
Exploring Intraspecific Variation in the Gut Communities of ...
224
Exploring Intraspecific Variation in the Gut Communities of Western Australian Endemic Termites (Isoptera, Termitidae) as a Foundation for Future Local Biofuel Initiatives Ghislaine Anne Marie Marguerite Platell (née Small) Bachelor of Science (Microbiology) with Honours This thesis is presented for the degree of Doctor of Philosophy of The University of Western Australia 2018 School of Chemistry and Biochemistry Centre for Integrative Bee Research Australian Research Council Centre of Excellence in Plant Energy Biology
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Exploring Intraspecific Variation in the Gut Communities of ...

Exploring Intraspecific Variation
in the Gut Communities of Western Australian Endemic
Termites (Isoptera, Termitidae) as a Foundation for Future Local
Biofuel Initiatives
Ghislaine Anne Marie Marguerite Platell (née Small)
Bachelor of Science (Microbiology) with Honours
This thesis is presented for the degree of Doctor of Philosophy of
The University of Western Australia
2018
School of Chemistry and Biochemistry Centre for Integrative Bee Research
Australian Research Council Centre of Excellence in Plant Energy Biology

ii
THESIS DECLARATION
I, Ghislaine Platell, certify that:
This thesis has been substantially accomplished during enrolment in the degree.
This thesis does not contain material which has been accepted for the award of any other degree or diploma in my name, in any university or other tertiary institution.
No part of this work will, in the future, be used in a submission in my name, for any other degree or diploma in any university or other tertiary institution without the prior approval of The University of Western Australia and where applicable, any partner institution responsible for the joint-award of this degree.
This thesis does not contain any material previously published or written by another person, except where due reference has been made in the text.
The work(s) are not in any way a violation or infringement of any copyright, trademark, patent, or other rights whatsoever of any person.
The following approvals were obtained from the Western Australia Department of Parks and Wildlife (formerly Department of Environment and Conversation) prior to commencing the relevant work described in this thesis: permits SF009435, SF009982, CE004974 and SF010459.
The work described in this thesis was funded by: a Research Collaboration Award and Discovery Early Career Researcher Award (DE120101117). Some of this research was supported by an Australian Government Research Training Program (RTP) Scholarship.
Part of the 16S rRNA gene sequencing data presented in Chapter 2 was organised by Andreas Brune and Aram Mikaelyan while at the Max Planck Institute for Terrestrial Microbiology, as described in the text. Paul Schmidt (University of Göttingen) undertook the molecular identification of my samples for Chapter 3.
This thesis does not contain work that I have published, nor work under review for publication, although publications are planned from Chapter 2 and 3 in particular and contributions to each chapter are listed on the next page.
Signature:
Date:

iii
CONTRIBUTIONS
Chapter 1 The General Introduction was entirely planned and authored by me and reviewed by all four of my supervisors.
Chapter 2 The experimental design was discussed with my supervisors and Andreas Brune from the Max Planck Institute for Terrestrial Microbiology. I undertook the experimental work under the supervision of Tamara Hartke (field work) and Kate Howell (laboratory work including in house sequencing) and a portion was conducted in Marburg by Aram Mikaelyan. I conducted the data analysis and wrote the chapter, which has been edited by Kate Howell, Tamara Hartke and Boris Baer.
Chapter 3 The experimental design was discussed with my supervisors and Theodore Evans, from UWA. I undertook the experimental work (including in house sequencing supervised by Kate Howell) except for the help of Paul Schmidt (University of Göttingen) who undertook the molecular identification of my samples and Michael Bunce and Nicole White (TrEnD Laboratory, Curtin University) who ran the pilot trnL sequencing. I conducted the data analysis, which included a random sampling program created by Ian Small. I wrote the chapter, which has been edited by Tamara Hartke and Boris Baer.
Chapter 4 I made the originally observation of protists in the higher termites included in this study (2014 dataset) and designed and supervised a short term secondary research project conducted in 2015 by Honours students Erika Eto and Steven Correia. William Orsi (Department of Earth and Environmental Sciences, Palaeontology & Geobiology, Ludwig Maximilian University of Munich) kindly helped identify the protists as flagellates. I wrote the chapter, which has been edited by Kate Howell, Tamara Hartke and Boris Baer.
Chapter 5 The use of cellulose binding probes was first suggested by Harvey Millar and these were prepared by Mitchell Hattie and Keith Stubbs, from UWA. Optimisation of protocols was primarily discussed with Harvey Millar, Catherine Colas des Francs and Ryan Dosselli from UWA. I conducted the work and wrote the chapter which has been edited by Harvey Millar, Boris Baer, Catherine Colas des Francs, Jahmila Parthenay and Kieran Mulroney.
Chapter 6 The General Discussion was planned and authored by me, and edited by Tamara Hartke, Harvey Millar and Boris Baer.

iv
ACKNOWLEDGEMENTS Thank you to my supervisors for supporting me through the ups and downs both in terms of this work and in each of our lives. Boris – thank you for taking on this termite project amongst the bees and for bringing valuable ideas to the project from a different perspective, even after relocating the US. Tamara – thank you for sharing your termite expertise with me; learning about and then falling in love with these fascinating mini social cockroaches has been a massive highlight of my Honours and PhD. Thank you for your continued support from Germany and for your (and my) productive visits. Kate – thank you for passing on laboratory and sequencing knowledge and in particular for teaching me to use the MiSeq. It allowed me to conduct every aspect of this project from the field work, to running the sequencer and the analysis. Harvey – Thank you for sharing your valuable protein knowledge, your experience with theses and your support as my last remaining supervisor at UWA. I would like to thank Andreas, Aram and Niclas for hosting me at the Max Planck Institute for Terrestrial Microbiology for one month, honing my dissection technique, teaching me valuable methods, passing on protocols, the great discussions and their famous DictDb. Thanks for sequencing some of my samples, which allowed me to make valuable comparisons and work on a revised pipeline for my next experiments. Thanks to Theo for the experimental design discussions since moving to Perth. I also want to extend a big thank you to PEB, CIBER and UWA for the space, the equipment and the lovely co-workers. In particular, thank you to Hayden for keeping the computing side of things running as smoothly as possible, to my students Nithin, Claire, Steven and Erika for their involvement and contributions, to Ryan for your help troubleshooting my (many) protein issues, to Ellen for a brilliant example to follow, to Rachael for pipeline discussions, to Karina for the outreach and extracurricular learning opportunities and to Sandi my office mate and Antarctica travel buddy! I must thank my fellow Homeward Bounders from across the globe for the continued contact, support and opportunities. I would love to thank my "maman" and dad which have been involved both as parents and co-workers. It was wonderful learning lab techniques from my mum and analysis tricks from dad. I am grateful for great friends and hobbies to keep me healthy. Thanks to the Games Workshop crew for a fun and rewarding work environment, to my rugby team the Divas for the motivation to exercise and practice my courage. Thank you to Buzz and Summer, my two puppies that were great company while I wrote and napped and last but certainly not least, to Peter, for your support while I have studied all these years.

v
ABBREVIATIONS ABPPs Activity-Based Proteomics Probes ANOVA ANalysis Of VAriance A. obeuntis Amitermes obeuntis B Beelu National Park BLAST Basic Local Alignment Search Tool CMC Carboxymethyl Cellulose CO2 Carbon dioxide COII Cytochrome c Oxidase subunit II C. acinaciformis Coptotermes acinaciformis raffrayi C:N Carbon-to-Nitrogen ratio C+ Aspergillus niger positive control for cellulase dbRDA distance-based RedunDancy Analysis DictDb Dictyopteran gut microbiota reference Database DMF Dimethylformamide DNA Deoxyribonucleic Acid DTT Dithiothreitol EDTA Ethylenediaminetetraacetic Acid EU European Union GH Glycoside Hydrolases GHGs Greenhouse Gases GH2d-ABP Glycoside Hydrolase Activity Based Probe 2d GL Gigalitre GPS Global Positioning System g/L Gram per Litre HEPES 4-(2-HydroxyEthyl)-1-PiperazineEthaneSulfonic acid hr Hour J John Forrest National Park kD Kilodalton M Molar mg Milligram min Minute ml Millilitre ML Megalitre mm Millimetre mM Millimolar MM301 Retsch Mixer Mill Mya Million years ago NaPO4 Sodium Phosphate NDSB201 Non-Detergents Sulfobetaine 201 nls Non-Linear Least-Squares nm Nanometer OTU Operational Taxonomic Unit O. occasus Occasitermes occasus PCoA Principal Coordinate Analysis PCR Polymerase Chain Reaction

vi
PERMANOVA PERmutational Multivariate ANalysis Of VAriance pH Potential of Hydrogen PhD Doctor of Philosophy PMSF Phenylmethylsulfonyl fluoride P3 Proctodeal segment 3 QIIME Quantitative Insights Into Microbial Ecology RE Renewable Energy rRNA Ribosomal Ribonucleic Acid SDS Sodium dodecyl sulfate RT Room Temperature TBTA tris[(1-benzyl-1H-1,2,3-triazol-4- yl)methyl] amine T. reesei Trichoderma reesei T. westraliensis Tumulitermes westraliensis UniFrac Unique Fraction metric US United States USB Universal Serial Bus UV Ultraviolet V Volt V4 V4 hypervariable region of the 16S rRNA gene V3-V4 V3 to V4 region of the 16S rRNA gene W Banyowla Regional Park WA Western Australia x g Times the force of gravity μl Microlitre μM Micromolar ° C Degrees Celsius

vii
ABSTRACT Climate change is arguably the biggest issue facing humanity today. Mitigation
strategies can reduce humankind's net greenhouse gas contributions and in
turn reduce our impact on further climate change. In particular, second
generation biofuel technologies produced from crop and forest residues, have
the benefits of decreased greenhouse gas emissions and low competition with
food needs, as well as the potential to reduce waste streams and improve
agricultural land quality. In Western Australia, the two most promising
substrates are wheat straw and eucalyptus mallee. Biofuels from wheat straw
would recycle crop "waste" that is currently being burnt, hence providing
improvements to two human activities that contribute to climate change.
The biggest barrier to the implementation of biofuel production from these
substrates is the development of a cost-effective cellulose hydrolysis process.
Higher termites (Isoptera, Termitidae) are a promising source of enzymes
because they harbour primarily bacterial consortia that efficiently degrade
various forms of lignocellulose, optimised over evolutionary time. In this
thesis, I have investigated short-term influences on the termite gut
community to determine whether members of the gut population of endemic
higher termites may warrant further study as part of optimised biofuel
production from wheat crop residue in Western Australia.
To my knowledge, I provide the first characterisation of gut communities of
three endemic Western Australian termites with broad diets, higher termites
Tumulitermes westraliensis, Amitermes obeuntis and the lower termite
Coptotermes acinaciformis raffrayi. I propose new standards for experimental
design in the study of termite gut communities, including the use of a
standardised 16S rRNA amplification strategy, increased replication, a
standard species core community definition and analysis method for accurate
core community calculations. My work evaluates potential factors shaping
intraspecific variation in the gut community of higher termites, providing a

viii
greater understanding of the vertically, horizontally and environmentally
acquired components of the bacterial and protistan microbiota and the
generation of intraspecific variation. Protein extraction and visualisation
protocols were optimised with the aim to improve the integrity of enzymes
recovered from termite guts and enhance binding of cellulase-specific probes,
with a future aim to isolate them for further testing.
My project confirms that diet affects termite gut core community composition
and abundance on a short time scale under field conditions. Intraspecific
variation occurring on a short time scale allows the manipulation of the gut
community to target substrates of interest, laying the foundation for future
local biofuel initiatives. Potential future studies are outlined and could focus
on the gut communities or enzymes produced by higher termites feeding
directly on biofuel substrates of interest, such as wheat and/or eucalyptus
mallee in Western Australian Wheatbelt fields.

ix
TABLE OF CONTENTS Thesis Declaration...................................................................................................ii Contributions..........................................................................................................iii Acknowledgements.................................................................................................iv Abbreviations...........................................................................................................v Abstract...................................................................................................................vii Table of Contents....................................................................................................ix
Chapter 1: General Introduction..............................................................................1 1. Renewable energy: a climate change mitigation strategy..................................2 2. Biofuel: a medium term mitigation strategy......................................................4 3. The quest for cellulase enzymes........................................................................10 4. Termites as a source of cellulases......................................................................14 5. Conclusion...........................................................................................................19 References...............................................................................................................21 Chapter 2: Colony Differences in the Gut Communities of Western Australian Termites Tumulitermes westraliensis and Coptotermes acinaciformis raffrayi.....................................................................................................................29 Introduction...........................................................................................................30 Methods..................................................................................................................33 Results.....................................................................................................................40 Discussion................................................................................................................51 References...............................................................................................................59 Supplementary figures...........................................................................................64 Chapter 3: Sampling Intensity, Scope and Scale Significantly Affect Estimation of Dietary Influence on the Gut Communities of Tumulitermes westraliensis and Amitermes obeuntis........................................................................................67 Introduction...........................................................................................................69 Methods..................................................................................................................72 Results.....................................................................................................................81 Discussion..............................................................................................................101 References..............................................................................................................112 Supplementary figures..........................................................................................116 Chapter 4: Observation of Flagellated Protists in the guts of Higher Termites Tumulitermes westraliensis and Amitermes obeuntis Following Rainfall.........137 Introduction..........................................................................................................138 Methods.................................................................................................................141 Results....................................................................................................................142 Discussion..............................................................................................................147 References.............................................................................................................150

x
Chapter 5: Accurate Characterisation of the Complete Termite Cellulase Profile Requires Selective Inhibition of Gut Proteases...................................................153 Introduction..........................................................................................................154 Protocols................................................................................................................157 Experiments...........................................................................................................161 Discussion..............................................................................................................183 References.............................................................................................................188 Chapter 6: General Discussion..............................................................................191 A: How much intraspecific variation exists in the gut communities of Western Australian endemic termites?..............................................................................192 B: How should the core community of a termite species be defined and accurately estimated?...........................................................................................196 C: Can the gut community of local higher termite species be modified in a field-based feeding experiment?.........................................................................199 D: Can enzymes relevant to breaking down a substrate of choice be identified using comparative enzymology?.........................................................................202 Broader Contributions.........................................................................................204 Conclusion............................................................................................................208 References.............................................................................................................210

1
CHAPTER 1: General Introduction

2
Overview:
Climate change is arguably the biggest issue facing humanity today. Mitigation
strategies can reduce humankind's net greenhouse gas contributions, to in
turn reduce our impact on climate change. This review will give an overview of
these issues and then focus on biofuel, a group of renewable energy
technologies that can lower the transport sector's emissions. In particular,
second generation biofuel production can be improved by the development of
a more cost effective cellulose hydrolysis process. Termites have been
identified as a promising source of enzymes or microbes to that end and are
the ultimate focus of this review. There are currently no commercial second
generation biofuel plants in Western Australia and local possibilities will be
explored, although the scope of this thesis remains in studying the gut
communities of local termite species with future possibilities to learn from
them in order to improve biofuel production.
1. Renewable energy: a climate change mitigation strategy
Anthropogenic climate change
The greenhouse effect, a process by which gases in the atmosphere trap heat,
makes the Earth habitable. Human activities such as deforestation, intensive
agriculture and burning of fossil fuels have and continue to increase the
concentration of major greenhouse gases (GHGs) in the atmosphere (Mitchell,
1989). While the naturally dominant GHG water vapour only persists in the
atmosphere for about a week, the major anthropogenic contributor carbon
dioxide (CO2) can persist for several centuries. Water vapour is replenished by
evaporation from the oceans, a process regulated by temperature (Bengtsson,
2010; Allwood et al., 2014). Hence anthropogenic GHG emissions cause an
amplification of warming in direct and indirect ways, leading to climate
change.

3
The scale of the change in Earth’s climate has started a new geological epoch.
Its suggested name "Anthropocene" represents the wide and increasing
impacts of human activity on the planet (Crutzen and Stoermer, 2000).
Climate change is arguably the biggest threat faced by humankind today,
particularly because it compounds with many other issues. For example,
effects of climate change on temperature and rainfall will impact food
production. Agriculture currently contributes about 30% of human GHG
emissions (Smith and Gregory, 2013). So not only will feeding the world's
growing population become more difficult, but producing more food using
current agricultural practices will intensify climate change.
Continuing the present course will lead to a much warmer climate, extensive
sea level rise and species impoverishment, with the risk of a sixth great
extinction event. Working toward a strong and rapid reduction in GHG
emissions should allow the climate to stabilise, although it will remain
significantly warmer than that of the Holocene (Steffen et al., 2016). Mitigation
strategies can be put in place to reduce the effects of climate change, while
adaptation strategies can prepare us for inevitable changes. This introductory
chapter focuses on one strategy for climate change mitigation, the use of
renewable energy, taking inspiration from natural cellulose-degrading systems
to improve energy production.
Renewable energy
Climate change mitigation requires human intervention to reduce the sources
of GHGs or enhance their sinks. One major mitigation strategy is the use of
renewable energy (RE), whose sources are replenished or added to by natural
processes (Allwood et al., 2014). RE also contributes to sustainable social and
economic development and leads to health benefits through the reduction of
air pollution, furthering its positive economic impact (IPCC, 2011; Mathiesen et
al., 2011).

4
The world's dependence on energy continues to increase with world
population and industrialisation, particularly in the developing world
(Wolfram et al., 2012). Non-renewable energy sources, such as oil and coal
cannot fill this need, due to their finite availability as well as the
environmental damage associated with their use. Indeed, half of the world's oil
supply may have already been accessed (an event called peak oil; Höök and
Tang, 2013; Chapman, 2014), underlining the urgent need to develop
replacement technologies. RE can complement and later replace oil to
mitigate both GHG emissions and oil depletion.
Electric vehicles are predicted to dominate future transport because of the
decreasing costs of electric batteries and the growth of renewable electricity
generation (Catenacci et al., 2013; Holland et al., 2016). However batteries
currently face reliability, size and range issues and the large scale deployment
of charging stations may impact existing distribution networks (Sims et al.,
2014; Haddadian et al., 2015). Until that gap is fully bridged, other technologies
are required to replace oil such as biofuels, which can be adopted to varying
extents with the current infrastructure (Fiorese et al., 2013; Sims et al., 2014).
Certain biofuels may play a longer term role in powering heavy machinery,
ships and planes (Buijs et al., 2013; Sims et al., 2014; Bharathiraja et al., 2017).
2. Biofuel: a medium term mitigation strategy
Biofuel overview
Biofuel is fuel derived from biomass, which is living or dead, non-fossilised
organisms or their by-products (Allwood et al., 2014; Brooksbank et al., 2014).
There are various technologies commercially available or in development to
produce biofuel. These are classified into four generations, all or most of
which will soon be required to meet transport fuel demands (Dutta et al.,
2014). Biofuel production does have environmental impacts, the extent of
which depends on the type of biomass, and how it is produced, harvested,

5
transported and processed. Caution needs to be used on a case-by-case basis
to assess the benefits and impacts of biofuel production (Koh and Ghazoul,
2008).
Bioethanol is the most common biofuel produced worldwide, followed by
biodiesel. Global production of bioethanol averaged approximately 103 GL per
year in 2016, 85% of which was produced by the United States (US) and Brazil
(Renewable Fuels Association, 2017). Biodiesel averaged 39 GL per year
(Renewable Fuels Association, 2017). By comparison, oil production averaged
5322 GL per year in 2015 (including crude oil, shale oil, oil sands and natural
gas liquids; BP, 2017). The European Union (EU) is the world's largest biodiesel
producer, with 13.5 GL produced in 2015, making up 80% of the EU's biofuel
production. Bioethanol production was estimated at 5.2 GL across the major
EU contributors in 2015 (Flach et al., 2016). Australia's biofuel production was
estimated at 250 ML of bioethanol and 50 ML of biodiesel in 2016 (Farrell,
2016). While biodiesel can be used in current vehicles with little modification,
bioethanol has shown to be corrosive to existing infrastructure and engines. It
requires specific transport and storage and to be used in most existing vehicles
as a low concentration blend (Jin et al. 2011).
Interest in so-called drop-in fuels that are similar enough to gasoline to be
distributed through existing gasoline infrastructure and be used almost
undiluted in existing engines has therefore grown (International Energy
Agency, 2011; Buijs et al., 2013; Bharathiraja et al., 2017). Biobutanol is a drop-in
fuel that would allow better mileage, less ignition problems, a safer use at high
temperatures, and easier storage and distribution than bioethanol. However
there are concerns over the low production yield of biobutanol from biomass
(Pfromm et al., 2010; Jin et al. 2011). One solution is to chemically convert
bioethanol to biobutanol (Ndaba et al., 2015). Regardless, bioethanol, biodiesel
and biobutanol can be produced from all four generations of biofuel
technology (Dutta et al., 2014) and will be covered in this review.

6
First generation biofuel: an ethical dilemma
First generation bioethanol production involves the fermentation of sugars or
starch, while biodiesel production is based on the esterification of edible oils
(Dutta et al., 2014). The US and Brazil primarily produce bioethanol from corn
and sugarcane respectively (Haq et al., 2016). In the EU, bioethanol is mainly
produced from wheat, corn and sugar beet and first generation biodiesel from
rapeseed oil and palm oil (Flach et al., 2016). In Australia, bioethanol is
produced from wheat starch, red sorghum and molasses from sugar
manufacture, while first generation biodiesel feedstocks include animal fats
(Farrell, 2016).
First generation biofuel production is cost effective but the feedstocks are
expensive and drive up the price of food (Dutta et al., 2014; Gasparatos et al.,
2013). In 2008, an international food crisis was declared as a result of the rising
prices of agricultural commodities, partly due to their use in fuel production
(Schmitz and Kavallari, 2009). Crops raised for first generation biofuel
production require large areas of prime farmland, which leads to deforestation
and therefore impacts biodiversity and GHG emissions (Gasparatos et al.,
2013). First generation biofuels are therefore not a sustainable and ethical
solution to singly meet growing transport fuel demands.
Second generation biofuel: a local solution
Second generation biofuels are fuels produced from non-food feedstocks
(Dutta et al., 2014). As of 2015, 67 biorefineries worldwide were producing
bioethanol, biodiesel, or aviation biofuel using second generation
technologies. Over a third of these were operating on a commercial scale,
while the rest were pilot projects (Nguyen et al., 2017). Second generation
biodiesel is produced from non-edible oil seeds or waste cooking oil using first
generation technology (Dutta et al., 2014; Farrell, 2016). Used cooking oil is the
second most used feedstock for biodiesel production in the EU and is
commonly used in Australia (Flach et al., 2016; Farrell, 2016).

7
Second generation bioethanol or biobutanol are produced from lignocellulose,
the main component of plant cell walls and therefore the most abundant
biomass on Earth (Dutta et al., 2014; Haq et al., 2016). Lignocellulosic
feedstocks include crop and forest residues, organic municipal solid waste and
purposely-grown energy crops such as grasses and short rotation forests
(Dutta et al., 2014). There are 17 operating lignocellulosic bioethanol plants in
the EU, four of which are on a commercial scale (Nguyen et al., 2017). There
are 14 operating lignocellulosic bioethanol plants in the US (9 of which are
commercial), and two pilot plants in Australia (Brooksbank et al., 2014;
Nguyen et al., 2017).
Lignocellulosic material is recalcitrant and more complex technology is
required for its use as a fuel source. The cellulose and hemicellulose
components must first be broken down into glucose to be fermented into
ethanol or butanol (Dutta et al., 2014; Haq et al., 2016). While lignocellulosic
feedstocks are cheap, the pre-treatment and hydrolysis processes required to
break down the substrate are not. Another barrier to widespread
commercialisation of lignocellulosic biofuel is the energy and monetary cost of
transporting biomass to the plant. Except for the case of dedicated energy
crops, the feedstock typically originates from multiple locations (Lange, 2007;
Ho et al., 2014). However, Searcy et al. (2007) showed that the cost of
transporting biomass is more than the cost of transporting its energy products
and that building power plants near the feedstock is a more cost effective
approach. For example, all four lignocellulosic bioethanol plants in Brazil use
sugarcane bagasse from local mills (Nguyen et al., 2017).
A local second generation bioethanol or biobutanol approach for Western
Australia (WA) would likely include wheat residue. WA contributes half of the
wheat production of Australia, with over 10 million tonnes produced in 2015
(Wilkinson, 2017). There are other uses for wheat residue such as animal feed
or bedding (Brooksbank et al., 2014; Giannoccaro et al., 2017). Fields also

8
benefit from the retention of crop stubble and mulched residue to limit
erosion and increase yield (Malinda, 1995; Erenstein, 2002; Brooksbank et al.,
2014). Yet of the 15 million tonnes of residue produced per annum, much of it
is currently being burnt (Taskforce, 2007). A 2014 WA Department of
Agriculture and Food report highlighted ten hubs in WA with the
infrastructure to support bioethanol plants from cereal straw. They estimated
that 149 to 1212 thousand tonnes of cereal straw would be available within 50
km of each of the ten hubs, taking into account alternative straw uses
(Brooksbank et al., 2014). As an example the Beta Renewables plant in
Crescentino is a commercial plant utilising second generation technology with
an input capacity of 200,000 t/y lignocellulosic crop residue including wheat
straw and an output capacity of 40,000 t/y (50.8 ML per year) of ethanol
(European Biofuels Technology Platform, 2016). All but two of the hubs
identified by Brooksbank et al. (2014) exceed the input capacity of the
Crescentino plant, suggesting there are multiple viable options for second
generation plants in Western Australia. Local farmers' interest would need to
be explored in those areas to determine the ultimate success of such a venture
(Giannoccaro et al., 2017).
Eucalyptus mallee has also been investigated as a potential substrate for
biofuel production in Western Australia, with diverse ecological benefits.
Mallee are multibranched short trees that can be harvested every 3-7 years,
providing frequently renewed biomass and carbon sequestration (Wu et al.,
2008; Yu et al., 2015). When planted between wheat fields, their root systems
rectify accumulation of groundwater and by extension the widespread issue of
dryland salinity in the Wheatbelt caused by intensive agriculture (Shepherd et
al., 2014; Yu et al., 2015). In addition, mallee plantations limit wind erosion,
foster local biodiversity (Wu et al., 2008), and are an almost carbon neutral
source of biomass (Yu et al., 2015). Second generation technologies therefore
have the benefits of decreased GHG emissions and decreased competition
with food needs, as well as the potential to reduce waste streams and improve
land quality (Wu et al., 2008; Brooksbank et al., 2014; Dutta et al., 2014).

9
However, further work is required to improve the cost efficiency of the
process. The difficult and costly hydrolysis step will be the focus of section 3 in
this review.
Third generation biofuel: a cost barrier
Third generation biofuel is produced from microalgae (Dutta et al., 2014).
Microalgae are a collection of single cells or simple multicellular organisms
from multiple kingdoms that live in a broad range of humid environments and
use sunlight to produce and store fixed carbon. The ease of cultivation of
microalgae and their higher lipid content than vascular plants make them
attractive for biodiesel production. They can be cultivated on non-arable land
using non-potable water and converted to biodiesel with much less waste than
first and second generation biodiesel feedstocks (Borowitzka and Moheimani,
2013; Leite et al., 2013). They can also be used to produce bioethanol or
biobutanol, in which case the lipid content is not important and different
species can be selected (Borowitzka and Moheimani, 2013; Dutta et al., 2014).
Despite these benefits, there are currently no commercial third generation
biofuel plants in operation, as it remains the most expensive type of biofuel to
produce (Dutta et al., 2014). Algae can be grown in highly controlled but
expensive photobioreactors. Open ponds are a lower cost alternative with a
lower energy input requirement, but have a higher risk of contamination and
lower productivity. Third generation biodiesel production is similar to first
generation technologies, with the addition of expensive harvesting procedures
to concentrate and dry the feedstock (Borowitzka and Moheimani, 2013; Leite
et al., 2013). Algal cultures do not scale very well and algal biodiesel is
projected to play a small part in meeting global energy demand (Borowitzka
and Moheimani, 2013).

10
Fourth generation biofuel and overcoming the cost barrier
Fourth generation biofuel technologies under development involve genetic
modification of microorganisms to improve fuel yield as a means to counteract
the cost barrier to commercialisation (Dutta et al., 2014; Vassilev and
Vassileva, 2016). Other uses for lignocellulosic and algal biomass are being
investigated to generate alternate money streams to justify second and third
generation biofuel production. These include the production of chemicals and
animal feed, as well as waste treatment (Mata et al., 2010; Kircher, 2015). A
combination of biofuels across multiple generations, with a localised
approach, is therefore the only way to meet transport fuel demands
sustainably, ethically and in economically viable ways.
3. The quest for cellulase enzymes
Cellulase: a collection of enzymes
The hydrolysis step of second generation lignocellulosic biofuel production
involves incubating the pre-treated plant fibre with commercial cellulase
enzymes. Three major types of glycoside hydrolases (GH) are required for the
breakdown of cellulose into glucose: endoglucanases, exoglucanases and β-
glucosidases, collectively referred to as cellulases (Figure 1). Naturally
occurring cellulose is insoluble but contains amorphous regions that can be
accessed by endoglucanases and subsequently cleaved. Exoglucanases, tunnel-
shaped enzymes, can then release cellobiose (made up of two glucose
subunits) from the ends of cellulose chains. Endoglucanases thus work
synergistically with exoglucanases by increasing the number of available ends
(Wilson, 2011). Finally, β-glucosidases act on cellobiose to complete the
digestion to glucose. Other accessory enzymes, for example hemicellulases,
aldo-keto reductases and laccases, facilitate this process by increasing access
to the cellulose component of lignocellulose (Coy et al., 2010; Mohanram et al.,
2013; Sethi et al. 2013).

11
Commercialised enzymes are simple mixtures
There are multiple deployment strategies for cellulases found in nature. The
two most common are 1) secretion of individual enzymes with one or multiple
catalytic domains, such as in fungi, bacteria and even termites (Brune, 2014;
Payne et al., 2015, Figure 1A); and 2) cell-bound complexes of up to hundreds
of enzymes called cellulosomes, primarily found in anaerobic bacteria (Bayer
et al., 2004; Artzi et al., 2017, Figure 1B). A rarer strategy involves direct
binding of bacteria to the substrate and importation of partially degraded
chains for further intracellular processing. This strategy has so far only been
described in phylum Fibrobacteres (Wilson, 2011; Ransom-Jones et al., 2012;
Abdul Rahman et al., 2016, Figure 1C). Different strategies can work
synergistically as demonstrated by Resch et al. (2013), suggesting that diversity
is key to an optimised cellulose-degrading approach.
However, most currently available commercial cellulases are mixtures derived
from Trichoderma or Aspergillus fungal species, in particular T. reesei
(Lambertz et al., 2014; Bischof et al., 2016). Fungi produce a high yield of
protein but the enzyme mixture cannot be modified to optimise the
breakdown of different substrates (Lambertz et al., 2014), which is necessary to
increase degradation efficiency (Banerjee et al., 2010; Mohanram et al., 2013).
Research has therefore focused on developing cellulose-degrading consortia,
as these can perform more complex functions, can be modulated to suit
different biofuel feedstocks and are more stable than monocultures (Zuroff
and Curtis, 2012; Brethauer and Studer, 2014). This reinforces the idea that
second generation biofuel production will benefit from an approach optimised
for locally available substrates.
Finding or engineering enzymes
There are two main approaches to optimising cellulase cocktails or consortia:
1) designing or modifying known cellulases and 2) finding more efficient
naturally occurring enzymes (Mohanram et al., 2013). Enzyme engineering can

12
take several forms: rational design by site-directed mutagenesis (Huang et al.,
2012), directed evolution using error-prone PCR (Liang et al., 2011), combining
enzymes into multifunctional chimeras (Morais et al., 2012) or designing
cellulosomes (Moraïs et al., 2010). This review will focus on the second
approach, enzyme discovery, as well as the idea that naturally occurring
enzymes or consortia can be naturally optimised to different substrates. For
example, the cellulosome of Clostridium thermocellum is naturally modified
when the bacterium is exposed to different substrates (Raman et al., 2009).
Novel cellulases can be found in certain free-living bacteria, fungi and protists.
Some animals, most of which are insects, also possess the ability to degrade
cellulose, commonly relying on symbioses with cellulose degrading gut
microbes (Oppert et al., 2010). Termites are particularly successful, and
produce their own (endogenous) cellulases, alongside those expressed by their
gut community (Brune, 2014). Termites therefore harbour consortia that could
be harnessed for more efficient commercial cellulose degradation.

13
Figure 1: The tree known deployment strategies for cellulases include (A)
secretion of individual enzymes, (B) cell-bound complexes of up to hundreds
of enzymes called cellulosomes and (C) direct binding to the substrate and
importation of partially degraded chains. All involve different classes of
cellulases working synergistically. Modelled on Ransom-Jones et al (2012).

14
4. Termites as a source of cellulases
Social ecosystem engineers
Termites are social insects of the Infraorder Isoptera (Krishna et al., 2013),
closely related to wood-dwelling cockroaches within the order Blattodea
(Inward et al., 2007; Figure 2). There are almost 3000 described living species
worldwide, of which only 12% are considered significant pest species (Krishna
et al., 2013). In Australia, there are at least 273 termite species from 41 genera
and 5 families (Krishna et al., 2013). Approximately 64% of these are endemic
to Australia (Eggleton, 2000). Pests species of economic importance have been
preferentially studied; and in the case of Australia include the world's earliest
branch termite lineage, Mastotermes darwiniensis (Veivers et al., 1983; Konig
et al., 2006; Scharf, 2015), the most damaging genus in Australia, Coptotermes
(Calaby and Gay, 1956; Peters and Fitzgerald, 2003; Evans and Gleeson, 2006;
Lee et al., 2015) and several species within the genus Nasutitermes (Hogan et
al., 1988; Lee et al., 2007; Webb and Mcclintock, 2015).
Although they are more renowned as pests, Australian termites primarily play
a key ecological role similar to that of earthworms in the Northern
hemisphere. Both animals are sometimes referred to as 'ecosystem engineers'
because they contribute to soil turnover and aeration, and nutrient cycling
(Lavelle et al., 1997; Black and Okwakol, 1997). Termites are particularly
successful cellulose-degraders, being able to digest up to 99% of the cellulose
they ingest (Esenther and Kirk, 1974). They produce their own (endogenous)
cellulases, but also take advantage of the cellulases expressed by the hundreds
of microorganisms in their intestinal tracts (Brune, 2014). Indeed, a termite
cannot survive on lignocellulose if its gut has been defaunated (Eutick et al.,
1978; Veivers et al., 1983), so their gut community is key to their success as
both pests and ecosystem engineers.

15
The successful higher termites
Termite families are classified as either lower or higher termites (Figure 2).
The lower termites include the basal families Mastotermitidae,
Hodotermitidae, Termopsidae, Kalotermitidae and Rhinotermitidae, which
primarily feed on wood and have gut communities dominated by cellulose-
degrading flagellates and their associated prokaryotes (Cleveland, 1923; Inoue
et al., 2000; Dolan, 2001). The cellulose-degrading flagellates are passed from
generation to generation by proctodeal trophallaxis (anus-to-mouth transfer;
Ohkuma et al., 2009). The gut community is established in the juveniles by
repeated transfer of gut fluid until the third instar, when they can maintain
the larger flagellates and become independent feeders, and again for up to two
weeks following any molt (Nalepa, 2015).
The higher termite family Termitidae appears to have lost these flagellates and
diversified its diet approximately 40-60 million years ago (Mya; Engel et al.,
2009; Bourguignon et al., 2016). This most derived family encompasses over
80% of all living termite species, each more or less specialised to consume
wood, grass, litter, humus, soil or fungus (Jones and Eggleton, 2011; Brune and
Ohkuma, 2011; Bignell, 2011; Poulsen, 2015; Figure 3). These substrates are
broken down by endogenous cellulases in the midgut, which are
complemented by microbial enzymes in the P3 hindgut segment (Brune, 2014;
Figure 3). Higher termites are thought to transmit their bacterial gut
community by trophallaxis (mouth-to-mouth) and coprophagy (excrement
ingestion) as proctodeal trophallaxis has never been reported (Diouf et al.,
2015). It is unclear to date whether the gut community of higher termites has
co-evolved with its host to the same extent as in the lower termites. The
potentially plastic bacterial gut community of the higher termites may thus be
the ideal source of enzymes or consortia suited to breaking down substrates of
interest to the biofuel industry.

16
Figure 2: Phylogenetic relationship of select Australian termite species, as
compared to two species of roaches and a mantis. This phylogeny was created
using Phylogeny.fr (Dereeper et al., 2008) based on cytochrome oxidase
subunit II (COII) sequences from Inward et al. (2007) and this study.
Figure 3: Gut structure and segments of Western Australian endemic higher
termites (A) Tumulitermes westraliensis, which feeds on grass and (B)
Amitermes obeuntis, which feeds on wood and soil. The gut outlines were
obtained by exaggerating the contrast of light microscopy photographs of each
gut, thinning the outline and colouring in the lumen. The two gut structures
are similar except for an enlarged P1 hindgut segment (blue) in A. obeuntis,
which has been found in all soil feeders and is thought to be involved in the
modification of humic acid (Brune, 2014). Cellulases produced by the host are
secreted in the midgut (red); whereas the bulk of the gut community resides
within the P3 hindgut segment (green), where microbial cellulases are
expressed. P2 (pink) is the enteric valve that separates the P1 and P3 hindgut
segments.

17
The importance of the core microbiome
Taxa that are consistently detected across samples from a predefined habitat
are known as the core community and are thought to be critical to the
functioning of that community. This concept allows scientists to define a
baseline community for that habitat and predict responses to perturbation and
manipulation (Shade and Handelsman, 2012). Each member of the core
community is thought to fill a niche in the termite gut that is consistently
present regardless of the location or food source of the host (Bignell et al.,
1980; Benjamino and Graf, 2016). Indeed, Huang et al. (2013) showed that
despite feeding the lower termite Reticulitermes flavipes either grassy or
woody diets, a core microbiome of 65% of commonly recovered taxa
(accounting for 95% of sequences) persisted across all diets. Yet the source of
the core and of the remainder of the gut community remains unclear (Scharf,
2015; Benjamino and Graf, 2016; Shapira, 2016).
Microbiota transplant experiments have shown that gut communities are
assembled primarily based on the available niches in the gut habitat. In
zebrafish, mice, termites and cockroaches, the newly established gut
community is made up of lineages from the community of origin, but their
relative abundances resemble that of the normal gut community of the
recipient host (Rawls et al., 2006; Mikaelyan et al., 2016). This implies that
bacterial phylotypes taken up from the environment or transferred by
nestmates may be amplified in the gut environment. Thus the core
microbiome within a termite species could be the result of passing on
particular phylotypes from generation to generation, consistently collecting
them from the environment or, most likely, a combination of both. Regardless
of its origin, studying the core microbiome indicates which community
members may consistently benefit the host and potential self-sustaining
consortium for industrial applications.
Core microbiomes for the termite gut habitat have been estimated in previous
studies using a variety of approaches, each defining which taxa should be

18
considered core in different ways (Huang et al., 2013; Dietrich et al., 2014;
Otani et al., 2014; Reid et al., 2014; Benjamino and Graf, 2016). Despite these
differences, the core community concept typically is used to draw evolutionary
conclusions about termite-microbe relationships based on a single point in
evolutionary time. The inconsistency of method and interpretation of the core
community concept in the literature affects the quality of these conclusions.
Modulating the gut community
Studying the non-core taxa is a way to measure plasticity in the termite gut
community. Variation in composition and structure (abundance of each
taxon) of gut communities may highlight microbial taxa relevant to a
particular set of conditions. For example, within colony differences are
thought to reflect taxa relevant to the different roles of castes and age groups
(Hongoh et al., 2006; Xiang et al., 2012; Li et al., 2016; Benjamino and Graf,
2016). Reid et al. (2014) found that each colony of the lower termite
Stolotermes ruficeps had a signature gut community, even when collected in
close proximity. They speculated that differences in diet, such as wood species
or degree of decay, may explain the variation. Understanding the extent of and
factors driving natural intraspecific gut community variation can suggest
appropriate levels of variation for commercialised consortia targeting different
substrates.
Changes in the gut community have been measured in response to forced
dietary changes for both lower and higher termites (Tanaka et al., 2006;
Miyata et al., 2007; Husseneder et al., 2009; Huang et al., 2013; Wang et al.,
2016). Boucias et al. (2013) reported no significant change in the gut
community of the lower termite Reticulitermes flavipes laboratory colonies
during a week-long exposure to filter paper and/or pinewood. On the other
hand, Huang et al. (2013) used a six-week time frame with the same termite
species, a greater variety of natural substrates and freshly collected termites.
They reported that the gut community could be differentiated based on the
recalcitrance of the diet, with diet affecting the community composition (with

19
taxa exclusively associated with certain diets) and and stucture (with many
taxa enriched under a particular diet).
Miyata et al. (2007) fed artificial diets to the higher termite Nasutitermes
takasagoensis over a three week period and reported large changes to the gut
community structure depending on the complexity of the diet. Wang et al.
(2016) also found changes following forced feeding of the higher termite
Mironasutitermes shangchengensis with corn stalks or filter paper for up to ten
days. However, they reported that the gut community seemed resilient
because the community composition and structure was most similar to the
control ten days following feeding, as compared to any other time point. For
example, the relative abundance of Spirochaetes was lower in groups fed for
four days as compared to the control, but similar to the control in groups fed
for seven and ten days. On the other hand, Firmicutes, Actinobacteria and
Acidobacteria were more abundant in groups fed for four and seven days, but
similar to the control in groups fed for ten days. Gut community manipulation
in termites requires further study to determine the benefits and duration of
gut community changes following a change in diet. Feeding times of multiple
weeks have generated greater changes in past studies but Wang et al. (2016)
note that keeping higher termites healthy in laboratory conditions is of greater
difficulty than with lower termites. Hence, field based feeding experiments
with substrates of interest may hold the key to optimising bacterial consortia
in higher termites for biofuel applications.
5. Conclusion
Climate change is the biggest issue facing humanity today and renewable
energy is one of the most effective mitigation strategies we can employ. A
combination of technologies will support this goal in the medium term, at
least, and second generation biofuel production is already playing a role. The
further implementation of this technology relies upon the development of

20
more efficient enzyme cocktails or bacterial consortia at a local level. Termites
are a promising source of enzymes or microbes since they successfully feed on
lignocellulose in various forms and therefore evolved highly optimised
cellulose-degrading consortia suited to different substrates. More work is
required to determine whether gut communities are also optimised on shorter
time scales to respond to changes in food sources.
Studying local higher termites with a predominantly bacterial gut population
may therefore lead to optimised bioethanol production from wheat crop
residue in Western Australia. With this information in mind, I studied the
following research questions during my PhD.
A: How much intraspecific variation exists in the gut communities of
Western Australian endemic termites?
Research I conducted to address this is covered in Chapters 2, 3 and 4.
B: How should the core community of a termite species be define and
accurately estimated?
Answers to this question are provided in Chapter 3.
C: Can the gut community of local higher termite species be modified under
field conditions?
Results from a field-based experiment to test this are provided in Chapter 3.
D: Can enzymes relevant to breaking down a substrate of choice be
identified using comparative enzymology?
Optimisation work to that end is presented in Chapter 6.

21
References:
Abdul Rahman, N., Parks, D. H., Vanwonterghem, I., Morrison, M., Tyson, G. W., &
Hugenholtz, P. (2016). A phylogenomic analysis of the bacterial phylum Fibrobacteres. Frontiers in Microbiology, 6, 5.
Allwood, J. M., Bosetti, V., Dubash, N. K., Gómez-Echeverri, L., & von Stechow, C. (2014). Glossary. In O. Edenhofer, R. Pichs-Madruga, Y. Sokona, E. Farahani, S. Kadner, K. Seyboth et al. (Eds.), Climate change 2014: Mitigation of climate change. Contribution of working group III to the fifth assessment report of the intergovernmental panel on climate change. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA.
Artzi, L., Bayer, E. A., & Moraïs, S. (2017). Cellulosomes: bacterial nanomachines for dismantling plant polysaccharides. Nature Reviews Microbiology, 15(2), 83-95.
Banerjee, G., Car, S., Scott-Craig, J., Borrusch, M., & Walton, J. (2010). Rapid optimization of enzyme mixtures for deconstruction of diverse pretreatment/biomass feedstock combinations. Biotechnology for Biofuels, 3(1), 22.
Bayer, E. A., Belaich, J.-P., Shoham, Y., & Lamed, R. (2004). The cellulosomes: multienzyme machines for degradation of plant cell wall polysaccharides. Annual Review of Microbiology, 58(1), 521-554.
Bengtsson, L. (2010). The global atmospheric water cycle. Environmental Research Letters.
Benjamino, J., & Graf, J. (2016). Characterization of the core and caste-specific microbiota in the termite, Reticulitermes flavipes. Frontiers in Microbiology, 7, 1.
Bharathiraja, B., Jayamuthunagai, J., Sudharsanaa, T., Bharghavi, A., Praveenkumar, R., Chakravarthy, M. et al. (2017). Biobutanol – an impending biofuel for future: a review on upstream and downstream processing techniques. Renewable and Sustainable Energy Reviews, 68, 788-807.
Bignell, D. E. (2011). Morphology, physiology, biochemistry and functional design of the termite gut: an evolutionary wonderland. In D. E. Bignell, Y. Roisin, & N. Lo (Eds.), Biology of Termites: a Modern Synthesis (pp. 375-412). Netherlands: Springer.
Bignell, D. E., Oskarsson, H., & Anderson, J. M. (1980). Specialization of the Hindgut Wall for the Attachment of Symbiotic Micro-Organisms in a Termite Procubitermes aburiensis (Isoptera, Termitidae, Termitinae). Zoomorphology, 96, 103-112.
Bischof, R. H., Ramoni, J., & Seiboth, B. (2016). Cellulases and beyond: the first 70 years of the enzyme producer Trichoderma reesei. Microbial Cell Factories, 15(1), 106.
Black, H. I. J., & Okwakol, M. J. N. (1997). Agricultural intensification, soil biodiversity and agroecosystem function in the tropics: the role of termites. Applied Soil Ecology, 6(1), 37-53.
Borowitzka, M. A., & Moheimani, N. R. (2013). Sustainable biofuels from algae. Mitigation and Adaptation Strategies for Global Change, 18(1), 13-25.
Boucias, D. G., Cai, Y., Sun, Y., Lietze, V.-U., Sen, R., Raychoudhury, R. et al. (2013). The hindgut lumen prokaryotic microbiota of the termite Reticulitermes flavipes and its responses to dietary lignocellulose composition. Molecular Ecology, 22(7), 1836-1853.
Bourguignon, T., Lo, N., Šobotník, J., Ho, S. Y. W., Iqbal, N., Coissac, E. et al. (2016). Mitochondrial Phylogenomics Resolves the Global Spread of Higher Termites, Ecosystem Engineers of the Tropics. Molecular Biology and Evolution, msw253.

22
BP (2017). BP Statistical Review of World Energy June 2017. Brethauer, S., & Studer, M. H. (2014). Consolidated bioprocessing of lignocellulose by
a microbial consortium. Energy & Environmental Science, 7(4), 1446. Brooksbank, K., Lever, M., Paterson, H., & Weybury, M. (2014). Biomass scoping study
(Bulletin 4862). Department of Agriculture and Food, Western Australia, Perth. Brune, A. (2014). Symbiotic digestion of lignocellulose in termite guts. Nature Reviews
Microbiology, 12(3), 168-180. Brune, A., & Ohkuma, M. (2011). Role of the termite gut microbiota in symbiotic
digestion. In D. E. Bignell, Y. Roisin, & N. Lo (Eds.), Biology of Termites: a Modern Synthesis (pp. 439-475). Netherlands: Springer.
Buijs, N. A., Siewers, V., & Nielsen, J. (2013). Advanced biofuel production by the yeast Saccharomyces cerevisiae. Current Opinion in Chemical Biology, 17(3), 480-488.
Calaby, J. H., & Gay, F. J. (1956). The distribution and biology of the genus Coptotermes (Isoptera) in Western Australia. Australian Journal of Zoology, 4(1), 19-39.
Catenacci, M., Verdolini, E., Bosetti, V., & Fiorese, G. (2013). Going electric: Expert survey on the future of battery technologies for electric vehicles. Energy Policy, 61, 403-413.
Chapman, I. (2014). The end of Peak Oil? Why this topic is still relevant despite recent denials. Energy Policy, 64, 93-101.
Cleveland, L. R. (1923). Symbiosis between termites and their intestinal protozoa. Proceedings of the National Academy of Sciences of the United States of America, 9(12), 424-428.
Coy, M. R., Salem, T. Z., Denton, J. S., Kovaleva, E. S., Liu, Z., Barber, D. S. et al. (2010). Phenol-oxidizing laccases from the termite gut. Insect Biochemistry and Molecular Biology, 40(10), 723-732.
Crutzen, P. J., & Stoermer, E. F. (2000). The Anthropocene. Global Change Newsletter, 41, 17-18.
Dereeper, A., Guignon, V., Blanc, G., Audic, S., Buffet, S., Chevenet, F. et al. (2008). Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Research, 36(Web Server issue), W465-9.
Dietrich, C., Köhler, T., & Brune, A. (2014). The cockroach origin of the termite gut microbiota: patterns in bacterial community structure reflect major evolutionary events. Applied and Environmental Microbiology.
Diouf, M., Roy, V., Mora, P., Frechault, S., Lefebvre, T., Hervé, V., Rouland-Lefèvre, C., Miambi, E. (2015). Profiling the succession of bacterial communities throughout the life stages of a higher termite Nasutitermes arborum (Termitidae, Nasutitermitinae) Using 16S rRNA Gene Pyrosequencing. PLoS One, 10, e0140014.
Dolan, M. F. (2001). Speciation of termite gut protists: the role of bacterial symbionts. International Microbiology, 4(4), 203-208.
Dutta, K., Daverey, A., & Lin, J.-G. (2014). Evolution retrospective for alternative fuels: first to fourth generation. Renewable Energy, 69, 114-122.
Eggleton, P. (2000). Global patterns of termite diversity. In T. Abe, D. E. Bignell, & M. Higashi (Eds.), Termites: Evolution, Sociality, Symbioses, Ecology (pp. 25-33). The Netherlands: Kluwer Academic Publishers.
Engel, M. S., Grimaldi, D. A., & Krishna, K. S. (2009). Termites (Isoptera): their phylogeny, classification, and rise to ecological dominance. American Museum Novitiates, 3650, 1-27.
Erenstein, O. (2002). Crop residue mulching in tropical and semi-tropical countries: An evaluation of residue availability and other technological implications. Soil

23
and Tillage Research, 67(2), 115-133. Esenther, G. R., & Kirk, T. K. (1974). Catabolism of aspen sapwood in Reticulitermes
flavipes (Isoptera: Rhinotermitidae). Annals of the Entomological Society of America, 67(6), 989-991.
Eutick, M. L., Veivers, P., O’Brien, R. W., & Slaytor, M. (1978). Dependence of the higher termite, Nasutitermes exitiosus and the lower termite, Coptotermes lacteus on their gut flora. Journal of Insect Physiology, 24(5), 363-368.
European Biofuels Technology Platform (2016). Biofuel Fact Sheet. Evans, T. A., & Gleeson, P. V. (2006). The effect of bait design on bait consumption in
termites (Isoptera: Rhinotermitidae). Bulletin of Entomological Research, 96(1), 85-90.
Fiorese, G., Catenacci, M., Verdolini, E., & Bosetti, V. (2013). Advanced biofuels: future perspectives from an expert elicitation survey. Energy Policy.
Flach, B., Lieberz, S., Rondon, M., Williams, B., & Wilson, C. (2016). EU Biofuels Annual 2016. Global Agricultural Information Network.
Gasparatos, A., Stromberg, P., & Takeuchi, K. (2013). Sustainability impacts of first-generation biofuels. Animal Frontiers, 3(2), 12-26.
Giannoccaro, G., de Gennaro, B. C., De Meo, E., & Prosperi, M. (2017). Assessing farmers’ willingness to supply biomass as energy feedstock: Cereal straw in Apulia (Italy). Energy Economics, 61, 179-185.
Haddadian, G., Khodayar, M., & Shahidehpour, M. (2015). Accelerating the global adoption of electric vehicles: barriers and drivers. The Electricity Journal, 28(10), 53-68.
Haq, F., Ali, H., Shuaib, M., Badshah, M., Hassan, S. W., Munis, M. F. H. et al. (2016). Recent progress in bioethanol production from lignocellulosic materials: A review. International Journal of Green Energy, 13(14), 1413-1441.
Ho, D. P., Ngo, H. H., & Guo, W. (2014). A mini review on renewable sources for biofuel. Bioresource Technology, 169, 742-749.
Hogan, M., Veivers, P. C., Slaytor, M., & Czolij, R. T. (1988). The site of cellulose breakdown in higher termites (Nasutitermes walkeri and Nasutitermes exitiosus). Journal of Insect Physiology, 34(9), 891-899.
Holland, S. P., Mansur, E. T., Muller, N. Z., & Yates, A. J. (2016). Are there environmental benefits from driving electric vehicles? The importance of local factors. American Economic Review, 106(12), 3700-3729.
Hongoh, Y., Ekpornprasit, L., Inoue, T., Moriya, S., Trakulnaleamsai, S., Ohkuma, M., Noparatnaraporn, N., Kudo, T. (2006). Intracolony variation of bacterial gut microbiota among castes and ages in the fungus-growing termite Macrotermes gilvus. Molecular Ecology, 15(2), 505-516.
Höök, M., & Tang, X. (2013). Depletion of fossil fuels and anthropogenic climate change—A review. Energy Policy, 52(0), 797-809.
Huang, J.-W., Cheng, Y.-S., Ko, T.-P., Lin, C.-Y., Lai, H.-L., Chen, C.-C. et al. (2012). Rational design to improve thermostability and specific activity of the truncated Fibrobacter succinogenes 1,3-1,4-β- d-glucanase. Applied Microbiology & Biotechnology, 94(1), 111-121.
Huang, X.-F., Bakker, M., Judd, T., Reardon, K., & Vivanco, J. (2013). Variations in diversity and richness of gut bacterial communities of termites (Reticulitermes flavipes) fed with grassy and woody plant substrates. Microbial Ecology, 65(3), 531-536.
Husseneder, C., Berestecky, J. M., & Grace, J. K. (2009). Changes in composition of culturable bacteria community in the gut of the formosan subterranean termite depending on rearing conditions of the host. Annals of the Entomological

24
Society of America, 102(3), 498-507. Wilkinson, I. (2017). Western Australian wheat industry. Retrieved 5/05/2017,
https://www.agric.wa.gov.au/grains-research-development/western-australian-wheat-industry.
Inoue, T., Kitade, O., Yoshimura, T., & Yamaoka, I. (2000). Symbiotic associations with protists. In T. Abe, D. E. Bignell, & M. Higashi (Eds.), Termites: Evolution, Sociality, Symbiosis, Ecology (pp. 275-288). Dordrecht: Kluwer Academic Publishers.
International Energy Agency (2011). Technology Roadmap, Biofuels for Transport. International Energy Agency (2017). IEA Oil Market Report. Inward, D., Beccaloni, G., & Eggleton, P. (2007). Death of an order: a comprehensive
molecular phylogenetic study confirms that termites are eusocial cockroaches. Biology Letters, 3(3), 331-335.
IPCC. (2011). Summary for Policymakers. In O. Edenhofer, R. Pichs-Madruga, Y. Sokona, K. Seyboth, P. Matschoss, S. Kadner et al. (Eds.), IPCC special report on renewable energy sources and climate change mitigation. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA.
Jin, C., Yao, M., Liu, H., Lee, C.-f. F., & Ji, J. (2011). Progress in the production and application of n-butanol as a biofuel. Renewable and Sustainable Energy Reviews, 15(8), 4080-4106.
Jones, D. T., & Eggleton, P. (2011). Global biogeography of termites: a compilation of sources. In D. E. Bignell, Y. Roisin, & N. Lo (Eds.), Biology of Termites: a Modern Synthesis (pp. 477-498). Netherlands: Springer.
Kircher, M. (2015). Sustainability of biofuels and renewable chemicals production from biomass. Curr Opin Chem Biol, 29, 26-31.
Koh, L. P., & Ghazoul, J. (2008). Biofuels, biodiversity, and people: understanding the conflicts and finding opportunities. Biological conservation, 141(10), 2450-2460.
Konig, H., Li, L., Wenzel, M., & Frohlich, J. (2006). Bacterial ectosymbionts which confer motility: Mixotricha paradoxa from the intestine of the Australian termite Mastotermes darwiniensis. Progress in Molecular & Subcellular Biology, 41, 77-96.
Krishna, K., Grimaldi, D. A., Krishna, V., & Engel, M. S. (2013). Treatise on the Isoptera of the world. Bulletin of the American Museum of Natural History, 1.
Lambertz, C., Garvey, M., Klinger, J., Heesel, D., Klose, H., Fischer, R. et al. (2014). Challenges and advances in the heterologous expression of cellulolytic enzymes: a review. Biotechnol Biofuels, 7(1), 135.
Lange, J.-P. (2007). Lignocellulose conversion: an introduction to chemistry, process and economics. Biofuels, Bioproducts and Biorefining, 1(1), 39-48.
Lavelle, P., Bignell, D., Lepage, M., Wolters, V., Roger, P., Ineson, P. et al. (1997). Soil function in a changing world: the role of invertebrate ecosystem engineers. European Journal of Soil Biology, 33(4), 159-193.
Lee, C.-Y., Charunee, V., & Lenz, M. (2007). Challenges to subterranean termite management of multi-genera faunas in Southeast Asia and Australia. Sociobiology, 50(1), 213-221.
Lee, T. R. C., Cameron, S. L., Evans, T. A., Ho, S. Y. W., & Lo, N. (2015). The origins and radiation of Australian Coptotermes termites: from rainforest to desert dwellers. Molecular Phylogenetics and Evolution, 82, 234-244.
Leite, G. B., Abdelaziz, A. E. M., & Hallenbeck, P. C. (2013). Algal biofuels: challenges and opportunities. Bioresource Technology, 145, 134-141.
Li, H., Dietrich, C., Zhu, N., Mikaelyan, A., Ma, B., Pi, R. et al. (2016). Age polyethism drives community structure of the bacterial gut microbiota in the fungus-

25
cultivating termite Odontotermes formosanus. Environmental Microbiology, 18(5), 1440-1451.
Liang, C., Fioroni, M., Rodríguez-Ropero, F., Xue, Y., Schwaneberg, U., & Ma, Y. (2011). Directed evolution of a thermophilic endoglucanase (Cel5A) into highly active Cel5A variants with an expanded temperature profile. Journal of Biotechnology, 154(1), 46-53.
Malinda, D. K. (1995). Factors in conservation farming that reduce erosion. Australian Journal of Experimental Agriculture, 35(7), 969-978.
Mata, T. M., Martins, A. A., & Caetano, N. S. (2010). Microalgae for biodiesel production and other applications: a review. Renewable and Sustainable Energy Reviews, 14(1), 217-232.
Mathiesen, B. V., Lund, H., & Karlsson, K. (2011). 100% Renewable energy systems, climate mitigation and economic growth. Applied Energy, 88(2), 488-501.
Mikaelyan, A., Thompson, C. L., Hofer, M. J., & Brune, A. (2016). Deterministic assembly of complex bacterial communities in guts of germ-free cockroaches. Appl Environ Microbiol, 82(4), 1256-1263.
Mitchell, J. (1989). The ”greenhouse” effect and global warming. Review of Geophysics. Miyata, R., Noda, N., Tamaki, H., Kinjyo, K., Aoyagi, H., Uchiyama, H., Tanaka, H.
Influence of feed components on symbiotic bacterial community structure in the gut of the wood-feeding higher termite Nasutitermes takasagoensis. Bioscience, Biotechnology, and Biochemistry, 71(5), 1244-1251.
Mohanram, S., Amat, D., Choudhary, J., Arora, A., & Nain, L. (2013). Novel perspectives for evolving enzyme cocktails for lignocellulose hydrolysis in biorefineries. Sustainable Chemical Processes, 1(1), 15.
Moraïs, S., Barak, Y., Caspi, J., Hadar, Y., Lamed, R., Shoham, Y. et al. (2010). Cellulase-xylanase synergy in designer cellulosomes for enhanced degradation of a complex cellulosic substrate. mBio, 1(5), e00285-10.
Morais, S., Barak, Y., Lamed, R., Wilson, D., Xu, Q., Himmel, M. et al. (2012). Paradigmatic status of an endo- and exoglucanase and its effect on crystalline cellulose degradation. Biotechnology for Biofuels, 5(1), 78.
Nalepa, C. A. (2015). Origin of termite eusociality: trophallaxis integrates the social, nutritional, and microbial environments. Ecological Entomology, 40(4), 323-335.
Ndaba, B., Chiyanzu, I., & Marx, S. (2015). n-Butanol derived from biochemical and chemical routes: A review. Biotechnology reports (Amsterdam, Netherlands), 8, 1-9.
Nguyen, Q., Bowyer, J., Howe, J., Bratkovich, S., & Groot…, H. (2017). Global production of second generation biofuels: trends and influences. Dovetail Partners, Inc.
Ohkuma, M., Noda, S., Hongoh, Y., Nalepa, C. A., & Inoue, T. (2009). Inheritance and diversification of symbiotic trichonymphid flagellates from a common ancestor of termites and the cockroach Cryptocercus. Proceedings of the Royal Society B: Biological Sciences, 276(1655), 239-245.
Ohkuma, M. (2003). Termite symbiotic systems: efficient bio-recycling of lignocellulose. Applied Microbiology and Biotechnology, 61(1), 1-9.
Oppert, C., Klingeman, W. E., Willis, J. D., Oppert, B., & Jurat-Fuentes, J. L. (2010). Prospecting for cellulolytic activity in insect digestive fluids. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology, 155(2), 145-154.
Otani, S., Mikaelyan, A., Nobre, T., Hansen, L. H., Kone, N. A., Sorensen, S. J., Aanen, D.K., Boomsma, J.J., Brune, A. Poulsen, M (2014). Identifying the core microbial community in the gut of fungus-growing termites. Molecular Ecology.

26
Payne, C. M., Knott, B. C., Mayes, H. B., Hansson, H., Himmel, M. E., Sandgren, M. et al. (2015). Fungal cellulases. Chemical Reviews, 115(3), 1308-1448.
Peters, B. C., & Fitzgerald, C. J. (2003). Field evaluation of the bait toxicant chlorfluazuron in eliminating Coptotermes acinaciformis (Froggatt) (Isoptera: Rhinotermitidae). Journal of Economic Entomology, 96(6), 1828-1831.
Pfromm, P. H., Amanor-Boadu, V., Nelson, R., Vadlani, P., & Madl, R. (2010). Bio-butanol vs. bio-ethanol: A technical and economic assessment for corn and switchgrass fermented by yeast or Clostridium acetobutylicum. Biomass and Bioenergy, 34(4), 515-524.
Poulsen, M. (2015). Towards an integrated understanding of the consequences of fungus domestication on the fungus-growing termite gut microbiota: Fungus-growing termite gut microbiota. Environmental Microbiology, 17(8), 2562-2572.
Raman, B., Pan, C., Hurst, G. B., Rodriguez Jr, M., McKeown, C. K., Lankford, P. K. et al. (2009). Impact of pretreated switchgrass and biomass carbohydrates on Clostridium thermocellum ATCC 27405 cellulosome composition: a quantitative proteomic analysis. PLoS ONE, 4(4), 1-13.
Ransom-Jones, E., Jones, D. L., McCarthy, A. J., & McDonald, J. E. (2012). The Fibrobacteres: an Important phylum of cellulose-degrading bacteria. Microbial Ecology, 63, 267-281.
Rawls, J. F., Mahowald, M. A., Ley, R. E., & Gordon, J. I. (2006). Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell, 127(2), 423-433.
Reid, N. M., Addison, S. L., West, M. A., & Lloyd-Jones, G. (2014). The bacterial microbiota of Stolotermes ruficeps (Stolotermitidae), a phylogenetically basal termite endemic to New Zealand. FEMS Microbiology Ecology, 90(3), 678-688.
Renewable Fuels Association, World Fuel Ethanol Production. Retrieved 5/05/2017, http://www.ethanolrfa.org/resources/industry/statistics/.
Resch, M. G., Donohoe, B. S., Baker, J. O., Decker, S. R., Bayer, E. A., Beckham, G. T. et al. (2013). Fungal cellulases and complexed cellulosomal enzymes exhibit synergistic mechanisms in cellulose deconstruction. Energy & Environmental Science, 6, 1858.
Farrell, R. (2016). Australia Biofuels Annual 2016. Global Agricultural Information Network.
Scharf, M. E. (2015). Omic research in termites: an overview and a roadmap. Frontiers in Genetics, 6, 76.
Schmitz, P. M., & Kavallari, A. (2009). Crop plants versus energy plants—On the international food crisis. Bioorganic & Medicinal Chemistry, 17(12), 4020-4021.
Searcy, E., Flynn, P., Ghafoori, E., & Kumar, A. (2007). The relative cost of biomass energy transport. Applied Biochemistry and Biotechnology, 137-140(1-12), 639-652.
Sethi, A., Slack, J. M., Kovaleva, E. S., Buchman, G. W., & Scharf, M. E. (2013). Lignin-associated metagene expression in a lignocellulose-digesting termite. Insect Biochemistry and Molecular Biology, 43, 91-101.
Shade, A., & Handelsman, J. (2012). Beyond the Venn diagram: the hunt for a core microbiome: The hunt for a core microbiome. Environmental Microbiology, 14(1), 4-12.
Shapira, M. (2016). Gut microbiotas and host evolution: scaling up symbiosis. Trends in Ecology & Evolution, 31(7), 539-549.
Shepherd, M., Bartle, J., Lee, D. J., Brawner, J., Bush, D., Turnbull, P. et al. (2014). Eucalypts as a biofuel feedstock. Biofuels, 2(6), 639-657.
Sims, R., Schaeffer, R., Creutzig, F., Cruz-Núñez, X., D’Agosto, M., Dimitriu, D. et al. (2014). Transport. In O. Edenhofer, R. Pichs-Madruga, Y. Sokona, E. Farahani, S.

27
Kadner, K. Seyboth et al. (Eds.), Climate change 2014: mitigation of climate change. contribution of working group III to the fifth assessment report of the intergovernmental panel on climate change. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA.
Smith, P., & Gregory, P. J. (2013). Climate change and sustainable food production. Proceedings of the Nutrition Society, 72(1), 21-28.
Steffen, W., Leinfelder, R., Zalasiewicz, J., Waters, C. N., Williams, M., Summerhayes, C. et al. (2016). Stratigraphic and Earth system approaches to defining the Anthropocene: defining the Anthropocene. Earth’s Future, 4(8), 324-345.
Tanaka, H., Aoyagi, H., Shiina, S., Doudou, Y., Yoshimura, T., Nakamura, R., Uchiyama, H. (2006). Influence of the diet components on the symbiotic microorganisms community in hindgut of Coptotermes formosanus Shiraki. Applied Microbiology & Biotechnology, 71(6), 907-917.
Tokuda, G., & Watanabe, H. (2007). Hidden cellulases in termites: revision of an old hypothesis. Biology Letters, 3(3), 336-339.
Vassilev, S. V., & Vassileva, C. G. (2016). Composition, properties and challenges of algae biomass for biofuel application: An overview. Fuel, 181, 1-33.
Veivers, P. C., O’Brien, R. W., & Slaytor, M. (1983). Selective defaunation of Mastotermes darwiniensis and its effect on cellulose and starch metabolism. Insect Biochemistry, 13(1), 95-101.
Wang, Y., Su, L., Huang, S., Bo, C., Yang, S., Li, Y. et al. (2016). Diversity and resilience of the wood-feeding higher termite Mironasutitermes shangchengensis gut microbiota in response to temporal and diet variations. Ecology and Evolution, 6(22), 8235-8242.
Webb, G. A., & Mcclintock, C. (2015). Elimination of the mound-building termite, Nasutitermes exitiosus (Isoptera: Termitidae) in South-Eastern Australia using bistrifluron bait. Journal of Economic Entomology, 108(6), 2702-2710.
Western Australia Biofuels Taskforce (2007). Report: April 2007. Wilson, D. B. (2011). Microbial diversity of cellulose hydrolysis. Current Opinion in
Microbiology, 14(3), 259-263. Wolfram, C., Shelef, O., & Gertler, P. (2012). How will energy demand develop in the
developing world? Journal of Economic Perspectives, 26(1), 119-138. Wu, H., Fu, Q., Giles, R., & Bartle, J. (2008). Production of mallee biomass in Western
Australia: energy balance analysis. Energy Fuels, 22(1), 190-198. Xiang, H., Xie, L., Zhang, J., Long, Y., Liu, N., Huang, Y., Wang, Q. (2012).
Intracolonial differences in gut bacterial community between worker and soldier castes of Coptotermes formosanus. Insect Science, 19, 86-95.
Yu, Y., Bartle, J., Mendham, D., & Wu, H. (2015). Site variation in life cycle energy and carbon footprints of mallee biomass production in Western Australia. Energy Fuels, 29(6), 3748-3752.
Zuroff, T. R., & Curtis, W. R. (2012). Developing symbiotic consortia for lignocellulosic biofuel production. Applied Microbiology and Biotechnology, 93(4), 1423-1435.

28

29
CHAPTER 2: Colony Differences in the Gut Communities of Western Australian Termites Tumulitermes westraliensis and Coptotermes acinaciformis raffrayi

30
Foreword:
The gut bacteria of termites are generally thought to be highly conserved, yet
the natural level of intraspecific variation has rarely been studied. In this
chapter I therefore begin to answer my first research question: How much
intraspecific variation exists in the gut communities of Western Australian
endemic termites? Sample replication in termite studies is typically low and
because there are no standardised primer pairs and PCR conditions across the
field, it is unclear whether datasets can be combined for greater explanatory
power. Two 16S rRNA gene amplification strategies were compared to measure
technical variation across datasets collected with different methods, before
comparing two local species to each other and to previous studies conducted
with the same methods.
Introduction:
Termites are eusocial insects that break down lignocellulose in various stages
of humification, with the use of both endogenous cellulases and symbioses
with cellulose-degrading microbes (reviewed by Brune, 2014). There are close
to 3,000 species of termites worldwide and these are typically classified as
either lower or higher termites (Krishna et al., 2013). Lower termites primarily
feed on wood and their gut is dominated by cellulose-degrading flagellates and
their associated prokaryotes (Cleveland, 1923; Inoue et al., 2000; Dolan, 2001).
Higher termites (Termitidae) appear to have lost these flagellates
approximately 60 million years ago, switching to a predominantly prokaryotic
gut community (Ohkuma, 2003; Tokuda and Watanabe, 2007; Brune, 2014).
They have become the most species-rich and ecologically diverse termite
family, containing over 80% of all termite species, each specialised to consume
wood, grass, litter, humus, soil or fungus (Jones and Eggleton, 2011; Brune and
Ohkuma, 2011; Bignell, 2011; Poulsen, 2015).

31
The bacterial gut community of termites is therefore key to their success, but
the drivers of community composition (the diversity of bacterial taxa present)
and community structure (the relative abundance of bacterial taxa) remain
poorly understood. Choosing case studies to examine the sources of natural
intraspecific variation would help to elucidate these community drivers.
Within colony differences may reflect different roles of castes and age groups
(Hongoh et al., 2006; Xiang et al., 2012; Diouf et al., 2015; Li et al., 2016;
Benjamino and Graf, 2016), but intercolony variation has rarely been studied.
To my knowledge, only four studies have discussed colony variation in mature
worker gut communities to date. Hongoh et al. (2005) found that the bacterial
community structure was conserved to some degree within the genera
Reticulitermes (lower termites) and Microcerotermes (higher termites), but
also found significant differences between certain sampling sites and termite
species within each genus. Boucias et al. (2013) found that the Reticulitermes
flavipes colony of origin had a stronger influence on the gut community
composition than the wood or paper diet they provided for a week. Benjamino
and Graf (2016) showed that the gut communities of termites from the same
colony were more similar to each other than between colonies, although they
shared a core community that accounted for most of the reads. Reid et al.
(2014) found that colonies of the lower termite Stolotermes ruficeps had
unique gut communities, even when collected in close proximity. They
speculated that differences in diet, in terms of wood species or humification
state, might explain these differences.
Intraspecies sample replication in termite studies to date is low and it is
unclear whether already published datasets can be combined for greater
explanatory power because there is no standardised method within the field.
Various PCR primers targeting different 16S rRNA gene hypervariable regions
have been used to characterise the gut community of termites (Miyata et al.,
2007; Köhler et al., 2012; He et al., 2013; Abdul Rahman et al., 2015). However,
it is known that the choice of PCR conditions always introduces bias in 16S
datasets (Sipos et al., 2007; Tremblay et al., 2015). I therefore tested two

32
different amplification strategies to measure the effect of such bias on the
recovered gut community to assess the comparability of datasets recovered
with different methods.
To investigate intraspecific variation in gut community composition and
structure, I chose a lower and a higher termite species known to have broad
diets. Coptotermes acinaciformis raffrayi is endemic to Western Australia (Lo
et al., 2006; Lee et al., 2015) and feeds broadly on the humification gradient,
from live to rotten wood (Greaves, 1962; Lee et al., 2015). Tumulitermes
westraliensis is a Western Australian endemic of the subfamily
Nasutitermitinae. It was first classified as a grass-feeder (Hill, 1921; Hill, 1925;
Hill, 1942) and its diet later refined to include dried grass, grass-tree
(Xanthorrhoea) debris and seeds (Abensperg-Traun and Milewski, 1995),
suggesting that T. westraliensis feeds on a wide variety of plant material with
varying degrees of lignification. This study investigates, for the first time, the
gut communities of these two termite species and moreover, is the first
examination of the gut community within the genus Tumulitermes.
The T. westraliensis gut community composition and structure was then
compared to those of previously studied higher termites obtained using the
same method (Dietrich et al., 2014; Mikaelyan et al., 2015a). I hypothesised that
the gut community of T. westraliensis would be colony specific and different
to that of true grass-feeders, reflecting its broader diet. Likewise, I compared
C. acinaciformis raffrayi to other lower termite species (Dietrich et al., 2014),
hypothesising that its gut community would be similar to other broadly
feeding species.

33
Methods:
Sample collection
T. westraliensis and C. acinaciformis raffrayi were collected from two different
areas near Perth, Western Australia (Figure 1). T. westraliensis samples were
collected from three mounds (T1, T2 and T3) in Banyowla Regional Park in the
Perth Hills. A small pickaxe was used to break into mounds and mature
workers, recognisable by their darker head capsules (Figure 2A), were
recovered with soft forceps. For colony T3, triplicate samples were collected
(T3a, T3b, T3c) to measure the level of intracolony variation (V4 dataset only,
see below). Food stores observed in T. westraliensis mounds during sample
collection included varying amounts of dried grass, dead grass tree leaves and
sections of woody stem (Figure 2D). Two C. acinaciformis raffrayi samples
were collected in the Gnangara pine plantation, one from a rotten pine log
(C1) and the other from a sound native tree species log (C2). Each sample was
collected at least 80 m from its nearest conspecific neighbour under the
assumption that these would be separate, independent colonies. Soldiers of
both species were also collected and stored in 70% EtOH to confirm species
identification using morphological characters and COII sequencing (with a
minimum of 99% sequence identity with previously published sequences;
Figure 2B).
Samples were transported to the laboratory in the dark with mound or woody
material from their colony. Dissections were performed on the day of
collection to ensure that the natural gut flora was sampled. Samples from
different colonies were dissected separately using sterilised equipment and
solutions, as follows. Workers were anaesthetised in a petri dish on ice for at
least ten minutes. To prevent contamination of samples during the dissection,
workers were then surfaced sterilised by immersion in a 5% hypochlorite
solution for 1 min, rinsed twice in distilled water and dried on filter paper on
ice (Rosengaus and Traniello, 1997). Whole intestinal tracts were dissected by

34
Fi
gure
1:
Sam
plin
g lo
catio
ns n
ear
Pert
h, W
este
rn A
ustr
alia
(A
). Tu
mul
iterm
es w
estr
alie
nsis
sam
ples
wer
e co
llect
ed i
n Ba
nyow
la
Regi
onal
Par
k (B
) an
d C
opto
term
es a
cina
cifo
rmis
raf
fray
i was
sam
pled
from
the
Gna
ngar
a St
ate
Fore
st (
C).
All
sam
ples
wer
e co
llect
ed
at l
east
80
m a
part
and
are
ass
umed
to
be s
epar
ate
colo
nies
. Red
: T. w
estr
alie
nsis
; yel
low
: C. a
cina
cifo
rmis
raf
fray
i. M
aps
mod
ified
fr
om G
PSV
isua
lizer
.com
.

35
Figure 2: Tumulitermes westraliensis, a Western Australian endemic species historically classified as a grass-feeder. Mature workers (A) with dark head capsules were used in preference to younger workers (marked with an asterisk), here photographed on clay. Soldiers (B) were used for species identification by morphology and COII sequencing. Whole worker guts (C) were pooled to profile the gut community; the gut includes the crop, midgut and hindgut segments (P1 to P5). A typical food store inside a mound (D) shows a mixture of woody (1), grassy (2) and grass tree leaf (3) material.

36
holding the head with forceps and pulling gently at the distal end of the
abdomen using Inox 5 Watchmaker forceps. Each excised intestinal tract (see
Figure 2C for an example) was placed in a 1.5 ml Eppendorf tube containing
100 μl of 1× phosphate buffered saline. The process was repeated to obtain a
pool of 10 guts for each colony.
To confirm species identity, DNA was extracted from whole individual soldiers
using the Zymo Research Tissue & Insect DNA MiniPrep™ Kit and the COII
gene amplified with the primers A-tLeu (5′-ATGGCAGATTAGTGCAATGG-
3′) and B-tLys (5′- GTTTAAGAGACCAGTACTTG-3′) as described by Liu and
Beckenbach (1992). Resulting amplicons were sequenced in both directions by
Macrogen (Korea). Paired-end sequences were aligned and trimmed to obtain
an identical overlapping region. These quality trimmed COII sequences were
compared within a species and to previously sequenced COII genes using the
nucleotide BLAST online tool (National Center for Biotechnology
Information).
Parallel sequencing and data analysis
DNA was extracted from pooled gut samples using a previously established
protocol (Paul et al., 2012) except that the bead-beating step was undertaken
with a Retsch Mixer Mill (MM301) for 3 min as detailed by Delmotte et al.
(2009). The V4 hypervariable region of the 16S rRNA gene (henceforth V4) was
amplified using the V4 primers recommended by Illumina at the time of data
collection in 2013, F515 (5′-GTGCCAGCMGCCGCGGTAA-3′) and R806 (5′-
GGACTACHVGGGTWTCTAAT-3′), as described by Caporaso et al. (2011).
Resulting amplicons were sequenced in-house as paired-end 150 bp reads on
an Illumina MiSeq using a 300 cycle Miseq Reagent Kit v2 according to the
manufacturer’s instructions. Aliquots of each DNA sample representing the 5
colonies (including only the first replicate of T3) were also amplified using V3-
V4 primers 343Fmod (5′-TACGGGWGGCWGCA-3′) and 784Rmod (5′-
GGGTMTCTAATCCBKTT-3′) designed by Köhler et al. (2012) for termite gut

37
community studies (henceforth V3-V4) and sequenced as described by
Mikaelyan et al. (2015a).
Data analysis utilised Usearch's UPARSE pipeline (Edgar, 2013), beginning
with assembling and quality filtering paired-end fastq reads, through to the
creation of an OTU (Operational Taxonomic Unit) table containing raw read
counts for each bacterial group. Quality filtering used the "stringent" method
recommended in Usearch, which removed reads with an expected error above
1. Singleton sequences were removed prior to clustering into OTUs. The
unfiltered trimmed dataset was used when mapping reads to the OTUs to
create an OTU table. This allowed the use of the full dataset while reducing
the likelihood of spurious OTUs.
During the quality-filtering step, the V4 reads were trimmed to 253 bp (the
expected length of the region targeted with the V4 primers), whereas the V3-
V4 reads were trimmed to 400 bp, to maximise read length while maintaining
read quality. The V3-V4 data was quality-filtered alongside 454 and MiSeq
higher termite data from previous studies (Dietrich et al., 2014; Mikaelyan et
al., 2015a) to allow for direct comparisons between the datasets. The V3-V4
dataset was filtered and trimmed a second time to match the V4 region
targeted by the V4 primers, to distinguish amplification bias from bias arising
during the taxonomy assignment step of the data analysis. In this case,
trimming involved removing the first 203 bases and shortening reads to 253
bp. This trimmed V3-V4 dataset was analysed together with the V4 dataset,
separately for each species, and will be hereafter referred to as the combined
V4 dataset.
OTUs were determined de novo for each of the three sub-analyses (V4, V3-V4
and combined V4) but all were clustered at 97% identity to represent bacterial
species. Taxonomy was assigned to the OTU tables in QIIME 1.9 (Caporaso et
al., 2010) using DictDb version 3.0, the curated 16S termite and cockroach
database created by Mikaelyan et al. (2015b), and the RDP classifier (Wang et

38
al., 2007), with a confidence value of 0.8. The datasets were rarefied to
different depths to normalise to the sample with the lowest number of reads in
each sub-analysis, as described below.
Amplification bias
Primer bias was predicted for both amplification strategies using a java script
(available upon request) to search for V4 and V3-V4 primer sequences within
reference sequences in DictDb version 3.0, taking into account all possible
versions of ambiguous bases in the primers but allowing no other mismatches.
The number of matches for each primer pair was compared separately for each
of the phyla represented in the database.
To characterise amplification bias in the biological dataset, the combined V4
dataset was summarised as a genus-level heatmap. This dataset was rarefied to
49,800 reads per sample, then OTUs were picked within each species and
clustered at the genus level using a relational database based on the DictDb
taxonomy assignments. Any OTUs not identified to the genus level were
removed (on average 22% of reads per sample for C. acinaciformis raffrayi and
38% for T. westraliensis). The raw read counts were then all increased by a
value of one (to eliminate zeros) and transformed to log 10 values. A phylum-
level summary was created by averaging across all colonies within each
species and including all reads identified to at least phylum level
(approximately 99% of reads). For ease of visualisation, only the 50 most
abundant genera in the gut of each species were included in the heatmap .
Heatmaps were created with Bioconductor's ComplexHeatmap package in R
(Gu, 2016). Paired t-tests conducted in R (R Development Core Team, 2016)
were used to contrast the abundance of each bacterial genus and phylum
measured using the V4 and V3-V4 methods.

39
Beta diversity
Two beta diversity metrics were applied to each sub-analysis in QIIME to
obtain distance values for each possible pairwise comparison. The metrics
used were unweighted UniFrac (which considers presence/absence of species
and their phylogenetic relationship, hereafter referred to as community
composition) and weighted UniFrac (which takes into account phylogenetic
relationships and relative abundance, hereafter community structure). To
visualise bias introduced by laboratory method in the V4 and the V3-V4
datasets, Principal Coordinate Analysis (PCoA) was conducted on the
combined V4 dataset in QIIME and plotted with ggplot2 (Wickham, 2009).
The V4 dataset containing replicates for colony T3 was rarefied to 500,000
reads per sample and used to measure intraspecific variation. The distance
measures were categorised as a) nestmates (pairs from the same colony; T3
only, n = 3), b) conspecifics (pairs from different colonies within a species, n =
8), or c) heterospecifics (pairs of T. westraliensis and C. acinaciformis raffrayi
colonies; n = 10). The difference in variation was tested within each metric
using the Kruskal-Wallis test and a post-hoc analysis with the Mann-Whitney-
Wilcoxon Test in R.
To compare the gut communities in T. westraliensis and C. acinaciformis
raffrayi to those reported in previous studies, OTUs were picked for the V3-V4
dataset for lower and higher termites separately and each sub-analysis rarefied
to 3,000 reads. PCoA was conducted as described above. For the higher
termites, the humus and litter groups were almost indistinguishable and
therefore consolidated as in Mikaelyan et al. (2015a). Mikaelyan et al. (2015a)
included Trinervitermes sp. with the wood-feeders and excluded Amitermes
meridionalis (from Dietrich et al., 2014). Here, they were both retained in a
separate grass-feeding group for comparison with T. westraliensis. Samples
were coloured by the termite host subfamily and points were shaped as per
their respective diet group (Gontijo and Domingos, 1991; Jones and Eggleton,
2011; Mikaelyan et al., 2015a), with the T. westraliensis samples forming an

40
"unknown" group. Finally, ellipses representing 95% confidence intervals for
each diet group containing at least four samples were superimposed on the
plots.
Alpha Diversity
Alpha diversity analysis was conducted in QIIME. The alpha diversity metrics
used were the number of observed OTUs (a measure of species richness) and
Simpson's Reciprocal Index (a measure of dominance). Rare OTUs have a
relatively low impact on the Index compared to more frequent OTUs, however
the uniformity in abundance caused by excluding rare OTUs would inflate the
Index. Non-parametric t-tests (with 999 Monte Carlo permutations) were
applied to each alpha diversity metric.
This analysis was performed on the combined V4 dataset, to determine the
effect of method choice on the detected bacterial species richness and
diversity. It was also used on all lower and higher termites making up the V3-
V4 dataset, to determine any correlations between diet and the species
richness in the gut.
Results:
The two amplification strategies yielded significantly different levels of
detection for particular bacterial groups in both gut communities, with a more
obvious bias in T. westraliensis. Signature gut communities were detected for
each colony, with greater differences between species, moderate differences
within a species, and minimal differences between nestmates. I found that
colonies of T. westraliensis had similar gut community structure to different
higher termites feeding on a variety of diets.

41
Technical biases in gut community sequencing
Comparison of both primer pairs to DictDb version 3.0 predicted primer bias
for all phyla in the database. Fewer than 640 sequences representing 15 phyla
(1.15% of 55,732 sequences from 62 phyla in DictDb) perfectly matched the V4
and/or V3-V4 primer pairs (Table 1). Within this subset, phylum Tenericutes
had seven matches with the V4 primer set and no matches with the V3-V4
primers. Candidate phylum TM7 had five matches with the V3-V4 primer set
and none with the V4 primers.
In practice, the two amplification strategies yielded significantly different
levels of detection for 14 out of 21 phyla in the gut of T. westraliensis (Figure
3C). When considering all reads identified to at least phylum level, the most
abundant phyla in the T. westraliensis gut were Spirochaetes, Firmicutes,
Bacteroidetes and Fibrobacteres in both datasets. However, Spirochaetes were
detected twice as frequently using the V4 method, while Firmicutes and
Bacteroidetes were over three times more frequent in the V3-V4 dataset. The
relative frequency for Fibrobacteres was not statistically different between the
two methods. Detection differed by more than 10:1 for two phyla, including an
average of 85 reads for Planctomycetes with the V4 primers and only 7 with
V3-V4 and an average of 204 for Synergistetes using V3-V4 primers and only 10
with V4. Cyanobacteria was only detected using the V4 method (46 reads)
while candidate phyla TM7 (958 reads) and SR1 (105) were only recorded in the
V3-V4 dataset (Table S1A; attached as Chapter2_TableS1.xlsx).
In the C. acinaciformis raffrayi gut, four out of 17 phyla were detected
differently between the methods (Figure 3D). The most abundant phyla were
Bacteroidetes, Spirochaetes, Firmicutes (significantly different), Elusimicrobia
(significantly different) and Proteobacteria in both datasets. However,
Spirochaetes was detected more than twice as frequently in the V4 dataset and
Firmicutes almost twice as frequently in the V3-V4 dataset. Elusimicrobia was
recorded four times more frequently in the V3-V4 dataset. Cyanobacteria,
Planctomycetes, Tenericutes were also all detected more frequently in the V4

42
Table 1: Phylum-level table containing numbers of sequences in the DictDb version 3.0 16S curated reference database that contain the forward and reverse primer sequences used in this study (allowing no mismatches and all possible versions of ambiguous bases) of the primer set targetting the V4 region of the 16S gene and the V3-V4 primer set. Phylum Tenericutes is biased against using the V3-V4 primer set, while TM7 is biased against with the V4 primers.
Phylum V4 matches V3-V4 matches Actinobacteria 8 8
Bacteroidetes 210 213
Candidate phylum TG3 2 2
Candidate phylum TM7 0 5
Chlorobi 2 2
Cyanobacteria 1 1
Deferribacteres 2 3
Elusimicrobia 25 21
Fibrobacteres 6 6
Firmicutes 186 191
Planctomycetes 10 10
Proteobacteria 53 51
Spirochaetes 90 89
Synergistetes 29 30
Tenericutes 7 0
Verrucomicrobia 4 7
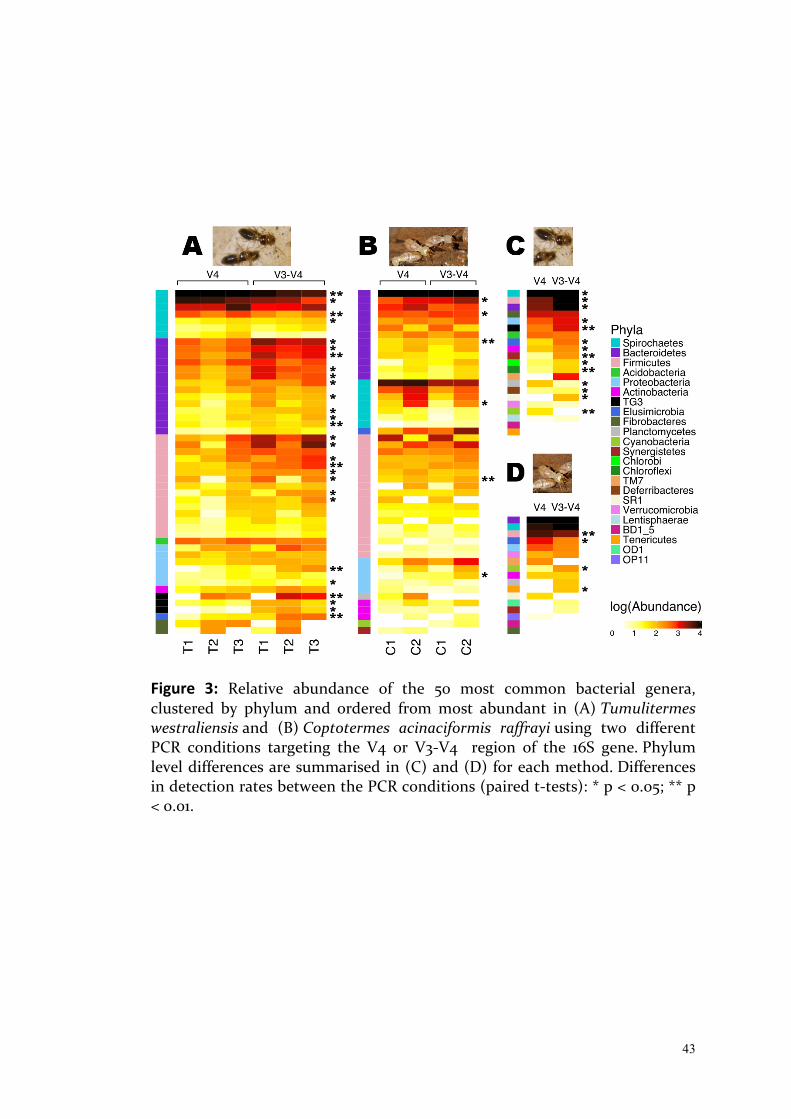
43
Figure 3: Relative abundance of the 50 most common bacterial genera, clustered by phylum and ordered from most abundant in (A) Tumulitermes westraliensis and (B) Coptotermes acinaciformis raffrayi using two different PCR conditions targeting the V4 or V3-V4 region of the 16S gene. Phylum level differences are summarised in (C) and (D) for each method. Differences in detection rates between the PCR conditions (paired t-tests): * p < 0.05; ** p < 0.01.

44
dataset, the latter two phyla represented by only 2 reads, on average, with the
V3-V4 method. Candidate phyla OD1 (14 reads) and Synergistetes (12) were
only detected using the V4 methods, while phyla SR1 (93 reads) and OP11 (5)
were only detected with the V3-V4 primer pair. TM7 was predominently
detected with the V3-V4 primer pair, resulting in 258 reads as compared to 2
with V4 (Table S1B).
Bias was also apparent at the genus level. Detection of 28 of the 50 genera in
Figure 3A was significantly different between methods in the T. westraliensis
gut, eight of which were highly significantly different (p < 0.01). These, in
order of appearance in Figure 3A, were: Treponema Ia and Treponema Ih
(Spirochaetes); M2PB4-61 termite group and Alisitpes IV (Bacteroidetes); Gut
cluster 7 (Firmicutes); Uncultured genus 2 (Proteobacteria); Subcluster IIIb
(TG3); and Endomicrobium (Elusimicrobia). Treponema Ia was the most
abundant genus in both datasets but was detected on average three times
more frequently in the V4 dataset, making up 39% of reads (Spirochaetes as a
whole made up 80% of V4 reads). M2PB4-61 termite group and Gut cluster 7
were detected five times more frequently using the V3-V4 method. Uncultured
genus 2 was recorded almost seven times, Subcluster IIIb four times and
Endomicrobium nine times more frequently in the V3-V4 dataset (Table S1A).
The vadinBC27 wastewater-sludge group (Bacteroidetes) and Termite
cockroach cluster (Synergistetes) were only detected using the V3-V4 primer
pair (Table S1A).
For C. acinaciformis raffrayi, only six of the 50 total genera represented in
Figure 3B were significantly different between the methods: Candidatus
Armantifilum, BCf9-17 termite group (Bacteroidetes), Termite Cockroach
cluster (Bacteroidetes, p < 0.01), Treponema II (Spirochaetes), Uncultured
genus 12 (Firmicutes, p < 0.01), and Uncultured genus 2 (Proteobacteria). The
gut was always found to be dominated by Candidatus Azobacteroides
(Bacteroidetes) with approximately 50% of reads assigned to this genus using
both methods. Treponema II was detected over three times more frequently in

45
the V4 dataset, while Uncultured genus 2 and Endomicrobium were recorded
four times as often in the V3-V4 dataset. Candidatus Hepatincola
(Proteobacteria) was the only other genus to be detected at least three times
more in one of the datasets, here in V3-V4. Termite cockroach cluster 2
(Planctomycetes) was recorded in the V4 dataset only, while Cluster TG2
(Cyanobacteria) was only detected in the V3-V4 dataset Table S1B).
Despite these differences in abundance, no significant differences were found
with either alpha diversity metric when comparing the two datasets (non-
parametric t-tests with 999 Monte Carlo permutations). A total of 1041
bacterial species were estimated in the T. westraliensis gut using the V3-V4
dataset, versus 982 in the V4 dataset (p = 0.826); and 385 estimated in the C.
acinaciformis raffrayi gut with V3-V4 versus 357 with V4 (p = 0.664). The
Simpson's Reciprocal Index averaged to 55.5 for T. westraliensis in the V3-V4
dataset and 21.3 for the V4 dataset (p = 0.141); the Index averaged 7.0 and 4.9 in
C. acinaciformis raffrayi respectively (p = 0.681).
PCoA was conducted on the combined V4 dataset to visualise any differences
caused by primer choice (Figure S1). With the unweighted UniFrac metric, PC1
explained 56% and 47% of the variation for C. acinaciformis raffrayi (Figure
S1B) and T. westraliensis (Figure S1A) respectively and samples clustered
primarily with their match from the alternate amplification strategy. PC2
separated the primers used, accounting for 45% and 20% of the variation.
With the weighted metric, samples clustered primarily by primer pair, with
PC1 describing 82% and 74% of the variation amongst C. acinaciformis raffrayi
and T. westraliensis samples. PC2 explained 18% and 14% of the variation,
clustering the samples by colony. The T. westraliensis colony 3 biological
replicates (T3a, T3b and T3c) clustered more closely within the V4 dataset
than the first replicate with its identical counterpart in the V3-V4 dataset. In
summary, a similar number of bacterial species was recovered regardless of
which primer pair was used, however the identity and relative abundances of
those bacteria species differed significantly between the two methods.

46
Colony differences
The V4 dataset containing replicates for colony T3 was used to estimate intra-
specific variation. Pairwise comparisons (beta diversity) were categorised as
nestmates, conspecifics or heterospecifics. Differences between the gut
communities were significantly greater for pairs of samples from different
species, followed by those from different colonies (conspecifics) and by
comparisons between nestmates (Kruskal-Wallis test p < 0.001; post-hoc
Mann-Whitney U tests Figure 4).
Intraspecific comparisons using the V3-V4 dataset found significant
differences between T. westraliensis colonies in relative frequencies of the
most abundant genera. The most abundant identified genus in the gut was
always Treponema Ia (Spirochaetes, a phylum associated with cellulose
degradation or accessory functions). However, nearly twice as many reads
were identified as Treponema Ia in colony T1, as compared to colony T3 (Table
S1A). Treponema Ic and If were the second and third most abundant
Spirochaete genera, If being most abundant in T1 and Ic in T3. Colony T2 had
the highest number of reads assigned to Spirochaetes overall (57% of the
data), with many reads belonging to unidentified genera. T1 was the only
colony with no known Candidate Phylum TG3 genera in its top 20 most
abundant . Subcluster IIIb (TG3) was the fourth and eighth most abundant
genus in T2 and T3 respectively, whereas only two reads were detected in T1.
Overall, four known TG3 genera were detected in the T. westraliensis gut
(Subclusters IIIa, IIIb, IVa and IVb). Approximately 1250 reads per sample were
assigned to unknown groups in the cellulose-degrading TG3 candidate
phylum. T2 was assigned most of the reads identified as known TG3 genera
but colony 3 contained the most reads assigned to TG3 overall (Table S1A).
T2 was the only colony with a member of the cellulose-degrading
Fibrobacteres genus (Subcluster Ia) in its top 20, while only one read
belonging to that genus was detected in T3. Fibrobacteres was more abundant
in T2, with two previously known genera, Subcluster Ia and Ib featuring more
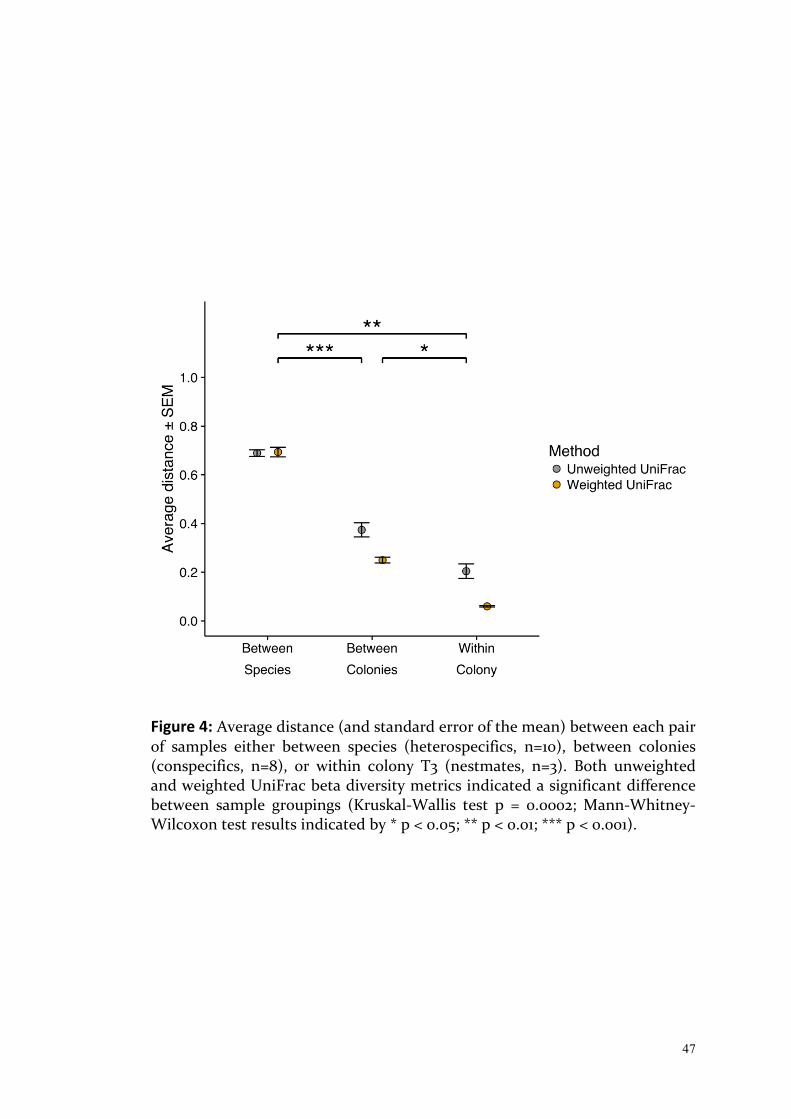
47
Figure 4: Average distance (and standard error of the mean) between each pair of samples either between species (heterospecifics, n=10), between colonies (conspecifics, n=8), or within colony T3 (nestmates, n=3). Both unweighted and weighted UniFrac beta diversity metrics indicated a significant difference between sample groupings (Kruskal-Wallis test p = 0.0002; Mann-Whitney-Wilcoxon test results indicated by * p < 0.05; ** p < 0.01; *** p < 0.001).

48
heavily in T2 (with 331 and 216 reads respectively in T2, 22 and three in T1 and
one and zero in T3). Unidentified Fibrobacteres reads (all belonging to the
Termite cluster family within Subphylum 2) were also more highly detected in
T2 with 3627 reads, followed by 71 reads in T3. T1 and T2 have Tannerella
(Bacteroidetes) and Treponema If (Spirochaetes) in second and third whereas
in T3 they rank fifth and eleventh respectively. Treponema Ic (Spirochaetes) is
fifth and fourth in T2 and T3 but tenth in T1, despite T3 recording twice as
many as either of the other two colonies. On the other hand, T1 has
Candidatus Armantifilum (Bacteroidetes) ranked eighth with double the
amount of reads as the other two colonies (where it ranked 14th and 15th;
Table S1).
The distance (beta diversity) values for the C. acinaciformis raffrayi colonies
were of the same magnitude as the T. westraliensis colonies. In the V3-V4
dataset, Gut cluster I (Firmicutes) was the third most abundant genus in C.
acinaciformis colony 1 (C1) with 40 times more reads than in C2.
Dysgonomonas (Bacteroidetes) was detected eight times more frequently in
C1, while Treponema Ig was detected eight times more and Treponema Il 16
times more in C2. C2 had over seven times more Endomicrobium and
Candidatus Hepatincola (Proteobacteria) than C1.
Species comparison
In the PCoA comparing gut community composition and structure of various
higher termite species (Figure 5), samples clustered primarily by diet,
regardless of the metric used (unweighted and weighted UniFrac in Figure 5A
and 5B respectively). The first two axes of the unweighted plot explained 26%
of the observed variation, while those of the weighted plot accounted for 52%
of variation. The metric used affected the relative position of each diet group.
The fungus-cultivating group was separated from all others in terms of
community composition while sharing a similar community structure with the
soil-feeders. The grass-feeders, containing only two species, did not form a
distinct group.

49
The T. westraliensis samples did not fit neatly into any of the feeding groups.
All colonies had a gut community composition comparable to the wood-
feeders and the grass-feeding Trinervitermes sp. (Figure 5A). However, two T.
westraliensis colonies had a similar community structure to the humus/litter
group, while colony T2 and Trinervitermes sp. sat outside of the confidence
interval of the wood-feeding group (Figure 5B). The second grass feeder, A.
meridionalis, had a unique community composition, most similar to the soil
and fungus groups, with more similarity to those groups in terms of
community structure.
Among the higher termites, the humus- and soil-feeding groups have the
highest number of bacterial species in the gut, with an average of 703 and 656
per sample, followed by termites consuming litter (519), fungus (388), grass
(386) and wood (265). The T. westraliensis samples averaged 332 species per
sample. Significantly different comparisons (p < 0.05) were between humus
and fungus, between soil and wood and between T. westraliensis and wood.
The only significantly different values for the Simpson's Reciprocal Index were
comparisons of wood and humus (p = 0.03) and wood and soil (p = 0.02). In
both cases, differences were only significant between the extremes but alpha
diversity values overall correlated with the humification gradient.
In the lower termites, sample clustering was best explained by PC1 in the
unweighted PCoA (explaining 26.8% of the variation) and PC2 in the weighted
PCoA (13.5%). Both separated the samples based on whether the termite hosts
feed broadly on the humification gradient or not. Coptotermes and
Mastotermes formed the bulk of the "broad" feeders group and the drywood,
dampwood and grass-feeding termites clustered together. Reticulitermes was
within the "broad" feeding group but was unique in terms of community
composition, and most similar to the grass feeders in community structure.
The Coptotermes samples shared similar community structures, but C. niger
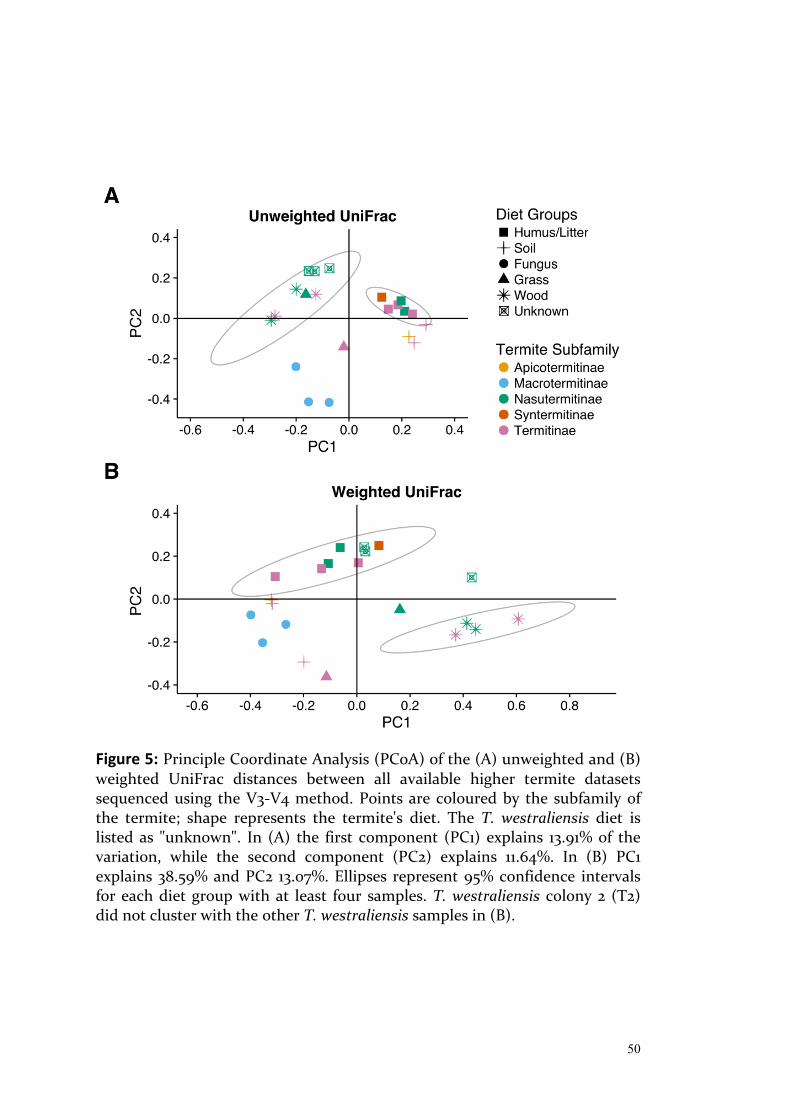
50
Figure 5: Principle Coordinate Analysis (PCoA) of the (A) unweighted and (B) weighted UniFrac distances between all available higher termite datasets sequenced using the V3-V4 method. Points are coloured by the subfamily of the termite; shape represents the termite's diet. The T. westraliensis diet is listed as "unknown". In (A) the first component (PC1) explains 13.91% of the variation, while the second component (PC2) explains 11.64%. In (B) PC1 explains 38.59% and PC2 13.07%. Ellipses represent 95% confidence intervals for each diet group with at least four samples. T. westraliensis colony 2 (T2) did not cluster with the other T. westraliensis samples in (B).

51
clustered with M. darwiniensis in terms of community composition (Figure
S2).
Alpha diversity analysis revealed significant differences amongst the lower
termites, in terms of the number of OTUs recovered from the guts of termites
from the "broad" (with an average of 83 OTUs per sample) and "narrow"
feeding groups. The drywood and dampwood termites and the grass-feeders
averaged 212, 345 and 188 bacterial species respectively (p = 0.008, non-
parametric t-test with 999 Monte Carlo permutations). The Simpson
Reciprocal Index was also significantly different between the two groups (p =
0.009), with an average of 7 for the "broad" diet and 45 (drywood), 65
(dampwood) and 27 (grass) for the "narrow" diet groups.
Discussion:
The current study has described, for the first time, the influence of primer
choice on estimates of gut community composition and structure in termites.
Using two endemic Western Australian termites, T. westraliensis and C.
acinaciformis raffrayi, I have demonstrated that colonies of both higher and
lower termites have signature gut communities, which limits the applicability
of previous single-colony studies to the question of core gut microbial
communities. I hypothesise that locally available substrates may drive the gut
community structure and composition at a colony level. Larger numbers of
biological replicates and the characterisation of colony specific food sources
would be required to confirm this.
Method comparison highlights the need for a standardised approach
PCR conditions (be it annealing temperature, primer choice or both)
significantly affected termite gut community profiles at both the phylum and
genus level. While certain differences could be predicted from the database
analysis, for example phyla Tenericutes and TM7 which were predicted to be

52
amplified unequally with each primer pair, the differences in the biological
datasets resulting from the two methods was much more extensive.
Overall the V3-V4 method yielded a higher species richness, i.e. more bacterial
groups were detected at the same depth, but genera were more evenly
distributed in the V4 dataset. The dominance of Treponema Ia and the
Spirochaetes in T. westraliensis, which appeared to amplify more efficiently
with the V4 primers, overshadowed lower abundance groups and increased
the Reciprocal Simpson Index for this dataset. The effect was not as
pronounced for the C. acinaciformis raffrayi data, possibly because its gut does
not contain as many bacterial species and because the most abundant genus
was not detected at significantly different levels with each method. The
increased species diversity detected in the V3-V4 dataset could be explained
by the lower annealing temperature recommended with this primer pair by
Köhler et al. (2012), which has been shown to limit some of the bias associated
with PCR (Sipos et al., 2007).
Because certain bacterial groups were only detected with one primer pair or
the other, it is not clear which method provides data that represents the gut
community most accurately. However it is clear that datasets collected with
each method cannot be directly compared and that caution must be exercised
when making generalised statements comparing biological findings across
studies. In the interest of collaboration, researchers should agree on a
standardised method to maximise re-use of publicly available datasets. The
method used on the widest range of termite species (across multiple families
and feeding groups) in the literature to date employs the V3-V4 primers
343Fmod and 784Rmod designed by Köhler et al. (2012) and the following PCR
conditions: initial denaturation for 3 min at 95°C, followed by 26 cycles of 20 s
at 95°C, 20 s at 48°C, and 30 s at 72°C and terminal extension for 3 min at 72°C.
I recommend that future termite gut studies use this amplification strategy
instead of, or in addition to other methods, as a means to standardise results
across the field.

53
Each colony has a signature gut microbiota
I hypothesised that there would be colony differences within each species in
terms of their gut microbiota, due to their broad diets. I did find significant
differences between the gut community composition and structure of
conspecific termites from different colonies, as compared to nestmates.
Previous studies have reported intercolony variation in the genera
Reticulitermes (Boucias et al. 2013; Benjamino and Graf, 2016) and
Microcerotermes (Hongoh et al., 2005), and in Stolotermes ruficeps (Reid et al.,
2014). Here, I extend these findings to T. westraliensis and C. acinaciformis.
Overall, I found that conspecific non-nestmates differed more in the types of
bacteria present than in their relative abundances. This suggests that different
colonies retained a core microbiome typical of the species or area, while
collecting bacteria from their direct environment, which could allow these
termites to optimise the digestion of local substrates (Otani et al., 2014; Brune
and Dietrich, 2015; Benjamino and Graf, 2016). Indeed, Miyata et al. (2007)
showed that altering the components of the diet of Nasutitermes
takasagoensis affected both the composition and structure of the gut
microbiota. In the lower termites, altering diet components or feeding of
different plant species has also been found to affect the gut community
(Tanaka et al., 2006; Husseneder et al., 2009; Huang et al., 2013). There
remains a baseline level of variation in the gut community within a colony,
even when controlling for caste and age, providing further support for the idea
that there are environmental drivers of the gut community in these termites.
In future, including replicate colonies sampled from different sites may reveal
even greater variability. Studying the variation in accessible food sources for
each colony would indicate if these differences could indeed be linked to diet.
Putative functional differences in gut communities support the possibility that
diversity and abundance differences are driven by dietary factors. T.
westraliensis colony 2 (T2) stood out from the other colonies in the weighted

54
PCoA, featuring high levels of known TG3 and Fibrobacteres genera and a
higher variety of abundant Spirochaetes. TG3, Fibrobacteres and certain
Spirochaete genera are associated with wood fibres in Nasutitermes species
(Mikaelyan et al. 2014) and Fibrobacteres and TG3 have both been associated
with cell-bound cellulose-degradation (Sorokin et al., 2014; Abdul et al., 2016),
suggesting a more highly lignified diet. The vast majority of Spirochaetes
belonged to Treponema cluster I, as previously reported in a range of termite
species (Lilburn et al., 1999). Interestingly, genus Treponema Ia, the most
abundant genus in the gut of T. westraliensis has not been associated with
fibre degradation, but rather enhancing cellulose-degradation by other
organisms and using their by-products (Kudo et al., 1987). Treponema Ic and
If, most abundant in T2, have been reported to associate with fibre and are the
most abundant genera in wood-feeding Nasutitermes (Dietrich et al., 2014;
Mikaelyan et al., 2014). This colony also clustered more closely with termite
species feeding on sound substrates (be it wood or grass). I therefore
hypothesise that T2 was consuming less humified and/or more highly lignified
substrate(s) than the other two colonies. Future studies could undertake a
detailed inventory of the storage chambers for each colony to determine
whether there are correlations between the lignification and humification level
of the stored material and the abundance of fibre-associated bacteria in the
gut community.
I found indications of intraspecific colony differences in C. acinaciformis
raffrayi, although replication was very low. The same genera were detected in
both colonies, but colony C1 had more Gut cluster I (Firmicutes) and
Dysgonomonas (Bacteroidetes, capable of varying degrees of cellulose
degradation; Pramono et al., 2015; Sun et al., 2015). More Treponema Ig,
Treponema II, Endomicrobium and Candidatus Hepatincola (Proteobacteria)
was detected in C2. Treponema Ig and II have been linked to protists in other
lower termites (Mikaelyan et al., 2015b; Benjamino and Graf, 2016) and were
not detected in T. westraliensis. Endomicrobium is a common endosymbiont of
lower termite gut protists, but has also been reported as a free-living

55
bacterium both in lower and higher termites (Stingl et al., 2005; Ohkuma et
al., 2007; Zheng and Brune, 2015). The differences in abundance of protist-
associated bacteria would suggest that colony differences in C. acinaciformis
raffrayi might in part be driven by different abundances of gut protists.
Because C1 was collected in rotten wood and C2 was collected in sound wood,
I hypothesise that protist numbers correlate with humification of the diet.
Niche breadth and variability of the gut microbiota of termites
The gut habitat (e.g. pH, oxygen concentration) shapes the microbiota in
universal ways by providing specific niches (Mikaelyan et al. 2017), and this
gut environment has an additive effect with diet on gut community structure,
due to the varying availability of plant fibre and nitrogen along the
humification gradient. Indeed, members of different higher termite
subfamilies share similarities in their gut community structure if they
naturally feed on the same diet (Mikaelyan et al. 2015a). I extended the results
of Mikaelyan et al. (2015a) by analysing their raw data with a different analysis
pipeline to show that diet also correlates with gut community composition.
Since vertical transfer of a symbiotic core microbiome facilitates co-evolution
of the gut community and its host species (Bignell, 2011; Brune and Dietrich,
2015), comparing the gut community composition and structure within and
between species offers a window into the relative contributions of inherited vs
environmentally acquired (ingested) microbes. Such an approach may also
allow us to make inferences about the diet of a species with sparse natural
history data, based on its gut community profile.
There was a correlation between the alpha diversity values for the higher
termites and the point on humification gradient at which they feed, with the
highest species richness and diversity in the guts of humus and soil feeders.
Litter-feeding termites were indistinguishable from the humus group when
comparing beta diversity values, but litter was linked to lower species richness,
presumably due to its less humified state. T. westraliensis had similar alpha
diversity values and gut community composition to the wood- and grass-

56
feeders, but the grouping of two out of three colonies with the humus/litter
group in terms of gut community structure had greater explanatory power. In
addition, the gut of T. westraliensis was shown to be dominated by
Spirochaetes like typical wood and grass feeders and Spirochaetes have been
associated with nitrogen fixation in higher termite guts (Warnecke et al., 2007;
Brune and Ohkuma, 2011; Mikaelyan et al., 2017). This reinforces our
observation that the diet of T. westraliensis may be more similar to litter than
grass, perhaps in terms of variety. However, it suggests that the diet is lower
on the humification gradient than litter, and hence lower in nitrogen. This
also underscores need for more extensive sampling within a species to capture
a more accurate picture of the plasticity of its microbial community and
dietary range.
The gut community of lower termites is shaped additionally by the presence of
cellulose-degrading protists that provide unique niches for bacteria. Certain
lineages of protist and bacteria have been shown to be co-evolving with the
host (Brune and Dietrich, 2015), but environmental uptake and diet still play a
significant role, especially for lower abundance phylotypes (Tai et al., 2015). I
found that the complexity of the diet might be driving the gut community
structure of lower termite guts more than their phylogeny.
Interestingly, lower termites which forage on broader diets were found to have
less diverse gut microbiota than specialist foraging or non-foraging termites at
the same sequencing depth. This result is in contrast to Waidele et al. (2017),
who recovered more OTUs from Reticulitermes (628 OTUs, also included in
this study) than Cryptotermes (476 OTUs, drywood termite closely related to
the one included in this study). They hypothesised that foraging-type species
with broad diets have a higher probability of encountering new
microorganisms, some of which may become resident in the gut.
They attempted to remove transient gut microbes by feeding the termites
sterile pine for six weeks, which should have decreased species richness in the
gut. It should be noted that community composition estimates are affected by

57
sequencing depth and community structure, as more abundant taxa may lead
to low abundance taxa remaining undetected at that depth. My comparative
analysis was conducted at a rarefied depth of 3,000 reads per sample,
compared to a depth of 32,824 reads in Waidele et al. (2017), which likely
influenced our respective results. Recent work applying the same V3-V4
strategy I used included more lower termite species (Bourguignon et al., 2018)
and may help resolve these inconsistencies. There, the sequencing
depth varied between 1,847 reads and 10,395 reads per sample for the lower
termites, so a new meta-analysis could be done with a subset of their data to
avoid rarefying below my current threshold.
Conclusion
I have shown that primer choice influences the estimation of gut community
composition and structure in termites and recommend that a standard
approach be applied using the primers 343Fmod and 784Rmod designed by
Köhler et al. (2012) that target the V3-V4 region of the 16S rRNA gene. The
recommended low annealing temperature would maximise data
reproducibility and utility across research groups.
There were colony differences within both T. westraliensis and C.
acinaciformis raffrayi. While their complex diets may be one explanation for
the existence of these intra-species differences, it also highlights the necessity
of extensive sampling. Future studies on T. westraliensis could quantify the
amount and condition of each plant type found within a mound's food store or
characterise the plant DNA recovered from the gut, to determine whether gut
community differences reflect colony-specific food availability. Replicated
biological sampling is necessary for future metagenomic studies to account for
variability of gut communities within a species and to further understand the
drivers of community structure and composition in termites.

58
The T. westraliensis gut community shares similarities with wood, grass, and
litter feeders, indicating that its diet may be more complex and less
specialised than previously thought. C. acinaciformis raffrayi shared
similarities with other lower termites that eat various species of wood at
different stages of decomposition. This suggests that the complexity of a diet
may have a larger impact on the gut community composition and structure of
termites than the plant species or humification level on their own.
The evolution of the dual-cellulolytic system in termites over 150 million years
allows us to learn from a fine-tuned natural process and to apply it to
renewable energy advancements such as biofuel (reviewed by König et al.,
2013; Ni and Tokuda, 2013; Brune, 2014). This apparent plasticity of the gut
community may allow for the development of cellulase enzyme cocktails
suited to biofuel feedstocks of interest. Bacterial enzymes are well suited to
industrial processes (Gronenberg et al., 2013), placing the spotlight on higher
termites. T. westraliensis' variable gut community makes it a potential model
species to study the drivers that shape the gut community structure in higher
termites, aside from host phylogeny.

59
References Abdul Rahman, N., Parks, D. H., Willner, D. L., Engelbrektson, A. L., Goffredi, S. K.,
Warnecke, F., Scheffrahn, R. H., Hugenholtz, P. (2015). A molecular survey of Australian and North American termite genera indicates that vertical inheritance is the primary force shaping termite gut microbiomes. Microbiome, 3, 5.
Abensperg-Traun, M., & Milewski, A. V. (1995). Abundance and diversity of termites (Isoptera) in imburnt versus burnt vegetation at the Barrens in Mediterranean Western Australia. Australian Journal of Ecology, 20(3), 413-417.
Benjamino, J., & Graf, J. (2016). Characterization of the core and caste-specific microbiota in the termite, Reticulitermes flavipes. Frontiers in Microbiology, 7, 1.
Bignell, D. E. (2011). Morphology, physiology, biochemistry and functional design of the termite gut: an evolutionary wonderland. In D. E. Bignell, Y. Roisin, & N. Lo (Eds.), Biology of Termites: a Modern Synthesis (pp. 375-412). Netherlands: Springer.
Boucias, D. G., Cai, Y., Sun, Y., Lietze, V.-U., Sen, R., Raychoudhury, R. et al. (2013). The hindgut lumen prokaryotic microbiota of the termite Reticulitermes flavipes and its responses to dietary lignocellulose composition. Molecular Ecology, 22(7), 1836-1853.
Bourguignon, T., Lo, N., Dietrich, C., Šobotník, J., Sidek, S., Roisin, Y., Brune, A., Evans, T. A. (2018). Rampant host switching shaped the termite gut microbiome. Current Biology, 28(4), 649-654.e2.
Brune, A. (2014). Symbiotic digestion of lignocellulose in termite guts. Nature Reviews Microbiology, 12(3), 168-180.
Brune, A., & Dietrich, C. (2015). The gut microbiota of termites: digesting the diversity in the light of ecology and evolution. Annual Review of Microbiology, 69(1), 145-166.
Brune, A., & Ohkuma, M. (2011). Role of the termite gut microbiota in symbiotic digestion. In D. E. Bignell, Y. Roisin, & N. Lo (Eds.), Biology of Termites: a Modern Synthesis (pp. 439-475). Netherlands: Springer.
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J. et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences, 108(Supplement 1), 4516-4522.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K. et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7, 335-336.
Cleveland, L. R. (1923). Symbiosis between termites and their intestinal protozoa. Proceedings of the National Academy of Sciences of the United States of America, 9(12), 424-428.
Colman, D. R., Toolson, E. C., & Takacs-Vesbach, C. D. (2012). Do diet and taxonomy influence insect gut bacterial communities? Molecular Ecology, 21, 5124-5137.
Delmotte, N., Knief, C., Chaffron, S., Innerebner, G., Roschitzki, B., Schlapbach, R. et al. (2009). Community proteogenomics reveals insights into the physiology of phyllosphere bacteria. Proceedings of the National Academy of Sciences, 106(38), 16428-16433.
Dietrich, C., Köhler, T., & Brune, A. (2014). The cockroach origin of the termite gut microbiota: patterns in bacterial community structure reflect major evolutionary events. Applied and Environmental Microbiology.
Diouf, M., Roy, V., Mora, P., Frechault, S., Lefebvre, T., Hervé, V., Rouland-Lefèvre,

60
C., Miambi, E. (2015). Profiling the succession of bacterial communities throughout the life stages of a higher termite Nasutitermes arborum (Termitidae, Nasutitermitinae) Using 16S rRNA Gene Pyrosequencing. PLoS One, 10, e0140014.
Dolan, M. F. (2001). Speciation of termite gut protists: the role of bacterial symbionts. International Microbiology, 4(4), 203-208.
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10(10), 996-998.
Gontijo, T. A., & Domingos, D. J. (1991). Guild distribution of some termites from cerrado vegetation in south-east Brazil. Journal of Tropical Ecology, 7(04), 523-529.
Greaves, T. (1962). Studies of foraging galleries and the invasion of living tres by Coptotermes acinaciformis and C. brunneus (Isoptera). Australian Journal of Zoology, 10, 630-651.
Gronenberg, L. S., Marcheschi, R. J., & Liao, J. C. (2013). Next generation biofuel engineering in prokaryotes. Current Opinion in Chemical Biology, 17(3), 462-471.
Gu, Z., Eils, R., & Schlesner, M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics, 32(18), 2847-2849.
He, S., Ivanova, N., Kirton, E., Allgaier, M., Bergin, C., Scheffrahn, R. H. et al. (2013). Comparative metagenomic and metatranscriptomic analysis of hindgut paunch microbiota in wood- and dung-feeding higher termites. PLoS ONE, 8(4), 1-14.
Hill, G. F. (1921). New and rare Australian termites, with notes on their biology. Proceedings of the Linnean Society of New South Wales, 46, 433-456.
Hill, G. F. (1925). Termites of the Australian region. Proceedings of The Royal Society of Victoria, 37, 206-229.
Hill, G. F. (1942). Termites (Isoptera) from the Australian region. Melbourne, Australia: Council for Scientific and Industrial Research.
Hongoh, Y., Ekpornprasit, L., Inoue, T., Moriya, S., Trakulnaleamsai, S., Ohkuma, M., Noparatnaraporn, N., Kudo, T. (2006). Intracolony variation of bacterial gut microbiota among castes and ages in the fungus-growing termite Macrotermes gilvus. Molecular Ecology, 15(2), 505-516.
Hongoh, Y., Deevong, P., Inoue, T., Moriya, S., Trakulnaleamsai, S., Ohkuma, M., Vongkaluang, C., Noparatnaraporn, N., Kudo, T. (2005). Intra- and interspecific comparisons of bacterial diversity and community structure support coevolution of gut microbiota and termite host. Applied & Environmental Microbiology, 71(11), 6590-6599.
Huang, X.-F., Bakker, M., Judd, T., Reardon, K., & Vivanco, J. (2013). Variations in diversity and richness of gut bacterial communities of termites (Reticulitermes flavipes) fed with grassy and woody plant substrates. Microbial Ecology, 65(3), 531-536.
Husseneder, C., Berestecky, J. M., & Grace, J. K. (2009). Changes in composition of culturable bacteria community in the gut of the formosan subterranean termite depending on rearing conditions of the host. Annals of the Entomological Society of America, 102(3), 498-507.
Inoue, T., Kitade, O., Yoshimura, T., & Yamaoka, I. (2000). Symbiotic associations with protists. In T. Abe, D. E. Bignell, & M. Higashi (Eds.), Termites: Evolution, Sociality, Symbiosis, Ecology (pp. 275-288). Dordrecht: Kluwer Academic Publishers.
Jones, D. T., & Eggleton, P. (2011). Global biogeography of termites: a compilation of sources. In D. E. Bignell, Y. Roisin, & N. Lo (Eds.), Biology of Termites: a Modern

61
Synthesis (pp. 477-498). Netherlands: Springer. Köhler, T., Dietrich, C., Scheffrahn, R. H., & Brune, A. (2012). High-resolution analysis
of gut environment and bacterial microbiota reveals functional compartmentation of the gut in wood-feeding higher termites (Nasutitermes spp.). Applied & Environmental Microbiology, 78(13), 4691-4701.
König, H., Li, L., & Fröhlich, J. (2013). The cellulolytic system of the termite gut. Applied Microbiology and Biotechnology, 97(18), 7943-7962.
Krishna, K., Grimaldi, D. A., Krishna, V., & Engel, M. S. (2013). Treatise on the Isoptera of the world. Bulletin of the American Museum of Natural History, 1.
Kudo, H., Cheng, K.-J., & Costerton, J. W. (1987). Interactions between and cellulolytic bacteria in the degradation of straw cellulose. Canadian Journal of Microbiology, 33(3), 244-248.
Lee, T. R. C., Cameron, S. L., Evans, T. A., Ho, S. Y. W., & Lo, N. (2015). The origins and radiation of Australian Coptotermes termites: From rainforest to desert dwellers. Molecular Phylogenetics and Evolution, 82, 234-244.
Li, H., Dietrich, C., Zhu, N., Mikaelyan, A., Ma, B., Pi, R. et al. (2016). Age polyethism drives community structure of the bacterial gut microbiota in the fungus-cultivating termite Odontotermes formosanus. Environmental Microbiology, 18(5), 1440-1451.
Lilburn, Schmidt, & Breznak. (1999). Phylogenetic diversity of termite gut Spirochaetes. Enviromental Microbiology, 1(4), 331-345.
Liu, H., & Beckenbach, A. T. (1992). Evolution of the mitochondrial cytochrome oxidase II gene among 10 orders of insects. Molecular Phylogenetics and Evolution, 1(1), 41-52.
Lo, N., Eldridge, R. H., & Lenz, M. (2006). Phylogeny of Australian Coptotermes (Isoptera: Rhinotermitidae) species inferred from mitochondrial COII sequences. Bulletin of Entomological Research, 96(4), 433-437.
Mikaelyan, A., Dietrich, C., Köhler, T., Poulsen, M., Sillam-Dusses, D., & Brune, A. (2015a). Diet is the primary determinant of bacterial community structure in the guts of higher termites. Molecular Ecology, 24(20), 5284-5295.
Mikaelyan, A., Köhler, T., Lampert, N., Rohland, J., Boga, H., Meuser, K. et al. (2015b). Classifying the bacterial gut microbiota of termites and cockroaches: A curated phylogenetic reference database (DictDb). Systematic and Applied Microbiology, 38(7), 472-482.
Mikaelyan, A., Meuser, K., & Brune, A. (2017). Microenvironmental heterogeneity of gut compartments drives bacterial community structure in wood-and humus-feeding higher termites. FEMS Microbiology Ecology, 93(1), fiw210.
Mikaelyan, A., Strassert, J. F. H., Tokuda, G., & Brune, A. (2014). The fibre‐associated cellulolytic bacterial community in the hindgut of wood‐feeding higher termites (Nasutitermes spp.). Environmental Microbiology, 16(9), 2711-2722.
Miyata, R., Noda, N., Tamaki, H., Kinjyo, K., Aoyagi, H., Uchiyama, H., Tanaka, H. Influence of feed components on symbiotic bacterial community structure in the gut of the wood-feeding higher termite Nasutitermes takasagoensis. Bioscience, Biotechnology, and Biochemistry, 71(5), 1244-1251.
National Center for Biotechnology Information. Basic Local Alignment Search Tool. 2017, from https://blast.ncbi.nlm.nih.gov/Blast.cgi.
Ni, J., & Tokuda, G. (2013). Lignocellulose-degrading enzymes from termites and their symbiotic microbiota. Biotechnology advances, 31(6), 838-850.
Ohkuma, M. (2003). Termite symbiotic systems: efficient bio-recycling of lignocellulose. Applied Microbiology and Biotechnology, 61(1), 1-9.
Ohkuma, M., Sato, T., Noda, S., Ui, S., Kudo, T., & Hongoh, Y. (2007). The candidate

62
phylum ‘Termite Group 1’ of bacteria: phylogenetic diversity, distribution, and endosymbiont members of various gut flagellated protists. FEMS Microbiology Ecology, 60(3), 467-476.
Otani, S., Mikaelyan, A., Nobre, T., Hansen, L. H., Kone, N. A., Sorensen, S. J., Aanen, D.K., Boomsma, J.J., Brune, A. Poulsen, M (2014). Identifying the core microbial community in the gut of fungus-growing termites. Molecular Ecology.
Paul, K., Nonoh, J. O., Mikulski, L., & Brune, A. (2012). “Methanoplasmatales,” Thermoplasmatales-related archaea in termite guts and other environments, are the seventh order of methanogens. Applied and Environmental Microbiology, 78(23), 8245-8253.
Poulsen, M. (2015). Towards an integrated understanding of the consequences of fungus domestication on the fungus-growing termite gut microbiota: fungus-growing termite gut microbiota. Environmental Microbiology, 17(8), 2562-2572.
Pramono, A. K., Sakamoto, M., Iino, T., Hongoh, Y., & Ohkuma, M. (2015). Dysgonomonas termitidis sp. nov., isolated from the gut of the subterranean termite Reticulitermes speratus. International Journal of Systematic and Evolutionary Microbiology, 65(Pt 2), 681-685.
R Development Core Team (2016). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
Reid, N. M., Addison, S. L., West, M. A., & Lloyd-Jones, G. (2014). The bacterial microbiota of Stolotermes ruficeps (Stolotermitidae), a phylogenetically basal termite endemic to New Zealand. FEMS Microbiology Ecology, 90(3), 678-688.
Rosengaus, R. & Traniello, J. (1997). Pathobiology and disease transmission in dampwood termites Zootermopsis angusticollis (Isoptera: Termopsidae) infected with the fungus Metarhizium anisopliae (Deuteromycotina: Hypomycetes). Sociobiology. 30. 185-195.
Sipos, R., Székely, A. J., Palatinszky, M., Révész, S., Márialigeti, K., & Nikolausz, M. (2007). Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis: PCR parameters influencing quantitative bias. FEMS Microbiology Ecology, 60(2), 341-350.
Sorokin, D. Y., Gumerov, V. M., Rakitin, A. L., Beletsky, A. V., Damsté, J. S. S., Muyzer, G. et al. (2014). Genome analysis of gen. nov., sp. nov., a novel extremely haloalkaliphilic anaerobic chitinolytic bacterium from the candidate phylum Termite Group 3 : Chitinolytic bacterium from the candidate phylum TG3. Environmental Microbiology, 16(6), 1549-1565.
Stingl, U., Radek, R., Yang, H., & Brune, A. (2005). “Endomicrobia”: cytoplasmic symbionts of termite gut protozoa form a separate phylum of prokaryotes. Applied and Environmental Microbiology, 71(3), 1473-1479.
Sun, X., Yang, Y., Zhang, N., Shen, Y., & Ni, J. (2015). Draft genome sequence of Dysgonomonas macrotermitis strain JCM 19375T, isolated from the gut of a termite. Genome Announcements, 3(4), e00963-e00915.
Tai, V., James, E. R., Nalepa, C. A., Scheffrahn, R. H., Perlman, S. J., & Keeling, P. J. (2015). The role of host phylogeny varies in shaping microbial diversity in the hindguts of lower termites. Applied and Environmental Microbiology, 81(3), 1059-1070.
Tanaka, H., Aoyagi, H., Shiina, S., Doudou, Y., Yoshimura, T., Nakamura, R., Uchiyama, H. (2006). Influence of the diet components on the symbiotic microorganisms community in hindgut of Coptotermes formosanus Shiraki. Applied Microbiology & Biotechnology, 71(6), 907-917.
Tokuda, G., & Watanabe, H. (2007). Hidden cellulases in termites: revision of an old

63
hypothesis. Biology Letters, 3(3), 336-339. Tremblay, J., Singh, K., Fern, A., Kirton, E. S., He, S., Woyke, T. et al. (2015). Primer
and platform effects on 16S rRNA tag sequencing. Frontiers in Microbiology, 6, 771.
Waidele, L., Korb, J., Voolstra, C. R., Künzel, S., Dedeine, F., & Staubach, F. (2017). differential ecological specificity of protist and bacterial microbiomes across a set of termite species. Frontiers in Microbiology, 8, 5.
Wang, Q., Garrity, G. M., Tiedje, J. M., & Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied & Environmental Microbiology, 73(16), 5261-5267.
Warnecke, F., Luginbuhl, P., Ivanova, N., Ghassemian, M., Richardson, T. H., Stege, J. T. et al. (2007). Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature, 450(7169), 560-565.
Watson, J. A. L., & McMahan, E. A. (1978). Polyethism in the Australian harvester termite Drepanotermes (Isoptera, Termitinae). Insectes Sociaux, 25(1), 53-62.
Wickham, H. (2009). ggplot2: Elegant Graphics for Data Analysis. New York: Springer-Verlag.
Xiang, H., Xie, L., Zhang, J., Long, Y., Liu, N., Huang, Y., Wang, Q. (2012). Intracolonial differences in gut bacterial community between worker and soldier castes of Coptotermes formosanus. Insect Science, 19, 86-95.
Zheng, H., & Brune, A. (2015). Complete genome sequence of Endomicrobium proavitum, a free- living relative of the intracellular symbionts of termite gut flagellates (Phylum Elusimicrobia). Genome Announcements, 4(3), e00679-e00615.
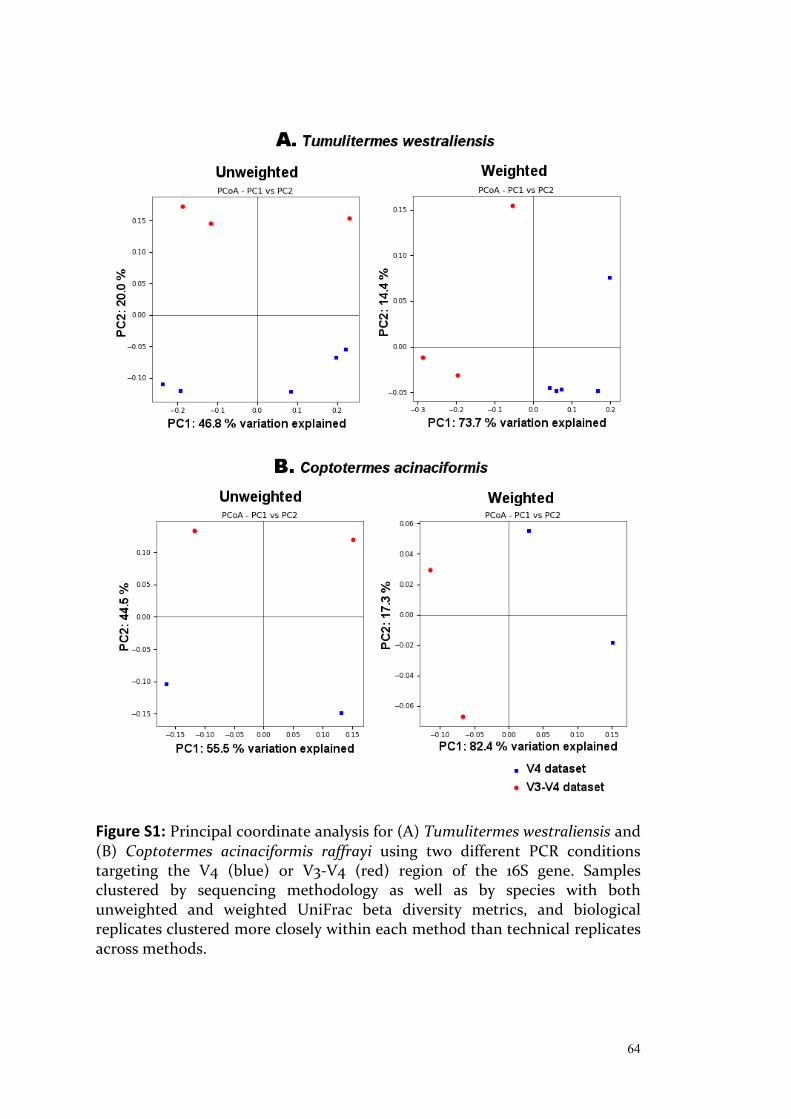
64
Figure S1: Principal coordinate analysis for (A) Tumulitermes westraliensis and (B) Coptotermes acinaciformis raffrayi using two different PCR conditions targeting the V4 (blue) or V3-V4 (red) region of the 16S gene. Samples clustered by sequencing methodology as well as by species with both unweighted and weighted UniFrac beta diversity metrics, and biological replicates clustered more closely within each method than technical replicates across methods.
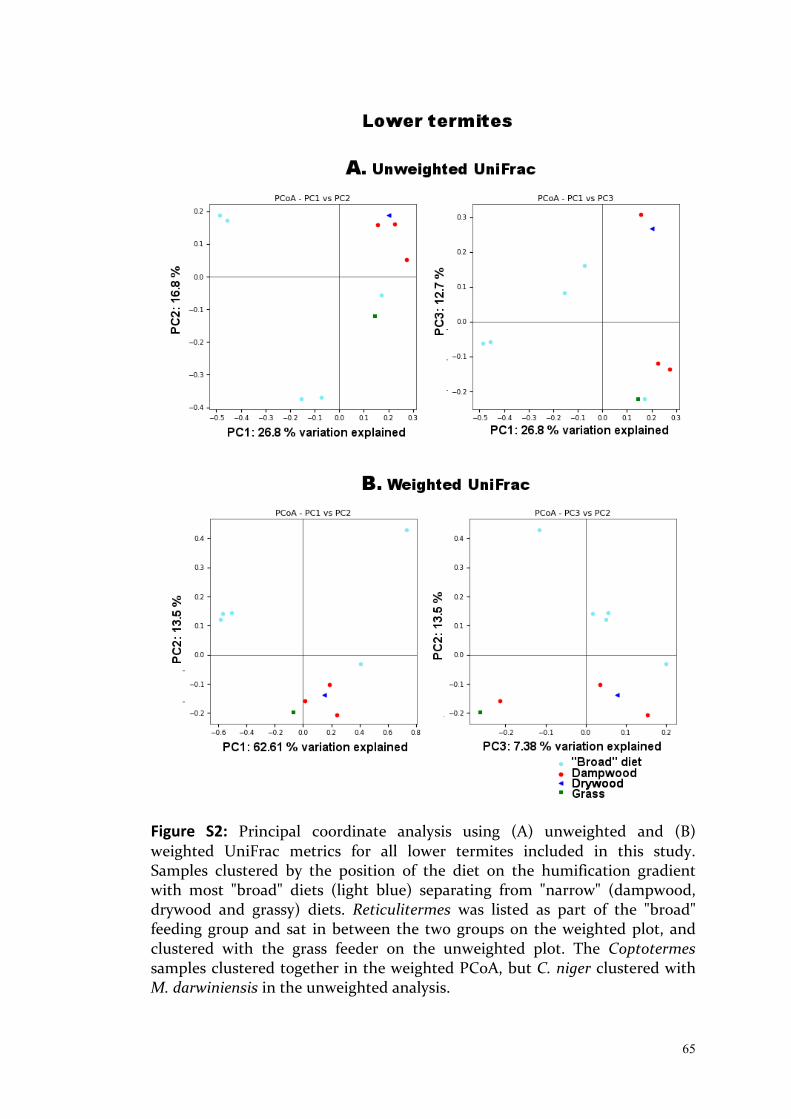
65
Figure S2: Principal coordinate analysis using (A) unweighted and (B) weighted UniFrac metrics for all lower termites included in this study. Samples clustered by the position of the diet on the humification gradient with most "broad" diets (light blue) separating from "narrow" (dampwood, drywood and grassy) diets. Reticulitermes was listed as part of the "broad" feeding group and sat in between the two groups on the weighted plot, and clustered with the grass feeder on the unweighted plot. The Coptotermes samples clustered together in the weighted PCoA, but C. niger clustered with M. darwiniensis in the unweighted analysis.

66

67
CHAPTER 3: Sampling Intensity, Scope and Scale Significantly Affect Estimation of Dietary Influence on the Gut Communities of Tumulitermes westraliensis and Amitermes obeuntis

68
Foreword:
In this chapter, I extend findings from Chapter 2 to further investigate my first
research question: How much intraspecific variation exists in the gut
communities of Western Australian endemic termites? Intraspecific variation in
the guts of two higher termites, Tumulitermes westraliensis and Amitermes
obeuntis, is characterised and I investigate local factors mediating horizontal
and environmental uptake which may contribute to this variation.
I also focus on identifying the portion of the gut community that is
consistently present in the gut of each species and likely to contain organisms
passed on from generation to generation through vertical transfer. This “core
community” has been reported in previous termite studies, but various
different methods have been applied, which may affect interpretation of
results across studies. This inconsistency led to my second research question,
also explored in this chapter: How should the core community of a termite
species be defined and accurately estimated? More specifically, I tested three
aspects related to sampling for termite core microbiome studies: scale, defined
as the geographic distribution of sampling sites; scope, the caste composition
of samples; and sampling intensity, the number of samples taken per unit of
interest.
Further, I report on a food-replacement experiment conducted with wheat and
lucerne straw to determine whether the gut communities become enriched in
taxa better suited to digesting these substrates. This experiment addresses my
third research question: Can the gut community of local higher termite species
be modified under field conditions? The core communities of each species, as
well as their shared core microbiome, were compared by size, abundance and
predicted functions. The results were compared between different sampling
intensities, scales and scopes.

69
Based on these results, the likelihood of vertical acquisition of common gut
community members from a common ancestor versus environmental or
horizontal transfer is then discussed. The source of termite gut community
members is a point of discussion in the literature, but this is the first direct
test of potential contributing factors under field conditions. My contributions
add to the broader discussion in the field, with implications for how we
understand the evolution and ecology of termite microbiomes.
Introduction:
Microbial communities from similar habitats contain consortia of taxa
consistently present in that habitat, a group defined as the core community or
core microbiome, in addition to transient or sample-specific taxa. Members of
the core microbiome are thought to be critical to the functioning of that
community; it is thus the baseline community for a particular habitat, from
which it should be possible to measure responses to perturbation and
manipulation (Shade and Handelsman, 2012). In practice, a core microbiome is
measured by determining which taxa, usually represented by highly similar
genetic sequences (operational taxonomic units, OTUs) are present in most or
all samples representing the habitat of interest (Shade and Handelsman, 2012).
Termite gut core microbiomes have previously been reported in different
termite species (Huang et al., 2013; Dietrich et al., 2014; Otani et al., 2014; Reid
et al., 2014; Benjamino and Graf, 2016). Benjamino and Graf (2016)
hypothesised that each member of the core microbiome occupies a specific
niche in the hindgut of a particular termite species, regardless of the location
or food source of the host. For example, Huang et al. (2013) found that the core
microbiome of the lower termite Reticulitermes flavipes comprised 65% of
common OTUs (95% of sequences) and persisted despite changes in diet to
either grassy or woody substrates. The diets did, however, significantly change
bacterial species richness and the relative abundance of bacterial taxa. The
authors concluded that the core microbiome has a role in lignocellulose

70
degradation and that differences in the recalcitrance of the substrates may
explain differences in the species richness of the gut, with more recalcitrant
food sources requiring more complex communities for efficient breakdown.
Comparison of termite gut microbiome studies is hindered by the fact that
different studies have used different sampling and bioinformatics methods to
search for and define the core microbiome. Previous approaches include 1)
using technical replicates of a single large pooled sample to calculate the core
community of a species (Su et al., 2016); 2) using single or replicated pooled
samples for multiple termite species to calculate a core microbiome across
several termite species (Dietrich et al., 2014; Otani et al., 2014; Abdul Rahman
et al., 2015, Su et al., 2016); 3) using replicate guts or pooled samples from
different colonies of a single termite species to calculate the core microbiome
(Reid et al., 2014; Benjamino and Graf, 2016); 4) using replicate guts from
different castes to calculate a core between each pair of castes in a colony
(Diouf et al., 2018) or 5) using pooled samples from one or more colonies fed
on different diets to infer a core community that persists on all diets (Huang
et al., 2013; Wang et al., 2016). In all of these studies the scope, i.e. caste
composition of samples, was limited to workers, except in Diouf et al. 2018
where five castes of Nasutitermes arborum were considered. The geographic
distribution of sampling sites (scale) was in most cases restricted to a single
colony, or a small portion of the species known range. Sampling intensity, the
number of samples taken per unit of interest, varied from 3 (Su et al., 2016) to
66 (Abdul Rahman et al., 2015). Finally, OTUs included in the core community
were required to be present in 40% (Reid et al., 2014) to 100% (Huang et al.,
2013; Otani et al., 2014; Abdul Rahman et al., 2015; Su et al., 2016; Wang et al.,
2016) of samples in these studies. These differences in sample selection and
bioinformatics methods are compounded by variation in laboratory methods
(see Chapter 2).
In termites, the core community shared across a species is generally thought
to be vertically transmitted from parent colony to new colonies through the

71
winged reproductives since they are the only link between generations of
colonies (Diouf et al., 2018). However the origin of the remainder of the gut
community (hereafter referred to as the non-core community, or in some
cases the group-specific core community) remains unclear. The prevalence of
core taxa across cockroaches, lower (basal) termites and higher (derived)
termites was specific to each host group in Dietrich et al. (2014) but no
evidence of co-speciation was found within each of these host groups,
suggesting that other factors, such as environmental uptake, shape bacterial
community composition within them. However, Otani et al. (2014) recovered a
core microbiome of 42 OTUs common to nine species of fungus-cultivating
termites (Macrotermitinae), which accounted for 56–68% of reads within each
species. The large number of shared taxa suggests vertical inheritance from a
common ancestor. Environmental factors seem to influence the relative
abundances of bacterial groups in these gut communities (i.e. community
structure), which more closely resembled omnivorous cockroaches than other
higher termites (Termitidae). In this case, the obligate association with
Termitomyces, consumed by the termites along with plant material on which
it is cultivated, likely influences the community structure in a consistent
manner.
To explore environmental influences on the gut microbiome and test how
varying numbers and types of samples affect inference of the core community,
I established a feeding experiment at three sites with two co-occurring, and
sometimes co-habiting, higher termite species. My study species have narrow,
nearly overlapping, distribution ranges (see Figure 1A) but different feeding
habits: Tumulitermes westraliensis feeds on dried plant debris stored in its
mound (Chapter 2) while Amitermes obeuntis consumes decaying wood (Perry
et al., 1985; Abensperg-Traun, 1992). I collected 16S gut metabarcoding data on
pooled and individual guts before and after the feeding experiment. The core
microbiome was defined as the OTUs shared by 100% of samples within a
given set of samples either within each species or across species. This cut-off
level was chosen because it is the most common used by previous studies, as

72
well as the most stringent. This data was then used in a sampling simulation
to test the effects of the three aspects of sample selection mentioned above:
scale, the geographic distribution of sampling sites; scope, the caste
composition of samples; and sampling intensity, the number of samples taken
per unit of interest. It was hypothesised that the choice of sampling scale,
scope and intensity all affect the estimation of the gut core microbiome. The
core microbiome calculated across all available samples and castes was used to
estimate a species-wide core community. Core communities were further
calculated and compared across locations, colonies and diets to further
elucidate potential sources of the gut microbial community and functional
differences.
Methods:
Feeding experiment
Six mounds were chosen in each of three locations in the Perth Hills, Western
Australia: John Forrest National Park, Hovea (J); Beelu National Park,
Mundaring (B); Banyowla Regional Park, Wattle Grove (W), covering a small
part of the species' ranges (Figure 1). Selected mounds were undamaged, at
least 2 m from any other mounds and a minimum 20 m from each other.
Where possible, I selected mounds housing both Tumulitermes westraliensis
and Amitermes obeuntis; otherwise the presence of T. westraliensis was
favoured due to its feeding habits. Chosen mounds were sampled as detailed
below prior to establishment of feeding treatments. Loose vegetation was
cleared in a 2 m radius around the mound, leaving exposed soil and live
vegetation. Two mounds per location were randomly assigned to one of three
diet treatments: wheat straw sourced from the monoculture treatment of a
long-term crop rotation trial (provided by Nathan Craig, UWA), lucerne hay
(grown locally, sourced from a pet store) or a control consisting of loose
cleared vegetation from the site (including dried leaves and grass, small sticks,
gumnuts). Twenty-five bags (hand made using UV resistant polyethylene

73
Figure 1: Species ranges (A) for T. westraliensis (pink) and A. obeuntis (orange) obtained from and created in The Atlas Of Living Australia Spatial Portal (https://spatial.ala.org.au/; points outside WA are questionable), with sampling locations in the Perth Hills (purple; B). Mound locations in John Forrest National Park (C), Beelu National Park (D) and Banyowla Regional Park (E), with purple: mound was sampled for T. westraliensis and A. obeuntis; pink: T. westraliensis only sampled from the mound; orange: A. obeuntis only sampled from the mound. Mounds provided with wheat are labelled in yellow, lucerne in green and control colonies in blue. Maps (B-E) created in GPSVisualizer.com and further modified.

74
netting, Diamond Networks, WA) filled with the diet were then secured
evenly across the cleared space. The contents were of similar volume for each
treatment but averaged 20 g for wheat, 30 g for lucerne and 40 g for controls.
After three weeks, termites were again sampled from the mounds and, when
possible, from the bags.
Sample collection
T. westraliensis and A. obeuntis were collected in March (before feeding) and
April 2016 (after feeding). An electric drill was used to create up to three
minimally invasive access points per mound and individuals of all available
castes were collected using a modified portable vacuum cleaner. At the end of
the experiment, a small pickaxe was also used, as necessary, to access greater
numbers of termites. Samples were transported to the laboratory in the dark
with mound material. Dissections were performed on the day of collection to
minimise possible effects of the sampling procedure on the gut flora.
Samples from different colonies were dissected separately using sterilised
equipment and solutions, as follows. Termites were anaesthetised on ice in a
petri dish for at least ten minutes and separated into the following caste
groups: workers (T. westraliensis workers were further separated into mature
workers with dark head capsules, pale young workers, as per Chapter 2 and a
third group of intermediate pigmentation), soldiers, alates (individuals with
fully developed wings) and nymphs (individuals with wing buds).
Whole intestinal tracts were dissected by first cutting off the head with a
scalpel, then holding the thorax and pulling gently at the distal end of the
abdomen using Inox 5 Watchmaker forceps. Each excised intestinal tract was
placed in a 1.5 ml Eppendorf tube containing 100 μl of 1× phosphate buffered
saline. The process was repeated to obtain up to five biological replicates of 10
guts for each caste. All nymph and alate samples were dissected individually
and young T. westraliensis workers were dissected in groups of up to ten, as

75
available. Ten individual T. westraliensis worker guts and three alate guts
collected in 2015 from mound TW1 (John Forrest National Park) and stored at -
20 °C were processed alongside the other samples to increase the total number
of alates and to measure the effect of sample pooling. In total, 260 T.
westraliensis samples (141 before and 119 after the field experiment; 5 alate, 15
nymph, 62 mature worker, 142 soldier and 36 other worker samples) and 61 A.
obeuntis samples (33 before and 28 after feeding; 4 alate, 3 nymph, 53 worker
and 1 soldier sample) were processed.
To confirm species identity, DNA was extracted from termite heads using a
Qiagen DNeasy Blood & Tissue Kit (Hilden, Germany) following the
manufacturer's protocol. All PCR reactions were performed in 25µl volumes
using HotStartMastermix 2x Polymerase (Genaxxon, Ulm, Germany) and the
primers Modified A-tLeu (5'-CAGATAAGTGCATTGGATTT-3') and TK-N-3785
(5'-GTTTAAGAGACCATTACTTA-3') from Dedeine et al. (2016). PCR products
were purified with the Gennaxon Purification Kit (Genaxxon; Ulm, Germany)
and sent to Seqlab (Sequence Laboratories Göttingen, Germany) for Sanger
sequencing. Sequences were assembled and proofread in Geneious version
8.1.8 (Kearse et al., 2012). These quality-trimmed COII sequences were
compared to previously sequenced COII genes using the nucleotide BLAST
online tool (National Center for Biotechnology Information).
Sequencing and data analysis
DNA was extracted from the gut samples using a previously established
protocol (Paul et al., 2012). The V3 and V4 hypervariable regions of the 16S
rRNA gene were amplified using the primers 343Fmod (5′-
TACGGGWGGCWGCA-3′) and 784Rmod (5′-GGGTMTCTAATCCBKTT-3′)
designed by Köhler et al. (2012) and recommended in Chapter 2. Resulting
amplicons were sequenced in-house as paired-end 300 bp reads on an Illumina
MiSeq using a 600 cycle Miseq Reagent Kit v3 and a Nextera XT Index Kit v2
according to the manufacturer’s instructions.

76
The reads were quality filtered as described in detail in Chapter 2 except that
reads were oriented against the DictDb reference database (Mikaelyan et al.,
2015) to reduce spurious OTUs. Reads were trimmed to 400 bp to maximise
read length while maintaining read quality. OTUs were determined de novo
both for each species separately, as well as together to allow direct
comparison; taxonomy was assigned as per Chapter 2. All datasets were
rarefied to an even sequencing depth of 11,400 reads per sample, to match the
sample with the lowest number of reads.
A pilot chloroplast DNA profiling analysis was also performed during this
study using trnL intron primers c and h (Taberlet et al., 2007) on a subsample
of ten gut extracts, to confirm that the termites had fed on the substrates
provided. The reads were quality filtered as in Chapter 2 except that they were
trimmed to 110 bp and OTUs clustered at 99%. A reference database
containing chloroplast genomes from RefSeq (O'Leary et al., 2016) as well as
unpublished choloroplast sequences from the Pilbara region of WA (Nevill et
al., 2017) was used to assign taxonomy to the reads.
Worker ages
To determine if T. westraliensis workers of different ages could be included in
the same downstream analyses, a UniFrac beta diversity analysis (both
unweighted and weighted) was conducted to measure pairwise distances
between the total reads recovered per worker age group per colony. Samples
were collapsed by colony in QIIME, which resulted in summing by OTU all
reads from workers collected during the first time point from each colony. The
beta diversity calculation was done at a depth of 11,400 reads. Distances were
visualised in a Principal Coordinate Analysis (PCoA) and the average distance
within each group (young n = 21 pairs; mature n = 153) and between ages (n =
126) was summarised and compared within each metric using the Kruskal-
Wallis test, followed by Mann-Whitney-Wilcoxon post-hoc tests with fdr
corrected p-values to account for multiple testing in R (version 3.5.0, R Core
Team, 2018).

77
Investigation of sample pooling, sampling intensity, scope and scale
The core microbiome was defined as the OTUs shared by 100% of samples
within a given set of samples either within each species or across species.
Further analysis was conducted in QIIME (version 1.9.1, Caporaso et al., 2010).
Core microbiomes were determined for every available sample in the
biologically relevant sample groupings listed in Table 1 (Biological category).
These samples represent one caste (low scope) and different levels of scale
(single colony, single location, multiple locations) and sampling intensity.
To test for the effect of sampling scope and intensity on the number and
abundance of the core taxa, these biologically relevant core communities were
compared to core communities generated from artificial datasets. 999 artificial
datasets were created containing 1, 2, 3, 5, 10, 20, 30 or 45 randomly selected
samples (Table 1, Random category), using simple random selection without
replacement from the pool of real samples. Note that these artificial datasets
will include multiple identical selections at a sampling intensity of 1 due to the
limited number of real samples in the pool, namely 52 for mature workers and
260 for all castes (including young workers and those in between ages) in T.
westraliensis. This will be increasingly unlikely at higher sampling intensities
(e.g. there are 1326 possible combinations for selections of 2 samples out of
52). Inverse models were fit to each random grouping using Non-Linear Least-
Squares (nls) in R.
To compare the core communities between simulated sampling intensities,
PCoA using unweighted and weighted Unifrac beta diversity metrics was
conducted on a subset of 20 randomly selected core communities per
sampling intensity for both mature workers and all castes. The samples
included in the analysis consisted of the total abundance of each core OTU
from the samples used in the core determination at each sampling intensity.
The average pairwise distances within each level of sampling intensity were
compared using a Kruskal-Wallis test followed by Mann-Whitney-Wilcoxon

78
Table 1: Summary of samples included in core microbiome analysis, before rarefaction Grouping
type Termite species Sample
number Replicates Sample type(s)
Biological T. westraliensis
A. obeuntis
1
1
29
26
Mature workers from the first time point
Biological Both, separately 3 4 Mature workers from a single colony and first time point
Biological Both, separately 5 3 Mature workers from a single location and first time point, collapsed by colony
Biological Both, separately 10 3 Mature workers from a single location and first time point
Biological T. westraliensis
A. obeuntis
29
26
1
1
All mature workers from a the first time point
Biological T. westraliensis
A. obeuntis
52
53
1
1
All mature workers from both time points
Random Both, separately 1 999 Mature workers/All castes
Random Both, separately 2 999 Mature workers/All castes
Random Both, separately 3 999 Mature workers/All castes
Random Both, separately 5 999 Mature workers/All castes
Random Both, separately 10 999 Mature workers/All castes
Random Both, separately 20 999 Mature workers/All castes
Random Both, separately 30 999 Mature workers/All castes
Random Both, separately 45 999 Mature workers/All castes
Random T. westraliensis 1 999 Mature workers and 10%, 30% or 50% soldiers
Random T. westraliensis 2 999 Mature workers and 10%, 30% or 50% soldiers
Random T. westraliensis 3 999 Mature workers and 10%, 30% or 50% soldiers
Random T. westraliensis 5 999 Mature workers and 10%, 30% or 50% soldiers
Random T. westraliensis 10 999 Mature workers and 10%, 30% or 50% soldiers
Random T. westraliensis 20 999 Mature workers and 10%, 30% or 50% soldiers
Random T. westraliensis 30 999 Mature workers and 10%, 30% or 50% soldiers
Random T. westraliensis 45 999 Mature workers and 10%, 30% or 50% soldiers

79
post-hoc tests with fdr corrected p-values. To test the effect of sample type, a
PCoA was used to compare 10 individual T. westraliensis worker guts from
mound TW1 to in-silico-pooled groups of five. Each randomly selected group
of five was offset by another group containing the remaining five samples.
Core Communities
Based on results of our random resampling analysis, only groupings with at
least 20 samples per analysed level were considered to have sufficient
replication to infer reliable core communities. Levels within each category of
interest (species, time point, location or diet) were rarefied to the same
number of samples prior to determination of the core community. The
resulting core microbiomes were compared by the number of OTUs and the
abundance of those taxa. For each species, separate core microbiomes were
calculated for each caste (including only mature workers for T. westraliensis)
individually and in all possible combinations. The number of shared OTUs
was visualised using the R VennDiagram package (version 1.6.20; Chen and
Boutros, 2011). Unreliable core communities obtained with less than 20
samples were greyed out. Pie charts (base R) were used to visualise the relative
number and abundance of OTUs in the core and non-core communities. As
above, all OTUs found in 100% of samples were considered the core
community and in both analyses, taxonomic groups were classified by
functions predicted from the literature (see Table S1 for full functions list and
sources).
Core communities were also calculated across all samples from both termite
species, as well as in triplicate for rarefied datasets including workers from
both species from all locations and within each location, in an attempt to tease
apart vertically transferred from environmentally acquired taxa. Rarefaction
was carried out at 27 samples in each dataset, the number of workers collected
in Beelu National Park, the unit of interest with the smallest number of
available samples. Core community sizes were compared using proportion
tests in R.

80
Effect of diet, location, colony of origin and co-habitation
The rarefied datasets described above were used in distance-based redundancy
analysis (dbRDA), a constrained form of PCoA, using unweighted and
weighted UniFrac beta diversity metrics. dbRDAs and ANOVA-like
permutation tests (PERMANOVA) were both conducted in QIIME (originally
from the R vegan package, Oksanen et al., 2018). Partial distance-based
redundancy analysis (dbRDA), which estimates the individual effects of
crossed factors, was conducted in the R vegan package (version 2.5-2, Oksanen
et al., 2018) to isolate the effects of location and diet, by removing the effect of
the other.
The geographical distance between all pairwise combinations of colonies was
calculated from their GPS coordinates, using the R package geosphere (version
1.5-7, Hijmans, 2017). The Pearson correlations and associated p-values were
calculated in R between physical distances and weighted or unweighted
UniFrac beta diversity distances for worker gut samples collected during the
first time point (natural diet), collapsed by colony in QIIME. The beta
diversity calculation was done at a depth of 11,400 reads as for previous
analyses. Workers from colony TW1 were not included in this analysis since
the GPS coordinates for the mound were not recorded. For each species, the
average pairwise weighted and unweighted UniFrac distances were also
compared a) within colony (pairs collected from the same colony, n = 18 and
23 pairs for T. westraliensis and A. obeuntis respectively), b) within location
(pairs of collapsed colonies from the same location, n = 40 and 23) and c)
between locations (pairs of collapsed colonies from different locations, n = 96
and 55) using a Kruskal-Wallis test followed by Mann-Whitney-Wilcoxon
post-hoc tests with fdr corrected p-values. Workers from colony TW1 were not
included as they were individual samples rather than pooled, although their
average pairwise beta diversity distance was compared to the within-location
average using Mann-Whitney-Wilcoxon tests.

81
Results:
Effect of sampling intensity and scope on the core community
The number of bacterial OTUs was compared in the core communities
obtained from our natural sample groupings to randomly generated groups of
samples of the same size (Figure 2). Increasing the number of samples used to
determine the core community decreased the number of bacterial OTUs in it.
This trend was modelled with an inverse function as y = 675.0/x+150.3 for T.
westraliensis and y = 1282.7/x+194.5 for A. obeuntis. In both cases, the average
core community size approached the asymptote (predicted "real" core
community size of 150 and 195 OTUs respectively) around 20 samples and
crossed it at 30 samples. Thus, for both species a minimum of 20 samples was
required to accurately estimate the core community, despite the gut
microbiome species richness of A. obeuntis being 1.5 times greater than that of
T. westraliensis. In T. westraliensis, where there were more replicates from
each caste, increasing scope by including multiple castes further decreased the
size of the core community (Figure 3A; y = 677.7/x+122.7). Interestingly,
changing the proportion of samples from each caste had no significant effect
on the slope or asymptote (Figure 3B; average equation y = 668.3/x+136.9).
As demonstrated in T. westraliensis, the sampling intensity used to determine
the core community affected the identity and abundance of OTUs in it
(weighted PCoA shown in Figure 4). Increasing sampling intensity also
decreased the variation between the replicate core communities. Indeed, both
when considering workers only or all castes, average UniFrac distances were
found to be highly significantly different by level of sampling intensity
(Kruskal-Wallis test, p < 2.2x10-16). All post-hoc comparisons (Mann-Whitney-
Wilcoxon tests) were highly significant except for pairwise tests between 20,
30 and 45 samples in the weighted analysis including all castes; all p-values are
listed in Table S2). Pooled samples were less variable than single-gut samples
in both bacterial OTU number and abundance, such that pools were always
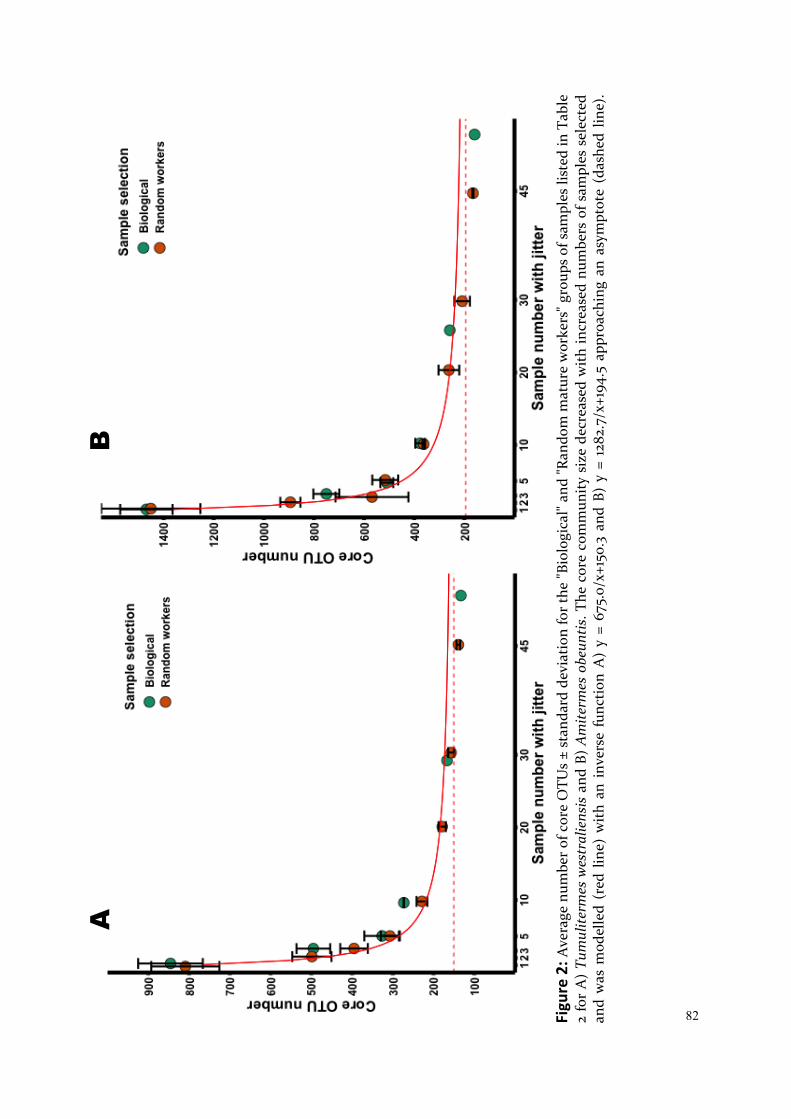
82
Fi
gure
2: A
vera
ge n
umbe
r of
cor
e O
TUs
± st
anda
rd d
evia
tion
for
the
"Bio
logi
cal"
and
"Ran
dom
mat
ure
wor
kers
" gro
ups
of s
ampl
es li
sted
in T
able
2
for
A)
Tum
ulite
rmes
wes
tral
iens
is a
nd B
) A
mite
rmes
obe
untis
. The
cor
e co
mm
unity
siz
e de
crea
sed
with
incr
ease
d nu
mbe
rs o
f sam
ples
sel
ecte
d an
d w
as m
odel
led
(red
lin
e) w
ith a
n in
vers
e fu
nctio
n A
) y
= 67
5.0/
x+15
0.3
and
B) y
= 1
282.
7/x+
194.
5 ap
proa
chin
g an
asy
mpt
ote
(das
hed
line)
.
A
B
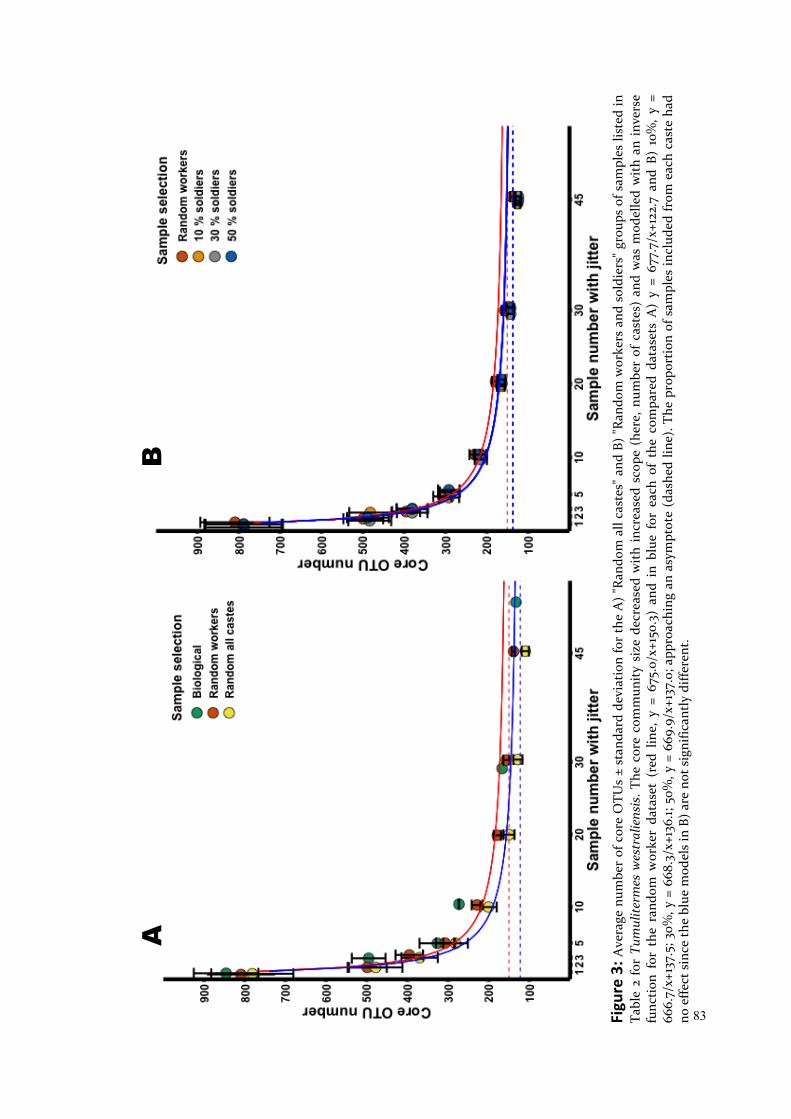
83
Fi
gure
3: A
vera
ge n
umbe
r of
cor
e O
TUs
± st
anda
rd d
evia
tion
for
the
A)
"Ran
dom
all
cast
es"
and
B) "
Rand
om w
orke
rs a
nd s
oldi
ers"
gro
ups
of s
ampl
es li
sted
in
Tabl
e 2
for
Tum
ulit
erm
es w
estr
alie
nsis
. The
cor
e co
mm
unit
y si
ze d
ecre
ased
wit
h in
crea
sed
scop
e (h
ere,
num
ber
of c
aste
s) a
nd w
as m
odel
led
wit
h an
inv
erse
fu
ncti
on f
or t
he r
ando
m w
orke
r da
tase
t (r
ed l
ine,
y =
675
.0/x
+150
.3)
and
in b
lue
for
each
of
the
com
pare
d da
tase
ts A
) y
= 67
7.7/
x+12
2.7
and
B) 1
0%,
y =
666.
7/x+
137.
5; 3
0%, y
= 6
68.3
/x+1
36.1;
50%
, y =
669
.9/x
+137
.0; a
ppro
achi
ng a
n as
ympt
ote
(das
hed
line)
. The
pro
port
ion
of s
ampl
es in
clud
ed fr
om e
ach
cast
e ha
d no
effe
ct s
ince
the
blue
mod
els
in B
) are
not
sig
nific
antl
y di
ffere
nt.
A
B

84 Fi
gure
4:
Prin
cipa
l C
oord
inat
e A
naly
sis
(PC
oA)
show
ing
wei
ghte
d U
niFr
ac d
ista
nces
bet
wee
n co
re c
omm
uniti
es c
alcu
late
d fr
om d
iffer
ent
num
bers
of r
ando
mly
sel
ecte
d sa
mpl
es fr
om th
e po
ol o
f A) m
atur
e T.
wes
tral
iens
is w
orke
rs B
) all
T. w
estr
alie
nsis
cas
tes.
Gro
upin
gs s
epar
ate
alon
g PC
1, ex
plai
ning
43%
of t
he v
aria
tion.
The
gro
ups
wer
e fo
und
to b
e si
gnifi
cant
ly d
iffer
ent
in b
oth
case
s, in
ter
ms
of t
he a
vera
ge p
airw
ise
dist
ance
w
ithin
eac
h gr
oup,
usi
ng a
Kru
skal
-Wal
lis t
est
(p <
2.2
x10-1
6 ). A
ll po
st-h
oc c
ompa
riso
ns (
Man
n-W
hitn
ey-W
ilcox
on t
ests
) w
ere
high
ly s
igni
fican
t ex
cept
for p
airw
ise
test
s in
B) b
etw
een
20, 3
0 an
d 45
sam
ples
.
A
B
Num
ber
of sa
mpl
es

85
more similar to each other than were the single-gut samples (Figure 5). The
average UniFrac distance was significantly lower (Mann-Whitney-Wilcoxon;
unweighted, p = 0.018; weighted, p = 1.912e-06) between pooled samples
collected from each T. westraliensis colony (unweighted, 0.326; weighted,
0.086), as compared to between the TW1 individuals (unweighted, 0.343;
weighted, 0.144).
Species and caste differences in gut and core communities
Gut communities of young workers were found to be distinct from mature
workers in T. westraliensis (Figure 6; Kruskal-Wallis test p < 4.287e-10). Their
gut communities were similar at the phylum level, but young workers had
twice as many Actinobacteria as mature workers, making up 3% of all young
worker reads (Table S3). Nymphs and alates were much more distinct. Overall,
44% of the gut microbiome of alates was Bacteroidetes as compared to 11% in
nymphs and 14-17% in other castes (soldiers and workers of various ages).
Spirochaetes were the most abundant phylum in nymphs, making up 44% of
all reads, but 21% in alates and 27-32% in other castes. Nymphs also had
almost four times more TM7 reads (alates 0.7%, other castes 0.5-0.9%) and 19
times more Chlorobi (alates 0.4%, other castes 0.1-0.3%).
The species-level core communities comprised 56 OTUs for T. westraliensis
and 116 for A. obeuntis (Figure 7A). Within each species, few OTUs (ranging
from 0 OTUs in A. obeuntis workers to 8 OTUs in T. westraliensis workers)
were found to be exclusive to, but universal within a single caste. When
categories with with less than 20 samples were included, such caste-specific
OTUs ranged from 19 OTUs in T. westraliensis nymphs to 84 OTUs in T.
westraliensis alates (Figure 7A).
Each species core was made up of a comparatively small number of highly
abundant taxa, forming about half of OTUs recovered from the gut
community (Figure 7C). The A. obeuntis core community was dominated by
Firmicutes, a phylum known to perform cellulose, hemicellulose and
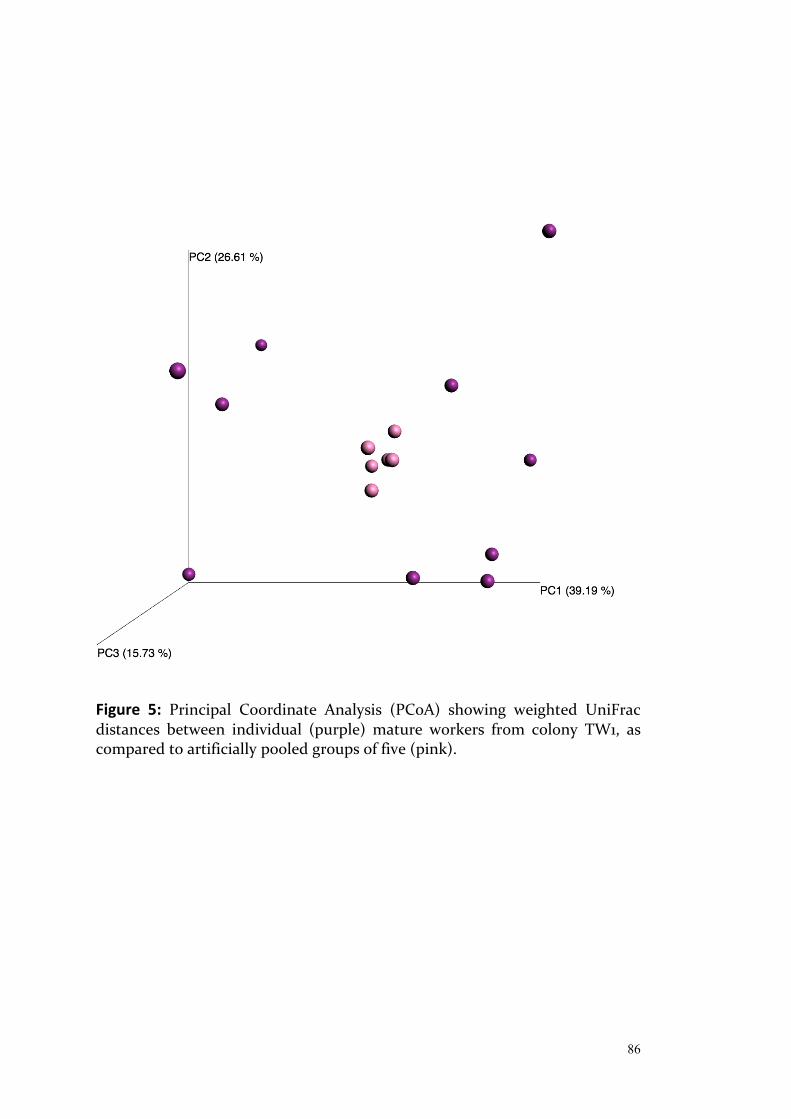
86
Figure 5: Principal Coordinate Analysis (PCoA) showing weighted UniFrac distances between individual (purple) mature workers from colony TW1, as compared to artificially pooled groups of five (pink).
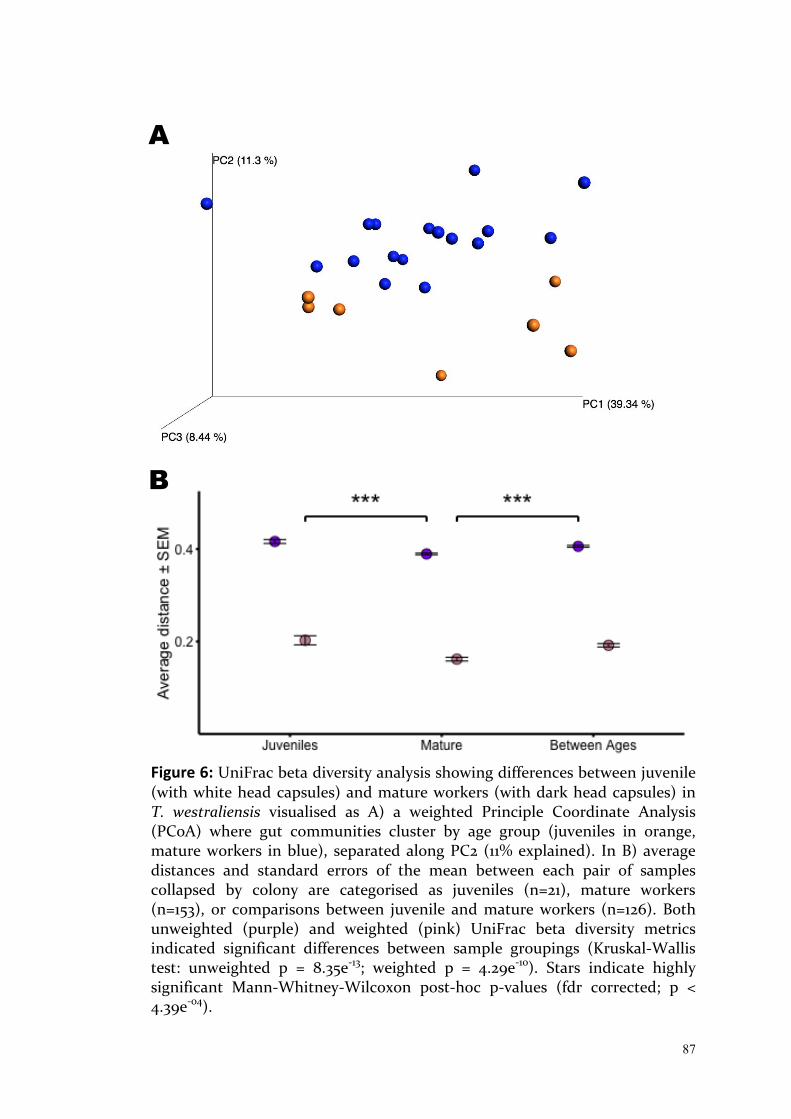
87
Figure 6: UniFrac beta diversity analysis showing differences between juvenile (with white head capsules) and mature workers (with dark head capsules) in T. westraliensis visualised as A) a weighted Principle Coordinate Analysis (PCoA) where gut communities cluster by age group (juveniles in orange, mature workers in blue), separated along PC2 (11% explained). In B) average distances and standard errors of the mean between each pair of samples collapsed by colony are categorised as juveniles (n=21), mature workers (n=153), or comparisons between juvenile and mature workers (n=126). Both unweighted (purple) and weighted (pink) UniFrac beta diversity metrics indicated significant differences between sample groupings (Kruskal-Wallis test: unweighted p = 8.35e-13; weighted p = 4.29e-10). Stars indicate highly significant Mann-Whitney-Wilcoxon post-hoc p-values (fdr corrected; p < 4.39e-04).
A
B
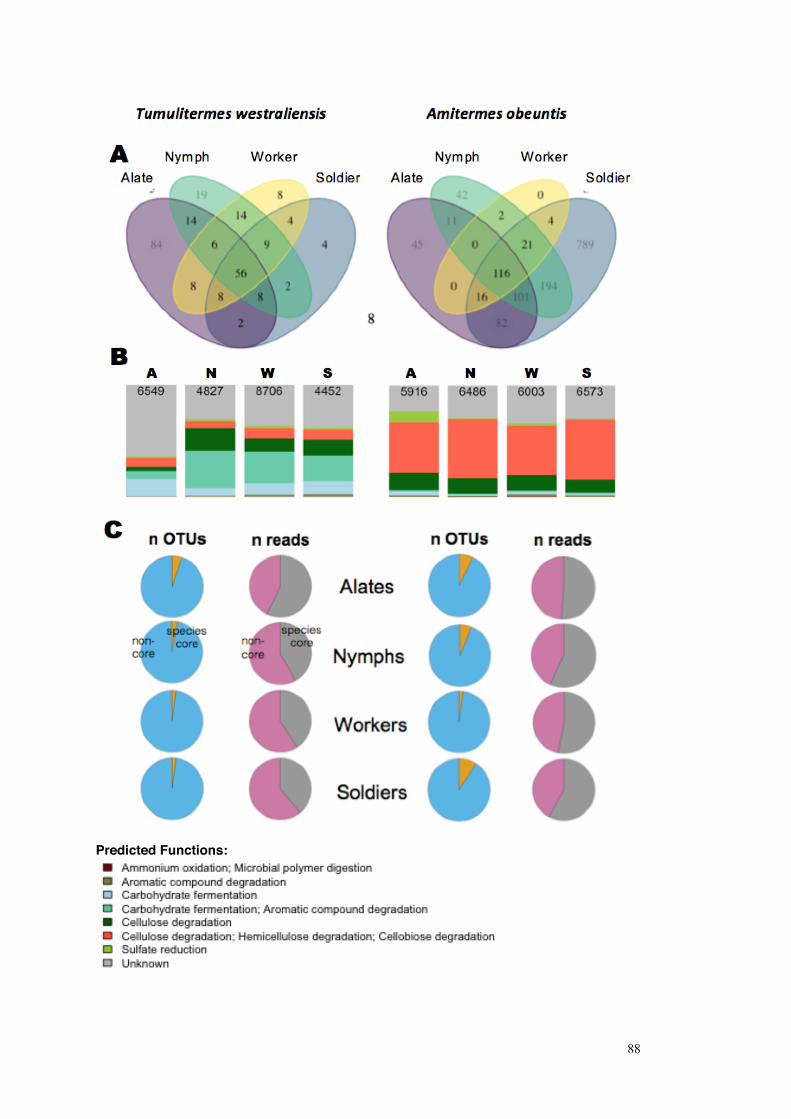
88
Predicted Functions:

89
Figure 7: Summary of the species level core for T. westraliensis and A. obeuntis. Venn diagrams (A) show that all T. westraliensis major castes (5 alate, 15 nymph, 52 mature worker, 142 soldier samples) shared 56 OTUs and 8 with A. obeuntis samples, which had a core made up of 116 OTUs (4 alate, 3 nymph, 53 worker and 1 soldier sample). Numbers shown in grey were calculated using less than the 20 samples recommended by this study and therefore are not thought to be typical of these castes (note a single A. obeuntis soldier pooled sample was included and hence a soldier core community was not calculated and the total number of OTUs not shared by other castes is shown in grey). Bar charts (B) show the predicted functions for the species core by average abundance within each caste. Pie charts (C) summarise the proportion of OTUs (orange) and reads (grey) made up on average by the species core in each caste.

90
cellobiose degradation (particularly family Ruminococcaceae, Table S1),
followed by taxa of unknown functions (Figure 7B). Compared to other castes,
alates had higher proportions of sulfate-reducing bacteria
(Deltaproteobacteria, Desulfovibrionaceae) and carbohydrate fermenters, and
fewer organisms of unknown functions. In T. westraliensis, a large proportion
of the species core community was of unknown function. This was particularly
pronounced in the alates, where two thirds of the reads could not be assigned
a function. The two major taxa whose functions could not be determined
were Bacteroidetes of the family Porphyromonadaceae, followed by
unclassified Treponema. Of those OTUs which could be assigned to functional
groups, taxa contributing to carbohydrate fermentation and aromatic
compound degradation (Spirochaetes, Treponema Ia) were the most abundant
in all castes except alates, where carbohydrate fermentation was more
common (Bacteroidetes, Rikenellaceae).
Eight OTUs were shared across all 321 samples collected from both species
(Figure 7A). Three of these were identified as members of the Firmicutes
family Ruminococcaceae, thought to contribute to cellulose degradation,
hemicellulose and cellobiose degradation. Other shared OTUs included
another cellulose degrader from Acidobacteria family Acidobacteriaceae, a
sulfate-reducer from family Desulfovibrionaceae (Proteobacteria), and an
aromatic compound degrading Peptococcaceae (Firmicutes). Two OTUs from
family Synergistaceae (Synergistetes) are of unknown function. Significant
differences in overall community composition were found between the
rarefied datasets of co-habiting and non-co-habiting workers within each
species (8 samples per species per group, unweighted Unifrac; dbRDA p-values
< 0.012 and PERMANOVA p-values < 0.009; Tables S4). Co-habiting termites
across both species (n=16) had 28 OTUs in common compared to 25 shared by
non-cohabiting colonies, 20 of which were common to both, which was not
significantly different (proportion test; p = 0.786; TableS4). Non-co-habiting
termites shared five exclusive OTUs as compared to the co-habiting group's
eight.

91
When rarefying to 27 samples with random inclusion of different castes, the
interspecies core community increased to 14-25 OTUs (triplicate core
calculations, Table S5). Similar to the simulation results, the more castes were
included, the smaller the resulting core community. In particular, core sizes of
23 and 25 OTUs were obtained when including four or five T. westraliensis
castes and a single A. obeuntis caste (workers). Adding in a further A. obeuntis
caste (alates) resulted in a drop to 14 OTUs. Randomly selecting 27 worker
samples from both species resulted in core community sizes of 15-18. Core
communities obtained for each location ranged from 14-17 OTUs for John
Forrest National Park, 21-23 for Banyowla Regional Park and 21 OTUs for Beelu
National Park, with 10 OTUs in common for all three. The size of each
location-specific core community was not significantly different to that of any
other location, or to cores calculated from worker samples selected from all
three locations (proportions tests, p-values > 0.2); however up to 2 OTUs were
unique to each location and universal within all replicate cores at those
locations. In Banyowla Regional Park, these belonged to the carbohydrate
fermenting genus Tannerella (family Porphyromonadaceae, phylum
Bacteroidetes) and family Synergistaceae. Members of Ruminococcaceae were
identified in both National Parks and a Lachnospiraceae OTU was additionally
identified in Beelu National Park.
Effect of sampling scale, location and diet
Significant effects of location were found in workers of both species, and in
soldiers of T. westraliensis using partial db-RDA (p-values < 0.036; Figure 8,
Table S6), dbRDA (p-values = 0.001) and PERMANOVA (p-values = 0.001;
Tables S4). Both species showed significant differences between average
UniFrac beta diversity distances among worker samples collected from the
same colony compared to workers collected from different colonies (Figure 9,
Kruskal-Wallis test: p < 6.70e-7) during the first time point. In T. westraliensis
only, there were significant differences detected between workers collected
from the same or different locations in the unweighted analysis only (Mann-
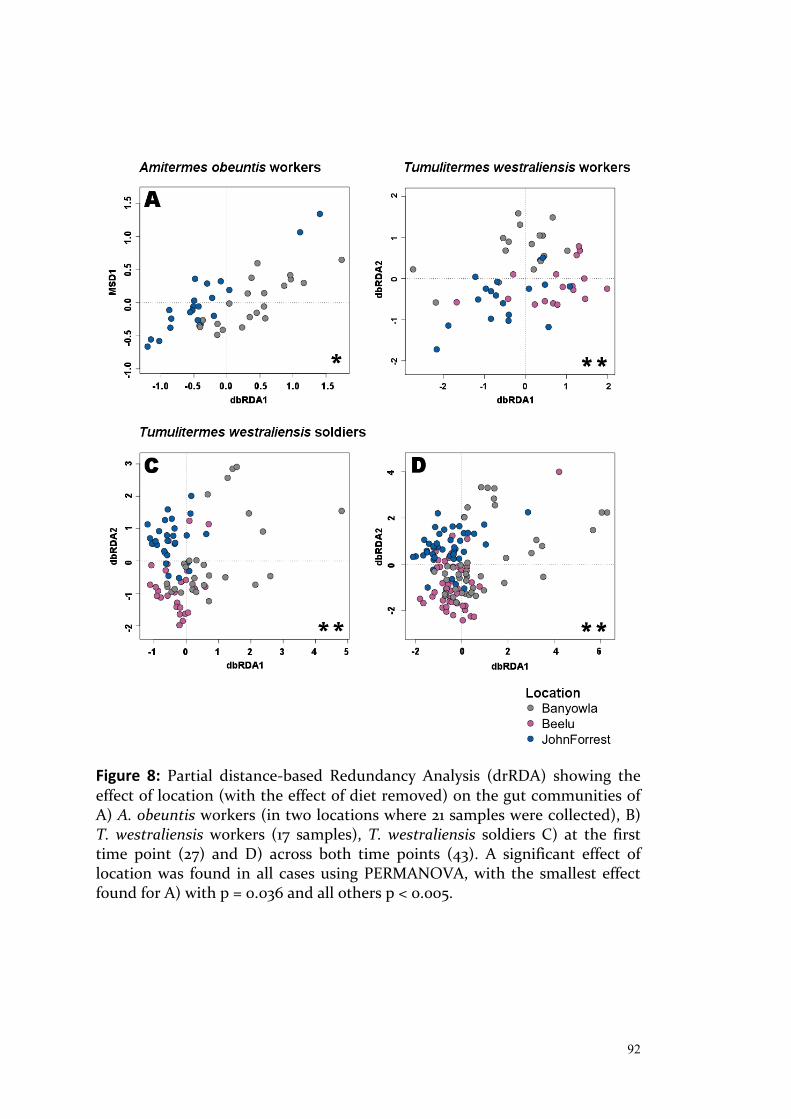
92
Figure 8: Partial distance-based Redundancy Analysis (drRDA) showing the effect of location (with the effect of diet removed) on the gut communities of A) A. obeuntis workers (in two locations where 21 samples were collected), B) T. westraliensis workers (17 samples), T. westraliensis soldiers C) at the first time point (27) and D) across both time points (43). A significant effect of location was found in all cases using PERMANOVA, with the smallest effect found for A) with p = 0.036 and all others p < 0.005.
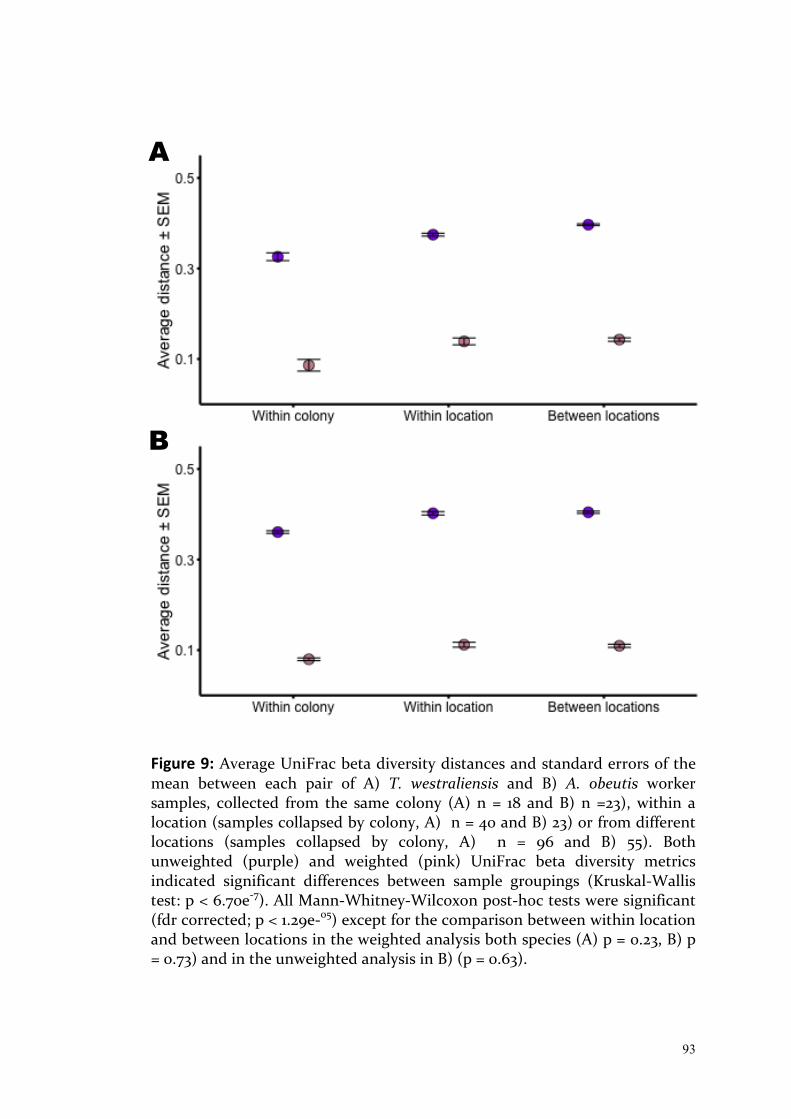
93
Figure 9: Average UniFrac beta diversity distances and standard errors of the mean between each pair of A) T. westraliensis and B) A. obeutis worker samples, collected from the same colony (A) n = 18 and B) n =23), within a location (samples collapsed by colony, A) n = 40 and B) 23) or from different locations (samples collapsed by colony, A) n = 96 and B) 55). Both unweighted (purple) and weighted (pink) UniFrac beta diversity metrics indicated significant differences between sample groupings (Kruskal-Wallis test: p < 6.70e-7). All Mann-Whitney-Wilcoxon post-hoc tests were significant (fdr corrected; p < 1.29e-05) except for the comparison between within location and between locations in the weighted analysis both species (A) p = 0.23, B) p = 0.73) and in the unweighted analysis in B) (p = 0.63).
A
B

94
Whitney-Wilcoxon; fdr corrected p = 3.70e-07). The relationship between
geographic distance between colonies and beta diversity distances between
worker gut communities was first compared overall, followed by comparisons
within a location and to increasingly distant locations (Table S7). Significant
positive correlations were found in T. westraliensis only, with the strongest
correlation between geographic distance and community composition
(unweighted analysis; r = 0.56, p = 8e-13). When community structure was
considered, the correlation remained significant albeit less strongly positive
(weighted analysis; r = 0.18, p = 0.03). No significant correlations were found
within a location, between mid-range locations or between distant locations,
except for the weighted analysis for mid-range locations in A. obeuntis, where
there was a strong negative correlation (r = -0.549; p = 0.034).
Significant effects of diet were found, but only when including at least half the
minimum recommended number of 20 samples per diet group using partial
db-RDA (p-values < 0.05; Figure 10, Table S6), dbRDA (p-values < 0.03) and
PERMANOVA (p-values < 0.03; Tables S4). However, it could not be
confirmed whether the termites fed on the substrates provided in the pilot
trnL dataset (data not shown). Lucerne could not be differentiated from local
native legumes and wheat was not detected in any of the three A. obeuntis and
one T. westraliensis samples tested expected to contain it. More sequences
remained unidentified in the samples provided with wheat as compared to the
others whereas the grass Family Poaceae, which includes wheat, was identified
in the crop of the “wood-feeding” Occasitermes occasus.
I examined the core communities in search of the taxa causing location and
diet effects (Figures 11 and 12; Tables S8-S17, attached as Chapter3_TablesS8-
S17.pdf). Overall core communities (shared between multiple locations or
diets) always constituted a larger proportion of the gut community in terms of
read abundance than core communities specific to a location or diet (made up
of taxa exclusive but universal to each location or diet). A. obeuntis overall
core communities (Figures 11A, 12A and B; Tables S8-S10) were dominated by

95
organisms classified as cellulose degraders also capable of hemicellulose and
cellobiose degradation. This group, the most abundant of which were family
Ruminococcaceae (Firmicutes), comprised about half of the reads assigned to
the core community. Ruminococcaceae was also the most abundant taxa in
half of the location or diet specific cores, while the other core microbiomes
contained larger proportions of the cellulose degrading Treponema Ic group.
Other core functions included carbohydrate fermentation, aromatic
compound degradation, sulfate reduction, ammonium oxidation and
microbial polymer digestion. Interestingly, one function identified in low
abundance in A. obeuntis only was "protist endosymbiont", due to the
presence of OTUs identified as order Rickettsiales (Alphaproteobcteria). It was
found in most group-specific cores, except in all location-specific cores (Table
S8; attached as Chapter3_TablesS8-S17.pdf) and the natural diet group in the
two diet comparison only (Table S10, i.e. was shared in the natural diet group
when including only six samples; Table S9) .
T. westraliensis overall core communities (for workers see Figures 11B, 12C-D,
Tables S11-S13; for soldiers Figures 11C-D, 12 E-F, Tables S14-S17) contained the
same functional groups as A. obeuntis cores, with cellulose, hemicellulose and
cellobiose degraders (Ruminococcaceae), cellulose degraders (Treponema Ic
and If), carbohydrate fermentation (Rikenellaceae and genus Tannerella,
Porphyromonadaceae) and carbohydrate fermenters (coupled with aromatic
compound degradation; Treponema Ia; the most abundant taxon in all T.
westraliensis overall cores) representing about a quarter of reads of known
functions each. Cellulose degradation coupled with hemicellulose and
cellobiose degradation was typically the most commonly assigned function (by
read abundance) in the group specific cores, due to the presence of large
abundances of Ruminococcaceae and/or Lachnospiraceae, depending on the
location or diet. In soldiers only, in Banyowla Regional Park, the
Lachnospiraceae were in similar abundance to the cellulose degrader
Treponema If. Overall, Ruminococcaceae more commonly dominated, but a
higher relative abundance of Lachnospiraceae was linked to the lucerne diet
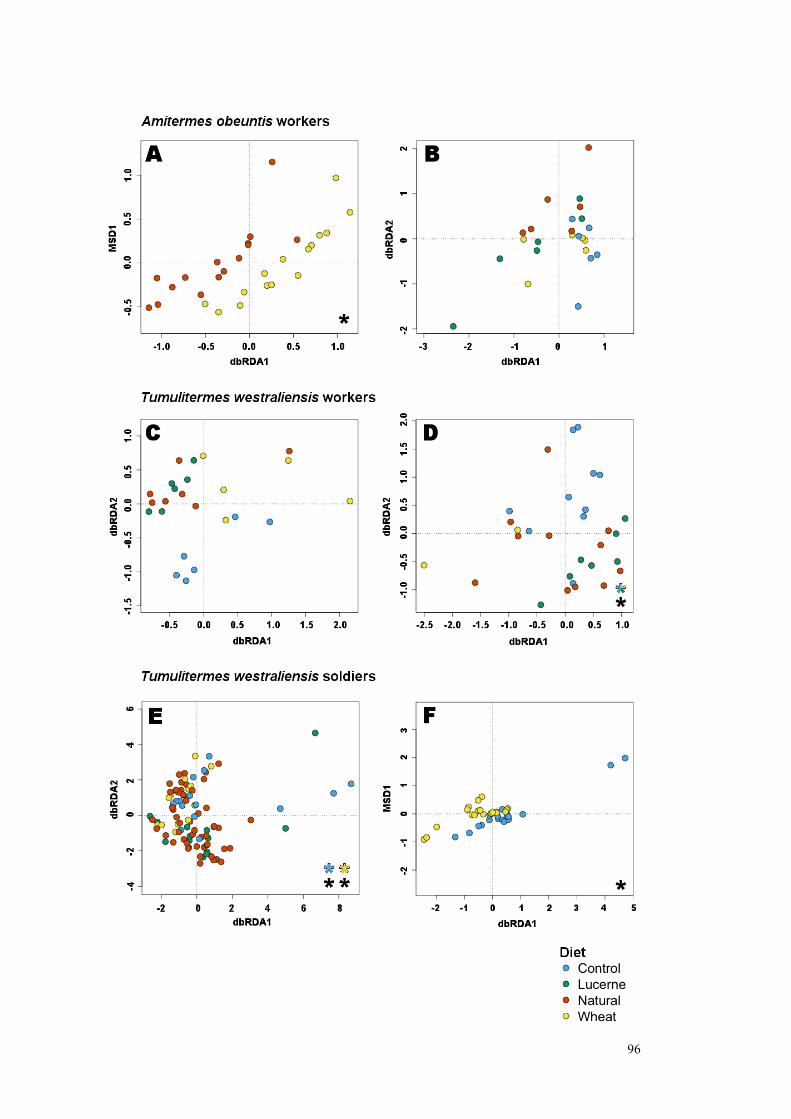
96

97
Figure 10: Partial distance-based Redundancy Analysis (drRDA) showing the effect of diet (with the effect of location removed) on the gut communities of A. obeuntis workers A) with two diets where the most samples were collected (15) and B) with six samples available for each diet; T. westraliensis workers C) with six samples available for each diet and D) with wheat and lucerne combined into a single group to increase sample numbers to 10; and finally T. westraliensis soldiers E) with all diets (14 samples) and F) two diets (19). A significant effect of diet was found in all cases using PERMANOVA, except where sample numbers were much lower than recommended. Post-hoc tests were conducted on D) and E) to determine whether introduced diets were significantly different to the control by using subsets of the datasets containing two diets at a time. The combined wheat and lucerne group was found to be significantly different to the control (p = 0.038) in D). In E) wheat was significantly different to the control (p = 0.001), lucerne was found to be marginally insignificant when compared to the control (p = 0.072) and an outlier resulted in an insignificant test result between wheat and lucerne (p = 0.245; data not shown).

98
Predicted Functions:
Figure 11: Barplots showing the abundance (number of reads shown in each plot) of the overall core community determined for each set of rarefied dataset, as well as the core specific to a location, coloured by function. Specifically, the A. obeuntis worker dataset was rarefied to 21 samples in A); the T. westraliensis worker dataset was rarefied to 17 samples (lower sampling intensity than the 20 sample minimum recommended by this study) in B); T. westraliensis soldier datasets were rarefied to 27 samples in C) and 43 in D). Taxonomic groups thought to undertake multiple functions were classified as a separated functional group, so as not to overestimate their abundance.

99

100
Predicted Functions:
Figure 12: Barplots showing the abundance (number of reads shown in each plot) of the overall core community determined for each set of rarefied datasets, as well as the core specific to a diet, coloured by function. Specifically, A. obeuntis worker datasets were rarefied to 15 samples in A) and 6 in B); T. westraliensis worker datasets were rarefied to 10 samples in C) and 6 in D); T. westraliensis soldier datasets were rarefied to 14 samples in E) and 19 in F), all lower sampling intensity than the 20 sample minimum recommended by this study. Taxonomic groups thought to undertake multiple functions were classified as a separated functional group, so as not to overestimate their abundance.

101
and to the soldier caste in general. Interestingly, workers had relatively more
reads from cellulose degrading bacteria (coupled with other functions) on
natural and wheat diets, whereas soldiers had more in the control and lucerne
diets.
Discussion:
Sampling recommendations for reliable conclusions
It had previously been suggested but never tested that increasingly smaller
termite gut core communities would be recovered across increasing
geographic ranges (Benjamino and Graf, 2016). Our comparisons of real and
simulated data at various sampling scales, scopes and intensities, indicate that
all three factors are important considerations. Sampling intensity, the number
of samples analysed per level of the factor of interest, had the largest influence
and is the easiest for researchers to implement. Pooled samples are a better
representation of a colony if the research question focuses on higher level
variation, whereas individuals allow the full capture of intracolony variation
and can increase sampling intensity if the number of available termites is low.
Overall core communities (shared between multiple locations or diets) always
constituted a larger proportion of the gut community in terms of read
abundance than core communities specific to a location or diet (made up of
taxa exclusive but universal to each location or diet). Functional groups in the
overall core had similar relative abundances in each location or diet specific
group, suggesting that there is a large and consistent portion of the gut
community that plays an important role. In the group-specific core
communities, the functional groups were not of consistent abundance,
suggesting that different functional combinations may be better suited to each
set of environmental conditions.

102
Sampling intensity
Sampling intensity had a bigger effect on the number of taxa included in the
core community than any other factor tested, with an inverse relationship
between sampling intensity and core community size. This suggests that
previous studies based on few samples systematically overestimate the number
of core gut community members. Increasing the number of samples used to
determine the core community decreased the number of core OTUs towards
an asymptote that represents a predicted "true" core size. Based on these
findings, a minimum of 20 samples and ideally 30 samples is required to
determine a core community accurately. At this point, the standard deviation
measured from the simulation overlaps with the asymptote indicating it is a
sufficient number of samples to approach the "true" core community size; any
further increase in sampling intensity has minimal impact on the core
community size. Increasing sampling intensity decreased the variation
between core communities, up to about 20 samples, suggesting that core
communities determined with larger numbers of samples also approach the
"true" core community in terms of composition and structure. These
limitations also apply to detecting treatment effects; increasing sampling
intensity returned more significant p-values when testing the effect of diet.
The simple random sampling method used in this chapter can be applied early
in the bioinformatic analysis phase to test whether sufficient sampling
intensity has been reached for the termite species, geographical range, and the
research question of interest.
Sampling scale
Differences in the gut community structure and composition, and the physical
distance between their colonies of origin were significantly positively
correlated in T. westraliensis only. This trend disappeared when comparing
within a location, or between mid-range or distant locations. In A. obeuntis, a
negative correlation was found in the mid-range colonies only. Significant
differences between average UniFrac beta diversity distances among worker

103
samples collected from the same colony as compared to workers collected
from different colonies were found in both species, however. In T.
westraliensis there was a further distinction in gut community composition
between workers collected from the same or different locations. The db-RDA
analysis resulted in significant effects of location in both termite species, even
at low sampling intensities. The core community size decreased from single
colony, to single location to multiple locations in comparison to the variability
expected from the simple random samples. This suggests that the effect of
location is not based solely on physical distance, but stems from other factors
that make a location unique, perhaps soil properties, available food sources,
the presence of heterospecific termites and/or phylogenetic divergence
between the host termites.
Each location and diet had a distinct set of characteristic OTUs which
appeared in every colony from that location or fed that specific diet. This
means that previous studies relying on samples from a single colony or
location likely also overestimate the number of taxa in the termite gut core
community by including taxa exclusive to that location and not shared across
the species. When the sampling area is limited, taxa considered to be part of
the species core community could be vertically inherited or common to the
environments from which the samples originated. By widening the sampling
scale, this study decreased the inclusion of environmentally acquired
organisms. These could be further limited by including samples from the
entire range of the species, although ruling out environmental acquisition may
remain difficult in species with small ranges like many Western Australian
endemics. I therefore recommend that future studies wishing to draw
conclusions about a species’ core gut microbiome include samples from across
the entire species range to minimise the effect of sampling location on the
recovered gut communities.
Microbiota transplant experiments show that gut communities are assembled
primarily around available niches in the gut habitat. In zebrafish with mouse

104
gut flora, mice with zebrafish flora, and cockroaches with mouse or termite
gut flora, the established gut community was made up of lineages from the
community of origin, but their relative abundances resembled those of the
normal gut community of the recipient host (Rawls et al., 2006; Mikaelyan et
al., 2016). Seedorf et al. (2014) found that the transplanted gut community
composition and structure more closely resembled the community of origin if
it was from a gut environment (human, zebrafish and termite gut) as
compared to a non-gut environment (human skin and tongue, soil, and
estuarine microbial mats). In this way, taxa present in the environment may
appear transiently in the gut community or establish and become resident in
the gut habitat when taken up with soil or food. This may be reflected in the
effects of location and diet described in the current study.
Sampling scope
In the inverse models calculated for T. westraliensis, increasing scope by
including multiple castes decreased core community size by 14 to 28 OTUs,
depending on the number of castes included. Changing the proportion of
samples from each caste had no effect. This has implications for the study of
the species core community. Assuming there is a vertically transmitted, co-
evolved portion of the gut community, I would expect it to include few but
abundant vital community members, so including as many castes as possible
(no matter their sampling intensities) more closely approaches the "true"
species core community. Since alates disperse to form new colonies, they must
carry the complete core community in their guts, presumably with minimal
extra taxa, to maximise the success of their future colonies. Unfortunately,
only a few alates were recovered during this study. The low sampling intensity,
a quarter of the recommended 20, suggests that the 84 OTUs found here as
exclusive but universal to that caste in T. westraliensis, is likely an
overestimation. In this study, alate gut communities were found to have lower
species diversity in both termite species than those of other castes (greater
only than soldiers in A. obeutis, although a single soldier sample was tested).
This is in marked contrast to Diouf et al. (2018), who recovered a greater

105
diversity of OTUs from the guts of 10 swarming alates (635 OTUs) than from
other castes of Nasutitermes arborum (244 to 449 OTUs from at least 25
individuals per caste). One possibility is that in this species, these taxa are
present in all castes but in lower abundance in alates, allowing the detection of
more taxa at the same sequencing depth. Future studies should aim to recover
at least 20 alates (or 20 pools) across the range of the species and maximise
sequencing depth to test how closely the alate gut community resembles the
species core community.
The relative abundances of core gut microbial taxa was similar in all castes of
A. obeuntis, but T. westraliensis alates differed from the other castes in terms
of the relative abundances of its core OTUs. In particular, the Bacteroidetes
family Porphyromonadaceae, of unknown function, dominated almost half of
the alate guts, whereas Spirochaetes and Firmicutes were much more
abundant in other castes. Porphyromonadaceae has been reported in various
other higher termites (Köhler, 2012; Dietrich, 2014; Diouf, 2015; Mikaelyan,
2017; Diouf, 2018) but its function has only been discussed in lower termites
where it is an ectobiont of protists, alongside Spirochaetes. There they may
play a role in consuming oxygen to protect their anaerobic hosts (Hongoh et
al., 2007b). Free-living Porphyromonadaceae in cattle are thought to have
varied functions related to cellulose degradation and carbohydrate
fermentation (Ziemer, 2014). Multiple OTUs within this family are shared by
all four major T. westraliensis castes and may contribute to several key
functions that are passed on across generations via the alates. Young workers
(with large bodies and pale heads) were found to harbour a significantly
different gut community to mature workers (with dark head capsules) in T.
westraliensis. I did not test this in A. obeuntis due to the difficulty in
consistently distinguishing worker instars. Li et al. (2016) concluded that
differences in the gut community structure of workers of different ages in the
fungus-cultivating termite Odontotermes formosanus were driven by changes
in both diet and the gut environment. Genera Candidatus Captivus and
Candidatus Hepatincola (order Rickettsiales, class Alphaproteobacteria) were

106
found in multiple A. obeuntis core communities but not in T. westraliensis.
These are typically protist endosymbionts, reported in at least ciliates and
amoebae (Szokoli et al., 2016). They may indicate the presence of protists in
the gut, not linked to any particular diet, which can be confirmed with the use
of eukaryotic primers (Pawlowski et al., 2012; Adl et al., 2014). The presence of
protists in higher termites is further discussed in Chapter 4.
Eight OTUs were shared across all samples collected from both species,
including three members of the lignocellulose-degrading Rumminococcaceae
(Firmicutes) and one member of Acidobacteriaceae (Acidobacteria; cellulose
degrader). A member of the sulfate-reducing family Desulfovibrionaceae
(Proteobacteria) may also consume oxygen and hydrogen, produce acetate and
fix nitrogen (Kuhnigk et al., 1996). Peptococcaceae (Firmicutes) was linked to
aromatic compound degradation and may play a role in the degradation of
lignin (Geib et al., 2008) or plant defensive secondary metabolites (Bennett
and Allsgrove, 1994). Two OTUs from family Synergistaceae (Synergistetes)
were of unknown function. Hongoh et al., (2007a) suggests that members of
this family may ferment amino acids into acetate and other products.
Importantly, cellulose degradation was a function represented in all core
communities (overall and group-specific) and the most common function for
OTUs shared across both species. Hence these taxa are good candidates for
further study. For example, taxon-specific primers could be designed based on
these 16S rRNA sequences to target cellulose degraders of interest with long-
read Pacific Biosciences sequencing (Orr et al., 2018) and recover cellulase
enzyme sequences to be cloned into culturable organims for testing.
The eight OTUs shared in all samples may be vertically transferred, acquired
from a common ancestor. Conversely, up to two OTUs per location were
found to be exclusive to each location but shared by workers from both
species (members of the carbohydrate fermenting genus Tannerella (family
Porphyromonadaceae, phylum Bacteroidetes), family Synergistaceae, and the
lignocellulose degraders Ruminococcaceae and Lachnospiraceae) and are

107
therefore likely to be environmentally acquired. However, due to the relatively
close proximity of the locations, it cannot be ruled out that additional OTUs
shared amongst multiple locations could be environmentally acquired. Indeed,
it is possible that the eight universal OTUs may be widespread taxa acquired
in all locations, since the two termite species share the whole geographic
range included in the study. Recent work suggests that members of
Ruminococcaceae gut cluster 8 (represented by two of these universal OTUs)
are generally environmentally and/or horizontally acquired (Bourguignon et
al., 2018). Significant differences between co-habiting and non-co-habiting
workers of both species demonstrate the strength of external influences on the
termite gut community, with co-habiting workers sharing eight exclusive
OTUs across species. Interestingly, five OTUs were found to be exclusive to
non-co-habiting termites, with a common source being more difficult to infer.
Future studies could investigate more closely related species, with similar
natural diets and partially overlapping geographic ranges to further tease apart
the source of gut community members. For example, Tumulitermes dalbiensis
also collects grass and leaf debris (Abensperg-Traun, 1992); its range
overlaping that of T. westraliensis but more widespread (Watson and Abbey,
1993). T. pastinator , like T. westraliensis, was classified as a grass-feeder by
Hill (1942) and is found in the northern half of Australia (Watson and Abbey,
1993). Taxa shared by all three species are likely to be vertically transferred and
the result of common ancestry, whereas taxa found exclusively in T.
westraliensis and T. dalbiensis where their range overlaps are likely to be
environmentally acquired. Profiling the microbial community from the
surrounding environment and available food sources may also indicate how
much of the gut community is free-living in the environment. However,
previous studies have shown that there is typically little overlap in
environmental and gut communities (Fall et al., 2007) presumably because
taxa found at non-detectable levels in the environment may establish in gut
niches (Seedorf et al. 2014).

108
Diet drives functional shift
I have shown that the termite gut community can be manipulated during
three weeks under field conditions. This method may allow the identification
of key organisms or enzymes relevant to breaking down substrates of interest,
all without introducing potentially traumatic relocation and artificial
conditions to colony members. When sampling intensity was sufficiently high,
a significant difference between diet groups was detected. A portion of the gut
community was exclusive and universal to each feeding group.
Changes in the gut community do not guarantee successful feeding on the
substrates provided, however. I could not confirm that the target colonies fed
on the provided substrates, although A. obeuntis was recovered from multiple
bags. Bags were weighed before and after the experiment, however I consider
this data unreliable due to the uneven loss of smaller food particles through
the nylon netting during transport and the presence of other termite species in
some bags. Any food that may have been collected by T. westraliensis would
be added to full stores in the mound, likely leading to a broadening of the diet
rather than a replacement. Natural food sources were disturbed while setting
up the experiment, so the diet has likely shifted regardless of whether the
colony fed on the substrates provided.
A pilot chloroplast trnL DNA profiling analysis (data not shown) conducted to
confirm that termites fed on the substrates provided lead to inconclusive
results. The termites were certainly not exclusively feeding on the plants of
interest and this diversity of reads and the low resolution of the trnL primers
meant I could not confidently differentiate lucerne from local native legumes.
Wheat was not detected, although more sequences remained unidentified in
samples expected to contain it as compared to the others. The grass Family
Poaceae, which includes wheat, was identified in one sample, the crop of the
“wood-feeding” Occasitermes occasus. This suggests wheat should have been
detected at least at family level, unless the sequences retained for this
substrate were not of sufficient length or quality for identification (Särkinen et

109
al., 2012). Analysing the crop contents and the P3 contents returned different
plant species, perhaps reflecting the time since ingestion or the level of
degradation of the target DNA. Primers targeting longer regions could result
in more accurate identification but may introduce further bias since the target
DNA is from dried material and being digested. One possibility would be to
optimise the DNA extraction protocol for dried plant samples (Särkinen et al.,
2012). Unpublished choloroplast sequences from the Pilbara region of WA
(Nevill et al., 2017) was used in this preliminary analysis; the addition of plants
found in the Perth Hills may help further define the diet of local species and
the success of field-based feeding experiments.
Significant differences between controls and lucerne or wheat-fed termites,
however, suggest each treatment had a different effect. Testing untouched
colonies on a natural diet at the second time point would have helped confirm
whether time, disturbance and the reshuffling of substrates caused by raking
the area around the colony, or the feeding treatment itself was responsible for
the observed changes.
Ruminococcaceae and Lachnospiraceae OTUs (Firmicutes) were commonly
shared across samples within a species and have both been shown to degrade
cellulose, as well as hemicellulose and cellobiose (Thompson et al., 2012),
establishing their ability to attack many components of the termites’ diet and
explaining their abundance in the gut. In fact, Firmicutes was the most
abundant phylum in all T. westraliensis castes except in the reproductives'
guts, where it was second most abundant in nymphs (after Spirochaetes) and
third most abundant in alates (after Bacteroidetes and Spirochaetes). Both
Bacteroidetes and Spirochaetes were primarily linked to carbohydrate
fermentation and Spirochaetes also to cellulose degradation (Mikaelyan et al.,
2014; Dietrich et al., 2014), although many of their functions remained
unknown. Multiple taxa from both phyla have also been linked to nitrogen
fixation (Lilburn, 2001; Inoue et al., 2015), an important contribution in the gut
due to the nitrogen poor diet of most termites and justifying their prevalence.

110
Sulfate reduction was a function found in all core communities. Kuhnigk et al.
(1996) suggested that sulfate-reducing bacteria of the genus Desulfovibrio
played a broader role in the termite gut, since their isolates had very high
respiration rates, consumed oxygen as well as hydrogen, and fixed nitrogen.
Hence they shifted digestive end products from sugars to acetate and
enhanced termites’ nitrogen poor diet. Planctomycetes were also commonly
identified in core microbiomes. Their role in the termite gut is still unknown
since they are all uncultivated lineages. They may degrade microbial polymers
found in decaying wood and humus, or if these are anaerobic organisms, they
could be oxidising ammonium (Köhler et al., 2008).
Due to difficulty in controlling access to other food sources, it is not clear from
these results whether field experiments will lead to the identification of
bacterial taxa specific to a substrate of interest. Future studies on harvesters
like T. westraliensis could focus on the plant species recovered from food
stores. Studies involving species such as A. obeuntis that feed directly at the
source could place the substrate directly on top of the mound to maximise
chances of it being consumed (Lambert and Power, 1999). This method was
successfully trialled during this project with plastic containers containing karri
wood or wheat straw to collect large numbers of A. obeuntis, Occasitermes
occasus or Coptotermes acinaciformis individuals. Evidence of feeding, ie holes
in the wood, was only found in the latter two species; wheat was not trialled
with these species.
Conclusion
A strong effect of location was found in this study, as well as an effect of diet
in a field setting for two Western Australian endemic species: Tumulitermes
westraliensis and Amitermes obeuntis. There is a relatively small number of
OTUs shared by all castes in each species. Together, they comprise
approximately half of reads recovered from the gut community. These OTUs
could form the core community passed on from generation to generation. In

111
future, comparing gut communities of termites within the same genus with
overlapping geographical ranges would help differentiate between vertical
transmission and environmental acquisition of gut community members. The
most abundant recurring taxa typically had multiple functions and cellulose
degradation was the most common function predicted in all core
communities. Widely shared and abundant taxa, as well as those universal but
exclusive to a diet group are good candidates for further genomic study to
isolate enzymes of interest for biofuel production, cloning in culturable hosts
and in-vitro study.
Based on the findings of this study, the following sampling guidelines are
recommended to obtain an "accurate" core community:
Scope: All relevant castes should be included, ie only workers if that is the
scope of the question, or all castes for a total species core community
estimation.
Scale: The maximum relevant range should be included, which could be a
single colony if that is the scope of the question, or multiple locations across
the entire species range for a total species core community estimation.
Intensity: A minimum of 20, ideally 30, samples per level of each unit of
interest should be included. This translates to at least 20 samples in total to
estimate a species core microbiome, or 20 samples per treatment, location or
colony as relevant to the scope of the research question.

112
References Abdul Rahman, N., Parks, D. H., Willner, D. L., Engelbrektson, A. L., Goffredi, S. K.,
Warnecke, F., Scheffrahn, R. H., Hugenholtz, P. (2015). A molecular survey of Australian and North American termite genera indicates that vertical inheritance is the primary force shaping termite gut microbiomes. Microbiome, 3, 5.
Abensperg-Traun, M. (1992). The effects of sheep-grazing on the subterranean termite fauna (Isoptera) of the Western Australian wheatbelt. Australian Journal of Ecology, 17, 425-432.
Adl, S. M., Habura, A., & Eglit, Y. (2014). Amplification primers of SSU rDNA for soil protists. Soil Biology and Biochemistry, 69(0), 328-342.
Benjamino, J., & Graf, J. (2016). Characterization of the core and caste-specific microbiota in the termite, Reticulitermes flavipes. Frontiers in Microbiology, 7, 1.
Bennett, R. N., & Allsgrove, R. M. W. (1994). Secondary metabolites in plant defence mechanisms. New Phytologist, 127, 617-633.
Bourguignon, T., Lo, N., Dietrich, C., Šobotník, J., Sidek, S., Roisin, Y., Brune, A., Evans, T. A. (2018). Rampant host switching shaped the termite gut microbiome. Current Biology, 28(4), 649-654.e2.
Brune, A., & Ohkuma, M. (2011). Role of the termite gut microbiota in symbiotic digestion. In D. E. Bignell, Y. Roisin, & N. Lo (Eds.), Biology of Termites: a Modern Synthesis (pp. 439-475). Netherlands: Springer.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K. et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7, 335-336.
Chen, H., & Boutros, P. C. (2011). VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics, 12, 35.
Dedeine, F., Dupont, S., Guyot, S., Matsuura, K., Wang, C., Habibpour, B. et al. (2016). Historical biogeography of Reticulitermes termites (Isoptera: Rhinotermitidae) inferred from analyses of mitochondrial and nuclear loci. Molecular Phylogenetics and Evolution, 94(Pt B), 778-790.
Dietrich, C., Köhler, T., & Brune, A. (2014). The cockroach origin of the termite gut microbiota: patterns in bacterial community structure reflect major evolutionary events. Applied and Environmental Microbiology.
Diouf, M., Hervé, V., Mora, P., Robert, A., Frechault, S., Rouland-Lefèvre, C., Miambi, E. (2018). Evidence from the gut microbiota of swarming alates of a vertical transmission of the bacterial symbionts in Nasutitermes arborum (Termitidae, Nasutitermitinae). Antonie Van Leeuwenhoek, 111(4), 573-587.
Diouf, M., Roy, V., Mora, P., Frechault, S., Lefebvre, T., Hervé, V., Rouland-Lefèvre, C., Miambi, E. (2015). Profiling the succession of bacterial communities throughout the life stages of a higher termite Nasutitermes arborum (Termitidae, Nasutitermitinae) Using 16S rRNA Gene Pyrosequencing. PLoS One, 10, e0140014.
Eutick, M. L., Veivers, P., O’Brien, R. W., & Slaytor, M. (1978). Dependence of the higher termite, Nasutitermes exitiosus and the lower termite, Coptotermes lacteus on their gut flora. Journal of Insect Physiology, 24(5), 363-368.
Fall, S., Hamelin, J., Ndiaye, F., Assigbetse, K., Aragno, M., Chotte, J. L. et al. (2007). Differences between bacterial communities in the gut of a soil-feeding termite (Cubitermes niokoloensis) and its mounds. Applied and Environmental Microbiology, 73(16), 5199-5208.
Geib, S. M., Filley, T. R., Hatcher, P. G., Hoover, K., Carlson, J. E., Jimenez-Gasco, M.

113
D. M. et al. (2008). Lignin degradation in wood-feeding insects. Proceedings of the National Academy of Sciences of the United States of America, 105(35), 12932-12937.
Hijmans, R. J. (2017). geosphere: Spherical Trigonometry. R package version 1.5-7. https://CRAN.R-project.org/package=geosphere.
Hongoh, Y., Sato, T., Dolan, M. F., Noda, S., Ui, S., Kudo, T. et al. (2007a). The motility symbiont of the termite gut flagellate Caduceia versatilis is a member of the “Synergistes” group. Applied and Environmental Microbiology, 73(19), 6270-6276.
Hongoh, Y., Sato, T., Noda, S., Ui, S., Kudo, T., & Ohkuma, M. (2007b). Candidatus Symbiothrix dinenymphae: bristle-like Bacteroidales ectosymbionts of termite gut protists. Environmental Microbiology, 9(10), 2631-2635.
Huang, X.-F., Bakker, M., Judd, T., Reardon, K., & Vivanco, J. (2013). Variations in diversity and richness of gut bacterial communities of termites (Reticulitermes flavipes) fed with grassy and woody plant substrates. Microbial Ecology, 65(3), 531-536.
Inoue, J.-i., Oshima, K., Suda, W., Sakamoto, M., Iino, T., Noda, S. et al. (2015). Distribution and evolution of nitrogen fixation genes in the phylum Bacteroidetes. Microbes and environments, 30(1), 44-50.
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S. et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics, 28(12), 1647-1649.
Köhler, T., Stingl, U., Meuser, K., & Brune, A. (2008). Novel lineages of Planctomycetes densely colonize the alkaline gut of soil-feeding termites (Cubitermes spp.). Environmental Microbiology, 10(5), 1260-1270.
Köhler, T., Dietrich, C., Scheffrahn, R. H., & Brune, A. (2012). High-resolution analysis of gut environment and bacterial microbiota reveals functional compartmentation of the gut in wood-feeding higher termites (Nasutitermes spp.). Applied & Environmental Microbiology, 78(13), 4691-4701.
Kuhnigk, T., Branke, J., Krekeler, D., Cypionka, H., & König, H. (1996). A feasible role of sulfate-reducing bacteria in the termite gut. Systematic and Applied Microbiology, 19(2), 139-149.
Lambert, C., & Power, V. (1999). Termite harvesting procedures. Perth Zoo’s Numbat Breeding Progamme.
Li, H., Dietrich, C., Zhu, N., Mikaelyan, A., Ma, B., Pi, R. et al. (2016). Age polyethism drives community structure of the bacterial gut microbiota in the fungus-cultivating termite Odontotermes formosanus. Environmental Microbiology, 18(5), 1440-1451.
Lilburn, T. G. (2001). Nitrogen fixation by symbiotic and free-living Spirochetes. Science, 292(5526), 2495-2498.
Mikaelyan, A., Köhler, T., Lampert, N., Rohland, J., Boga, H., Meuser, K. et al. (2015). Classifying the bacterial gut microbiota of termites and cockroaches: A curated phylogenetic reference database (DictDb). Systematic and Applied Microbiology, 38(7), 472-482.
Mikaelyan, A., Meuser, K., & Brune, A. (2017). Microenvironmental heterogeneity of gut compartments drives bacterial community structure in wood-and humus-feeding higher termites. FEMS Microbiology Ecology, 93(1), fiw210.
Mikaelyan, A., Strassert, J. F. H., Tokuda, G., & Brune, A. (2014). The fibre‐associated cellulolytic bacterial community in the hindgut of wood‐feeding higher termites (Nasutitermes spp.). Environmental Microbiology, 16(9), 2711-2722.

114
Mikaelyan, A., Thompson, C. L., Hofer, M. J., & Brune, A. (2016). Deterministic assembly of complex bacterial communities in guts of germ-free cockroaches. Applied and Environmental Microbiology, 82(4), 1256-1263.
National Center for Biotechnology Information. Basic Local Alignment Search Tool. 2017, from https://blast.ncbi.nlm.nih.gov/Blast.cgi.
Nevill, P. G., Zhong, X., Tonti-Filippini, J., Byrne, M., Thiele, K., Leeuwen, S. V. et al. (2017). Plastids of the Pilbara. https://s3-ap-southeast-2.amazonaws.com/pilbara-cpt-website/index.html.
Ohkuma, M., & Brune, A. (2011). Diversity, structure, and evolution of the termite gut microbial community. In D. E. Bignell, Y. Roisin, & N. Lo (Eds.), Biology of Termites: a Modern Synthesis (pp. 413-438). Netherlands: Springer.
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D. et al. (2018). vegan: community ecology package. R package version 2.5-2. https://CRAN.R-project.org/package=vegan.
O’Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., Haddad, D., McVeigh, R. et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res, 44(D1), D733-D745.
Orr, R. J. S., Zhao, S., Klaveness, D., Yabuki, A., Ikeda, K., Watanabe, M. M. et al. (2018). Enigmatic Diphyllatea eukaryotes: culturing and targeted PacBio RS amplicon sequencing reveals a higher order taxonomic diversity and global distribution. BMC Evolutionary Biology, 18(1), 115.
Otani, S., Mikaelyan, A., Nobre, T., Hansen, L. H., Kone, N. A., Sorensen, S. J., Aanen, D.K., Boomsma, J.J., Brune, A. Poulsen, M (2014). Identifying the core microbial community in the gut of fungus-growing termites. Molecular Ecology.
Paul, K., Nonoh, J. O., Mikulski, L., & Brune, A. (2012). “Methanoplasmatales,” Thermoplasmatales-related archaea in termite guts and other environments, are the seventh order of methanogens. Applied and Environmental Microbiology, 78(23), 8245-8253.
Pawlowski, J., Audic, S., Adl, S., Bass, D., Belbahri, L., Berney, C. et al. (2012). CBOL protist working group: barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms. PLoS. Biol., 10(11).
Perry, D. H., Watson, J. A. L., Buun, S. E., & Black, R. (1985). Guide to the termites (Isoptera) from the extreme south-west of western Australia. Journal of the Royal Society of Western Australia, 67(part 2), 66-78.
R Core Team (2018). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
Rawls, J. F., Mahowald, M. A., Ley, R. E., & Gordon, J. I. (2006). Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell, 127(2), 423-433.
Reid, N. M., Addison, S. L., West, M. A., & Lloyd-Jones, G. (2014). The bacterial microbiota of Stolotermes ruficeps (Stolotermitidae), a phylogenetically basal termite endemic to New Zealand. FEMS Microbiology Ecology, 90(3), 678-688.
Rosengaus, R. B., Zecher, C. N., Schultheis, K. F., Brucker, R. M., & Bordenstein, S. R. (2011). Disruption of the termite gut microbiota and its prolonged consequences for fitness. Applied and Environmental Microbiology, 77(13), 4303-4312.
Seedorf, H., Griffin, N. W., Ridaura, V. K., Reyes, A., Cheng, J., Rey, F. E. et al. (2014). Bacteria from diverse habitats colonize and compete in the mouse gut. Cell, 159(2), 253-266.
Shade, A., & Handelsman, J. (2012). Beyond the Venn diagram: the hunt for a core microbiome: The hunt for a core microbiome. Environmental Microbiology,

115
14(1), 4-12. Shimada, K., Lo, N., Kitade, O., Wakui, A., & Maekawa, K. (2013). Cellulolytic protist
numbers rise and fall dramatically in termite queens and kings during colony foundation. Eukaryotic Cell, 12(4), 545-550.
Su, L., Yang, L., Huang, S., Su, X., Li, Y., Wang, F. et al. (2016). Comparative gut microbiomes of four species representing the higher and the lower termites. Journal of Insect Science, 16(1).
Szokoli, F., Castelli, M., Sabaneyeva, E., Schrallhammer, M., Krenek, S., Doak, T. G. et al. (2016). Disentangling the taxonomy of Rickettsiales and description of two novel symbionts (”Candidatus Bealeia paramacronuclearis” and “Candidatus Fokinia cryptica”) sharing the cytoplasm of the ciliate protist Paramecium biaurelia. Applied and Environmental Microbiology, 82(24), 7236-7247.
Taberlet, P., Coissac, E., Pompanon, F., Gielly, L., Miquel, C., Valentini, A. et al. (2007). Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Research, 35(3), e14.
Thompson, C. L., Vier, R., Mikaelyan, A., Wienemann, T., & Brune, A. (2012). ‘Candidatus Arthromitus’ revised: segmented filamentous bacteria in arthropod guts are members of Lachnospiraceae: Arthromitus filaments in arthropod guts. Environmental Microbiology, 14(6), 1454-1465.
Wang, Y., Su, L., Huang, S., Bo, C., Yang, S., Li, Y. et al. (2016). Diversity and resilience of the wood-feeding higher termite Mironasutitermes shangchengensis gut microbiota in response to temporal and diet variations. Ecology and Evolution, 6(22), 8235-8242.
Watson, J. A. L., & Abbey, . H. M. (1993). Atlas of Australian Termites. Ziemer, C. J. (2014). Newly cultured bacteria with broad diversity isolated from eight-
week continuous culture enrichments of cow feces on complex polysaccharides. Applied and Environmental Microbiology, 80(2), 574-585.

116 Ta
ble
S1: F
unct
iona
l gro
ups
pred
icte
d fr
om th
e lit
erat
ure
(with
ref
eren
ces)
for c
ore
taxa
iden
tifie
d in
the
guts
of T
. wes
tral
iens
is a
nd
A. o
beun
tis b
ased
on
taxo
nom
y as
sign
men
ts m
ade
usin
g th
e rd
p cl
assi
fier i
n Q
IIM
E an
d th
e D
ictD
b v3
refe
renc
e da
taba
se
Phy
lum
C
lass
O
rder
Fa
mil
y G
enus
Fu
ncti
on
Sour
ce
Unk
now
n
U
nkno
wn
Aci
doba
cter
ia
Aci
doba
cter
ia
Aci
doba
cter
iale
s A
cido
bact
eria
ceae
U
ncul
ture
d_23
C
ellu
lose
de
grad
atio
n C
ampb
ell
(201
4)
Aci
doba
cter
ia
Hol
opha
gae
Hol
opha
gale
s H
olop
haga
ceae
U
ncul
ture
d_3
Aro
mat
ic
com
poun
d de
grad
atio
n C
ampb
ell
(201
4)
Act
inob
acte
ria
Act
inob
acte
ria
U
nkno
wn
Act
inob
acte
ria
Act
inob
acte
ria
Cor
ioba
cter
iale
s C
orio
bact
eria
ceae
Car
bohy
drat
e fe
rmen
tatio
n G
upta
et a
l. (2
013)
Act
inob
acte
ria
Act
inob
acte
ria
Fran
kial
es
Aci
doth
erm
acea
e A
cido
ther
mus
C
ellu
lose
de
grad
atio
n N
orm
and
et a
l. (2
015)
Act
inob
acte
ria
Act
inob
acte
ria
Mic
roco
ccal
es_1
Sa
ngui
bact
erac
eae
Sang
uiba
cter
C
arbo
hydr
ate
ferm
enta
tion
Schu
man
n &
St
acke
bran
dt
(201
4)
Act
inob
acte
ria
Act
inob
acte
ria
Mic
roco
ccal
es_2
Pr
omic
rom
onos
pora
ceae
X
ylan
imon
as
Cel
lulo
se
degr
adat
ion;
H
emic
ellu
lose
de
grad
atio
n Ri
vas
et a
l. (2
003)
Ba
cter
oide
tes
Unk
now
n
Bact
eroi
dete
s Ba
cter
oidi
a Ba
cter
oida
les
Unk
now
n
Bact
eroi
dete
s Ba
cter
oidi
a Ba
cter
oida
les
Mar
inila
biac
eae
Alk
alifl
exus
C
arbo
hydr
ate
The
Edito
rial
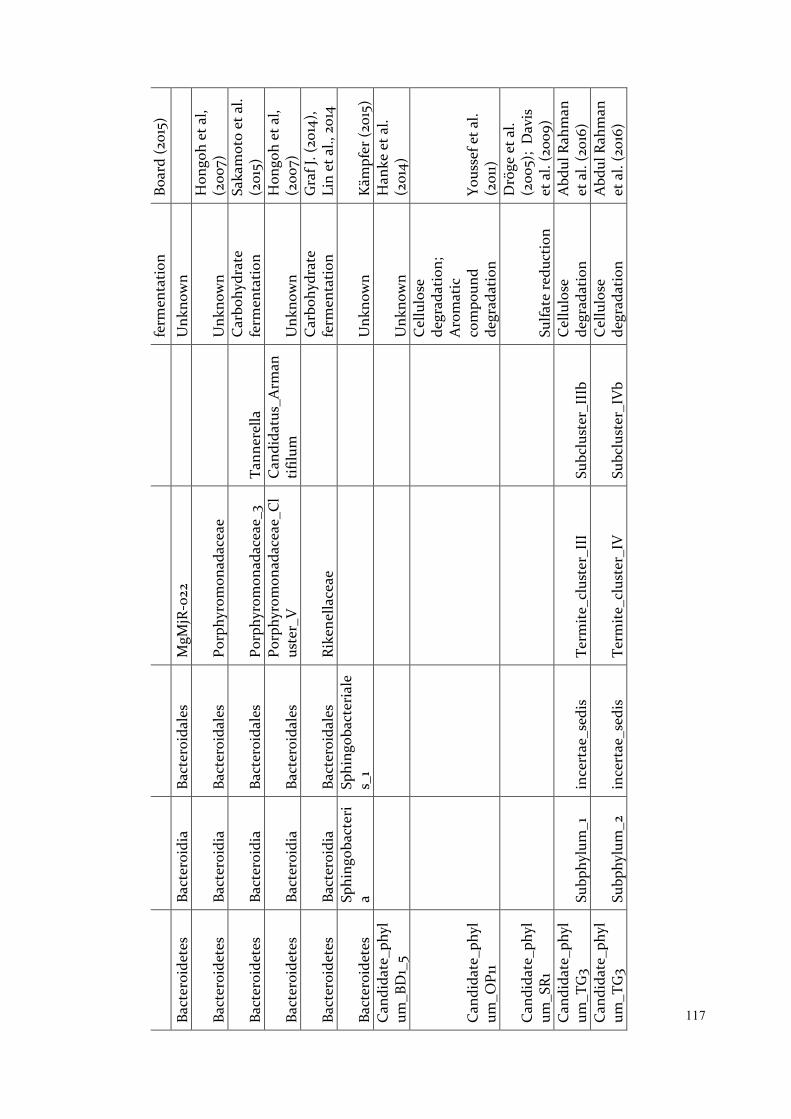
117
ferm
enta
tion
Boar
d (2
015)
Ba
cter
oide
tes
Bact
eroi
dia
Bact
eroi
dale
s M
gMjR
-022
Unk
now
n
Bact
eroi
dete
s Ba
cter
oidi
a Ba
cter
oida
les
Porp
hyro
mon
adac
eae
U
nkno
wn
Hon
goh
et a
l, (2
007)
Bact
eroi
dete
s Ba
cter
oidi
a Ba
cter
oida
les
Porp
hyro
mon
adac
eae_
3 Ta
nner
ella
C
arbo
hydr
ate
ferm
enta
tion
Saka
mot
o et
al.
(201
5)
Bact
eroi
dete
s Ba
cter
oidi
a Ba
cter
oida
les
Porp
hyro
mon
adac
eae_
Cl
uste
r_V
C
andi
datu
s_A
rman
tifilu
m
Unk
now
n H
ongo
h et
al,
(200
7)
Bact
eroi
dete
s Ba
cter
oidi
a Ba
cter
oida
les
Rike
nella
ceae
Car
bohy
drat
e fe
rmen
tatio
n G
raf J
. (20
14),
Lin
et a
l., 2
014
Bact
eroi
dete
s Sp
hing
obac
teri
a Sp
hing
obac
teri
ale
s_1
Unk
now
n K
ämpf
er (2
015)
C
andi
date
_phy
lum
_BD
1_5
Unk
now
n H
anke
et a
l. (2
014)
Can
dida
te_p
hyl
um_O
P11
Cel
lulo
se
degr
adat
ion;
A
rom
atic
co
mpo
und
degr
adat
ion
Yous
sef e
t al.
(201
1)
Can
dida
te_p
hyl
um_S
R1
Sulfa
te re
duct
ion
Drö
ge e
t al.
(200
5);
Dav
is
et a
l. (2
009)
C
andi
date
_phy
lum
_TG
3 Su
bphy
lum
_1
ince
rtae
_sed
is
Term
ite_c
lust
er_I
II
Subc
lust
er_I
IIb
Cel
lulo
se
degr
adat
ion
Abd
ul R
ahm
an
et a
l. (2
016)
C
andi
date
_phy
lum
_TG
3 Su
bphy
lum
_2
ince
rtae
_sed
is
Term
ite_c
lust
er_I
V
Subc
lust
er_I
Vb
Cel
lulo
se
degr
adat
ion
Abd
ul R
ahm
an
et a
l. (2
016)

118 C
andi
date
_phy
lum
_TM
7
U
nkno
wn
C
hlor
obi
Chl
orob
ia
Chl
orob
iale
s O
PB56
Te
rmite
_clu
ster
U
nkno
wn
C
hlor
obi
Chl
orob
ia
Chl
orob
iale
s SJ
A-2
8
Unk
now
n
Chl
orof
lexi
va
dinB
A26
Unk
now
n G
arri
ty e
t al.
(200
1)
Cya
noba
cter
ia
Cya
noba
cter
ia
Cya
noba
cter
iale
s C
0d-2
Nitr
ogen
fixa
tion
Flor
es e
t al.
(201
5)
Cya
noba
cter
ia
Cya
noba
cter
ia
Cya
noba
cter
iale
s M
L635
J-21
C
lust
er_T
G2
Nitr
ogen
fixa
tion
Flor
es e
t al.
(201
5)
Def
erri
bact
eres
D
efer
riba
cter
es
Def
erri
bact
eral
es
Def
erri
bact
erac
eae
Muc
ispi
rillu
m
Unk
now
n
Elus
imic
robi
a El
usim
icro
bia
Elus
imic
robi
ales
Li
neag
e_II
I
Car
bohy
drat
e fe
rmen
tatio
n;
Nitr
ogen
fixa
tion
Zhen
g &
Bru
ne
(201
5)
Elus
imic
robi
a El
usim
icro
bia
Endo
mic
robi
ales
En
dom
icro
biac
eae
Endo
mic
robi
um
Car
bohy
drat
e fe
rmen
tatio
n;
Nitr
ogen
fixa
tion
Zhen
g &
Bru
ne
(201
5)
Fibr
obac
tere
s Su
bphy
lum
_2
Inse
ct_c
lust
er
Cel
lulo
se
degr
adat
ion
Abd
ul R
ahm
an
et a
l. (2
016)
Fi
rmic
utes
U
nkno
wn
Firm
icut
es
Baci
lli
Lact
obac
illal
es
Car
bohy
drat
e fe
rmen
tatio
n Sa
lvet
ti et
al.
(201
3)
Firm
icut
es
Baci
lli
Lact
obac
illal
es
Stre
ptoc
occa
ceae
Car
bohy
drat
e fe
rmen
tatio
n Lo
ry (2
014)
Firm
icut
es
Baci
lli
Lact
obac
illal
es
Stre
ptoc
occa
ceae
La
ctov
um
Car
bohy
drat
e fe
rmen
tatio
n D
rake
(201
4)
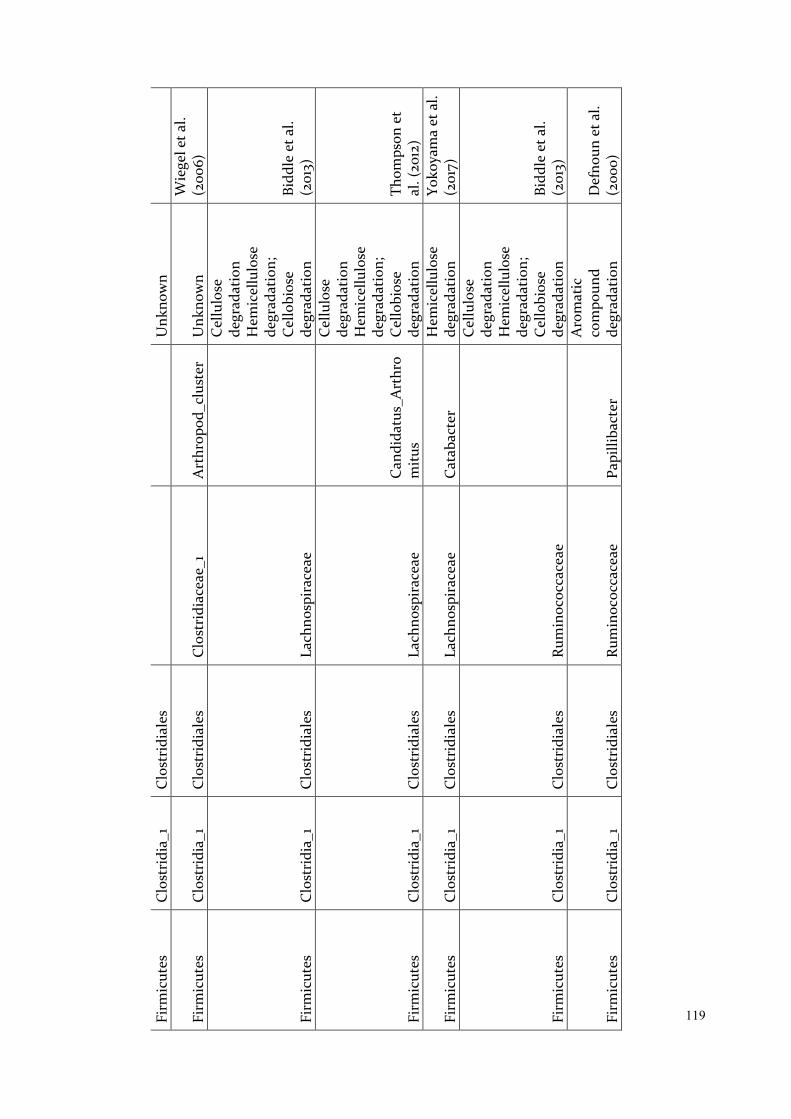
119 Fi
rmic
utes
C
lost
ridi
a_1
Clo
stri
dial
es
Unk
now
n
Firm
icut
es
Clo
stri
dia_
1 C
lost
ridi
ales
C
lost
ridi
acea
e_1
Art
hrop
od_c
lust
er
Unk
now
n W
iege
l et a
l. (2
006)
Firm
icut
es
Clo
stri
dia_
1 C
lost
ridi
ales
La
chno
spir
acea
e
Cel
lulo
se
degr
adat
ion
Hem
icel
lulo
se
degr
adat
ion;
C
ello
bios
e de
grad
atio
n Bi
ddle
et a
l. (2
013)
Firm
icut
es
Clo
stri
dia_
1 C
lost
ridi
ales
La
chno
spir
acea
e C
andi
datu
s_A
rthr
om
itus
Cel
lulo
se
degr
adat
ion
Hem
icel
lulo
se
degr
adat
ion;
C
ello
bios
e de
grad
atio
n Th
omps
on e
t al
. (20
12)
Firm
icut
es
Clo
stri
dia_
1 C
lost
ridi
ales
La
chno
spir
acea
e C
atab
acte
r H
emic
ellu
lose
de
grad
atio
n Yo
koya
ma
et a
l. (2
017)
Firm
icut
es
Clo
stri
dia_
1 C
lost
ridi
ales
Ru
min
ococ
cace
ae
Cel
lulo
se
degr
adat
ion
Hem
icel
lulo
se
degr
adat
ion;
C
ello
bios
e de
grad
atio
n Bi
ddle
et a
l. (2
013)
Firm
icut
es
Clo
stri
dia_
1 C
lost
ridi
ales
Ru
min
ococ
cace
ae
Papi
lliba
cter
Aro
mat
ic
com
poun
d de
grad
atio
n D
efno
un e
t al.
(200
0)
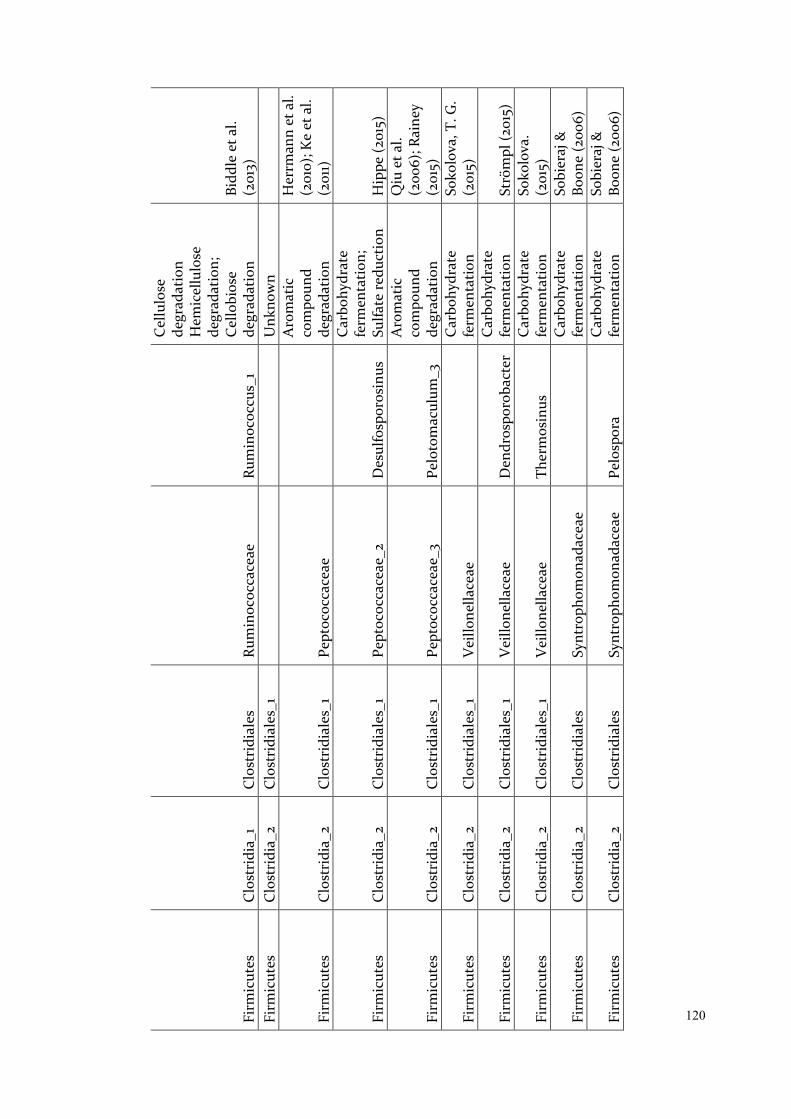
120 Fi
rmic
utes
C
lost
ridi
a_1
Clo
stri
dial
es
Rum
inoc
occa
ceae
Ru
min
ococ
cus_
1
Cel
lulo
se
degr
adat
ion
Hem
icel
lulo
se
degr
adat
ion;
C
ello
bios
e de
grad
atio
n Bi
ddle
et a
l. (2
013)
Fi
rmic
utes
C
lost
ridi
a_2
Clo
stri
dial
es_1
U
nkno
wn
Firm
icut
es
Clo
stri
dia_
2 C
lost
ridi
ales
_1
Pept
ococ
cace
ae
Aro
mat
ic
com
poun
d de
grad
atio
n
Her
rman
n et
al.
(201
0); K
e et
al.
(201
1)
Firm
icut
es
Clo
stri
dia_
2 C
lost
ridi
ales
_1
Pept
ococ
cace
ae_2
D
esul
fosp
oros
inus
Car
bohy
drat
e fe
rmen
tatio
n;
Sulfa
te re
duct
ion
Hip
pe (2
015)
Firm
icut
es
Clo
stri
dia_
2 C
lost
ridi
ales
_1
Pept
ococ
cace
ae_3
Pe
loto
mac
ulum
_3
Aro
mat
ic
com
poun
d de
grad
atio
n
Qiu
et a
l. (2
006)
; Rai
ney
(201
5)
Firm
icut
es
Clo
stri
dia_
2 C
lost
ridi
ales
_1
Vei
llone
llace
ae
C
arbo
hydr
ate
ferm
enta
tion
Soko
lova
, T. G
. (2
015)
Firm
icut
es
Clo
stri
dia_
2 C
lost
ridi
ales
_1
Vei
llone
llace
ae
Den
dros
poro
bact
er
Car
bohy
drat
e fe
rmen
tatio
n St
röm
pl (2
015)
Firm
icut
es
Clo
stri
dia_
2 C
lost
ridi
ales
_1
Vei
llone
llace
ae
Ther
mos
inus
C
arbo
hydr
ate
ferm
enta
tion
Soko
lova
. (2
015)
Firm
icut
es
Clo
stri
dia_
2 C
lost
ridi
ales
Sy
ntro
phom
onad
acea
e
Car
bohy
drat
e fe
rmen
tatio
n So
bier
aj &
Bo
one
(200
6)
Firm
icut
es
Clo
stri
dia_
2 C
lost
ridi
ales
Sy
ntro
phom
onad
acea
e Pe
losp
ora
Car
bohy
drat
e fe
rmen
tatio
n So
bier
aj &
Bo
one
(200
6)
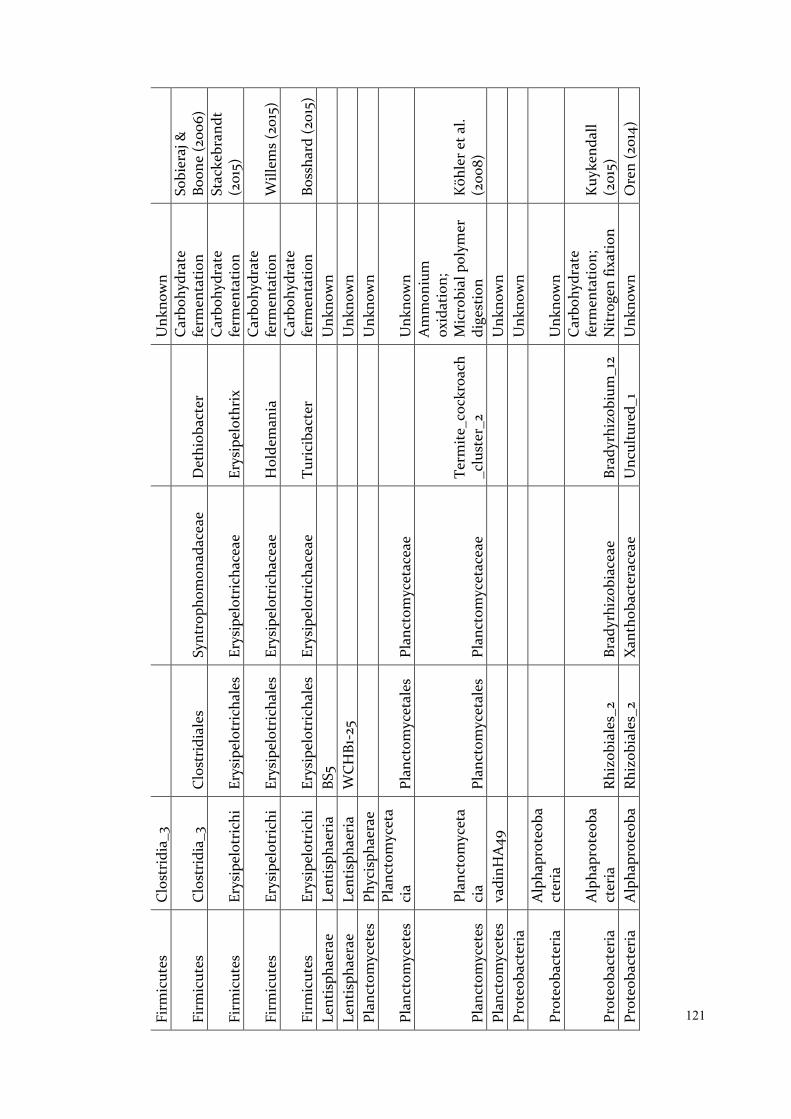
121 Fi
rmic
utes
C
lost
ridi
a_3
U
nkno
wn
Firm
icut
es
Clo
stri
dia_
3 C
lost
ridi
ales
Sy
ntro
phom
onad
acea
e D
ethi
obac
ter
Car
bohy
drat
e fe
rmen
tatio
n So
bier
aj &
Bo
one
(200
6)
Firm
icut
es
Erys
ipel
otri
chi
Erys
ipel
otri
chal
es
Erys
ipel
otri
chac
eae
Erys
ipel
othr
ix
Car
bohy
drat
e fe
rmen
tatio
n St
acke
bran
dt
(201
5)
Firm
icut
es
Erys
ipel
otri
chi
Erys
ipel
otri
chal
es
Erys
ipel
otri
chac
eae
Hol
dem
ania
C
arbo
hydr
ate
ferm
enta
tion
Will
ems
(201
5)
Firm
icut
es
Erys
ipel
otri
chi
Erys
ipel
otri
chal
es
Erys
ipel
otri
chac
eae
Turi
ciba
cter
C
arbo
hydr
ate
ferm
enta
tion
Boss
hard
(201
5)
Lent
isph
aera
e Le
ntis
phae
ria
BS5
Unk
now
n
Lent
isph
aera
e Le
ntis
phae
ria
WC
HB1
-25
Unk
now
n
Plan
ctom
ycet
es
Phyc
isph
aera
e
Unk
now
n
Plan
ctom
ycet
es
Plan
ctom
ycet
aci
a Pl
anct
omyc
etal
es
Plan
ctom
ycet
acea
e
Unk
now
n
Plan
ctom
ycet
es
Plan
ctom
ycet
aci
a Pl
anct
omyc
etal
es
Plan
ctom
ycet
acea
e Te
rmite
_coc
kroa
ch_c
lust
er_2
Am
mon
ium
ox
idat
ion;
M
icro
bial
pol
ymer
di
gest
ion
Köh
ler e
t al.
(200
8)
Plan
ctom
ycet
es
vadi
nHA
49
U
nkno
wn
Pr
oteo
bact
eria
U
nkno
wn
Prot
eoba
cter
ia
Alp
hapr
oteo
bact
eria
Unk
now
n
Prot
eoba
cter
ia
Alp
hapr
oteo
bact
eria
Rh
izob
iale
s_2
Brad
yrhi
zobi
acea
e Br
adyr
hizo
bium
_12
Car
bohy
drat
e fe
rmen
tatio
n;
Nitr
ogen
fixa
tion
Kuy
kend
all
(201
5)
Prot
eoba
cter
ia
Alp
hapr
oteo
baRh
izob
iale
s_2
Xan
thob
acte
race
ae
Unc
ultu
red_
1 U
nkno
wn
Ore
n (2
014)

122
cter
ia
Prot
eoba
cter
ia
Alp
hapr
oteo
bact
eria
Rh
odos
piri
llale
s_1
Rhod
ospi
rilla
ceae
U
ncul
ture
d_2
Unk
now
n
Prot
eoba
cter
ia
Alp
hapr
oteo
bact
eria
Rh
odos
piri
llale
s_2
DA
111
U
nkno
wn
Prot
eoba
cter
ia
Alp
hapr
oteo
bact
eria
Ri
cket
tsia
les
Can
dida
tus_
Cap
tivus
C
andi
datu
s_C
aptiv
us
Prot
ist
endo
sym
bion
t Sz
okol
i et a
l (2
016)
Prot
eoba
cter
ia
Alp
hapr
oteo
bact
eria
Ri
cket
tsia
les
Can
dida
tus_
Hep
atin
cola
C
andi
datu
s_H
epat
inc
ola
Prot
ist
endo
sym
bion
t Sz
okol
i et a
l (2
016)
Prot
eoba
cter
ia
Beta
prot
eoba
cter
ia
Burk
hold
eria
les
Unk
now
n
Prot
eoba
cter
ia
Beta
prot
eoba
cter
ia
Burk
hold
eria
les
Oxa
loba
cter
acea
e
Unk
now
n Ba
ldan
i et a
l. (2
014)
Prot
eoba
cter
ia
Beta
prot
eoba
cter
ia
Rhod
ocyc
lale
s Rh
odoc
ycla
ceae
Unk
now
n O
ren
(201
4b)
Prot
eoba
cter
ia
Beta
prot
eoba
cter
ia
SC-I
-84
Inse
ct_c
lust
er_I
I
Unk
now
n
Prot
eoba
cter
ia
Del
tapr
oteo
bac
teri
a Bd
ello
vibr
iona
les
Bact
erio
vora
ceae
Unk
now
n
Prot
eoba
cter
ia
Del
tapr
oteo
bac
teri
a D
esul
farc
ulal
es
Des
ulfa
rcul
acea
e D
esul
farc
ulus
Su
lfate
redu
ctio
n Su
n et
al.
(201
0)
Prot
eoba
cter
ia
Del
tapr
oteo
bac
teri
a D
esul
foba
cter
ales
D
esul
fobu
lbac
eae
Des
ulfo
bulb
us
Sulfa
te re
duct
ion
Kue
ver e
t al.
(201
5)
Prot
eoba
cter
ia
Del
tapr
oteo
bac
teri
a D
esul
fovi
brio
nale
s D
esul
fovi
brio
nace
ae
Su
lfate
redu
ctio
n Su
n et
al.
(201
0)
Prot
eoba
cter
ia
Del
tapr
oteo
bac
Des
ulfo
vibr
iona
leD
esul
fovi
brio
nace
ae
Des
ulfo
vibr
io_5
Su
lfate
redu
ctio
n K
ueve
r et a
l.
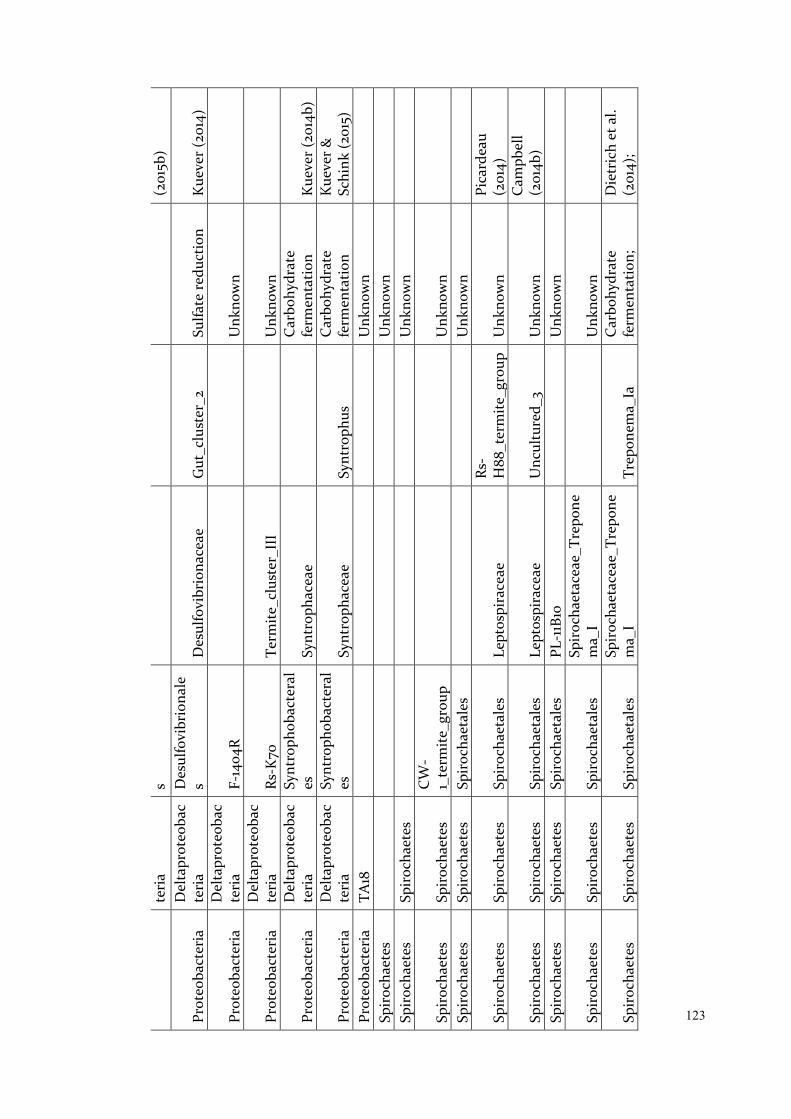
123
teri
a s
(201
5b)
Prot
eoba
cter
ia
Del
tapr
oteo
bac
teri
a D
esul
fovi
brio
nale
s D
esul
fovi
brio
nace
ae
Gut
_clu
ster
_2
Sulfa
te re
duct
ion
Kue
ver (
2014
)
Prot
eoba
cter
ia
Del
tapr
oteo
bac
teri
a F-
1404
R
U
nkno
wn
Prot
eoba
cter
ia
Del
tapr
oteo
bac
teri
a Rs
-K70
Te
rmite
_clu
ster
_III
Unk
now
n
Prot
eoba
cter
ia
Del
tapr
oteo
bac
teri
a Sy
ntro
phob
acte
ral
es
Synt
roph
acea
e
Car
bohy
drat
e fe
rmen
tatio
n K
ueve
r (20
14b)
Prot
eoba
cter
ia
Del
tapr
oteo
bac
teri
a Sy
ntro
phob
acte
ral
es
Synt
roph
acea
e Sy
ntro
phus
C
arbo
hydr
ate
ferm
enta
tion
Kue
ver &
Sc
hink
(201
5)
Prot
eoba
cter
ia
TA18
Unk
now
n
Spir
ocha
etes
U
nkno
wn
Sp
iroc
haet
es
Spir
ocha
etes
Unk
now
n
Spir
ocha
etes
Sp
iroc
haet
es
CW
-1_
term
ite_g
roup
U
nkno
wn
Sp
iroc
haet
es
Spir
ocha
etes
Sp
iroc
haet
ales
U
nkno
wn
Spir
ocha
etes
Sp
iroc
haet
es
Spir
ocha
etal
es
Lept
ospi
race
ae
Rs-
H88
_ter
mite
_gro
up
Unk
now
n Pi
card
eau
(201
4)
Spir
ocha
etes
Sp
iroc
haet
es
Spir
ocha
etal
es
Lept
ospi
race
ae
Unc
ultu
red_
3 U
nkno
wn
Cam
pbel
l (2
014b
) Sp
iroc
haet
es
Spir
ocha
etes
Sp
iroc
haet
ales
PL
-11B
10
U
nkno
wn
Spir
ocha
etes
Sp
iroc
haet
es
Spir
ocha
etal
es
Spir
ocha
etac
eae_
Trep
one
ma_
I
Unk
now
n
Spir
ocha
etes
Sp
iroc
haet
es
Spir
ocha
etal
es
Spir
ocha
etac
eae_
Trep
one
ma_
I Tr
epon
ema_
Ia
Car
bohy
drat
e fe
rmen
tatio
n;
Die
tric
h et
al.
(201
4);
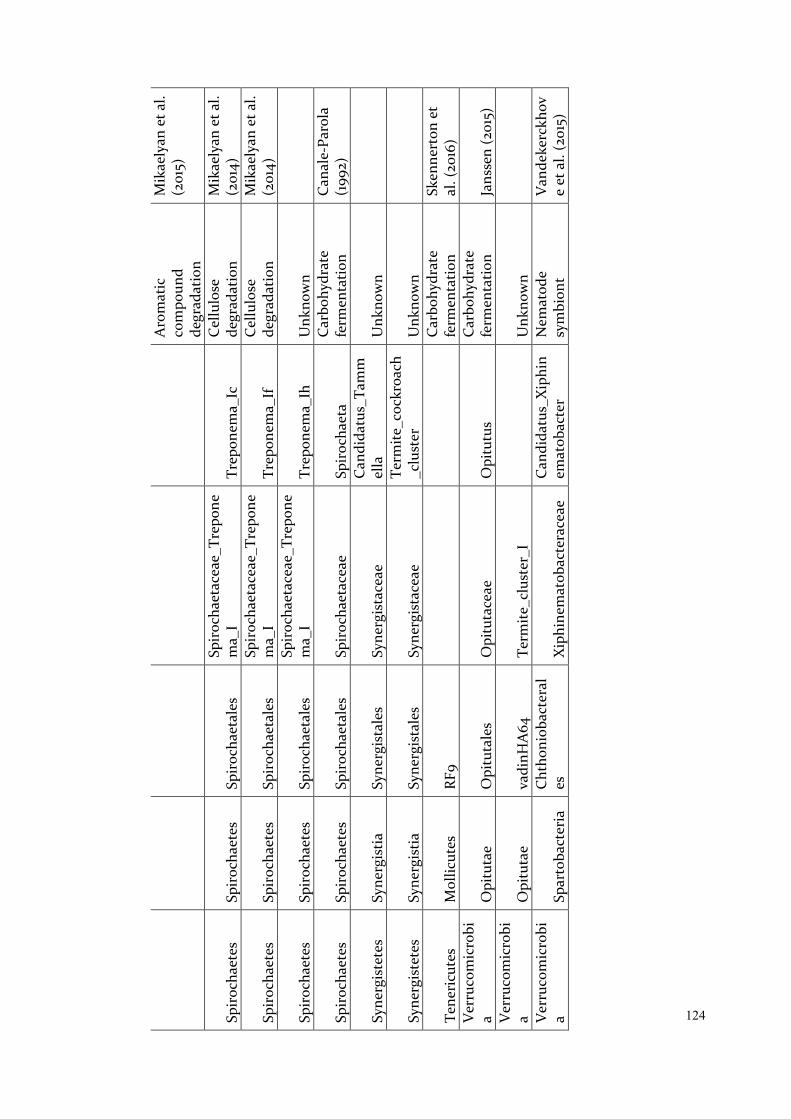
124
Aro
mat
ic
com
poun
d de
grad
atio
n
Mik
aely
an e
t al.
(201
5)
Spir
ocha
etes
Sp
iroc
haet
es
Spir
ocha
etal
es
Spir
ocha
etac
eae_
Trep
one
ma_
I Tr
epon
ema_
Ic
Cel
lulo
se
degr
adat
ion
Mik
aely
an e
t al.
(201
4)
Spir
ocha
etes
Sp
iroc
haet
es
Spir
ocha
etal
es
Spir
ocha
etac
eae_
Trep
one
ma_
I Tr
epon
ema_
If C
ellu
lose
de
grad
atio
n M
ikae
lyan
et a
l. (2
014)
Spir
ocha
etes
Sp
iroc
haet
es
Spir
ocha
etal
es
Spir
ocha
etac
eae_
Trep
one
ma_
I Tr
epon
ema_
Ih
Unk
now
n
Spir
ocha
etes
Sp
iroc
haet
es
Spir
ocha
etal
es
Spir
ocha
etac
eae
Spir
ocha
eta
Car
bohy
drat
e fe
rmen
tatio
n C
anal
e-Pa
rola
(1
992)
Syne
rgis
tete
s Sy
nerg
istia
Sy
nerg
ista
les
Syne
rgis
tace
ae
Can
dida
tus_
Tam
mel
la
Unk
now
n
Syne
rgis
tete
s Sy
nerg
istia
Sy
nerg
ista
les
Syne
rgis
tace
ae
Term
ite_c
ockr
oach
_clu
ster
U
nkno
wn
Tene
ricu
tes
Mol
licut
es
RF9
Car
bohy
drat
e fe
rmen
tatio
n Sk
enne
rton
et
al. (
2016
) V
erru
com
icro
bia
Opi
tuta
e O
pitu
tale
s O
pitu
tace
ae
Opi
tutu
s C
arbo
hydr
ate
ferm
enta
tion
Jans
sen
(201
5)
Ver
ruco
mic
robi
a O
pitu
tae
vadi
nHA
64
Term
ite_c
lust
er_I
Unk
now
n
Ver
ruco
mic
robi
a Sp
arto
bact
eria
C
htho
niob
acte
ral
es
Xip
hine
mat
obac
tera
ceae
C
andi
datu
s_X
iphi
nem
atob
acte
r N
emat
ode
sym
bion
t V
ande
kerc
khov
e et
al.
(201
5)
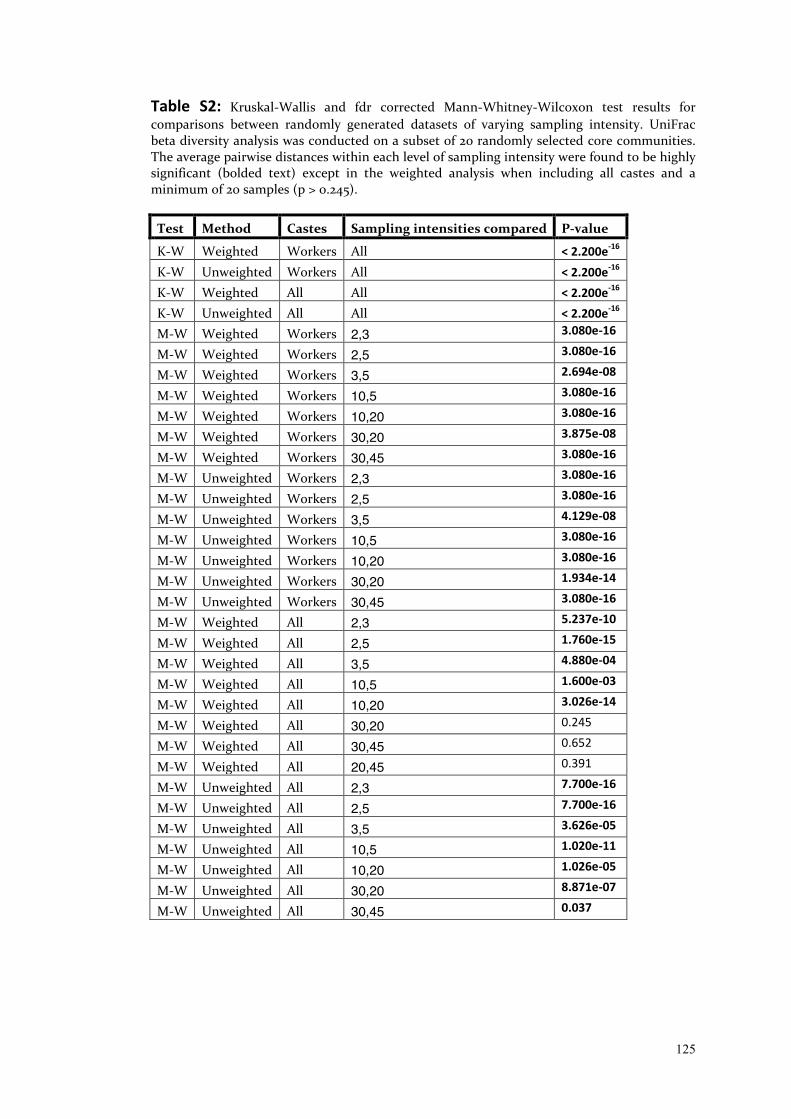
125
Table S2: Kruskal-Wallis and fdr corrected Mann-Whitney-Wilcoxon test results for comparisons between randomly generated datasets of varying sampling intensity. UniFrac beta diversity analysis was conducted on a subset of 20 randomly selected core communities. The average pairwise distances within each level of sampling intensity were found to be highly significant (bolded text) except in the weighted analysis when including all castes and a minimum of 20 samples (p > 0.245).
Test Method Castes Sampling intensities compared P-value
K-W Weighted Workers All < 2.200e-16 K-W Unweighted Workers All < 2.200e-16 K-W Weighted All All < 2.200e-16 K-W Unweighted All All < 2.200e-16 M-W Weighted Workers 2,3 3.080e-16
M-W Weighted Workers 2,5 3.080e-16
M-W Weighted Workers 3,5 2.694e-08
M-W Weighted Workers 10,5 3.080e-16
M-W Weighted Workers 10,20 3.080e-16
M-W Weighted Workers 30,20 3.875e-08
M-W Weighted Workers 30,45 3.080e-16
M-W Unweighted Workers 2,3 3.080e-16
M-W Unweighted Workers 2,5 3.080e-16
M-W Unweighted Workers 3,5 4.129e-08
M-W Unweighted Workers 10,5 3.080e-16
M-W Unweighted Workers 10,20 3.080e-16
M-W Unweighted Workers 30,20 1.934e-14
M-W Unweighted Workers 30,45 3.080e-16
M-W Weighted All 2,3 5.237e-10
M-W Weighted All 2,5 1.760e-15
M-W Weighted All 3,5 4.880e-04
M-W Weighted All 10,5 1.600e-03
M-W Weighted All 10,20 3.026e-14
M-W Weighted All 30,20 0.245
M-W Weighted All 30,45 0.652
M-W Weighted All 20,45 0.391
M-W Unweighted All 2,3 7.700e-16
M-W Unweighted All 2,5 7.700e-16
M-W Unweighted All 3,5 3.626e-05
M-W Unweighted All 10,5 1.020e-11
M-W Unweighted All 10,20 1.026e-05
M-W Unweighted All 30,20 8.871e-07
M-W Unweighted All 30,45 0.037
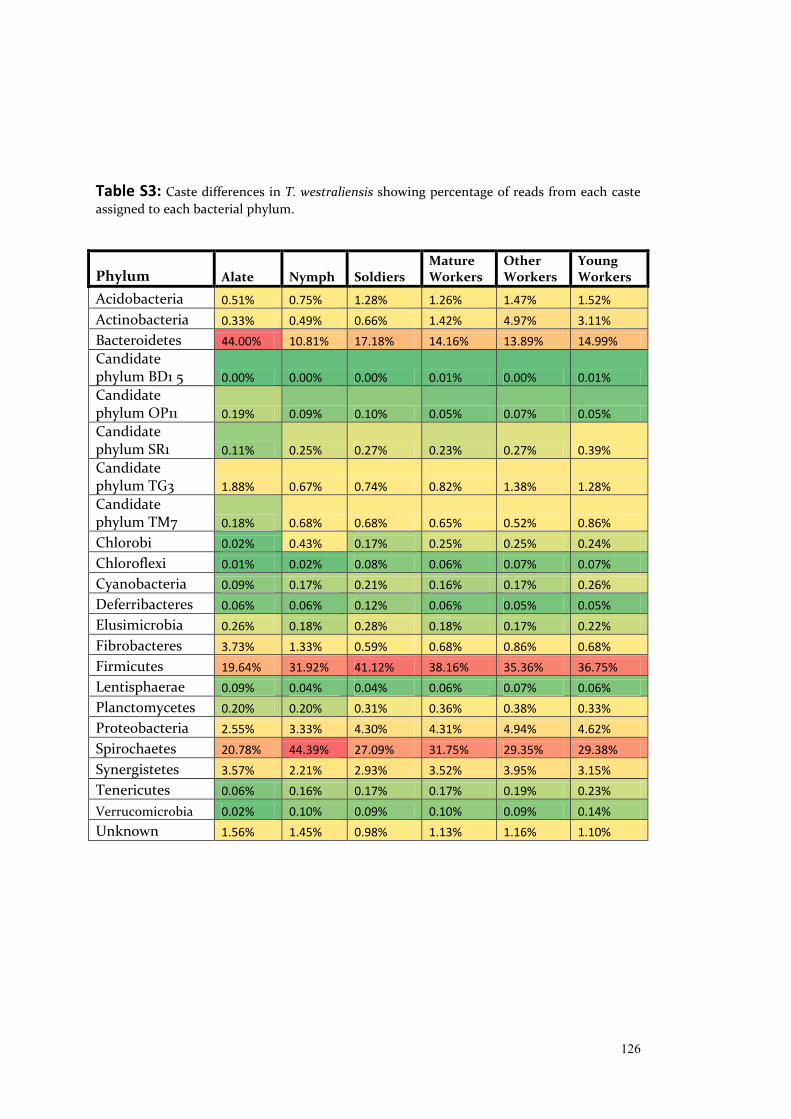
126
Table S3: Caste differences in T. westraliensis showing percentage of reads from each caste assigned to each bacterial phylum.
Phylum Alate Nymph Soldiers Mature Workers
Other Workers
Young Workers
Acidobacteria 0.51% 0.75% 1.28% 1.26% 1.47% 1.52%
Actinobacteria 0.33% 0.49% 0.66% 1.42% 4.97% 3.11%
Bacteroidetes 44.00% 10.81% 17.18% 14.16% 13.89% 14.99%
Candidate phylum BD1 5 0.00% 0.00% 0.00% 0.01% 0.00% 0.01%
Candidate phylum OP11 0.19% 0.09% 0.10% 0.05% 0.07% 0.05%
Candidate phylum SR1 0.11% 0.25% 0.27% 0.23% 0.27% 0.39%
Candidate phylum TG3 1.88% 0.67% 0.74% 0.82% 1.38% 1.28%
Candidate phylum TM7 0.18% 0.68% 0.68% 0.65% 0.52% 0.86%
Chlorobi 0.02% 0.43% 0.17% 0.25% 0.25% 0.24%
Chloroflexi 0.01% 0.02% 0.08% 0.06% 0.07% 0.07%
Cyanobacteria 0.09% 0.17% 0.21% 0.16% 0.17% 0.26%
Deferribacteres 0.06% 0.06% 0.12% 0.06% 0.05% 0.05%
Elusimicrobia 0.26% 0.18% 0.28% 0.18% 0.17% 0.22%
Fibrobacteres 3.73% 1.33% 0.59% 0.68% 0.86% 0.68%
Firmicutes 19.64% 31.92% 41.12% 38.16% 35.36% 36.75%
Lentisphaerae 0.09% 0.04% 0.04% 0.06% 0.07% 0.06%
Planctomycetes 0.20% 0.20% 0.31% 0.36% 0.38% 0.33%
Proteobacteria 2.55% 3.33% 4.30% 4.31% 4.94% 4.62%
Spirochaetes 20.78% 44.39% 27.09% 31.75% 29.35% 29.38%
Synergistetes 3.57% 2.21% 2.93% 3.52% 3.95% 3.15%
Tenericutes 0.06% 0.16% 0.17% 0.17% 0.19% 0.23%
Verrucomicrobia 0.02% 0.10% 0.09% 0.10% 0.09% 0.14%
Unknown 1.56% 1.45% 0.98% 1.13% 1.16% 1.10%
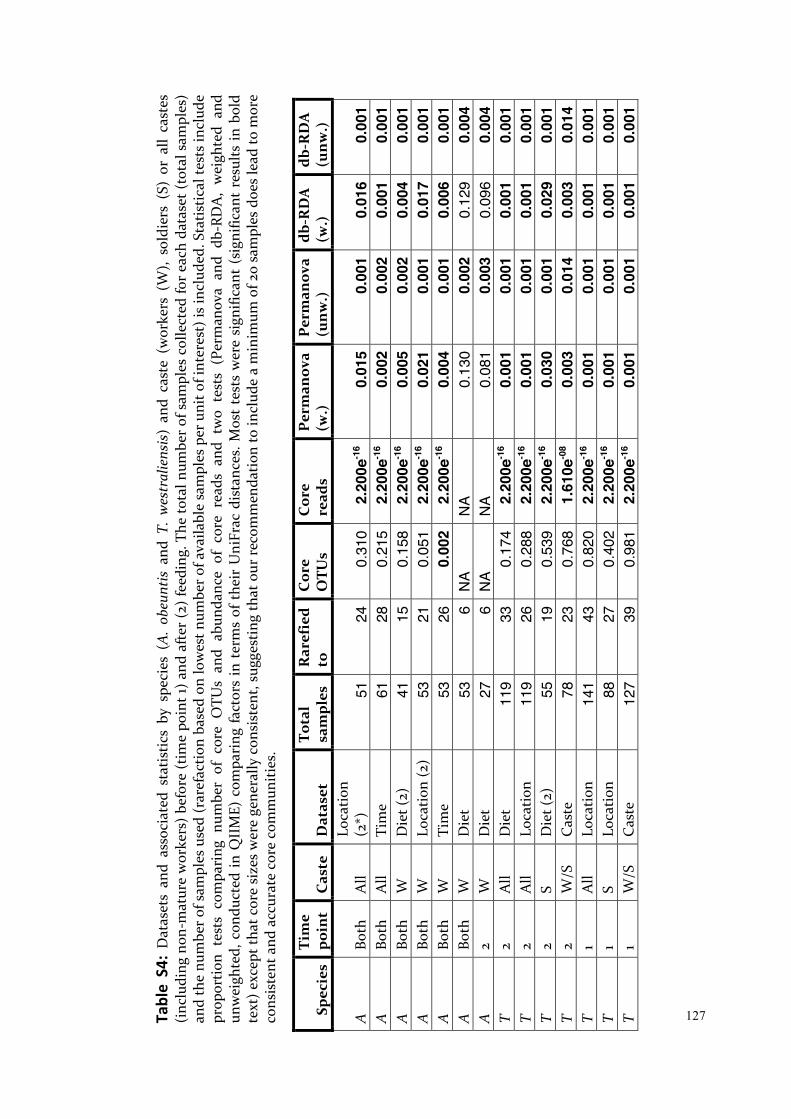
127 Ta
ble
S4:
Dat
aset
s an
d as
soci
ated
sta
tistic
s by
spe
cies
(A
. ob
eunt
is a
nd T
. w
estr
alie
nsis
) an
d ca
ste
(wor
kers
(W
), so
ldie
rs (
S) o
r al
l ca
stes
(i
nclu
ding
non
-mat
ure
wor
kers
) be
fore
(tim
e po
int 1
) an
d af
ter
(2)
feed
ing.
The
tota
l num
ber
of s
ampl
es c
olle
cted
for
each
dat
aset
(to
tal s
ampl
es)
and
the
num
ber o
f sam
ples
use
d (r
aref
actio
n ba
sed
on lo
wes
t num
ber o
f ava
ilabl
e sa
mpl
es p
er u
nit o
f int
eres
t) is
incl
uded
. Sta
tistic
al te
sts
incl
ude
prop
ortio
n te
sts
com
pari
ng n
umbe
r of
cor
e O
TUs
and
abun
danc
e of
cor
e re
ads
and
two
test
s (P
erm
anov
a an
d db
-RD
A,
wei
ghte
d an
d un
wei
ghte
d, c
ondu
cted
in Q
IIM
E) c
ompa
ring
fact
ors
in t
erm
s of
the
ir U
niFr
ac d
ista
nces
. Mos
t te
sts
wer
e si
gnifi
cant
(si
gnifi
cant
res
ults
in b
old
text
) exc
ept t
hat c
ore
size
s w
ere
gene
rally
con
sist
ent,
sugg
estin
g th
at o
ur r
ecom
men
datio
n to
incl
ude
a m
inim
um o
f 20
sam
ples
doe
s le
ad to
mor
e co
nsis
tent
and
acc
urat
e co
re c
omm
uniti
es.
Spec
ies
Tim
e po
int
Cas
te
Dat
aset
To
tal
sam
ples
R
aref
ied
to
Cor
e O
TUs
Cor
e re
ads
Per
man
ova
(w.)
P
erm
anov
a (u
nw.)
db
-RD
A
(w.)
db
-RD
A
(unw
.)
A
Both
A
ll Lo
catio
n (2
*)
51
24
0.31
0 2.
200e
-16
0.01
5 0.
001
0.01
6 0.
001
A
Both
A
ll Ti
me
61
28
0.
215
2.20
0e-1
6 0.
002
0.00
2 0.
001
0.00
1 A
Bo
th
W
Die
t (2)
41
15
0.
158
2.20
0e-1
6 0.
005
0.00
2 0.
004
0.00
1 A
Bo
th
W
Loca
tion
(2)
53
21
0.05
1 2.
200e
-16
0.02
1 0.
001
0.01
7 0.
001
A
Both
W
Ti
me
53
26
0.
002
2.20
0e-1
6 0.
004
0.00
1 0.
006
0.00
1 A
Bo
th
W
Die
t 53
6
NA
NA
0.13
0 0.
002
0.12
9 0.
004
A
2 W
D
iet
27
6 N
A N
A 0.
081
0.00
3 0.
096
0.00
4 T
2 A
ll D
iet
119
33
0.17
4 2.
200e
-16
0.00
1 0.
001
0.00
1 0.
001
T 2
All
Loca
tion
119
26
0.28
8 2.
200e
-16
0.00
1 0.
001
0.00
1 0.
001
T 2
S D
iet (
2)
55
19
0.53
9 2.
200e
-16
0.03
0 0.
001
0.02
9 0.
001
T 2
W/S
C
aste
78
23
0.
768
1.61
0e-0
8 0.
003
0.01
4 0.
003
0.01
4 T
1 A
ll Lo
catio
n 14
1 43
0.
820
2.20
0e-1
6 0.
001
0.00
1 0.
001
0.00
1 T
1 S
Loca
tion
88
27
0.40
2 2.
200e
-16
0.00
1 0.
001
0.00
1 0.
001
T 1
W/S
C
aste
12
7 39
0.
981
2.20
0e-1
6 0.
001
0.00
1 0.
001
0.00
1

128 T
Both
A
ll D
iet
260
33
0.14
2 2.
200e
-16
0.00
1 0.
001
0.00
1 0.
001
T Bo
th
All
Loca
tion
260
79
0.33
3 2.
200e
-16
0.00
1 0.
001
0.00
1 0.
001
T Bo
th
All
Tim
e
260
119
0.41
2 2.
200e
-16
0.00
3 0.
001
0.00
2 0.
001
T Bo
th
S D
iet
143
14
0.28
1 2.
200e
-16
0.02
9 0.
001
0.03
1 0.
001
T Bo
th
S Lo
catio
n 14
3 43
0.
799
2.14
0e-1
1 0.
001
0.00
1 0.
001
0.00
1 T
Both
S
Tim
e
143
55
0.93
9 2.
200e
-16
0.24
0 0.
025
0.23
9 0.
032
T Bo
th
W
Loca
tion
62
17
0.46
0 2.
200e
-16
0.00
1 0.
001
0.00
1 0.
001
T Bo
th
W
Tim
e
62
23
0.50
1 2.
200e
-16
0.23
9 0.
019
0.23
4 0.
018
T Bo
th
W
Die
t 62
6
NA
NA
0.17
4 0.
003
0.19
0.
005
T Bo
th
W
Die
t (W
+L)†
62
10
NA
NA
0.32
3 0.
002
0.34
2 0.
002
T 2
W
Die
t 23
6
NA
NA
0.14
0 0.
005
0.12
9 0.
006
T 2
W
Die
t (W
+L)
23
10
NA
NA
0.37
8 0.
003
0.38
0.
006
T Bo
th
W/S
C
aste
20
5 62
1.
000
2.20
0e-1
6 0.
001
0.00
1 0.
001
0.00
1 Bo
th
1 A
ll Sp
ecie
s 17
4 33
3.
040e
-4
2.20
0e-1
6 0.
001
0.00
1 0.
001
0.00
1 Bo
th
1 W
Sp
ecie
s 65
26
0.
624
2.20
0e-1
6 0.
001
0.00
1 0.
001
0.00
1 Bo
th
Both
A
ll Sp
ecie
s 32
1 61
1.
086e
-4
0.62
2 0.
001
0.00
1 0.
001
0.00
1
Both
Bo
th
W
Co-
habi
tati
on
34
16
0.78
6 0.
725
0.76
8 0.
444
NA
NA
Bo
th
Both
W
Sp
ecie
s 11
5 53
0.
005
2.20
0e-1
6 0.
001
0.00
1 0.
001
0.00
1
Both
(T)
Both
W
C
o-ha
bita
tion
17
8
NA
NA
0.12
4 0.
009
0.2
70
0.0
12
Both
(A)
Both
W
C
o-ha
bita
tion
17
8
NA
NA
0.22
5 0.
005
0.2
31
0.0
07
*Num
ber
of g
roup
s in
clud
ed to
max
imis
e nu
mbe
r of
sam
ples
in r
aref
ied
grou
ps
† Whe
at a
nd L
ucer
ne d
iet g
roup
s co
mbi
ned
to m
axim
ise
num
ber
of s
ampl
es in
rar
efie
d gr
oups
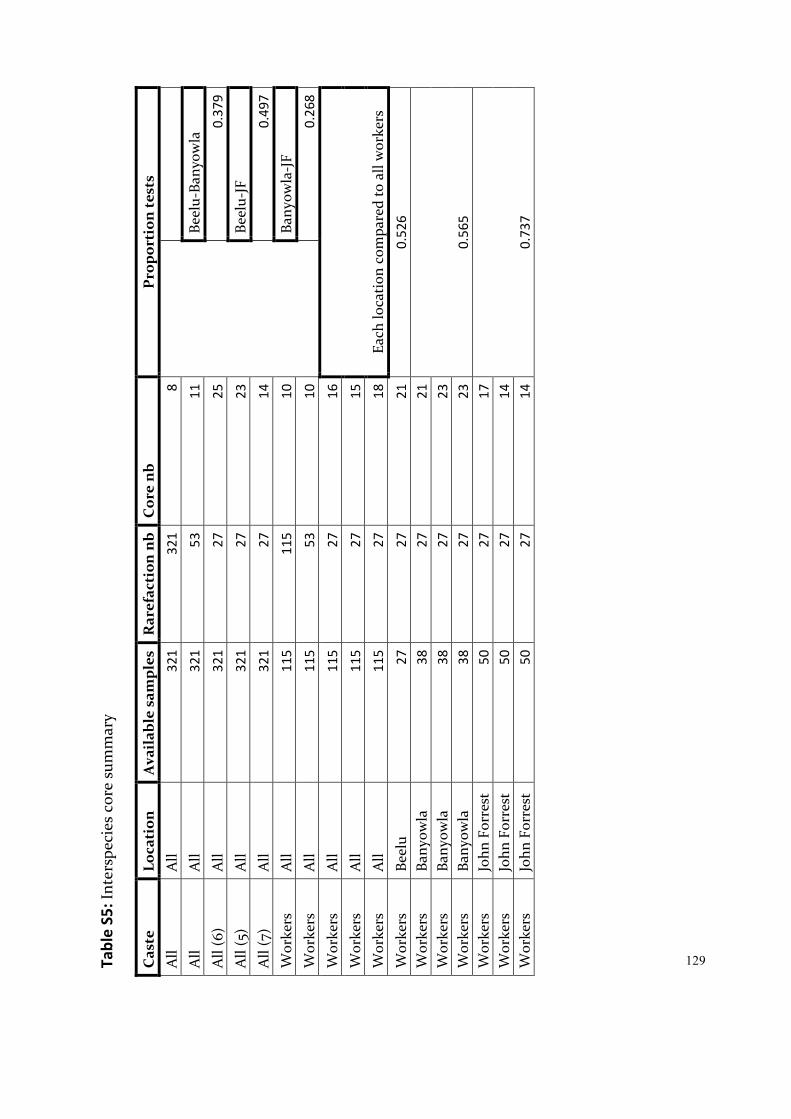
129 Ta
ble
S5: I
nter
spec
ies
core
sum
mar
y C
aste
Lo
cati
on
Ava
ilab
le s
ampl
es
Rar
efac
tion
nb
Cor
e n
b P
ropo
rtio
n te
sts
All
All
321
3
21
8
All
All
321
5
3 1
1
Beel
u-Ba
nyow
la
All
(6)
All
321
2
7 2
5
0.3
79
All
(5)
All
321
2
7 2
3
Beel
u-JF
A
ll (7
) A
ll 3
21
27
14
0
.49
7
Wor
kers
A
ll 1
15
115
1
0
Bany
owla
-JF
Wor
kers
A
ll 1
15
53
10
0
.26
8
Wor
kers
A
ll 1
15
27
16
Each
loca
tion
com
pare
d to
all
wor
kers
W
orke
rs
All
115
2
7 1
5
Wor
kers
A
ll 1
15
27
18
Wor
kers
Be
elu
27
27
21
0.5
26
Wor
kers
Ba
nyow
la
38
27
21
0.5
65
Wor
kers
Ba
nyow
la
38
27
23
Wor
kers
Ba
nyow
la
38
27
23
Wor
kers
Jo
hn F
orre
st
50
27
17
0.7
37
Wor
kers
Jo
hn F
orre
st
50
27
14
Wor
kers
Jo
hn F
orre
st
50
27
14
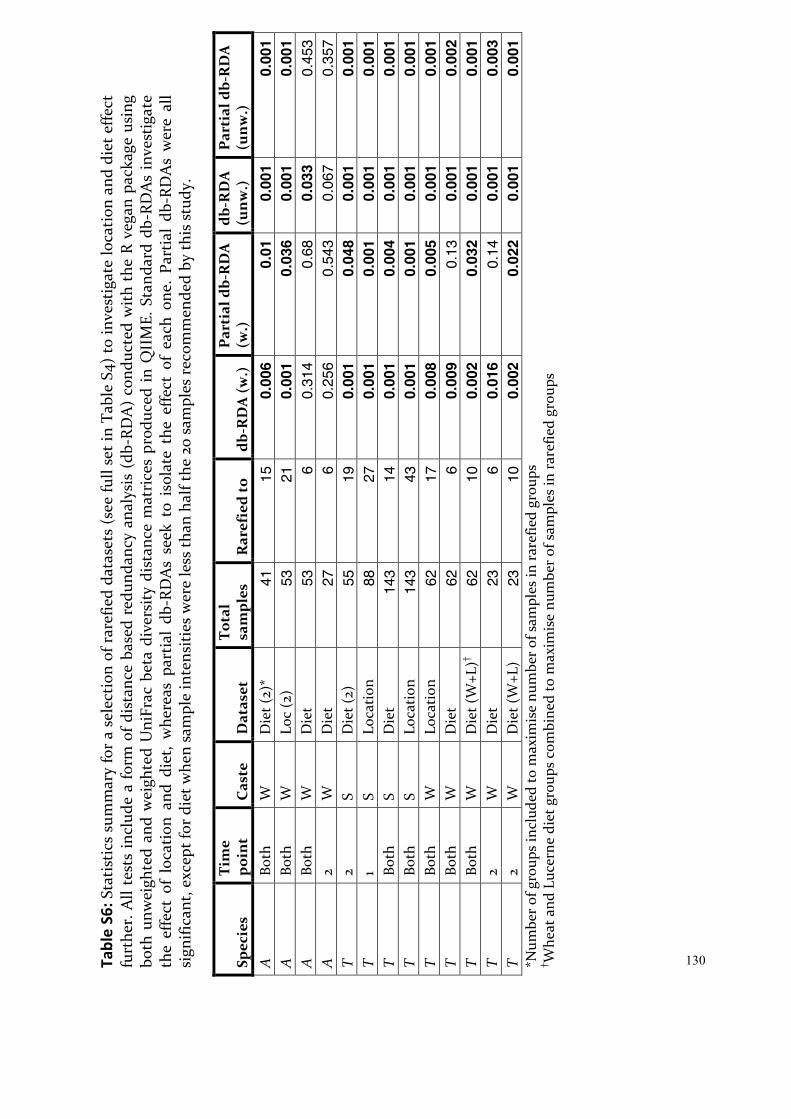
130 Ta
ble
S6: S
tati
stic
s su
mm
ary
for
a se
lect
ion
of r
aref
ied
data
sets
(se
e fu
ll se
t in
Tabl
e S4
) to
inve
stig
ate
loca
tion
and
die
t effe
ct
furt
her.
All
test
s in
clud
e a
form
of d
ista
nce
base
d re
dund
ancy
ana
lysi
s (d
b-RD
A)
cond
ucte
d w
ith t
he R
veg
an p
acka
ge u
sing
bo
th u
nwei
ghte
d an
d w
eigh
ted
Uni
Frac
bet
a di
vers
ity d
ista
nce
mat
rice
s pr
oduc
ed in
QII
ME.
Sta
ndar
d db
-RD
As
inve
stig
ate
the
effe
ct o
f lo
catio
n an
d di
et,
whe
reas
par
tial
db-
RDA
s se
ek t
o is
olat
e th
e ef
fect
of
each
one
. Pa
rtia
l db
-RD
As
wer
e al
l si
gnifi
cant
, exc
ept f
or d
iet w
hen
sam
ple
inte
nsiti
es w
ere
less
than
hal
f the
20
sam
ples
rec
omm
ende
d by
this
stu
dy.
Spec
ies
Tim
e po
int
Cas
te
Dat
aset
To
tal
sam
ples
R
aref
ied
to
db-R
DA
(w.)
P
arti
al d
b-R
DA
(w
.)
db-R
DA
(u
nw.)
P
arti
al d
b-R
DA
(u
nw.)
A
Bo
th
W
Die
t (2)
* 41
15
0.
006
0.01
0.
001
0.00
1 A
Bo
th
W
Loc
(2)
53
21
0.00
1 0.
036
0.00
1 0.
001
A
Both
W
D
iet
53
6 0.
314
0.68
0.
033
0.45
3 A
2
W
Die
t 27
6
0.25
6 0.
543
0.06
7 0.
357
T 2
S D
iet (
2)
55
19
0.00
1 0.
048
0.00
1 0.
001
T 1
S Lo
catio
n 88
27
0.
001
0.00
1 0.
001
0.00
1 T
Both
S
Die
t 14
3 14
0.
001
0.00
4 0.
001
0.00
1 T
Both
S
Loca
tion
143
43
0.00
1 0.
001
0.00
1 0.
001
T Bo
th
W
Loca
tion
62
17
0.00
8 0.
005
0.00
1 0.
001
T Bo
th
W
Die
t 62
6
0.00
9 0.
13
0.00
1 0.
002
T Bo
th
W
Die
t (W
+L)†
62
10
0.00
2 0.
032
0.00
1 0.
001
T 2
W
Die
t 23
6
0.01
6 0.
14
0.00
1 0.
003
T 2
W
Die
t (W
+L)
23
10
0.00
2 0.
022
0.00
1 0.
001
*Num
ber o
f gro
ups
incl
uded
to m
axim
ise
num
ber o
f sam
ples
in ra
refie
d gr
oups
† W
heat
and
Luc
erne
die
t gro
ups
com
bine
d to
max
imis
e nu
mbe
r of s
ampl
es in
rare
fied
grou
ps

131 Ta
ble
S7:
Stat
istic
s su
mm
ary
for
the
rela
tions
hip
betw
een
geog
raph
ic d
ista
nce
betw
een
colo
nies
and
the
bet
a di
vers
ity
dist
ance
bet
wee
n w
orke
r gu
t co
mm
uniti
es c
olle
cted
dur
ing
the
first
tim
e po
int.
Thre
e m
ain
grou
ping
s of
com
pari
sons
wer
e ob
tain
ed, o
ne c
onta
inin
g co
mpa
riso
ns w
ithin
a lo
catio
n, fo
llow
ed b
y co
mpa
riso
ns to
incr
easi
ngly
dis
tant
loca
tion
s. S
igni
fican
t po
siti
ve c
orre
latio
ns w
ere
foun
d ac
ross
all
loca
tions
in
T. w
estr
alie
nsis
onl
y, w
ith t
he s
tron
gest
cor
rela
tion
indi
catin
g th
at
incr
ease
d di
stan
ce b
etw
een
colo
nies
was
pri
mar
ily li
nked
to
incr
ease
d di
ffere
nces
in t
erm
s of
the
bac
teri
al s
peci
es p
rese
nt in
th
e gu
t. H
owev
er w
hen
focu
sing
on
indi
vidu
al g
roup
ings
(co
mpa
riso
ns w
ithin
a l
ocat
ion,
bet
wee
n m
id-r
ange
loc
atio
ns o
r di
stan
t loc
atio
ns) n
o si
gnifi
cant
tren
ds w
ere
foun
d.
Spec
ies
Met
ric
Acr
oss
all l
ocat
ions
W
ithi
n lo
cati
on
Bet
wee
n m
id-r
ange
lo
cati
ons
Bet
wee
n di
stan
t lo
cati
ons
Cor
rela
tio
n
P-v
alue
C
orre
lati
on
P
-va
lue
Cor
rela
tion
P
-val
ue
Cor
rela
tion
P
-val
ue
T U
nwei
ghte
d 0.
564
8.88
3e-
13
-0.2
36
0.14
2 -0
.181
0.
291
-0.2
01
0.12
3 T
Wei
ghte
d 0.
183
0.03
3 -0
.004
0.
979
-0.0
94
0.58
7 -0
.192
0.
143
A
Unw
eigh
ted
0.18
6 0.
103
0.00
2 0.
992
-0.4
91
0.06
3 -0
.193
0.
232
A
Wei
ghte
d -0
.047
0.
684
0.25
4 0.
242
-0.5
49
0.03
4 -0
.271
0.
091

132
References Abdul Rahman, N., Parks, D. H., Vanwonterghem, I., Morrison, M., Tyson, G. W., &
Hugenholtz, P. (2016). A phylogenomic analysis of the bacterial phylum Fibrobacteres. Frontiers in Microbiology, 6, 5.
Baldani J.I., Rouws L., Cruz L.M., Olivares F.L., Schmid M., Hartmann A. (2014) The family Oxalobacteraceae. In: Rosenberg E., DeLong E.F., Lory S., Stackebrandt E., Thompson F. (eds) The Prokaryotes. Springer, Berlin, Heidelberg.
Biddle, A., Stewart, L., Blanchard, J., & Leschine, S. (2013). Untangling the genetic basis of fibrolytic specialization by Lachnospiraceae and Ruminococcaceae in diverse gut communities. Diversity, 5(3), 627-640.
Bosshard, P. P. (2015). Turicibacter. In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Campbell B.J. (2014) The Family Acidobacteriaceae. In: Rosenberg E., DeLong E.F., Lory S., Stackebrandt E., Thompson F. (eds) The Prokaryotes. Springer, Berlin, Heidelberg.
Campbell B.J. (2014b) The Family Acidobacteriaceae. In: Rosenberg E., DeLong E.F., Lory S., Stackebrandt E., Thompson F. (eds) The Prokaryotes. Springer, Berlin, Heidelberg.
Canale-Parola E. (1992) Free-living saccharolytic Spirochetes: The Genus Spirochaeta. In: Balows A., Trüper H.G., Dworkin M., Harder W., Schleifer KH. (eds) The Prokaryotes. Springer, New York, NY.
Davis, J. P., Youssef, N. H., & Elshahed, M. S. (2009). Assessment of the diversity, abundance, and ecological distribution of members of candidate division SR1 reveals a high level of phylogenetic diversity but limited morphotypic diversity. Applied and Environmental Microbiology, 75(12), 4139-4148.
Defnoun, S., Labat, M., Ambrosio, M., Garcia, J.-L., & Patel, B. K. C. (2000). Papillibacter cinnamivorans gen. nov., sp. nov., a cinnamate-transforming bacterium from a shea cake digester. International Journal of Systematic and Evolutionary Microbiology, 50, 1221-1228.
Dietrich, C., Köhler, T., & Brune, A. (2014). The cockroach origin of the termite gut microbiota: patterns in bacterial community structure reflect major evolutionary events. Applied and Environmental Microbiology.
Drake, H. L. (2014). The genus Lactovum. In Lactic acid bacteria (eds W. H. Holzapfel and B. J. Wood).
Dröge, S., Limper, U., Emtiazi, F., Schönig, I., Pavlus, N., Drzyzga, O. et al. (2005). In vitro and in vivo sulfate reduction in the gut contents of the termite Mastotermes darwiniensis and the rose-chafer Pachnoda marginata. The Journal of General and Applied Microbiology, 51(2), 57-64.
Flores, E. , López‐Lozano, A. and Herrero, A. (2015). Nitrogen fixation in the oxygenic phototrophic prokaryotes (Cyanobacteria): the fight against oxygen. In Biological Nitrogen Fixation, F. J. de Bruijn (Ed.).
Garrity G.M., Holt J.G., Castenholz R.W., Pierson B.K., Keppen O.I., Gorlenko V.M. (2001) Phylum BVI. Chloroflexi phy. nov.. In: Boone D.R., Castenholz R.W., Garrity G.M. (eds) Bergey’s manual of systematic bacteriology. Springer, New York, NY.
Graf J. (2014) The Family Rikenellaceae. In: Rosenberg E., DeLong E.F., Lory S., Stackebrandt E., Thompson F. (eds) The Prokaryotes. Springer, Berlin, Heidelberg.
Gupta, R. S., Chen, W. J., Adeolu, M., & Chai, Y. (2013). Molecular signatures for the

133
class Coriobacteriia and its different clades; proposal for division of the class Coriobacteriia into the emended order Coriobacteriales, containing the emended family Coriobacteriaceae and Atopobiaceae fam. nov., and Eggerthellales ord. nov., containing the family Eggerthellaceae fam. nov. International Journal of Systematic and Evolutionary Microbiology, 63(Pt 9), 3379-3397.
Hanke, A., Hamann, E., Sharma, R., Geelhoed, J. S., Hargesheimer, T., Kraft, B. et al. (2014). Recoding of the stop codon UGA to glycine by a BD1-5/SN-2 bacterium and niche partitioning between Alpha- and Gammaproteobacteria in a tidal sediment microbial community naturally selected in a laboratory chemostat. Frontiers in Microbiology, 5, 533.
Herrmann, S., Kleinsteuber, S., Chatzinotas, A., Kuppardt, S., Lueders, T., Richnow, H.-H. et al. (2010). Functional characterization of an anaerobic benzene-degrading enrichment culture by DNA stable isotope probing. Environmental Microbiology, 12(2), 401-411.
Hippe, H. and Stackebrandt, E. (2015). Desulfosporosinus. In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Hongoh, Y., Sato, T., Noda, S., Ui, S., Kudo, T., & Ohkuma, M. (2007b). Candidatus Symbiothrix dinenymphae: bristle-like Bacteroidales ectosymbionts of termite gut protists. Environmental Microbiology, 9(10), 2631-2635.
Janssen, P. H. (2015). Opitutus. In Bergey's Manual of Systematics of Archaea and Bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Kämpfer, P. (2015). Sphingobacteriales ord. nov. In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Ke, J., Singh, D., & Chen, S. (2011). Aromatic compound degradation by the wood-feeding termite Coptotermes formosanus (Shiraki). International Biodeterioration & Biodegradation, 65(6), 744-756.
Köhler, T., Stingl, U., Meuser, K., & Brune, A. (2008). Novel lineages of Planctomycetes densely colonize the alkaline gut of soil-feeding termites (Cubitermes spp.). Environmental Microbiology, 10(5), 1260-1270.
Kuykendall, L. D. (2015). Bradyrhizobium. In Bergey's Manual of Systematics of Archaea and Bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Kuever J. (2014) The Family Desulfovibrionaceae. In: Rosenberg E., DeLong E.F., Lory S., Stackebrandt E., Thompson F. (eds) The Prokaryotes. Springer, Berlin, Heidelberg.
Kuever J. (2014b) The Family Syntrophaceae. In: Rosenberg E., DeLong E.F., Lory S., Stackebrandt E., Thompson F. (eds) The Prokaryotes. Springer, Berlin, Heidelberg.
Kuever, J. , Rainey, F. A. and Widdel, F. (2015). Desulfobulbus. In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh)
Kuever, J. , Rainey, F. A. and Widdel, F. (2015b). Desulfovibrio. In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Kuever, J. and Schink, B. (2015). Syntrophus. In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).

134
Lin, S. Y., Hsu, Y. H., Liu, Y. C., Hung, M. H., Hameed, A., Lai, W. A. et al. (2014). Rhizobium straminoryzae sp. nov., isolated from the surface of rice straw. International Journal of Systematic and Evolutionary Microbiology, 64(Pt 9), 2962-2968.
Lory S. (2014) The Family Streptococcaceae. In: Rosenberg E., DeLong E.F., Lory S., Stackebrandt E., Thompson F. (eds) The Prokaryotes. Springer, Berlin, Heidelberg.
Mikaelyan, A., Köhler, T., Lampert, N., Rohland, J., Boga, H., Meuser, K. et al. (2015). Classifying the bacterial gut microbiota of termites and cockroaches: A curated phylogenetic reference database (DictDb). Systematic and Applied Microbiology, 38(7), 472-482.
Mikaelyan, A., Strassert, J. F. H., Tokuda, G., & Brune, A. (2014). The fibre‐associated cellulolytic bacterial community in the hindgut of wood‐feeding higher termites (Nasutitermes spp.). Environmental Microbiology, 16(9), 2711-2722.
Normand, P. , Berry, A. and Benson, D. R. (2015). Acidothermus. In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Oren A. (2014) The Family Xanthobacteraceae. In: Rosenberg E., DeLong E.F., Lory S., Stackebrandt E., Thompson F. (eds) The Prokaryotes. Springer, Berlin, Heidelberg.
Oren A. (2014b) The Family Rhodocyclaceae. In: Rosenberg E., DeLong E.F., Lory S., Stackebrandt E., Thompson F. (eds) The Prokaryotes. Springer, Berlin, Heidelberg.
Picardeau M. (2014) The Family Leptospiraceae. In: Rosenberg E., DeLong E.F., Lory S., Stackebrandt E., Thompson F. (eds) The Prokaryotes. Springer, Berlin, Heidelberg. Qiu, Y.-L., Sekiguchi, Y., Hanada, S., Imachi, H., Tseng, I.-C., Cheng, S.-S. et al.
(2006). Pelotomaculum terephthalicum sp. nov. and Pelotomaculum isophthalicum sp. nov.: two anaerobic bacteria that degrade phthalate isomers in syntrophic association with hydrogenotrophic methanogens. Archives of Microbiology, 185(3), 172-182.
Rainey, F. A. (2015). Pelotomaculum. In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Rivas, R., Sánchez, M., Trujillo, M. E., Zurdo-Piñeiro, J. L., Mateos, P. F., Martínez-Molina, E. et al. (2003). Xylanimonas cellulosilytica gen. nov., sp. nov., a xylanolytic bacterium isolated from a decayed tree (Ulmus nigra). International Journal of Systematic and Evolutionary Microbiology, 53(Pt 1), 99-103.
Sakamoto, M. , Tanner, A. C. and Benno, Y. (2015). Tannerella. In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Salvetti, E., Fondi, M., Fani, R., Torriani, S., & Felis, G. E. (2013). Evolution of lactic acid bacteria in the order Lactobacillales as depicted by analysis of glycolysis and pentose phosphate pathways. Systematic and Applied Microbiology, 36(5), 291-305.
Schumann P., Stackebrandt E. (2014) The Families Sanguibacteraceae and Rarobacteraceae. In: Rosenberg E., DeLong E.F., Lory S., Stackebrandt E., Thompson F. (eds) The Prokaryotes. Springer, Berlin, Heidelberg.
Skennerton, C. T., Haroon, M. F., Briegel, A., Shi, J., Jensen, G. J., Tyson, G. W. et al. (2016). Phylogenomic analysis of Candidatus ‘Izimaplasma’ species: free-living

135
representatives from a Tenericutes clade found in methane seeps. ISME Journal, 10(11), 2679-2692.
Sobieraj M., Boone D.R. (2006) Syntrophomonadaceae. In: Dworkin M., Falkow S., Rosenberg E., Schleifer KH., Stackebrandt E. (eds) The Prokaryotes. Springer, New York, NY.
Sokolova, T. G. (2015). Thermosinus. In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Stackebrandt, E. (2015). Erysipelothrix. In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Strömpl, C. (2015). Dendrosporobacter. In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Sun, H., Spring, S., Lapidus, A., Davenport, K., Del Rio, T. G., Tice, H. et al. (2010). Complete genome sequence of Desulfarculus baarsii type strain (2st14T). Standards in Genomic Sciences., 3, 276-284.
Szokoli, F., Castelli, M., Sabaneyeva, E., Schrallhammer, M., Krenek, S., Doak, T. G. et al. (2016). Disentangling the taxonomy of Rickettsiales and description of two novel symbionts (”Candidatus Bealeia paramacronuclearis” and “Candidatus Fokinia cryptica”) sharing the cytoplasm of the ciliate protist Paramecium biaurelia. Applied and Environmental Microbiology, 82(24), 7236-7247.
The Editorial Board (2015). Alkaliflexus . In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Thompson, C. L., Vier, R., Mikaelyan, A., Wienemann, T., & Brune, A. (2012). ‘Candidatus Arthromitus’ revised: segmented filamentous bacteria in arthropod guts are members of Lachnospiraceae: Arthromitus filaments in arthropod guts. Environmental Microbiology, 14(6), 1454-1465.
Vandekerckhove, T. T., Navarro, J. B., Coomans, A. and Hedlund, B. P. (2015). “Candidatus Xiphinematobacter”. In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Wiegel, J., Tanner, R., & Rainey, F. A. (2006). An Introduction to the Family Clostridiaceae. The Prokaryotes, 654-678.
Willems, A. (2015). Holdemania. In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Yokoyama, H. , Wagner, I. D. and Wiegel, J. (2017). Caldicoprobacter. In Bergey's manual of systematics of archaea and bacteria (eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, B. Hedlund and S. Dedysh).
Youssef, N. H., Blainey, P. C., Quake, S. R., & Elshahed, M. S. (2011). Partial genome assembly for a candidate division OP11 single cell from an anoxic spring (Zodletone Spring, Oklahoma). Applied and Environmental Microbiology, 77(21), 7804-7814.
Zheng, H., & Brune, A. (2015). Complete genome sequence of, a free-living relative of the intracellular symbionts of termite gut flagellates (phylum Elusimicrobia). Genome Announcements, 3(4), 11.

136

137
CHAPTER 4: Observation of Flagellated Protists in the guts of Higher Termites Tumulitermes westraliensis and Amitermes obeuntis Following Rainfall

138
Foreword:
I have thus far focused on the bacterial community in the hindgut of termites
and factors that may affect the community structure and composition. One
factor that is usually considered for lower termites only, is the presence of a
protistan community in the gut. It is widely accepted that protists occur in
lower termites, have coevolved with the host and play a role in shaping the gut
community by providing extra niches. There have been multiple historical
sightings in higher termite guts but they remain rarely acknowledged in recent
publications. As part of my first research question – How much intraspecific
variation exists in the gut communities of Western Australian endemic
termites? – the presence of protists was investigated in three local termites
species, including two higher termites.
Introduction:
Phylogenetic studies have shown that termites are closely related to the
cockroaches and should be nested within their order Blattodea (Lo et al., 2000;
Inward et al., 2007; Engel et al., 2009). Furthermore, a symbiosis with
cellulose-degrading flagellated protists developed in a common ancestor to
the lower termites and their sister group, the wood-feeding roaches
(Cryptocercus). These flagellates are transferred vertically to the offspring by
proctodeal trophallaxis (anus-to-mouth) and have been shown to have co-
evolved with their host (Ikeda-Ohtsubo and Brune, 2009; Ohkuma et al., 2009;
Brune and Dietrich, 2015). The establishment of the gut community is
sequential, with the offspring requiring repeated transfer of gut fluid until the
third instar, when they can maintain the larger flagellates and become
independent feeders. In the lower termites, proctodeal trophallaxis is also
important in the two weeks following any molt to re-establish the gut
community (Nalepa, 2015).

139
The flagellate community is crucial to the survival of lower termites on a
cellulose-based diet (Cleveland, 1923; Veivers et al., 1983). The termite secretes
endoglucanases that randomly cleave cellulose chains in the midgut, which
are endocytosed by the larger flagellates in the hindgut. These produce endo-
and exoglucanases, as well as β-glucosidases and accessory enzymes to
synergistically break down cellulose into glucose (Watanabe et al., 1998;
Ohkuma, 2008; Brune, 2014). This product is fermented into acetate within the
protists and taken up by the termite as its primary carbon and energy source
(Leadbetter et al., 1999; Brune, 2014). The flagellates make up the bulk of the
gut volume, creating niches for bacteria to colonise. Bacterial ectosymbionts
coat the protists and confer motility beyond that of the flagella, allowing the
host cell to navigate the currents of the gut and access wood particles
(Ohkuma, 2008). The flagellates have also been shown to produce hydrogen,
used by certain endosymbionts to produce more acetate (Leadbetter et al.,
1999). Protists in lower termites therefore play key roles in metabolism and
shape the bacterial community structure.
These flagellates are generally thought to have been lost in the lineage leading
to the higher termites around 40-60 Mya (family Termitidae; Engel et al.,
2009; Bourguignon et al., 2016). Cellulose degradation is undertaken by the
host and its gut bacteria (Tokuda and Watanabe, 2007; Brune and Dietrich,
2015). Proctodeal trophallaxis has not been reported in the higher termites,
which are thought to share their gut contents by trophallaxis (mouth-to-
mouth) and coprophagy (excrement ingestion; Diouf et al., 2015). The higher
termites underwent a diversification of their diet, with species specialised in
feeding on wood, but also grass, litter, soil or fungus. Termitidae now makes
up more than 70% of all termite species with over 2000 species (Krishna et al.,
2013). The shift from a protist-dominated to a bacterial gut community has
likely contributed to the success of the family Termitidae, in terms of its
diversity and abundance (Engel et al., 2009; Brune, 2014).

140
However, there have been multiple reports of protists found in higher
termites. Cleveland (1923) observed protists in one species of Nasutitermes and
two species of Mirotermes and concluded that protists are strongly linked to
wood-feeding termite species. Kirby (1927) described flagellates and amoebae
in the guts of higher termites. The most abundant were amoebae in the guts of
the wood-feeding Mirotermes. Kirby (1932) also observed three flagellates, one
amoeba and one ciliate species across multiple Amitermes species. Inoue et al.
(1998, unpublished) correlated cellulase activity in the hindgut of the
Australian termite species Ephelotermes melanoma, Macrognathotermes
sunteri and Xylochomitermes melvillensis with the presence of amoeba
(Slaytor, 2000). It was theorised that amoeba were responsible for cellulose-
degradation in certain higher termites. However, for the last decade, the focus
has been on bacterial cellulases (Tokuda and Watanabe, 2007; Brune and
Dietrich, 2015) and the irregular appearance of protists in higher termite guts
has not been extensively investigated.
Rahman et al. (2015) detected protists with molecular methods in four out of
eight pooled samples from the higher termite Gnathamitermes, collected in
Arizona (United States of America). The protist rRNA sequence most closely
resembled that of the ciliate Clevelandella, previously reported as
endosymbionts of wood-feeding roaches (Kidder, 1937; Lynn and Wright,
2013). Metaclevelandella of the same protistan family was first described in the
gut of the higher termite Capritermes incola Wasm (Uttangi and Desai, 1963).
Rahman et al. (2015) concluded that protists may have been retained or
reacquired in low abundance in certain higher termite taxa, but there are only
rare reports of non-flagellated protists in lower termites (Dobell, 1910; de
Mello, 1941).
Protists in higher termites may be low abundance symbionts or transient and
regardless their presence likely impacts the bacterial gut community, either
through functional competition, predation, or through the presence of protist-
associated bacteria. It is therefore important to characterise the function and

141
prevalence of protists in higher termites. In the present study, protists were
observed in the guts of two Western Australian higher termites: Tumulitermes
westraliensis and Amitermes obeuntis. A systematic approach was undertaken
to determine the prevalence of protists in T. westraliensis using light
microscopy.
Methods:
In September 2014, Tumulitermes westraliensis and Amitermes obeuntis
workers were freshly collected from John Forrest National Park (Western
Australia), as described in Chapter 2. Whole guts were prepared as squash
mounts, viewed under a compound light microscope with a 100x oil-
immersion objective lens and filmed with an AM4023X Dino-Eye USB digital
microscope eyepiece. Protists were identified as motile cells with clear
contents, typically larger in size to the gut bacteria (longer than 4 µm).
To determine the prevalence of protists in T. westraliensis, a systematic
approach was used in 2015. The lower termite Coptotermes acinaciformis,
known to harbour flagellates was used as a positive control for the protist
examination method. Three to five T. westraliensis workers were collected
from eight mounds located at two different sites in April and May 2015. Five of
the colonies were located in John Forrest National Park whereas the remaining
three colonies were located at Banyowla Regional Park, Western Australia.
The guts were dissected as described in Chapter 3. The crop was separated
from the remainder of the gut, both prepared as squash mounts and observed
separately. The crop was examined for a minimum of five minutes and the rest
of the gut (referred to as the hindgut hereafter) for a minimum of fifteen
minutes. Protists were filmed as before.

142
Results:
Protists were observed in both A. obeuntis and T. westraliensis individuals
collected in September 2014. Two flagellated oval-shaped protists (one 10 µm
in length and 4 µm in width and the other 5 µm by 3 µm) were found in a
single A. obeuntis worker (Figure 1A, ProtistsAobeSep2014.mp4). An oval
protist (4 µm by 4 µm) containing a dark circular structure was found in
another A. obeuntis individual, as well as a minimum of five elongated protists
(approximately 6-15 µm in length and 1-2 µm in width; Figure 1B).
Oval protists (4-10 µm in length and 3-5 µm in width) were present in multiple
T. westraliensis individuals (Figure 2; ProtistsTwestSep2014.mp4). Two
contained dark rounded structures, arranged in a circular fashion (Figure 2A
and D), while others were clear (Figure 3B and C). Clear elongated protists
(approximately 6-15 µm in length and 1-2 µm in width) were present in large
numbers and protists were found over multiple days in the same batch of T.
westraliensis.
Protists were found in the crop and in the hindgut of the C. acinaciformis
controls in April 2015. No protist activity was examined within either the crop
or the hindgut in 26 T. westraliensis specimens collected in April 2015. No
protists were found in the crop of specimens collected in May 2015. However,
elongated protists (approximately 6-15 µm in length and 1-2 µm in width) were
found in the hindgut of all 11 individuals examined in May (from three
colonies; Figure 3). Two out of five individuals collected from one colony
harboured oval protists as well (4-10 µm in length and 3-5 µm in width; Figures
3 and 4; ProtistsTwestMay2015.mp4). Two oval protists contained dark
rounded structures, but these were scattered throughout the cell (Figure 4A
and B).
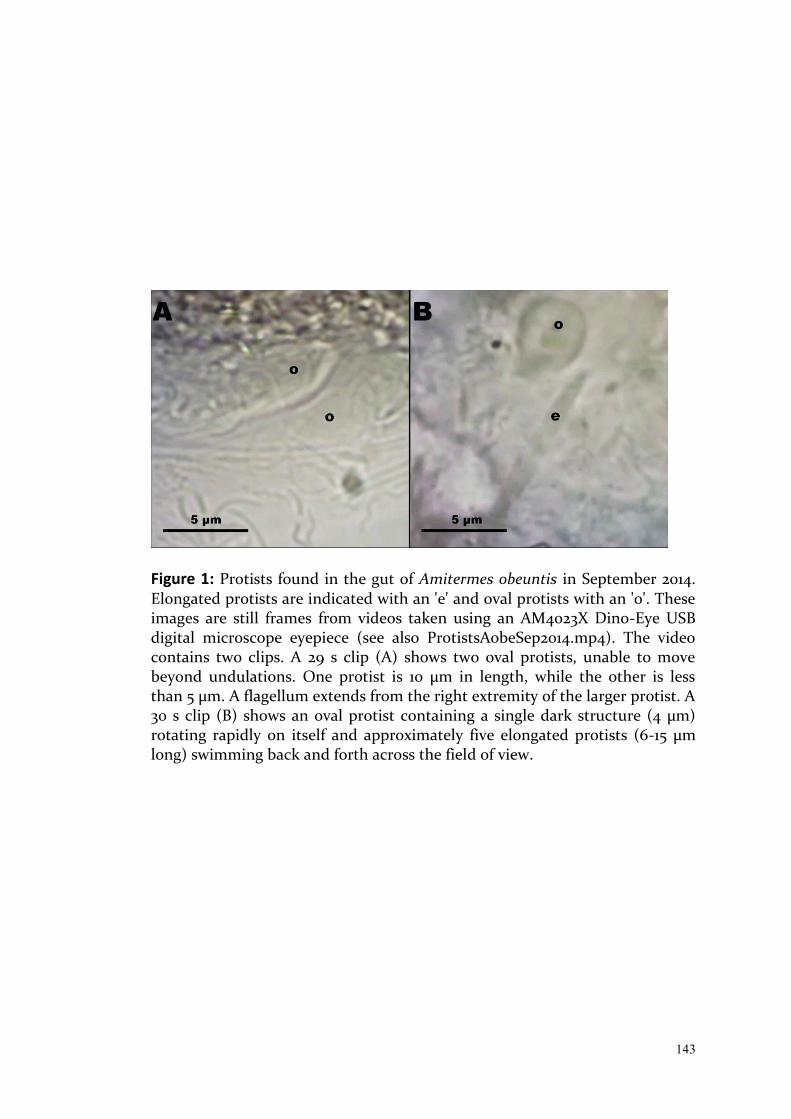
143
Figure 1: Protists found in the gut of Amitermes obeuntis in September 2014. Elongated protists are indicated with an 'e' and oval protists with an 'o'. These images are still frames from videos taken using an AM4023X Dino-Eye USB digital microscope eyepiece (see also ProtistsAobeSep2014.mp4). The video contains two clips. A 29 s clip (A) shows two oval protists, unable to move beyond undulations. One protist is 10 µm in length, while the other is less than 5 µm. A flagellum extends from the right extremity of the larger protist. A 30 s clip (B) shows an oval protist containing a single dark structure (4 µm) rotating rapidly on itself and approximately five elongated protists (6-15 µm long) swimming back and forth across the field of view.
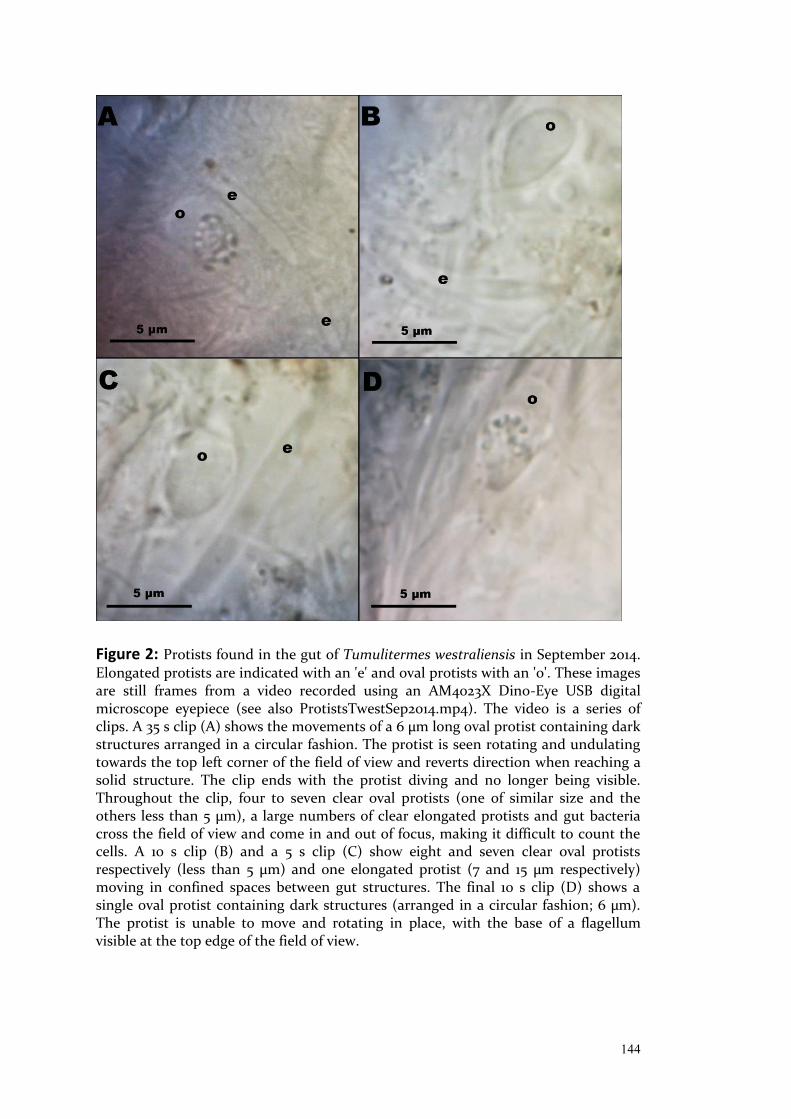
144
Figure 2: Protists found in the gut of Tumulitermes westraliensis in September 2014. Elongated protists are indicated with an 'e' and oval protists with an 'o'. These images are still frames from a video recorded using an AM4023X Dino-Eye USB digital microscope eyepiece (see also ProtistsTwestSep2014.mp4). The video is a series of clips. A 35 s clip (A) shows the movements of a 6 µm long oval protist containing dark structures arranged in a circular fashion. The protist is seen rotating and undulating towards the top left corner of the field of view and reverts direction when reaching a solid structure. The clip ends with the protist diving and no longer being visible. Throughout the clip, four to seven clear oval protists (one of similar size and the others less than 5 µm), a large numbers of clear elongated protists and gut bacteria cross the field of view and come in and out of focus, making it difficult to count the cells. A 10 s clip (B) and a 5 s clip (C) show eight and seven clear oval protists respectively (less than 5 µm) and one elongated protist (7 and 15 µm respectively) moving in confined spaces between gut structures. The final 10 s clip (D) shows a single oval protist containing dark structures (arranged in a circular fashion; 6 µm). The protist is unable to move and rotating in place, with the base of a flagellum visible at the top edge of the field of view.
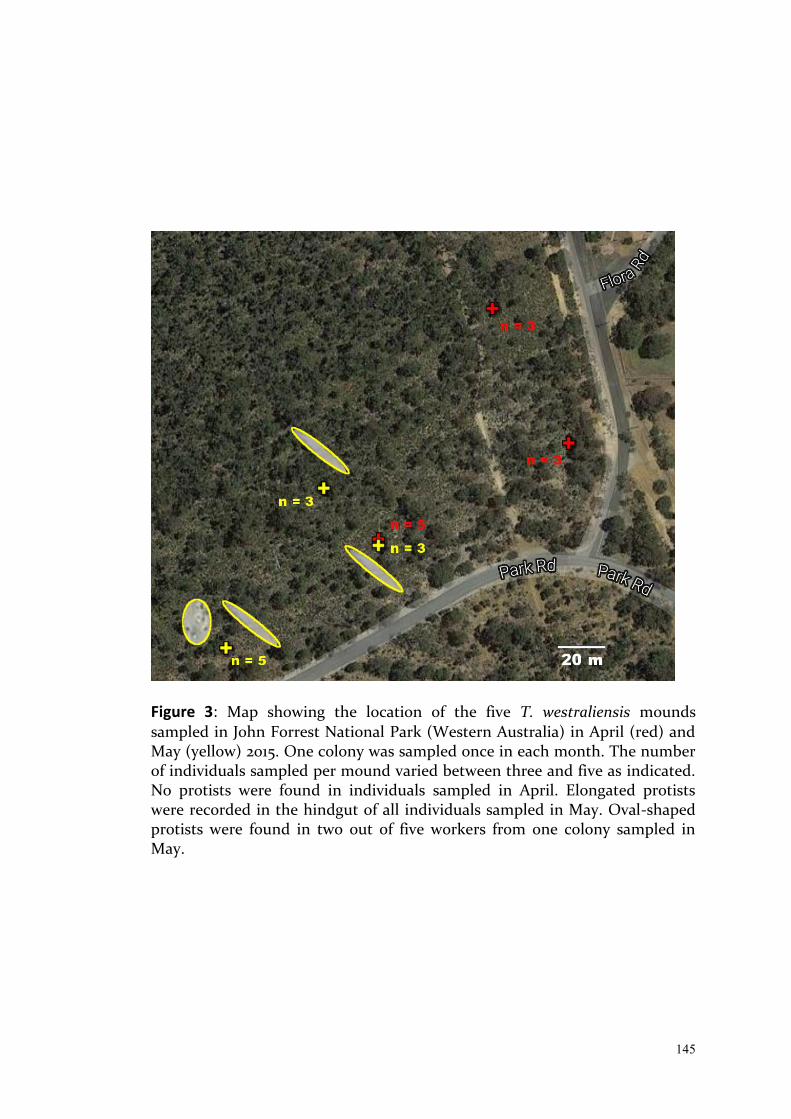
145
Figure 3: Map showing the location of the five T. westraliensis mounds sampled in John Forrest National Park (Western Australia) in April (red) and May (yellow) 2015. One colony was sampled once in each month. The number of individuals sampled per mound varied between three and five as indicated. No protists were found in individuals sampled in April. Elongated protists were recorded in the hindgut of all individuals sampled in May. Oval-shaped protists were found in two out of five workers from one colony sampled in May.
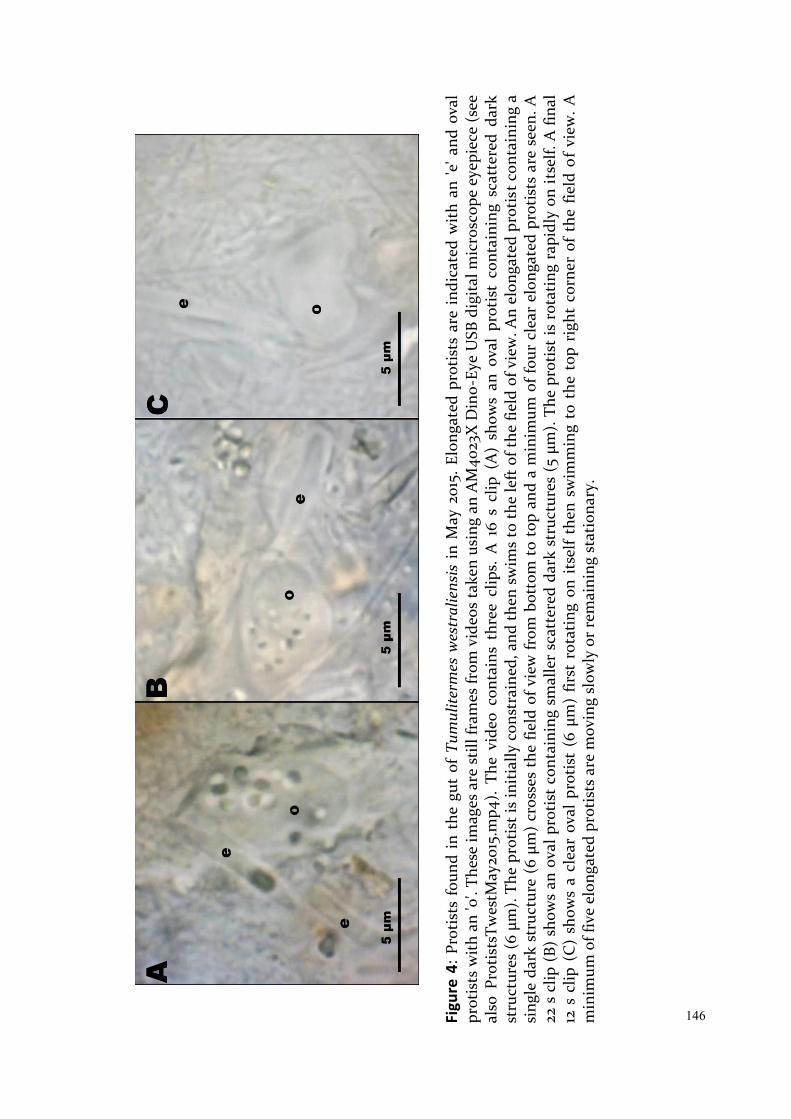
146
Fi
gure
4:
Prot
ists
fou
nd i
n th
e gu
t of
Tum
ulite
rmes
wes
tral
iens
is i
n M
ay 2
015.
Elo
ngat
ed p
rotis
ts a
re i
ndic
ated
with
an
'e' a
nd o
val
prot
ists
with
an
'o'.
Thes
e im
ages
are
stil
l fra
mes
from
vid
eos
take
n us
ing
an A
M40
23X
Din
o-Ey
e U
SB d
igita
l mic
rosc
ope
eyep
iece
(see
al
so P
rotis
tsTw
estM
ay20
15.m
p4).
The
vide
o co
ntai
ns t
hree
clip
s. A
16
s cl
ip (
A)
show
s an
ova
l pr
otis
t co
ntai
ning
sca
tter
ed d
ark
stru
ctur
es (6
µm
). Th
e pr
otis
t is
init
ially
con
stra
ined
, and
then
sw
ims
to th
e le
ft o
f the
fiel
d of
vie
w. A
n el
onga
ted
prot
ist c
onta
inin
g a
sing
le d
ark
stru
ctur
e (6
µm
) cr
osse
s th
e fie
ld o
f vie
w fr
om b
otto
m t
o to
p an
d a
min
imum
of f
our
clea
r el
onga
ted
prot
ists
are
see
n. A
22
s c
lip (
B) s
how
s an
ova
l pro
tist
con
tain
ing
smal
ler
scat
tere
d da
rk s
truc
ture
s (5
µm
). Th
e pr
otis
t is
rot
atin
g ra
pidl
y on
itse
lf. A
fina
l 12
s c
lip (
C)
show
s a
clea
r ov
al p
roti
st (
6 µm
) fir
st r
otat
ing
on i
tsel
f th
en s
wim
min
g to
the
top
rig
ht c
orne
r of
the
fie
ld o
f vi
ew. A
m
inim
um o
f fiv
e el
onga
ted
prot
ists
are
mov
ing
slow
ly o
r re
mai
ning
sta
tiona
ry.

147
Discussion:
Protists were found in the guts of T. westraliensis and A. obeuntis in
September 2014 and in T. westraliensis in May 2015. They were smaller in size
and in lower numbers than in C. acinaciformis. There were two main shapes of
protists observed (oval and elongated), some containing dark rounded
structures. Some of the oval protists were seen to be flagellated and all other
protists are thought to be flagellated because they moved in a similar fashion
(Orsi, personal communication, 11/10/16). The oval-shaped protists were
similar in size and appearance to flagellates described in Amitermes by Kirby
(1932).
The use of an inverted microscope and a higher resolution camera would
confirm the presence of any flagella and cilia. The use of Toluidine blue stain
could establish whether any protists contain plant matter and thus if they are
cellulose-degraders like the lower termite symbionts (Parker et al., 1982). A
molecular approach would be a more systematic and high throughput way of
determining the prevalence of protists in the guts of higher termites. As there
are no universal barcoding primer pair currently available for protists, a
combination of primers or a two-step barcoding approach with universal
eukaryotic primers should be used to investigate the presence and diversity of
protists in higher termites (Pawlowski et al., 2012; Adl et al., 2014).
The protists were not found at all time points or uniformly in all colonies.
Rahman (2015) reported the presence of the ciliate Clevelandella in half of their
Gnathamitermes samples. Clevelandella have been found in cockroach guts,
pointing to a symbiont-host interaction (Kidder, 1937; Lynn and Wright, 2013).
This hypothesis of low-level persistence or reacquisition of protists as
symbionts in higher termites is not supported by this dataset, as it would not
explain the time-dependent presence (or dramatic changes in abundance).
Indeed, protists were found in May 2015 in all three individuals collected from

148
a colony, when they had not been detected in five individuals recovered from
the same colony in April. Another possibility is horizontal transfer of protists
through interactions with lower termites (Radek et al., 2018) or environmental
acquisition for example through soil. Indeed, Clevelandella has since been
detected in soil (Gabilondo et al., 2015). Soil contains many protists which are
often endemic to an area and not well studied (Adl et al., 2014). Using
molecular approaches on gut contents from multiple termite species with
overlapping distributions and the neighbouring soil would help confirm
whether protists in higher termites are environmentally or horizontally
acquired.
In the current study, protist presence seemed to be linked to rainfall. Protists
were first observed in termites collected early in September 2014, following
heavy rain on the 30th August (70.8 mm in Mundaring, Western Australia;
http://www.bom.gov.au/climate/data/). No protists were found in three
colonies sampled in April 2015, when the maximum daily rainfall was 24.0 mm.
Elongated protists were recorded in all three John Forrest National Park
colonies sampled in May and oval protists in one of these colonies, one week
after heavier rain of 42.2 mm. Protists were only detected following the
substantial rain event in the colony sampled twice. Termites examined by
Kirby were collected in Panama in "summer" (1927) and August and in
California in "fall" (1932) and the presence of protozoa was not uniform. All of
these sampling times make up part of the respective locations' typically rainy
seasons, supporting the link between increased rainfall and the observation of
protists in higher termite guts.
Moisture availability is the dominant factor determining the biogeographic
patterns of soil protists (Bates et al., 2013; Adl et al., 2014). In dry weather,
protists may encyst and lay dormant, until the conditions become more
favourable (Adl and Gupta, 2006). Sampling repeatedly from several colonies
over multiple seasons would confirm whether the presence of protists in the
gut is linked to rainfall events and how long they persist in the gut. Such a

149
study would confirm whether the presence of protists in the gut of higher
termites is due to seasonal environmental uptake of active terrestrial protists.
Microscopy would be key to differentiate between active protists and cysts, as
both will be detected with molecular methods (Santos et al., 2015).
This study confirms that at least some higher termites can harbour live
flagellated protists in their guts. The inconsistent nature of protist presence
reported in higher termites in the literature and the current study suggests
that they are likely to be transient, taken up seasonally from the environment
following rain, rather than in a symbiosis with the host. Soil protists can be
cellulose-degrading or bactericidal (Habte and Alexander, 1975; Kramer et al.,
2016), indicating there could be significant impacts caused by the presence of
these protists in higher termite guts through functional competition or
predation. Additionally, certain soil protists have been reported to have
associated bacteria (Bland J. Finlay and Fenchel, 1991, Atef Omara et al., 2017),
which would contribute to a 16S rRNA gene profiling dataset. I did find
protist-associated genera Candidatus Captivus and Candidatus Hepatincola
(order Rickettsiales, class Alphaproteobacteria) in A. obeuntis samples, as
reported in Chapter 3, during a rainy period. The presence of protists may
hence seasonally affect the bacterial community within the gut and should be
taken into account going forward.

150
References: Abdul Rahman, N., Parks, D. H., Willner, D. L., Engelbrektson, A. L., Goffredi, S. K.,
Warnecke, F., Scheffrahn, R. H., Hugenholtz, P. (2015). A molecular survey of Australian and North American termite genera indicates that vertical inheritance is the primary force shaping termite gut microbiomes. Microbiome, 3, 5.
Adl, M. S., & Gupta, V. V. S. R. (2006). Protists in soil ecology and forest nutrient cycling. Canadian Journal of Forest Research, 36(7), 1805-1817.
Adl, S. M., Habura, A., & Eglit, Y. (2014). Amplification primers of SSU rDNA for soil protists. Soil Biology and Biochemistry, 69(0), 328-342.
Bates, S. T., Clemente, J. C., Flores, G. E., Walters, W. A., Parfrey, L. W., Knight, R., Fierer, N. (2013). Global biogeography of highly diverse protistan communities in soil. ISME J, 7(3), 652-659.
Bland J. Finlay, & Fenchel, T. (1991). An anaerobic protozoon, with symbiotic methanogens, living in municipal landfill material. FEMS Microbiology Ecology, 85, 169-180.
Bourguignon, T., Lo, N., Šobotník, J., Ho, S. Y. W., Iqbal, N., Coissac, E. et al. (2016). Mitochondrial Phylogenomics Resolves the Global Spread of Higher Termites, Ecosystem Engineers of the Tropics. Molecular Biology and Evolution, msw253.
Brune, A. (2014). Symbiotic digestion of lignocellulose in termite guts. Nature Reviews Microbiology, 12(3), 168-180.
Brune, A., Dietrich, C. (2015). The gut microbiota of termites: digesting the diversity in the light of ecology and evolution. Annual Review of Microbiology. 69(1). 145-166.
Cleveland, L. R. (1923). Symbiosis between termites and their intestinal protozoa. Proceedings of the National Academy of Sciences of the United States of America, 9(12), 424-428.
de Mello, F. (1941). First record of an amœba parasite of an Indian termite. Proceedings of the Indian Academy of Sciences - Section B, 14, 68-73.
Diouf, M., Roy, V., Mora, P., Frechault, S., Lefebvre, T., Hervé, V., Rouland-Lefèvre, C., Miambi, E. (2015). Profiling the succession of bacterial communities throughout the life stages of a higher termite Nasutitermes arborum (Termitidae, Nasutitermitinae) Using 16S rRNA Gene Pyrosequencing. PLoS One, 10, e0140014.
Dobell, C. (1910). On some parasitic protozoa from Ceylon. Spolia Zeylan, 7, 65-87. Engel, M. S., Grimaldi, D. A., Krishna, K. S. (2009). Termites (Isoptera): their phylogeny, classification, and rise to ecological dominance. American Museum Novitiates, 3650, 1-27. Gabilondo, R., Fernández-Montiel, I., García-Barón, I., & Bécares, E. (2015). The
effects of experimental increases in underground carbon dioxide on edaphic protozoan communities. International Journal of Greenhouse Gas Control, 41, 11-19.
Habte, M., & Alexander, M. (1975). Protozoa as agents responsible for the decline of Xanthomonas campestris in soil. Appl Microbiol, 29(2), 159-164.
Ikeda-Ohtsubo, W., & Brune, A. (2009). Cospeciation of termite gut flagellates and their bacterial endosymbionts: species and ‘Endomicrobium trichonymphae’. Molecular Ecology, 18(2), 332-342.
Inward, D., Beccaloni, G., & Eggleton, P. (2007). Death of an order: a comprehensive molecular phylogenetic study confirms that termites are eusocial cockroaches. Biology Letters, 3(3), 331-335.

151
Kidder, G. W. (1937). The intestinal protozoa of the wood-feeding roach Panesthia. Parasitology, 29(02), 163.
Kirby, H. (1927). Studies on some amoebae from the termite Mirotermes, with notes on some other Protozoa from the Termitidae. Quarterly Journal of Microscopical Science,
Kirby, H. (1932). Protozoa in termites of the genus Amitermes. Parasitology, 24(03), 289.
Kramer, S., Dibbern, D., Moll, J., Huenninghaus, M., Koller, R., Krueger, D. et al. (2016). Resource Partitioning between Bacteria, Fungi, and Protists in the Detritusphere of an Agricultural Soil. Frontiers in Microbiology, 7, 1524.
Krishna, K., Grimaldi, D. A., Krishna, V., & Engel, M. S. (2013). Treatise on the Isoptera of the world. Bulletin of the American Museum of Natural History, 1.
Leadbetter, J. R., Schmidt, T. M., Graber, J. R., & Breznak, J. A. (1999). Acetogenesis from H2 plus CO2 by spirochetes from termite guts. Science, 283(5402), 686-689.
Lo, N., Tokuda, G., Watanabe, H., Rose, H., Slaytor, M., Maekawa, K. et al. (2000). Evidence from multiple gene sequences indicates that termites evolved from wood-feeding cockroaches. Current Biology, 10(13), 801-804.
Lynn, D. H., & Wright, A. D. (2013). Biodiversity and molecular phylogeny of Australian Clevelandella species (class Armophorea, order Clevelandellida, family Clevelandellidae), intestinal endosymbiotic ciliates in the wood-feeding roach Panesthia cribrata Saussure, 1864. Journal of Eukaryotic Microbiology, 60(4), 335-341.
Nalepa, C. A. (2015). Origin of termite eusociality: trophallaxis integrates the social, nutritional, and microbial environments: Origin of termite eusociality. Ecological Entomology, 40(4), 323-335.
Ohkuma, M. (2008). Symbioses of flagellates and prokaryotes in the gut of lower termites. Trends in Microbiology, 16(7), 345-352.
Ohkuma, M., Noda, S., Hongoh, Y., Nalepa, C. A., & Inoue, T. (2009). Inheritance and diversification of symbiotic trichonymphid flagellates from a common ancestor of termites and the cockroach Cryptocercus. Proceedings of the Royal Society B: Biological Sciences, 276(1655), 239-245.
Omar, A., Zhang, Q., Zou, S., & Gong, J. (2017). Morphology and Phylogeny of the Soil Ciliate Metopus yantaiensis n. sp. (Ciliophora, Metopida), with Identification of the Intracellular Bacteria. Journal of Eukaryotic Microbiology, 64, 792-805.
Orsi, W (Department of Earth and Environmental Sciences, Palaeontology & Geobiology, Ludwig Maximilian University of Munich). (11/10/16). Personal communication.
Parker, A. J., Haskins, E. F., & Deyrup-Olsen, I. (1982). Toluidine blue: a simple, effective stain for plant tissues. The American Biology Teacher, 487-489.
Pawlowski, J., Audic, S., Adl, S., Bass, D., Belbahri, L., Berney, C. et al. (2012). CBOL protist working group: barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms. PLoS. Biology, 10(11).
Radek, R., Meuser, K., Strassert, J. F. H., Arslan, O., Teßmer, A., Šobotník, J. et al. (2018). Exclusive Gut Flagellates of Serritermitidae Suggest a Major Transfaunation Event in Lower Termites: Description of Heliconympha glossotermitis gen. nov. spec. nov. Journal of Eukaryotic Microbiology, 65(1), 77-92. Santos, S. S., Nielsen, T. K., Hansen, L. H., & Winding, A. (2015). Comparison of three DNA extraction methods for recovery of soil protist DNA. Journal of Microbiological Methods, 115, 13-19.
Slaytor, M. (2000). Energy metabolism in the termite and its gut microbiota. In T.

152
Abe, D. E. Bignell, & M. Higashi (Eds.), Termites: Evolution, Sociality, Symbioses, Ecology (pp. 307-332). Dordrecht, The Netherlands: Kluwer Academic Publishers.
Tokuda, G., & Watanabe, H. (2007). Hidden cellulases in termites: revision of an old hypothesis. Biology Letters, 3(3), 336-339.
Uttangi, J. C., & Desai, R. N. (1963). Metaclevelandella termitis, a new genus and species of Heterotrichous ciliate (family Clevelandellidae) found in the Indian termite Capritermes incola Wasm. Parasitology, 53(1-2), 39.
Veivers, P. C., O’Brien, R. W., & Slaytor, M. (1983). Selective defaunation of Mastotermes darwiniensis and its effect on cellulose and starch metabolism. Insect Biochemistry, 13(1), 95-101.
Watanabe, H., Noda, H., Tokuda, G., & Lo, N. (1998). A cellulase gene of termite origin. Nature, 394(6691), 330-331.

153
CHAPTER 5: Accurate Characterisation of the Complete Termite Cellulase Profile Requires Selective Inhibition of Gut Proteases

154
Foreword:
The long term goal of this research is to isolate enzymes from higher termite
guts relevant to second generation biofuel production in Western Australia. A
shorter term goal was to establish if the gut community of local higher
termites can be optimised for breaking down substrates of interest through
feeding experiments and this has been the primary focus of previous thesis
chapters. In this chapter, I begin to answer the final research question – Can
enzymes relevant to breaking down a substrate of choice be identified using
comparative enzymology?
I report on the optimisation of protein extraction and gel based protein
visualisation, enzyme assays and cellulase-binding probe methods for whole
and segmented termite guts. I do not cover in tube cellulase assays or protein
concentration experiments that were also undertaken. The chapter includes a
chronological rationale for each relevant optimisation step to highlight
obstacles to this research, followed by recommendations for future work.
Introduction:
Higher termites are able to efficiently degrade cellulose due to the production
of endogenous enzymes in the midgut and the synergistic action of their gut
bacteria ( reviewed in Brune, 2014). Three major types of glycoside hydrolases
(GHs, collectively referred to as cellulases) are required for the breakdown of
cellulose into glucose. Cellulose is typically insoluble but contains amorphous
regions that can be accessed by endoglucanases (also known as
endocellulases). These cleave the cellulose chain randomly, producing more
ends for exoglucanases (or exocellulases) to act. There are two types of
exoglucanases, allowing these tunnel-shaped enzymes to cleave cellobiose
(made up of two glucose subunits) from either end of the cellulose chain
(reviewed in Wilson, 2011). Finally, β-glucosidase acts on cellobiose to

155
complete the digestion to glucose. Other accessory enzymes, such as
hemicellulases, laccases and aldo-keto reductase, facilitate this process by
increasing access to the cellulose chain ( Coy et al., 2010; Mohanram et al.,
2013; Sethi et al., 2013).
The deployment strategies for these cellulases differ amongst different
organisms. The termite itself secretes soluble endoglucanases in the midgut,
as well as β-glucosidases to break down any resulting cellobiose and prevent
enzyme inhibition (as reviewed by Brune, 2014). The hindgut microbiota
employs a variety of strategies, from secreting soluble enzymes, producing
cell-bound enzymes (typically clustered together in a cellulosome), to binding
directly to the substrate and importing partially degraded chains for further
intracellular processing (Fibrobacteres; Wilson, 2011; Ransom-Jones et al.,
2012). Quantifying and characterising the activity of these enzymes must
therefore be adapted to each enzyme deployment strategy. Indeed, until
methods were developed to target the cell-bound enzymes that dominate the
hindgut of many of the higher termites, the consensus was that endogenous
enzymes drove cellulose digestion in higher termites (Schulz et al., 1986;
Hogan et al., 1988). In 2007, Tokuda and Watanabe showed that cell-bound
cellulases could be accessed following the breakdown of the bacterial cell walls
and centrifugation. This method allows the separation of soluble (so-called
crude extract) and cell-bound (pellet extract) enzymes. Separating the gut
segments also allows for the comparison of the endogenous and symbiont
derived cellulases (O'Brien et al., 1979; McEwen et al., 1980; Tokuda and
Watanabe, 2007).
Cellulase zymography is a commonly used electrophoretic method to visualise
endoglucanase activity on commercially available amorphous cellulose
(carboxymethyl cellulose (CMC); Schwarz et al., 1987; Tokuda and Watanabe,
2007; Oppert et al., 2010). Endoglucanases can be extracted from the resulting
gel in different ways and identified using mass spectrometry: 1) Proteins can
be extracted directly from the zymogram by excising the clear zone that

156
signals cellulase activity (Nakashima et al., 2002; Ni et al., 2014), 2) they can be
extracted following silver staining of the zymogram, allowing the distinction
between proteins of similar sizes (Willis et al., 2010), 3) they can be extracted
from the zymogram using a standard acrylamide gel run in parallel as a guide
(Tarayre et al., 2015), or 4) from the guide acrylamide gel itself (Park et al.,
2002).
Activity-based proteomics probes (ABPPs) can be used to detect and identify
active enzymes of a class of interest. Probes are synthesised to resemble a
substrate or inhibitor of the target enzyme and make use of a reporter group
to visualise or enrich the protein (Stubbs, 2014). Several cellulase-binding
probes have been designed for use in microbial cultures (Chauvigné-Hines et
al., 2012). The use of click chemistry allows for the probes to be coupled with a
fluorescent reporter group for gel visualisation, or biotin for subsequent
protein concentration and identification using mass spectrometry.
I conducted a series of experiments with the aim to visualise cellulases from
the Western Australian termite species Tumulitermes westraliensis and
Amitermes obeuntis and their activity. First, total protein was visualised for
quality control purposes and then the commonly used zymography method
applied, for comparison of endoglucanase activity with previously studied
species. Finally, a cellulase-binding probe method was applied to termite gut
extracts, to allow for the identification of a broad range of cellulases
(Chauvigné-Hines et al., 2012; Anderson et al., 2013).

157
Protocols:
Protocols were optimised throughout the study and relevant versions have
been numbered below. These versions will be referred to in the experiments
section.
Protein Extraction (PE)
PE1: Working on ice, termite gut segments of interest were pooled into 2 mL
bead-beating tubes containing 0.7 g of 0.1 mm zirconia/silica beads and 1 ml
extraction buffer (25 mM Tris pH 8.5, 2% SDS, 10 mM DTT, Roche cOmplete™
Protease Inhibitor Cocktail as instructed). Samples were homogenised with a
FastPrep®-24 (MP Biomedicals) for 45 s at 6.5 m/s, heated at 99° C for 3 min
and centrifuged at 20,000 × g for 10 min at 4° C.
PE2: Working on ice, termite gut segments of interest were pooled into 2 mL
bead-beating tubes containing 0.2 g of 0.1 mm zirconia/silica beads and 200 µl
extraction buffer (25 mM Tris pH 8.5, 2% SDS, 10 mM DTT, Roche cOmplete™
Protease Inhibitor Cocktail as instructed). Phenylmethylsulfonyl fluoride
(PMSF, Sigma Aldrich) was prepared at 100 mM in ethanol and 10 µl added to
samples after the collection of gut segments, as detailed below. Samples were
homogenised with a FastPrep24 for 45 s at 6.5 m/s and centrifuged at 20,000 ×
g for 10 min at 4° C.
PE3: Working on ice, termite gut segments of interest were pooled into 1.5 ml
tubes in 100 µl Tris buffer (pH 7.6, 0.42 µM SDS, as per Chauvigné-Hines et al.
(2012), with Roche cOmplete™ Protease Inhibitor Cocktail). A positive control
for cellulase activity was included, made up of 1 mg/ml cellulase (Aspergillus
niger, Sigma Aldrich) in Tris buffer. 10 µl of 100 mM PMSF was added to each
sample after the dissections were completed. Samples were homogenised with
a micropestle (cleaned between each sample with ethanol) and centrifuged at
20,000 × g for 10 min at 4° C. The supernatant will be referred to as the crude

158
extract and were stored at 4° C until their same-day use (Tokuda et al., 2005).
The pellet was washed three times in Tris buffer, resuspended in 100 µl
CelLytic B (Sigma), vigorously mixed for 15 s and incubated on ice for 10 min.
Samples were centrifuged at 20,000 × g for 10 min at 4° C; the supernatant will
be referred to as the pellet extract (Tokuda and Watanabe, 2007).
PE4: Working on ice, termite gut segments of interest were pooled into 1.5 ml
tubes in 150 µl 50 mM HEPES buffer (pH 7.6, with 1M NDSB201, 20 mM DTT,
50 mM EDTA and 2x Halt™ Protease and Phosphatase Inhibitor Cocktail,
EDTA-free (ThermoFisher Scientific). A positive control for cellulase activity
was included, made up of 30 µg/µl cellulase (Aspergillus niger) in HEPES
buffer. Samples were homogenised with a micropestle (sterilised between each
sample by washing in ethanol) and centrifuged at 20,000 × g for 10 min at 4° C.
The supernatant will be referred to as the crude extract and were stored at 4°
C until their same-day use (Tokuda et al., 2005). The pellet was washed three
times and resuspended in 150 µl of the HEPES buffer with 0.5% Triton x-100.
Homogenates were transferred to bead-beating tubes containing 0.2 g of 0.1
mm zirconia/silica beads and processed three times in a FastPrep24 at 6 m/s
for 40 s, incubating samples on ice for 5 min in between each run. Samples
were centrifuged at 20,000 × g for 5 min at 4° C, transferred to new tubes and
centrifuged once more for 10 min to remove the beads. The supernatant will
be referred to as the pellet extract (Tokuda and Watanabe, 2007).
Protein Precipitation (PP)
PP1: Four volumes of -20° C acetone were added to each extract, and these
were mixed and incubated at -20° C overnight. The next day, they were
centrifuged at 20,000 × g for 20 min at 4° C, the supernatant discarded and the
pellets air-dried. Pellets were resuspended in 30 µL of 2 × Laemmli sample
buffer (0.0625 M Tris pH 6.8; 2 % SDS; 10 % glycerol; 0.001 % Bromophenol
blue) with freshly added 0.4% β-mercaptoethanol (Sigma Aldrich) and heated
at 65° C for 5 min.

159
PP2: Each extract was mixed with 4 volumes methanol, then with 1 volume
chloroform and finally with 2.5 volumes water (All diluents were chilled on ice
prior to use). These were centrifuged at 20,000 × g for 5 min and the aqueous
phase discarded carefully from the top, leaving the proteins in the interphase.
The proteins were mixed once more with 2.5 volumes of methanol and
centrifuged at 20,000 × g for 10 min. The supernatant was discarded and the
pellets air dried and stored at -80 °C until use. Pellets were resuspended in 20
µl 50 mM HEPES, 2% SDS, 1M NDSB201, and 1x Halt™ Protease and
Phosphatase Inhibitor Cocktail, EDTA-free (ThermoFisher Scientific).
Acrylamide gel electrophoresis
Samples in Laemmli sample buffer were centrifuged at 20,000 × g for 3 min at
ambient temperature. Up to 25 µl (avoiding any pellets formed post
centrifugation) were loaded in each well of a 12% acrylamide gel, along with 5
µl of Precision Plus Protein™ Dual Color Standard (BioRad). Gel
electrophoresis was conducted at 90 V until the samples had migrated out of
the stacking gel, whereupon the voltage was increased to 130 V, until the dye
reached the bottom of the gel. For Coomassie staining, SimplyBlue™ SafeStain
(Invitrogen) was applied using the recommended microwave method, which
involves heating the gel 3 x 1 min in distilled water on high and once in the
stain, shaking the gel for 1 min between each stage and 5 min in the stain. The
gel was then rinsed in distilled water on a shaker for several minutes prior to
scanning.
Cellulase zymography
This method was modified from previous studies (Schwarz et al., 1987; Tokuda
and Watanabe, 2007; Oppert et al., 2010). Samples in Laemmli sample buffer
were centrifuged at 20,000 × g for 3 min at RT and 10 µl were loaded in each
well of a 12% acrylamide gel containing 0.1% carboxymethylcellulose (CMC,
Sigma Aldritch), along with 5 µl of MW standard. Gel electrophoresis was
conducted at 90 V until the samples had migrated out of the stacking gel,

160
whereupon the voltage was increased to 130 V, until the dye reached the
bottom of the gel. The gel was washed in distilled water for 5 min, soaked
three times in 0.1 M NaPO4 buffer (pH 6.5) for 20 min, incubated for 60 min in
0.1 M NaPO4 buffer at 28° C. The gel was stained in 1 g/L Congo red solution
(prepared in ethanol) for 30 min and destained in 1M NaCl for 30 min. The
contrast of the stain was increased by adding 5% glacial acetic acid
(Waeonukul, 2007) and the gel photographed on a light box.
Cellulase-binding Probe (CP)
CP1: Aliquots of 30 µl of each enzyme extract were labelled with 2 μl of 75 µM
GH2d-ABP probe (prepared by Mitchell Hattie and Dr Keith Stubbs;
Chauvigné-Hines et al., 2012) and incubated at 60° C for 3 h with mild
agitation (300 rpm) on a Thermomixer Comfort (Eppendorf). Samples were
precipitated with acetone as per protocol PP1. The precipitated samples were
centrifuged at 20,000 × g for 20 min, the pellet dried at ambient temperature
and resuspended in 15 µl Tris pH 7.6 (containing 2% SDS, 50 mM DTT and
Roche cOmplete™ Protease Inhibitor Cocktail). Samples containing probes
were mixed with 6 µl of a labelling mastermix containing Fluor 488-alkyne
(Sigma Aldrich), DTT, TRIS[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl] amine
(TBTA, Sigma Aldrich, prepared in dimethylformamide, DMF) and copper
sulfate as described by Chauvigné-Hines et al. (2012). These were incubated for
1 hr in the dark at ambient temperature. Following gel electrophoresis (see
protocol 1 above, with Coomassie staining deferred) the probe gel was fixed in
40% methanol, 10% acetic acid for 2 × 15 min, rinsed in water and visualised on
a GE Typhoon Trio™ with the green-excitation mode (532 nm) and the 526 nm
filter. The gel was then stained with Coomassie blue.
CP2: Aliquots of 30 µl of each enzyme extract were labelled with 2 μl of 75 µM
GH2d-ABP and incubated at 4° C for 30 min with mild agitation. Samples were
precipitated using protocol PP2. Resuspended samples were mixed with 6 µl of
a labelling mastermix containing Fluor 488-alkyne (Sigma Aldrich), DTT,
tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl] amine (TBTA, prepared in DMF)

161
and copper sulfate as described by Chauvigné-Hines et al. (2012). These were
incubated for 30 min in the dark at ambient temperature. Following gel
electrophoresis (see protocol above, with Coomassie blue staining done the
following day) the probe gel was visualised on a GE Typhoon Trio™ with the
green-excitation mode (532 nm) and the 526 nm filter. Finally, gels were fixed
in 40% methanol, 10% acetic acid for 2 × 15 min, rinsed in water overnight and
stained with Coomassie blue.
Experiments:
Below is the optimisation process into visualising termite gut cellulases
undergone throughout this study. The rationale behind each experiment and
the protocols used is followed by each experiment's results, so as to better
explain the reasoning behind the next experiment. The results are discussed
further in a separate section.
Total protein visualisation of termite gut sample extracts
Rationale
Gut samples from a variety of sources were treated minimally and compared at
a total protein level to test standard protein techniques on termite gut samples
and check for any obvious differences between different termite species, diets
(supplied substrates and mound material) or time in captivity.
Methods
Single whole worker guts, in triplicate, were dissected as summarised in Table
1. Total protein was visualised on acrylamide gels stained with Coomassie blue
as per extraction PE1, precipitation PP1 and gel electrophoresis protocols.
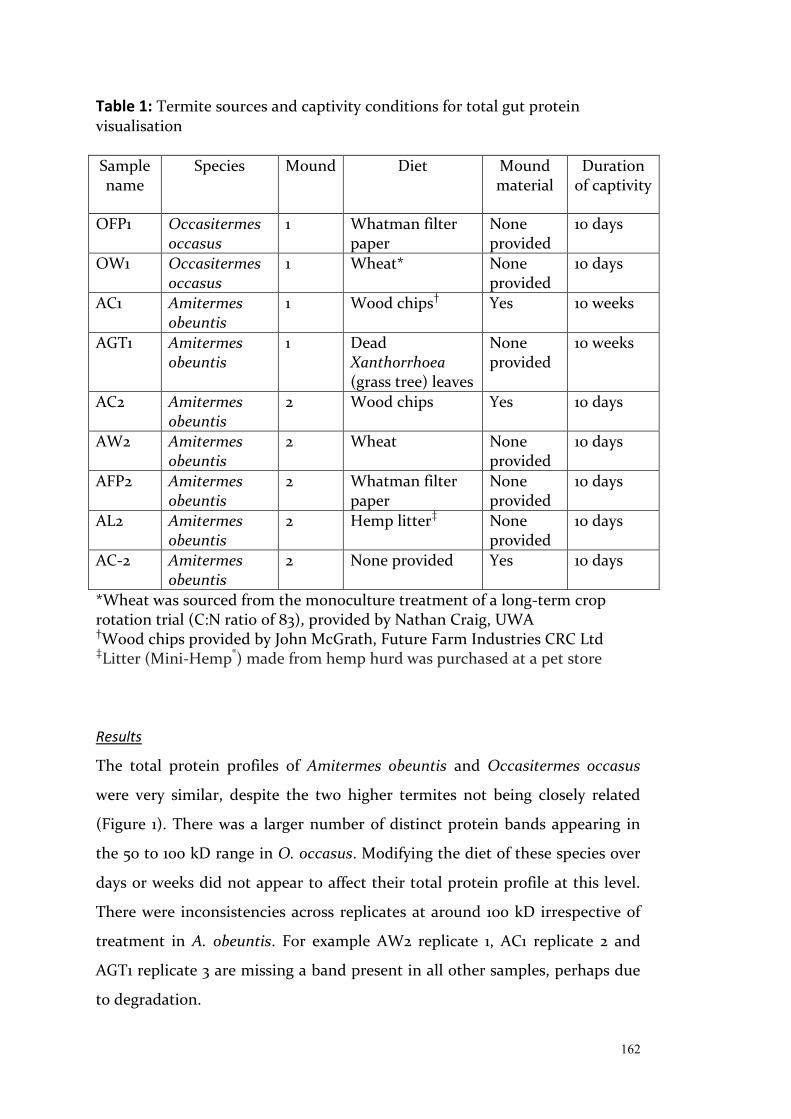
162
Table 1: Termite sources and captivity conditions for total gut protein visualisation Sample name
Species Mound Diet Mound material
Duration of captivity
OFP1 Occasitermes occasus
1 Whatman filter paper
None provided
10 days
OW1 Occasitermes occasus
1 Wheat* None provided
10 days
AC1 Amitermes obeuntis
1 Wood chips† Yes 10 weeks
AGT1 Amitermes obeuntis
1 Dead Xanthorrhoea (grass tree) leaves
None provided
10 weeks
AC2 Amitermes obeuntis
2 Wood chips Yes 10 days
AW2 Amitermes obeuntis
2 Wheat None provided
10 days
AFP2 Amitermes obeuntis
2 Whatman filter paper
None provided
10 days
AL2 Amitermes obeuntis
2 Hemp litter‡
None provided
10 days
AC-2 Amitermes obeuntis
2 None provided Yes 10 days
*Wheat was sourced from the monoculture treatment of a long-term crop rotation trial (C:N ratio of 83), provided by Nathan Craig, UWA †Wood chips provided by John McGrath, Future Farm Industries CRC Ltd ‡Litter (Mini-Hemp®) made from hemp hurd was purchased at a pet store Results
The total protein profiles of Amitermes obeuntis and Occasitermes occasus
were very similar, despite the two higher termites not being closely related
(Figure 1). There was a larger number of distinct protein bands appearing in
the 50 to 100 kD range in O. occasus. Modifying the diet of these species over
days or weeks did not appear to affect their total protein profile at this level.
There were inconsistencies across replicates at around 100 kD irrespective of
treatment in A. obeuntis. For example AW2 replicate 1, AC1 replicate 2 and
AGT1 replicate 3 are missing a band present in all other samples, perhaps due
to degradation.

163
Figure 1: Total protein from whole Amitermes obeuntis and Occasitermes occasus guts, following captivity and feeding, as summarised in Table 1. Biological triplicates were processed as described in version 1 of the protein extraction, precipitation and gel electrophoresis protocols. A larger number of proteins appear in the 50 to 100 kD range in O. occasus and a 100 kD band appears inconsistently across replicates in A. obeuntis (missing bands are indicated by arrows).

164
Conclusions
Any differences between the proteomes of termites fed on different diets could
not be identified by visualising total protein profiles in gel and more specific
methods are required. However, they do provide a good method to monitor
overall quality of the extracts and in this case indicated some possible protein
degradation.
Contribution of termite-derived proteins to cellulase profiles
Rationale
This experiment tested whether the addition of Phenylmethylsulfonyl fluoride
(PMSF) would further prevent protein degradation and result in more
consistent protein profiles (without the potentially missing bands seen in the
previous experiment). It also tested whether heating the samples prior to gel
loading was a step that damaged or improved the resulting protein profiles.
Finally, it aimed to compare whole guts to the remaining gutless bodies, as gut
proteomes were expected to contain more cellulases (of interest), but also
more proteases (with potential to disrupt the work).
Methods
Five freshly collected Tumulitermes westraliensis workers were pooled for each
treatment as indicated in Table 2. Protein was extracted as described in
extraction PE2 and 70 µl aliquots from each sample were heated at 99° C for 3
min. Total protein was visualised on acrylamide gels via gel electrophoresis
and cellulase zymography. A positive control for cellulase activity was
included, made up of 1 mg/ml cellulase (Aspergillus niger) in Tris buffer.
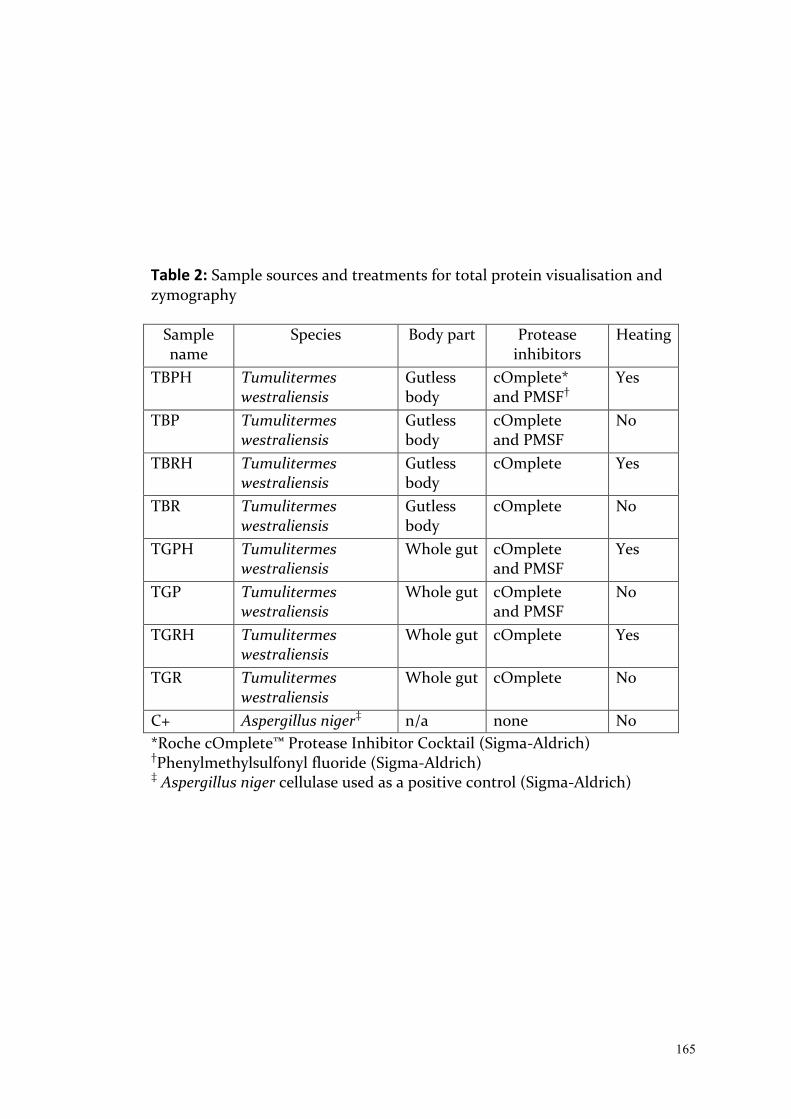
165
Table 2: Sample sources and treatments for total protein visualisation and zymography
Sample name
Species Body part Protease inhibitors
Heating
TBPH Tumulitermes westraliensis
Gutless body
cOmplete* and PMSF†
Yes
TBP Tumulitermes westraliensis
Gutless body
cOmplete and PMSF
No
TBRH Tumulitermes westraliensis
Gutless body
cOmplete
Yes
TBR Tumulitermes westraliensis
Gutless body
cOmplete
No
TGPH Tumulitermes westraliensis
Whole gut cOmplete and PMSF
Yes
TGP Tumulitermes westraliensis
Whole gut cOmplete and PMSF
No
TGRH Tumulitermes westraliensis
Whole gut cOmplete
Yes
TGR Tumulitermes westraliensis
Whole gut cOmplete
No
C+ Aspergillus niger‡ n/a none No *Roche cOmplete™ Protease Inhibitor Cocktail (Sigma-Aldrich) †Phenylmethylsulfonyl fluoride (Sigma-Aldrich) ‡ Aspergillus niger cellulase used as a positive control (Sigma-Aldrich)

166
Results
Gutless termite bodies always resulted in more abundant and distinct bands in
their protein profiles than their corresponding whole guts (Figure 2A).
Endoglucanase activity was more abundant in the gut samples, but still
detected in gutless termites (Figure 2B). Heating at 99° C for 3 min prior to gel
electrophoresis resulted in less abundant profiles for gutless termites and less
distinct bands in gut profiles.
Heating also resulted in a major loss of endoglucanase activity. In unheated
samples, proteins just larger than 50 kD produced the bulk of the
endoglucanase activity (see white arrows in Figure 2B). An equivalent band
was present in all total protein profiles, however was more abundant in the
gutless bodies and inconsistent in the gut samples (see black arrows in Figure
2B).
The addition of PMSF did not lead to consistent improvement of the protein
profile. Indeed, proteins larger than 37 kD in TBPH (which included PMSF,
Figure 2A) are in lower abundance than in all other gutless termite samples,
whereas a 50 kD protein in sample TGP (with PMSF) is not visible in TGR (no
PMSF).
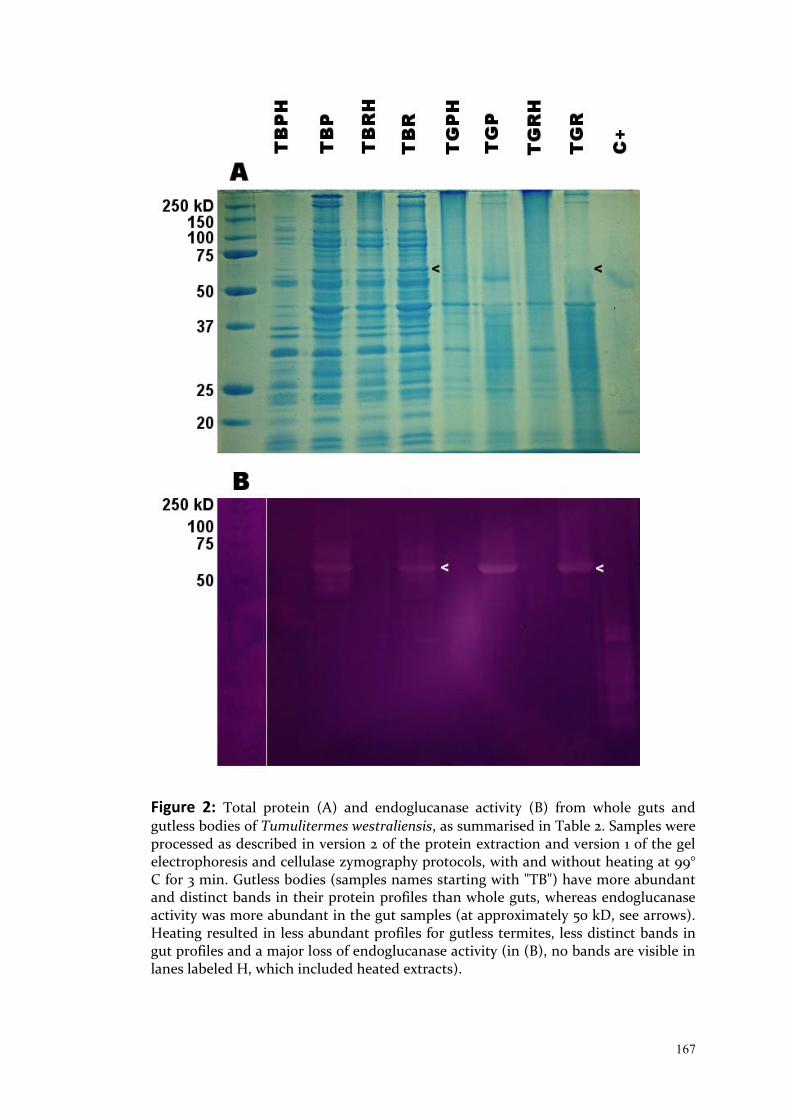
167
Figure 2: Total protein (A) and endoglucanase activity (B) from whole guts and gutless bodies of Tumulitermes westraliensis, as summarised in Table 2. Samples were processed as described in version 2 of the protein extraction and version 1 of the gel electrophoresis and cellulase zymography protocols, with and without heating at 99° C for 3 min. Gutless bodies (samples names starting with "TB") have more abundant and distinct bands in their protein profiles than whole guts, whereas endoglucanase activity was more abundant in the gut samples (at approximately 50 kD, see arrows). Heating resulted in less abundant profiles for gutless termites, less distinct bands in gut profiles and a major loss of endoglucanase activity (in (B), no bands are visible in lanes labeled H, which included heated extracts).

168
Conclusions
The comparative quality of the gutless body protein profiles indicated that the
gut extracts were impacted by greater protease activity, not suitably controlled
for by our chosen additives.
Heating samples prior to gel electrophoresis offered no benefits in the context
of this study and was removed from later protocols, in the interest of handling
samples at a minimum.
PMSF did not consistently offer benefits but should not in theory negatively
impact samples and was added in in later experiments as a precaution.
Comparison of total protein and cellulase profiles between species and gut
segments
Rationale
This experiment aimed to visualise T. westraliensis and A. obeuntis gut protein
profiles among different gut segments. The foregut/midgut primarily contains
endogenous enzymes, whereas the hindgut (P1-P5 sections) is dominated by
microbial enzymes (Brune, 2014). Sample sizes were increased to 20 gut
segments per sample, to account for the small gut segment sizes and increase
the amount of protein to visualise.
Methods
Twenty freshly collected T. westraliensis and A. obeuntis gut segments were
pooled for each treatment, as indicated in Table 3. Total protein and
endoglucanase activity (assay conducted in duplicate) was visualised on
acrylamide gels as per extraction PP2 and cellulase zymography. A positive
control for cellulase activity was included, made up of 1 mg/ml cellulase
(Aspergillus niger) in Laemmli sample buffer.

169
Table 3: Gut segment origin for comparison by total protein visualisation and zymography Sample name Species Gut segment
AFM Amitermes obeuntis Foregut/midgut
AH Amitermes obeuntis Hindgut
TFM Tumulitermes westraliensis Foregut/midgut
TH Tumulitermes westraliensis Hindgut
C+ Aspergillus niger _
Results
Separating the guts into foregut/midgut and hindgut (P1-P5) resulted in
distinct profiles for each gut section, which were similar for T. westraliensis
and A. obeuntis (Figure 3A). In both species the foregut/midgut profile is
smeared more heavily and contains few distinct bands, particularly at high
molecular weights.
The cellulase zymogram (Figure 3B) on the other hand identified clear
differences between the two species. A. obeuntis has two major activity bands
in each lane, either side of the 50 kD marker, which do not obviously
correspond with bands of a similar size on the total protein gel. There are
smaller bands (less than 37 kD) that may correspond to those seen in T.
westraliensis. T. westraliensis has a stronger activity in both gut segments,
making the number of bands difficult to determine, however major bands are
smaller than 50 kD.

170
Figure 3: Total protein (A) and endoglucanase activity (B; which includes 2 technical replicates for each treatment) from foregut/midgut and hindgut portions of Tumulitermes westraliensis and Amitermes obeuntis guts (see Table 3), visualised as described in version 2 of the protein extraction and version 1 of the gel electrophoresis and cellulase zymography protocols. T. westraliensis and A. obeuntis have similar total protein profiles, with smears in the foregut/midgut lanes. They have similar sized endoglucanases but these are more active in T. westraliensis.

171
Conclusions
I confirmed that separating guts into foregut/midgut and hindgut sections
provided distinct proteomes. The foregut/midgut section likely contains
greater concentrations of proteases that were not adequately prevented from
affecting the extracts.
Despite the apparent protein degradation, the cellulase zymogram identified
relatively distinct activity bands, specific to each species with endocellulase
activity being stronger in the foregut/midgut and in T. westraliensis, perhaps
due to its more sound and hence more recalcitrant diet. This indicates that the
zymogram method is very sensitive but results likely underestimate the level
of activity in the gut.
Termite cellulase extraction and visualisation using cellulase-binding probes
Rationale
This experiment aimed to replicate the crude and pellet extracts protocol from
Tokuda et al. (2005) to further differentiate microbial proteins as either
secreted or cell bound. A cellulase-binding probe method was also tested to
highlight potential cellulases. Probe GH2d-ABP (Chauvigné-Hines et al., 2012)
was chosen for its affinity to a wide range of GH families and because it was
used by the authors for a subsequent study (Anderson et al., 2013).
Methods
Two replicates of 30 guts each from freshly collected A. obeuntis were divided
into the midgut, the P1 and the P3. Crude and pellet extracts were obtained
(PE3), as summarised in Table 4. CP1 binding and gel electrophoresis protocols
were followed. Aliquots of each enzyme extract were stored following protein
extraction, protein precipitation or following probe binding and precipitation.
Precipitation was deemed required for this protocol to concentrate the
enzyme extracts.

172
Table 4: Sample sources and extract types for cellulase-binding probe test
Sample name
Species Gut segment Enzyme extract
AMC1 Amitermes obeuntis
Midgut Crude
AH1C1 Amitermes obeuntis
P1
Crude
AH3C1 Amitermes obeuntis
P3 Crude
AMC2 Amitermes obeuntis
Midgut Crude
AH1C2 Amitermes obeuntis
P1
Crude
AH3C2 Amitermes obeuntis
P3 Crude
AMPe1 Amitermes obeuntis
Midgut Pellet
AH1Pe1 Amitermes obeuntis
P1
Pellet
AH3Pe1 Amitermes obeuntis
P3 Pellet
AMPe2 Amitermes obeuntis
Midgut Pellet
AH1Pe2 Amitermes obeuntis
P1
Pellet
AH3Pe2 Amitermes obeuntis
P3 Pellet
C+ Aspergillus niger N/A Commercial preparation (soluble extract)

173
Results
The extracts had very similar total protein profiles before and after
precipitation (Figure 4A and B), although the bands look more concentrated
in B. Aliquots of extracts incubated with GH2d-ABP (Figure 4C) have a
different total protein profile after compared to prior the incubation. Select
bands are more visible than they were prior to the incubation, while others
disappear. For example, there is a group of three bands between 37 and 50 kD
in AH3C1 and AH3C2 in Figure 4A that almost entirely disappear in C
(indicated by black brackets in Figure 4A and 4C). Two bands level with these
appear in AMC1 and AMC2 in C (white brackets), as do two thick bands
smaller than 37 kD in the Aspergillus niger control (grey brackets).
The P1 hindgut segment lanes contained only small amounts of protein
(Figure 4A, B and C). The midgut crude extract lanes contained very few
proteins larger than 50 kD. Smearing suggests this may be evidence of protein
degradation. Many of the wells and lanes of the stacking gels can also be seen
to contain (likely aggregated) protein in Figure 4A, B and C.
Fluorescent labelling of proteins bound to GH2d-ABP was largely unsuccessful
with most of the fluorescence detected at the very top or bottom of the gel, as
well as a small amount bound to the largest band featuring in the positive
control, between 50 and 75 kD.
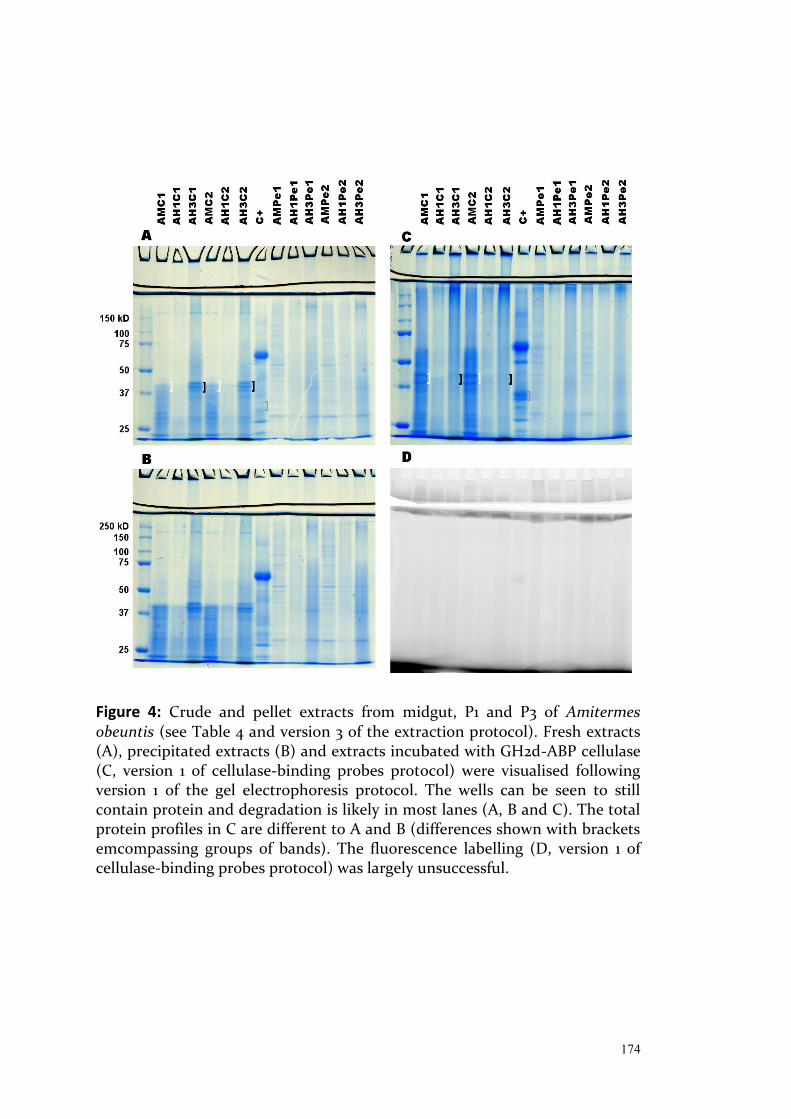
174
Figure 4: Crude and pellet extracts from midgut, P1 and P3 of Amitermes obeuntis (see Table 4 and version 3 of the extraction protocol). Fresh extracts (A), precipitated extracts (B) and extracts incubated with GH2d-ABP cellulase (C, version 1 of cellulase-binding probes protocol) were visualised following version 1 of the gel electrophoresis protocol. The wells can be seen to still contain protein and degradation is likely in most lanes (A, B and C). The total protein profiles in C are different to A and B (differences shown with brackets emcompassing groups of bands). The fluorescence labelling (D, version 1 of cellulase-binding probes protocol) was largely unsuccessful.

175
Conclusions
Many of the wells and lanes of the stacking gels were found to contain protein,
suggesting aggregation. Further additives such as non-detergent
sulphobetaines (NSDB) may assist in solubilisation of these proteins.
The P1 hindgut segment lanes contained only small amounts of protein and
are deemed of lesser interest, at least during the optimisation stages of these
methods.
Fluorescent binding was unsuccessful and the presence of EDTA and DTT
(chelating agents; Krȩżel et al., 2001) in the resuspension buffer likely
inhibited click chemistry with the fluorescent probes by making the required
copper catalyst inert. Removal of these agents may result in further protein
degradation.
Determining the effect of temperature on the use of cellulase-binding probes
Rationale
Non-detergent sulphobetaines (NSDB) have been shown to have a protective
effect against protein denaturation while assisting in protein solubilisation
(Vuillard et al., 1995). Due to persistent issues with potential protein
degradation and aggregation, the addition of the 3-(1-Pyridinio)-1-
propanesulfonate (NDSB201, Sigma Aldritch) and the precipitation of samples
using chloroform/methanol, better suited to membrane-associated proteins
(Wessel and Flügge, 1984) were tested.
Due to the likely effect of EDTA on the click chemistry step, the resuspension
buffer was modified to include 1 × Halt™ Protease and Phosphatase Inhibitor
Cocktail (ThermoFisher Scientific), EDTA-free.

176
In the interest of limiting protein degradation, this experiment aimed to
determine whether the probe binding conditions described by Chauvigné-
Hines et al. (2012) could be conducted in shorter and cooler conditions.
Methods
Cellulase from Aspergillus niger only was used in this experiment and the
temperature and duration of probe binding modulated, as summarised in
Table 5 (10 combinations were chosen to fill a 20 well gel with replication,
starting with the 3 hour, 60°C step described by Chauvigné-Hines et al.). Each
reaction contained 0.2 mg cellulase in 50 mM HEPES, 1M NDSB201 and 1 ×
Halt™ Protease and Phosphatase Inhibitor Cocktail (EDTA-free) and was
labelled with 2 μl of 75 µM GH2d-ABP. Samples were precipitated using PP2
and visualised using CP1.
Table 5: Treatments to find range of successful probe binding to cellulase positive control
Time Temperature (°C) 30 min 1 hr 2 hr 3 hr 30 - - - √ 40 √ √ √ √ 50 √ √ √ √ 60 √ √ √ √ Results
The total protein profile of cellulase from Aspergillus niger was visibly
impacted with increasing temperature, both at 50 and 60° C (Figure 5A and C).
The effect worsened with increasing time at 50° C and was constantly poor at
60° C. Treatments at 40° C did not decline visibly. GH2d-ABP binding
occurred at all temperatures and durations, with the fluorescent profiles
largely matching the Coomassie staining (Figure 5B and D).
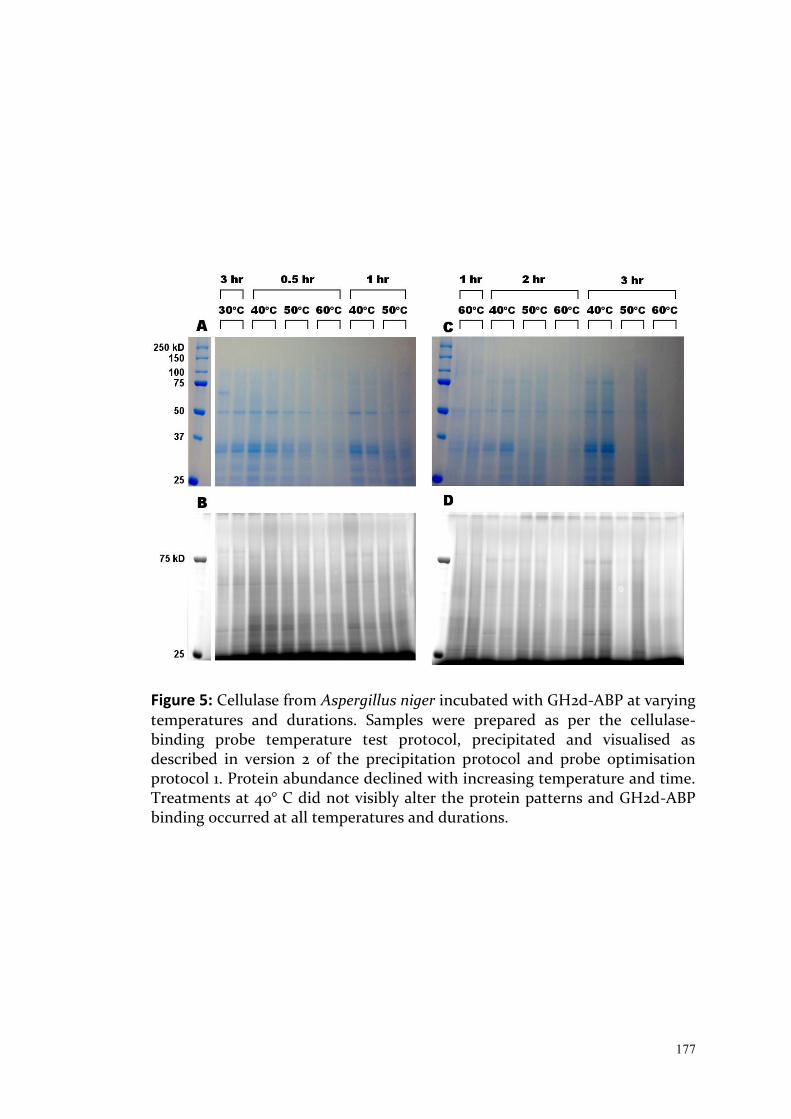
177
Figure 5: Cellulase from Aspergillus niger incubated with GH2d-ABP at varying temperatures and durations. Samples were prepared as per the cellulase-binding probe temperature test protocol, precipitated and visualised as described in version 2 of the precipitation protocol and probe optimisation protocol 1. Protein abundance declined with increasing temperature and time. Treatments at 40° C did not visibly alter the protein patterns and GH2d-ABP binding occurred at all temperatures and durations.

178
Conclusions
GH2d-ABP binding occurred at all temperatures and durations, however the
quality of the protein profile decreased with increasing temperature.
Since no difference was noticed between 30 and 40° C, a further experiment
trialed binding at lower temperatures (data not shown). Binding was
successful at 4° C for 30 min, which may allow the maintenance of protein
quality during the experiment.
During this follow up experiment, issues were encountered with the
precipitation of the detergent (SDS) at 4° C. The replacement of SDS with
Triton x-100 was then tested to rectify this.
Finalised cellulase-binding probe protocol
Rationale
Based on the results obtained from the previous experiments, a final protocol
was developed for binding of the GH2d-ABP cellulase-binding probe,
involving Triton x-100 instead of SDS (to prevent precipitation at cold
temperatures) and probe binding at 4° C for 30 min.
Methods
This final protocol was trialled with both T. westraliensis and A. obeuntis over
several days. Up to three replicates with 30 gut segments were used, based on
available harvested material and volume recovered for the pellet extract, as
indicated in Table 6. It involved PE4, CP2 and associated precipitation and gel
electrophoresis protocols.
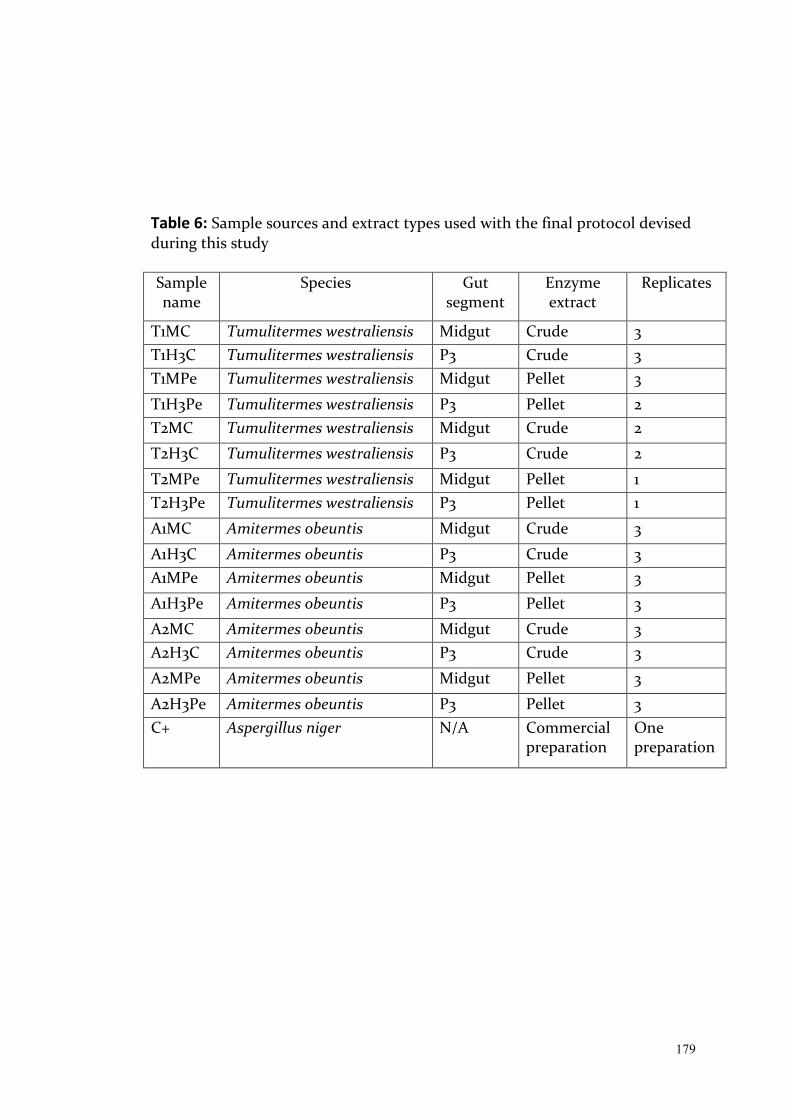
179
Table 6: Sample sources and extract types used with the final protocol devised during this study Sample name
Species Gut segment
Enzyme extract
Replicates
T1MC Tumulitermes westraliensis Midgut Crude 3 T1H3C Tumulitermes westraliensis P3 Crude 3 T1MPe Tumulitermes westraliensis Midgut Pellet 3
T1H3Pe Tumulitermes westraliensis P3 Pellet 2 T2MC Tumulitermes westraliensis Midgut Crude 2
T2H3C Tumulitermes westraliensis P3 Crude 2
T2MPe Tumulitermes westraliensis Midgut Pellet 1 T2H3Pe Tumulitermes westraliensis P3 Pellet 1
A1MC Amitermes obeuntis Midgut Crude 3
A1H3C Amitermes obeuntis P3 Crude 3 A1MPe Amitermes obeuntis Midgut Pellet 3
A1H3Pe Amitermes obeuntis P3 Pellet 3
A2MC Amitermes obeuntis Midgut Crude 3 A2H3C Amitermes obeuntis P3 Crude 3
A2MPe Amitermes obeuntis Midgut Pellet 3
A2H3Pe Amitermes obeuntis P3 Pellet 3 C+ Aspergillus niger N/A Commercial
preparation One preparation

180
Results
The P3 hindgut segment of Tumulitermes westraliensis contained large
amounts of protein, resulting in an overload of proteins in these lanes. The
fluorescent profiles all contain distinct bands, except for T2H3Pe (E). Across
the two T. westraliensis colonies (Figure 6), the fluorescent profiles (Figure 6B
and E) are more consistent than their associated Coomassie stained profiles (A
and D). Two bands between 75 and 100 kD in T1H3C (A) are not present in
T2H3C (D) and not visible in either under fluorescence. On the other hand,
two bands between 50 and 75 kD in P3 crude extracts in B and E are not clearly
seen in A and D. In the midgut, one band around 25 kD is much more defined
under fluorescence in T1MC (B) and T2MC (E). The same is true of a 50 kD
band in T1MPe (B) and T2MPe (E).
Endoglucanase activity was detected in both gut segments (Figure 6C and F).
Most of the activity was detected in crude extracts and in particular a band
was recorded at 50 kD in at least the P3 crude extract and the midgut pellet
extract. It is not possible to confirm the presence of this band in the midgut
crude extract due to the intense activity seen in this gut segment. Both crude
extracts contain proteins smaller than 50 kD and the hindgut pellet extract
contains a small amount of activity just above 50 kD.
There are proteins potentially responsible for this activity visualised in Figure
6B and E, particularly a distinct band slightly larger than 50 kD in T1H3Pe. In
many cases it is very difficult to match bands across gels, due to the large
number of weak bands and the appearance of the molecular weight marker
under fluorescence and on the cellulase zymogram.
In both Amitermes obeuntis colonies (Figure 7), very few bands were detected
larger than 37 kD in all gut segments both with Coomassie staining and under
fluorescence. In contrast, all of the endoglucanase activity detected was from
proteins larger than 37 kD, around 50 kD (Figure 7C and F).

181
Figure 6: Total protein profiles (A, D) and endoglucanase activity (C, F, cellulase zymography protocol 1) of the crude and pellet extracts from the midgut and P3 of two Tumulitermes westraliensis colonies (see Table 6) recovered as per extraction protocol version 4. (B) and (E) show the fluorescent labelling of GH2d-ABP, as described in probe binding protocol 2 (and associated precipitation and gel electrophoresis protocols).

182
Figure 7: Total protein profiles (A, D) and endoglucanase activity (C, F, cellulase zymography protocol 1) of the crude and pellet extracts from the midgut and P3 of two Amitermes obeuntis colonies (see Table 6) recovered as per extraction protocol version 4. (B) and (E) show the fluorescent labelling of GH2d-ABP, as described in probe binding protocol 2 (and associated precipitation and gel electrophoresis protocols).

183
The fluorescent profile of the second colony (E) was more consistent across
replicates than the first (B), particularly between 25 and 37 kD where two
proteins vary dramatically in abundance.
Conclusions
Concern over the small size of gut segments and protein degradation resulted
in overloading of certain lanes, although the fluorescent probe resulted in
more distinct profiles. These profiles differed slightly from the Coomassie
stained versions, indicating limited specificity of the binding.
In both termite species, more endoglucanase activity was detected in the
midgut, particularly in the crude extracts. This midgut crude extract most
likely represents termite-derived endocellulases and distinct bands from the
hindgut indicate the presence of microbial endoglucanases.
Discussion:
The set of experiments presented in this chapter aimed to apply a cellulase
zymography method, as well as adapt the use of cellulase-binding probe
GH2d-ABP (Chauvigné-Hines et al., 2012) to the Western Australian termite
species Tumulitermes westraliensis and Amitermes obeuntis. The goal is to
ultimately extract proteins of interest from acrylamide gels resulting from
both methods and to identify the cellulases of interest with mass
spectrometry. My work revealed that this aim was challenging to achieve with
high quality extracts, which may have been overlooked by previous studies if
total protein was not visualised (data not shown in previous studies), leading
to a potential underestimation of cellulase capabilities in termites. The issues
encountered are discussed below alongside recommendations for future
studies.

184
The cellulase zymography method was successfully applied to both termite
species, however I found that protein bands could not be reliably excised from
the zymogram or the corresponding Coomassie stained gel. The difficult to
match the activity bands to protein bands was based on three reasons: 1) the
sites of activity overlapped with multiple protein bands; 2) endoglucanase
activity was observed at higher molecular weights than where most visible
protein bands appeared and 3) the molecular weight marker was not always
visible (and did not migrate equally) on the cellulose-containing gel. The high
sensitivity of the enzyme activity staining reaction meant that too low a
concentration of proteins was added to allow elution from the zymogram. One
possible solution may be to heat the samples to partially denature the
endoglucanases, decreasing their activity and allowing more protein to be
loaded. Schwarz et al. (1987) showed that heating clostridial cellulases for 10
min at 70-80° C in the presence of SDS resulted in partial denaturation and
more distinct zymograms. Oppert et al. (2010) heated extracts from various
insects at 70° C for 20 min, and observed reduced smearing as well. They also
reported not detecting protein bands in corresponding Coomassie-stained
gels. In the current study, heating at 99° C for 3 min irreversibly denatured the
cellulases, whereas cellulase activity from marine shipworm bacteria was not
destroyed in harsher conditions (10 min boiling in SDS and ß-
mercaptoethanol; Imam et al., 1993). Further testing is required to optimise
the partial denaturation of the endoglucanases of T. westraliensis and A.
obeuntis in a method similar to that described when testing probe binding
across a temperature range.
Further to point 2) above, it is not clear if endoglucanases were present in
small abundance in the biological samples, or were partially lost during
sample manipulations. Protocol optimisation aimed to minimise protein
aggregation and degradation, but no consistent improvements were achieved
with the addition of protease and phosphatase inhibitor cocktails, PMSF,
NDSB21, Triton x-100, or with heating or chloroform-methanol precipitation.
It could be that not all proteases were inhibited. For example,

185
metalloproteases have been described in termite guts (Sethi et al., 2011; Lima
et al., 2014) and would have still been active following precipitation in
extraction PE4, as EDTA was omitted due to its potential interaction with
fluorescent labelling of the probes. There was no difference in the quality of
the protein profiles following the removal of EDTA with precipitation, but the
potential for other non-inhibited proteases remains. Measurements of
protease activity following incubation of enzyme extracts with several class-
specific inhibitors (Lima et al., 2014) should be undertaken with each termite
species to determine what could cause protein degradation, prior to
investigating the cellulase enzymes.
The zymography method used in this study relied on enzymes renaturing after
migration on a denaturing gel (Schwarz et al., 1987). So the second possible
explanation for the inconsistent protein profiles is that protein degradation is
controlled throughout sample processing, but proteases were able to renature
on the acrylamide gel itself. This is supported by the fact that degradation
seemed to occur primarily in the top half of the gel and always in the midgut
(and occasionally in other gut segments, especially for A. obeuntis). SDS-
resistant proteases have previously been reported and in particular Blumentals
(1990) reported smearing in half of a protease zymogram, caused by a 66 kD
serine protease. In this study, heating at 99° C for 3 min did not seem to
inhibit the proteases, while irreversibly denaturing the cellulases.
Nasutitermes corniger worker proteases have been shown to remain active
following heating at 90° C for 30 min (Lima et al., 2014), suggesting that
heating alone may not be sufficient. Optimising conditions that will
irreversibly denature all enzyme activity in termite guts would allow for a
complete protein profile recovery.
Zymograms showed endoglucanase activity ranged from less than 37 to 50 kD
in T. westraliensis and between 37 and 60 kD in A. obeuntis. Activity bands
ranging from 24 to 40 kD have been reported in a wide range of insect gut
fluids, including the termite Reticulitermes hageni Banks (Oppert et al., 2010).

186
Endoglucanases smaller than 50 kD were reported in the midguts of higher
termites Nasutitermes takasagoensis, N. walkeri and Macrotermes barneyi
(Tokuda and Watanabe, 2007; Wu et al., 2012; Ni et al., 2014). Larger
endoglucanases were reported in the hindguts of N. takasagoensis and N.
walkeri with bands ranging from less than 50 to 120 kD (Tokuda and
Watanabe, 2007). Endoglucanases described in this study are well aligned with
previous findings, particularly in the midgut. Decreasing the protein load or
partial denaturation of the enzymes to limit smearing would confirm the
presence or absence of larger endoglucanases in the hindgut.
Chauvigné-Hines et al. (2012) prescribed harsh conditions of 60° C for 3 hr for
the probe binding. It was shown that probe binding could occur at 4° C over
30 min, to maximise protein quality. A large number of proteins were found to
bind to GH2d-ABP, regardless of the conditions. GHs exist in a wide range of
sizes, from 20 to over 100 kD, if they exist in aggregates (Stütz and Wrodnigg,
2011). The GH profiles obtained in the study could be due to a wide range of
cellulases and other GHs in termite guts, or due to a lack of probe specificity.
This could be determined by adapting the biotin click-chemistry method
described by Chauvigné-Hines et al. (2012) to termite gut extracts to obtain a
proteome and check whether it is primarily made up of GHs.
In summary, I concluded from my experiments that the integrity of the
proteins extracted is the first obstacle that will need to be addressed in the
future to study cellulase activity from termite gut extracts. Active proteases are
likely to result in an underestimated total cellulase activity. In particular, in-
gel assays may underestimate or not detect cellulases that are of similar mass
to proteases that remain active. Optimising a method to partially denature the
endoglucanase activity for more distinct zymograms in combination with a
method to irreversibly denature all enzymes for complete protein profiles
would allow for the largest recovery for endoglucanases of interest. This
strategy is not appropriate for use with cellulase-binding probes, as GHs need
to be in an active form to bind with the probes. A better solution in this case is

187
to test for effective protease inhibitors and use probes with biotin reporter
groups, for protein enrichment and proteomics.
The overarching goal of this work was to identify enzymes of interest to
biofuel production in Western Australia and study their activity. By using
cellulase-binding probes, I sought to identify enzymes in numerous
unsequenced termites and bacterial genomes at once and to study their
abundance and localisation in real time (Stubbs, 2014). Genetic approaches
would then be required to obtain sequences for the most promising enzymes
(for example rapid amplification of cDNA ends or RACE; Ni et al., 2014), which
could then be cloned into culturable organisms (since most organisms in the
gut cannot be cultured; Small et al. 2012) and further tested. This path, if
protein quality can be successfully maintained, would allow targetting of the
most abundant and/or active enzymes, which are of most interest for biofuel
applications.

188
References: Anderson, L. N., Culley, D. E., Hofstad, B. A., Chauvigne-Hines, L. M., Zink, E. M.,
Purvine, S. O., Smith, R. D., Callister, S. J., Magnuson, J. M., Wright, A. T. (2013). Activity-based protein profiling of secreted cellulolytic enzyme activity dynamics in Trichoderma reesei QM6a, NG14, and RUT-C30. Molecular BioSystems, 9(12), 2992-3000.
Blumentals, I. I., Robinson, A. S., & Kelly, R. M. (1990). Characterization of sodium dodecyl sulfate-resistant proteolytic activity in the hyperthermophilic archaebacterium Pyrococcus furiosus. Applied and Environmental Microbiology, 56(7), 1992.
Brune, A. (2014). Symbiotic digestion of lignocellulose in termite guts. Nature Reviews Microbiology, 12(3), 168-180.
Chauvigné-Hines, L. M., Anderson, L. N., Weaver, H. M., Brown, J. N., Koech, P. K., Nicora, C. D., Hofstad, B. A., Smith, R. D., Wilkins, M. J., Callister, S. J., Wright, A. T. (2012). Suite of activity-based probes for cellulose-degrading enzymes. Journal of the American Chemical Society, 134(50), 20521-20532.
Coy, M. R., Salem, T. Z., Denton, J. S., Kovaleva, E. S., Liu, Z., Barber, D. S. et al. (2010). Phenol-oxidizing laccases from the termite gut. Insect Biochemistry and Molecular Biology, 40(10), 723-732.
Hogan, M., Veivers, P. C., Slaytor, M., & Czolij, R. T. (1988). The site of cellulose breakdown in higher termites (Nasutitermes walkeri and Nasutitermes exitiosus). Journal of Insect Physiology, 34(9), 891-899.
Imam, S. H., Greene, R. V., & Hockridge, M. E. (1993). Zymographic analyses of carboxymethylcellulases secreted by the bacterium from wood-boring marine shipworms. Biotechnology Techniques, 7(8), 579-584.
Krȩżel, A., Leśniak, W., Jeżowska-Bojczuk, M., Młynarz, P., Brasuñ, J., Kozłowski, H. et al. (2001). Coordination of heavy metals by dithiothreitol, a commonly used thiol group protectant. Journal of Inorganic Biochemistry, 84(1-2), 77-88.
Lima, T. A., Pontual, E. V., Dornelles, L. P., Amorim, P. K., Sá, R. A., Coelho, L. C. et al. (2014). Digestive enzymes from workers and soldiers of termite Nasutitermes corniger. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology, 176, 1-8.
McEwen, S. E., Slaytor, M., & O’Brien, R. W. (1980). Cellobiase activity in three species of Australian termites. Insect Biochemistry, 10(5), 563-567.
Mohanram, S., Amat, D., Choudhary, J., Arora, A., & Nain, L. (2013). Novel perspectives for evolving enzyme cocktails for lignocellulose hydrolysis in biorefineries. Sustainable Chemical Processes, 1(1), 15.
Nakashima, K., Watanabe, H., Saitoh, H., & Tokuda, G., Azuma, J.I. (2002). Dual cellulose-digesting system of the wood-feeding termite, Coptotermes formosanus Shiraki. Insect Biochemistry and Molecular Biology, 32(7), 777-784.
Ni, J., Wu, Y., Yun, C., Yu, M., & Shen, Y. (2014). cDNA cloning and heterologous expression of an endo-β-1,4-glucanase from the fungus-growing termite Macrotermes barneyi. Archives of Insect Biochemistry and Physiology, 86(3), 151-164.
O’Brien, G. W., Veivers, P. C., McEwen, S. E., Slaytor, M., & O’Brien, R. W. (1979). The origin and distribution of cellulase in the termites, Nasutitermes exitiosus and Coptotermes lacteus. Insect Biochemistry, 9(6), 619-625.
Oppert, C., Klingeman, W. E., Willis, J. D., Oppert, B., & Jurat-Fuentes, J. L. (2010). Prospecting for cellulolytic activity in insect digestive fluids. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology, 155(2),

189
145-154. Park, S. G., Kho, C. W., Cho, S., Lee, D. H., Kim, S. H., & Park, B. C. (2002). A
functional proteomic analysis of secreted fibrinolytic enzymes from Bacillus subtilis 168 using a combined method of two-dimensional gel electrophoresis and zymography. Proteomics, 2(2), 206-211.
Ransom-Jones, E., Jones, D. L., McCarthy, A. J., & McDonald, J. E. (2012). The Fibrobacteres: an important phylum of cellulose-degrading bacteria. Microbial Ecology, 63, 267-281.
Schulz, M. W., Slaytor, M., Hogan, M., & O’Brien, R. W. (1986). Components of cellulase from the higher termite, Nasutitermes walkeri. Insect Biochemistry, 16(6), 929-932.
Schwarz, W. H., Bronnenmeier, K., Gräbnitz, F., & Staudenbauer, W. L. (1987). Activity staining of cellulases in polyacrylamide gels containing mixed linkage β-glucans. Analytical Biochemistry, 164(1), 72-77.
Sethi, A., Slack, J. M., Kovaleva, E. S., Buchman, G. W., & Scharf, M. E. (2013). Lignin-associated metagene expression in a lignocellulose-digesting termite. Insect Biochemistry and Molecular Biology, 43, 91-101.
Sethi, A., Xue, Q. G., La Peyre, J. F., Delatte, J., & Husseneder, C. (2011). Dual origin of gut proteases in Formosan subterranean termites (Coptotermes formosanus Shiraki) (Isoptera: Rhinotermitidae). Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology, 159(3), 261-267.
Small, G., Hartke, T., Peacock, C., Baer B., & Sutton, D. (2012). Comparative study of two endemic Australian termite species and their symbiotic bacteria. University of Western Australia.
Stubbs, K. A. (2014). Activity-based proteomics probes for carbohydrate-processing enzymes: current trends and future outlook. Carbohydrate research, 390, 9-19.
Stütz, A. E., & Wrodnigg, T. M. (2011). Imino sugars and glycosyl hydrolases: historical context, current aspects, emerging trends. Advances in Carbohydrate Chemistry and Biochemistry, 66, 187-298.
Tarayre, C., Bauwens, J., Brasseur, C., Mattéotti, C., Millet, C., Guiot, P. A. et al. (2015). Isolation and cultivation of xylanolytic and cellulolytic Sarocladium kiliense and Trichoderma virens from the gut of the termite Reticulitermes santonensis. Environmental Science and Pollution Research (international), 22(6), 4369-4382.
Tokuda, G., Lo, N., & Watanabe, H. (2005). Marked variations in patterns of cellulase activity against crystalline‐vs. carboxymethyl‐cellulose in the digestive systems of diverse, wood‐feeding termites. Physiological Entomology, 30(4), 372-380.
Tokuda, G., & Watanabe, H. (2007). Hidden cellulases in termites: revision of an old hypothesis. Biology Letters, 3(3), 336-339.
Vuillard, L., Braun-Breton, C., & Rabilloud, T. (1995). Non-detergent sulphobetaines: a new class of mild solubilization agents for protein purification. Biochemical Journal, 305(Pt 1), 337-343.
Waeonukul, R. , Kya, K. L., Ratanakhanokchai, K. (2007). Multiple cellulases and xylanases from Bacillus circulans B6 during growth on avicel under aerobic condition. Thai Journal for Biotechnology, 8, 27-32.
Wessel, D., & Flügge, U. I. (1984). A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Analytical biochemistry, 138(1), 141-143.
Willis, J. D., Klingeman, W. E., Oppert, C., Oppert, B., & Jurat-Fuentes, J. L. (2010). Characterization of cellulolytic activity from digestive fluids of Dissosteira carolina (Orthoptera: Acrididae). Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology, 157(3), 267-272.

190
Wilson, D. B. (2011). Microbial diversity of cellulose hydrolysis. Current Opinion in Microbiology, 14(3), 259-263.
Wu, Y., Chi, S., Yun, C., Shen, Y., Tokuda, G., Ni, J. (2012). Molecular cloning and characterization of an endogenous digestive β-glucosidase from the midgut of the fungus-growing termite Macrotermes barneyi. Insect Molecular Biology, 21(6), 604-614.

191
CHAPTER 6: General Discussion

192
As I covered in detail in Chapter 1, higher termites are a promising source of
enzymes or microbes for the production of second-generation lignocellulosic
biofuel, since they efficiently degrade various forms of lignocellulose and have
optimised their primarily bacterial cellulose-degrading consortia over
evolutionary time. In this thesis, I have investigated influences on the gut
community over shorter time scales, to determine whether certain taxa in the
bacterial gut population of endemic higher termites (Termitidae) may warrant
further study as part of optimised biofuel production from wheat crop residue
in Western Australia.
To my knowledge, I provide the first characterisation of gut communities of
three endemic Western Australian termites with broad diets, Tumulitermes
westraliensis, Amitermes obeuntis and Coptotermes acinaciformis raffrayi, as
well as propose standardised techniques for the field of termite gut
microbiology. The findings, implications and limitations of my project are
described below under each of the four research questions, in the context of
improving biofuel production, followed by a discussion on the contributions of
this thesis to the broader understanding of microbial gut communities.
A: How much intraspecific variation exists in the gut communities of
Western Australian endemic termites?
Boucias et al. (2013) and Benjamino and Graf (2016) previously demonstrated
that gut communities of the lower termite Reticulitermes flavipes from the
same colony are more similar to each other than to the gut flora of other
colonies. Reid et al. (2014) found that colonies of the lower termite Stolotermes
ruficeps have unique gut communities, even when collected from colonies in
close proximity to each other, and speculated that differences in diet, such as
wood species or humification state, might explain these differences. In
Chapter 2, I extended these results to two endemic Western Australian
termites, the lower termite C. acinaciformis raffrayi and the higher termite T.

193
westraliensis. I found that colonies of both higher and lower termites have
signature gut communities and suggested that colony differences may be
explained by the complexity of their diets.
There is a correlation between species richness in the guts of higher termites
and the point on the humification gradient at which they feed, with the
highest species richness and diversity in the guts of humus and soil feeders. I
found that the T. westraliensis gut community shared similarities with wood,
grass, and litter feeders, indicating that its diet may be more complex and less
specialised than previously reported by Hill (1942). The gut community of
lower termites is also shaped by the presence of cellulose-degrading protists
that provide unique niches for bacteria (Brune and Dietrich, 2015). My work
uncovered a difference in the abundance of protist-associated bacteria
between colonies of C. acinaciformis raffrayi, suggesting that colony
differences might in part be driven by different abundances of gut protists.
Because one colony was sampled from rotten wood and the other from sound
wood, protist numbers could correlate with humification level of the diet,
although this needs to be tested on a larger dataset. Overall, C. acinaciformis'
gut community composition and structure is similar to other lower termites
that eat various species of wood at different stages of decomposition. I
concluded that locally available substrates may drive gut community structure
and composition at the colony level and that the complexity of a diet, i.e.
number of different food sources, may have a larger impact on gut community
composition and structure in termites than the plant species or humification
level alone. These findings support the idea that the gut community can be
manipulated in the search for optimised consortia to degrade substrates of
interest.
Chapter 2 also highlighted the necessity for extensive sampling to account for
variability of gut communities within a species and to further understand the
drivers of gut microbial community structure and composition in termites. To
that end, Chapter 3 included increased sampling of two higher termites

194
species, T. westraliensis and A. obeuntis, to extend the findings of intraspecific
variation. In particular, Chapter 3 confirmed the finding of intercolony
differences and added findings of caste differences and effects of location and
diet on gut communities. Within colony differences have previously been
investigated between castes of the lower termites C. formosanus (Xiang et al.,
2012) and R. flavipes (Benjamino and Graf, 2016) and between worker age
groups in the higher fungus-cultivating termites Macrotermes gilvus (Hongoh
et al., 2006) and Odontotermes formosanus (Li et al., 2016). Here, caste
differences were described in T. westraliensis and A. obeuntis. Young workers
of T. westraliensis were found to harbour significantly different gut
communities than older workers, the first such reported case in a non fungus-
cultivating species.
In my work, the location of a colony had a strong effect on the gut community
of both termite species and these differences remained significant even at low
sampling intensities. The size of the core community was found to decrease as
sampling intensity increased from single colony, to single location to multiple
locations, in comparison to the variability expected from simple random
sampling. In T. westraliensis, significant differences in gut community
composition were also found between workers collected from the same or
different locations. Hongoh et al. (2005) also found significant differences
between certain sampling sites in the genera Reticulitermes (lower termites)
and Microcerotermes (higher termites). I concluded that the effect of location
is based on various factors that make a location unique, perhaps soil
properties, available food sources, or presence of heterospecifics and their
respective microbial communities.
Chapter 3 also reported the presence of Alphaproteobacteria order
Rickettsiales in most guts of the higher termite A. obeuntis. This group are
known symbionts of protists (Szokoli et al., 2016), suggesting by extrapolation
the presence of their protist hosts. Protists are well characterised in lower
termites and key to their success (Ohkuma and Brune, 2011), while they have

195
been rarely reported in higher termites. Chapter 4 confirmed that at least
some individual T. westraliensis and A. obeuntis workers harbour live
flagellated protists in their guts at certain times. This and other reports of
inconsistent protist presence in guts of higher termites (Cleveland, 1923; Kirby,
1927; Abdul Rahman et al., 2015) suggest that they are likely to be transient,
taken up seasonally from the environment, rather than co-evolving with the
host as in lower termites (Tai et al., 2015; Poinar, 2009). Indeed, soil moisture
content has been found to determine the structure of soil protist communities
(Bates et al., 2013; Adl et al., 2014) and preliminary observations in Chapter 4
suggest they may be taken up following rainfall. This may have a seasonal
effect on the bacterial community and lead to further intraspecific variation. A
more systematic and high throughput molecular approach is needed to
determine the prevalence of protists in the guts of higher termites. As there is
no universal barcoding primer pair currently available for protists, a
combination of primers or a two-step barcoding approach with universal
eukaryotic primers should be used to investigate the presence and diversity of
protists in higher termites (Pawlowski et al., 2012; Adl et al., 2014).
Alongside biological variation, Chapter 2 described the influence of the 16S
rRNA amplication strategy on estimates of gut community composition and
structure in termites. Certain bacterial groups were only detected with one
primer pair or the other, however the V3-V4 method yielded a higher species
richness than the V4 method tested at the same depth. This increased species
diversity could be explained by the lower annealing temperature
recommended with the V3-V4 primer pair by Köhler et al. (2012), which has
been shown to limit some of the bias associated with PCR (Sipos et al., 2007). I
therefore recommend that a standard approach be applied using the primers
343Fmod and 784Rmod designed by (Köhler et al., 2012) to target the V3-V4
region of the 16S rRNA gene, along with a low annealing temperature of 48°C.
This method has already been used on the widest range of termite species
(across all families and many feeding groups) in the literature to date, allowing
broader comparisons (e.g. Bourgignon et al. 2018). If used more broadly, this

196
approach would maximise data reproducibility and utility across research
groups.
B: How should the core community of a termite species be defined and
accurately estimated?
The identification of the core community of a habitat is important, because
those taxa are critical to the functioning of that community (Shade and
Handelsman, 2012). I chose to define the core community as the list of
Operational Taxonomic Units (OTUs) clustered at 97% similarity and present
in 100% of samples in the grouping of interest. One limitation of this approach
is the arbitrary threshold of 97% similarity. Although frequently chosen to
represent a bacterial species, it is not necessarily the best approximation of the
number of bacterial species actually present (Nguyen et al., 2016). Indeed, it
has been shown that even bacteria with identical 16S sequences could
represent multiple species (Antony-Babu et al., 2017) and that many bacterial
species harbour multiple copies of the 16S rRNA gene, which may not all be
identical (Větrovský and Baldrian, 2013). This has implications for the
conclusions drawn about relative abundances of taxa in the gut, as well as
species richness estimations and by extension the size of the core community.
Despite this, low numbers of OTUs were shared across species as compared to
within a species, suggesting that this method is still useful in the investigation
of the factors shaping the presence of OTUs, if not bacterial species, in the gut.
In spite of these limitations, I chose the 97% similarity OTU clustering cut-off
because it has been used in the majority of termite gut meta-barcoding studies
to date (e.g. Otani et al. 2014, Abdul Rahman et al., 2015, Tai et al. 2015,
Benjamino and Graf, 2016, Mikaelyan et al., 2017). In the future, comparing the
clustering of OTUs at 97% to 99% or even 100% is warranted, as
recommended by multiple studies (Nguyen et al., 2016; De Filippis et al., 2018;
Edgar, 2018). As the accessibility of single-molecule real-time sequencing (e.g.
Pacific Biosciences, Oxford Nanopore) or Whole Genome Shotgun Sequencing

197
(WGS) increases, bacterial species can be better distinguished (Franzén et al.,
2015; Ranjan et al., 2016). The caveat is that the datasets would have a different
set of biases than when obtained with V3-V4 primers and Illumina sequencing
(Ross et al., 2013), making comparisons with existing studies more difficult.
The definition of a core community has differed substantially between termite
microbiome studies (Huang et al., 2013; Dietrich et al., 2014; Otani et al., 2014;
Reid et al., 2014; Abdul Rahman et al., 2015, Benjamino and Graf, 2016; Su et al.,
2016; Wang et al., 2016). The caste composition of samples (scope) was usually
limited to workers, the geographic distribution of sampling sites (scale) in
most cases restricted to a single location or small portion of the species known
range, and the number of samples taken per unit of interest (sampling
intensity) varied widely (see Chapter 3 for further details). The recovery cut-
offs for inclusion of a bacterial OTU in the core community varied between
40-100% of samples in these studies. To test the effects of modifying sampling
intensity, scale and scope, a stringent cut-off of 100% for the inclusion of
OTUs in the core community was chosen. Some taxa that are in fact members
of the core community may not have been included if they were below
detection in certain or all samples with the even sequencing depth of 11,400
reads achieved in Chapter 3. However, false positives were excluded by
ensuring that taxa were shared in all samples. The field as a whole would
benefit from a standardised definition of the core community and methods for
its determination and my aim in Chapter 3 was to propose a way forward.
I hypothesised that sampling scale, scope and intensity would all affect core
community estimations. I found that sampling intensity had the largest effect
on the outcome, which is fortunately the easiest factor for researchers to
control. Scale and scope also had significant effects and hence, all three form
important considerations when designing a study. I have shown that a
minimum of 20 samples but ideally 30 per factor of interest should be included
in any core community calculation, in order to draw statistically meaningful
conclusions. I developed a simple random sampling method which future

198
researchers can use on their own data sets to test whether sufficient sampling
intensity has been reached. The many factors contributing to intraspecific
variation (Research Question A, above) highlight how previous studies relying
on samples from a single colony or location have likely overestimated the
number of taxa in the termite gut core community. I therefore recommend
that future studies include samples from the maximum relevant range to
minimise the possibility of spurious results due to unintended location effects.
I found that increasing scope by including multiple castes decreased core
community size but changing the proportion of samples from each caste had
no effect. Hence all relevant castes should be included, even if very few
samples are available for some castes. In practice, this means sampling only
workers if the question is exclusive to them or using all castes for a species
core estimation.
Including all castes in a core community calculation should better represent
the vertically transmitted gut microbiome passed from parent colonies to new
colonies via the alates (founding queen and king), and shared from the
founders to all future colony members by the workers. It should be noted that
soldiers are effectively a dead end, as their gut community is not shared with
others (Noirot and Darlington, 2000). I chose a cautious and more stringent
approach by including them since soldier guts are also inoculated via the
workers (Diouf et al., 2015) and the conditions in soldier guts may foster taxa
that are present but less abundant in workers. I found a core community
shared by all castes, made up of 56 and 116 OTUs for T. westraliensis and A.
obeuntis respectively. In T. westraliensis I had sufficient samples to show that
there is a community of 14 OTUs exclusive and universal to the reproductive
castes (nymphs and alates). Because of the alates' role in vertical transmission,
I would expect the alate gut community to most closely resemble the species
core community. This suggests that alate-exclusive OTUs are likely be present
at low levels in other castes. I typically recovered a smaller number of OTUs
from alates in both termite species than from other castes, but Diouf et al.
(2018) found a higher species diversity in the swarming alate gut compared to

199
any other caste of Nasutitermes arborum. It has been suggested that alates
purge their gut prior to flight (Nutting, 1969), which could result in lower,
more even abundances of gut bacteria and recovery of a greater number of
OTUs at the same sequencing depth. In the future, comparisons of pre- and
post-flight alates will reveal whether they have distinct gut communities and
may provide insights into pre-swarming behaviour.
Taxa exclusive to a location or feeding group are likely to be environmentally
acquired, ingested with food or soil. The most abundant taxa in most core
communities have multiple functions, with cellulose degradation being the
most common function predicted in both species. Taxa exclusive but universal
to termites consuming a diet of interest may degrade it most efficiently, or
more effectively counter the plant's defensive mechanisms (Bennett and
Allsgrove, 1994; Sugio et al., 2015). Such a list is obtained by calculating the
core community for a group feeding on the substrate of interest (including at
least 20 samples, as shown above) and excluding taxa found in the core
community of a larger group. For example, this larger group may be the entire
species (species core community) or termites sampled across multiple feeding
groups, as relevant. These exclusive taxa may in turn be a source of enzymes
well suited to breaking down the substrate of choice and hence have industrial
applications.
C: Can the gut community of local higher termite species be modified in a
field-based feeding experiment?
Feeding experiments have most commonly been conducted with lower
termites and in laboratory conditions (e.g. Tanaka et al., 2006; Husseneder et
al., 2009; Huang et al., 2013), where several reported that altering diet
components or feeding different plant species has been found to affect the gut
community. Miyata et al. (2007) showed that altering the components of the
diet of the higher termite N. takasagoensis in laboratory conditions affected

200
both the composition and structure of the gut microbiota. Wang et al. (2016)
found that the gut community of the wood-feeding higher termite
Mironasutitermes shangchengensis reverted to its original state after feeding
on corn stalks and filter paper for 10 days and noted poor survival of higher
termites in laboratory conditions (with a maximum of 10 days). Preliminary
work conducted during my PhD in which T. westraliensis, A. obeuntis and
Occasitermes occasus were kept under controlled laboratory conditions met
with inconsistent success; survival ranged from a minimum of one day in T.
westraliensis to a maximum of around one month in all species. Activity of the
termites declined over this time and presence of mites increased. Presence of
mites has been linked to deteriorating nest conditions and decreased activity
of termites in other species, including Cryptotermes secundus (Korb & Fuchs,
2006). Due to the limitations of laboratory-based experiments in higher
termites, a field-based study was conducted and described in Chapter 3.
The field study's primary benefit was to minimise the impact of experimental
procedures on the termites, which, in laboratory conditions, could have
resulted in changes in the gut community beyond those induced simply by a
change in diet. A three-week field study resulted in a significant effect of
multiple diet treatments on the gut communities of both species. These
effects, including significant differences between the control diet and lucerne
or wheat-fed termites, could only be detected at greater sampling intensities.
While underscoring the importance of adequate replication in gut microbiome
studies (Research Question B, above), this result demonstrates that
manipulating the gut community under field conditions is possible, potentially
allowing the identification of key organisms or enzymes relevant to breaking
down substrates of interest without relocation and artificial conditions.
However, there are two main limitations to a field-based study. I could not
differentiate between transient and resident gut members (Waidele et al.,
2017) and importantly, confining the termites to the provided food source
remains difficult. In this study, I could not guarantee that the change was

201
solely caused by the provided diets rather than the relocation of natural food
sources during the experimental setup. Future studies should include
additional control colonies that remain undisturbed throughout the
experiment, isolated enough to guarantee they are not affected by the
substrates available at other mounds. Further optimisation of methods to
characterise the actual diet using chloroplast gene sequencing may help to
confirm whether this was successful. Investigating the microbial community
introduced alongside the provided substrates would also be beneficial.
Another approach would be to target species like A. obeuntis that feed directly
at the source by placing the substrate directly on top of the mound to
potentially enhance feeding, as well as providing easy means to collect the
feeding termites (Lambert and Power, 1999).
An approach with more direct applications would be to study higher termites
collected directly from Western Australian Wheatbelt fields (feeding on wheat
or the eucalyptus mallee plantations surrounding the fields; Yu et al., 2015) or
crop residue. The idea to feed termites biofuel feedstocks was reviewed by
Scharf and Boucias (2010) and attempted in Reticulitermes flavipes with corn
stover and soybean residue (Rajarapu et al., 2015; Rajarapu and Scharf, 2017).
They reported a loss of protists likely due to plant defensive mechanisms, as
well as small changes in enzyme expression between treatments. However as
discussed previously, higher termites naturally feed on a wider variety of
substrates and offer primarily bacterial enzymes, which are more applicable to
industrial processes. Wheat and/or eucalyptus mallee would also be more
relevant substrates for a WA specific biofuel approach, warranting further
study.
Overall, it was shown that it is possible to influence the gut community of
local higher termites over a three week period in field conditions.
Environmental uptake may in part explain the effects of diet and location
measured in Chapter 3. Co-habitation, i.e. the two species sharing a mound,
was found to have a significant effect on the gut communities, presumably due

202
to horizontal transfer of taxa between the species, either through ingestion of
heterospecific corpses, shared mound material or faecal matter (Bourguignon
et al, 2018).
D: Can enzymes relevant to breaking down a substrate of choice be
identified using comparative enzymology?
The long-term goal of this work is to identify enzymes of interest to biofuel
production in Western Australia and study their biochemical activity.
Knowing that the microbial gut communities, which produce enzymes of
interest, can be influenced on a short time scale, future studies could focus on
enzyme-related work to lead to more practical applications. In Chapter 5,
protein extraction and visualisation protocols were optimised to improve the
integrity of enzymes recovered from termite guts, an important obstacle that
was not consistently achieved. The biggest challenge was preventing protein
degradation while maintaining cellulase activity. Previous studies on termite
gut cellulases have not reported the effect of proteases, except for Lima et al.
(2014), and zymograms are rarely shown in conjunction with total protein gels.
If denaturation occurred during sample preparation, it is likely that this was
not an issue exclusive to my work and may have gone unnoticed in previous
studies, leading to an underestimation of cellulase abundance and activity in
the guts of termites.
As I have outlined in Chapter 5, the inconsistent protein profiles obtained on
acrylamide gels could also be caused by renaturation of proteases during
electrophoresis, since degradation occurred primarily in the top half of the gel.
Indeed, Blumentals (1990) reported smearing caused by an SDS-resistant
serine protease in the top half of a protease zymogram. In Chapter 5, heating
extracts at 99° C for 3 min did not inhibit the proteases, but did irreversibly
denature the cellulases of interest. Furthermore, Nasutitermes corniger worker
proteases have been shown to remain active following heating at 90° C for 30

203
min (Lima et al., 2014), suggesting that heating alone is not an appropriate
treatment to inactivate termite gut proteases. Further work is required to find
ways to control these proteases without interfering with probe binding or
cellulase activity. One way to achieve this could be to measure protease
activity following incubation of enzyme extracts with several class-specific
inhibitors (Lima et al., 2014), with the aim to optimise a protective cocktail.
The endoglucanases described in Chapter 5 were regardless well aligned with
previous findings. In both T. westraliensis and A. obeuntis, more
endoglucanase activity was detected in the midgut, where the crude extract
should represent termite derived endocellulases (Tokuda et al., 2005; Tokuda
& Watanabe, 2007). Different bands were found in hindgut extracts, indicating
the presence of distinct microbial endoglucanases. Endoglucanase activity
bands ranged from less than 37 to 50 kD in T. westraliensis and between 37
and 60 kD in A. obeuntis, similar to sizes reported in a wide range of insect gut
fluids, including the termite Reticulitermes hageni Banks (24 to 40 kD; Oppert
et al., 2010). Endoglucanases smaller than 50 kD were reported in the midguts
of higher termites Nasutitermes takasagoensis, N. walkeri and Macrotermes
barneyi (Tokuda and Watanabe, 2007; Wu et al., 2012; Ni et al., 2014). Larger
endoglucanases were reported in the hindguts of N. takasagoensis and N.
walkeri with bands ranging from less than 50 to 120 kD (Tokuda and
Watanabe, 2007).
I then focused on cellulase-binding probes to visualise enzymes of interest,
with the aim of isolating them as outlined by Chauvigné-Hines et al. (2012).
Chauvigné-Hines et al. (2012) prescribed harsh conditions of 60° C for 3 hr for
the probe binding step. It was shown in Chapter 5 that GH2d-ABP binding
could occur at 4° C over 30 min, which might be better conditions to maximise
protein integrity. A large number of proteins were found to bind to this probe,
regardless of the conditions. This could indicate the presence of a wide range
of cellulases and glycoside hydrolases (GH) since they have been reported as
ranging from 20 to over 100 kD in aggregate forms (Stütz and Wrodnigg, 2011).

204
Another possibility is a lack of probe specificity, which could be determined by
using biotin reporter groups instead of fluorescent groups (Chauvigné-Hines
et al., 2012) to obtain a proteome and check whether it is primarily made up of
GHs. Genetic approaches would then be required to obtain sequences for the
most promising enzymes, which could then be cloned into culturable
organisms for further testing (Ni et al., 2014).
Another option to circumvent protein degradation issues would be to use
genomic approaches to find candidate enzymes. This could be achieved with
WGS or a more tailored approach would be to design taxon-specific primers
based on 16S rRNA data to target cellulose degraders of interest, followed by
long-read sequencing (Orr et al., 2018). A difficulty with this strategy is that
cellulases have evolved multiple times independently with unrelated
sequences and structures, and hence each cellulase-containing protein family
would need to be searched for independently (Sukharnikov et al., 2011).
Further research would then be necessary to clone sequences of interest into
culturable hosts and test for functionality, optimal conditions and specificity
of the identified cellulases (Willis et al., 2010). In fact metagenome studies
have had a poor record of predicting enzymes recovered in proteomic studies
in termite guts (Scharf, 2015). The appeal of the approach tested in Chapter 5
is that targeted enzymes would be known to be functional and efficient in
vivo, although their optimal conditions, specificity and efficiency in the new
host would still need to be evaluated.
Broader Contributions
In the context of local optimisation of biofuel production from lignocellulose,
my work provides a roadmap for discovering and studying enzymes from
termite guts, and other microbial habitats. My investigation into primer bias
reinforces the need for standardised techniques and increased replication in
the field of gut metagenomics, to allow for valid comparisons between studies.
My stringent core community analysis method allows the most accurate

205
species core community estimation in termites to date, also contributing to
reproducibility across studies. Subseting the gut community into groups
shared by all species members or within groups of interest (colony, location,
diet) facilitates a better understanding of functional roles and eventually
sources of enzymes. Finally, my work into optimising protein extraction and
enzyme visualisation highlighted the need for greater quality control in this
field, to more accurately measure the abundance and activity of enzymes of
interest. These advances change the way past studies are interpreted and will
have an impact on how future research on microbial communities is
conducted.
The gut communities of three endemic Western Australian termites, higher
termites T. westraliensis and A. obeunits and the lower termite C.
acinaciformis raffrayi were described for the first time. I have shown in the
process that termite gut microbiomes are more complex than previously
thought. Bacterial communities specific to each colony, location and diet were
characterised, highlighting the plasticity of the gut community and building
upon the more recent idea that the bacterial community as a whole is not in
fact highly conserved (Brune and Dietrich, 2015; Tai et al., 2015; Bourguignon
et al., 2018). To my knowledge, the alate bacterial gut community has been
investigated in only three other studies to date (Hongoh et al., 2006;
Benjamino and Graf, 2016; Diouf et al. 2018) despite being the vehicle for
vertically transferred organisms. The alate gut community warrants further
study for evidence of co-speciation between gut symbionts and the termite
host. The inconsistent presence of protists in two higher termite species was
supported by both observational and sequencing data in this study, indicating
another, perhaps seasonal, factor shaping the gut community of higher
termites.
My work has implications for how we understand the evolution and ecology of
termite microbiomes, particularly the factors shaping intraspecies variation.

206
The mechanism likely to generate intraspecific differences in the termite gut
community are summarised in Figure 1. A key point is the mixing of vertically
and horizontally acquired organisms to create unique, locally adapted gut
communities. Microbiota transplant experiments have shown that gut
communities are assembled primarily around available niches in the gut
habitat, with the final gut community composed of lineages from the
community of origin, but the community structure resembling the normal gut
community of the recipient host (Rawls et al., 2006; Mikaelyan et al., 2016). An
example niche in a higher termite gut are prominent cuticular spines
extending about halfway the width of the gut lumen described in
Procubitermes aburiensis by Bignell et al. (1980). These are more extensive
than in most other insect species, providing surfaces for the attachment of
microorganisms. Seedorf et al. (2014) found that the transplanted gut
community composition and structure more closely resembled the community
of origin if it was from a gut environment than a non-gut environment,
suggesting that these gut specific niches suit particular taxonomic groups.
Although these transplants were artificial, termites naturally receive
microbiome transplants via trophallaxis every time they moult, because the
lining of the gut is shed (Xing et al., 2013). In lower termites at least, symbionts
are shed ahead of molting and die in the process (Raina et al., 2008; Napela,
2017), so that gut microbes must be obtained from other workers.
Recently, Bourguignon et al. (2018) compared 94 termite species across all six
termite families and all eight Termitidae subfamilies, with a similar
sequencing depth to mine (11,509 reads per sample compared to my 11,400).
They suggest the gut community has been shaped by a ‘‘mixed-mode’’
transmission combining vertical transfer with more extensive horizontal
transfer between termite lineages than previously expected. They compared
representative sequences for "genus level" OTUs to the most closely related
published environmental sequences. Often the termite-derived sequences
clustered together, indicating that most of the taxa had a strong host
specificity for termites and may be vertically or horizontally acquired. Other

207
Fi
gure
1: F
acto
rs in
fluen
cing
ter
mite
gut
com
mun
ity c
ompo
sitio
n an
d le
adin
g to
intr
aspe
cific
var
iatio
n. D
ispe
rsin
g re
prod
uctiv
es in
ocul
ate
the
new
ly f
ound
ed c
olon
y w
ith o
rgan
ism
s br
ough
t fr
om t
heir
par
ent
colo
nies
. Fur
ther
diff
eren
ces
wit
hin
a sp
ecie
s m
ay r
esul
t fr
om e
nvir
onm
enta
l ac
quis
ition
via
con
tact
with
foo
d, s
oil,
mou
nd m
ater
ial,
or h
oriz
onta
l tr
ansf
er b
y co
nsum
ing
hete
rosp
ecifi
c co
rpse
s or
fae
cal
mat
ter.
The
se
acqu
ired
org
anis
ms
may
be
tran
sien
t or
bec
ome
resi
dent
in
the
gut
com
mun
ity.
The
re-e
stab
lishm
ent
of t
he g
ut c
omm
unity
in
mou
lted
indi
vidu
als
may
pro
vide
opp
ortu
nitie
s fo
r co
loni
satio
n of
a g
iven
nic
he b
y a
diffe
rent
mic
robe
. Th
e gu
t co
mm
unity
is
exch
ange
d be
twee
n ge
nera
tions
of c
olon
y m
embe
rs, b
efor
e in
ocul
atin
g di
sper
sing
rep
rodu
ctiv
es. T
his
mix
ing
of v
ertic
ally
tra
nsm
itted
and
env
iron
men
tally
acq
uire
d or
gani
sms
gene
rate
s un
ique
gut
com
mun
ities
in in
divi
dual
s an
d at
the
colo
ny le
vel.

208
clusters were interspersed with environmental sequences suggesting
horizontal transfer with other animal groups or environmental uptake.
Such environmental uptake may in part explain the effects of diet and location
measured in Chapter 3. Diet was shown to influence the gut community both
over evolutionary time (Chapter 2) and on a shorter time scale (Chapter 3).
Co-habitation, which increases the opportunity for horizontal transfer, was
also found to have a significant effect on the gut communities of T.
westraliensis and A. obeuntis in Chapter 3; certain OTUs were shared between
the species only when cohabitating. Taxa present in the environment may
appear transiently in the gut community or establish and be amplified in the
gut habitat when taken up from soil, food or other termites. This mixed-mode
transmission could foster adaptive flexibility of the gut community to the
environmental conditions and food sources of the colony (Shapira, 2016).
Presumably a sample of all gut microbes, whether vertically transferred,
environmentally and/or horizontally acquired, is shared with post-moult
nestmates (Figure 1). All of these organisms, whether established or transient
in the donor, would have the opportunity to establish in the unpopulated
lining of the gut, increasing colony-level differences.
Conclusion
My project confirms that diet affects termite gut core community composition
and abundance on a short time scale under field conditions. Future studies
could focus on the enzymes produced by the gut communities of higher
termites directly feeding on biofuel substrates of interest, e.g. crops or crop
residue in Western Australian Wheatbelt fields, including wheat and/or
eucalyptus mallee. Continued effort is warranted: certain biofuels are the best
renewable energy option currently available for niche applications such as
powering large ships and mining equipment (Sims et al., 2014) and second
generation biofuel production would recycle crop "waste" that is currently
being burnt. Biofuels from wheat straw would thus result in harnessing CO2

209
currently produced from crop residue burning, to diminish the need for
burning of fossil fuels, two activities that contribute to climate change.
I have also proposed new standards for experimental design in the study of
termite gut communities, including the use of a standardised 16S rRNA
amplification strategy, increased replication, a standard species core
community definition and analysis method for accurate calculations. The
communities of three endemic Western Australian termites were described for
the first time and factors shaping intraspecific variation were further
characterised. This has provided a greater understanding of the vertically,
horizontally and environmentally acquired components of the gut community
and the generation of intraspecific variation. Hence, intraspecific variation
occuring on a short time scale allows the manipulation of the gut community
to target substrates of interest, laying the foundation for future local biofuel
initiatives.

210
References Abdul Rahman, N., Parks, D. H., Willner, D. L., Engelbrektson, A. L., Goffredi, S. K.,
Warnecke, F., Scheffrahn, R. H., Hugenholtz, P. (2015). A molecular survey of Australian and North American termite genera indicates that vertical inheritance is the primary force shaping termite gut microbiomes. Microbiome, 3, 5.
Adl, S. M., Habura, A., & Eglit, Y. (2014). Amplification primers of SSU rDNA for soil protists. Soil Biology and Biochemistry, 69(0), 328-342.
Antony-Babu, S., Stien, D., Eparvier, V., Parrot, D., Tomasi, S., & Suzuki, M. T. (2017). Multiple Streptomyces species with distinct secondary metabolomes have identical 16S rRNA gene sequences. Sci Rep, 7(1), 66.
Bates, S. T., Clemente, J. C., Flores, G. E., Walters, W. A., Parfrey, L. W., Knight, R., Fierer, N. (2013). Global biogeography of highly diverse protistan communities in soil. ISME J, 7(3), 652-659.
Benjamino, J., & Graf, J. (2016). Characterization of the core and caste-specific microbiota in the termite, Reticulitermes flavipes. Frontiers in Microbiology, 7, 1.
Bennett, R. N., & Allsgrove, R. M. W. (1994). Secondary metabolites in plant defence mechanisms. New Phytologist, 127, 617-633.
Blumentals, I. I., Robinson, A. S., & Kelly, R. M. (1990). Characterization of sodium dodecyl sulfate-resistant proteolytic activity in the hyperthermophilic archaebacterium Pyrococcus furiosus. Applied and Environmental Microbiology, 56(7), 1992.
Bourguignon, T., Lo, N., Dietrich, C., Šobotník, J., Sidek, S., Roisin, Y., Brune, A., Evans, T. A. (2018). Rampant host switching shaped the termite gut microbiome. Current Biology, 28(4), 649-654.e2.
Brune, A., & Dietrich, C. (2015). The gut microbiota of termites: digesting the diversity in the light of ecology and evolution. Annual Review of Microbiology, 69(1), 145-166.
Chauvigné-Hines, L. M., Anderson, L. N., Weaver, H. M., Brown, J. N., Koech, P. K., Nicora, C. D., Hofstad, B. A., Smith, R. D., Wilkins, M. J., Callister, S. J., Wright, A. T. (2012). Suite of activity-based probes for cellulose-degrading enzymes. Journal of the American Chemical Society, 134(50), 20521-20532.
Cleveland, L. R. (1923). Symbiosis between termites and their intestinal protozoa. Proceedings of the National Academy of Sciences of the United States of America, 9(12), 424-428.
Costa-Leonardo, A. M., Laranjo, L. T., Janei, V., & Haifig, I. (2013). The fat body of termites: functions and stored materials. Journal of Insect Physiology, 59(6), 577-587.
De Filippis, F., Parente, E., Zotta, T., & Ercolini, D. (2018). A comparison of bioinformatic approaches for 16S rRNA gene profiling of food bacterial microbiota. International Journal of Food Microbiology, 265, 9-17.
Dietrich, C., Köhler, T., & Brune, A. (2014). The cockroach origin of the termite gut microbiota: patterns in bacterial community structure reflect major evolutionary events. Applied and Environmental Microbiology.
Diouf, M., Hervé, V., Mora, P., Robert, A., Frechault, S., Rouland-Lefèvre, C., Miambi, E. (2018). Evidence from the gut microbiota of swarming alates of a vertical transmission of the bacterial symbionts in Nasutitermes arborum (Termitidae, Nasutitermitinae). Antonie Van Leeuwenhoek, 111(4), 573-587.
Diouf, M., Roy, V., Mora, P., Frechault, S., Lefebvre, T., Hervé, V., Rouland-Lefèvre, C., Miambi, E. (2015). Profiling the succession of bacterial communities

211
throughout the life stages of a higher termite Nasutitermes arborum (Termitidae, Nasutitermitinae) Using 16S rRNA Gene Pyrosequencing. PLoS One, 10, e0140014.
Edgar, R. C. (2018). Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics, 34(14), 2371-2375.
Franzén, O., Hu, J., Bao, X., Itzkowitz, S. H., Peter, I., & Bashir, A. (2015). Improved OTU-picking using long-read 16S rRNA gene amplicon sequencing and generic hierarchical clustering. Microbiome, 3, 43.
Hill, G. F. (1942). Termites (Isoptera) from the Australian Region. Melbourne, Australia: Council for Scientific and Industrial Research.
Hongoh, Y., Ekpornprasit, L., Inoue, T., Moriya, S., Trakulnaleamsai, S., Ohkuma, M., Noparatnaraporn, N., Kudo, T. (2006). Intracolony variation of bacterial gut microbiota among castes and ages in the fungus-growing termite Macrotermes gilvus. Molecular Ecology, 15(2), 505-516.
Hongoh, Y., Deevong, P., Inoue, T., Moriya, S., Trakulnaleamsai, S., Ohkuma, M., Vongkaluang, C., Noparatnaraporn, N., Kudo, T. (2005). Intra- and interspecific comparisons of bacterial diversity and community structure support coevolution of gut microbiota and termite host. Applied & Environmental Microbiology, 71(11), 6590-6599.
Huang, X.-F., Bakker, M., Judd, T., Reardon, K., & Vivanco, J. (2013). Variations in diversity and richness of gut bacterial communities of termites (Reticulitermes flavipes) fed with grassy and woody plant substrates. Microbial Ecology, 65(3), 531-536.
Husseneder, C., Berestecky, J. M., & Grace, J. K. (2009). Changes in composition of culturable bacteria community in the gut of the formosan subterranean termite depending on rearing conditions of the host. Annals of the Entomological Society of America, 102(3), 498-507.
Kirby, H. (1927). Studies on some amoebae from the termite Mirotermes, with notes on some other Protozoa from the Termitidae. Quarterly Journal of Microscopical Science.
Köhler, T., Dietrich, C., Scheffrahn, R. H., & Brune, A. (2012). High-resolution analysis of gut environment and bacterial microbiota reveals functional compartmentation of the gut in wood-feeding higher termites (Nasutitermes spp.). Applied & Environmental Microbiology, 78(13), 4691-4701.
Korb, J., & Fuchs, A. (2006). Termites and mites - adaptive behavioural responses to infestation? Behaviour, 143(7), 891-907.
Li, H., Dietrich, C., Zhu, N., Mikaelyan, A., Ma, B., Pi, R. et al. (2016). Age polyethism drives community structure of the bacterial gut microbiota in the fungus-cultivating termite Odontotermes formosanus. Environmental Microbiology, 18(5), 1440-1451.
Lima, T. A., Pontual, E. V., Dornelles, L. P., Amorim, P. K., Sá, R. A., Coelho, L. C. et al. (2014). Digestive enzymes from workers and soldiers of termite Nasutitermes corniger. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology, 176, 1-8.
Mikaelyan, A., Meuser, K., & Brune, A. (2017). Microenvironmental heterogeneity of gut compartments drives bacterial community structure in wood-and humus-feeding higher termites. FEMS Microbiology Ecology, 93(1), fiw210.
Mikaelyan, A., Thompson, C. L., Hofer, M. J., & Brune, A. (2016). Deterministic assembly of complex bacterial communities in guts of germ-free cockroaches. Applied and Environmental Microbiology, 82(4), 1256-1263.
Miyata, R., Noda, N., Tamaki, H., Kinjyo, K., Aoyagi, H., Uchiyama, H., Tanaka, H.

212
Influence of feed components on symbiotic bacterial community structure in the gut of the wood-feeding higher termite Nasutitermes takasagoensis. Bioscience, Biotechnology, and Biochemistry, 71(5), 1244-1251.
Nalepa, C. A. (2017). What kills the hindgut flagellates of lower termites during the host molting cycle. Microorganisms, 5(4).
Nguyen, N. P., Warnow, T., Pop, M., & White, B. (2016). A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Microbiomes, 2, 16004.
Ni, J., Wu, Y., Yun, C., Yu, M., & Shen, Y. (2014). cDNA cloning and heterologous expression of an endo-β-1,4-glucanase from the fungus-growing termite Macrotermes barneyi. Archives of Insect Biochemistry and Physiology, 86(3), 151-164.
Ohkuma, M., & Brune, A. (2011). diversity, structure, and evolution of the termite gut microbial community. In D. E. Bignell, Y. Roisin, & N. Lo (Eds.), Biology of Termites: a Modern Synthesis (pp. 413-438). Netherlands: Springer.
Oppert, C., Klingeman, W. E., Willis, J. D., Oppert, B., & Jurat-Fuentes J. L. (2010). Prospecting for cellulolytic activity in insect digestive fluids. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology, 155(2), 145-154.
Otani, S., Mikaelyan, A., Nobre, T., Hansen, L. H., Kone, N. A., Sorensen, S. J., Aanen, D.K., Boomsma, J.J., Brune, A. Poulsen, M (2014). Identifying the core microbial community in the gut of fungus-growing termites. Molecular Ecology.
Pawlowski, J., Audic, S., Adl, S., Bass, D., Belbahri, L., Berney, C. et al. (2012). CBOL protist working group: barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms. PLoS. Biol., 10(11).
Poinar, G. O. (2009). Description of an early Cretaceous termite (Isoptera: Kalotermitidae) and its associated intestinal protozoa, with comments on their co-evolution. Parasites and Vectors, 2(1), 12.
Raina, A., Park, Y. I., & Gelman, D. (2008). Molting in workers of the Formosan subterranean termite Coptotermes formosanus. Journal of Insect Physiology, 54(1), 155-161.
Rajarapu, S. P., & Scharf, M. E. (2017). Saccharification of agricultural lignocellulose feedstocks and protein-level responses by a termite gut-microbe bioreactor. Frontiers in Energy Research, 5, 186.
Rajarapu, S. P., Shreve, J. T., Bhide, K. P., Thimmapuram, J., & Scharf, M. E. (2015). Metatranscriptomic profiles of Eastern subterranean termites, Reticulitermes flavipes (Kollar) fed on second generation feedstocks. BMC Genomics, 16, 332.
Ranjan, R., Rani, A., Metwally, A., McGee, H. S., & Perkins, D. L. (2016). Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochemical and Biophysical Research Communications, 469(4), 967-977.
Rawls, J. F., Mahowald, M. A., Ley, R. E., & Gordon, J. I. (2006). Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell, 127(2), 423-433.
Reid, N. M., Addison, S. L., West, M. A., & Lloyd-Jones, G. (2014). The bacterial microbiota of Stolotermes ruficeps (Stolotermitidae), a phylogenetically basal termite endemic to New Zealand. FEMS Microbiology Ecology, 90(3), 678-688.
Ross, M. G., Russ, C., Costello, M., Hollinger, A., Lennon, N. J., Hegarty, R., Nusbaum, C., Jaffe, D.B. (2013). Characterizing and measuring bias in sequence data. Genome Biology, 14(5), R51.
Scharf, M. E., & Boucias, D. G. (2010). Potential of termite-based biomass pre-

213
treatment strategies for use in bioethanol production: Termite-based biomass pre-treatment strategies. Insect Science, 17(3), 166-174.
Seedorf, H., Griffin, N. W., Ridaura, V. K., Reyes, A., Cheng, J., Rey, F. E. et al. (2014). Bacteria from diverse habitats colonize and compete in the mouse gut. Cell, 159(2), 253-266.
Shade, A., & Handelsman, J. (2012). Beyond the Venn diagram: the hunt for a core microbiome: The hunt for a core microbiome. Environmental Microbiology, 14(1), 4-12.
Scharf, M. E. (2015). Omic research in termites: an overview and a roadmap. Frontiers in Genetics, 6, 76.
Sims, R., Schaeffer, R., Creutzig, F., Cruz-Núñez, X., D’Agosto, M., Dimitriu, D. et al. (2014). Transport. In O. Edenhofer, R. Pichs-Madruga, Y. Sokona, E. Farahani, S. Kadner, K. Seyboth et al. (Eds.), Climate Change 2014: Mitigation of Climate Change. Contribution of Working Group III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA.
Sipos, R., Székely, A. J., Palatinszky, M., Révész, S., Márialigeti, K., & Nikolausz, M. (2007). Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis: PCR parameters influencing quantitative bias. FEMS Microbiology Ecology, 60(2), 341-350.
Su, L., Yang, L., Huang, S., Su, X., Li, Y., Wang, F. et al. (2016). comparative gut microbiomes of four species representing the higher and the lower termites. Journal of Insect Science, 16(1).
Sugio, A., Dubreuil, G., Giron, D., & Simon, J.-C. (2015). Plant-insect interactions under bacterial influence: ecological implications and underlying mechanisms. Journal of Experimental Botany, 66(2), 467-478.
Sukharnikov, L. O., Cantwell, B. J., Podar, M., & Zhulin, I. B. (2011). Cellulases: ambiguous nonhomologous enzymes in a genomic perspective. Trends in Biotechnology, 29(10), 473-479.
Szokoli, F., Castelli, M., Sabaneyeva, E., Schrallhammer, M., Krenek, S., Doak, T. G. et al. (2016). Disentangling the taxonomy of Rickettsiales and description of two novel symbionts (”Candidatus Bealeia paramacronuclearis” and “Candidatus Fokinia cryptica”) sharing the cytoplasm of the ciliate protist Paramecium biaurelia. Applied and Environmental Microbiology, 82(24), 7236-7247.
Tai, V., James, E. R., Nalepa, C. A., Scheffrahn, R. H., Perlman, S. J., & Keeling, P. J. (2015). The role of host phylogeny varies in shaping microbial diversity in the hindguts of lower termites. Applied and Environmental Microbiology, 81(3), 1059-1070.
Tanaka, H., Aoyagi, H., Shiina, S., Doudou, Y., Yoshimura, T., Nakamura, R., Uchiyama, H. (2006). Influence of the diet components on the symbiotic microorganisms community in hindgut of Coptotermes formosanus Shiraki. Applied Microbiology & Biotechnology, 71(6), 907-917.
Tokuda, G., Lo, N., & Watanabe, H. (2005). Marked variations in patterns of cellulase activity against crystalline‐vs. carboxymethyl‐cellulose in the digestive systems of diverse, wood‐feeding termites. Physiological Entomology, 30(4), 372-380.
Tokuda, G., & Watanabe, H. (2007). Hidden cellulases in termites: revision of an old hypothesis. Biology Letters, 3(3), 336-339.
Větrovský, T., & Baldrian, P. (2013). The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS ONE, 8(2), e57923.

214
Waidele, L., Korb, J., Voolstra, C. R., Künzel, S., Dedeine, F., & Staubach, F. (2017). Differential ecological specificity of protist and bacterial microbiomes across a set of termite species. Frontiers in Microbiology, 8, 5.
Wang, Y., Su, L., Huang, S., Bo, C., Yang, S., Li, Y. et al. (2016). Diversity and resilience of the wood-feeding higher termite Mironasutitermes shangchengensis gut microbiota in response to temporal and diet variations. Ecology and Evolution, 6(22), 8235-8242.
Willis, J. D., Oppert, C., & Jurat-Fuentes, J. L. (2010). Methods for discovery and characterization of cellulolytic enzymes from insects. Insect Science, 17(3), 184-198.
Wu, Y., Chi, S., Yun, C., Shen, Y., Tokuda, G., Ni, J. (2012). Molecular cloning and characterization of an endogenous digestive β-glucosidase from the midgut of the fungus-growing termite Macrotermes barneyi. Insect Molecular Biology, 21(6), 604-614.
Yu, Y., Bartle, J., Mendham, D., & Wu, H. (2015). Site variation in life cycle energy and carbon footprints of mallee biomass production in Western Australia. Energy Fuels, 29(6), 3748-3752.
Xiang, H., Xie, L., Zhang, J., Long, Y., Liu, N., Huang, Y., Wang, Q. (2012). Intracolonial differences in gut bacterial community between worker and soldier castes of Coptotermes formosanus. Insect Science, 19, 86-95.
Xing, L., Chouvenc, T., & Su, N.-Y. (2013). Molting process in the Formosan subterranean termite (Isoptera: Rhinotermitidae). Annals of the Entomological Society of America, 106(5), 619-625.




















