
Functional characterization of splicing and ligand‐binding domain variants in the LDL receptor
12
RESEARCH ARTICLE OFFICIAL JOURNAL www.hgvs.org Functional Characterization of Splicing and Ligand-Binding Domain Variants in the LDL Receptor Aitor Etxebarria, 1 Lourdes Palacios, 2 Marianne Stef, 2 Diego Tejedor, 2 Kepa B. Uribe, 1 Amalia Oleaga, 3 Luis Irigoyen, 4 Beatriz Torres, 4 Helena Ostolaza, 1 and Cesar Martin 1 ∗ 1 Unidad de Biof ´ ısica (Centro Mixto CSIC-UPV/EHU) and Departamento de Bioqu ´ ımica, Universidad del Pa ´ ıs Vasco, Apdo. 644, 48080 Bilbao, Spain; 2 Progenika Biopharma S.A., Derio, Spain; 3 Endocrinolog ´ ıa y Nutrici ´ on, Hospital de Basurto, Bilbao, Spain; 4 Sevicio de Endocrinolog ´ ıa y Nutrici ´ on, Hospital Santiago Apostol, Vitoria, Spain Communicated by Bruce R. Gottlieb Received 9 May 2011; accepted revised manuscript 26 September 2011. Published online 11 October 2011 in Wiley Online Library (www.wiley.com/humanmutation).DOI: 10.1002/humu.21630 ABSTRACT: Familial hypercholesterolemia (FH) is an au- tosomal dominant disorder mostly caused by mutations in the LDLR gene. Although the detection of functional mutations in the LDLR gene provides an unequivocal diagnosis of the FH condition, there are many variants whose pathogenicity is still unknown. The aims of this study were to set up a rapid method to determine the effect of LDLR mutations, thereby providing an accu- rate diagnosis of FH, and to functionally characterize six LDLR mutations detected at high frequency by the LIPOchip R platform (Progenika Biopharma, Spain) in the Spanish population. LDLR expression and activity were analyzed by one-single-step flow cytometry assay and confocal microscopy. Splicing effects were determined by sequencing reverse transcription polymerase chain reac- tion products. The analysis of three heterozygous vari- ants with a single point mutation within the low-density lipoprotein binding domain allowed us to classify the c.806G>A variant as nonpathogenic, and c.862G>A and c.895G>A variants as causative of FH. The results ob- tained for three variants affecting donor splice sites of the LDLR mRNA, c.313+2dupT, c.1186+5G>A, and c.1845+1G>C, demonstrated that these mutations are pathogenic. These results expand our knowledge of muta- tions responsible for FH, providing an accurate diagnosis and leading to early treatment to reduce the risk of pre- mature cardiovascular events. Hum Mutat 33:232–243, 2012. C 2011 Wiley Periodicals, Inc. KEY WORDS: LDL receptor; familial hypercholes- terolemia; LDL uptake assay Additional Supporting Information may be found in the online version of this article. ∗ Correspondence to: Cesar Martin, Unidad de Biof´ ısica (Centro Mixto CSIC- UPV/EHU) and Departamento de Bioqu´ ımica, Universidad del Pa´ ıs Vasco, Apdo. 644, 48080 Bilbao, Spain. E-mail: [email protected] Contract grant sponsor: Plan +Euskadi09 (UE09+/10) program of the Basque Govern- ment; Postdoctoral contract from the Basque Government (to A.E.); Bizkaia Biophysics Foundation (to A.E. and K.B.U.). Introduction Familial hypercholesterolemia (FH; MIM# 143890) is a frequent autosomal dominant disorder characterized by increased plasma low-density lipoprotein cholesterol (LDL-C), tendon xanthomas, and premature coronary heart disease [Goldstein et al., 2001]. FH usually results from mutations in the low-density lipoprotein recep- tor (LDLR; MIM# 606945), which is responsible for the uptake of cholesterol-containing particles into cells [Goldstein et al., 2001]; in its ligand, apolipoprotein B-100 (APOB; MIM# 107730) [Pullinger et al., 1995]; or in the proprotein convertase subtilisin/kexin type 9 gene (PCSK9; MIM# 607786), which promotes LDLR degradation [Abifadel et al., 2003]. In the general population, FH is observed with a heterozygous frequency of 1:500 and a homozygous frequency of 1:1,000,000 [Goldstein et al., 2001]. To date, more than 1,200 different mutations in the LDLR gene, located on chromosome 19p, have been identified worldwide. These mutations are distributed along 18 exons, introns, and the promoter region of the LDLR gene [Leigh et al., 2008]; these include point mutations (both substitutions and indels), as well as copy number variations such as duplications and deletions, which can differen- tially affect residual LDLR activity. Mutations can be divided into five different classes according to the nature and location of the mutations within the LDLR gene, as well as their phenotypic ef- fects on the protein: Class 1: no detectable LDLR protein [Hobbs et al., 1988]; Class 2: defective LDLR transport, either completely blocked (class 2A) or partially blocked (class 2B or leaky LDLRs) [Goldstein et al., 1985]; Class 3: defective LDL binding [Hobbs et al., 1992]; Class 4: defective clustering in clathrin-coated pits [Davis et al., 1986; Lehrman et al., 1985]; and Class 5: recycling-defective receptors [Beglova et al., 2004]. Heterozygous FH is diagnosed based on high LDL-C levels (usu- ally >250 mg/dl) in untreated adults, as well as the presence of tendon xanthomas and arcus cornealis. However, it is often diffi- cult to establish an early diagnosis in younger patients because the physical manifestations of FH develop late in life. Several sets of diagnostic criteria have been extensively used in the clinic, includ- ing criteria established by the Dutch Lipid Clinic Network (DLCN). The DLCN uses a numeric score whereby a higher score indicates a more reliable diagnosis of FH [Defesche, 2000]; patients with scores of 8 or higher are classified as definite FH patients. Initiating FH detection and treatment as early as possible, as recommended by the World Health Organization, can maximize the health benefit obtained through treatment [WHO 1999]. As long as the identification of functional mutations in the LDLR gene provides an unequivocal FH diagnosis, there is a need for C 2011 WILEY PERIODICALS, INC.
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Functional characterization of splicing and ligand‐binding domain variants in the LDL receptor

RESEARCH ARTICLEOFFICIAL JOURNAL
www.hgvs.org
Functional Characterization of Splicing and Ligand-BindingDomain Variants in the LDL Receptor
Aitor Etxebarria,1 Lourdes Palacios,2 Marianne Stef,2 Diego Tejedor,2 Kepa B. Uribe,1 Amalia Oleaga,3 Luis Irigoyen,4
Beatriz Torres,4 Helena Ostolaza,1 and Cesar Martin1∗1Unidad de Biofısica (Centro Mixto CSIC-UPV/EHU) and Departamento de Bioquımica, Universidad del Paıs Vasco, Apdo. 644, 48080 Bilbao, Spain;2Progenika Biopharma S.A., Derio, Spain; 3Endocrinologıa y Nutricion, Hospital de Basurto, Bilbao, Spain; 4Sevicio de Endocrinologıa y Nutricion,Hospital Santiago Apostol, Vitoria, Spain
Communicated by Bruce R. GottliebReceived 9 May 2011; accepted revised manuscript 26 September 2011.Published online 11 October 2011 in Wiley Online Library (www.wiley.com/humanmutation).DOI: 10.1002/humu.21630
ABSTRACT: Familial hypercholesterolemia (FH) is an au-tosomal dominant disorder mostly caused by mutationsin the LDLR gene. Although the detection of functionalmutations in the LDLR gene provides an unequivocaldiagnosis of the FH condition, there are many variantswhose pathogenicity is still unknown. The aims of thisstudy were to set up a rapid method to determine theeffect of LDLR mutations, thereby providing an accu-rate diagnosis of FH, and to functionally characterizesix LDLR mutations detected at high frequency by theLIPOchip R© platform (Progenika Biopharma, Spain) inthe Spanish population. LDLR expression and activitywere analyzed by one-single-step flow cytometry assay andconfocal microscopy. Splicing effects were determined bysequencing reverse transcription polymerase chain reac-tion products. The analysis of three heterozygous vari-ants with a single point mutation within the low-densitylipoprotein binding domain allowed us to classify thec.806G>A variant as nonpathogenic, and c.862G>A andc.895G>A variants as causative of FH. The results ob-tained for three variants affecting donor splice sites ofthe LDLR mRNA, c.313+2dupT, c.1186+5G>A, andc.1845+1G>C, demonstrated that these mutations arepathogenic. These results expand our knowledge of muta-tions responsible for FH, providing an accurate diagnosisand leading to early treatment to reduce the risk of pre-mature cardiovascular events.Hum Mutat 33:232–243, 2012. C© 2011 Wiley Periodicals, Inc.
KEY WORDS: LDL receptor; familial hypercholes-terolemia; LDL uptake assay
Additional Supporting Information may be found in the online version of this article.∗Correspondence to: Cesar Martin, Unidad de Biofısica (Centro Mixto CSIC-
UPV/EHU) and Departamento de Bioquımica, Universidad del Paıs Vasco, Apdo. 644,
48080 Bilbao, Spain. E-mail: [email protected]
Contract grant sponsor: Plan +Euskadi09 (UE09+/10) program of the Basque Govern-
ment; Postdoctoral contract from the Basque Government (to A.E.); Bizkaia Biophysics
Foundation (to A.E. and K.B.U.).
IntroductionFamilial hypercholesterolemia (FH; MIM# 143890) is a frequent
autosomal dominant disorder characterized by increased plasmalow-density lipoprotein cholesterol (LDL-C), tendon xanthomas,and premature coronary heart disease [Goldstein et al., 2001]. FHusually results from mutations in the low-density lipoprotein recep-tor (LDLR; MIM# 606945), which is responsible for the uptake ofcholesterol-containing particles into cells [Goldstein et al., 2001]; inits ligand, apolipoprotein B-100 (APOB; MIM# 107730) [Pullingeret al., 1995]; or in the proprotein convertase subtilisin/kexin type 9gene (PCSK9; MIM# 607786), which promotes LDLR degradation[Abifadel et al., 2003]. In the general population, FH is observed witha heterozygous frequency of 1:500 and a homozygous frequency of1:1,000,000 [Goldstein et al., 2001].
To date, more than 1,200 different mutations in the LDLR gene,located on chromosome 19p, have been identified worldwide. Thesemutations are distributed along 18 exons, introns, and the promoterregion of the LDLR gene [Leigh et al., 2008]; these include pointmutations (both substitutions and indels), as well as copy numbervariations such as duplications and deletions, which can differen-tially affect residual LDLR activity. Mutations can be divided intofive different classes according to the nature and location of themutations within the LDLR gene, as well as their phenotypic ef-fects on the protein: Class 1: no detectable LDLR protein [Hobbset al., 1988]; Class 2: defective LDLR transport, either completelyblocked (class 2A) or partially blocked (class 2B or leaky LDLRs)[Goldstein et al., 1985]; Class 3: defective LDL binding [Hobbs et al.,1992]; Class 4: defective clustering in clathrin-coated pits [Daviset al., 1986; Lehrman et al., 1985]; and Class 5: recycling-defectivereceptors [Beglova et al., 2004].
Heterozygous FH is diagnosed based on high LDL-C levels (usu-ally >250 mg/dl) in untreated adults, as well as the presence oftendon xanthomas and arcus cornealis. However, it is often diffi-cult to establish an early diagnosis in younger patients because thephysical manifestations of FH develop late in life. Several sets ofdiagnostic criteria have been extensively used in the clinic, includ-ing criteria established by the Dutch Lipid Clinic Network (DLCN).The DLCN uses a numeric score whereby a higher score indicates amore reliable diagnosis of FH [Defesche, 2000]; patients with scoresof 8 or higher are classified as definite FH patients. Initiating FHdetection and treatment as early as possible, as recommended bythe World Health Organization, can maximize the health benefitobtained through treatment [WHO 1999].
As long as the identification of functional mutations in the LDLRgene provides an unequivocal FH diagnosis, there is a need for
C© 2011 WILEY PERIODICALS, INC.
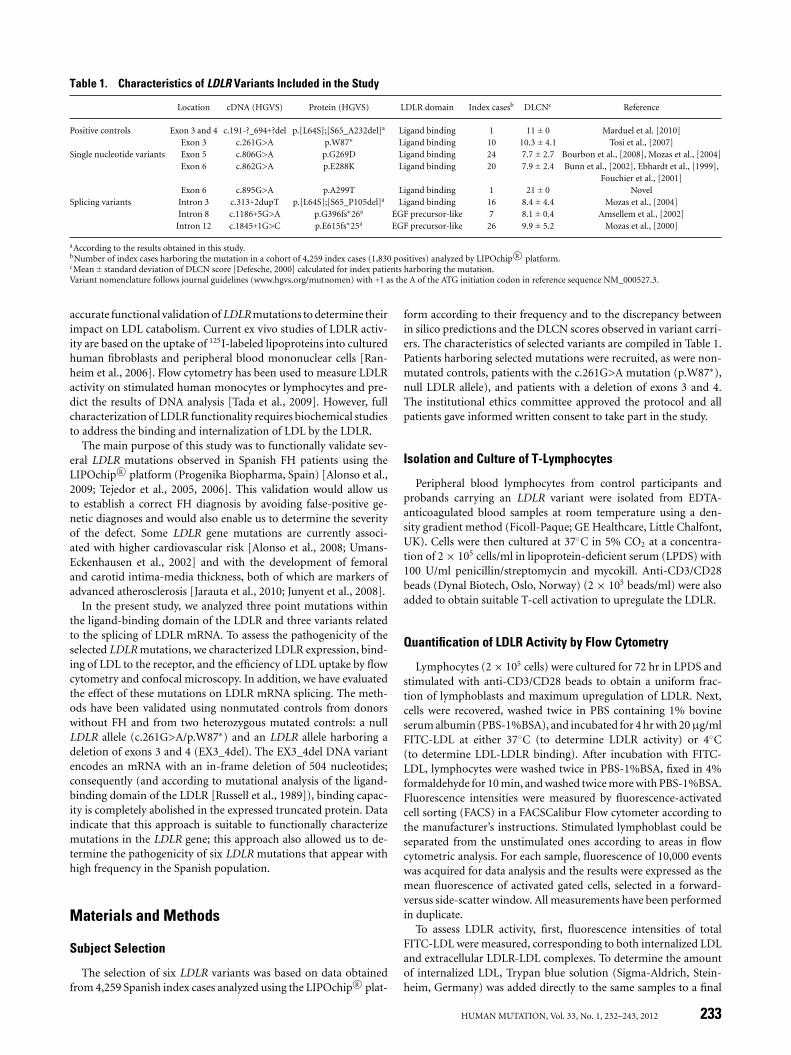
Table 1. Characteristics of LDLR Variants Included in the Study
Location cDNA (HGVS) Protein (HGVS) LDLR domain Index casesb DLCNc Reference
Positive controls Exon 3 and 4 c.191-?_694+?del p.[L64S];[S65_A232del]a Ligand binding 1 11 ± 0 Marduel et al. [2010]Exon 3 c.261G>A p.W87∗ Ligand binding 10 10.3 ± 4.1 Tosi et al., [2007]
Single nucleotide variants Exon 5 c.806G>A p.G269D Ligand binding 24 7.7 ± 2.7 Bourbon et al., [2008], Mozas et al., [2004]Exon 6 c.862G>A p.E288K Ligand binding 20 7.9 ± 2.4 Bunn et al., [2002], Ebhardt et al., [1999],
Fouchier et al., [2001]Exon 6 c.895G>A p.A299T Ligand binding 1 21 ± 0 Novel
Splicing variants Intron 3 c.313+2dupT p.[L64S];[S65_P105del]a Ligand binding 16 8.4 ± 4.4 Mozas et al., [2004]Intron 8 c.1186+5G>A p.G396fs∗26a EGF precursor-like 7 8.1 ± 0.4 Amsellem et al., [2002]
Intron 12 c.1845+1G>C p.E615fs∗25a EGF precursor-like 26 9.9 ± 5.2 Mozas et al., [2000]
aAccording to the results obtained in this study.bNumber of index cases harboring the mutation in a cohort of 4,259 index cases (1,830 positives) analyzed by LIPOchip R© platform.cMean ± standard deviation of DLCN score [Defesche, 2000] calculated for index patients harboring the mutation.Variant nomenclature follows journal guidelines (www.hgvs.org/mutnomen) with +1 as the A of the ATG initiation codon in reference sequence NM_000527.3.
accurate functional validation of LDLR mutations to determine theirimpact on LDL catabolism. Current ex vivo studies of LDLR activ-ity are based on the uptake of 125I-labeled lipoproteins into culturedhuman fibroblasts and peripheral blood mononuclear cells [Ran-heim et al., 2006]. Flow cytometry has been used to measure LDLRactivity on stimulated human monocytes or lymphocytes and pre-dict the results of DNA analysis [Tada et al., 2009]. However, fullcharacterization of LDLR functionality requires biochemical studiesto address the binding and internalization of LDL by the LDLR.
The main purpose of this study was to functionally validate sev-eral LDLR mutations observed in Spanish FH patients using theLIPOchip R© platform (Progenika Biopharma, Spain) [Alonso et al.,2009; Tejedor et al., 2005, 2006]. This validation would allow usto establish a correct FH diagnosis by avoiding false-positive ge-netic diagnoses and would also enable us to determine the severityof the defect. Some LDLR gene mutations are currently associ-ated with higher cardiovascular risk [Alonso et al., 2008; Umans-Eckenhausen et al., 2002] and with the development of femoraland carotid intima-media thickness, both of which are markers ofadvanced atherosclerosis [Jarauta et al., 2010; Junyent et al., 2008].
In the present study, we analyzed three point mutations withinthe ligand-binding domain of the LDLR and three variants relatedto the splicing of LDLR mRNA. To assess the pathogenicity of theselected LDLR mutations, we characterized LDLR expression, bind-ing of LDL to the receptor, and the efficiency of LDL uptake by flowcytometry and confocal microscopy. In addition, we have evaluatedthe effect of these mutations on LDLR mRNA splicing. The meth-ods have been validated using nonmutated controls from donorswithout FH and from two heterozygous mutated controls: a nullLDLR allele (c.261G>A/p.W87∗) and an LDLR allele harboring adeletion of exons 3 and 4 (EX3_4del). The EX3_4del DNA variantencodes an mRNA with an in-frame deletion of 504 nucleotides;consequently (and according to mutational analysis of the ligand-binding domain of the LDLR [Russell et al., 1989]), binding capac-ity is completely abolished in the expressed truncated protein. Dataindicate that this approach is suitable to functionally characterizemutations in the LDLR gene; this approach also allowed us to de-termine the pathogenicity of six LDLR mutations that appear withhigh frequency in the Spanish population.
Materials and Methods
Subject Selection
The selection of six LDLR variants was based on data obtainedfrom 4,259 Spanish index cases analyzed using the LIPOchip R© plat-
form according to their frequency and to the discrepancy betweenin silico predictions and the DLCN scores observed in variant carri-ers. The characteristics of selected variants are compiled in Table 1.Patients harboring selected mutations were recruited, as were non-mutated controls, patients with the c.261G>A mutation (p.W87∗),null LDLR allele), and patients with a deletion of exons 3 and 4.The institutional ethics committee approved the protocol and allpatients gave informed written consent to take part in the study.
Isolation and Culture of T-Lymphocytes
Peripheral blood lymphocytes from control participants andprobands carrying an LDLR variant were isolated from EDTA-anticoagulated blood samples at room temperature using a den-sity gradient method (Ficoll-Paque; GE Healthcare, Little Chalfont,UK). Cells were then cultured at 37◦C in 5% CO2 at a concentra-tion of 2 × 105 cells/ml in lipoprotein-deficient serum (LPDS) with100 U/ml penicillin/streptomycin and mycokill. Anti-CD3/CD28beads (Dynal Biotech, Oslo, Norway) (2 × 105 beads/ml) were alsoadded to obtain suitable T-cell activation to upregulate the LDLR.
Quantification of LDLR Activity by Flow Cytometry
Lymphocytes (2 × 105 cells) were cultured for 72 hr in LPDS andstimulated with anti-CD3/CD28 beads to obtain a uniform frac-tion of lymphoblasts and maximum upregulation of LDLR. Next,cells were recovered, washed twice in PBS containing 1% bovineserum albumin (PBS-1%BSA), and incubated for 4 hr with 20 μg/mlFITC-LDL at either 37◦C (to determine LDLR activity) or 4◦C(to determine LDL-LDLR binding). After incubation with FITC-LDL, lymphocytes were washed twice in PBS-1%BSA, fixed in 4%formaldehyde for 10 min, and washed twice more with PBS-1%BSA.Fluorescence intensities were measured by fluorescence-activatedcell sorting (FACS) in a FACSCalibur Flow cytometer according tothe manufacturer’s instructions. Stimulated lymphoblast could beseparated from the unstimulated ones according to areas in flowcytometric analysis. For each sample, fluorescence of 10,000 eventswas acquired for data analysis and the results were expressed as themean fluorescence of activated gated cells, selected in a forward-versus side-scatter window. All measurements have been performedin duplicate.
To assess LDLR activity, first, fluorescence intensities of totalFITC-LDL were measured, corresponding to both internalized LDLand extracellular LDLR-LDL complexes. To determine the amountof internalized LDL, Trypan blue solution (Sigma-Aldrich, Stein-heim, Germany) was added directly to the same samples to a final
HUMAN MUTATION, Vol. 33, No. 1, 232–243, 2012 233

concentration of 0.2%, eliminating the extracellular signal due tothe noninternalized LDLR-LDL complexes. This procedure allowsus to remove extracellular fluorescence by quenching and to deter-mine the intensity of the remaining fluorescent particles inside thecells (which is unaffected by the external quencher).
Quantification of LDLR Expression by Flow Cytometry
Lymphocytes (2 × 105) were cultured in LPDS with anti-CD3/CD28 beads for 72 hr, then resuspended in PBS-1%BSA andblocked 30 min with PBS containing 10% fetal bovine serum (PBS-10%FBS) and washed twice with PBS-1%BSA. Cells were incubatedwith a mouse anti-LDLR primary antibody (1:100; 2.5 mg/l; ProgenBiotechnik GmbH) for 1 hr at room temperature, washed twice withPBS-1%BSA, and incubated with an Alexa Fluor 488-conjugatedgoat antimouse secondary IgG antibody (1:100; Invitrogen, Molec-ular Probes, Carlsbad, USA).
For each sample, fluorescence of 10,000 events was acquired fordata analysis and the results were expressed as the mean fluorescenceof activated gated cells, selected in a forward- versus side-scatterwindow. All measurements have been performed in duplicate.
Confocal Laser Scanning Microscopy
Confocal laser scanning microscopy (CLSM) was used to de-termine LDLR-LDL binding rates and cell surface expression ofLDLR. Briefly, cells were plated on polylysine-coated glass slidesand cultured for 72 hr at 37◦C in 5% CO2 in LPDS with 100 U/mlpenicillin/streptomycin, mycokill, and anti-CD3/CD28 beads.
After culturing, the medium was removed and glass slides werewashed twice with PBS-1%BSA. Nonlabeled lipoproteins (20 μg/mlLDL) were added and cells were incubated at 37◦C for an addi-tional 4 hr. Cells were fixed with 4% paraformaldehyde for 10 minand washed three times with PBS-1%BSA, then blocked in PBS-10%FBS for 1 hr and washed in PBS-1%BSA three times. Next,samples were incubated with the appropriate primary antibodies(mAb anti-C7 IgG for LDLR, and rabbit polyclonal raised againstAPOB) for 1 hr, followed by incubation with FITC- or Texas Red R©-conjugated secondary antibodies to visualize LDLR and LDL, re-spectively. Coverslips were mounted onto glass slides and sampleswere visualized using a confocal microscope (Olympus IX 81) withsequential excitation and capture image acquisition with a digitalcamera (Axiocam NRc5, Zeiss). Images were processed with Flu-oview v.50 software. Image analysis to quantify the fluorescenceintensities was accomplished using the public domain software Im-ageJ (available at http://rsb.info.nih.gov/ij) on a standard PC.
Reverse Transcription Polymerase Chain Reaction
Reverse transcription-polymerase chain reaction (RT-PCR) wasperformed using 0.5 μg total RNA and 5 pmol primers followingthe Qiagen Onestep RT-PCR kit protocol (Qiagen). Primers weredesigned to produce overlapping PCR products corresponding toexons 1–8, exons 3–10, exons 7–14, exons 11–17, and exons 13–18 ofthe LDLR open reading frame, as described previously [Tveten et al.,2006]. Thermal cycling conditions were 50◦C for 30 min; 95◦C for15 min; and 40 cycles of 95◦C for 30 sec, 60◦C for 30 sec, and 72◦Cfor 60 sec. The amplified products were analyzed on Tris-bufferedEDTA 2% agarose gels, which were stained with SYBR (Invitrogen).Two additional primers inside intron 8 and intron 12 were designedfor use with primers in exons 7 and 11, respectively. The bands
were extracted with a gel/PCR extraction kit (ATP Biotech) andsequenced.
In Silico Prediction of the Effect of Molecular Eventson the LDL Receptor
Three different software packages were used to predict thepossible impact of amino acid substitutions on the structureand function of c.806G>A/p.G269D, c.862G>A/p.E288K, andc.895G>A/p.A299T variants: Align GVGD (http://agvgd.iarc.fr)[Mathe et al., 2006; Tavtigian et al., 2006], Polyphen-2(http://genetics.bwh.harvard.edu/pph2) [Adzhubei et al., 2010],and SIFT (http://sift.jcvi.org) [Ng and Henikoff, 2003]. GVGDand SIFT analysis were performed by comparing amino acidsubstitutions in the human LDLR peptide sequence (P01130)against LDLR amino acid sequences from nine species: Pantroglodytes (XP_001167447.1), Macaca mulatta (NP_001028078.1),Mus musculus (P35951.1), Rattus norvegicus (P35952.1), Orycto-lagus cuniculus (P20063.1), Sus scrofa (Q28832.2), Equus caballus(XP_001490793.2), Xenopus laevis (AAI70345.1), and Danio rerio(NP_001025454.1).
Predictions of the possible effects caused by alternative splicingin the variants included in the study were estimated with fourdifferent software packages: NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html) [Reese et al., 1997], SpliceSiteFinder-like (http://www.genet.sickkids.on.ca/˜ali/splicesitefinder.html)[Zhang, 1998],MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html) [Yeo and Burge, 2004], and HumanSplicing Finder for splicing predictions (http://www.umd.be/HSF)[Desmet et al., 2009]. The reference sequence used for LDLR wasNM_000527.3 (NCBI RefSeq).
Conservation Analysis
Nucleotide and amino acid conservation analyses among specieswere conducted using data from the University of California, SantaCruz (UCSC) genome browser (http://genome.ucsc.edu), based onmultiz alignments of 46 vertebrate species sequences [Blanchetteet al., 2004] and expressed as the number of sequences with thehuman residue divided by the total number of available sequences.
Statistical Analysis
All measurements were performed at least three times, with n = 3unless otherwise stated, and results are presented as mean ± standarddeviation (SD). Levels of significance were determined using a two-tailed Student’s t-test, and a confidence level of greater than 95%(P < 0.05) was used to establish statistical significance.
Supp. Methods and techniques are described further in the onlineSupporting Information.
Results
Validation of the Methods with Nonmutated Controls,EX3_4del Probands, and c.261G>A/p.W87∗ Probands
In order to set up different approaches to validate LDLR func-tionality, three different controls were used: nonmutated controlsfrom donors without FH (wt), and two heterozygous mutated con-trols, EX3_4del and c.261G>A. The latter two variants were usedas internal controls because their theoretical effects on binding and
234 HUMAN MUTATION, Vol. 33, No. 1, 232–243, 2012
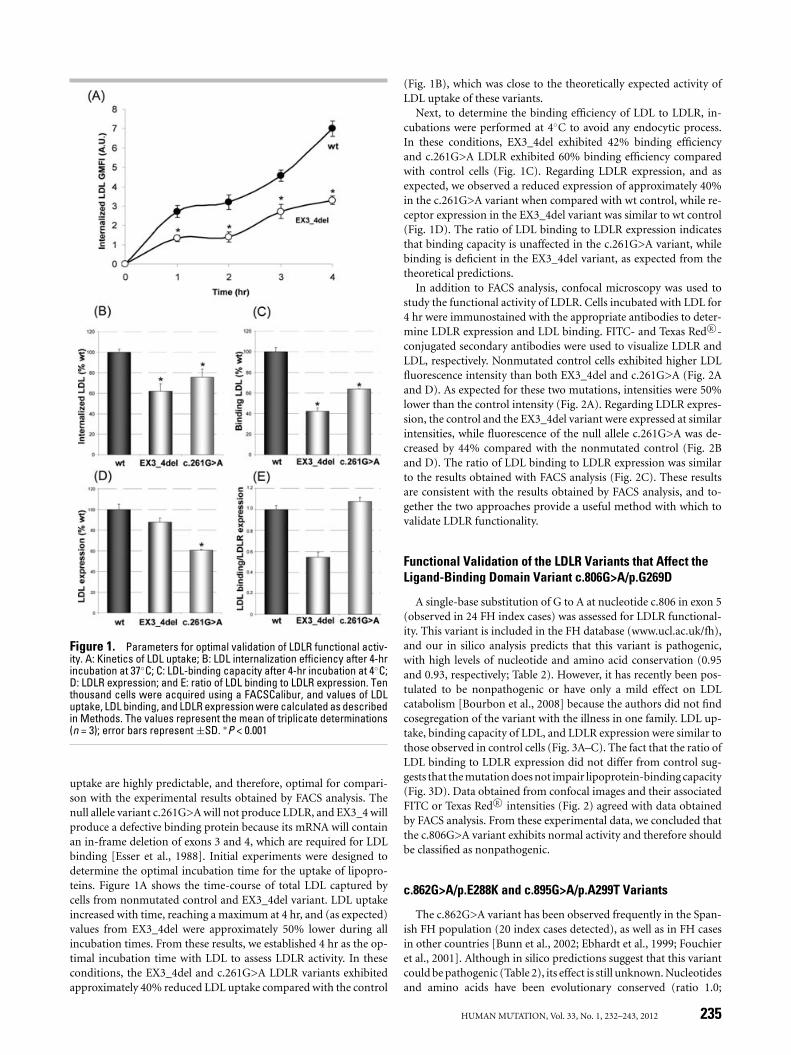
Figure 1. Parameters for optimal validation of LDLR functional activ-ity. A: Kinetics of LDL uptake; B: LDL internalization efficiency after 4-hrincubation at 37◦C; C: LDL-binding capacity after 4-hr incubation at 4◦C;D: LDLR expression; and E: ratio of LDL binding to LDLR expression. Tenthousand cells were acquired using a FACSCalibur, and values of LDLuptake, LDL binding, and LDLR expression were calculated as describedin Methods. The values represent the mean of triplicate determinations(n = 3); error bars represent ±SD. ∗P < 0.001
uptake are highly predictable, and therefore, optimal for compari-son with the experimental results obtained by FACS analysis. Thenull allele variant c.261G>A will not produce LDLR, and EX3_4 willproduce a defective binding protein because its mRNA will containan in-frame deletion of exons 3 and 4, which are required for LDLbinding [Esser et al., 1988]. Initial experiments were designed todetermine the optimal incubation time for the uptake of lipopro-teins. Figure 1A shows the time-course of total LDL captured bycells from nonmutated control and EX3_4del variant. LDL uptakeincreased with time, reaching a maximum at 4 hr, and (as expected)values from EX3_4del were approximately 50% lower during allincubation times. From these results, we established 4 hr as the op-timal incubation time with LDL to assess LDLR activity. In theseconditions, the EX3_4del and c.261G>A LDLR variants exhibitedapproximately 40% reduced LDL uptake compared with the control
(Fig. 1B), which was close to the theoretically expected activity ofLDL uptake of these variants.
Next, to determine the binding efficiency of LDL to LDLR, in-cubations were performed at 4◦C to avoid any endocytic process.In these conditions, EX3_4del exhibited 42% binding efficiencyand c.261G>A LDLR exhibited 60% binding efficiency comparedwith control cells (Fig. 1C). Regarding LDLR expression, and asexpected, we observed a reduced expression of approximately 40%in the c.261G>A variant when compared with wt control, while re-ceptor expression in the EX3_4del variant was similar to wt control(Fig. 1D). The ratio of LDL binding to LDLR expression indicatesthat binding capacity is unaffected in the c.261G>A variant, whilebinding is deficient in the EX3_4del variant, as expected from thetheoretical predictions.
In addition to FACS analysis, confocal microscopy was used tostudy the functional activity of LDLR. Cells incubated with LDL for4 hr were immunostained with the appropriate antibodies to deter-mine LDLR expression and LDL binding. FITC- and Texas Red R©-conjugated secondary antibodies were used to visualize LDLR andLDL, respectively. Nonmutated control cells exhibited higher LDLfluorescence intensity than both EX3_4del and c.261G>A (Fig. 2Aand D). As expected for these two mutations, intensities were 50%lower than the control intensity (Fig. 2A). Regarding LDLR expres-sion, the control and the EX3_4del variant were expressed at similarintensities, while fluorescence of the null allele c.261G>A was de-creased by 44% compared with the nonmutated control (Fig. 2Band D). The ratio of LDL binding to LDLR expression was similarto the results obtained with FACS analysis (Fig. 2C). These resultsare consistent with the results obtained by FACS analysis, and to-gether the two approaches provide a useful method with which tovalidate LDLR functionality.
Functional Validation of the LDLR Variants that Affect theLigand-Binding Domain Variant c.806G>A/p.G269D
A single-base substitution of G to A at nucleotide c.806 in exon 5(observed in 24 FH index cases) was assessed for LDLR functional-ity. This variant is included in the FH database (www.ucl.ac.uk/fh),and our in silico analysis predicts that this variant is pathogenic,with high levels of nucleotide and amino acid conservation (0.95and 0.93, respectively; Table 2). However, it has recently been pos-tulated to be nonpathogenic or have only a mild effect on LDLcatabolism [Bourbon et al., 2008] because the authors did not findcosegregation of the variant with the illness in one family. LDL up-take, binding capacity of LDL, and LDLR expression were similar tothose observed in control cells (Fig. 3A–C). The fact that the ratio ofLDL binding to LDLR expression did not differ from control sug-gests that the mutation does not impair lipoprotein-binding capacity(Fig. 3D). Data obtained from confocal images and their associatedFITC or Texas Red R© intensities (Fig. 2) agreed with data obtainedby FACS analysis. From these experimental data, we concluded thatthe c.806G>A variant exhibits normal activity and therefore shouldbe classified as nonpathogenic.
c.862G>A/p.E288K and c.895G>A/p.A299T Variants
The c.862G>A variant has been observed frequently in the Span-ish FH population (20 index cases detected), as well as in FH casesin other countries [Bunn et al., 2002; Ebhardt et al., 1999; Fouchieret al., 2001]. Although in silico predictions suggest that this variantcould be pathogenic (Table 2), its effect is still unknown. Nucleotidesand amino acids have been evolutionary conserved (ratio 1.0;
HUMAN MUTATION, Vol. 33, No. 1, 232–243, 2012 235

Tabl
e2.
Des
crip
tion
ofth
eVa
rian
tsSt
udie
d,N
ucle
otid
ean
dA
min
oA
cid
Cons
erva
tion,
and
InSi
lico
Pred
ictio
ns
Spli
cem
odifi
cati
onp
red
icti
onP
ath
ogen
icit
yp
red
icti
on
cDN
A(H
GV
S)P
rote
in(H
GV
S)N
TC
ons.
bA
AC
ons.
cSP
LIC
ESI
TE
FIN
DE
RLI
KE
MA
XE
NT
SCA
NN
NSP
LIC
EH
um
ansp
lici
ng
fin
der
Ali
gnG
VG
DP
olyp
hen
-2SI
FT
c.80
6G>
Ap.
G26
9D0.
950.
93N
och
ange
No
chan
geN
och
ange
No
chan
geA
ffec
tin
g(C
65)
Poss
ibly
dam
agin
g(0
.736
)N
otto
lera
ted
(0.0
)c.
862G
>A
p.E
288K
1.00
1.00
No
chan
geN
och
ange
No
chan
geN
och
ange
Aff
ecti
ng
(C55
)P
roba
bly
dam
agin
g(0
.999
)N
otto
lera
ted
(0.0
)c.
895G
>A
p.A
299T
0.36
0.65
No
chan
geN
och
ange
No
chan
geN
och
ange
Not
affe
ctin
g(C
0)B
enin
g(0
.007
)To
lera
ted
(0.1
9)c.
313+
2du
pTp.
[L64
S];[
S65_
P10
5del
]a0.
75N
ALo
ssof
don
orsi
teLo
ssof
don
orsi
teLo
ssof
don
orsi
teLo
ssof
don
orsi
teN
AN
AN
Ac.
1186
+5G
>A
p.G
396f
s∗ 26a
0.97
NA
Loss
ofdo
nor
site
Loss
ofdo
nor
site
Loss
ofdo
nor
site
No
chan
geN
AN
AN
Ac.
1845
+1G
>C
p.E
615f
s∗ 25a
1.00
NA
Loss
ofdo
nor
site
Loss
ofdo
nor
site
Loss
ofdo
nor
site
Loss
ofdo
nor
site
NA
NA
NA
aA
ccor
din
gto
the
resu
lts
obta
ined
inth
isst
udy
.bN
ucl
eoti
deco
nse
rvat
ion
.c A
min
oac
idco
nse
rvat
ion
.
Table 2), suggesting they have an important role in protein function.The c.895G>A variant has not been described before and we haveonly observed a unique index case with a severe FH phenotype.Nevertheless, all algorithms predict that it is nonpathogenic andits conservation among species is quite low (ratios 0.36 and 0.65for nucleotide and amino acid conservation, respectively; Table 2).Because there was no experimental evidence that these mutationscause FH, we followed the described strategy to study their possibleeffect on the LDLR. First, LDLR activity was determined; LDL in-ternalization in both variants was significantly decreased comparedwith control cells (Fig. 3A). LDL-binding capacity was also reducedcompared with controls in both variants, while LDLR expressionwas unaffected (Fig. 3B and C). The LDL binding to LDLR expres-sion ratio also confirmed the lower binding capacity of these LDLRvariants (Fig. 3D). Confocal microscopy analysis and fluorescence-associated intensities of immunolabeled LDLR and LDL confirmedthe impaired binding capacity of the c.862G>A and c.895G>A vari-ants (Fig. 2). The experimentally obtained data demonstrate thepathogenicity of these LDLR alterations, whose effect was similar tothe effect induced by the EX3_4del variant.
Functional Validation of the LDLR Variants that AffectmRNA Splicing
We evaluated the effects of three mutations observed in the Span-ish population on mRNA splicing: c.313+2dupT (n = 16 index cases),c.1186+5G>A (n = 7 index cases), and c.1845+1G>C (n = 26 indexcases). First, as with the previous LDLR variants, we character-ized in what manner these mutations affect LDLR expression, LDLbinding, and LDL internalization. When cells were incubated withLDL for 4 hr and analyzed by FACS, efficiency of LDL uptake bythese receptors was diminished by 35% in c.313+2dupT, 39% inc.1186+5G>A, and 68% in c.1845+1G>C compared with wt control(Fig. 3A). LDL-binding capacity was lower in all three variants thanin wt controls, being diminished by 40% in c.313+2dupT, 36% inc.1186+5G>A, and 39% in c.1845+1G>C (Fig. 3B). Receptor expres-sion was reduced by 49% in c.1186+5G>A and 67% in c.1845+1G>C,while the c.313+2dupT variant did not exhibit significant reductioncompared with control cells (Fig. 3C). The LDL binding to LDLRexpression ratio also showed that the receptors expressed in thec.1186+5G>A and c.1845+1G>C variants were functional, while40% of 313+2dupT receptors were not functional (Fig. 3D). Wefurther confirmed these results by confocal microscopy, and CLSMresults agreed with FACS data (Fig. 2). LDLR expression was re-duced by 38% in c.1186+5G>A, by 40% in c.1845+1G>C, and wasnot reduced in c.313+2dupT. The resulting LDL binding was re-duced by 37% in c.313+2dupT, 36% in c.1186+5G>A, and 42% inc.1845+1G>C compared with wt control (Fig. 3). Based on this ex-perimental characterization, we can classify these three variants aspathogenic: c.1186+5G>A and c.1845+1G>C are Class 1 mutationsbecause they lack LDLR expression, and 313+2dupT is a Class 3mutation because it affects ligand binding.
Mutations’ Effects on mRNA Splicing
The possible effects of mutations on mRNA splicing were ana-lyzed in silico using four different software packages (Table 2). Thepredictions of all software packages were similar, and no change waspredicted for the exonic variants; however, for the three intronicvariants a “loss of donor site” was predicted by all software pack-ages except for Human Splicing Finder, which did not predict anychange for the 1186+5G>A variant. To determine the effect of the
236 HUMAN MUTATION, Vol. 33, No. 1, 232–243, 2012
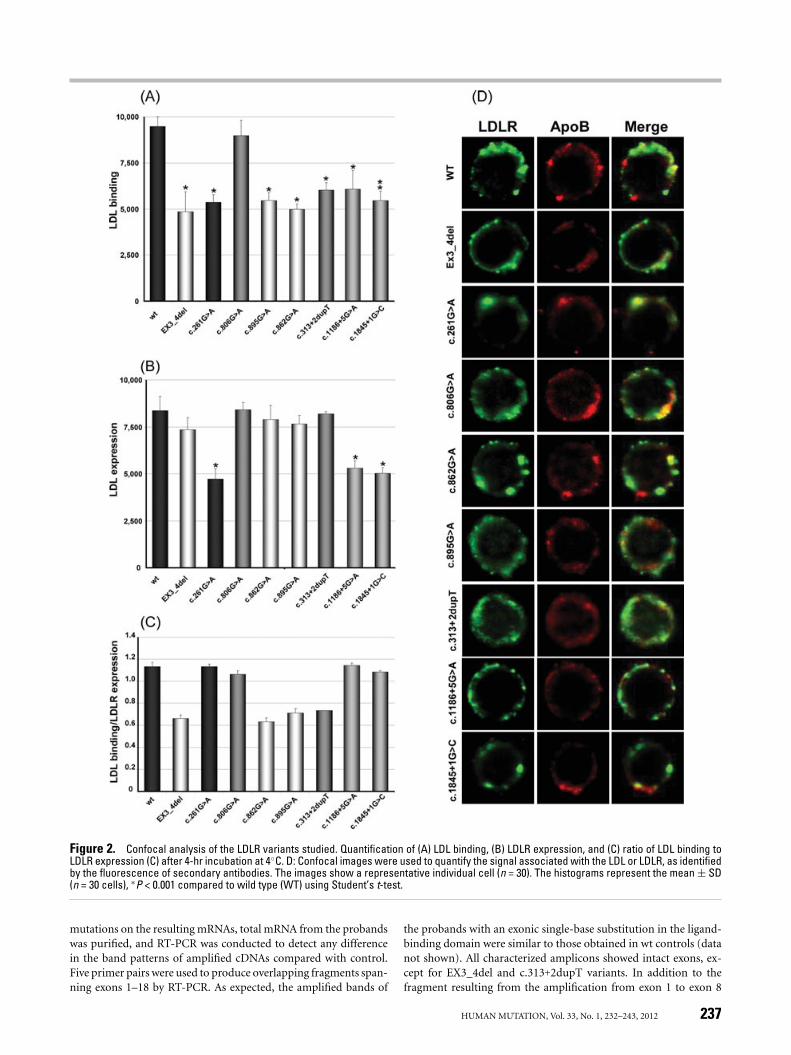
Figure 2. Confocal analysis of the LDLR variants studied. Quantification of (A) LDL binding, (B) LDLR expression, and (C) ratio of LDL binding toLDLR expression (C) after 4-hr incubation at 4◦C. D: Confocal images were used to quantify the signal associated with the LDL or LDLR, as identifiedby the fluorescence of secondary antibodies. The images show a representative individual cell (n = 30). The histograms represent the mean ± SD(n = 30 cells), ∗P < 0.001 compared to wild type (WT) using Student’s t-test.
mutations on the resulting mRNAs, total mRNA from the probandswas purified, and RT-PCR was conducted to detect any differencein the band patterns of amplified cDNAs compared with control.Five primer pairs were used to produce overlapping fragments span-ning exons 1–18 by RT-PCR. As expected, the amplified bands of
the probands with an exonic single-base substitution in the ligand-binding domain were similar to those obtained in wt controls (datanot shown). All characterized amplicons showed intact exons, ex-cept for EX3_4del and c.313+2dupT variants. In addition to thefragment resulting from the amplification from exon 1 to exon 8
HUMAN MUTATION, Vol. 33, No. 1, 232–243, 2012 237
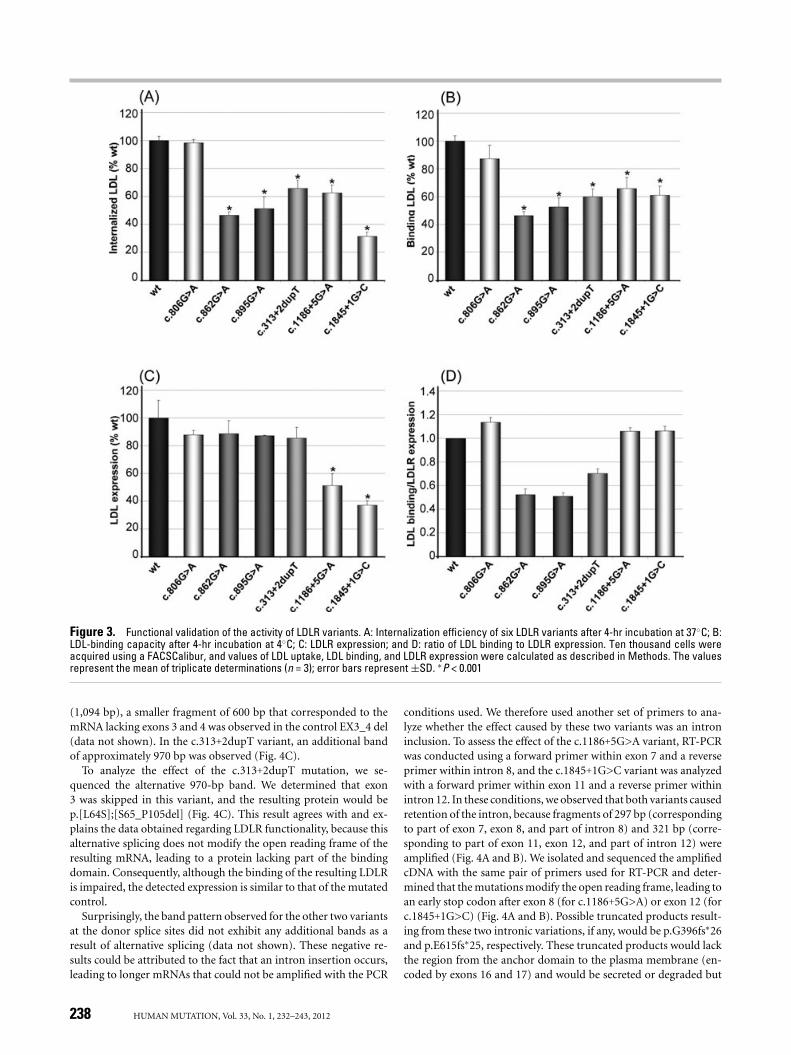
Figure 3. Functional validation of the activity of LDLR variants. A: Internalization efficiency of six LDLR variants after 4-hr incubation at 37◦C; B:LDL-binding capacity after 4-hr incubation at 4◦C; C: LDLR expression; and D: ratio of LDL binding to LDLR expression. Ten thousand cells wereacquired using a FACSCalibur, and values of LDL uptake, LDL binding, and LDLR expression were calculated as described in Methods. The valuesrepresent the mean of triplicate determinations (n = 3); error bars represent ±SD. ∗P < 0.001
(1,094 bp), a smaller fragment of 600 bp that corresponded to themRNA lacking exons 3 and 4 was observed in the control EX3_4 del(data not shown). In the c.313+2dupT variant, an additional bandof approximately 970 bp was observed (Fig. 4C).
To analyze the effect of the c.313+2dupT mutation, we se-quenced the alternative 970-bp band. We determined that exon3 was skipped in this variant, and the resulting protein would bep.[L64S];[S65_P105del] (Fig. 4C). This result agrees with and ex-plains the data obtained regarding LDLR functionality, because thisalternative splicing does not modify the open reading frame of theresulting mRNA, leading to a protein lacking part of the bindingdomain. Consequently, although the binding of the resulting LDLRis impaired, the detected expression is similar to that of the mutatedcontrol.
Surprisingly, the band pattern observed for the other two variantsat the donor splice sites did not exhibit any additional bands as aresult of alternative splicing (data not shown). These negative re-sults could be attributed to the fact that an intron insertion occurs,leading to longer mRNAs that could not be amplified with the PCR
conditions used. We therefore used another set of primers to ana-lyze whether the effect caused by these two variants was an introninclusion. To assess the effect of the c.1186+5G>A variant, RT-PCRwas conducted using a forward primer within exon 7 and a reverseprimer within intron 8, and the c.1845+1G>C variant was analyzedwith a forward primer within exon 11 and a reverse primer withinintron 12. In these conditions, we observed that both variants causedretention of the intron, because fragments of 297 bp (correspondingto part of exon 7, exon 8, and part of intron 8) and 321 bp (corre-sponding to part of exon 11, exon 12, and part of intron 12) wereamplified (Fig. 4A and B). We isolated and sequenced the amplifiedcDNA with the same pair of primers used for RT-PCR and deter-mined that the mutations modify the open reading frame, leading toan early stop codon after exon 8 (for c.1186+5G>A) or exon 12 (forc.1845+1G>C) (Fig. 4A and B). Possible truncated products result-ing from these two intronic variations, if any, would be p.G396fs∗26and p.E615fs∗25, respectively. These truncated products would lackthe region from the anchor domain to the plasma membrane (en-coded by exons 16 and 17) and would be secreted or degraded but
238 HUMAN MUTATION, Vol. 33, No. 1, 232–243, 2012
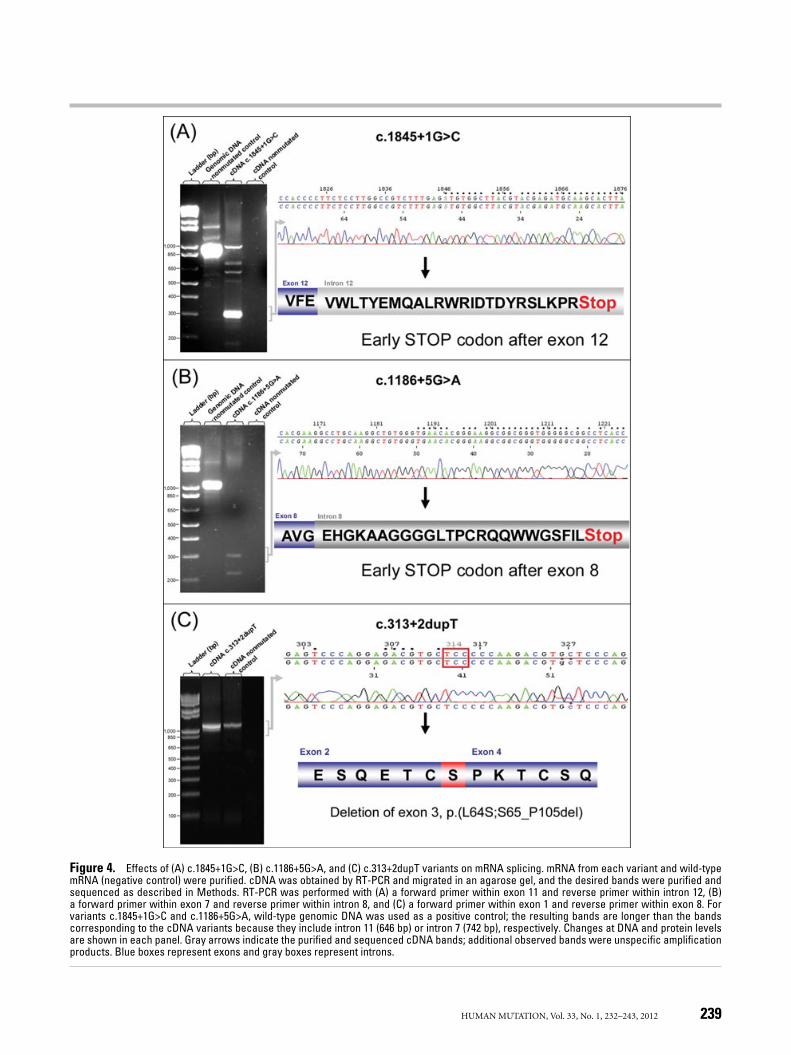
Figure 4. Effects of (A) c.1845+1G>C, (B) c.1186+5G>A, and (C) c.313+2dupT variants on mRNA splicing. mRNA from each variant and wild-typemRNA (negative control) were purified. cDNA was obtained by RT-PCR and migrated in an agarose gel, and the desired bands were purified andsequenced as described in Methods. RT-PCR was performed with (A) a forward primer within exon 11 and reverse primer within intron 12, (B)a forward primer within exon 7 and reverse primer within intron 8, and (C) a forward primer within exon 1 and reverse primer within exon 8. Forvariants c.1845+1G>C and c.1186+5G>A, wild-type genomic DNA was used as a positive control; the resulting bands are longer than the bandscorresponding to the cDNA variants because they include intron 11 (646 bp) or intron 7 (742 bp), respectively. Changes at DNA and protein levelsare shown in each panel. Gray arrows indicate the purified and sequenced cDNA bands; additional observed bands were unspecific amplificationproducts. Blue boxes represent exons and gray boxes represent introns.
HUMAN MUTATION, Vol. 33, No. 1, 232–243, 2012 239

not properly localized. Therefore, they would not interfere with thebinding or uptake of LDL arising from the nonmutated allele. Thephenotype of these LDLR variants would then be similar to a nullallele, as has been observed in FACS and CLSM experiments.
DiscussionTo date, the majority of FH-causing variations within the LDLR
gene have been investigated by DNA screening, and most of thepossible damaging effects at the protein level are predicted by insilico analysis [Fouchier et al., 2005; Leigh et al., 2008]. However,algorithms predicting the potential effects of these variants can-not always accurately predict pathogenicity [Leigh et al., 2008], andexperimental support is required to demonstrate reduced LDLR ac-tivity and provide a conclusive FH diagnosis. The currently availabletools to experimentally assess whether a mutation affects LDLR ac-tivity include ex vivo studies from patients’ cells [Tada et al., 2009]and expression of the mutant gene in heterologous transfected cells[Ranheim et al., 2006]. In this work, we have functionally character-ized LDLR variants in cultured cells derived from patients, providinga feasible approach that does not require time-consuming methodssuch as mutagenesis and in vitro expression of the mutant allele inheterologous cells. LDLR activity, assessed by FACS analysis coupledwith the use of FITC-labeled LDL and Trypan blue as an externalquencher of the noninternalized LDLR-LDL complexes, provides aunique advantage over the use of 125I-LDL and other methods. Inaddition to ease of handling and disposal, LDLR activity is moreaccurately determined in the same cell population and the likeli-hood of internal error due to the method used is diminished. Theuse of confocal microscopy also allows one to determine the classof the mutated receptors and the level at which the variant is inter-fering. These two frequently used techniques can be easily used byany group conducting genetic diagnosis of FH to validate in silicopredictions and to avoid reporting false positives. However, inter-pretation of the effects of mutations on receptor protein functionin cells from heterozygous patients expressing both the normal anda mutant allele is difficult, because expression of the normal alleleinterferes with the effects of mutant allele expression. Therefore,the effects of the mutation must be carefully interpreted. In spite ofthis limitation, these methods are biologically relevant because theyreflect effects that may be occurring in vivo.
In this study, we have functionally validated six mutations in theLDLR gene. Three of these variants are located within the LDL-binding domain, and the other three affect donor splice sites in themRNA.
We have determined the activity of the c.806G>A/p.G269D vari-ant because of its high prevalence in the Spanish population. Thec.806G>A variant has also been reported by Bourbon et al. [2008],who observed no cosegregation of this variant with hypercholes-terolemia in one family. Their finding is in agreement with theresults obtained by our FACS and confocal microscopy analyses(Figure 2 and 3), in which LDLR expression and activity are similarto those observed in nonmutated control cells. These results arealso supported by a study reported by Mozas et al. [2004], in whichthis variant was observed in cis with the c.1045delC/p.Q349Sfs∗21variant, which is likely to be the pathogenic variant in the individualreported. Therefore, the c.806G>A variant should be classified asnonpathogenic. The c.806G>A variant is highly conserved amongspecies. In fact, only three of the 43 species in which this LDLR re-gion has been sequenced have a different amino acid in this position.Interestingly, in two of the three, the substituted amino acid is also
an aspartate, supporting the notion that this specific amino acidchange is tolerated and the variant does not affect LDLR activity.
Similarly to the c.806G>A variant, the c.862G>A/p.E288K substi-tution is frequently detected in the Spanish population. The affectednucleotide and amino acid residue are completely conserved amongspecies (Table 2), indicating their importance in both the gene andthe protein. In the c.862G>A/p.E288K variant, the ratio of LDLbinding to LDLR expression demonstrated that the LDLR exhibitedreduced binding capacity in approximately 50% of cells comparedwith wt control (Fig. 3), demonstrating its pathogenicity.
The c.895G>A/p.A299T substitution was the unique LDLR vari-ant observed in an FH patient presenting with a high DLCN score,that is, clinically diagnosed as definite FH. Nevertheless, bioinfor-matic predictions classify this variant as nonpathogenic and it ismildly conserved among species (Table 2), although none of theorthologous proteins carry a threonine in this position and it ispreferentially replaced by a valine residue. The c.895G>A/p.A299Tsubstitution caused the same effect as the c.862G>A mutation, whichexhibited an impairment in LDL binding (Fig. 3). The substitutionof alanine 299 with a threonine in the human LDLR affects theprotein’s function, making it a pathogenic variant.
These substitutions were analyzed by three different programs:Align-GVGD, SIFT, and Polyphen-2. All three programs predictedthat the two most conserved substitutions analyzed (c.806G>A andc.862G>A) would be pathogenic, while the less conserved c.895G>Avariant was predicted to be nonpathogenic. These results are inagreement with the mode of operation of these three programs,each of which bases its analysis on protein conservation and thephysicochemical differences between the wild type and the mutatedamino acid, among other variables [Adzhubei et al., 2010; Matheet al., 2006; Ng and Henikoff, 2003; Tavtigian et al., 2006]. Thelarger Grantham distance [Grantham, 1974] in c.806G>A/p.G269Dversus c.862G>A/p.E288K (94 and 56, respectively) could then ex-plain the difference in the classification obtained using Align-GVGD,because the first change was predicted slightly more likely to bepathogenic. Polyphen-2 predicted that the p.G269D variant waspossibly pathogenic, and that the p.E288K variant was probablypathogenic. This difference is most likely due to the presence atthe analyzed position of various aspartate in the same position fororthologous and paralogous protein sequences used for the align-ments against p.G269D, while the p.E288K variant amino acid isnot present in any of the protein sequences at the analyzed po-sition (data not shown). The predicted nonpathogenicity of thec.895G>A/p.A299T variant by the three programs used in this studycould be explained either by the low conservation of this aminoacid, or by the relatively small physicochemical difference betweenthe wild type and mutated amino acids.
The different effects of the c.806G>A/p.G269D, c.862G>A/p.E288K, and c.895G>A/p.A299T variants on pathogenicity may de-pend on the location of the amino acid substitution within theLDL-binding domain. The LDL-binding domain of the LDLR con-sists of seven cystein-rich repeats (R1–R7) that contain two loopsstabilized by three disulfide bridges and one Ca2+ ion [Fass et al.,1997]. LDLR binds LDL with 1:1 stoichiometry [van Driel et al.,1989] and it has been shown that modules R2–R7 play a key role inbinding [Nguyen et al., 2006; Russell et al., 1989]. Ca2+ coordinationallows the establishment of a compact structure that forms patchesof conserved acidic motifs within the surface of the R modules thatwould bind to positively charged regions of lipoproteins. However,the extent of the interactions between LDLR and LDL is unclear, andthe surface contours of individual modules differ markedly [Northand Blacklow, 2000], indicating that the R modules are unlikely toshare a single criterion for binding.
240 HUMAN MUTATION, Vol. 33, No. 1, 232–243, 2012

The most remarkable in silico prediction error occurs in thec.895G>A/p.A299T variant, which is predicted to be nonpathogenic.At this position, the alanine is not highly conserved among speciesand is preferentially replaced by a valine residue. A more detailedanalysis of the R7 module reveals that replacement of the alanine atposition 299 with a threonine may destabilize the structure of theCa2+ coordination site because the hydroxyl group in the threoninecould interact with the surrounding groups necessary for Ca2+ co-ordination, destabilizing the R7 structure, and preventing properfolding of the module (Supp. Fig. S1). Ca2+ is specifically requiredfor both the acquisition and maintenance of native LDLR structurein the endoplasmic reticulum, specifically for the proper foldingof the ligand-binding domains in R1, R3, R5, and R7 [Pena et al.,2010]. These electrostatic interactions of the threonine hydroxylgroup with the surrounding groups do not occur with either ala-nine or valine; therefore, a correct Ca2+ coordination site is allowed,leading to an optimal LDLR conformation for LDL binding (Supp.Fig. S1).
A mutation known as p.E240K, similar to the mutation observedin the c.862G>A/p.E288K variant, has been described near theamino-terminal end of the sixth module. In p.E240K, a hydrogenbond existing between the carboxylate side chain of glutamate 240and the backbone amide proton of arginine 237 would be disrupted,resulting in a protein folding defect [North and Blacklow, 2000].However, in keeping with predictions, the c.862G>A/p.E288K vari-ant this mutation maintains the same hydrogen bond and doesnot modify the folding of the module (Supp. Fig. S2A and B). Inthe c.862G>A/p.E288K variant, the observed pathogenicity wouldbe due to an increase in the positive electrostatic potential ofthe surface (Supp. Fig. S2C and D). As a result, the electrostaticcomponent required for LDL binding would be disturbed due torepulsion forces, resulting in diminished binding activity (Supp.Fig. S2E and F).
Given the domain and structure in which the mutation in thec.806G>A/p.G269D variant is located (Supp. Fig. S3), we can deter-mine that the amino acid substitution does not modify the pattern ofpolar interactions that is maintained with the rest of the protein. Inaddition, a modification within this region, which links two mod-ules, does not significantly alter the protein structure. Therefore,this change does not appear to affect LDL binding.
In silico, analysis does not consider the consequences of missensemutations on the structure of the R modules or the interaction ofthe LDLR protein with the LDL, which would explain the differencesbetween in silico and in vitro data observed in the present study. Theadditional analysis presented above may help accurately predict theimpact of missense mutations on protein function and may therebyensure a better fit between in silico and in vitro data. Nevertheless,additional studies are necessary to characterize the role of eachresidue within its module.
In the present study, we have also included some variants thataffect donor splice sites in the LDLR mRNA. Variants that affecthighly conserved GT/AG dinucleotides at the splice sites are likely tobe pathogenic because these nucleotides form part of the recognitionsignals at the respective donor and acceptor splice sites [Burset et al.,2000]. As expected, the c.1845+1G>C variant abolishes donor splicesite. As a consequence, intron 12 is included in the mature mRNAbut exon 12 is not skipped, as expected [Houdayer et al., 2008;Krawczak et al., 2007].
The c.1186+5G>A variant destroys the 5′ donor splice site ofexon 8 and, such as c.1845+1G>C, does not cause exon skippingbut does cause intron 8 inclusion in the mature mRNA. In thedonor sites, point mutations of the conserved G at position +5are frequently observed and appear to lead to aberrant splicing
[Buratti et al., 2007; Krawczak et al., 2007]. Nevertheless, not everyvariant at position +5 abolishes the splice site. For instance, LDLRc.2140+5G>A is demonstrably nonpathogenic [Bourbon et al., 2009;Holla et al., 2009a]. It has been postulated that the effect of changesat nucleotide +5 can be defined by the “weakness” of the donor splicesite. Sequences with nucleotides that differ from the conserved AGat the positions –2 and –1, respectively, would be more affected bychanges of the G at position +5 [ Buratti et al., 2007; Carmel et al.,2004], which may explain why the c.2140+5G>A variant (whichhas AG as the consensus dinucleotide) is nonpathogenic, while thec.1186+5G>A variant (which has GG as the consensus dinucleotide)causes an aberrant 5′ splice site.
The intron insertions caused by c.1186+5G>A and c.1845+1G>Ceach produce an early stop codon, with supposed subsequent acti-vation of the nonsense-mediated mRNA decay system and mutatedmRNA decay [Holla et al., 2009b]. Although mRNA expression isnot completely abolished, the truncated proteins that are producedwould not be properly located in the membrane because they lackthe anchoring domain (encoded by exons 16 and 17). In both vari-ants, we have confirmed by FACS analysis and confocal microscopythat both LDLR expression and LDL uptake are reduced comparedwith control cells. The results obtained above led us to concludethat any truncated proteins resulting from the mutations would beunable to accomplish their function, and would behave as thoughthey were null alleles.
Although the c.313+2dupT variant causes exon 3 skipping, theopen reading frame of the mRNA is not altered. The resulting LDLreceptor lacks the second calcium-binding repeat and therefore ex-hibits reduced LDL-binding capacity. Although in silico predictionsfor this variant were similar to the other two splicing mutationsstudied (Table 2), this variant induces the skipping of an exon, butnot the inclusion of an intron. This effect is the most frequent con-sequence when an aberrant splice site is created [Houdayer et al.,2008; Krawczak et al., 2007]. The inclusion of a T at position +3 dis-places the next nucleotides, leading to respective G-to-A, and T-to-Gchanges at positions +5 and +6. The resulting change c.313+5G>Aresults in the skipping of exon 3 and has been shown to cause FH[Liguori et al., 2001]. Notably, another variant, c.313+6T>C, alsocauses exon skipping [Bourbon et al., 2009]. Therefore, positions +5and +6 after exon 3 would be important for correct splicing. Indeed,in many other genes, the position +5, which is highly conserved atdonor consensus domains, seems to be prone to aberrant splicing[Buratti et al., 2007; Krawczak et al., 2007]. Taking into account thatLDLR position 313+5 is completely conserved among species, whileposition 313+3 is less conserved, the nucleotide change responsiblefor the alternative splicing is likely to be the +5G>A that resultsfrom displacement, similar to a situation described by Liguori et al.[2001], and perhaps aggravated by the +6 position change. Becausethe exon-terminal dinucleotide is TC instead of the highly conservedAG [Burset et al., 2000], this splice site would be more susceptiblethan other splice sites to any change in the surrounding nucleotides.
Several studies have demonstrated the important function ofLDLR modules R2–R7 in LDL binding, showing that LDLR variantslacking R3, R4, R5, R6, or R7 cannot bind LDL, and the deletion ofR2 reduces LDL binding [Nguyen et al., 2006; Russell et al., 1989].The results obtained in the c.313+2dupT variant suggest that thedeletion of R2, the second calcium-binding repeat, leads to an effectsimilar to that caused by EX3_4del. Indeed, Spanish index casesharboring the c.313+2dupT variant present a severe phenotype. Ithas been reported that c.313+1G>A produces two alternative splic-ing results, the skipping of exon 3 and the inclusion of intron 3[Cameron et al., 2009]. No LDLR can be produced from any tran-script that includes intron 3, which introduces an early stop codon
HUMAN MUTATION, Vol. 33, No. 1, 232–243, 2012 241

into the mRNA. Meanwhile, a transcript in which exon 3 is skippedproduces a receptor with markedly reduced ability to internalizeLDL. Our results demonstrate that c.313+2dupT only causes exon3 skipping because LDLR expression is not altered, and its effect issimilar to that of EX3_4del. Based on presented results, and similarlyto findings reported by Cameron et al. [2009], the functionality ofLDLR receptor lacking exon 3 appears to be lower than has beenobserved in transfection experiments using heterologous cells [Esseret al., 1988; Russell et al., 1989]. One possibility is that the single ser-ine residue inserted at the new splice site in the naturally occurringreceptor, which was not present in the receptor resulting from site-directed mutagenesis to remove exon 3 [Esser et al., 1988; Russellet al., 1989]), would destabilize the protein in some way. Anotherpossibility is that the R2 calcium-binding repeat is also crucial forLDL binding, at least in vivo.
The loss of donor sites has been well predicted by the four soft-ware packages used, except for the failure of the Human SplicingFinder software to detect the effect of the c.1186+5G>A mutation(Table 2). Nevertheless, all four software packages produced con-sensus predictions for two LDLR variants: c.2140+5G>A (whichproduces no effect) and c.1186+5G>A (which results in the loss ofa donor site).
In silico predictions for splice sites determine the probability ofcreating or losing a splicing donor or acceptor site; however, theydo not predict how the mRNA will be spliced given the creationor loss of a site, or how such a modification will affect the proteinproduced. In this study, for variants with identical predictions, weobserved two different splicing effects that also produce differentconsequences: intron inclusion results in a null allele, while exonskipping results in a binding-defective protein.
Although these bioinformatics tools are useful to provide firstimpressions, validation studies are needed to assure the pathogenic-ity of any change, because (as several studies have demonstrated)software accuracy and sensitivity are not 100% [Buratti et al., 2007;Desmet et al., 2010; Hicks et al., 2011].
FH is usually diagnosed on the basis of clinical and laboratoryfindings that are often obtained later in life, which makes earlydiagnosis in young patients difficult. Genetic diagnosis can detectFH at any age, before symptoms appear; however, a complete ge-netic diagnosis should include all existing evidence regarding thepathogenicity of the detected genetic change. In this sense, only val-idated variants can be reported as pathogenic; other variants shouldbe reported as variants with unknown pathogenicity [Richards et al.,2008]. The methods used in this work allow one to characterize thephenotypic effects of LDLR variants and establish a correct classi-fication of the variants as causing or not causing FH. In fact, thevariant whose nonpathogenicity has been demonstrated, c.806G>A,presented the lowest DLCN score, while all variants classified aspathogenic presented definite diagnosis (DLCN score >8) (Table 1).It is also useful to distinguish whether the mutation affects LDLRexpression, the capacity of the LDLR to bind LDL, or LDLR inter-nalization.
In conclusion, this work demonstrates how important it is tovalidate LDLR function to establish an accurate diagnosis of FH.We can conclude from the data obtained that any FH diagnosisinferred from in silico analysis alone is inconclusive; a functionalcharacterization of LDLR activity is required to avoid false-positivediagnoses. The methods used in this study to characterize LDLRvariants have enabled us to classify the c.806G>A variant as non-pathogenic, and to classify five variants that were not previouslyfunctionally characterized as pathogenic. The studied variants weredetected by LIPOchip R© v9.1 DNA array [Alonso et al., 2009; Tejedoret al., 2005, 2006], which, together with the methods used in this
work, can corroborate genetic analysis, providing an unequivocaldiagnosis and allowing the initiation of treatment early in life.
Acknowledgments
We thank all index patients and relatives for participating in this study. Wesincerely thank the SGIKER at the University of Basque Country, Prof. A.Gomez-Munoz for flow cytometry facilities, and Rocio Alonso for excellenttechnical assistance.
References
Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, Cruaud C,Benjannet S, Wickham L, Erlich D, Derre A, Villeger L, Farnier M, Beucler I,Bruckert E, Chambaz J, Chanu B, Lecerf JM, Luc G, Moulin P, Weissenbach J, PratA, Krempf M, Junien C, Seidah NG, Boileau C. 2003. Mutations in PCSK9 causeautosomal dominant hypercholesterolemia. Nat Genet 34:154–156.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, KondrashovAS, Sunyaev SR. 2010. A method and server for predicting damaging missensemutations. Nat Methods 7:248–249.
Alonso R, Defesche JC, Tejedor D, Castillo S, Stef M, Mata N, Gomez-Enterria P,Martinez-Faedo C, Forga L, Mata P. 2009. Genetic diagnosis of familial hyperc-holesterolemia using a DNA-array based platform. Clin Biochem 42:899–903.
Alonso R, Mata N, Castillo S, Fuentes F, Saenz P, Muniz O, Galiana J, Figueras R,Diaz JL, Gomez-Enterrıa P, Mauri M, Piedecausa M, Irigoyen L, Aguado R, MataP; Spanish Familial Hypercholesterolaemia Group. 2008. Cardiovascular diseasein familial hypercholesterolaemia: influence of low-density lipoprotein receptormutation type and classic risk factors. Atherosclerosis 200:315–321.
Amsellem S, Briffaut D, Carrie A, Rabes JP, Girardet JP, Fredenrich A, Moulin P,Krempf M, Reznik Y, Vialettes B, de Gennes JL, Brukert E, Benlian P. 2002.Intronic mutations outside of Alu-repeat-rich domains of the LDL receptor geneare a cause of familial hypercholesterolemia. Hum Genet 111:501–510.
Beglova N, Jeon H, Fisher C, Blacklow SC. 2004. Cooperation between fixed and lowpH-inducible interfaces controls lipoprotein release by the LDL receptor. Mol Cell16:281–292.
Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosen-bloom K, Clawson H, Green ED, Haussler D, Miller W. 2004. Aligning multiplegenomic sequences with the threaded blockset aligner. Genome Res 14:708–715.
Bourbon M, Alves AC, Medeiros AM, Silva S, Soutar AK; Investigators of Portuguese FHStudy. 2008. Familial hypercholesterolaemia in Portugal. Atherosclerosis 196:633–642.
Bourbon M, Duarte MA, Alves AC, Medeiros AM, Marques L, Soutar AK. 2009. Geneticdiagnosis of familial hypercholesterolaemia: the importance of functional analysisof potential splice-site mutations. J Med Genet 46:352–357.
Bunn CF, Lintott CJ, Scott RS, George PM. 2002. Comparison of SSCP and DHPLC forthe detection of LDLR mutations in a New Zealand cohort. Hum Mutat 19:311–315.
Buratti E, Chivers M, Kralovicova J, Romano M, Baralle M, Krainer AR, Vorechovsky I.2007. Aberrant 5’ splice sites in human disease genes: mutation pattern, nucleotidestructure and comparison of computational tools that predict their utilization.Nucleic Acids Res 35:4250–4263.
Burset M, Seledtsov IA, Solovyev VV. 2000. Analysis of canonical and non-canonicalsplice sites in mammalian genomes. Nucleic Acids Res 28:4364–4375.
Cameron J, Holla ØL, Kulseth MA, Leren TP, Berge KE. 2009. Splice-site mutationc.313+1, G>A in intron 3 of the LDL receptor gene results in transcripts withskipping of exon 3 and inclusion of intron 3. Clin Chim Acta 403:131–135.
Carmel I, Tal S, Vig I, Ast G. 2004. Comparative analysis detects dependencies amongthe 5’ splice-site positions. RNA 10:828–840.
Davis CG, Lehrman MA, Russell DW, Anderson RG, Brown MS, Goldstein JL. 1986.The J.D. mutation in familial hypercholesterolemia: amino acid substitution incytoplasmic domain impedes internalization of LDL receptors. Cell 45:15–24.
Defesche J. 2000. Familial hypercholesterolemia. In: Betteridge DJ, editor. Lipids andvascular disease. London: Martin Dunitz Ltd. p. 65–76.
Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. 2009.Human Splicing Finder: an online bioinformatics tool to predict splicing signals.Nucleic Acid Res 37:e67.
Desmet FO, Hamroun D, Collod-Beroud G, Beroud C. 2010. Bioinformatics identi-fication of splice site signals and prediction of mutation effects. In: Mohan RM,editor. Research advances in nucleic acid research. Kerala: Global research network(1–14).
Ebhardt M, Schmidt H, Doerk T, Tietge U, Haas R, Manns MP, Schmidtke J, StuhrmannM. 1999. Mutation analysis in 46 German families with familial hypercholes-terolemia: identification of 8 new mutations. Hum Mutat 13:257–262.
242 HUMAN MUTATION, Vol. 33, No. 1, 232–243, 2012

Esser V, Limbird LE, Brown MS, Goldstein JL, Russell DW. 1988. Mutational analysisof the ligand binding domain of the low density lipoprotein receptor. J Biol Chem263:13282–13290.
Fass D, Blacklow S, Kim PS, Berger JM. 1997. Molecular basis of familial hypercholes-terolaemia from structure of LDL receptor module. Nature 388:691–693.
Fouchier SW, Defesche JC, Umans-Eckenhausen MW, Kastelein JP. 2001. The molecu-lar basis of familial hypercholesterolemia in the Netherlands. Hum Genet 109:602–615.
Fouchier SW, Kastelein JJ, Defesche JC. 2005. Update of the molecular basis of familialhypercholesterolemia in the Netherlands. Hum Mutat 26:550–556.
Grantham R. 1974. Amino acid difference formula to help explain protein evolution.Science 185:862–864.
Goldstein JL, Brown MS, Anderson RG, Russell DW, Schneider WJ. 1985. Receptor-mediated endocytosis: concepts emerging from the LDL receptor system. AnnuRev Cell Biol 1:1–39.
Goldstein JL, Hobbs HH, Brown MS. 2001. Familial hypercholesterolemia. In: ScriverCR, Beaudet AL, Sly WS, Valle D, Editors. The metabolic basis of inherited disease.New York: McGraw-Hill. p. 2863–2913.
Hicks S, Wheeler DA, Plon SE, Kimmel M. 2011. Prediction of missense mutationfunctionality depends on both the algorithm and sequence alignment employed.Hum Mutat 32:661–668.
Hobbs HH, Brown MS, Goldstein JL. 1992. Molecular genetics of the LDL receptorgene in familial hypercholesterolemia. Hum Mutat 1:445–466.
Hobbs HH, Leitersdorf E, Goldstein JL, Brown MS, Russell DW. 1988. Multiple crm-mutations in familial hypercholesterolemia. Evidence for 13 alleles, including fourdeletions. J Clin Invest 81:909–917.
Holla ØL, Kulseth MA, Berge KE, Leren TP, Ranheim T. 2009b. Nonsense-mediateddecay of human LDL receptor mRNA. Scand J Clin Lab Invest 69:409–417.
Holla ØL, Nakken S, Mattingsdal M, Ranheim T, Berge KE, Defesche JC, Leren TP.2009a. Effects of intronic mutations in the LDLR gene on pre-mRNA splicing:comparison of wet-lab and bioinformatics analyses. Mol Genet Metab 96:245–252.
Houdayer C, Dehainault C, Mattler C, Michaux D, Caux-Moncoutier V, Pages-BerhouetS, d’Enghien CD, Lauge A, Castera L, Gauthier-Villars M, Stoppa-Lyonnet D. 2008.Evaluation of in silico splice tools for decision-making in molecular diagnosis.Hum Mutat 29:975–982.
Jarauta E, Mateo-Gallego R, Bea A, Burillo E, Calmarza P, Civeira F. 2010. Carotidintima-media thickness in subjects with no cardiovascular risk factors. Rev EspCardiol 63:97–102.
Junyent M, Tucker KL, Smith CE, Lane JM, Mattei J, Lai CQ, Parnell LD, Or-dovas JM. 2008. Femoral atherosclerosis in heterozygous familial hypercholes-terolemia: influence of the genetic defect. Arterioscler Thromb Vasc Biol 28:580–586.
Krawczak M, Thomas NS, Hundrieser B, Mort M, Wittig M, Hampe J, Cooper DN. 2007.Single base-pair substitutions in exon-intron junctions of human genes: nature,distribution, and consequences for mRNA splicing. Hum Mutat 28:150–158.
Lehrman MA, Goldstein JL, Brown MS, Russell DW, Schneider WJ. 1985.Internalization-defective LDL receptors produced by genes with nonsense andframeshift mutations that truncate the cytoplasmic domain. Cell 41:735–743.
Leigh SE, Foster AH, Whittall RA, Hubbart CS, Humphries SE. 2008. Update andanalysis of the University College London low density lipoprotein receptor familialhypercholesterolemia database. Ann Hum Genet 72:485–498.
Liguori R, Bianco AM, Argiriou A, Pauciullo P, Giannino A, Rubba P, De SimoneV. 2001. LDL receptor cDNA sequence analysis in familial hypercholesterolemiapatients: 5 novel mutations with high prevalence in families originating fromsouthern Italy. Hum Mutat 17:433–436.
Marduel M, Carrie A, Sassolas A, Devillers M, Carreau V, Di Filippo M, Erlich D,Abifadel M, Marques-Pinheiro A, Munnich A, Junien C; French ADH ResearchNetwork, Boileau C, Varret M, Rabes JP, Farnier M, Luc G, Lecerf JM, BruckertE, Bonnefont-Rousselot D, Giral P, Kalopissis A, Girardet JP, Polak M, Chanu B,Moulin P, Perrot L, Krempf M, Zaır Y, Bonnet J, Ferrieres J, Ferrieres D, Bongard V,Cournot M, Reznick Y, Schlienger JL, Fredenrich A, Durlach V. 2010. Molecularspectrum of autosomal dominant hypercholesterolemia in France. Hum Mutat31:E1811–E1824.
Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. 2006. Computa-tional approaches for predicting the biological effect of p53 missense mutations: acomparison of three sequence analysis based methods. Nucleic Acids Res 34:1317–1325.
Mozas P, Castillo S, Tejedor D, Reyes G, Alonso R, Franco M, Saenz P, Fuentes F,Almagro F, Mata P, Pocovı M. 2004. Molecular characterization of familial hyper-cholesterolemia in Spain: identification of 39 novel and 77 recurrent mutations inLDLR. Hum Mutat 24:187–199.
Mozas P, Cenarro A, Civeira F, Castillo S, Ros E, Pocovi M. 2000. Mutation analysis in36 unrelated Spanish subjects with familial hypercholesterolemia: identificationof 3 novel mutations in the LDL receptor gene. Hum Mutat 15:483–484.
Ng PC, Henikoff S. 2003. SIFT: predicting amino acid changes that affect proteinfunction. Nucleic Acids Res 31:3812–3814.
Nguyen AT, Hirama T, Chauhan V, Mackenzie R, Milne R. 2006. Binding characteristicsof a panel of monoclonal antibodies against the ligand binding domain of thehuman LDLr. J Lipid Res 47:1399–1405.
North CL, Blacklow SC. 2000. Solution structure of the sixth LDL-A module of theLDL receptor. Biochemistry 39:2564–2571.
Pena F, Jansens A, van Zadelhoff G, Braakman I. 2010. Calcium as a crucial cofactorfor low density lipoprotein receptor folding in the endoplasmic reticulum. J BiolChem. 285:8656–8664.
Pullinger CR, Hennessy LK, Chatterton JE, Liu W, Love JA, Mendel CM, Frost PH,Malloy MJ, Schumaker VN, Kane JP. 1995. Familial ligand-defective apolipopro-tein B. Identification of a new mutation that decreases LDL receptor bindingaffinity. J Clin Invest 95:1225–1234.
Ranheim T, Kulseth MA, Berge KE, Leren TP. 2006. Model system for phenotypiccharacterization of sequence variations in the LDL receptor gene. Clin Chem52:1469–1479.
Reese MG, Eeckman FH, Kulp D, Haussler D. 1997. Improved splice site detection ingenie. J Comp Biol 4:311–323.
Russell DW, Brown MS, Goldstein JL. 1989. Different combinations of cysteine-rich re-peats mediate binding of low density lipoprotein receptor to two different proteins.J Biol Chem 264:21682–21688.
Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE,Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee.2008. ACMG recommendations for standards for interpretation and reporting ofsequence variations: revisions 2007. Genet Med 10:294–300.
Tada H, Kawashiri MA, Noguchi T, Mori M, Tsuchida M, Takata M, Nohara A, InazuA, Kobayashi J, Yachie A, Mabuchi H, Yamagishi M. 2009. A novel method fordetermining functional LDL receptor activity in familial hypercholesterolemia:application of the CD3/CD28 assay in lymphocytes. Clin Chim Acta 400:42–47.
Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, de Silva D,Zharkikh A, Thomas A. 2006. Comprehensive statistical study of 452 BRCA1 mis-sense substitutions with classification of eight recurrent substitutions as neutral. JMed Genet 43:295–305.
Tejedor D, Castillo S, Mozas P, Jimenez E, Lopez M, Tejedor MT, Artieda M, Alonso R,Mata P, Simon L, Martınez A, Pocovı M; Spanish FH Group. 2005. Reliable low-density DNA array based on allele-specific probes for detection of 118 mutationscausing familial hypercholesterolemia. Clin Chem 51:1137–1144.
Tejedor D, Castillo S, Mozas P, Jimenez E, Lopez M, Tejedor MT, Artieda M, Alonso R,Mata P, Simon L, Martınez A, Pocovı M. 2006. Comparison of DNA array platformvs DNA sequencing as genetic diagnosis tools for familial hypercholesterolemia.Clin Chem 52:1971–1972.
Tosi I, Toledo-Leiva P, Neuwirth C, Naoumova RP, Soutar AK. 2007. Genetic defectscausing familial hypercholesterolaemia: identification of deletions and duplica-tions in the LDL-receptor gene and summary of all mutations found in patientsattending the Hammersmith Hospital Lipid Clinic. Atherosclerosis 194:102–111.
Tveten K, Ranheim T, Berge KE, Leren TP, Kulseth MA. 2006. Analysis of alternativelyspliced isoforms of human LDL receptor mRNA. Clin Chim Acta 373:151–157.
Umans-Eckenhausen MA, Sijbrands EJ, Kastelein JJ, Defesche JC. 2002. Low-densitylipoprotein receptor gene mutations and cardiovascular risk in a large geneticcascade screening population. Circulation 106:3031–3036.
Van Driel IR, Brown MS, Goldstein JL.1989. Stoichiometric binding of low densitylipoprotein (LDL) and monoclonal antibodies to LDL receptors in a solid phaseassay. J Biol Chem 264:9533–9538.
Wang J, Ban MR, Hegele RA. 2005. Multiplex ligation-dependent probe amplificationof LDLR enhances molecular diagnosis of familial hypercholesterolemia. J LipidRes 46:366–372.
Yeo G, Burge C.B. 2004. Maximum entropy modeling of short sequence motifs withapplications to RNA splicing signals. J Comput Biol 11:377–394.
Zhang MQ. 1998. Statistical features of human exons and their flanking regions. HumMol Genet 7:919–932.
HUMAN MUTATION, Vol. 33, No. 1, 232–243, 2012 243




















