
G6PC3 mutations are associated with a major defect of glycosylation: a novel mechanism for...
11
G6PC3 mutations are associated with a major defect of glycosylation: a novel mechanism for neutrophil dysfunction Bu’Hussain Hayee 2,3,* , Aristotelis Antonopoulos 4,* , Emma J Murphy 2 , Farooq Z Rahman 2 , Gavin Sewell 2 , Bradley N Smith 5 , Sara McCartney 3 , Mark Furman 6 , Georgina Hall 7 , Stuart L Bloom 3 , Stuart M Haslam 4 , Howard R Morris 4 , Kaan Boztug 8,9 , Christoph Klein 8 , Bryan Winchester 10 , Edgar Pick 11 , David C Linch 5 , Rosemary E Gale 5 , Andrew M Smith 2 , Anne Dell 1,4 , and Anthony W Segal 1,2 2 Department of Molecular Medicine, and 3 Research Department of Haematology, University College London, London WC1E 6BT, UK; 4 Division of Molecular Biosciences, Imperial College London, London SW7 2AZ, UK; 5 Department of Gastroenterology, UCLH NHS Foundation Trust, London NW1 2BU, UK; 6 Department of Paediatric Gastroenterology, Royal Free Hampstead NHS Trust, London WC1N 3JH, UK; 7 Paediatric Haematology/Oncology Unit, Oxford Children’ s Hospital, John Radcliffe Hospital, Oxford, UK; 8 Department of Pediatric Hematology/Oncology, Hannover Medical School, Hannover, Germany; 9 Center for Molecular Medicine (CeMM) of the Austrian Academy of Sciences, A-1080 Vienna, Austria; 10 Biochemistry Research Group, UCL Institute of Child Health, London WC1N 1EH, UK; and 11 Julius Friedrich Cohnheim Laboratory of Phagocyte Research, Sackler School of Medicine,Tel Aviv University, Tel Aviv, Israel Received on December 4, 2010; revised on February 3, 2011; accepted on March 2, 2011 Glucose-6-phosphatase, an enzyme localized in the endo- plasmic reticulum (ER), catalyzes the hydrolysis of glucose-6-phosphate (G6P) to glucose and inorganic phos- phate. In humans, there are three differentially expressed glucose-6-phosphatase catabolic genes (G6PC1–3). Recently, it has been shown that mutations in the G6PC3 gene result in a syndrome associating congenital neutrope- nia and various organ malformations. The enzymatic function of G6PC3 is dependent on G6P transport into the ER, mediated by G6P translocase (G6PT). Mutations in the gene encoding G6PT result in glycogen storage disease type-1b (GSD-1b). Interestingly, GSD-1b patients exhibit a similar neutrophil dysfunction to that observed in G6PC3- deficient patients. To better understand the causes of neu- trophil dysfunction in both diseases, we have studied the neutrophil nicotinamide adenine dinucleotide phosphate (NADPH) oxidase of patients with G6PC3 and G6PT syndromes. Unexpectedly, sodium dodecyl sulfate– polyacrylamide gel electrophoresis experiments indicated hypo-glycosylation of gp91 phox , the electron-transporting component of the NADPH oxidase, in all of these patients. Rigorous mass spectrometric glycomic profiling showed that most of the complex-type antennae which characterize the neutrophil N-glycome of healthy individuals were severely truncated in the patients’ neutrophils. A compar- able truncation of the core 2 antenna of the O-glycans was also observed. This aberrant neutrophil glycosylation is predicted to have profound effects on the neutrophil func- tion and merit designation of both syndromes as a new class of congenital disorders of glycosylation. Keywords: G6PC3 / glycogen storage disease type-1b / neutrophil / glycosylation / respiratory burst Introduction Glucose-6-phosphatase (EC 3.1.3.9) catalyzes the hydrolysis of glucose-6-phosphate (G6P) to glucose and inorganic phos- phate. It is located in the endoplasmic reticulum (ER) mem- brane with its active site facing the ER lumen (Pan et al. 1998). In humans, there are three differentially expressed glucose-6-phosphatase genes. Glucose-6-phosphatase cata- bolic-1 (G6PC1) is expressed in the major gluconeogenic organs (liver, kidney and small intestine; Mithieux et al. 1996; Rajas et al. 1999) and plays a key role in overall glucose homeostasis (Mithieux 1997; Mithieux et al. 2004; Pocai et al. 2005). Mutations in G6PC1 cause glycogen storage disease type-1a (GSD-1a) that is characterized by growth retardation, hypoglycemia, hepatomegaly, kidney enlargement, hyperlipidemia, hyperuricemia and lactic acidemia (Lei et al. 1993; Chou et al. 2010). G6PC2 is exclusively expressed in pancreatic islet cells (Arden et al. 1999; Martin et al. 2001) and may be involved in glucose-dependent insulin secretion by controlling free glucose levels (Petrolonis et al. 2004). The functions of the third family member, G6PC3, are poorly understood, although it is known to be ubiquitously expressed (Martin et al. 2002; Guionie et al. 2003). Recently, studies of diseases that involve neutrophil dys- function have begun to cast some light on the role of G6PC3. Significantly, it has been found that G6PC3 is the defective *Co-first authors 1 To whom correspondence should be addressed: Tel: +44-207-679-6170; Fax: +44-207-225-0458; e-mail: [email protected] (A.W.S.); Tel: +44-207-594- 5219; Fax: +44-207-679-0967; e-mail: [email protected] (A.D.) Glycobiology vol. 21 no. 7 pp. 914–924, 2011 doi:10.1093/glycob/cwr023 Advance Access publication on March 8, 2011 © The Author 2011. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: [email protected] 914
Transcript of G6PC3 mutations are associated with a major defect of glycosylation: a novel mechanism for...

G6PC3 mutations are associated with a major defect ofglycosylation: a novel mechanism for neutrophildysfunction
Bu’Hussain Hayee2,3,*, Aristotelis Antonopoulos4,*, EmmaJ Murphy2, Farooq Z Rahman2, Gavin Sewell2, BradleyN Smith5, Sara McCartney3, Mark Furman6,Georgina Hall7, Stuart L Bloom3, Stuart M Haslam4,Howard R Morris4, Kaan Boztug8,9, Christoph Klein8,Bryan Winchester10, Edgar Pick11, David C Linch5,Rosemary E Gale5, Andrew M Smith2, Anne Dell1,4,and Anthony W Segal1,2
2Department of Molecular Medicine, and 3Research Department ofHaematology, University College London, London WC1E 6BT, UK;4Division of Molecular Biosciences, Imperial College London, London SW72AZ, UK; 5Department of Gastroenterology, UCLH NHS Foundation Trust,London NW1 2BU, UK; 6Department of Paediatric Gastroenterology,Royal Free Hampstead NHS Trust, London WC1N 3JH, UK; 7PaediatricHaematology/Oncology Unit, Oxford Children’s Hospital, John RadcliffeHospital, Oxford, UK; 8Department of Pediatric Hematology/Oncology,Hannover Medical School, Hannover, Germany; 9Center for MolecularMedicine (CeMM) of the Austrian Academy of Sciences, A-1080 Vienna,Austria; 10Biochemistry Research Group, UCL Institute of Child Health,London WC1N 1EH, UK; and 11Julius Friedrich Cohnheim Laboratory ofPhagocyte Research, Sackler School of Medicine, Tel Aviv University,Tel Aviv, Israel
Received on December 4, 2010; revised on February 3, 2011; accepted onMarch 2, 2011
Glucose-6-phosphatase, an enzyme localized in the endo-plasmic reticulum (ER), catalyzes the hydrolysis ofglucose-6-phosphate (G6P) to glucose and inorganic phos-phate. In humans, there are three differentially expressedglucose-6-phosphatase catabolic genes (G6PC1–3).Recently, it has been shown that mutations in the G6PC3gene result in a syndrome associating congenital neutrope-nia and various organ malformations. The enzymaticfunction of G6PC3 is dependent on G6P transport into theER, mediated by G6P translocase (G6PT). Mutations inthe gene encoding G6PT result in glycogen storage diseasetype-1b (GSD-1b). Interestingly, GSD-1b patients exhibit asimilar neutrophil dysfunction to that observed in G6PC3-deficient patients. To better understand the causes of neu-trophil dysfunction in both diseases, we have studied theneutrophil nicotinamide adenine dinucleotide phosphate
(NADPH) oxidase of patients with G6PC3 and G6PTsyndromes. Unexpectedly, sodium dodecyl sulfate–polyacrylamide gel electrophoresis experiments indicatedhypo-glycosylation of gp91phox, the electron-transportingcomponent of the NADPH oxidase, in all of these patients.Rigorous mass spectrometric glycomic profiling showedthat most of the complex-type antennae which characterizethe neutrophil N-glycome of healthy individuals wereseverely truncated in the patients’ neutrophils. A compar-able truncation of the core 2 antenna of the O-glycans wasalso observed. This aberrant neutrophil glycosylation ispredicted to have profound effects on the neutrophil func-tion and merit designation of both syndromes as a newclass of congenital disorders of glycosylation.
Keywords: G6PC3 / glycogen storage disease type-1b /neutrophil / glycosylation / respiratory burst
Introduction
Glucose-6-phosphatase (EC 3.1.3.9) catalyzes the hydrolysisof glucose-6-phosphate (G6P) to glucose and inorganic phos-phate. It is located in the endoplasmic reticulum (ER) mem-brane with its active site facing the ER lumen (Pan et al.1998). In humans, there are three differentially expressedglucose-6-phosphatase genes. Glucose-6-phosphatase cata-bolic-1 (G6PC1) is expressed in the major gluconeogenicorgans (liver, kidney and small intestine; Mithieux et al. 1996;Rajas et al. 1999) and plays a key role in overall glucosehomeostasis (Mithieux 1997; Mithieux et al. 2004; Pocaiet al. 2005). Mutations in G6PC1 cause glycogen storagedisease type-1a (GSD-1a) that is characterized by growthretardation, hypoglycemia, hepatomegaly, kidney enlargement,hyperlipidemia, hyperuricemia and lactic acidemia (Lei et al.1993; Chou et al. 2010). G6PC2 is exclusively expressed inpancreatic islet cells (Arden et al. 1999; Martin et al. 2001)and may be involved in glucose-dependent insulin secretionby controlling free glucose levels (Petrolonis et al. 2004). Thefunctions of the third family member, G6PC3, are poorlyunderstood, although it is known to be ubiquitously expressed(Martin et al. 2002; Guionie et al. 2003).Recently, studies of diseases that involve neutrophil dys-
function have begun to cast some light on the role of G6PC3.Significantly, it has been found that G6PC3 is the defective*Co-first authors
1To whom correspondence should be addressed: Tel: +44-207-679-6170; Fax:+44-207-225-0458; e-mail: [email protected] (A.W.S.); Tel: +44-207-594-5219; Fax: +44-207-679-0967; e-mail: [email protected] (A.D.)
Glycobiology vol. 21 no. 7 pp. 914–924, 2011doi:10.1093/glycob/cwr023Advance Access publication on March 8, 2011
© The Author 2011. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: [email protected] 914

gene in a subset of patients with severe congenital neutrope-nia (Boztug et al. 2009). The neutrophils of these patientsdisplay enhanced ER stress and increased rates of apoptosis(Aróstegui et al. 2009; Boztug et al. 2009). Moreover, patientswith this syndrome have short stature and cardiac defects.They do not, however, have the metabolic disorders associatedwith GSD-1a, presumably because their G6PC1 is normalimplying that their major gluconeogenic organs have a fullyfunctioning glucose-6-phosphatase. A recent study identifiedthat homozygous G6PC3 G260R mutation abolishes enzy-matic function causing neutropenia but without a failure ofneutrophil production (McDermott et al. 2010).G6P, the substrate for glucose-6-phosphatase, is transported
into the ER by G6P translocase (G6PT; Pan et al. 1999).G6PT and G6PC appear to work in concert to maintainglucose homeostasis in gluconeogenic organs (Lei et al. 1996;Chou et al. 2002; Boztug and Klein 2009). Mutations in thegene encoding G6PT (Annabi et al. 1998; Chou et al. 2002)cause GSD-1b (Narisawa et al. 1986; Melis et al. 2005),which shares the GSD-1a symptoms. However, in contrast toGSD-1a, GSD-1b additionally exhibits congenital neutropeniaand neutrophil dysfunction resulting in impaired neutrophilrespiratory burst, defective bacterial killing and consequentsusceptibility to infection. Moreover, mice lacking glucose-6-phosphatase-β (equivalent to G6PC3) display a similar neutro-phil dysfunction and phenotypic features (Cheung et al.2007). Responsible for the latter neutrophil respiratory burst isthe multimeric enzyme complex nicotinamide adenine dinu-cleotide phosphate (NADPH) oxidase which contains bothmembrane-bound (gp91phox and p22phox; cytochrome b558)and various cytosolic proteins (Lambeth 2004).In order to better understand the causes of neutrophil dys-
function in the G6PC3 and G6PT syndromes, we have under-taken an investigation of the NADPH oxidase in theneutrophils of five patients with G6PC3 mutations and twowith defects in the gene for G6PT. Unexpectedly, we foundthat gp91phox, the electron-transporting subunit of the neutro-phil NADPH oxidase, ran aberrantly on sodium dodecylsulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gelsconsistent with hypo-glycosylation. In order to better under-stand this hypo-glycosylation, we employed high-sensitivitymass spectrometric (MS) methodologies to profile the N- andO-glycomes of the patients’ neutrophils. Remarkably, these
analyses showed that the complex-type antennae of most ofthe N- and O-glycans were severely truncated. This aberrantglycosylation was found in all patients and is likely to be themajor cause of neutrophil dysfunction.
ResultsG6PC3 mutation analysisFive patients from four unrelated families were studied. Therewas a common phenotype of short stature, neutropenia andsusceptibility to bacterial infection (Table I). In addition totwo previously reported mutations in G6PC3, two novelmutations were identified, resulting in amino acid substitution(Pro44Ser; patient C; Figure 1A) or deletion (Thr64-Ile70,siblings A and B; Figure 1A and B). All mutations were pre-dicted to involve transmembrane helices of G6PC3(Figure 1C).
The neutrophil respiratory burst and cell-free assayIn keeping with observations in glucose-6-phosphataseβ-deficient mice (Cheung et al. 2007), superoxide production(in nmol of O2
−/106 cells min−1; Figure 2A) was diminished inall G6PC3-deficient patients studied (patient A, 1.78; patientB, 1.54; patient C, 3.11) and in GSD-1b patients (patient X,2.66; patient Y, 1.42) compared with the mean ± SEM (stan-dard error of the mean) result in healthy controls (HCs: 7.17± 0.29; n = 5). Findings were similar in peripheral bloodmonocyte (PBMC)-derived macrophages (Figure 2B), wherethe production of hydrogen peroxide in response to phorbol-myristyl acetate (PMA) was diminished in patient cells com-pared with HCs. Neutrophil superoxide production in theparents of G6PC3-deficient patients (heterozygote carriers ofthe corresponding mutation in their offspring) was normalcompared with HCs (6.07 ± 0.67, P = 0.10). This was con-firmed by flow cytometry-based measurement of the (intra-cellular) respiratory burst (Figure 2C and D).In the cell-free assay (Figure 2E), neutrophil membranes
from patient A displayed comparable levels of oxidase activityin molO�
2 =s=mol cytochrome b558 (83.57 ± 5.94) to positivecontrols (77.03 ± 1.29), when reconstituted with recombinantcytosolic components of the NADPH oxidase.
Table I. Patient characteristics
Pt Sex Age (years) Ethnic origin Infections Other phenotypic features Genotype
A M 28 P Bronchiectasis, Aspergillus pneumonia(11 years), recurrent bacterial pneumonia,PA
Short stature, type 2 ASD, IBD c.[190_210]del + [190_210]del
B F 16 P Recurrent bacterial pneumonia, PA Short stature, IBD c.[190_210]del + [190_210]del
C M 20 P Recurrent oral ulceration — c.[130C>T] + [130C>T]D M 6 T Neonatal sepsis Type 2 ASD, cryptorchidism, PSVP c.[758G>A] + [758G>A]E F 12 T Pneumonia, sepsis Type 2 ASD, PV stenosis, panniculitis,
PSVPc.[554T>C] + [554T>C]
M, male; F, female; P, Pakistani; T, Turkish; PA, perianal abscesses; ASD, atrial septal defect; PV, pulmonary valve; IBD, inflammatory bowel disease; PSVP,prominent superficial venous pattern; patients D and E correspond to patients 4 and 6, respectively, in Boztug et al. (2009).
G6PC3 mutations are associated with a major defect of glycosylation
915

Hexose-monophosphate shunt activityThe reduced neutrophil respiratory burst seen in GSD-1b isthought to arise as a result of impaired hexose-monophosphate shunt (HMPS) activity, which leads to a rela-tive lack of NADPH as a substrate for the enzyme NADPHoxidase (Lange et al. 1980; Narisawa et al. 1986; Bashanet al. 1988; Potashnik et al. 1990; Kuijpers et al. 2003). Alack of NADPH is indicated by blunted HMPS activity inresponse to methylene blue. Although HMPS activity inG6PC3-deficient neutrophils was blunted in response toPMA, a normal response was observed after the addition of
methylene blue (Figure 3). This response has also beendescribed in chronic granulomatous disease (Holmes et al.1967), where the dysfunction of the NADPH oxidasecomplex underlies the abnormal respiratory burst.
NADPH oxidase subunit analysisWestern blotting for NADPH oxidase components revealed anaberrant band for gp91phox with an abnormally low apparentmolecular weight of �65 kDa in all patients with G6PC3mutations and in two unrelated patients with GSD-1b(Figure 4A and B). Expression of p67, p47 and p22phox inpatient neutrophils was normal as was the cDNA sequence forthe CYBB gene encoding gp91phox in patient A (data notshown). The latter data suggested that incompleteN-glycosylation was responsible for the abnormally lowapparent molecular weight of gp91phox seen in patient neutro-phils. Normal amounts of gp91phox were expressed on the cellsurface in patients’ neutrophils, as determined by immunor-eactivity on intact neutrophils (Figure 4C). In addition,reduced-minus-oxidized difference spectroscopy (Figure 4D)revealed a normal level of cytochrome b558 in patient (0.35µM) compared with HC neutrophils (0.25 µM).
G6PC3 mutation induces major alterations in neutrophilN- and O-glycomesThe observation of an abnormally low molecular weight forgp91phox in patients indicated impaired glycosylation of thismolecule. In order to determine whether global defects inneutrophil glycosylation are associated with G6PC3mutations, we performed MS profiling of healthy and patientneutrophils.The glycomes of the healthy neutrophil sample were in
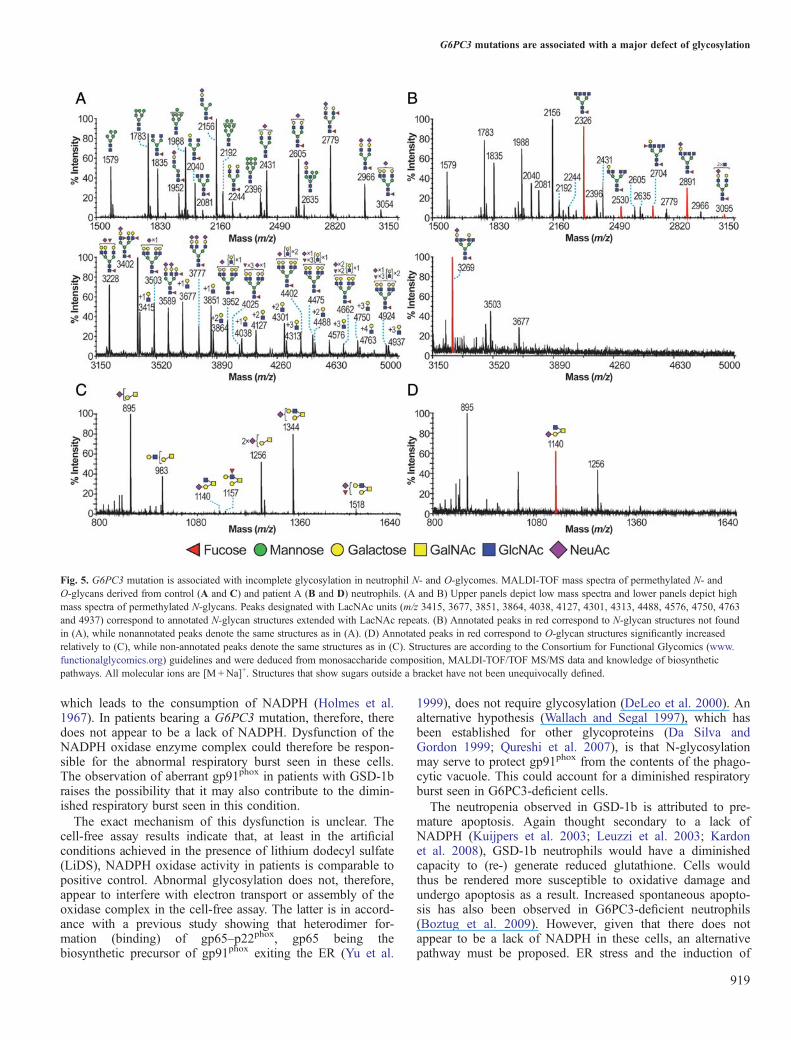
agreement with earlier studies on pooled neutrophils (Babuet al. 2009; see also data on the Consortium for FunctionalGlycomics website: www.functionalglycomics.org). Briefly,N-glycans included high mannose and complex bi-, tri- andtetra-antennary structures, of which the latter were core-fucosylated, nonbisected and terminated mainly with sialicacid and LewisX epitopes (Figure 5A). AgalactosylatedN-glycan structures were found to be minor and restrictedonly to core-fucosylated bi- and tri-antennary structures (m/z1835 and 2081; Figure 5A, upper panel). A great number ofcomplex N-glycans were extended with N-acetyllactosamine(LacNAc) units, resulting in multibranchedhigh-molecular-weight glycans (Figure 5A, lower panel). Incontrast, neutrophil N-glycomes of patient A (Figure 5B)showed a dramatic reduction in high-molecular-weightglycans, which appears to be caused by a failure to incorpor-ate galactose (Gal) into the majority of the complex-typeglycans. Consequently, many of the complex glycans hadtruncated antennae. Of particular note is the dominant signalat m/z 2326 which is the molecular ion for the agalactosylatedtetra-antennary N-glycan shown in the cartoon annotation.Other N-glycans deficient in Gal were observed at m/z 2530,2704, 2891, 3095 and 3269 (Figure 5B). None of these weredetected in the HC. The O-glycan profiles also exhibiteddefects in galactosylation, although to a lesser extent. Thus,the healthy O-glycome is comprised of sialylated core 1 (m/z895 and 1256) and core 2 (m/z 983, 1344 and 1518) structures
Fig. 1. G6PC3 mutation analysis. (A) Pedigrees of two unrelated,consanguineous Pakistani families with novel mutations in G6PC3. Theinheritance of patients with previously described mutations is detailedelsewhere (Boztug et al. 2009). (B) The linear deduced peptide sequence ofG6PC3 shows residues defined as being of key catalytic importance shaded ingrey. Amino acid substitution (with substituted residue above) or deletionresulting from the mutations in patients studied are underlined and annotatedfor each patient (A–E). (C) All mutations were predicted to occur intransmembrane helices of the folded protein (strictly- and semi-conservedresidues shown in black and grey, respectively; adapted from Guionie et al.2003).
B Hayee et al.
916

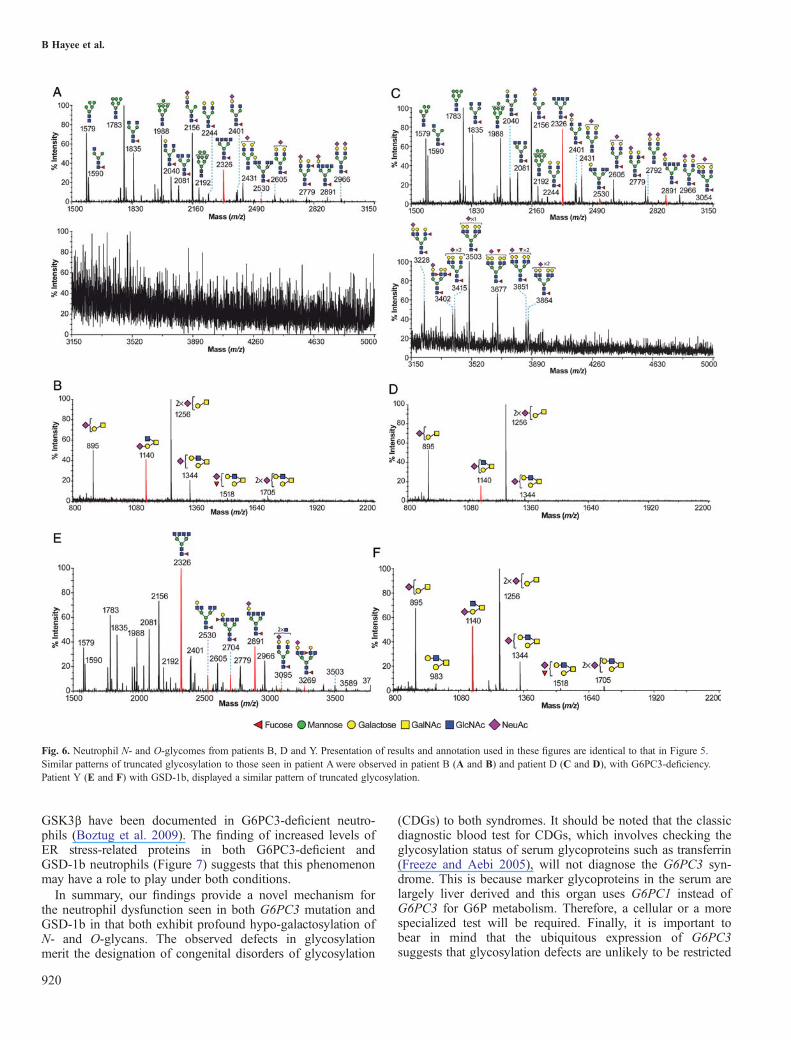
(Figure 5C), whereas the patient lacks Gal on the core 2antenna (m/z 1140; Figure 5D), but retains normal galactosy-lation of core 1 sequences (m/z 895 and 1256). Takentogether, these data suggest that the G6PC3 mutation induceda profound alteration on the N- and O-glycosylation profilesof patient’s neutrophils, characterized by the almost completeabsence of tetra-antennary and LacNAc extended N-glycansand mature core 2 O-glycans. Similarly, abnormal glycomicprofiles were also demonstrated in patient B (the sibling ofpatient A), an unrelated patient with G6PC3 mutation (patientD) and a patient with GSD-1b (patient Y; Figure 6). Theabsence of glycans with mature antennae in the glycomic pro-files was fully compensated for by the presence of abundant
peaks for glycans with truncated antennae. Also, the overallquality of the glycomic data obtained from the patient’s neu-trophils was in accord with the normal levels of N- andO-glycan occupancy on their glycoproteins.
ER stress-related protein expression in neutrophilsExpression of the ER stress-related proteins Grp78 andpEIF2α was increased both in patient A with G6PC3deficiency and in an unrelated patient (Y) with GSD-1a(Figure 7).
Discussion
In this study, we have made the remarkable discovery thatboth G6PC3 mutations and the GSD-1b syndrome are associ-ated with a major defect of neutrophil glycosylation. Thus,glycomic analysis of patient neutrophils showed truncated gly-cosylation of the majority of their complex-type N-glycans, aswell as the core 2 antennae of their O-glycans. Glycans werearrested at an “immature” stage and were profoundly deficientin the sialylated and LewisX epitopes, characteristic of thenormal neutrophil glycome (Haslam et al. 2006; Babu et al.2009). The antennae of the abnormal N-glycans wereobserved as GlcNAc “stubs”, indicating a failure of galactosy-lation. We hypothesize that this could be the result of insuffi-cient levels of uridine diphosphate (UDP)-Gal in the Golgiapparatus. The formation of complex N-glycans involves thestrict, stepwise addition of sugar residues (Spiro 2002), and alack of UDP-Gal would lead to truncation at the stageobserved in our patients. Such truncation would also result in
Fig. 2. NADPH oxidase function is diminished in G6PC3-deficient phagocytes. (A) Superoxide production in neutrophils and (B) hydrogen peroxide productionin macrophages after stimulation with PMAwere measured in patients with G6PC3 mutations (G6PC3−/−), their parents (G6PC3+/−) and patients with GSD-1band were compared with HCs (n = 5). (C) FACS analysis of the intracellular reduction in dihydrorhodamine by PMA-stimulated neutrophils detected twopopulations of cells at 15 min that had mounted a robust (N2) or weak (N1) respiratory burst compared with unstimulated cells (open histograms). (D) This resultwas used to calculate the “stimulation index” (Vowells et al. 1995) and expressed as a percentage of the mean HC response (n = 5). A–C and P1–P4 correspondto Figure 1 and Table I. **P = 0.009 vs HCs. All experiments were conducted in triplicate and on at least two separate occasions. (E) Superoxide production inthe cell-free assay (triplicate experiments) with membranes from patient A (filled bars) was comparable with that of positive controls (open bars; M, activity withmembrane alone; M + C, reconstituted activity with membrane and cytosolic components).
Fig. 3. HMPS activity in neutrophils. Neutrophils of Patient A (filled bars)were compared with HCs (n = 5; open bars). HMPS activity based on theliberation of 14CO2 from radiolabeled 14C1 glucose was diminished inresponse to PMA but normal after the addition of methylene blue. **P =0.01; ns, P= 0.57; compared with HCs. All experiments were conducted intriplicate and on at least two separate occasions.
G6PC3 mutations are associated with a major defect of glycosylation
917

a reduction in glycan-binding protein epitopes for functionallyimportant lectins such as selectins, siglecs and galectins withprofound implications for cellular function.Hypo-galactosylation of the O-glycans of the patients’ neutro-phils was observed on their core 2 antennae. In contrast,galactosylation to form the core 1 precursor was found to benormal, implying that UDP-Gal levels are sufficient for thefirst galactosylation step in O-glycan biosynthesis but not forevents in later Golgi compartments. However, our data do notexclude the possibility that entire glycan chains are absentfrom NADPH oxidase or other glycoproteins. Complex ER–Golgi interactions may underlie what appear to be partial andselective absence of Gal residues.The underlying biochemical causes of hypo-galactosylation
remain enigmatic, and further work is needed to establishhow impaired hydrolysis of G6P affects Gal metabolism.Interestingly, we observe the normal levels of capping withsialic acid or fucose of the small minority of glycans whoseantennae had been successfully galactosylated, suggesting thatthe metabolism of these sugars is not impaired.The electron-transporting subunit of NADPH oxidase,
gp91phox, has a 58 kDa (unglycosylated) “protein backbone”(Kleinberg et al. 1989) and is N-glycosylated at Asn-132,-149 and -240 (Wallach and Segal 1997; Taylor et al. 2006).In neutrophil lysates from all five patients with G6PC3mutations and the two patients with GSD-1b, western blotsfor gp91phox demonstrated that much of this glycoprotein hadan apparent mass of �65 kDa, rather than the �90 kDa exhib-ited by normal gp91phox. The latter has been reported to carry
complex N-glycans at three sites (Harper et al. 1985; Tayloret al. 2006) and its apparent molecular weight is �15 kDahigher than predicted for typical neutrophil N-glycosylation.However, this is not surprising, as it is well known that highlyglycosylated proteins exhibit anomalous SDS–PAGE behavior(Segrest et al. 1971). On the other hand, the observed massdifference of �7 kDa between the patients’ gp91phox and theunglycosylated backbone of gp91phox is fully consistent withthe occupancy of all three glycosylation sites with the abnor-mal glycans observed in our glycomic analysis of the patients’neutrophils.The diminished neutrophil respiratory burst seen in
GSD-1b is thought to be due to a lack of NADPH as a conse-quence of attenuated HMPS activity (Chou et al. 2002).However, the HMPS is a cytosolic cascade (Wamelink et al.2008) dependent on the supply of glucose-1-phosphate which,in neutrophils, is primarily derived from stored glycogen(Stossel et al. 1970)—a process which would not appear to bedependent on glucose transport and metabolism in the ER.The rate-limiting enzyme of the HMPS, G6P dehydrogenase,is allosterically induced by NADP and inhibited by NADPH.In this way, normal activity of the NADPH oxidase in neutro-phils stimulates the HMPS and in the presence of oxidasedysfunction, it is suppressed by accumulating NADPH. In thelatter situation, HMPS activity may be stimulated by anothermeans of NADPH consumption. We demonstrated that HMPSactivity was diminished in G6PC3-deficient neutrophils inresponse to PMA (an activator of the NADPH oxidase), butnormal in response to methylene blue—an oxidizing agent
Fig. 4. Expression and characterization of NADPH oxidase components in neutrophils. (A) Western blots for NADPH oxidase components are shown in patientA compared with normal (HCs). (B) Western blots for gp91phox for patients with mutations in G6PC3 (A–E), their parents (P1–P4) and two unrelated patientswith GSD-1b (X and Y). The aberrant gp91phox with an apparent molecular weight of �65 kDa was expressed in all patients A–E, X and Y. (C) Surfaceimmunoreactivity for gp91phox on intact neutrophils by FACS (patient A, open histogram; control, grey). (D) Reduced-minus-oxidized difference spectra ofneutrophils, showing gp91phox as cytochrome b558 with peaks at 559 and 428 nm [patient A, solid line; HCs (n = 3), dashed line; unstimulated cells, dotted line].Experiments were conducted on two separate occasions.
B Hayee et al.
918
which leads to the consumption of NADPH (Holmes et al.1967). In patients bearing a G6PC3 mutation, therefore, theredoes not appear to be a lack of NADPH. Dysfunction of theNADPH oxidase enzyme complex could therefore be respon-sible for the abnormal respiratory burst seen in these cells.The observation of aberrant gp91phox in patients with GSD-1braises the possibility that it may also contribute to the dimin-ished respiratory burst seen in this condition.The exact mechanism of this dysfunction is unclear. The
cell-free assay results indicate that, at least in the artificialconditions achieved in the presence of lithium dodecyl sulfate(LiDS), NADPH oxidase activity in patients is comparable topositive control. Abnormal glycosylation does not, therefore,appear to interfere with electron transport or assembly of theoxidase complex in the cell-free assay. The latter is in accord-ance with a previous study showing that heterodimer for-mation (binding) of gp65–p22phox, gp65 being thebiosynthetic precursor of gp91phox exiting the ER (Yu et al.
1999), does not require glycosylation (DeLeo et al. 2000). Analternative hypothesis (Wallach and Segal 1997), which hasbeen established for other glycoproteins (Da Silva andGordon 1999; Qureshi et al. 2007), is that N-glycosylationmay serve to protect gp91phox from the contents of the phago-cytic vacuole. This could account for a diminished respiratoryburst seen in G6PC3-deficient cells.The neutropenia observed in GSD-1b is attributed to pre-
mature apoptosis. Again thought secondary to a lack ofNADPH (Kuijpers et al. 2003; Leuzzi et al. 2003; Kardonet al. 2008), GSD-1b neutrophils would have a diminishedcapacity to (re-) generate reduced glutathione. Cells wouldthus be rendered more susceptible to oxidative damage andundergo apoptosis as a result. Increased spontaneous apopto-sis has also been observed in G6PC3-deficient neutrophils(Boztug et al. 2009). However, given that there does notappear to be a lack of NADPH in these cells, an alternativepathway must be proposed. ER stress and the induction of
Fig. 5. G6PC3 mutation is associated with incomplete glycosylation in neutrophil N- and O-glycomes. MALDI-TOF mass spectra of permethylated N- andO-glycans derived from control (A and C) and patient A (B and D) neutrophils. (A and B) Upper panels depict low mass spectra and lower panels depict highmass spectra of permethylated N-glycans. Peaks designated with LacNAc units (m/z 3415, 3677, 3851, 3864, 4038, 4127, 4301, 4313, 4488, 4576, 4750, 4763and 4937) correspond to annotated N-glycan structures extended with LacNAc repeats. (B) Annotated peaks in red correspond to N-glycan structures not foundin (A), while nonannotated peaks denote the same structures as in (A). (D) Annotated peaks in red correspond to O-glycan structures significantly increasedrelatively to (C), while non-annotated peaks denote the same structures as in (C). Structures are according to the Consortium for Functional Glycomics (www.functionalglycomics.org) guidelines and were deduced from monosaccharide composition, MALDI-TOF/TOF MS/MS data and knowledge of biosyntheticpathways. All molecular ions are [M +Na]+. Structures that show sugars outside a bracket have not been unequivocally defined.
G6PC3 mutations are associated with a major defect of glycosylation
919
GSK3β have been documented in G6PC3-deficient neutro-phils (Boztug et al. 2009). The finding of increased levels ofER stress-related proteins in both G6PC3-deficient andGSD-1b neutrophils (Figure 7) suggests that this phenomenonmay have a role to play under both conditions.In summary, our findings provide a novel mechanism for
the neutrophil dysfunction seen in both G6PC3 mutation andGSD-1b in that both exhibit profound hypo-galactosylation ofN- and O-glycans. The observed defects in glycosylationmerit the designation of congenital disorders of glycosylation
(CDGs) to both syndromes. It should be noted that the classicdiagnostic blood test for CDGs, which involves checking theglycosylation status of serum glycoproteins such as transferrin(Freeze and Aebi 2005), will not diagnose the G6PC3 syn-drome. This is because marker glycoproteins in the serum arelargely liver derived and this organ uses G6PC1 instead ofG6PC3 for G6P metabolism. Therefore, a cellular or a morespecialized test will be required. Finally, it is important tobear in mind that the ubiquitous expression of G6PC3suggests that glycosylation defects are unlikely to be restricted
Fig. 6. Neutrophil N- and O-glycomes from patients B, D and Y. Presentation of results and annotation used in these figures are identical to that in Figure 5.Similar patterns of truncated glycosylation to those seen in patient Awere observed in patient B (A and B) and patient D (C and D), with G6PC3-deficiency.Patient Y (E and F) with GSD-1b, displayed a similar pattern of truncated glycosylation.
B Hayee et al.
920

to neutrophils in patients with the G6PC3 syndrome. Furtherstudies are needed to define the extent of aberrant glycosyla-tion in these patients and to establish how alterations in glyco-sylation contribute to this multisystem syndrome.
Materials and MethodsParticipantsHeparinized (6 U mL−1) blood samples were obtained frompatients, relatives and controls with informed consent withapproval from the joint UCL/UCLH committees on the ethicsof human research. Patient details are summarized in Table I.The fraternal twin of patient A has sensorineural deafness butotherwise normal phenotype. Cell samples from patients Dand E were kindly provided by K.B. and C.K. and frompatient C by R.K.G. Patients with GSD-1b were unrelated,with previously characterized mutations in SLC37A4 (per-formed by the Sheffield Molecular Genetics Service, UK;Veiga-da-Cunha et al. 1998). All patients were sampled aftertreatment with granulocyte colony-stimulating factor had beencommenced. The attenuated respiratory burst in patient A wasunaffected by treatment (data not shown), but peripheralblood neutrophil counts corrected to normal range.
Macrophage culture and neutrophil isolationPBMCs were isolated from whole blood by centrifugationthrough Lymphoprep® (Axis Shield, Norway) and culturedfor 5 days in an RPMI-1640 medium (Gibco, UK) sup-plemented with 10% fetal calf serum (Sigma, UK) with100 U mL−1 penicillin and 100 µg mL−1 streptomycin(Gibco). Adherent cells were regarded as differentiated asdescribed previously (Smith et al. 2009). After the removalof monocytes from the above centrifugation, neutrophilswere isolated by sedimentation with 1% dextran and hypo-tonic lysis of red blood cells. Cells were counted andre-suspended in phosphate-buffered saline (PBS; Gibco) forall subsequent investigations.
Respiratory burst assaysThe respiratory burst was studied in neutrophils and macro-phages. First, 2 × 106 neutrophils were stimulated with 1 µgmL−1 PMA (Sigma) in a temperature-controlled (37°C)spectrophotometer cuvette assay based on the reductionin cytochrome c (Sigma). The color change produced bysuperoxide dismutase-inhibitable cytochrome c reduction wasdetected at 550 nm and the maximal rate of change in absor-bance was compared with a control cuvette containing cells
and cytochrome c without the addition of PMA. The maximalrate of change in absorbance (nm min−1) was converted tonmol O2/10
6 cells min−1 using a previously published formula(Dahlgren and Karlsson 1999). Triplicated experiments, againbased on the reduction in cytochrome c (Teufelhofer et al.2003), were conducted with neutrophils using a temperature-controlled (37°C) microplate reader (Omega FLUOStar, BMGLabtech, UK). Second, the intracellular reduction in dihydror-hodamine (Sigma) was measured in response to 1 µg mL−1
PMA by fluorescence-activated cell sorter (FACS) analysis,using a modification of a previously published method wherepurified neutrophils were used (as opposed to whole bloodsamples; Vowells et al. 1995). Mean fluorescence in the gatedpopulations of cells indicated (N2) was used to calculate the“stimulation index” as a ratio of the result produced by unsti-mulated cells in paired reactions. For patients, this wasexpressed as a percentage of the mean response from a studyof five HC volunteers. The macrophage respiratory burst wasstudied in a microplate assay (Rahman et al. 2009) inresponse to PMA using the Amplex Red reagent (Invitrogen)to produce a color change as the consequence of productionof hydrogen peroxide.
Cell-free assay (Molshanski-Mor et al. 2007)Neutrophil membranes were purified from whole-cell lysates,in the presence of protease inhibitors, by gradient centrifu-gation and diluted to achieve 1 pmol of cytochrome b558 perwell. NADPH oxidase activity was measured by cytochromec reduction in a 96-well plate format from wells containingmembrane alone or after the addition of the cytosolic com-ponents of the NADPH oxidase (all purified human recombi-nant proteins): p47phox (100 nM), p67phox (100 nM) and Rac1(Q61L)-GTP (100 nM). Incubation with 130 µmol of LiDSwas carried out for 90 s prior to the addition of 238 µMNADPH. Positive control was obtained from solubilizedguinea-pig macrophage membranes.
HMPS activityAbout 1 × 106 neutrophils were stimulated with either 1 µgmL−1 PMA or 300 µM methylene blue (BDHPharmaceuticals), in a modification of a previously publishedmethod (Holmes et al. 1967). Briefly, reactions were con-ducted in sealed containers in 1 mL of PBS containing 1 mMglucose and 0.5 µCi (0.00185 Mbq) 14C1 glucose(Perkin-Elmer). 14CO2 released by the activity of the HMPS(Wamelink et al. 2008) was collected onto a filter paper satu-rated with 20% sodium hydroxide and suspended above thereaction mixture within the closed container. After 1 h, thefilter paper was removed into 5 mL scintillation fluid (UltimaGold®, Perkin Elmer) and counted immediately in a scintil-lation counter calibrated for 14C (Tricarb 2100TR, PerkinElmer). Results thus obtained were expressed as counts perminute. All experiments were repeated on at least twice.“Protein lysates” were prepared using a previously pub-
lished method (Alterman et al. 1990), and 20 µg of proteinper lane was separated using SDS–PAGE (8% gel for gp91and p67, and 10% for p47 and p22). Protein lysates fromother patients with G6PC3 mutations were also kindly sup-plied by C.K. MoAb48 (gift from Prof Dirk Roos,
Fig. 7. ER stress in neutrophils from G6PC3-deficient and GSD1b patients.Western blots of lysates from resting neutrophils showing increased levels ofER stress-related proteins Grp78 and pEIF2α in cell lysates from patient A(G6PC3−/−) and patient X (GSD-1b) compared with three unrelated HCs.
G6PC3 mutations are associated with a major defect of glycosylation
921

Amsterdam, the Netherlands) was used to detect gp91,whereas other NADPH oxidase subunit antibodies were pre-pared “in-house”. Antibodies to Grp78, pEiF2α and actin(Abcam) were used according to the manufacturer’s protocols.
Surface expression of gp91phox and spectral analysisPurified neutrophils were suspended in PBS (Gibco) contain-ing 0.1% bovine serum albumin (Merck) and 0.01% sodiumazide (Sigma). FACS analysis for gp91phox was performedusing a commercially available fluorescein isothiocyanate-conjugated antibody (7D5; MBL International). Thereduced-minus-oxidized UV/visible difference spectra ofintact neutrophils were recorded 10 min after the addition ofsodium dithionite to 3 × 107 cells stimulated with 1 µg mL−1
PMA, in PBS at pH 7.3 and at 37°C. The concentration ofcytochrome b558 was calculated from the difference betweenthe height of the 559 nm peak and the depth at 540 nm, usingan extinction coefficient of ε559 = 21.6 mM−1 cm−1 (Robertset al. 1982).
Glycomic profile analysisSnap-frozen neutrophils (107) purified as above were treatedas described previously (Jang-Lee et al. 2006; Sutton-Smithand Dell 2006). Briefly, all samples were subjected to hom-ogenization using a 130 W Vibra-Cell ultrasonic processor(VC 130 PB, Sonics & Materials) within a sound-abatingenclosure in an extraction buffer (25 mM Tris, 150 mM NaCl,5 mM EDTA and 1% (3-[(3-cholamidopropyl)dimethyl-ammonio]-1-propane sulfonate, at pH 7.4), reduction in 4 Mguanidine–HCl (Pierce), carboxymethylation and trypsindigestion. The digested glycoproteins were purified byC18-Sep-Pak (Waters Corp). N-Glycans were released byPNGaseF (EC 3.5.1.52, Roche Applied Science) digestion,whereas O-glycans were released by reductive elimination. N-and O-glycans were then permethylated using the sodiumhydroxide procedure, and finally, the permethylated N- andO-glycans were purified by C18-Sep-Pak.All permethylated samples were dissolved in 10 μL of
methanol and 1 μL of dissolved sample was premixed with1 μL of matrix (20 mg mL−1 2,5-dihydroxybenzoic acid in70%, v/v, aqueous methanol), spotted onto a target plate (2 ×0.5 μL) and dried under vacuum. MS data were acquiredusing a Voyager-DE STR matrix-assisted laser desorption/ionization-time of flight mass spectrometer (MALDI-TOF;Applied Biosystems). MS/MS data were acquired using a4800 MALDI-TOF/TOF (Applied Biosystems) mass spec-trometer. The collision energy was set to 1 kV, and argon wasused as a collision gas. The 4700 Calibration Standard Kit,Calmix (Applied Biosystems), was used as the external cali-brant for the MS mode of both instruments, and [Glu1] fibri-nopeptide B human (Sigma) was used as an external calibrantfor the MS/MS mode of the MALDI-TOF/TOF instrument.The MS and MS/MS data were processed using Data
Explorer 4.9 Software (Applied Biosystems). The spectrawere subjected to manual assignment and annotation with theaid of the glycobioinformatics tool GlycoWorkBench (Ceroniet al. 2008). The proposed assignments for the selected peakswere based on 12C isotopic composition together with knowl-edge of the biosynthetic pathways. The proposed structures
were then confirmed by data obtained from MS/MSexperiments.
Mutation analysisDNAwas extracted from patient’s peripheral blood leucocytesusing standard procedures. All patients were screened formutations in ELA2, HAX1 and WAS (none identified). All sixcoding exons and flanking introns of the G6PC3 gene(RefSeq NM_138387) were amplified via the polymerasechain reaction (PCR) using the high-fidelity enzyme Phusion(New England Biolabs) according to manufacturer’s instruc-tions. Primer sequences are available on request. PCR pro-ducts were screened for mutations by denaturinghigh-performance liquid chromatography using the WAVEplatform (Transgenomic). Homozygous G6PC3 mutationswere identified by mixing equal amounts of patient PCRproduct with a control PCR product known to be homozygouswild type, prior to denaturation to allow the heteroduplex for-mation. Patient samples with abnormal WAVE chromatogramswere sequenced using the DTCS Quick Start Kit andCEQ8000 DNA Genetic Analysis System (Beckman Coulter).Effects on protein sequence were predicted using Ensemblrelease 55 (©Wellcome Trust Sanger Institute/EuropeanBioinformatics Institute).
Statistical analysisError bars in all figures represent the SEM. Comparisons(using Prism v4.0, GraphPad) were made between groupsusing Student’s t-test if data were normally distributed (byShapiro-Wilk normality test) or two-tailed Mann–WhitneyU-test if not. Significance was assumed at P < 0.05.
Funding
This work was supported by the Biotechnology andBiological Sciences Research Council (grant numberBBF0083091 to A.D., H.R.M. and S.M.H.) and the WellcomeTrust (to A.W.S.).
Conflict of interest
None declared.
Abbreviations
CDG, congenital disorder of glycosylation; ER, endoplasmicreticulum; FACS, fluorescence-activated cell sorting; G6P,glucose-6-phosphate; G6PC, glucose-6-phosphatase catabolic;G6PT1, glucose-6-phosphate translocase-1; GSD-1a, glyco-gen storage disease type-1a; HC, healthy control; HMPS,hexose-monophosphate shunt; LacNAc, N-acetyllactosamine;LiDS, lithium dodecyl sulfate; MALDI-TOF, matrix-assistedlaser desorption/ionization-time of flight mass spectrometer;MS, mass spectrometric; NADPH, nicotinamide adenine dinu-cleotide phosphate; PBS, phosphate-buffered saline; PBMC,peripheral blood monocyte; PCR, polymerase chain reaction;PMA, phorbol-myristyl acetate; PNGaseF,
B Hayee et al.
922

peptide-N-glycosidase F; SDS–PAGE, sodium dodecylsulfate–polyacrylamide gel electrophoresis; SEM, standarderror of the mean; UDP, uridine diphosphate.
References
Alterman LA, Crispe IN, Kinnon C. 1990. Characterization of the murineheat-stable antigen: An hematolymphoid differentiation antigen defined bythe J11d, M1/69 and B2A2 antibodies. Eur J Immunol. 20:1597–1602.
Annabi B, Hiraiwa H, Mansfield BC, Lei KJ, Ubagai T, Polymeropoulos MH,Moses SW, Parvari R, Hershkovitz E, Mandel H, et al. 1998. The gene forglycogen-storage disease type 1b maps to chromosome 11q23. Am J HumGenet. 62:400–405.
Arden SD, Zahn T, Steegers S, Webb S, Bergman B, O’Brien RM, HuttonJC. 1999. Molecular cloning of a pancreatic islet-specific glucose-6-phosphatase catalytic subunit-related protein. Diabetes. 48:531–542.
Aróstegui JI, de Toledo JS, Pascal M, García C, Yagüe J. 2009. A novelG6PC3 homozygous 1-bp deletion as a cause of severe congenital neutro-penia. Blood. 114:1718–1719.
Babu P, North SJ, Jang-Lee J, Chalabi S, Mackerness K, Stowell SR,Cummings RD, Rankin S, Dell A, Haslam SM. 2009. Structural character-isation of neutrophil glycans by ultra sensitive mass spectrometric glyco-mics methodology. Glycoconj J. 26:975–986.
Bashan N, Hagai Y, Potashnik R, Moses SW. 1988. Impaired carbohydratemetabolism of polymorphonuclear leukocytes in glycogen storage diseaseIb. J Clin Invest. 81:1317–1322.
Boztug K, Appaswamy G, Ashikov A, Schäffer AA, Salzer U, Diestelhorst J,Germeshausen M, Brandes G, Lee-Gossler J, Noyan F, et al. 2009. A syn-drome with congenital neutropenia and mutations in G6PC3. N Engl JMed. 360:32–43.
Boztug K, Klein C. 2009. Novel genetic etiologies of severe congenital neu-tropenia. Curr Opin Immunol. 21:472–480.
Ceroni A, Maass K, Geyer H, Geyer R, Dell A, Haslam SM. 2008.GlycoWorkbench: A tool for the computer-assisted annotation of massspectra of glycans. J Proteome Res. 7:1650–1659.
Cheung YY, Kim SY, Yiu WH, Pan CJ, Jun HS, Ruef RA, Lee EJ, WestphalH, Mansfield BC, Chou JY. 2007. Impaired neutrophil activity andincreased susceptibility to bacterial infection in mice lackingglucose-6-phosphatase-beta. J Clin Invest. 117:784–793.
Chou JY, Jun HS, Mansfield BC. 2010. Glycogen storage disease type I andG6Pase-β deficiency: Etiology and therapy. Nat Rev Endocrinol. 6:676–688.
Chou JY, Matern D, Mansfield BC, Chen YT. 2002. Type I glycogen storagediseases: Disorders of the glucose-6-phosphatase complex. Curr Mol Med.2:121–143.
Da Silva RP, Gordon S. 1999. Phagocytosis stimulates alternative glycosyla-tion of macrosialin (mouse CD68), a macrophage-specific endosomalprotein. Biochem J. 338:687–694.
Dahlgren C, Karlsson A. 1999. Respiratory burst in human neutrophils.J Immunol Methods. 232:3–14.
DeLeo FR, Burritt JB, Yu L, Jesaitis AJ, Dinauer MC, Nauseef WM. 2000.Processing and maturation of flavocytochrome b558 Include incorporationof heme as a prerequisite for heterodimer assembly. J Biol Chem.275:13986–13993.
Freeze HH, Aebi M. 2005. Altered glycan structures: The molecular basis ofcongenital disorders of glycosylation. Curr Opin Struct Biol. 15:490–498.
Guionie O, Clottes E, Stafford K, Burchell A. 2003. Identification and charac-terisation of a new human glucose-6-phosphatase isoform. FEBS Lett.551:159–164.
Harper AM, Chaplin MF, Segal AW. 1985. Cytochrome b-245 from humanneutrophils is a glycoprotein. Biochem J. 227:783–788.
Haslam SM, North SJ, Dell A. 2006. Mass spectrometric analysis of N- andO-glycosylation of tissues and cells. Curr Opin Struct Biol. 16:584–591.
Holmes B, Page AR, Good RA. 1967. Studies of the metabolic activity ofleukocytes from patients with a genetic abnormality of phagocytic function.J Clin Invest. 46:1422–1432.
Jang-Lee J, North SJ, Sutton-Smith M, Goldberg D, Panico M, Morris H,Haslam S, Dell A. 2006. Glycomic profiling of cells and tissues by massspectrometry: Fingerprinting and sequencing methodologies. MethodsEnzymol. 415:59–86.
Kardon T, Senesi S, Marcolongo P, Legeza B, Bánhegyi G, Mandl J, FulceriR, Benedetti A. 2008. Maintenance of luminal NADPH in the endoplasmic
reticulum promotes the survival of human neutrophil granulocytes. FEBSLett. 582:1809–1815.
Kleinberg ME, Rotrosen D, Malech HL. 1989. Asparagine-linked glycosyla-tion of cytochrome b558 large subunit varies in different human phagocy-tic cells. J Immunol. 143:4152–4157.
Kuijpers TW, Maianski NA, Tool AT, Smit GP, Rake JP, Roos D, Visser G.2003. Apoptotic neutrophils in the circulation of patients with glycogenstorage disease type 1b (GSD1b). Blood. 101:5021–5024.
Lambeth JD. 2004. Nox enzymes and the biology of reactive oxygen. NatRev Immunol. 4:181–189.
Lange AJ, Arion WJ, Beaudet AL. 1980. Type Ib glycogen storage disease iscaused by a defect in the glucose-6-phosphate translocase of the micro-somal glucose-6-phosphatase system. J Biol Chem. 255:8381–8384.
Lei KJ, Chen H, Pan CJ, Ward JM, Mosinger B, Jr, Lee EJ, Westphal H,Mansfield BC, Chou JY. 1996. Glucose-6-phosphatase dependent substratetransport in the glycogen storage disease type-1a mouse. Nat Genet.13:203–209.
Lei KJ, Shelly LL, Pan CJ, Sidbury JB, Chou JY. 1993. Mutations in theglucose-6-phosphatase gene that cause glycogen storage disease type 1a.Science. 262:580–583.
Leuzzi R, Bánhegyi G, Kardon T, Marcolongo P, Capecchi PL, Burger HJ,Benedetti A, Fulceri R. 2003. Inhibition of microsomal glucose-6-phosphate transport in human neutrophils results in apoptosis: A potentialexplanation for neutrophil dysfunction in glycogen storage disease type 1b.Blood. 101:2381–2387.
Martin CC, Bischof LJ, Bergman B, Hornbuckle LA, Hilliker C, Frigeri C,Wahl D, Svitek CA, Wong R, Goldman JK, et al. 2001. Cloning and charac-terization of the human and rat islet-specific glucose-6-phosphatase catalyticsubunit-related protein (IGRP) genes. J Biol Chem. 276:25197–25207.
Martin CC, Oeser JK, Svitek CA, Hunter SI, Hutton JC, O’Brien RM. 2002.Identification and characterization of a human cDNA and gene encoding aubiquitously expressed glucose-6-phosphatase catalytic subunit-relatedprotein. J Mol Endocrinol. 29:205–222.
McDermott DH, De Ravin SS, Jun HS, Liu Q, Long Priel DA, Noel P,Takemoto CM, Ojode T, Paul SM, Dunsmore KP, et al. 2010. Severe con-genital neutropenia resulting from G6PC3 deficiency with increased neutro-phil CXCR4 expression and myelokathexis. Blood. 116:2793–2802.
Melis D, Fulceri R, Parenti G, Marcolongo P, Gatti R, Parini R, Riva E, DellaCasa R, Zammarchi E, Andria G, et al. 2005. Genotype/phenotype corre-lation in glycogen storage disease type 1b: A multicentre study and reviewof the literature. Eur J Pediatr. 164:501–508.
Mithieux G. 1997. New knowledge regarding glucose-6 phosphatase geneand protein and their roles in the regulation of glucose metabolism. Eur JEndocrinol. 136:137–145.
Mithieux G, Rajas F, Gautier-Stein A. 2004. A novel role for glucose 6-phos-phatase in the small intestine in the control of glucose homeostasis. J BiolChem. 279:44231–44234.
Mithieux G, Vidal H, Zitoun C, Bruni N, Daniele N, Minassian C. 1996.Glucose-6-phosphatase mRNA and activity are increased to the sameextent in kidney and liver of diabetic rats. Diabetes. 45:891–896.
Molshanski-Mor S, Mizrahi A, Ugolev Y, Dahan I, Berdichevsky Y, Pick E.2007. Cell-free assays: The reductionist approach to the study of NADPHoxidase assembly, or “all you wanted to know about cell-free assays butdid not dare to ask”. Methods Mol Biol. 412:385–428.
Narisawa K, Ishizawa S, Okumura H, Tada K, Kuzuya T. 1986. Neutrophilmetabolic dysfunction in genetically heterogeneous patients with glycogenstorage disease type 1b. J Inherit Metab Dis. 9:297–300.
Pan C-J, Lei K-J, Annabi B, Hemrika W, Chou JY. 1998. Transmembrane top-ology of glucose-6-phosphatase. J Biol Chem. 273:6144–6148.
Pan C-J, Lin B, Chou JY. 1999. Transmembrane topology of human glucose6-phosphate transporter. J Biol Chem. 274:13865–13869.
Petrolonis AJ, Yang Q, Tummino PJ, Fish SM, Prack AE, Jain S, Parsons TF,Li P, Dales NA, Ge L, et al. 2004. Enzymatic characterization of the pan-creatic islet-specific glucose-6-phosphatase-related protein (IGRP). J BiolChem. 279:13976–13983.
Pocai A, Obici S, Schwartz GJ, Rossetti L. 2005. A brain-liver circuit regu-lates glucose homeostasis. Cell Metab. 1:53–61.
Potashnik R, Moran A, Moses SW, Peleg N, Bashan N. 1990. Hexose uptakeand transport in polymorphonuclear leukocytes from patients with glyco-gen storage disease Ib. Pediatr Res. 28:19–23.
Qureshi OS, Paramasivam A, Yu JC, Murrell-Lagnado RD. 2007. Regulationof P2X4 receptors by lysosomal targeting, glycan protection and exocyto-sis. J Cell Sci. 120:3838–3849.
G6PC3 mutations are associated with a major defect of glycosylation
923

Rahman FZ, Hayee B, Chee R, Segal AW, Smith AM. 2009. Impaired macro-phage function following bacterial stimulation in chronic granulomatousdisease. Immunology. 128:253–259.
Rajas F, Bruni N, Montano S, Zitoun C, Mithieux G. 1999. The glucose-6phosphatase gene is expressed in human and rat small intestine:Regulation of expression in fasted and diabetic rats. Gastroenterology.117:132–139.
Roberts PJ, Cross AR, Jones OT, Segal AW. 1982. Development of cyto-chrome b and an active oxidase system in association with maturation of ahuman promyelocytic (HL-60) cell line. J Cell Biol. 95:720–726.
Segrest JP, Jackson RL, Andrews EP, Marchesi VT. 1971. Human erythrocytemembrane glycoprotein: A re-evaluation of the molecular weight as deter-mined by SDS polyacrylamide gel electrophoresis. Biochem Biophys ResCommun. 44:390–395.
Smith AM, Rahman FZ, Hayee BH, Graham SJ, Marks DJ, Sewell GW,Palmer CD, Wilde J, Foxwell BM, Gloger IS, et al. 2009. Disorderedmacrophage cytokine secretion underlies impaired acute inflammation andbacterial clearance in Crohn’s disease. J Exp Med. 206:1883–1897.
Spiro RG. 2002. Protein glycosylation: Nature, distribution, enzymatic for-mation, and disease implications of glycopeptide bonds. Glycobiology.12:43R–56R.
Stossel TP, Murad F, Mason RJ, Vaughan M. 1970. Regulation of glycogenmetabolism in polymorphonuclear leukocytes. J Biol Chem.245:6228–6234.
Sutton-Smith M, Dell A. 2006. Analysis of carbohydrates/glycoproteins bymass spectrometry. In: Celis JE, editor. Cell Biology: A LaboratoryHandbook. Amsterdam: Academic Press. p. 415–425.
Taylor RM, Baniulis D, Burritt JB, Gripentrog JM, Lord CI, Riesselman MH,Maaty WS, Bothner BP, Angel TE, Dratz EA, et al. 2006. Analysis ofhuman phagocyte flavocytochrome b(558) by mass spectrometry. J BiolChem. 281:37045–37056.
Teufelhofer O, Weiss RM, Parzefall W, Schulte-Hermann R,Micksche M, Berger W, Elbling L. 2003. Promyelocytic HL60 cellsexpress NADPH oxidase and are excellent targets in a rapid spectrophoto-metric microplate assay for extracellular superoxide. Toxicol Sci.76:376–383.
Veiga-da-Cunha M, Gerin I, Chen YT, de Barsy T, de Lonlay P, Dionisi-ViciC, Fenske CD, Lee PJ, Leonard JV, Maire I, et al. 1998. A gene onchromosome 11q23 coding for a putative glucose- 6-phosphate translocaseis mutated in glycogen-storage disease types Ib and Ic. Am J Hum Genet.63:976–983.
Vowells SJ, Sekhsaria S, Malech HL, Shalit M, Fleisher TA. 1995. Flow cyto-metric analysis of the granulocyte respiratory burst: A comparison study offluorescent probes. J Immunol Methods. 178:89–97.
Wallach TM, Segal AW. 1997. Analysis of glycosylation sites on gp91phox,the flavocytochrome of the NADPH oxidase, by site-directed mutagenesisand translation in vitro. Biochem J. 321:583–585.
Wamelink MM, Struys EA, Jakobs C. 2008. The biochemistry, metabolismand inherited defects of the pentose phosphate pathway: A review.J Inherit Metab Dis. 31:703–717.
Yu L, DeLeo FR, Biberstine-Kinkade KJ, Renee J, Nauseef WM, DinauerMC. 1999. Biosynthesis of flavocytochrome b558. gp91(phox) is syn-thesized as a 65-kDa precursor (p65) in the endoplasmic reticulum. J BiolChem. 274:4364–4369.
B Hayee et al.
924




















