with elevated ALT[70].
People wi thout the necessar y mot ivat ion and compliance to therapy should be considered as untreated. Costs of treatment and severe side-effects must be weighed against the low efficacy of therapy in this group[14,34,71,72].
There are some absolute and re lat ive medica l contraindications for treatment with IFN, PEG-IFN and/or ribavirin. Since ribavirin is teratogenic, in both males and females, anticonception is recommended during therapy and at least 6 mo after end of therapy[73].
Also breast feeding should be avoided. People with cardiac problems rarely develop reversible arrhythmia[74]
or cardiomyopathy[75] under interferon therapy. But for patients with significant cardiac disease, death from cardiac failure is more likely than from chronic hepatitis C. Therefore, within this patient group anti-viral therapy can be dispensed.
Further contraindications for treatment with interferon and/or ribavirin are hepatic decompensation and renal failure. Interferon and ribavirin can enhance liver failure[76] and, as mentioned above, close monitoring is required. Some hepatic comorbidities, additional to chronic HCV infection, are discussed later. Ribavirin undergoes renal elimination. Therefore, renal impairment leads to elevated serum levels of ribavirin, enhancing side-effects such as hemolysis. The same is true for INF, while PEG-IFN, due to its molecular size, is better tolerated in patients with end stage renal disease. PEG-IFN alpha-2a does not result in elevated serum levels even with creatinine clearance < 30 mL/min. Since kidney diseases often do not limit life expectancy, antiviral therapy must be considered in patients with both chronic HCV and end-stage renal disorder[77,78]. Available data suggests higher SVR rates in patients with renal disease, using IFN monotherapy, compared to patients with normal renal function[79,80]. Few studies have dealt with very low dose ribavirin application in these patients[81,82], alone or in combination with PEG-INF. PEG-INF a-2a should be reduced from 180 μg to 135 μg, while the recommendation for PEG-INFa-2b is dose reduction by 50%[83].
The immune system is heavily influenced by both ribavirin and interferon and this is the probable mechanism of their antiviral activity. But in HCV patients with comorbid autoimmune disease, both drugs could worsen the disease. Therefore, application of antiviral therapy must be considered dangerous as long as the autoimmune disease is not controlled.
The most common side effects of treatment with PEG-IFN are fatigue, muscle aches and psychological disorders such as depression, irritability, anxiety and sleep disturbance. Interferon further induces pancytopenia through its bone marrow depressing activity[15,16,84]. The most common adverse effect of ribavirin is haemolysis and anaemia. Therefore, patients treated with the combination therapy suffer from anaemia, with the lowest haemoglobin 4 wk after initiation of treatment[84]. Patients with ischaemic problems should be monitored closely and, if necessary, should have blood components transfused. The application of growth factors, such as erythropoetin, G-CSF or GM-CSF, cannot generally be recommended considering the cost-benefit-ratio, but growth factors may be useful in some patient populations. Significantly higher risk for bacterial infection has not been demonstrated during treatment with ribavirin and/or PEG-IFN[15,16,85,86]. The most common autoimmune reaction to therapy is the development of autoimmune thyroiditis[15,16,84,85,87]. Flu symptoms caused by interferon can be treated with paracetamol or similar drugs[84], and thyroid disorders by hormone application. More severe side effects are mood changes and depression[15,16,84,88]. The later is especially true in people already suffering from instable psychiatric
Weigand K et al . Treatment of HCV infection 1899
www.wjgnet.com

disorders prior to therapy. Mild symptoms can be covered by selective serotonin reuptake inhibitors (SSRI)[89], while development of severe depression or suicidal tendencies are clear indications to discontinue therapy.
Rare side-effects are hearing impairment, hair thinning and loss, insomnia, visual disorders, interstitial pneumonia, pancreatitis, colitis and exacerbation of inflammatory diseases[83,84]. Every patient should be informed sufficiently about potential adverse events before therapy is started.
TREATMENT OF HEPATITIS C INPATIENTS WITH LIVER CIRRHOSISMortality from hepatitis C is mainly due to manifest cirrhosis. Liver biopsy studies indicate that hepatic fibrosis may regress under therapy with PEG-IFN and ribavirin[90,91]. The recommendation today is to treat patients with compensated cirrhosis with the standard combination therapy[92,93]. While patients with Child-Pugh A and B cirrhosis respond and tolerate this therapy relatively well[15,16,40,87,94], it is unclear whether a drug reduction in Child C cirrhosis should be recommended[95-97]. In general, antiviral therapy in decompensated liver cirrhosis is not recommended[98].
There is data indicating that reduction of body weight in obese patients is associated with reduction in hepatic fat[99,100] and in some cases of fibrosis[101], resulting in a better response to antiviral treatment.
A study car r ied out on a Japanese populat ion demonstrated the prevention of hepatocellular carcinoma (HCC) in individuals receiving antiviral therapy[102]. The same effect may be found in Caucasians[103,104]. Successful antiviral therapy also reduces the rate of hepatic decompensation.
Overall, even if liver transplantation in end-stage disease is not prevented by antiviral therapy, the data suggest that recurrence of disease after transplantation is significantly lowered if treated previously[76].
TREATMENT OF HEPATITIS C IN PATIENTS AFTER LIVER TRANSPLANTATIONChronic HCV infection with end-stage liver disease or hepatocellular carcinoma due to HCV is currently the most common indication for liver transplantation[105,106]. All transplanted patients become reinfected with HCV[107-111] and, combined with immune suppressive therapy to avoid rejection of the organ, some patients develop rapid progressive hepatitis[109,112-115]. In addition, acute rejection seems more frequent in patients with liver transplantation for hepatitis C[116,117].
Before transplantation, the patient should be treated for HCV infection, if possible (see above) [96,97,118]. Unfortunately, most patients in need of transplantation have decompensated disease, limiting effective antiviral therapy before transplantation[76]. Even when IFN combined with ribavirin was previously used[119-121], after transplantation the standard treatment should be PEG-IFN and ribavirin[122-128]. Nevertheless, SVR rates post-transplantation were significantly worse compared to
antiviral treatment in the non-transplantation setting. Overall, SVR rates post-transplantation are only about 20%[119,123,127,129-131]. Furthermore, in a high percentage of patients, interferon and ribavirin are poorly tolerated after liver transplantation and therapy must be considered carefully[83,131].
TREATMENT OF HEPATITIS C IN PATIENTS CO-INFECTED WITH HIVThere is a high rate of patients co-infected with HCV and HIV due to the same transmission route in high-risk populations. An estimated 25%-30% of patients with HIV are co-infected with HCV in Europe and the United States[132]. In these patients, HCV progression to end-stage liver disease is almost doubled[133,134]. Therefore, it is favourable to treat these patients as early as possible, if they have no other contraindications.
Since therapy for HCV does not significantly increase HIV RNA levels, it is recommended to use standard treatment with PEG-IFN and ribavirin. SVR rates in co-infected patients are sl ightly lower than in the mono-infected population[135,136]. For genotype 1 (and 4), SVR ranges from 14% to 29%, while for genotype 2 and 3 it ranges from 44% to 73%[137-139]. Patients on antiretroviral therapy are more likely to develop the side-effects of ribavirin[139-141]. These patients must be monitored closely and, in the case of severe side effects, ribavirin should be reduced or discontinued.
If HIV infection itself is not stable (CD4 count < 200 cells/mm), highly active antiretroviral therapy (HAART) should be initiated first and secondarily, after stabilisation, treatment for HCV should be started[142-144].
NEW THERAPEUTIC IDEAS FOR PATIENTS INFECTED WITH HEPATITIS C Since the number of non-responders or patients with relapse is increasing, many new drugs are being tested briefly. Meta-analysis of several controlled trials for amantadin, for example, showed a significantly better SVR with amantadin and IFN alpha compared to IFN alpha monotherapy[19,20,145]. Further, a German study showed an increase of SVR using a triple therapy of INF alpha plus ribavirin plus amantadin compared to INF alpha plus ribavirin plus placebo[18]. Still missing are trials with PEG-INF, ribavirin and amantadin, as well as studies looking at long-term outcomes.
Further, alternative interferon types and ribavirin analogues, for example viramidine, which is a ribavirin prodrug cleaved to ribavirin in the liver, were tested to reduce side effects and were found to vary in success[146-148]. Most promising are specific inhibitors, such as specific HCV protease and HCV polymerase inhibitors, which were tested in experimental settings or phase 1-2 studies currently[21,26]. In phase 1 and 2 trials, several HCV specific protease inhibitors, such as BILN 2061, VX 950, SCH 503034, demonstrated a reduction of HCV RNA levels from 2 to 4.4 log10 from baseline[149-153]. Due to cardiac toxicity, further development of BILN 2061 has been
1900 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

stopped. VX 950, which is another protease inhibitor, significantly lowers HCV-RNA levels more than 4 log10 units within the first 14 d[154]. Unfortunately, in the follow-up, the concentration increased again, probably due to mutational resistance. Nevertheless, since RVR is a strong positive predictive factor, a combination of standard therapy together with VX 950 could increase SVR rates. Similar results are true for SCH 503 034 with a HCV-RNA drop of 2 to 3 log10 units. Also, specific polymerase inhibitors and nucleosid analogues have been tested for reduction of HCV RNA levels[155,156]. They result in a reduction of HCV-RNA concentrations, but they seem to be less effective in their antiviral activity than protease inhibitors[157]. Studies combining polymerase inhibitors with standard therapy are ongoing and it is most likely that polymerase and protease inhibitors will be combined with PEG-IFN in the future.
Additional concepts are the use of toll-like receptor agonists and RNA-based therapy[26], as well as drugs like thymosin-a, inosinmonophaphatedehydrogenase inhibitors, anti oxidants, glucosidase inhibitors, cytokines, inhibitors of the internal ribosomal entry site (IRES) and fusion proteins. Silymarin, which in some cases resulted in a drop of the transaminases, does not produce an effect on HCV-RNA concentration and liver histology in the trials presented to date. Most eagerly, many approaches involve a search to find a vaccine to prevent HCV infection[24,25,158].
ACKNOWLEDGMENTSWe gratefully thank Kerstin Stein, MD and Christoph Eisenbach, MD of our department for proof reading and critical comments on the manuscript.
REFERENCES1 Alter MJ. Hepatitis C virus infection in the United States. J
Hepatol 1999; 31 Suppl 1: 88-912 Murphy EL, Bryzman SM, Glynn SA, Ameti DI, Thomson
RA, Williams AE, Nass CC, Ownby HE, Schreiber GB, Kong F, Neal KR, Nemo GJ. Risk factors for hepatitis C virus infection in United States blood donors. NHLBI Retrovirus Epidemiology Donor Study (REDS) Hepatology 2000; 31: 756-762
3 Conry-Cantilena C, VanRaden M, Gibble J, Melpolder J, Shakil AO, Viladomiu L, Cheung L, DiBisceglie A, Hoofnagle J, Shih JW. Routes of infection, viremia, and liver disease in blood donors found to have hepatitis C virus infection. N Engl J Med 1996; 334: 1691-1696
4 Conte D, Fraquelli M, Prati D, Colucci A, Minola E. Prevalence and clinical course of chronic hepatitis C virus (HCV) infection and rate of HCV vertical transmission in a cohort of 15,250 pregnant women. Hepatology 2000; 31: 751-755
5 Haley RW , F i scher RP . Commerc ia l t a t too ing as a potentially important source of hepatitis C infection. Clinical epidemiology of 626 consecutive patients unaware of their hepatitis C serologic status. Medicine (Baltimore) 2001; 80: 134-151
6 Akahane Y, Kojima M, Sugai Y, Sakamoto M, Miyazaki Y, Tanaka T, Tsuda F, Mishiro S, Okamoto H, Miyakawa Y, Mayumi M. Hepatitis C virus infection in spouses of patients with type C chronic liver disease. Ann Intern Med 1994; 120: 748-752
7 Kenny-Walsh E. Clinical outcomes after hepatitis C infection from contaminated anti-D immune globulin. Irish Hepatology Research Group. N Engl J Med 1999; 340: 1228-1233
8 Di Bisceglie AM. Natural history of hepatitis C: its impact on clinical management. Hepatology 2000; 31: 1014-1018
9 Barrera JM, Bruguera M, Ercilla MG, Gil C, Celis R, Gil MP, del Valle Onorato M, Rodés J, Ordinas A. Persistent hepatitis C viremia after acute self-limiting posttransfusion hepatitis C. Hepatology 1995; 21: 639-644
10 Alter MJ, Kruszon-Moran D, Nainan OV, McQuillan GM, Gao F, Moyer LA, Kaslow RA, Margolis HS. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N Engl J Med 1999; 341: 556-562
11 Jaeckel E, Cornberg M, Wedemeyer H, Santantonio T, Mayer J, Zankel M, Pastore G, Dietrich M, Trautwein C, Manns MP. Treatment of acute hepatitis C with interferon alfa-2b. N Engl J Med 2001; 345: 1452-1457
12 Wiegand J, Buggisch P, Boecher W, Zeuzem S, Gelbmann CM, Berg T, Kauffmann W, Kallinowski B, Cornberg M, Jaeckel E, Wedemeyer H, Manns MP. Early monotherapy with pegylated interferon alpha-2b for acute hepatitis C infection: the HEP-NET acute-HCV-II study. Hepatology 2006; 43: 250-256
13 Gerlach JT, Diepolder HM, Zachoval R, Gruener NH, Jung MC, Ulsenheimer A, Schraut WW, Schirren CA, Waechtler M, Backmund M, Pape GR. Acute hepatitis C: high rate of both spontaneous and treatment-induced viral clearance. Gastroenterology 2003; 125: 80-88
14 National Institutes of Health Consensus Development Conference Statement: Management of hepatitis C: 2002-June 10-12, 2002. Hepatology 2002; 36: S3-S20
15 Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M, Albrecht JK. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 2001; 358: 958-965
16 Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Gonçales FL, Häussinger D, Diago M, Carosi G, Dhumeaux D, Craxi A, Lin A, Hoffman J, Yu J. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 2002; 347: 975-982
17 Strader DB, Wright T, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C. Hepatology 2004; 39: 1147-1171
18 Berg T, Kronenberger B, Hinrichsen H, Gerlach T, Buggisch P, Herrmann E, Spengler U, Goeser T, Nasser S, Wursthorn K, Pape GR, Hopf U, Zeuzem S. Triple therapy with amantadine in treatment-naive patients with chronic hepatitis C: a placebo-controlled trial. Hepatology 2003; 37: 1359-1367
19 Brillanti S, Levantesi F, Masi L, Foli M, Bolondi L. Triple antiviral therapy as a new option for patients with interferon nonresponsive chronic hepatitis C. Hepatology 2000; 32: 630-634
20 Teuber G, Pascu M, Berg T, Lafrenz M, Pausch J, Kullmann F, Ramadori G, Arnold R, Weidenbach H, Musch E, Junge U, Wiedmann KH, Herrmann E, Zankel M, Zeuzem S. Randomized, controlled trial with IFN-alpha combined with ribavirin with and without amantadine sulphate in non-responders with chronic hepatitis C. J Hepatol 2003; 39: 606-613
21 De Francesco R, Migliaccio G. Challenges and successes in developing new therapies for hepatitis C. Nature 2005; 436: 953-960
22 Pawlotsky JM, McHutchison JG. Hepatitis C. Development of new drugs and clinical trials: promises and pitfalls. Summary of an AASLD hepatitis single topic conference, Chicago, IL, February 27-March 1, 2003. Hepatology 2004; 39: 554-567
23 Gale M, Foy EM. Evasion of intracellular host defence by hepatitis C virus. Nature 2005; 436: 939-945
24 Arvin AM, Greenberg HB. New viral vaccines. Virology 2006; 344: 240-249
25 Houghton M, Abrignani S. Prospects for a vaccine against the hepatitis C virus. Nature 2005; 436: 961-966
26 McHutchison JG, Bartenschlager R, Patel K, Pawlotsky JM. The face of future hepatitis C antiviral drug development: recent biological and virologic advances and their translation to drug development and clinical practice. J Hepatol 2006; 44: 411-421
27 Orland JR, Wright TL, Cooper S. Acute hepatitis C. Hepatology 2001; 33: 321-327
Weigand K et al . Treatment of HCV infection 1901
www.wjgnet.com

28 Alter HJ, Seeff LB. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin Liver Dis 2000; 20: 17-35
29 Nomura H, Sou S, Tanimoto H, Nagahama T, Kimura Y, Hayashi J, Ishibashi H, Kashiwagi S. Short-term interferon-alfa therapy for acute hepatitis C: a randomized controlled trial. Hepatology 2004; 39: 1213-1219
30 Alberti A, Boccato S, Vario A, Benvegnù L. Therapy of acute hepatitis C. Hepatology 2002; 36: S195-S200
31 Hofer H, Watkins-Riedel T, Janata O, Penner E, Holzmann H, Steindl-Munda P, Gangl A, Ferenci P. Spontaneous viral clearance in patients with acute hepatitis C can be predicted by repeated measurements of serum viral load. Hepatology 2003; 37: 60-64
32 Kaplan M, Gawrieh S, Cotler SJ, Jensen DM. Neutralizing antibodies in hepatitis C virus infection: a review of immunological and clinical characteristics. Gastroenterology 2003; 125: 597-604
33 Fattovich G, Giustina G, Favarato S, Ruol A. A survey of adverse events in 11,241 patients with chronic viral hepatitis treated with alfa interferon. J Hepatol 1996; 24: 38-47
34 Peters MG, Terrault NA. Alcohol use and hepatitis C. Hepatology 2002; 36: S220-S225
35 Wedemeyer H, Jäckel E, Wiegand J, Cornberg M, Manns MP. Whom? When? How? Another piece of evidence for early treatment of acute hepatitis C. Hepatology 2004; 39: 1201-1203
36 Kamal SM, Ismail A, Graham CS, He Q, Rasenack JW, Peters T, Tawil AA, Fehr JJ, Khalifa Kel S, Madwar MM, Koziel MJ. Pegylated interferon alpha therapy in acute hepatitis C: relation to hepatitis C virus-specific T cell response kinetics. Hepatology 2004; 39: 1721-1731
37 Kamal SM, Fouly AE, Kamel RR, Hockenjos B, Al Tawil A, Khalifa KE, He Q, Koziel MJ, El Naggar KM, Rasenack J, Afdhal NH. Peginterferon alfa-2b therapy in acute hepatitis C: impact of onset of therapy on sustained virologic response. Gastroenterology 2006; 130: 632-638
38 Marcellin P, Boyer N, Gervais A, Martinot M, Pouteau M, Castelnau C, Kilani A, Areias J, Auperin A, Benhamou JP, Degott C, Erlinger S. Long-term histologic improvement and loss of detectable intrahepatic HCV RNA in patients with chronic hepatitis C and sustained response to interferon-alpha therapy. Ann Intern Med 1997; 127: 875-881
39 McHutchison JG, Davis GL, Esteban-Mur R. Durability of sustained virologic response in patients with chronic hepatitis C after treatment with interferon-2b alone or in combination with ribavirin. Hepatology 2001; 34: 244A
40 Hadziyannis SJ, Sette H, Morgan TR, Balan V, Diago M, Marcellin P, Ramadori G, Bodenheimer H, Bernstein D, Rizzetto M, Zeuzem S, Pockros PJ, Lin A, Ackrill AM. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med 2004; 140: 346-355
41 Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med 2006; 355: 2444-2451
42 Lau JY, Tam RC, Liang TJ, Hong Z. Mechanism of action of ribavirin in the combination treatment of chronic HCV infection. Hepatology 2002; 35: 1002-1009
43 Di Bisceglie AM, Conjeevaram HS, Fried MW, Sallie R, Park Y, Yurdaydin C, Swain M, Kleiner DE, Mahaney K, Hoofnagle JH. Ribavirin as therapy for chronic hepatitis C. A randomized, double-blind, placebo-controlled trial. Ann Intern Med 1995; 123: 897-903
44 Bodenheimer HC, Lindsay KL, Davis GL, Lewis JH, Thung SN, Seeff LB. Tolerance and efficacy of oral ribavirin treatment of chronic hepatitis C: a multicenter trial. Hepatology 1997; 26: 473-477
45 von Wagner M, Huber M, Berg T, Hinrichsen H, Rasenack J, Heintges T, Bergk A, Bernsmeier C, Häussinger D, Herrmann E, Zeuzem S. Peginterferon-alpha-2a (40KD) and ribavirin for 16 or 24 weeks in patients with genotype 2 or 3 chronic hepatitis C. Gastroenterology 2005; 129: 522-527
46 Mangia A, Santoro R, Minerva N, Ricci GL, Carretta V, Persico M, Vinelli F, Scotto G, Bacca D, Annese M, Romano M, Zechini
F, Sogari F, Spirito F, Andriulli A. Peginterferon alfa-2b and ribavirin for 12 vs. 24 weeks in HCV genotype 2 or 3. N Engl J Med 2005; 352: 2609-2617
47 Keating GM, Curran MP. Peginterferon-alpha-2a (40kD) plus ribavirin: a review of its use in the management of chronic hepatitis C. Drugs 2003; 63: 701-730
48 Scott LJ, Perry CM. Interferon-alpha-2b plus ribavirin: a review of its use in the management of chronic hepatitis C. Drugs 2002; 62: 507-556
49 Carithers RL, Emerson SS. Therapy of hepatitis C: meta-analysis of interferon alfa-2b trials. Hepatology 1997; 26: 83S-88S
50 Buti M, Sanchez-Avila F, Lurie Y, Stalgis C, Valdés A, Martell M, Esteban R. Viral kinetics in genotype 1 chronic hepatitis C patients during therapy with 2 different doses of peginterferon alfa-2b plus ribavirin. Hepatology 2002; 35: 930-936
51 Davis GL, Esteban-Mur R, Rustgi V, Hoefs J, Gordon SC, Trepo C, Shiffman ML, Zeuzem S, Craxi A, Ling MH, Albrecht J. Interferon alfa-2b alone or in combination with ribavirin for the treatment of relapse of chronic hepatitis C. International Hepatitis Interventional Therapy Group. N Engl J Med 1998; 339: 1493-1499
52 Cammà C, Bruno S, Schepis F, Lo Iacono O, Andreone P, Gramenzi AG, Mangia A, Andriulli A, Puoti M, Spadaro A, Freni M, Di Marco V, Cino L, Saracco G, Chiesa A, Crosignani A, Caporaso N, Morisco F, Rumi MG, Craxì A. Retreatment with interferon plus ribavirin of chronic hepatitis C non-responders to interferon monotherapy: a meta-analysis of individual patient data. Gut 2002; 51: 864-869
53 Cheng SJ, Bonis PA, Lau J, Pham NQ, Wong JB. Interferon and ribavirin for patients with chronic hepatitis C who did not respond to previous interferon therapy: a meta-analysis of controlled and uncontrolled trials. Hepatology 2001; 33: 231-240
54 S h i f f m a n M L . M a n a g e m e n t o f i n t e r f e r o n t h e r a p y nonresponders. Clin Liver Dis 2001; 5: 1025-1043
55 Zeuzem S, Herrmann E, Lee JH, Fricke J, Neumann AU, Modi M, Colucci G, Roth WK. Viral kinetics in patients with chronic hepatitis C treated with standard or peginterferon alpha2a. Gastroenterology 2001; 120: 1438-1447
56 Zeuzem S , Lee JH, Franke A, Rüster B, Prümmer O, Herrmann G, Roth WK. Quantification of the initial decline of serum hepatitis C virus RNA and response to interferon alfa. Hepatology 1998; 27: 1149-1156
57 Davis GL, Wong JB, McHutchison JG, Manns MP, Harvey J, Albrecht J. Early virologic response to treatment with peginterferon alfa-2b plus ribavirin in patients with chronic hepatitis C. Hepatology 2003; 38: 645-652
58 Davis GL. Monitoring of viral levels during therapy of hepatitis C. Hepatology 2002; 36: S145-S151
59 Wong JB, Davis GL, McHutchinson JG. Clinical implications of testing viral response during ribavirin and peginterferon alfa-2b treatment for chronic hepatitis C. Hepatology 2002; 36: 281A
60 Zeuzem S, Buti M, Ferenci P, Sperl J, Horsmans Y, Cianciara J, Ibranyi E, Weiland O, Noviello S, Brass C, Albrecht J. Efficacy of 24 weeks treatment with peginterferon alfa-2b plus ribavirin in patients with chronic hepatitis C infected with genotype 1 and low pretreatment viremia. J Hepatol 2006; 44: 97-103
61 Shiffman ML, Pappas SC, Bacon B, Godofsky E, Nelson D, Harley H, Diago M, Lin A, Hooper G, Zeuzem S. Utility of virological response at weeks 4 and 12 in the prediction of SVR rates in genotype 2/3 patients treated with PEGInterferon alfa-2A (40KD) plus ribavirin: findings from accelerate. Hepatology 2006; 44: 97A340
62 Vogt M, Lang T, Frösner G, Klingler C, Sendl AF, Zeller A, Wiebecke B, Langer B, Meisner H, Hess J. Prevalence and clinical outcome of hepatitis C infection in children who underwent cardiac surgery before the implementation of blood-donor screening. N Engl J Med 1999; 341: 866-870
63 Jacobson IM, Ahmed F, Russo MW, Brown RS, Lebovics E, Min A, Esposito S, Brau N, Tobias H, Klion F, Bini E, Brodsky N, Rovner D, Brass C NP-ISG. Pegylated interferon alfa-2b plus ribavirin in patients with chronic hepatitis C. A trial in prior nonresponders to interferon monotherapy or combination
1902 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

therapy and in combination therpay relapsers: final results (abstr). Gastroenterology 2003; 124 Suppl 1: A714
64 Jonas MM. Children with hepatitis C. Hepatology 2002; 36: S173-S178
65 Wirth S, Lang T, Gehring S, Gerner P. Recombinant alfa-interferon plus ribavirin therapy in children and adolescents with chronic hepatitis C. Hepatology 2002; 36: 1280-1284
66 Alberti A, Benvegnù L. Management of hepatitis C. J Hepatol 2003; 38 Suppl 1: S104-S118
67 Jacobson KR, Murray K, Zellos A, Schwarz KB. An analysis of published trials of interferon monotherapy in children with chronic hepatitis C. J Pediatr Gastroenterol Nutr 2002; 34: 52-58
68 González-Peralta RP, Kelly DA, Haber B, Molleston J, Murray KF, Jonas MM, Shelton M, Mieli-Vergani G, Lurie Y, Martin S, Lang T, Baczkowski A, Geffner M, Gupta S, Laughlin M. Interferon alfa-2b in combination with ribavirin for the treatment of chronic hepatitis C in children: efficacy, safety, and pharmacokinetics. Hepatology 2005; 42: 1010-1018
69 EASL International Consensus Conference on hepatitis C. Paris, 26-27 February 1999. Consensus statement. J Hepatol 1999; 31 Suppl 1: 3-8
70 Zeuzem S, Diago M, Gane E, Reddy KR, Pockros P, Prati D, Shiffman M, Farci P, Gitlin N, O'Brien CB, Lamour F, Lardelli P. Peginterferon alfa-2a (40 kilodaltons) and ribavirin in patients with chronic hepatitis C and normal aminotransferase levels. Gastroenterology 2004; 127: 1724-1732
71 Edlin BR. Prevention and treatment of hepatitis C in injection drug users. Hepatology 2002; 36: S210-S219
72 Edlin BR, Seal KH, Lorvick J, Kral AH, Ciccarone DH, Moore LD, Lo B. Is it justifiable to withhold treatment for hepatitis C from illicit-drug users? N Engl J Med 2001; 345: 211-215
73 Heathcote J, Main J. Treatment of hepatitis C. J Viral Hepat 2005; 12: 223-235
74 Deyton LR, Walker RE, Kovacs JA, Herpin B, Parker M, Masur H, Fauci AS, Lane HC. Reversible cardiac dysfunction associated with interferon alfa therapy in AIDS patients with Kaposi's sarcoma. N Engl J Med 1989; 321: 1246-1249
75 Angulo MP , Navajas A, Galdeano JM, Astigarraga I , Fernández-Teijeiro A. Reversible cardiomyopathy secondary to alpha-interferon in an infant. Pediatr Cardiol 1999; 20: 293-294
76 Forns X, García-Retortillo M, Serrano T, Feliu A, Suarez F, de la Mata M, García-Valdecasas JC, Navasa M, Rimola A, Rodés J. Antiviral therapy of patients with decompensated cirrhosis to prevent recurrence of hepatitis C after liver transplantation. J Hepatol 2003; 39: 389-396
77 Gentil MA, Rocha JL, Rodríguez-Algarra G, Pereira P, López R, Bernal G, Muñoz J, Naranjo M, Mateos J. Impaired kidney transplant survival in patients with antibodies to hepatitis C virus. Nephrol Dial Transplant 1999; 14: 2455-2460
78 Strader DB. Understudied populations with hepatitis C. Hepatology 2002; 36: S226-S236
79 Russo MW , Goldsweig CD, Jacobson IM, Brown RS. Interferon monotherapy for dialysis patients with chronic hepatitis C: an analysis of the literature on efficacy and safety. Am J Gastroenterol 2003; 98: 1610-1615
80 Fabrizi F, Poordad FF, Martin P. Hepatitis C infection and the patient with end-stage renal disease. Hepatology 2002; 36: 3-10
81 Bruchfeld A, Lindahl K, Ståhle L, Söderberg M, Schvarcz R. Interferon and ribavirin treatment in patients with hepatitis C-associated renal disease and renal insufficiency. Nephrol Dial Transplant 2003; 18: 1573-1580
82 Gupta SK, Pittenger AL, Swan SK, Marbury TC, Tobillo E, Batra V, Sack M, Glue P, Jacobs S, Affrime M. Single-dose pharmacokinetics and safety of pegylated interferon-alpha2b in patients with chronic renal dysfunction. J Clin Pharmacol 2002; 42: 1109-1115
83 Dienstag JL, McHutchison JG. American Gastroenterological Association technical review on the management of hepatitis C. Gastroenterology 2006; 130: 231-264; quiz 214-217
84 Fried MW. Side effects of therapy of hepatitis C and their management. Hepatology 2002; 36: S237-S244
85 Russo MW, Fried MW. Side effects of therapy for chronic hepatitis C. Gastroenterology 2003; 124: 1711-1719
86 Soza A, Everhart JE, Ghany MG, Doo E, Heller T, Promrat K, Park Y, Liang TJ, Hoofnagle JH. Neutropenia during combination therapy of interferon alfa and ribavirin for chronic hepatitis C. Hepatology 2002; 36: 1273-1279
87 McHutchison JG, Gordon SC, Schiff ER, Shiffman ML, Lee WM, Rustgi VK, Goodman ZD, Ling MH, Cort S, Albrecht JK. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy Group. N Engl J Med 1998; 339: 1485-1492
88 Zeuzem S, Feinman SV, Rasenack J, Heathcote EJ, Lai MY, Gane E, O'Grady J, Reichen J, Diago M, Lin A, Hoffman J, Brunda MJ. Peginterferon alfa-2a in patients with chronic hepatitis C. N Engl J Med 2000; 343: 1666-1672
89 Musselman DL, Lawson DH, Gumnick JF, Manatunga AK, Penna S, Goodkin RS, Greiner K, Nemeroff CB, Miller AH. Paroxetine for the prevention of depression induced by high-dose interferon alfa. N Engl J Med 2001; 344: 961-966
90 Poynard T, McHutchison J, Manns M, Trepo C, Lindsay K, Goodman Z, Ling MH, Albrecht J. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology 2002; 122: 1303-1313
91 Fontana RJ, Everson GT, Tuteja S, Vargas HE, Shiffman ML. Controversies in the management of hepatitis C patients with advanced fibrosis and cirrhosis. Clin Gastroenterol Hepatol 2004; 2: 183-197
92 Wright TL. Treatment of patients with hepatitis C and cirrhosis. Hepatology 2002; 36: S185-S194
93 Schalm SW, Weiland O, Hansen BE, Milella M, Lai MY, Hollander A, Michielsen PP, Bellobuono A, Chemello L, Pastore G, Chen DS, Brouwer JT. Interferon-ribavirin for chronic hepatitis C with and without cirrhosis: analysis of individual patient data of six controlled trials. Eurohep Study Group for Viral Hepatitis. Gastroenterology 1999; 117: 408-413
94 Heathcote EJ, Shiffman ML, Cooksley WG, Dusheiko GM, Lee SS, Balart L, Reindollar R, Reddy RK, Wright TL, Lin A, Hoffman J, De Pamphilis J. Peginterferon alfa-2a in patients with chronic hepatitis C and cirrhosis. N Engl J Med 2000; 343: 1673-1680
95 Everson GT. Long-term outcome of patients with chronic hepatitis C and decompensated liver disease treated with the LADR protocol. Hepatology 2002; 36: 297A
96 Everson GT. Treatment of patients with hepatitis C virus on the waiting list. Liver Transpl 2003; 9: S90-S94
97 Everson GT, Trotter J, Forman L, Kugelmas M, Halprin A, Fey B, Ray C. Treatment of advanced hepatitis C with a low accelerating dosage regimen of antiviral therapy. Hepatology 2005; 42: 255-262
98 Crippin JS, McCashland T, Terrault N, Sheiner P, Charlton MR. A pilot study of the tolerability and efficacy of antiviral therapy in hepatitis C virus-infected patients awaiting liver transplantation. Liver Transpl 2002; 8: 350-355
99 Poynard T, Ratziu V, McHutchison J, Manns M, Goodman Z, Zeuzem S, Younossi Z, Albrecht J. Effect of treatment with peginterferon or interferon alfa-2b and ribavirin on steatosis in patients infected with hepatitis C. Hepatology 2003; 38: 75-85
100 Bressler BL, Guindi M, Tomlinson G, Heathcote J. High body mass index is an independent risk factor for nonresponse to antiviral treatment in chronic hepatitis C. Hepatology 2003; 38: 639-644
101 Hickman IJ, Clouston AD, Macdonald GA, Purdie DM, Prins JB, Ash S, Jonsson JR, Powell EE. Effect of weight reduction on liver histology and biochemistry in patients with chronic hepatitis C. Gut 2002; 51: 89-94
102 Yoshida H, Shiratori Y, Moriyama M, Arakawa Y, Ide T, Sata M, Inoue O, Yano M, Tanaka M, Fujiyama S, Nishiguchi S, Kuroki T, Imazeki F, Yokosuka O, Kinoyama S, Yamada G, Omata M. Interferon therapy reduces the risk for hepatocellular carcinoma: national surveillance program of cirrhotic and noncirrhotic patients with chronic hepatitis C in Japan. IHIT Study Group. Inhibition of Hepatocarcinogenesis
Weigand K et al . Treatment of HCV infection 1903
www.wjgnet.com

by Interferon Therapy. Ann Intern Med 1999; 131: 174-181103 Cammà C, Giunta M, Andreone P, Craxì A. Interferon and
prevention of hepatocellular carcinoma in viral cirrhosis: an evidence-based approach. J Hepatol 2001; 34: 593-602
104 Papatheodoridis GV, Papadimitropoulos VC, Hadziyannis SJ. Effect of interferon therapy on the development of hepatocellular carcinoma in patients with hepatitis C virus-related cirrhosis: a meta-analysis. Aliment Pharmacol Ther 2001; 15: 689-698
105 Seaberg EC, Belle SH, Beringer KC, Schivins JL, Detre KM. Liver transplantation in the United States from 1987-1998: updated results from the Pitt-UNOS Liver Transplant Registry. Clin Transpl, 1998:17-37.
106 Brown RS. Hepatitis C and liver transplantation. Nature 2005; 436: 973-978
107 Forman LM, Lewis JD, Berlin JA, Feldman HI, Lucey MR. The association between hepatitis C infection and survival after orthotopic liver transplantation. Gastroenterology 2002; 122: 889-896
108 Féray C, Caccamo L, Alexander GJ, Ducot B, Gugenheim J, Casanovas T, Loinaz C, Gigou M, Burra P, Barkholt L, Esteban R, Bizollon T, Lerut J, Minello-Franza A, Bernard PH, Nachbaur K, Botta-Fridlund D, Bismuth H, Schalm SW, Samuel D. European collaborative study on factors influencing outcome after liver transplantation for hepatitis C. European Concerted Action on Viral Hepatitis (EUROHEP) Group. Gastroenterology 1999; 117: 619-625
109 Neumann UP, Berg T, Bahra M, Seehofer D, Langrehr JM, Neuhaus R, Radke C, Neuhaus P. Fibrosis progression after liver transplantation in patients with recurrent hepatitis C. J Hepatol 2004; 41: 830-836
110 Charlton M. Hepatitis C infection in liver transplantation. Am J Transplant 2001; 1: 197-203
111 Berenguer M. Natural history of recurrent hepatitis C. Liver Transpl 2002; 8: S14-S18
112 Samonakis DN, Triantos CK, Thalheimer U, Quaglia A, Leandro G, Teixeira R, Papatheodoridis GV, Sabin CA, Rolando N, Davies S, Dhillon AP, Griffiths P, Emery V, Patch DW, Davidson BR, Rolles K, Burroughs AK. Immuno-suppression and donor age with respect to severity of HCV recurrence after liver transplantation. Liver Transpl 2005; 11: 386-395
113 Berenguer M, Prieto M, San Juan F, Rayón JM, Martinez F, Carrasco D, Moya A, Orbis F, Mir J, Berenguer J. Contribution of donor age to the recent decrease in patient survival among HCV-infected liver transplant recipients. Hepatology 2002; 36: 202-210
114 Lake JR, Shorr JS, Steffen BJ, Chu AH, Gordon RD, Wiesner RH. Differential effects of donor age in liver transplant recipients infected with hepatitis B, hepatitis C and without viral hepatitis. Am J Transplant 2005; 5: 549-557
115 Everson GT. Impact of immunosuppressive therapy on recurrence of hepatitis C. Liver Transpl 2002; 8: S19-S27
116 McTaggart RA, Terrault NA, Vardanian AJ, Bostrom A, Feng S. Hepatitis C etiology of liver disease is strongly associated with early acute rejection following liver transplantation. Liver Transpl 2004; 10: 975-985
117 Prieto M, Berenguer M, Rayón JM, Córdoba J, Argüello L, Carrasco D, García-Herola A, Olaso V, De Juan M, Gobernado M, Mir J, Berenguer J. High incidence of allograft cirrhosis in hepatitis C virus genotype 1b infection following transplantation: relationship with rejection episodes. Hepatology 1999; 29: 250-256
118 Thomas RM , Brems JJ , Guzman-Hartman G, Yong S, Cavaliere P, Van Thiel DH. Infection with chronic hepatitis C virus and liver transplantation: a role for interferon therapy before transplantation. Liver Transpl 2003; 9: 905-915
119 Samuel D , Bizollon T, Feray C, Roche B, Ahmed SN, Lemonnier C, Cohard M, Reynes M, Chevallier M, Ducerf C, Baulieux J, Geffner M, Albrecht JK, Bismuth H, Trepo C. Interferon-alpha 2b plus ribavirin in patients with chronic hepatitis C after liver transplantation: a randomized study. Gastroenterology 2003; 124: 642-650
120 Lavezzo B, Franchello A, Smedile A, David E, Barbui A, Torrani M, Ottobrelli A, Zamboni F, Fadda M, Bobbio A, Salizzoni M, Rizzetto M. Treatment of recurrent hepatitis C in liver transplants: efficacy of a six versus a twelve month course of interferon alfa 2b with ribavirin. J Hepatol 2002; 37: 247-252
121 Berenguer M, Prieto M, Palau A, Carrasco D, Rayón JM, Calvo F, Berenguer J. Recurrent hepatitis C genotype 1b following liver transplantation: treatment with combination interferon-ribavirin therapy. Eur J Gastroenterol Hepatol 2004; 16: 1207-1212
122 Mukherjee S, Rogge J, Weaver L, Schafer DF. Pilot study of pegylated interferon alfa-2b and ribavirin for recurrent hepatitis C after liver transplantation. Transplant Proc 2003; 35: 3042-3044
123 Chalasani N, Manzarbeitia C, Ferenci P, Vogel W, Fontana RJ, Voigt M, Riely C, Martin P, Teperman L, Jiao J, Lopez-Talavera JC. Peginterferon alfa-2a for hepatitis C after liver transplantation: two randomized, controlled trials. Hepatology 2005; 41: 289-298
124 Bahr MJ, Manns MP. Changing faces-natural course and treatment of hepatitis C after liver transplantation. J Hepatol 2004; 40: 699-701
125 Dumortier J, Scoazec JY, Chevallier P, Boillot O. Treatment of recurrent hepatitis C after liver transplantation: a pilot study of peginterferon alfa-2b and ribavirin combination. J Hepatol 2004; 40: 669-674
126 Garcia-Retortillo M, Forns X. Prevention and treatment of hepatitis C virus recurrence after liver transplantation. J Hepatol 2004; 41: 2-10
127 Rodriguez-Luna H, Khatib A, Sharma P, De Petris G, Williams JW, Ortiz J, Hansen K, Mulligan D, Moss A, Douglas DD, Balan V, Rakela J, Vargas HE. Treatment of recurrent hepatitis C infection after liver transplantation with combination of pegylated interferon alpha2b and ribavirin: an open-label series. Transplantation 2004; 77: 190-194
128 Shergill AK, Khalili M, Straley S, Bollinger K, Roberts JP, Ascher NA, Terrault NA. Applicability, tolerability and efficacy of preemptive antiviral therapy in hepatitis C-infected patients undergoing liver transplantation. Am J Transplant 2005; 5: 118-124
129 Gane EJ, Lo SK, Riordan SM, Portmann BC, Lau JY, Naoumov NV, Williams R. A randomized study comparing ribavirin and interferon alfa monotherapy for hepatitis C recurrence after liver transplantation. Hepatology 1998; 27: 1403-1407
130 Sheiner PA. Hepatitis C after liver transplantation. Semin Liver Dis 2000; 20: 201-209
131 Gane E. Treatment of recurrent hepatitis C. Liver Transpl 2002; 8: S28-S37
132 Sherman KE, Rouster SD, Chung RT, Rajicic N. Hepatitis C Virus prevalence among patients infected with Human Immunodeficiency Virus: a cross-sectional analysis of the US adult AIDS Clinical Trials Group. Clin Infect Dis 2002; 34: 831-837
133 Goedert JJ, Eyster ME, Lederman MM, Mandalaki T, De Moerloose P, White GC, Angiolillo AL, Luban NL, Sherman KE, Manco-Johnson M, Preiss L, Leissinger C, Kessler CM, Cohen AR, DiMichele D, Hilgartner MW, Aledort LM, Kroner BL, Rosenberg PS, Hatzakis A. End-stage liver disease in persons with hemophilia and transfusion-associated infections. Blood 2002; 100: 1584-1589
134 Benhamou Y, Di Martino V, Bochet M, Colombet G, Thibault V, Liou A, Katlama C, Poynard T. Factors affecting liver fibrosis in human immunodeficiency virus-and hepatitis C virus-coinfected patients: impact of protease inhibitor therapy. Hepatology 2001; 34: 283-287
135 Thomas DL. Hepatitis C and human immunodeficiency virus infection. Hepatology 2002; 36: S201-S209
136 Pérez-Olmeda M, Núñez M, Romero M, González J, Castro A, Arribas JR, Pedreira J, Barreiro P, García-Samaniego J, Martín-Carbonero L, Jiménez-Nácher I, Soriano V. Pegylated IFN-alpha2b plus ribavirin as therapy for chronic hepatitis C in HIV-infected patients. AIDS 2003; 17: 1023-1028
1904 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

137 Chung RT, Andersen J, Volberding P, Robbins GK, Liu T, Sherman KE, Peters MG, Koziel MJ, Bhan AK, Alston B, Colquhoun D, Nevin T, Harb G, van der Horst C. Peginterferon Alfa-2a plus ribavirin versus interferon alfa-2a plus ribavirin for chronic hepatitis C in HIV-coinfected persons. N Engl J Med 2004; 351: 451-459
138 Torriani FJ, Rodriguez-Torres M, Rockstroh JK, Lissen E, Gonzalez-García J, Lazzarin A, Carosi G, Sasadeusz J, Katlama C, Montaner J, Sette H, Passe S, De Pamphilis J, Duff F, Schrenk UM, Dieterich DT. Peginterferon Alfa-2a plus ribavirin for chronic hepatitis C virus infection in HIV-infected patients. N Engl J Med 2004; 351: 438-450
139 Carrat F, Bani-Sadr F, Pol S, Rosenthal E, Lunel-Fabiani F, Benzekri A, Morand P, Goujard C, Pialoux G, Piroth L, Salmon-Céron D, Degott C, Cacoub P, Perronne C. Pegylated interferon alfa-2b vs standard interferon alfa-2b, plus ribavirin, for chronic hepatitis C in HIV-infected patients: a randomized controlled trial. JAMA 2004; 292: 2839-2848
140 Bräu N, Rodriguez-Torres M, Prokupek D, Bonacini M, Giffen CA, Smith JJ, Frost KR, Kostman JR. Treatment of chronic hepatitis C in HIV/HCV-coinfection with interferon alpha-2b+ full-course vs. 16-week delayed ribavirin. Hepatology 2004; 39: 989-998
141 Lafeuillade A , Hittinger G, Chadapaud S. Increased mitochondrial toxicity with ribavirin in HIV/HCV coinfection. Lancet 2001; 357: 280-281
142 Qurishi N , Kreuzberg C, Lüchters G, Effenberger W, Kupfer B, Sauerbruch T, Rockstroh JK, Spengler U. Effect of antiretroviral therapy on liver-related mortality in patients with HIV and hepatitis C virus coinfection. Lancet 2003; 362: 1708-1713
143 Kottilil S, Polis MA, Kovacs JA. HIV Infection, hepatitis C infection, and HAART: hard clinical choices. JAMA 2004; 292: 243-250
144 Chung RT, Evans SR, Yang Y, Theodore D, Valdez H, Clark R, Shikuma C, Nevin T, Sherman KE. Immune recovery is associated with persistent rise in hepatitis C virus RNA, infrequent liver test flares, and is not impaired by hepatitis C virus in co-infected subjects. AIDS 2002; 16: 1915-1923
145 Mangia A, Leandro G, Helbling B, Renner EL, Tabone M, Sidoli L, Caronia S, Foster GR, Zeuzem S, Berg T, Di Marco V, Cino N, Andriulli A. Combination therapy with amantadine and interferon in naïve patients with chronic hepatitis C: meta-analysis of individual patient data from six clinical trials. J Hepatol 2004; 40: 478-483
146 Nelson D, Rustgi V, Balan V, McHutchinson JG, Davis GL, Lambiase L. A phase 2 study of albuferon in combination with ribavirin in nonresponders to prior interferon therapy for chronic hepatitis C. Hepatology 2005; 42: 278A
147 Wu JZ, Walker H, Lau JY, Hong Z. Activation and deactivation of a broad-spectrum antiviral drug by a single enzyme: adenosine deaminase catalyzes two consecutive deamination reactions. Antimicrob Agents Chemother 2003; 47: 426-431
148 Gish RG, Nelson D, Arora S, Fried MW, Reddy KR, Xu Y. Virologic response and safety outcomes in therapy-naive patients treated for chronic hepatitis C with viramidine in combination
with pegylated interferon alfa-2a. J Hepatol 2005; 42: 39A149 Lamarre D, Anderson PC, Bailey M, Beaulieu P, Bolger G,
Bonneau P, Bös M, Cameron DR, Cartier M, Cordingley MG, Faucher AM, Goudreau N, Kawai SH, Kukolj G, Lagacé L, LaPlante SR, Narjes H, Poupart MA, Rancourt J, Sentjens RE, St George R, Simoneau B, Steinmann G, Thibeault D, Tsantrizos YS, Weldon SM, Yong CL, Llinàs-Brunet M. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 2003; 426: 186-189
150 Hinrichsen H , Benhamou Y, Wedemeyer H, Reiser M, Sentjens RE, Calleja JL, Forns X, Erhardt A, Crönlein J, Chaves RL, Yong CL, Nehmiz G, Steinmann GG. Short-term antiviral efficacy of BILN 2061, a hepatitis C virus serine protease inhibitor, in hepatitis C genotype 1 patients. Gastroenterology 2004; 127: 1347-1355
151 Reiser M , Hinrichsen H, Benhamou Y, Reesink HW, Wedemeyer H, Avendano C, Riba N, Yong CL, Nehmiz G, Steinmann GG. Antiviral efficacy of NS3-serine protease inhibitor BILN-2061 in patients with chronic genotype 2 and 3 hepatitis C. Hepatology 2005; 41: 832-835
152 Reesink HW, Zeuzem S, Weegink CJ, Forestier N, van Vliet A, van de Wetering de Rooij J. Final results of a phase 1B, multiple-dose study of VX-950, a hepatitis C virus protease inhibitor. Hepatology 2005; 42: 234A
153 Zeuzem S, Sarrazin C, Rouzier R, Tarral A, Brion N, Forestier N. Anti-viral activity of SCH 503034, a HCV protease inhibitor, administered as monotherapy in hepatitis C genotype-1 (HCV-1) patients refractory to pegylated interferon (PEG-INF-a). Hepatology 2005; 42: 233A
154 Kieffer T, Sarrazin C, Miller J, Traver S, Zhou Y, Bartels D, Hanzelka B, Müh U, Lin C, Reesink HW, Kwong A, Zeuzem S. Combination of Telaprevir (VX-950) and PEG-IFN-ALFA suppresses both wild-type virus and resistance variants in HCV genotype 1-infected patients in a 14-day phase 1B study. Hepatology 2006; 44: 222A92
155 Afdahl N, Godofsky E, Dienstag JL, Rustgi V, Schick L, McEniry D. Final phaseⅠ/Ⅱ trial results for NM283, a new polymerase inhibitor for hepatitis C: antiviral efficacy and tolerance in patients with HCV-1 infection, including previous interferon failures. Hepatology 2004; 40: 726A
156 O’Brien C, Godofsky E, Rodriguez-Torres M, Afdahl N, Pappas SC, Pockros P. Randomized trial of valopicitabine (NM283), alone or with peg-interferon, vs. retreatment with peg-interferon plus ribavirin (pegINF/RBV) in hepatitis C patients with previous non-response to PegINF/RBV: first interim results. Hepatology 2005; 42: 234A
157 Lawitz E, Nguyen T, Younes Z, Santoro J, Gitlin N, McEniry D, Chasen R, Goff J. Knox S, Kleber K, Belanger B, Brown NA, Dieterich D, Valopicitabine (NM283) plus PEG-Interferon in treatment-naive hepatitis C patients with HCV genotype-1 infection: HCV RNA clearance during 24 weeks of treatment. Hepatology 2006; 44: 223A93
158 Ahlen G, Nystrom J, Pult I, Frelin L, Hultgren C, Sallberg M. In vivo clearance of hepatitis C virus nonstructural 3/4A-expressing hepatocytes by DNA vaccine-primed cytotoxic T lymphocytes. J Infect Dis 2005; 192: 2112-2116
S- Editor Liu Y L- Editor Lutze M E- Editor Che YB
Weigand K et al . Treatment of HCV infection 1905
www.wjgnet.com

Clinical characteristics of idiopathic portal hypertension
Ozgur Harmanci, Yusuf Bayraktar
www.wjgnet.com
Yusuf Bayraktar, MD, Series Editor
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1906-1911www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
Ozgur Harmanci, Yusuf Bayraktar, Hacettepe University Faculty of Medicine, Department of Gastroenterology, Sihhiye 06100, Ankara, TurkeyCorrespondence to: Ozgur Harmanci, MD, Hacettepe University Faculty of Medicine, Department of Gastroenterology, Sihhiye 06100, Ankara, Turkey. [email protected]: + 90-312-4439428 Fax: +90-312-4429429Received: 2006-10-10 Accepted: 2006-10-25
AbstractIdiopathic portal hypertension is one of the interesting causes of portal hypertension. Even in very developed medical centers, this disorder is st i l l one of the most important misdiagnoses of clinical practice. To inexperienced physicians, presenting esophageal varices and upper gastrointestinal bleeding usually prompt an unfortunate diagnosis of cirrhosis. A heterogenous clinical presentation and progression of this disorder should be recognized by physicians, and management should be directed towards some specific problems confined to this disorder. Although a genetic basis and other factors are implicated in its pathogenesis, exact underlying mechanism(s) is (are) unknown. In this review, we discuss the heterogeneity of idiopathic portal hypertension, its etiopathogenesis, clinical presentation and management issues. With the expectation of an excellent prognosis, a practicing gastroenterologist should be aware that “not all varices mean cirrhosis”.
© 2007 The WJG Press. All rights reserved.
Key words: Idiopathic portal hypertension; Non-cirrhotic portal fibrosis; Hepatoportal sclerosis; Portal vein thrombosis
Harmanci O, Bayraktar Y. Clinical characteristics of idiopathic portal hypertension. World J Gastroenterol 2007; 13(13): 1906-1911
http://www.wjgnet.com/1007-9327/13/1906.asp
INTRODUCTIONNon-cirrhotic portal hypertension is a constellation of liver disorders in which liver cirrhosis is not present and the main clinicopathologic findings are encountered in the portal venous system. Portal vein thrombosis,
portal vein cavernous transformation, parasitic diseases (schistosomiasis), peliosis hepatis, nodular regenerative hyperplasia of the liver, congenital hepatic fibrosis and veno-occlusive liver disease constitute the major sub-categories. The most confusing and complex disorder is idiopathic portal hypertension (IPH). IPH is characterized by non-pathognomonic pathological changes (with the absence of cirrhosis) of the liver in addition to findings of portal hypertension. Study groups (mostly originating from Japan and India) have not reached a conclusion on the nomenclature for IPH. Indian study groups prefer to use the term non-cirrhotic portal fibrosis, while Japanese researchers call the disorder IPH. American study groups named the disorder as hepatoportal sclerosis[1]. Regardless of the nomenclature used, IPH is a distinct clinical entity with characteristic features including interesting and complicated pathogenetic mechanisms.
In this review we will discuss the pathogenesis, clinicopathologic findings, treatment and prognosis of this disorder under the light of current knowledge.
PATHOGENESISIPH has a non-sporadic character in some parts of the world. It is one of the most important causes of portal hypertension and esophageal variceal bleeding. The latter case is encountered in Indian continent. In India, IPH causes up to 25% of all esophageal variceal bleeding[2]. Such a heterogeneous distribution is most probably due to possible underlying etiological factors. The pathogenesis of this disorder is still not understood and there is some controversy about this subject.
Theories on the pathogenesis of IPHAlthough there are some theories on the pathogenesis of IPH, unfortunately none have proved to be a single factor fully explaining the pathogenesis. These theories include the trace element-chemical theory, autoimmunity theory, infection theory, thrombosis theory and genetic theory.
IPH seems to be a multifactorial disease in which two or more etiological factors may play a role.
Trace element-chemical theoryChronic exposure to arsenic[3] or vinyl chemicals[4] has been incriminated in the development of IPH. Exposure to these chemical substances for a long time may result in histological findings resembling hepatoportal sclerosis. Although it is tempting to incriminate these chemicals as primary etiological factors, most patients never report such

Harmanci O et al. Idiopathic portal hypertension 1907
www.wjgnet.com
exposure. Some drugs and pharmaceutical agents may also result in clinicopathologic findings mimicking IPH. Vitamin A toxicity, methotrexate and 6-mercaptopurine may result in such a clinical picture[5].
Autoimmunity theoryBased on several reports in the literature, there is some evidence that IPH may be related to autoimmune reactions and immunological abnormalities. It is not known whether this immunity connection is the primary underlying disorder or a coincidental phenomenon. However, it is widely accepted that autoimmune diseases (especially connective tissue diseases) increases the prevalence of IPH in certain patient groups. The most important IPH associated diseases are mixed connective tissue disease (6 cases[6]), systemic sclerosis (20 cases[7]) and systemic lupus erythematosus (16 cases[8]). In addition, a Japanese survey[9] found that hypergammaglobulinemia is the most common presenting autoimmune dysfunction (26% of all included IPH patients), along with chronic thyroiditis as the most common autoimmune disease. Although there may be a coincidental relationship between these disorders, some evidence supports autoimmunity having some effect over IPH in selected patients. For example, platelet-derived growth factor (PDGF), connective tissue growth factor (CTGF) and other cytokines are found to play role in the development of such disorders. These cytokines may result in pathological changes resembling IPH[6], such as peri-biliary fibrosis in the portal tracts and increased myofibroblastic spindle cell activity in the portal tracts. As previously noted by Nakanuma et al[10], the increased expression of adhesion molecules in portal venous endothelial lining cells may also reflect the secondary effects of these autoimmune diseases.
Infection theoryChronic or recurrent infections are incriminated as an etiological factor for the development of IPH. The insidious clinical nature of IPH supports this theory. Chronic exposure to antigenemia of intestinal origin may result in mild portal inflammation. When repetitive antigenemia occurs for a long time, these successive inflammatory reactions in portal tracts may trigger the pathological changes to eventually result in IPH. The cornerstone studies that first showed the importance of portal antigenemia in the development of IPH-like pathological changes were animal studies performed in rabbits and dogs[11-13]. The profound high prevalence of IPH in India is attributed to increased prevalence of abdominal and intestinal infections[14].
Thrombosis theoryThe most controversial issue in IPH is the contribution of portal tract thrombosis in the pathogenesis of IPH. As stated previously by Okuda[15], the Japanese Research Committee on IPH argues against the thrombosis theory based on the following: (1) the insidious onset of IPH, (2) increased splenic blood flow in IPH (splenic blood flow is expected to decrease in portal vein thrombosis), (3) no evidence of increased thrombophilia in IPH cases,
and (4) biopsy examinations of IPH cases[16] reveal a very low percentage of portal vein thrombosis (2, 3%). However, some of these points conflict. In our opinion, the thrombosis theory must be expanded as “repetitive micro-thrombosis theory”. We believe that in the very early stages, clinically undetectable micro-thrombosis in the small intrahepatic branches of the portal vein eventually result in periportal fibrosis-like reconstruction. The disease then becomes clinically evident when portal hypertension develops, either as splenomegaly (related to anemia or found incidentally) or variceal bleeding. In the very late stages, overt portal vein thrombosis in the major branches is observed. These suggestions are supported by other studies (discussed later). In our own unpublished data, the prevalence of thrombophilic factors (either genetic or acquired) is found to be increased. In our IPH patient population, protein C-S deficiencies were found in 13%, the factor-Ⅱ mutation was found in 4.3%, the factor-Ⅴ Leiden mutation in 13%, paroxysmal nocturnal hemoglobinuria in 4.3% and systemic lupus erythematosus in 4.3% of all IPH patients. We believe that the increased prevalence of thrombophilic factors in this patient population is beyond the point of coincidence.
Therefore, extrahepatic portal vein obstruction secondary to thrombosis is a distinct clinical entity and should not be confused with the thrombosis theory. Secondly, there are studies that suggest thrombosis of the portal vein occurs during the course of IPH. In 2005, Matsutani et al[17] showed that 2 of 22 IPH patients had partial portal vein thrombosis at the time of IPH diagnosis, and another 7 patients (who were free of thrombosis at the time of IPH diagnosis) developed portal vein thrombosis in a mean period of 10.5 years. The authors believe that frequent follow up ultrasonography screening may be helpful in identifying thrombosis attacks. The IPH patients with portal vein thrombosis in this study were found to have a significantly higher degree of decreased liver functions, refractory ascites, hypersplenism and poor general status. In 2002, Hillaire et al[18] investigated a group of IPH patients for prothrombotic conditions. They initially included 42 patients with predominant pathological findings of IPH but excluded 11 patients with concurrent portal vein thrombosis. Additionally, during follow up of the final 28 patients studied, 13 of the patients developed portal vein thrombosis and 9 of them were also positive for an overt prothrombotic condition. The authors concluded that in 50% of the IPH cases, a prothrombotic disorder was found (which is similar to our patient population) and patients with portal vein thrombosis were given a poor prognosis, indicating advanced disease and poor liver capacity.
T hese two s tud ies were per for med a f te r the comprehensive review by Nakanuma et al[10] who suggested a staging system for IPH cases. They proposed that 4 stages are observed and identifiable in IPH. Stage-1 indicates the earliest stage of IPH where the liver is free of apparent atrophy, and stage-4 consistes of advanced liver atrophy together with portal vein thrombosis, which is clinically detectable and indicates a poor prognosis. This classification system also defines the stages of the natural

history of IPH. We believe that undetectable micro-portal venule thrombosis is one of the major contributors to the development of IPH and detectable macro-portal vein thrombosis is an indicator of advanced IPH with poor prognosis. This topic is very controversial and is open for prospective follow-up studies to investigate the contribution of thrombosis in the pathogenesis of IPH. Additionally, there is no prospective study in the literature that shows clinicopathologic and histologic progression of a total thrombosis in the main portal vein or intrahepatic portal vein branches to IPH.
Genetic theoryIn the literature, there is a lack of genetic studies in IPH. In 1987, Sarin et al[19] found the association of a high degree of HLA-DR3 aggregation in family members with IPH. Interestingly, in 2005 and 2006, two case reports[20-22] associated IPH with a genetic syndrome called “Adams-Oliver” syndrome. It is characterized by skeleton abnormalities (i.e. limp and extremity malformations, brachydactyly, poly and oligodactyly, hypoplastic nails, scalp and skull defects) and congenital heart defects. Extrahepatic portal vein thrombosis is not accepted as a component of the syndrome. This syndrome is mostly inherited in an autosomal dominant fashion and is related to undefined genes that regulate vascular development. Among these 4 cases, two of them were found to have portal vein thrombosis along with a diagnosis of IPH. The authors believe that this vascular genetic disorder predisposes to thrombosis and IPH. Interestingly, the only genetic link to IPH also seems to be dependent on the thrombosis theory.
Single factor or multifactorial disease?The l i terature includes conf l ict ing studies about the etiopathogenesis of IPH, which has resulted in controversy. We believe that this is a multifactorial disease that includes a possible genetic background and onset may
occur at any time in life. Thrombosis throughout the portal system probably plays a key role in both the initiation and progression of the disease. Our proposal about the etiopathogenesis of IPH is shown in Figure 1.
PATHOLOGICAL FINDINGS IN IPHThe pathological findings in IPH are very heterogeneous and have been studied in detail by various groups[10,18,23,24]. This heterogeneity is most probably due to changes occurring with the progression of the disease and changes in hepatic blood flow dynamics.
Grossly, the liver may be atrophic and/or nodular. Liver atrophy is a later finding in the course of the disease due to collapse of the peripheral liver architecture, along with ischemia related hepatocyte drop-out via apoptosis. In histological examination (Figure 2), the classical findings of IPH can be divided into two aspects. The primary finding directly related to IPH is intimal fibroelastic thickening of medium and small branches of the intrahepatic portal vein. The second aspect, resulting secondarily to obliterated portal branches, are aberrant neo-vascular formations, sinusoidal dilatation and hepatocellular nodular hyperplasia. (NOTE: Neo-vascular formations are sometimes termed “herniation of portal vein” and these neo-vessels are thought to arise from vasa septalis or inlet venules. These vessels play an important role in shunting blood flow from the obliterated portal segment towards unaffected sites).
At autopsy, there is a high degree of thrombosis in medium to large portal branches. Indian and Japanese authors are in agreement on this topic. In advanced cases, liver examination at autopsy revealed this common finding in the previously mentioned studies. Additionally, at various anatomic segments of the portal tract, depending on the biopsy site (e.g. wedge biopsy, needle biopsy and autopsy findings), thrombosis in the portal venous system was also found[10,18,25]. These findings support the staging system suggested by Nakanuma et al[10] in which grossly evident portal thrombosis is accepted as an advanced stage finding. In Figure 3, splenoportography of a patient with IPH (who was diagnosed and followed in our hospital) is shown. The patient developed a portal vein thrombosis during follow up. Although there are 2 major sites of thrombosis in the
Increased risk of thrombophilia Genetic backgroud Genetic risks of thrombophilia Other unknown predisposing factors? Acquired thrombophilic factors
Internal triggering factors of thrombosis? Autoimmune diseases Subclinical infections Repeated portal antigenemia Repeated portal microthrombi
External triggering factors of thrombosis? Chemical toxicity Drugs Pica Specific infectious pathology? (Unknown viral hepatitis)
Repetetive microthrombosis in the portal venous system resulting in periportal fibrosis
Increased hepatic fibrosis mainly around portal tracts, increased portal hypertension and finally overt iph
Figure 1 Our proposed theory about etiopathogenesis of idiopathic portal hypertension.
Figure 2 The histological examination of the portal vein shows periportal thickening (marked by asterisks) found in an IPH patient.
1908 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com
major portal veins, the portal vein at the hepatic hilum is found to be dilated despite a severely occluded portal vein. This finding is very suggestive of concomitant IPH (Figures 3 and 4).
CLINICAL FINDINGS IN IPHA cl inical definit ion of IPH is “a state of por tal hypertension in which no liver disease is found”. Thus IPH is usually the final diagnosis of an exclusion process. Usually IPH patients are in good condition, have near normal or normal liver functions (including normal albumin levels and prothrombin time) but present with variceal bleeding that is detected in investigations of hypersplenism. Ascites is almost always a finding of advanced cases in which the liver atrophies and residual capacity is limited.
There is no known male or female preponderance of IPH; both sexes are believed to be equally affected. There is a slight female preponderance in some studies[23,26] and this may be related to autoimmune disease, which is a condition mostly observed in the female population.
Clinical presentation has been studied in two large Indian studies. Sarin et al[14] reported that 13.5% of patients had splenomegaly, 84.5% of patients had a history of upper gastrointestinal bleeding, 92% of patients had esophageal varices, and 22.3% of patients had gastric varices. When they compared these patients with portal vein thrombosis patients, the IPH group was found to
have larger spleens but lower prevalence of ascites, gastric varices, history of upper gastrointestinal bleeding and almost no jaundice. In another Indian study, Dhiman et al[23] reported that 96.7% of patients had splenomegaly and 64 . 9% o f pa t i en t s had a h i s to r y o f uppe r gastrointestinal bleeding. Interestingly, the durations of patient presentation to a medical unit after onset of symptoms were within 12 mo (36.4%), 13-60 mo (27.2%), 61-120 mo (20.5%) and more than 120 mo (15.9%). This is possibly a reflection of medical care problems in a developing country. Another study by Okuda et al[26] reported different results. In 1984, they reported 86 cases of IPH. The main presenting symptom of these patients were anemia related symptoms (26.2%), hematemesis (23.7%) and splenomegaly (18.4% of all patients, mostly detected and referred by a physician). They detected varices in 84% of all cases. The clear difference in presentation patterns suggests that there are two, non homogenous IPH groups in these countries. But we believe that this important clue has a very important and basic message: the Indian IPH population is formed of more chronic and advanced IPH cases than the Japanese and Western cases. We believe this is one of the main causes of conflict in the IPH literature.
COMPLICATIONS AND TREATMENTThe major complications of IPH can be summarized as esophageal varices and hypersplenism. Although portal hypertension is very evident (Figure 5), the absence of liver dysfunction (except for very chronic and late stages of the disease) is a protective factor against these aforementioned complications. Ascites, hepatic encephalopathy, abnormal liver function tests and jaundice are rarely observed and all of these findings are transient if occurring at all. Interestingly, in contrast to this judgment, the studies from India state that 70% of IPH patients present with a history of major variceal bleeding[27]. This might not be confusing because, as previously stated, the Indian patient population harbors chronologically older IPH disease and patients present in later stages.
Esophageal varicesThe esophageal varices (EV) that form during the course
Figure 3 Sp lenopo-r tography shows two major sites of thrombosis in portal venous system (shown by aster isks). Note the dilated portal ve in at hepat ic h i lum d e s p i t e p o r t a l v e i n thrombosis.
Figure 4 The computed tomography of the same patient in figure-3. Note the thrombosis in the portal vein (Marked by arrow).
Figure 5 Splenoportography shows patent portal vein with severe dilation and collateral circulation. Note the small caliber of intrahepatic portal vein branches compared to extrahepatic portal vein.
Harmanci O et al. Idiopathic portal hypertension 1909
www.wjgnet.com

of IPH have some distinctive features. The walls of these variceal veins are relatively thicker than the varices observed in cirrhosis. They rarely harbor “red-spots” that herald a variceal bleeding. They are simply dilated veins that rarely complicate, and are relatively easy to treat compared to cirrhosis. Varices with varying degrees are found in almost all of the patients with IPH. In some surveys, the EV is reported to be found in 90% of the IPH patients[2].
One of the important issues in management of EV in IPH is the mode of treatment. The portal hypertension that occurs in IPH is not accompanied by liver synthetic dysfunction. The mechanisms that play a major role in cirrhotic patients are not observed in IPH due to a liver that remains functioning. The hyper-dynamic mesenteric circulation and imbalance in vasoactive mediators are not observed in IPH. Therefore, conventional medical treatment regimens, such as beta-blockers and nitrates, can be accepted as ineffective due to the lack of these mentioned changes. This is supported by a recent study by Sarin et al[28] in which medical treatment was found to be inferior to endoscopic variceal ligation (EVL) in patients with IPH in reducing the rebleeding rate. One of the major disadvantages of this report is the low number of patients studied. Another uncontrolled study from India found that variceal sclerotherapy reduces the monthly rebleeding rate 14-fold[27].
Another management problem is the development of portal hypertensive gastropathy and gastric varices. Although these two conditions are rarely observed in IPH cases, EVL may have serious adverse effects in that EVL may induce their formation. Once formed, gastric varices should be treated by intravariceal glue-injection therapy, which is an effective modality of eradication. Portal gastropathy in IPH cases are usually transient. The formation of new spontaneous shunts after a successful eradication program is an expected finding and is protective against varices formation.
HypersplenismLevels of all blood elements begin to decrease as the condition worsens and severe hypersplenism is now considered as one of the most important indications for splenectomy in this g roup of patients. In our personal experience, it may be good practice to select patients according to their co-morbid conditions, degree of hypersplenism and the condition of varices. Also splenectomy has been found to be more complicated in patients with massive splenomegaly, and these patients must be followed carefully after this surgery.
PROGNOSISThe overall prognosis for IPH is almost always excellent. This is mostly related to and dependent on preserved liver synthetic functions. The most important factor is the follow up of EV and management of EV by EVL or sclerotherapy. One important follow-up study from Japan reported very important findings about the prognosis of
these patients[29]. Thirty per cent of all deaths were due to esophageal bleeding while 25% were due to hepatic insufficiency. The major adverse prognostic factor were sex and age of onset. Male patients showed a shorter life-span than females, and the patients who had onset of disease before 40 years of age showed poorer prognoses. These data (despite the small size of the study) suggest that early onset of disease results in earlier completion of the natural course. The disease resembles a cascade in which liver atrophy is the main rate-limiting step in the course of IPH. The overall prognosis can be placed between the normal population and cirrhotic patients.
CONCLUSIONIPH is a heterogenous disorder with varying clinical pictures, including asymptomatic splenomegaly and recurrent variceal bleeding. Its etiology is still unknown, but some evidence and epidemiological studies suggest that it is a multifactorial disease with a genetic basis. Current knowledge indicates that some triggers are intra-abdominal infections, autoimmune processes, and chemical toxicity. Although the prognosis is excellent, careful follow up and management of patients, with extra attention to treatment esophageal varices, is required. Further studies are needed in order to clarify the issues of etiology and possible genetic background.
REFERENCES1 Mikkelsen WP, Edmondson HA, Peters RL, Redeker AG,
Reynolds TB. Extra- and intrahepatic portal hypertension without cirrhosis (hepatoportal sclerosis). Ann Surg 1965; 162: 602-620
2 Sarin SK, Aggarwal SR. Idiopathic portal hypertension. Digestion 1998; 59: 420-423
3 Huet PM, Guillaume E, Cote J, Légaré A, Lavoie P, Viallet A. Noncirrhotic presinusoidal portal hypertension associated with chronic arsenical intoxication. Gastroenterology 1975; 68: 1270-1277
4 Thomas LB, Popper H, Berk PD, Selikoff I, Falk H. Vinyl-chloride-induced liver disease. From idiopathic portal hypertension (Banti's syndrome) to Angiosarcomas. N Engl J Med 1975; 292: 17-22
5 Sarin SK. Non-cirrhotic portal fibrosis. Gut 1989; 30: 406-4156 Rai T, Ohira H, Fukaya E, Abe K, Yokokawa J, Takiguchi
J, Shishido S, Sato Y. A case of merged idiopathic portal hypertension in course of mixed connective tissue disease. Hepatol Res 2004; 30: 51-55
7 Takagi K, Nishio S, Akimoto K, Yoshino T, Kawai S. A case of systemic sclerosis complicated by idiopathic portal hypertension: case report and literature review. Mod Rheumatol 2006; 16: 183-187
8 Inagaki H , Nonami T, Kawagoe T, Miwa T, Hosono J, Kurokawa T, Harada A, Nakao A, Takagi H, Suzuki H, Sakamoto J. Idiopathic portal hypertension associated with systemic lupus erythematosus. J Gastroenterol 2000; 35: 235-239
9 Saito K, Nakanuma Y, Takegoshi K, Ohta G, Obata Y, Okuda K, Kameda H. Non-specific immunological abnormalities and association of autoimmune diseases in idiopathic portal hypertension. A study by questionnaire. Hepatogastroenterology 1993; 40: 163-166
10 Nakanuma Y , Tsuneyama K, Ohbu M, Katayanagi K. Pathology and pathogenesis of idiopathic portal hypertension with an emphasis on the liver. Pathol Res Pract 2001; 197: 65-76
1910 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

11 Sugita S , Ohnishi K, Sai to M, Okuda K. Splanchnic hemodynamics in portal hypertensive dogs with portal fibrosis. Am J Physiol 1987; 252: G748-G754
12 Kono K, Ohnishi K, Omata M, Saito M, Nakayama T, Hatano H, Nakajima Y, Sugita S, Okuda K. Experimental portal fibrosis produced by intraportal injection of killed nonpathogenic Escherichia coli in rabbits. Gastroenterology 1988; 94: 787-796
13 Kathayat R, Pandey GK, Malhotra V, Omanwar S, Sharma BK, Sarin SK. Rabbit model of non-cirrhotic portal fibrosis with repeated immunosensitization by rabbit splenic extract. J Gastroenterol Hepatol 2002; 17: 1312-1316
14 Sarin SK. Non-cirrhotic portal fibrosis. J Gastroenterol Hepatol 2002; 17 Suppl 3: S214-S223
15 Okuda K. Non-cirrhotic portal hypertension: why is it so common in India? J Gastroenterol Hepatol 2002; 17: 1-5
16 Okudaria M. Report of the subcommittee for the study of intrahepatic portal thrombosis in IPH. In: Futagawa S, editor. 1994 Report of the Research Committee on Aberrant Portal Hemodynamics. Japan: Japanese Ministry of Health and Welfare, 1995: 69-70
17 Matsutani S, Maruyama H, Akiike T, Kobayashi S, Yoshizumi H, Okugawa H, Fukuzawa T, Kimura K, Saisho H. Study of portal vein thrombosis in patients with idiopathic portal hypertension in Japan. Liver Int 2005; 25: 978-983
18 Hillaire S, Bonte E, Denninger MH, Casadevall N, Cadranel JF, Lebrec D, Valla D, Degott C. Idiopathic non-cirrhotic intrahepatic portal hypertension in the West: a re-evaluation in 28 patients. Gut 2002; 51: 275-280
19 Sarin SK, Mehra NK, Agarwal A, Malhotra V, Anand BS, Taneja V. Familial aggregation in noncirrhotic portal fibrosis: a report of four families. Am J Gastroenterol 1987; 82: 1130-1133
20 Girard M, Amiel J, Fabre M, Pariente D, Lyonnet S, Jacquemin E. Adams-Oliver syndrome and hepatoportal sclerosis: occasional association or common mechanism? Am J Med
Genet A 2005; 135: 186-18921 Pouessel G , Dieux-Coeslier A, Wacrenier A, Fabre M,
Gottrand F. Association of Adams-Oliver syndrome and hepatoportal sclerosis: an additional case. Am J Med Genet A 2006; 140: 1028-1029
22 Swartz EN, Sanatani S, Sandor GG, Schreiber RA. Vascular abnormalities in Adams-Oliver syndrome: cause or effect? Am J Med Genet 1999; 82: 49-52
23 Dhiman RK, Chawla Y, Vasishta RK, Kakkar N, Dilawari JB, Trehan MS, Puri P, Mitra SK, Suri S. Non-cirrhotic portal fibrosis (idiopathic portal hypertension): experience with 151 patients and a review of the literature. J Gastroenterol Hepatol 2002; 17: 6-16
24 Abramowsky C , Romero R, Heffron T. Pathology of noncirrhotic portal hypertension: clinicopathologic study in pediatric patients. Pediatr Dev Pathol 2003; 6: 421-426
25 Willner IR, Waters B, Payton JD. Hepatoportal sclerosis: clinical features and association with hypercoagulable states. Hepatology 1997; 26: 204A
26 Okuda K, Kono K, Ohnishi K, Kimura K, Omata M, Koen H, Nakajima Y, Musha H, Hirashima T, Takashi M. Clinical study of eighty-six cases of idiopathic portal hypertension and comparison with cirrhosis with splenomegaly. Gastroenterology 1984; 86: 600-610
27 Chawla YK, Dilawari JB, Dhiman RK, Goenka MK, Bhasin DK, Kochhar R, Singh K, Kaur U. Sclerotherapy in noncirrhotic portal fibrosis. Dig Dis Sci 1997; 42: 1449-1453
28 Sarin SK, Wadhawan M, Gupta R, Shahi H. Evaluation of endoscopic variceal ligation (EVL) versus propanolol plus isosorbide mononitrate/nadolol (ISMN) in the prevention of variceal rebleeding: comparison of cirrhotic and noncirrhotic patients. Dig Dis Sci 2005; 50: 1538-1547
29 Ichimura S, Sasaki R, Takemura Y, Iwata H, Obata H, Okuda H, Imai F. The prognosis of idiopathic portal hypertension in Japan. Intern Med 1993; 32: 441-444
S- Editor Liu Y L- Editor Lutze M E- Editor Zhou T
Harmanci O et al. Idiopathic portal hypertension 1911
www.wjgnet.com

Hepatic venous outflow obstruction: Three similar syndromes
Ulas Darda Bayraktar, Soley Seren, Yusuf Bayraktar
www.wjgnet.com
Yusuf Bayraktar, MD, Series Editor
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1912-1927www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
Ulas Darda Bayraktar, Department of Internal Medicine, Interfaith Medical Center, Brooklyn, NY 11230, United StatesSoley Seren, Department of Internal Medicine, Wayne State University, Detroit, MI, United StatesYusuf Bayraktar, Division of Gastroenterology, Hacettepe University School of Medicine, Ankara, TurkeyCorrespondence to: Ulas Darda Bayraktar, MD, Department of Internal Medicine, Interfaith Medical Center, 229 Parkville Ave Apt# 4B, Brooklyn, NY 11230, United States. [email protected]: +1-718- 4314657 Received: 2006-10-11 Accepted: 2006-11-14
AbstractOur goal is to provide a detailed review of veno-occlusive disease (VOD), Budd-Chiari syndrome (BCS), and congestive hepatopathy (CH), all of which results in hepatic venous outflow obstruction. This is the first article in which all three syndromes have been reviewed, enabling the reader to compare the characteristics of these disorders. The histological findings in VOD, BCS, and CH are almost identical: sinusoidal congestion and cell necrosis mostly in perivenular areas of hepatic acini which eventually leads to bridging fibrosis between adjacent central veins. Tender hepatomegaly with jaundice and ascites is common to all three conditions. However, the clinical presentation depends mostly on the extent and rapidity of the outflow obstruction. Although the etiology and treatment are completely different in VOD, BCS, and CH; the similarities in clinical manifestations and liver histology may suggest a common mechanism of hepatic injury and adaptation in response to increased sinusoidal pressure.
© 2007 The WJG Press. All rights reserved.
Key words: Veno-occlusive disease; Sinusoidal obstru-ction syndrome; Budd-Chiari syndrome; Congestive hepatopathy
Bayraktar UD, Seren S, Bayraktar Y. Hepatic venous outflow obstruction: Three similar syndromes. World J Gastroenterol 2007; 13(13): 1912-1927
http://www.wjgnet.com/1007-9327/13/1912.asp
INTRODUCTIONAlthough the liver makes up < 3% of the total body
weight, it receives one-quarter of the total cardiac output through the hepatic artery and portal vein[1]. Blood is drained from the hepatic acini via central veins into sublobular veins and then into the right, left, and middle hepatic veins, the inferior vena cava and the right atrium[2]. An obstruction to the blood flow out of the liver can result in a spectrum of clinical abnormalities ranging from acute hepatic failure to passive hepatic congestion, depending on the acuity and level of obstruction.
Hepatic venous outflow obstruction (HVOO) can be divided into three categories according to the level of obstruction: (1) Veno-occlusive disease (VOD): at the level of sinusoids and terminal venules, (2) Budd-Chiari syndrome (BCS): from hepatic veins to the superior end of inferior vena cava, and (3) Venous obstruction at the level of heart referred to as congestive hepatopathy (CH) (Figure 1).
The etiology of VOD, BCS, and CH are entirely different (Table 1). It should be noted that VOD develops within three weeks of an acute insult to the sinusoidal endothel ia l cel ls while BCS and CH may develop within a few days or may take several years of venous thrombosis and heart failure, respectively. The variations in the ‘acuteness’ of venous obstruction leads to subtle differences in the clinical presentations of VOD, BCS, and CH. Although patients with HVOO generally present with abdominal pain due to hepatomegaly, and jaundice and ascites due to portal hypertension; chronic BCS patients may first present with cirrhosis and its complications. BCS patients with inferior vena cava obstruction may also have leg edema and venous collaterals over the trunk. Additionally, signs and symptoms of heart failure such as jugular venous distention, leg edema, and dyspnea may be seen in patients with CH. Nonetheless, the histological findings in all three syndromes are almost identical and include sinusoidal congestion and hepatocyte necrosis predominating in perivenular areas of hepatic acini which eventually leads to bridging fibrosis between adjacent central veins (Table 1)[1,3,4].
The laboratory findings in VOD, BCS, and CH are also very similar. Hyperbilirubinemia is universal in HVOO except in patients with constrictive pericarditis[5], and is believed to be due to hepatocellular dysfunction, hemolysis, and biliary canalicular obstruction secondary to distended hepatic veins[6]. Serum aminotransferase levels may be mildly elevated except in fulminant BCS and CH with severe cardiac output impairment causing hepatic ischemia when the levels may exceed 1000 U/L. Alkaline phosphatase levels may also be mildly elevated.

Bayraktar UD et al. Hepatic venous outflow obstruction 1913
www.wjgnet.com
New markers for diagnosis of VOD such as plasminogen activator inhibitor-1 are under investigation.
The gold standard for the diagnosis of VOD is liver biopsy. However, because of the risks of liver biopsy in thrombocytopenic patients, the diagnosis is primarily based on clinical criteria. CH is usually diagnosed by routine laboratory tests in patients with symptomatic heart failure. However, physicians may miss the diagnosis of CH in patients with constrictive pericarditis who do not have overt symptoms. Echocardiogram and invasive cardiac hemodynamic evaluation may be necessary in patients with a strong suspicion of CH. Diagnosis of BCS, unlike VOD and CH, is greatly dependant on radiological studies, although a careful history and physical examination is crucial in order not to overlook this protean syndrome. Doppler sonogram followed by venogram are the first line tests in patients with suspected BCS (Table 2).
The treatment and prognosis of VOD, BCS, and CH are summarized in Table 2. It should be noted that despite
new promising therapies, prevention is still the mainstay of VOD management because of its high mortality and the limited efficacy of current therapeutic modalities. Treatment of BCS depends on the site and extent of the obstruction, and varies from sole anticoagulation to TIPS and liver transplantation. Treatment of the underlying heart disease is the basis of the management of CH.
Following are the detailed reviews of VOD, BCS, and CH.
HepaTIC VeNO-OCClUsIVe DIsease (sINUsOIDal ObsTRUCTION syNDROme)Hepatic VOD is characterized by tender hepatomegaly, fluid retention, weight gain, and jaundice. This condition is seen typically after hematopoietic stem cell transplantation (HSCT)[7], high dose abdominal radiation therapy[8], use of certain chemotherapeutic agents[9], ingestion of pyrrolizidine alkaloids[10,11], and liver transplantation[12]. In 1954, Bras et al [13] first described veno-occlusive disease in Jamaican children who developed occlusion of the tributaries of the hepatic vein with subsequent centrilobular or non-portal fibrosis, associated with poisoning with Senecio type alkaloids. The term, sinusoidal obstruction syndrome was introduced in 2002 by deLeve et al[14] to replace VOD based on their studies identifying the primary site of injury as the sinusoidal endothelial cells, which results in fibrosis and obstruction of hepatic venous outflow. VOD after HSCT will be the focus of our review since it is the most common and studied form of VOD.
Clinical presentationThe reported incidence of VOD after HSCT varies greatly from 5% to 50% largely due to differences in the chemotherapeutic conditioning regimens[15]. Most patients develop signs and symptoms of VOD within the first three
Congestive HepatopathyIncreased right atrial pressureCongestive heart failureCor pulmonalePericardial disease
Budd-Chiari syndromeFrom hepatic veins to right atriumHepatic vein thrombosisInferior vena caval webs
Veno-occlusive diseaseObstruction in sinusoids and central veins
Figure 1 Site of venous obstruction in veno-occlusive disease, Budd-Chiari syndrome, and congestive hepatopathy.
Table 1 Definition, etiology and histology of VOD, BCS, and CH
VOD: Veno-occlusive disease, BCS: Budd-Chiari Syndrome, CH: Congestive hepatopathy, IVC: Inferior vena cava, HSCT: Hematopoietic stem cell transplantation, CHF: Congestive heart failure, CAD: Coronary artery disease, COPD: Chronic obstructive pulmonary disease, ILD: Interstitial lung disease, SEC: Sinusoidal endothelial cell, PVT: Portal vein thrombosis.
VOD BCS CHSite of venous obstruction Hepatic sinusoids and terminal venules From hepatic veins to the superior end
of IVCHeart
Etiology Sinusoidal endothelial injury due to HSCT, chemotherapy, abdominal radiotherapy, and pyrrolizidine alkaloids
Hepatic vein thrombosis, IVC webs, compression of hepatic veins or IVC by tumor, cyst, or abscess
Increased right atrial pressure due to CHF (CAD, cardiomyopathies, valve abnormalities), cor pulmonale (COPD, ILD, pulmonary HTN), and pericardial disease (constrictive pericarditis, pericardial tamponade)
Histology Changes predominantly in perivenular areas
Gaps in SEC barrier leading to subendothelial edema
Narrowing of central veins and sinusoids with sinusoidal congestion and hepatocellular necrosis
Collagen accumulation in sinusoids and veins leading to bridging fibrosis between central veins
Predominantly in perivenular areas except in presence of concomitant PVT.
Sinusoidal congestion followed by ischemic cell necrosis and bridging fibrosis between central veins
Caudate lobe hypertrophy, with fibrosis and atrophy in the rest of liver
Predominantly in perivenular areasSinusoidal congestion and hepatocellular necrosisBridging fibrosis between central veins leading to cardiac fibrosis in chronic cases

weeks after HSCT. The initial signs are sudden weight gain and development of hepatomegaly[16]. Weight gain is believed to be due to renal retention of water and sodium. Hyperbilirubinemia is seen after a few days, sometimes followed by increase in serum transaminase and alkaline phosphatase levels. The first symptom and generally the only one reported by patients with VOD is right upper quadrant pain, likely due to acute congestion of liver[17]. Physical exam reveals tender hepatomegaly and ascites in 83% and 39% patients with histologically proven VOD respectively[18].
Patients with VOD generally require more platelet transfusions than patients without liver disease[19]. This might be because of ongoing thrombotic process in the liver sinusoids or increased splenic sequestration due to portal hypertension[17]. Patients with VOD may develop renal failure as a result of hepatorenal syndrome. The prevalence of renal, cardiac, and pulmonary failure was noted to be higher in patients with VOD than in those without VOD in a report of 355 patients who underwent HSCT[16]. Coagulation factor deficiencies and prolonged prothrombin time can be encountered as the hepatic function declines.
The overall mortality is reported to be between 3% and 67%[20,21]. The severity of VOD can be classified as mild (requiring no treatment and with complete resolution), moderate (requiring treatment with diuretics and pain medications and with complete resolution), and severe (requiring treatment and no resolution before death or by transplant d 100). In one large series, the mortality rate was reported to be 98%, 23%, and 9% in patients with severe, moderate, and mild VOD, respectively[16]. Patients with severe VOD differed from those with mild or moderate VOD in the amount of weight gained, increment speed of total serum bilirubin level, and frequency of edema and ascites. One drawback of this classification is that it can be used only for research purposes because of its retrospective assessment. Therefore, in 1994 Bearman
et al[22] formulated a mathematical model to predict the severity of VOD based on alterations in weight and serum bilirubin level.
Risk factorsSeveral pretransplant and transplant-associated risk factors have been identified in different studies.
One of the most important independent risk factors of VOD is the presence of liver injury prior to HSCT. In a cohort of 1652 patients, the relative risk of VOD in the presence of pre-transplant elevated AST levels and prior radiotherapy to the abdomen were 2.4 and 2.9, respectively[15]. In addition, the presence of hepatic metastases of solid tumors was associated with an increased risk of VOD[21]. Although hepatitis C virus (HCV) infection was reported to increase the risk of VOD in one study[23], in another study on 355 patients the risk for VOD was found not to be increased unless the pretransplant AST levels were elevated[24].
Advanced age[25] and previous HSCT may be associated with higher incidence of VOD[16,17]. Additionally, Factor V Leiden and prothrombin G20210A mutation may be associated with VOD[26,27].
Medications used before, during or after HSCT may increase the risk of VOD. Prior therapy with gemtuzumab ozogamicin, a calicheamicin-conjugated monoclonal CD33 antibody used in the treatment of acute myelogenous leukemia, significantly increases the risk of VOD, regardless of the dose and serum AST levels[28]. The use of vancomycin or acyclovir in the pre-transplant period has been associated with the development of VOD. However, it is unclear whether the increased risk of VOD is due to antibiotics or the underlying infections that necessitated their use. Additionally, women who received norethisterone to prevent menstruation during HSCT have been found to have an increased incidence of VOD[29,30].
One of the most important risk factors for the development of VOD is the type and dose of the
Table 2 Radiological findings, treatment, and prognosis in VOD, BCS, and CH
VOD BCS CHRadiological findings Ultrasonography to rule out other liver
disordersDoppler: Abnormal flow in a hepatic vein; large intrahepatic collateral vessels; enlarged, stenotic, or tortuous hepatic veins
Dilatation of all three hepatic veins on sonogram
Doppler may show reverse blood flow in the portal vein
MRI: Large intrahepatic comma shaped collaterals. Hepatic venography: Spider web venous network pattern
ECHO: Increased pulmonary artery pressure, dilatation of right side of heart, TR, abnormal diastolic ventricular filling due to pericardial disease
Treatment (1) Prevention: UDCA, heparin, LMWH, and defibrotide
(1) Prevention of thrombus extension: Anticoagulation with heparin and warfarin
Treatment of the underlying heart disease
(2) Treatment: Symptomatic care, defibrotide, tPA, AT-Ⅲ concentrate
(2) Restoration of blood flow: Thrombolytic therapy, percutaneous, angioplasty, TIPS, or shunt surgery
Pericardiectomy in constrictive pericarditis
(3)TIPS and liver transplantation in selected cases
(3) Liver transplantation
Prognosis Mortality rate between 9% to 98% depending on the severity
Five-year survival rate 42% to 89% in hepatic vein thrombosis and 25% in IVC obstruction
Liver disease rarely contributes to mortality in these patients
MRI: Magnetic resonance imaging, TR: Tricuspid regurgitation, ECHO: Transthoracic echocardiogram UDCA: Ursodeoxycholic acid, LMWH: Low-molecular weight heparin, tPA: Tissue plasminogen activator, AT-Ⅲ: Anti-thrombin Ⅲ, TIPS: Transjugular intrahepatic portosystemic shunt.
1914 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

conditioning regimens of HSCT. The incidence of VOD was found to be 3 times higher in patients who received cyclophosphamide in combination with busulfan or total body irradiation (TBI) (> 12 Gy) or carmustine and etoposide[15]. The mechanism is thought to involve acrolein, the inactive toxic metabolite of cyclophosphamide[31] which is detoxified by conjugation with glutathione[32]. Hepatic glutathione reserve is decreased by busulfan, carmustine and TBI, thus explaining the increased risk of VOD by these drug combinations.
It is widely believed that the incidence of VOD is higher after allogeneic stem cell transplantation than after autologous stem cell transplantation. However, some studies have failed to show any difference[16,33]. Furthermore, the differences seen in other studies[15] may be due to higher intensity conditioning regimens used in allogeneic transplantation. Additionally, patients receiving stem cells from unrelated or mismatched donors have higher risk of developing VOD[16].
DiagnosisThe gold standard for the diagnosis of VOD is liver histology. However, because of the risks of liver biopsy in thrombocytopenic patients, diagnosis is primarily based on clinical findings. The Seattle and Baltimore clinical criteria (Table 3) are generally used for the diagnosis of VOD[33,34]. Carreras et al[18] reported that the diagnosis of VOD could be confirmed histologically in 41%, 91%, and 91% of patients satisfying two and three features of Seattle criteria, and Baltimore criteria, respectively. Sensitivity of latter two was 56%.
Liver specimens for histological examination can be obtained through percutaneous, laparoscopic, and transvenous (transjugular or femoral) biopsies. The latter method has the advantage of lower risk of bleeding and the opportunity to measure wedge hepatic venous pressures at the same time. Although transvenous liver biopsy specimens tend to be small, with modern techniques and equipment a representative sample can be obtained[35]. Transfemoral approach often results in crush artifact in the tissues obtained so that the transjugular method is generally preferred. It should be noted that the histological abnormalities may be patchy, especially in the early stages and could lead to false negative biopsy results. In one series, 2 of the 29 patients (7%) died due to intraperitoneal bleeding after transvenous liver biopsies[35]
whereas Carreras et al[18,36] reported no procedure-related deaths in two series of 71 and 30 patients. Hepatic venous pressure gradient of > 10 mmHg was reported to have > 80 % sensitivity and specificity in these series.
Ultrasonography is useful in excluding other disorders that mimic VOD. Yoshimoto et al [37] suggested that detection of reverse blood flow in a segment of portal vein by color-Doppler sonogram is useful for early diagnosis of VOD.
Plasma plasminogen activator inhibitor-1 (PAI-1) level above 100 ng/mL was found to be 100% sensitive and specific for VOD in a series of 31 posttransplant patients with bilirubin levels > 3 mg/dL[38]. Additionally, serum N-terminal peptide of type Ⅲ procollagen and hyaluronic acid levels may also have a diagnostic role[39,40].
A variety of clinical conditions can mimic VOD (Table 4) and it is difficult to differentiate these from VOD in the early stages. The main causes of intrahepatic cholestasis after HSCT are acute graft-versus-host disease (GVHD), cholangitis lenta (sepsis related cholestasis), cyclosporine-induced cholestasis, and VOD. Compared to VOD, GVHD generally develops at a later stage after the appearance of intestinal and cutaneous manifestations and moreover does not cause hepatomegaly or ascites. Although hyperbilirubinemia and increased alkaline phosphatase levels can be seen in sepsis, other features of VOD are rare[17,41,42].
Histology and pathophysiologyDeLeve et al[43] reported that the initial histological change in murine models of VOD was the loss of sinusoidal endothelial cell (SEC) fenestrae and appearance of gaps in SEC barrier. Other early histological findings were narrowing of the sublobular and central veins due to subendothelial edema, likely secondary to disruption of SEC barrier and congestion of hepatic sinusoids surrounded by pale necrotic hepatocytes[3]. Fragmented red blood cells, fibrinogen, and Factor Ⅷ/von Willebrand factor can be demonstrated in the subendothelial space of central veins and perivenular zones of hepatic acini[44]. In the later stages, sinusoidal and venous lumen become obliterated by typeⅠ, Ⅲ, and Ⅳ collagen accompanied by an increase in the stellate cells that line the sinusoids[44,45] (Figure 2). In some cases, fibrous bridges between central venules were observed[14].
One hypothesis for the pathogenesis of VOD is depletion of liver glutathione reserve resulting in diminished ability to detoxify acrolein, an inactive but hepatotoxic metabolite of cyclophosphamide. SECs are especially prone to toxic effects of acrolein because of their lower glutathione level compared to hepatocytes[31,46].
Table 3 Diagnostic criteria for veno-occlusive disease
Seattle criteriaDevelopment of at least 2 of the following 3 clinical features before d 30 after transplantation
Jaundice Hepatomegaly with right upper quadrant pain Ascites and/or unexplained weight gainBaltimore criteria
Development of hyperbilirubinemia with serum bilirubin > 2 mg/dL within 21 d after transplantation and at least 2 of the following clinical signs and symptoms
Hepatomegaly, which may be painful Weight gain > 5% from baseline Ascites
Table 4 Differential diagnosis of veno-occlusive disease
Cholangitis lenta (sepsis-related cholangitis)Drug/parenteral nutrition induced hepatotoxicity/cholestasisAcute graft-versus-host diseaseFungal infectionViral hepatitisCongestive heart failure
Bayraktar UD et al. Hepatic venous outflow obstruction 1915
www.wjgnet.com

Most of the drug metabolism in liver occurs in zone 3 hepatocytes which are rich in p-450 enzymes, explaining why the initial morphological changes are seen in SECs in zone 3. Several findings support this hypothesis. Continuous infusion of glutathione into the portal vein prevents VOD in murine models that were injected with monocrotaline to induce VOD[47]. Additionally, polymorphisms of glutathione S-transferase M1 gene were associated with increased risk of VOD in thalassemic patients undergoing HSCT[48]. Glutathione may also suppress expression of matrix metalloproteinase-9 that was shown to increase in VOD and whose inhibition completely prevents VOD in murine models[14].
Presence of fibrinogen, Factor Ⅷ/von Willebrand factor in the subendothelial space and the finding of low plasma levels of Protein C and antithrombin Ⅲ suggest involvement of the coagulation system in the pathogenesis of VOD[49]. However, it is unclear if coagulation plays a primary role or is a secondary event. Finally, an immunologic mechanism may have a pathogenetic a role in VOD. Increased risk of VOD in mismatched and unrelated transplants, and decreased risk in T-cell depleted HSCT supports this hypothesis[42,50].
PreventionIn the absence of an established effective therapy, prevention of VOD is a priority. Non-myeloablative conditioning regimens may be used in eligible patients with a high risk of VOD. In patients who require myeloablation, several measures that may protect against the development of VOD have been recommended. These include: administering busulfan intravenously and adjusting its dose, increasing the interval between cytotoxic drugs and TBI, reducing the total dose and dose-rate of TBI, shielding the liver during TBI, fractionating TBI and carmustine dose, and administering busulfan after the other agents have been given[42].
Administration of ursodeoxycholic acid (UDCA) prior and during the HSCT has been shown to decrease the incidence of VOD in two randomized, prospective, placebo-controlled studies[51,52]. However, two more recent studies showed no decrease in the incidence of VOD in UDCA-treated patients, although one of them reported lower incidence of grade Ⅲ and Ⅳ acute graft-versus-host
disease. UDCA is well tolerated, and therefore is used in many transplantation centers for prophylaxis.
Low-dose continuous infusion of heparin is associated with decreased incidence of VOD in a few trials[53,54]. However, other studies have failed to show any beneficial effect[15,29,55]; additionally heparin may increase the risk of bleeding. Low-molecular weight heparin is safer and easier to administer, and may have a preventive role in VOD[56,57], and moreover may accelerate platelet engraftment[58]. However, well designed, randomized controlled studies are needed to confirm its efficacy.
Other agents that have been used in the prevention of VOD with varying results are: glutamine infusion, prostaglandin-E1, and pentoxyfilline[59-63]. Defibrotide, is another promising agent found to be effective in the prevention of VOD[64,65].
TreatmentIn most patients the only treatment needed is the use of diuretics and sodium restriction for the management of water and sodium balance. Repeated paracenteses may be required if ascites causes abdominal discomfort or pulmonary compromise. Renal perfusion and intravascular volume should be maintained while avoiding extravascular f luid accumulation[42]. Hepatotoxic drugs should be avoided and infections should be identified and treated promptly. Further treatment approaches vary greatly in different institutions, both in the choice of intervention and choice of patients to be treated.
A modest response to recombinant human tissue plasminogen activator (γ-tPA) and continuous heparin infusion has been reported in several studies[66,67]. However, thrombolytic therapy is limited by its high risk of fatal bleeding[68,69]. In a series of 42 patients, 12 patients responded to γ-TPA and heparin while 10 patients experienced severe bleeding, at least three of which were fatal[70].
Admin i s t r a t ion o f an t i - th rombin Ⅲ (AT-Ⅲ) concentrate might decrease the mortality in patients with severe VOD, without any associated serious side effects[71,72]. In one report of 10 patients with severe VOD, clinical improvement was seen in all patients after 5 d of AT-Ⅲ infusion[73].
There are several reports suggesting a beneficial effect of prostaglandin E1 administration combined with heparin infusion in severe VOD[74-76]. However severe adverse effects limits its routine use. There are anecdotal reports on the successful use of charcoal hemofiltration[77], N-acetylcysteine[78] and glutamine/vitamin E[79,80].
Defibrotide, the most promising drug currently available for the treatment of VOD, is a single-stranded polydeoxyribonucleotide with anti-ischemic, anti-thrombotic and thrombolytic properties. It lacks systemic anticoagulant activity, an advantage in severely thrombocytopenic patients after HSCT[81]. Its mechanism of action is poorly understood but likely involves adenosine receptors[82]. Richardson et al[83] reported that 32 (36%) of 88 patients with VOD with a predicted risk of ≥ 30% of developing severe disease had a complete response to defibrotide treatment. Thirty-one of the 32 patients survived beyond 100 d. Adverse effects were limited to nausea, transient
Figure 2 Histological examination demonstrates partial central vein occlusion in a patient with veno-occlusive disease.
1916 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

systolic hypotension, fever, abdominal cramping, and vasomotor symptoms such as hot flushes. In another clinical trial of defibrotide, complete response was achieved in 22 (55%) of 40 VOD patients[84].
I so la ted case repor t s have shown benef i t o f transjugular intrahepatic portosystemic shunt (TIPS) in patients with VOD[85-87]. Although it can control portal hypertension, it is not clear whether TIPS can alter the course of the disease. Orthotopic liver transplantation has been attempted successfully in a few cases, however few institutions are capable of performing it on patients undergoing HSCT[88-90].
bUDD-CHIaRI syNDROmeBudd-Chiari Syndrome (BCS) is an uncommon but potentially life-threatening disorder caused by obstruction of the hepatic venous outflow at any level from the small hepatic veins to the junction of the inferior vena cava (IVC) with the right atrium[91].
EtiologyBCS can be classified as primary (due to intrinsic intraluminal thrombosis or webs) or secondary (due to intraluminal invasion by a parasite or malignant tumor or due to extraluminal compression by an abscess, cyst, or solid tumor)[92]. Intravascular thrombosis, mostly seen in primary myeloproliferative disorders, is the most common mechanism leading to the obstruction of the hepatic venous system. At least one hereditary or acquired procoagulative disorder can be identified in 75% of patients with BCS (Table 5)[93]. Polycythemia vera is found in between 10%-40% of patients, whereas essential
thrombocythemia and myelofibrosis are less prevalent causes[94,95]. Of note, hepatic vein thrombosis occurs in up to 12% of patients with paroxysmal nocturnal hemoglobinuria and is the leading cause of mortality in this disorder[96,97]. As many as 30% of all cases of BCS are found to have factor Ⅴ Leiden mutation which is present in the majority of pregnancy- or oral contraceptive-related cases of hepatic vein thrombosis[98,99]. Protein C, Protein S and antithrombin levels may be decreased nonspecifically due to impaired hepatic synthesis in patients with BCS, but levels below 20% of normal suggest inherited deficiency of these proteins[100]. Patients with BCS, as well as their relatives, should be counseled and investigated for hereditary thrombophilias.
Although less common in western countries, primary membranous obstruction of the inferior vena cava (IVC) is the most common cause of BCS in South Africa and Asia, and is thought to be a consequence of IVC thrombosis[101]. For unknown reasons, 45-50% patients with known membranous occlusion ultimately develop hepatocellular carcinoma (HCC), even in the absence of cirrhosis[102]. On the other hand, HCC has not been reported to be a complication of hepatic vein thrombosis, except in Behcet’s disease-associated BCS[103].
In about 10% of patients with BCS, an underlying etiology cannot be identified[95]. Recently, endothelial dysfunction and decreased fibrinolytic activity have been suggested as contributing factors in patients with idiopathic BCS[104].
PathologyThe parenchymal hepatic damage and the histologic abnormalities are variable, depending on the acuity and extent of hepatic venous outflow obstruction. Rapid occlusion of all 3 hepatic veins or 2 veins including the right hepatic vein leads to diffuse hepatic congestion and enlargement. The result is ischemic necrosis followed by fibrosis, predominantly in the perivenular areas (Figure 3). However, in patients with concomitant portal vein thrombosis, fibrosis prevails in the periportal zone[6,105]. In chronic disease, direct blood flow from the caudate lobe to the IVC compensates for hepatic outflow obstruction. Over time, the caudate lobe becomes hypertrophic while cirrhosis and atrophy develops in the rest of the liver[106,107].
Compensatory nodular regenerative hyperplasia is common in areas of hepatic parenchyma that have an adequate blood supply, with progression to fibrosis and
Table 5 Causes of the Budd-Chiari Syndrome
Common causesHypercoagulable states Inherited thrombophilic disorders Antithrombin Ⅲ deficiency Protein C deficiency Protein S deficiency Factor Ⅴ Leiden mutation Prothrombin gene mutationAcquired procoagulative disorders Myeloproliferative disorders (overt and occult) Paroxysmal nocturnal hemoglobinuria Antiphospholipid syndrome Cancer Pregnancy Use of oral contraceptivesUncommon causes Tumoral invasion Hepatocellular carcinoma Renal cell carcinoma Adrenal carcinoma Miscellaneous Aspergillosis Behcet’s syndrome Inferior vena caval webs Trauma Inflammatory bowel syndrome Dacarbazine therapy Idiopathic
Figure 3 Histological examina t i on shows sinusoidal dilatation and congestion.
Bayraktar UD et al. Hepatic venous outflow obstruction 1917
www.wjgnet.com

cirrhosis at a later stage of the disease[6,108]. Also, in the advanced stages, it is common to see areas of infarction secondary to concomitant thrombosis of the intrahepatic and extrahepatic portal veins[109].
Clinical featuresThere is a wide clinical spectrum, ranging from a fulminant picture to an asymptomatic condition, depending on the location, extent, and rapidity of the obstructive process and on whether the portal vein is thrombosed or not. Obstruction of a single main hepatic vein is clinically silent[110], whereas sudden occlusion of all major hepatic veins may result in fulminant hepatic failure.
BCS is more common in women, and usually presents in the third or fourth decade of life[111]. The most common signs and symptoms are ascites, hepatomegaly, and abdominal pain. In a series of 237 patients, obstruction was found in hepatic veins, IVC or both in 62%, 7%, and 31% patients respectively[112].
For descriptive purposes only, the syndrome can be classified as asymptomatic, fulminant, acute, subacute, or chronic[113]. However, it should be stressed that the correlation between clinical history and the duration of the occlusive process is weak and this classification’s prognostic significance is not clear[114].
Patients with the asymptomatic variant are usually discovered on investigation of abnormal liver function tests. These patients constitute between 5%-20% of BCS cases[112,113]. Absence of ascites and abdominal pain may be attributed to large intrahepatic and portosystemic venous collaterals or patency of one large hepatic vein[93].
Pat ients with acute BCS develop severe RUQ abdominal pain, hepatomegaly, jaundice, and intractable ascites within a few weeks. The acute form is observed in 20% of BCS patients[113]. Serum aminotransferase levels are elevated due to ischemic hepatocellular damage. Serum alkaline phosphatase level is elevated, usually between 300 and 400 IU/L. Liver functions can deteriorate quickly leading to fulminant hepatic failure.
Fortunately, the fulminant form is uncommon and typically follows rapid and complete occlusion of all major hepatic veins. Patients rapidly develop hepatic encephalopathy, renal failure and coagulopathy. Serum aminotransferase levels are markedly elevated. Unlike the findings in fulminant viral hepatitis, the liver is enlarged and tender[93].
The subacute form has a more insidious presentation, spread over several months. Patients may have minimal ascites, splenomegaly, hepatomegaly, and vague RUQ discomfort. Jaundice is either absent or mild.
The chronic form is seen in 60% of patients with BCS. Patients have signs or symptoms for more than six months and generally present with complications of cirrhosis including variceal bleeding, encephalopathy, coagulopathy, and hepatorenal syndrome. Additionally, hepatopulmonary syndrome has been described in up to 28% patients[115]. Renal impairment is present in one-half of the cases[93].
There is currently no consensus on the association of disease severity with disease duration. Furthermore, the duration of symptoms do not correlate with the degree of histologic damage to the liver. It has been observed
that 58% of patients with an acute clinical onset may have marked liver fibrosis, suggesting a long preclinical course. These findings can be explained by the development of recent thrombosis superimposed on previous lesions[110,116].
IVC compression or thrombosis generally presents with less severe symptoms compared with hepatic vein thrombosis and is characterized by leg edema or venous collaterals over the trunk in addition to hepatomegaly, ascites, and abdominal pain. The clinical course is chronic, with repeated acute episodes eventually leading to congestive cirrhosis[117,118].
Portal venous system is usually affected in patients with BCS because: 1) increased sinusoidal pressure decreases flow through the portal system, 2) hypertrophied caudate lobe may compress the main portal branches and the intrahepatic portal venules. These changes can result in stasis and vulnerability to thrombosis in the portal vein, further depleting liver perfusion[119]. However, in our experience the clinical picture in patients with both hepatic and portal vein thrombosis is better compared to that in patients with hepatic vein thrombosis only. This may be due to decreased blood flow to liver resulting in reduced hepatic congestion.
Prognosis and survivalThe natural history of BCS is poorly understood, mainly because most patients receive some form of treatment. The spectrum of clinical outcome ranges from progressive deterioration of the general condition to a few reports of spontaneous regression of the acute manifestations[120]. After the institution of anticoagulation therapy and early recognition of asymptomatic cases, mortality rates have decreased over time[113]. In a cohort of 120 patients, survival rates of 77%, 65% and 57% were found at 1, 5 and 10 years, respectively[121]. Murad et al[112] developed a model predicting survival in BCS, classifying patients into three categories with statistically different 5-year survival rates of 89%, 74%, and 42% based on the presence of ascites, encephalopathy, and prothrombin time and serum bilirubin levels.
Histologic abnormalities of the liver were not found to determine the prognosis[112,121,122] except in one study in which advanced fibrosis was associated with increased mortality[123]. One reason for this discrepancy can be explained by biopsy specimens being not representative of the whole liver due to uneven distribution of the hepatic lesions.
IVC obstruction has a good short term prognosis but there is limited data regarding long term prognosis. In a study from Japan, there was a 25% mortality rate over 15 years in patients with obliterative cavopathy. The main causes of death were liver failure, variceal bleeding and HCC[101,124].
In a series of 237 patients, portal vein thrombosis (PVT) with and without spleno-mesenteric vein thrombosis was identified in 18 (9%) and 15 (7%) patients respectively (total 33 patients, 16%). The five-year survival was 59% in patients with BCS and PVT and 85% in isolated BCS[125].
DiagnosisThe diagnosis of BCS requires a high index of suspicion.
1918 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

It should be considered in all patients presenting with ascites, hepatomegaly and RUQ abdominal pain or when intractable ascites occurs in the presence of mildly altered liver function tests. Standard laboratory investigations are rarely helpful in establishing the diagnosis, because abnormal liver function tests are not specific for BCS. Edema of the lower extremities and presence of venous collaterals on the back suggests IVC occlusion. Ascitic fluid analysis adds little specificity to the diagnosis and ascitic fluid albumin content can differ according to the stage of the BCS[126]. Thus, the diagnosis is largely made by imaging studies. Real-time and Doppler sonography: Real-time or color and pulsed Doppler should be the initial investigation for BCS, with a sensitivity and specificity of nearly 85%[127]. Specific findings for hepatic vein obstruction are abnormal flow in a hepatic vein, large intrahepatic or subcapsular collateral vessels and inability to visualize the junction of major hepatic veins with IVC. Moreover, the presence of intrahepatic venous to venous spider web collaterals is highly suggestive of BCS[128,129] (Figure 4) Acute venous occlusion is identified by enlarged, stenotic, or tortuous hepatic veins, whereas in patients with chronic disease the major hepatic veins may not be readily visualized. Additional findings include: compression of IVC by hypertrophic caudate lobe or obstruction by thrombus, tumor, or membranes[130,131].Computed tomography (CT): Failure to visualize the hepatic veins or IVC is suggestive of BCS. Contrast enhancement may be seen more centrally than peripherally
resulting in a patchy flea-bitten appearance (Figure 5). CT also allows assessment of the extent of hepatic parenchymal disease, ascites, and splenomegaly[132]. However, the role of CT scan is limited because of false-positive and indeterminate results in almost one-half of the patients[128].Magnetic resonance imaging (MRI): In patients with an unremarkable ultrasound examination but in whom the suspicion for BCS is high, MRI with intravenous gadolinium is a reasonable option (Figure 6). It allows excellent visualization of hepatic veins and IVC. Large intrahepatic, comma-shaped collaterals can be seen in patients with chronic obstruction. The major drawback of MRI is that, unlike ultrasonography, it does not provide the direction of hepatic venous blood flow[133,134].Hepatic venography: Although not considered essential for the diagnosis of BCS, venography provides useful information such as the extent of the thrombosis and caval pressures which help in determining the optimal therapy. Iodinated contrast can be injected into the hepatic veins either by transhepatic puncture or retrograde cannulation through the IVC (Figure 7). Abnormalities specific for BCS include a “spider-web” venous network, a coarse network of collaterals arching outward from the catheter tip, and a patent vein upstream from a stricture. A distorted appearance of hepatic veins that is observed in cirrhosis should not be confused with BCS. Another advantage of venography is that the portal vein can be assessed at the same sitting and if found to be occluded can be dilated with or without stenting in order to decompress the liver. Pressure measurements obtained during venography provide useful information before attempting surgical decompression[135,136]. Also, pressure measurements are useful in the post-surgical follow-up of these patients.Liver biopsy: Liver biopsy may demonstrate congestion, hepatocyte necrosis, and fibrosis in the centrilobular
Figure 4 Abdominal u l t r asonog raphy reveals thick and obliterated middle hepatic veins.
Figure 5 Computed tomography showing c e n t r a l c o n t r a s t enhancement in the liver.
Ivk
Cor > Tra 13
10 cm
Figure 6 Magnetic resonance imaging of the liver with contrasting agent shows that inferior vena cava is compressed by hypertrophy of caudate lobe and gross lobulation of the liver.
Figure 7 Almost completely obstructed IVC and shows collateral drainage through azygous system.
Bayraktar UD et al. Hepatic venous outflow obstruction 1919
www.wjgnet.com

area, findings that are similar to those seen in congestive hepatopathy. Venular thrombosis is usually not evident in biopsy specimens. All patients under consideration for surgery should undergo liver biopsy in conjunction with angiography to determine whether the patient will benefit more from a shunt procedure or a liver transplant[135,137].
ManagementThe goals of treatment are to alleviate venous obstruction, prevent extension of thrombosis in the hepatic veins and preserve hepatic function by decreasing the centrilobular congestion. Diagnostic workup to identify the underlying cause should be considered at the time of the initial diagnosis.Medical therapy: Medical therapy for BCS consists of efforts to control ascites, to prevent the extension of thrombosis with anticoagulation therapy, to dissolve the clot and relieve the hepatic congestion with thrombolysis and to treat the complications. Additionally, the underlying cause of BCS should be investigated and treated.
Ascites is managed by low-sodium diet, diuretics, and therapeutic paracentesis when needed. The type and duration of optimal anticoagulation therapy has not been established, however most centers give lifelong anticoagulation, initially with heparin followed by warfarin titrated to maintain the INR between 2 and 3[100,126]. During active variceal bleeding, balloon tamponade or endoscopic therapy is preferred to vasoconstrictor agents because the reduction in splanchnic blood flow induced by vasoconstrictors can theoretically precipitate mesenteric and portal vein thrombosis[138].
Medical therapy alone can be a good option for patients in whom there is no ongoing hepatic necrosis, as indicated by the presence of minimal symptoms, relatively normal liver function results, and ascites that is easily controlled[139]. Patients on medical therapy alone should be monitored closely for disease progression and to detect progressive necrosis by serial upper endoscopies and liver biopsies. Restoration of hepatic blood flow: Relief of venous obstruction can be attempted by thrombolytic therapy, percutaneous angioplasty, transjugular intrahepatic portosystemic shunt (TIPS), or shunt surgery. In patients with an acute presentation, especially when angiography shows presence of a fresh thrombus, thrombolytic therapy can be administered systemically or directly into the thrombosed hepatic vein. The best results are obtained when the thrombolytic agent is administered early directly into the vein, although promising data has been reported with thrombolysis carried out two to three weeks after the onset of symptoms[140,141].
Balloon angioplasty has been used to relieve hepatic outflow obstruction secondary to caval webs or short-segment hepatic vein stenosis[142]. Although short-term results are excellent, the sustained patency rate is only 50% at two years after the procedure[143]. However, the use of intraluminal stents has been shown to increase the long-term patency rates to nearly 90%[144-146]. Once inserted, the stents cannot be removed, and placement of a stent above the intrahepatic IVC may interfere with liver transplantation. Failure of angioplasty or stenting, should
prompt consideration for a surgical portosystemic shunt or TIPS[147].
TIPS decompresses the liver by creating an alternative venous outflow tract. It is particularly useful, either alone or as a bridge to liver transplantation, in patients with an acute presentation such as those with variceal bleeding, and patients with fulminant hepatic failure in whom thrombolysis and angioplasty were unsuccessful[148,149]. TIPS may be preferred over surgical shunting because it avoids laparotomy and has less periprocedure mortality and morbidity, its efficacy is not affected by caudate lobe hypertrophy, and it can be done in patients with PVT[150-152]. Although long-term patency rate is only about 50%, patients in whom stent stenosis occurs do not generally worsen, perhaps because the shunt allows time for collateral circulation to develop[153]. The recently introduced, polytetrafluoroethylene-covered stents may improve the patency rates[154]. However, extension of the stent into the suprahepatic IVC may preclude liver transplantation and should be avoided.
Surgical shunts decompress the liver by creating hepatofugal flow through the portal vein. Patients with a non-fulminant presentation and those who have a chronic presentation without significant hepatic fibrosis should be considered for surgical shunting, providing the portal vein is patent[150]. The five-year survival rate after surgical shunting ranges between 57% and 94% and absence of IVC occlusion is associated with better outcomes[155-157].
A pressure difference of at least 10 mmHg between the portal vein and intrahepatic IVC is essential for portocaval and mesocaval shunts to function[123]. Acute hepatic decompensation may occur postoperatively, therefore shunt operations should be performed in centers where rescue liver transplantation can take place.
The type of surgical shunt depends on the anatomy of the obstruction. Side-to-side portacaval shunts have the highest patency rate but may make subsequent liver transplantation more difficult. Side-to-side mesocaval shunts are technically less demanding and do not interfere with liver transplantation but when placed with a synthetic graft, have a higher thrombosis rate than a side-to-side portacaval shunt[147]. Mesoatrial shunt should be considered if there is IVC compression resulting in low portocaval pressure gradient. Mesoatrial shunt has a high rate of thrombosis due to relatively low flow rate in the prosthetic graft[126]. Side-to-side portocaval shunt and cavoatrial shunt have been described for patients with both caval and hepatic venous thrombosis[136,157,158].
In addition to shunts, other surgical procedures can also provide adequate decompression in selected patients. Transatrial membranotomy can be effective in patients with IVC membranes[136,157,159,160]. Dorsocranial resection of the liver is the only feasible procedure if there is both portal and superior mesenteric vein obstruction[161].Liver transplantation: Patients who have cirrhosis, fulminant hepatic failure or biochemical evidence of advanced liver dysfunction are best managed with liver transplantation[157]. Long term survival rates range from 50% to 95%[155,162-164]. Development of postoperative portal vein or hepatic artery thrombosis occurs in about
1920 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

12% of patients. Liver transplantation can cure almost all hereditary thrombophilias, however thrombosis can still occur and anticoagulation is necessary[150].
CONgesTIVe HepaTOpaTHyCongestive hepatopathy refers to hepatic manifestations attributable to passive hepatic congestion resulting from right-sided heart failure of any cause including and not limited to constrictive pericarditis (CP), tricuspid regurgitation, cor pulmonale, and cardiomyopathies. Development of hepatic ischemia and infarction secondary to left-sided heart failure which usually coincides with congestive hepatopathy will not be discussed here. However, it should be kept in mind that the clinical picture in congestive hepatopathy varies greatly depending on both the degree and acuity of congestion, and presence or absence of hepatic ischemia and infarction[2].
Clinical ManifestationsLiver dysfunction in congestive hepatopathy is usually mild and asymptomatic and often detected incidentally on routine biochemical testing. Symptomatic patients usually present with mild jaundice which is characteristically absent in patients with CP. In patients with severe heart failure, jaundice may be so deep as to suggest cholestasis[165]. Right upper quadrant discomfort due to stretching of liver capsule and ascites may also be present.
Occasionally, the clinical picture may resemble that of acute viral hepatitis, when jaundice is accompanied by elevated aminotransferase levels due to acute hepatic ischemia[166,167]. Several cases of fulminant hepatic failure resulting in death have been reported due to congestive heart failure. However, most of these patients had characteristics of both hepatic congestion and ischemia[165,168,169].
On physical examination, patients may have tender hepatomegaly, sometimes massive, with a firm and smooth liver edge. Splenomegaly is uncommon, and like ascites is due to transmitted elevated central venous pressure. Jugular venous distention and hepatojugular reflux may be present and are helpful in differentiating congestive hepatopathy from BCS and primary liver diseases. Liver may be pulsatile especially in patients with tricuspid regurgitation. Loss of hepatic pulsatility suggests progression to cardiac cirrhosis. The most common laboratory finding is elevation of total serum bilirubin level, most of which is unconjugated. Hyperbilirubinemia occurs in up to 70% of patients with congestive hepatopathy[170]. Severe hyperbilirubinemia may
develop in patients with severe, usually acute, right-sided heart failure. Serum bilirubin level shows a correlation with right atrial pressure but not with cardiac output[6]. Even in the presence of deep jaundice, serum alkaline phosphatase level is generally only mildly elevated, which helps to distinguish congestive hepatopathy from obstructive jaundice. Serum aminotransferase levels also show a mild elevation unless cardiac output is impaired. In patients with severe acute heart failure, aminotransferase levels may be extremely high secondary to hepatic ischemia and the degree of elevation correlates with the extent of necrosis as seen on liver biopsy specimens. Additionally, the prothrombin time may be mildly abnormal[171], albumin level may be decreased[172], and serum ammonia level may be elevated[173].
DiagnosisIncreasing specialization in medicine has caused physicians to overlook diseases that cross the boundaries of two specialties. Congestive hepatopathy may be missed in patients with heart failure and mild hepatic congestion, and in patients with overt hepatic congestion and vague cardiac symptoms[174,175]. Physicians should consider right-sided heart failure in patients with hepatomegaly with or without jaundice. CP is especially difficult to differentiate from primary liver cirrhosis and BCS because of its relatively nonspecific clinical features.
A careful history and a thorough physical examination are crucial in the diagnosis of congestive hepatopathy. Symptoms such as exertional dyspnea, orthopnea, and angina, and physical findings like jugular venous distention, heart murmurs, and rales may help physicians to differentiate congestive hepatopathy from primary liver diseases. In addition to liver biochemical testing, viral hepatitis serology, and abdominal ultrasound with Doppler studies of the liver; EKG and echocardiogram should be performed when congestive hepatopathy is suspected. However, a normal echocardiogram does not always rule out congestive hepatopathy. Upon the finding of dilatation of all three main hepatic veins on abdominal sonogram (Figure 8), physicians should look for a cardiac cause such as constrictive pericarditis more vigorously with cardiac angiogram or other radiological methods.
Diagnostic paracentesis in patients with congestive hepatopathy usually reveals a high ascitic fluid protein content and a serum to ascites albumin gradient > 1.1 g/dL reflecting the contribution of “hepatic lymph” and portal hypertension to the ascites[176].
Improvement in liver biochemical tests with treatment of the underlying cardiac disease supports the diagnosis. Liver biopsy may help confirm the diagnosis in equivocal cases.
PathologyThe characteristic gross appearance of liver in congestive hepatopathy is referred to as the nutmeg liver. This appearance is the result of contrasting areas of red caused by sinusoidal congestion and bleeding in the necrotic regions surrounding the enlarged hepatic veins and yellow due to the normal or fatty liver tissue. Histologically, sinusoidal engorgement and hemorrhagic necrosis is
Figure 8 Ultrasound of the liver shows gross dilatation in the inferior vena cava and hepatic Ⅴ.
Bayraktar UD et al. Hepatic venous outflow obstruction 1921
www.wjgnet.com

apparent in perivenular areas of hepatic acini. Variable degrees of cholestasis with occasional bile thrombi in the canaliculi may also be apparent. With chronic heart failure, fibrosis develops in perivenular areas, ultimately causing bridging fibrosis between adjacent central veins. This process results in cardiac fibrosis, inappropriately referred as cardiac cirrhosis, which is distinct from primary liver cirrhosis in which fibrous bands tend to link adjacent portal areas. The regeneration of periportal hepatocytes in cardiac fibrosis may result in nodular regenerative hyperplasia. If heart failure is treated successfully, the early histologic changes of congestive hepatopathy may resolve, and even cardiac fibrosis may regress histologically and clinically[2].
Treatment and PrognosisTreatment of the underlying heart disease is fundamental to the management of congestive hepatopathy and is outside the scope of this review. Jaundice and ascites usually respond significantly to diuresis. However, excessive diuresis may decrease cardiac output and result in hepatic ischemia in patients with severe congestive heart failure.
Most patients with congestive hepatopathy die of cardiac causes and liver disease rarely contributes to the morbidity or mortality in these patients. Unlike patients with liver cirrhosis, those with cardiac fibrosis rarely develop serious complications such as variceal bleeding[177]. Hepatocellular carcinoma may rarely complicate cardiac fibrosis[178]. However, the incidence of hepatocellular carcinoma and liver failure due to congestive hepatopathy is likely to increase as survival is prolonged with advances in the treatment of heart failure.
CONsTRICTIVe peRICaRDITIs (Cp)CP deserves mention because its diagnosis can be easily missed and its clinical manifestations are different from those seen in congestive hepatopathy due to other causes. CP is the result of scarring and loss of elasticity of the pericardial sac which restricts cardiac filling. Tuberculosis, cardiac surgery, radiation therapy, and connective tissue disorders are among common causes, however most of the cases are idiopathic or viral[179-181].
Patients with CP may present with symptoms of fluid overload such as edema and ascites, or symptoms of diminished cardiac output like exertional dyspnea and fatigue. Hepatomegaly, massive ascites, and peripheral edema are common findings. For reasons that are unclear, patients with CP typically do not develop jaundice[5] and very rarely may develop chylous ascites[182-183]. Because its manifestations are protean, it is easy to mistake CP for BCS and liver cirrhosis[184]. Jugular venous distention which was noted in 93% patients in a large series is a critical finding in the diagnosis of CP[180]. Although rarely observed, Kussmaul’s sign, pericardial knock, and pulsus paradoxus may offer additional clues to the diagnosis.
EKG may show a low voltage and nonspecific ST and T wave changes. Chest X-ray may demonstrate pericardial calcification. Echocardiography is essential in the diagnosis of CP, but right and left heart catheterization with
hemodynamic evaluation may be required to confirm the diagnosis[185-187].
Cardiac fibrosis develops more frequently and rapidly in CP than in other causes of right-sided heart failure, perhaps due to higher hepatic venous pressures leading to severe zone 3 congestion and necrosis[2]. Pericardiectomy is the standard of treatment and curative if performed early.
CONClUsIONThe clinical manifestations and liver histology in VOD, BCS, and congestive hepatopathy are similar because of the common underlying mechanism of hepatic injury and adaptation in response to increased sinusoidal pressure. Further animal and in-vitro studies are needed to better understand the pathways involved in sinusoidal injury in response to increased venous pressure. The severity of clinical picture depends mostly on the rapidity and extent of the obstructive process. Physicians should consider congestive hepatopathy and BCS in all patients with tender hepatomegaly and ascites. Presence of jugular venous distention is an invaluable clue to differentiate congestive hepatopathy from BCS and other primary liver diseases. Veno-occlusive disease should be kept in mind in patients who rapidly develop jaundice and ascites with hepatomegaly after HSCT. The advent of interventional radiology and new medical therapies has improved survival in patients with hepatic venous outflow obstruction, although prevention is still mainstay in the management of VOD.
ReFeReNCes1 Lautt WW, Greenway CV. Conceptual review of the hepatic
vascular bed. Hepatology 1987; 7: 952-9632 Rosenberg PM, Friedman LS. The liver in circulatory failure.
In: Schiff ER, Sorrell MF, Maddrey WC. Schiff’s diseases of the liver. 9th ed. Philadelphia: Lippincott Williams & Wilkins, 2004, 1327-1340
3 Shulman HM, McDonald GB, Matthews D, Doney KC, Kopecky KJ, Gauvreau JM, Thomas ED. An analysis of hepatic venocclusive disease and centrilobular hepatic degeneration following bone marrow transplantation. Gastroenterology 1980; 79: 1178-1191
4 Tanaka M, Wanless IR. Pathology of the liver in Budd-Chiari syndrome: portal vein thrombosis and the histogenesis of veno-centric cirrhosis, veno-portal cirrhosis, and large regenerative nodules. Hepatology 1998; 27: 488-496
5 Arora A, Tandon N, Sharma MP, Acharya SK. Constrictive pericarditis masquerading as Budd-Chiari syndrome. J Clin Gastroenterol 1991; 13: 178-181
6 Sherlock S. The liver in heart failure; relation of anatomical, functional, and circulatory changes. Br Heart J 1951; 13: 273-293
7 Berk PD, Popper H, Krueger GR, Decter J, Herzig G, Graw RG. Veno-occlusive disease of the liver after allogeneic bone marrow transplantation: possible association with graft-versus-host disease. Ann Intern Med 1979; 90: 158-164
8 Fajardo LF, Colby TV. Pathogenesis of veno-occlusive liver disease after radiation. Arch Pathol Lab Med 1980; 104: 584-588
9 King PD , Perry MC. Hepatotoxicity of chemotherapy. Oncologist 2001; 6: 162-176
10 Tandon BN, Tandon HD, Tandon RK, Narndranathan M, Joshi YK. An epidemic of veno-occlusive disease of liver in central India. Lancet 1976; 2: 271-272
11 Mohabbat O, Younos MS, Merzad AA, Srivastava RN, Sediq GG, Aram GN. An outbreak of hepatic veno-occlusive disease in north-western Afghanistan. Lancet 1976; 2: 269-271
1922 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

12 Sebagh M, Debette M, Samuel D, Emile JF, Falissard B, Cailliez V, Shouval D, Bismuth H, Reynès M. "Silent" presentation of veno-occlusive disease after liver transplantation as part of the process of cellular rejection with endothelial predilection. Hepatology 1999; 30: 1144-1150
13 Bras G, Jelliffe DB, Stuart KL. Veno-occlusive disease of liver with nonportal type of cirrhosis, occurring in Jamaica. AMA Arch Pathol 1954; 57: 285-300
14 DeLeve LD, Shulman HM, McDonald GB. Toxic injury to hepatic sinusoids: sinusoidal obstruction syndrome (veno-occlusive disease). Semin Liver Dis 2002; 22: 27-42
15 Carreras E , Bertz H, Arcese W, Vernant JP, Tomás JF, Hagglund H, Bandini G, Esperou H, Russell J, de la Rubia J, Di Girolamo G, Demuynck H, Hartmann O, Clausen J, Ruutu T, Leblond V, Iriondo A, Bosi A, Ben-Bassat I, Koza V, Gratwohl A, Apperley JF. Incidence and outcome of hepatic veno-occlusive disease after blood or marrow transplantation: a prospective cohort study of the European Group for Blood and Marrow Transplantation. European Group for Blood and Marrow Transplantation Chronic Leukemia Working Party. Blood 1998; 92: 3599-3604
16 McDonald GB, Hinds MS, Fisher LD, Schoch HG, Wolford JL, Banaji M, Hardin BJ, Shulman HM, Clift RA. Veno-occlusive disease of the liver and multiorgan failure after bone marrow transplantation: a cohort study of 355 patients. Ann Intern Med 1993; 118: 255-267
17 Kumar S, DeLeve LD, Kamath PS, Tefferi A. Hepatic veno-occlusive disease (sinusoidal obstruction syndrome) after hematopoietic stem cell transplantation. Mayo Clin Proc 2003; 78: 589-598
18 Carreras E, Grañena A, Navasa M, Bruguera M, Marco V, Sierra J, Tassies MD, García-Pagán JC, Martí JM, Bosch J. On the reliability of clinical criteria for the diagnosis of hepatic veno-occlusive disease. Ann Hematol 1993; 66: 77-80
19 Rio B, Andreu G, Nicod A, Arrago JP, Dutrillaux F, Samama M, Zittoun R. Thrombocytopenia in venocclusive disease after bone marrow transplantation or chemotherapy. Blood 1986; 67: 1773-1776
20 Méresse V, Hartmann O, Vassal G, Benhamou E, Valteau-Couanet D, Brugieres L, Lemerle J . Risk factors for hepatic veno-occlusive disease after high-dose busulfan-containing regimens followed by autologous bone marrow transplantation: a study in 136 children. Bone Marrow Transplant 1992; 10: 135-141
21 Ayash LJ , Hunt M, Antman K, Nadler L, Wheeler C, Takvorian T, Elias A, Antin JH, Greenough T, Eder JP. Hepatic venoocclusive disease in autologous bone marrow transplantation of solid tumors and lymphomas. J Clin Oncol 1990; 8: 1699-1706
22 Bearman SI, Anderson GL, Mori M, Hinds MS, Shulman HM, McDonald GB. Venoocclusive disease of the liver: development of a model for predicting fatal outcome after marrow transplantation. J Clin Oncol 1993; 11: 1729-1736
23 Frickhofen N, Wiesneth M, Jainta C, Hertenstein B, Heymer B, Bianchi L, Dienes HP, Koerner K, Bunjes D, Arnold R. Hepatitis C virus infection is a risk factor for liver failure from veno-occlusive disease after bone marrow transplantation. Blood 1994; 83: 1998-2004
24 Strasser SI, Myerson D, Spurgeon CL, Sullivan KM, Storer B, Schoch HG, Kim S, Flowers ME, McDonald GB. Hepatitis C virus infection and bone marrow transplantation: a cohort study with 10-year follow-up. Hepatology 1999; 29: 1893-1899
25 Rozman C, Carreras E, Qian C, Gale RP, Bortin MM, Rowlings PA, Ash RC, Champlin RE, Henslee-Downey PJ, Herzig RH, Hinterberger W, Klein JP, Prentice HG, Reiffers J, Zwaan FE, Horowitz MM. Risk factors for hepatic veno-occlusive disease following HLA-identical sibling bone marrow transplants for leukemia. Bone Marrow Transplant 1996; 17: 75-80
26 Ertem M, Akar N. Factor V Leiden mutation as a predisposing factor for veno-occlusive disease following BMT. Bone Marrow Transplant 2000; 25: 1110-1111
27 Duggan C, Schmidt M, Lawler M, White B, Cusack S, McCann S, Smith O. The prothrombin gene variant G20210A but not
factor V leiden may be associated with veno-occlusive disease following BMT. Bone Marrow Transplant 1999; 24: 693-694
28 Wadleigh M, Richardson PG, Zahrieh D, Lee SJ, Cutler C, Ho V, Alyea EP, Antin JH, Stone RM, Soiffer RJ, DeAngelo DJ. Prior gemtuzumab ozogamicin exposure significantly increases the risk of veno-occlusive disease in patients who undergo myeloablative allogeneic stem cell transplantation. Blood 2003; 102: 1578-1582
29 Hägglund H , Remberger M, Klaesson S, Lönnqvist B, Ljungman P, Ringdén O. Norethisterone treatment, a major risk-factor for veno-occlusive disease in the liver after allogeneic bone marrow transplantation. Blood 1998; 92: 4568-4572
30 Kalayoglu-Besisik S, Yenerel MN, Caliskan Y, Ozturk S, Besisik F, Sargin D. Time-related changes in the incidence, severity, and clinical outcome of hepatic veno-occlusive disease in hematopoietic stem cell transplantation patients during the past 10 years. Transplant Proc 2005; 37: 2285-2289
31 DeLeve LD. Cellular target of cyclophosphamide toxicity in the murine liver: role of glutathione and site of metabolic activation. Hepatology 1996; 24: 830-837
32 Shulman HM, Hinterberger W. Hepatic veno-occlusive disease- l iver toxic i ty syndrome af ter bone marrow transplantation. Bone Marrow Transplant 1992; 10: 197-214
33 M c D o n a l d G B , S h a r m a P , M a t t h e w s D E , S h u l m a n HM, Thomas ED. Venocclusive disease of the liver after bone marrow transplantation: diagnosis, incidence, and predisposing factors. Hepatology 1984; 4: 116-122
34 Jones RJ, Lee KS, Beschorner WE, Vogel VG, Grochow LB, Braine HG, Vogelsang GB, Sensenbrenner LL, Santos GW, Saral R. Venoocclusive disease of the liver following bone marrow transplantation. Transplantation 1987; 44: 778-783
35 Shulman HM, Gooley T, Dudley MD, Kofler T, Feldman R, Dwyer D, McDonald GB. Utility of transvenous liver biopsies and wedged hepatic venous pressure measurements in sixty marrow transplant recipients. Transplantation 1995; 59: 1015-1022
36 Carreras E, Grañena A, Navasa M, Bruguera M, Marco V, Sierra J, Tassies MD, García-Pagán JC, Martí JM, Bosch J. Transjugular liver biopsy in BMT. Bone Marrow Transplant 1993; 11: 21-26
37 Yoshimoto K, Ono N, Okamura T, Sata M. Recent progress in the diagnosis and therapy for veno-occlusive disease of the liver. Leuk Lymphoma 2003; 44: 229-234
38 Salat C, Holler E, Kolb HJ, Reinhardt B, Pihusch R, Wilmanns W, Hiller E. Plasminogen activator inhibitor-1 confirms the diagnosis of hepatic veno-occlusive disease in patients with hyperbilirubinemia after bone marrow transplantation. Blood 1997; 89: 2184-2188
39 Rio B, Bauduer F, Arrago JP, Zittoun R. N-terminal peptide of type III procollagen: a marker for the development of hepatic veno-occlusive disease after BMT and a basis for determining the timing of prophylactic heparin. Bone Marrow Transplant 1993; 11: 471-472
40 Fried MW, Duncan A, Soroka S, Connaghan DG, Farrand A, Peter J, Strauss RM, Boyer TD, McDonald GB. Serum hyaluronic acid in patients with veno-occlusive disease following bone marrow transplantation. Bone Marrow Transplant 2001; 27: 635-639
41 Strasser SI, McDonald GB. Hepatobiliary complications of hematopoietic cell transplantation. In: Schiff ER, Sorrell MF, Maddrey WC. Schiff’s diseases of the liver. 9th ed. Philadelphia: Lippincott Williams & Wilkins, 2004, 1635-1658
42 Carreras E . Veno-occlusive disease of the l iver after hemopoietic cell transplantation. Eur J Haematol 2000; 64: 281-291
43 DeLeve LD, McCuskey RS, Wang X, Hu L, McCuskey MK, Epstein RB, Kanel GC. Characterization of a reproducible rat model of hepatic veno-occlusive disease. Hepatology 1999; 29: 1779-1791
44 Shulman HM , Gown AM, Nugent DJ. Hepatic veno-occlusive disease after bone marrow transplantation. Immunohistochemical identification of the material within
Bayraktar UD et al. Hepatic venous outflow obstruction 1923
www.wjgnet.com

occluded central venules. Am J Pathol 1987; 127: 549-55845 Sato Y, Asada Y, Hara S, Marutsuka K, Tamura K, Hayashi
T, Sumiyoshi A. Hepatic stellate cells (Ito cells) in veno-occlusive disease of the liver after allogeneic bone marrow transplantation. Histopathology 1999; 34: 66-70
46 DeLeve LD, Wang X, Kuhlenkamp JF, Kaplowitz N. Toxicity of azathioprine and monocrotaline in murine sinusoidal endothelial cells and hepatocytes: the role of glutathione and relevance to hepatic venoocclusive disease. Hepatology 1996; 23: 589-599
47 Wang X , Kanel GC, DeLeve LD. Support of sinusoidal endothelial cell glutathione prevents hepatic veno-occlusive disease in the rat. Hepatology 2000; 31: 428-434
48 Srivastava A, Poonkuzhali B, Shaji RV, George B, Mathews V, Chandy M, Krishnamoorthy R. Glutathione S-transferase M1 polymorphism: a risk factor for hepatic venoocclusive disease in bone marrow transplantation. Blood 2004; 104: 1574-1577
49 Lee JH, Lee KH, Kim S, Lee JS, Kim WK, Park CJ, Chi HS, Kim SH. Relevance of proteins C and S, antithrombin III, von Willebrand factor, and factor VIII for the development of hepatic veno-occlusive disease in patients undergoing allogeneic bone marrow transplantation: a prospective study. Bone Marrow Transplant 1998; 22: 883-888
50 Soiffer RJ, Dear K, Rabinowe SN, Anderson KC, Freedman AS, Murray C, Tarbell NJ, Mauch P, Nadler LM, Ritz J. Hepatic dysfunction following T-cell-depleted allogeneic bone marrow transplantation. Transplantation 1991; 52: 1014-1019
51 Ohashi K, Tanabe J, Watanabe R, Tanaka T, Sakamaki H, Maruta A, Okamoto S, Aotsuka N, Saito K, Nishimura M, Oh H, Matsuzaki M, Takahashi S, Yonekura S. The Japanese multicenter open randomized trial of ursodeoxycholic acid prophylaxis for hepatic veno-occlusive disease after stem cell transplantation. Am J Hematol 2000; 64: 32-38
52 Essell JH, Schroeder MT, Harman GS, Halvorson R, Lew V, Callander N, Snyder M, Lewis SK, Allerton JP, Thompson JM. Ursodiol prophylaxis against hepatic complications of allogeneic bone marrow transplantation. A randomized, double-blind, placebo-controlled trial. Ann Intern Med 1998; 128: 975-981
53 Attal M, Huguet F, Rubie H, Huynh A, Charlet JP, Payen JL, Voigt JJ, Brousset P, Selves J, Muller C. Prevention of hepatic veno-occlusive disease after bone marrow transplantation by continuous infusion of low-dose heparin: a prospective, randomized trial. Blood 1992; 79: 2834-2840
54 Rosenthal J, Sender L, Secola R, Killen R, Millerick M, Murphy L, Cairo MS. Phase II trial of heparin prophylaxis for veno-occlusive disease of the liver in children undergoing bone marrow transplantation. Bone Marrow Transplant 1996; 18: 185-191
55 Marsa-Vila L, Gorin NC, Laporte JP, Labopin M, Dupuy-Montbrun MC, Fouillard L, Isnard F, Najman A. Prophylactic heparin does not prevent liver veno-occlusive disease following autologous bone marrow transplantation. Eur J Haematol 1991; 47: 346-354
56 Or R , Nagler A, Shpi lberg O, E lad S , Napars tek E , Kapelushnik J, Cass Y, Gillis S, Chetrit A, Slavin S, Eldor A. Low molecular weight heparin for the prevention of veno-occlusive disease of the liver in bone marrow transplantation patients. Transplantation 1996; 61: 1067-1071
57 Forrest DL, Thompson K, Dorcas VG, Couban SH, Pierce R. Low molecular weight heparin for the prevention of hepatic veno-occlusive disease (VOD) after hematopoietic stem cell transplantation: a prospective phase II study. Bone Marrow Transplant 2003; 31: 1143-1149
58 Or R, Elad S, Shpilberg O, Eldor A. Low molecular weight heparin stimulates megakaryocytopoiesis in bone-marrow transplantation patients. Am J Hematol 1996; 53: 46-48
59 Brown SA, Goringe A, Fegan C, Davies SV, Giddings J, Whittaker JA, Burnett AK, Poynton CH. Parenteral glutamine protects hepatic function during bone marrow transplantation. Bone Marrow Transplant 1998; 22: 281-284
60 Gluckman E, Jolivet I, Scrobohaci ML, Devergie A, Traineau R, Bourdeau-Esperou H, Lehn P, Faure P, Drouet L. Use
of prostaglandin E1 for prevention of liver veno-occlusive disease in leukaemic patients treated by allogeneic bone marrow transplantation. Br J Haematol 1990; 74: 277-281
61 Bearman SI, Shen DD, Hinds MS, Hill HA, McDonald GB. A phase I/II study of prostaglandin E1 for the prevention of hepat ic venocclus ive disease af ter bone marrow transplantation. Br J Haematol 1993; 84: 724-730
62 Attal M, Huguet F, Rubie H, Charlet JP, Schlaifer D, Huynh A, Laurent G, Pris J. Prevention of regimen-related toxicities after bone marrow transplantation by pentoxifylline: a prospective, randomized trial. Blood 1993; 82: 732-736
63 Clift RA, Bianco JA, Appelbaum FR, Buckner CD, Singer JW, Bakke L, Bensinger WI, Bowden RA, McDonald GB, Schubert M. A randomized controlled trial of pentoxifylline for the prevention of regimen-related toxicities in patients undergoing allogeneic marrow transplantation. Blood 1993; 82: 2025-2030
64 Chalandon Y, Roosnek E, Mermillod B, Newton A, Ozsahin H, Wacker P, Helg C, Chapuis B. Prevention of veno-occlusive disease with defibrotide after allogeneic stem cell transplantation. Biol Blood Marrow Transplant 2004; 10: 347-354
65 Versluys B, Bhattacharaya R, Steward C, Cornish J, Oakhill A, Goulden N. Prophylaxis with defibrotide prevents veno-occlusive disease in stem cell transplantation after gemtuzumab ozogamicin exposure. Blood 2004; 103: 1968
66 Bearman SI , Shuhart MC, Hinds MS, McDonald GB. Recombinant human tissue plasminogen activator for the treatment of established severe venocclusive disease of the liver after bone marrow transplantation. Blood 1992; 80: 2458-2462
67 Leahey AM , Bunin NJ . Recombinant human t i ssue plasminogen activator for the treatment of severe hepatic veno-occlusive disease in pediatric bone marrow transplant patients. Bone Marrow Transplant 1996; 17: 1101-1104
68 Ringdén O, Wennberg L, Ericzon BG, Kallman R, Aström M, Duraj F, Söderdahl G, Tydén G, Groth CG. Alteplase for hepatic veno-occlusive disease after bone marrow transplantation. Lancet 1992; 340: 546-547
69 Hágglund H, Ringdén O, Ericzon BG, Duraj F, Ljungman P, Lönnqvist B, Winiarski J, Tydén G. Treatment of hepatic venoocclusive disease with recombinant human tissue plasminogen activator or orthotopic liver transplantation after allogeneic bone marrow transplantation. Transplantation 1996; 62: 1076-1080
70 Bearman SI, Lee JL, Barón AE, McDonald GB. Treatment of hepatic venocclusive disease with recombinant human tissue plasminogen activator and heparin in 42 marrow transplant patients. Blood 1997; 89: 1501-1506
71 Ibrahim RB, Peres E, Dansey R, Abidi MH, Abella EM, Klein J. Anti-thrombin III in the management of hematopoietic stem-cell transplantation-associated toxicity. Ann Pharmacother 2004; 38: 1053-1059
72 Mertens R, Brost H, Granzen B, Nowak-Göttl U. Antithrombin treatment of severe hepatic veno-occlusive disease in children with cancer. Eur J Pediatr 1999; 158 Suppl 3: S154-S158
73 Morris JD, Harris RE, Hashmi R, Sambrano JE, Gruppo RA, Becker AT, Morris CL. Antithrombin-III for the treatment of chemotherapy-induced organ dysfunction following bone marrow transplantation. Bone Marrow Transplant 1997; 20: 871-878
74 Higashigawa M, Watanabe M, Nishihara H, Tabata N, Azuma E, Ido M, Ito M, Sakurai M. Successful treatment of an infant with veno-occlusive disease developed after allogeneic bone marrow transplantation by tissue plasminogen activator, heparin and prostaglandin E1. Leuk Res 1995; 19: 477-480
75 Ibrahim A, Pico JL, Maraninchi D, Zambon E, Attal M, Brault P, Tilly H, Blaise D, Hayat M. Hepatic veno-occlusive disease following bone marrow transplantation treated by prostaglandin E1. Bone Marrow Transplant 1991; 7 Suppl 2: 53
76 S c h l e g e l P G , H a b e r H P , B e c k J , K r ü m p e l m a n n S , Handgretinger R, Bader P, Bierings M, Niethammer D, Klingebiel T. Hepatic veno-occlusive disease in pediatric stem cell recipients: successful treatment with continuous infusion of prostaglandin E1 and low-dose heparin. Ann Hematol
1924 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

1998; 76: 37-4177 Tefferi A, Kumar S, Wolf RC, Lacy MQ, Inwards DJ, Gloor JM,
Albright RC, Kamath PS, Litzow MR. Charcoal hemofiltration for hepatic veno-occlusive disease after hematopoietic stem cell transplantation. Bone Marrow Transplant 2001; 28: 997-999
78 Ringdén O, Remberger M, Lehmann S, Hentschke P, Mattsson J, Klaesson S, Aschan J. N-acetylcysteine for hepatic veno-occlusive disease after allogeneic stem cell transplantation. Bone Marrow Transplant 2000; 25: 993-996
79 Goringe AP, Brown S, O'Callaghan U, Rees J, Jebb S, Elia M, Poynton CH. Glutamine and vitamin E in the treatment of hepatic veno-occlusive disease following high-dose chemotherapy. Bone Marrow Transplant 1998; 21: 829-832
80 Nattakom TV, Charlton A, Wilmore DW. Use of vitamin E and glutamine in the successful treatment of severe veno-occlusive disease following bone marrow transplantation. Nutr Clin Pract 1995; 10: 16-18
81 P a l m e r K J , G o a K L . D e f i b r o t i d e . A r e v i e w o f i t s pharmacodynamic and pharmacokinetic properties, and therapeutic use in vascular disorders. Drugs 1993; 45: 259-294
82 Bianchi G, Barone D, Lanzarotti E, Tettamanti R, Porta R, Moltrasio D, Cedro A, Salvetti L, Mantovani M, Prino G. Defibrotide, a single-stranded polydeoxyribonucleotide acting as an adenosine receptor agonist. Eur J Pharmacol 1993; 238: 327-334
83 Richardson PG, Murakami C, Jin Z, Warren D, Momtaz P, Hoppensteadt D, Elias AD, Antin JH, Soiffer R, Spitzer T, Avigan D, Bearman SI, Martin PL, Kurtzberg J, Vredenburgh J, Chen AR, Arai S, Vogelsang G, McDonald GB, Guinan EC. Multi-institutional use of defibrotide in 88 patients after stem cell transplantation with severe veno-occlusive disease and multisystem organ failure: response without significant toxicity in a high-risk population and factors predictive of outcome. Blood 2002; 100: 4337-4343
84 Chopra R, Eaton JD, Grassi A, Potter M, Shaw B, Salat C, Neumeister P, Finazzi G, Iacobelli M, Bowyer K, Prentice HG, Barbui T. Defibrotide for the treatment of hepatic veno-occlusive disease: results of the European compassionate-use study. Br J Haematol 2000; 111: 1122-1129
85 Fried MW, Connaghan DG, Sharma S, Martin LG, Devine S, Holland K, Zuckerman A, Kaufman S, Wingard J, Boyer TD. Transjugular intrahepatic portosystemic shunt for the management of severe venoocclusive disease following bone marrow transplantation. Hepatology 1996; 24: 588-591
86 Azoulay D, Castaing D, Lemoine A, Hargreaves GM, Bismuth H. Transjugular intrahepatic portosystemic shunt (TIPS) for severe veno-occlusive disease of the liver following bone marrow transplantation. Bone Marrow Transplant 2000; 25: 987-992
87 Annaloro C, Robbiolo L, Pozzoli E, Ibatici A, Nicolini A, Salerno F, Deliliers GL. Four-year survival after trans-jugular intrahepatic porto-systemic shunt for veno-occlusive disease following autologous bone marrow transplantation. Leuk Lymphoma 2004; 45: 1485-1487
88 Nimer SD , Milewicz AL, Champlin RE, Busuttil RW. Successful treatment of hepatic venoocclusive disease in a bone marrow transplant patient with orthotopic liver transplantation. Transplantation 1990; 49: 819-821
89 Rapoport AP , Doyle HR, Starzl T, Rowe JM, Doeblin T, DiPersio JF. Orthotopic liver transplantation for life-threatening veno-occlusive disease of the liver after allogeneic bone marrow transplant. Bone Marrow Transplant 1991; 8: 421-424
90 Kim ID, Egawa H, Marui Y, Kaihara S, Haga H, Lin YW, Kudoh K, Kiuchi T, Uemoto S, Tanaka K. A successful liver transplantation for refractory hepatic veno-occlusive disease originating from cord blood transplantation. Am J Transplant 2002; 2: 796-800
91 Okuda K , Kage M, Shrestha SM. Proposal of a new nomenclature for Budd-Chiari syndrome: hepatic vein thrombosis versus thrombosis of the inferior vena cava at its hepatic portion. Hepatology 1998; 28: 1191-1198
92 Bogin V, Marcos A, Shaw-Stiffel T. Budd-Chiari syndrome: in
evolution. Eur J Gastroenterol Hepatol 2005; 17: 33-3593 Valla DC. Hepatic vein thrombosis (Budd-Chiari syndrome).
Semin Liver Dis 2002; 22: 5-1494 Valla D, Casadevall N, Lacombe C, Varet B, Goldwasser
E, Franco D, Maillard JN, Pariente EA, Leporrier M, Rueff B. Primary myeloproliferative disorder and hepatic vein thrombosis. A prospective study of erythroid colony formation in vitro in 20 patients with Budd-Chiari syndrome. Ann Intern Med 1985; 103: 329-334
95 Denninger MH, Chaït Y, Casadevall N, Hillaire S, Guillin MC, Bezeaud A, Erlinger S, Briere J, Valla D. Cause of portal or hepatic venous thrombosis in adults: the role of multiple concurrent factors. Hepatology 2000; 31: 587-591
96 Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med 1995; 333: 1253-1258
97 Socié G, Mary JY, de Gramont A, Rio B, Leporrier M, Rose C, Heudier P, Rochant H, Cahn JY, Gluckman E. Paroxysmal nocturnal haemoglobinuria: long-term follow-up and prognostic factors. French Society of Haematology. Lancet 1996; 348: 573-577
98 Deltenre P, Denninger MH, Hillaire S, Guillin MC, Casadevall N, Brière J, Erlinger S, Valla DC. Factor V Leiden related Budd-Chiari syndrome. Gut 2001; 48: 264-268
99 Mahmoud AE, Elias E, Beauchamp N, Wilde JT. Prevalence of the factor V Leiden mutation in hepatic and portal vein thrombosis. Gut 1997; 40: 798-800
100 Valla DC. The diagnosis and management of the Budd-Chiari syndrome: consensus and controversies. Hepatology 2003; 38: 793-803
101 Okuda K. Inferior vena cava thrombosis at its hepatic portion (obliterative hepatocavopathy). Semin Liver Dis 2002; 22: 15-26
102 Simson IW. Membranous obstruction of the inferior vena cava and hepatocellular carcinoma in South Africa. Gastroenterology 1982; 82: 171-178
103 Bayraktar Y, Egesel T, Sağlam F, Balkanci F, Van Thiel DH. Does hepatic vein outflow obstruction contribute to the pathogenesis of hepatocellular carcinoma? J Clin Gastroenterol 1998; 27: 67-71
104 Harmanci O, Buyukasik Y, Kirazli S, Balkanci F, Bayraktar Y. Does endothelium agree with the concept of idiopathic hepatic vessel thrombosis? World J Gastroenterol 2006; 12: 1273-1277
105 Henrion J . Ischemia/reperfusion injury of the l iver: pathophysiologic hypotheses and potential relevance to human hypoxic hepatitis. Acta Gastroenterol Belg 2000; 63: 336-347
106 Olzinski AT, Sanyal AJ. Treating Budd-Chiari Syndrome: making rational choices from a myriad of options. J Clin Gastroenterol 2000; 30: 155-161
107 Tavill AS, Wood EJ, Kreel L, Jones EA, Gregory M, Sherlock S. The Budd-Chiari syndrome: correlation between hepatic scintigraphy and the clinical, radiological, and pathological findings in nineteen cases of hepatic venous outflow obstruction. Gastroenterology 1975; 68: 509-518
108 Wanless IR . Micronodular transformation (nodular regenerative hyperplasia) of the liver: a report of 64 cases among 2,500 autopsies and a new classification of benign hepatocellular nodules. Hepatology 1990; 11: 787-797
109 Cazals-Hatem D, Vilgrain V, Genin P, Denninger MH, Durand F, Belghiti J, Valla D, Degott C. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37: 510-519
110 Parker RG. Occlusion of the hepatic veins in man. Medicine (Baltimore) 1959; 38: 369-402
111 Mahmoud AE, Mendoza A, Meshikhes AN, Olliff S, West R, Neuberger J, Buckels J, Wilde J, Elias E. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89: 37-43
112 Darwish Murad S, Valla DC, de Groen PC, Zeitoun G, Hopmans JA, Haagsma EB, van Hoek B, Hansen BE, Rosendaal FR, Janssen HL. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39: 500-508
Bayraktar UD et al. Hepatic venous outflow obstruction 1925
www.wjgnet.com

113 Hadengue A, Poliquin M, Vilgrain V, Belghiti J, Degott C, Erlinger S, Benhamou JP. The changing scene of hepatic vein thrombosis: recognition of asymptomatic cases. Gastroenterology 1994; 106: 1042-1047
114 Dilawari JB, Bambery P, Chawla Y, Kaur U, Bhusnurmath SR, Malhotra HS, Sood GK, Mitra SK, Khanna SK, Walia BS. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73: 21-36
115 De BK, Sen S, Biswas PK, Mandal SK, Das D, Das U, Guru S, Bandyopadhyay K. Occurrence of hepatopulmonary syndrome in Budd-Chiari syndrome and the role of venous decompression. Gastroenterology 2002; 122: 897-903
116 Singh V, Sinha SK, Nain CK, Bambery P, Kaur U, Verma S, Chawla YK, Singh K. Budd-Chiari syndrome: our experience of 71 patients. J Gastroenterol Hepatol 2000; 15: 550-554
117 Kage M, Arakawa M, Kojiro M, Okuda K. Histopathology of membranous obstruction of the inferior vena cava in the Budd-Chiari syndrome. Gastroenterology 1992; 102: 2081-2090
118 Shrestha SM, Okuda K, Uchida T, Maharjan KG, Shrestha S, Joshi BL, Larsson S, Vaidya Y. Endemicity and clinical picture of liver disease due to obstruction of the hepatic portion of the inferior vena cava in Nepal. J Gastroenterol Hepatol 1996; 11: 170-179
119 Bayraktar Y, Harmanci O. Etiology and consequences of thrombosis in abdominal vessels. World J Gastroenterol 2006; 12: 1165-1174
120 Brinson RR, Curtis WD, Schuman BM, Mills LR. Recovery from hepatic vein thrombosis (Budd-Chiari syndrome) complicating ulcerative colitis. Dig Dis Sci 1988; 33: 1615-1620
121 Zeitoun G, Escolano S, Hadengue A, Azar N, El Younsi M, Mallet A, Boudet MJ, Hay JM, Erlinger S, Benhamou JP, Belghiti J, Valla D. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30: 84-89
122 Tang TJ, Batts KP, de Groen PC, van Hoek B, Haagsma EB, Hop WC, Janssen HL. The prognostic value of histology in the assessment of patients with Budd-Chiari syndrome. J Hepatol 2001; 35: 338-343
123 Henderson JM , Warren WD, Mill ikan WJ, Galloway JR, Kawasaki S, Stahl RL, Hertzler G. Surgical options, hematologic evaluation, and pathologic changes in Budd-Chiari syndrome. Am J Surg 1990; 159: 41-48; discussion 48-50
124 Okuda H, Yamagata H, Obata H, Iwata H, Sasaki R, Imai F, Okudaira M, Ohbu M, Okuda K. Epidemiological and clinical features of Budd-Chiari syndrome in Japan. J Hepatol 1995; 22: 1-9
125 Darwish Murad S, Valla DC, de Groen PC, Zeitoun G, Haagsma EB, Kuipers EJ, Janssen HL. Pathogenesis and treatment of Budd-Chiari syndrome combined with portal vein thrombosis. Am J Gastroenterol 2006; 101: 83-90
126 Wadhawan M , Kumar N. Budd-Chiari syndrome. Trop Gastroenterol 2003; 24: 3-7
127 Bolondi L, Gaiani S, Li Bassi S, Zironi G, Bonino F, Brunetto M, Barbara L. Diagnosis of Budd-Chiari syndrome by pulsed Doppler ultrasound. Gastroenterology 1991; 100: 1324-1331
128 Gupta S, Barter S, Phillips GW, Gibson RN, Hodgson HJ. Comparison of ultrasonography, computed tomography and 99mTc liver scan in diagnosis of Budd-Chiari syndrome. Gut 1987; 28: 242-247
129 Millener P, Grant EG, Rose S, Duerinckx A, Schiller VL, Tessler FN, Perrella RR, Ragavendra N. Color Doppler imaging findings in patients with Budd-Chiari syndrome: correlation with venographic findings. AJR Am J Roentgenol 1993; 161: 307-312
130 Ralls PW, Johnson MB, Radin DR, Boswell WD, Lee KP, Halls JM. Budd-Chiari syndrome: detection with color Doppler sonography. AJR Am J Roentgenol 1992; 159: 113-116
131 Lim JH, Park JH, Auh YH. Membranous obstruction of the inferior vena cava: comparison of findings at sonography, CT, and venography. AJR Am J Roentgenol 1992; 159: 515-520
132 Mathieu D, Vasile N, Menu Y, Van Beers B, Lorphelin JM, Pringot J. Budd-Chiari syndrome: dynamic CT. Radiology 1987;
165: 409-413133 Stark DD, Hahn PF, Trey C, Clouse ME, Ferrucci JT. MRI of
the Budd-Chiari syndrome. AJR Am J Roentgenol 1986; 146: 1141-1148
134 Soyer P, Rabenandrasana A, Barge J, Laissy JP, Zeitoun G, Hay JM, Levesque M. MRI of Budd-Chiari syndrome. Abdom Imaging 1994; 19: 325-329
135 Mitchell MC, Boitnott JK, Kaufman S, Cameron JL, Maddrey WC. Budd-Chiari syndrome: etiology, diagnosis and management. Medicine (Baltimore) 1982; 61: 199-218
136 Wang ZG, Jones RS. Budd-Chiari syndrome. Curr Probl Surg 1996; 33: 83-211
137 Millikan WJ, Henderson JM, Sewell CW, Guyton RA, Potts JR, Cranford CA, Cramer AR, Galambos JT, Warren WD. Approach to the spectrum of Budd-Chiari syndrome: which patients require portal decompression? Am J Surg 1985; 149: 167-176
138 Brearley S, Hawker PC, Dykes PW, Keighley MR. A lethal complication of peripheral vein vasopressin infusion. Hepatogastroenterology 1985; 32: 224-225
139 Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350: 578-585
140 Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69: 877-881
141 Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91: 1262-1263
142 Sparano J, Chang J, Trasi S, Bonanno C. Treatment of the Budd-Chiari syndrome with percutaneous transluminal angioplasty. Case report and review of the literature. Am J Med 1987; 82: 821-828
143 Xu K, He FX, Zhang HG, Zhang XT, Han MJ, Wang CR, Kaneko M, Takahashi M, Okawada T. Budd-Chiari syndrome caused by obstruction of the hepatic inferior vena cava: immediate and 2-year treatment results of transluminal angioplasty and metallic stent placement. Cardiovasc Intervent Radiol 1996; 19: 32-36
144 Zhang CQ, Fu LN, Xu L, Zhang GQ, Jia T, Liu JY, Qin CY, Zhu JR. Long-term effect of stent placement in 115 patients with Budd-Chiari syndrome. World J Gastroenterol 2003; 9: 2587-2591
145 Pisani-Ceretti A, Intra M, Prestipino F, Ballarini C, Cordovana A, Santambrogio R, Spina GP. Surgical and radiologic treatment of primary Budd-Chiari syndrome. World J Surg 1998; 22: 48-53; discussion 53-54
146 Witte AM, Kool LJ, Veenendaal R, Lamers CB, van Hoek B. Hepatic vein stenting for Budd-Chiari syndrome. Am J Gastroenterol 1997; 92: 498-501
147 Janssen HL, Garcia-Pagan JC, Elias E, Mentha G, Hadengue A, Valla DC. Budd-Chiari syndrome: a review by an expert panel. J Hepatol 2003; 38: 364-371
148 Ryu RK, Durham JD, Krysl J, Shrestha R, Shrestha R, Everson GT, Stephens J, Kam I, Wachs M, Kumpe DA. Role of TIPS as a bridge to hepatic transplantation in Budd-Chiari syndrome. J Vasc Interv Radiol 1999; 10: 799-805
149 Ganger DR, Klapman JB, McDonald V, Matalon TA, Kaur S, Rosenblate H, Kane R, Saker M, Jensen DM. Transjugular intrahepatic portosystemic shunt (TIPS) for Budd-Chiari syndrome or portal vein thrombosis: review of indications and problems. Am J Gastroenterol 1999; 94: 603-608
150 Senzolo M, Cholongitas EC, Patch D, Burroughs AK. Update on the classification, assessment of prognosis and therapy of Budd-Chiari syndrome. Nat Clin Pract Gastroenterol Hepatol 2005; 2: 182-190
151 Mancuso A, Fung K, Mela M, Tibballs J, Watkinson A, Burroughs AK, Patch D. TIPS for acute and chronic Budd-Chiari syndrome: a single-centre experience. J Hepatol 2003; 38: 751-754
152 Mancuso A, Watkinson A, Tibballs J, Patch D, Burroughs AK. Budd-Chiari syndrome with portal, splenic, and superior mesenteric vein thrombosis treated with TIPS: who dares wins. Gut 2003; 52: 438
153 Perelló A, García-Pagán JC, Gilabert R, Suárez Y, Moitinho E,
1926 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

Cervantes F, Reverter JC, Escorsell A, Bosch J, Rodés J. TIPS is a useful long-term derivative therapy for patients with Budd-Chiari syndrome uncontrolled by medical therapy. Hepatology 2002; 35: 132-139
154 Hernández-Guerra M, Turnes J, Rubinstein P, Olliff S, Elias E, Bosch J, García-Pagán JC. PTFE-covered stents improve TIPS patency in Budd-Chiari syndrome. Hepatology 2004; 40: 1197-1202
155 Slakey DP, Klein AS, Venbrux AC, Cameron JL. Budd-Chiari syndrome: current management options. Ann Surg 2001; 233: 522-527
156 Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232: 340-352
157 Ringe B , Lang H, Oldhafer KJ, Gebel M, Flemming P, Georgii A, Borst HG, Pichlmayr R. Which is the best surgery for Budd-Chiari syndrome: venous decompression or liver transplantation? A single-center experience with 50 patients. Hepatology 1995; 21: 1337-1344
158 Eid A, Rahamimov R, Ilan Y, Tur-Kaspa R, Berlatzky Y. Cavoatrial shunt: a graft salvage procedure for suprahepatic caval anastomosis obstruction after liver transplantation. Liver Transpl Surg 1998; 4: 239-240
159 Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160: 128-133
160 Chang CH, Lee MC, Shieh MJ, Chang JP, Lin PJ. Transatrial membranotomy for Budd-Chiari syndrome. Ann Thorac Surg 1989; 48: 409-412
161 Senning A. Transcaval posterocranial resection of the liver as treatment of the Budd-Chiari syndrome. World J Surg 1983; 7: 632-640
162 Jamieson NV, Williams R, Calne RY. Liver transplantation for Budd-Chiari syndrome, 1976-1990. Ann Chir 1991; 45: 362-365
163 Srinivasan P, Rela M, Prachalias A, Muiesan P, Portmann B, Mufti GJ, Pagliuca A, O'Grady J , Heaton N. Liver transplantation for Budd-Chiari syndrome. Transplantation 2002; 73: 973-977
164 Campbell DA, Rolles K, Jamieson N, O'Grady J, Wight D, Williams R, Calne R. Hepatic transplantation with perioperative and long term anticoagulation as treatment for Budd-Chiari syndrome. Surg Gynecol Obstet 1988; 166: 511-518
165 Moussavian SN, Dincsoy HP, Goodman S, Helm RA, Bozian RC. Severe hyperbilirubinemia and coma in chronic congestive heart failure. Dig Dis Sci 1982; 27: 175-180
166 Logan RG, Mowry FM, Judge RD. Cardiac failure simulating viral hepatitis. Three cases with serum transaminase levels above 1,000. Ann Intern Med 1962; 56: 784-788
167 Cohen JA, Kaplan MM. Left-sided heart failure presenting as hepatitis. Gastroenterology 1978; 74: 583-587
168 Nouel O, Henrion J, Bernuau J, Degott C, Rueff B, Benhamou JP. Fulminant hepatic failure due to transient circulatory failure in patients with chronic heart disease. Dig Dis Sci 1980; 25: 49-52
169 Kisloff B, Schaffer G. Fulminant hepatic failure secondary to congestive heart failure. Am J Dig Dis 1976; 21: 895-900
170 Dunn GD, Hayes P, Breen KJ, Schenker S. The liver in congestive heart failure: a review. Am J Med Sci 1973; 265: 174-189
171 Richman SM, Delamn AJ, Grob D. Alterations in indices
of liver function in congestive heart failure with particular reference to serum enzymes. Am J Med 1961; 30: 211-225
172 Jafri SM. Hypercoagulability in heart failure. Semin Thromb Hemost 1997; 23: 543-545
173 Bessman AN, Evans JM. The blood ammonia in congestive heart failure. Am Heart J 1955; 50: 715-719
174 Lowe MD, Harcombe AA, Grace AA, Petch MC. Lesson of the week: restrictive-constrictive heart failure masquerading as liver disease. BMJ 1999; 318: 585-586
175 Denis C, De Kerguennec C, Bernuau J, Beauvais F, Cohen Solal A. Acute hypoxic hepatitis ( 'liver shock'): still a frequently overlooked cardiological diagnosis. Eur J Heart Fail 2004; 6: 561-565
176 Runyon BA . Cardiac ascites: a characterization. J Clin Gastroenterol 1988; 10: 410-412
177 Naschitz JE, Slobodin G, Lewis RJ, Zuckerman E, Yeshurun D. Heart diseases affecting the liver and liver diseases affecting the heart. Am Heart J 2000; 140: 111-120
178 Ho SS, Brown R, Fitzgibbon B. Hepatocellular carcinoma with cardiac cirrhosis. Med J Aust 1990; 152: 553-554
179 Cameron J, Oesterle SN, Baldwin JC, Hancock EW. The etiologic spectrum of constrictive pericarditis. Am Heart J 1987; 113: 354-360
180 Ling LH, Oh JK, Schaff HV, Danielson GK, Mahoney DW, Seward JB, Tajik AJ. Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy. Circulation 1999; 100: 1380-1386
181 Bertog SC, Thambidorai SK, Parakh K, Schoenhagen P, Ozduran V, Houghtaling PL, Lytle BW, Blackstone EH, Lauer MS, Klein AL. Constrictive pericarditis: etiology and cause-specific survival after pericardiectomy. J Am Coll Cardiol 2004; 43: 1445-1452
182 Marshall JB, Pola L. Chylous ascites secondary to constrictive pericarditis. A discussion of pathophysiologic mechanisms. Dig Dis Sci 1982; 27: 84-87
183 Riza Altiparmak M, Avsar S, Yanik S. Chylous ascites and chylothorax due to constrictive pericarditis in a patient undergoing haemodialysis. Neth J Med 2004; 62: 59-61
184 Solano FX, Young E, Talamo TS, Dekker A. Constrictive pericarditis mimicking Budd-Chiari syndrome. Am J Med 1986; 80: 113-115
185 Cheitlin MD, Armstrong WF, Aurigemma GP, Beller GA, Bierman FZ, Davis JL, Douglas PS, Faxon DP, Gillam LD, Kimball TR, Kussmaul WG, Pearlman AS, Philbrick JT, Rakowski H, Thys DM, Antman EM, Smith SC , Alpert JS, Gregoratos G, Anderson JL, Hiratzka LF, Hunt SA, Fuster V, Jacobs AK, Gibbons RJ, Russell RO. ACC/AHA/ASE 2003 guideline update for the clinical application of echocardiography: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/ASE Committee to Update the 1997 Guidelines for the Clinical Application of Echocardiography). Circulation 2003; 108: 1146-1162
186 Talreja DR, Edwards WD, Danielson GK, Schaff HV, Tajik AJ, Tazelaar HD, Breen JF, Oh JK. Constrictive pericarditis in 26 patients with histologically normal pericardial thickness. Circulation 2003; 108: 1852-1857
187 Troughton RW, Asher CR, Klein AL. Pericarditis. Lancet 2004; 363: 717-727
S- Editor Wang J L- Editor Anand BS E- Editor Che YB
Bayraktar UD et al. Hepatic venous outflow obstruction 1927
www.wjgnet.com

Is portal vein cavernous transformation a component of congenital hepatic fibrosis?
Ozlem Yonem, Yusuf Bayraktar
www.wjgnet.com
Yusuf Bayraktar, MD, Series Editor
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1928-1929www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
Ozlem Yonem, Yusuf Bayraktar, Hacettepe University Faculty of Medicine, Department of Gastroenterology, Sihhiye 06100, Ankara, Turkey Correspondence to: Yusuf Bayraktar, MD, Hacettepe University Faculty of Medicine, Department of Gastroenterology, Sihhiye 06100, Ankara, Turkey. [email protected] Telephone: +90-312-4439428 Fax: +90-312-4429429 Received: 2006-10-11 Accepted: 2006-11-14
AbstractCongenital hepatic fibrosis (CHF) is an autosomal recessive disorder that belongs to the family of fibropolycystic liver diseases. This family includes a spectrum of disorders which are usually found in combination with each other and are usually inherited. Clinically fibropolycystic diseases have three effects being present in different proportions, those of a space occupying lesion, of portal hypertension and of cholangitis. In most patients, the first manifestations of CHF are signs and symptoms related to portal hypertension such as splenomegaly and varices. Portal hypertension in these patients has been attributed to the hypoplasia or compression of the portal vein radicles in the fibrous bands. Cavernous transformation of the portal vein (CTPV) is a relatively rare condition resulting from extrahepatic portal vein obstruction with recanalization or collateral vein formation to bypass the obstruction. It has been found that patients with CHF having an accompanying CTPV have relatively large splenomegaly and suffers more frequent episodes of bleeding from esophageal varices.We believe that CTPV is a congenital component of CHF and also one of the important causative factors of portal hypertension in these patients.
© 2007 The WJG Press. All rights reserved.
Key words: Congenital hepatic fibrosis; Cavernous transformation of portal vein; Portal hypertension
Yonem O, Bayrak ta r Y . I s por ta l ve in cavernous transformation a component of congenital hepatic fibrosis? World J Gastroenterol 2007; 13(13): 1928-1929
http://www.wjgnet.com/1007-9327/13/1928.asp
INTRODUCTIONLiver diseases characterized by some degree of dilatation of segments of intrahepatic bile ducts with associated fibrosis are named as fibropolycystic liver diseases[1]. It is now clear with the help of improved methods of imaging that these diseases do not exist as single entities, but as members of a family. The members are found in various combinations[2]. They consist of congenital hepatic fibrosis, Caroli’s disease, autosomal dominant polycystic kidney disease, autosomal recessive polycystic kidney disease, Von Meyenburg complex and mesenchymal hamartoma[3]. Since their original recognition, many reports on the pathogenesis, genetics and combinations with other morphological alterations of these groups of diseases have been published[4]. Congenital hepatic fibrosis (CHF) belongs to this group, is an autosomal recessive inherited condition and is characterized by a destructive type of cholangiopathy of variable speed and duration associated with scarring fibrosis[5].
ClINICal ChaRaCTeRIsTICs Of Chf wITh aN emphasIs ON pORTal hypeRTeNsIONTypically, patients with CHF do not have cirrhosis and maintain normal hepatic lobular architecture with normal hepatic function. Onset of symptoms and signs is highly variable and ranges from early childhood to the 5th or 6th decades of life, although this disorder is diagnosed in most patients during adolescence or young adulthood[6].
In most patients, the first manifestations of the disease are signs or symptoms related to portal hypertension especially spelenomegaly and varices- often with spontaneous gastrointestinal bleeding[7]. Portal hypertension, a very common clinical feature of CHF, has been attributed to the compression of portal vein radicles in the fibrous bands and to an anomaly in the branching pattern of the portal vein, giving rise to hypoplastic and involutive branches[8].
embRyOlOgICal explaNaTION Of The lINk beTweeN pORTal veIN aND ChfIn view of the embryology, it is not surprising that anomaly of the vessels and of the intrahepatic bile ducts

Yonem O et al. The importance of portal vein thrombosis in CHF 1929
www.wjgnet.com
are usually combined because a close relationship exists between vascular and bile ductal development[9]. The liver develops in the fourth week of gestation on the hepatic diverticulum. Intrahepatic ducts develop from primitive hepatocytes around branches of the portal vein. The layer of cells surrounding the portal vein transforms in to a double-walled cylinder with a slit like lumen and is termed the ductal plate. A progressive progress termed remodelling starts during the 12th wk of gestation. It is the lack of remodelling of the ductal plate that results in persistence of an excess of embryonic duct structures. This abnormality has been termed the ductal plate malformation (DPM) and consists of persistence of the ductal plate with an increase in ducts elements and an increase in portal fibroid tissue[1,9] DPM is frequently associated with abnormalities in the ramification pattern of the portal vein, referred to as a “pollard willow” pattern, resulting in too many, too small, and too closely spaced branches. A transverse section through such a pollard willow area will appear as an enlarged portal tract (in fact, a fusion of several portal tracts) containing several ductal plates (or remnants of incompletely remodelled ductal plates) surrounding several hypoplastic or even obliterated portal vein branches. The ductal plates appear around the mesenchyme surrounding the developing portal vein branches, whereas a similar phenomenon does not occur around the draining hepatic veins. The portal vein clearly has a dominant role in determining the three-dimensional configuration of the biliary tree, in as much as it has a critical role in the development of the liver as a whole[1]. Also Terada et al conducted a detailed study of the development of the human prebiliary capillary plexus and observed that the development and maturation of the prebiliary capillary plexus progress parallel to that of the intrahepatic bile ducts. They hypothesized that the development of both these structures is regulated by common substances on growth factors, which have yet to be determined[3].
CaveRNOUs TRaNsfORmaTION Of The pORTal veIN aND ChfCavernous transformation of portal vein (CTPV) in adults is an uncommon finding, whose cause cannot usually be identified. Some researches consider this condition to be congenital, while others accept that it is a consequence of neonatal umbilical vein sepsis or a complication of an inflammatory neoplastic process occurring later in life[10]. CTPV is frequently associated with congenital anomalies (Figure 1). Among these abnormalities the most frequent
are atrial septal defects or malformations of the biliary tract or of the inferior vena cava. Much less common is the association of prehepatic portal hypertension with intestinal telangiectasia, cutaneous hemangioma, valvular aortic stenosis, left renal agenesis, oesophageal atresia or pyloric stenosis. In one study Bayraktar et al[11]. have found that almost 50% of the cases of congenital hepatic fibrosis had CTPV. In this study also; it was found that congenital hepatic fibrosis patients having CTPV had relatively larger splenomegaly than those who had no CTPV and they also suffered more frequent bleeding episodes from esophageal varices.
CONClUsIONIn conclusion, CHF is an uncommon but important cause of portal hypertension[12]. As a close relationship exists between the development of portal vein and bile ducts; we think that this association between CHF and CTPV is not just a coincidence but rather one of the congenital components of this clinical condition. For this reason we believe that when CTPV is found in a patient with CHF, serious complications of portal hypertension such as bleeding should be expected.
RefeReNCes1 Desmet VJ. Ludwig symposium on biliary disorders-part I.
Pathogenesis of ductal plate abnormalities. Mayo Clin Proc 1998; 73: 80-89
2 Sherlock S, Dooley J. Diseases of the liver and billiary system. 11th ed. Milano: Blackwell Sci Pub, 2002: 583
3 Awasthi A, Das A, Srinivasan R, Joshi K. Morphological and immunohistochemical analysis of ductal plate malformation: correlation with fetal liver. Histopathology 2004; 45: 260-267
4 Teufel J, Farack UM. Hepatobiliary fibropolycystic diseases. Two cases of Caroli's disease. Scand J Gastroenterol Suppl 1987; 139: 76-80
5 Giouleme O, Nikolaidis N, Tziomalos K, Patsiaoura K, Vassiliadis T, Grammatikos N, Papanikolaou V, Eugenidis N. Ductal plate malformation and congenital hepatic fibrosis Clinical and histological findings in four patients. Hepatol Res 2006; 35: 147-150
6 Zeitoun D, Brancatelli G, Colombat M, Federle MP, Valla D, Wu T, Degott C, Vilgrain V. Congenital hepatic fibrosis: CT findings in 18 adults. Radiology 2004; 231: 109-116
7 Mindikoglu AL, Regev A, O'Sullivan MJ, Schiff ER. Multiple normal deliveries in a woman with severe portal hypertension due to congenital hepatic fibrosis: the importance of preserved hepatocellular function. Am J Gastroenterol 2005; 100: 2359-2361
8 El-Youssef M, Mu Y, Huang L, Stellmach V, Crawford SE. Increased expression of transforming growth factor-beta1 and thrombospondin-1 in congenital hepatic fibrosis: possible role of the hepatic stellate cell. J Pediatr Gastroenterol Nutr 1999; 28: 386-392
9 Desmet VJ. Pathogenesis of ductal plate malformation. J Gastroenterol Hepatol 2004; 19: S356-S360
10 Bayraktar Y, Tuncer ZS, Kabukçu A, Uzunalimoğlu B, Ayhan A. Pregnancy complicated by congenital hepatic fibrosis with cavernous transformation of the portal vein: a case report. Am J Obstet Gynecol 1997; 177: 459-461
11 Bayraktar Y, Balkanci F, Kayhan B, Uzunalimoglu B, Ozenc A, Ozdemir A, Dündar S, Arslan S, Sivri B, Telatar H. Congenital hepatic fibrosis associated with cavernous transformation of the portal vein. Hepatogastroenterology 1997; 44: 1588-1594
12 Poddar U, Thapa BR, Vashishta RK, Girish CS, Singh K. Congenital hepatic fibrosis in Indian children. J Gastroenterol Hepatol 1999; 14: 1192-1196
S- Editor Wang J L- Editor Zhu LH E- Editor Che YB
Figure 1 Arterial portography shows the cavernous transformation in portal vein.

Clinical characteristics of Caroli’s disease
Ozlem Yonem, Yusuf Bayraktar
www.wjgnet.com
Yusuf Bayraktar, MD, Series Editor
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1930-1933www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
Ozlem Yonem, Yusuf Bayraktar, Hacettepe University Faculty of Medicine, Department of Gastroenterology, Sihhiye 06100, Ankara, TurkeyCorrespondence to: Yusuf Bayraktar, MD, Hacettepe University Faculty of Medicine, Department of Gastroenterology, Sihhiye 06100, Ankara, Turkey. [email protected]: +90-312-4439428 Fax: +90-312-4429429Received: 2006-10-11 Accepted: 2006-12-20
AbstractCaroli’s disease is a rare congenital condition chara-cterized by non-obstructive saccular or fusiform dilatation of larger intrahepatic bile ducts. Cholangitis, liver cirrhosis, and cholangiocarcinoma are its potential complicat ions. The diagnosis of Carol i ’s disease depends on demonstrating that the cystic lesions are in continuity with the biliary tree which can be showed by ultrasonography, computerized tomography, endoscopic retrograde cholangiopancreatography, percutaneous transhepatic cholangiography or magnetic resonance cholangiopancreatography. Treatment of Caroli’s disease relies on the location of the biliary abnormalities. While localized forms confined to one lobe can be treated with surgery, liver transplantation is the only effective modality for diffuse forms. Although a rare disorder; Caroli’s disease should always be considered in the differential diagnosis of chronic cholestasis of unknown cause.
© 2007 The WJG Press. All rights reserved.
Key words: Caroli’s disease; Liver transplantation; Endoscopic retrograde cholangiopancreatography
Yonem O, Bayraktar Y. Clinical characteristics of Caroli’s disease. World J Gastroenterol 2007; 13(13): 1930-1933
http://www.wjgnet.com/1007-9327/13/1930.asp
INTRODUCTIONCaroli’s disease (CD) was first described by Caroli as a congenital malformation of intrahepatic bile ducts, characterized by segmental cystic dilatation of the intrahepatic ducts; increased incidence of biliary lithiasis, cholangitis and liver abscesses; absence of cirrhosis and portal hypertension; and association of renal tubular
ectasia or similar renal cystic disease[1]. Mode of inheritance is still unclear but in majority of cases it is transmitted in autosomal recessive fashion[2]. Review of the literature suggests that there are no large series of this disease[3]. Although present from birth, the disease usually remains asymptomatic during the first 20 years, and may also remain so throughout life. However when symptomatic, a significant number of these patients present significant loss in their quality of life and their clinical course frequently worsen due to the repeated episodes of cholangitis with the presence of intrahepatic calculi, intrahepatic abscesses and sepsis[4].
EPIDEMIOLOGYUntil 1984, 162 cases of Caroli disease had been reported[1]. The estimated incidence of Caroli’s disease is 1 in 1 000 000 population[5]. Males and females are equally affected and more than 80% of patients present before 30 years of age[1].
PATHOGENESISThe cause of CD is unknown but occasional familial clustering suggests that some cases are inherited, in particular when CD occurs with Polycystic kidney disease[6]. In one study liver biopsies of patients with Caroli’s disease were cytogenetically analyzed and an unbalanced translocation between chromosome 3 and 8 was detected and suggested that loss of distal 3p and/or gain of 8q could be of pathogenetic importance in CD[7]. The likely mechanism involves an in utero event that makes a derangement in the normal embryologic remodelling of ducts and causes varying degrees of destructive inflammation and segmental dilatation[8].
CLINICAL PRESENTATIONThe clinical course can be asymptomatic for the first 5-20 years or symptoms can occur infrequently throughout the patient’s life[9]. Intrahepatic ductal ectasia predisposes to stagnation of the bile which may lead to the formation of stones and favour infections such as cholangitis, abscess or even septicaemia. However this disease is frequently noted by recurrent fever, jaundice and/or pain in the right hypochondrium. A literature review found recurrent acute cholangitis as the main mode of presentation in 64% of the patients[5]. The notion of isolated choledochal lithiasis in the absence of gallbladder lithiasis should lead us think

Yonem O et al. Caroli’s disease 1931
www.wjgnet.com
about an intrahepatic lithogenic focus and, consequently about Caroli’s disease[4].
Patients with CD and congenital hepatic fibrosis can present with portal hypertension and its sequelae such as ascites and esophageal varice hemorrhage. Histological intra hepatic bile duct ectasia and proliferation are associated with severe periportal fibrosis. On physical examination the liver is frequently enlarged and the spleen becomes palpable as portal hypertension develops.
Laboratory studies typically show an elevation of serum alkaline phosphatase, direct bilirubin and a leucocytosis with a predominance of neutrophils. Hepatic synthetic function is well-preserved initially, but may be affected by progressive liver damage due to recurrent cholangitis and biliary obstruction. Coagulopathy from vitamin K malabsorbtion may occur in cholestatic patients[10].
DIAGNOSISHistologically, the main macroscopic and microscopic features of CD are: non-obstructive, localized dilatation of the bile ducts, intraluminal bulbar protrusions of the ductal wall and intra ductal vascular tracts containing patent portal venous and hepatic arterial channels that traverse the true lumen and terminate within the lumen[11]. The diagnosis of CD therefore rests on demonstrating that the cystic lesions are in continuity with the biliary tree. It can be done by imaging studies such as abdominal USG, CT scan, and isotope scan, ERCP, PTC and MRCP. The classical finding of CD is finding of a cold area on 99 m TC sulphur colloid scan becoming hot on 99 m Tc DISIDA scan[12].
USG and CT studies may visualize liver cysts and possible intrahepatic lithiasis and provide information on the common bile duct, but differentiation from other liver cysts such as polycystic liver disease is often difficult[6].
CT scan shows central dot signs in CD patients. The fibrovascular bundles containing portal vein radical and a branch of hepatic artery bridging the saccule appear as central dots or linear streak. This central dot sign described on CT scan is suggested as a pathognomic finding in CD and it can also be demonstrated on USG. ERCP is the method with the highest sensitivity in the diagnosis of CD. The cholangiographic features of Caroli’s disease are
well established as saccular or fusiform dilatation of the intrahepatic bile ducts. These findings can also be seen on computed tomography (Figure 1). Irregular bile duct walls, strictures and stones may be present. Therefore, direct cholangiography is considered the method of choice for an accurate diagnosis of Caroli’s disease. However serious complications (sepsis, bile leak, bleeding and death) may occur with both ERCP and PTC, with an overall incidence of approximately 3%. MRCP is a noninvasive technique that has shown good correlation with ERCP in diagnosing biliary tract abnormalities without any of the risks seen with invasive procedures. The ability to demonstrate the intrahepatic ductal anatomy makes MRCP a reliable diagnostic tool in Caroli’s disease, providing that acceptable image quality can be maintained[13].
DIFFERENTIAL DIAGNOSISThe differential diagnosis for patients exhibiting the symptoms of CD is broad and includes PSC, recurrent pyogenic cholangitis, polycystic liver disease, a choledochal cysts, biliary papillomotosis and occasionally obstructive biliary dilatation. PSC and recurrent pyogenic cholangitis may be associated with ductal dilatation, stenosis, intrahepatic calculi and malignancy. However the ductal dilatation in PSC is rarely saccular and is typically more isolated and fusiform, which is not characteristic of CD. Additionally 70% of white patients with PSC have coexisting inflammatory bowel disase. Patients with polycystic liver disease may present with both hepatic and renal cysts. However, the hepatic cysts of polycystic liver only rarely communicate with the bile ducts. These patients have typically intrinsically normal bile ducts.
Recurrent pyogenic cholangitis is the most difficult diagnosis to exclude because patients with pyogenic cholangitis present with sepsis and have intra- and extrahepatic biliary dilatation. The presence of saccular dilatation favors the diagnosis of Caroli’s disease because saccular dilatation is unusual in recurrent pyogenic cholangitis[8]. Both Caroli’s disease and choledochal cyst are inherited anomalies of the bile ducts but Caroli’s disease involves the congenital dilation of the segmental intrahepatic biliary tree, whereas choledochal cysts involve the cystic dilation of the common bile duct[1].
ASSOCIATED CONDITIONSCaroli’s disease often coexists with congenital hepatic fibrosis and is then designated Caroli’s syndrome. Both result from malformations of the embryonic ductal plate at different levels of the biliary tree[14]. If the defective remodelling involves the larger intrahepatic ducts Caroli’s disease develops, but when the entire intrahepatic bilary tree is involved the condition is Caroli’s syndrome[15]. Incidence of Caroli’s syndrome is more than Caroli’s disease[2]. In addition, various renal disorders may be ssen in conjunction with these hepatic diseases, including autosomal polycystic kidney disease (both dominant and recessive forms), medullary sponge kidney and medullary cystic disease[7]. Between the times 1971-2005 we have followed-up 10 patients with Caroli’s disease. Of these, 4
Figure 1 Abdominal computed tomography shows cystic dilations of the biliary system.
R L

had isolated Caroli’s disease while the remaining 6 patients had Caroli’s syndrome. Among the patients with Caroli’s syndrome; 2 patients had Cavernomatous transformation of portal vein and 2 had polycystic kidney disease as an associated condition. One of these patients died due to sepsis and two of them underwent liver transplantation because of the development of secondary bi l iar y cirrhosis[16].
COMPLICATIONSDespite the advances made in medical, endoscopic and surgical treatments over the last 3 decades, they have not yet been sufficiently effective to allow sustained recovery from the symptoms of CD. The evolution of CD is dominated by the occurrence of repetitive bouts of cholangitis, the frequency of which may vary importantly from one patient to another. In case of frequent episodes of cholangitis, the long term prognosis is poor and these patients graduately lose their quality of life at a usually productive age[6]. Caroli’s disease can be complicated by the formation of liver abscesses, intra- and extrahepatic lithiasis and even cholangiocarcinoma[17].
The incidence of cancer associated with congenital cysts of bile ducts has been reported to be between 2.5% and 16% and appears to be associated with biliary stasis, the action of the carcinogenic substances contained in the bile and chronic inflammation of the biliary epithelium, leading to dysplastic diseases[4]. Amyoidosis is also described as another complication of Caroli’s disease. Death is related to septicaemia and hepatic abscesses[18].
TREATMENTThe treatment of CD depends on the clinical features and the location of the biliary abnormalities. The localized forms, which involve the whole of the left half of the liver, or the right half of the liver, are curable by surgery. They should be treated by hemi-hepatectomy with associated treatment of any problem affecting the common duct[19]. Diffuse involvement of both lobes can be treated with conservative management, (Appropriate antibiotics for cholangitis and ursodeoxycolic acid therapy for litholysis in case of intrahepatic cholelithiasis), endoscopic therapy (sphincterotomy for clearance of intrahepatic stones) and internal biliary bypass procedure[15].
Surgery can offer a definite therapy, with an acceptable morbidity in localized CD[20]. Symptomatic relief after partial hepatectomy is often complete and permanent. Bilateral disease complicated by recurrent cholangitis, cirrhosis or both do not find the same solution and is often difficult to manage. Emergency surgery in the presence of acute cholangitis and deteriorating liver function is associated with high mortality (20%-40%) and morbidity (44%-80%). So it seems that liver transplantation is the only and ultimate management option for these patients even if they are asymptomatic[6]. Survival after liver transplantation was found to be poor in patients with congenital hepatic fibrosis (Caroli’s syndrome) and in those who had cholangitis at the time of the ortotopic liver transplantation in one study[21].
The role of liver transplantation for cholangiocarcinoma whether associated with CD or not is controversial. Because of the high rate of recurrence published previously, most centers have abandoned cholangiocarcinoma as an indication for liver transplantation[6].
CONCLUSIONAs a result CD is an inherited disorder which may cause severe, life threatening cholangitis or which may lead to hepatobiliary degeneration[17]. But CD can be misdiagnosed due to the lack of experience in low volume hospitals where the symptoms of this disease have been repeatedly treated and where many of the diverse modern imaging techniques are unavailable. For this reason despite its rare incidence; Caroli’s disease should not be forgotten in the differential diagnosis of recurrent cholangitis.
REFERENCES1 Lu SC, Debian KA. Cystic diseases of the biliary tract. In:
Yamada T, Alpers DH, Kaplowitz N, Laine L, Owyang C, Powell DW. Textbook of Gastroenterology. Philadelphia: Lippincott Williams and Wilkins, 2003: 2225-2233
2 Gupta AK, Gupta A, Bhardwaj VK, Chansoria M. Caroli's disease. Indian J Pediatr 2006; 73: 233-235
3 Madjov R, Chervenkov P, Madjova V, Balev B. Caroli's disease. Report of 5 cases and review of literature. Hepatogastroenterology 2005; 52: 606-609
4 Waechter FL , Sampaio JA, Pinto RD, Alvares-da-Silva MR, Cardoso FG, Francisconi C, Pereira-Lima L. The role of liver transplantation in patients with Caroli's disease. Hepatogastroenterology 2001; 48: 672-674
5 Giovanardi RO. Monolobar Caroli's disease in an adult. Case report. Hepatogastroenterology 2003; 50: 2185-2187
6 Kassahun WT, Kahn T, Wittekind C, Mössner J, Caca K, Hauss J, Lamesch P. Caroli's disease: liver resection and liver transplantation. Experience in 33 patients. Surgery 2005; 138: 888-898
7 Parada LA, Hallén M, Hägerstrand I, Tranberg KG, Johansson B. Clonal chromosomal abnormalities in congenital bile duct dilatation (Caroli's disease). Gut 1999; 45: 780-782
8 Levy AD, Rohrmann CA, Murakata LA, Lonergan GJ. Caroli's disease: radiologic spectrum with pathologic correlation. AJR Am J Roentgenol 2002; 179: 1053-1057
9 Wu KL, Changchien CS, Kuo CM, Chuah SK, Chiu YC, Kuo CH. Caroli's disease-a report of two siblings. Eur J Gastroenterol Hepatol 2002; 14: 1397-1399
10 Suchy FJ. Caroli’s disease. Uptodate 2006 Version 14.111 Keramidas DC, Kapouleas GP, Sakellaris G. Isolated Caroli's
disease presenting as an exophytic mass in the liver. Pediatr Surg Int 1998; 13: 177-179
12 Sharma R, Mondal A, Taneja V, Rawat HS. Radionuclide scintigraphy in Caroli's disease. Indian J Pediatr 1997; 64: 105-107
13 Asselah T, Ernst O, Sergent G, L'herminé C, Paris JC. Caroli's disease: a magnetic resonance cholangiopancreatography diagnosis. Am J Gastroenterol 1998; 93: 109-110
14 Sherlock S, Dooley J. Diseases of the liver and billiary system. 11th ed. Milano: Blackwell Sci Pub, 2002: 583
15 Desmet VJ. Pathogenesis of ductal plate malformation. J Gastroenterol Hepatol 2004; 19: S356-S360
16 Yönem O, Ozkayar N, Balkanci F, Harmanci O, Sökmensüer C, Ersoy O, Bayraktar Y. Is congenital hepatic fibrosis a pure liver disease? Am J Gastroenterol 2006; 101: 1253-1259
17 De Kerckhove L, De Meyer M, Verbaandert C, Mourad M, Sokal E, Goffette P, Geubel A, Karam V, Adam R, Lerut J. The place of liver transplantation in Caroli's disease and syndrome.
1932 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

Transpl Int 2006; 19: 381-38818 Patil S, Das HS, Desai N, Manjunath SM, Thakur BS, Sawant P.
Caroli's syndrome-a rare cause of portal hypertension. J Assoc Physicians India 2004; 52: 261
19 Yilmaz S, Kirimlioglu H, Kirimlioglu V, Isik B, Coban S, Yildirim B, Ara C, Sogutlu G, Yilmaz M. Partial hepatectomy is curative for the localized type of Caroli's disease: a case
report and review of the literature. Surgeon 2006; 4: 101-10520 Bockhorn M, Malagó M, Lang H, Nadalin S, Paul A, Saner F,
Frilling A, Broelsch CE. The role of surgery in Caroli's disease. J Am Coll Surg 2006; 202: 928-932
21 Habib S, Shakil O, Couto OF, Demetris AJ, Fung JJ, Marcos A, Chopra K. Caroli's disease and orthotopic liver transplanta-tion. Liver Transpl 2006; 12: 416-421
S- Editor Wang J L- Editor Lutze M E- Editor Che YB
Yonem O et al. Caroli’s disease 1933
www.wjgnet.com

Clinical characteristics of Caroli’s syndrome
Ozlem Yonem, Yusuf Bayraktar
www.wjgnet.com
Yusuf Bayraktar, MD, Series Editor
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1934-1937www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
Ozlem Yonem, Yusuf Bayraktar, Hacettepe University Faculty of Medicine, Department of Gastroenterology, Sihhiye 06100, Ankara, TurkeyCorrespondence to: Yusuf Bayraktar, MD, Hacettepe University Faculty of Medicine, Department of Gastroenterology, Sihhiye 06100, Ankara, Turkey ayrakhacettepeedutr ayrakhacettepeedutrayrakhacettepeedutrTelephone: +9019191999 Fax: +90199+90199901991991999999Received: 0061011 Accepted: 00619
AbstractCaroli’s syndrome is characterized by multiple segmental cystic or saccular dilatations of intrahepatic bile ducts associated with congenital hepatic fibrosis. The clinical features of this syndrome reflect both the characteristics of congenital hepatic fibrosis such as portal hypertension and that of Carol i ’s disease named as recurrent cholangitis and cholelithiasis. The diagnosis depends on both histology and imaging methods which can show the communication between the sacculi and the bile ducts. Treatment consists of symptomatic treatment of cholangitis attacks by antibiotics, some endoscopic, radiological and surgical drainage procedures and surgery. Liver transplantation seems the ultimate treatment for this disease. Prognosis is fairly good unless recurrent cholangitis and renal failure develops.
© 2007 The WJG Press. All rights reserved.
Key words: Caroli's syndrome; Liver transplantation; Endoscopic retrograde cholangiopancreatography
Yonem O, Bayraktar Y. Clinical characteristics of Caroli'sClinical characteristics of Caroli's syndrome. World J Gastroenterol 2007; 13(13): 1934-1937
http://www.wjgnet.com/1007-9327/13/1934.asp
INTRODUCTIONCystic lesions of the liver and bile ducts are increasingly being diagnosed. It is now clear that fibropolycystic diseases do not exist as single entities but as members of a family. The members are found in various combinations and are usually inherited. They consist of polycystic liver disease, microhamartoma, congenital hepatic fibrosis, Caroli’s disease and choledochal cysts[1].
Caroli’s syndrome consists of Caroli’s disease and congenital hepatic fibrosis. The term congenital hepatic
fibrosis refers to a unique congenital liver histology characterized by bland portal fibrosis, hyperproliferation of interlobular bile ducts within the portal areas with variable shapes and sizes of bile ducts, and preservation of normal lobular architecture[2]. Caroli’s disease is however characterized by multiple segmental cystic dilatations of the intrahepatic bile ducts[3]. Many authors believe that the two conditions are actually different stages of the same disease characterized by periportal fibrosis and ductal dilatation[4].
EPIDEMIOLOGYThe major published reports concerning bile duct cysts have come from Great Britain, France, Japan and the United States[5]. More than 200 cases of Caroli’s disease have been reported in the literature[3] and the incidence of Caroli’s syndrome is more than the pure form of Caroli’s disease[6].
PATHOGENESISCaroli’s syndrome is a developmental anomaly and there are many theories explaining its pathogenesis. The most acceptable theory is related to ductal plate malformation at different levels of the intrahepatic biliary tree. Intrahepatic bile ducts develop from bipotential liver progenitor cells in contact with the mesenchyme of the portal vein which form from the ductal plates. The ductal plates are then remodeled into mature tubular ducts[7]. This process is regulated by factors influencing epithelial proliferation and apoptosis, the surrounding mesenchymal tissue, as well as the portal veins and various adhesion molecules[8]. Just as the formation of ductal plates follows the branching growth of portal vein from the hilus of the liver to its periphery, so does the remodelling process of the ductal plates beginning from the larger ducts to the smaller ducts. So the hereditary factors causing Caroli's syndrome seem to exert their influence not only during the early embryologic period of large intrahepatic bile duct formation, but also later during the development of the more proximal intralobular ducts related with congenital hepatic fibrosis[9]. It appears to be inherited in an autosomal recessive manner because mutations in PKHD1, the gene linked to adult recessive polycystic kidney disease (ARPKD) have also been identified in patients with Caroli's syndrome.
CLINICAL FEATURESCaroli's syndrome presents a clinical syndrome which is

Yonem O et al. Caroli's syndrome 1935
www.wjgnet.com
a combination of Caroli's disease (bouts of cholangitis, hepatolithiasis, and gallbladder stones) and those of congenital hepatic fibrosis (portal hypertension). Clinical progression and presentation of Caroli's syndrome is highly variable and symptoms may appear early or late during life. The main consequences of congenital hepatic fibrosis are portal hypertension and the development of oesophageal varices. Portal hypertension may result in hematemesis or melena. In the majority of patients, portal hypertension will not be present or will appear only later in the disease evolution[10]. The late appearance of these symptoms in patients with Caroli's disease suggests that congenital hepatic fibrosis in Caroli's disease is dynamic and progressive[11]. Bile stagnation and hepatolithiasis explain the recurrent cholangitis which dominates the clinical course and which is the principal cause of morbidity and mortality. Chronic abdominal pain, pancreatitis resulting from biliary stones and liver abscess are other disease complications[10].
DIAGNOSISThe laboratory findings are non-specific. Transaminase levels may be slightly elevated. The complete blood count may reveal thrombocytopenia and leukopenia if portal hypertension and hypersplenism are present. Elevated white blood cell count or erythrocyte sedimentation rate may indicate cholangitis. BUN and creatinine values should be obtained to detect associated renal disease.
Demonstration of communication between sacculi and bile ducts is important in showing the Caroli's disease component of Caroli's syndrome which can be achieved by USG, CT, MRCP or ERCP. The sonographic appearance in Caroli's disease is intrahepatic cystic anechoic areas in which fibrovascular bundles, stones and linear bridging or septum may be present. The fibrovascular bundles are composed of portal vein and hepatic arteries, which can be demonstrated by Doppler ultrasonography. Gorka mentioned that Doppler sonographic monitoring of portal vein in the fibrovascular bundle was useful in following the progression of congenital hepatic fibrosis component of Caroli's disease[3]. MR imaging is a non-invasive technique which can confirm the diagnosis especially in the large or small cystic patterns. The use of gadolinium enhanced sequences may allow the visualization of the dot sign which seems to be very specific of the malformation of the ductal plate. MR imaging can also suggest
accompanying abnormalities such as portal hypertension, cirrhosis and renal involvement[12]. The diagnosis is more difficult to establish in the case of fusiform dilatations of the biliary tracts and ERCP is the gold standard in this situation. ERCP shows communication between the sacculi and bile ducts and diverticulum-like sacculi of the intrahepatic biliary tree[3] (Figure 1). Intra or extra hepatic cysts can easily be detected by these methods; whereas congenital hepatic fibrosis is a histopathological diagnosis. Histopathological intrahepatic bile duct ectasia and proliferation are associated with severe periportal fibrosis and confirm the congenital hepatic fibrosis component of Caroli's syndrome (Figure 2).
DIFFERENTIAL DIAGNOSISThe appearance of ductal dilatation related to the Caroli’s disease component of Caroli's syndrome on CT/MRI can be confused with polycystic liver disease or obstructive bile duct dilatation. Whereas the cystic spaces in Caroli’s are irregular in shape and communicate with biliary tree, cysts in polycystic liver disease are rounder and smoother and they deform but do not communicate with bile ducts[13]. Portal hypertension is rare in polycystic liver disease[2].
In Caroli’s disease dilated bile ducts have a random bizarre pattern and there are focal areas of cystic ectasia. This differs from appearance in obstructive bile duct dilatation where dilatation is most marked centrally, tapers towards periphery in an organized pattern and lacks focal areas of cystic dilatation. Other diseases that should be considered in the differential diagnosis are primary sclerosing cholangitis, biliary papillomatosis and a choledochal cyst. The cholangiographic features of Caroli’s disease are well established as saccular or fusiform dilatation of the intrahepatic bile ducts. The association of Caroli's disease with extrahepatic bile duct dilatation is mentioned throughout the literature, as is the coexistence of Caroli's disease with choledochal cysts. The exact incidence of extrahepatic duct involvement in Caroli's disease is not known. A literature review of 46 cases found extrahepatic dilatation in 21% of patients. In the series of Levy including 17 patients (5 with Caroli's syndrome) this ratio is even higher with 53% of patients having extrahepatic duct dilatation. Recognition of this feature is important because Caroli's disease should be considered in the differential diagnosis for patients with intra- and extrahepatic biliary dilatation. Repeated bouts of
Figure 1 The ERCP image shows multiple cystic dilatations throughout the biliary tract.
Figure 2 Severe peri-portal fibrotic bands upon histological exa-mination.

cholangitis, stone formation and stone passage may explain extrahepatic duct dilatation in some patients with Caroli's disease or Caroli's syndrome. In the series of Levy, the presence of diffuse fusiform dilatation of the extrahepatic duct measuring 3 cm or less in diameter combined with the characteristic intrahepatic ductal findings has been suggested to be useful in differentiating patients with Caroli's disease from patients with a choledochal cyst and biliary dilatation[13].
ASSOCIATED CONDITIONSAs Caroli's syndrome is a member of the fibropolycystic disease family and its members are usually found in various combinations, presence of an association with any member should be sought.
Caroli's syndrome is associated with renal involvement in up to 60% of patients and implies a dilatation of the collecting renal tubules. Kidney lesions include renal tubular ectasia (medullary sponge kidney, cortical cyst) lesions of adult recessive polycystic kidney disease or rarely autosomal dominant polycystic kidney disease. The association between congenital hepatic fibrosis and ARKPD is caused by mutations in PKHD1 gene that encodes fibrocystin, a protein of primary cilia. Genetic defects in fibrocystin cause ciliary dysfunction, presently considered a major pathogenic event in cystogenesis[10]. In our study; between the time period 1971-2003 we followed-up 6 cases of Caroli’s syndrome in whom, 2 patients had cavernomatous transformation of portal vein and 2 had polycystic kidney disease as an associated condition. One of these patients died due to sepsis and two of them underwent liver transplantataion because of the development of secondary biliary cirrhosis[14].
Over the past years there has been an increasing recognition of the association of congenital hepatic fibrosis with not only fibropolycystic disease family related diseases but also with abnormalities in other organs such as pulmonary fibrosis, congenital heart disease, pulmonary hypertension with arteriovenous fistula, cavernomatous transformation of the portal vein and with Joubert’s syndrome[15-17].
COMPLICATIONSComplications from Caroli’s syndrome are cholangitis, sepsis, choledocholithiasis, hepatic abscess, cholangio-carcinoma and portal hypertension[18]. After cholangitis occurs, a large number of patients die within 5-10 years. Caroli's syndrome may progress to cholangiocarcinoma[3].The occurence of hepatobiliary malignant transformation explained by chronic inflammation of the biliary tree, has been reported in 7%-14% of patients. Death is related to liver failure or complications of portal hypertension[18,19].
TREATMENTMedical treatment of Caroli's syndrome using antibiotics may stabilize the acute cholangitis. Drainage procedures with ERCP or PTC are important and sphincterotomy can aid biliary drainage and stone removal or subsequent
passage and may decrease bouts of cholang i t i s. Radiological, endoscopical and surgical biliary drainage procedures are however palliative treatments that may improve temporarily bile drainage but that become finally inefficient in relation to disease progression. Repeated palliative treatments should be avoided because of the prolongation of the symptomatic period and the increased risk of malignant transformation.
Imaging is essential in planning the surgical treatment which can consist of enterostomy, segmental or lobar hepatic resection, or liver transplantation[12] if the process is confined to one lobe, lobectomy completely relieves the symptoms. Internal surgical bypass procedures (choledochojejunostomy, Roux-en-Y hepaticojejunostomy) i s he lpful in d i f fuse for ms of the d isease. Liver transplantation represents the only curative treatment for symptomatic Caroli's disease or Caroli's syndrome. LT should be offered early in case of recurrent cholangitis and (suspicion of) early malignant transformation of the bilary tract. In case of associated renal disease, both renal and liver diseases should be documented in order to optimize the decision to perform sequential or combined hepatorenal transplantation[10].
As for portal hypertension, therapy for acute variceal haemorrhage includes stabilization with blood and blood products, vasopressin or somatostatin, early endoscopy sclerotherapy, and band ligation. Portocaval shunts are usually received as salvage treatment when sclerotherapy/banding ligation fails[20]. In cases when progression to biliary fibrosis might occur, transplantation has also to be considered. Early surgery with splenorenal or portocaval shunting may be required in recurrent bleeding episodes not available to sclerotherapy, oesophageal tamponade together with supportive medical treatment[15]. Transjugular intrahepatic portosystemic shunts are considered for patients not available to sclerotherapy. The inheritance of the disease seems to be autosomally recessive; therefore genetic counselling for the family may be important[21].
CONCLUSIONAs a result, Caroli’s syndrome is an autosomal recessive disorder having diverse manifestations related to its association with the family of fibropolycystic diseases. Cholestasis can be observed in patients with Caroli's syndrome without cholangitis. This is in part due to the anatomical abnormality in small bile ducts related with Caroli's disease and also due to the small bile duct proliferation in the portal area related with congenital hepatic fibrosis which are both components of this syndrome. Another explanation for this situation is that stones can remain silent without causing cholangitis.
In conclusion, because of the slow and usually silent progress of Caroli’s syndrome along with its rarity and fatal complications, it should be considered in the differential diagnosis of recurrent cholangitis of unknown cause.
REFERENCES1 Sherlock S, Dooley J. Diseases of the liver and billiary system.
11th ed. Milano: Blackwell Sci Pub, 2002: 5832 Lu SC, Debian KA. Cystic diseases of the biliary tract. In:
1936 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

Yamada T, Alpers DH, Kaplowitz N, Laine L, Owyang C, Powell DW. Textbook of Gastroenterology. Philadelphia: Lippincott Williams and Wilkins, 2003: 2225-2233
3 Wu KL, Changchien CS, Kuo CM, Chuah SK, Chiu YC, Kuo CH. Caroli's disease-a report of two siblings. Eur J Gastroenterol Hepatol 2002; 14: 1397-1399
4 Keramidas DC, Kapouleas GP, Sakellaris G. Isolated Caroli's disease presenting as an exophytic mass in the liver. Pediatr Surg Int 1998; 13: 177-179
5 Madjov R, Chervenkov P, Madjova V, Balev B. Caroli's disease. Report of 5 cases and review of literature. Hepatogastroenterology 2005; 52: 606-609
6 Gupta AK, Gupta A, Bhardwaj VK, Chansoria M. Caroli's disease. Indian J Pediatr 2006; 73: 233-235
7 Kim JT, Hur YJ, Park JM, Kim MJ, Park YN, Lee JS. Caroli's syndrome with autosomal recessive polycystic kidney disease in a two month old infant. Yonsei Med J 2006; 47: 131-134
8 Awasthi A, Das A, Srinivasan R, Joshi K. Morphological and immunohistochemical analysis of ductal plate malformation: correlation with fetal liver. Histopathology 2004; 45: 260-267
9 Desmet VJ. Pathogenesis of ductal plate malformation. J Gastroenterol Hepatol. 2004; 19: S356-S360
10 De Kerckhove L, De Meyer M, Verbaandert C, Mourad M, Sokal E, Goffette P, Geubel A, Karam V, Adam R, Lerut J. The place of liver transplantation in Caroli's disease and syndrome. Transpl Int 2006; 19: 381-388
11 Gorka W, Lewall DB. Value of Doppler sonography in the assessment of patients with Caroli's disease. J Clin Ultrasound 1998; 26: 283-287
12 Guy F, Cognet F, Dranssart M, Cercueil JP, Conciatori L,
Krausé D. Caroli's disease: magnetic resonance imaging features. Eur Radiol 2002; 12: 2730-2736
13 Levy AD, Rohrmann CA, Murakata LA, Lonergan GJ. Caroli's disease: radiologic spectrum with pathologic correlation. AJR Am J Roentgenol 2002; 179: 1053-1057
14 Yönem O, Ozkayar N, Balkanci F, Harmanci O, Sökmensüer C, Ersoy O, Bayraktar Y. Is congenital hepatic fibrosis a pure liver disease? Am J Gastroenterol 2006; 101: 1253-1259
15 Abdullah AM, Nazer H, Atiyeh M, Ali MA. Congenital hepatic fibrosis in Saudi Arabia. J Trop Pediatr 1991; 37: 240-243
16 Bayraktar Y, Balkanci F, Kayhan B, Uzunalimoglu B, Ozenc A, Ozdemir A, Dündar S, Arslan S, Sivri B, Telatar H. Congenital hepatic fibrosis associated with cavernous transformation of the portal vein. Hepatogastroenterology 1997; 44: 1588-1594
17 Dahlstrom JE, Cookman J, Jain S. Joubert syndrome: an affected female with bilateral colobomata. Pathology 2000; 32: 283-285
18 Patil S, Das HS, Desai N, Manjunath SM, Thakur BS, Sawant P. Caroli's syndrome-a rare cause of portal hypertension. J Assoc Physicians India 2004; 52: 261
19 Kassahun WT, Kahn T, Wittekind C, Mössner J, Caca K, Hauss J, Lamesch P. Caroli's disease: liver resection and liver transplantation. Experience in 33 patients. Surgery 2005; 138: 888-898
20 Shun A, Delaney DP, Martin HC, Henry GM, Stephen M. Portosystemic shunting for paediatric portal hypertension. J Pediatr Surg 1997; 32: 489-493
21 Yüce A, Koçak N, Akhan O, Gürakan F, Ozen H. Caroli's syndrome in two siblings. Am J Gastroenterol 2002; 97 : 1855-1856
S- Editor Wang J L- Editor Zhu LH E- Editor Che YB
Yonem O et al. Caroli's syndrome 1937
www.wjgnet.com

with rvAdCMV/NK4 (2537.4 ± 892.3 mm3) compared to those treated by either PBS (5175.2 ± 1228.6 mm3) or Ad-LacZ (5578.8 ± 1955.7 mm3) (P < 0.05). The tumor growth inhibition rate was as high as 51%. Immunohistochemical staining revealed a similar PCNA labeling index (75.1% ± 11.2% in PBS group vs 72.8% ± 7.6% in Ad-LacZ group vs 69.3% ± 9.4% in rvAdCMV/NK4 group) in al l groups, but signif icantly lower microvessel density (10.7 ± 2.4 in rvAdCMV/NK4 group vs 25.6 ± 3.8 in PBS group or 21.3 ± 3.5 in Ad-LacZ group, P < 0.05), and a markedly higher apoptotic index (7.3% ± 2.4% in rvAdCMV/NK4 group vs 2.6 ± 1.1% in PBS group or 2.1% ± 1.5% in Ad-LacZ group, P < 0.05) in the rvAdCMV/NK4 group compared to the two control groups. In the tumor metastasis model, the number and weight of disseminated tumors of mice treated with rvAdCMV/NK4 were much lower than those of the control groups (tumor number: 6.2 ± 3.3 in rvAdCMV/NK4 group vs 22.9 ± 7.6 in PBS group or 19.8 ± 8.5 in Ad-LacZ group, P < 0.05; tumor weight: 324 ± 176 mg in rvAdCMV/NK4 group vs 962 ± 382 mg in PBS group or 1116 ± 484 mg in Ad-LacZ group, P < 0.05).
CONCLUSION: The recombinant adenovirus, rvAdCMV/NK4, can attenuate the growth of colon cancer in vivo , probably by suppressing angiogenesis and inducing tumor cell apoptosis, but not by direct suppression of tumor cell proliferation. Moreover, rvAdCMV/NK4 may inhibit peritoneal dissemination of colon cancer cells in a murine tumor metastasis model. These findings indicate that NK4 gene transfer may be an effective tool for the treatment of colon cancer.
© 2007 The WJG Press. All rights reserved.
Key words: Human colon cancer; NK4; Hepatocyte growth factor; Adenoviral vector; Gene therapy
Jie JZ, Wang JW, Qu JG, Hung T. Suppression of human colon tumor growth by adenoviral vector-mediated NK4 expression in an athymic mouse model. World J Gastroenterol 2007; 13(13): 1938-1946
http://www.wjgnet.com/1007-9327/13/1938.asp
INTRODUCTIONAlthough it is one of the most common malignant tumors in the digestive tract, the therapeutic tractability of colon
COLORECTAL CANCER
Suppression of human colon tumor growth by adenoviral vector-mediated NK4 expression in an athymic mouse model
Jian-Zheng Jie, Jian-Wei Wang, Jian-Guo Qu, Tao Hung
www.wjgnet.com
Jian-Zheng Jie, Jian-Wei Wang, State Key Laboratory of Molecular Virology and Genetic Engineering, Beijing 100730, ChinaJian-Zheng Jie, Jian-Wei Wang, Jian-Guo Qu, Tao Hung, Natinoal Institute for Viral Disease Control and Prevention, China CDC, Beijing 100052, ChinaJian-Zheng Jie, Department of General Surgery, China-Japan Friendship Hospital, Beijing 100029, ChinaCorrespondence to: Dr. Jian-Wei Wang, State Key Laboratory of Molecular Virology and Genetic Engineering, 9# Dong Dan San Tiao, Dong Cheng Qu, Beijing 100730, China. [email protected] Telephone: +86-10-65105188 Fax: +86-10-65105188Received: 2006-11-26 Accepted: 2007-03-18
AbstractAIM: To investigate the suppressive effects of adenoviral vector-mediated expression of NK4, an antagonist of hepatocyte growth factor (HGF), on human colon cancer in an athymic mouse model to explore the possibility of applying NK4 to cancer gene therapy.
METHODS: A human colon tumor model was developed by subcutaneous implantation of tumor tissue formed by LS174T cells grown in athymic mice. Fifteen tumor-bearing mice were randomized into three groups (n = 5 in each group) at d 3 after tumor implantation and mice were injected intratumorally with phosphate-buffered saline (PBS) or with recombinant adenovirus expressing β-galactosidase (Ad-LacZ) or NK4 (rvAdCMV/NK4) at a 6-d interval for total 5 injections in each mouse. Tumor sizes were measured during treatment to draw a tumor growth curve. At d 26 after the first treatment, all animals were sacrificed and the tumors were removed to immunohistochemically examine proliferating cell nuclear antigen (PCNA), microvessel density (represented by CD31), and apoptotic cells. In a separate experiment, 15 additional athymic mice were employed to develop a tumor metastasis model by intraperitoneal injection (ip) of LS174T cells. These mice were randomized into 3 groups (n = 5 in each group) at d 1 after injection and were treated by ip injection of PBS, or Ad-LacZ, or rvAdCMV/NK4 at a 6-d interval for total two injections in each mouse. All animals were sacrificed at d 14 and the numbers and weights of disseminated tumors within the abdominal cavity were measured.
RESULTS: Growth of human colon tumors were significantly suppressed in the athymic mice treated
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1938-1946www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.

cancer remains unsatisfying and the failure of treatment is mainly attributed to tumor metastasis to the liver. Previous studies demonstrated that 15%-35% of the patients with primary colon cancer had metastasis in their liver when diagnosed; even after surgical resection, distant metastases still occurred in 25% of patients[1]. Therefore, it is of utmost importance to both inhibit tumor growth and prevent metastasis to increase therapeutic efficacy and improve the prognosis of patients with colon cancer.
Hepatocyte growth factor (HGF), originally identified and cloned as a potent hepatocyte mitogen[2,3], is a heterodimer consisting of 728 amino acids with a 69-ku α-chain and a 34-ku β-chain linked by a pair of disulfide bonds. As a multi-functional growth factor derived from interstitial or matrix cells, HGF has been recognized as a mitogen, motogen, and morphogen through interaction with the transmembrane receptor c-Met, a product of the proto-oncogene c-Met and the only known receptor for HGF[4]. Several in vitro experiments have validated that HGF powerfully promotes angiogenesis through stimulating proliferation and motility of the endothelial cells, eventually resulting in vessel growth[5-7]. Accumulating evidence has shown that HGF plays an important role in tumor-stroma interactions[8-11], promotes motility of tumor cells, stimulates cancer cells escape through the extracellular matrix, thereby leading to invasion and metastasis of tumor cells in a variety of tumors[12-14]. Since over-expression of the c-Met receptor in many tumors derived from epithelial cells[15-17], including colon cancer[18], has been confirmed, studies on blocking the HGF/c-Met signaling pathway to inhibit growth, invasion and metastasis indicate that this is a promising direction in current tumor treatment investigations[19].
NK4, an antagonist of HGF discovered by Date et al[19] in 1997, is a fragment of the HGF molecule consisting of 447 residues. It comprises the N-terminal hairpin and subsequent four-kringle domains of HGF, but lacks the 16 amino acids at the C-terminus of the α-chain and the whole β-chain. NK4 binds to the c-Met receptor competitively with HGF by virtue of common interactions of the HGF binding domain, but does not activate tyrosine phosphorylation of c-Met because of the absence of the β-chain. Consequently, the competitive binding of NK4 to c-Met interrupts the HGF/c-Met signaling pathway, and thereby suppresses HGF-induced invasion and metastasis of tumor cells[20]. Moreover, as an HGF antagonist, NK4 can inhibit angiogenesis induced not only by HGF, but also by other pro-angiogenic factors, such as basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF)[21]. By such mechanisms, NK4 robustly inhibits tumors through suppression of invasion, metastasis, and angiogenesis. Many lines of evidence have demonstrated the tremendous potential of NK4 in the treatment of tumors, such as glioblastoma[22], pancreatic cancer[23], gastric cancer[24], gallbladder cancer[25], and liver cancer[26].
Gene therapy is a revolutionary approach in cancer therapy. To date, there are two types of adenoviral vectors-based gene therapy drugs have been licensed in the world. In order to exploit NK4 in the development of
Jie JZ et al. Adenoviral-mediated NK4 gene therapy for colon cancer 1939
www.wjgnet.com
novel strategies against human colon tumors and based on previous in vitro experiments[27], we have chosen to employ rvAdCMV/NK4, an established non-replicating recombinant adenovirus containing the foreign NK4 gene, for gene therapy for human colon cancer cells in an athymic mouse model.
MATERIALS AND METHODSVirus and cells culture rvAdCMV/NK4 and Ad-LacZ [8.9 × 109 infection focus units (ifu)/mL)], the first generation of non-replicating recombinant adenoviruses expressing NK4 or β-galactosidase, were generated, verified, and propagated in HEK293 cells (American Type Culture Collection, Manassas, VA) as previously described[27,28]. The human colon cancer cell line LS174T, purchased from Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, was cultured in RPMI-1640 media (Invitrogen, Carsbad, CA) containing 100 mL/L heat-inactivated fetal bovine serum, 100 U/mL peniciliin, 100 μg/mL streptomycin, 2 mmol/L glutamine and 1.5 g/L sodium bicarbonate (Invitrogen) and incubated at 37 in a humidified atmosphere consisting of 50 mL/L CO2 in air. The expression of c-Met in LS174T cells and NK4 transduction by the recombinant adenovirus were confirmed by Western blot as previously described[27].
Establishment of a human colon cancer model in athymic mouse Four to six-week-old female athymic mice (BALB/c-nu/nu), weighing 18-22 g, were purchased from the Institute of Laboratory Animal Sciences, Chinese Academy of Medical Sciences (ILAS, CAMS) and were bred in homeothermic (25-27), constantly humidified (45%-50%), dust-free fresh air in a sterilized, specific pathogen-free (SPF) environment. All procedures for animal experiments were approved and supervised by the Animal Protection and Usage Committee of ILAS, CAMS and institutional guidelines for animal welfare and experimental conduct were followed.
LS174T cells in the logarithmic growth phase were treated with 2.5 g/L trypsin (Invitrogen), centrifuged, and resuspended in phosphate-buffered saline (PBS, pH7.4). After assessment for cell survival rate over 95% by trypan blue, the cell concentration was adjusted to 5 × 107/mL with PBS and 200 μL of the cell suspension (1 × 107 cells) was subcutaneously injected to the subscapular region of an athymic mouse using a 6-gauge needle. The tumors were not recovered until the diameter had increased to nearly 1 cm. After fixation in formalin, routine embedding in paraffin wax, cut into 5-μm thick sections, and staining with hematoxylin and eosin (HE), the tissue property of the tumors was confirmed by light microscopy.
In vivo tumor treatmentAfter the tumors were continuously passaged for 4 generations in mice, the athymic mice carrying productive tumors without ulcer were anesthetized by ethoxyethane and sacrificed. The tumors were removed under sterile

conditions, put into sterile plates on ice, de-enveloped, and cut into pieces of about 1 mm3. The tumor pieces were then implanted into the right forelimb subscapular region of mice usin 18-gauge trochars. All 15 animals were randomly divided into 3 groups, each group containing 5 mice, when the tumor volume grew to approximately 100 mm3 (the 3rd d after implantation) as fol lows: PBS group (blank control); Ad-LacZ group (negative control); and rvAdCMV/NK4 group (treatment group). Subsequently, 100 μL of PBS alone, or a suspension of adenovirus (108 ifu) in 100 μL of PBS was administered directly into the tumor-bearing mice in each group every 6 d for a total of 5 injections per mouse.
Long diameter (a) and short diameter (b) were measured every 3 to 4 d and volumes (V) of the tumors were calculated according to the formula V = 1/2ab2 and a tumor growth curve was drawn. Inhibition of tumor growth was determined using the formula as follows: Tumor inhibition rate (%) = (volume of tumors in the control group-volume of tumors in the treatment group)/ volume of tumors in control group × 100%.
At d 3 following the final treatment, all 15 mice were sacrificed. The tumors were removed, fixed in 100 mL/L formalin for 24 h, embedded in paraffin wax, and serially sectioned (4-μm thick). Immunohistochemical staining was performed as described below.
Tumor proliferation assayProliferating tumor cells were detected by SP immu-nohistochemical method using an antibody against proliferating cell nuclear antigen (PCNA) according to the manufacturer’s (Maxin Biotech) instructions. Four sections were observed for each mouse. To determine the PCNA Labeling Index (PCNA LI), five representative PCNA-positive visual fields were selected under a low power microscope (× 100) in each section, then 200 tumor cells were counted in each visual field under a high power microscope (× 200), finally the percentage of labeled nuclei was calculated (%).
Evaluation of microvessel density The microvessel density (MVD) in each tumor was evaluated by counting CD31-positive vessels. The expression of CD31 was detected by immunohistochemical methods as aforementioned. The primary antibody was a rat-anti-mouse CD31 monoclonal antibody (BD Biosciences, 1:200 dilution) and the secondary antibody was a biotinylated anti-rat IgG (Maxin Biotech, 1:100). For the microvessel counting, positive stainings for MVD in the 3 most highly vascularized areas (“hot spots”) in each slide, were counted in 200 × fields and MVD was expressed as the average of the microvessel count in these areas[29]. Any brown endothelial cell or cluster of endothelial cells were counted as one microvessel as long as it was separated from other neighboring microvessels, tumor cells, and connective tissues, while vessels with a thick muscular layer or diameter over 8 erythrocytes were not counted.
TUNEL assayApoptotic cells were detected using a terminal deoxynu-
cleotidyl transferase (TdT)-mediated nick-end labeling (TUNEL) assay. The assay was performed according to the manufacturer’s instructions (Boehringer Mannheim). The apoptotic index (AI) was determined by selecting five representative TUNEL-positive fields under low power microscopy (× 100) in each section, then 200 tumor cells were counted in each field under high power microscopy (× 200). The number of positively labeled nuclei per total nuclei in those fields was expressed as the apoptotic index (AI). AI = (the number of apoptotic cells/total number of tumor cells) × 100%.
Treatment of peritoneal tumor disseminationLS174T cells (1 × 107 in 200 μL PBS) were injected intraperitoneally (ip) into 15 mice at d 0, and mice were randomly divided into 3 groups (n = 5 in each group) as described above. Then 100 μL of PBS alone or a suspension of adenovirus (108 ifu) in 100 μL of PBS was ip injected in each group at an interval of 6 d for a total of two injections per mouse. All animals were sacrificed at d 15 to determine intraperitoneal tumor dissemination by direct visual inspection in terms of number, site and weight of the metastases.
Statistical analysisData were expressed as the mean ± SD. Statistical comparisons were made using the Student’s t test for the measurement data and Mann-Whitney U test for enumeration data. P < 0.05 was considered statistically significant.
RESULTSEstablishment of a human colon cancer model in athymic mice The human colon cancer cell line LS174T robustly formed tumors. Primary tumors growth was apparent in the subdermal tissue of athymic mice at 1 wk after implantation. The tumor grew rapidly into an oval shape with a smooth and glossy surface at early stages and an irregular shape with surface nodosity at later periods. Thus, it is an ideal tumor model for colon cancer research. Macroscopically, the subcutaneous tumors in athymic mice were silver-gray and looked like fish flesh when dissected. HE staining confirmed the tumors as colon adenocarcinoma, as expected. Microscopically, tumor cells were seen in a ball-like distribution with larger cell bodies and obvious heteromorphism; the nuclei were large and irregular and the nucleoli were obviously visible. Without necrotic changes, typical micro-cavity-like structures were seen separately and multi-nuclear giant cells were occasionally found in the tumor tissues. All of the aforementioned features are consistent with the characterization of a poorly differentiated human colon adenocarcinoma (Figure 1).
Inhibitory effect of rvAdCMV/NK4 on tumor growth Subcutaneous injection of LS174T cells resulted in tumor formation in all 15 athymic mice (tumor formation rate 100%) and the tumors grew a large enough size to be treated with PBS, or Ad-LacZ, or rvAdCMV/NK4 from
1940 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

the 3rd d after tumor cell implantation. No animals died during the treatment and all of them grew well without dyscrasia. There was no significant difference in body weight among three groups before and after treatment (data not shown). No metastases in organs of the thoracic or abdominal cavity were found. Tumor growth curves showed that the average size of tumors in the rvAdCMV/NK4 group was significantly smaller compared to the two control groups (2537.4 ± 892.3 mm3 in rvAdCMV/NK4 group vs 5175.2 ± 1228.6 mm3 in PBS group or 5578.8 ± 1955.7 mm3 in Ad-LacZ group, P < 0.05) (Figure 2). The tumor growth inhibit ion rate was 51% in the rvAdCMV/NK4 group when compared with the Ad-LacZ group, while no significant difference was observed between the Ad-LacZ and PBS groups (5578.8 ± 1955.7 mm3 vs 5175.2 ± 1228.6 mm3). These findings indicate that the rvAdCMV/NK4 treatment can inhibit tumor growth effectively.
Because PCNA is mainly expressed during S, G1, and early G2 phase of the cell-cycle in proliferating cells, it is accepted as a marker of cell proliferation and malignancy of tumors[30]. In order to assess the inhibitory effects of the rvAdCMV/NK4 treatment on human colon cancer cells, we immunohistochemically examined the PCNA level in the tumors. The results showed that there was no significant difference in PCNA LI among the three groups (75.1% ± 11.2% in PBS group vs 72.8% ± 7.6% in Ad-LacZ group vs 69.3% ± 9.4% in rvAdCMV/NK4 group) (Figure 3), indicating that the rvAdCMV/NK4 treatment did not inhibit proliferation of tumor cells and the inhibitory effects on tumor growth clearly do not rely on
direct inhibition of tumor cell proliferation. These results are also identical to those obtained in previous in vitro experiments and similar with the reports of others[27,31,32].
Inhibition of angiogenesis in tumor by rvAdCMV/NK4 In order to demonstrate the inhibitor y effect of
Figure 1 Tumors established in athymic BALB/c mice by subcutaneous injection of LS174T cells. A: Photography of subcutaneous tumor of LS174T in athymic mice; B: The tumor sections (HE, × 200).
8000
7000
6000
5000
4000
3000
2000
1000
0
PBS
Ad-LacZ
rvAdCMV/NK4
3 7 12 16 21 25 28
Days after tumor inoculation
Mea
n tu
mor
vol
ume
(mm
3 )
A
B
Figure 2 Suppression of LS174T tumor growth in athymic mice by intratumoral administration of rvAdCMV/NK4. A: Tumor growth curves. Data are presented as mean ± SD (n = 5/group); B: PBS control; C: Ad-LacZ; D: rvAdCMV/NK4.
A
B
C
D
Jie JZ et al. Adenoviral-mediated NK4 gene therapy for colon cancer 1941
www.wjgnet.com

rvAdCMV/NK4 treatment on tumor angiogenesis in the athymic mice model, tumor MVDs were quantified by immunohistochemical staining for CD31. The results indicated a marked reduction in MVD in the rvAdCMV/NK4-treated tumors (10.7 ± 2.4) compared to the PBS-treated (25.6 ± 3.8) and Ad-LacZ-treated (21.3 ± 3.5) tumors (P < 0.05) (Figure 4), indicating that NK4
may suppress tumor growth via inhibition of tumor angiogenesis.
rvAdCMV/NK4-induced apoptosis In order to further elucidate the mechanisms underlying the suppression of human colon cancer growth by adenovirus vector-mediated NK4-transduction, we
A
B
C
100
80
60
40
20
0
a
PBS Ad-LacZ rvAdCMV/NK4
PCN
A in
dex
(%)
D
Figure 3 PCNA expression in LS174T tumors after rvAdCMV/NK4 treatment. A, B, C: Immmunohistochemical staining of PCNA in tumor tissues (× 200), A: PBS; B: Ad-LacZ; C: rvAdCMV/NK4; D: PCNA labeling index in different groups (mean ± SD). aР > 0.05 vs PBS/Ad-LacZ.
A
B
C
30
25
20
0
a
PBS Ad-LacZ rvAdCMV/NK4
Num
ber
of v
esse
ls/h
pf
D
Figure 4 Microvessel density of LS174T tumors in athymic mice after rvAdCMV/NK4 treatment. A, B, C: Immmunohistochemical staining of CD31 in tumor tissues (× 100), A: PBS; B: Ad-LacZ; C: rvAdCMV/NK4; D: Number of vessels in tumor tissues after treatment (mean ± SD). aР < 0.05 vs PBS or Ad-LacZ.
35
15
10
5
1942 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com
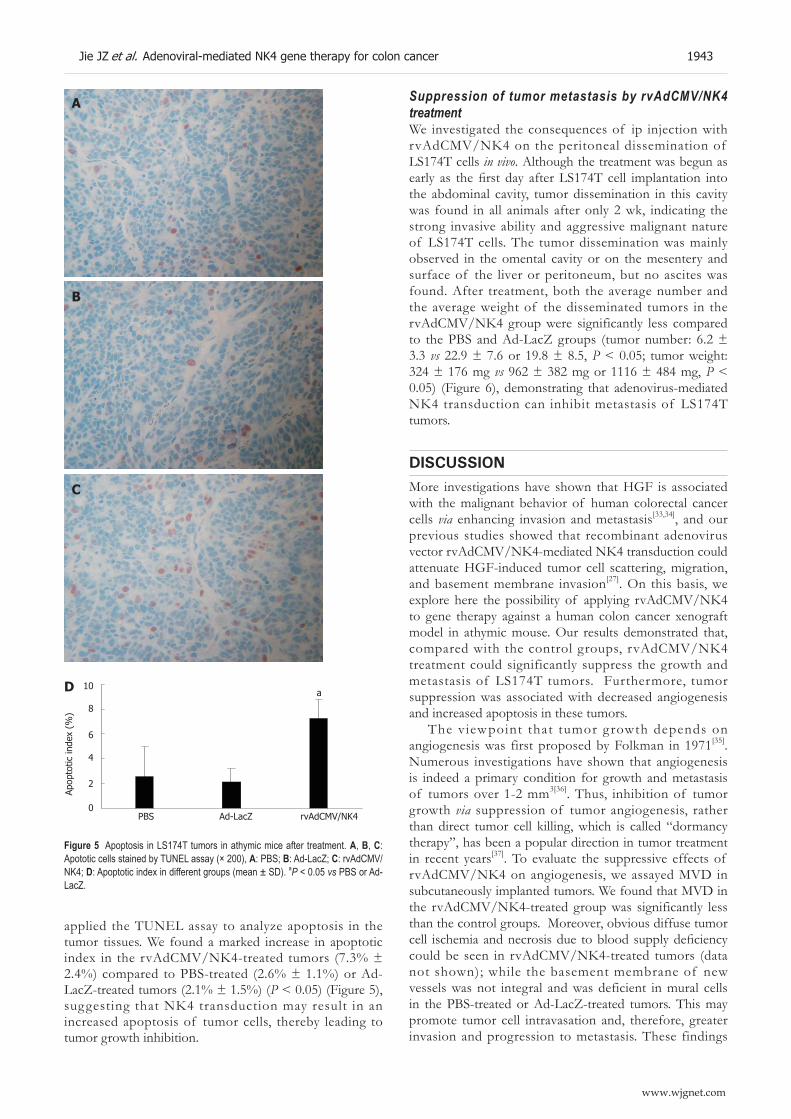
applied the TUNEL assay to analyze apoptosis in the tumor tissues. We found a marked increase in apoptotic index in the rvAdCMV/NK4-treated tumors (7.3% ± 2.4%) compared to PBS-treated (2.6% ± 1.1%) or Ad-LacZ-treated tumors (2.1% ± 1.5%) (P < 0.05) (Figure 5), suggesting that NK4 transduction may result in an increased apoptosis of tumor cells, thereby leading to tumor growth inhibition.
Suppression of tumor metastasis by rvAdCMV/NK4 treatmentWe investigated the consequences of ip injection with rvAdCMV/NK4 on the peritoneal dissemination of LS174T cells in vivo. Although the treatment was begun as early as the first day after LS174T cell implantation into the abdominal cavity, tumor dissemination in this cavity was found in all animals after only 2 wk, indicating the strong invasive ability and aggressive malignant nature of LS174T cells. The tumor dissemination was mainly observed in the omental cavity or on the mesentery and surface of the liver or peritoneum, but no ascites was found. After treatment, both the average number and the average weight of the disseminated tumors in the rvAdCMV/NK4 group were significantly less compared to the PBS and Ad-LacZ groups (tumor number: 6.2 ± 3.3 vs 22.9 ± 7.6 or 19.8 ± 8.5, P < 0.05; tumor weight: 324 ± 176 mg vs 962 ± 382 mg or 1116 ± 484 mg, P < 0.05) (Figure 6), demonstrating that adenovirus-mediated NK4 transduction can inhibit metastasis of LS174T tumors.
DISCUSSIONMore investigations have shown that HGF is associated with the malignant behavior of human colorectal cancer cells via enhancing invasion and metastasis[33,34], and our previous studies showed that recombinant adenovirus vector rvAdCMV/NK4-mediated NK4 transduction could attenuate HGF-induced tumor cell scattering, migration, and basement membrane invasion[27]. On this basis, we explore here the possibility of applying rvAdCMV/NK4 to gene therapy against a human colon cancer xenograft model in athymic mouse. Our results demonstrated that, compared with the control groups, rvAdCMV/NK4 treatment could significantly suppress the growth and metastasis of LS174T tumors. Furthermore, tumor suppression was associated with decreased angiogenesis and increased apoptosis in these tumors.
The viewpoint that tumor g rowth depends on angiogenesis was first proposed by Folkman in 1971[35]. Numerous investigations have shown that angiogenesis is indeed a primary condition for growth and metastasis of tumors over 1-2 mm3[36]. Thus, inhibition of tumor growth via suppression of tumor angiogenesis, rather than direct tumor cell killing, which is called “dormancy therapy”, has been a popular direction in tumor treatment in recent years[37]. To evaluate the suppressive effects of rvAdCMV/NK4 on angiogenesis, we assayed MVD in subcutaneously implanted tumors. We found that MVD in the rvAdCMV/NK4-treated group was significantly less than the control groups. Moreover, obvious diffuse tumor cell ischemia and necrosis due to blood supply deficiency could be seen in rvAdCMV/NK4-treated tumors (data not shown); while the basement membrane of new vessels was not integral and was deficient in mural cells in the PBS-treated or Ad-LacZ-treated tumors. This may promote tumor cell intravasation and, therefore, greater invasion and progression to metastasis. These findings
A
B
C
Figure 5 Apoptosis in LS174T tumors in athymic mice after treatment. A, B, C: Apototic cells stained by TUNEL assay (× 200), A: PBS; B: Ad-LacZ; C: rvAdCMV/NK4; D: Apoptotic index in different groups (mean ± SD). aР < 0.05 vs PBS or Ad-LacZ.
10
8
6
4
2
a
PBS Ad-LacZ rvAdCMV/NK4
Apop
totic
inde
x (%
)
D
0
Jie JZ et al. Adenoviral-mediated NK4 gene therapy for colon cancer 1943
www.wjgnet.com

suggest that ischemia and necrosis due to blood supply deficiency caused by rvAdCMV/NK4 treatment-induced anti-angiogenesis might be one of the primary reasons for tumor growth inhibition.
Apoptosis plays an important role in the inhibition of oncogenesis, tumor development and growth. Balance between proliferation and apoptosis drives a tumor to a silent or dormant state, which may inhibit clinical recurrence and metastasis caused by mini-tumors, placing the patients, especially those who received an operation, in a sub-clinical therapy state[38]. Thus, apoptosis in a tumor usually indicates a relatively good clinical prognosis. The majority of chemotherapeutic drugs at present suppress tumor growth through induction of apoptosis[39]. In our study, the AI of the rvAdCMV/NK4-treated group was much higher compared to the control groups, indicating its potential value for clinical therapy of cancer. The mechanism by which rvAdCMV/NK4 treatment promotes apoptosis in LS174T tumors is yet unknown. However, it is unlikely to be caused by direct suppression of cell proliferation, because the treatment did not lead to a significant decrease of PCNA LI. In consideration of the established linkage between inhibition of angiogenesis and tumor apoptosis, as well as the potent anti-angiogenic activity of NK4, it is reasonable to attribute the markedly increased apoptosis to the signif icantly inhibited angiogenesis. Hence, inhibition of angiogenesis may play a central role in the tumor suppression mediated by NK4.
At present, surgical resection remains the leading therapeutic approach for colon cancer. However, surgery has risks contributing to tumor cell dissemination around operation fields and metastasis in the abdominal cavity. In order to investigate whether rvAdCMV/NK4 may inhibit tumor dissemination, we established disseminating tumor models in athymic mice through injection of LS174T cells into the abdominal cavity. For modeling clinical patients who receive systemic therapy from the first day after the operation, we treated the animals from the first day after injection of LS174T cells. Our results suggest that rvAdCMV/NK4 may effectively inhibit tumor dissemination. Both the size and weight of tumors in rvAdCMV/NK4-treated group were lower than those in the control groups. Tumor implantation in the abdominal cavity is presently regarded to be caused by adherence between tumor cells and interstitial cells, allowing tumor cells to eventually break through the basement membrane. According to previous investigations, HGF released from fibroblasts and interstitial cells may cause the morphological changes of stromal cells and promote tumor cell invasion into the basal layer[40]. Thus, HGF may play a role in tumor dissemination, in addition to the effects of transforming growth factor-β (TGF-β), etc. In our study, rvAdCMV/NK4 inhibited the dissemination of LS174T cells in the abdominal cavity via HGF-antagonist activity of the NK4 protein, thereby inhibiting adherence between tumor cells and matrix. At the same time, the anti-angiogenic and pro-apoptotic activity of NK4 suppressed the growth of implanted tumors.
In summary, we have applied the first generation non-replicating recombinant adenovirus vector, rvAdCMV/NK4, to effectively treat human colon cancer in an in
A
B
C
30
20
15
10
0
a
PBS Ad-LacZ rvAdCMV/NK4
Num
ber
of
diss
emin
ated
foc
i
D
Figure 6 Suppression of peritoneal dissemination by intraperitoneal injection of rvAdCMV/NK4. A, B, C: Macroscopic appearance of peritoneal dissemination on d 14 after the treatment following peritoneal implantation of 1 × 106 LS174T cells (n = 5/group), A: PBS; B: Ad-LacZ; C: rvAdCMV/NK4. Arrowheads indicate disseminated tumor nodules; D: Number of disseminated foci (mean ± SD); E: Total weight of disseminated foci (mean ± SD). aР < 0.05 vs PBS or Ad-LacZ.
35
25
5
a
PBS Ad-LacZ rvAdCMV/NK4
Tota
l wei
ght
of
diss
emin
ated
foc
i (m
g)
E 2000
1500
1000
500
0
1944 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

vivo athymic mouse model. The findings are in agreement with previous studies reported on various human cancers, such as pancreatic cancer[23], hepatocellular cancer[26], gastric cancer[41], etc. Although our preliminary results are encouraging, showing the relative efficacy of this type of treatment in human colon cancer cells, one must keep in mind that the efficacy of gene therapy as the sole treatment may be hampered by several factors, such as immunity against the viral vector, the duration of gene expression, and the specificity of cancer targeting, etc. Such therapy should be combined with currently available conventional therapies, including surgical resection, chemotherapy, and radiotherapy to achieve better therapeutic success. In future studies, we will examine the synergistic effects of combination with chemotherapy and/or radiotherapy, as well as any sensitizing effects on chemotherapy and/or radiotherapy.
REFERENCES1 Calaluce R, Miedema BW, Yesus YW. Micrometastasis in
colorectal carcinoma: a review. J Surg Oncol 1998; 67: 194-202 2 Nakamura T, Nawa K, Ichihara A. Partial purification and
characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem Biophys Res Commun 1984; 122: 1450-1459
3 Nakamura T, Nishizawa T, Hagiya M, Seki T, Shimonishi M, Sugimura A, Tashiro K, Shimizu S. Molecular cloning and expression of human hepatocyte growth factor. Nature 1989; 342: 440-443
4 Matsumoto K, Nakamura T. NK4 (HGF-antagonist/angiogenesis inhibitor) in cancer biology and therapeutics. Cancer Sci 2003; 94: 321-327
5 Mart in TA, Hard ing KG, J i ang WG. Regula t ion o f angiogenesis and endothelial cell motility by matrix-bound fibroblasts. Angiogenesis 1999; 3: 69-76
6 Sugawara J, Fukaya T, Murakami T, Yoshida H, Yajima A. Hepatocyte growth factor stimulated proliferation, migration, and lumen formation of human endometrial epithelial cells in vitro. Biol Reprod 1997; 57: 936-942
7 Jiang WG, Hiscox SE, Parr C, Martin TA, Matsumoto K, Na-kamura T, Mansel RE. Antagonistic effect of NK4, a novelAntagonistic effect of NK4, a novel hepatocyte growth factor variant, on in vitro angiogenesis of human vascular endothelial cells. Clin Cancer Res 1999; 5: 3695-3703
8 Nagy J , Curry GW, Hil lan KJ , McKay IC, Mal lon E, Purushotham AD, George WD. Hepatocyte growth factor/scatter factor expression and c-met in primary breast cancer. Surg Oncol 1996; 5: 15-21
9 Longati P, Comoglio PM, Bardelli A. Receptor tyrosine kinases as therapeutic targets: the model of the MET oncogene. Curr Drug Targets 2001; 2: 41-55
10 Jeffers M, Koochekpour S, Fiscella M, Sathyanarayana BK, Vande Woude GF. Signaling requirements for oncogenic forms of the Met tyrosine kinase receptor. Oncogene 1998; 17: 2691-2700
11 Jeffers M, Fiscella M, Webb CP, Anver M, Koochekpour S, Vande Woude GF. The mutationally activated Met receptor mediates motility and metastasis. Proc Natl Acad Sci USA 1998; 95: 14417-14422
12 Seslar SP, Nakamura T, Byers SW. Regulation of fibroblastRegulation of fibroblast hepatocyte growth factor/scatter factor expression by human breast carcinoma cell lines and peptide growth factors. Cancer Res 1993; 53: 1233-1238
13 Nakamura T, Matsumoto K, Kiritoshi A, Tano Y, Nakamura T. Induction of hepatocyte growth factor in fibroblasts by tumor-derived factors affects invasive growth of tumor cells: in vitro analysis of tumor-stromal interactions. Cancer Res 1997; 57: 3305-3313
14 Jiang W, Hiscox S, Matsumoto K, Nakamura T. Hepatocyte growth factor/scatter factor, its molecular, cellular and clinical implications in cancer. Crit Rev Oncol Hematol 1999; 29: 209-248
15 Di Renzo MF, Narsimhan RP, Olivero M, Bretti S, Giordano S, Medico E, Gaglia P, Zara P, Comoglio PM. Expression of theExpression of the Met/HGF receptor in normal and neoplastic human tissues. Oncogene 1991; 6: 1997-2003
16 Kaji M, Yonemura Y, Harada S, Liu X, Terada I, Yamamoto H. Participation of c-met in the progression of human gastric cancers: anti-c-met oligonucleotides inhibit proliferation or invasiveness of gastric cancer cells. Cancer Gene Ther 1996; 3: 393-404
17 Maehara N, Matsumoto K, Kuba K, Mizumoto K, Tanaka M, Nakamura T. NK4, a four-kringle antagonist of HGF, inhibits spreading and invasion of human pancreatic cancer cells. Br J Cancer 2001; 84: 864-873
18 Di Renzo MF, Olivero M, Giacomini A, Porte H, Chastre E, Mirossay L, Nordlinger B, Bretti S, Bottardi S, Giordano S. Overexpression and amplification of the met/HGF receptor gene during the progression of colorectal cancer. Clin Cancer Res 1995; 1: 147-154
19 Date K, Matsumoto K, Shimura H, Tanaka M, Nakamura T. HGF/NK4 is a specific antagonist for pleiotrophic actions of hepatocyte growth factor. FEBS Lett 1997; 420: 1-6
20 Matsumoto K, Nakamura T. Mechanisms and significance of bifunctional NK4 in cancer treatment. Biochem Biophys Res Commun 2005; 333: 316-327
21 Kuba K, Matsumoto K, Date K, Shimura H, Tanaka M, Nakamura T. HGF/NK4, a four-kringle antagonist of hepatocyte growth factor, is an angiogenesis inhibitor that suppresses tumor growth and metastasis in mice. Cancer Res 2000; 60: 6737-6743
22 Brockmann MA, Papadimitriou A, Brandt M, Fillbrandt R, Westphal M, Lamszus K. Inhibition of intracerebral glioblastoma growth by local treatment with the scatter factor/hepatocyte growth factor-antagonist NK4. Clin Cancer Res 2003; 9: 4578-4585
23 Ogura Y, Mizumoto K, Nagai E, Murakami M, Inadome N, Saimura M, Matsumoto K, Nakamura T, Maemondo M, Nukiwa T, Tanaka M. Peritumoral injection of adenovirus vector expressing NK4 combined with gemcitabine treatment suppresses growth and metastasis of human pancreatic cancer cells implanted orthotopically in nude mice and prolongs survival. Cancer Gene Ther 2006; 13: 520-529
24 Fujiwara H, Kubota T, Amaike H, Inada S, Takashima K, Atsuji K, Yoshimura M, Maemondo M, Narumi K, Nukiwa T, Matsumoto K, Nakamura T, Hagiwara A, Yamagishi H. Suppression of peritoneal implantation of gastric cancer cells by adenovirus vector-mediated NK4 expression. Cancer Gene Ther 2005; 12: 206-216
25 Tanaka T, Shimura H, Sasaki T, Narumi K, Maemondo M, Nukiwa T, Matsumoto K, Nakamura T, Ikeda S. Gallbladder cancer treatment using adenovirus expressing the HGF/NK4 gene in a peritoneal implantation model. Cancer Gene Ther 2004; 11: 431-440
26 Son G, Hirano T, Seki E, Iimuro Y, Nukiwa T, Matsumoto K, Nakamura T, Fujimoto J. Blockage of HGF/c-Met system by gene therapy (adenovirus-mediated NK4 gene) suppresses hepatocellular carcinoma in mice. J Hepatol 2006; 45: 688-695
27 Jie JZ , Wang JW, Qu JG, Wang W, Hung T. Effects of adenovira l -mediated gene t ransduct ion of NK4 on proliferation, movement, and invasion of human colonic LS174T cancer cells in vitro. World J Gastroenterol 2006; 12: 3983-3988
28 Jie JZ, Wang JW, Wang W, Jiang XL, Wei Q, Hung T. Expression and characterization of HGF mutant NK4 in adenovirus vector. Zhongguo Shiyan Zhenduanxue Zazhi 2003; 7: 397-400
29 Weidner N. Current pathologic methods for measuring intratumoral microvessel density within breast carcinoma and other solid tumors. Breast Cancer Res Treat 1995; 36: 169-180
30 McCormick D, Hall PA. The complexities of proliferating cell
Jie JZ et al. Adenoviral-mediated NK4 gene therapy for colon cancer 1945
www.wjgnet.com

nuclear antigen. Histopathology 1992; 21: 591-59431 Saimura M, Nagai E, Mizumoto K, Maehara N, Okino H,
Katano M, Matsumoto K, Nakamura T, Narumi K, Nukiwa T, Tanaka M. Intraperitoneal injection of adenovirus-mediated NK4 gene suppresses peritoneal dissemination of pancreatic cancer cell line AsPC-1 in nude mice. Cancer Gene Ther 2002; 9: 799-806
32 Kubota T, Fujiwara H, Amaike H, Takashima K, Inada S, Atsuji K, Yoshimura M, Matsumoto K, Nakamura T, Yamagi-shi H. Reduced HGF expression in subcutaneous CT26 tumor genetically modified to secrete NK4 and its possible relation with antitumor effects. Cancer Sci 2004; 95: 321-327
33 Hiscox SE, Hallett MB, Puntis MC, Nakamura T, Jiang WG. Expression of the HGF/SF receptor, c-met, and its ligand in human colorectal cancers. Cancer Invest 1997; 15: 513-521
34 Empereur S, Djelloul S, Di Gioia Y, Bruyneel E, Mareel M, Van Hengel J, Van Roy F, Comoglio P, Courtneidge S, Paraskeva C, Chastre E, Gespach C. Progression of familial adenomatous polyposis (FAP) colonic cells after transfer of the src or polyoma middle T oncogenes: cooperation between src and HGF/Met in invasion. Br J Cancer 1997; 75: 241-250
35 Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med 1971; 285: 1182-1186
36 Carmeliet P, Jain RK. Angiogenesis in cancer and other
diseases. Nature 2000; 407: 249-25737 Ramanujan S, Koenig GC, Padera TP, Stoll BR, Jain RK.
Local imbalance of proangiogenic and antiangiogenic factors: a potential mechanism of focal necrosis and dormancy in tumors. Cancer Res 2000; 60: 1442-1448
38 Drixler TA, Borel Rinkes IH, Ritchie ED, van Vroonhoven TJ, Gebbink MF, Voest EE. Continuous administration of angiostatin inhibits accelerated growth of colorectal liver metastases after partial hepatectomy. Cancer Res 2000; 60: 1761-1765
39 Fulda S, Los M, Friesen C, Debatin KM. Chemosensitivity of solid tumor cells in vitro is related to activation of the CD95 system. Int J Cancer 1998; 76: 105-114
40 Yashiro M, Chung YS, Inoue T, Nishimura S, Matsuoka T, Fujihara T, Sowa M. Hepatocyte growth factor (HGF) produced by peritoneal fibroblasts may affect mesothelial cell morphology and promote peritoneal dissemination. Int J Cancer 1996; 67: 289-293
41 Ueda K, Iwahashi M, Matsuura I, Nakamori M, Nakamura M, Ojima T, Naka T, Ishida K, Matsumoto K, Nakamura T, Yamaue H. Adenoviral-mediated gene transduction of the hepatocyte growth factor (HGF) antagonist, NK4, suppresses peritoneal metastases of gastric cancer in nude mice. Eur J Cancer 2004; 40: 2135-2142
S- Editor Liu Y L- Editor Kumar M E- Editor Ma WH
1946 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1947-1952www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
Celecoxib attenuates 5-fluorouracil-induced apoptosis in HCT-15 and HT-29 human colon cancer cells
Yun Jeong Lim, Jong Chul Rhee, Young Mee Bae, Wan Joo Chun
www.wjgnet.com
BASIC RESEARCH
Yun Jeong Lim, Department of Internal Medicine, Dongguk University International Hospital, Dongguk University College of Medicine, Goyang, KoreaJong Chul Rhee, Division of Gastroenterology, Department of Internal Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, KoreaYoung Mee Bae, Department of Parasitology, Seoul National University College of Medicine, Seoul, Korea Wan Joo Chun, Department of Pharmacology, Kangwon National University College of Medicine, Chuncheon, KoreaCorrespondence to: Jong Chul Rhee, Division of Gastroen-terology, Department of Internal Medicine, Samsung Medical Center, 50 Irwon-dong, Gangnam-gu, Seoul, 135-710, Korea. [email protected]: +82-2-34103409 Fax: +82-2-34103849Received: 2006-11-12 Accepted: 2006-12-28
AbstractAIM: To investigate the combined chemotherapeutic effects of celecoxib when used with 5-FU in vitro .
METHODS: Two human colon cancer cell lines (HCT-15 and HT-29) were treated with 5-FU and celecoxib, alone and in combination. The effects of each drug were evaluated using the MTT [3- (4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] assay, flow cytometry, and western blotting.
RESULTS: 5-FU and ce lecox ib showed a dose-dependent cytotoxic effect. When treated with 10-3 mol/L 5-FU (IC50) and celecoxib with its concentration ranging from 10-8 mol/L to 10-4 mol/L of celecoxib, cel ls showed reduced cytotoxic effect than 5-FU (10-3 mol/L) alone. Flow cytometry showed that celecoxib attenuated 5-FU induced accumulation of cells at subG1 phase. Western blot analyses for caspase-3 and poly (ADP-ribose) polymerase (PARP) cleavage showed that celecoxib attenuated 5-FU induced apoptosis. Western blot analyses for cell cycle molecules showed that G2/M arrest might be possible cause of 5-FU induced apoptosis and celecoxib attenuated 5-FU induced apoptosis via blocking of cell cycle progression to the G2/M phase, causing an accumulation of cells at the G1/S phase.
CONCLUSION: We found that celecoxib attenuated cytotoxic effect of 5-FU. Celecoxib might act via inhibition of cell cycle progression, thus preventing apoptosis induced by 5-FU.
© 2007 The WJG Press. All rights reserved.
Key words: Antagonism; Celecoxib; Colorectal neoplasms; Fluorouracil
Lim YJ, Rhee JC, Bae YM, Chun WJ. Celecoxib attenuates 5-fluorouracil-induced apoptosis in HCT-15 and HT-29 human colon cancer cells. World J Gastroenterol 2007; 13(13): 1947-1952
http://www.wjgnet.com/1007-9327/13/1947.asp
INTRODUCTIONColorectal cancer is the second leading cause of cancer death in the United States[1]. Despite advances in medical practices and chemotherapeutic protocols, survival rates in cases of colorectal cancer have changed little over the last 20 years. In the treatment of colorectal cancer, 5-fluorouracil (5-FU), a potent inhibitor of thymidylate synthesis during DNA synthesis, is one of the most commonly used chemotherapeutic agents, but the overall response rate is only around 15% when used in adjuvant chemotherapy[2]. 5-FU is also used to treat various cancers, but results are not satisfactory. To increase the response rate, 5-FU is usually used in combination with other drugs, such as cyclophosphamide and methotrexate (breast cancer)[3], cisplatin (head and neck cancer)[4], or leucovorin (colorectal cancer)[5,6]. 5-FU has also been tested in combination chemotherapy with other drugs, which act via different mechanisms[7-9].
Thun et al[10] suggested that NSAIDs reduce the risk of colorectal cancer, and several investigators tested the application of NSAIDs as a new chemopreventive agent[11,12]. Celecoxib, a cyclooxygenase-2 (COX-2) specific inhibitor known to have antiproliferative effects on colorectal cancer[13,14], was also tested as a chemotherapeutic agent[15]. Milas et al[16] combined radiation therapy with a COX-2 inhibitor in a mouse cancer model, and observed an enhanced therapeutic response. In a recent clinical trial that incorporated the use of celecoxib as a combined chemotherapeutic drug, Altorki et al[17] tested celecoxib as a combined chemotherapeutic agent with paclitaxel and carboplatin on the early-stage non-small cell lung cancer. In their report, celecoxib showed additive or synergistic effect.
In this paper, we tested celecoxib as a possible candidate for combined chemotherapeutic agent to be used with 5-FU in the treatment of colorectal cancer. Thus, we treated HCT-15 and HT-29 human colon cancer cell lines

with 5-FU and celecoxib and assessed their effects by MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] assay, flow cytometry and western blotting.
MATERIALS AND METHODSCell cultureThe HCT-15 and HT-29 human colon cancer cel l lines were purchased from the American Type Culture Collection (ATCC, Rockville, MD) and cultured in RPMI1640 medium supplemented with 10% fetal bovine serum (FBS) at 37 in a 5% CO2 atmosphere. To examine the effects of 5-FU and celecoxib alone, the cells were treated with 10-6 mol/L, 10-5 mol/L, 10-4 mol/L, 10-3 mol/L, and 10-2 mol/L 5-FU and celecoxib for 24 h, respectively. To address the effects of 5-FU and celecoxib co-treatment, various concentrations of celecoxib were added immediately after treating cells with 10-3 mol/L 5-FU.
MTT assayThe MTT assay was performed as previously described[18]. In brief, HCT-15 and HT-29 human colon cancer cells were cultured in a 24-well plate (Corning Inc., Corning, NY) at a density of 5 × 104 cells per well. The cells were then treated with varying concentrations of 5-FU, or celecoxib, or both drugs. After 48 h, the cells were washed and treated with MTT. Plates were incubated in the dark for 4 h, and the absorbances were measured at 570 nm using a microtiter plate reader (Bio-Tek, Winooski, VT). To determine cell viability, percent viability was calculated as [(absorbance of drug-treated) sample/(control absorbance)] × 100.
Flow cytometry Apoptosis detection and analysis of cell cycle distribution were perfor med by f low cytometr y, as descr ibed previously[19,20]. Briefly, cells were incubated for 24 h in a medium without FBS to synchronize the cell cycle. Cells were then treated for 48 h in the medium containing 10% FBS with celecoxib, 5-FU or both, respectively. Cells were harvested by trypsinization, washed twice with PBS, incubated with 0.125% Triton X-100, and stained with propidium iodide (PI) in PBS containing 0.2 mg/mL RNase A. Stained cells were analyzed using a FACS calibur (Becton Dickinson, San Jose, CA, USA). For each sample, cells were counted until the count reached 10 000 cells in a predefined G1-gate. The percentages of cells in the subG1, G0/G1, S, and G2/M phases were determined using the CELLQUEST software.
Western blotting Cells were incubated for 48 h with 10-5 mol/L celecoxib, 10-3 mol/L 5-FU or both drugs. Cells were harvested in cold phosphate-buffered saline (PBS), collected by centrifugation, resuspended in a homogenizing buffer (50 mmol/L Tris-HCl, pH 7.5; 150 mmol/L NaCl; 1 mmol/L EDTA; 0.1 mmol/L phenylmethylsulfonyl fluoride (PMSF); and 10 µg/ml of each of aprotinin, leupeptin, and pepstatin), and sonicated on ice. Protein concentrations of the homogenates were determined using the bicinochoninic acid (BCA) method (Pierce).
Homogenates were diluted to a final concentration of 2 mg/ml with 2X reducing stop buffer (0.25 mol/L Tris-HCl, pH 6.8, 5 mmol/L EDTA, 5 mmol/L EGTA, 25 mmol/L dithiothreitol, 2% SDS, and 10% glycerol with bromophenol blue as the tracking dye). Samples (50 µg of protein) were resolved on SDS-polyacrylamide gels, and transferred to nitrocellulose. Blots were blocked in 5% nonfat dried milk in TBST (20 mmol/L Tris-HCl, pH 7.6, 137 mmol/L NaCl, 0.1% Tween 20) for 1 h at room temperature. Blots were then incubated with the respective primary antibodies directed against poly(ADP-ribose) polymerase (PARP) (1:1000, Cell Signaling Technology), active caspase-3 (1:1000, Cell Signaling Technology), cdk1 (1:1000, Neomarkers), cdk2 (1:1000, Neomarkers), cyclin A (1:1000, Neomarkers), and cyclin B (1:1000, Neomarkers), in the same buffer overnight at 4. The membranes were then washed three times with TBST and incubated with HRP-conjugated goat anti-mouse IgG (1:2000) monoclonal antibody or with HRP- conjugated goat anti-rabbit IgG (1:2000) polyclonal antibody for 2 h at room temperature. The membranes were then rinsed three times for 30 min with TBST, four times quickly with distilled water, and developed using the enhanced chemiluminescence method (ECL) (Amersham).
Reagents Cell culture reagents were purchased from Gibco (Carlsbad, CA). Celecoxib was obtained as a generous gift from Dr. JH Chung (Kyung Hee Univ) and 5-FU was obtained from Sigma (St. Louis, MO). The stock solution of celecoxib was prepared by dissolving it in DMSO (Sigma). All other chemicals were purchased from Sigma unless otherwise stated.
RESULTSMTT assay To investigate the effects of 5-FU and celecoxib in isolation, various concentrations of 5-FU or celecoxib were added to HCT-15 or HT-29 cells. When treated with 10-6 mol/L, 10-5 mol/L, 10-4 mol/L, 10-3 mol/L or 10-2 mol/L 5-FU, the viabilities of HCT-15 cells were 83.6% ± 2.9%, 65.0% ± 4.1%, 69.9% ± 1.3%, 53.6% ± 3.6%, and 21.6% ± 3.7%, respectively. In HT-29 cells, the viabilities of cells for the same concentrations were 89.0% ± 2.0%, 83.8% ± 1.8%, 65.3% ± 4.7%, 43.1% ± 2.7%, and 24.5% ± 1.2%, respectively (Figure 1).
When treated with 10-7 mol/L, 10-6 mol/L, 10-5 mol/L, 10-4 mol/L, or 10-3 mol/L celecoxib, the viabilities of HCT-15 cells were 89.37% ± 3.7%, 85.0% ± 7.8%, 76.4% ± 5.2%, 61.2% ± 3.3%, and 19.1% ± 2.0%, respectively, and of HT-29 cells were 95.6% ± 6.7%, 90.2% ± 4.6%, 78.9% ± 3.3%, 51.1% ± 2.7%, and 19.1% ± 0.8%, respectively (Figure 2).
Based on the MTT results, we opted for 10-3 mol/L 5-FU to be used as the drug-treated standard. Therefore, cells were treated with 10-3 mol/L 5-FU and various concentrations of celecoxib. Cells treated with celecoxib ranging in the concentration of 10-8 mol/L to 10-4 mol/L showed antagonistic effects on the 10-3 mol/L 5-FU-treated cells. But 10-3 mol/L celecoxib showed synergistic
1948 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com
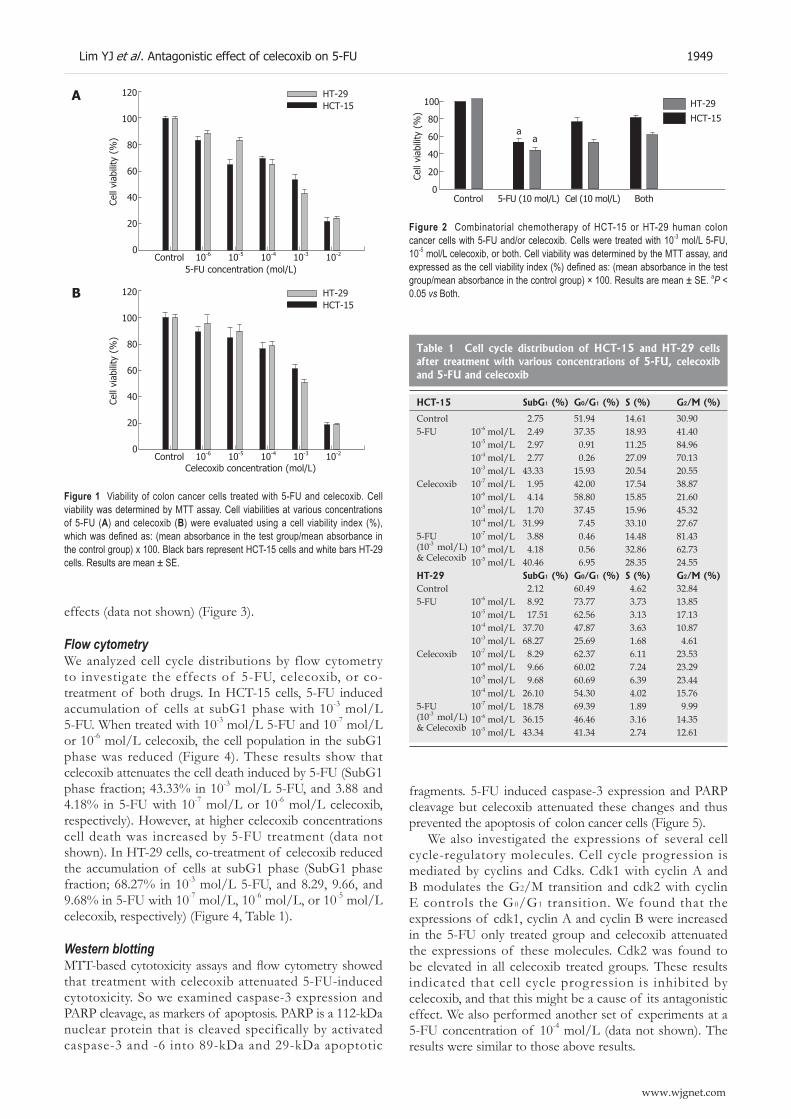
effects (data not shown) (Figure 3).
Flow cytometry We analyzed cell cycle distributions by flow cytometry to investigate the effects of 5-FU, celecoxib, or co-treatment of both drugs. In HCT-15 cells, 5-FU induced accumulation of cells at subG1 phase with 10-3 mol/L 5-FU. When treated with 10-3 mol/L 5-FU and 10-7 mol/L or 10-6 mol/L celecoxib, the cell population in the subG1 phase was reduced (Figure 4). These results show that celecoxib attenuates the cell death induced by 5-FU (SubG1 phase fraction; 43.33% in 10-3 mol/L 5-FU, and 3.88 and 4.18% in 5-FU with 10-7 mol/L or 10-6 mol/L celecoxib, respectively). However, at higher celecoxib concentrations cell death was increased by 5-FU treatment (data not shown). In HT-29 cells, co-treatment of celecoxib reduced the accumulation of cells at subG1 phase (SubG1 phase fraction; 68.27% in 10-3 mol/L 5-FU, and 8.29, 9.66, and 9.68% in 5-FU with 10-7 mol/L, 10-6 mol/L, or 10-5 mol/L celecoxib, respectively) (Figure 4, Table 1).
Western blotting MTT-based cytotoxicity assays and flow cytometry showed that treatment with celecoxib attenuated 5-FU-induced cytotoxicity. So we examined caspase-3 expression and PARP cleavage, as markers of apoptosis. PARP is a 112-kDa nuclear protein that is cleaved specifically by activated caspase-3 and -6 into 89-kDa and 29-kDa apoptotic
fragments. 5-FU induced caspase-3 expression and PARP cleavage but celecoxib attenuated these changes and thus prevented the apoptosis of colon cancer cells (Figure 5).
We also investigated the expressions of several cell cycle-regulatory molecules. Cell cycle progression is mediated by cyclins and Cdks. Cdk1 with cyclin A and B modulates the G2/M transition and cdk2 with cyclin E controls the G0/G1 transition. We found that the expressions of cdk1, cyclin A and cyclin B were increased in the 5-FU only treated group and celecoxib attenuated the expressions of these molecules. Cdk2 was found to be elevated in all celecoxib treated groups. These results indicated that cell cycle progression is inhibited by celecoxib, and that this might be a cause of its antagonistic effect. We also performed another set of experiments at a 5-FU concentration of 10-4 mol/L (data not shown). The results were similar to those above results.
A 120
100
80
60
40
20
0Control 10-6 10-5 10-4 10-3 10-2
5-FU concentration (mol/L)
Cell
viab
ility
(%
)
HT-29HCT-15
B 120
100
80
60
40
20
0Control 10-6 10-5 10-4 10-3 10-2
Celecoxib concentration (mol/L)
Cell
viab
ility
(%
)
HT-29HCT-15
Figure 1 Viability of colon cancer cells treated with 5-FU and celecoxib. Cell viability was determined by MTT assay. Cell viabilities at various concentrations of 5-FU (A) and celecoxib (B) were evaluated using a cell viability index (%), which was defined as: (mean absorbance in the test group/mean absorbance in the control group) x 100. Black bars represent HCT-15 cells and white bars HT-29 cells. Results are mean ± SE.
Figure 2 Combinatorial chemotherapy of HCT-15 or HT-29 human colon cancer cells with 5-FU and/or celecoxib. Cells were treated with 10-3 mol/L 5-FU, 10-5 mol/L celecoxib, or both. Cell viability was determined by the MTT assay, and expressed as the cell viability index (%) defined as: (mean absorbance in the test group/mean absorbance in the control group) × 100. Results are mean ± SE. aP < 0.05 vs Both.
100
80
60
40
20
0Control 5-FU (10 mol/L) Cel (10 mol/L) Both
Cell
viab
ility
(%
)
HT-29
HCT-15a
a
Lim YJ et al . Antagonistic effect of celecoxib on 5-FU 1949
www.wjgnet.com
Table 1 Cell cycle distribution of HCT-15 and HT-29 cells after treatment with various concentrations of 5-FU, celecoxib and 5-FU and celecoxib
HCT-15 SubG1 (%) G0/G1 (%) S (%) G2/M (%)
Control 2.75 51.94 14.61 30.905-FU 10-6 mol/L 2.49 37.35 18.93 41.40
10-5 mol/L 2.97 0.91 11.25 84.9610-4 mol/L 2.77 0.26 27.09 70.1310-3 mol/L 43.33 15.93 20.54 20.55
Celecoxib 10-7 mol/L 1.95 42.00 17.54 38.8710-6 mol/L 4.14 58.80 15.85 21.6010-5 mol/L 1.70 37.45 15.96 45.3210-4 mol/L 31.99 7.45 33.10 27.67
5-FU (10-3 mol/L)& Celecoxib
10-7 mol/L 3.88 0.46 14.48 81.4310-6 mol/L 4.18 0.56 32.86 62.7310-5 mol/L 40.46 6.95 28.35 24.55
HT-29 SubG1 (%) G0/G1 (%) S (%) G2/M (%) Control 2.12 60.49 4.62 32.845-FU 10-6 mol/L 8.92 73.77 3.73 13.85
10-5 mol/L 17.51 62.56 3.13 17.1310-4 mol/L 37.70 47.87 3.63 10.8710-3 mol/L 68.27 25.69 1.68 4.61
Celecoxib 10-7 mol/L 8.29 62.37 6.11 23.5310-6 mol/L 9.66 60.02 7.24 23.2910-5 mol/L 9.68 60.69 6.39 23.4410-4 mol/L 26.10 54.30 4.02 15.76
5-FU (10-3 mol/L) & Celecoxib
10-7 mol/L 18.78 69.39 1.89 9.9910-6 mol/L 36.15 46.46 3.16 14.3510-5 mol/L 43.34 41.34 2.74 12.61
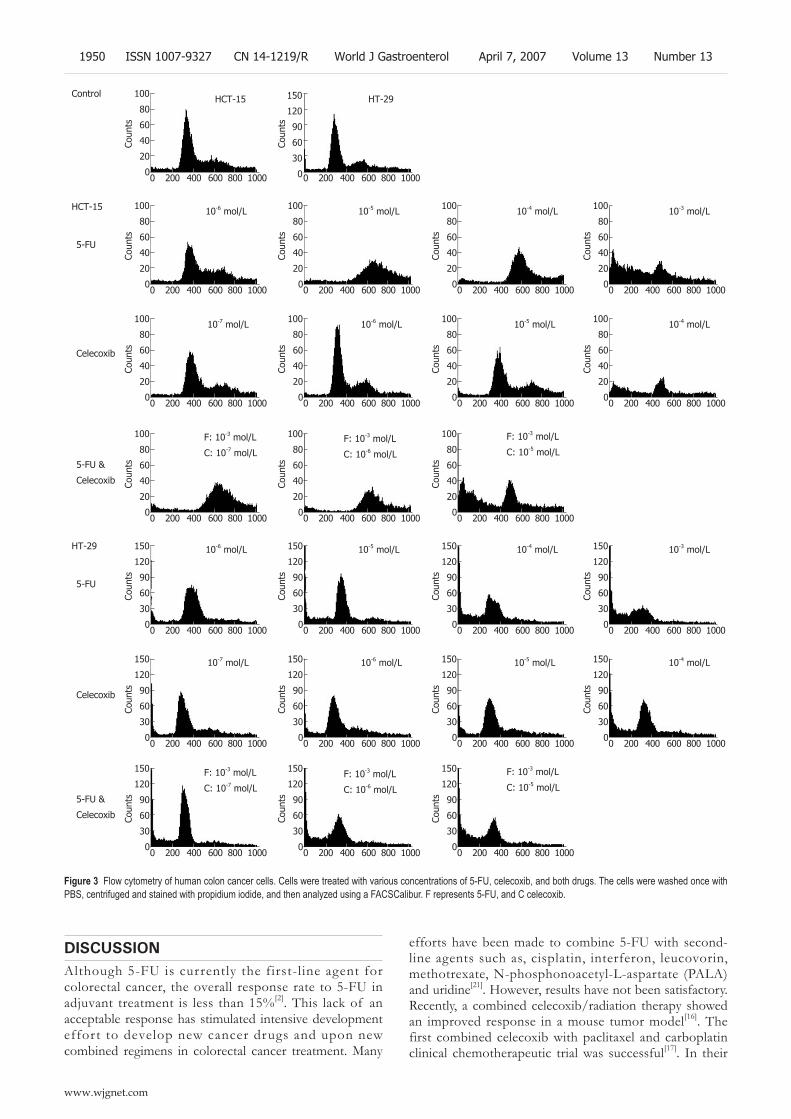
DISCUSSIONAlthough 5-FU is currently the first-line agent for colorectal cancer, the overall response rate to 5-FU in adjuvant treatment is less than 15%[2]. This lack of an acceptable response has stimulated intensive development effort to develop new cancer drugs and upon new combined regimens in colorectal cancer treatment. Many
efforts have been made to combine 5-FU with second-line agents such as, cisplatin, interferon, leucovorin, methotrexate, N-phosphonoacetyl-L-aspartate (PALA) and uridine[21]. However, results have not been satisfactory. Recently, a combined celecoxib/radiation therapy showed an improved response in a mouse tumor model[16]. The first combined celecoxib with paclitaxel and carboplatin clinical chemotherapeutic trial was successful[17]. In their
100
80
60
40
20
00 200 400 600 800 1000
HCT-15
Coun
ts
0 200 400 600 800 1000
HT-29
Coun
ts
100
80
60
40
20
00 200 400 600 800 1000
10-6 mol/L
Coun
ts
100
80
60
40
20
00 200 400 600 800 1000
10-5 mol/L
Coun
ts
100
80
60
40
20
00 200 400 600 800 1000
10-4 mol/L
Coun
ts
100
80
60
40
20
00 200 400 600 800 1000
10-3 mol/L
Coun
ts
100
80
60
40
20
00 200 400 600 800 1000
10-4 mol/L
Coun
ts
100
80
60
40
20
00 200 400 600 800 1000
10-5 mol/L
Coun
ts
100
80
60
40
20
00 200 400 600 800 1000
10-6 mol/L
Coun
ts
100
80
60
40
20
00 200 400 600 800 1000
10-7 mol/L
Coun
ts
100
80
60
40
20
00 200 400 600 800 1000
Coun
ts100
80
60
40
20
00 200 400 600 800 1000
Coun
ts
100
80
60
40
20
00 200 400 600 800 1000
Coun
ts
150
120
90
60
30
00 200 400 600 800 1000
Coun
ts
150
120
90
60
30
00 200 400 600 800 1000
Coun
ts
150
120
90
60
30
00 200 400 600 800 1000
Coun
ts
150
120
90
60
30
00 200 400 600 800 1000
Coun
ts
150
120
90
60
30
00 200 400 600 800 1000
F: 10-3 mol/L
C: 10-5 mol/L
Coun
ts
150
120
90
60
30
00 200 400 600 800 1000
F: 10-3 mol/L
C: 10-6 mol/L
Coun
ts
150
120
90
60
30
00 200 400 600 800 1000
F: 10-3 mol/L
C: 10-7 mol/L
Coun
ts
150
120
90
60
30
00 200 400 600 800 1000
Coun
ts
150
120
90
60
30
00 200 400 600 800 1000
Coun
ts
150
120
90
60
30
00 200 400 600 800 1000
Coun
ts
150
120
90
60
30
00 200 400 600 800 1000
Coun
ts
F: 10-3 mol/L
C: 10-5 mol/LF: 10-3 mol/L
C: 10-6 mol/L
F: 10-3 mol/L
C: 10-7 mol/L
10-6 mol/L 10-5 mol/L 10-4 mol/L 10-3 mol/L
10-4 mol/L10-5 mol/L10-6 mol/L10-7 mol/L
Control
5-FU
Celecoxib
5-FU &
Celecoxib
5-FU
Celecoxib
5-FU &
Celecoxib
Figure 3 Flow cytometry of human colon cancer cells. Cells were treated with various concentrations of 5-FU, celecoxib, and both drugs. The cells were washed once with PBS, centrifuged and stained with propidium iodide, and then analyzed using a FACSCalibur. F represents 5-FU, and C celecoxib.
HCT-15
HT-29
150
120
90
60
30
0
1950 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

study, celecoxib was given concomitantly with paclitaxel and carboplatin to non-small-cell lung cancer patients preoperatively. The results obtained were very promising.
In the present study, we examined the effect of celecoxib on 5-FU-induced cell death in two human colon cancer lines. Interestingly, the MTT assay showed that celecoxib antagonized the cytotoxic effect of 10-3 mol/L 5-FU. So we investigated the mechanism underlying this antagonism by flow cytometry and western blot. First, we focused apoptosis signal and cell cycle regulation. By flow cytometry, 5-FU treatment increased cell accumulation at subG1 phase, which indicate apoptosis in these cells. Celecoxib decreased apoptotic cells at subG1 phase. In western blot analyses, antagonistic effect of celecoxib was proved. Co-treatment of celecoxib with 5-FU attenuated the caspase-3 expression and PARP cleavage. These results well correspondence with flow cytometry data.
It is reported that down-regulation of cyclin B1 expression is observed after treatment of celecoxib and p21 is up-regulated[22]. These changes were not dependent on p53. HCT-15 and HT-29 cells have mutant p53. Mutated p53 is often expressed in variety of human tumors and contribute to malignant process by loss of tumor suppressor function and gain of novel functions that enhance transformed properties of cells[23]. Swamy et al[24] reported that celecoxib increases nuclear localization of active p53. The treatment of celecoxib in HCT-15 and HT-29 cells might involve inhibition of function of mutated p53.
Although we examined the cell cycle distribution by flow cytometry, these results cannot fully explain involvement of cell cycle arrest as a mechanism of antagonistic action of celecoxib. So we also studied the cell cycle regulatory protein expressions. In western blot analyses, cyclin A, cyclin B and cdk1 expressions were increased, which indicates that cell cycle progression was arrested at the G2/M phase. Celecoxib treatment inhibited cell cycle progression at the G2/M phase, and reduced the expressions of cyclin A, cyclin B and cdk1, but the expression of cdk2, which regulates the G1/S transition, was increased. Our results are similar to study by Peng et al[25]. According to the studies by Peng et al[25], celecoxib down-regulated the expression of CDK2 and CDK4 expression in HT-29 cells. Although we do not conduct on the experiment of CDK4, the results from CDK2 was
similar to their study. In this study, we investigated whether celecoxib
has the potential to be used with 5-FU as a combined chemotherapeutic agent for the treatment of colorectal cancer. However, unfortunately, celecoxib was found to antagonize the effect of 5-FU in human colon cancer cells. Although our results are based on an in vitro study and sometimes in vitro data does not correspond with results in vivo results, the results of the present study indicate that celecoxib cannot be used with a cell cycle-arresting agents like 5-FU. Further studies are needed to clarify our results and studies in clinical setting focusing on the usefulness of celecoxib as a combined chemotherapeutic agent are also needed.
REFERENCES1 Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor
A, Feuer EJ, Thun MJ. Cancer statistics, 2005. CA Cancer J Clin 2005; 55: 10-30
2 Yoshikawa R, Kusunoki M, Yanagi H, Noda M, Furuyama JI, Yamamura T, Hashimoto-Tamaoki T. Dual antitumor effects of 5-fluorouracil on the cell cycle in colorectal carcinoma cells: a novel target mechanism concept for pharmacokinetic modulating chemotherapy. Cancer Res 2001; 61: 1029-1037
3 Norum J . Adjuvant cyclophosphamide, methotrexate, fluorouracil (CMF) in breast cancer--is it cost-effective? Acta Oncol 2000; 39: 33-39
4 de Mulder PH. The chemotherapy of head and neck cancer. Anticancer Drugs 1999; 10 Suppl 1: S33-S37
5 de Gramont A, Louvet C, André T, Tournigand C, Krulik M. A review of GERCOD trials of bimonthly leucovorin plus 5-fluorouracil 48-h continuous infusion in advanced colorectal cancer: evolution of a regimen. Groupe d'Etude et de Recherche sur les Cancers de l'Ovaire et Digestifs (GERCOD). Eur J Cancer 1998; 34: 619-626
6 Wolmark N, Rockette H, Fisher B, Wickerham DL, Redmond C, Fisher ER, Jones J, Mamounas EP, Ore L, Petrelli NJ. The benefit of leucovorin-modulated fluorouracil as postoperative adjuvant therapy for primary colon cancer: results from National Surgical Adjuvant Breast and Bowel Project protocol C-03. J Clin Oncol 1993; 11: 1879-1887
7 Mełeń-Mucha G . Effects of short term treatment with pentagastrin, proglumide, tamoxifen given separately or together with 5-fluorouracil on the growth in the murine transplantable Colon 38 cancer. Neoplasma 2001; 48: 133-138
19 kDa17 kDa
115 kDa
89 kDaPARP
Cleavedcaspase-3
HCT-15 HT-29
A B C D A B C D
Figure 4 Western blotting of apoptosis molecules. A: Control; B: 10-3 mol/L 5-FU; C: 10-5 mol/L Celecoxib; D: 5-FU (10-3 mol/L) and celecoxib (10-5 mol/L). HCT-15 and HT-29 human colon cancer cell lines were treated with 10-3 mol/L 5-FU, 10-5 mol/L celecoxib, and 5-FU (10-3 mol/L) and celecoxib (10-5 mol/L). Western blotting for apoptosis-related molecules was performed as described in ‘Materials and Methods’. Co-treatment with celecoxib attenuated the caspase-3 expression and PARP cleavage.
60 kDa
34 kDa
62 kDa
33 kDa
Cyclin A
Cdk-1
Cyclin B
Cdk-2
HCT-15 HT-29
Figure 5 Western blotting of cell cycle-regulatory molecules. A: Control; B: 10-3 mol/L 5-FU; C: 10-5 mol/L Celecoxib; D: 5-FU (10-3 mol/L ) and celecoxib (10-5 mol/L ). HCT-15 and HT-29 human colon cancer cells were treated with 10-3 mol/L 5-FU, 10-5 mol/L celecoxib, and the two in combination. Western blotting for cdk1, cyclin A, cyclin B, and cdk2 were performed as described in ‘Materials and Methods’. Celecoxib reduced G2/M phase accumulation, and increased the G1/S phase protein, cdk-2.
A B C D A B C D
Lim YJ et al . Antagonistic effect of celecoxib on 5-FU 1951
www.wjgnet.com

8 Johnson KR, Young KK, Fan W. Antagonistic interplay between antimitotic and G1-S arresting agents observed in experimental combination therapy. Clin Cancer Res 1999; 5: 2559-2565
9 Chen XX, Lai MD, Zhang YL, Huang Q. Less cytotoxicity to combination therapy of 5-fluorouracil and cisplatin than 5-fluorouracil alone in human colon cancer cell lines. World J Gastroenterol 2002; 8: 841-846
10 Thun MJ, Namboodiri MM, Heath CW. Aspirin use and reduced risk of fatal colon cancer. N Engl J Med 1991; 325: 1593-1596
11 Giovannucci E, Egan KM, Hunter DJ, Stampfer MJ, Colditz GA, Willett WC, Speizer FE. Aspirin and the risk of colorectal cancer in women. N Engl J Med 1995; 333: 609-614
12 Kubba AK. Non steroidal anti-inflammatory drugs and colorectal cancer: is there a way forward? Eur J Cancer 1999; 35: 892-901
13 Arico S, Pattingre S, Bauvy C, Gane P, Barbat A, Codogno P, Ogier-Denis E. Celecoxib induces apoptosis by inhibiting 3-phosphoinositide-dependent protein kinase-1 activity in the human colon cancer HT-29 cell line. J Biol Chem 2002; 277: 27613-27621
14 Koki AT, Masferrer JL. Celecoxib: a specific COX-2 inhibitor with anticancer properties. Cancer Control 2002; 9: 28-35
15 Masferrer JL, Leahy KM, Koki AT, Zweifel BS, Settle SL, Woerner BM, Edwards DA, Flickinger AG, Moore RJ, Seibert K. Antiangiogenic and antitumor activities of cyclooxygenase-2 inhibitors. Cancer Res 2000; 60: 1306-1311
16 Milas L, Kishi K, Hunter N, Mason K, Masferrer JL, Tofilon PJ. Enhancement of tumor response to gamma-radiation by an inhibitor of cyclooxygenase-2 enzyme. J Natl Cancer Inst 1999; 91: 1501-1504
17 Altorki NK, Keresztes RS, Port JL, Libby DM, Korst RJ, Flieder DB, Ferrara CA, Yankelevitz DF, Subbaramaiah K, Pasmantier
MW, Dannenberg AJ. Celecoxib, a selective cyclo-oxygenase-2 inhibitor, enhances the response to preoperative paclitaxel and carboplatin in early-stage non-small-cell lung cancer. J Clin Oncol 2003; 21: 2645-2650
18 Yoo YM, Yim SV, Kim SS, Jang HY, Lea HZ, Hwang GC, Kim JW, Kim SA, Lee HJ, Kim CJ, Chung JH, Leem KH. Melatonin suppresses NO-induced apoptosis via induction of Bcl-2 expression in PGT-beta immortalized pineal cells. J Pineal Res 2002; 33: 146-150
19 Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, Traganos F. Features of apoptotic cells measured by flow cytometry. Cytometry 1992; 13: 795-808
20 Grösch S, Tegeder I, Niederberger E, Bräutigam L, Geisslinger G. COX-2 independent induction of cell cycle arrest and apoptosis in colon cancer cells by the selective COX-2 inhibitor celecoxib. FASEB J 2001; 15: 2742-2744
21 Calabresi P, Chabner BA. Chemotherapy of neoplastic diseases. In Goodman&Gilman’s The pharmacological basis of therapeutics, 10th ed, New York: McGraw-Hill Companies, 2001
22 Dvory-Sobol H, Cohen-Noyman E, Kazanov D, Figer A, Birkenfeld S, Madar-Shapiro L, Benamouzig R, Arber N. Celecoxib leads to G2/M arrest by induction of p21 and down-regulation of cyclin B1 expression in a p53-independent manner. Eur J Cancer 2006; 42: 422-426
23 Pugacheva EN, Ivanov AV, Kravchenko JE, Kopnin BP, Levine AJ, Chumakov PM. Novel gain of function activity of p53 mutants: activation of the dUTPase gene expression leading to resistance to 5-fluorouracil. Oncogene 2002; 21: 4595-4600
24 Swamy MV, Herzog CR, Rao CV. Inhibition of COX-2 in colon cancer cell lines by celecoxib increases the nuclear localization of active p53. Cancer Res 2003; 63: 5239-5242
25 Peng J, Zhang GY, Xiao ZQ. Effects of celecoxib on the proliferation and apoptosis of human colorectal cancer cell line HT-29. Zhongnan Daxue Xuebao Yixueban 2004; 29: 261-265
S-Editor Wang J L- Editor BS Anand E- Editor Chin GJ
1952 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

but had no effect on the level of endothelial NO synthase (eNOS) after reperfusion in liver. The 7 d survival rate was significantly higher in C + I/R group than in I/R group.
CONCLUSION: Curcumin has protective effects against hepatic I/R injury. Its mechanism might be related to the overexpression of Hsp70 and antioxidant enzymes.
© 2007 The WJG Press. All rights reserved.
Key words: Ischemia; Reperfusion injury; Curcumin; Liver; Protection
Shen SQ, Zhang Y, Xiang JJ, Xiong CL. Protective effect ofProtective effect of curcumin against liver warm ischemia/reperfusion injury in rat model is associated with regulation of heat shock protein and antioxidant enzymes. World J Gastroenterol 2007; 13(13): 1953-1961
http://www.wjgnet.com/1007-9327/13/1953.asp
INTRODUCTIONLiver warm ischemia/reperfusion (I/R) injury leads to liver cell (parenchymal and sinusoidal cells) damage, retards liver cell regeneration and predisposes to liver failure. Since I/R injury resulting in liver failure poses severe clinical problems, it is important for surgeons to prevent or ameliorate it.
Several factors contribute to liver warm I/R injury. Hepatic ischemic injury is caused by oxygen deficiency. During reperfusion, hypoxia organ damage is aggravated following recovery of blood flow and oxygen delivery. During the ischemic period, hypoxia causes mitochondrial adenosine triphosphate depletion, alters H+, Na+, and Ca2+ homeostasis, activates hydrolytic enzymes, and impairs cell-volume regulation. After reperfusion, Kupffer cell activation is a principal pathophysiologic mechanism of early liver reperfusion injury. Activated Kupffer cells release reactive oxygen species (ROS), nitric oxide, and several proinflammatory cytokines. These cytokines, together with the increased expression of adhesion molecules by sinusoidal endothelial cells, promote liver neutrophil infiltration, thus contributing to the progression
BASIC RESEARCH
Protective effect of curcumin against liver warm ischemia/reperfusion injury in rat model is associated with regulation of heat shock protein and antioxidant enzymes
Shi-Qiang Shen, Yuan Zhang, Jin-Jian Xiang, Cheng-Long Xiong
www.wjgnet.com
Shi-Qiang Shen, Yuan Zhang, Cheng-Long Xiong, Department of General Surgery, Renmin Hospital of Wuhan University, 238 Jiefang Road, Wuhan 430060, Hubei Province, ChinaJin-Jian Xiang, Department of Hepatobiliary Surgery, Xiamen First Hospital Affiliated to Fujian Medical University, Xiamen 361004, Fujian Province, ChinaCorrespondence to: Dr. Shi-Qiang Shen, Department of General Surgery, Renmin Hospital of Wuhan University, 238 Jiefang Road, Wuhan 430060, Hubei Province, China. [email protected]: +86-27-88041919-2212 Fax: +86-27-88042292Received: 2006-11-01 Accepted: 2007-01-09
AbstractAIM: To investigate the hypothesis that the protective effects of curcumin in hepatic warm ischemia/reperfusion (I/R) injury are associated with increasing heat shock protein 70 (Hsp70) expression and antioxidant enzyme activity.
METHODS: Sixty Sprague-Dawley male rats were randomly divided into sham, I/R, C + I/R groups. The model of reduced-size liver warm ischemia and reperfusion was used. Curcumin (50 mg/kg) was administered by injection through a branch of superior mesenteric vein at 30 min before ischemia in C + I/R group. Five rats were used to investigate the survival during 1 wk after operation in each group. Blood samples and liver tissues were obtained in the remaining animals after 3, 12, and 24 h of reperfusion to assess serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), l iver tissue NO2
- + NO3-,
malondialdehyde (MDA) content, superoxide dismutase (SOD), catalase (CAT), nitricoxide synthase (NOS) and myeloperoxidase (MPO) activity, Hsp70 expression and apoptosis ratio.
RESULTS: Compared with I/R group, curcumin pretreatment group showed less ischemia/reperfusion-induced injury. CAT and SOD activity and Hsp70 expression increased significantly. A higher rate of apoptosis was observed in I/R group than in C + I/R group, and a significant increase of MDA, NO2
- + NO3-
and MPO level in liver tissues and serum transaminase concentration was also observed in I/R group compared to C + I/R group. Curcumin also decreased the activity of inducible NO synthase (iNOS) in liver after reperfusion,
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1953-1961www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.

of parenchymal injury. Efforts to attenuate I/R injury have been focused on blocking the events associated with inconvertible liver damage.
Curcumin [1, 6-heptadiene, 3-5-dione, 1, 7-bis (4-hydroxy-3-methoxyphenyl) (C21H20O6)], a yellow-orange dye extracted from the spice tumeric, possessing both anti-inflammatory antioxidant and anti-carcinogenic effect[1], is a potent stimulator of the stress-induced expression of heat shock protein 70 (Hsp70). It was recently reported that administration of curcumin could ameliorate I/R injury in rat kidney, myocardium, and nervous tissue[2-6]. We aimed to study whether curcumin can ameliorate I/R injury, inhibit hepatocyte apoptosis, and induce Hsp over-expression in hepatic I/R injury.
MATERIALS AND METHODSExperimental animalsMale Sprague-Dawley rats (8-10 wk old) weighing 220-300 g were purchased from the Center of Experimental Animals, Medical College of Wuhan University. This project was approved by the Committee of Experimental Animals of Wuhan University. All experimental procedures complied with the Guide for the Care and Use of Laboratory Animals.
Surgical procedures and experimental designFasted (10-14 h) rats were anesthetized with 20% urethane solution (5 mL/kg). After midline laparotomy, liver warm ischemia was induced by clamping the left branches of both portal vein and hepatic artery to prevent portal congestion for 45 min (ischemia of about 70% liver). After 45 min of ischemia, the clip was released to allow reperfusion of the liver, and the non-ischemic lobes were immediately excised, leaving the remaining two reperfusion lobes. The abdominal cavity was closed, and the animals were allowed to recover with free access to food and water. The rats were divided into three groups: sham group in which the rats were laparotomized for 5 min without hepatectomy or vascular clamping, I/R group and C + I/R group (ischemia/reperfusion + curcumin infusion group) (20 animals per group). The rats in C + I/R group were injected with 50 mg/kg curcumin (Sigma, St. Louis, MO) (dissolved in 1 mL dimethyl sulfoxide solution) into a branch of superior mesenteric vein 30 min before the left branches of both portal vein and hepatic artery were clamped. The rats in I/R group were infused with only 1 mL dimethyl sulfoxide 30 min before the left branches of vessels were clamped. Five rats in each group were used to investigate the survival in 1 wk. Five rats in each group were killed at 3, 12, or 24 h after the start of reperfusion, and the left ischemic lobe of the liver was removed, weighed, snap frozen in liquid nitrogen and stored at -70 for biochemical analysis or stored in 4% PBS-buffered paraformaldehyde for histopathological analysis. Blood samples were collected via the inferior vena cava.
Blood analysis Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) concentrations were measured
using standard techniques with a serum analyzer (Olympus Automated Chemistry Analyzer AU2700, Japan).
Malondialdehyde, catalase, superoxide dismutase, nitrico-xide, nitricoxide synthase, and myeloperoxidase in liver homogenate determination Liver homogenate malondialdehyde (MDA) concentration was measured using the thiobarbituric acid (TBA) method. The amount of lipid peroxides (LPO) was measured as the production of MDA, which in combination with TBA forms a pink chromogen compound whose absorbance at 532 nm was measured. The result was expressed as nmol/mg protein.
Total superoxide dismutase (SOD) activity in hepatic homogenate was measured through the inhibition of nitroblue tetrazolium (NBT) reduction by O2
- generated by the xanthine/xanthine oxidase system. One SOD activity unit was defined as the enzyme amount causing 50% inhibition in 1 mL reaction solution per milligram tissue protein and the result was expressed as U/mg protein.
Catalase (CAT) activity in liver homogenate was detected using ammonium molybdate method by measuring the intensity of a yellow complex formed by molybdate and H2O2 at 405 nm, after ammonium molybdate was added to terminate the H2O2 degradation reaction catalyzed by CAT. An enzyme activity unit was defined as the degradation of 1 mmol H2O2 per second per mg tissue protein and the enzyme activity was expressed as U/mg protein.
Liver homogenate NO products nitrite/nitrate (NO2- +
NO3-) were detected using a colorimetric NO detection kit,
which reflected the concentration of NO in the liver.Total nitricoxide synthase (NOS), endothelial NO
synthase (eNOS) and inducible NO synthase (iNOS) activity in hepatic homogenate was also detected using a spectrophotometric method assay kit.
Myeloperoxidase (MPO) activity in hepatice tissue was detected using a spectrophotometric method, reflecting the number of polymorphonuclear neutrophils (PMN) in the liver. This method uses 3, 3’, 5, 5’-tetramethyl benzidine (TMB) as an oxidizable dye, and the reaction was started by adding hydrogen peroxide (H2O2) in the medium.
Total protein concentrations in liver homogenate samples were determined with Coomassie blue method.
All assay kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Determination of Hsp70 mRNA and Hsp70 expression Hsp70 mRNA was determined in liver tissues using semi-quantitative RT-PCR at 3, 12, and 24 h after surgery. Total RNA was isolated from liver tissue by using Trizol reagent (GIBCO BRL, Gaithersburg, MD). Five μg of RNA was reverse-transcribed to complementary DNA (cDNA) by using M-MuLV reverse transcriptase (Applied Biosystems, Foster City, CA) at 42 for 90 min, and cDNA was then amplified by using the following primers for the rat Hsp70mRNA gene (sense: 5'-TGCCCGCCTACTTCAACGA-3' (614-632 bp) and anti-sense: 5'-CCGCGTGATGGACGTGTAG-3' (1074-1056 bp), product of the reaction was 461 bp. Thermal cycle conditions were 94
1954 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

for 30 s, 58 for 30 s, and 72 for 30 s, for a total of 30 cycles. The final cycle was followed with 5 min incubation at 72. Amplification of a house keeping gene (β-actin) was performed by the same reaction program using the following primers for the rat β-actin mRNA gene (sense: 5’-TGGAGAAGATTTGGCACC-3’ (54-61 bp) and anti-sense: 5’-TACGACCAGAGGCATACAGG-3’ (222-241 bp) (all primers were synthesized by SBS Genetech, Shanghai, China), product of the reaction was 188bp. Seven point five μL of RT-PCR products was subjected to electrophoresis on 2% agarose gel containing 0.05% ethidium bromide and analyzed by densitometry using BIO-RAD Quantity One 4.4.0 software (BIO-RAD, Hercules, CA). Hsp70 mRNA expression was illustrated by determining the ratio of band intensity of Hsp70 and β-actin mRNA.
Hsp70 protein expression in liver tissue was detected us ing a commercia l Hsp70 ELISA ki t (Stressgen Bioreagents, Ann Arbor, MI). The protein was isolated from liver tissue homogenate by using protein extraction reagent. According to the assay procedure supplemented with the kit, Hsp70 standards and samples were added to the Hsp70 immunoassay plate, incubated and washed, then anti-Hsp70 biotin conjugate was added, incubated and washed, finally avidin-HRP conjugate was added, incubated and washed. The plant was developed with 3, 3', 5, 5' tetramethylbenzidine. We added acid solution to the plant, and measured its absorbance at 450 nm. Hsp70 in samples was calculated from a curve of absorbance vs log of the standards. Tissue total protein concentration was also measured with Coomassie blue method (assay kit was purchased from Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Histological analysisExcised liver specimens were fixed in 4% PBS-buffered paraformaldehyde and embedded in paraffin. Five-micrometer liver sections stained with hematoxylin and eosin were evaluated at × 200 magnification by a point-counting method for severity of hepatic injury using the ordinal scale: grade 0 = minimal or no evidence of injury; grade 1 = mild injury with cytoplasm vacuolization and focal nuclear pyknosis; grade 2 = moderate to severe injury with extensive nuclear pyknosis, loss of intercellular borders, and mild to moderate neutrophil infiltration; grade 3 = severe injury with disintegration of hepatic cords, hemorrhage, and severe PMN infiltration. Statistical evaluation was made using this scale. An average of 100 adjacent points on 1 mm2 grid was graded for each specimen.
Detection of apoptosis in liver cells with in situ terminal deoxynucleotidyl transferase-mediated biotin-dUTP nick end labeling methodLiver sample was fixed in 4% PBS-buffered paraforma-ldehyde and embedded in paraffin. Apoptosis was detected by using the terminal deoxynucleotidyl transferase-mediated biotin-dUTP nick end-labeling (TUNEL) technique with in situ apoptosis detection kit (Beijing Zhongshan Biotechnology Co. Ltd, Beijing, China)
according to the manufacturer’s instructions. The number of TUNEL-positive hepatocytes was counted by 2 blinded observers under microscope (× 400 magnification). Five different visual fields were counted in each section. Hepatocytes with brown-stained nuclei were TUNEL-positive. The percentage of TUNEL-positive cells (apoptosis index) was calculated by the number of apoptotic cells divided by the total cells.
Flow cytometry for apoptosisFresh liver tissue samples were taken under aseptic conditions. Small vessels and connective tissue were removed. They were placed on a sterile 250 μm brass mesh over a conical tube, and gently teased with sterile forceps. Cells were rinsed through the brass mesh with pre-cold RPMI-1640 (Invitrogen, Carlsbad, CA), and washed twice at a concentration of 300 × g for 4 min.
Apoptosis was detected using the Annexin V-FITC kit (BD-Pharmingen, San Diego, CA). Liver cell samples were washed twice with cold PBS and resuspended in 1 × binding buffer at a concentration of 1 × 106 cells/mL. One hundred μL of the cell suspension was incubated with Annexin V-FITC (5 μL) and propidium iodide (5 μL) in binding buffer on ice for 10 min in dark, then 150 μL of ice-cold (diluted in 1 × binding buffer) was added to the cell samples. Early apoptotic cells (Annexin V positive and propidium iodide negative) were quantitated by flow cytometry.
Statistical analysisAll values were expressed as mean ± SE. Statistical significance between two groups of parametric data was evaluated by ANOVA. Survival curves for 7 d were analyzed with the Kaplan-Meier method, and the differences in survival rates were evaluated by log-rank test. All statistical analyses were performed with the SPSS statistical package (SPSS 14.0 for Windows; SPSS, Inc, Chicago, IL). P < 0.05 was considered statistically significant.
RESULTSChanges in serum AST and ALT concentrations after operation in each group are shown in Figure 1. Serum ALT and AST levels increased significantly after reperfusion in I/R group compared with sham group. Pretreatment with curcumin significantly decreased AST and AlT levels compared with I/R group.
The MDA content as an index of lipid peroxidation was significantly higher after reperfusion in I/R group than in sham group. Pretreatment with curcumin significantly decreased MDA content after 12 and 24 h of reperfusion (P < 0.05). CAT and SOD activity in hepatic tissue decreased markedly after reperfusion as compared with the sham group. In contrast, a significant decrease in the activity of CAT and SOD was observed in I/R group compared with curcumin pretreatment group (Figure 2).
Changes in nitric oxide of hepatic tissue measured as total NO2
- + NO3- improved in the I/R group compared
with the sham group after I/R. Pretreatment with
Shen SQ et al. Curcumin against liver warm I/R injury 1955
www.wjgnet.com

curcumin significantly decreased total NO2- + NO3
-
levels when compared with I/R group without curcumin pretreatment (Figure 2).
Changes in iNOS, eNOS, and MPO activity after operation in each group are shown in Figure 3. MPO and iNOS activity increased in liver samples from animals subjected to ischemia-reperfusion as compared with the sham group. Curcumin pretreatment significantly reduced the MPO and iNOS activity compared with I/R group. No significant difference in eNOS activity was observed between curcumin pretreatment group and other groups after operation.
The expressions of β-actin mRNA and Hsp70mRNA are shown in Figure 4. Hsp70mRNA expression was not detected in sham group. No obvious alteration in β-actin mRNA expression was found in all groups. Hsp70mRNA expression in C + I/R group was significantly higher than that in I/R group after reperfusion. The expression of Hsp70 is shown in Figure 5.
Analysis of apoptosis by TUNEL method showed that the apoptosis index was lower in C + I/R group than in I/R group. Very few TUNEL-positive hepatocytes were detected in sham group. TUNEL-positive hepatocytes remarkably increased to about 36% of total hepatocytes
b
b,d b
b,d
b,d
b
Sham
C + I/R
I/R
3 12 24 t (after operation)/h
ALT
(U/L
)
5000.00
4000.00
3000.00
2000.00
1000.00
0.00
Sham
C + I/R
I/R
3 12 24 t (after operation)/h
AST
(U/L
)
5000.00
4000.00
3000.00
2000.00
1000.00
0.00
b
b,d
b,d
b,d
b
b
A B Figure 1 S ign i f i cant ly increased serum conce-ntrations of ALT (A) and AST (B) after reperfusion a s c o m p a r e d w i t h t h e sham group . Curcumin pretreatment significantly reduced the hepa t i c I /R-related elevation of ALT and AST levels. bP < 0.01 vs Sham group; dP < 0.01 vs C + I/R group. Error bars represent the 95% CI.
120.00
100.00
80.00
60.00
40.00
20.00
0.00
Sham
C + I/R
I/R
b
b,d
b
b
b,d b,d
SOD
(U
/mgp
rot)
A
b
b,d
b
b
b,db,d
50.00
40.00
30.00
20.00
10.00
0.00 3 12 24
CAT
(U/m
gpro
t)
B
t (after operation)/h 3 12 24
t (after operation)/h
Sham
C + I/R
I/R
Sham
C + I/R
I/R
Sham
C + I/R
I/R
25.00
20.00
15.00
10.00
5.00
0.00
MD
A (n
mol
/mgp
rot)
C
3 12 24 t (after operation)/h
b
b,d b
bb,d
b,d 0.600
NO
2- + N
O3- (
μmol
/gpr
ot)
D
3 12 24 t (after operation)/h
b
a,d0.500
0.400
0.300
0.200
0.100
0.000
b
da
d
Figure 2 The results of SOD ac t i v i t y ( A ) , CAT activity (B), MDA content (C), and total NO2
- + NO3-
level (D) in l iver t issue. Curcumin pre t reatment significantly reduced MDA content and total NO2
- + NO3
- levels, and increased CAT and SOD activity as compared to I/R group. aP < 0.05, bP < 0.01 vs Sham group; dP < 0.01 vs C + I/R group. Error bars represent the 95% CI.
1956 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

in I/R group at 24 h after reperfusion, the increase in TUNEL-positive hepatocytes was significantly inhibited by about 25% in the C + I/R group at the same time point (Figures 6 and 7). Flow cytometry showed a similar difference between C + I/R group and I/R group
(Figure 8). No significant changes were found in sham group at
light microscopy. At 3 h after reperfusion, cytoplasmic vacuolation and focal nuclear pyknosis were seen in I/R group. At 12 and 24 h after reperfusion, extensive karyopycnosis, severe necrosis and loss of intercellular borders were seen in I/R group. At these time points, hepatocyte damage was slighter in curcumin treatment group than in I/R group (Table 1). These results indicated that hepatic injury induced by ischemia-reperfusion was
2.00
1.50
1.00
0.50
0.00
Sham
C + I/R
I/R
MPO
(U
/g)
3 12 24 t (after operation)/h
b,d b,d
b,d
a
b
Sham
C + I/R
I/R
Sham
C + I/R
I/R
A
1.00
0.20
0.00
iNO
S (U
/mgp
rot)
3 12 24 t (after operation)/h
b,d
B
0.40
0.60
0.80
b,db,d
b
bb
0.100
0.020
0.000
eNO
S (U
/mgp
rot)
3 12 24 t (after operation)/h
C
0.040
0.060
0.080
Figure 3 Activity of MPO (A), iNOS (B), and eNOS (C) in liver tissue. aP < 0.05, bP < 0.01 vs sham group; dP < 0.01 vs C + I/R group. Error bars represent the 95% CI.
β-actinmRNA
Hsp70mRNA
6 5 4 3 2 1
Figure 4 Expression of β-actin mRNA and Hsp70 mRNA in I/R group and C + I/R group. 6: 24 h after I/R in I/R group; 5: 24 h after I/R in C + I/R group; 4:12 h after I/R in I/R group; 3: 12 h after I/R in C + I/R group; 2: 3 h after I/R in I/R group; 1: 3 h after I/R in C + I/R group.
C + I/R
I/R1.2500
1.0000
0.7500
0.5000
0.2500
0.0000
b
b
b
3 12 24 t (after operation)/h
Hsp
70m
RNA
A
C + I/R
I/R
b
b
b
Hsp
70 (
pg/m
gpro
t)
1500.0000
1000.0000
500.0000
0.0000 3 12 24
t (after operation)/h
B
Figure 5 Expression of Hsp70mRNA (A) and Hsp70 (B) in C + I/R group increased after liver reperfusion as compared with I/R group. bP < 0.01 vs C + I/R group. Error bars represent the 95% confidence intervals.
Shen SQ et al. Curcumin against liver warm I/R injury 1957
www.wjgnet.com
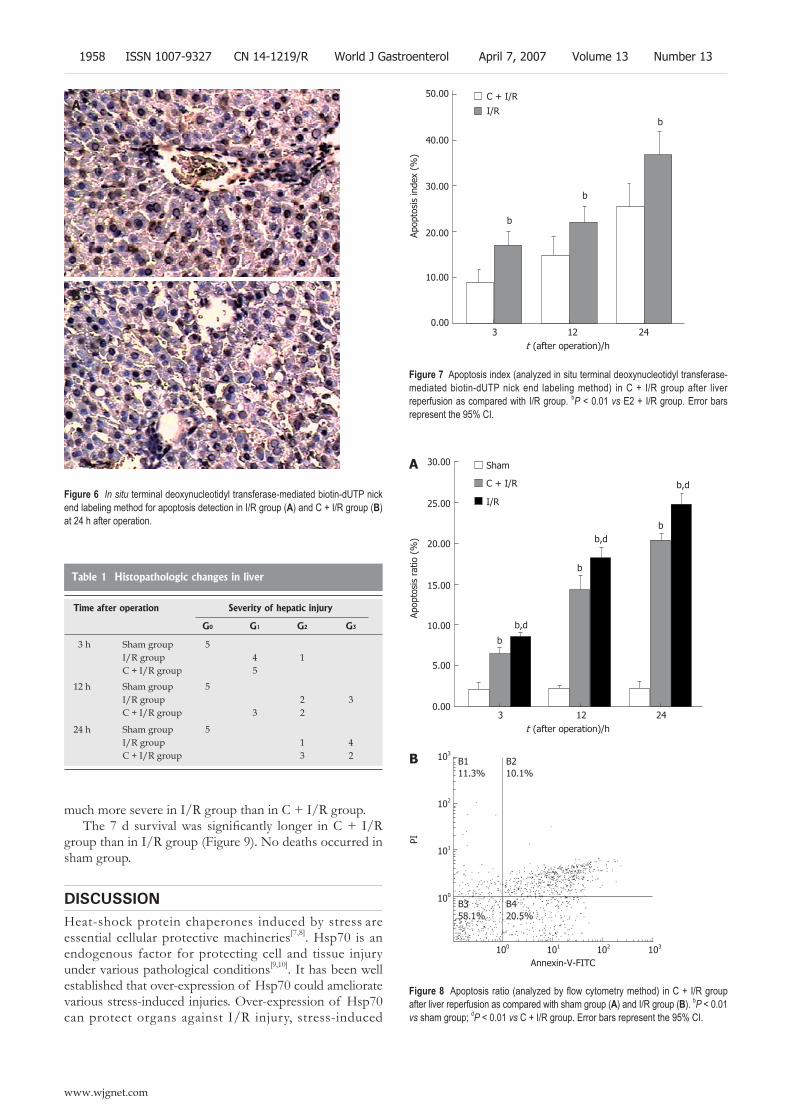
much more severe in I/R group than in C + I/R group. The 7 d survival was significantly longer in C + I/R
group than in I/R group (Figure 9). No deaths occurred in sham group.
DISCUSSIONHeat-shock protein chaperones induced by stress are essential cellular protective machineries[7,8]. Hsp70 is an endogenous factor for protecting cell and tissue injury under various pathological conditions[9,10]. It has been well established that over-expression of Hsp70 could ameliorate various stress-induced injuries. Over-expression of Hsp70 can protect organs against I/R injury, stress-induced
B
A
Figure 6 In situ terminal deoxynucleotidyl transferase-mediated biotin-dUTP nick end labeling method for apoptosis detection in I/R group (A) and C + I/R group (B) at 24 h after operation.
C + I/R
I/R
b
Apop
tosi
s in
dex
(%)
3 12 24 t (after operation)/h
A
Figure 7 Apoptosis index (analyzed in situ terminal deoxynucleotidyl transferase-mediated biotin-dUTP nick end labeling method) in C + I/R group after liver reperfusion as compared with I/R group. bP < 0.01 vs E2 + I/R group. Error bars represent the 95% CI.
b
b
50.00
40.00
30.00
20.00
10.00
0.00
Sham
C + I/R
I/R
b
b,d
b
bb,d
b,d
30.00
25.00
20.00
15.00
10.00
5.00
0.00
Apop
tosi
s ra
tio (
%)
3 12 24 t (after operation)/h
Figure 8 Apoptosis ratio (analyzed by flow cytometry method) in C + I/R group after liver reperfusion as compared with sham group (A) and I/R group (B). bP < 0.01 vs sham group; dP < 0.01 vs C + I/R group. Error bars represent the 95% CI.
103
102
101
100
100 101 102 103
B111.3%
B210.1%
B358.1%
B420.5%
Annexin-V-FITC
PI
Table 1 Histopathologic changes in liver
Time after operation Severity of hepatic injury
G0 G1 G2 G3
3 h Sham group 5I/R group 4 1C + I/R group 5
12 h Sham group 5I/R group 2 3C + I/R group 3 2
24 h Sham group 5I/R group 1 4C + I/R group 3 2
1958 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com
B
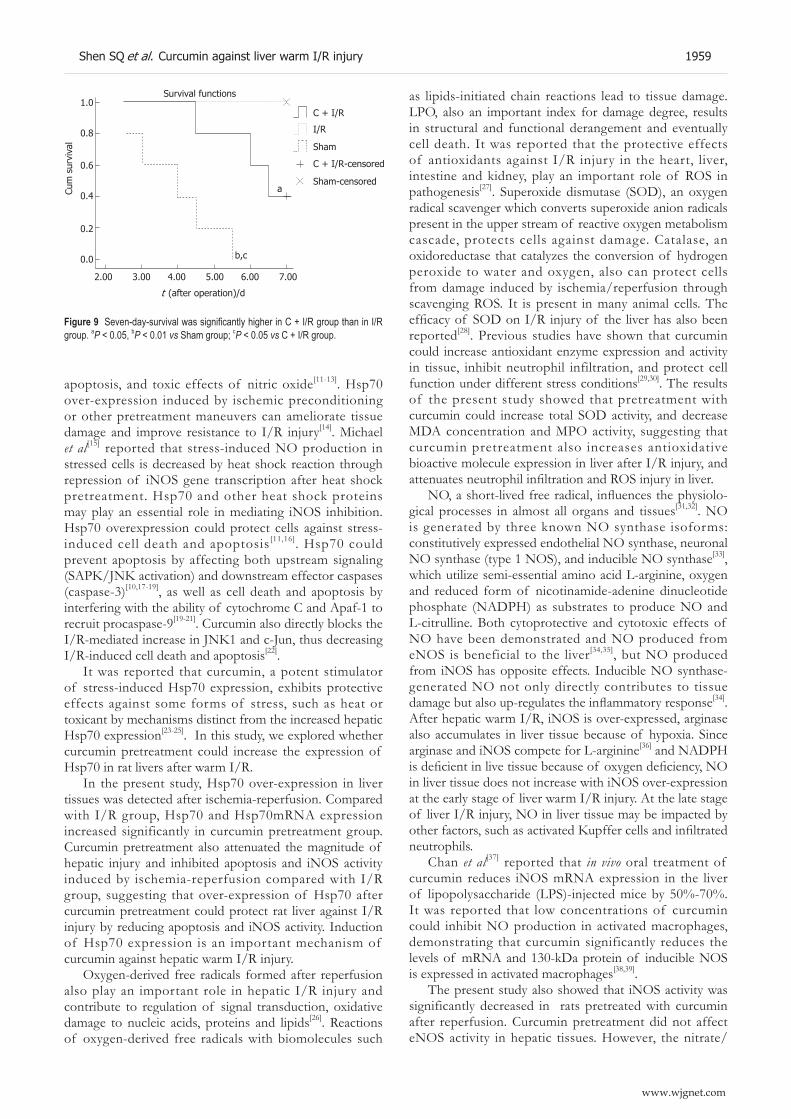
apoptosis, and toxic effects of nitric oxide[11-13]. Hsp70 over-expression induced by ischemic preconditioning or other pretreatment maneuvers can ameliorate tissue damage and improve resistance to I/R injury[14]. Michael et al[15] reported that stress-induced NO production in stressed cells is decreased by heat shock reaction through repression of iNOS gene transcription after heat shock pretreatment. Hsp70 and other heat shock proteins may play an essential role in mediating iNOS inhibition. Hsp70 overexpression could protect cells against stress-induced cell death and apoptosis[11,16]. Hsp70 could prevent apoptosis by affecting both upstream signaling (SAPK/JNK activation) and downstream effector caspases (caspase-3)[10,17-19], as well as cell death and apoptosis by interfering with the ability of cytochrome C and Apaf-1 to recruit procaspase-9[19-21]. Curcumin also directly blocks the I/R-mediated increase in JNK1 and c-Jun, thus decreasing I/R-induced cell death and apoptosis[22].
It was reported that curcumin, a potent stimulator of stress-induced Hsp70 expression, exhibits protective effects against some forms of stress, such as heat or toxicant by mechanisms distinct from the increased hepatic Hsp70 expression[23-25]. In this study, we explored whether curcumin pretreatment could increase the expression of Hsp70 in rat livers after warm I/R.
In the present study, Hsp70 over-expression in liver tissues was detected after ischemia-reperfusion. Compared with I/R group, Hsp70 and Hsp70mRNA expression increased significantly in curcumin pretreatment group. Curcumin pretreatment also attenuated the magnitude of hepatic injury and inhibited apoptosis and iNOS activity induced by ischemia-reperfusion compared with I/R group, suggesting that over-expression of Hsp70 after curcumin pretreatment could protect rat liver against I/R injury by reducing apoptosis and iNOS activity. Induction of Hsp70 expression is an important mechanism of curcumin against hepatic warm I/R injury.
Oxygen-derived free radicals formed after reperfusion also play an important role in hepatic I/R injury and contribute to regulation of signal transduction, oxidative damage to nucleic acids, proteins and lipids[26]. Reactions of oxygen-derived free radicals with biomolecules such
as lipids-initiated chain reactions lead to tissue damage. LPO, also an important index for damage degree, results in structural and functional derangement and eventually cell death. It was reported that the protective effects of antioxidants against I/R injury in the heart, liver, intestine and kidney, play an important role of ROS in pathogenesis[27]. Superoxide dismutase (SOD), an oxygen radical scavenger which converts superoxide anion radicals present in the upper stream of reactive oxygen metabolism cascade, protects cells against damage. Catalase, an oxidoreductase that catalyzes the conversion of hydrogen peroxide to water and oxygen, also can protect cells from damage induced by ischemia/reperfusion through scavenging ROS. It is present in many animal cells. The efficacy of SOD on I/R injury of the liver has also been reported[28]. Previous studies have shown that curcumin could increase antioxidant enzyme expression and activity in tissue, inhibit neutrophil infiltration, and protect cell function under different stress conditions[29,30]. The results of the present study showed that pretreatment with curcumin could increase total SOD activity, and decrease MDA concentration and MPO activity, suggesting that curcumin pretreatment also increases antioxidative bioactive molecule expression in liver after I/R injury, and attenuates neutrophil infiltration and ROS injury in liver.
NO, a short-lived free radical, influences the physiolo-gical processes in almost all organs and tissues[31,32]. NO is generated by three known NO synthase isoforms: constitutively expressed endothelial NO synthase, neuronal NO synthase (type 1 NOS), and inducible NO synthase[33], which utilize semi-essential amino acid L-arginine, oxygen and reduced form of nicotinamide-adenine dinucleotide phosphate (NADPH) as substrates to produce NO and L-citrulline. Both cytoprotective and cytotoxic effects of NO have been demonstrated and NO produced from eNOS is beneficial to the liver[34,35], but NO produced from iNOS has opposite effects. Inducible NO synthase- generated NO not only directly contributes to tissue damage but also up-regulates the inflammatory response[34]. After hepatic warm I/R, iNOS is over-expressed, arginase also accumulates in liver tissue because of hypoxia. Since arginase and iNOS compete for L-arginine[36] and NADPH is deficient in live tissue because of oxygen deficiency, NO in liver tissue does not increase with iNOS over-expression at the early stage of liver warm I/R injury. At the late stage of liver I/R injury, NO in liver tissue may be impacted by other factors, such as activated Kupffer cells and infiltrated neutrophils.
Chan et al[37] reported that in vivo oral treatment of curcumin reduces iNOS mRNA expression in the liver of lipopolysaccharide (LPS)-injected mice by 50%-70%. It was reported that low concentrations of curcumin could inhibit NO production in activated macrophages, demonstrating that curcumin significantly reduces the levels of mRNA and 130-kDa protein of inducible NOS is expressed in activated macrophages[38,39].
The present study also showed that iNOS activity was significantly decreased in rats pretreated with curcumin after reperfusion. Curcumin pretreatment did not affect eNOS activity in hepatic tissues. However, the nitrate/
1.0
0.8
0.6
0.4
0.2
0.0 b,c
a
Survival functions
2.00 3.00 4.00 5.00 6.00 7.00
t (after operation)/d
Cum
sur
viva
l
C + I/R
I/R
Sham
Sham-censored
C + I/R-censored
Figure 9 Seven-day-survival was significantly higher in C + I/R group than in I/R group. aP < 0.05, bP < 0.01 vs Sham group; cP < 0.05 vs C + I/R group.
Shen SQ et al. Curcumin against liver warm I/R injury 1959
www.wjgnet.com

nitrite levels were substantially decreased, thus significantly reducing organ damage in C + I/R group when compared with I/R group.
In summary, curcumin pretreatment protects liver from warm I/R injury through multiple pathways, regulates multiple bioactive molecule expression and activity, and inhibits cell apoptosis and neutrophil infiltration. Over-expression of Hsp70 and antioxidant enzymes after pretreatment with curcumin may play an essential role in protecting liver against warm I/R injury.
COMMENTSBackgroundLiver warm ischemia-reperfusion (I/R) injury leads to liver cell (parenchymal and sinusoidal cells) damage, retards liver cell regeneration and predisposes to liver failure. Since I/R injury resulting in liver function failure poses significant clinical problems, it is important for surgeons to prevent or ameliorate I/R-induced liver injury. Curcumin [1, 6-heptadiene, 3-5-dione, 1, 7-bis (4-hydroxy-3-methoxyphenyl) (C21H20O6)], a yellow-orange dye extracted from spice tumeric, possessing both anti-inflammatory antioxidant and anti-carcinogenic effect, is a potent stimulator of stress-induced expression of heat shock protein 70 (Hsp70). It has been reported that administration of curcumin attenuates I/R injury in rat kidney, myocardium, and nervous tissue. In this study, we aimed to study whether curcumin could ameliorate I/R injury, inhibit hepatocyte apoptosis, and induce Hsp and antioxidant enzymes over-expression in hepatic I/R injury.
Research frontiersCurcumin has antioxidant and anti-carcinogenic properties. It has been reported that administration of curcumin attenuates I/R injury in rat kidney, myocardium, and nervous tissue, but no research has investigated its protective effects against liver warm I/R injury.
Innovations and breakthroughsThe objective of this study was to examine the hypothesis that the protective effects of curcumin against hepatic warm I/R injury are associated with increasing Hsp70 expression and antioxidant enzyme activity. Our data demonstrate that pretreatment with curcumin could protect rat liver from warm I/R injury through multiple pathways, such as expression of Hsps, decreased NO, enhanced antioxidant enzyme activities, and neutrophil infiltration.
Applications Curcumin can be used in the treatment of hepatic warm I/R injury.
Peer reviewThe authors demonstrated that curcumin treatment significantly reduced I-R-induced liver injury by modulating several pathways. These findings included several novel findings which are interested to readers in this field.
REFERENCES1 Barthelemy S, Vergnes L, Moynier M, Guyot D, Labidalle S,
Bahraoui E. Curcumin and curcumin derivatives inhibit Tat-mediated transactivation of type 1 human immunodeficiency virus long terminal repeat. Res Virol 1998; 149: 43-52
2 Shahed AR, Jones E, Shoskes D. Quercetin and curcumin up-regulate antioxidant gene expression in rat kidney after ureteral obstruction or ischemia/reperfusion injury. Transplant Proc 2001; 33: 2988
3 Yeh CH, Chen TP, Wu YC, Lin YM, Jing Lin P. Inhibition of NFkappaB activation with curcumin attenuates plasma inflammatory cytokines surge and cardiomyocytic apoptosis following cardiac ischemia/reperfusion. J Surg Res 2005; 125: 109-116
4 Wang Q , Sun AY, Simonyi A, Jensen MD, Shelat PB, Rottinghaus GE, MacDonald RS, Miller DK, Lubahn DE, Weisman GA, Sun GY. Neuroprotective mechanisms of
curcumin against cerebral ischemia-induced neuronal apoptosis and behavioral deficits. J Neurosci Res 2005; 82: 138-148
5 Thiyagarajan M, Sharma SS. Neuroprotective effect of curcumin in middle cerebral artery occlusion induced focal cerebral ischemia in rats. Life Sci 2004; 74: 969-985
6 Gaddipati JP, Sundar SV, Calemine J, Seth P, Sidhu GS, Maheshwari RK. Differential regulation of cytokines and transcription factors in liver by curcumin following hemorrhage/resuscitation. Shock 2003; 19: 150-156
7 Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell 1998; 92: 351-366
8 Frydman J, Nimmesgern E, Ohtsuka K, Hartl FU. Folding ofFolding of nascent polypeptide chains in a high molecular mass assembly with molecular chaperones. Nature 1994; 370: 111-117
9 Tacchini L, Radice L, Pogliaghi G, Bernelli-Zazzera A. Differential activation of heat shock and nuclear factor kappaB transcription factors in postischemic reperfused rat liver. Hepatology 1997; 26: 186-191
10 Lin YH, Chiu JH, Tung HH, Tsou MT, Lui WY, Wu CW. Preconditioning somatothermal stimulation on right seventh intercostal nerve territory increases hepatic heat shock protein 70 and protects the liver from ischemia-reperfusion injury in rats. J Surg Res 2001; 99: 328-334
11 Jäättelä M, Wissing D, Kokholm K, Kallunki T, Egeblad M. Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO J 1998; 17: 6124-6134
12 Jaattela M. Heat shock proteins as cellular lifeguards. Ann Med 1999; 31: 261-271
13 Mosser DD, Caron AW, Bourget L, Denis-Larose C, Massie B. Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol Cell Biol 1997; 17: 5317-5327
14 Schoeniger LO, Andreoni KA, Ott GR, Risby TH, Bulkley GB, Udelsman R, Burdick JF, Buchman TG. Induction of heat-shock gene expression in postischemic pig liver depends on superoxide generation. Gastroenterology 1994; 106: 177-184
15 de Vera ME, Kim YM, Wong HR, Wang Q, Billiar TR, Geller DA. Heat shock response inhibits cytokine-inducible nitricHeat shock response inhibits cytokine-inducible nitric oxide synthase expression in rat hepatocytes. Hepatology 1996; 24: 1238-1245
16 Mosser DD, Caron AW, Bourget L, Meriin AB, Sherman MY, Morimoto RI, Massie B. The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol Cell Biol 2000; 20: 7146-7159
17 Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Høyer-Hansen M, Weber E, Multhoff G, Rohde M, Jäättelä M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med 2004; 200: 425-435
18 Musch MW, Sugi K, Straus D, Chang EB. Heat-shock proteinHeat-shock protein 72 protects against oxidant-induced injury of barrier function of human colonic epithelial Caco2/bbe cells. Gastroenterology 1999; 117: 115-122
19 Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2000; 2: 469-475
20 Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol 2000; 2: 476-483
21 Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, Zamzami N, Mak T, Jäättelä M, Penninger JM, Garrido C, Kroemer G. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol 2001; 3: 839-843
22 Sato M, Cordis GA, Maulik N, Das DK. SAPKs regulation of ischemic preconditioning. Am J Physiol Heart Circ Physiol 2000; 279: H901-H907
23 Ishida T, Taketoh J, Nakatsune E, Kan-o S, Naito E, Takeda S, Mutoh J, Ishii Y, and Yamada H. Curcumin anticipates the suppressed body weight gain with 2,3,7,8-tetrachlorodibenzo-
1960 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com
COMMENTS

p-dioxin in mice. J Health Sci 2004; 50: 474-482 24 Sood A, Mathew R, Trachtman H. Cytoprotective effect of
curcumin in human proximal tubule epithelial cells exposed to shiga toxin. Biochem Biophys Res Commun 2001; 283: 36-41
25 Kato K, Ito H, Kamei K, Iwamoto I. Stimulation of the stress-induced expression of stress proteins by curcumin in cultured cells and in rat tissues in vivo. Cell Stress Chaperones 1998; 3: 152-160
26 Rauen U, Viebahn R, Lauchart W, de Groot H. The potential role of reactive oxygen species in liver ischemia/reperfusion injury following liver surgery. Hepatogastroenterology 1994; 41: 333-336
27 Jaeschke H, Mitchell JR. Use of isolated perfused organs in hypoxia and ischemia/reperfusion oxidant stress. Methods Enzymol 1990; 186: 752-759
28 Yuan GJ , Ma JC, Gong ZJ, Sun XM, Zheng SH, Li X. Modulation of liver oxidant-antioxidant system by ischemic preconditioning during ischemia/reperfusion injury in rats. World J Gastroenterol 2005; 11: 1825-1828
29 Ghoneim AI, Abdel-Naim AB, Khalifa AE, El-Denshary ES. Protective effects of curcumin against ischaemia/reperfusion insult in rat forebrain. Pharmacol Res 2002; 46: 273-279
30 Madan B, Ghosh B. Diferuloylmethane inhibits neutrophil infiltration and improves survival of mice in high-dose endotoxin shock. Shock 2003; 19: 91-96
31 Taylor BS, Alarcon LH, Billiar TR. Inducible nitric oxide synthase in the liver: regulation and function. Biochemistry (Mosc) 1998; 63: 766-781
32 Taylor BS, Geller DA. Molecular regulation of the human inducible nitric oxide synthase (iNOS) gene. Shock 2000; 13:
413-42433 Billiar TR, Curran RD, Harbrecht BG, Stuehr DJ, Demetris
AJ, Simmons RL. Modulation of nitrogen oxide synthesis in vivo: NG-monomethyl-L-arginine inhibits endotoxin-induced nitrate/nitrate biosynthesis while promoting hepatic damage. J Leukoc Biol 1990; 48: 565-569
34 Harbrecht BG, Wu B, Watkins SC, Marshall HP, Peitzman AB, Billiar TR. Inhibition of nitric oxide synthase duringInhibition of nitric oxide synthase during hemorrhagic shock increases hepatic injury. Shock 1995; 4: 332-337
35 Li J, Billiar TR. Nitric Oxide. IV. Determinants of nitric oxide protection and toxicity in liver. Am J Physiol 1999; 276: G1069-G1073
36 Hecker M , Nematollahi H, Hey C, Busse R, Racké K. Inhibition of arginase by NG-hydroxy-L-arginine in alveolar macrophages: implications for the utilization of L-arginine for nitric oxide synthesis. FEBS Lett 1995; 359: 251-254
37 Chan MM, Huang HI, Fenton MR, Fong D. In vivo inhibition of nitric oxide synthase gene expression by curcumin, a cancer preventive natural product with anti-inflammatory properties. Biochem Pharmacol 1998; 55: 1955-1962
38 Brouet I, Ohshima H. Curcumin, an anti-tumour promoter and anti-inflammatory agent, inhibits induction of nitric oxide synthase in activated macrophages. Biochem Biophys Res Commun 1995; 206: 533-540
39 Pan MH, Lin-Shiau SY, Lin JK. Comparative studies on the suppression of nitric oxide synthase by curcumin and its hydrogenated metabolites through down-regulation of IkappaB kinase and NFkappaB activation in macrophages. Biochem Pharmacol 2000; 60: 1665-1676
S- Editor Liu Y L- Editor Wang XL E- Editor Ma WH
Shen SQ et al. Curcumin against liver warm I/R injury 1961
www.wjgnet.com

PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1962-1965www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
Lactobacillus plantarum inhibits epithelial barrier dysfunction and interleukin-8 secretion induced by tumor necrosis factor-a
Jae Sung Ko, Hye Ran Yang, Ju Young Chang, Jeong Kee Seo
www.wjgnet.com
RAPID COMMUNICATION
Jae Sung Ko, Hye Ran Yang, Ju Young Chang, Jeong Kee Seo, Department of Pediatrics, Seoul National University College of Medicine, Seoul 110-744, KoreaSupported by grant No. 0520050040 from the Seoul National University Hospital Research Fund and by KT&G Reserach FundCorrespondence to: Jeong Kee Seo, Department of Pediatrics, Seoul National University Children’s Hospital, 28 Yongon-dong, Chongro-gu, Seoul 110-744, Korea. [email protected]: +82-2-20723627 Fax: +82-2-7433455Received: 2007-02-05 Accepted: 2007-03-19
AbstractAIM: To determine whether Lactobacillus plantarum can modify the deleterious effects of tumor necrosis factor-a (TNF-a) on intestinal epithelial cells.
METHODS: Caco-2 cells were incubated with TNF-a alone or in the presence of L. plantarum. Transepithelial electrical resistance was used to measure epithelial barrier function. Interleukin 8 (IL-8) secretion by intestinal epithelial cells was measured using an ELISA. Cellular lysate proteins were immunoblotted using the anti-extracellular regulated kinase (ERK), anti-phospho-ERK and anti-IκB-a.
RESULTS: A TNF-a-induced decrease in transepithelial electrical resistance was inhibited by L. plantarum. TNF-a-induced IL-8 secretion was reduced by L. plantarum. L. plantarum inhibited the activation of ERK and the degradation of IκB-a in TNF-a-treated Caco-2 cells.
CONCLUSION: Induction of epithelial barrier dysfunction and IL-8 secretion by TNF-a is inhibited by L. plantarum. Probiotics may preserve epithelial barrier function and inhibit the inflammatory response by altering the signal transduction pathway.
© 2007 The WJG Press. All rights reserved.
Key words: Lactobacillus plantarum; Tumor necrosis factor-a; Epithelial barrier; Interleukin-8; ERK; IκB-a
Ko JS, Yang HR, Chang JY, Seo JK. Lactobacillus plantarum inhibits epithelial barrier dysfunction and interleukin-8 secretion induced by tumor necrosis factor-a. World J Gastroenterol 2007; 13(13): 1962-1965
http://www.wjgnet.com/1007-9327/13/1962.asp
INTRODUCTIONProbiotics are defined as living microorganisms that exert beneficial effects on human health[1]. They are effective in shortening the duration of infectious diarrhea in children, and preventing antibiotics-associated diarrhea[2,3]. Probiotics have been shown to prevent a relapse of postoperative pouchitis in ulcerative colitis[4].
Tumor necrosis factor-a (TNF-a) is a proinflamma-tory cytokine and plays a central role in intestinal inflammation in Crohn disease. TNF-a levels in serum, stool and intestinal tissues are elevated in patients with Crohn’s disease[5,6]. In Crohn’s disease, the elevation in epithelial permeability of the ileal mucosa may be mediated by TNF-a [7]. Treatment with anti-TNF-a antibody is effective in cases of intractable Crohn’s disease[8].
As disturbance of the intestinal microflora plays an impor tant role in the pathogenesis of murine experimental colitis and human inflammatory bowel disease [3], probiotics have been used to modify the bacterial f lora of the gut. Lactobacil lus plantarum is isolated from Kimchi, a traditional Korean food made from fermented vegetables [9]. L. plantarum attenuates intestinal inflammation in the interleukin (IL) 10 gene-deficient mouse model, which spontaneously develops enterocolitis[10].
The mechanisms of action of probiotics include improvement of ep i the l i a l bar r ier funct ion and immunoregulatory effects[11]. Each probiotic species may have an individual mechanism of action. The combination probiotic, VSL3 contains L. plantarum and enhances human intestinal epithelial barrier function[12]. Intestinal epithelial cells release potent neutrophil attractant chemokines such as IL-8 when stimulated by TNF-a. Secretion of IL-8 by epithelial cells has been suggested to be important in the pathogenesis of inflammatory bowel diseases, because IL-8 induces migration of inflammatory cells into the mucosa. Some lactobacilli inhibit the induction of IL-8 production by TNF-a in human intestinal epithelial cells[13-15]. TNF-a-stimulated IL-8 secretion by intestinal epithelial cells is mediated by extracellular signal-regulated kinase (ERK) and nuclear factor κB (NF-κB)[16].
The aim of this study was to determine whether L. plantarum reverses the deleterious effects of TNF-a on intestinal epithelial cells. We performed an in vitro study in which Caco-2 cells were treated with TNF-a alone or with TNF-a plus L. plantarum. We investigated the effect of

Ko JS et al . Effect of L. plantarum on epithelial barrier 1963
www.wjgnet.com
L. plantarum on TNF-a-induced alteration of epithelial barrier function, IL-8 production, and ERK/NF-κB pathway dynamics.
MATERIALS AND METHODSCell linesCaco-2 cells, an established cell line model for mature differentiated enterocytes, were obtained from the American Type Culture Collection (ATCC). Cell lines were cultured in 25 mmol/L glucose-Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 100 U/mL penicillin, 100 µg/mL streptomycin, 1% nonessential amino acids, and 4 mmol/L glutamine. Cultures were maintained at 37 in an incubator containing an atmosphere of 5% CO2. Cells were used within 14 d of seeding or within five days of confluency. The Caco-2 cell culture medium was replaced with antibiotic-free culture medium 24 h before experiments.
ProbioticsL. plantarum (ATCC 8014) was incubated in Lactobacillus MRS broth at 37 for 24 h, then diluted in MRS broth to a density of 0.5 absorbance units at a wavelength of 600 nm. Then, 1 × 107 colony-forming units of L. plantarum per mL were added at a multiplicity of 10:1 to the Caco-2 cells. Untreated cells were used as controls in all experiments.
Electrical resistance measurementsCaco-2 cells were grown as polarized monolayers on 6.5 mm transwell plates (0.4 µm pores; Corning Incorporated, Acton, MA, USA). Caco-2 monolayers with epithelial res istance g reater than 500 Ωcm 2 were used, and L. p lan tarum was added apica l ly to the polar ized monolayers. TNF-a (10 ng/mL) was simultaneously added to the basolateral side of the cell monolayers. Electrical resistance across the monolayers was measured at various times using an epithelial volt-ohm meter (World Precision Instruments, Sarasota, FL, USA). Measurements were expressed in Ωcm2 after subtracting mean values for resistance obtained from cell-free inserts.
ELISA for IL-8 measurementTNF-a (10 ng/mL) and L. p lantarum were added simultaneously to Caco-2 cells and incubated for 5 h. Culture medium was collected and centrifuged for 10 min to pellet residual bacteria. The supernatant was collected for determination of IL-8 concentration using an ELISA (Pierce, Rockford, IL, USA). Cytokine concentrations were determined using 96-well plates as described by the manufacturer.
Western blottingTNF-a (10 ng/mL) and L. p lantarum were added simultaneously to Caco-2 cells. The treated and untreated cells were washed with PBS and scraped into cell lysis buffer (20 mmol/L HEPES, 0.1% SDS, 1% Triton X-100, phosphatase inhibitor and protease inhibitor cocktail). Thirty minutes after treatment, the lysate was centrifuged at 15 000 r/min for 15 min at 4. The protein content
of the supernatant was determined using Bio-Rad DC reagents (Bio-Rad, Hercules, CA, USA). For western blotting, equal amounts of cellular lysate protein were mixed with Laemmli sample buffer and separated by SDS-PAGE. Separated proteins were transferred to PVDF membranes, which were blocked and then immunoblotted with anti-phospho-ERK, anti-ERK and anti-IκB-a (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The blot was then developed using horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence.
Statistical analysisAll data are expressed as means ± SD. Data comparisons were made with Student’s t test. Differences were considered significant at P < 0.05.
RESULTSTransepithelial electrical resistanceTo determine the effect of L. plantarum on TNF-a-induced epithelial barrier dysfunction, Caco-2 cells were basolaterally incubated with TNF-a alone or with TNF-a plus L. plantarum, which was administered apically. Transepithelial electrical resistance was monitored for 36 h. The monolayer resistance of TNF-a treated cells did not change until 12 h had elapsed. TNF-a caused a decline in transepithelial resistance 24 h after treatment. L. plantarum inhibited TNF-a-induced decrease in transepithelial electrical resistance at 24 h and 36 h after treatment (P < 0.05) (Figure 1). The epithelial barrier function of TNF-a-stimulated Caco-2 cells was thus preserved by L. plantarum.
IL-8 inductionThe secre t ion of IL-8 into cu l ture medium was measured to determine the effect of L. plantarum on the inflammatory response of Caco-2 cells to TNF-a. IL-8 concentrations in media of Caco-2 cells cultured with L. plantarum were not significantly different from those of the controls. When TNF-a (10 ng/mL) was incubated with the cells for 5 h, IL-8 secretion was increased to
900
800
700
600
500
4000 12 24 36
Resi
stan
ce (
ohm
cm
2 )
TNF + LP ControlTNF
t /h
Figure 1 Effect of L. plantarum on transepithelial resistance. TNF-a decreased Caco-2 monolayer resistance. L. plantarum reversed TNF-a-induced decreases in transepithelial resistance. aP < 0.05, compared with TNF-a. TER: transepithelial electrical resistance; LP: L. plantarum; TNF, TNF-a.
a a
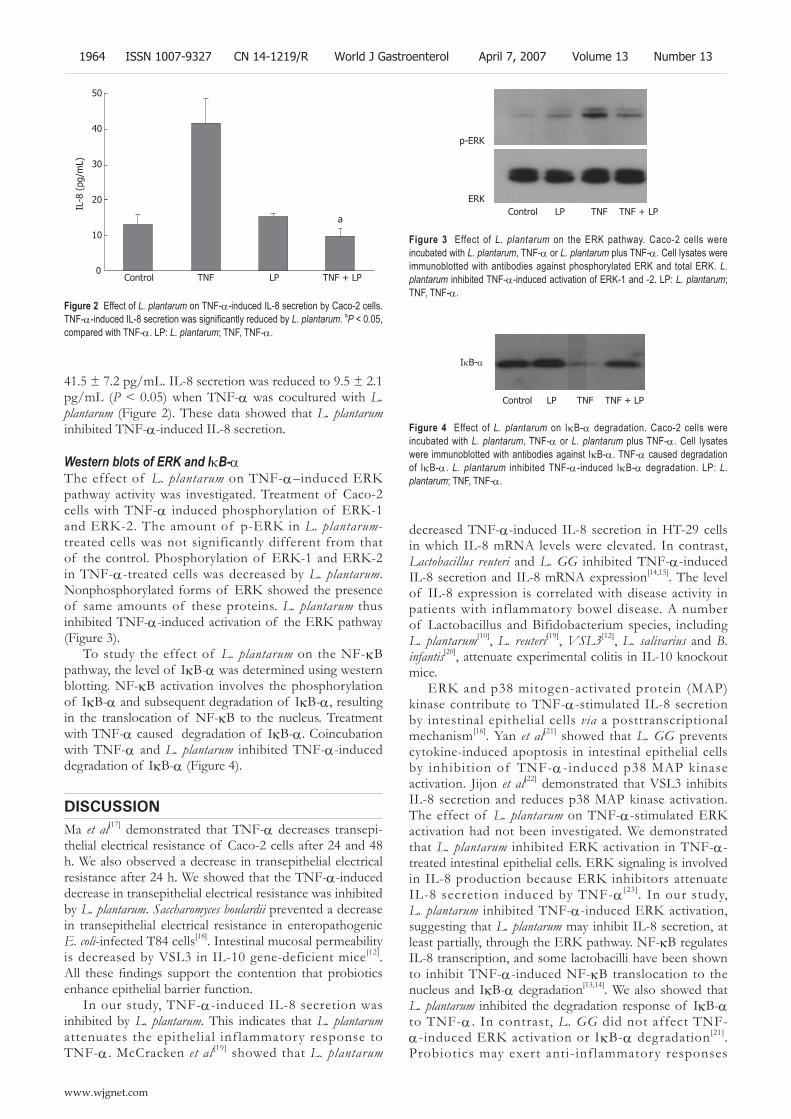
a
41.5 ± 7.2 pg/mL. IL-8 secretion was reduced to 9.5 ± 2.1 pg/mL (P < 0.05) when TNF-a was cocultured with L. plantarum (Figure 2). These data showed that L. plantarum inhibited TNF-a-induced IL-8 secretion.
Western blots of ERK and IκB-aThe effect of L. plantarum on TNF-a–induced ERK pathway activity was investigated. Treatment of Caco-2 cells with TNF-a induced phosphorylation of ERK-1 and ERK-2. The amount of p-ERK in L. plantarum-treated cells was not significantly different from that of the control. Phosphorylation of ERK-1 and ERK-2 in TNF-a-treated cells was decreased by L. plantarum. Nonphosphorylated forms of ERK showed the presence of same amounts of these proteins. L. plantarum thus inhibited TNF-a-induced activation of the ERK pathway (Figure 3).
To study the effect of L. plantarum on the NF-κB pathway, the level of IκB-a was determined using western blotting. NF-κB activation involves the phosphorylation of IκB-a and subsequent degradation of IκB-a, resulting in the translocation of NF-κB to the nucleus. Treatment with TNF-a caused degradation of IκB-a. Coincubation with TNF-a and L. plantarum inhibited TNF-a-induced degradation of IκB-a (Figure 4).
DISCUSSIONMa et al[17] demonstrated that TNF-a decreases transepi-thelial electrical resistance of Caco-2 cells after 24 and 48 h. We also observed a decrease in transepithelial electrical resistance after 24 h. We showed that the TNF-a-induced decrease in transepithelial electrical resistance was inhibited by L. plantarum. Saccharomyces boulardii prevented a decrease in transepithelial electrical resistance in enteropathogenic E. coli-infected T84 cells[18]. Intestinal mucosal permeability is decreased by VSL3 in IL-10 gene-deficient mice[12]. All these findings support the contention that probiotics enhance epithelial barrier function.
In our study, TNF-a-induced IL-8 secretion was inhibited by L. plantarum. This indicates that L. plantarum attenuates the epithelial inf lammatory response to TNF-a. McCracken et al [19] showed that L. plantarum
decreased TNF-a-induced IL-8 secretion in HT-29 cells in which IL-8 mRNA levels were elevated. In contrast, Lactobacillus reuteri and L. GG inhibited TNF-a-induced IL-8 secretion and IL-8 mRNA expression[14,15]. The level of IL-8 expression is correlated with disease activity in patients with inflammatory bowel disease. A number of Lactobacillus and Bifidobacterium species, including L. plantarum[10], L. reuteri[19], VSL3[12], L. salivarius and B. infantis[20], attenuate experimental colitis in IL-10 knockout mice.
ERK and p38 mitogen-activated protein (MAP) kinase contribute to TNF-a-stimulated IL-8 secretion by intestinal epithelial cells via a posttranscriptional mechanism[16]. Yan et al[21] showed that L. GG prevents cytokine-induced apoptosis in intestinal epithelial cells by inhibition of TNF-a-induced p38 MAP kinase activation. Jijon et al[22] demonstrated that VSL3 inhibits IL-8 secretion and reduces p38 MAP kinase activation. The effect of L. plantarum on TNF-a-stimulated ERK activation had not been investigated. We demonstrated that L. plantarum inhibited ERK activation in TNF-a-treated intestinal epithelial cells. ERK signaling is involved in IL-8 production because ERK inhibitors attenuate IL-8 secretion induced by TNF-a [23]. In our study, L. plantarum inhibited TNF-a-induced ERK activation, suggesting that L. plantarum may inhibit IL-8 secretion, at least partially, through the ERK pathway. NF-κB regulates IL-8 transcription, and some lactobacilli have been shown to inhibit TNF-a-induced NF-κB translocation to the nucleus and IκB-a degradation[13,14]. We also showed that L. plantarum inhibited the degradation response of IκB-a to TNF-a . In contrast, L. GG did not affect TNF-a-induced ERK activation or IκB-a degradation [21]. Probiotics may exert anti-inf lammatory responses
50
40
30
20
10
0Control TNF LP TNF + LP
IL-8
(pg
/mL)
Figure 2 Effect of L. plantarum on TNF-a-induced IL-8 secretion by Caco-2 cells. TNF-a-induced IL-8 secretion was significantly reduced by L. plantarum. aP < 0.05, compared with TNF-a. LP: L. plantarum; TNF, TNF-a.
p-ERK
ERKControl LP TNF TNF + LP
Figure 3 Effect of L. plantarum on the ERK pathway. Caco-2 cells were incubated with L. plantarum, TNF-a or L. plantarum plus TNF-a. Cell lysates were immunoblotted with antibodies against phosphorylated ERK and total ERK. L. plantarum inhibited TNF-a-induced activation of ERK-1 and -2. LP: L. plantarum; TNF, TNF-a.
IκB-a
Control LP TNF TNF + LP
Figure 4 Effect of L. plantarum on IκB-a degradation. Caco-2 cells were incubated with L. plantarum, TNF-a or L. plantarum plus TNF-a. Cell lysates were immunoblotted with antibodies against IκB-a. TNF-a caused degradation of IκB-a. L. plantarum inhibited TNF-a-induced IκB-a degradation. LP: L. plantarum; TNF, TNF-a.
1964 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

by modifying the signal transduction pathway. The mechanisms involved may depend on the species of probiotics.
Epithelial barrier functions are modulated by the NF-κB and MAP kinase pathways. A TNF-a-induced increase in intestinal tight junction permeability was shown to be mediated by NF-κB activation[17]. The increase in transepithelial resistance induced by VSL3 is mediated in part via the ERK pathway[24]. The effect of L. plantarum on monolayer resistance appears to be mediated by NF-κB and the ERK pathway. Although in vitro models are useful for evaluating mechanisms by which probiotics exert beneficial effects and provide a rationale for the therapeutic use of probiotics, the beneficial health effects of probiotics should also be determined by double- blinded placebo-controlled trials.
In summary, L. plantarum inhibits epithelial barrier dysfunction, IL-8 secretion, ERK activation, and IκB-a degradation in TNF-a-stimulated Caco-2 cells. Our findings suggest that probiotics may preserve epithelial barrier function and inhibit the inflammatory response by affecting the signal transduction pathway in human intestinal epithelium.
REFERENCES1 Sullivan A, Nord CE. Probiotics and gastrointestinal diseases.
J Intern Med 2005; 257: 78-92 2 Guandalini S, Pensabene L, Zikri MA, Dias JA, Casali
LG, Hoekstra H, Kolacek S, Massar K, Micetic-Turk D, Papadopoulou A, de Sousa JS, Sandhu B, Szajewska H, Weizman Z. Lactobacillus GG administered in oral rehydration solution to children with acute diarrhea: a multicenter European trial. J Pediatr Gastroenterol Nutr 2000; 30: 54-60
3 Fedorak RN , Madsen KL. Probiotics and prebiotics in gastrointestinal disorders. Curr Opin Gastroenterol 2004; 20: 146-155
4 Gionchetti P, Rizzello F, Venturi A, Brigidi P, Matteuzzi D, Bazzocchi G, Poggioli G, Miglioli M, Campieri M. Oral bacteriotherapy as maintenance treatment in patients with chronic pouchitis: a double-blind, placebo-controlled trial. Gastroenterology 2000; 119: 305-309
5 Braegger CP, Nicholls S, Murch SH, Stephens S, MacDonald TT. Tumour necrosis factor alpha in stool as a marker of intestinal inflammation. Lancet 1992; 339: 89-91
6 Van Deventer SJ. Tumour necrosis factor and Crohn's disease. Gut 1997; 40: 443-448
7 Söderholm JD, Streutker C, Yang PC, Paterson C, Singh PK, McKay DM, Sherman PM, Croitoru K, Perdue MH. Increased epithelial uptake of protein antigens in the ileum of Crohn's disease mediated by tumour necrosis factor alpha. Gut 2004; 53: 1817-1824
8 Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, Rachmilewitz D, Wolf DC, Olson A, Bao W, Rutgeerts P. Maintenance infliximab for Crohn's disease: the ACCENT I randomised trial. Lancet 2002; 359: 1541-1549
9 Rhee CH, Park HD. Three glycoproteins with antimutagenic activity identified in Lactobacillus plantarum KLAB21. Appl Environ Microbiol 2001; 67: 3445-3449
10 Schultz M, Veltkamp C, Dieleman LA, Grenther WB, Wyrick PB, Tonkonogy SL, Sartor RB. Lactobacillus plantarum 299V in the treatment and prevention of spontaneous colitis in interleukin-10-deficient mice. Inflamm Bowel Dis 2002; 8: 71-80
11 Mack DR, Lebel S. Role of probiotics in the modulation of intestinal infections and inflammation. Curr Opin Gastroenterol 2004; 20: 22-26
12 Madsen K, Cornish A, Soper P, McKaigney C, Jijon H, Yachimec C, Doyle J, Jewell L, De Simone C. Probiotic bacteria enhance murine and human intestinal epithelial barrier function. Gastroenterology 2001; 121: 580-591
13 Bai AP, Ouyang Q, Zhang W, Wang CH, Li SF. Probiotics inhibit TNF-alpha-induced interleukin-8 secretion of HT29 cells. World J Gastroenterol 2004; 10: 455-457
14 Ma D , Forsythe P, Bienenstock J . Live Lactobacil lus rhamnosus [corrected] is essential for the inhibitory effect on tumor necrosis factor alpha-induced interleukin-8 expression. Infect Immun 2004; 72: 5308-5314
15 Zhang L, Li N, Caicedo R, Neu J. Alive and dead Lactobacillus rhamnosus GG decrease tumor necrosis factor-alpha-induced interleukin-8 production in Caco-2 cells. J Nutr 2005; 135: 1752-1756
16 Jijon HB, Panenka WJ, Madsen KL, Parsons HG. MAP kinases contribute to IL-8 secretion by intestinal epithelial cells via a posttranscriptional mechanism. Am J Physiol Cell Physiol 2002; 283: C31-C41
17 Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol 2004; 286: G367-G376
18 Czerucka D , Dahan S, Mograbi B, Rossi B, Rampal P. Saccharomyces boulardii preserves the barrier function and modulates the signal transduction pathway induced in enteropathogenic Escherichia coli-infected T84 cells. Infect Immun 2000; 68: 5998-6004
19 McCracken VJ, Chun T, Baldeón ME, Ahrné S, Molin G, Mackie RI, Gaskins HR. TNF-alpha sensitizes HT-29 colonic epithelial cells to intestinal lactobacilli. Exp Biol Med (Maywood) 2002; 227: 665-670
20 McCarthy J, O'Mahony L, O'Callaghan L, Sheil B, Vaughan EE, Fitzsimons N, Fitzgibbon J, O'Sullivan GC, Kiely B, Collins JK, Shanahan F. Double blind, placebo controlled trial of two probiotic strains in interleukin 10 knockout mice and mechanistic link with cytokine balance. Gut 2003; 52: 975-980
21 Yan F, Polk DB. Probiotic bacterium prevents cytokine-induced apoptosis in intestinal epithelial cells. J Biol Chem 2002; 277: 50959-50965
22 Jijon H, Backer J, Diaz H, Yeung H, Thiel D, McKaigney C, De Simone C, Madsen K. DNA from probiotic bacteria modulates murine and human epithelial and immune function. Gastroenterology 2004; 126: 1358-1373
23 Yu Y, Zeng H, Lyons S, Carlson A, Merlin D, Neish AS, Gewirtz AT. TLR5-mediated activation of p38 MAPK regulates epithelial IL-8 expression via posttranscriptional mechanism. Am J Physiol Gastrointest Liver Physiol 2003; 285: G282-G290
24 Otte JM, Podolsky DK. Functional modulation of enterocytes by gram-positive and gram-negative microorganisms. Am J Physiol Gastrointest Liver Physiol 2004; 286: G613-G626
S- Editor Liu Y L- Editor Alplini GD E- Editor Chin GJ
Ko JS et al . Effect of L. plantarum on epithelial barrier 1965
www.wjgnet.com

Fu CY, Yeh CN, Hsu JT, Jan YY, Hwang TL. Timing of mortality in severe acute pancreatitis: Experience from 643 patients. World J Gastroenterol 2007; 13(13): 1966-1969
http://www.wjgnet.com/1007-9327/13/1966.asp
INTRODUCTIONAcute pancreatitis is a common disorder ranging in severity from mild disease to multiple organ failure (MOF) and sepsis. Severe acute pancreatitis (SAP) has a 20% mortality rate[1,2]. SAP reveals its progresses into two phases. The early phase refers to the first 7 to 14 d after onset of acute pancreatitis complicated with systemic inflammatory response syndrome (SIRS) and MOF due to release of large amount of cytokine[3,4]. In the late phase (14 to 28 d after onset of acute pancreatitis), the disease may be complicated by infection of pancreatic necrosis and secondary MOF[5]. Although two peak mortality rates are observed in the course of SAP[6], death occurs primarily in the early or late phase remains unclear. The causes of death in early and late phase of SAP varied in several reports[3,7-14]. This study analyzes the time of death in a large series of patients with SAP and compares clinical features of early and late mortality to fully clarify this disease entity.
MATERIALS AND METHODSSubjectsMedical charts and computerized records for all patients with acute pancreatitis treated at the Division of General Surgery of Chang Gung Memorial Hospital from July 1996 to June 2005 were retrospectively reviewed. During this period, 3250 episodes of acute pancreatitis were recorded in 2248 patients (1431 males and 817 females; median age, 55.6 years; range, 18-97 years). Acute pancreatitis was diagnosed if clinical features and imaging studies revealed elevated serum amylase and lipase (at least three times higher than the normal level). The definition of severe acute pancreatitis (SAP) was based on the Atlanta classification[5].
MethodsThe following general characteristics were recorded: demographic data, etiological factors, early prognostic signs (Ranson’s and Acute Physiology and Chronic Health
RAPID COMMUNICATION
Timing of mortality in severe acute pancreatitis: Experience from 643 patients
Chih-Yuan Fu, Chun-Nan Yeh, Jun-Te Hsu, Yi-Yin Jan, Tsann-Long Hwang
www.wjgnet.com
Chih-Yuan Fu, Chun-Nan Yeh, Jun-Te Hsu, Yi-Yin Jan, Tsann-Long Hwang, Department of General Surgery, Chang Gung Memorial Hospital, Chang Gung University, 5 Fu-shin Street, Kweishan, Taoyuan, Taiwan, ChinaCorrespondence to: Dr. Tsann-Long Huang, Department of General Surgery, Chang Gung Memorial Hospital, Chang Gung University, 5 Fu-shin Street, Kweishan, Taoyuan, Taiwan, China. [email protected]: +886-3-3281200-3219 Fax: +886-3-3285818Received: 2007-01-03 Accepted: 2007-01-25
AbstractAIM: To determine the timing of mortality after onset of severe acute pancreatitis (SAP) and the course of the disease in a large series of patients.
METHODS: From July 1996 to June 2005, all patients diagnosed with acute pancreatitis at Chang Gung Memorial Hospital, Taipei, Taiwan were retrospectively studied. Three thousand two hundred and fifty episodes of acute pancreatitis were recorded in 2248 patients (1431 males and 817 females; median age, 55.6 years; range, 18-97 years). Mortality was divided into two groups: early death (≤ 14 d after admission), and late death (> 14 d after admission). The clinical features of patients in these two groups were compared.
RESULTS: Although the overall mortality rate of acute pancreatitis was 3.8% (123/3250), mortality rate of SAP was as high as 16.3% (105/643). Of those 105 SAP mortalities, 44 (41.9%) deaths occurred within the first 14 d after admission and 61 (58.1%) occurred after14 d. Incidence of early death did not significantly differ from that of late death. The co-morbidities did not contribute to the timing of death. Early deaths mainly resulted from multiple organ failure. Late deaths were mainly caused by secondary complication of infected necrosis. Intra-abdominal bleeding significantly caused higher mortality in late death.
CONCLUSION: Approximately half (42%) of SAP deaths occur within 14 d and most were due to multiple organ failure. The late deaths of SAP were mostly due to infected necrosis.
© 2007 The WJG Press. All rights reserved.
Key words: Severe acute pancreatitis; Mortality; Multiple organ failure
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1966-1969www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.

Evaluation Ⅱ score)[6] at 48 h after admission, body mass index (BMI) and presence or lack of necrosis. Necrosis was deemed to be present upon observation of aseptic or infected necrosis. The aforementioned factors were compared between early and late deaths. Instances of comorbidity and cause of death were recorded. Pancreatic necrosis was defined by the character of abnormalities on contrast-enhanced computerized tomography scan[3,5] or the findings of surgical operation. Infection was considered present when necrotic tissue obtained from percutaneous fine-needle aspiration or surgical specimen was positive for bacteria culture.
Early deaths were defined as deaths occurring within 14 d after admission, and late deaths were defined as deaths occurring more than 14 d after admission. Comorbidity was defined as presentation of a pre-existing disease before SAP which became an active problem. Chronic obstructive pulmonary disease, cardiac insufficiency (New York Heart Association; NYHA class Ⅲ or Ⅳ), renal insufficiency, liver cirrhosis, diabetes mellitus, and malignant disease diagnosed within three years before the current episode (immunological disease or chronic immunosuppressive medication) were included.
All patients received treatment from the same physicians throughout the study. However, treatments varied in duration. In cases of biliary pancreatitis, endoscopic retrograde cholangiopancreatography (ERCP) with sphincterotomy was performed. Several patients received intravenous continuous infusion of antiproteases (gabexate mesilate). Antibiotics were routinely administered for infected pancreatitis. Surgery was performed upon infection of necrotic tissue. However, in rare cases, hemorrhage, pancreatic ascites, perforation, pseudocyst rupture or its rapid increase in size were indications of surgery.
Fu CY et al. Timing of mortality in severe acute pancreatitis 1967
www.wjgnet.com
Statistical analysisAll data were presented as percentage of patients or mean ± SD. Numerical data were compared using independent two-sample t tests. Nominal data were compared using χ2 test, Mann-Whitney U-test, and Mantel-Haenszel linear-by-linear association when appropriate. All statistical analyses were performed using the SPSS computer software package (Version 11.0, Chicago, IL, USA). P < 0.05 was considered statistically significant.
RESULTSThe present study included 3250 episodes of acute pancreatitis observed in 2248 patients at our facility. Presence of SAP was observed in 643 (19.8%) patients based on characteristics of pancreatic necrosis. The other 2607 (80.2%) episodes were mild forms of acute pancreatitis. Although the overall mortality rate of acute pancreatitis was 3.8% (123/3250), mortality rate of SAP was as high as 16.3% (105/643) All deaths involved patients experiencing their first episode of pancreatitis. With equal distribution, of those 105 SAP mortalities, 44 (41.9%) patients died within 14 d of admission, and 61 (58.1%) died after 14 d of admission. The two groups did not differ in gender, age, Ranson’s and APACHE Ⅱ score, BMI, or co-morbidity. However, early death significantly differed from late death in etiology (Table 1). Higher percentage of biliary pancreatitis patients developed more late deaths than early deaths. Forty-two of the 44 (95.5%) patients with early mortality had necrotizing pancreatitis. Similarly, 58 of the 61 (96.2%) patients with late mortality presented necrotizing pancreatitis. A significantly higher (P < 0.0001) number of patients died from infected necrosis in the late death group (Table 2). On the other hand, sterile necrosis occurred more frequently in early deaths than late deaths (P = 0.008). Forty of the 44 (90.9%) patients suffered early deaths related to MOF (Table 2). Late deaths occurred post-operatively in 37 patients with infected necrosis (23 also presented MOF). A higher incidence of
Table 1 Characteristics of severe acute pancreatitis patients with early (≤ 14 d after admission) and late death (>14 d after admission)
Early death Late death P value (n = 44) (n = 61)
Age, yr (range) 54.7 (34-93) 56.3 (30-84) 0.904Gender 0.147Females 32 (72.7%) 36 (59.0%)Males 12 (27.3%) 25 (41.0%)EtiologyBiliary 13 (29.5%) 32 (52.5%) 0.019Alcoholic 13 (29.5%) 11 (18.0%) 0.166Idiopathic 12 (27.3%) 7 (27.9%) 0.946Other 6 (13.7%) 1 (1.6%)Ranson’s score 3.9 (1-9) 4.1 (1-8) 0.676APACHE-II score 22.4 (9-31) 24.5(11-32) 0.732BMI, (Kg/m2) 24.5 (18-39) 23.6 (19-48) 0.814Comorbidity 0.557No comorbidity 9 (20.5%) 9 (32.1%)1-2 organ systems 22 (50.0%) 22 (50.0%)3-4 organ systems 8 (18.2%) 17 (27.9%)> 4 organ systems 5 (11.3%) 4 (6.6%)
BMI: Body mass index.
Table 2 Pancreatic necrosis and causes of death in severe acute pancreatitis patients with early and late mortality n (%)
Pathology and cause of death
Early deaths Late deaths P value(n = 44) (n = 61)
PathologyEdematous 2 (4.5) 3 (4.9) 0.95Sterile necrosis 21 (42.7) 14 (23.0) 0.008Infected necrosis 6 (13.6) 31 (50.8) < 0.0001Necrosis without microbiological data
15 (34.2) 13 (21.3) 0.144
Cause of deathMOF 40 (90.9) 9 (14.8) < 0.0001Infected necrosis 0 (0.0) 14 (23.0) 0.001Infected necrosis + MOF 1 (2.3) 22 (36.1) < 0.0001Intra-abdominal bleeding 1 (2.3) 12 (19.7) 0.008Heart failure 2 (4.5) 2 (3.3) 0.738Cerebral stroke 0 (0.0) 2 (3.3) 0.225
MOF: Multi-organ failure.

hemorrhagic complication from intra-abdominal bleeding was noted in the late death group than in the early death group (P = 0.008). Clearly, MOF was more prevalent in the early death group, and death due to infection was more common in late deaths. In addition to the pancreatic pathologic complications, co-morbidities, such as stroke or heart failure, did not significantly differ between the two groups.
DISCUSSIONAccording to previous studies, the pattern of morbidity and mortality in SAP varied[8-10,12,14-19]. The primary causes of death in SAP remain controversial. A literature review reveals early mortality rates ranging from 0 to 80% (mean value, 53.9%, Table 3). In present study, the early mortality rate was 42%. The cause of the wide range in reported early mortality rates is unclear. Aggressive restoration of multiple organ failure (MOF) may be important to reduce early mortality rate. On the other hand, as shown in this study, the studied populations have consisted of groups of patients from a single department or ward rather than from the entire hospital population. This fact may explain the lower early mortality rate. In addition, time of admission may also be related to variations in SAP early mortality rates[12]. The other explanation is that the early surgical debridement was avoided in this study. It could prevent the frequent conversion of sterile necrosis to infected necrosis which resulted in increased early SAP mortality rate[19].
The present study revealed that approximately half of the deaths (41.9%) occurred within two weeks; the major cause was MOF. Extended pancreatic necrosis, especially the extent of intra-pancreatic parenchyma necrosis, leads to a high incidence of early organ failure[19]. In addition, if early organ failure occurs at the time of admission in SAP, the risk of ongoing and progressive organ failure remains extremely high[20]. According to recent reports, if organ failure worsens in the first week of admission, mortality rate increases significantly[21,22]. In our study, once the disease has been initiated by the pathogenetic mechanism, the course and outcome were not influenced by underlying etiological factors, demographic features, physical features or co-morbidity. Even some reports indicated the importance of co-morbid medical problems in SIRS and MOF in elderly SAP patients, in addition to the effect of infected necrosis[6,14]. The present study revealed no significant differences in age or pre-existing disease. Early prognostic factors, such as Ranson’s score and APACHE Ⅱ score, did not differentiate patient risk
of early mortality or late mortality in SAP. However, some reports suggest that obesity increases MOF rate and SAP mortality[14,23]. Similar to other studies[9,12,24], BMI values were not associated with differences in early or late death in SAP in the present study.
The present study demonstrated vital role of MOF occurrence in the outcome of SAP. Considering all SAP cases were fatal (105 patients), a high incidence of MOF was observed in this group. Death caused by MOF was observed in 72 (68.6%) patients. In particular, 24 of the 38 patients with infected necrosis suffered at least two organ failures directly related to the fatal outcome. According to previous reports[3-6,25-28], the standard treatment for infected pancreatic necrosis, including antibiotics and surgical resection or drainage procedure, is widely accepted. However, in the present study, the results of MOF management remained poor in SAP cases[6,25,27]. As a result, the goal of therapeutic planning was based on continuous resuscitation and restoration of failed organs. However, McKay et al [10] documented that even after intensive treatment, 40% of patients died 3 d after hospital admission.
This study demonstrated that MOF occurred more frequently in the early death group than in the late death group, being the major cause of early death in SAP. However, the incidence of infected necrosis was higher in the late death group than in the early death group. Other causes of death, including stroke or heart disease, were rare and statistically insignificant. Therefore, in the first two weeks of admission, the goals of treatment should be aggressive resuscitation, restoration of failed organ systems and inhibition of MOF progression. In the late phase, controlling infection and preventing infected necrosis is vital. The importance of co-morbidity control is not significant in elderly patients. SAP remains a serious medical problem. Effective control of early MOF and treatment of systemic complications associated with the infected necrosis require innovative strategies.
In conclusion, approximately half (42%) of SAP deaths occur within two weeks and most are due to multiple organ failure. The data revealed the significance of multiple organ failure in the outcome of SAP. The late deaths of SAP are mostly due to infected necrosis.
REFERENCES1 Banks PA. Infected necrosis: morbidity and therapeutic
consequences. Hepatogastroenterology 1991; 38: 116-1192 Büchler , Gloor B, Müller CA, Friess H, Seiler CA, Uhl W.
Acute necrotizing pancreatitis: treatment strategy according to the status of infection. Ann Surg 2000; 232: 619-626
3 Steinberg , Tenner S. Acute pancreatitis. N Engl J Med 1994; 330: 1198-1210
4 Toouli J, Brooke-Smith M, Bassi C, Carr-Locke D, Telford J, Freeny P, Imrie C, Tandon R. Guidelines for the management of acute pancreatitis. J Gastroenterol Hepatol 2002; 17 Suppl: S15-S39
5 Bradley EL. A clinically based classification system for acute pancreatitis. Summary of the International Symposium on Acute Pancreatitis, Atlanta, Ga, September 11 through 13, 1992. Arch Surg 1993; 128: 586-590
6 United Kingdom guidelines for the management of acute pancreatitis. British Society of Gastroenterology. Gut 1998; 42 Suppl 2: S1-S13
Table 3 The incidence of early death in severe acute pancreatitis in this study and in the literature
Total number Early death (%)
Renner et al, 1985[8] 405 60Wilson et al, 1988[15] 126 55.5Lankisch et al, 1996[17] 37 56.7Isenmann et al, 2001[19] 36 55.5Present study 105 42
1968 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

7 ann DV, Hershman MJ, Hittinger R, Glazer G. Multicentre audit of death from acute pancreatitis. Br J Surg 1994; 81: 890-893
8 Renner IG, Savage WT, Pantoja JL, Renner VJ. Death due toDeath due to acute pancreatitis. A retrospective analysis of 405 autopsy cases. Dig Dis Sci 1985; 30: 1005-1018
9 Talamini G, Bassi C, Falconi M, Sartori N, Frulloni L, Di Fran-cesco V, Vesentini S, Pederzoli P, Cavallini G. Risk of death from acute pancreatitis. Role of early, simple routine data.Role of early, simple routine data. Int J Pancreatol 1996; 19: 15-24
10 cKay CJ, Evans S, Sinclair M, Carter CR, Imrie CW. High early mortality rate from acute pancreatitis in Scotland, 1984-1995. Br J Surg 1999; 86: 1302-1305
11 Lowham A , Lavelle J , Leese T. Mortali ty from acute pancreatitis. Late septic deaths can be avoided but some early deaths still occur. Int J Pancreatol 1999; 25: 103-106
12 utinga , Rosenbluth A, Tenner SM, Odze RR, Sica GT, Banks PA. Does mortality occur early or late in acute pancreatitis? Int J Pancreatol 2000; 28: 91-95
13 Rau B, Uhl W, Buchler MW, Beger HG. Surgical treatment ofSurgical treatment of infected necrosis. World J Surg 1997; 21: 155-161
14 Gloor B, Müller CA, Worni M, Martignoni ME, Uhl W, Büchler MW. Late mortality in patients with severe acuteLate mortality in patients with severe acute pancreatitis. Br J Surg 2001; 88: 975-979
15 ilson C, Imrie CW, Carter DC. Fatal acute pancreatitis. Gut 1988; 29: 782-788
16 de Beaux AC, Palmer KR, Carter DC. Factors influencingFactors influencing morbidity and mortality in acute pancreatitis; an analysis of 279 cases. Gut 1995; 37: 121-126
17 Lankisch PG, Burchard-Reckert S, Petersen M, Lehnick D, Schirren CA, Köhler H, Stöckmann F, Peiper HJ, Creutzfeldt W. Morbidity and mortality in 602 patients with acute pancreatitis seen between the years 1980-1994. Z Gastroenterol 1996; 34: 371-377
18 Uhl , Büchler MW, Malfertheiner P, Beger HG, Adler G, Gaus W. A randomised, double blind, multicentre trial of octreotide in moderate to severe acute pancreatitis. Gut 1999; 45: 97-104
19 Isenmann R, Rau B, Beger HG. Early severe acute pancreatitis:Early severe acute pancreatitis: characteristics of a new subgroup. Pancreas 2001; 22: 274-278
20 Bradley EL. Indications for debridement of necrotizing pancreatitis. Pancreas 1996; 13: 219-223
21 Isenmann R, Rau B, Beger HG. Bacterial infection and extentBacterial infection and extent of necrosis are determinants of organ failure in patients with acute necrotizing pancreatitis. Br J Surg 1999; 86: 1020-1024
22 Lankisch PG, Pflichthofer D, Lehnick D. Acute pancreatitis: which patient is most at risk? Pancreas 1999; 19: 321-324
23 Funnell IC, Bornman PC, Weakley SP, Terblanche J, Marks IN. Obesity: an important prognostic factor in acute pancreatitis. Br J Surg 1993; 80: 484-486
24 Tenner S, Sica G, Hughes M, Noordhoek E, Feng S, Zinner M, Banks PA. Relationship of necrosis to organ failure in severe acute pancreatitis. Gastroenterology 1997; 113: 899-903
25 Baron TH, Morgan DE. Acute necrotizing pancreatitis. N Engl J Med 1999; 340: 1412-1417
26 Bradley EL. Indications for surgery in necrotizing pancreatitis--a millennial review. JOP 2000; 1: 1-3
27 Uhl , Warshaw A, Imrie C, Bassi C, McKay CJ, Lankisch PG, Carter R, Di Magno E, Banks PA, Whitcomb DC, Dervenis C, Ulrich CD, Satake K, Ghaneh P, Hartwig W, Werner J, McEntee G, Neoptolemos JP, Büchler MW. IAP Guidelines for the Surgical Management of Acute Pancreatitis. Pancreatology 2002; 2: 565-573
28 Uomo G. Inflammatory pancreatic diseases in older patients: recognition and management. Drugs Aging 2003; 20: 59-70
29 Ogawa . Acute pancreatitis and cytokines: "second attack" by septic complication leads to organ failure. Pancreas 1998; 16: 312-315
S- Editor Wang J L- Editor Kumar M E- Editor Ma WH
Fu CY et al. Timing of mortality in severe acute pancreatitis 1969
www.wjgnet.com

PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1970-1974www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
Perioperative management of primary liver cancer
Lu-Nan Yan, Xiao-Li Chen, Zhi-Hui Li, Bo Li, Shi-Chun Lu, Tian-Fu Wen, Yong Zeng, Hui-Hua Yiao, Jia-Yin Yang, Wen-Tao Wang, Ming-Qing Xu
www.wjgnet.com
RAPID COMMUNICATION
Lu-Nan Yan, Xiao-Li Chen, Zhi-Hui Li, Bo Li, Shi-Chun Lu, Tian-Fu Wen, Yong Zeng, Hui-Hua Yiao, Jia-Yin Yang, Wen-Tao Wang, Ming-Qing Xu, Department of General Surgery, West China Hospital, Sichuan University, Chengdu 610041, Sichuan Province, ChinaCorrespondence to: Lu-Nan Yan, MD, Department of General Surgery, West China Hospital, Sichuan University, Chengdu 610041, Sichuan Province, China. [email protected] Telephone: +86-28-85422476 Fax: +86-28-85423724Received: 2006-11-23 Accepted: 2007-03-12
AbstractAIM: To minimize the complications and mortality and improve the survival in primary liver cancer (PLC) patients undergoing hepatic resection.
METHODS: We conducted a retrospective analysis of 2143 PLC patients treated from January 1990 to January 2004. The patients were divided into two groups using January 1997 as a cut-off. Small tumor size (< 5 cm), preoperative redox tolerance index (RTI), vascular control method, and postoperative arterial ketone body ratio (AKBR) were used as indicators of surgical outcome.
RESULTS: Small tumors had less complications and lower mortality and higher overall survival rate. Use of RTI for selecting patients and types of hepatectomy, reduced complications (21.1% vs 11.0%) and mortality (1.6% vs 0.3%). The half liver vascular occlusion protocol (n = 523) versus the Pringle method (n = 476) showed that the former significantly reduced the postoperative complications (25.8% vs 11.9%) and mortality (2.3% vs 0.6%) respectively, and cut mean hospital stay was 3.5 d. Postoperative AKBR was a reliable indicator of the energy status in survivors.
CONCLUSION: RTI is of value in predicting hepatic functional reserve, half liver occlusion could protect the residual liver function, and AKBR measurement is a simple and accurate means of assessing the state of postoperative metabolism. Optimal perioperative management is an important factor for minimizing complications and mortality in patients undergoing hepatic resection.
© 2007 The WJG Press. All rights reserved.
Key words: L iver cancer; Hepatectomy; Optimal
perioperative management; Arterial ketone body ratio; Redox tolerance index
Yan LN, Chen XL, Li ZH, Li B, Lu SC, Wen TF, Zeng Y, Yiao HH, Yang JY, Wang WT, Xu MQ. Perioperative management of primary liver cancer. World J Gastroenterol 2007; 13(13): 1970-1974
http://www.wjgnet.com/1007-9327/13/1970.asp
INTRODUCTIONPrimary liver cancer (PLC) is one of the most common malignant tumors in China. Its prognosis has been greatly improved due to the major advances in its clinical treatment and basic research since 1950’s[1]. Surgical resection is still the primary means of achieving long-term survival of PLC patients[2-4]. Massive bleeding and liver failure are the leading risk factors in hepatic resection of PLC patients. The development of various effective vascular control measures[5-7], thanks to the recent technical innovations, has reduced the number of patients with intraoperative massive hemorrhage. However, postoperative liver failure is still a major impediment to hepatic resection, especially in patients with compromised liver. Although a variety of methods for estimating hepatic function have been described, accurate prediction of hepatic functional reserve and assessment of postoperative risk remains a challenge[8]. To establish a simple and practical system for assessing hepatic functional reserve to guide the decision-making process in treating PLC patients, we have conducted a systematic clinical study on hepatic energy metabolism since 1980s. This hepatic functional reserve predictive system is of value in terms of minimizing unnecessary invasive hepatic excision and designing optimal strategies for the perioperative and postoperative management of individual PLC patients.
MATERIALS AND METHODSPatientsWe reviewed the hospital records of all the hepatic tumor patients treated at the Department of General Surgery, West China Hospital, Sichuan University between January 1990 and January 2004. A retrospective analysis 2143 patients with intact records who were clinically diagnosed as PLC and underwent surgical treatment was conducted. The review covered sex, age, time of operation, diagnosis,

Yan LN et al . Management of liver cancer 1971
www.wjgnet.com
liver function test, kidney function test, hepatitis marker, tumor marker, ar terial ketone body ratio (AKBR), redox tolerance index (RTI), Child-Pugh classification, preoperative management, preoperative evaluation, operation approach, postoperative measurement of AKBR, and therapy protocols. Postoperative clinical course, calculation of incidence of complications, operation mortality and evaluation of follow-up visits were also included. Operation mortality was defined as death during operation or within one month after operation.
Preoperative measurement of RTIEach patient received 75 g glucose orally in the morning. Arterial blood samples were collected respectively at 30, 60, 90, and 120 min before and after the glucose intake. The following biochemical parameters including the levels of acetoacetate and β-hydroxybutyrate, AKBR, blood glucose, insulin and serum free fatty acids were measured as previously described[9-12]. The corresponding data obtained were used to plot the oral glucose tolerance test (OGTT) and AKBR curve. The area covered by the respective curve (∆OGTT and ∆AKBR) was calculated. The value of RTI was determined according to the following equation: ∆RTI = ∆AKBR/∆OGTT × 5.6The extent of liver cirrhosis and hepatic functional reserve were judged based on the measured value of RTI. A value of RTI > 0.65 predicted that damage to the liver was mild and the patient had a relatively good hepatic functional reserve and could tolerate various types of hepatectomy. A value of RTI < 0.65 indicated that damage to the liver was either moderate or severe and that the hepatic functional reserve of the patient was apparently compromised. For PLC patients undergoing hepatic resection in this category, special consideration needed to be taken with regard to the extent of hepatic resection. In case of RTI < 0.50, the patient would not be a suitable candidate for more than half liver resection and meticulous perioperative management care should be implemented.
Blood inflow occlusion of the half live The procedure began with excision of the abdomen, and subsequent exposure of the hepatoduodenal ligament, followed by explorations proceeding upwards along the common hepatic duct until reaching the confluence of the left and right duct. On the visceral envelope overlying the confluence, a small hole was made using a sharp blade and a right-angle forceps was inserted to gently mobilize within liver parenchyma outside the Glisson sheath. To avoid damage to the portal vein and the small branches of caudate portal vein, the right-angle forceps should mobilize in the liver parenchyma towards the caudate lobe in such a way that there was no feel of resistance. Finally, the sharp ends of the forceps were passed through the confluence of the posterior branching part of portal vein and the caudate lobe, and an 8 gauge catheter was introduced. Vascular exclusion to the left or right half of the liver was achieved by tightening the catheter during hepatic resection[13].
For hepatic resection involving no more than half the liver, the half liver occlusion technique was routinely
employed to keep the blood supply to the contralateral half and to minimize postoperative liver failure and complications[5]. In cases that big liver tumors involving more than half the liver or tumors located in the middle part of, or crossing the left- and the right half liver or tumors encroaching the hepatoduodenal ligament or patients with apparent anatomic variations of the hilum hepatis, the whole liver vascular exclusion was used (Pringle method).
Regular postoperative measurement of AKBR Measurement of AKBR and routine hepatic function tests were performed postoperatively on d 1, 3, 5, 7 until stabilization of the patient’s condition. For patients developing postoperative complications, the monitoring time course was extended until stabilization of the patient’s condition or death. The levels of AKBR were determined using a modified Mellamby approach[5]. Briefly, all patients received intravenous injection of 10% glucose to allow the blood glucose level to reach at 6.72-11.2 mmol/L one hour later, followed by collection of 4 mL of arterial blood from each patient. The blood samples were centrifuged and 1 mL of blood plasma was collected, then 2 mL of 0.15 mol/L Ba(OH)2 and 2 mL of 5% H2SO4 were gradually added. After mixing, the suspensions were centrifuged at 3000 r/min for 10 min and the supernatants were collected, then 0.15 mol/L Ba(OH)2 was continuously added and thoroughly mixed until the solution became alkaline. The resultant solutions were put inside the modified distiller and distillates collected later. The amounts of acetoacetate and β-hydroxybutyrate in the collected distillates were respectively determined by salicylic acid colorimetric method and the arterial ketone body ratio was calculated[14,15].
Patients were classified into three groups: group A including patients with a consistent postoperative AKBR > 0.7, group B with AKBR in the range of 0.4-0.7, group C with the level of AKBR gradually dropped to below 0.4 after operation. For postoperative therapy, standard treatment was applied to group A patients. Because of impaired hepatic function, group B patients received treatment with side chain-containing amino acids and long chain fatty acids and middle- and long chain fatty-containing cream, and large dose of Danshen injection (90 mL/d), as well as liver protection therapy to enhance the recovery of microcirculation in the impaired liver. Since patients in group C could utilize neither glucose nor fatty acids as substrates for energy production because of their severe liver dysfunction and deteriorated energy metabolism, they might ultimately die of the dysfunction of multiple organs. The patients in this group were difficult to treat and liver transplantation or temporal support by artificial liver could be a viable option.
Statistical analysisSmall tumor size, preoperative RTI, vascular control method, and postoperative AKBR were compared by the Wilcoxon tests in two groups. Survival analysis was performed to evaluate the impact of different variables. Cox model was used for univariate analysis,

which incorporates the quantitative variables assumed to determine the outcome. Statistical analysis was performed with SPSS software package.
RESULTSGeneral characteristics of the patientsOf the 2143 PLC patients undergoing surgical treatment between January 1990 to January 2004 in our institution, 1869 were men and 274 were women (male/female ratio: 6.8) with a median age of 46.3 (range 12 to 84) years. In 2037 cases of hepatocellular carcinoma (95.1%), 82 were found to have cholangiocellular carcinoma (3.8%), and 24 mixed type of both carcinomas (1.1%). In 1724 cases of cirrhosis (80.0%), 72% were HBsAg positive, 56.6% had their AFP ≥ 100 μg/L. A total of 1289 patients (60.1%) underwent hepatic tumor resection with an operative mortality of 1.6% (21/1289), and postoperative complications of 21.1% (272/1289). A total of 1198 (92.9%) patients were followed up for 6 mo -10.5 years. The 1-, 3-, and 5-year survival rate was 82.1%, 59.4%, and 37.6%, respectively. The remaining 854 cases underwent non-resection surgical treatment with a mortality of 2.1% (18/854), and complications were found in 19.9% (170/854). Operation approaches for all the patients are listed in Table 1.
Comparison of patients in two groupsUsing January 1997 as a cut-off, the patients were assigned to two groups[16]. The earlier period group (January 1990 to January 1997) contained 731 patients including 647 men and 84 women, their age ranged from 16 to 84 years (median 47.4). The tumor size was ≤ 5 cm in diameter in 52 cases (7.1%), and ≤ 3 cm in diameter in 19 cases (2.6%). A total of 312 patients (42.7%)underwent hepatic resection
and 419 (57.3%) received non-resection treatment. The later period group (January 1997 to January 2004) included 1412 cases (1222 men, 190 women, male/female: 6.4; age range 12 to 78 years, median 46.9). The tumor size was ≤5 cm in diameter in 298 (21.1%) patients and ≤ 3 cm in diameter in 183 (13.0%) patients. A total of 977 (69.2%) patients underwent hepatic resection, 435 (30.8%) received non-resection treatment. Operation approaches inr the two groups of patients are listed in Table 1. Mortality, complications, and the 1-, 3- and 5-year survival rates are listed in Table 2. The mortality and complication rates after non-resection treatment were 2.1% and 22.6% in the first group, and 2.1% and 17.2% in the second group.
Measurement of RTIRTI was measured in 336 patients, including 288 men and 48 women (age range 28-65 years, median 48.1). Operation approaches included hepatic lobectomy inr 287 patients (left or right trisectionectomy in 9, left or right hepatectomy in 14, sectionectomy 112, segmentectomy in 152), hepatic artery ligation or pump in 49 cases. Of the 253 (75.3%) patients with a measured value of RTI ≥ 0.65, 9 underwent left or right trisectionectomy, 14 left or right hepatectomy, 223 sectionectomy or segmentectomy, and 7 hepatic artery ligation or pump when the tumors were unresectable during operation. Postoperative complication rate was 10.3% (26/253), with no death occurred during operation. Of the 83 patients with a RTI < 0.65, 41 underwent sectionectomy or segmentectomy, 42 hepatic artery ligation or pump. The postoperative complication rate was 13.3% (11.83) and one patient died of liver failure on d 29. The overall complication rate was 11.0% (37/336) and mortality rate during operation was 0.3%.
Half liver vascular occlusionThe half liver vascular occlusion protocol was employed in 523 (40.6%) of 1289 patients undergoing hepatic lobectomy. Of these 523 patients, 105 underwent either left or right hepatectomy, 418 sectionectomy or segmentectomy. The duration of the half liver occlusion ranged from 12 to 63 min, with a mean of 25.2 min. The amount of blood transfusion was 0 to 3200 mL, averaged of 306 mL. Three patients died after operation (1 due to hepatic failure, and 2 due to infection and MOF). The postoperative mortality was 0.6%, complication rate was 11.9% (62/523), postoperative hospital stay was 8-45 d (mean 18.2 d).
Of 476 patients undergoing hepatic resection using the routine Pringle procedure, 55 received either left or right trisectiontomy, 173 left or right hepatpectomy, 42 middle sectionectomy, and 206 sectionectomy or segmentectomy.
Operative approach Earlier period group (%)
Later period group (%)
Resection groupRight trisectionectomy 6 (0.8) 19 (1.3)Right hepatectomy 82 (11.2) 73 (5.2)Right posterior sectionectomy 26 (3.6) 125 (8.9)Right anterior sectionectomy 13 (1.8) 131 (9.3)Middle sectionectomy 11 (1.5) 44 (3.1)Left trisectsanectomy 8 (1.1) 24 (1.7)Left hepatectomy 27 (3.7) 129 (9.1)Left lateral sectionectomy 31 (4.2) 88 (6.2)Candate lobectomy 5 (0.7) 38 (2.7)Segmentectomy 103 (14.1) 306 (21.7)Total 312 (42.7) 977 (69.2)Non-resection groupHepatic artery ligation or pump 251 (34.3) 143 (10.1)Intro arterial 131I resin form embolization 96 (13.1) 49 (3.5)Intro-arterial 32P-GMS embolization1 28 (3.8) 36 (2.5)Cryosurgical ablation 24 (3.3) 82 (5.8)Radiofrequancy ablation 0 (0) 102 (7.2)Laparotomy 20 (2.7) 23 (1.6)Total 419 (57.3) 435 (30.8)
Table 1 Operation approach for patients in two groups
132P-GMS: Phosphorus 32 glass microspheres; ∆RFA: Radiofrequancy ablation.
Group Operative mortality
Complication rate Survival rate1 yr 3 yr 5 yr
Earlier period group 2.2 23.7 73.1 54.2 34Later period group 1.4 20.3 84.9 61.1 38.7
Table 2 Comparison of mortality, complication, and survival rate in two groups after hepatectomy (%)
1972 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

The duration of blood occlusion was 11-48 min. Occlusion was performed twice in 37 cases, the mean duration was 24.8 min, the amount of blood transfusion was 0-3600 mL, averaged 383 mL. Postoperative complications were found in 123 cases (25.8%). The postoperative mortality was 2.3% (11/476), and postoperative stay was 8-45 d, averaged 18.2 d.
Regular postoperative measurement of AKBR was conducted in 635 cases (49.3%) undergoing hepatic resection. Based on the AKBR, 472 (74.5%) cases were assigned to group A, 149 (23.5%) to group B, and 13 (2.0%) to group C. The complication rate was 5.9% in group A (n = 28), 71.8% in group B (n = 107) with 1 patient died due to MOF (0.9%), and 100% in group C with a mortality rate of 69.2% due to infection and MOF.
DISCUSSIONPLC accounted for 95.1% of the 2143 cases, and 80.4% of patients were associated with cirrhosis, 72.0% were HBsAg positive, and only 56.6% were AFP positive, which are consistent with the reported results in China. Based on the outcome of this series, surgical treatment is still the primary choice of treatment for patients with liver cancer. The major progress made in PLC could be attributed to the following factors: (1) early diagnosis of hepatic tumor, as reflected by the higher percentage of cases with small tumor (< 5 cm in diameter) in the later period group than in the earlier period group (21.1% vs 7.1%); (2) increased percentage of patients with complete resection as evidenced by 42.7% in the earlier group compared to 69.2% in the later group; (3) long-term survival of liver cancer patients due to the improved perioperative management and refined surgical techniques; (4) improved safety and efficacy due to the implementation of standardized treatment protocol and development of non-resection treatment such as intra-arterial embolization, cryosurgical ablation, radiofrequency ablation.
Liver failure is the leading cause of postoperative death[17]. There is still no reliable way for estimating hepatic functional reserve in diseased liver[18-21]. Routine hepatic function test, assay of metabolic proteins and amino acids, clearance capacity test, excretion function test, liver blood in-flow measurement, etc, can only detect the specific function of liver and cannot be used to determine the tolerance of diseased liver to surgical resection. Since any activity of the liver requires consumption of energy and insufficient energy metabolism and compromise the liver function, our previous studies[22,23] showed that OGTT and AKBR can be used to determine the energy metabolism status in the liver and thus are good indicators of hepatic function reserve. Therefore, we adopted the redox tolerance test (RIT), which reflects the changing pattern of OGTT and AKBR, to compare the diseased liver tissue preoperatively and postoperatively[9,10], and the data of our current series further confirmed that RTI could reflect the hepatic functional reserve. Our predictive system for hepatic functional reserve is based on the following criteria: (1) when RTI > 0.65, the liver function is considered to be good or only mildly affects functional reserve and should be capable of tolerating
more than half the liver resection; (2) a value of RTI < 0.65 suggests either a moderate or severe liver tissue damage; (3) if RTI < 0.5, the hepatic function is judged to be substantially compromised, and the patients should not be suitable for more than half the liver resection. In this series, the system was used for estimating the residual hepatic functional reserve in 366 cases and for guiding the choice of resection type, resulting in marked improvement in complications from 21.1% to 11.0%, and mortality from 1.6% to 0.3, demonstrating that RTI is a valid indictor of hepatic functional reserve.
The Pringle maneuver under normothermic condition is still routinely employed in hepatic resection[6], which tends to compromise the function of spared liver tissue, especially in patients with cirrhosis, eventually leading to high occurrence of liver failure[17]. To overcome this drawback, based on observations through B ultrasonic wave and anatomical study of the hilum, we devised the half-liver vascular occlusion protocol in 1991[5,13]. The subsequent use of this procedure in 63 patients undergoing hepatectomy, as opposed to 42 cases using the Pringle protocol, showed that half liver occlusion method being was superior to the whole Pringle procedure in terms of diminished adverse effects on energy metabolism in the liver and less invasive nature, and rapid postoperative recovery[24]. The half liver blood occlusion does not require the dissection of porta hepatis, and a right-angle forceps is passed into the liver to introduce a catheter around the Glisson sheath to achieve vascular occlusion of the half liver, thus this procedure is simple, safe and can be easily adopted. Its benefit was better in 523 cases than that of the Pringle maneuver in 476 cases, as demonstrated by significant reduction in postoperative mortality (0.6% vs 2.3%) and complications (11.9% vs 25.8%) in patients undergoing hepatectomy. However, it was not until 2001 that Horgan et al[7] proposed a similar protocol for partial vascular control, termed the half Pringle technique.
AKBR measurement and liver function tests were done in 93 patients after undergoing hepatectomy in our previous study[15]. Based on the measured value of AKBR, the 93 patients were classified into 3 groups. Group A (AKBR > 0.7) had a postoperative complication rate of 4.8%, group B (AKBR 0.4-0.7) 39%, and group C (AKBR < 0.4) consisting of only 3 patients who died of MOF. However, routine live function test failed to show any significant differences in patients of the three groups, indicating that AKBR cannot reveal the degree of damage to the liver. In contrast, AKBR can reflect the redox status in liver mitochondria. The patients in group A had normal functional mitochondria and could utilize glucose for energy production, and metabolism was normal. The patients in group B with severely damaged liver function combined with the subsequent surgical stress, had impaired mitochondria and therefore could not efficiently utilize glucose for the entire energy need and could compensate for the insufficiency when fatty acids were used for energy production, dropping AKBR to 0.4-0.7. With proper management, the mitochondrial function could gradually recover and AKBR could increase to above 0.7 and the patients would recover gradually. If the function of mitochondria fails to recover, AKBR would drop to below
Yan LN et al . Management of liver cancer 1973
www.wjgnet.com

0.4, suggesting that damage to the mitochondria is severe and the oxidative phosphorylation process would stop and patients could use neither glucose nor fatty acids as energy substrates, and the energy metabolism would deteriorate, ultimately leading to multiorgan failure, MOF and death.
In th is s tudy, we cont inuously monitored the postoperative AKBR in 635 patients and the complication rate in groups A-C was 5.9%, 71.8%, and 100%, and the postoperative mortality was 0%, 0.9% and 69.2% respectively, demonstrating that continuous monitoring of AKBR levels can reveal the energy metabolism status and is a simple, sensitive and practical indicator of hepatic functional reserve and a valuable tool for the management and prevention of liver failure.
REFERENCES1 Tang ZY. Tang Zhaoyou clinical hepatic oncology. Shanghai:
Shanghai scientific and educational publishing house, 2001: 5-6
2 MC Wu. Current review and perspective on integrated. Zhonghua Waike Zazhi 2004: 19: 13
3 Jievaltas M, Stoskuviene L, Petrenkiene V, Barauskas G, Pundzius J. Results of treatment of primary liver cancer at Kaunas University of Medicine Hospital. Medicina (Kaunas) 2004; 40: 127-134
4 Gasztonyi B, Pár A, Battyány I, Hegedüs G, Molnár TF, Horváth L, Mózsik G. Multimodality treatment resulting in long-term survival in hepatocellular carcinoma. J Physiol Paris 2001; 95: 413-416
5 Yan LN, Yuan CX, Zhang ZD. Report on the application of half-liver occlusion in hepatic lobectomy on 29 patients. Zhonghua Waike Zazhi 1994; 32: 35-36
6 Huang ZQ. Blood Occlusion techniques in Hepatectomy. In: Huang ZQ, editor. Hepatic Surgery. 1st ed. Beijing: People’s Military Publishing House, 1996: 69-80
7 Horgan PG, Leen E. A simple technique for vascular control during hepatectomy: the half-Pringle. Am J Surg 2001; 182: 265-267
8 Linet MS, Gridley G, Nyrén O, Mellemkjaer L, Olsen JH, Keehn S, Adami HO, Fraumeni JF. Primary liver cancer, other malignancies, and mortality risks following porphyria: a cohort study in Denmark and Sweden. Am J Epidemiol 1999; 149: 1010-1015
9 Yan LN, Meng XY, Wu YT. The utility of redox tolerance test on the assessment of the hepatic functional reserve of patients
with liver cancer. Zhonghua Yixue Zazhi 1993; 73: 17510 Yan LN, Li YL, Wu YT. Redox tolerance test: an indicator
of preoperative hepatic functional reserve in liver diseases. Gandanyi Waike Zazhi 1997; 4: 249
11 Mellamby J, Williamson DH. Acetoacetate. In: Bergmeyer HV, editor. Methods of Enzymatic analysis. New York: Academic Press, 1974: 1840-1843
12 Yamamoto R. Changes in arterial ketone body ratio after transcatheter arterial embolization for hepatocellular carcinoma-clinical and experimental studies. Nihon Shokakibyo Gakkai Zasshi 1990; 87: 1401-1409
13 Yan LN, Li B, Zeng Y, Wen TT, Zhao JC, Wang WT, Yang JY, Xu MQ, Ma YK, Chen ZY, Wu H. Adult-to-adult living donor liver transplantation. Sichuan Daxue Xuebao Yixueban 2006; 37: 88-92
14 Yan LN, Ozawa K, Kobayashi N. Changes in hepatic energy metabolism in experimental acute pancreatitis. Chin Med J (Engl) 1992; 105: 684-688
15 Yan LN, Wu YT, Zheng GQ. Analysis of postoperative prognosis in relation to blood ketone body ratio in surgical patients with liver disease. Asain J Surg 1994; 1: 156
16 Yan LN, Zheng Y, Wen TF. Surgical treatment for 1038 patients with primary liver cancer. Zhonghua Waike Zazhi 2000; 38: 520
17 Chen XP, Chen H, Meng CY. Observations on the 81 cases with hepatectomy using blood exclusion under normal thermo conditions. Zhonghua Waike Zazhi 1991; 29: 84
18 Zhou WP, Chen H, Wu MC. Method and practical value of preoperative estimation of hepatic functional reserve. Gandan Waike Zazhi 2003; 11: 245
19 Geller DA, Tsung A, Marsh JW, Dvorchik I, Gamblin TC, Carr BI. Outcome of 1000 liver cancer patients evaluated at the UPMC Liver Cancer Center. J Gastrointest Surg 2006; 10: 63-68
20 Ikai I, Arii S, Ichida T, Okita K, Omata M, Kojiro M, Takayasu K, Nakanuma Y, Makuuchi M, Matsuyama Y, Yamaoka Y. Report of the 16th follow-up survey of primary liver cancer. Hepatol Res 2005; 32: 163-172
21 Park JW. Hepatocellular carcinoma in Korea: introduction and overview. Korean J Gastroenterol 2005; 45: 217-226
22 Wen TF, Zheng GQ, Meng XY. The predictive value of sugar tolerance test and insulin secretion assay for risk assessment of surgical treatment for patient with hepatic carcinoma. Zhonghua Waike Zazhi 1989; 27: 597
23 Yan LN, Kobayabhi N, Ozawa K. Dynamic observations of hepatic energy metabolism in patients with acute pancreatic necrosis. Zhonghua Waike Zazhi 1990; 28: 295
24 Yan LN, Li ZW, Liu ZP. Studies on the changes of hepatic energy metabolism during the surgical procedure using half-liver blood occlusion. Zhonghua Waike Zazhi 1995; 33: 56
S- Editor Liu Y L- Editor Wang XL E- Editor Zhou T
1974 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

in Eastern China. World J Gastroenterol 2007; 13(13): 1975-1979
http://www.wjgnet.com/1007-9327/13/1975.asp
INTRODUCTIONIt is estimated that more than 4 billion people worldwide have been infected with hepatitis B virus (HBV)[1]. The infection is persistent in over 350 million individuals with 20%-30% risk of death from HBV-related liver failure or liver cirrhosis[2]. HBV has a circular and partial double-strand DNA genome of 3.2 kb containing four overlapping open reading frames[3]. Based on nucleotide differences of 8% or more in the complete genome, HBV has been classified into eight genotypes from A to H[4-6]. HBV genotype C (HBV/C) induces more severe liver diseases, including hepatocellular carcinoma (HCC), than HBV/B in Asia [7]. Furthermore, patients with HBV genotypes C and D have a lower response rate to interferon alpha (IFN-α) compared with genotypes A and B[8]. Therefore, the analysis of HBV genotype infecting a patient may assist clinical and therapeutic decisions.
Although many HBV genotyping methods exist, there is no standardized or commercially available method for direct molecular typing of the HBV genome. An HBV genotype assay based on the oligonucleotide chip was developed for standardization and potential diagnostic use. This method is based on the reverse hybridization principle so that Cy5-labeled amplicons hybridize to genotype-specific oligonucleotide probes that are immobilized on slides.
In Eastern China, HBV infection is highly prevalent and is an important cause of liver diseases. However, the HBV genotype distribution of the whole area in Eastern China has not been reported systemically. In this study, 400 HBV DNA-positive serum samples collected from patients of the eight cities, including Nanjing, Zhengjiang, Changzhou, Xuzhou, Shanghai, Ningbo, Pingxiang, and Suzhou, in Eastern China were tested using the oligonucleotide chip. In addition, part of results was compared with sequencing.
MATERIALS AND METHODSPatientsA total of 400 HBV DNA-positive sera were collected from patients of the eight cities, including Nanjing,
RAPID COMMUNICATION
Detection of hepatitis B virus genotypes using oligonucleotide chip among hepatitis B virus carriers in Eastern China
Xiang-Rong Tang, Ji-Shen Zhang, Hui Zhao, Yu-Hua Gong, Yong-Zhong Wang, Jian-Long Zhao
www.wjgnet.com
Xiang-Rong Tang, Ji-Shen Zhang, Hui-Zhao, Jian-Long Zhao, Shanghai Institute of Microsystem and Information Technology, CAS, Shanghai 200050, ChinaYu-Hua Gong, The Third People’s Hospital of Zhenjiang, Zhenjiang 212003, Jiangsu Province, ChinaYong-Zhong Wang, The Third People’s Hospital of Changzhou, Changzhou 213001, Jiangsu Province, ChinaSupported by Chinese Academy of Sciences Intellective Innovation Great Project, No. KSCX-06Correspondence to: Jian-Long Zhao, PhD, Shanghai Institute of Microsystem and Information Technology, CAS, No. 865, Changning Road, Shanghai 200050, China. [email protected]: +86-21-62511070-8707 Fax: +86-21-62128934Received: 2006-12-21 Accepted: 2007-03-19
AbstractAIM: To determine the genotype distribution of hepatitis B virus (HBV) with a newly oligonucleotide chip assay among the HBV carriers in Eastern China.
METHODS: An assay using oligonucleotide chip was developed for detection of HBV genotypes in serum samples from HBV DNA-positive patients in Eastern China. This method is based on the principle of reverse hybridization with Cy5-labeled amplicons hybridizing to type-specific oligonucleotide probes that are immobilized on slides. The results of 80 randomly chosen sera were confirmed by direct sequencing.
RESULTS: HBV genotype B, C and mixed genotype were detected in 400 serum samples, accounting for 8.3% (n = 33), 83.2% (n = 333), and 8.5% (n = 34), respectively. The evaluation of the oligonucleotide assay showed 100% concordance with the amplicon phylogenetic analysis except 9 mixed genotype infections undetected by sequencing.
CONCLUSION: The study indicates that HBV genotype C and B prevail in the Eastern China. It is suggested that the oligonucleotide chip is a reliable and convenient tool for the detection of HBV genotyping.
© 2007 The WJG Press. All rights reserved.
Key words: Oligonucleotide chip; Hepatitis B virus; Genotype
Tang XR, Zhang JS, Zhao H, Gong YH, Wang YZ, Zhao JL. Detect ion of hepat it is B virus genotypes using oligonucleotide chip among hepatitis B virus carriers
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1975-1979www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.

Zhengjiang, Changzhou, Xuzhou, Shanghai, Ningbo, Pingxiang, and Hangzhou, in Eastern China from October 2004 to May 2006. Serum samples were taken from 44 asymptomatic HBV carriers (ASC) and 356 subjects with liver disease, consisting of 18 acute hepatitis (AH), 251 chronic hepatitis (CH), 74 liver cirrhosis (LC) and 13 hepatocellular carcinoma (HCC) patients. ASC usually had normal liver function test and no physical signs and symptoms. CH had chronic inflammatory reaction continuously. LC and HCC were diagnosed by ultrasonography and alpha-fetoprotein (AFP) level. All samples came from inpatient and all were known to have positive surface antigen (HbsAg) for more than 6 mo. The demographic data of the patients are listed in Table 1. Sera were tested for ALT, HbsAg, HbsAb, HbeAg, HbeAb, HbcAb using the related kits (Shanghai Kehua Bio-engineering Co. Ltd, Shanghai, China). HBV DNA was quantified using the kit produced by Shenzhen PG Biotech Co. Ltd. The sera were stored at -80 until analysis.
HBV DNA preparation and amplificationViral DNA was extracted from 200 μL of serum samples by using a QIAamp DNA Blood Mini kit (Qiagen Inc., Germany). The target DNA for hybridization was amplified by a nested PCR. A 418-bp product was amplified with the outer primers (sense primer, 5’- ACGYAGCGCCTCATTTTGTG-3’; antisense primer, 5’-CACTGCATGGCCTGAGGAT-3’) which were specific for the pre-S region of HBV. The second stage amplification generated 270-bp product with inner primers (sense primer, 5’-GGGTCACCATATTCTTGGGAA-3’; antisense primer, 5’-Cy5-TGAGGGCTCCACCCCA-3’). Each PCR mixture (40 μL) contained 4 μL of template DNA, 1 × PCR buffer, 1.5 mmol MgCl2, 200 μmol/L of each deoxynucleoside triphosphate, 20 pmol of each
primer, and 2 U of Taq DNA polymerase (all primers and reagents were supplied by TaKaRa Biotechnology Inc., Dalian, China). Both two round PCR were performed with a thermocycle (PCR Express; Thermohybaid, UK), and the samples were amplified through 35 cycles, each amplification cycle consisting of denaturation at 94 for 30 s, primer annealing at 55 for 30 s, and extension at 72 for 4 min. Cycles were preceded by incubation at 94 for 4 min to ensure full denaturation of the target gene and were followed by an extra incubation at 72 for 10 min to ensure full extension of the products.
Design of genotype-specific probes, PC probe and QC probeA total of 128 sequences of whole HBV genome from GenBank providing clear branching of the eight genotypes with A (n = 19), B (n = 21), C (n = 30), D (n = 21), E (n = 12), F (n = 13), G (n = 6), and H (n = 6) were aligned by using DNA-Star software (Lasergene, Madison, Wis). Eight consensuses of different genotypes were obtained by the alignments of each genotype. When comparing the alignment of each genotype with the alignment of eight consensuses, the result showed that there was a consensus sequence specific to each genotype at the same nucleotide positions in pre-S region among different isolates for each genotype. According to this result, genotype-specific probes were designed for different genotypes of HBV with DNA-Star software (Table 2). Based on the most conservative sequence of pre-S region, a positive control probe (PC probe) was designed for HBV DNA detection. To evaluate the quality of oligonucleotide chip fabrication and hybridization, a quality control probe (QC probe) was designed which reversely complemented the inner anti-sense primer. Probes were synthesized by TaKaRa Biotechnology Inc. All probes contained amino linkers at 5’ ends so that they could covalently attach to aldehyde-coated slides (CEL Associates Inc., Texas). A space arm with a poly (T) 16-mer was inserted between the probe sequence and the 5’ amino linker to decrease steric interference on the chips.
Oligonucleotide chips fabrication The probes were diluted to a final concentration of 10 μmol/L with spotting solution (TeleChem, USA) and spotted onto aldehyde-coated slides with a robot (Cartesian Pixsys 7500, USA). Four replicate spots of each probe were aligned in the same rows. QC and PC probes were spotted on the first and second left column on the chip (Figure 1). After the spotting process, the oligonucleotide chips were fixed at 25 for 48 h and dried for further use.
Table 1 Clinical and demographic data of 400 HBV-infected patients
Gender (male/female) Age (yr) HBV DNA (copies/mL) HbeAg+/HbeAg - ALT (IU/L)
Acute infection (n = 18) 13/5 33.1 ± 9.8 1.5 × 107 (3.6 × 104 - 9.6 × 109) 14/4 317.1 ± 630.4Asymptomatic carrier (n = 44) 27/17 28.7 ± 10.2 4.6 × 104 (1.2 × 103 - 5.6 × 106) 14/30 15.4 ± 8.7Chronic hepatitis (n = 251) 190/61 39.1 ± 10.1 1.4 × 106 (1.3 × 103 - 7.5 × 109) 165/86 219.6 ± 312.4Liver cirrhosis (n = 74) 47/27 49.7 ± 16.2 2.2 × 105 (5.3 × 103 - 4.7 × 106) 33/41 49.3 ± 59.8HCC (n = 13) 10/3 54.3 ± 15.4 5.6 × 104 (1.2 × 103 - 3.8 × 106) 7/6 192 ± 327.1
ALT: Alanine aminotransferase; HBeAg: Hepatitis B e antigen; HCC: Hepatocellular carcinoma.
Table 2 Probe sequence used for HBV genotyping
Genotype Oligonucleotide sequence Tm ()
A 5’-NH2-(T)16-AAC CCC (A/G)TC AAG GAC CAC TG-3’ 56B 5’-NH2-(T)16-CCC TGC ATT CAA AGC CAA CTC-3’ 56C 5’-NH2-(T)16-CTT CAA CCC CAA CAA GGA TCA-3’ 54D 5’-NH2-(T)16-CAA TCC CAA CAA GGA CAC CTG-3’ 55E 5’-NH2-(T)16-GAC CAC AAT CCC AAC AAA GAC C-3’ 55F 5’-NH2-(T)16-TGG CCA ATG GCA AAC AAG G-3’ 54G 5’-NH2-(T)16-CAA TCC CAA AAA GGA CCC TTG-3’ 54H 5’-NH2-(T)16-CTC TCA ACG GCG AGA AGG G-3’ 54QC probe 5’-NH2-(T)16-TGG GGT GGA GCC CTC AG-3’ 59PC probe 5’-NH2-(T)16- ATC CTC AGG CCA TGC AGT G-3’ 58
1976 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

Hybridization and scanning Two microliters of Cy5-labeled unpurified PCR product was mixed with 10 μL of DIG Easy Hyb (Roche Molecular Biochemicals, Canada). The mixture was denatured for 2 min at 95 and then chilled on ice for 5 min. The hybridization mixture was applied to the array and covered with a glass cover slip to prevent evaporation of the mixture during the hybridization. The oligonucleotide chips were incubated at 40 for 30 min in a moist incubator. After hybridization, the washing procedure was carried out as described in our previous research[9]. The oligonucleotide chip images were obtained with a f luorescent scanner (GenePix 4000B; Axon Instruments, Inc., Calif).
Sequencing and phylogenetic analysis The products of the first-round PCR were monitored on agarose gel to ensure that sufficient DNA was available for sequencing. Eighty products were randomly selected for the phylogenetic analysis. The outer sense primer also was used for DNA sequencing. The sequence was determined by Shanghai Bioasia Biotechnology Ltd. with ABI Prism 3730 genetic analyzer (Applied Biosystems, Inc.). The sequence datum of each selected specimen was aligned with pre-S sequences of eight consensuses for different genotypes which were obtained from the alignment of 128 complete HBV genome sequences in GenBank. Alignment and phylogenetic analysis were performed with DNA-Star software by using Clustal W.
RESULTSGenotype distribution detected by oligonucleotide chipTwo HBV genotypes (B, C) and a mixed type (B and C) were detected among the 400 samples using our
oligonucleotide chips. The layout of oligonucleotide chip and the representative fluorescent scanning of genotyping detection are shown in Figure 1. Genotype distribution in Eastern China in our study was finally determined as follows: genotype C, 333 (83.2%); genotype B, 33 (8.3%); and mixed genotype (B and C), 34 (8.5%). The other genotypes were not detected in this study.
Comparison of HBV genotype determined by sequencing and the oligonucleotide chipGenotype of the HBV DNA in 80 specimens was performed by sequencing and phylogenetic analysis of the pre-S region, and these genotypes were compared to the genotypes determined by the oligonucleotide chip for the same specimens. Phylogenetic analysis of pre-S sequence for each specimen by using the eight consensus sequences of different genotypes for comparison provided clear genotyping determination for each sample (data not shown). The results of the two methods for single genotypes were 100% concordant. The oligonucleotide chip detected n ine mixed genotypes which were determined by sequencing in a single genotype. Four of them were selected for clonal analysis. The PCR products of the four samples were ligated into pGEM-T easy vector (Promega), then the ligated products were transformed into E. coli DH5α (Clontech). Individual colonies were tested by the oligonucleotide chip at first, and then were sent to be sequenced. Clonal analysis confirmed that all the four specimens were mixed genotypes.
DISCUSSIONThere are eight genotypes of HBV designated A to H based on greater than 8% nucleotide variation over the entire genome. The eight genotypes of HBV show a distinct geographical distribution and influence the course of disease and the efficacy of treatment. Genotype A is prevalent in Northern and Central Europe, but is also common in North America and sub-Saharan Africa. Genotypes B and C are confined to Asia, genotype D is widespread but is the predominant genotype in the Mediterranean region, while genotype E is found mainly in West Africa[10]. Genotype F shows the highest divergence among the genotypes and is indigenous to aboriginal populations of the Americas[11]. Genotype G is found in USA, France, and Germany[5, 12, 13], genotype H in the Central America[6]. Thus, the variability of HBV genotypes can influence the interpretation of diagnostic data and the therapeutic decisions.
Our results showed that genotype B, C and mixed genotype exist in Eastern China. Genotype C is the major genotype in this area (accounting for 83.2%), which is similar with HBV genotype distribution in China in previous reports[7,14-16]. Genotype B was more frequently found in patients with mild hepatic inflammation (ASC), while genotype C in those with advanced liver diseases (CH and LC) (P < 0.05) (Table 3). Although there was no significant difference, infection with genotype C showed highest frequency (92.3%) in the patients with HCC. This
Genotype A
Genotype C
Genotype E
Genotype G
Genotype B
Genotype D
Genotype F
Genotype HPosi
tive
cont
rol p
robe
s
Qua
lity
cont
rol p
robe
sA
Fluorescent intensity
B Quality control C Genotype B
D Genotype C E Mixed genotype (B and C)
Figure 1 Layout of probes and fluorescence images of results. Probes layout on the oligonucleotide chip (A). Following hybridization of Cy5-labeled fragmented DNA, as described in Materials and Methods, the fluorescent signals of different HBV genotypes were detected as follows: Genotype B (C), genotype C (D), mixed genotype (E). Blank template amplified by PCR also was hybridized to the chip and the image of the result is shown in (B).
Tang XR et al. Determine the genotype distribution of HBV 1977
www.wjgnet.com
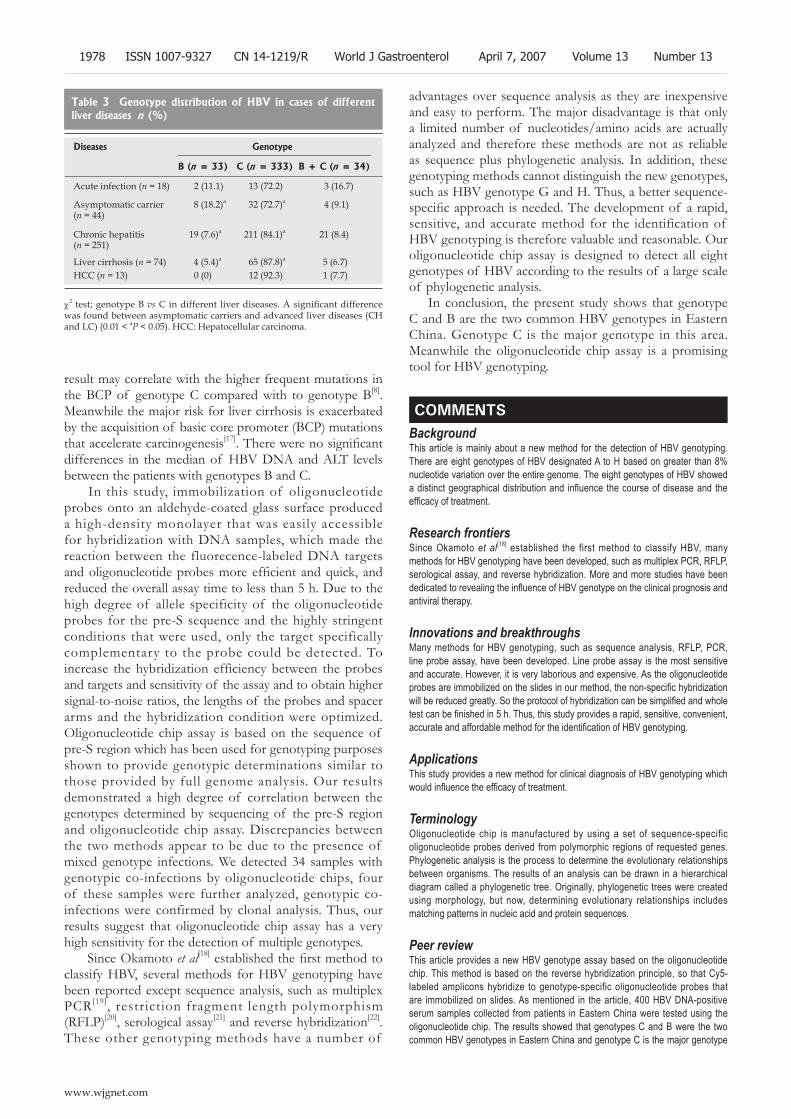
result may correlate with the higher frequent mutations in the BCP of genotype C compared with to genotype B[8]. Meanwhile the major risk for liver cirrhosis is exacerbated by the acquisition of basic core promoter (BCP) mutations that accelerate carcinogenesis[17]. There were no significant differences in the median of HBV DNA and ALT levels between the patients with genotypes B and C.
In this study, immobilization of oligonucleotide probes onto an aldehyde-coated glass surface produced a high-density monolayer that was easily accessible for hybridization with DNA samples, which made the reaction between the fluorecence-labeled DNA targets and oligonucleotide probes more efficient and quick, and reduced the overall assay time to less than 5 h. Due to the high degree of allele specificity of the oligonucleotide probes for the pre-S sequence and the highly stringent conditions that were used, only the target specifically complementary to the probe could be detected. To increase the hybridization efficiency between the probes and targets and sensitivity of the assay and to obtain higher signal-to-noise ratios, the lengths of the probes and spacer arms and the hybridization condition were optimized. Oligonucleotide chip assay is based on the sequence of pre-S region which has been used for genotyping purposes shown to provide genotypic determinations similar to those provided by full genome analysis. Our results demonstrated a high degree of correlation between the genotypes determined by sequencing of the pre-S region and oligonucleotide chip assay. Discrepancies between the two methods appear to be due to the presence of mixed genotype infections. We detected 34 samples with genotypic co-infections by oligonucleotide chips, four of these samples were further analyzed, genotypic co-infections were confirmed by clonal analysis. Thus, our results suggest that oligonucleotide chip assay has a very high sensitivity for the detection of multiple genotypes.
Since Okamoto et al[18] established the first method to classify HBV, several methods for HBV genotyping have been reported except sequence analysis, such as multiplex PCR[19], restriction fragment length polymorphism (RFLP)[20], serological assay[21] and reverse hybridization[22]. These other genotyping methods have a number of
advantages over sequence analysis as they are inexpensive and easy to perform. The major disadvantage is that only a limited number of nucleotides/amino acids are actually analyzed and therefore these methods are not as reliable as sequence plus phylogenetic analysis. In addition, these genotyping methods cannot distinguish the new genotypes, such as HBV genotype G and H. Thus, a better sequence-specific approach is needed. The development of a rapid, sensitive, and accurate method for the identification of HBV genotyping is therefore valuable and reasonable. Our oligonucleotide chip assay is designed to detect all eight genotypes of HBV according to the results of a large scale of phylogenetic analysis.
In conclusion, the present study shows that genotype C and B are the two common HBV genotypes in Eastern China. Genotype C is the major genotype in this area. Meanwhile the oligonucleotide chip assay is a promising tool for HBV genotyping.
COMMENTSBackgroundThis article is mainly about a new method for the detection of HBV genotyping. There are eight genotypes of HBV designated A to H based on greater than 8% nucleotide variation over the entire genome. The eight genotypes of HBV showed a distinct geographical distribution and influence the course of disease and the efficacy of treatment.
Research frontiers Since Okamoto et al[18] established the first method to classify HBV, many methods for HBV genotyping have been developed, such as multiplex PCR, RFLP, serological assay, and reverse hybridization. More and more studies have been dedicated to revealing the influence of HBV genotype on the clinical prognosis and antiviral therapy.
Innovations and breakthroughsMany methods for HBV genotyping, such as sequence analysis, RFLP, PCR, line probe assay, have been developed. Line probe assay is the most sensitive and accurate. However, it is very laborious and expensive. As the oligonucleotide probes are immobilized on the slides in our method, the non-specific hybridization will be reduced greatly. So the protocol of hybridization can be simplified and whole test can be finished in 5 h. Thus, this study provides a rapid, sensitive, convenient, accurate and affordable method for the identification of HBV genotyping.
Applications This study provides a new method for clinical diagnosis of HBV genotyping which would influence the efficacy of treatment.
TerminologyOligonucleotide chip is manufactured by using a set of sequence-specific oligonucleotide probes derived from polymorphic regions of requested genes. Phylogenetic analysis is the process to determine the evolutionary relationships between organisms. The results of an analysis can be drawn in a hierarchical diagram called a phylogenetic tree. Originally, phylogenetic trees were created using morphology, but now, determining evolutionary relationships includes matching patterns in nucleic acid and protein sequences.
Peer reviewThis article provides a new HBV genotype assay based on the oligonucleotide chip. This method is based on the reverse hybridization principle, so that Cy5-labeled amplicons hybridize to genotype-specific oligonucleotide probes that are immobilized on slides. As mentioned in the article, 400 HBV DNA-positive serum samples collected from patients in Eastern China were tested using the oligonucleotide chip. The results showed that genotypes C and B were the two common HBV genotypes in Eastern China and genotype C is the major genotype
Table 3 Genotype distribution of HBV in cases of different liver diseases n (%)
Diseases Genotype
B (n = 33) C (n = 333) B + C (n = 34)
Acute infection (n = 18) 2 (11.1) 13 (72.2) 3 (16.7)
Asymptomatic carrier (n = 44)
8 (18.2)a 32 (72.7)a 4 (9.1)
Chronic hepatitis (n = 251)
19 (7.6)a 211 (84.1)a 21 (8.4)
Liver cirrhosis (n = 74)HCC (n = 13)
4 (5.4)a
0 (0) 65 (87.8)a
12 (92.3) 5 (6.7) 1 (7.7)
c2 test; genotype B vs C in different liver diseases. A significant difference was found between asymptomatic carriers and advanced liver diseases (CH and LC) (0.01 < aP < 0.05). HCC: Hepatocellular carcinoma.
1978 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com
COMMENTS

in this area. Meanwhile the oligonucleotide chip assay is a promising tool for HBV genotyping.
REFERENCES1 Lai CL, Ratziu V, Yuen MF, Poynard T. Viral hepatitis B.
Lancet 2003; 362: 2089-20942 Kao JH, Chen DS. Global control of hepatitis B virus infection.
Lancet Infect Dis 2002; 2: 395-4033 Magnius LO, Norder H. Subtypes, genotypes and molecular
epidemiology of the hepatitis B virus as reflected by sequence variability of the S-gene. Intervirology 1995; 38: 24-34
4 Norder H, Couroucé AM, Magnius LO. Complete genomes, phylogenetic relatedness, and structural proteins of six strains of the hepatitis B virus, four of which represent two new genotypes. Virology 1994; 198: 489-503
5 Stuyver L, De Gendt S, Van Geyt C, Zoulim F, Fried M, Schinazi RF, Rossau R. A new genotype of hepatitis B virus: complete genome and phylogenetic relatedness. J Gen Virol 2000; 81: 67-74
6 Arauz-Ruiz P, Norder H, Robertson BH, Magnius LO. Genotype H: a new Amerindian genotype of hepatitis B virus revealed in Central America. J Gen Virol 2002; 83: 2059-2073
7 Ding X, Mizokami M, Ge X, Orito E, Iino S, Ueda R, Nakanishi M. Different hepatitis B virus genotype distributions among asymptomatic carriers and patients with liver diseases in Nanning, southern China. Hepatol Res 2002; 22: 37-44
8 Kao JH, Chen PJ, Lai MY, Chen DS. Hepatitis B genotypes correlate with clinical outcomes in patients with chronic hepatitis B. Gastroenterology 2000; 118: 554-559
9 Zhou W, Du W, Cao H, Zhao J, Yang S, Li W, Shen Y, Zhang S, Du W, Zhang X. Detection of gyrA and parC mutations associated with ciprofloxacin resistance in Neisseria gonorrhoeae by use of oligonucleotide biochip technology. J Clin Microbiol 2004; 42: 5819-5824
10 Norder H , Hammas B, Lee SD, Bile K, Couroucé AM, Mushahwar IK, Magnius LO. Genetic relatedness of hepatitis B viral strains of diverse geographical origin and natural variations in the primary structure of the surface antigen. J Gen Virol 1993; 74 (Pt 7): 1341-1348
11 Sánchez LV, Maldonado M, Bastidas-Ramírez BE, Norder H, Panduro A. Genotypes and S-gene variability of Mexican hepatitis B virus strains. J Med Virol 2002; 68: 24-32
12 Kato H, Orito E, Gish RG, Sugauchi F, Suzuki S, Ueda R,
Miyakawa Y, Mizokami M. Characteristics of hepatitis B virus isolates of genotype G and their phylogenetic differences from the other six genotypes (A through F). J Virol 2002; 76: 6131-6137
13 Vieth S, Manegold C, Drosten C, Nippraschk T, Günther S. Sequence and phylogenetic analysis of hepatitis B virus genotype G isolated in Germany. Virus Genes 2002; 24: 153-156
14 Ding X, Mizokami M, Yao G, Xu B, Orito E, Ueda R, Nakanishi M. Hepatitis B virus genotype distribution among chronic hepatitis B virus carriers in Shanghai, China. Intervirology 2001; 44: 43-47
15 Yuen MF , Sablon E, Tanaka Y, Kato T, Mizokami M, Doutreloigne J, Yuan HJ, Wong DK, Sum SM, Lai CL. Epidemiological study of hepatitis B virus genotypes, core promoter and precore mutations of chronic hepatitis B infection in Hong Kong. J Hepatol 2004; 41: 119-125
16 Fang ZL, Zhuang H, Wang XY, Ge XM, Harrison TJ. Hepatitis B virus genotypes, phylogeny and occult infection in a region with a high incidence of hepatocellular carcinoma in China. World J Gastroenterol 2004; 10: 3264-3268
17 Kuang SY, Jackson PE, Wang JB, Lu PX, Muñoz A, Qian GS, Kensler TW, Groopman JD. Specific mutations of hepatitis B virus in plasma predict liver cancer development. Proc Natl Acad Sci USA 2004; 101: 3575-3580
18 Okamoto H, Tsuda F, Sakugawa H, Sastrosoewignjo RI, Imai M, Miyakawa Y, Mayumi M. Typing hepatitis B virus by homology in nucleotide sequence: comparison of surface antigen subtypes. J Gen Virol 1988; 69 (Pt 10): 2575-2583
19 Mizokami M, Nakano T, Orito E, Tanaka Y, Sakugawa H, Mukaide M, Robertson BH. Hepatitis B virus genotype assignment using restriction fragment length polymorphism patterns. FEBS Lett 1999; 450: 66-71
20 Naito H, Hayashi S, Abe K. Rapid and specific genotyping system for hepatitis B virus corresponding to six major genotypes by PCR using type-specific primers. J Clin Microbiol 2001; 39: 362-364
21 Usuda S, Okamoto H, Tanaka T, Kidd-Ljunggren K, Holland PV, Miyakawa Y, Mayumi M. Differentiation of hepatitis B virus genotypes D and E by ELISA using monoclonal antibodies to epitopes on the preS2-region product. J Virol Methods 2000; 87: 81-89
22 Osiowy C, Giles E. Evaluation of the INNO-LiPA HBV genotyping assay for determination of hepatitis B virus genotype. J Clin Microbiol 2003; 41: 5473-5477
S- Editor Liu Y L- Editor Kumar M E- Editor Ma WH
Tang XR et al. Determine the genotype distribution of HBV 1979
www.wjgnet.com

INTRODUCTIONAcquired rectovestibular fistula (RVF) is always resulted from anal infection in newborn period. Following the healing of the acute infection, a well-epithelialized tract between the vestibular and anal canal develops, passing feces from the vestibular perforation. As the stool becomes solid, the leakage stops, and ARF does not give any remarkable inconvenience on children’s quality of life, growth and development[1]. The indication of operation is mainly about psychological concern of parents and children. However, malpractice of the surgical treatment of rectovestibular fistula, including lay-open or string treatment of fistula, will certainly cause total perineal disruption resulting to vestibular cosmetic defect and remarkable fecal incontinence. From June 1996 to April 2006, 15 girls with such defects were admitted to Beijing Children’s Hospital, and treated successfully by anterior perineal anorectoplasty.
MATERIALS AND METHODSRecords of fifteen girls, age ranged from 3 to 15 (average 7.5) years, were enrolled in this study. All of them came with a perineal defect and fecal incontinence due to malpractice in the treatment of RVF by lay-open or string treatment performed in other hospitals half to five years ago. These children had no normal perineal skin between the vestibular fossa and anus or underneath muscles, but had a mucosal patch between the vaginal orifice and anterior wall of the rectum. Sphincters ani was divided and its broken ends shrank backward to both sides and behind the rectum; consequently, it lost the normal appearance of perineum (Figure 1). All children were suffering from fecal soiling and some degree of anal incontinence partly with loose stool, which was graded Ⅲ-Ⅳ by Li Zheng’s score[2].
Operative procedureMetronidazole tablet (10 mg/kg) and oral garamycine (4 mg/kg) were given pre-operatively for three days. Food but water was restricted one day prior to operation. Saline enemas were given in the night before and on the morning of operation. The children were laid in lithotomy position. Almost half roll of sterile bandage lubricated with liquid paraffin, with the tail soaked with iodophor, was inserted and stacked in the rectum to prevent rectal
RAPID COMMUNICATION
Recto-vestibular disruption defect resulted from the malpractice in the treatment of the acquired recto-vestibular fistula in infants
Ting-Chong Zhang, Wen-Bo Pang, Ya-Jun Chen, Jin-Zhe Zhang
www.wjgnet.com
Ting-Chong Zhang, Wen-Bo Pang, Ya-Jun Chen, Jin-Zhe Zhang, Department of Surgery, Beijing Children’s Hospital affiliated to Capital Medical University, Beijing 100045, ChinaCorrespondence to: Dr. Ya-Jun Chen, MD, Department of Surgery, Beijing Children’s Hospital affiliated to Capital Medical University, Beijing 100045, China. [email protected]: +86-10-68028401-2403Received: 2007-01-05 Accepted: 2007-03-14
AbstractAIM: To explore the pathogenesis of the rectovestibular disruption (RVD) defect and to recommend a successful repair, and prevention of it.
METHODS: Clinical records of 15 girls, age ranged from 3 to 15 (median, 7.5) years, with acquired rectovestibular fistula (RVF) mistreated before were retrospectively reviewed. All of them presented an abnormal appearance of perineum and were suffering from some degree of fecal incontinence, and those were graded Ⅲ to Ⅳ by Li Zheng’s Score. Repair of anal sphincters and reconstruction of perineum body and skin by anterior perineal rectoanoplasty were performed in all cases.
RESULTS: Operation in all cases was successful. The perineum looked practically normal and fecal continence score rose up to VI by Li Zheng’s Score.
CONCLUSION: The convent ional treatment for anal fistula, lay-open or string-treatment, should be considered as malpractice of RVF, and certainly leads to the RVD defect, and the anterior perineal rectoanoplasty could cure it satisfactorily.
© 2007 The WJG Press. All rights reserved.
Key words: Rectovestibular fistula; Acquired; Disruption; Children
Zhang TC, Pang WB, Chen YJ, Zhang JZ. Recto-vestibular disruption defect resulted from the malpractice in the treatment of the acquired recto-vestibular fistula in infants. World J Gastroenterol 2007; 13(13): 1980-1982
http://www.wjgnet.com/1007-9327/13/1980.asp
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1980-1982www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.

discharge soiling out. The bandage tail was tied by a long heavy thread, which was left outside the anus for easier removal after operation. The embedded broken ends of the sphincter ani externus were detected by electric nerve stimulator. Four to six stay sutures were applied to the mucosal patch at mucocutaneous junction around vagina and anus, respectively. The stay sutures were stretched, and a half-circular incision anterior to the anus up to the level of two broken underneath ends of the external sphincter was made by sharp point of Bovie. Inward dissection along the bilateral wings of disrupted perineum was carried to separate vagina from rectum completely for about 5 cm in length. And the two broken ends of the sphincter ani externus were found by means of electric nerve stimulator. The transversus perinei, levator ani, and broken sphincter ani externus were approximately sutured layer by layer, the skin cut edge was approximately sutured around the rectal opening and behind the vagina orifice, and the skin cut edge between the anus and vagina was sutured vertically (anterior-posteriorly) interruptedly with 5-0 Dexon. Thus, the perineal body sphincter as well as skin bridge were all re-established (Figure 2). Routinely, food but water was restricted for three days. Antibiotics were given intravenously for 3 or 5 d. Perianal area was kept clean and dry by warm ventilation three to five times a day for some five days. The daily anal dilation began two weeks after the operation, and continued for 6 mo.
RESULTSDuring operation, vagina was injured and repaired immediately in two of the cases, but incision healed by first intention in all 15 cases. All children were followed up for 3 mo to 9.5 years (average 7.5 years). In two children, anal dilation was difficult at beginning, because their sphincter ani externus was too tight and the anus was too small after operation. Perineal appearance of all children resumed normal, and the bowel control was good, with bowel movement one or two times a day, without soiling, incontinence or constipation, with grade Ⅵ on Li Zheng’s score.
DISCUSSIONRVF with normal anus is considered acquired, in spite of
Zhang TC et al. Treatment of RVD defect 1981
www.wjgnet.com
a lot of disputes[3-6], as secondary to perineal infection in neonatal period. In recent 10 years, 319 cases, averaged to 30 per year, were admitted to the Surgical Department of Beijing Chidren’s Hospital. Clinically, the fistula had perfect mucosal lining, but none of the patients had a definite history of fistula at birth. Pathological section never showed normal histology of mucosa, submucosa or continuation of smooth muscle in the fistula tract[1,7]. Anyway, the opening was not big enough to allow solid stool leakage, and these patients could have normal marriage and childbirth, so the treatment should be safe and simple.
The conventional treatment of anal fistula is lay-open by perpendicular cutting of sphincters and the tract at one site, or string-treatment by gradual cutting through by a rubber band. In rectovestibular fistula, the external opening is in the vestibule and inner rectum opening lies in the anterior wall of rectum at the dentate line, with the tract crossing beneath the sphincter ani. Dividing the wall of fistula tract and the sphincter by lay-open or by string-treatment, the cut ends of sphincter will retract apart, leaving a big mucosal patch between the rectum and vestibule. The defect will not heal. Loss of normal perineal appearance and normal function undoubtedly makes the mother and child eager to have it repaired. Among our 15 cases, five resulted from lay-open, and ten from string-treatment. Nine were suffering from fecal soiling or fecal incontinence graded Ⅲ-Ⅳ by Li Zheng’s score. Anorectal surgeons should be alert to avoid the malpractices of lay-open and string treatment in acquired rectovestibular fistula.
In our series of 319 cases in the last 10 years, one stage repairing the fistula through either transrectal approach or through vestibular opening had obtained successful rate above 90%[8,9].
With anterior perineal anorectoplasty conventionally used for imperforated anus, and rectovestibular fistula with normal anus[10-12], we recommand it here for rectovestibular disruption (RVD). It provided a good exposure of operation field to separate the rectovaginal septum, and to find the broken ends of sphincter ani externus for reunion. It has been successfully proved in all our 15 cases. Key points for successful operation include: (1) bandage packing in the rectum to prevent fecal contamination of
Figure 1 Mucosal patch, no skin or muscles between the vaginal and anal orifice, sphincters ani broken and shrank backward.
Figure 2 Sphincters ani repaired and perineal body rebuilt.

operation field; (2) sharp dissection of natural clearance by Bovie to prevent bleed and creat a clean operation field; (3) bipolar coagulation of the two main arterioles 1 cm inward at 1 and 11 o’clock to lessen active oozing; (4) no dead space left between the vagina and rectum; (5) repair of the vagina wall or rectum wall immediately if injured during operation, as much as possible not injuring the rectum wall. In our group, vaginal wall was injured in two cases which were immediately sutured interruptedly with 5-0 Dexon. The wound healed well without any complication.
All children in this group were older than three year of age. All of them had satisfactory result. It proved, at least, that anterior perineal anorectoplasty is preferable for children older than 3 years. Besides, the anorectoplasty ought to be performed 6 mo to three years after the injured wound of perineum healed up. If delayed, the broken sphincter ani might be atrophic and contracted back the anus canal. Two girls in this series had anorectoplasty three and half years and four years, respectively, after the string-treatment. Both of them had a small anal orifice after the operation, and had difficult dilation for certain time.
Diverting colostomy has been a routine for better perineum hygiene in Western World [13], but for the suffering in RVD is minor, it seems not worthwhile to make two additional abdominal operations, which will increase the operation risk as well as the economic burden. In our 15 cases of anterior perineal anorectoplasty for RVD, diverting colostomy seems unnecessary, provided we pay more attention to lessen the operative trauma and contamination, and to avoid early bowel movement.
REFERENCES1 ZhangJZ. Anorectal Diseases Among Children. First edition.
Beijing: International Academic Publishers, 1993: 239-242 2 WangHZ, Li Z. Primary advice on standard of anal function
measuring after anoplasty. Zhonghua Xiao’er Waike Zazhi 1985; 6: 116-117
3 BremH, Guttman FM, Laberge JM, Doody D. Congenital anal fistula with normal anus. J Pediatr Surg 1989; 24: 183-185
4 Bianchini MA, Fava G, Cortese MG, Vinardi S, Costantino S, Canavese F. A rare anorectal malformation: a very large H-type fistula. Pediatr Surg Int 2001; 17: 649-651
5 YazlclM, Etensel B, Gürsoy H, Ozklsaclk S. Congenital H-type anovestibuler fistula. World J Gastroenterol 2003; 9: 881-882
6 ZhangJZ. Etiological and pathological study of fistula-in-ano in children. Zhonghua Xiao’er Waike Zazhi 1988; 9: 111-112
7 SunL, Wang YX, Liu Y. Histopathological study of fistula-in-ano in female. Zhonghua Xiao’er Waike Zazhi 1995; 16: 136-137
8 Chen YJ, Niu ZY, Zhang JZ, Liu YT, Li L, Wang DY. Transperineal approach to repair acquired rectovestibular fistula in female. Shiyong Erke Linchuang Zazhi 2001; 16: 242-243
9 Chen YJ , Zhang TC, Zhang JZ. Transanal approach in repairing acquired rectovestibular fistula in females. World J Gastroenterol 2004; 10: 2299-2300
10 OkadaA, Kamata S, Imura K, Fukuzawa M, Kubota A, Yagi M, Azuma T, Tsuji H. Anterior sagittal anorectoplasty for rectovestibular and anovestibular fistula. J Pediatr Surg 1992; 27: 85-88
11 Wakhlu A, Pandey A, Prasad A, Kureel SN, Tandon RK, Wakhlu AK. Anterior sagittal anorectoplasty for anorectal malformations and perineal trauma in the female child. J Pediatr Surg 1996; 31: 1236-1240
12 AzizMA, Banu T, Prasad R, Khan AR. Primary anterior sagittal anorectoplasty for rectovestibular fistula. Asian J Surg 2006; 29: 22-24
13 Mirza I, Zia-ul-Miraj M. Management of perineal canal anomaly. Pediatr Surg Int 1997; 12: 611-612
S-EditorLiu Y L-Editor Kumar M E-EditorMa WH
1982 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1983-1988www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
Pioglitazone attenuates the severity of sodium taurocholate-induced severe acute pancreatitis
Ping Xu, Xiao-Jiang Zhou, Ling-Quan Chen, Jiang Chen, Yong Xie, Long-Hua Lv, Xiao-Hua Hou
www.wjgnet.com
RAPID COMMUNICATION
Ping Xu, Xiao-Hua Hou, Department of Gastroenterology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, Hubei Province, ChinaPing Xu, Xiao-jiang Zhou, Ling-quan Chen, Jiang Chen, Yong Xie, Long-hua Lv, Department of Gastroenterology, First Affiliated Hospital, Nanchang University, Nanchang 330006, Jiangxi Province, ChinaSupported by Health Bureau Foundation of Jiangxi Province, No.20045019 and Natural Science Foundation of Jiangxi Province, No.0640069 Correspondence to: Xiao-Hua Hou, Department of Gastroente-rology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, Hubei Province, China. [email protected]: +82-27-85726930 Fax: +82-27-85726930Received: 2007-02-08 Accepted: 2007-03-12
AbstractAIM: To determine the effect of pioglitazone, a specific peroxisome proliferator-activated receptor-γ (PPARγ) ligand, on development of severe acute pancreatitis (SAP) and expression of nuclear factor-kappa B (NF-κB) and intercellular adhesion molecule-1 (ICAM-1) in the pancreas.
METHODS: Male Sprague-Dawley (SD) rats (160-200 g) were randomly allocated into three groups (n = 18 in each group): severe acute pancreatit is group, pioglitazone group, sham group. SAP was induced by retrograde infusion of 1 mL/kg body weight 5% sodium taurocholate (STC) into the biliopancreatic duct of male SD rats. Pioglitazone was injected intraperitoneally two hours piror to STC infusion. Blood and ascites were obtained for detecting amylase and ascit ic capacity. Pancreatic wet/dry weight ratio, expression of NF-κB and ICAM-1 in pancreatic tissues were detected by immunohistochemical staining. Pancreatic tissue samples were stained with hematoxylin and eosin (HE) for routine optic microscopy.
RESULTS: Sham group displayed normal pancreatic structure. SAP group showed diffuse hemorrhage, necrosis and severe edema in focal areas of pancreas. There was obvious adipo-saponification in abdominal cavity. Characteristics such as pancreatic hemorrhage, necrosis, severe edema and adipo-saponification were found in pioglitazone group, but the levels of those injuries were lower in pioglitazone group than those in SAP group. The wet/dry pancreatic weight ratio, ascetic capacity, serum and ascitic activities of anylase
in the SAP group were significantly higher than those in the sham group and pioglitazone group respectively (6969.50 ± 1368.99 vs 2104.67 ± 377.16, 3.99 ± 1.22 vs 2.48 ± 0.74, P < 0.01 or P < 0.05). According to Kusske criteria, the pancreatic histologic score showed that interstitial edema, inflammatory infiltration, parenchyma necrosis and parenchyma hommorrhage in SAP group significantly differed from those in the sham group and pioglitazone group (7.17 ± 1.83 vs 0.50 ± 0.55, 7.67 ± 0.82 vs 6.83 ± 0.75, P < 0.01, P < 0.05. The expression of NF-κB and ICAM-1 in sham group was lower than that in SAP group and pioglitazone group (0.50 ± 0.55 vs 33 ± 1.21, P < 0.01). There was a significant difference in the expression of NF-κB and ICAM-1 between SAP group and pioglitazone group (7.50 ± 1.05 vs 11.33 ± 1.75, 0.80 ± 0.53 vs 1.36 ± 0.54, P < 0.01 or P < 0.05) at 12 h after the induction of pancreatitis.
CONCLUSION: Pioglitazone attenuates the severity of SAP. The beneficial effect of pioglitazone is multifactorial due to its anti-inflammatory activities, most likely through the inhibition of ICAM-1 expression and NF-κB activation. Specific ligands of PPARγ may represent the novel and effective means of clinical therapy for SAP.
© 2007 The WJG Press. All rights reserved.
Key words: Sodium taurocholate; Severe acute pancreatitis; Peroxisome proliferators-activated receptor-γ ligand; Nuclear transcription factor-κB; Intercellular adhesion molecule-1
Xu P, Zhou XJ, Chen LQ, Chen J, Xie Y, Lv LH, Hou XH. Pioglitazone attenuates the severity of sodium taurocholate-induced severe acute pancreatitis. World J Gastroenterol 2007; 13(13): 1983-1988
http://www.wjgnet.com/1007-9327/13/1983.asp
INTRODUCTIONThe morbidity and mortality of acute pancreatitis (AP) are high and the clinical course AP is unpredictable. Approximately 25% of AP patients develop severe inflammation with pancreatic and peripancreatic fat necrosis that requires intensive care. Effective treatment strategies are lack due to a relatively poor understanding of its exact pathogenesis[1].

The pathophysiology of acute pancreatitis involves a number of inflammatory mediators, which contribute to the initiation and progression of both local pancreatic destruction and systemic manifestat ions. Various proinflammatory cytokines, such as interleukin-1β (IL-1β), IL-6, and tumor necrosis factor-α (TNF-α), appear to play a major role in acute pancreatitis[2,3]. The inhibition of cytokine production may decrease the severity of pancreatitis[4,5].
It was recently reported that peroxisome proliferator-activated receptors (PPARs) play a modulatory role in the inflammatory response of different organs[6]. The PPAR family consists of at least three different isoforms; PPARα, PPARδ, and PPARγ[7]. PPARγ is predominantly detectable in adipose tissue, but it is also expressed in other tissues, including pancreatic tissue. When activated by a specific ligand, PPARs heterodimerize with retinoid X receptor and then bind to peroxisome proliferator response element leading to changes in the transcription of target genes involved in lipid metabolism, glucose homeostasis, cell proliferation and differentiation, and inflammatory response. PPARs are activated by natural ligands such as fatty acids, eicosanoids, oxidized fatty acids, and pharmacological compounds such as glitazones. Pioglitazone is a member of glitazones and has been used to increase the sensitivity to insulin in clinical practice[8].
Recent experimental studies appeared to have shed some light on the intracellular signaling pathway in the inflammatory cascade of AP[9,10]. For example, there is evidence that NF-κB plays a crucial role in the initiation of AP, not only in pancreatic acinar cells and monocytes/macrophages but also in specific distant organs, such as the lung. NF-κB is able to mediate a variety of inflammatory mediators involved in AP, including cytokines and adhesion molecules[11-13].
In addition, intercellular adhesion molecule-1 (ICAM-1) has been reported to be up-regulated and involved in the evolution of acute pancreatitis by recruiting leukocytes into the area of inf lammation[14,15]. NF-κB activity increases with pancreatitis. The inhibition of NF-κB has been shown to ameliorate the inflammatory effects of pancreatitis[16-19].
PPARγ plays a critical role in adipogenesis and glucose metabolism[20,21]. In addition to these effects, it has been reported that PPARγ ligands possess in vitro anti-inflammatory properties. For instance, PPARγ ligands reduce the secretion of IL-1β, IL-6, and TNF-α in monocytes and macrophages[22], and the expression of ICAM-1 in endothelial cells[23], in part by antagonizing the activities of transcription factors such as AP-1 and NF-κB[22,24].
S eve r a l r e po r t s have demons t r a t ed an an t i -inflammatory action of the specific ligands of PPARγ. Activators of PPARγ inhibit the generation of pro-inflammatory cytokines such as interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α) and interleukin-6[22] and this effect seems to be dependent on the inhibition of NF-κB pathway[25].
The aim of the present study was to determine the effect of pioglitazone, a specific PPARγ ligand, on
development of sodium taurocholate (STC)-induced SAP, to assess the effect of pretreatment with pioglitazone on expression of ICAM-1 inpanreas, and to evaluate the effect of pretreatment with pioglitazone on expression of NF-κB in pancreas.
In this study, we made a well-characterized secre-tagogue-induced murine model of pancreatit is to investigate the anti-inflammatory effects of pioglitazone, a PPARγ ligand. Pretreatment with pioglitazone markedly decreased the severity of pancreatit is, most l ikely through the inhibition of NF-κB activation and ICAM-1 expression.
Our results suggest that PPARγ can be used in the treatment of acute pancreatitis. Moreover, these findings further demonstrate NF-κB and ICAM-1 play a role in the pathogenesis of pancreatitis.
MATERIALS AND METHODSExperimental model of pancreatitisFifty-four male Sprague-Dawley (SD) rats (Animal Center, Medical College of Nanchang University), weighing 160-200 g, were used in this experiment. Animals were housed in cages under standard conditions at room temperature in a 12-h light-dark cycle. Prior to the experiments, rats were deprived of food with free access to water. All procedures including ketamine hydrochloride intra-peritoneal injection (10 mg/100 g) for anesthesia were performed under sterile conditions. SD rats were divided into severe acute pancreatitis (SAP) group, pretreatment group with pioglitazone (PP) and sham group. One mL/kg body weight of sodium taurocholate (STC, 50 g/L, Sigma) was retrograde injected into the biliopancreatic duct of the rats to induce SAP, 10% dimethyl sulphoxide (DMSO, 1 mL/100 g) was injected intraperitoneally two hours prior to STC injection[26]. PP group received pioglitazone replacing 10% DMSO administered intraperitoneally two hours prior to STC injection (2 mg/100 g). Rats in the sham group underwent operation with nothing infused. The pancreas was flipped and striked gently three times. After operation, rats were fasted with free access to water and killed by abdominal aorta exsanguination 3, 6 and 12 h after the induction of pancreatitis.
Serum and ascites were obtained to measure amylase levels and ascitic capacity. Pancreata were quickly removed and fixed in 10% formalin for morphologic studies. Portions of the pancreas were freshly processed for determining pancreatic water contents.
Macroscopic assessment of pancreasEdema, hemorrhage and necrosis of the pancreas were each graded from 0 to 3 as follows as previously described[27]: pancreatic edema: 0 = absent, 1 = mild gland swelling with no cysts, 2 = moderate gland swelling with cysts in the duodenal region, 3 = severe gland swelling with multiple cysts extending to the splenic region; pancreatic fat necrosis: 0 = absent, 1 = confined to the pancreas, 2 = extending to the omentum, 3 = widespread in the retroperitoneum, pancreatic hemorrhage: 0 = absent, 1 = periductal punctate, 2 = diffuse in pancreas, 3 = peripancreatic or pancreatic blood with clots.
1984 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

Histological scoring of pancreasPancreas was removed from each rat and fixed in 10% buffered formalin at 4 overnight. The pancreas was then embedded in paraffin. Full-length (4-μm) sections were taken and stained with hematoxylin and eosin for histologic evaluation. Edema, inflammation, hemorrhage and necrosis of the pancreas were each graded from 0 to 3 as follows as previously described[28]: edema: 0 = absent, 1 = focally increased between lobules, 2 = diffusely increased between lobules, 3 = tense acini and widely separated lobules, 4 = gross lobular separation; inflammation: 0 = absent, 1 = around ductal margins, 2 = in parenchyma (< 50% of lobules), 3 = in parenchyma (51%-75% of lobules), 4 = massive collections and abscesses; hemorrhage: 0 = absent, 1 = blood in parenchyma(< 25%), 2 = blood in parenchyma (25%-50%), 3 = blood in parenchyma (50%-75%), 4 = blood in 100% of lobules; necrosis: 0 = absent, 1 = periductal parenchymal destruction, 2 = focal parenchymal necrosis (< 20%), 3 = diffuse loss of lobules (20%-50%), 4 = severe loss of lobules (> 50%).
Biochemical assaySerum amylase levels were measured with the CNPG3 amylase test and spectrophotometrically according to the manufacturer’s instructions.
Pancreatic water contentThe wet/dry weight ratio of the pancreas was obtained when the freshly prepared pancreatic tissue was dried for 24 h at 90.
NF-κB p65 in pancreatic tissueNF-κB p65 in pancreat ic t issues was detected by immunohistochemistry. In brief, endogenous peroxidase was blocked for 30 min with 0.3% methanol-H2O2. After several washes, samples were incubated overnight in a humidified chamber at 4 with the rabbit and mouse antibody against NF-κB p65 diluted 1:100, followed by non-biotinylated second antibody diluted 1:200 for 30 min. Negative controls were carried out by omission of
the primary antibody. Several sections were counterstained with hematoxylin.
ICAM-1 in pancreatic tissueICAM-1 in pancreat ic t i ssue was deter mined by immunohistochemistry. In brief, endogenous peroxidase was blocked for 30 min with 0.3% H2O2. After several washes, samples were incubated overnight in a humidified chamber at 4 with rabbit antibody against ICAM-1 diluted 1:50, followed by non-biotinylated second antibody diluted 1:200 for 30 min. Several sections were counterstained with hematoxylin.
Statistical analysisThe values were expressed as mean ± SE. As the values in some data sets were not normally distributed or exhibited unequal standard deviations, the nonparametric Wilcoxon rank sum test was used to test for statistical differences between groups. P < 0.05 was considered statistically significant.
RESULTSAmylase assay and ascitesAfter the establishment of the model, in SAP group, the serum level of amylase was significantly higher in SAP group and PP group than that in sham group at different time points (P < 0.05, Table 1). The serum levels of amylase in PP group were significantly lower than those in SAP group at 12 h after the establishment of the studied model (P < 0.01, Table 1).
No ascites was found in sham group. No difference in ascites was found between SAP group and PP group at all time points (P > 0.05, Table 2).
Morphological examinationAfter infusion of STC, the pancreas was enlarged with a visible collection of edematous fluid and the pancreatic mass was almost doubled. Three and six hours after establishment of the studied model, typical pathological changes in SAP, such as a large number of inflammatory cells, edema, hemorrhage, necrosis, a large amount of ascites was found. The same pathological changes were observed in PP group (Table 3, Figure 1-3).
Histological examinationUnder light microscope, interlobular and intralobular edema, vacuolization of acinar cells, and moderate perivascular and scare diffuse leukocyte infiltration were observed. Inflammatory changes in the pancreas were
Table 1 Levels of amylase in three subgroups (mean ± SE)
Group Amylase (IU/L)
3 6 12Sham 2376.00 ± 413.88 2104.67 ± 377.16 1879.83 ± 528.42PP 5364.83 ± 14082bc 5645.67 ± 1907.8b 2979.50 ± 1080.12SAP 5053.33 ± 2632.1c 6969.50 ± 1368.9b 7598.00 ± 1071.92d
aP < 0.05, bP < 0.01 vs sham, cP < 0.05 vs 12 h, dP < 0.01 vs PP, fP < 0.01 vs 12 h.
Table 2 Ascites in three subgroups (mean ± SE)
Group Ascites (mL)
3 6 12Sham 0.00 ± 0.00 0.00 ± 0.00 0.00 ± 0.00PP 4.42 ± 1.85a 4.87 ± 1.04a 5.70 ± 1.87a
SAP 6.70 ± 1.70b 7.67 ± 2.47b 8.37 ± 1.19b
aP < 0.05, bP < 0.01 vs sham.
Table 3 Morphological score (mean ± SE)
Group Time point3 h 6 h 12 h
Sham 0.50 ± 0.55 0.17 ± 0.41 0.17 ± 0.41PP 5.50 ± 1.64b 6.83 ± 0.75b 4.50 ± 2.07ab
SAP 7.17 ± 1.83b 7.67 ± 0.82b 7.17 ± 1.47bd
aP < 0.05 vs PP at 6 h; bP < 0.01 vs Sham, dP < 0.01 vs PP at 12 h.
Xu P et al . Anti-inflammatory role of pioglitazone in severe acute pancreatitis 1985
www.wjgnet.com

significantly less in PP group than in SAP group (Table 4).
Pancreatic water contentThe extent of pancreatic edema and water content was estimated as the wet/dry weight ratio of the tissue (Table 5). In sham group and PP group, the pancreatic water content was reduced. However, it was increased in SAP group (P < 0.05 vs the PP group and sham group at 12 h, *P < 0.05 vs PP group at 3 h).
Expression of NF-κBp65The expression of NF-κB p65 at 3, 6 and 12 h was determined by immunohistochemistry. In sham group, no expression of NF-κB p65 in pancreas was found in
pancreas at 6 h (Figure 3A). However, the expression of NF-κB p65 in pancreas was significantly increased in SAP and PP groups compared to sham group at all time points (P < 0.01). In SAP group, the expression of NF-κB p65 in pancreas was up-regulated and differed significantly at 6, 12 and 3 h (P < 0.01, Figure 3B). In PP group, the expression of NF-κB p65 in pancreas was the highest at 6 h (Figure 3C), but was significantly lower than that in SAP group. The expression of NF-κB p65 in pancreas in PP group was significantly lower at 12 h than that in SAP group (P < 0.01).
Expression of ICAM-1In sham group, mild expression of ICAM-1 in pancreas was found only at 3 h. The expression of ICAM-1 in pancreas was significantly higher in SAP group than in sham group at all time points (P < 0.01). In SAP group, the expression of ICAM-1 in pancreas was up-regulated and differed significantly at 12 and 3 h (P < 0.01). However, in PP group, the expression of ICAM-1 in pancreas was the highest at 6 h, but was significantly lower than that in SAP group. The expression of ICAM-1 in pancreas differed significantly between PP group and SAP group at 12 h (P < 0.05, Table 6).
DISCUSSIONAlthough previous reports indicated that PPARγ agonists exhibit anti-inflammatory effects on AP in vitro and
Figure 1 Normal pancreas in sham group after the establishment of the studied model.
Table 4 Histological score (mean ± SE)
Group Time point 3 6 12
Sham 1.67 ± 2.16 0.50 ± 0.55 0.50 ± 0.55PP 8.83 ± 1.94b 7.83 ± 0.75b 7.50 ± 1.05b
SAP 9.67 ± 1.37b 10.33 ± 1.21bd 11.33 ± 1.75bd
bP < 0.01 vs sham, dP < 0.01 vs PP.
Table 5 Pancreatic water content (mean ± SE)
aP < 0.05 vs PP at 3 h, cP < 0.05 vs PP and sham at 12 h.
Group ww/dw3 h 6 h 12 h
Sham 3.10 ± 1.06 2.70 ± 0.78 2.44 ± 0.68PP 4.01 ± 1.32 2.88 ± 0.96 2.48 ± 0.74a
SAP 3.22 ± 0.81 3.65 ± 1.72 3.99 ± 1.22c
Table 6 ICAM-1 expression in acinar cells of the pancreas (mean ± SE)
aP < 0.05, bP < 0.01 vs sham, cP < 0.05 vs PP, eP < 0.05 vs 3 h.
Group Time point
3 h 6 h 12 hSham 0.19 ± 0.10 0.24 ± 0.21 0.11 ± 0.12PP 0.57 ± 0.21b 0.86 ± 0.40b 0.80 ± 0.53a
SAP 0.73 ± 0.27b 0.93 ± 0.41b 1.36 ± 0.54bce
Figure 3 Expression of p65 in sham group (A), SAP group (B), and PP group (C) after the establishment of the studied model (10 × 400).
C
BA
Figure 2 Edema, hemorrhage, necrosis, large amount of ascites in SAP group (A) and PP group (B) after the establishment of the studied model.
A B
1986 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

in vivo [22,29], the effect of PPARγ on AP has not been fully elucidated. This anti-inflammatory effect seems to be related to multiple mechanisms. Ricote et al [24] demonstrated that PPARγ ligands inhibit macrophage activation and inflammatory cytokine production. In our study, pancreatic PPARγ was induced and activated in STC-induced pancreatitis, suggesting that PPARγ plays a potential role in regulating acute inflammation.
In the present study, the serum levels of amylase and ascites decreased significantly in STC- induced pancreatitis after petreatment with pioglitazone. The pathological changes were less in pacreas such as the number of inflammatory cells, edema, hemorrhage, necrosis after pretreatment with pioglitazone. The reason for this is not entirely known, but it may be associated with an altered population of activated macrophages in the pancreas. The initiation of acute pancreatitis involves intrapancreatic activation of digestive zymogens and subsequent autodigestion, which triggers the sequestial activation of monocytes/macrophages within the pancreas and the overproduction of macrophages-derived inflammatory mediators, which result in local pancreatic injury[30,31].
Furthermore, PPARγ activators are negative regulators of macrophage activation[24] and induce macrophage apoptos i s by in ter fer ing wi th the ant i apoptot ic NF-κB pathway[32]. Therefore, we postulate that the anti-inflammatory effect of pioglitazone in STC-induced pancreatitis may be due, in part, to the inhibition of macrophage activation. Further studies are required to determine whether pioglitazone affects pancreatic macrophage activation and PPARγ expression.
Pro-inflammatory cytokines play a main role in acute pancreatitis and its complications[33,34]. Failure of different organ systems is a frequent problem in SAP. The majority of fatalities in patients with SAP are associated with the failure of at least one or more organ systems. Organ failure in SAP has to be regarded as part of the inflammatory response following the liberation of activated enzymes from the pancreas.
NF-κB plays a key role in the transcriptional regulation of adhesion molecules, enzymes and cytokines involved in acute inflammatory diseases. NF-κB is a key transcription factor for the expression of various proinflammatory molecules such as chemokines, cytokines and adhesion molecules. This notion has recently been verified by adenoviral-mediated gene transfer of an active subunit, RelA/p65, into acinar cells into the rat pancreatic duct[35]. Over-expressed RelA/p65 protein transactivates NF-κB and induces pancrea t i c in f l ammat ion , a pathological state very similar to acute pancreatitis. In addition to the importance of NF-κB in the pathogenesis of acute pancreatitis, it was also reported that NF-κB activation in macrophages is a key event in the development of generalized complications and a cause of the high mortality of SAP. Studies also showed that glucocorticoid is one of the most potent inhibitors of NF-κB, which might be attributable to the inhibition of this key transcription factor both in acinar cells and in inflammatory cells such as monocytes/macrophages. Because the activation of NF-κB is an early event in acute pancreatitis, corticosteroid has beneficial effects on acute pancreatitis[36].
The transcription factor NF-kB is activated in the course of acute pancreatitis[16,37] and regulates several inflammatory processes by mediating the induction of numerous inf lammator y mediators, including proinflammatory cytokines and ICAM-1[15,38,39]. Our results suggest that pioglitazone could attenuate the severity of STC-induced pancreatitis by inhibiting NF-κB. Therefore, the observed beneficial effects of pioglitazone could be mediated, in part, by inhibiting NF-κB activity.
Similarly, other studies have demonstrated that nonspecific NF-κB inhibitors ameliorate the course of experimental pancreatitis[16-18]. Our findings suggest that pioglitazone can inhibit NF-κB activity. The deleterious role of ICAM-1 in inducing acute pancreatitis has been reported[40]. Cerulein induced pancreatitis in ICAM-1 knockout mice showed reduced severity of pancreatitis and of pancreatitis-associated lung injury[14]. ICAM-1, present not only in endothelial cells but also in acinar cells of the pancreas, is up-regulated by cerulein and mediates direct binding of neutrophils to acinar cells[15].
Our present study demonstrated that the expression of ICAM-1 was increased in acinar cells of pancreas with STC-induced pancreatitis, which is consistent with previous reports[14,15,40]. Decreased expression of ICAM-1 in pancreas after treatment with pioglitazone provides a possible explanation for the decreased pancreatic infiltration of inflammatory cells, especially neutrophils.
In summary, pioglitazone, a specific PPARγ ligand, ameliorates significantly the severity of STC-induced SAP. The expression of NF-κB and ICAM-1 in pancreas can be significantly inhibited by pioglitazone. Treatment of acute pancreatitis with pioglitazone may offer a new therapeutic option.
REFERENCES1 Chiang DT, Anozie A, Fleming WR, Kiroff GK. Comparative
study on acute pancreatitis management. ANZ J Surg 2004; 74: 218-221
2 Saluja AK. Pathophysiology of pancreatitis. Role of cytokines and other mediators of inflammation. Digestion 1999; 60 Suppl 1: 27-33
3 Norman J. The role of cytokines in the pathogenesis of acute pancreatitis. Am J Surg 1998; 175: 76-83
4 Rongione AJ, Kusske AM, Kwan K, Ashley SW, Reber HA, McFadden DW. Interleukin 10 reduces the severity of acute pancreatitis in rats. Gastroenterology 1997; 112: 960-967
5 Norman J, Franz M, Messina J, Riker A, Fabri PJ, Rosemurgy AS, Gower WR. Interleukin-1 receptor antagonist decreases severity of experimental acute pancreatitis. Surgery 1995; 117: 648-655
6 Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors and inflammation: from basic science to clinical applications. Int J Obes Relat Metab Disord 2003; 27 Suppl 3: S41-S45
7 Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell 1995; 83: 835-839
8 Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med 2002; 53: 409-435
9 Frossard JL, Pastor CM, Hadengue A. Effect of hyperthermia on NF-kappaB binding activity in cerulein-induced acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 2001; 280: G1157-G1162
Xu P et al . Anti-inflammatory role of pioglitazone in severe acute pancreatitis 1987
www.wjgnet.com

10 Kim H , Seo JY, Kim KH. NF-kappaB and cytokines in pancreatic acinar cells. J Korean Med Sci 2000; 15 Suppl: S53-S54
11 Jaffray C, Yang J, Carter G, Mendez C, Norman J. Pancreatic elastase activates pulmonary nuclear factor kappa B and inhibitory kappa B, mimicking pancreatitis-associated adult respiratory distress syndrome. Surgery 2000; 128: 225-231
12 Rakonczay Z, Jármay K, Kaszaki J, Mándi Y, Duda E, Hegyi P, Boros I, Lonovics J, Takács T. NF-kappaB activation is detrimental in arginine-induced acute pancreatitis. Free Radic Biol Med 2003; 34: 696-709
13 Altavilla D, Famulari C, Passaniti M, Campo GM, Macrì A, Seminara P, Marini H, Calò M, Santamaria LB, Bono D, Venuti FS, Mioni C, Leone S, Guarini S, Squadrito F. Lipid peroxidation inhibition reduces NF-kappaB activation and attenuates cerulein-induced pancreatitis. Free Radic Res 2003; 37: 425-435
14 Frossard JL, Saluja A, Bhagat L, Lee HS, Bhatia M, Hofbauer B, Steer ML. The role of intercellular adhesion molecule 1 and neutrophils in acute pancreatitis and pancreatitis-associated lung injury. Gastroenterology 1999; 116: 694-701
15 Zaninovic V, Gukovskaya AS, Gukovsky I, Mouria M, Pandol SJ. Cerulein upregulates ICAM-1 in pancreatic acinar cells, which mediates neutrophil adhesion to these cells. Am J Physiol Gastrointest Liver Physiol 2000; 279: G666-G676
16 Gukovsky I, Gukovskaya AS, Blinman TA, Zaninovic V, Pandol SJ. Early NF-kappaB activation is associated with hormone-induced pancreatitis. Am J Physiol 1998; 275 : G1402-G1414
17 Satoh A, Shimosegawa T, Fujita M, Kimura K, Masamune A, Koizumi M, Toyota T. Inhibition of nuclear factor-kappaB activation improves the survival of rats with taurocholate pancreatitis. Gut 1999; 44: 253-258
18 Vaquero E, Gukovsky I, Zaninovic V, Gukovskaya AS, Pandol SJ. Localized pancreatic NF-kappaB activation and inflammatory response in taurocholate-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol 2001; 280: G1197-G1208
19 Ethridge RT, Hashimoto K, Chung DH, Ehlers RA, Rajaraman S, Evers BM. Selective inhibition of NF-kappaB attenuates the severity of cerulein-induced acute pancreatitis. J Am Coll Surg 2002; 195: 497-505
20 Chawla A , Schwarz EJ, Dimaculangan DD, Lazar MA. Peroxisome proliferator-activated receptor (PPAR) gamma: adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology 1994; 135: 798-800
21 Nolan JJ , Ludvik B, Beerdsen P, Joyce M, Olefsky J . Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N Engl J Med 1994; 331: 1188-1193
22 Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998; 391: 82-86
23 Pasceri V, Wu HD, Willerson JT, Yeh ET. Modulation of vascular inflammation in vitro and in vivo by peroxisome proliferator-activated receptor-gamma activators. Circulation 2000; 101: 235-238
24 Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998; 391:
79-8226 Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh
SA, Flanigan A, Murthy S, Lazar MA, Wu GD. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J Clin Invest 1999; 104: 383-389
27 Goldacre MJ, Roberts SE. Hospital admission for acute pancreatitis in an English population, 1963-98: database study of incidence and mortality. BMJ 2004; 328: 1466-1469
28 Hughes CB, Grewal HP, Gaber LW, Kotb M, El-din AB, Mann L, Gaber AO. Anti-TNFalpha therapy improves survival and ameliorates the pathophysiologic sequelae in acute pancreatitis in the rat. Am J Surg 1996; 171: 274-280
29 Kusske AM, Rongione AJ, Ashley SW, McFadden DW, Reber HA. Interleukin-10 prevents death in lethal necrotizing pancreatitis in mice. Surgery 1996; 120: 284-288; discussion 289
30 Hashimoto K, Ethridge RT, Saito H, Rajaraman S, Evers BM. The PPARgamma ligand, 15d-PGJ2, attenuates the severity of cerulein-induced acute pancreatitis. Pancreas 2003; 27: 58-66
32 Norman JG, Fink GW, Denham W, Yang J, Carter G, Sexton C, Falkner J, Gower WR, Franz MG. Tissue-specific cytokine production during experimental acute pancreatitis. A probable mechanism for distant organ dysfunction. Dig Dis Sci 1997; 42: 1783-1788
33 Bhatia M, Brady M, Shokuhi S, Christmas S, Neoptolemos JP, Slavin J. Inflammatory mediators in acute pancreatitis. J Pathol 2000; 190: 117-125
34 Chinetti G, Griglio S, Antonucci M, Torra IP, Delerive P, Majd Z, Fruchart JC, Chapman J, Najib J, Staels B. Activation of proliferator-activated receptors alpha and gamma induces apoptosis of human monocyte-derived macrophages. J Biol Chem 1998; 273: 25573-25580
35 Osman MO, Gesser B, Mortensen JT, Matsushima K, Jensen SL, Larsen CG. Profiles of pro-inflammatory cytokines in the serum of rabbits after experimentally induced acute pancreatitis. Cytokine 2002; 17: 53-59
36 Bidarkundi GK, Wig JD, Bhatnagar A, Majumdar S. Clinical relevance of intracellular cytokines IL-6 and IL-12 in acute pancreatitis, and correlation with APACHE III score. Br J Biomed Sci 2002; 59: 85-89
37 Chen X, Ji B, Han B, Ernst SA, Simeone D, Logsdon CD. NF-kappaB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology 2002; 122: 448-457
38 Takaoka K, Kataoka K, Sakagami J. The effect of steroid pulse therapy on the development of acute pancreatitis induced by closed duodenal loop in rats. J Gastroenterol 2002; 37: 537-542
39 Steinle AU, Weidenbach H, Wagner M, Adler G, Schmid RM. NF-kappaB/Rel activation in cerulein pancreatitis. Gastroenterology 1999; 116: 420-430
40 Ghosh S. Regulation of inducible gene expression by the transcription factor NF-kappaB. Immunol Res 1999; 19: 183-189
41 Mercurio F, Manning AM. NF-kappaB as a primary regulator of the stress response. Oncogene 1999; 18: 6163-6171
42 Sun W, Watanabe Y, Wang ZQ. Expression and significance of ICAM-1 and its counter receptors LFA-1 and Mac-1 in experimental acute pancreatitis of rats. World J Gastroenterol 2006; 12: 5005-5009
S- Editor Wang J L- Editor Wang XL E- Editor Chin GJ
1988 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

combination of MK-AS and ADM has a much better in vitro and in vivo synergism.
© 2007 The WJG Press. All rights reserved.
Key words: Antisense oligonucleotide; Midkine; Carci-noma; Hepatocellular; Combination therapy
Dai LC, Wang X, Yao X, Lu YL, Ping JL, He JF. Enhanced therapeutic effects of combined chemotherapeutic drugs and midkine antisense oligonucleotides for hepatocellular carcinoma. World J Gastroenterol 2007; 13(13): 1989-1994
http://www.wjgnet.com/1007-9327/13/1989.asp
INTRODUCTIONHepatocellular carcinoma (HCC) is one of the malignant diseases with a high incidence and mortality in the world, which is often diagnosed at an advanced stage when most potentially curative therapies such as resection, transplantation or percutaneous and transar terial interventions are of limited efficacy[1-4]. It is accepted that there is no satisfactory treatment available for patients with HCC and chemotherapy has been extremely disappointing[5-7]. The poor prognosis of patients with HCC and the lack of satisfactory therapy for advanced cases indicate a need for more effective therapeutic options. Recent insights into the biology of HCC suggest that certain pathways and molecular alterations are likely to play essential roles in HCC development by promoting cell growth and survival. Dysregulation of growth factors, receptors and their downstream signaling pathway components represent a central pro-tumorigenic principle in human hepatocarcinogenesis.
Growth factors and the downstream signaling system are often overexpressed in tumors, and become the target of their treatment. It has been reported that MK, a heparin-binding growth factor or cytokine, is usually overexpressed in various malignant tumors, such as lung, breast, esophageal, gastric, colorectal, liver, pancreatic, ovarian, urinary bladder, prostatic, cerebral and renal malignancies[8-16], whereas in normal adult tissues, MK is low or undetectable[2,16]. Midkine (MK) can promote the growth, survival, and migration of various target cells[17].
RAPID COMMUNICATION
Enhanced therapeutic effects of combined chemotherapeutic drugs and midkine antisense oligonucleotides for hepatocellular carcinoma
Li-Cheng Dai, Xiang Wang, Xing Yao, Yong-Liang Lu, Jin-Liang Ping, Jian-Fang He
www.wjgnet.com
Li-Cheng Dai, Xiang Wang, Jian-Fang He, Huzhou Key Laboratory of Molecular Medicine, Huzhou Central Hospital, Huzhou 313000, Zhejiang Province, ChinaXing Yao, Yong-Liang Lu, Department of General Surgery, Huzhou Central Hospital, Huzhou 313000, Zhejiang Province, ChinaJin-Liang Ping, Department of Pathology, Huzhou Central Hospital, Huzhou 313000, Zhejiang Province, ChinaSupported by grants from the Zhejiang Province Medicine and Health Research Fund, No. 2003A077 and Huzhou Natural Science Foundation, No. 2004SZX07-11, ChinaCorrespondence to: Li-Cheng Dai, Professor, Huzhou Key Laboratory of Molecular Medicine, Huzhou Central Hospital, Huzhou 313000, Zhejiang Province, China. [email protected]: +86-572-2033020 Fax: +86-572-2033020Received: 2007-01-11 Accepted: 2007-03-15
AbstractAIM: To evaluate the effect of combined antisense ol igonucleotides targeting midkine (MK-AS) and chemotherapeutic drugs [cisplatin(DDP), 5-fluorouracil (5-FU) and adriamycin (ADM)] on inhibition of HepG2 cell proliferation, and to analyze the efficacy of MK-AS used in combined ADM in in situ human hepatocellular carcinoma (HCC) model.
METHODS: HepG2 cells were treated with MK-AS and/or chemotherapeutic drugs mediated by Lipofectin, and cell growth activity was determined by MTS assay. An in situ HCC model was used in this experiment. MK-AS, ADM and MK-AS + ADM were given intravenously for 20 d, respectively. The animal body weight and their tumor weight were measured to assess the effect of the combined therapy in vivo .
RESULTS: Combined treatment with MK-AS reduced the IC50 of DDP, 5-FU and ADM in HepG2 cells. MK-AS significantly increased the inhibition rate of DDP, 5-FU and ADM. Additionally, synergism (Q 1.15) occurred at a lower concentration of ADM, 5-FU and DDP with combined MK-AS. Combined treatment with MK-AS and ADM resulted in the more growth inhibition on in situ human HCC model compared with treatment with chemotherapeutic drugs alone.
CONCLUSION: MK-AS increases the chemosensitivity in HepG2 cells and in situ human HCC model, and the
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1989-1994www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.

The level of midkine expression correlates negatively with the patients’ prognosis. It is reported that antisense oligo (DNA) targeting MK suppresses the growth of tumors in nude mice[18,19]. Addtionally, siRNA or antisense oligo (DNA) targeting against MK inhibits neointima formation[20] and renal injury after ischemia[21]. All of these studies suggested that midkine may play an important role in carcinogenesis, development and metastasis of tumors, and that it could serve as a novel tumor marker.
A novel approach to cancer therapy is the integration of new cytotoxic agents with multiple selective inhibitors of relevant signaling pathways. Such a strategy indicates that it may be possible to use agents that cooperate at low doses and/or through simple administration modalities to increase tumor targeting and patient compliance and reduce toxicity. We intend to explore the potential of the antisense targeting against MK (MK-AS) combined with chemotherapeutic drugs (ADM, DDP and 5-FU) in inhibition of HCC growth.
MATERIALS AND METHODSOligonucleotides synthesis and drugs Antisense phosphorothioate ol igonucleotide (5’- CCCCGGGCCGCCCTTCTTCA-3’) were synthesized by an Applied Biosystems Model 391 DNA synthesizer on solid supports using Oligo Pilot Ⅱ DNA (Amersham-Pharmacia, Piscataway, NJ, USA) and purified by HPLC Prep 4000 (Waters Delta, USA) with SOURCE 15Q (Amersham-Pharmacia, USA). Chemotherapeutic drugs (DDP, 5-FU and ADM) were purchased from the Shanghai Donghaipu Pharmaceuticals Company (Shanghai, China).
Cell culture conditionsHuman HCC cell (HepG2) was obtained from the Cancer Institute, Chinese Academy of Medical Sciences. HepG2 cells were cultured in DMEM (GIBCO BRL, Grand Island, NY, USA), supplemented with 10% FCS (GIBCO BRL), 100 U/mL penicillin and 100 U/mL streptomycin. Tumor cells were kept at 37 in a humidified atmosphere containing 50 mL/L CO2.
MK-AS and chemotherapeutic treatment3 × 103 cells were seeded in 96-well microtiter plate and allowed to attach overnight. MK-AS was then tranfected into these cells mediated by Lipofectin (Invitrogen, USA). After incubation for 6 h, the cultural medium was replaced with 100 μL of cell culture medium with different chemotherapeutic drugs. The concentrations were used in this experiment around the IC50 of each chemotherapeutic drug (DDP: 0.25, 0.5, 1, 2, 4 mg/L; 5-FU: 1.0, 2.5, 5, 10, 20 mg/L; and ADM: 0.025, 0.05, 0.1, 0.2, 0.4 mg/L). Additionally, the concentrations of each anticancer drug were combined with three concentrations of MK-AS (0.1, 0.2, 0.4 μmol/L) respectively. After 48 h incubation, 20 μL of MTS (Promega, USA) was added into each well, followed by a 90 min incubation at 37. MTS assay was then used to assess the cell proliferation with a Victor 1420 Multilable Counter (WALLAC, USA). Zheng-Jun Jin’s methods[22] was used to analyze the combined effect (the antagonistic effects: Q < 0.85; the additive effect: 0.85 ≤
Q < 1.15; and the synergistic effects: Q ≥ 1.15).
Analysis of combined effects The Balb/c nude mice (virgin female) used in these experiments were obtained from the Academy of Military Medical Sciences (Beijing, China). The human HCC model used in this experiment was described previously[23]. Briefly, the HCM-Y89 tumor was cut into 1 mm × 1 mm× 1 mm tissues, and implanted into the liver of mice. Two days after development of in situ HCC models, mice were injected intravenously with saline (saline alone used as control), MK-AS of 50 mg/kg per day and/or ADM of 10 mg/kg per day for 20 d. The body weight and general physical status of the animals were recorded daily. Mice were killed at different time points by cervical dislocation and the tumors were removed and weighed.
Western blotting and RT-PCR The total RNA was extracted and RT-PCR reaction was performed using an RT-PCR kit (Promega, Madison, WI, USA). PCR products were analyzed using 2.0% agarose gel and visualized by ethidium bromide staining. For Western-blotting, the tumor tissues were lysed with lysis buffer (50 mmol/L Tris-HCl, pH 7.4, 0.5 mmol/L EDTA, 0.5% NP40, and 150 mmol/L NaCl) in the presence of protease inhibitors. The lysates were then centrifuged at 15 000 ×g for 15 min to remove debris. Protein samples (60 μg) were separated by 12% SDS-PAGE gel and transferred onto PVDF membranes (Hybond-polyvinylidene difluoride membranes, Amersham Biosciences). The reactive band was visualized with an ECL-plus Detection Kit (Amersham Biosciences, Piscataway, NJ) and scanned by Gel Doc 1000 (Bio-Rad CA, USA). β-actin was used as a control.
Statistical analysisData were expressed as means ± SD, statistical analysis was carried out using Student’s t test (two tailed), and P < 0.05 indicates statistical significance.
RESULTSMK-AS transfer increases the cytotoxicity of DDP, 5-Fu and ADM in HepG2After transfection with MK-AS, cells were treated with 5-FU, ADM or DDP at different concentrations. Transfection of MK-AS was found to enhance the cytotoxicity of 5-FU, ADM and DDP significantly. As shown in Figure 1A, the IC50 of ADM alone is 0.109 mg/L. However, combined ADM and MK-AS (0.1 μmol/L) decreased the IC50 from 0.109 mg/L to 0.0517 mg/L. Meanwhile, we also observed that 0.1 μmol/L MK-AS decreased the IC50 of 5-FU from 5.6147 mg/L to 2.61 mg/L (Figure 1B), and the IC50 of DDP from 1.048 mg/L to 0.594 mg/L. All these results indicated that MK-AS transfer increased the chemosensitivity in HepG2 cells.
MK-AS synergistically interacts with chemotherapeutic drugs in HepG2Furthermore, we used Zheng-Jun Jin’s method to analyze the antagonism, additivity or synergy of the
1990 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com
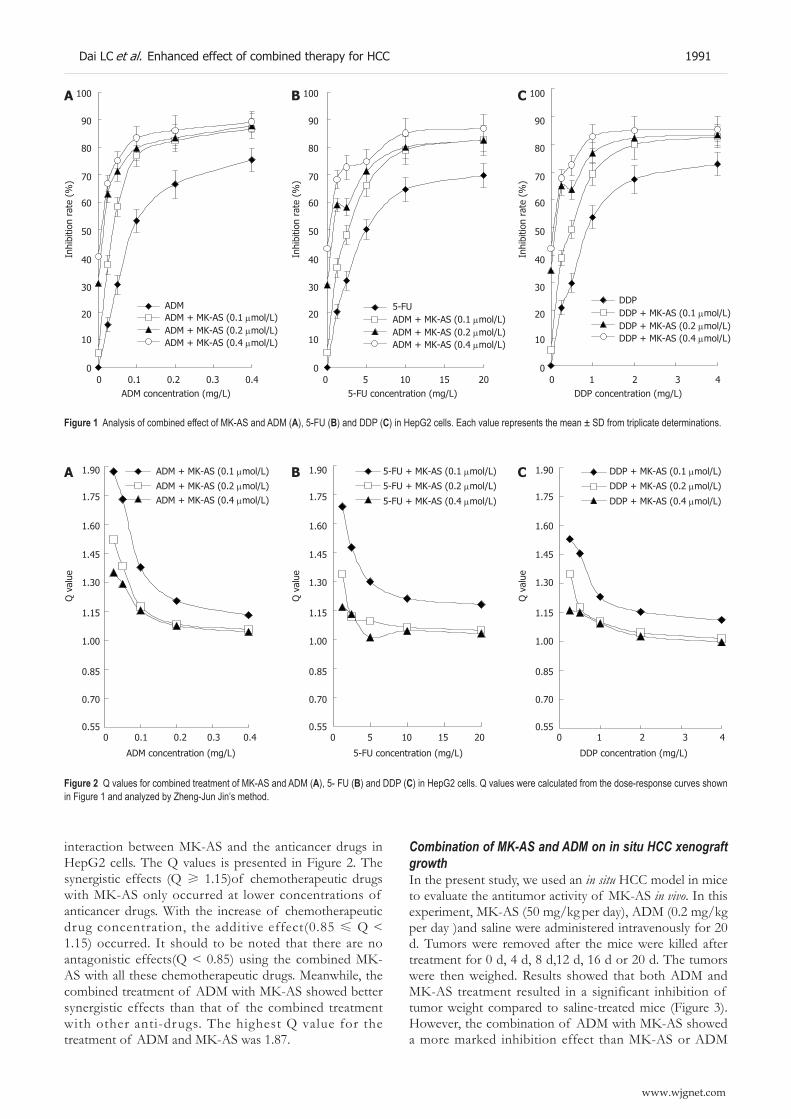
interaction between MK-AS and the anticancer drugs in HepG2 cells. The Q values is presented in Figure 2. The synergistic effects (Q ≥ 1.15)of chemotherapeutic drugs with MK-AS only occurred at lower concentrations of anticancer drugs. With the increase of chemotherapeutic drug concentration, the additive effect(0.85 ≤ Q < 1.15) occurred. It should to be noted that there are no antagonistic effects(Q < 0.85) using the combined MK-AS with all these chemotherapeutic drugs. Meanwhile, the combined treatment of ADM with MK-AS showed better synergistic effects than that of the combined treatment with other anti-drugs. The highest Q value for the treatment of ADM and MK-AS was 1.87.
Combination of MK-AS and ADM on in situ HCC xenograft growthIn the present study, we used an in situ HCC model in mice to evaluate the antitumor activity of MK-AS in vivo. In this experiment, MK-AS (50 mg/kg per day), ADM (0.2 mg/kg
per day )and saline were administered intravenously for 20 d. Tumors were removed after the mice were killed after treatment for 0 d, 4 d, 8 d,12 d, 16 d or 20 d. The tumors were then weighed. Results showed that both ADM and MK-AS treatment resulted in a significant inhibition of tumor weight compared to saline-treated mice (Figure 3). However, the combination of ADM with MK-AS showed a more marked inhibition effect than MK-AS or ADM
100
90
80
70
60
50
40
30
20
10
0 0 0.1 0.2 0.3 0.4
ADMADM + MK-AS (0.1 μmol/L)ADM + MK-AS (0.2 μmol/L)ADM + MK-AS (0.4 μmol/L)
ADM concentration (mg/L)
Inhi
bitio
n ra
te (
%)
A 100
90
80
70
60
50
40
30
20
10
0
Inhi
bitio
n ra
te (
%)
B
0 5 10 15 205-FU concentration (mg/L)
5-FUADM + MK-AS (0.1 μmol/L)ADM + MK-AS (0.2 μmol/L)ADM + MK-AS (0.4 μmol/L)
100
90
80
70
60
50
40
30
20
10
0
Inhi
bitio
n ra
te (
%)
C
0 1 2 3 4DDP concentration (mg/L)
DDPDDP + MK-AS (0.1 μmol/L)DDP + MK-AS (0.2 μmol/L)DDP + MK-AS (0.4 μmol/L)
Figure 1 Analysis of combined effect of MK-AS and ADM (A), 5-FU (B) and DDP (C) in HepG2 cells. Each value represents the mean ± SD from triplicate determinations.
1.90
1.75
1.60
1.45
1.30
1.15
1.00
0.85
0.70
0.55 0 0.1 0.2 0.3 0.4
ADM + MK-AS (0.1 μmol/L)
ADM + MK-AS (0.2 μmol/L)ADM + MK-AS (0.4 μmol/L)
ADM concentration (mg/L)
Q v
alue
A 1.90
1.75
1.60
1.45
1.30
1.15
1.00
0.85
0.70
0.55 0 5 10 15 20
5-FU + MK-AS (0.1 μmol/L)
5-FU + MK-AS (0.2 μmol/L)
5-FU + MK-AS (0.4 μmol/L)
5-FU concentration (mg/L)
Q v
alue
B 1.90
1.75
1.60
1.45
1.30
1.15
1.00
0.85
0.70
0.55 0 1 2 3 4
DDP + MK-AS (0.1 μmol/L)
DDP + MK-AS (0.2 μmol/L)
DDP + MK-AS (0.4 μmol/L)
DDP concentration (mg/L)
Q v
alue
C
Figure 2 Q values for combined treatment of MK-AS and ADM (A), 5- FU (B) and DDP (C) in HepG2 cells. Q values were calculated from the dose-response curves shown in Figure 1 and analyzed by Zheng-Jun Jin’s method.
Dai LC et al. Enhanced effect of combined therapy for HCC 1991
www.wjgnet.com

treatment alone. Additionally, the mean body weight of the mice was not significantly different between the groups from the beginning to the end (Figure 4), suggesting that all the treatment groups showed no remarkable toxicity in animal.
MK-AS treatment decreased MK mRNA and protein levelWe analyzed the effect of ADM or MK-AS treatment in MK mRNA and protein in HepG2 cells (Figure 5). Results showed that MK treatment alone or combination with ADM significantly downregulated MK expression including the protein and mRNA level in in situ HCC
model in mice. However, ADM treatment alone has little effect on MK expression.
DISCUSSIONMK is a heparin-binding growth factor identified as a product of a retinoic acid response gene[24,25]. The pathophysiological effects of MK include an enhanced plasminogen activity[26], oncogenic transformation of fibroblasts[27], antiapoptotic activity[28], and angiogenic activity[29]. MK and pleiotrophin (also known as the heparin-binding growth-associated molecule) comprise a family of heparin-binding growth/differentiation factors, different from other heparin-binding growth factors such as fibroblast growth factor and hepatocyte growth factor[2-4]. It was reported that MK activates mitogen-activated protein kinase pathways through inducing the phosphorylation of p44/42 MAPK, subsequently promoting cell growth[30]. Moreover, MK also activates extracellular signal-regulated kinases 1 and 2, which are well known as signal transducers[28]. The activated MAPK pathway was believed to downregulate caspase-3 activity in neurons[30]. In addition, MK can induce phosphorylation of protein kinase B. The phosphorylated AKT can promote a series of anti-apoptosis pathways in cells. Recently, HCC tumor cells were found to overexpress MK[31]. These findings suggest that MK could be a potential target for HCC therapy.
At present, although surgery and chemotherapy arealthough surgery and chemotherapy are effective in patients with localized tumors, the prognosis of patients with advanced or metastatic tumors is not ideal. Therefore, novel treatment approaches to the cancer are urgently needed. We analyzed the combination of MK-AS with chemotherapeutic drugs. The results showed that MK-AS transfer increased the anti-cancer effect of chemotherapeutic drugs. Furthermore, we also observed the synergism at a lower concentration of all these chemotherapeutic drugs. This result implied that combination of MK-AS with chemotherapeutic drugs will possibly provide a more marked therapeutic effect. ADM is a reagent that leads to DNA break in cells. However,
23
22
21
20
19
18
17
16
MK-AS
ADMMK-AS + ADM
CK
0 4 8 12 16 20t (treatment)/d
Anim
al w
eigh
t (g
)
Figure 4 The human HCC model animal weight after treatment with MK-AS and/or ADM. CK: Treatment with saline; ADM: Treatment with ADM; MK-AS: Treatment with MK-AS; ADM + MK-AS: Combination of ADM with MK-AS. Each value represents the mean ± SD from 7 determinations.
2000
1600
12001000750
500
250
100
GAPDHMK
A
B CK ADM MK-AS MK-AS + ADM
MK
β-actin
Figure 5 Effects of MK-AS and ADM on MK expression in in situ human hepatocellular carcinoma (HCC) model. A: Electrophoresis of RT-PCR products of MK gene and GAPDH gene, GAPDH is used as control; B: The total protein were separated by SDS gel electrophoresis, transferred to PVDF membranes, and blotted with anti-MK body. β-actin rabbit polyclonal antibodies was used as a loading control. CK: Treatment with saline; ADM: Treatment with ADM; MK-AS: Treatment with MK-AS; ADM + MK-AS: Combination of ADM with MK-AS.
Figure 3 Tumor growth in situ human HCC model after treatment with MK-AS and/or ADM. CK: Treatment with saline; ADM: Treatment with ADM; MK-AS: Treatment with MK-AS; ADM + MK-AS: Combination of ADM with MK-AS. Each value represents the mean ± SD from 7 determinations.
MK-AS
ADM
MK-AS + ADM
CK
1.6
1.4
1.2
1.0
0.8
0.6
0.4
0.2
0.0 0 4 8 12 16 20
Tum
or w
eigh
t (g
)
t (treatment)/d
Marker CK ADM MK-AS MK-AS + ADM
1992 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

MK-AS can inhibit MK growth factor expression. Taken together, we supposed that this good synergism might be due to the different antitumor functions.
Considering the in vitro and in vivo difference, we then analyzed the combination of MK-AS with ADM in in situ human HCC model. Both the MK-AS and ADM administration showed a remarkable inhibition of tumor weight. Interestingly, the combination decreased the tumor weight more than MK-AS or ADM treatment alone. Our experimental model of HCC has some advantages compared with the inoculated tumor model[23]. The tumor originated from human HCC maintained a series of characteristics of human HCC tissues, such as AFP secretion and drug sensitivity. Additionally, the pathological evidence also suggests that this model shows various features in clinical HCC patients. Therefore, the results obtained from this model can reflect the true clinical picture of patients to some degree. We also observed that MK-AS suppressed MK expression, including the mRNA and protein. However, ADM treatment showed little effect on MK expression, suggesting that the suppressed tumor effect of MK-AS is due to the inhibition of MK expression.
In summary, our results suggest that MK-AS can increase the therapeutic effect of chemotherapeutic drugs both in vivo and in vitro. The combined ADM and MK-AS showed a better synergism at a low concentration, which will provide another possible strategy for cancer treatment.
REFERENCES1 Kadomatsu K, Tomomura M, Muramatsu T. cDNA cloning
and sequencing of a new gene intensely expressed in early differentiation stages of embryonal carcinoma cells and in mid-gestation period of mouse embryogenesis. Biochem Biophys Res Commun 1988; 151: 1312-1318
2 Li YS, Milner PG, Chauhan AK, Watson MA, Hoffman RM, Kodner CM, Milbrandt J, Deuel TF. Cloning and expression of a developmentally regulated protein that induces mitogenic and neurite outgrowth activity. Science 1990; 250: 1690-1694
3 Merenmies J, Rauvala H. Molecular cloning of the 18-kDa growth-associated protein of developing brain. J Biol Chem 1990; 265: 16721-16724
4 Muramatsu T. Midkine (MK), the product of a retinoic acid responsive gene, and pleiotrophin constitute a new protein family regulating growth and differentiation. Int J Dev Biol 1993; 37: 183-188
5 Minemura M, Tanimura H, Tabor E. Overexpression of multidrug resistance genes MDR1 and cMOAT in human hepatocellular carcinoma and hepatoblastoma cell lines. Int J Oncol 1999; 15: 559-563
6 Huang M, Liu G. The study of innate drug resistance of human hepatocellular carcinoma Bel7402 cell line. Cancer Lett 1999; 135: 97-105
7 Nies AT, König J, Pfannschmidt M, Klar E, Hofmann WJ, Keppler D. Expression of the multidrug resistance proteins MRP2 and MRP3 in human hepatocellular carcinoma. Int J Cancer 2001; 94: 492-499
8 Garver RI, Chan CS, Milner PG. Reciprocal expression of pleiotrophin and midkine in normal versus malignant lung tissues. Am J Respir Cell Mol Biol 1993; 9: 463-466
9 Garver RI, Radford DM, Donis-Keller H, Wick MR, Milner PG. Midkine and pleiotrophin expression in normal andMidkine and pleiotrophin expression in normal and malignant breast tissue. Cancer 1994; 74: 1584-1590
10 Aridome K, Tsutsui J, Takao S, Kadomatsu K, Ozawa M,
Aikou T, Muramatsu T. Increased midkine gene expression in human gastrointestinal cancers. Jpn J Cancer Res 1995; 86: 655-661
11 Nakanishi T , Kadomatsu K, Okamoto T, Tomoda Y, Muramatsu T. Expression of midkine and pleiotropin in ovarian tumors. Obstet Gynecol 1997; 90: 285-290
12 O’Brien T, Cranston D, Fuggle S, Bicknell R, Harris AL. The angiogenic factor midkine is expressed in bladder cancer, and overexpression correlates with a poor outcome in patients with invasive cancers. Cancer Res 1996; 56: 2515-2518
13 Konishi N , Nakamura M, Nakaoka S , Hiasa Y, Cho M, Uemura H, Hirao Y, Muramatsu T, Kadomatsu K. Immunohistochemical analysis of midkine expression in human prostate carcinoma. Oncology 1999; 57: 253-257
14 Mishima K, Asai A, Kadomatsu K, Ino Y, Nomura K, Narita Y, Muramatsu T, Kirino T. Increased expression of midkine during the progression of human astrocytomas. Neurosci Lett 1997; 233: 29-32
15 Nakagawara A, Milbrandt J, Muramatsu T, Deuel TF, Zhao H, Cnaan A, Brodeur GM. Differential expression of pleiotrophin and midkine in advanced neuroblastomas. Cancer Res 1995; 55: 1792-1797
16 Tsutsui J, Kadomatsu K, Matsubara S, Nakagawara A, Hamanoue M, Takao S, Shimazu H, Ohi Y, Muramatsu T. A new family of heparin-binding growth/differentiation factors: increased midkine expression in Wilms’ tumor and other human carcinomas. Cancer Res 1993; 53: 1281-1285
17 Muramatsu T. Midkine and pleiotrophin: two related proteins involved in development, survival, inflammation and tumorigenesis. J Biochem 2002; 132: 359-371
18 Takei Y, Kadomatsu K, Matsuo S, Itoh H, Nakazawa K, Kubota S, Muramatsu T. Antisense oligodeoxynucleotide targeted to Midkine, a heparin-binding growth factor, suppresses tumorigenicity of mouse rectal carcinoma cells. Cancer Res 2001; 61: 8486-8491
19 Takei Y, Kadomatsu K, Goto T, Muramatsu T. Combinational antitumor effect of siRNA against midkine and paclitaxel on growth of human prostate cancer xenografts. Cancer 2006; 107: 864-873
20 Banno H, Takei Y, Muramatsu T, Komori K, Kadomatsu K. Controlled release of small interfering RNA targeting midkine attenuates intimal hyperplasia in vein grafts. J Vasc Surg 2006; 44: 633-641
21 Sato W , Takei Y, Yuzawa Y, Matsuo S, Kadomatsu K, Muramatsu T. Midkine antisense oligodeoxyribonucleotide inhibits renal damage induced by ischemic reperfusion. Kidney Int 2005; 67: 1330-1339
22 Jin ZJ. Addition in drug combination (author's transl). Zhongguo Yaoli Xuebao 1980; 1: 70-76
23 Lin RX, Tuo CW, Lü QJ, Zhang W, Wang SQ. Inhibition of tumor growth and metastasis with antisense oligonucleotides (Cantide) targeting hTERT in an in situ human hepatocellular carcinoma model. Acta Pharmacol Sin 2005; 26: 762-768
24 Tomomura M, Kadomatsu K, Matsubara S, Muramatsu T. A retinoic acid-responsive gene, MK, found in the teratocarcinoma system. Heterogeneity of the transcript and the nature of the translation product. J Biol Chem 1990; 265: 10765-10770
25 Ka do m a t su K , H u a n g R P , Su g a n u ma T , Mu r a t a F , Muramatsu T. A retinoic acid responsive gene MK found in the teratocarcinoma system is expressed in spatially and temporally controlled manner during mouse embryogenesis. J Cell Biol 1990; 110: 607-616
26 Kojima S, Inui T, Muramatsu H, Kimura T, Sakakibara S, Muramatsu T. Midkine is a heat and acid stable polypeptide capable of enhancing plasminogen activator activity and neurite outgrowth extension. Biochem Biophys Res Commun 1995; 216: 574-581
27 Kadomatsu K, Hagihara M, Akhter S, Fan QW, Muramatsu H, Muramatsu T. Midkine induces the transformation of NIH3T3 cells. Br J Cancer 1997; 75: 354-359
Dai LC et al. Enhanced effect of combined therapy for HCC 1993
www.wjgnet.com

28 Owada K, Sanjo N, Kobayashi T, Mizusawa H, Muramatsu H, Muramatsu T, Michikawa M. Midkine inhibits caspase-dependent apoptosis via the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase in cultured neurons. J Neurochem 1999; 73: 2084-2092
29 Choudhuri R, Zhang HT, Donnini S, Ziche M, Bicknell R. An angiogenic role for the neurokines midkine and pleiotrophin
in tumorigenesis. Cancer Res 1997; 57: 1814-181930 Sandra F, Harada H, Nakamura N, Ohishi M. Midkine
induced growth of ameloblastoma through MAPK and Akt pathways. Oral Oncol 2004; 40: 274-280
31 Kato M, Shinozawa T, Kato S, Awaya A, Terada T. Increased midkine expression in hepatocellular carcinoma. Arch Pathol Lab Med 2000; 124: 848-852
S- Editor Liu Y L- Editor Ma JY E- Editor Ma WH
1994 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1995-1997www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
A case of eosinophilic cholangitis: Imaging findings ofcontrast-enhanced ultrasonography, cholangioscopy, andintraductal ultrasonography
Naoki Matsumoto, Kiyoshi Yokoyama, Kazuhiko Nakai, Toshiki Yamamoto, Takeshi Otani, Masahiro Ogawa, Naohide Tanaka, Ariyoshi Iwasaki, Yasuyuki Arakawa, Masahiko Sugitani
www.wjgnet.com
CASE REPORT
Naoki Matsumoto, Kiyoshi Yokoyama, Kazuhiko Nakai, Toshiki Yamamoto, Takeshi Otani, Masahiro Ogawa, Naohide Tanaka, Ariyoshi Iwasaki, Yasuyuki Arakawa, Department of Gastroenterology and Hepatology, Nihon University Surugadai Hospital, 1-8-13, Kandasurugadai, Chiyoda-ku, Tokyo 101-8309, JapanMasahiko Sugitani, Department of Pathology, Nihon University School of Medicine, 30-1, Oyaguchikamimachi, Itabashi-ku, Tokyo 101-8309, JapanCorrespondence to: Dr. Naoki Matsumoto, Department of Gastroenterology and Hepatology, Nihon University Surugadai Hospital, 1-8-13, Kandasurugadai, Chiyoda-ku, Tokyo 101-8309, Japan. [email protected]: +81-3-32931711 Fax: +81-3-32936078 Received: 2007-02-04 Accepted: 2007-03-23
AbstractA 38-year-old woman was referred to our institution due to epigastralgia. She presented with obstructive jaundice and eosinophilia. Endoscopic retrograde cholangiopancreatography showed diffuse narrowing from the distal common bile duct to the bifurcation of the hepatic ducts. An endoscopic plastic biliary stent was inserted; the specimen obtained from the common bile duct wall revealed dense infiltration by eosinophils. Treatment was started with prednisolone 60 mg daily. The patient’s biliary stenosis and eosinophilia gradually improved. Eosinophilic infiltration in the lungs or stomach is relatively common, but it is rare in the common bile duct. Most of the reported cases of eosinophilic cholangitis presented with eosinophilia; our patient’s eosinophil count was over 1000/mm3. Since our patient had allergies to pollen and house dust, a relationship between the allergies and the eosinophilic cholangitis was suspected, but no cause was identified.
© 2007 The WJG Press. All rights reserved.
Key words: Eos inophi l ic cholangit is; Sc leros ing cholangitis; Contrast-enhanced ultrasonography; Cholangioscopy; Intraductal ultrasonography
Matsumoto N, Yokoyama K, Nakai K, Yamamoto T, Otani T, Ogawa M, Tanaka N, Iwasaki A, Arakawa Y, Sugitani M. A case of eosinophilic cholangitis: Imaging findings of contrast-enhanced ultrasonography, cholangioscopy, and intraductal ultrasonography. World J Gastroenterol 2007;
13(13): 1995-1997
http://www.wjgnet.com/1007-9327/13/1995.asp
INTRODUCTIONMalignancy should be ruled out in all patients with obstructive jaundice. However, benign disorders, such as primary sclerosing cholangitis or autoimmune pancreatitis-associated sclerosing cholangitis, can present with obstructive jaundice. Eosinophilic cholangiopathy can cause either obstructive jaundice or cholecystitis. We report a case of eosinophilic cholangitis successfully treated with endoscopic therapy and steroids.
CASE REPORTA 38-year-old woman was referred to our hospital due to epigastralgia and back pain of 3 d duration. On admission, physical examination revealed jaundice and epigastric tenderness. For 4 mo, the patient had been taking salazosulfapyridine (SASP) and teprenone for the treatment of suspected rheumatoid arthritis. The total leukocyte count was 4700/mm3 (44.8% neutrophils, 15.3% eosinophils). Blood chemistry showed: aspartate aminotransferase (AST), 151 U/L; alanine aminotransferase (ALT), 400 U/L; alkaline phosphatase, 945 U/L; total bilirubin, 6.63 mg/dL, with direct bilirubin, 4.48 mg/dL; and IgG, 812 mg/dL (normal, 870-1700). Tumor marker results were: CA19-9, 125.9 mg/L (normal, ≤ 37). Antinuclear antibodies and P-ANCA were negative. Contrast-enhanced computed tomography (CECT) showed a dilated bile duct from the porta hepatis to the right and left lobes. A solid lesion was seen in the extrahepatic bile duct, which was slightly enhanced by the contrast medium. The gallbladder did not appear swollen, and paraaortic lymphadenopathy was noted. Ultrasound (US) examination revealed an isoechoic solid lesion in the common hepatic duct, with irregular wall thickening of the left hepatic duct. Contrast-enhanced ultrasonography (CEUS), using Levovist (Schering, Berlin, Germany), demonstrated that the wall of the intrahepatic bile ducts was thickened from the hilar region to the periphery with dilatation; a well-enhanced lesion was also seen (Figure 1). Endoscopic retrograde cholangiopancreatography
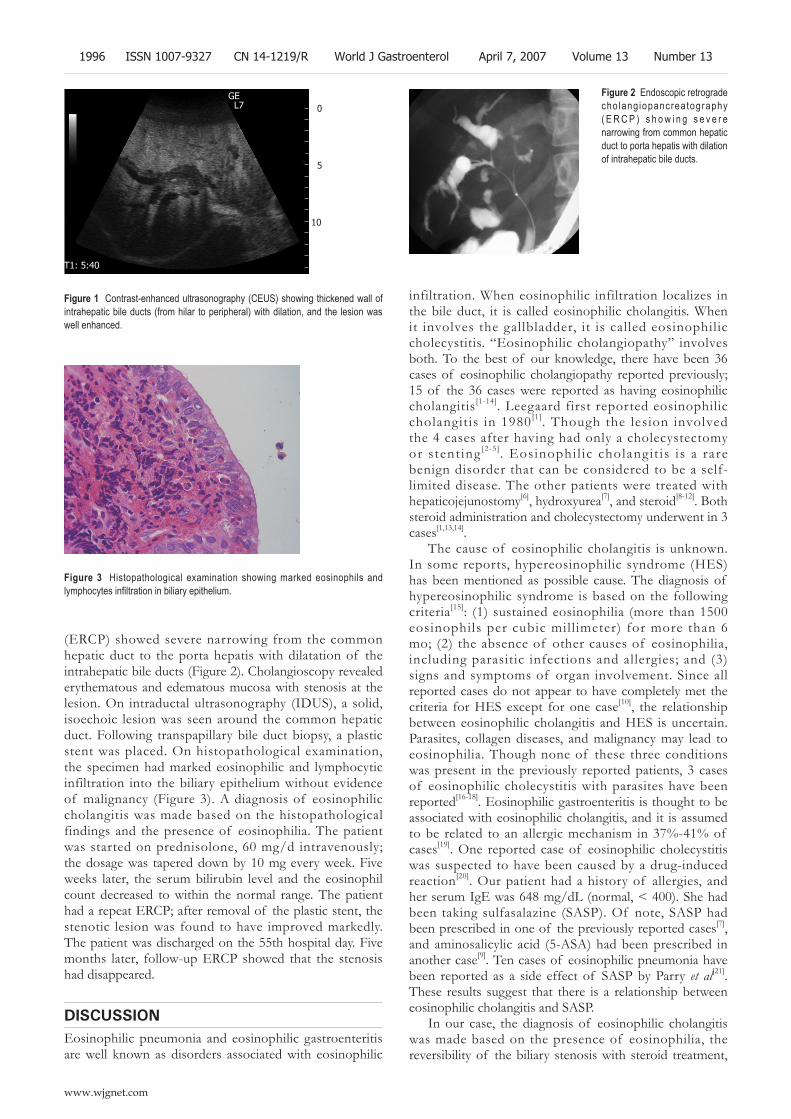
(ERCP) showed severe narrowing from the common hepatic duct to the porta hepatis with dilatation of the intrahepatic bile ducts (Figure 2). Cholangioscopy revealed erythematous and edematous mucosa with stenosis at the lesion. On intraductal ultrasonography (IDUS), a solid, isoechoic lesion was seen around the common hepatic duct. Following transpapillary bile duct biopsy, a plastic stent was placed. On histopathological examination, the specimen had marked eosinophilic and lymphocytic infiltration into the biliary epithelium without evidence of malignancy (Figure 3). A diagnosis of eosinophilic cholangitis was made based on the histopathological findings and the presence of eosinophilia. The patient was started on prednisolone, 60 mg/d intravenously; the dosage was tapered down by 10 mg every week. Five weeks later, the serum bilirubin level and the eosinophil count decreased to within the normal range. The patient had a repeat ERCP; after removal of the plastic stent, the stenotic lesion was found to have improved markedly. The patient was discharged on the 55th hospital day. Five months later, follow-up ERCP showed that the stenosis had disappeared.
DISCUSSIONEosinophilic pneumonia and eosinophilic gastroenteritis are well known as disorders associated with eosinophilic
infiltration. When eosinophilic infiltration localizes in the bile duct, it is called eosinophilic cholangitis. When it involves the gallbladder, it is called eosinophilic cholecystitis. “Eosinophilic cholangiopathy” involves both. To the best of our knowledge, there have been 36 cases of eosinophilic cholangiopathy reported previously; 15 of the 36 cases were reported as having eosinophilic cholangitis[1-14]. Leegaard first reported eosinophilic cholangitis in 1980[1]. Though the lesion involved the 4 cases after having had only a cholecystectomy or stent ing [2-5]. Eosinophi l ic cholangit is i s a rare benign disorder that can be considered to be a self-limited disease. The other patients were treated with hepaticojejunostomy[6], hydroxyurea[7], and steroid[8-12]. Both steroid administration and cholecystectomy underwent in 3 cases[1,13,14].
The cause of eosinophilic cholangitis is unknown. In some reports, hypereosinophilic syndrome (HES) has been mentioned as possible cause. The diagnosis of hypereosinophilic syndrome is based on the following criteria[15]: (1) sustained eosinophilia (more than 1500 eosinophils per cubic mill imeter) for more than 6 mo; (2) the absence of other causes of eosinophilia, including parasitic infections and allergies; and (3) signs and symptoms of organ involvement. Since all reported cases do not appear to have completely met the criteria for HES except for one case[10], the relationship between eosinophilic cholangitis and HES is uncertain. Parasites, collagen diseases, and malignancy may lead to eosinophilia. Though none of these three conditions was present in the previously reported patients, 3 cases of eosinophilic cholecystitis with parasites have been reported[16-18]. Eosinophilic gastroenteritis is thought to be associated with eosinophilic cholangitis, and it is assumed to be related to an allergic mechanism in 37%-41% of cases[19]. One reported case of eosinophilic cholecystitis was suspected to have been caused by a drug-induced reaction[20]. Our patient had a history of allergies, and her serum IgE was 648 mg/dL (normal, < 400). She had been taking sulfasalazine (SASP). Of note, SASP had been prescribed in one of the previously reported cases[7], and aminosalicylic acid (5-ASA) had been prescribed in another case[9]. Ten cases of eosinophilic pneumonia have been reported as a side effect of SASP by Parry et al[21]. These results suggest that there is a relationship between eosinophilic cholangitis and SASP.
In our case, the diagnosis of eosinophilic cholangitis was made based on the presence of eosinophilia, the reversibility of the biliary stenosis with steroid treatment,
Figure 2 Endoscopic retrograde cho lang iopancreatography ( E R C P ) s h o w i n g s e v e r e narrowing from common hepatic duct to porta hepatis with dilation of intrahepatic bile ducts.
Figure 3 Histopathological examination showing marked eosinophils and lymphocytes infiltration in biliary epithelium.
GE L7
T1: 5:40
Figure 1 Contrast-enhanced ultrasonography (CEUS) showing thickened wall of intrahepatic bile ducts (from hilar to peripheral) with dilation, and the lesion was well enhanced.
0
5
10
1996 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

and the eosinophilic infiltration in the biliary epithelium. 11/15 previously reported cases of eosinophilic cholangitis had eosinophilia. However, eosinophilia may not be necessary for diagnosing eosinophilic cholangitis, since not all cases of eosinophilic gastroenteritis present with eosinophilia. In our opinion, the following findings are important for making the diagnosis: (1) wall thickening or stenosis of the biliary system; (2) histopathological findings of eosinophilic infiltration; and (3) reversibility of biliary abnormalities without treatment or following steroid treatment. Abdominal pain was present in 12/18 cases and should be taken into account when making the diagnosis.
Song et al[5] suggested that bile duct wall thickening was a characteristic finding of eosinophilic cholangitis on US and CECT. Irregular narrowing of the bile duct can be seen on magnetic resonance cholangiopancreatography (MRCP)[22]. In our case, the common bile duct was slightly enhanced, and cystic duct wall thickening on CECT. However, it is difficult to make a diagnosis based on only these imaging findings, since CT, ERCP, and MRCP have limited spatial resolution. Therefore, CEUS, cholangioscopy, and IDUS were done. This is the first reported case of eosinophilic cholangitis to have been fully investigated using all these modalities. CEUS has a high spatial resolution and can detect blood flow[23]. In our patient, a parenchymal echo was visualized in the common bile duct on CEUS; staining of the lesion along the intrahepatic bile ducts was considered to be a characteristic finding of eosinophilic cholangitis in this case. This finding enabled us to exclude malignancy. On cholangioscopy, mucosal erythema and stenosis were found. Intraductal ultrasonography (IDUS) revealed hypertrophic stenosis of the extrahepatic bile duct. Though concentric wall thickening was similar to autoimmune pancreatitis-associated sclerosing cholangitis, the stenosis was more extensive in our case. After prednisolone treatment, the patient’s biliary stenosis improved. The ERCP done 5 wk later still showed a mild localized stenosis of the intrahepatic bile ducts, and on histopathological examination of the biopsy specimen obtained at that time, slight eosinophilic infiltration was noted. Given the possibility of recurrence in other organs[3,9], the patient requires long-term follow-up. It is usually difficult to diagnose benign biliary stenosis. Since the patient’s quality of life after discharge depends on whether medical therapy or surgery is chosen, therapy should be chosen carefully.
REFERENCES1 Leegaard M. Eosinophilic cholecystitis. Acta Chir Scand 1980;
146: 295-2962 Butler TW, Feintuch TA, Caine WP. Eosinophilic cholangitis,
lymphadenopathy, and peripheral eosinophilia: a case report. Am J Gastroenterol 1985; 80: 572-574
3 Platt ML, Kiesling VJ, Vaccaro JA. Eosinophilic ureteritis
associated with eosinophilic cholangitis: a case report. J Urol 1990; 144: 127-129
4 Rosengart TK , Rotterdam H, Ranson JH. Eosinophilic cholangitis: a self-limited cause of extrahepatic biliary obstruction. Am J Gastroenterol 1990; 85: 582-585
5 Song HH, Byun JY, Jung SE, Choi KH, Shinn KS, Kim BK. Eosinophilic cholangitis: US, CT, and cholangiography findings. J Comput Assist Tomogr 1997; 21: 251-253
6 Shanti CM, Lucas CE, Tyburski JG, Soulen RL, Lucas DR. Eosinophilic abscess and eosinophilic pseudotumor presenting as bile duct masses: a report of 2 cases. Surgery 2001; 130: 104-108
7 Scheurlen M, Mörk H, Weber P. Hypereosinophilic syndrome resembling chronic inflammatory bowel disease with primary sclerosing cholangitis. J Clin Gastroenterol 1992; 14: 59-63
8 Grauer L, Padilla VM, Bouza L, Barkin JS. Eosinophilic sclerosing cholangitis associated with hypereosinophilic syndrome. Am J Gastroenterol 1993; 88: 1764-1769
9 Schoonbroodt D, Horsmans Y, Laka A, Geubel AP, Hoang P. Eosinophilic gastroenteritis presenting with colitis and cholangitis. Dig Dis Sci 1995; 40: 308-314
10 Vauthey JN , Loyer E, Chokshi P, Lahoti S . Case 57: eosinophilic cholangiopathy. Radiology 2003; 227: 107-112
11 Jimenez-Saenz M, Villar-Rodriguez JL, Torres Y, Carmona I, Salas-Herrero E, Gonzalez-Vilches J, Herrerias-Gutierrez JM. Biliary tract disease: a rare manifestation of eosinophilic gastroenteritis. Dig Dis Sci 2003; 48: 624-627
12 Duseja A, Nada R, Dhiman RK, Chawla YK, Kalra N, Prashad S, Karwasra RK. Eosinophilic cholangiopathy--a case report. Dig Dis Sci 2005; 50: 1422-1425
13 Tenner S, Roston A, Lichtenstein D, Brooks D, Herlihy E, Carr-Locke D. Eosinophilic cholangiopathy. Gastrointest Endosc 1997; 45: 307-309
14 al-Abdulla NA, Schulick RD, Regan F. Hypereosinophilic sclerosing cholangitis: findings using half-Fourier magnetic resonance imaging. Hepatogastroenterology 2000; 47: 359-361
15 Chusid MJ, Dale DC, West BC, Wolff SM. The hypere-osinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine (Baltimore) 1975; 54: 1-27
16 Alfaro Torres J, Fernández Fernández L, Hörndler Argárate C, Ruiz Liso JM, Sanz Anquela JM, López Carreira M, Pellicer Espligares L.Eosinophilic cholecystitis associated with rupture of hepatic hydatid cyst of the bile ducts. Rev Esp Enferm Dig 1995; 87: 899-902
17 Russell CO, Dowling JP, Marshall RD. Acute eosinophilic cholecystitis in association with hepatic echinococcosis. Gastroenterology 1979; 77: 758-760
18 Kim YH. Eosinophilic cholecystitis in association with clonorchis sinensis infestation in the common bile duct. Clin Radiol 1999; 54: 552-554
19 Ureles AL, Alschibaja T, Lodico D, Stabins SJ. Idiopathic eosinophilic infiltration of the gastrointestinal tract, diffuse and circumscribed; a proposed classification and review of the literature, with two additional cases. Am J Med 1961; 30: 899-909
20 Felman RH , Sutherland DB, Conklin JL, Mitros FA. Eosinophilic cholecystitis, appendiceal inflammation, pericarditis, and cephalosporin-associated eosinophilia. Dig Dis Sci 1994; 39: 418-422
21 Parry SD, Barbatzas C, Peel ET, Barton JR. Sulphasalazine and lung toxicity. Eur Respir J 2002; 19: 756-764
22 Vitellas KM, Keogan MT, Freed KS, Enns RA, Spritzer CE, Baillie JM, Nelson RC. Radiologic manifestations of sclerosing cholangitis with emphasis on MR cholangiopancreatography. Radiographics 2000; 20: 959-975; quiz 1108-1109, 1112
23 Leen E, Moug SJ, Horgan P. Potential impact and utilization of ultrasound contrast media. Eur Radiol 2004; 14 Suppl 8: P16-P24
S- Editor Liu Y L- Editor Negro F E- Editor Che YB
Matsumoto N et al . A case of eosinophilic cholangitis 1997
www.wjgnet.com

PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 1998-2001www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
An unhappy triad: Hemochromatosis, porphyria cutanea tarda and hepatocellular carcinoma-A case report
Martina T Mogl, Andreas Pascher, Sabine J Presser, Michael Schwabe, Peter Neuhaus, Natascha C Nuessler
www.wjgnet.com
CASE REPORT
Martina T Mogl, Andreas Pascher, Sabine J Presser, Michael Schwabe, Peter Neuhaus, Natascha C Nuessler, Department of Surgery, Charité Campus Virchow-Klinikum, Augustenburger Platz 1, Berlin 13353, GermanyCorrespondence to: Martina T Mogl, Department of Surgery, Charité Campus Virchow-Klinikum, Augustenburger Platz 1, Berlin 13353, Germany. [email protected]: +49-30- 450552001 Fax: +49-30-450552900Received: 2007-01-27 Accepted: 2007-03-22
AbstractLiver fibrosis and cirrhosis are predisposing factors for the development of hepatocellular carcinoma (HCC). Hemosiderosis has also been described to trigger carcinogenesis. A significant iron overload, as found in hereditary hemochromatosis (HHC), is a risk factor for HCC and may also promote the symptoms of porphyria cutanea tarda (PCT). A 68-year old male patient presented to our clinic with a suspected HCC, elevated alpha-fetoprotein but normal liver function tests. He reported a 25 year-old history of vitiligo upon exposure to sunlight. The patient underwent an extended left hemihepatectomy, and the recovery was uneventful, with the exception of a persistent hyperbilirubinemia. Perfusion problems and extrahepatic cholestasis were ruled out by CT-scan with angiography and MR-cholangiopancreatography. However, MRI showed an iron overload. Histology confirmed the HCC (pT3, pN0, G3, R0) and revealed a portal fibrosis and hemosiderosis. Based on the skin lesions we suspected a PCT that was confirmed by laboratory tests showing elevated porphyrin, uroporphyrin, coproporphyrin and porphobilinogen. Concurrently, molecular diagnostics revealed homozygosity for the C282Y mutation within the hemochromatosis HFE gene. After phlebotomy and normalization of liver function tests the patient was discharged. This is the first case ever showing the unusual combination of HCC in a fibrotic liver with HHC and PCT. This diagnosis not only warrants oncological follow-up but also symptomatic therapy to normalize iron metabolism and thereby improve liver function and alleviate the symptoms of HHC and PCT. Thus progression of fibrosis may be prevented and liver regeneration supported.
© 2007 The WJG Press. All rights reserved.
Key words: Hepatocellular carcinoma; Hemochromatosis; Porphyria cutanea tarda
Mogl MT, Pascher A, Presser SJ, Schwabe M, Neuhaus P, Nuessler NC. An unhappy triad: Hemochromatosis, porphyria cutanea tarda and hepatocellular carcinoma-A case report. World J Gastroenterol 2007; 13(13): 1998-2001
http://www.wjgnet.com/1007-9327/13/1998.asp
INTRODUCTION Liver fibrosis and cirrhosis can lead to the development of hepatocellular carcinoma (HCC). The incidence of HCC in cirrhotic patients is 10-fold higher than among non-cirrhotic patients. Other risk factors for HCC are male gender, age above 50 years and elevated alpha-fetoprotein (AFP).
A relatively infrequent cause of cirrhosis is hereditary hemochromatosis (HHC), which belongs to a group of iron-storage diseases due to mutations in the HFE gene. All forms of HHC present with massive iron accumulation in hepatocytes, ultimately leading to cirrhosis. However, the clinical course of patients with HHC may vary considerably. HHC-induced hepatic iron overload may not only lead to cirrhosis, but may also trigger the manifestation of porphyria cutanea tarda (PCT). PCT is a metabolic disorder characterised by an increased amount of circulating porphyrins due to disrupted heme biosynthesis. The porphyrins accumulate particularly in the skin where they induce cytotoxic effects. PCT occurs both as hereditary (autosomal dominant inheritance) and acquired (sporadic) disease and can be triggered by iron overload, alcohol, estrogens and viral infections. The induction of PCT by iron overload also explains the association of this disease with hemochromatosis gene mutations.
We report about a patient who presented with HCC occurring in a fibrotic liver due to a hitherto unrecognized homozygous hemochromatosis and PCT.
CASE REPORTA 68-year old male patient was referred to our clinic with a 5 cm mass in the left liver lobe detected by a CT-scan. The elevated AFP levels (434 μg/L) were highly suggestive of HCC. There were no morphological or biochemical signs of liver cirrhosis and preoperative laboratory values were normal (bilirubin 0.9 mg/dL, albumin 4.0 g/dL, INR 1.09, AST 52 IU/L). The patient presented in good clinical

Mogl MT et al . Coincidence of HCC with HHC and PCT 1999
www.wjgnet.com
condition without fever, weight loss or pain. Additional diagnoses were atrial fibrillation, arterial hypertension, type 2 diabetes mellitus and hypothyroidism. Physical examination revealed vitiligous lesions on sun-exposed skin. The patient recalled them to be associated with “winegrowers’ disease” 25 years earlier.
An extended left hemihepatectomy was performed without surgical complications. The postoperative course was uneventful. Liver enzymes returned to normal values within the first week, but a persistent hyperbilirubinemia, with bilirubin levels around 3 mg/dL, was noticed. In order to rule out cholestasis or impaired liver perfusion, Doppler-ultrasound, CT-scan and MR-cholangiography were performed. These exams failed to reveal significant pathologies, apart from alterations in the MRI, suggesting an iron overload.
His to log y conf i r med a modera te ly to poor ly differentiated HCC (Figure 1) and described a hitherto unrecognized periportal and initial bridging fibrosis with marked hemosiderosis (Figure 2). A transferrin saturation > 95% raised the suspicion of HHC. Molecular testing revealed a homozygous C282Y mutation within the HFE gene, confirming the diagnosis of HHC. Elevated urinary porphyrin levels (porphyrin 1638 μg/d, coproporphyrin 264 μg/d and uroporphyrin 624 μg/d) confirmed the diagnosis of PCT. Symptomatic therapy, including phlebotomies, resulted in normalization of transferrin saturation and iron levels. The hyperbilirubinemia resolved several weeks after discharge. Maintenance therapy with chloroquine has not been necessary so far. After one year follow-up no recurrence of HCC has been detected.
DISCUSSIONWe present the unhappy triad of HCC, HHC and PCT. HHC is one of the most frequent autosomal recessive hereditary diseases in European Caucasians with a prevalence of 1:200 to 1:500. Patients show extensive deregulation of their iron metabolism with elevated transferrin saturation and high ferritin levels. Later, they develop clinical symptoms due to the iron overload of internal organs. The mutation responsible for HHC is based on a 854G → A transition which leads to a cys282tyr substitution in the HFE gene on chromosome 6p21.3[1]. The specific effect of this mutation on the
iron metabolism has not been entirely clarified. The favoured model describes the iron uptake at the basolateral membrane of duodenal crypt cells via the transferrin 1-receptor (TfR1) that needs the interaction with the wild-type HFE-molecule to guarantee the balance between absorption, storage and consumption[2,3]. Further allelic variants in the HFE gene have been described but seem to lack clinical relevance[4-6].
Our patient presented with an additional hereditary disease, PCT. PCT belongs to the non-acute porphyric diseases that are mostly hereditary and only rarely sporadic. Most porphyrias become clinically apparent after exposure to triggering factors such as alcohol, UV radiation, hormones, viral hepatitis and porphyrogenic drugs. The prominent feature of non-acute porphyrias is an increased photosensitivity with lesions on sun-exposed skin. Our patient’s cutaneous lesions led to hypothesize a non-acute hepatic porphyria. The so-called “winegrowers’ disease”, he erroneously recalled, may have been caused by arsenic poisoning. The German word is “Winzer” disease, which sounds like the word “Windsor” disease, a term that was used synonymously in spoken language for acute intermittent porphyria, a hereditary defect in the British royal family. PCT is the most common porphyric disease worldwide. It is caused by a mutation of the uroporphyrinogen-decarboxylase gene (URO-D) on chromosome 1p34[7]. URO-D is the fifth enzyme of heme biosynthesis and decarboxylates uroporphyrinogen to coproporphyrinogen[8].
About 80% of all PCT patients carry the sporadic mutation that is s ingularly associated with a 50% decrease in URO-D act iv i ty of the l iver [9]. Only approximately 20% of all patients suffer from the autosomal dominantly inherited form of PCT that causes a 50% reduction of the URO-D-activity in all tissues[10]. Due to the malfunctioning URO-D, oxidized uroporphyrinogen accumulates in the liver, is excreted in urine and feces and becomes deposited in skin. Here, the photoactivation of the porphyrines by UV radiation leads to the typical lesions with blisters, erosions and scarring. URO-D function is also influenced by iron. Iron directly blocks URO-D and produces peroxides and free radicals that oxidize uroporphyrinogen to porphyrin, which by itself blocks URO-D function. As soon as the iron content of the liver is substantially decreased by
Figure 1 Histology of HCC with hemosiderosis, Hematoxylin and eosin staining. Figure 2 Histology of hemosiderosis, Berlin blue staining.

phlebotomy, the URO-D activity is partially restored[11]. The latter interaction constitutes the link between
porphyrias and hemochromatosis. HHC, as an iron storage disease, precipitates the clinical manifestation of porphyrias through various mechanisms. The excessive amount of iron, on the one hand, catalyses the formation of reactive oxygen-species that oxidize uroporphyrinogen to uroporphyrin. On the other hand, iron inhibits URO-D indirectly with oxidized metabolites that block the enzyme. Additionally, induction of the δ-ALA-synthase increases the amount of δ-aminolevulinic acid, the primary product of the porphyrin synthesis pathway, thus accumulating intermediate products which provoke the symptoms of PCT. The high frequency of homo- and heterozygous allele carriers for the C282Y- and H63D-mutations of HHC among patients with sporadic PCT, as opposed to healthy controls, was demonstrated in several countries, such as Germany[12], Great-Britain[13], the Netherlands[14] and Australia[15]. Further studies confirmed the HFE mutations as a risk factor for PCT as well as additional risk factors like alcohol consumption, smoking and hepatitis C virus infection[16-18].
HHC and PCT may have triggered the development of HCC in our patient. HCC is one of the few cancers with well-defined risk factors. Among these, liver cirrhosis is believed to be the strongest predisposing factor[19] and also fibrotic changes seem to induce carcinogenesis. Other risk factors differ regionally, such as hepatitis B virus and aflatoxin B1 exposure in Asia and Africa[20]. Further predisposing factors are male gender, age above 50 years, elevated AFP and alcohol[21]. Concerning HHC, only few data exist. Homozygosity for the C282Y mutation seems to increase the risk for HCC in cirrhotic patients, but neither heterozygous nor compound heterozygous (C282Y/H63D) mutations are unequivocally identified risk factors[22,23]. In 2001, PCT was described to be a risk factor for the development of malignant liver tumours, confirming similar observations reported over 30 years earlier[24-26].
Combining these statements we describe the unu-sual combination of HCC in a non-cirrhotic patient with two rather rare conditions, HHC and PCT. Both hereditary diseases are representing minor risk factors for carcinogenesis, but, taken together, may synergistically increase the risk of developing HCC. HHC, as iron-storage disease, on the one hand, promoted the symptoms of PCT, but, on the other hand, may also have triggered carcinogenesis, even with only minimal parenchymal destruction. PCT itself, via the iron overload of HHC, should be considered as carcinogenic risk factor of equal importance, although only few reports underlined this fact so far. We state that both conditions in our case must be considered equally important for the development of HCC, as no additional risk factors could be identified.
In conclusion, the case presented here with HCC, HHC and PCT is special in several ways. Histology revealed a fibrosis with pronounced hemosiderosis suggesting a HHC, which was confirmed by genetic assay. Nevertheless, this constituted an unusual risk profile for the development of HCC, as approximately 80% of all HCCs develop in cirrhosis due to HCV infection and alcoholism, while
HHC only ranks as cofactor of carcinogenesis. However, the hemochromatosis amplified the manifestations of a hitherto unrecognized PCT, another risk factor for carcinogenesis.
To the best of our knowledge, this is the first report ever of the unhappy triad of HCC in a fibrotic liver with PCT and homozygous HHC. It may shed light on the importance of both risk factors, HHC and PCT, for the carcinogenesis of HCC and certainly has to be taken into account in the medical care of these patients.
REFERENCES1 Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA,
Basava A, Dormishian F, Domingo R, Ellis MC, Fullan A, Hinton LM, Jones NL, Kimmel BE, Kronmal GS, Lauer P, Lee VK, Loeb DB, Mapa FA, McClelland E, Meyer NC, Mintier GA, Moeller N, Moore T, Morikang E, Prass CE, Quintana L, Starnes SM, Schatzman RC, Brunke KJ, Drayna DT, Risch NJ, Bacon BR, Wolff RK. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996; 13: 399-408
2 Fleming RE, Sly WS. Mechanisms of iron accumulation in hereditary hemochromatosis. Annu Rev Physiol 2002; 64: 663-680
3 Waheed A, Parkkila S, Saarnio J, Fleming RE, Zhou XY, Tomatsu S, Britton RS, Bacon BR, Sly WS. Association of HFE protein with transferrin receptor in crypt enterocytes of human duodenum. Proc Natl Acad Sci USA 1999; 96: 1579-1584
4 Best LG, Harris PE, Spriggs EL. Hemochromatosis mutations C282Y and H63D in 'cis' phase. Clin Genet 2001; 60: 68-72
5 Mura C, Raguenes O, Férec C. HFE mutations analysis in 711 hemochromatosis probands: evidence for S65C implication in mild form of hemochromatosis. Blood 1999; 93: 2502-2505
6 Pointon JJ, Wallace D, Merryweather-Clarke AT, Robson KJ. Uncommon mutations and polymorphisms in the hemochromatosis gene. Genet Test 2000; 4: 151-161
7 Frank J, Christiano AM. The genetic bases of the porphyrias. Skin Pharmacol Appl Skin Physiol 1998; 11: 297-309
8 Roméo PH , Raich N, Dubart A, Beaupain D, Pryor M, Kushner J, Cohen-Solal M, Goossens M. Molecular cloning and nucleotide sequence of a complete human uroporphyrinogen decarboxylase cDNA. J Biol Chem 1986; 261: 9825-9831
9 Felsher BF, Carpio NM, Engleking DW, Nunn AT. Decreased hepatic uroporphyrinogen decarboxylase activity in porphyria cutanea tarda. N Engl J Med 1982; 306: 766-769
10 de Verneuil H, Beaumont C, Deybach JC, Nordmann Y, Sfar Z, Kastally R. Enzymatic and immunological studies of uroporphyrinogen decarboxylase in familial porphyria cutanea tarda and hepatoerythropoietic porphyria. Am J Hum Genet 1984; 36: 613-622
11 Elder GH , Urquhart AJ, De Salamanca RE, Munoz JJ, Bonkovsky HL. Immunoreact ive uroporphyrinogen decarboxylase in the liver in porphyria cutanea tarda. Lancet 1985; 2: 229-233
12 Tannapfel A, Stölzel U, Köstler E, Melz S, Richter M, Keim V, Schuppan D, Wittekind C. C282Y and H63D mutation of the hemochromatosis gene in German porphyria cutanea tarda patients. Virchows Arch 2001; 439: 1-5
13 Roberts AG, Whatley SD, Morgan RR, Worwood M, Elder GH. Increased frequency of the haemochromatosis Cys282Tyr mutation in sporadic porphyria cutanea tarda. Lancet 1997; 349: 321-323
14 Santos M, Clevers HC, Marx JJ. Mutations of the hereditary hemochromatosis candidate gene HLA-H in porphyria cutanea tarda. N Engl J Med 1997; 336: 1327-1328
15 Stuart KA, Busfield F, Jazwinska EC, Gibson P, Butterworth LA, Cooksley WG, Powell LW, Crawford DH. The C282Y mutation in the haemochromatosis gene (HFE) and hepatitis C virus infection are independent cofactors for porphyria cutanea tarda in Australian patients. J Hepatol 1998; 28: 404-409
2000 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

16 Egger NG, Goeger DE, Payne DA, Miskovsky EP, Weinman SA, Anderson KE. Porphyria cutanea tarda: multiplicity of risk factors including HFE mutations, hepatitis C, and inherited uroporphyrinogen decarboxylase deficiency. Dig Dis Sci 2002; 47: 419-426
17 Skowron F, Bérard F, Grézard P, Wolf F, Morel Y, Perrot H. Role of the hemochromatosis gene in prophyria cutanea tarda. Prospective study of 56 cases Ann Dermatol Venereol 2001; 128: 600-604
18 Fargion S , Piperno A, Cappell ini MD, Sampietro M, Fracanzani AL, Romano R, Caldarelli R, Marcelli R, Vecchi L, Fiorelli G. Hepatitis C virus and porphyria cutanea tarda: evidence of a strong association. Hepatology 1992; 16: 1322-1326
19 Macías Rodríguez MA, Rendón Unceta P, Tejada Cabrera M, Infante Hernández JM, Correro Aguilar F, Díaz García F, Benítez Rodríguez E, Mangas Rojas A, Martín Herrera L. Risk factors for hepatocellular carcinoma in patients with liver cirrhosis. Rev Esp Enferm Dig 2000; 92: 458-469
20 Tsukuma H, Hiyama T, Tanaka S, Nakao M, Yabuuchi T, Kitamura T, Nakanishi K, Fujimoto I, Inoue A, Yamazaki H. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N Engl J Med 1993; 328: 1797-1801
21 Niederau C, Fischer R, Sonnenberg A, Stremmel W, Trampisch
HJ, Strohmeyer G. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N Engl J Med 1985; 313: 1256-1262
22 Cauza E, Peck-Radosavljevic M, Ulrich-Pur H, Datz C, Gschwantler M, Schöniger-Hekele M, Hackl F, Polli C, Rasoul-Rockenschaub S, Müller C, Wrba F, Gangl A, Ferenci P. Mutations of the HFE gene in patients with hepatocellular carcinoma. Am J Gastroenterol 2003; 98: 442-447
23 Boige V, Castéra L, de Roux N, Ganne-Carrié N, Ducot B, Pelletier G, Beaugrand M, Buffet C. Lack of association between HFE gene mutations and hepatocellular carcinoma in patients with cirrhosis. Gut 2003; 52: 1178-1181
24 Fracanzani AL, Taioli E, Sampietro M, Fatta E, Bertelli C, Fiorelli G, Fargion S. Liver cancer risk is increased in patients with porphyria cutanea tarda in comparison to matched control patients with chronic liver disease. J Hepatol 2001; 35: 498-503
25 Kordac V . Frequency of occurrence of hepatocellular carcinoma in patients with porphyria cutanea tarda in long-term follow-up. Neoplasma 1972; 19: 135-139
26 Si e r se m a PD , t e n K a t e F J , Mu l de r P G , W i l so n J H . Hepatocellular carcinoma in porphyria cutanea tarda: frequency and factors related to its occurrence. Liver 1992; 12: 56-61
S- Editor Liu Y L- Editor Negro F E- Editor Che YB
Mogl MT et al . Coincidence of HCC with HHC and PCT 2001
www.wjgnet.com

PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 2002-2003www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
Treatment of pediatric Ogilvie’s syndrome with low-dose erythromycin: A case report
Da-Peng Jiang, Zhao-Zhu Li, Sheng-Yang Guan, Yu-Bo Zhang
www.wjgnet.com
CASE REPORT
Da-Peng Jiang, Zhao-Zhu Li, Sheng-Yang Guan,Yu-Bo Zhang, Department of Pediatric Surgery, Second Affiliated Hospital of Harbin Medical University, Harbin 150086, Heilongjiang Province, ChinaCorrespondence to: Da-Peng Jiang, Department of Pediatric Surgery, Second Affiliated Hospital of Harbin Medical University, #246 Xuefu Road, Harbin 150086, Heilongjiang Province, China. [email protected] Telephone: +86-451-86605247 Received: 2007-01-18 Accepted: 2007-03-05
AbstractAcute colonic pseudo-obstruction is a poorly understood syndrome, characterized by the signs, symptoms and radiological pattern of a large bowel obstruction without evidence for a mechanical obstruction. We report a case of a 2-year old boy who presented with progressive abdominal distention, vomiting and abdominal pain on postoperative d 3. Plain abdominal Χ-ray showed markedly dilated large bowel. Mechanical colonic obstruction was ruled out with hypaque enema. Ogilvie’ssyndrome was suspected. The pat ient received treatment with oral erythromycin which had an immediate beneficial effect. During the 6 mo follow-up, no recurrences of symptoms were observed. We provide a safe and effective therapy for Ogilvie’s syndrome in pediatric individuals.
© 2007 The WJG Press. All rights reserved.
Key words: Acute co lon ic pseudo-obst ruc t ion; Erythromycin; Abdominal distention; Prokinetic agent
Jiang DP, Li ZZ, Guan SY, Zhang YB. Treatment of pediatric Ogilvie’s syndrome with low-dose erythromycin: A case report. World J Gastroenterol 2007; 13(13): 2002-2003
http://www.wjgnet.com/1007-9327/13/2002.asp
INTRODUCTIONAcute colonic pseudo-obstruction (ACPO), also known as Ogilvie’s syndrome, is a disorder characterized by massive dilation of the colon in the absence of mechanical obstruction. William H Ogilvie first observed and reported this rare ileus[1]. It typically occurs in patients with serious illnesses, trauma, burns, surgery and infection. Most cases
of it recover after conservative management, but the patient with distended abdomen is at risk of developing cecal perforation, peritonitis and nutritional depletion[2]. We present here the successful treatment of Ogilvie’s syndrome with low-dose erythromycin in a child.
CASE REPORTA 2-year old boy was admitted with diagnosis of tuberculosis located at the first lumbar vertebra. After removal of tuberculous foci an acellular bone was fixed. The procedure was performed under general anesthesia and the operation was successful. On postoperative d 3 the patient developed progressive abdominal distention, vomiting and abdominal pain. Physical examination showed that he had a temperature of 37.9, a regular pulse of 118 beats/min and poor nutrition. His abdomen was distended and tympanitic to percussion but soft with no tenderness, rebound or guarding. Bowel sounds were present. Laboratory findings were as follows: 10.7 × 109/L white blood cells, 267 × 109/L platelets, 108 g/L hemoglobin, 34 IU/L alanine aminotransferase, 4.3 mmol/L K+, 140 mmol/L Na+, 2.4 mmol/L Ca2, 0.92 mmol/L Mg2+. Plain abdominal Χ-ray showed markedly dilated large bowel (Figure 1). Mechanical colonic obstruction was ruled out with hypaque enema. The diagnosis of Ogilvie’s syndrome was established according to the clinical and radiographic findings.
The patient was treated with intravenous benzyl penicillin, nasogastric decompression and intravenous fluids. Drugs affecting colonic motility were discontinued. When the patient did not respond to 30 h of conservative therapy, nasogastr ic tube was occluded and ora l erythromycin (50 mg four times a day) was started. Within 36 h, the patient began to pass flatus and large volumes of stool with rapid resolution of his abdominal distention. Further radiography and physical examination showed resolution of the colonic pseudo-obstruction. Erythromycin was continued for 3 d with no recurrence of colonic dilation. He recovered with no complication and was discharged in excellent condition. During the 6 mo follow-up, he was well with no gastrointestinal symptoms.
DISCUSSIONOgilvie’s syndrome is a severe form of colonic ileus without evidence for mechanical obstruction[3]. Although Ogilvie’s syndrome has been extensively described in the literature, the precise mechanisms underlying acute pseudo-

Jiang DP et al . Treatment of Ogilvie’s syndrome with erythromycin 2003
www.wjgnet.com
obstruction are still controversial[4]. It often arises in patients with severe infections, retroperitoneal hemorrhage during spinal or pelvic surgery, trauma, burns and narcotic administration[2]. Spinal surgery might contribute to the pathogenesis of the syndrome in our patient. Nausea, vomiting, abdominal pain and abdominal distension are the most common features of Ogilvie’s syndrome. Its diagnosis is sometimes delayed in children, so that many patients are still not properly treated and have a significant mortality[4].
In most cases, conservative management consisting of nasogastric decompression, intravenous fluid, and correction of electrolyte abnormalities can resolve ileus. If unsuccessful, the patient is at risk of developing perforation and colonoscopic decompression is indicated. This method fails in approximately 12%-27% of patients and has a recurrence rate of 18%-33%[5]. It is necessary to find a safe and effective therapy for Ogilvie’s syndrome.
Erythromycin, a macrolide antibiotic, is known to stimulate gastric and small bowel motor activity by binding to the motilin receptor and inducing smooth muscle contraction through a nifedipine-sensitive mechanism[6,7]. However, the short half-life of erythromycin and the rapid onset of tachyphylaxis hamper its broad application in treatment of Ogilvie’s syndrome. There are only a few reports on the efficacy of erythromycin in Ogilvie’s syndrome in adults [8,9]. No recur rence has been
observed in these described cases. Neostigmine, an acetyl-cholinesterase inhibitor, has emerged as an effective pharmacyologic alternative for pseudo-obstruction[2]. However, patients eligible for neostigmine must have their potentially mechanical obstruction ruled out. Neostigmine should not be used if a recently sealed-off duodenal or colonic perforation is unplugged by strong peristalsis. The side-effects of cholinesterase inhibitors include salivation, nausea, vomiting, abdominal pain, bradycardia, hypotension and bronchospasm. During the infusion of neostigmine, patients should undergo cardiac monitoring, and atropine should be available. Finally, severely impaired renal function prolongs the elimination of neostigmine.
In conclusion, this report introduces the first successful treatment of Ogilvie’s syndrome with low-dose erythro-mycin in a child. It may be a safe and effective therapy for children with this disorder.
REFERENCES1 Ogilvie WH. William Heneage Ogilvie 1887-1971. Large-
intestine colic due to sympathetic deprivation. A new clinical syndrome. Dis Colon Rectum 1987; 30: 984-987
2 Gmora S, Poenaru D, Tsai E. Neostigmine for the treatment of pediatric acute colonic pseudo-obstruction. J Pediatr Surg 2002; 37: E28
3 Singh S, Nadgir A, Bryan RM. Post-cesarean section acute colonic pseudo-obstruction with spontaneous perforation. Int J Gynaecol Obstet 2005; 89: 144-145
4 De Giorgio R, Barbara G, Stanghellini V, Tonini M, Vasina V, Cola B, Corinaldesi R, Biagi G, De Ponti F. Review article: the pharmacological treatment of acute colonic pseudo-obstruction. Aliment Pharmacol Ther 2001; 15: 1717-1727
5 Saunders MD, Cappell MS. Endoscopic management of acute colonic pseudo-obstruction. Endoscopy 2005; 37: 760-763
6 Curry JI, Lander TD, Stringer MD. Review article: eryth-romycin as a prokinetic agent in infants and children. Aliment Pharmacol Ther 2001; 15: 595-603
7 Costalos C, Gounaris A, Varhalama E, Kokori F, Alexiou N, Kolovou E. Erythromycin as a prokinetic agent in preterm infants. J Pediatr Gastroenterol Nutr 2002; 34: 23-25
8 Armstrong DN, Ballantyne GH, Modlin IM. Erythromycin for reflex ileus in Ogilvie's syndrome. Lancet 1991; 337: 378
9 Rovira A, López A, Cambray C, Gimeno C. Acute colonic pseudo-obstruction (Ogilvie's syndrome) treated with erythromycin. Intensive Care Med 1997; 23: 798
S- Editor Liu Y L- Editor Wang XL E- Editor Che YB
Figure 1 Plain abdominal X-ray of the patient showing massive dilatation of the large bowel.

department with complaints of nausea, dyspepsia and abdominal pain at the left lower quadrant. On physical examination, tenderness at the left lower quadrant was observed. Complete blood count revealed a mild leukocytosis (11 600/mm3) and blood chemistry revealed an elevated serum alkaline phosphatase level (167 mg/L). Patient was clinically diagnosed to have acute sigmoid diverticulitis. Ultrasonography (US) and computed tomography (CT) of the abdomen revealed a duplicated gallbladder, with thickened wall and considerable sludge in one gallbladder, while tortuous cystic duct but normal wall and luminal content in another gallbladder (Figures 1 and 2). Endoscopic retrograde cholangiopancreatography (ERCP) showed two cystic ducts, normal common hepatic duct, and no luminal opacification (Figure 3). Although her bowel and paracolic spaces had normal appearance, the patient was put on medical treatment for acute diverticulitis. Her symptoms were subsequently improved. Follow-up US examination in the third month revealed almost similar findings to the initial scan; one of the gallbladders had slightly tortuous wall but normal luminal content, while the other one had a 4-mm thickened wall and contained a small amount of sludge.
Cholecystectomy was performed to treat the chronic cholecystitis. Two gallbladders that were demonstrated during the radiological workup had normal walls and contained clear fluid (Figure 4). A third gallbladder was unexpectedly discovered during the operation. It had a thick wall and was filled with sludge, and its cystic duct was shorter than others. All gallbladders were dissected after the ligation of their cystic ducts and accompanying arteries. No stone was detected in any of the gallbladders. Pathological diagnosis revealed gallbladder triplication; two of them were normal and the other one showed ulcerated active chronic cholecystitis.
DISCUSSIONMultiple gallbladders are thought to be caused by the rudimentary bile ducts which failed to regress during embryological development. The persistence of these cellular out-pocketings along the common bile duct and their growth lead to this anomaly. The relationship of the gallbladder to the cystic duct and to the common bile duct seems to be determined by the location of persistent rudimentary duct buds: if the persistent buds originate from the hepatic duct or common bile duct, gallbladders will have separate cystic ducts; if they arise from the cystic duct, the gallbladders share single common cystic duct draining into a normal common bile duct. Based on the anatomic configuration of the cystic ducts, three distinct types are described. The first type is characterized by
CASE REPORT
An incidental case of triple gallbladder
Banu Alicioglu
www.wjgnet.com
Banu Alicioglu, Department of Radiology, Trakya University School of Medicine, Edirne 22030, TurkeyCorrespondence to: Dr. Banu Alicioglu, Department of Radiology, Trakya University School of Medicine, Fatih mah. 4.cad 43.sok, Edirne 22030, Turkey. [email protected]: +90-284-2363089 Fax: +90-284-2352730Received: 2006-12-12 Accepted: 2007-01-04
AbstractTriplication of the gallbladder is a very rare congenital anomaly of the biliary tract; there are only eleven reported cases to date. Gallbladder multiplications are not likely to be discovered unless associated with cholelithiasis, sludge, cholecystitis and carcinoma. Here we report an incidentally diagnosed triplicate gallbladder in a patient with sigmoid diverticulitis; two of the triplicate gallbladder were demonstrated with ultrasound and computed tomography, and an additional galballder was found at surgery.
© 2007 The WJG Press. All rights reserved.
Key words: Biliary system; Biliary tract imaging; Multiple gallbladders; Triple gallbladder
Alicioglu B. An incidental case of triple gallbladder. World J Gastroenterol 2007; 13(13): 2004-2006
http://www.wjgnet.com/1007-9327/13/2004.asp
INTRODUCTIONVesica fellae triplex, also known as the triplication of the gallbladder, is a very rare congenital anomaly of the biliary tract. To our knowledge, only 11 cases have been reported in the literature to date. The first case was described by Huber in a human cadaver in 1752[1]. Multiple gallbladder anomalies are generally discovered if they are associated with symptoms caused by cholelithiasis, sludge, cholecystitis or carcinoma. We report a case of triple gallbladder which was incidentally diagnosed in a patient with sigmoid diverticulitis. Radiological workup revealed a duplication of gallbladder and chronic cholecystitis with sludge formation. However, a triplication was found during surgery.
CaSe RepORTA 54-year-old woman was admitted to emergency
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 2004-2006www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
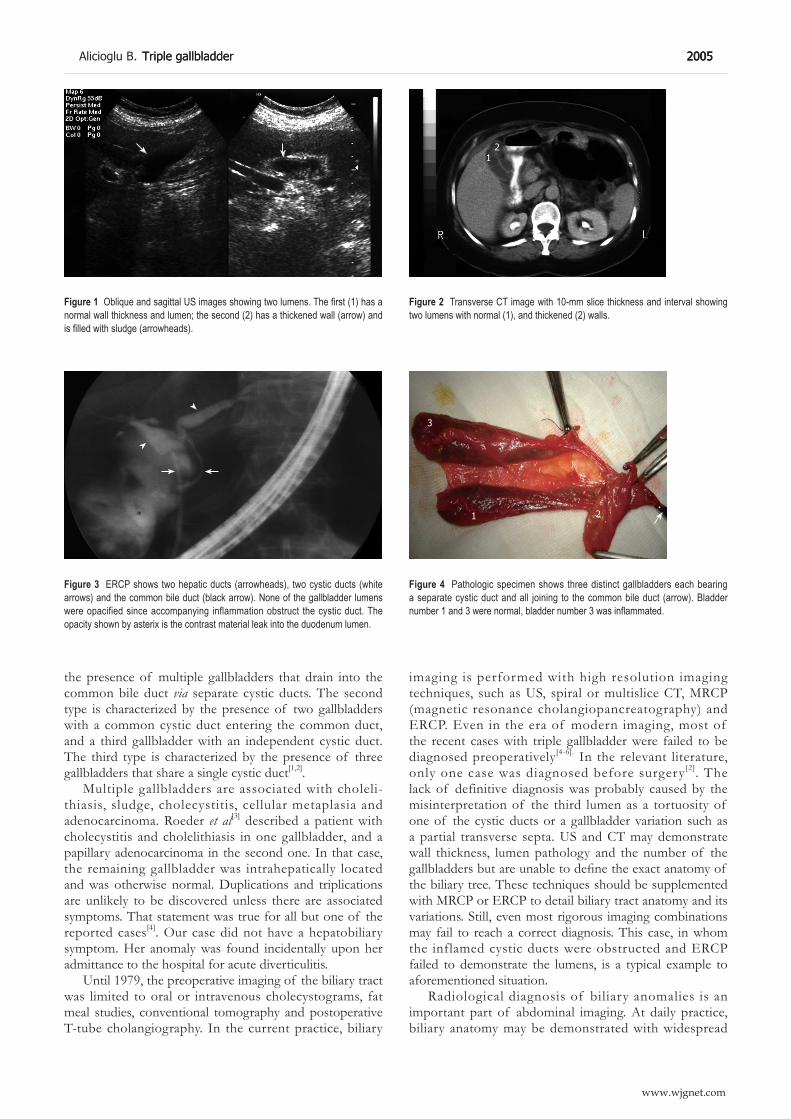
the presence of multiple gallbladders that drain into the common bile duct via separate cystic ducts. The second type is characterized by the presence of two gallbladders with a common cystic duct entering the common duct, and a third gallbladder with an independent cystic duct. The third type is characterized by the presence of three gallbladders that share a single cystic duct[1,2].
Multiple gallbladders are associated with choleli-thiasis, sludge, cholecystitis, cellular metaplasia and adenocarcinoma. Roeder et al[3] described a patient with cholecystitis and cholelithiasis in one gallbladder, and a papillary adenocarcinoma in the second one. In that case, the remaining gallbladder was intrahepatically located and was otherwise normal. Duplications and triplications are unlikely to be discovered unless there are associated symptoms. That statement was true for all but one of the reported cases[4]. Our case did not have a hepatobiliary symptom. Her anomaly was found incidentally upon her admittance to the hospital for acute diverticulitis.
Until 1979, the preoperative imaging of the biliary tract was limited to oral or intravenous cholecystograms, fat meal studies, conventional tomography and postoperative T-tube cholangiography. In the current practice, biliary
Alicioglu B . Triple gallbladder 200Triple gallbladder 200 200
www.wjgnet.com
imaging is performed with high resolution imaging techniques, such as US, spiral or multislice CT, MRCP (magnetic resonance cholangiopancreatography) and ERCP. Even in the era of modern imaging, most of the recent cases with triple gallbladder were failed to be diagnosed preoperatively[4-6]. In the relevant literature, only one case was diagnosed before surgery[2]. The lack of definitive diagnosis was probably caused by the misinterpretation of the third lumen as a tortuosity of one of the cystic ducts or a gallbladder variation such as a partial transverse septa. US and CT may demonstrate wall thickness, lumen pathology and the number of the gallbladders but are unable to define the exact anatomy of the biliary tree. These techniques should be supplemented with MRCP or ERCP to detail biliary tract anatomy and its variations. Still, even most rigorous imaging combinations may fail to reach a correct diagnosis. This case, in whom the inflamed cystic ducts were obstructed and ERCP failed to demonstrate the lumens, is a typical example to aforementioned situation.
Radiological diagnosis of biliary anomalies is an important part of abdominal imaging. At daily practice, biliary anatomy may be demonstrated with widespread
Figure 1 Oblique and sagittal US images showing two lumens. The first (1) has a normal wall thickness and lumen; the second (2) has a thickened wall (arrow) and is filled with sludge (arrowheads).
Figure 2 Transverse CT image with 10-mm slice thickness and interval showing two lumens with normal (1), and thickened (2) walls.
12
Figure 3 ERCP shows two hepatic ducts (arrowheads), two cystic ducts (white arrows) and the common bile duct (black arrow). None of the gallbladder lumens were opacified since accompanying inflammation obstruct the cystic duct. The opacity shown by asterix is the contrast material leak into the duodenum lumen.
Figure 4 Pathologic specimen shows three distinct gallbladders each bearing a separate cystic duct and all joining to the common bile duct (arrow). Bladder number 1 and 3 were normal, bladder number 3 was inflammated.
* 1
3
2

techniques, such as high resolution US, multislice CT and MRCP. Preoperative awareness of any variation may minimize the chance of an unexpected course during cholecystectomy, and helps to avoid any unwanted damage to the biliary tract. It also has a paramount importance in preventing the postcholecystectomy syndrome due to a retained accessory gallbladder.
aCKNOWLeDGMeNTSI would like to express my gratitude to Ozlem Karagozler and Hakki Muammer Karakas for proof reading the article in English.
ReFeReNCeS1 Kurzweg FT, Cole PA. Triplication of the gallbladder: review
of literature and report of a case. Am Surg 1979; 45: 410-41241222 Barnes S, Nagar H, Levine C, Santo M, Sold A, Mercer D,
Kessler A. Triple gallbladder: preoperative sonographic diag-nosis. J Ultrasound Med 2004; 23: 1399-14021402402
3 Roeder WJ, Mersheimer WL, Kazarian KK. Triplication of the gallbladder with cholecystitis, cholelithiasis, and papillary adenocarcinoma. Am J Surg 1971; 121: 746-7474
4 Ross RJ, Sachs MD. Triplication of the gallbladder. Am J Ro-entgenol Radium Ther Nucl Med 196; 104: 656-66166161
5 Schroeder C, Draper KR. Laparoscopic cholecystectomy for triple gallbladder. Surg Endosc 2003; 17: 1322
6 Nanthakumaran S, King PM, Sinclair TS. Laparoscopic excisi-on of a triple gallbladder. Surg Endosc 2003; 17: 1323
S- Editor Wang J L- Editor Kumar M E- Editor Ma WH
2006 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 2007-2010www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
Pure red cell aplasia due to parvovirus B19 infection after liver transplantation: A case report and review of the literature
Ting-Bo Liang, Dong-Lin Li, Jun Yu, Xue-Li Bai, Liang Liang, Shi-Guo Xu, Wei-Lin Wang, Yan Shen, Min Zhang, Shu-Sen Zheng
www.wjgnet.com
CASE REPORT
Ting-Bo Liang, Dong-Lin Li, Jun Yu, Xue-Li Bai, Liang Liang, Shi-Guo Xu, Wei-Lin Wang, Yan Shen, Min Zhang, Shu-Sen Zheng, Department of Hepatobiliary and Pancreatic Surgery, Key Laboratory of Combined Multi-organ Transplantation, Ministry of Public Health, the First Affiliated Hospital of Medical College, Zhejiang University, Hangzhou 310003, Zhejiang Province, ChinaCorrespondence to: Shu-Sen Zheng, MD, PhD, Department of Hepatobiliary and Pancreatic Surgery, Key Laboratory of Combined Multi-organ Transplantation, Ministry of Public Health, the First Affiliated Hospital of Medical College, Zhejiang University, Hangzhou 310003, Zhejiang Province, China. [email protected]: +86-571-87236601 Fax: +86-571-87236678Received: 2007-02-02 Accepted: 2007-03-22
AbstractPure red cell aplasia (PRCA) due to parvovirus B19 (PVB19) infection after solid organ transplantation has been rarely reported and most of the cases were renal transplant recipients. Few have been described after liver transplantation. Moreover, little information on the management of this easily recurring disease is available at present. We describe the first case of a Chinese liver transplant recipient with PVB19-induced PRCA during immunosuppressive therapy. The patient suffered from progressive anemia with the lowest hemoglobin level of 21 g/L. Bone marrow biopsy showed selectively inhibited erythropoiesis with giant pronormoblasts. Detection of PVB19-DNA in serum with quantitative polymerase chain reaction (PCR) revealed a high level of viral load. After 2 courses of intravenous immunoglobulin (IVIG) therapy, bone marrow erythropoiesis recovered with his hemoglobin level increased to 123 g/L. He had a low-level PVB19 load for a 5-mo follow-up period without recurrence of PRCA, and finally the virus was cleared. Our case indicates that clearance of PVB19 by IVIG in transplant recipients might be delayed after recovery of anemia.
© 2007 The WJG Press. All rights reserved.
Key words: Pure red cell aplasia; Parvovirus B19; Intravenous immunoglobul in; Recurrence; Liver transplantation
Liang TB, Li DL, Yu J, Bai XL, Liang L, Xu SG, Wang WL, Shen Y, Zhang M, Zheng SS. Pure red cell aplasia due to parvovirus B19 infection after liver transplantation: A case
report and review of the literature. World J Gastroenterol 2007; 13(13): 2007-2010
http://www.wjgnet.com/1007-9327/13/2007.asp
INTRODUCTIONPure red cell aplasia (PRCA) is a relatively rare disease character ized by the inhibi t ion of bone mar row erythropoiesis with multiple factors involved in its development[1]. Solid organ transplantation-associated PRCA may be attributed to immunosuppressants and parvovirus B19 (PVB19) infection. An increasing number of reports on PRCA caused by PVB19 after renal transplantation are available[2], but there are very few cases describing liver transplant recipients. Furthermore, this severe complication usually responds to high-dose intravenous immunoglobulin (IVIG) therapy with recovery of erythropoiesis, but relapses are common and experience in dealing with this rare and easily recurring disease is insufficient. We describe the first case of a Chinese liver transplant recipient with severe PRCA due to PVB19 infection and show our experience in managing this disease. Accidentally, the patient’s blood group was preoperatively identified as Rho (D)-negative that is extremely rare in China and he received a Rho (D)-incompatible liver transplantation, which made the severe anemia embarrassing. We also made a review of the literature and discussed several key points of PVB19-induced PRCA.
CASE REPORTA 38-year old Chinese man was diagnosed as hepatocellular carcinoma with hepatitis B in a cirrhosis background. The tumor was within 3 cm in diameter without extrahepatic metastasis. Peripheral blood cell counts were all normal. Blood group was A and Rho (D)-negative. In August 2005, he received orthotopic liver transplantation (OLT) from a donor. The donor’s blood group was A and Rho (D)-positive. Packed red blood cells transfused intraoperatively were all Rho (D)-negative. The patient recovered uneventfully after the operation, and a triple-immunosuppressant protocol consisting of tacrolimus (FK506), mycophenolate mofetil (MMF) and prednisolone was adopted.
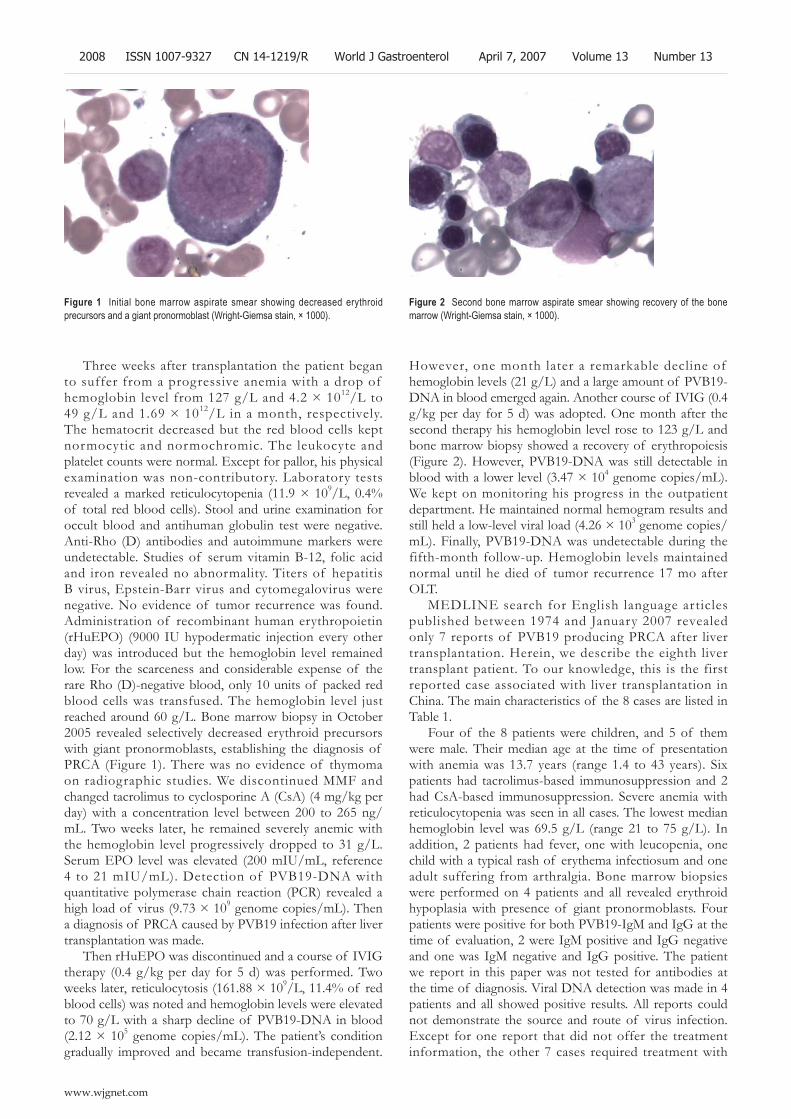
Three weeks after transplantation the patient began to suffer from a progressive anemia with a drop of hemoglobin level from 127 g/L and 4.2 × 1012/L to 49 g/L and 1.69 × 1012/L in a month, respectively. The hematocrit decreased but the red blood cells kept normocytic and normochromic. The leukocyte and platelet counts were normal. Except for pallor, his physical examination was non-contributory. Laboratory tests revealed a marked reticulocytopenia (11.9 × 109/L, 0.4% of total red blood cells). Stool and urine examination for occult blood and antihuman globulin test were negative. Anti-Rho (D) antibodies and autoimmune markers were undetectable. Studies of serum vitamin B-12, folic acid and iron revealed no abnormality. Titers of hepatitis B virus, Epstein-Barr virus and cytomegalovirus were negative. No evidence of tumor recurrence was found. Administration of recombinant human erythropoietin (rHuEPO) (9000 IU hypodermatic injection every other day) was introduced but the hemoglobin level remained low. For the scarceness and considerable expense of the rare Rho (D)-negative blood, only 10 units of packed red blood cells was transfused. The hemoglobin level just reached around 60 g/L. Bone marrow biopsy in October 2005 revealed selectively decreased erythroid precursors with giant pronormoblasts, establishing the diagnosis of PRCA (Figure 1). There was no evidence of thymoma on radiographic studies. We discontinued MMF and changed tacrolimus to cyclosporine A (CsA) (4 mg/kg per day) with a concentration level between 200 to 265 ng/mL. Two weeks later, he remained severely anemic with the hemoglobin level progressively dropped to 31 g/L. Serum EPO level was elevated (200 mIU/mL, reference 4 to 21 mIU/mL). Detection of PVB19-DNA with quantitative polymerase chain reaction (PCR) revealed a high load of virus (9.73 × 109 genome copies/mL). Then a diagnosis of PRCA caused by PVB19 infection after liver transplantation was made.
Then rHuEPO was discontinued and a course of IVIG therapy (0.4 g/kg per day for 5 d) was performed. Two weeks later, reticulocytosis (161.88 × 109/L, 11.4% of red blood cells) was noted and hemoglobin levels were elevated to 70 g/L with a sharp decline of PVB19-DNA in blood (2.12 × 105 genome copies/mL). The patient’s condition gradually improved and became transfusion-independent.
However, one month later a remarkable decline of hemoglobin levels (21 g/L) and a large amount of PVB19-DNA in blood emerged again. Another course of IVIG (0.4 g/kg per day for 5 d) was adopted. One month after the second therapy his hemoglobin level rose to 123 g/L and bone marrow biopsy showed a recovery of erythropoiesis (Figure 2). However, PVB19-DNA was still detectable in blood with a lower level (3.47 × 104 genome copies/mL). We kept on monitoring his progress in the outpatient department. He maintained normal hemogram results and still held a low-level viral load (4.26 × 103 genome copies/mL). Finally, PVB19-DNA was undetectable during the fifth-month follow-up. Hemoglobin levels maintained normal until he died of tumor recurrence 17 mo after OLT.
MEDLINE search for English language articles published between 1974 and January 2007 revealed only 7 reports of PVB19 producing PRCA after liver transplantation. Herein, we describe the eighth liver transplant patient. To our knowledge, this is the first reported case associated with liver transplantation in China. The main characteristics of the 8 cases are listed in Table 1.
Four of the 8 patients were children, and 5 of them were male. Their median age at the time of presentation with anemia was 13.7 years (range 1.4 to 43 years). Six patients had tacrolimus-based immunosuppression and 2 had CsA-based immunosuppression. Severe anemia with reticulocytopenia was seen in all cases. The lowest median hemoglobin level was 69.5 g/L (range 21 to 75 g/L). In addition, 2 patients had fever, one with leucopenia, one child with a typical rash of erythema infectiosum and one adult suffering from arthralgia. Bone marrow biopsies were performed on 4 patients and all revealed erythroid hypoplasia with presence of giant pronormoblasts. Four patients were positive for both PVB19-IgM and IgG at the time of evaluation, 2 were IgM positive and IgG negative and one was IgM negative and IgG positive. The patient we report in this paper was not tested for antibodies at the time of diagnosis. Viral DNA detection was made in 4 patients and all showed positive results. All reports could not demonstrate the source and route of virus infection. Except for one report that did not offer the treatment information, the other 7 cases required treatment with
Figure 1 Initial bone marrow aspirate smear showing decreased erythroid precursors and a giant pronormoblast (Wright-Giemsa stain, × 1000).
Figure 2 Second bone marrow aspirate smear showing recovery of the bone marrow (Wright-Giemsa stain, × 1000).
2008 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com
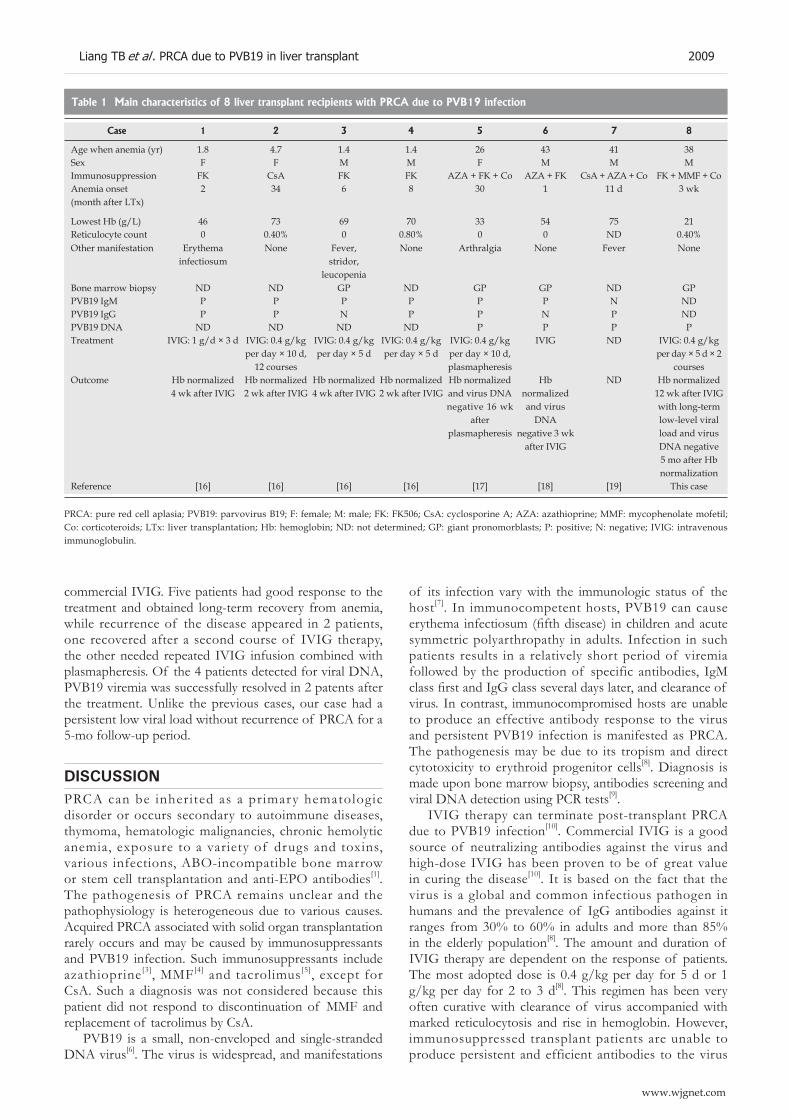
commercial IVIG. Five patients had good response to the treatment and obtained long-term recovery from anemia, while recurrence of the disease appeared in 2 patients, one recovered after a second course of IVIG therapy, the other needed repeated IVIG infusion combined with plasmapheresis. Of the 4 patients detected for viral DNA, PVB19 viremia was successfully resolved in 2 patents after the treatment. Unlike the previous cases, our case had a persistent low viral load without recurrence of PRCA for a 5-mo follow-up period.
DISCUSSIONPRCA can be inherited as a primary hematologic disorder or occurs secondary to autoimmune diseases, thymoma, hematologic malignancies, chronic hemolytic anemia, exposure to a variety of drugs and toxins, various infections, ABO-incompatible bone marrow or stem cell transplantation and anti-EPO antibodies[1]. The pathogenesis of PRCA remains unclear and the pathophysiology is heterogeneous due to various causes. Acquired PRCA associated with solid organ transplantation rarely occurs and may be caused by immunosuppressants and PVB19 infection. Such immunosuppressants include azathioprine[3], MMF[4] and tacrolimus[5], except for CsA. Such a diagnosis was not considered because this patient did not respond to discontinuation of MMF and replacement of tacrolimus by CsA.
PVB19 is a small, non-enveloped and single-stranded DNA virus[6]. The virus is widespread, and manifestations
of its infection vary with the immunologic status of the host[7]. In immunocompetent hosts, PVB19 can cause erythema infectiosum (fifth disease) in children and acute symmetric polyarthropathy in adults. Infection in such patients results in a relatively short period of viremia followed by the production of specific antibodies, IgM class first and IgG class several days later, and clearance of virus. In contrast, immunocompromised hosts are unable to produce an effective antibody response to the virus and persistent PVB19 infection is manifested as PRCA. The pathogenesis may be due to its tropism and direct cytotoxicity to erythroid progenitor cells[8]. Diagnosis is made upon bone marrow biopsy, antibodies screening and viral DNA detection using PCR tests[9].
IVIG therapy can terminate post-transplant PRCA due to PVB19 infection[10]. Commercial IVIG is a good source of neutralizing antibodies against the virus and high-dose IVIG has been proven to be of great value in curing the disease[10]. It is based on the fact that the virus is a global and common infectious pathogen in humans and the prevalence of IgG antibodies against it ranges from 30% to 60% in adults and more than 85% in the elderly population[8]. The amount and duration of IVIG therapy are dependent on the response of patients. The most adopted dose is 0.4 g/kg per day for 5 d or 1 g/kg per day for 2 to 3 d[8]. This regimen has been very often curative with clearance of virus accompanied with marked reticulocytosis and rise in hemoglobin. However, immunosuppressed transplant patients are unable to produce persistent and efficient antibodies to the virus
Case 1 2 3 4 5 6 7 8
Age when anemia (yr) 1.8 4.7 1.4 1.4 26 43 41 38Sex F F M M F M M MImmunosuppression FK CsA FK FK AZA + FK + Co AZA + FK CsA + AZA + Co FK + MMF + CoAnemia onset (month after LTx)
2 34 6 8 30 1 11 d 3 wk
Lowest Hb (g/L) 46 73 69 70 33 54 75 21Reticulocyte count 0 0.40% 0 0.80% 0 0 ND 0.40%Other manifestation Erythema
infectiosumNone Fever,
stridor,leucopenia
None Arthralgia None Fever None
Bone marrow biopsy ND ND GP ND GP GP ND GPPVB19 IgM P P P P P P N NDPVB19 IgG P P N P P N P NDPVB19 DNA ND ND ND ND P P P PTreatment IVIG: 1 g/d × 3 d IVIG: 0.4 g/kg
per day × 10 d, 12 courses
IVIG: 0.4 g/kg per day × 5 d
IVIG: 0.4 g/kg per day × 5 d
IVIG: 0.4 g/kg per day × 10 d, plasmapheresis
IVIG ND IVIG: 0.4 g/kg per day × 5 d × 2
coursesOutcome Hb normalized
4 wk after IVIGHb normalized 2 wk after IVIG
Hb normalized 4 wk after IVIG
Hb normalized 2 wk after IVIG
Hb normalized and virus DNA negative 16 wk
after plasmapheresis
Hb normalized and virus
DNA negative 3 wk
after IVIG
ND Hb normalized 12 wk after IVIG with long-term low-level viral load and virus DNA negative 5 mo after Hb normalization
Reference [16] [16] [16] [16] [17] [18] [19] This case
Table 1 Main characteristics of 8 liver transplant recipients with PRCA due to PVB19 infection
PRCA: pure red cell aplasia; PVB19: parvovirus B19; F: female; M: male; FK: FK506; CsA: cyclosporine A; AZA: azathioprine; MMF: mycophenolate mofetil; Co: corticoteroids; LTx: liver transplantation; Hb: hemoglobin; ND: not determined; GP: giant pronomorblasts; P: positive; N: negative; IVIG: intravenous immunoglobulin.
Liang TB et al . PRCA due to PVB19 in liver transplant 2009
www.wjgnet.com

and the inadequate clearance of virus after cessation of IVIG may easily induce recurrence. Geetha[11] identified a 10% recurrence rate in 22 cases of PRCA due to PVB19 infection in transplant recipients treated with IVIG through reviewing the literature. Recurrence of viraemia and anemia may require repeated courses of IVIG infusion.
The patient we described here was faced with the same situation and the relapse of PRCA was successfully cured after a repeated course of IVIG without any maintenance therapy. However, unlike the previous cases he maintained a long-term low-level viremia without recurrence of PRCA during the 5-mo follow-up period. For all the literature offered sparse information on alteration of viral DNA during the period of recovery, little is known about the viral clearance after treatment. Our results indicate that the clearance of PVB19 by IVIG therapy in immunosuppressed transplant recipients might be delayed for a relatively long period after the reversion of anemia. We therefore recommend that it would seem more reasonable to simply follow the serial monitoring of viral DNA and hemoglobin levels instead of preventative maintenance therapy with high-dose IVIG in such patients. Re-administration of IVIG would be considered if increase of viral load or drop of hemoglobin levels was observed.
In addition, it is notable that some side effects of high-dose IVIG infusion, such as fever, headache, myalgia, hypertension and acute renal failure, have been reported in some patients receiving IVIG therapy[12]. Furthermore, IVIG has been reported to be contaminated by PVB19 and might be a source of infection[13]. All these reports may raise important questions concerning the safety of IVIG therapy. Though such effects were absent in the 8 liver transplant recipients, attention is needed when we perform the therapy.
No proven specific strategy for preventing PVB19 infection is available. Transmission of the virus infection may occur through the respiratory tract, blood-derived products[8] and donated organs containing the virus[10]. Accordingly, avoiding PVB19 exposure to patients, virus screening of blood products before transfusion and testing organ donors prior to transplantation may be considered as preventive strategies. Since transmission via blood products is rare, universal blood screening is not recommended, though the risk of PVB19 infection is of great concern for blood and blood product suppliers[14]. Strategies for reducing the viral load in the manufacture plasma pool by discarding PVB19-DNA-positive donations and developing new strong virus inactivation methods are recommended to these suppliers. Finally, PVB19 vaccines are being explored[15]. Results achieved appear promising, and following researches are on the way.
In conclusion, liver transplant recipients are also at risk of developing PRCA due to PVB19 infection after transplantation. PRCA can be reversed by IVIG therapy but easily recurs. Little is known about the host-pathogen interaction after therapy which is related to recurrence.
Our case signifies that clearance of PVB19 by IVIG infusion in transplant recipients might be delayed for a relatively long time after recovery of anemia. Further researches are needed to find out most effective modalities of therapy and preventative strategies.
REFERENCES1 Djaldetti M, Blay A, Bergman M, Salman H, Bessler H. Pure
red cell aplasia--a rare disease with multiple causes. Biomed Pharmacother 2003; 57: 326-332
2 Vales-Albertos LJ, García-Cárdenas M, Chávez-Becerra S, Gómez-Navarro B, Monteón-Ramos F, Cueto-Manzano AM. Pure red cell aplasia associated with parvovirus B19 infection in renal transplantation: the first case report in Mexico. Transplantation 2005; 79: 739
3 Agrawal A, Parrott NR, Riad HN, Augustine T. Azathioprine-induced pure red cell aplasia: case report and review. Transplant Proc 2004; 36: 2689-2691
4 Hodo Y , Tsuji K, Mizukoshi E, Yamashita T, Sakai A, Nakamoto Y, Honda M, Kaneko S. Pure red cell aplasia associated with concomitant use of mycophenolate mofetil and ribavirin in post-transplant recurrent hepatitis C. Transpl Int 2006; 19: 170-171
5 Gregoor PS, Weimar W. Tacrolimus and pure red-cell aplasia. Am J Transplant 2005; 5: 195-196
6 Kaufmann B, Simpson AA, Rossmann MG. The structure of human parvovirus B19. Proc Natl Acad Sci USA 2004; 101: 11628-11633
7 Young NS, Brown KE. Parvovirus B19. N Engl J Med 2004; 350: 586-597
8 Heegaard ED, Brown KE. Human parvovirus B19. Clin Microbiol Rev 2002; 15: 485-505
9 Zerbini M, Gallinella G, Cricca M, Bonvicini F, Musiani M. Diagnostic procedures in B19 infection. Pathol Biol (Paris) 2002; 50: 332-338
10 Mouthon L, Guillevin L, Tellier Z. Intravenous immun-oglobulins in autoimmune- or parvovirus B19-mediated pure red-cell aplasia. Autoimmun Rev 2005; 4: 264-269
11 Geetha D, Zachary JB, Baldado HM, Kronz JD, Kraus ES. Pure red cell aplasia caused by Parvovirus B19 infection in solid organ transplant recipients: a case report and review of literature. Clin Transplant 2000; 14: 586-591
12 Cantú TG, Hoehn-Saric EW, Burgess KM, Racusen L, Scheel PJ. Acute renal failure associated with immunoglobulin therapy. Am J Kidney Dis 1995; 25: 228-234
13 Hayakawa F, Imada K, Towatari M, Saito H. Life-threatening human parvovirus B19 infection transmitted by intravenous immune globulin. Br J Haematol 2002; 118: 1187-1189
14 Brown KE, Young NS, Alving BM, Barbosa LH. Parvovirus B19: implications for transfusion medicine. Summary of a workshop. Transfusion 2001; 41: 130-135
15 Ballou WR, Reed JL, Noble W, Young NS, Koenig S. Safety and immunogenicity of a recombinant parvovirus B19 vaccine formulated with MF59C.1. J Infect Dis 2003; 187: 675-678
16 Nour B, Green M, Michaels M, Reyes J, Tzakis A, Gartner JC, McLoughlin L, Starzl TE. Parvovirus B19 infection in pediatric transplant patients. Transplantation 1993; 56: 835-838
17 Ramage JK, Hale A, Gane E, Cohen B, Boyle M, Mufti G, Williams R. Parvovirus B19-induced red cell aplasia treated with plasmapheresis and immunoglobulin. Lancet 1994; 343: 667-668
18 Chang FY, Singh N, Gayowski T, Marino IR. Parvovirus B19 infection in a liver transplant recipient: case report and review in organ transplant recipients. Clin Transplant 1996; 10: 243-247
19 Gallinella G, Manaresi E, Venturoli S, Grazi GL, Musiani M, Zerbini M. Occurrence and clinical role of active parvovirus B19 infection in transplant recipients. Eur J Clin Microbiol Infect Dis 1999; 18: 811-813
S- Editor Liu Y L- Editor Wang XL E- Editor Zhou T
2010 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

rare neoplasms of the gastrointestinal tract, which a re charac ter ized by a spec i f i c h i s to log ica l and immunohistochemical pattern. These neoplasms have been recently defined as a discrete molecular and pathological entity. The management of GIST has not been clearly established[1].
In some rare cases coexistence of GIST with other malignancies has been reported[2-6]. Here we describe a case of a 74-year old male with GIST who developed multiple myeloma (MM) during the course of GIST treatment. To the best of our knowledge, the co-existence of GIST and multiple myeloma has not been reported so far.
Case reportA 74-year old male was presented complaining of dyspepsia. Clinical examination revealed a palpable epigastric mass, with no other abnormal clinical findings. Computed tomography of the abdomen showed a 15 cm × 16 cm solid mass in the right side of abdomen, deriving from the gastric wall. A submucosal non-metastatic tumor was suspected (Figure 1A). Endoscopy showed normal mucosa of the gastrointestinal tract. Laboratory tests were normal. During the surgery, a 5 cm × 11 cm mass of the posterior wall of the gastric antrum protruding from the inner muscularis propria to the mucosa was found. The mass was completely resected and the histological exami-nation demonstrated whirling sheets of spindle cells which were stained positively for CD 117 (c-kit) and CD34, mitotic index > 10/50 HPF, while smooth muscle actin (SMA) and vimentin were focally positive, and keratine, desmin, S-100 protein were negative. This specific immu-nophenotype characterized GIST. No further therapy was applied. Physical examination, laboratory tests and com-puted tomography were performed when the patient was followed up at 3-month intervals.
Fourteen months after resection, a CT scan of the abdomen showed liver metastases (Figure 1B) and imatinib mesylate at a dose of 400 mg once daily, as the appropriate second line therapy, was administered. After one month of treatment, the patient reported persistent skeletal pain, especially on the thorax, accompanied with weakness and fatigue. X-ray bone survey showed lytic lesions. Laboratory examination revealed normocytic, normochromic anemia (Hb: 10.6 gr/dL), hypercalciemia (Ca: 11.7 mg/dL) and abnormal ESR (140 mm/h). Serum protein electrophoresis and immunofixation showed an
CASE REPORT
Development of multiple myeloma in a patient with gastrointestinal stromal tumor treated with imatinib mesylate: a case report
D Tzilves, A Gatopoulou, K Zervas, E Katodritou, F Patakiouta, A Tarpagos, I Katsos
www.wjgnet.com
D Tzilves, A Gatopoulou, A Tarpagos, I Katsos, Department of Gastroenterology, Theagenio Cancer Hospital, 2 Al. Simeonidi 54007, Thessaloniki, GreeceK Zervas, E Katodritou, Department of Hematology, Theagenio Cancer Hospital, 2 Al. Simeonidi 54007, Thessaloniki, GreeceF Patakiouta, Department of Pathology, Theagenio Cancer Hospital, 2 Al. Simeonidi 54007, Thessaloniki, GreeceCorrespondence to: Dr. Anthia Gatopoulou, Theagenio Cancer Hospital, 2, Al. Simeonidi 54007, Thessaloniki, Greece. [email protected]: +30-2310-898412 Fax: +30-2310-898408Received: 2007-02-01 Accepted: 2007-03-08
AbstractGastrointestinal stromal tumors (GISTs) are rare tumors, which represent approximately 1% of the neoplasms of the gastrointestinal tract. These tumors rarely give extra-abdominal metastases. However, their clinical outcome is potentially adverse. In some rare cases, co-existance of GISTs with other malignancies has been reported. Here we present a case of a 74-year old male with GIST, which was managed by surgical resection. Fourteen months later, the patient presented with liver metastases and imatinib mesylated was administered. During treatment, the patient reported skeletal pain and plane X-rays revealed osteolytic bone lesions. Further investigation revealed the presence of multiple myeloma. To the best of our knowledge, this is the first report of the co-existence of multiple myeloma (MM) with GIST.
© 2007 The WJG Press. All rights reserved.
Key words: Gastrointestinal stromal tumor; Multiple myeloma; Imatinib mesylate
Tzilves D, Gatopoulou A, Zervas K, Katodritou E, Patakiouta F, Tarpagos A, Katsos I. Development of multiple myeloma in a patient with gastrointestinal stromal tumor treated with imatinib mesylate: A case report. World J Gastroenterol 2007; 13(13): 2011-2013
http://www.wjgnet.com/1007-9327/13/2011.asp
INtroDUCtIoNGastrointestinal stromal tumors (GISTs) represent
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 2011-2013www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.
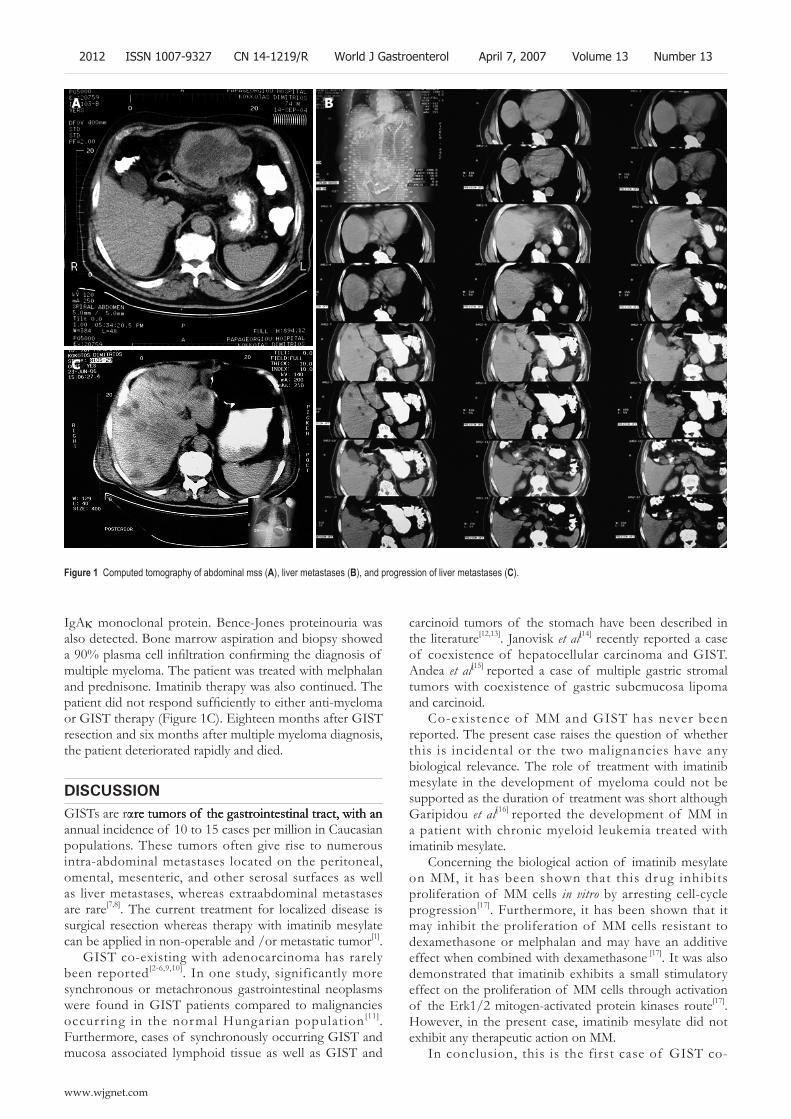
IgAκ monoclonal protein. Bence-Jones proteinouria was also detected. Bone marrow aspiration and biopsy showed a 90% plasma cell infiltration confirming the diagnosis of multiple myeloma. The patient was treated with melphalan and prednisone. Imatinib therapy was also continued. The patient did not respond sufficiently to either anti-myeloma or GIST therapy (Figure 1C). Eighteen months after GIST resection and six months after multiple myeloma diagnosis, the patient deteriorated rapidly and died.
DIsCUssIoNGISTs are rre tumors of the gastrointestinal tract, with anre tumors of the gastrointestinal tract, with anre tumors of the gastrointestinal tract, with an annual incidence of 10 to 15 cases per million in Caucasian populations. These tumors often give rise to numerous intra-abdominal metastases located on the peritoneal, omental, mesenteric, and other serosal surfaces as well as liver metastases, whereas extraabdominal metastases are rare[7,8]. The current treatment for localized disease is surgical resection whereas therapy with imatinib mesylate can be applied in non-operable and /or metastatic tumor[1].
GIST co-existing with adenocarcinoma has rarely been reported[2-6,9,10]. In one study, significantly more synchronous or metachronous gastrointestinal neoplasms were found in GIST patients compared to malignancies occurring in the normal Hungarian population[11]. Furthermore, cases of synchronously occurring GIST and mucosa associated lymphoid tissue as well as GIST and
carcinoid tumors of the stomach have been described in the literature[12,13]. Janovisk et al[14] recently reported a case of coexistence of hepatocellular carcinoma and GIST. Andea et al[15] reported a case of multiple gastric stromal tumors with coexistence of gastric subcmucosa lipoma and carcinoid.
Co-existence of MM and GIST has never been reported. The present case raises the question of whether this is incidental or the two malignancies have any biological relevance. The role of treatment with imatinib mesylate in the development of myeloma could not be supported as the duration of treatment was short although Garipidou et al[16] reported the development of MM in a patient with chronic myeloid leukemia treated with imatinib mesylate.
Concerning the biological action of imatinib mesylate on MM, it has been shown that this drug inhibits proliferation of MM cells in vitro by arresting cell-cycle progression[17]. Furthermore, it has been shown that it may inhibit the proliferation of MM cells resistant to dexamethasone or melphalan and may have an additive effect when combined with dexamethasone [17]. It was also demonstrated that imatinib exhibits a small stimulatory effect on the proliferation of MM cells through activation of the Erk1/2 mitogen-activated protein kinases route[17]. However, in the present case, imatinib mesylate did not exhibit any therapeutic action on MM.
In conclusion, this is the first case of GIST co-
a B
C
Figure 1 Computed tomography of abdominal mss (A), liver metastases (B), and progression of liver metastases (C).
2012 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13
www.wjgnet.com

existing with multiple myeloma. Although the existence of a second malignancy in a patient with GIST is very uncommon, such a possibility should not be ignored. Long-term observation of GIST patients may prove a causal relationship between GIST and other malignancies.
reFereNCes1 Blay JY, Bonvalot S, Casali P, Choi H, Debiec-Richter M,
Dei Tos AP, Emile JF, Gronchi A, Hogendoorn PC, Joensuu H, Le Cesne A, McClure J, Maurel J, Nupponen N, Ray-Coquard I, Reichardt P, Sciot R, Stroobants S, van Glabbeke M, van Oosterom A, Demetri GD. Consensus meeting for the management of gastrointestinal stromal tumors. Report of the GIST Consensus Conference of 20-21 March 2004, under the auspices of ESMO. Ann Oncol 2005; 16: 566-578
2 Bircan S , Candir O, Aydin S, Başpinar S , Bülbül M, Kapucuoğlu N, Karahan N, Ciriş M. Synchronous primary adenocarcinoma and gastrointestinal stromal tumor in the stomach: a report of two cases. Turk J Gastroenterol 2004; 15: 187-191
3 Moschos J, Tzilves D, Paikos D, Tagarakis G, Pilpilidis I, Antonopoulos Z, Kadis S, Katsos I, Tarpagos A. Large mesenteric gastrointestinal stromal tumor in a patient with familial adenomatous polyposis syndrome. Wien Klin Wochenschr 2006; 118: 355-357
4 Rauf F, Ahmad Z, Muzzafar S, Hussaini AS. Synchronous occurrence of gastrointestinal stromal tumor and gastric adenocarcinoma: a case report. J Pak Med Assoc 2006; 56: 184-186
5 Melis M, Choi EA, Anders R, Christiansen P, Fichera A. Synchronous colorectal adenocarcinoma and gastrointestinal stromal tumor (GIST). Int J Colorectal Dis 2007; 22: 109-114
6 Al-Brahim N, Radhi J, Gately J. Synchronous epithelioid stromal tumour and lipoma in the stomach. Can J Gastroenterol 2003; 17: 374-375
7 Tran T , Davila JA, El-Serag HB. The epidemiology of malignant gastrointestinal stromal tumors: an analysis of 1,458 cases from 1992 to 2000. Am J Gastroenterol 2005; 100: 162-168
8 Joensuu H. Gastrointestinal stromal tumor (GIST). Ann Oncol 2006; 17 Suppl 10: x280-x286
9 Lin YL, Tzeng JE, Wei CK, Lin CW. Small gastrointestinalSmall gastrointestinal stromal tumor concomitant with early gastric cancer: a case report. World J Gastroenterol 2006; 12: 815-817
10 Liu SW, Chen GH, Hsieh PP. Collision tumor of the stomach:Collision tumor of the stomach: a case report of mixed gastrointestinal stromal tumor and adenocarcinoma. J Clin Gastroenterol 2002; 35: 332-334
11 Kalmár K, Tornóczky T, Pótó L, Illényi L, Kalmár Nagy K, Kassai M, Kelemen D, Horváth OP. Gastrointestinal stromal tumours in a single institute: is there an association to other gastrointestinal malignancies? Magy Seb 2004; 57: 251-256
12 Kaffes A, Hughes L, Hollinshead J, Katelaris P. Synchronous primary adenocarcinoma, mucosa-associated lymphoid tissue lymphoma and a stromal tumor in a Helicobacter pylori-infected stomach. J Gastroenterol Hepatol 2002; 17: 1033-1036
13 B u r a g a s M , K i d d M , M o d l i n I M , C h a C . M u l t i p l e gastrointestinal stromal tumors and synchronous ileal carcinoids. Nat Clin Pract Oncol 2005; 2: 166-170; quiz 1 p following 170
14 Jaworski R , Jastrzebski T, Swierblewski M, Drucis K, Kobierska-Gulida G. Coexistence of hepatocellular carcinoma and gastrointestinal stromal tumor: a case report. World J Gastroenterol 2006; 12: 665-667
15 Andea AA, Lucas C, Cheng JD, Adsay NV. Synchronous occurrence of epithelial and stromal tumors in the stomach. Arch Pathol Lab Med 2001; 125: 318-319
16 Garipidou V, Vakalopoulou S, Tziomalos K. Development of multiple myeloma in a patient with chronic myeloid leukemia after treatment with imatinib mesylate. Oncologist 2005; 10: 457-458
17 Pandiella A, Carvajal-Vergara X, Tabera S, Mateo G, Gutiérrez N, San Miguel JF. Imatinib mesylate (STI571) inhibits multiple myeloma cell proliferation and potentiates the effect of common antimyeloma agents. Br J Haematol 2003; 123: 858-868
S- Editor Wang J L- Editor Wang XL E- Editor Ma WH
Tzilves D et al. Multiple myeloma in a patient with GIST 2013
www.wjgnet.com

Acknowledgments to Reviewers of World Journal of Gastroenterology
Akira Andoh, MDDepartment of Internal Medicine, Shiga University of Medical Science, Seta Tukinowa, Otsu 520-2192, Japan
Flavia Bortolotti, DrClinica Medica 5, Via Giustiniani 2, 35100 Padova. Italy
Lee Bouwman, DrLeiden University Medical Centre, department of surgery, Albinusdreef 2 PO Box 9600, 230 RC Leiden, The Netherlands
Giuseppe Brisinda, MDDepartment of Surgery, Catholic School of Medicine “Agostino Gemelli”, Largo Agostino Gemelli 8-00168 Rome, Italy
Jose Castellote, PhDUniversitari de Bellvitge. L’Hospitalet de Llobregat Barcelona. C/ Feixa Llarga S/N, L’hospitalet de Llobregat Barcelona 08023, Spain
Dario Conte, ProfessorGI Unit-IRCCS Osp. Maggiore, Chair of Gastroenterology, Via F. Sforza, 35, Milano 20122, Italy
Paolo Del Poggio, DrHepatology Unit, Department of Internal Medicine, Treviglio Hospital, Piazza Ospedale 1, Treviglio Bg 24047, Italy
Kazuma Fujimoto, ProfessorDepartment of Internal Medicine, Saga Medical School, Nabeshima, Saga, Saga 849-8501, Japan
Nikolaus Gassler, Professor Institute of Pathology, University Hospital RWTH Aachen, Pauwelsstrasse 30, 52074 Aachen, Germany
Marek HartlebGastroenterologii, CSK, ul. Medyków 14, 40-752 Katowice, Poland
Martin HennenbergDipl-Biol, Medizinische Klinik & Poliklinik I, Uni-Klinik Bonn, Sigmund-Freud Str. 25, 53105 Bonn, Germany
Yik-Hong Ho, ProfessorDepartment of Surgery, School of Medicine, James Cook University, Townsville 4811, Australia
Shunji Ishihara, MDDepartment of Gastroenterology and Hepatology, Shimane University, School of Medicine, 89-1, Enya-cho, Izumo 693-8501, Japan
Ryuichi Iwakiri, DrDepartment of Medicine and Gastrointestinal Endoscopy, Saga Medical School, 5-1-1 Nabeshima, Saga 849-8501, Japan
Ioannis E Koutroubakis, Assistant Professor
Ioannis Koutroubakis, Gastroenterology, University Hospital Heraklion, PO BOX: 1352 Heraklion, 71110 Crete, Greece
Richard A Kozarek, MDDepartment of Gastroenterology, Virginia Mason Medical Center, 1100 Ninth Avenue, PO Box 900, Seattle 98111-0900, United States
James YW LauDepartment of Surgery, Prince of Wales Hospital, the Chinese University of Hong Kong, Hong Kong , China
Shou-Dong Lee, ProfessorDepartment of Medicine, Taipei Veterans General Hospital, 201 Shih-Pai Road, Sec. 2. Taipei 112, Taiwan, China
Yasushi Matsuzaki, Associated ProfessorDivision of Gastroenterology and Hepatology, Graduate School of Comprehensive Human Sciences and University Hospital, 1-1-1, Tennodai, Tsukuba 305-8575, Japan
Yuji Naito, ProfessorKyoto Prefectural University of Medicine, Kamigyo-ku, Kyoto 602-8566, Japan
CS Pitchumoni, ProfessorRobert Wood Johnson School of Medicine, Robert Wood Johnson School of Medicine, New Brunswiuc NJ D8903, United States
Ian C Roberts-Thomson, ProfessorDepartment of Gastroenterology and Hepatology, The Queen Elizabeth Hospital, 28 Woodville Road, Woodville South 5011, Australia
Luis Rodrigo, ProfessorGastroenterology Service, Hospital Central de Asturias, c/ Celestino Villamil, s.n., Oviedo 33.006, Spain
Spiros SgourosNaypaktias 5, Agia Paraskevi, Athens 15341, Greece
Shivendra Shukla, ProfessorDepartment of Medical Pharmacology and Physiology, University of Missouri School of Medicine, 1 Hospital Drive, M530 Medical Sciences Bldg., Columbia MO 65212, United States
Spring Kevin John Spring, DrConjoint Gastroenterology, RBWHF-CRC & QIMR, PO Royal Brisbane Hospital, Herston, Brisbane 4029, Australia
Seyed Alireza Taghavi, Associate ProfessorDepartment of Internal Medicine, Nemazee Hospital, No.23, 59th Alley, Ghasrodasht St., Shiraz 71838-95453, Iran
Takuji Torimura, MDSecond Department of Medicine, Kurume University School of Medicine, 67 Asahi-machi, Kurume City, Fukuoka 830-0011, Japan
Akihito Tsubota, Assistant ProfessorInstitute of Clinical Medicine and Research , Jikei University School of Medicine, 163-1 Kashiwa-shita, Kashiwa, Chiba 277-8567, Japan
Tung-Yu Tsui, DrDepartment of Surgery, University of Regensburg Medical Centre, Franz-Josef-Strauss-Allee 11, 93053 Regensburg, Germany
Silvana Zanlungo, ProfessorDepartamento de Gastroenterología, Pontificia Universidad Católica de Chile, Marcoleta 367, Casilla 114-D, Santiago, Chile
ACKNOWLEDGMENTS
Many reviewers have contributed their expertise and time to the peer review, a critical process to ensure the quality of World Journal of Gastroenterology. The editors and authors of the articles submitted to the journal are grateful to the following reviewers for evaluating the articles (including those were published and those were rejected in this issue) during the last editing period of time.
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 2014www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.

2-6 November 2007 Boston-MA www.aasld.org
Gastro 2009, World Congress of Gas-troenterology and Endoscopy Lon-don, United Kingdom 2009
Meeting Falk Symposium 160:Pathogenesis and Clinical Practice inGastroenterology 15-16 June 2007 Portoroz [email protected]
Meeting ILTS 13th AnnualInternational Congress 20-23 June 2007 Rio De Janeiro www.ilts.org
Meeting 9th World Congress onGastrointestinal Cancer 27-30 June 2007 Barcelona [email protected]
Meeting 15th International Congressof the European Association forEndoscopic Surgery 4-7 July 2007 Athens [email protected]/
Meeting 39th Meeting of theEuropean Pancreatic Club 4-7 July 2007 Newcastle www.e-p-c2007.com
Meeting XXth InternationalWorkshop on Heliobacter andrelated bacteria in cronic degistiveinfl ammation20-22 September 2007 Istanbul www.heliobacter.org
Meeting Falk Workshop: Mechanisms of Intestinal Infl ammation 10 October 2007Dresden [email protected]
Meeting Falk Symposium 161: Future Perspectives in Gastroenterology 11-12 October 2007 Dresden [email protected]
Meeting Falk Symposium 162: LiverCirrhosis - From Pathophysiology toDisease Management 13-14 October 2007 Dresden [email protected]
American College ofGastroenterology Annual Scientifi cMeeting 12-17 October 2007 Pennsylvania Convention Center Philadelphia, PA
Meeting APDW 2007 - Asian Pacific Digestive Disease Week 2007 15-18 October 2007 Kobe [email protected]
15th United EuropeanGastroenterology Week, UEGW 27-31 October 2007 Le Palais des Congrès de Paris, Paris, France
Meeting The Liver Meeting® 2007 - 57th Annual Meeting of the AmericanAssociation for the Study of LiverDiseases
Gastroenterology 15-16 June 2007 Portoroz [email protected]
Meeting ILTS 13th AnnualInternational Congress 20-23 June 2007 Rio De Janeiro www.ilts.org
Meeting 9th World Congress onGastrointestinal Cancer27-30 June 2007 Barcelona [email protected]
EVENTS AND MEETINGS IN 2007 Meeting Falk Research Workshop:Morphogenesis and Cancerogenesisof the Liver 25-26 January 2007 Goettingen [email protected]
Meeting Canadian Digestive Diseases Week (CDDW) 16-20 February 2007 Banff-ABcagoffi [email protected] www.cag-acg.org/cddw/cddw2007.htm
Meeting Falk Symposium 158:Intestinal Infl ammation andColorectal Cancer 23-24 March 2007 Sevilla [email protected]
Meeting BSG Annual Meeting 26-29 March 2007Glasgow www.bsg.org.uk/
Meeting 42nd Annual Meeting of the European Association for the Study of the Liver 11-15 April 2007 Barcelona [email protected]/liver-meeting/
Meeting Falk Symposium 159: IBD 2007 - Achievements in Research and Clinical Practice 4-5 May 2007 Istanbul [email protected]
Meeting European Society forPaediatric Gastroenterology,Hepatology and Nutrition Congress2007 9-12 May 2007 Barcelona [email protected]
Meeting Gastrointestinal Endoscopy Best Practices: Today and Tomorrow, ASGE Annual Postgraduate Course at DDW 23-24 May 2007 Washington-DC [email protected]
Meeting ESGAR 2007 18th Annual Meeting and Postgraduate Course 12-15 June 2007 Lisbon [email protected]
Meetings
MAJOR MEETINGS COMING UPMeeting Falk Research Workshop:Morphogenesis and Cancerogenesisof the Liver 25-26 January 2007 Goettingen [email protected]
Meeting Canadian Digestive Diseases Week (CDDW) 16-20 February 2007 Banff-AB cagoffi [email protected]/cddw/cddw2007.htm
Meeting Falk Symposium 158:Intestinal Infl ammation andColorectal Cancer 23-24 March 2007 Sevilla [email protected]
Meeting BSG Annual Meeting26-29 March 2007Glasgowwww.bsg.org.uk/
NEXT 6 MONTHSMeeting 42nd Annual Meeting of theEuropean Association for the Studyof the Liver 11-15 April 2007 Barcelona [email protected]/liver-meeting/
Meeting Falk Symposium 159: IBD2007 - Achievements in Research andClinical Practice 4-5 May 2007 Istanbul [email protected]
Meeting European Society forPaediatric Gastroenterology,Hepatology and Nutrition Congress2007 9-12 May 2007 Barcelona [email protected]
Digestive Disease Week 19-24 May 2007 Washington Convention Center,Washington DC
Meeting Gastrointestinal EndoscopyBest Practices: Today and Tomorrow,ASGE Annual Postgraduate Courseat DDW 23-24 May 2007 Washington-DC [email protected]
Meeting ESGAR 2007 18th AnnualMeeting and Postgraduate Course 12-15 June 2007 Lisbon [email protected]
Meeting Falk Symposium 160:Pathogenesis and Clinical Practice in
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 2015www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.

www.wjgnet.com
Instructions to authorsGENERAL INFORMATIONWorld Journal of Gastroenterology (WJG, World J Gastroenterol ISSN 1007-9327 CN 14-1219/R) is a weekly journal of more than 48 000 circulation, published on the 7th, 14th, 21st and 28th of every month.
Original Research, Clinical Trials, Reviews, Comments, and Case Reports in esophageal cancer, gastric cancer, colon cancer, liver cancer, viral liver diseases, etc., from all over the world are welcome on the condition that they have not been published previously and have not been submitted simultaneously elsewhere.
Indexed and abstracted inCurrent Contents®/Clinical Medicine, Science Citation Index Expanded (also known as SciSearch®) and Journal Citation Reports/Science Edition, Index Medicus, MEDLINE and PubMed, Chemical Abstracts, EMBASE/Excerpta Medica, Abstracts Journals, Nature Clinical Practice Gastroenterology and Hepatology, CAB Abstracts and Global Health. ISI JCR 2003-2000 IF: 3.318, 2.532, 1.445 and 0.993.
Published byThe WJG Press
SUBMISSION OF MANUSCRIPTSManuscripts should be typed double-spaced on A4 (297 mm × 210 mm) white paper with outer margins of 2.5 cm. Number all pages consecutively, and start each of the following sections on a new page: Title Page , Abstract, Introduction, Materials and Methods, Results, Discussion, acknowledgements, References, Tables, Figures and Figure Legends. Neither the editors nor the Publisher is responsible for the opinions expressed by contributors. Manuscripts formally accepted for publication become the permanent property of The WJG Press, and may not be reproduced by any means, in whole or in part without the written permission of both the authors and the Publisher. We reserve the right to put onto our website and copy-edit accepted manuscripts. Authors should also follow the guidelines for the care and use of laboratory animals of their institution or national animal welfare committee.
Authors should retain one copy of the text, tables, photographs and illustrations, as rejected manuscripts will not be returned to the author(s) and the editors will not be responsible for the loss or damage to photographs and illustrations in mailing process.
Online submissionOnline submission is strongly advised. Manuscripts should be submitted through the Online Submission System at: http://www.wjgnet.com/index.jsp. Authors are highly recommended to consult the ONLINE INSTRUCTIONS TO AUTHORS (http://www.wjgnet.com/wjg/help/instructions.jsp) before attempting to submit online. Authors encountering problems with the Online Submission System may send an email you describing the problem to [email protected] for assistance. If you submit your manuscript online, do not make a postal contribution. A repeated online submission for the same manuscript is strictly prohibited.
Postal submissionSend 3 duplicate hard copies of the full-text manuscript typed double-spaced on A4 (297 mm × 210 mm) white paper together with any original photographs or illustrations and a 3.5 inch computer diskette or CD-ROM containing an electronic copy of the manuscript including all the fi gures, graphs and tables in native Microsoft Word format or *.rtf format to:
Editorial Offi ce World Journal of GastroenterologyEditorial Department: Apartment 1066, Yishou Garden,58 North Langxinzhuang Road,PO Box 2345, Beijing 100023, ChinaE-mail: [email protected]: //www.wjgnet.comTelephone: +86-10-85381892Fax: +86-10-85381893
MANUSCRIPT PREPARATIONAll contributions should be written in English. All articles must be submitted using a word-processing software. All submissions must be typed in 1.5
line spacing and in word size 12 with ample margins. The letter font is Tahoma. For authors from China, one copy of the Chinese translation of the manuscript is also required (excluding references). Style should conform to our house format. Required information for each of the manuscript sections is as follows:
Title pageFull manuscript title, running title, all author(s) name(s), affiliations, institution(s) and/or department(s) where the work was accomplished, disclosure of any financial support for the research, and the name, full address, telephone and fax numbers and email address of the corresponding author should be included. Titles should be concise and informative (removing all unnecessary words), emphasize what is new, and avoid abbreviations. A short running title of less than 40 letters should be provided. List the author(s)’ name(s) as follows: initial and/or first name, middle name or initial(s) and full family name.
AbstractAn informative, structured abstract of no more than 250 words should accompany each manuscript. Abstracts for original contributions should be structured into the following sections: AIM: Only the purpose should be included. METHODS: The materials, techniques, instruments and equipments, and the experimental procedures should be included. RESULTS: The observatory and experimental results, including data, effects, outcome, etc. should be included. Authors should present P value where necessary, and the signifi cant data should accompany. CONCLUSION: Accurate view and the value of the results should be included.
The format of structured abstracts is at: http://www.wjgnet.com/wjg/help/11.doc
Key wordsPlease list 5-10 key words that could refl ect content of the study mainly from Index Medicus.
TextFor most article types, the main text should be structured into the following sections: INTRODUCTION, MATERIALS AND METHODS, RESULTS and DISCUSSION, and should include in appropriate Figures and Tables. Data should be presented in the body text or in Figures and Tables, but not in both.
IllustrationsFigures should be numbered as 1, 2, 3 and so on, and mentioned clearly in the main text. Provide a brief title for each fi gure on a separate page. No detailed legend should be involved under the fi gures. This part should be added into the text where the fi gures are applicable. Digital images: black and white photographs should be scanned and saved in TIFF format at a resolution of 300 dpi; color images should be saved as CMYK (print fi les) but not as RGB (screen-viewing fi les). Place each photograph in a separate fi le. Print images: supply images of size no smaller than 126 mm × 85 mm printed on smooth surface paper; label the image by writing the Figure number and orientation using an arrow. Photomicrographs: indicate the original magnifi cation and stain in the legend. Digital Drawings: supply fi les in EPS if created by freehand and illustrator, or TIFF from photoshops. EPS fi les must be accompanied by a version in native fi le format for editing purposes. Existing line drawings should be scanned at a resolution of 1200 dpi and as close as possible to the size where they will appear when printed. Please use uniform legends for the same subjects. For example: Figure 1 Pathological changes of atrophic gastritis after treatment. A: ...; B: ...; C: ...; D: ...; E: ...; F: ...; G: ...
TablesThree-line tables should be numbered as 1, 2, 3 and so on, and mentioned clearly in the main text. Provide a brief title for each table. No detailed legend should be included under the tables. This part should be added into the text where the tables are applicable. The information should complement but not duplicate that contained in the text. Use one horizontal line under the title, a second under the column heads, and a third below the Table, above any footnotes. Vertical and italic lines should be omitted.
Notes in tables and illustrationsData that are not statistically significant should not be noted. aP<0.05, bP<0.01 should be noted (P>0.05 should not be noted). If there are other series of P values, cP<0.05 and dP<0.01 are used. Third series of P values can be expressed as eP<0.05 and fP<0.01. Other notes in tables or under
PO Box 2345, Beijing 100023, China World J Gastroenterol 2007 April 7; 13(13): 2016-2018www.wjgnet.com World Journal of Gastroenterology ISSN [email protected] © 2007 The WJG Press. All rights reserved.

illustrations should be expressed as 1F, 2F, 3F; or some other symbols with a superscript (Arabic numerals) in the upper left corner. In a multi-curve illustration, each curve should be labeled with , , , , , , etc. in a certain sequence.
AcknowledgmentsBrief acknowledgments of persons who have made genuine contributions to the manuscripts and who endorse the data and conclusions are included. Authors are responsible for obtaining written permission to use any copyrighted text and/or illustrations.
REFERENCESCoding systemThe author should code the references according the citation order in text in Arabic numerals, put references codes in square brackets, superscript it at the end of citation content or the author name of the citation. For those citation content as the narrate part, the coding number and square brackets should be typeset normally. For example, Crohn’s disease (CD) is associated with increased intestinal permeability[1,2]. If references are directly cited in the text, they would be put together with the text, for example, from references [19,22-24 ], we know that... When the authors code the references, please ensure that the order in text is the same as in reference part and also insure the spelling accuracy of the fi rst author’s name. Do not code the same citation twice.
PMID requirementPMID roots in the abstract serial number indexed by PubMed (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=PubMed). The author should supply the PMID for journal citation. For those references that have not been indexed by PubMed, a printed copy of the fi rst page of the full reference should be submitted. The accuracy of the information of the journal citations is very important. Through reference testing system, the authors and editor could check the authors name, title, journal title, publication date, volume number, start page, and end page. We will interlink all references with PubMed in ASP file so that the readers can read the abstract of the citations online immediately.
Style for journal referencesAuthors: the fi rst author should be typed in bold-faced letter. The surname of all authors should be typed with the initial letter capitalized and followed by their name in abbreviation (For example, Lian-Sheng Ma is abbreviated as Ma LS, Bo-Rong Pan as Pan BR). Title of the cited article and italicized journal title (Journal title should be in its abbreviation form as shown in PubMed), publication date, volume number (in black), start page, and end page [PMID: 11819634] Note: The author should test the references through reference testing system (http://www.wjgnet.com/cgi-bin/index.pl)
Style for book referencesAuthors: the fi rst author should be typed in bold-faced letter. The surname of all authors should be typed with the initial letter capitalized and followed by their name in abbreviation (For example, Lian-Sheng Ma is abbreviated as Ma LS, Bo-Rong Pan as Pan BR) Book title. Publication number. Publication place: Publication press, Year: start page and end page.
FormatJournals English journal article (list all authors and include the PMID where applicable)1 Grover VP, Dresner MA, Forton DM, Counsell S, Larkman DJ, Patel N,
Thomas HC, Taylor-Robinson SD. Current and future applications of magnetic resonance imaging and spectroscopy of the brain in hepatic encephalopathy. World J Gastroenterol 2006; 12: 2969-2978 [PMID: 16718775]
Chinese journal article (list all authors and include the PMID where applicable)2 Lin GZ, Wang XZ, Wang P, Lin J, Yang FD. Immunologic effect of
Jianpi Yishen decoction in treatment of Pixu-diarrhoea. Shijie Huaren Xiaohua Zazhi 1999; 7: 285-287
In press3 Tian D, Araki H, Stahl E, Bergelson J, Kreitman M. Signature of
balancing selection in Arabidopsis. Proc Natl Acad Sci U S A 2006; In press
Organization as author4 Diabetes Prevention Program Research Group. Hypertension,
insulin, and proinsulin in participants with impaired glucose tolerance. Hypertension 2002; 40: 679-686 [ PMID: 12411462 ]
Both personal authors and an organization as author 5 Vallancien G, Emberton M, Harving N, van Moorselaar RJ; Alf-One
Study Group. Sexual dysfunction in 1, 274 European men suffering from lower urinary tract symptoms. J Urol 2003; 169: 2257-2261 [PMID: 12771764]
No author given6 21st century heart solution may have a sting in the tail. BMJ 2002; 325:
184 [PMID: 12142303]Volume with supplement7 Geraud G, Spierings EL, Keywood C. Tolerability and safety of
frovatriptan with short- and long-term use for treatment of migraine and in comparison with sumatriptan. Headache 2002; 42 Suppl 2: S93-99 [PMID: 12028325]
Issue with no volume8 Banit DM, Kaufer H, Hartford JM. Intraoperative frozen section
analysis in revision total joint arthroplasty. Clin Orthop Relat Res 2002; (401): 230-238 [ PMID: 12151900 ]
No volume or issue9 Outreach: bringing HIV-positive individuals into care. HRSA Careaction
2002; 1-6 [PMID: 12154804 ]Books Personal author(s)10 Sherlock S, Dooley J. Diseases of the liver and billiary system. 9th ed.
Oxford: Blackwell Sci Pub, 1993: 258-296Chapter in a book (list all authors)11 Lam SK. Academic investigator’s perspectives of medical treatment for
peptic ulcer. In: Swabb EA, Azabo S. Ulcer disease: investigation and basis for therapy. New York: Marcel Dekker, 1991: 431-450
Author(s) and editor(s)12 Breedlove GK, Schorfheide AM. Adolescent pregnancy. 2nd ed.
Wieczorek RR, editor. White Plains (NY): March of Dimes Education Services, 2001: 20-34
Conference proceedings13 Harnden P, Joffe JK, Jones WG, editors. Germ cell tumours V.
Proceedings of the 5th Germ Cell Tumour Conference; 2001 Sep 13-15; Leeds, UK. New York: Springer, 2002: 30-56
Conference paper14 Christensen S, Oppacher F. An analysis of Koza's computational effort
statistic for genetic programming. In: Foster JA, Lutton E, Miller J, Ryan C, Tettamanzi AG, editors. Genetic programming. EuroGP 2002: Proceedings of the 5th European Conference on Genetic Programming; 2002 Apr 3-5; Kinsdale, Ireland. Berlin: Springer, 2002: 182-191
Electronic journal (list all authors) Morse SS. Factors in the emergence of infectious diseases. Emerg
Infect Dis serial online, 1995-01-03, cited 1996-06-05; 1(1): 24 screens. Available from: URL: http//www.cdc.gov/ncidod/EID/eid.htm
Patent (list all authors)16 Pagedas AC, inventor; Ancel Surgical R&D Inc., assignee. Flexible
endoscopic grasping and cutting device and positioning tool assembly. United States patent US 20020103498. 2002 Aug 1
Inappropriate referencesAuthors should always cite references that are relevant to their article, and avoid any inappropriate references. Inappropriate references include those that are linked with a hyphen and the difference between the two numbers at two sides of the hyphen is more than 5. For example, [1-6], [2-14] and [1, 3, 4-10, 22] are all considered as inappropriate references. Authors should not cite their own unrelated published articles.
Statistical dataPresent as mean ± SD or mean ± SE.
Statistical expressionExpress t test as t (in italics), F test as F (in italics), chi square test as χ2 (in Greek), related coeffi cient as r (in italics), degree of freedom as γ (in Greek), sample number as n (in italics), and probability as P (in italics).
UnitsUse SI units. For example: body mass, m (B) = 78 kg; blood pressure, p (B) = 16.2/12.3 kPa; incubation time, t (incubation) = 96 h, blood glucose concentration, c (glucose) 6.4 ± 2.1 mmol/L; blood CEA mass concentration, p (CEA) = 8.6 24.5 μg/L; CO2 volume fraction, 50 mL/L CO2 not 5% CO2; likewise for 40 g/L formaldehyde, not 10% formalin; and mass fraction, 8 ng/g, etc. Arabic numerals such as 23, 243, 641 should be read 23 243 641.
The format about how to accurately write common units and quantum is at: http://www.wjgnet.com/wjg/help/15.doc
Instructions to authors 2017

AbbreviationsStandard abbreviations should be defi ned in the abstract and on fi rst mention in the text. In general, terms should not be abbreviated unless they are used repeatedly and the abbreviation is helpful to the reader. Permissible abbreviations are listed in Units, Symbols and Abbreviations: A Guide for Biological and Medical Editors and Authors (Ed. Baron DN, 1988) published by The Royal Society of Medicine, London. Certain commonly used abbreviations, such as DNA, RNA, HIV, LD50, PCR, HBV, ECG, WBC, RBC, CT, ESR, CSF, IgG, ELISA, PBS, ATP, EDTA, mAb, can be used directly without further mention.
ItalicsQuantities: t time or temperature, c concentration, A area, l length, m mass, V volume.Genotypes: gyrA, arg 1, c myc, c fos, etc.Restriction enzymes: EcoRI, HindI, BamHI, Kbo I, Kpn I, etc.Biology: H pylori, E coli, etc.
SUBMISSION OF THE REVISED MANUSCRIPTS AFTER ACCEPTEDPlease revise your article according to the revision policies of WJG. The revised version including manuscript and high-resolution image fi gures (if any) should be copied on a fl oppy or compact disk. Author should send the revised manuscript, along with printed high-resolution color or black and white photos, copyright transfer letter, the fi nal check list for authors, and responses to reviewers by a courier (such as EMS) (submission of revised manuscript by e-mail or on the WJG Editorial Offi ce Online System is NOT available at present).
Language evaluation The language of a manuscript will be graded before sending for revision. (1) Grade A: priority publishing; (2) Grade B: minor language polishing; (3) Grade C: a great deal of language polishing; (4) Grade D: rejected. The revised articles should be in grade B or grade A.
Copyright assignment formPlease downloaded CAF from http://www.wjgnet.com/wjg/help/9.doc. We certify that the material contained in this manuscript:Ms:
Title:
is original, except when appropriately referenced to other sources, and that written permission has been granted by any existing copyright holders. We agree to transfer to WJG all rights of our manuscript, including: (1) all copyright ownership in all print and electronic formats; (2) the right to grant permission to republish or reprint the stated material in whole or in part, with or without a fee; (3) the right to print copies for free distribution or sale; (4) the right to republish the stated material in a collection of articles or in any other format. We also agree that our article be put on the Internet.Criteria for authorship: The WJG requests and publishes information about contributions of each author named to the submitted study. Authorship credit should be based on (1)direct participation in the study, including substantial contributions to conception and design of study, or acquisition of data, or analysis and interpretation of data; (2) manuscript writing, including drafting the article, or revising it critically for important intellectual content; (3)supportive work, including statistical analysis of data, or acquisition of funding, or administration, technology and materials support, or supervision, or supportive contributions. Authors should meet at least one of the three conditions. The WJG does not publish co-fi rst authors and co-corresponding authors.
We hereby assign copyright transfer to WJG if this paper is accepted.Author Name in full (Full names should be provided, with fi rst name
fi rst, followed by middle names and family name at the last, eg, Eamonn MM Quigley). Handwritten names are not accepted.
Author Name in abbreviation (Family name is put fi rst in full, followed by middle names and fi rst name in abbreviation with fi rst letter in capital, eg, Quigley EMM). Handwritten names are not accepted.
1 Full Name: Abbreviation Name: Signed: Date:
2 Full Name: Abbreviation Name: Signed: Date:
3 Full Name: Abbreviation Name: Signed: Date:
4 Full Name: Abbreviation Name: Signed: Date:
5 Full Name: Abbreviation Name: Signed: Date:
6 Full Name: Abbreviation Name: Signed: Date:
7 Full Name: Abbreviation Name: Signed: Date:
8 Full Name: Abbreviation Name: Signed: Date:
9 Full Name: Abbreviation Name: Signed: Date:
10 Full Name: Abbreviation Name: Signed: Date:
Final check list for authorsThe format is at: http://www.wjgnet.com/wjg/help/13.doc
Responses to reviewersPlease revise your article according to the comments/suggestions of reviewers. The format for responses to the reviewers’ comments is at: http://www.wjgnet.com/wjg/help/10.doc
Proof of fi nancial supportFor paper supported by a foundation, authors should provide a copy of the document and serial number of the foundation.
Publication feeAuthors of accepted articles must pay publication fee.EDITORIAL and LETTERS TO THE EDITOR are free of change.
2018 ISSN 1007-9327 CN 14-1219/R World J Gastroenterol April 7, 2007 Volume 13 Number 13




















