
Bioactivity of anti-angiogenic ribozymes targeting Flt1 and KDR mRNA
Upload
independentCategory
view
4download
0
Evidence for the Involvement of miRNA in Redox RegulatedAngiogenic Response of Human Microvascular
Endothelial CellsShani Shilo, Sashwati Roy, Savita Khanna, Chandan K. Sen
Objective—A Dicer knockdown approach was used to test the significance of miRNA in regulating the redox state andangiogenic response of human microvascular endothelial cells (HMECs).
Methods and Results—Lowering of miRNA content by Dicer knockdown induced vascular endothelial growth factorexpression but diminished the angiogenic response of HMECs as determined by cell migration and Matrigel tubeformation. Such impairment of angiogenic response in the Matrigel was rescued by exogenous low micromolar H2O2.Dicer knockdown HMECs demonstrated lower inducible production of reactive oxygen species (ROS) when activatedwith either phorbol ester, tumor necrosis factor-�, or vascular endothelial growth factor. Limiting the production of ROSby antioxidant treatment or NADPH oxidase knockdown approaches impaired angiogenic responses. Experiments toidentify how ROS production is limited by Dicer knockdown identified lower expression of p47phox protein in thesecells. This lowering of cellular miRNA content induced expression of the transcription factor HBP1, a suppressortranscription factor that negatively regulates p47phox expression. Knockdown of HBP1 restored the angiogenicresponse of miRNA-deficient HMECs.
Conclusion—This study provides the first evidence that redox signaling in cells is subject to regulation by miRNA.Specifically, p47phox of the NADPH oxidase complex has been identified as one target that regulates the angiogenicproperties of endothelial cells. (Arterioscler Thromb Vasc Biol. 2008;28:471-477)
Key Words: free radicals/free-radical scavengers � gene expression � hypoxia � oxygen � vascular biology� redox � angiogenesis � endothelial function
miRNA represent a class of endogenous small (�22nt)RNA molecules that can repress protein synthesis.1 It is
estimated that there are more than 600 miRNAs in mammaliancells, and that about 30% of all genes are regulated by miRNA.2
The key protein responsible for miRNA maturation is thecytosolic enzyme Dicer.3 Arrest of Dicer activity represents aproductive approach to evaluate the overall functional signifi-cance of miRNA in any specific biological paradigm.4–6
Several studies have demonstrated a central role ofNADPH oxidase–derived reactive oxygen species (ROS) assignaling messengers in driving angiogenesis.7–11 Whethersuch redox control of angiogenesis is subject to regulation bymiRNA remains unknown. In this study, we used a Dicerknockdown approach to test the significance of miRNA inregulating the redox state and angiogenic response of humanmicrovascular endothelial cells (HMECs).
Materials and MethodsCells and Cell CultureHMECs were grown under standard culture conditions (at 37°C in ahumidified atmosphere consisting of 95% air and 5% CO2) in
MCDB-131 growth medium supplemented with 10% FBS, 100IU/mL penicillin, 0.1 mg/mL streptomycin, 10 mmol/L L-glutamine(GIBCO-BRL) as described previously.12,13
siRNA Delivery to CellsHMECs (0.2�106 cells per well in 12-well plate) were seeded inantibiotic-free medium 24 hours before transfection. Dharma-FECTTM 1 transfection reagent (Dharmacon RNA Technologies)was used to transfect cells with 100 nmol/L siRNA smart pool, forhuman Dicer, p47phox, or HBP1 as required. Transfection ofnontargeting siRNA (Dharmacon RNA Technologies) was per-formed for the control group. The first transfection was performedfor 72 hours as described previously (single transfection).14 Inaddition, a second transfection was performed for lowering cellularmiRNA content. After 48 hours of the first transfection, cells werereseeded in 12-well plates and retransfected the following day for anadditional 72 hours. After transfection, cells were collected orassayed for miRNA content. siRNA transfections for the knockdownof HBP1 were performed during the process of second siDicertransfection.
Quantification of mRNA Transcription LevelsTotal RNA was isolated from cells using the Absolutely RNAMiniprep kit (Stratagene). RNA (1 �g) was reverse transcribed into
Original received August 30, 2007; final version accepted December 30, 2007.From the Comprehensive Wound Center, Department of Surgery, Davis Heart & Lung Research Institute, The Ohio State University Medical Center,
Columbus.Correspondence to Professor Chandan K. Sen, 512 Davis Heart & Lung Research Institute, 473 West 12th Avenue, The Ohio State University Medical
Center, Columbus, Ohio 43210. E-mail [email protected]© 2008 American Heart Association, Inc.
Arterioscler Thromb Vasc Biol is available at http://atvb.ahajournals.org DOI: 10.1161/ATVBAHA.107.160655
471
Cell Biology/Signaling
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
by guest on Septem
ber 23, 2016http://atvb.ahajournals.org/
Dow
nloaded from
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
by guest on Septem
ber 23, 2016http://atvb.ahajournals.org/
Dow
nloaded from
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
by guest on Septem
ber 23, 2016http://atvb.ahajournals.org/
Dow
nloaded from
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
by guest on Septem
ber 23, 2016http://atvb.ahajournals.org/
Dow
nloaded from
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
by guest on Septem
ber 23, 2016http://atvb.ahajournals.org/
Dow
nloaded from
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
by guest on Septem
ber 23, 2016http://atvb.ahajournals.org/
Dow
nloaded from
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
by guest on Septem
ber 23, 2016http://atvb.ahajournals.org/
Dow
nloaded from
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
by guest on Septem
ber 23, 2016http://atvb.ahajournals.org/
Dow
nloaded from
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
by guest on Septem
ber 23, 2016http://atvb.ahajournals.org/
Dow
nloaded from
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
by guest on Septem
ber 23, 2016http://atvb.ahajournals.org/
Dow
nloaded from
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
by guest on Septem
ber 23, 2016http://atvb.ahajournals.org/
Dow
nloaded from
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
by guest on Septem
ber 23, 2016http://atvb.ahajournals.org/
Dow
nloaded from
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
by guest on Septem
ber 23, 2016http://atvb.ahajournals.org/
Dow
nloaded from
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
by guest on Septem
ber 23, 2016http://atvb.ahajournals.org/
Dow
nloaded from
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
by guest on Septem
ber 23, 2016http://atvb.ahajournals.org/
Dow
nloaded from

cDNA using a Superscript III First-Strand synthesis system (Invitro-gen). The transcription levels of mRNA for Dicer, vascular endo-thelial growth factor (VEGF)-A, HBP1, and �-Actin were quantifiedusing real-time polymerase chain reaction (PCR) using SYBRgreen-I (Applied Biosystems). See details of primer sets in supple-mental Table I (available online at http://atvb.ahajournals.org). Therelative quantification approach was used to analyze the results.
Quantification of miRNA Expression LevelTotal RNA including the miRNA fraction was isolated usingmirVana miRNA isolation kit, according to the manufactures proto-col (Ambion). miRNA levels were then quantified using specificTaqman assays for miRNA (Applied Biosystems) and mirVanaqRT-PCR miRNA RT Kit (Applied Biosystems) using real-timePCR system and Taqman universal master mix. miRNA levels werequantified with the relative quantification method using �-actin ascontrol.
Nuclear Protein ExtractionNuclear protein extracts of HMECs were prepared using NuclearExtraction Kit (Active Motif) according to the manufacturer’sprotocol.
Protein DetectionFor details of Western Blot and ELISA procedures used seesupplemental Table II.
Cell ProliferationTransfected cells were assayed for cell proliferation using CyQUANTcell proliferation assay kit (Invitrogen).
Cell ViabilityTransfected cells were washed with PBS, centrifuged (500g, 5minutes), resuspended in PBS, and cell membrane integrity wasdetected using a flow cytometer (FACSort, BD). For this assay, thenonpermeant DNA-intercalating dye propidium iodide (PI), which isexcluded by viable cells, was used.15
In Vitro Angiogenic ResponseFor the methods used for Matrigel endothelial tube formation assayand endothelial cell migration assay see supplemental Table III.
Determination of Intracellular ROSSee supplemental Table IV.
ImmunocytochemistrySee supplemental Table V.
Statistical AnalysesData reported represent means�SD of at least 3 independentexperiments. Difference between 2 means was tested by Student ttest. P�0.05 was considered statistically significant.
ResultsDicer siRNA Double Transfection in HMECsSilences Dicer Expression and EffectivelyDecreases miRNA Expression LevelsOur goal was to lower miRNA content in HMECs byarresting the function of Dicer. This approach would enableus to examine the significance of miRNA in the angiogenicproperties of HMECs. We noted that standard siRNA trans-fection procedure (single transfection, 72 hour) to knock-down Dicer resulted in lower abundance of Dicer mRNA andprotein (supplemental Figure IA and IB). However, theprotocol was insufficient to significantly lower the content ofabundant16 endothelial cell miRNAs such as Hsa-miR-222
and Hsa-miR-18a (supplemental Figure IC and ID). Thesingle transfection procedure did not significantly influencecell proliferation as well (supplemental Figure IE). To obtainsignificant lowering of miRNA content in the HMECs, weperformed a double transfection procedure. This procedureincluded a second transfection of the same cells after 72 hoursof the first transfection. After the second transfection, cellswere maintained for an additional 72 hours. This additionaltime was allowed for the miRNA pool to be depleted whereasmaturation of new miRNA was arrested by Dicer knockdown.Using this approach we were able to obtain not only signif-icant lowering of Dicer mRNA and protein in HMECs butalso significant lowering of both Has-miR-222 and Has-miR-18a (supplemental Figure IF through II). The double trans-fection approach also significantly lowered the rate of cellproliferation (supplemental Figure IJ). For all subsequentexperiments, the double transfection approach was used.
Lowering of miRNA Content by Dicer KnockdownInduced VEGF ExpressionTo determine the general effect of miRNA depletion inHMECs on its angiogenic pathways, the expression ofVEGF-A mRNA and proteins was determined in Dicerknockdown HMECs. Compared with siControl transfectedcells, we noted induction of VEGF-A transcript as well asprotein in miRNA-deficient HMECs (supplemental FigureII). These observations suggest the presence of a repressivecontrol of VEGF by miRNA in HMECs.
Lowering of miRNA Content by Dicer KnockdownDiminished the Angiogenic Response of HMECs asDetermined by Tube Formation in MatrigelThe ability to form tubes in a Matrigel is one of the majorangiogenic characteristics of endothelial cells. After doubletransfection with siDicer or siControl, cells were counted andreseeded on Matrigel for 24 hours. Although miRNA-deficient Dicer knockdown cells produced more VEGF-A(supplemental Figure II), interestingly these cells demon-strated limited ability for tube formation in Matrigel com-pared with the corresponding siControl treated cells (Figure1A). Objective analysis of this observation was performedusing standardized quantitation of tube length based onspecific criteria described previously17 (Figure 1B).
Lowering of miRNA Content by Dicer KnockdownLimited Angiogenic Response of HMECs asDetermined by the Analysis of Cell MigrationEndothelial cell migration represents an integral componentof the angiogenic response and is driven by inducibleNADPH oxidase derived ROS.18,19 To further elucidate therole of miRNA in regulating the angiogenic properties ofHMECs, the migratory response of Dicer knockdownHMECs to scratch was investigated. This represents a stan-dard model to investigate endothelial cell migration.20 Resultsof HMEC migration were recorded every 2 hours (supple-mental Figure III). Dicer knockdown HMECs, with lowermiRNA content, demonstrated compromised migratory prop-erties (Figure 1C). Compared with siControl transfected cells,
472 Arterioscler Thromb Vasc Biol March 2008
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from

the rate of migration of siDicer transfected cells was cut downby half (Figure 1D).
Lowering of miRNA Content by Dicer Knockdowndid not Influence Cell ViabilityCompared with their corresponding controls, the viability ofDicer knockdown HMECs was not statistically different(Figure 1E). This result excludes the possibility that thelimited angiogenic response of miRNA depleted cells wasbecause of compromised cell viability of Dicer knockdownHMECs.
Lowering of miRNA Content by Dicer KnockdownLimited ROS Production in HMECsROS-dependent signaling is required for the angiogenicbehavior of endothelial cells.7–11 NADPH-oxidase derivedROS are required for endothelial cell migration and tubeformation.18,21,22 We sought to test the effect of miRNAdepletion in HMECs on inducible ROS production. Thephorbol ester PMA (phorbol myristate acetate) is known toinduce ROS production and facilitate angiogenic response.23
When activated with PMA for 30 minutes, Dicer knockdownHMECs demonstrated lower DHE fluorescence indicatinglower superoxide production in response to PMA activation(Figure 2A). Consistent results were obtained when DCF wasused as the fluorescent probe for the detection of cellularROS. Additionally, we noted that miRNA depletion does notonly dampen inducible ROS production but also lowersresting production of ROS by HMECs (Figure 2B). Addi-tional study of cellular ROS levels using a microscopicapproach produced consistent results demonstrating lowerROS levels in Dicer knockdown HMECs (supplementalFigure IVA). To test whether the observed effects of miRNAdepletion on lower cellular ROS was limited to the phorbolester pathway we tested the effects of tumor necrosis factor(TNF)� as well as that of VEGF on HMECs. TNF� is knownto induce ROS in endothelial cells and stimulate angiogenicbehavior.24 –27 miRNA depletion in Dicer knockdownHMECs significantly lowered TNF� induced ROS produc-tion (Figure 2C). VEGF induces oxidant production inendothelial cells and drives angiogenic responses via redoxsignaling.9 miRNA depletion in Dicer knockdown HMECssignificantly lowered VEGF-induced ROS production (Fig-ure 2D and supplemental Figure IVB). Thus, results fromphorbol ester, TNF�, as well as VEGF-activated HMECsconsistently demonstrate that miRNA depletion impairedboth resting as well as inducible cellular ROS production.
Significance of Cellular ROS in the AngiogenicResponse of HMECsDecomposition of cellular ROS by the treatment of HMECswith the antioxidant N-acetylcysteine (NAC) significantlyslowed cell migration (supplemental Figure VA) and im-paired tube formation on the Matrigel (supplemental FigureVB). Of note, compromise in Matrigel tube formation of
Figure 1. Dicer knockdown induced miRNA deficiency impairedthe angiogenic response of human microvascular endothelial cells.A-B, Matrigel® tube formation assay: A, Matrigel® tube formationvisualized by phase contrast microscopy. Representative image ofat least 3 independent experiments; B, measurement of tube curvelength (�M); Results are mean � S.D. * p�0.05 compared withcontrol. C, cell migration, quantitative data of the observationshown in Supplementary figure III; D, Cell migration, as determinedby the slope of the line between the 4h and 6h time points (seesupplementary figure III), was diminished by 50% in response toDicer knockdown; E, Cell viability as measured by propidiumiodide exclusion following double transfection for Dicer knock-down. Results are mean � S.D. * p�0.05 compared with control.
Figure 2. Dicer knockdown induced miRNA deficiency limited ROS production under basal and activation conditions. A, HMEC doubletransfected with siDicer or siControl were activated with 100 nM PMA for 30 min. DHE fluorescence was measured using a flow cytom-eter; B, Dicer knockdown HMEC were activated with 100 nM PMA for 4 h. DCF fluorescence was measured using a flow cytometer; C,Dicer knockdown HMEC were activated with 50 ng TNF� for 90 min. DCF fluorescence was measured as in B; D, HMEC double-transfected with siDicer or siControl were activated with 50 ng/ml VEGF for 1 h. Fluorescence microscopic images (see supplementaryfigure IV) were collected. Quantitation of DCF green fluorescence. Results are mean � S.D. * p�0.05 compared with correspondingsiControl.
Shilo et al miRNA Regulation of Redox Signaling 473
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from
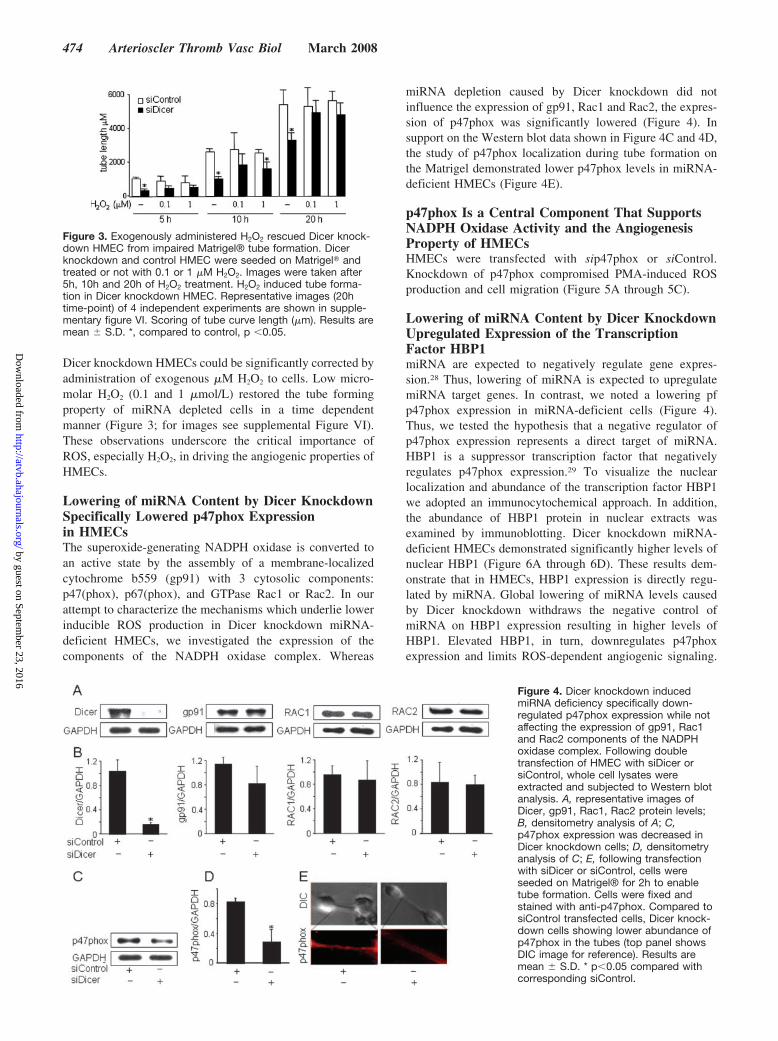
Dicer knockdown HMECs could be significantly corrected byadministration of exogenous �M H2O2 to cells. Low micro-molar H2O2 (0.1 and 1 �mol/L) restored the tube formingproperty of miRNA depleted cells in a time dependentmanner (Figure 3; for images see supplemental Figure VI).These observations underscore the critical importance ofROS, especially H2O2, in driving the angiogenic properties ofHMECs.
Lowering of miRNA Content by Dicer KnockdownSpecifically Lowered p47phox Expressionin HMECsThe superoxide-generating NADPH oxidase is converted toan active state by the assembly of a membrane-localizedcytochrome b559 (gp91) with 3 cytosolic components:p47(phox), p67(phox), and GTPase Rac1 or Rac2. In ourattempt to characterize the mechanisms which underlie lowerinducible ROS production in Dicer knockdown miRNA-deficient HMECs, we investigated the expression of thecomponents of the NADPH oxidase complex. Whereas
miRNA depletion caused by Dicer knockdown did notinfluence the expression of gp91, Rac1 and Rac2, the expres-sion of p47phox was significantly lowered (Figure 4). Insupport on the Western blot data shown in Figure 4C and 4D,the study of p47phox localization during tube formation onthe Matrigel demonstrated lower p47phox levels in miRNA-deficient HMECs (Figure 4E).
p47phox Is a Central Component That SupportsNADPH Oxidase Activity and the AngiogenesisProperty of HMECsHMECs were transfected with sip47phox or siControl.Knockdown of p47phox compromised PMA-induced ROSproduction and cell migration (Figure 5A through 5C).
Lowering of miRNA Content by Dicer KnockdownUpregulated Expression of the TranscriptionFactor HBP1miRNA are expected to negatively regulate gene expres-sion.28 Thus, lowering of miRNA is expected to upregulatemiRNA target genes. In contrast, we noted a lowering pfp47phox expression in miRNA-deficient cells (Figure 4).Thus, we tested the hypothesis that a negative regulator ofp47phox expression represents a direct target of miRNA.HBP1 is a suppressor transcription factor that negativelyregulates p47phox expression.29 To visualize the nuclearlocalization and abundance of the transcription factor HBP1we adopted an immunocytochemical approach. In addition,the abundance of HBP1 protein in nuclear extracts wasexamined by immunoblotting. Dicer knockdown miRNA-deficient HMECs demonstrated significantly higher levels ofnuclear HBP1 (Figure 6A through 6D). These results dem-onstrate that in HMECs, HBP1 expression is directly regu-lated by miRNA. Global lowering of miRNA levels causedby Dicer knockdown withdraws the negative control ofmiRNA on HBP1 expression resulting in higher levels ofHBP1. Elevated HBP1, in turn, downregulates p47phoxexpression and limits ROS-dependent angiogenic signaling.
Figure 3. Exogenously administered H2O2 rescued Dicer knock-down HMEC from impaired Matrigel® tube formation. Dicerknockdown and control HMEC were seeded on Matrigel� andtreated or not with 0.1 or 1 �M H2O2. Images were taken after5h, 10h and 20h of H2O2 treatment. H2O2 induced tube forma-tion in Dicer knockdown HMEC. Representative images (20htime-point) of 4 independent experiments are shown in supple-mentary figure VI. Scoring of tube curve length (�m). Results aremean � S.D. *, compared to control, p �0.05.
Figure 4. Dicer knockdown inducedmiRNA deficiency specifically down-regulated p47phox expression while notaffecting the expression of gp91, Rac1and Rac2 components of the NADPHoxidase complex. Following doubletransfection of HMEC with siDicer orsiControl, whole cell lysates wereextracted and subjected to Western blotanalysis. A, representative images ofDicer, gp91, Rac1, Rac2 protein levels;B, densitometry analysis of A; C,p47phox expression was decreased inDicer knockdown cells; D, densitometryanalysis of C; E, following transfectionwith siDicer or siControl, cells wereseeded on Matrigel® for 2h to enabletube formation. Cells were fixed andstained with anti-p47phox. Compared tosiControl transfected cells, Dicer knock-down cells showing lower abundance ofp47phox in the tubes (top panel showsDIC image for reference). Results aremean � S.D. * p�0.05 compared withcorresponding siControl.
474 Arterioscler Thromb Vasc Biol March 2008
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from

Such conclusion is supported by the observation that HBP1knockdown improved the angiogenic outcome in HMECssubjected to Dicer knockdown (Figure 6E through 6G).
DiscussionDicer is the key enzyme controlling miRNA biogenesis.Silencing Dicer in vivo causes embryonic lethality because ofimpairments in angiogenesis.30 Silencing Dicer in vitro im-pairs the angiogenic response of human umbilical veinendothelial cells (HUVECs) and EA.hy.926 endothelialcells.16,31 Although these observations indicate the possibleregulation of angiogenesis by miRNA, the underlying mech-anisms remain unknown. Majority of human miRNA loci arelocated within intronic regions and are transcribed by RNApolymerase II as part of their hosting transcription units. Theprimary transcripts are cleaved by Drosha to release approx-imately 70-nt pre-miRNAs that are subsequently processedby Dicer to generate mature approximately 22-nt miRNAs.32
Endogenous miRNAs have a long half-life.16,33 As such, it isunderstandable why the long double-transfection approachwas more effective in lowering endogenous miRNA levels.
Our observation that Dicer knockdown miRNA-deficientHMECs show impaired tube formation on the Matrigel isconsistent with a recent report demonstrating that addingVEGF to the cell culture media of Dicer knockdown cells donot restore the ability of HUVECs to form tubes on Matri-gel.16 Results of the current study display that impaired tubeformation in Dicer knockdown HMECs is noted despiteelevated VEGF expression. This finding is consistent with theobservation that Dicer-deficient embryos contain higher lev-els of VEGF yet impaired angiogenesis.30 These lines ofevidence lead to the notion that Dicer-dependent miRNAsregulate the angiogenic response of endothelial cells down-stream of VEGF.
ROS have been implicated to serve as signaling moleculesin numerous mechanisms including angiogenesis.34–37 VEGFstimulates proliferation, migration, and tube formation ofendothelial cells primarily through the VEGF receptor type 2(VEGFR2). Ligation of VEGF to VEGFR2 activates NADPHoxidase which in turn produces ROS to support the angio-genic response of endothelial cells.9 Activation of VEGFthrough ROS mediates 20-hydroxyeicosatetraenoic acid-
Figure 5. Knockdown of p47phox impaired cell migration anddecreased inducible ROS production. HMEC were transfectedwith siRNA against p47phox for 72h. A, Western blot; B, follow-ing activation with 100 nM PMA for 4h, p47phox knockdowncells showed decreased levels of ROS production as detectedby DCF staining and flow cytometry; C, following transfectionwith sip47phox, cells were seeded and scratched the followingday. Cell migration (2h) was impaired in p47phox knockdowncells; Results are mean � S.D. * p�0.05 compared with corre-sponding siControl.
Figure 6. Dicer silencing up-regulated the expression of thetranscription factor HBP1 which played a central role in impair-ing the angiogenic response of HMEC. Following siDicer orsiControl double transfection, HMEC were re-seeded on coverslips and stained with antibodies against Dicer and HBP1. DAPIwas used for staining of nuclei. A, Dicer knockdown lowered theabundance of cellular Dicer protein levels; B, Dicer knockdownelevated the expression of HBP1; C, Quantitation of HBP1 redfluorescence shown in B; D, Western blot of nuclear extracts; E,co-transfection of siDicer with siRNA against HBP1 decreasedHBP1 mRNA levels; F-G, co-transfection of siDicer with siRNAagainst HBP1 significantly released the inhibitory effects of Dicerknockdown on Matrigel® tube formation. increased the cellsangiogenic response shown by tube formation on Matrigel; G,Quantitation of tube length. * p�0.05 compared with corre-sponding siDicer transfected controls.
Shilo et al miRNA Regulation of Redox Signaling 475
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from

induced endothelial cell proliferation.38 ROS seems to be acommon denominator signaling mediator for angiogenesisbeyond that caused by VEGF. Hydrogen peroxide producedby angiopoietin-1 mediates angiogenesis.34 Endothelial cellmigration is regulated by localized production of ROS at theleading edge and at the site of injury.18 Decomposition ofROS stalls angiogenesis both in vivo as well as in vitro.10,18
The current finding that Dicer knockdown miRNA-deficientHMECs produce lower levels of basal ROS demonstrates thatnonphagocytic oxidases and other sources of ROS in cellsunder standard culture conditions are subject to regulation byendogenous miRNAs. Two lines of evidence in the currentstudy support a central role of ROS in facilitating theangiogenic behavior of HMECs. First, that downregulation ofcellular ROS by the antioxidant NAC impairs Matrigel tubeformation. Second, that impaired Matrigel tube formation inmiRNA-deficient HMECs can be corrected by exogenousH2O2. The latter line of evidence also indicates that the effectof miRNA on cellular redox state is functionally significantwith respect to determining angiogenic outcomes. NADPHoxidase activity represents a major pathway that contributesto cellular ROS in response to activation by phorbol ester,TNF�, and VEGF.9,39,40 Our observation that miRNA-deficientHMECs show lower NADPH oxidase activity, both basal aswell as inducible, provide first pointer indicating that miRNAare critically important in regulating the redox signalingnetwork that are now known to be implicated in numerousphysiological as well pathophysiological processes.35,41–43
NADPH oxidase is one of the major sources of ROS invasculature. It consists of a catalytic subunit (Nox1, Nox2,Nox3, Nox4, or Nox5), p22phox, p47phox, p67phox, and thesmall GTPase Rac1 and Rac2. The phagocytic NADPHoxidase consists of a membrane-localized glycosylated, cat-alytic subunit, gp91phox (also known as Nox2), along with asecond membrane-associated subunit, p22phox. gp91Phoxand p22phox stabilize one another in a tightly associatedheterodimer which mediates the transfer of electrons fromNADPH to molecular oxygen to generate superoxide anionradicals. The interaction of the membrane components withcytosolic regulatory subunits is important for the activation ofelectron flow. p22Phox plays a central role in this process, viainteraction with p47phox. Cytosolic small GTPase proteinsRac1 and Rac2 support optimal activity of the NADPHoxidase complex.44 Findings of the current study demonstratethat whereas miRNA deficiency caused by Dicer knockdowndoes not affect the expression of gp91phox, Rac1, or Rac2, itspecifically downregulates both p47phox expression as wellas ROS production. Serine phosphorylation of p47phox andits enhanced binding to p22phox is involved in NADPHoxidase function in endothelial cells.45 p47Phox-deficientmice exhibit impaired wound angiogenesis.10 Consistently,results of this study demonstrate that knockdown of p47phoximpaired ROS production and cell migration.
Given that the primary function of miRNA is to interferewith the expression of gene products, downregulation of p47expression in miRNA-deficient cells indicated secondaryregulation of p47phox by one or more repressors which mayhave been upregulated in response to the withdrawal of thenegative control of genes by miRNA. Upregulation of HBP1
(3-hydroxy-3-methylglutaryl [HMG] box-containing protein1) in miRNA-deficient HMECs proved that hypothesis. Thetranscriptional repressor HBP1 regulates the gene for thep47phox regulatory subunit of the NADPH oxidase. HBP1represses growth regulatory genes (eg, N-Myc, c-Myc, andcyclin D1) and is an inhibitor of G1 progression. Thepromoter of the p47phox gene contains 6 tandem high-affinity HBP1 DNA-binding elements at positions �1243 to�1318 bp from the transcriptional start site which wererequired for repression. Furthermore, HBP1 represses theexpression of the endogenous p47phox gene throughsequence-specific binding. Thus, HBP1 downregulatesNADPH oxidase-dependent superoxide production throughtranscriptional repression of the p47phox gene.29,46 The ob-servation that HBP1 is upregulated in miRNA-deficientHMECs is consistent with computational predictions ofTargetScan47 and miRbase.48 These databases list HBP1 asbeing highly susceptible to miRNA regulation. The family ofhsa-miR-29 (a, b, and c) seem likely to regulate HBP1expression for 2 reasons. First, they were predicted with thehighest scores in both computational models. Second, hsa-miR-29c and hsa-miR-29a have been recently known to beabundant in endothelial cells.31
In summary, this study provides the first evidence thatredox signaling in cells is subject to regulation by miRNA.Specifically, p47phox of the NADPH oxidase complex hasbeen identified as one functional target. Although the focus ofthe current study has been on angiogenesis, it is plausible thatother redox-sensitive aspects of human cell biology aresubject to control of endogenous miRNA.
Sources of FundingThis work was supported by NIH awards GM 077185 and GM069589 to C.K.S.
DisclosuresNone.
References1. Hammond SM. RNAi, microRNAs, and human disease. Cancer Chemother
Pharmacol. 2006;58 Suppl 1:s63–s68.2. Rajewsky N. microRNA target predictions in animals. Nat Genet.
2006;38 Suppl:S8–S13.3. Lund E, Dahlberg JE. Substrate selectivity of exportin 5 and Dicer in the
biogenesis of microRNAs. Cold Spring Harb Symp Quant Biol. 2006;71:59–66.
4. Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ,Mills AA, Elledge SJ, Anderson KV, Hannon GJ. Dicer is essential formouse development. Nat Genet. 2003;35:215–217.
5. Murchison EP, Stein P, Xuan Z, Pan H, Zhang MQ, Schultz RM, HannonGJ. Critical roles for Dicer in the female germline. Genes Dev. 2007;21:682–693.
6. Tang KF, Wang Y, Wang P, Chen M, Chen Y, Hu HD, Hu P, Wang B,Yang W, Ren H. Upregulation of PHLDA2 in Dicer knockdown HEK293cells. Biochim Biophys Acta. 2007;1770:820–825.
7. Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S,Shah AM. NADPH oxidases in cardiovascular health and disease.Antioxid Redox Signal. 2006;8:691–728.
8. Arbiser JL, Petros J, Klafter R, Govindajaran B, McLaughlin ER, BrownLF, Cohen C, Moses M, Kilroy S, Arnold RS, Lambeth JD. Reactiveoxygen generated by Nox1 triggers the angiogenic switch. Proc NatlAcad Sci U S A. 2002;99:715–720.
9. Ushio-Fukai M. VEGF signaling through NADPH oxidase-derived ROS.Antioxid Redox Signal. 2007;9:731–739.
476 Arterioscler Thromb Vasc Biol March 2008
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from

10. Roy S, Khanna S, Nallu K, Hunt TK, Sen CK. Dermal wound healing issubject to redox control. Mol Ther. 2006;13:211–220.
11. Sen CK, Khanna S, Babior BM, Hunt TK, Ellison EC, Roy S. Oxidant-induced vascular endothelial growth factor expression in human kerati-nocytes and cutaneous wound healing. J Biol Chem. 2002;277:33284–33290.
12. Mason JC, Yarwood H, Sugars K, Morgan BP, Davies KA, Haskard DO.Induction of decay-accelerating factor by cytokines or the membrane-attack complex protects vascular endothelial cells against complementdeposition. Blood. 1999;94:1673–1682.
13. Roy S, Khanna S, Shah H, Rink C, Phillips C, Preuss H, Subbaraju GV,Trimurtulu G, Krishnaraju AV, Bagchi M, Bagchi D, Sen CK. Humangenome screen to identify the genetic basis of the anti-inflammatoryeffects of Boswellia in microvascular endothelial cells. DNA Cell Biol.2005;24:244–255.
14. Khanna S, Roy S, Park HA, Sen CK. Regulation of c-Src Activity inGlutamate-induced Neurodegeneration. J Biol Chem. 2007;282:23482–23490.
15. Sen CK, Roy S, Han D, Packer L. Regulation of cellular thiols in humanlymphocytes by alpha-lipoic acid: a flow cytometric analysis. Free RadicBiol Med. 1997;22:1241–1257.
16. Suarez Y, Fernandez-Hernando C, Pober JS, Sessa WC. Dicer dependentmicroRNAs regulate gene expression and functions in human endothelialcells. Circ Res. 2007;100:1164–1173.
17. Roy S, Khanna S, Alessio HM, Vider J, Bagchi D, Bagchi M, Sen CK.Anti-angiogenic property of edible berries. Free Radic Res. 2002;36:1023–1031.
18. Ikeda S, Yamaoka-Tojo M, Hilenski L, Patrushev NA, Anwar GM,Quinn MT, Ushio-Fukai M. IQGAP1 regulates reactive oxygenspecies-dependent endothelial cell migration through interacting withNox2. Arterioscler Thromb Vasc Biol. 2005;25:2295–2300.
19. Yamaoka-Tojo M, Ushio-Fukai M, Hilenski L, Dikalov SI, Chen YE,Tojo T, Fukai T, Fujimoto M, Patrushev NA, Wang N, Kontos CD,Bloom GS, Alexander RW. IQGAP1, a novel vascular endothelial growthfactor receptor binding protein, is involved in reactive oxygen species–dependent endothelial migration and proliferation. Circ Res. 2004;95:276–283.
20. Tressel SL, Huang RP, Tomsen N, Jo H. Laminar shear inhibits tubuleformation and migration of endothelial cells by an angiopoietin-2dependent mechanism. Arterioscler Thromb Vasc Biol. 2007;27:2150–2156.
21. El-Remessy AB, Al-Shabrawey M, Platt DH, Bartoli M, Behzadian MA,Ghaly N, Tsai N, Motamed K, Caldwell RB. Peroxynitrite mediatesVEGF’s angiogenic signal and function via a nitration-independentmechanism in endothelial cells. Faseb J. 2007;21:2528–2539.
22. Gordillo GM, Sen CK. Revisiting the essential role of oxygen in woundhealing. Am J Surg. 2003;186:259–263.
23. Eligini S, Stella Barbieri S, Cavalca V, Camera M, Brambilla M, DeFranceschi M, Tremoli E, Colli S. Diversity and similarity in signalingevents leading to rapid Cox-2 induction by tumor necrosis factor-alphaand phorbol ester in human endothelial cells. Cardiovasc Res. 2005;65:683–693.
24. Chen JX, Chen Y, DeBusk L, Lin W, Lin PC. Dual functional roles ofTie-2/angiopoietin in TNF-alpha-mediated angiogenesis. Am J PhysiolHeart Circ Physiol. 2004;287:H187–H195.
25. Rajashekhar G, Willuweit A, Patterson CE, Sun P, Hilbig A, Breier G,Helisch A, Clauss M. Continuous endothelial cell activation increasesangiogenesis: evidence for the direct role of endothelium linking angio-genesis and inflammation. J Vasc Res. 2006;43:193–204.
26. Corda S, Laplace C, Vicaut E, Duranteau J. Rapid reactive oxygenspecies production by mitochondria in endothelial cells exposed to tumornecrosis factor-alpha is mediated by ceramide. Am J Respir Cell Mol Biol.2001;24:762–768.
27. Mukherjee TK, Mukhopadhyay S, Hoidal JR. The role of reactive oxygenspecies in TNFalpha-dependent expression of the receptor for advanced
glycation end products in human umbilical vein endothelial cells. BiochimBiophys Acta. 2005;1744:213–223.
28. Sen CK, Roy S. miRNA: licensed to kill the messenger. DNA Cell Biol.2007;26:193–194.
29. Berasi SP, Xiu M, Yee AS, Paulson KE. HBP1 repression of the p47phoxgene: cell cycle regulation via the NADPH oxidase. Mol Cell Biol.2004;24:3011–3024.
30. Yang WJ, Yang DD, Na S, Sandusky GE, Zhang Q, Zhao G. Dicer isrequired for embryonic angiogenesis during mouse development. J BiolChem. 2005;280:9330–9335.
31. Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S. Role of Dicer andDrosha for endothelial microRNA expression and angiogenesis. Circ Res.2007;101:59–68.
32. Kim YK, Kim VN. Processing of intronic microRNAs. Embo J. 2007;26:775–783.
33. Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. NatRev Mol Cell Biol. 2005;6:376–385.
34. Kim YM, Kim KE, Koh GY, Ho YS, Lee KJ. Hydrogen peroxideproduced by angiopoietin-1 mediates angiogenesis. Cancer Res. 2006;66:6167–6174.
35. Stone JR, Yang S. Hydrogen peroxide: a signaling messenger. AntioxidRedox Signal. 2006;8:243–270.
36. Chen JX, Zeng H, Tuo QH, Yu H, Meyrick B, Aschner JL. NADPHoxidase modulates myocardial Akt, ERK1/2 activation, and angiogenesisafter hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. 2007;292:H1664–H1674.
37. Huang SS, Zheng RL. Rosmarinic acid inhibits angiogenesis and itsmechanism of action in vitro. Cancer Lett. 2006;239:271–280.
38. Guo AM, Arbab AS, Falck JR, Chen P, Edwards PA, Roman RJ, ScicliAG. Activation of vascular endothelial growth factor through reactiveoxygen species mediates 20-hydroxyeicosatetraenoic acid-induced endo-thelial cell proliferation. J Pharmacol Exp Ther. 2007;321:18–27.
39. Chen XL, Zhang Q, Zhao R, Medford RM. Superoxide, H2O2, and ironare required for TNF-alpha-induced MCP-1 gene expression in endothe-lial cells: role of Rac1 and NADPH oxidase. Am J Physiol Heart CircPhysiol. 2004;286:H1001–H1007.
40. Li JM, Mullen AM, Yun S, Wientjes F, Brouns GY, Thrasher AJ, ShahAM. Essential role of the NADPH oxidase subunit p47(phox) in endo-thelial cell superoxide production in response to phorbol ester and tumornecrosis factor-alpha. Circ Res. 2002;90:143–150.
41. Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and theregulation of hydrogen peroxide signaling. Science. 2003;300:650–653.
42. Fox GC, Shafiq M, Briggs DC, Knowles PP, Collister M, Didmon MJ,Makrantoni V, Dickinson RJ, Hanrahan S, Totty N, Stark MJ, Keyse SM,McDonald NQ. Redox-mediated substrate recognition by Sdp1 defines anew group of tyrosine phosphatases. Nature. 2007;447:487–492.
43. Lander HM, Milbank AJ, Tauras JM, Hajjar DP, Hempstead BL,Schwartz GD, Kraemer RT, Mirza UA, Chait BT, Burk SC, Quilliam LA.Redox regulation of cell signalling. Nature. 1996;381:380–381.
44. Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duoxenzymatic activity and expression. Free Radic Biol Med. 2007;43:319–331.
45. Li JM, Shah AM. Mechanism of endothelial cell NADPH oxidase acti-vation by angiotensin II. Role of the p47phox subunit. J Biol Chem.2003;278:12094–12100.
46. Yee AS, Paulson EK, McDevitt MA, Rieger-Christ K, Summerhayes I,Berasi SP, Kim J, Huang CY, Zhang X. The HBP1 transcriptionalrepressor and the p38 MAP kinase: unlikely partners in G1 regulation andtumor suppression. Gene. 2004;336:1–13.
47. Hsu PW, Lin LZ, Hsu SD, Hsu JB, Huang HD. ViTa: prediction of hostmicroRNAs targets on viruses. Nucleic Acids Res. 2007;35:D381–D385.
48. Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ.miRBase: microRNA sequences, targets and gene nomenclature. NucleicAcids Res. 2006;34:D140–D144.
Shilo et al miRNA Regulation of Redox Signaling 477
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from

Shani Shilo, Sashwati Roy, Savita Khanna and Chandan K. SenHuman Microvascular Endothelial Cells
Evidence for the Involvement of miRNA in Redox Regulated Angiogenic Response of
Print ISSN: 1079-5642. Online ISSN: 1524-4636 Copyright © 2008 American Heart Association, Inc. All rights reserved.
Greenville Avenue, Dallas, TX 75231is published by the American Heart Association, 7272Arteriosclerosis, Thrombosis, and Vascular Biology
doi: 10.1161/ATVBAHA.107.1606552008;28:471-477; originally published online February 7, 2008;Arterioscler Thromb Vasc Biol.
http://atvb.ahajournals.org/content/28/3/471World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://atvb.ahajournals.org/content/suppl/2008/02/26/ATVBAHA.107.160655.DC1.htmlData Supplement (unedited) at:
http://atvb.ahajournals.org//subscriptions/
at: is onlineArteriosclerosis, Thrombosis, and Vascular Biology Information about subscribing to Subscriptions:
http://www.lww.com/reprints
Information about reprints can be found online at: Reprints:
document. Question and AnswerPermissions and Rightspage under Services. Further information about this process is available in the
which permission is being requested is located, click Request Permissions in the middle column of the WebCopyright Clearance Center, not the Editorial Office. Once the online version of the published article for
can be obtained via RightsLink, a service of theArteriosclerosis, Thrombosis, and Vascular Biologyin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on September 23, 2016
http://atvb.ahajournals.org/D
ownloaded from

SUPPLEMENTARY INFORMATION (SI): Legends for Figures I to VI.
Fig. I. Double, but not single, transfection of HMEC with Dicer siRNA successfully down-
regulated miRNA content. Single transfection of HMEC with Dicer siRNA (72h) decreased
Dicer mRNA transcription (A), lowered Dicer protein expression (western blot), C+D. did not
reduce significantly hsa-miR-222 and hsa-miR-18a expression levels (real-time PCR), E. cell
proliferation was not effected in 24 h and 48 h, F. double Dicer siRNA transfection (48+72h)
decreased Dicer mRNA transcription (real-time PCR), G. silenced Dicer protein expression
(western blot), H+I. significant reduction of hsa-miR-222 and hsa-miR-18a expression (real-time
PCR); J. cell proliferation is decreased; Results are mean ± S.D. * p<0.05 compared with
control.
Fig. II. Dicer knockdown induced miRNA deficiency increased VEGF expression. Dicer
knockdown increased VEGF gene (A) and protein (B) expression. VEGF protein was measured
from cell culture media. Results are mean ± S.D. * p<0.05 compared with control.
Fig. III. Dicer knockdown induced miRNA deficiency impaired human microvascular
endothelial cell migration. Following double transfection with control and Dicer siRNA, cells
were re-seeded, scratched the next day and cell migration was measured every 2 h using phase
contrast microscopy. miRNA deficient HMEC showed impaired cell migration. Representative
image of at least 3 independent experiments. For quantitative results see Fig. 1D.

Fig. IV. Dicer knockdown induced miRNA deficiency limited ROS production under basal
and activation conditions. Dicer knockdown HMEC were activated with 100 nM PMA for 4 h.
A, Fluorescence microscopic images of DCF treated HMEC. DAPI staining shows nuclei, image
representative of 3 independent experiments; B, HMEC double-transfected with siDicer or
siControl were activated with 50 ng/ml VEGF for 1 h. Fluorescence microscopic images of DCF
(green, ROS) and DAPI (blue, nuclei) were collected. Images shown are representative of three
independent experiments. For quantitative results see Fig. 2D.
Fig. V. Significance of ROS in the angiogenic response of HMEC. A. Cells were seeded and
scratched the following day. Percentage of the gap covered by migrating cells was monitored by
time lapse microscopy. Treatment of HMEC with the antioxidant NAC limited cell migration. B.
Cells were seeded on Matrigel® and tube formation was determined. Results are mean ± S.D. *
p<0.05 compared with control.
Fig. VI. Exogenously administered H2O2 rescued Dicer knockdown HMEC from impaired
Matrigel® tube formation. Dicer knockdown and control HMEC were seeded on Matrigel®
and treated or not with 0.1 or 1 µM H2O2. Images were taken after 5h, 10h and 20h of H2O2
treatment. H2O2 induced tube formation in Dicer knockdown HMEC. Representative images
(20h time-point) of 4 independent experiments are shown. For quantitative scoring see Figure 3.

Supplementary Table III. Methods to assay angiogenic responses in vitro
In vitro angiogenic response: Matrigel® endothelial tube formation assay
Four well plates were coated with 100 µl Matrigel® (Cultrex® Basement Membrane Extract
reduced growth factor; R&D Systems, Minneapolis, MN) and let to solidify for 30 min at 37oc.
Next, transfected HMEC were seeded (10,000 cells/well) on top of the solidified Matrigel® and
maintained in a cell culture incubator. Endothelial tube formation was observed and digitally
photographed 24h post seeding under an inverted light microscope at 10 X magnification
(Axiovert 200M; Zeiss, Oberkochen, Germany) as described previously 1.
In vitro angiogenic response: Endothelial cell migration assay
After transfection, HMEC were reseeded in 4-well plates (0.2 x 106/500µl in each well). The
next day, the monolayers were scratched with a 1-10 µl size pipette tip. The distance of the gap
caused was measured under a 10x phase objective of a light microscope and recorded every 2 h
(Axiovert 200M; Zeiss, Oberkochen, Germany) 2.
References 1. Roy S, Khanna S, Alessio HM, Vider J, Bagchi D, Bagchi M, Sen CK. Anti-angiogenic
property of edible berries. Free Radic Res. 2002;36:1023-1031. 2. Wang A, Nomura M, Patan S, Ware JA. Inhibition of protein kinase Calpha prevents
endothelial cell migration and vascular tube formation in vitro and myocardial neovascularization in vivo. Circ Res. 2002;90:609-616.

Supplementary Table IV. Methods to determine intracellular reactive oxygen species (ROS)
Detection of ROS was performed using dichlorodihydrofluorescein diacetate (H2DCF-DA)
(Molecular Probes, Invitrogen, Carlsbad, CA). This probe has high reactivity to hydrogen
peroxide and low reactivity to superoxide anions 1. After transfection, the cells were washed with
PBS, centrifuged (500g, 5 min), resuspended in PBS and incubated with 10 µM H2DCF-DA for
20 min at 37°C. To detect cellular fluorescence, the fluorochrome-loaded cells were excited
using a 488-nm argon-ion laser in a flow cytometer. The dichlorofluorocein (DCF) emission was
recorded at 530 nm. Data were collected using a flow-cytometer from at least 5000 cells.
Alternatively dihydroethidium (DHE) (Molecular Probes, Invitrogen, Carlsbad, CA)
fluorescence was recorded using the same protocol with excitation at 488 nm and emission at
575 nm. As additional approach, the Image-iT live green ROS detection system (Molecular
Probes, Invitrogen, Carlsbad, CA) was used to visualize ROS in live cells. Fluorescent carboxy-
H2DCF-DA permeates live cells and is deacetylated by non-specific intracellular esterases. In the
presence of ROS, the reduced fluorescein compound is oxidized and emits bright green
fluorescence. Fluorescence microscopy (10x, Axiovert 200M; Zeiss, Oberkochen, Germany) was
performed to capture images of nuclei (blue fluorescence; Hoechst 33342) and oxidized
fluorescein 2. Transfected cells were activated with either PMA (Sigma-Aldrich, St. Louis, MO),
TNFα or VEGF165 (R&D Systems, Minneapolis, MN). Cells were serum-starved for 16 h with
0.5 % FBS prior to treatment with VEGF.
References 1. Shilo S, Aharoni-Simon M, Tirosh O. Selenium attenuates expression of MnSOD and
uncoupling protein 2 in J774.2 macrophages: molecular mechanism for its cell-death and antiinflammatory activity. Antioxid Redox Signal. 2005;7:276-286.
2. Khanna S, Roy S, Parinandi NL, Maurer M, Sen CK. Characterization of the potent neuroprotective properties of the natural vitamin E alpha-tocotrienol. J Neurochem. 2006;98:1474-1486.

Supplementary Table V. Immunohostochemical methods used.
Following transfection of HMEC, cells (0.5x106/well) were seeded on cover slips in 6-well
plates. The following day, cells were washed twice with ice-cold PBS and then fixed in 10%
buffered formalin for 20 min. Next, the cells were washed thrice with PBS followed by
permeabilization using 0.1% Triton X-100/PBS for 15 min. The cells were then washed 3 times
with PBS and incubated with 10% goat or horse serum (Vector Laboratories) for 1h at room
temperature. Next, the cells were incubated with anti-Dicer (1:100, in 10% goat serum) or anti-
HBP1, (Santa Cruz Biotechnologies, Santa Cruz, CA; 1:75, in 10% horse serum) overnight in
4ºC. After incubation with primary antibodies, cells were washed thrice with PBS and incubated
with an Alexa-fluor mouse (green) for Dicer and Alexa-fluor goat (red) for HBP1 for 1h at room
temperature. After three washes with PBS and incubation with DAPI (1:10,000) for 2 min, the
endothelial cells were washed with PBS and mounted in gelmount (aqueous mount, Vector
Laboratories) for microscopic imaging. For immunocytochemistry during tube formation, cells
were seeded on Matrigel® for tube formation assay and fixed after 2 h. Next, the same staining
assay was performed using goat serum, anti-p47phox and Alexa-fluor mouse (red).

Supplementary Table VI. Detailed Figure Legends for Illustrations Appearing in Hard-
Fig. 1. Dicer knockdown induced miRNA deficiency impaired the angiogenic response of
human microvascular endothelial cells. A-B, Matrigel® tube formation assay: A, Matrigel®
tube formation visualized by phase contrast microscopy. Representative image of at least 3
independent experiments; B, measurement of tube curve length (µM); Results are mean ± S.D. *
p<0.05 compared with control. C, cell migration, quantitative data of the observation shown in
Supplementary figure III; D, Cell migration, as determined by the slope of the line between the
4h and 6h time points (see Supplementary Fig. III), was diminished by 50% in response to Dicer
knockdown; E, Cell viability as measured by propidium iodide exclusion following double
transfection for Dicer knock-down. Results are mean ± S.D. * p<0.05 compared with control.
Fig. 2. Dicer knockdown induced miRNA deficiency limited ROS production under basal
and activation conditions. A, HMEC double transfected with siDicer or siControl were
activated with 100 nM PMA for 30 min. DHE fluorescence was measured using a flow
cytometer; B, Dicer knockdown HMEC were activated with 100 nM PMA for 4 h. DCF
fluorescence was measured using a flow cytometer; C, Dicer knockdown HMEC were activated
with 50 ng TNFα for 90 min. DCF fluorescence was measured as in B; D, HMEC double-
transfected with siDicer or siControl were activated with 50 ng/ml VEGF for 1 h. Fluorescence
microscopic images (see Supplementary Fig. IV) were collected. Quantitation of DCF green
fluorescence. Results are mean ± S.D. * p<0.05 compared with corresponding siControl.

Fig. 3.Exogenously administered H2O2 rescued Dicer knockdown HMEC from impaired
Matrigel® tube formation. Dicer knockdown and control HMEC were seeded on Matrigel®
and treated or not with 0.1 or 1 µM H2O2. Images were taken after 5h, 10h and 20h of H2O2
treatment. H2O2 induced tube formation in Dicer knockdown HMEC. Representative images
(20h time-point) of 4 independent experiments are shown in supplementary Fig. VI. Scoring of
tube curve length (µm). Results are mean ± S.D. *, compared to control, p <0.05.
Fig. 4. Dicer knockdown induced miRNA deficiency specifically down-regulated p47phox
expression while not affecting the expression of gp91, Rac1 and Rac2 components of the
NADPH oxidase complex. Following double transfection of HMEC with siDicer or siControl,
whole cell lysates were extracted and subjected to Western blot analysis. A, representative
images of Dicer, gp91, Rac1, Rac2 protein levels; B, densitometry analysis of A; C, p47phox
expression was decreased in Dicer knockdown cells; D, densitometry analysis of C; E, following
transfection with siDicer or siControl, cells were seeded on Matrigel® for 2h to enable tube
formation. Cells were fixed and stained with anti-p47phox. Compared to siControl transfected
cells, Dicer knockdown cells showing lower abundance of p47phox in the tubes (top panel shows
DIC image for reference). Results are mean ± S.D. * p<0.05 compared with corresponding
siControl.
Fig. 5. Knockdown of p47phox impaired cell migration and decreased inducible ROS
production. HMEC were transfected with siRNA against p47phox for 72h. A, Western blot; B,
following activation with 100 nM PMA for 4h, p47phox knockdown cells showed decreased
levels of ROS production as detected by DCF staining and flow cytometry; C, following
transfection with sip47phox, cells were seeded and scratched the following day. Cell migration
(2h) was impaired in p47phox knockdown cells; Results are mean ± S.D. * p<0.05 compared
with corresponding siControl.

Fig. 6. Dicer silencing up-regulated the expression of the transcription factor HBP1 which
played a central role in impairing the angiogenic response of HMEC. Following siDicer or
siControl double transfection, HMEC were re-seeded on cover slips and stained with antibodies
against Dicer and HBP1. DAPI was used for staining of nuclei. A, Dicer knockdown lowered the
abundance of cellular Dicer protein levels; B, Dicer knockdown elevated the expression of
HBP1; C, Quantitation of HBP1 red fluorescence shown in B; D, Western blot of nuclear
extracts; E, co-transfection of siDicer with siRNA against HBP1 decreased HBP1 mRNA levels;
F-G, co-transfection of siDicer with siRNA against HBP1 significantly released the inhibitory
effects of Dicer knockdown on Matrigel® tube formation. increased the cells angiogenic
response shown by tube formation on Matrigel; G, Quantitation of tube length. * p<0.05
compared with corresponding siDicer transfected controls.

0
0.2
0.4
0.6
0.8
1
1.2
1.4
Dicer mRNA (fold change)
siControlsiDicer
+
+
_
_
A
0
0.2
0.4
0.6
0.8
1
1.2
1.4
miR-18a (fold change)
+
+
_
_
B D
Dicer
GAPDH
Figure I
*
E
0
5
10
15
20
25
30
siControlsiDicer
24 h 48 h
cell proliferation (A.U)
+ _
+
_
siControlsiDicer
+
+
_
_ +
+
_
_ + _
+
_ Dicer mRNA (fold change)
0
0.2
0.4
0.6
0.8
1
1.2
1.4
*
Dicer
GAPDH
miR-18a (fold change) J
24 h 48 h
cell proliferation (A.U)
siControlsiDicer
*
0
5
10
15
20
25
30F G
0
0.2
0.4
0.6
0.8
1
1.2
1.4
*
I
+ _
0
0.2
0.4
0.6
0.8
1
1.2
miR-222 (fold change)
+ _
C
0
0.2
0.4
0.6
0.8
1
1.2
+ _ +
_
miR-222 (fold change)
*
H

Figure II
0
0.2
0.4
0.6
0.8
1
1.2
1.4
0
20
40
60
80
100
120
140V
EG
F m
RN
A (
fold
chan
ge)
siControlsiDicer
A
+
+
_
_
B V
EG
F (
ng/µ
gpro
tein
)
siControlsiDicer
+
+
_
_
*
*

siControl siDicer
2 h
4 h
6 h
0 h
Figure III

A
DCF
DAPI
siControl siDicer
PMA 4 h (100 nM)
B
no treatment
VEGF (50 ng/m
l)
siControl siDicer
Figure IV

0
10
20
30
40
50
*
4 h 2 h
*
_ + NAC
A
% cell migration
B
+ NAC
Figure V
_

Figure VI
siControl
siDicer
no treatment 0.1 µM H2O2
1 µM H2O2

Supplementary Table I. Primer sets used for real-time PCR
h Dicer,
F: 5'- TGC CAG TTG GGA AAG AGA CTG TTA A-3'
R: 5'- TAT GGG TTT GGC CGT CAG TAT T-3';
h VEGF A,
F: 5'- TGC CCA CTG AGG AGT CCA ACA T-3'
R: 5'- CAC GTC TGC GGA TCT TGT ACA AAC A-3'
h HBP1,
F: 5'- GAATTTGCCATCTTCACCTGGATAT-3'
R: 5'- CTGAGGTATTTTGGTTCCAAGATGA-3'
h β-Actin,
F: 5'- TCA ACT CCA TCA TGA AGT GTG ACG T-3'
R: 5'- TTC TGC ATC CTG TCG GCA AT-3'.

Supplementary Table II. Methods for the detection of proteins
Western blot
After protein extraction, protein concentrations were determined using BCA protein reagents
(Pierce, Rockford, IL). Samples (20-35 µg of protein/lane for whole cell lysates; 15 µg of
protein/lane for nuclear extracts) were separated on 8%-12.5% SDS-polyacrylamide gel
electrophoresis, and probed with the following antibodies: anti-Dicer (Abcam, Cambridge, MA),
anti-gp91, anti-RAC1, anti-RAC2 (Upstate Biotechnology, Inc., Lake Placid, NY), anti-p47phox
(BD transduction laboratories, San Jose, CA) or anti-HMG box containing protein-1 (anti-HBP1,
Santa Cruz Biotechnologies, Santa Cruz, CA). The secondary antibodies used were as follows:
anti-mouse and anti-rabbit (Amersham Biosciences, Buckinghamshire, UK) and anti-goat
(Sigma-Aldrich, St. Louis, MO). To evaluate the loading efficiency, membranes were probed
with anti-GAPDH-HRP (Sigma-Aldrich, St. Louis, MO) or with anti-Lamin A (Sigma-Aldrich,
St. Louis, MO) antibody.
Determination of VEGF Protein
Following transfection, cell media was collected. VEGF level in the medium was determined
using commercially available ELISA kit (R&D Systems, Minneapolis, MN). Results were
adjusted against total protein concentration in the samples.
Copyright © 2022 FDOKUMEN




















