
Expression of prostaglandin endoperoxide H synthase-2 induced by nitric oxide in conditionally...
14
Expression of prostaglandin endoperoxide H synthase-2 induced by nitric oxide in conditionally immortalized murine colonic epithelial cells JAY M. MEI,* ,1,2 NORMAN G. HORD,* ,2,3 DOLORES F. WINTERSTEIN § , STEVEN P. DONALD,* AND JAMES M. PHANG* ,1 *Metabolism and Cancer Susceptibility Section, Basic Research Laboratory, Division of Basic Sciences, National Cancer Institute and § Intramural Research Support Program, SAIC-Frederick, NCI-FCRDC, Frederick, Maryland ABSTRACT Increased expression of prostaglandin endoperoxide H synthase-2 (PGHS-2) has been im- plicated in pathological conditions such as inflamma- tory bowel diseases and colon cancer. Recently, it has been demonstrated that inducible nitric oxide syn- thase (NOS II) expression and nitric oxide (NO) production are up-regulated in these diseases as well. However, the apparent link between PGHS-2 and NOS II has not been thoroughly investigated in nontransformed and nontumorigenic colonic epithe- lial cells. In the present study, we examined the concomitant expression of PGHS-2 and NOS II as well as the production of prostaglandin E2 (PGE2) and NO in conditionally immortalized mouse colonic epithelial cells, namely YAMC (Apc 1/1 ). We found that the induction of PGHS-2 and generation of PGE2 in these cells by IFN-g and lipopolysaccharide (LPS) were greatly reduced by two selective NOS II inhibitors, L-NIL and SMT. To ascertain the effect of NO on PGHS-2 overexpression, we tested NO- releasing compounds, NOR-1 and SNAP, and found that they caused PGHS-2 expression and PGE2 pro- duction. This effect was abolished by hemoglobin, a NO scavenger. Using electrophoretic mobility shift assays, we found that both NOR-1 and SNAP caused b-catenin/LEF-1 DNA complex formation. Super- shift by anti-b-catenin antibody confirmed the pres- ence of b-catenin in the complex. Cell fractionation studies indicated that NO donors caused an increase in free soluble cytoplasmic b-catenin. This is further corroborated by the immunocytochemistry data showing the redistribution of b-catenin from the predominantly membrane localization into the cyto- plasm and nucleus after treatment with NO donors. To further explore the possible connection between PGHS-2 expression and b-catenin/LEF-1 DNA com- plex formation, we studied IMCE (Apc Min/1 ) cells, a sister cell line of YAMC with similar genetic back- ground but differing in Apc genotype and, conse- quently, their b-catenin levels. We found that IMCE cells, in comparison with YAMC cells, had markedly higher b-catenin/LEF-1 DNA complex formation under both resting conditions as well as after induc- tion with NO. In parallel fashion, IMCE cells ex- pressed significantly higher levels of PGHS-2 mRNA and protein, and generated more PGE2. Overall, this study suggests that NO may be involved in PGHS-2 overexpression in conditionally immortalized mouse colonic epithelial cells. Although the molecular mechanism of the link is still under investigation, this effect of NO appears directly or indirectly to be a result of the increase in free soluble b-catenin and the formation of nuclear b-catenin/LEF-1 DNA com- plex.—Mei, J. M., Hord, N. G., Winterstein, D. F., Donald, S. P., and Phang, J. M. Expression of prostaglandin endoperoxide H synthase-2 induced by nitric oxide in conditionally immortalized murine colonic epithelial cells. FASEB J. 14, 1188 –1201 (2000) Key Words: PGHS-2 z NOS II z nitric oxide z colonic epithelial cells Increased expression of prostaglandin endoperox- ide H synthase-2 (PGHS-2) is associated with inflam- matory bowel diseases and colon cancer (1–5). More- over, high PGHS-2 expression in human colon cancer cells is closely associated with increased met- astatic potential (6). In contrast, PGHS-1, a constitu- tively expressed PGHS family member with high- sequence homology to PGHS-2, is not induced during colorectal neoplastic transformation, sup- porting the hypothesis that the inducible expression of the PGHS-2 isozyme may be important in the 1 Correspondence: Metabolism and Cancer Susceptibility Section, Basic Research Laboratory, Division of Basic Sci- ences, Rm 12–90, Bldg. 560, National Cancer Institute, Fred- erick, MD 21702, USA. E-mail: [email protected]; [email protected] 2 These authors contributed equally to this work. 3 Present address: Department of Food Science and Human Nutrition, Michigan State University, 2100 Anthony Hall, East Lansing, MI 48824-1225. 1188 0892-6638/00/0014-1188/$02.25 © FASEB
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Expression of prostaglandin endoperoxide H synthase-2 induced by nitric oxide in conditionally...

Expression of prostaglandin endoperoxide H synthase-2induced by nitric oxide in conditionally immortalizedmurine colonic epithelial cells
JAY M. MEI,*,1,2 NORMAN G. HORD,*,2,3 DOLORES F. WINTERSTEIN§,STEVEN P. DONALD,* AND JAMES M. PHANG*,1
*Metabolism and Cancer Susceptibility Section, Basic Research Laboratory, Division of BasicSciences, National Cancer Institute and §Intramural Research Support Program, SAIC-Frederick,NCI-FCRDC, Frederick, Maryland
ABSTRACT Increased expression of prostaglandinendoperoxide H synthase-2 (PGHS-2) has been im-plicated in pathological conditions such as inflamma-tory bowel diseases and colon cancer. Recently, it hasbeen demonstrated that inducible nitric oxide syn-thase (NOS II) expression and nitric oxide (NO)production are up-regulated in these diseases as well.However, the apparent link between PGHS-2 andNOS II has not been thoroughly investigated innontransformed and nontumorigenic colonic epithe-lial cells. In the present study, we examined theconcomitant expression of PGHS-2 and NOS II aswell as the production of prostaglandin E2 (PGE2)and NO in conditionally immortalized mouse colonicepithelial cells, namely YAMC (Apc1/1). We foundthat the induction of PGHS-2 and generation ofPGE2 in these cells by IFN-g and lipopolysaccharide(LPS) were greatly reduced by two selective NOS IIinhibitors, L-NIL and SMT. To ascertain the effectof NO on PGHS-2 overexpression, we tested NO-releasing compounds, NOR-1 and SNAP, and foundthat they caused PGHS-2 expression and PGE2 pro-duction. This effect was abolished by hemoglobin, aNO scavenger. Using electrophoretic mobility shiftassays, we found that both NOR-1 and SNAP causedb-catenin/LEF-1 DNA complex formation. Super-shift by anti-b-catenin antibody confirmed the pres-ence of b-catenin in the complex. Cell fractionationstudies indicated that NO donors caused an increasein free soluble cytoplasmic b-catenin. This is furthercorroborated by the immunocytochemistry datashowing the redistribution of b-catenin from thepredominantly membrane localization into the cyto-plasm and nucleus after treatment with NO donors.To further explore the possible connection betweenPGHS-2 expression and b-catenin/LEF-1 DNA com-plex formation, we studied IMCE (ApcMin/1) cells, asister cell line of YAMC with similar genetic back-ground but differing in Apc genotype and, conse-quently, their b-catenin levels. We found that IMCEcells, in comparison with YAMC cells, had markedlyhigher b-catenin/LEF-1 DNA complex formation
under both resting conditions as well as after induc-tion with NO. In parallel fashion, IMCE cells ex-pressed significantly higher levels of PGHS-2 mRNAand protein, and generated more PGE2. Overall, thisstudy suggests that NO may be involved in PGHS-2overexpression in conditionally immortalized mousecolonic epithelial cells. Although the molecularmechanism of the link is still under investigation, thiseffect of NO appears directly or indirectly to be aresult of the increase in free soluble b-catenin andthe formation of nuclear b-catenin/LEF-1 DNA com-plex.—Mei, J. M., Hord, N. G., Winterstein, D. F.,Donald, S. P., and Phang, J. M. Expression ofprostaglandin endoperoxide H synthase-2 inducedby nitric oxide in conditionally immortalized murinecolonic epithelial cells. FASEB J. 14, 1188–1201(2000)
Key Words: PGHS-2 z NOS II z nitric oxide z colonic epithelialcells
Increased expression of prostaglandin endoperox-ide H synthase-2 (PGHS-2) is associated with inflam-matory bowel diseases and colon cancer (1–5). More-over, high PGHS-2 expression in human coloncancer cells is closely associated with increased met-astatic potential (6). In contrast, PGHS-1, a constitu-tively expressed PGHS family member with high-sequence homology to PGHS-2, is not inducedduring colorectal neoplastic transformation, sup-porting the hypothesis that the inducible expressionof the PGHS-2 isozyme may be important in the
1 Correspondence: Metabolism and Cancer SusceptibilitySection, Basic Research Laboratory, Division of Basic Sci-ences, Rm 12–90, Bldg. 560, National Cancer Institute, Fred-erick, MD 21702, USA. E-mail: [email protected];[email protected]
2 These authors contributed equally to this work.3 Present address: Department of Food Science and Human
Nutrition, Michigan State University, 2100 Anthony Hall, EastLansing, MI 48824-1225.
1188 0892-6638/00/0014-1188/$02.25 © FASEB

development of colon adenoma and cancer. Non-steroid anti-inflammatory drugs that inhibit PGHSactivity, such as aspirin and the newly developedselective PGHS-2 inhibitors, have well-documentedpreventive effects against precancerous colon polypformation and subsequent malignant transformation(7–9). Furthermore, it has been reported that intes-tinal polyp formation was dramatically reduced byablation of the PGHS-2 gene (10). Although PGHS-2protein catalyzes the first step in the production of ahost of bioactive arachidonate metabolites, the pro-duction of prostaglandin E2 (PGE2) is probably themost important in carcinogenesis. This prostanoidhas been shown to have antiapoptotic and mitogenicactivities that may enhance neoplastic progression(11, 12).
The role of the adenomatous polyposis coli gene(Apc) as a ‘gate-keeper’ in colonic carcinogenesisand its link with the downstream transcription regu-lator b-catenin/Tcf-LEF complexes has been re-cently established (13–18). It has been hypothesized,although not proven, that PGHS-2 may be one of thegenes responsive to b-catenin accumulation andpossibly the b-catenin/Tcf-LEF transcription path-way (19, 20). PGHS-2 mRNA and protein are highlyoverexpressed in human and mouse colonic adeno-mas compared with normal tissue (3–5). However,evidence is mounting to support the contributoryrole for inducible nitric oxide synthase (NOS II)expression in colorectal tumorigenesis. Overexpres-sion of NOS II frequently occurs in inflammatorybowel diseases that predispose the subjects to coloncancer (21, 22). Like PGHS-2, NOS II mRNA andprotein are overexpressed in colonic adenomas com-pared with normal tissue (23). These studies, takentogether with in vitro studies of PGHS-2 catalysis byNO, link the production of NO to the regulation ofPGHS-2 catalytic activity as well as transcriptionalactivation of the PGHS-2 gene. Indeed, NO-releasingdrugs have been shown to either activate the catalyticfunction of PGHS-2 or transcription of the PGHS-2gene, resulting in accumulation of PGHS-2 protein(24–27). But, it has not yet been established whetherNOS II and PGHS-2 expression are coincident, tem-porally distinct, or causally related biological events,and the possible mechanism(s) involved remain un-known.
To gain insight into the interconnection betweenPGHS-2 and NOS II and the potential mechanismsinvolved, we examined the expression of PGHS-2and NOS II as well as the production of PGE2 andNO in the nontransformed and nontumorigenicmurine colonic epithelial cells. These conditionallyimmortal cells are designated YAMC (Apc1/1) de-rived from a SV40LT antigen parental mouse; IMCE(ApcMin/1) are derived from the F1 hybrids resultingfrom the mating of ApcMin/1 and SV40LT antigen
transgenic mice. This pair of nontransformed mu-rine colon epithelial cell lines of similar geneticbackground includes one that carries the ApcMin/1
mutation and consequently expresses higher b-cate-nin levels. Because both YAMC and IMCE expressthe heat-labile SV40LT antigen that allows them toproliferate at 33°C, they revert to a nontransformedphenotype at the restrictive temperature of 39°C, atwhich the proliferation of these cells ceases (28–30).The epithelial nature of these cells was demonstratedby staining with anti-keratin antisera. The genotypeand expression of adematous polyposis coli (APC) pro-tein of these cell types have been confirmed byallele-specific polymerase chain reaction and by west-ern immunoblotting, respectively (28, 29). Further-more, these cells have been used to demonstrate thatthe ApcMin/1 mutation in IMCE cells can cooperatewith stably transduced oncogenic ras to produce thetransformed, tumorigenic phenotype (e.g., growthin soft agar; tumor formation in athymic mice) (31).In contrast to most transformed cells or cell linesderived from colon tumors, these cells have unde-tectable (YAMC) or little (IMCE) constitutivePGHS-2 expression by northern and Western blotanalysis. On stimulation with IFN-g/LPS or NOdonors at 39°C, they express high levels of PGHS-2and generate PGE2. This stimulation by LPS andIFN-g or NO mimics the exposure of colonic epithe-lial cells to products of bacterial metabolism andimmune cells in vivo. Data from the present studysuggest that the presence of NO caused by IFN-g/LPS stimulation or NO-releasing drugs may play animportant contributory role in PGHS-2 overexpres-sion in these conditionally immortalized, nontumor-derived, colonic epithelial cells under nontransform-ing conditions. Furthermore, this overexpression ofPGHS-2 is associated, either directly or indirectly,with the accumulation of b-catenin in the cytoplasmand nucleus, and possibly the formation of theb-catenin/LEF-1 DNA complex.
MATERIALS AND METHODS
Reagents
Anti-NOS II, anti-PGHS-2, anti-b-catenin, anti-E-cadherin,and anti-mouse immunoglobulin G (IgG) horseradish perox-idase monoclonal antibodies were purchased from Transduc-tion (Lexington, Ky.). Anti-b-catenin polyclonal antibody wasfrom Sigma (St. Louis, Mo.). Anti-actin monoclonal antibodywas from Boehringer Mannheim (Indianapolis, Ind.). MurinecDNA PGHS-2 probe was purchased from Oxford Biomedical(Oxford, Mich.). For cell culture, the following products wereused and purchased from their respective sources: RPMI 1640media and mouse IFN-g from Life Technologies-BRL (GrandIsland, N.Y.); neonatal calf serum from Gemini (Calabasas,Calif.), ITS1 from Collaborative (Bedford, Mass.). An ELISA(enzyme-linked immunosorbent assay) test kit for the quan-titative analysis of PGE2 was purchased from Cayman (Ann
1189NITRIC OXIDE INDUCES PGHS-2

Arbor, Mich.). NOS II inhibitors S-methylisothiourea sulfate(SMT), and L-N6-(1-iminoethyl) lysine (L-NIL) and NOdonors (E)-methyl-2-[(E)-hydroxyimino]-5-nitro-6-methoxy-3-hexeneamide (NOR-1) and S-nitroso-N-acetylpenicillamine(SNAP) were purchased from Calbiochem (San Diego,Calif.). Bovine hemoglobin was purchased from Sigma andthen prepared by dissolving in H2O and reacting with 1 molexcess of sodium dithionite, followed by purification withgel-chromatography (Sephadex G 25, Pharmacia, Piscataway,N.J.). All other chemicals and reagents were purchased fromSigma unless indicated otherwise.
Cell culture
Experiments were carried out using the conditionally immor-talized murine colonic epithelial cell YAMC (young adultmouse colon, gift of Dr. Robert Whitehead at Ludwig Insti-tute for Cancer Research, Melbourne, Australia). Both YAMCand IMCE cells express the heat-labile SV40 large T antigenwith an IFN-g-inducible promoter (28, 29, 31). The temper-ature-sensitive SV40 large T antigen with IFN-g-induciblepromoter is active only at 33°C. It becomes inactivated andnonfunctional once cells are transferred to 39°C. All cellswere grown on 75 cm2 culture flask coated with type Icollagen (5 mg/cm2) in RPMI 1640 media supplemented with5% neonatal calf serum, ITS1 (insulin 6.25 mg/ml, trans-ferrin 6.25 mg/ml, selenious acid 6.25 ng/ml, linoleic acid5.35 mg/ml, and bovine serum albumin 1.25 mg/ml), 5IU/ml of murine IFN-g, 100,000 IU/l penicillin and 100 mg/lstreptomycin. They were cultured under transforming (per-missive) conditions in a 33°C incubator with 5% CO2 plus allthe aforementioned supplements in the media. All cells werethen transferred on attaining confluency into a 39°C incuba-tor under nontransforming (nonpermissive) conditions inserum-free and IFN-g-free media for 72 h before each exper-iment. For NOS II and PGHS-2 induction by inflammatorystimuli, cells in the treatment groups were stimulated byincubation in media containing LPS (1 mg/ml) and IFN-g(100 IU/ml) for the desired period of time as indicated ineach experiment.
Nitrite assay
Culture media from treated cells were incubated for 60 min at25°C in the presence of 3.5 mU/ml of nitrate reductase and0.18 nM NADPH to reduce nitrate to nitrite. Nitrite levelswere then measured in culture media by adding equalvolumes of Griess reagent (1% sulfanilamide, 0.1% N-(1-naphthyl)-ethylenediamine dihydrochloride, 2.5% phospho-ric acid) and absorbance was read at 550 nm with a micro-plate reader (Molecular, Sunnyvale, Calif.). Sodium nitritewas used as the standard.
Analysis of PGE2
An ELISA test kit (Cayman) for the quantitative analysis ofPGE2 was used that operates on the basis of competitionbetween the enzyme conjugate and PGE2 in the sample for alimited number of binding sites on the monoclonal antibody-coated plate. The sample (culture media from treated cells)or standard solution was first added to the microplate. Next,the diluted enzyme conjugate was added, and the mixture wasshaken and incubated at room temperature for 1 h. The platewas then washed, removing all unbound material. The boundenzyme conjugate was detected by the addition of K-BlueSubstrate, which generated color intensity after 30 min.Quantitative test results were obtained by measuring andcomparing the absorbance reading of the sample wells
against the standards with a microplate reader at 650 nm. Theextent of color development was inversely proportional to theamount of PGE2 in the sample or standard.
Northern blotting
As described previously (30), total RNA was prepared fromYAMC cells by the Trizol method (Life Technologies-BRL).We electrophoresed 10 mg RNA on a 1% agarose/6.66%formaldehyde gel at 25V for 18 h with 13 MOPS buffercontaining 0.2 M MOPS (pH 7.0), 0.08 M sodium acetate, and0.01 M ethylenediaminetetraacetate (EDTA) (pH 8.0). TheRNA was transferred onto Nytran (Schleicher & Schuell,Keene, N.H.) membrane in 203 SSC overnight. The murinecDNA of PGHS-2 (1.9 kb) probe was radiolabeled by randompriming using Pharmacia Labeling Beads (dCTP) (Pharma-cia). The probe was then hybridized to the RNA blot using2 3 106 cpm/ml in ExpressHyb (Clontech, Palo Alto, Calif.)hybridization solution at 65°C for 1 h. Blots were washed at astringency of 50°C in 0.23 SSC/0.1% sodium dodecyl sulfate(SDS).
Western blotting
Cells were washed twice with cold phosphate-buffered saline(PBS) and harvested under either denaturing conditions byscraping in boiling 23 Laemmli sample buffer (Bio-Rad,Hercules, Calif.) or nondenaturing conditions by using aRIPA Buffer Set (Boehringer Mannheim). For total celllysates under denaturing conditions, samples were heated atboiling temperature for an additional 5 min. Homogenateswere then prepared in the Laemmli sample buffer by sonica-tion (1 min each). After centrifugation at 2000 g for 5 min,the supernatants were used as the protein source. To makeprotein preparations that contain only soluble cytosolic frac-tions, cells were lysed in RIPA buffer by a micro-homogenizer(Sonifier, Branson, Danbury, Conn.) under nondenaturingconditions at 4°C. The homogenized soluble supernatant wasthen prepared by centrifugation at 100,000 g for 30 min at4°C. The protein concentration was determined by the BCAmethod (Pierce, Rockford, Ill.). Electrophoresis samples wereprepared by mixing the respective protein preparations with23 Laemmli sample buffer. To each well of SDS-polyacryl-amide gel, 15–30 ml of cell lysate (;15–30 mg total protein)was applied and electrophoresed in 0.75 mm thick 7.5%Tris-glycine gel (Bio-Rad). Transfer to nitrocellulose mem-brane was done using a semi-dry blotter (Bio-Rad) at 15V for30 min. Blots were then probed with the respective primaryantibodies at the manufacturers’ suggested dilution, followedby a secondary anti-mouse IgG antibody conjugated to horse-radish peroxidase (1:2000). Detection was done using an ECLkit (Amersham, Arlington Heights, Ill.). Blots were routinelystripped by Encore Blot Stripping Kit (Novus, San Diego,Calif.) and reprobed with anti-actin monoclonal antibody toserve as controls.
Preparation of nuclear extracts and electrophoretic mobilityshift assays
Nuclear extracts were prepared from YAMC cells according tothe method by Dignam et al. with modifications (30, 32).Cells were rinsed once with cold PBS, followed by trypsiniza-tion. After centrifugation at 1000 g for 5 min, they wereresuspended in 5 pellet volumes of hypotonic buffer contain-ing 0.2 mM PMSF and 0.5 mM DTT. They were then chilledon ice for 10 min, followed by lysis with a PT 3000 Polytron(Brinkmann, Littau, Switzerland) for 30 s and centrifuged at4000 g for 15 min. The pellet was resuspended in 0.5 pellet
1190 Vol. 14 June 2000 MEI ET AL.The FASEB Journal

volumes of low-salt buffer. An equal volume of high-salt bufferwas added dropwise to the gently stirred suspension. Thenuclear extracts were subjected to centrifugation at 16,000 gfor 30 min, followed by dialysis overnight. We added 5 mgnuclear protein to a 20 ml reaction mix containing 300 ngpoly dI-dC; binding buffer (10 mM Hepes, pH 7.6; 60 mMKCl; 1 mM EDTA; 1 mM DTT; 12% glycerol); with or withoutdouble-stranded mouse LEF-1 oligonucleotide (Life Technol-ogies-BRL), CACCCTTTGAAGCTC with 59 overhang, as aspecific competitor. Samples were incubated on ice for 10min. Then, LEF-1 oligonucleotide, radio-labeled using T4kinase (Life Technologies-BRL) and g32P ATP (NEN, Boston,Mass.), was added at 1.5–2 3 104 cpm per reaction andincubated at RT for 30 min. DNA loading dye (QualityBiological, Gaithersburg, Md.) was added to stop the reac-tion. Samples were run on a 4% polyacrylamide (37.5:1)(Protogel, National Diagnostics, Atlanta, Ga.) gel at 189V for2.5 h in 0.53 TBE running buffer. Gels were dried andexposed to XAR-5 film (Kodak). For super-shift studies, 3–5mg nuclear lysate was mixed in a 20 ml reaction mixture asdescribed for electrophoretic mobility shift assays (EMSA)and incubated on ice for 10 min. Antibodies, 12 mg polyclonalanti-b-catenin and 500 ng monoclonal anti-E-cadherin, orpreimmune IgG, were then added to the respective reactiontubes. Reactions were incubated on ice for 15 min. 32P-labeled murine LEF-1 oligonucleotide probe was added at1.5–2 3 104 cpm per reaction and incubated at RT for 30 min.
Immunocytochemical staining
Cells grown on Lab-Tec chambered coverslips (Nunc, Naper-ville, Ill.) were fixed in 95% Ethanol at room temperature for10 min and air-dried. Immunocytochemical staining wasperformed following the protocol from a Vectastain ABC kit(Vector, Burlingame, Calif.). Coverslips were rinsed in TBSfor 10 min before being blocked in 2% nonimmune mouseserum for 20 min. Negative control coverslips were kept inPBS without exposure to the primary antibody, monoclonalmouse anti-b-catenin antibody (Transduction), whereas theremaining coverslips were incubated for 30 min with theprimary antibody at 1:50 dilution. The coverslips were thenincubated with 0.5% mouse biotinylated secondary antibodyfor 30 min and stained with biotin-avidin reagents (Vec-torstain Elite ABC kit, Vector) for 30 min, followed by3,39-diaminobenzidine tetrahydrochloride staining for 5 min.The coverslips were then counterstained with Gill’s hematox-ylin for 6 s.
RESULTS
Expression of PGHS-2 and NOS II induced byinflammatory stimuli IFN-g and LPS
The expression of PGHS-2 and NOS II was assessedby Western blot in YAMC cells in response to IFN-g(100 IU/ml) and LPS (1 mg/ml), the classic inflam-matory stimuli for PGHS-2 and NOS II induction(Fig. 1A). Actin served as a control. We found thatnot only PGHS-2 was induced, as previously reported(30), but also NOS II was induced by the samestimulation. To ascertain the functionality of theseproteins, their enzymatic products PGE2 and nitrite(the stable derivative of NO) were measured (Fig.1B). Furthermore, the generation of PGE2 and NO
Figure 1. Expression of PGHS-2 and NOS II as well as produc-tion of PGE2 and nitrite in YAMC cells. YAMC were cultured innonpermissive conditions for 72 h before each experimentthroughout this study. The nonpermissive conditions are de-fined as incubation at 39°C in the absence of serum, ITS1, andIFN-g. A) Expression of PGHS-2 and NOS II induced by IFN-gand LPS. YAMC cells were stimulated with IFN-g (100 IU/ml)and LPS (1 mg/ml) for 24 h. PGHS-2 and NOS II proteins wereanalyzed by Western blot with respective monoclonal antibodies.The same blot was then stripped and probed for actin as acontrol. This is a representative blot from at least five experi-ments. B) The generation of PGE2 and nitrite in response toIFN-g (100 IU) and LPS (1 mg/ml). The culture media fromcells treated with IFN-g and LPS were collected for PGE2 andnitrite measurement. Specific inhibitors of PGHS-2 and NOS II,sulindac sulfide (5 mM) and L-NIL (10 mM) or SMT (10 mM),respectively, were used to co-treat the cells and to block theproduction of PGE2 and nitrite. Data are the mean 6 sd oftriplicate determinations and were analyzed by one-way ANOVAwith the Duncan test. *P,0.01, compared basal levels with theinduced production of PGE2 or nitrite. **P,0.01, comparedthe induced production of PGE2 or nitrite in the absence andpresence of respective inhibitors.
1191NITRIC OXIDE INDUCES PGHS-2

was inhibited by the respective inhibitors of PGHS-2(sulindac sulfide) and NOS II (L-NIL and SMT),demonstrating the specific functional activity of theinduced PGHS-2 and NOS II.
Attenuation of PGHS-2 expression and PGE2generation by selective NOS II inhibitors
The close interconnection between NO and theenzymatic activity of PGHS-2 has been reported inthe literature (24–27). NO has been found to stim-ulate the generation of PGE2 through the activationof PGHS-2 activity. To examine this effect of endog-enously produced NO in YAMC, we tested two selec-tive NOS II inhibitors, L-NIL and SMT, on theexpression of PGHS-2 and the generation of PGE2induced by IFN-g and LPS. We found that theinducible expression of PGHS-2 was greatly reducedby both L-NIL and SMT (Fig. 2A, B). As expected,L-NIL and SMT also significantly decreased PGE2generation by blocking endogenous NO production(Fig. 2C). This experiment not only confirmed thatNO increases the enzymatic activity of PGHS-2 butalso implied that NO may play a role in regulatingthe expression of PGHS-2.
Induction of PGHS-2 by NO-releasing compoundsNOR-1 and SNAP
The above findings suggest that NO may exert aninduction effect on PGHS-2 overexpression in re-sponse to the stimulation of IFN-g and LPS. In anattempt to determine whether exogenous NO couldgenerate the same effect, NO donors, NOR-1 andSNAP, were used at several different concentrationsto treat these cells. The expression of PGHS-2 wasthen examined by northern and western blotting(Figs. 3, 4). In addition, the enzymatic activity ofPGHS-2 was studied by measuring PGE2 production(Fig. 5). As demonstrated in these experiments, bothNOR-1 and SNAP induced PGHS-2 mRNA expres-sion in YAMC cells in a time-dependent manner (Fig.3). NOR-1 and SNAP also caused a dose-dependentinduction of PGHS-2 protein (Fig. 4). These effectswere specifically caused by NO released spontane-ously from these two chemically distinct compoundsas they were abolished in the presence of hemoglo-bin, a potent NO scavenger (Figs. 4, 5). Theseexperiments demonstrated that NO, either endog-enously produced or exogenously supplied, cangreatly enhance the expression of PGHS-2 in colonicepithelial cells.
Formation of DNA/b-catenin/LEF-1 complexinduced by NO-releasing compounds NOR-1 andSNAP
It has been suggested that the promoter region ofthe PGHS-2 gene contains Tcf-LEF binding sites.
Therefore, PGHS-2 has been proposed to be underthe control of the b-catenin/Tcf-LEF complex,although definitive proof is still lacking (19). Wefound that the mouse PGHS-2 gene promoter doesindeed contain potential binding sites of the con-sensus sequence for Tcf-LEF (see Discussion). Weset out to explore the possibility that the expres-sion of PGHS-2 by NO donors in these colonicepithelial cells may be associated with, directly orindirectly, the b-catenin/Tcf-LEF DNA complex
Figure 2. Inhibition of PGHS-2 expression and PGE2 gener-ation by L-NIL and SMT. YAMC cells were stimulated withIFN-g (100 IU/ml) and LPS (1 mg/ml) for 24 h in theabsence or presence of specific NOS II inhibitors, L-NIL (10mM) and SMT (10 mM). Culture media were collected forPGE2 analysis while total cell lysates were prepared forWestern blot. Attenuation of PGHS-2 expression by L-NIL (A)and SMT (B). Total cell lysates were analyzed by Western blotusing the monoclonal anti-PGHS-2 antibody. The same blotwas then stripped and reprobed for actin as a control. This isa representative blot from at least three experiments. C)Inhibition of PGE2 generation by L-NIL and SMT. Data arethe mean 6 sd of triplicate determinations and were analyzedby one-way ANOVA with the Duncan test. *P,0.01 comparedbasal levels with the induced production of PGE2. **P,0.01,compared the induced production of PGE2 in the absenceand presence of respective inhibitors.
1192 Vol. 14 June 2000 MEI ET AL.The FASEB Journal

formation. Unlike cell lines derived from colontumors that often express constitutively high levelsof b-catenin/Tcf-LEF activity, the nontransformedand nontumorigenic YAMC cells express very littleb-catenin/LEF-1 complex when cultured undernontransforming conditions (see Materials andMethods). As shown in Fig. 6, the formation of theb-catenin/LEF-1 complex was greatly enhanced bytwo NO donors, NOR-1 and SNAP. Furthermore,the amount of b-catenin/LEF-1 complex inducedby both NOR-1 (1, 5, and 10 mM) and SNAP (5, 10,and 20 mM) appeared to be concentration-depen-dent.
Time-dependence of DNA/b-catenin/LEF-1complex formation induced by NO-releasingcompounds NOR-1 and SNAP
To further confirm that NO released by NOR-1 andSNAP specifically caused DNA/b-catenin/LEF-1complex formation in YAMC cells, a time coursestudy was carried out. Cells were treated with eitherNOR-1 (5 mM) or SNAP (10 mM) for 10, 30, 60, and120 min (Figs. 7A, B). The formation of b-catenin/LEF-1 DNA complex induced by NOR-1 and SNAPoccurred in a time-dependent fashion.
Super-shifting by anti-b-catenin antibody of theDNA/b-catenin/LEF-1 complexes induced byNOR-1 or SNAP
The suggested participation of b-catenin in the DNAbinding complex was confirmed by a series of super-
shift assays using an anti-b-catenin antibody (Figs.8A, B). Cells were first treated with either NOR-1 (5mM) or SNAP (10 mM) for 30 min before nuclearextracts were harvested. Nonspecific antibodies,namely, rabbit preimmune IgG and anti-E-cadherinantibody, were used as controls in the super-shiftassays and did not cause any super-shifting of thebands. This study directly demonstrates the presenceof b-catenin in the DNA binding complex in con-junction with LEF-1.
Increase of free b-catenin in the soluble cytosolicfraction in response to NOR-1 or SNAP treatment
The localization of b-catenin is normally associatedwith the transmembrane glycoprotein E-cadherin.The resting b-catenin level remains very low becauseof the constant degradation by APC and GSK-3b inconjunction with Axin (34, 35). We attempted toexplore the source of the increased nuclear b-cate-
Figure 3. Induction of PGHS-2 mRNA by NO donors, NOR-1and SNAP. A) YAMC cells were treated with either NOR-1 (5mM) or SNAP (10 mM) for various periods of time (0, 0.5, 2,and 5 h). Total RNA was extracted and analyzed by Northernblot. PGHS-2 (4.3 kb) was probed and found to be induced ina time-dependent fashion. B) The same blot with ethidiumbromide stained total RNA was used for loading control. Thisis a representative blot from three experiments.
Figure 4. Expression of PGHS-2 protein induced by NOdonors, NOR-1 and SNAP. YAMC cells were treated witheither NOR-1 or SNAP for 24 h at various concentrationsbefore cells were harvested. The total cell lysates were thenprepared and analyzed by Western blot. A) NOR-1 inducedexpression of PGHS-2 in a dose-dependent fashion. Theexpression of PGHS-2 protein was analyzed by Western blot inresponse to NOR-1 treatment at concentrations of 0, 1, 5, and10 mM. As a positive control, LPS plus IFN-g (L1I) was usedto induce PGHS-2 expression. Hemoglobin (Hb) (1 mM), anavid scavenger of NO, was used to block the NOR-1 (5 mM)effect. The same blot was then stripped and probed for actinas a control. This is a representative blot from three experi-ments. B) SNAP induced expression of PGHS-2 in a dose-dependent fashion. The expression of PGHS-2 protein wasanalyzed by Western blot in response to SNAP treatment atconcentrations of 0, 5, 10, and 20 mM. As a positive control,LPS plus IFN-g (L1I) was used to induce PGHS-2 expression.Hemoglobin (Hb) (1 mM) was used to block SNAP (10 mM)effect. The same blot was then stripped and probed for actinas a control. This is a representative blot from three experi-ments.
1193NITRIC OXIDE INDUCES PGHS-2

nin present in the DNA binding complex. Theamount of total b-catenin in whole cell lysate wasexamined and did not change after various treat-ments with NO donors, NOR-1 (5 mM) and SNAP(10 mM) (Fig. 9A). In contrast, the amount of freeb-catenin in the soluble cytosolic fraction showed adramatic increase from nondetectable in control
cells to easily appreciated levels in cells treated withNO donors for 2 h (Fig. 9B). Although this findingdoes not reveal the mechanism responsible for theincrease in the free cytoplasmic pool of b-catenin, itimplies that NO donors may facilitate the nuclearb-catenin/LEF-1 complex formation by increasingthe amount of free b-catenin available for its nucleartranslocation.
Subcellular localization and redistribution of b-catenin
b-Catenin was primarily localized in the membranein YAMC cells after culturing under nontransform-
Figure 5. Generation of PGE2 in response to NO donors,NOR-1 or SNAP. YAMC cells were stimulated as in Fig. 4 witheither NOR-1 (5 mM) or SNAP (10 mM) for 24 h. The culturemedia were collected and analyzed for PGE2 generation.Hemoglobin (Hb) (1 mM) was used to block the effect ofeither NOR-1 or SNAP. Data are the mean 6 sd of triplicatedeterminations and were analyzed by one-way ANOVA withthe Duncan test. *P,0.01 compared basal levels with theinduced production of PGE2 by either NOR-1 or SNAP.**P,0.01 compared the induced production of PGE2 by NOdonors with or without concurrent treatment of hemoglobin.
Figure 6. Analysis by electrophoretic mobility shift assay(EMSA) of the formation of DNA/b-catenin/LEF-1 transcrip-tion complex by NO donors, NOR-1 or SNAP. The formationof DNA/b-catenin/LEF-1 complex was greatly enhanced by a2 h treatment with either NOR-1 (0, 1, 5, 10 mM) or SNAP (0,5, 10, 20 mM) in a concentration-dependent manner. This isa representative blot from at least five different experiments.
Figure 7. Time course of DNA/b-catenin/LEF-1 complexformation induced by NOR-1 or SNAP. YAMC cells weretreated with either (A) NOR-1 (5 mM) or (B) SNAP (10 mM)for various periods of time (0, 10, 30, 60, 120 min). DNA/b-catenin/LEF-1 complex formation was induced by eitherNOR-1 or SNAP in a time-dependent fashion. UnlabeledLEF-1 probe was used as a competitor and effectively com-peted away the DNA/b-catenin/LEF-1 complex. This is arepresentative blot from at least three different experiments.
1194 Vol. 14 June 2000 MEI ET AL.The FASEB Journal

ing condition for 48 h, presumably bound to thetransmembrane protein E-cadherin (Fig. 10A).There was very little, if any, staining of b-catenin inthe nucleus and cytoplasm. However, pronouncednuclear staining of b-catenin was detected when cellswere exposed to NOR-1 (5 mM) overnight (16 h)
(Fig. 10B), indicating a redistribution and nucleartranslocation of subcellular b-catenin in response toNO treatment. Corroborating the findings demon-strated in Fig. 9A, B, immunocytochemical studiesshowed the increase of b-catenin in cytoplasm (Fig.10B). Similar findings were observed in the redistri-bution of b-catenin with SNAP treatment (data notshown).
Differential formation of b-catenin/LEF-1 DNAbinding complex and induction of PGHS-2 mRNAand protein as well as production of PGE2 inYAMC and IMCE cells contrasting in Apc genotype
Although the findings with NO treatment suggesteda link between PGHS-2 expression and the formation
Figure 8. Super-shifting by anti-b-catenin antibody of theDNA/b-Catenin/LEF-1 complexes induced by NOR-1 orSNAP. The suggested participation of b-catenin in the DNAbinding complex was confirmed by super-shift assays using apolyclonal anti-b-catenin antibody. Cells were first treatedwith either NOR-1 (5 mM) (A) or SNAP (10 mM) (B) for 30min before nuclear extracts were collected and analyzed byEMSA. The anti-b-catenin antibody caused a concentration-dependent increase of the amount of super-shift with thefollowing dilutions from stock: 1:12.5, 1:25, and 1:50. Non-specific antibodies, namely rabbit pre-immune IgG and anti-E-cadherin antibody, were used as controls in the super-shiftassays and did not cause any super-shifting of the complex.This is a representative blot from at least four differentexperiments.
Figure 9. Analysis by Western blot of the redistribution ofb-catenin into a soluble cell fraction in response to NOdonors, NOR-1 or SNAP. YAMC cells were stimulated witheither NOR-1 (5 mM) or SNAP (10 mM) for 2 h. Cells werethen harvested and fractionated into either total cell lysate orsoluble fraction preparation. b-catenin was analyzed by West-ern blot. A) b-catenin was detected by Western blot in thetotal cell lysate and remained constant. We applied 30 mg oftotal protein into each lane of the pre-cast Bio-Rad 7.5%Tris-HCl gels. b-catenin was detected by an anti-b-cateninmonoclonal antibody. The same blot was then stripped andreprobed with actin as a control. This is a representative blotfrom at least three different experiments. B) b-catenin wasonly detected in the soluble fraction from cells treated for 2 hwith either NOR-1 (5 mM) or SNAP (10 mM), but not thatfrom the untreated cells. The same blot was then stripped andreprobed for actin as a control. This is a representative blotfrom at least three different experiments.
1195NITRIC OXIDE INDUCES PGHS-2

of b-catenin/LEF-1 DNA binding complex, the ef-fects of NO on PGHS-2 may be mediated by distinctNO-activated pathways. To implicate the participa-tion of b-catenin in the NO effect on PGHS-2, wecompared YAMC and IMCE cells differing in b-cate-nin degradation because of their genetic differencein Apc. We found that IMCE cells expressed mark-edly higher b-catenin/LEF-1 DNA binding complexthan YAMC cells (Fig. 11A). The difference in b-cate-nin/LEF-1 DNA complex between these two celllines and the corresponding differential expressionof PGHS-2 mRNA and protein were both amplifiedafter treatment with SNAP (Fig. 11B, C). Further-more, PGE2 production in IMCE was significantlyhigher than that in YAMC cells (Fig. 11D). Althoughthis is not definitive proof that b-catenin/LEF DNAcomplex is directly responsible for the expression of
PGHS-2, it strongly indicates that they are associatedeither directly or indirectly.
DISCUSSION
The conditionally immortal murine colonic epithe-lial cells have been used to elucidate the role of NOin the overexpression of PGHS-2. These nontrans-formed and nontumorigenic cells provide an excel-lent model system in which to study the early eventsin colorectal carcinogenesis, such as the inducibleexpression of PGHS-2 and NOS II. They have anaverage life span of ;10 days under nontransform-ing conditions at 39°C when the temperature-sensi-tive SV40 large T antigen becomes inactive andnonfunctional (28–30). Using the classical stimulifor NOS II and PGHS-2, IFN-g and LPS, we usedthese cells to investigate the association between NOgeneration and PGHS-2 expression as well as thepossible roles of soluble free b-catenin accumulationand b-catenin/Tcf-LEF complex formation. It is im-portant to note that the effect of IFN-g and LPSduring the treatment is unrelated to the IFN-ginducible promoter activity because the tempera-ture-sensitive mutant SV40 large T antigen is inacti-vated and remains nonfunctional at 39°C during theexperiment (data not shown) (28–30). The linkbetween PGHS-2 and NO was suggested by thefinding that selective NOS II inhibitors, L-NIL andSMT, greatly reduced the overproduction of PGE2induced by IFN-g and LPS under the above-de-scribed conditions (Fig. 2C). It is unlikely that thesetwo chemically distinct selective NOS inhibitors non-specifically inhibited the activity of PGHS-2. Instead,it was likely that perturbations of NO generationmodulated PGHS-2 catalytic activity as previouslyreported (24–27). However, to our surprise, wefound that both L-NIL and SMT decreased theexpression of PGHS-2 induced by IFN-g and LPS(Fig. 2A, B). To confirm this apparent connectionbetween endogenous NO and PGHS-2 expression,we used two NO-releasing compounds, NOR-1 andSNAP, which do not share any structural similarity;both released NO spontaneously when dissolved inaqueous solution. We found that not only did NOR-1and SNAP increase PGE2 production (Fig. 5), theyalso induced the expression of PGHS-2 at bothmRNA and protein levels in YAMC cells (Figs. 3, 4).When these cells were co-treated with hemoglobin,an avid scavenger of NO, the induction of PGHS-2and generation of PGE2 by NO was abolished,indicating the specificity of NO-induced PGHS-2expression (Figs. 4, 5). Furthermore, the inductionof PGHS-2 by either NOR-1 or SNAP appeared bothtime and concentration dependent (Figs. 3, 4). Toexplore the possible role of the b-catenin/Tcf-LEF
Figure 10. Demonstration by immunohistochemical stainingof the relocalization of b-catenin from the membrane to thecytosol and nucleus. A) b-catenin was primarily localized inthe membranes of the cell-to-cell borders in YAMC cells thatwere cultured in nontransforming conditions for 72 h, As acontrol, no positive staining was observed in sections whenthe primary anti-b-catenin antibody was not applied (data notshown). B) Relocalization of b-catenin in YAMC cells. Cyto-solic and nuclear staining for b-catenin was prominentlyobserved after treatment with NOR-1 (5 mM) for 16 h. 3400.
1196 Vol. 14 June 2000 MEI ET AL.The FASEB Journal
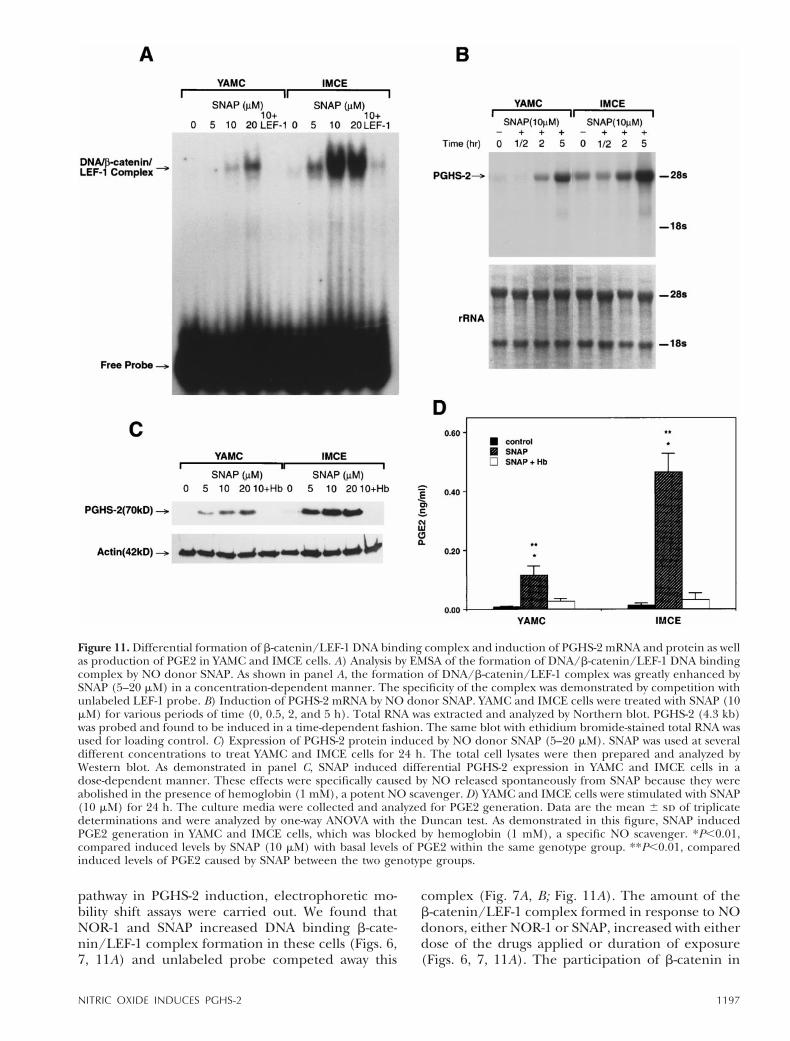
pathway in PGHS-2 induction, electrophoretic mo-bility shift assays were carried out. We found thatNOR-1 and SNAP increased DNA binding b-cate-nin/LEF-1 complex formation in these cells (Figs. 6,7, 11A) and unlabeled probe competed away this
complex (Fig. 7A, B; Fig. 11A). The amount of theb-catenin/LEF-1 complex formed in response to NOdonors, either NOR-1 or SNAP, increased with eitherdose of the drugs applied or duration of exposure(Figs. 6, 7, 11A). The participation of b-catenin in
Figure 11. Differential formation of b-catenin/LEF-1 DNA binding complex and induction of PGHS-2 mRNA and protein as wellas production of PGE2 in YAMC and IMCE cells. A) Analysis by EMSA of the formation of DNA/b-catenin/LEF-1 DNA bindingcomplex by NO donor SNAP. As shown in panel A, the formation of DNA/b-catenin/LEF-1 complex was greatly enhanced bySNAP (5–20 mM) in a concentration-dependent manner. The specificity of the complex was demonstrated by competition withunlabeled LEF-1 probe. B) Induction of PGHS-2 mRNA by NO donor SNAP. YAMC and IMCE cells were treated with SNAP (10mM) for various periods of time (0, 0.5, 2, and 5 h). Total RNA was extracted and analyzed by Northern blot. PGHS-2 (4.3 kb)was probed and found to be induced in a time-dependent fashion. The same blot with ethidium bromide-stained total RNA wasused for loading control. C) Expression of PGHS-2 protein induced by NO donor SNAP (5–20 mM). SNAP was used at severaldifferent concentrations to treat YAMC and IMCE cells for 24 h. The total cell lysates were then prepared and analyzed byWestern blot. As demonstrated in panel C, SNAP induced differential PGHS-2 expression in YAMC and IMCE cells in adose-dependent manner. These effects were specifically caused by NO released spontaneously from SNAP because they wereabolished in the presence of hemoglobin (1 mM), a potent NO scavenger. D) YAMC and IMCE cells were stimulated with SNAP(10 mM) for 24 h. The culture media were collected and analyzed for PGE2 generation. Data are the mean 6 sd of triplicatedeterminations and were analyzed by one-way ANOVA with the Duncan test. As demonstrated in this figure, SNAP inducedPGE2 generation in YAMC and IMCE cells, which was blocked by hemoglobin (1 mM), a specific NO scavenger. *P,0.01,compared induced levels by SNAP (10 mM) with basal levels of PGE2 within the same genotype group. **P,0.01, comparedinduced levels of PGE2 caused by SNAP between the two genotype groups.
1197NITRIC OXIDE INDUCES PGHS-2

the DNA binding complex was confirmed by super-shifting with an anti-b-catenin antibody, but not byeither preimmune IgG or the anti-E-cadherin anti-body (Fig. 8). Further studies indicate that NO maypromote b-catenin/LEF-1 DNA binding complexformation by increasing the level of free solubleb-catenin but not the total amount of whole cellb-catenin (Fig. 9). This finding is corroborated byimmunocytochemistry studies showing the redistri-bution of b-catenin into the nucleus and cytoplasm(Fig. 10).
Our demonstration that NO is involved in theaccumulation of b-catenin and activation of theb-catenin/LEF-1 DNA binding in nontransformedmouse colonic epithelial cells may help elucidate theearly molecular events during colorectal carcinogen-esis. Several investigators have emphasized the im-portance of Apc mutations and their effects onb-catenin in the early phase of colorectal tumordevelopment (13–18, 35–37). From transfectionstudies in cultured cells and analysis of normal andneoplastic human colorectal tissues, a model hasemerged (19, 20, 34, 35). APC, along with interact-ing proteins GSK-3b and Axin, actively degradesb-catenin and maintains cytoplasmic b-catenin atvery low levels. Direct or indirect disruption of thedegradation process resulting in accumulation ofcytoplasmic b-catenin leads to the formation of atranscriptionally active b-catenin/Tcf-LEF complex(38). These activities have been associated with thetransformation of normal colonic epithelium to ad-enomas and adenocarcinomas (40–43). Althoughthe genes that are controlled by the b-catenin/Tcf-LEF pathway have not been fully elucidated, the roleof b-catenin in the early stage of colon cancerdevelopment is now well demonstrated (35–38, 43–45). Thus, our demonstration that NO may activatethe b-catenin/LEF-1 complex may be of consider-able importance.
Although the free b-catenin is being kept at a verylow level in the cytoplasm and nucleus as a result ofAPC-mediated degradation, an abundant amount ofb-catenin is normally associated with the transmem-brane protein E-cadherin. The E-cadherin–boundb-catenin is also called the ‘insoluble’ b-cateninbecause it is present in the cytoplasm and nucleus atvery low or undetectable levels (Figs. 9, 10). It hasbeen reported, however, that the dissociation ofb-catenin from E-cadherin can increase free solubleb-catenin in the cytoplasm. As a matter of fact, theassociation of b-catenin and E-cadherin can be af-fected by a variety of signals directly or indirectly(46–48). Our data, as demonstrated in Figs. 9 and10, suggest that NO may be able to facilitate suchdissociation and increase free b-catenin in the cyto-plasm and nucleus. The morphological changes ofNOR-1-treated cells (Fig. 10), namely, the enlarge-
ment as well as swelling and blebbing of the cells,could be because of the dissociation of the b-cateninand E-cadherin complex at the membrane and thesubsequent disruption of the adherens junctions atthe cell–cell border (49–50). But, it remains un-known how NO works to dissociate b-catenin fromthe membrane-bound cadherins.
It is clearly demonstrated that NO is involved inthe release and/or accumulation of b-catenin intothe cytosol and the formation of b-catenin/LEF-1DNA binding complex in the nucleus. But, theassociation of b-catenin either directly or indirectlyto the expression of PGHS-2 required additionalevidence. The use of an oncogenic b-catenin con-struct in transfection studies was attractive, but wewere unsuccessful in transfecting these nontrans-formed, nonmalignant colonic epithelial cells withthe constructs we tried. Therefore, we took advan-tage of cells differing in Apc genotype. IMCE cells,with their truncated, nonfunctional APC, are defec-tive in their degradation of b-catenin. Thus, onewould expect higher levels of b-catenin under rest-ing and stimulated conditions. Our finding thatIMCE expressed markedly higher b-catenin/LEF-1DNA binding complex formation than YAMC is amanifestation of the genotypic difference betweenthese two cell lines (Fig. 11A). The amplification ofthe difference after treatment with NO is consistentwith the defective degradation machinery for b-cate-nin in IMCE cells. The differential formation ofb-catenin/LEF-1 DNA binding complex correspondsto the differential expression of PGHS-2 mRNA andprotein as well as the generation of PGE2 (Fig.11B–D). Although NO may also cause PGHS-2 ex-pression and b-catenin/LEF-1 DNA complex forma-tion by other independent pathways, our data sug-gests that these two events are functionallyconnected.
Both PGHS-2 and NOS II genes share a commonregulatory pathway in their transcriptional expres-sion; both are under the regulatory control of NFkBmediated response, either through inflammatorystimulation or hypoxia (51–53). Recently, it has beensuggested that the up-regulation of PGHS-2 in colonpolyp and adenoma as well as adenocarcinoma maybe a result of activation of the b-catenin/Tcf-LEFpathway (19). Although the evidence is still incon-clusive regarding the precise role of b-catenin/Tcf-LEF, it clearly indicates that Apc through the Wnt-signaling pathway may be involved in the expressionof PGHS-2 (20). We found that the promoter regionof murine PGHS-2 gene (Accession Number s82456,GenBank) does contain several potential bindingsites for LEF-1 with close homology (seven out ofnine bases as compared with the murine LEF-1consensus sequence CCTTTGAAG). Two of thesetentative LEF-1 binding sites are identical to the
1198 Vol. 14 June 2000 MEI ET AL.The FASEB Journal

consensus sequence within the seven core bases atthe center (positions 21963 to 21955: ACTTT-GAAT; 22974 to 22966: TCTTTGAAT). Althoughfurther studies are needed to confirm that the b-cate-nin/Tcf-LEF heterodimeric complex directly modu-lates transcriptional activation of the PGHS-2 gene,data presented in this paper support that b-catenin/LEF-1 is at least indirectly involved in this process.Furthermore, this study indicates that PGHS-2 ex-pression may be regulated by NO, presumablythrough the Apc/b-catenin pathway, a feature thatmay represent a level of integration in epithelialcells. Although, in circulating immune cells, NFkB isknown as the primary transcriptional regulator forinflammatory response, the overexpression ofPGHS-2 that is often seen in precancerous adenomasas well as malignant carcinomas, may be the result ofboth pathways. This may help explain the findingsreported in animal studies that NOS II inhibitorsare potent chemopreventive agents against cancer(54–57).
The expression of both NOS II and PGHS-2 hasbeen associated with vascular injury, inflammation,and cellular proliferation in pathological colonicconditions (58–62). More specifically, in addition toits widespread detection in various malignancies,NOS II is known to be highly expressed in invasiveinflammatory bowel diseases and precancerous co-lonic adenomas as well as carcinomas (21–23). NOcan cause DNA damage directly or indirectlythrough the formation of carcinogenic compoundsin the body (59, 60). Selective NOS II inhibitors havebeen demonstrated to exert anti-tumor and chemo-preventive effects in various animal models (54–57).NOS II expression is relatively decreased in colonadenocarcinomas compared with adenomas, indicat-ing that NO may contribute to colon cancer progres-sion at the transition of colon adenoma to carcinomain situ (23). This evidence for a temporally distinctexpression pattern in humans shows that the role ofNOS II in colon tumor progression may be stage-specific.
The role of PGHS-2 in the cellular inflammatoryresponse and carcinogenesis also has been estab-lished. Increased PGHS-2 expression is closely corre-lated with the malignancies of the gastrointestinaltract, colonic adenoma and carcinoma in particular(3–5). Aspirin, a nonselective inhibitor of bothPGHS-1 and PGHS-2, has shown a preventive effectagainst polyp formation in FAP patients. Recently,the newly developed selective PGHS-2 inhibitorshave shown potent chemopreventive effects againstcolon tumor growth (7–9). The chemopreventiveactivity of these drugs can be explained, at leastpartially, by the finding that PGHS-2 and its eico-sanoid products have potent anti-apoptotic, mito-genic, and proliferative effects (11, 12, 62).
Our data indicate that one of the consequencesassociated with elevated NOS II expression in mu-rine colon epithelial cells is the NO-associated induc-tion of PGHS-2 and accumulation of PGE2. Evidencepresented in this study supports the notion thatthese events may be causally linked. Furthermore,they are probably associated with, albeit indirectly,the accumulation of free soluble b-catenin in thecytoplasm and the formation of b-catenin/Tcf-LEFDNA binding complex in the nucleus. This is thefirst direct demonstration in nontransformed andnontumorigenic colonic epithelial cells that the in-duction of NOS II activity and subsequent NO gen-eration contributes to PGHS-2 overexpression andPGE2 accumulation in response to inflammatorystimuli. Our study suggests that the expression ofNOS II and the presence of NO under these patho-logical conditions may contribute to the overexpres-sion of PGHS-2 that has been frequently observed incolonic epithelium of precancerous adenomas aswell as adenocarcinomas. Taken together, these find-ings lend biological plausibility to the hypothesis thatNOS II-derived NO may play an important role inthe early stages of colorectal carcinogenesis.
The authors acknowledge the National Cancer Institute forallocation of computing time and staff support at the Freder-ick Biomedical Supercomputing Center of the FrederickCancer Research and Development Center.
REFERENCES
1. Singer, I. I., Kawka, D. W., Schloemann, S., Tessner, T., Riehl,T., and Stenson, W. F. (1998) Cyclooxygenase 2 is induced incolonic epithelial cells in inflammatory bowel disease. Gastroen-terology 115, 297–306
2. Hendel, J., and Nielsen, O. H. (1997) Expression of cyclooxy-genase-2 mRNA in active inflammatory bowel disease. Am. J.Gastroenterol. 92, 1170–1173
3. Kutchera, W., Jones, D. A., Matsunami, N., Groden, J., McIntyre,T. M., Zimmerman, G. A., White, R. L., and Prescott, S. M.(1996) Prostaglandin H synthase 2 is expressed abnormally inhuman colon cancer: evidence for a transcriptional effect. Proc.Natl. Acad. Sci. USA 93, 4816–4820
4. DuBois, R. N., Radhika, A., Reddy, B. S., and Entingh, A. J.(1996) Increased cyclooxygenase-2 levels in carcinogen-inducedrat colonic tumors. Gastroenterology 110, 1259–1262
5. Williams, C. S., Luongo, C., Radhika, A., Zhang, T., Lamps,L. W., Nanney, L. B, Beachamp, R. D., and DuBois, R. N. (1996)Elevated cyclooxygenase-2 levels in Min mouse adenomas. Gas-troenterology 111, 1134–1140
6. Tsujii, M., Kawano, S., and DuBois, R. N. (1997) Cyclooxygen-ase-2 expression in human colon cancer cells increases meta-static potential. Proc. Natl. Acad. Sci. USA 94, 3336–3340
7. Jacoby, R. F., Marshall, D. J., Newton, M. A., Novakovic, K.,Tutsch, K., Cole, C. E., Lubet, R. A., Kelloff, G. J., Verma, A.,Moser, A. R., and Dove, W. F. (1996) Chemoprevention ofspontaneous intestinal adenomas in the Apc Min mouse modelby the nonsteroidal anti-inflammatory drug piroxicam. CancerRes. 56, 710–714
8. Boolbol, S. K., Dannerberg, A. J., Chadburn, A., Martucci, C.,Guo, X., Ramonetti, J. T., Abreu-Goris, M., Newmark, H. L.,Lipkin, M. L., DeCosse, J. J., and Bertagnolli, M. M. (1996)Cyclooxygenase-2 overexpression and tumor formation areblocked by sulindac in a murine model of familial adenomatouspolyposis. Cancer Res. 56, 2556–2560
1199NITRIC OXIDE INDUCES PGHS-2

9. Subbaramaiah, K., Zakim, D., Weksler, B. B., and Dannenberg,A. J. (1997) Inhibition of cyclooxygenase: a novel approach tocancer prevention. Proc. Soc. Exp. Biol. Med. 216, 201–210
10. Oshima, M., Dinchuk, J. E., Kargman, S. L., Oshima, H.,Hancock, B., Kwong, E., Trzaskos, J. M., Evans, J. F., and Taketo,M. M. (1996) Suppression of intestinal polyposis in Apc del-ta716 knockout mice by inhibition of cyclooxygenase-2. Cell 87,803–809
11. Tsujii, M., and DuBois, R. N. (1995) Alterations in cellularadhesion and apoptosis in epithelial cells overexpressing pros-taglandin endoperoxide synthase 2. Cell 83, 493–501
12. Sheng, H., Shao, J., Morrow, J. D., Beauchamp, R. D., andDuBois, R. N. (1998) Bcl-2 expression by prostaglandin E2 inmodulation of apoptosis human colon cancer cells. Cancer Res.58, 362–366
13. Kinzler. K. W., and Vogelstein, B. (1996) Lessons from heredi-tary colorectal cancer. Cell 87, 159–170
14. Groden, J., Thliveris, A., Samowitz, W., Carlson, M., Gelbert, L.,Albertsen, H., Josyln, G., Stevens, J., Spirio, L., Robertson, M.,Sargeant, L., Krapcho, K., Wolff, E., Burt, R., Hughes, J. P.,Warrington, J., McPherson, J. P., Wasmuth, J., Paslier, D. L.,Abderrahim, H., Cohen, D., Leppert, M., and White, R. (1991)Identification and characterization of the familial adenomatouspolyposis coli gene. Cell 66, 589–600
15. Oshima, M., Oshima, H., Kitagawa, K., Kobayashi, M., Itakura,C., and Taketo, M. (1995) Loss of Apc heterozygosity andabnormal tissue building in nascent intestinal polyps in micecarrying a truncated Apc gene. Proc. Natl. Acad. Sci. USA 92,4482–4486
16. Moser, A. R., Luongo, C., Gould, K. A., McNeley, M. K.,Shoemaker, A. R., and Dove, W. F. (1995) ApcMin: a mousemodel for intestinal and mammary tumorigenesis. Eur. J. Cancer31, 1061–1066
17. Munemitsu, S., Albert, I., Souza, B., Rubinfeld, B., and Polakis,P. (1995) Regulation of intracellular beta-catenin levels by theadenomatous polyposis coli (APC) tumor-suppressor protein.Proc. Natl. Acad. Sci. USA 92, 3046–3050
18. Morin, P. J., Sparks, A. B., Krinek, V., Barker, N., Clevers, H.,Vogelstein, B., and Kinzler, K. W. (1997) Activation of beta-cateni-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275, 1787–1790
19. Prescott, S. M., and White, R. L. (1996) Self-promotion? Inti-mate connections between APC and prostaglandin H syn-thase-2. Cell 87, 783–786
20. Howe, L. R., Subbaramaiah, K., Chung, W. J., Dannenberg, A. J.,and Brown, A. M. C. (1999) Transcriptional activation ofcyclooxygenase-2 in Wnt-1-transformed mouse mammary epi-thelial cells. Cancer Res. 59, 1572–1577
21. ter Steege, J., Buurman, W., Arends, J. W., and Forget, P. (1997)Presence of inducible nitric oxide synthase, nitrotyrosine, CD68,and CD14 in the small intestine in celiac disease. Lab. Invest. 77,29–36
22. Singer, I. I., Kawka, D. W., Scott, S., Weidner, J. R., Mumford,R. A., Riehl, T. E., and Stenson, W. F. (1996) Expression ofinducible nitric oxide synthase and nitrotyrosine in colonicepithelium in inflammatory bowel disease. Gastroenterology 111,871–885
23. Ambs, S., Merriam, W. G., Bennett, W. P., Felley-Bosco, E.,Ogunfusika, M. O., Oser, S., Klein, S., Shields, P. G., Billiar,T. R., and Harris, C. C. (1998) Frequent nitric oxide synthase-2expression in human colon adenomas: implication for tumorangiogenesis and colon cancer progression. Cancer Res. 58,334–341
24. Salvemini, D., Misko, T. P., Masferrer, J. L., Seibert, K., Currie,M. G., and Needleman, P. (1993) Nitric oxide activates cycloox-ygenase enzymes. Proc. Natl. Acad. Sci, USA 90, 7240–7244
25. von Knethen, A., and Brune, B. (1997) Cyclooxygenase-2: anessential regulator of NO-mediated apoptosis. FASEB J. 11,887–895
26. Landino, L. M., Crews, B. C., Timmons, M. D., Morrows, J. D.,and Marnett, L. J. (1996) Peroxynitrite, the coupling product ofnitric oxide and superoxide, activates prostaglandin biosynthe-sis. Proc. Natl. Acad. Sci. USA 93, 15069-15074
27. Salvemini, D. (1997) Regulation of cyclooxygenase enzymes bynitric oxide. Cell. Mol. Life Sci. 53, 576–582
28. Whitehead, R. H., and Joseph, J. L. (1994) Derivation ofconditionally immortalized cell lines containing the Min muta-
tion from the normal colonic mucosa and other tissues of an“Immortomouse”/Min hybrid. Epith. Cell Biol. 3, 119–125
29. Whitehead, R. H., VanEeden, P. E., Nobel, M. D., Ataliotis, P.,and Jat, P. S. (1993) Establishment of conditionally immortal-ized epithelial cell lines from both colon and small intestine ofadult H-2Kb-tsA58 transgenic mice. Proc. Natl. Acad. Sci. USA 90,587–591
30. Mei, J. M., Hord, N. G., Winterstein, D. F., Donald, S. P., andPhang, J. M. (1999) Differential expression of prostaglandinendoperoxide H synthase-2 and formation of activated beta-catenin-LEF-1 transcription complex in mouse colonic epithe-lial cells contrasting in Apc. Carcinogenesis 20, 737–740
31. D’Abaco, G. M., Whitehead, R. H., and Burgess, A. W. (1996)Synergy between Apc min and an activated ras mutation issufficient to induce colon carcinomas. Mol. Cell. Biol. 3, 884–891
32. Dignam, J. D., Lebovitz, R. M., and Roeder, R. G. (1983)Accurate transcription initiation by RNA polymerase II in asoluble extract from isolated mammalian nuclei. Nucleic AcidsRes. 11, 1475–1489
33. Sakanaka, C., Weiss, J. B., and Williams, L. T. (1998) Bridging ofbeta-catenin and glycogen synthase kinase-3 beta by axin andinhibition of beta-catenin-mediated transcription. Proc. Natl.Acad. Sci. USA 95, 3020–3023
34. Rubinfeld, B., Souza, B., Albert, I., Muller, O., Chamberlain,S. H., Masiarz, F. R., Munemitsu, S., and Polakis, P. (1993)Association of the Apc gene product with beta-catenin. Science262, 1731–1734
35. Korinek, V., Barker, N., Morin, P. J., Van Wichen, D., De Weger,R., Kinzler, K. W., Vogelstein, B., and Clevers, H. (1997)Constitutive transcriptional activation by beta-catenin-Tcf com-plex in APC2/2 colon carcinoma. Science 275, 1784–1787
36. Kinzler, K. W., and Vogelstein, B. (1997) Cancer-susceptibilitygene: gatekeepers and caretakers. Nature (London) 386, 761–762
37. Rubinfeld, B., Albert, I., Porfiri, E., Munemitsu, S., and Polakis,P. (1997) Loss of beta-catenin regulation by the APC tumorsuppressor protein correlates with loss of structure due tocommon somatic mutations of the gene. Cancer Res. 57, 4624–4630
38. Porfiri, E., Rubinfeld, B., Albert, I., Hovanes, K., Waterman, M.,and Polakis, P. (1997) Induction of a beta-catenin-LEF-1 com-plex by wnt-1 and transforming mutations of beta-catenin.Oncogene 15, 2833–2839
39. Shoemaker, A. R., Luongo, C., Moser, A. R., Marton, L. J., andDove, W. F. (1997) Somatic mutational mechanisms involved inintestinal tumor formation in Min mice. Cancer Res. 57, 1999–2006
40. Shoemaker, A. R., Gould, K. A., Luongo, C., Moser, A. R., andDove, W. F. (1997) Studies of neoplasia in the Min mouse.Biochim. Biophys. Acta 1332, F25–F48
41. Moser, A. R., Pitot, H. C., and Dove, W. F. (1990) A dominantmutation that predisposes to multiple intestinal neoplasia in themouse. Science 247, 322–324
42. Jacoby, R. F., Marshall, D. J., Kailas, S., Schlack, S., Harms, B.,and Love, R. (1995) Genetic instability associated with adenomato carcinoma progression in hereditary nonpolyposis coloncancer. Gastroenterology 109, 73–82
43. Behrens, J., Von Kries, J. P., Kuhl, M., Bruhn, L., Wedlich, D.,Grosschedl, R., and Birchmeier, W. (1996) Functional interac-tion of beta-catenin with the transcription factor LEF-1. Nature(London) 382, 638–642
44. Huber, O., Korn, R., McLaughlin, J., Ohsugi, M., Herrmann,B. G., and Kemler, R. (1996) Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech.Dev. 59, 3–10
45. Peifer, M. (1997) Beta-catenin as oncogene: the smoking gun.Science 275, 1752–1753
46. Muller, T., Choidas, A., Reichmann, E., and Ullrich, A. (1999)Phosphorylation and Free pool of beta-catenin are regulated bytyrosine kinases and tyrosine phosphatases during epithelial cellmigration. J. Biol. Chem. 274, 10173–10183
47. Kuroda, S., Fukata, M., Nakagawa, M., Fujii, K., Nakamura, T.,Ookubo, T., Izawa, I., Nagase, T., Nomura, N., Tani, H., Shoji,I., Matsuura, Y., Yonehara, S., and Kaibuchi, K. (1998) Role ofIQGAP1, a target of the small GTPases Cdc42 and Rac1, inregulation of E-cadherin-mediated cell-cell adhesion. Science281, 832–835
1200 Vol. 14 June 2000 MEI ET AL.The FASEB Journal

48. Ryuto, M., Jimi, S., Ono, M., Naito, S., Nakayama, Y., Yamada, Y.,Komiyama, S., and Kuwano, M. (1997) All-trans-retinoic acid-dependent inhibition of E-cadherin-based cell adhesion withconcomitant dephosphorylation of beta-catenin in metastatichuman renal carcinoma cells. Jpn. J. Cancer Res. 88, 982–991
49. Aberle, H., Schwarts, H., and Kemler, R. (1996) Cadherin-catenin complex: protein interactions and their implications forcadherin function. J. Cell. Biochem. 61, 514–523
50. Barth, A. I., Nathke, I. S., and Nelson, W. J. (1997) Cadherins,catenins and APC protein: interplay between cytoskeletal com-plexes and signaling pathways. Curr. Opin. Cell Biol. 9, 683–690
51. Melillo, G., Musso, T., Sica, A. Taylor, L. S., Cox, G. W., andVaresio, L. (1995) A hypoxia-responsive element mediates anovel pathway of activation of the inducible nitric oxide syn-thase promoter. J. Exp. Med. 182, 1683–1693
52. Schmedtje, J. F., Yan-Shan, J., Liu, W. L., DuBois, R. N., andRunge, M. S. (1997) Hypoxia induces cyclooxygenase-2 via theNF-kappaB p65 transcription factor in human vascular endothe-lial cells. J. Biol. Chem. 272, 601–608
53. MacMicking, J., Xie, Q., and Nathan, C. (1997) Nitric oxide andmacrophage function. Annu. Rev. Immunol. 15, 323–350
54. Thomsen, L. L., Scott, J. M., Topley, P., Knowles, R. G., Keerie,A. J., and Frend, A. J. (1997) Selective inhibition of induciblenitric oxide synthase inhibits tumor growth in vivo: studies with1400W, a novel inhibitor. Cancer Res. 57, 3300–3304
55. Rao, C. V., Kawamori, T., Hamid, R., and Reddy, S. (1999)Chemoprevention of colonic aberrant crypt foci by an induciblenitric oxide synthase-selective inhibitor. Carcinogenesis 20, 641–644
56. Brouet, I., and Ohshima, H. (1995) Curcumin, an anti-tumorpromotor and anti-inflammatory agent, inhibits induction ofnitric oxide synthase in activated macrophages. Biochem. Biophys.Res. Commun. 206, 533–540
57. Rao, C. V., Rivenson, A., Simi, B., and Reddy, B. S. (1995)Chemoprevention of colon carcinogenesis by dietary curcumin,a naturally occurring plant phenolic compound. Cancer Res. 55,259–266
58. Ohshima, H., and Bartsch, H. (1994) Chronic infections andinflammatory processes as cancer risk factors: possible role ofnitric oxide in carcinogenesis. Mutat. Res. 305, 253–264
59. Liu, J. H., and Hotchkiss, R. H. (1995) Potential genotoxicity ofchronically elevated nitric oxide: a review. Mutat. Res. 339,73–89
60. Lander, H. M., Jacovina, A. T., Davis, R. J., and Tauras, J. M.(1996) Differential activation of mitogen-activated protein ki-nases by nitric oxide-related species. J. Biol. Chem. 271, 19705–19709
61. Herschman, H. R. (1996) Prostaglandin synthase-2. Biochim.Biophys. Acta 1299, 125–140
62. DuBois, R. N., Shao, J., Tsujii, M., Sheng, H., and Beauchamp,D. (1996) G1 delay in cells overexpressing prostaglandin endo-peroxide synthase-2. Cancer Res. 56, 733–737
Received for publication August 3, 1999.Revised for publication November 18, 1999.
1201NITRIC OXIDE INDUCES PGHS-2




















