Studying miRNA-mRNA interactions in Neuro-developmental disorders
30
1 Studying miRNA-mRNA interactions in Neuro- developmental disorders Thesis Submitted in partial fulfillment of the requirements of BITS C421T By Biswa Prasanna Mishra 2008B1A1609H Under the supervision of Dr. Savitha Govardhan Assistant Professor, Department of Biological Sciences BIRLA INSTITUTE OF TECHNOLOGY AND SCIENCE, PILANI HYDERABAD CAMPUS 28 November, 2012
-
Upload
bits-hyderabad -
Category
Documents
-
view
0 -
download
0
Transcript of Studying miRNA-mRNA interactions in Neuro-developmental disorders

1
Studying miRNA-mRNA interactions in Neuro-developmental disorders
Thesis
Submitted in partial fulfillment of the requirements of BITS C421T
By
Biswa Prasanna Mishra 2008B1A1609H
Under the supervision of Dr. Savitha Govardhan
Assistant Professor, Department of Biological Sciences
BIRLA INSTITUTE OF TECHNOLOGY AND SCIENCE, PILANI HYDERABAD CAMPUS
28 November, 2012

2
Acknowledgements
I would like to thank Dr. Savitha Govardhan, my Thesis Instructor,
Department of Biological Sciences, who guided me through the
course of this thesis and for giving valuable suggestions regarding
the same.
I am greatly indebted to Ms. Priyanka Purkayastha, RS in
Department of Biological Sciences for her continued guidance
and assistance during this thesis. I am also thankful to other
Research Scholars for their support.

3
CERTIFICATE
This is to certify that the thesis entitled, “Studying miRNA-mRNA interactions in Neuro-developmental disorders” and submitted by Biswa Prasanna Mishra, ID No. 2008B1A1609H, in partial fulfillment of the requirements of BITS C421T Thesis, embodies the work done by him/her under my supervision.
Signature of the Supervisor:
Date:
Name:
Designation:

4
Abstract
A neuro-developmental disorder is an impairment of the growth and development of the brain or central nervous system. It usually refers to a disorder of brain function that affects emotion, learning ability and memory and that unfolds as the individual grows. The major ones are Schizophrenia and Autism. In this dissertation, the causes and genetic information for Schizophrenia have been studied. RNA interference and subsequent miRNA-mRNA interactions are looked into from a genetic point of view. Certain important web-servers like miRanda and TargetScan have been used to predict target genes specific to the aforementioned interactions and then looking into conserved and partly conserved families.

5
Table of Contents
Acknowledgements…………………………………………………………… 2
Certificate………………………………………………………………………. 3
Abstract………………………………………………………………………… 4
Chapter 1: Introduction…………………………………………………......... 6
Chapter 2: Neuro-developmental disorders………………………………... 7
Chapter 3: RNA Interference (RNAi)………………………………………. 15
Chapter 4: Prediction of microRNA targets……………………………….. 16
Chapter 5: Results and Discussion…………………………………………19
Conclusion……………………………………………………………………..28
References……………………………………………………………………. 29

6
Chapter 1: Introduction MicroRNAs (miRNAs) are endogenous short single-stranded RNAs, acting as post-transcriptional modulators of gene expression. Animal miRNAs interact with target mRNAs via partially complementary base pairing. In most of the cases, this results in the repression of mRNA expression through destabilization and/or translational repression. miRNAs are estimated to comprise almost 1% of animal genes and this makes them one of the largest classes of regulators. Computational approaches, as well as large-scale transcriptomic and proteomic approaches, have revealed that 1 miRNA can potentially regulate 100s of mRNAs, by globally modulating their expression at a rather small scale. Thus, they are often considered as fine-tuning regulators or buffering agents for genetic networks. They are as such attractive candidates for harboring genetic variations which lead to non-lethal human diseases. This is particularly relevant in disorders of the human nervous system. Here, subtle modulations of the brain physiology may not affect the organism as a whole, but may have a tremendous impact on cognitive functions or societal interactions.
The miRNA machinery has been shown to influence almost every cellular and developmental process investigated so far. What remains a challenge is the understanding of the functions of single or subnetworks of miRNAs in specific cellular contexts, the biological relevance of their predicted miRNA-mRNAs interactions. Various research groups have now undertaken this task. Specific miRNAs have been conclusively implicated at each and every step of brain development and maturation, but only a few of the miRNAs have been studied.
Currently, there are roughly 470 odd miRNAs in humans, although there might be many more. miRNAs are expected to have multiple targets but till now only 66 have been confirmed experimentally. It is vital that computational techniques are used to unravel their regulatory effects and implications for diseases and diagnostic purposes. The prediction of miRNA targets has started since the 3’ untranslated regions (3’UTRs) of transcripts have been determined to contain binding sites for them. The efficiency of computational approaches to locate and rank potential genomic binding sites is supported by the relatively higher degree of miRNA complementarity to the experimentally determined binding sites. In this project, I have tried to study the interactions by indentifying the possible target sites using a few web-servers like miRanda and TargetScan.

7
Chapter 2: Neuro-developmental disorders A neurodevelopmental disorder is essentially an impairment of the growth and development of the brain or CNS. It refers to a disorder of brain function that affects emotion, learning ability and memory, and unfolds as the individual grows. Major ones include Autism, Schizophrenia, Asperger syndrome, Fetal alcohol spectrum disorder, fragile-X syndrome and Down syndrome. These disorders are associated with varying degrees of mental, emotional, physical and economic burden to individuals, families and societies in general.
Causes
Now, there are many causes of neurodevelopmental disorder, ranging from deprivation, genetic and metabolic diseases, immune disorders, infectious diseases, nutritional factors, physical trauma, and toxic and environmental factors. Some like autism and schizophrenia are considered to be multifactorial syndromes, i.e., they have many causes but more specific neurodevelopmental manisfestation. However others like PANDAS are presently thought to have a more primary cause and a more specific manifestation.
Deprivation: Babies and children require emotional nurture from caregivers. Thus, there is a variety of disorders arising from the lack of it. Hospitalism, the most sever one, is a wasting away to the point of death. A sublethal form, anaclithic depression, was first described in the 1940s. it occurred infants over the age of 6 months who suffered the loss of their mothers, who then became depressed and thus showed behavioral retardation. This form of retardation has been observed in emotionally deprived children living with their families. A common example of sensory deprivation due to biological factors is blindness. Untreated blind infants may lead to severe autistic-like behaviors.
Genetic disorders: A prominent example is Trisomy 21 also called Down syndrome, which usually results from an extra chromosome 21. It is characterized by short stature, eyelid folds, abnormal fingerprints and palmprints, heart defects, poor muscle tone and mental retardation. Less common disorders include Fragile X syndrome, Rett syndrome and Williams’s syndrome.
Immune dysfunction: During pregnancy, immune reactions in both maternal and developing child can produce neurodevelopmental disorders. Prominent examples are PANDAS (Pediatric Autoimmune Neuropsychiatric Disorders Associated with Streptococcal infection) and Sydenham’s chorea. The former produces abnormal body movements, emotional disturbance and obsessive compulsive disorder symptoms while the latter results in more abnormal movements of the body.

8
Infectious diseases: A number of infectious diseases can be transmitted either by congenitally or in the early childhood, causing serious neurodevelopmental disorders such as Schizophrenia. Congenital toxoplasmosis may result in formation of cysts in the brain and other organs. This causes a variety of neurological deficits. Congenital syphilis may progress to neurosyphilis, if untreated.
Metabolic disorders: They are present in either the mother or the child, causing neurodevelopmental disorders. Two examples are diabetes mellitus and phenylketonuria. A lot of such inherited diseases may directly affect the child’s metabolism and neural development, but they can also affect the child during gestation.
Nutrition: Nutrition deficits may cause neurodevelopmental disorders such as Spina bifida and anencephaly, which is rare. Both are neural tube defects with malformation and dysfunction of the nervous system and supporting structures, leading to some serious physical disabilities as well as emotional sequellae. The most common cause of neural tube defects is maternal deficiency of folic acid. Iodine deficiency can produce a spectrum of neurodevelopmental disorders ranging from mild emotional disturbance to severe mental retardation.
Trauma: Brain trauma in the developing brain is a common cause of neurodevelopmental syndromes. It may be subdivided into 2 major categories, congenital injury and injury occurring in infancy or childhood. In industrial nations, the most common causes of childhood brain trauma are falls and transportation-related incidents. Child maltreatment can also produce disorders along with blindness,, neuromotor deficits and cognitive impairment.
Toxic and environmental factors
The first evidence for a proper functional role of miRNAs in the nevous system came from the genetic studies pointing to a role of miRNAs in neural cell fate speciation. The best characterized example is the asymmetric specification of the two taste receptor neurons, ASER (right) and ASEL (left) in Caenorhabditis elegans. These 2 neurons express a different set of chemoreceptor genes, which are associated with a functional
: Prenatal exposure to any amount of alcohol can also result in fetal alcohol spectrum disorder. Heavy-metal poisoning like mercury-poisoning can cause Minamata disease. Developmental mercury poisoning can cause a variety of problems from mild impairment of emotional development to full blown syndromes of nerve damage, visual impairment, impaired co-ordination and ambulation, hallucinations, mental retardation, derpression and death.
MicroRNAs and the Nervous system

9
lateralization. 2 miRNAs, lsy-6 and miR-273 are specifically expressed in ASEL and ASER respectively.
The miRNAs, miR-9a and miR-7, play a role in cell fate specification in Drosophila developing sensory organs. Sensory organs in flies develop from the divisions of a single sensory organ precursor cell (SOP). SOP selection from the ectoderm begins with the delimitation of proneural clusters, competence groups of cells expressing proneural genes. In 1 cell of the cluster, the level of such genes will be set a tad bit higher, through a lateral inhibition mechanism involving the Notch pathway. This is specified as SOP. The flies mutant for miR-9a develop supernumerary SOP cells. Epistasis experiments suggest that miR-9a, expressed in non-SOP cells, acts via direct targeting of the transcription factor Senseless. In turn, miR-7 targets Enhancer-of-split factors, negative regulators of proneural genes expression, thereby further increasing proneural gene expression in SOP cells. They appear to provide accuracy to the developmental programme via buffering gene expression levels. Whether stabilization and canalization of cell fate specification is a recurrent function of miRNAs remains to be assessed.
MicroRNAs and brain patterning
The spatio-temporally restricted expression of miRNAs in the developing vertebrate central nervous system suggests an involvement of brain patterning. The first clear functional implication of the miRNA patway in brain development and morphogenesis comes from the analysis of MZdicer zebrafish mutant cells. These are defective for both maternal and zygotic Dicer activity and display severe defects in brain morphogenesis and neuronal differentiation. Most miRNAs may be actually dispensable for early tissue fate establishment invertebrates. But in Drosophila, miRNA knock-downs lead to severe blastoderm patterning defects. The reason is probably that the plasticity of early developmental processes in vertebrates makes them less sensitive to subtle modulations in the timing, expression pattern or expression levels of patterning regulators.
MicroRNAs and neurogenesis
The fact that miRNAs facilitate the transition between developmental states renders them attractive candidate regulators of neurogenesis progression. Most mammalian in vitro culture systems suggest that some miRNAs are indeed required for the transition between a progenitor state and differentiated neurons, through inhibition of progenitor factors. The highly enriched miRNAs are induced upon ES cells neuronal differentiation. They block the cells’ function during this process, which decreases neuronal differentiation at the expense of the astrocytes. The in vivo implication of miRNAs in neurogenesis has recently been started. Conditional knock-out mice, in which Dicer is

10
ablated in neuroepithilial cells of the dorsal telencephalon, display a severe deficit in neuronal production in the neocortex. Neurons that escape also show defective expression of neuronal markers. This suggests that miRNAs are essential for proper neuronal differentiation.
MicroRNAs and brain physiology
Some miRNAs and components of the RISC complex are enriched at synapses and associated with mouse brain polysomes at dendritic spines. Biochemical evidence obtained in both Drosophila and mammals further demonstrate that the Fragile-X Mental Retardation Protein, which controls translation at the synapse, interacts with the RISC factors including Dicer and Argonaute.
Autism It is a disorder of neural development characterized by impaired social interaction and communication, and by restricted and repetitive behavior. Autism affects information processing in the brain by altering how nerve cells and their synapses connect and organize. It has a strong genetic bias, although autism genetics are complex and it is unclear whether to explain them by rare mutations or by rare combinations of common genetic variants. A number of other possible causes have been suspected, but not proven. They are namely diet, digestive tract changes, mercury poisoning, and the body’s inability to properly use vitamins and minerals, and vaccine sensitivity.
Symptoms
Children with autism typically have difficulties in pretending to play, social interactions and verbal and nonverbal communication. They may be overly sensitive in sight, hearing, touch, smell or taste, have unusual distress when routines are changed, perform unusual body movements and show unusual attachments to objects. Communication problems may be many. They cannot start or maintain a social conversation, communicate with gestures instead of words, develop language slowly or not at all, does not adjust gaze to look at objects that others are looking at, does not refer to self correctly or maybe repeat words or memorized passages.
There may be various social interactions too. Not making friends, not playing interactive games, being withdrawn, not responding to eye contact or smiles or avoid eye contact, treating others as objects and showing a lack of empathy or emotion are common ones.

11
Signs and tests
If a child fails to meet any of the following language milestones, he/she is autistic:
• Babbling by 12 months
• Gesturing (pointing, waving bye-bye) by 12 months
• Saying single words by 16 months
• Saying two-word spontaneous phrases by 24 months (not just echoing)
• Losing any language or social skills at any age
Genetics of Autism
The Autism Genome Project Consortium performed linkage analysis on 10,000 SNP markers genotyped in 1,180 odd families. With this powerful combination of marker density and sample size, this group was able to identify a single linkage peak at chromosome 11p12—13 that exceeds the threshold value for genome-wide suggestive linkage, particularly in families that have at least one female with ASD (Autism Spectrum Disorder). Only modest support was observed for the previously identified linkage peaks on chromosomes 2q and 7q. The results of this study suggest that rare genetic variants on chromosome 11p12—13 may contribute to ASD risk. The most recent attempt at linkage analysis in ASD was using technology that genotyped 500,000 SNPs in 878 families. Since marker densities that are too high can create statistical problems in linkage analyses, the authors pruned the number of markers used in their analyses to 16,311 highly polymorphic, high-quality SNPs. They found significant genome-wide linkage on chromosome 20p13 and suggestive evidence for linkage on chromosome 6q27. These linkage data provided no evidence in support of the previously identified linkage peaks on chromosomes 2q, 7q, and 17q. Neither chromosome 20p13 nor chromosome 6q27 was suggestive for linkage in the Autism Genome Project Consortium study. A Genome-wide association study (GWAS) of ASD found evidence of a genome-wide significant association signal on chromosome 5p14, outside any previously reported linkage peak for ASD. The significant association signal lies between the genes encoding cadherin 9 (CDH9) and cadherin 10 (CDH10), both of which encode cell adhesion molecules that are expressed in the developing brain. The genetic evidence is extremely compelling and convincing: the original genome-wide significant association signal in 780 families was replicated in a large case-control sample. Also, the association peaked at a single SNP that was flanked by several other markers that also showed evidence of association, suggesting that the association here was not due to a technical artifact. The strongest ASD candidate genes are those for which convergent evidence exists. Ideally speaking, linkage analysis would implicate the chromosomal region, candidate

12
gene association studies would describe replicated association of alleles, GWAS studies would implicate the gene, rare Copy Number Variations (CNVs) of the gene would be identified, and there would be some functional evidence that the gene is involved in ASD risk. No candidate gene meets all of these criteria. However, there are five genes that have convergent evidence for contributing to ASD risk, which are mentioned below. MET The MET gene encodes the MET receptor tyrosine kinase, a key regulator of neuronal migration and synapse formation in the brain. Most linkage studies implicate the chromosome 7q31 region in which the MET gene lies. The results of all genetic association studies reported to date indicate the positive association of MET gene variants. The MET promoter variant rs1858830 C allele has been associated in 5 independent samples. The MET rs1858830 C allele is functional and it decreases transcription 2-fold due to altered binding of transcription factor complexes. Expression of MET protein is decreased by a factor of 2 in postmortem brains of individuals with ASD. GABRB3 The GABRB3 gene encodes the GABAA receptor β3 subunit protein, a critical component of inhibitory signaling in the brain. Some, but not all, linkage studies of ASD implicated the chromosome 15q11—13 region in ASD. Association of the GABRB3 marker 155CA-2 has been observed in 2 samples but not replicated in 3 other samples. However, there is now overwhelming evidence in favor of chromosome 15q11—13 duplication contributing to ASD risk in ∼1% of families. A functional rare variant, also present in ∼1% of families, is associated with ASD risk. The ASD-associated mutant form of the protein causes a decreased expression of the receptor on the cell surface. Therefore, despite the absence of a clear association of common genetic variants, accumulating evidence indicates that rare variation of the GABRB3 gene contributes to a subset of cases of ASD. EN2 The EN2 gene encodes ENGRAILED 2, a transcription factor involved in cerebellar development. Many linkage studies have implicated the chromosome 7q36 location of the EN2 gene. Association of 2 alleles, the rs1861972 A allele and rs1861973 C allele, in the only intron of the EN2 gene was first observed in 2 samples and then was replicated in 4 additional samples. A study of 210 Chinese Han families did not precisely replicate the association of rs1861972 and rs1861973 but instead found association of another SNP in the EN2 intron, rs3824068. These results have implicated the EN2 gene in ASD risk, but suggested that the functional variant had not been identified. However, recent functional studies indicate that the alleles of rs1861972 and rs1861973 bind

13
different transcription factor complexes and cause a low (∼20%) but significant change in transcriptional efficiency. SLC6A The SLC6A4 gene encodes the serotonin transporter, a critical regulator of the neurotransmitter serotonin in both the brain and peripheral tissues. Since increased platelet serotonin is one of the few biomarkers that identify a subset of patients with ASD, the serotonin transporter is a plausible biological candidate for ASD risk. Multiple linkage studies found evidence for linkage of the chromosome 17q11.1—12 region at which the SLC6A4 gene resides. The short allele of the SLC6A4 promoter variant is functional and it decreases transcription efficiency, resulting in decreased gene expression and serotonin uptake activity. OXTR The OXTR gene encodes the oxytocin receptor, a known modulator of social behavior. Intranasal oxytocin administration has been shown to improve the ability of individuals with ASD to recognize emotions, emphasizing the biological plausibility of OXTR contribution to ASD risk. A genome-wide linkage analysis highlighted the chromosome 3p24—26 region of OXTR. Association of the rs2254298 A allele has been replicated in a case-control sample. A recent report described deletion of the region including OXTR and 4 neighboring genes in 1 of 120 families, altered methylation of the OXTR promoter in individuals with ASD, and decreased expression of OXTR in postmortem brains of individuals with ASD. Thus, there may be multiple modes of disrupting OXTR that result in decreased oxytocin receptor and an increased risk for ASD.
Treatment
A variety of therapies are available, including Applied behavior analysis (ABA), medications, occupational therapy, physical therapy and speech-language therapy.
Schizophrenia It is a mental disorder characterized by a breakdown of thought processes and by poor emotional responsiveness. It affects both men and women equally, beginning usually in the teen years or young adulthood, but it may also begin later in life. Childhood-onset schizophrenia begins after 5. It is rare and can be hard to tell apart from other developmental problems in childhood, such as autism.
Symptoms
Common symptoms include auditory hallucinations, paranoid or bizarre delusions, or disorganized speech and thinking, and is mostly accompanied by significant social

14
or occupational dysfunction. The onset of symptoms typically occurs in young adulthood, with a global lifetime prevalence of about 0.3-0.7%.
Classification of Schizophrenia
The schizophrenic disorder has played a pivotal role in the development of a system of classification of behavioral disorders. It has long been a prototype of the group of serious disorders of thought processes termed as psychoses, lesser serious disorders of life adjustment are termed neuroses. This disease afflicts more than 1% of the population and is both chronic and progressive accounting for more than half of the resident mental hospital population. The effectiveness of chlorpromazine in the treatment of schizophrenia set the stage for the development of realistic models of organic causes of mental disease. The primary symptoms of the disorder include disturbance of thought patterns, disturbance or affective reactions and autism or withdrawal. They represent a loss of contact with reality. The patients show very atypical responses to their social situations. They tend to make up words and sentence structures, their responses to verbal communication may bear little or no relationship to the topic at hand and frequent odd behavior. Secondary symptoms include hallucinations, delusions and paranoia. A strong evidence for a biological basis of schizophrenia is the strong genetic pattern of occurrence, the strongest relationship occurs between monozygotic twins.
Genes
Detecting specific genes on specific pathways is a first step to identifying more specific targets for improved drug treatments. The genes CACNA1B and DOC2A are known to be the important ones in schizophrenia. They carry the codes for proteins that use Calcium signals to help control how neurotransmitters are released in the brain. Two other genes RET and RIT2, are members of another signaling gene family known to be involved in brain development. Mapping studies in various mammalian species have shown the association of individual SNPs and SNP haplotypes in NRG1 (neuregulin-1, 8p21-p12). NRG1 is a glycoprotein with a variety of isoforms that bind to the ErbB family of tyrosine kinase transmembrane receptors. The association of schizophrenia to SNPs and SNP haplotypes on chromosome 6p22.3 have been reported which implicate the DTNBP1 (dysbindin) gene, in the linkage region. Dysbindin binds to dystrobrevin, part of the protein complex involved in the pathogenesis of muscular dystrophy. The proteins have diverse functions related to neurotransmitter signal transduction. Schizophrenia is associated to SNPs in PRODH2 (proline dehydroenase, 22q11.21), particularly in childhood cases and in adults with age at onset below the age of 18 years. It is a mitochondrial enzyme involved in transferring redox potential across the mitochondrial membrane. Catechol-o-methyltransferase (COMT, 22q11.21) is one of the major degradative pathways for catecholamines include dopamine. COMT sequence variation modiefies cognition through effects on dopaminergic transmission in prefrontal cortex.
15
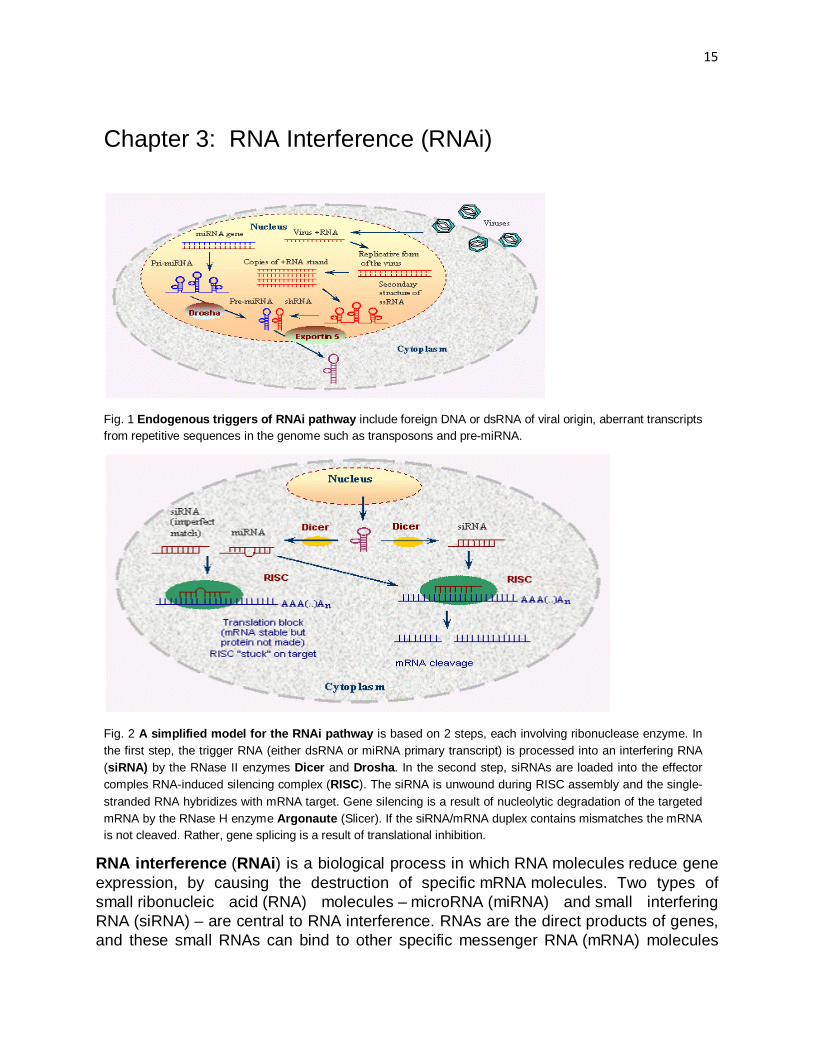
Chapter 3: RNA Interference (RNAi)
Fig. 1 Endogenous triggers of RNAi pathway include foreign DNA or dsRNA of viral origin, aberrant transcripts from repetitive sequences in the genome such as transposons and pre-miRNA.
Fig. 2 A simplified model for the RNAi pathway is based on 2 steps, each involving ribonuclease enzyme. In the first step, the trigger RNA (either dsRNA or miRNA primary transcript) is processed into an interfering RNA (siRNA) by the RNase II enzymes Dicer and Drosha. In the second step, siRNAs are loaded into the effector comples RNA-induced silencing complex (RISC). The siRNA is unwound during RISC assembly and the single-stranded RNA hybridizes with mRNA target. Gene silencing is a result of nucleolytic degradation of the targeted mRNA by the RNase H enzyme Argonaute (Slicer). If the siRNA/mRNA duplex contains mismatches the mRNA is not cleaved. Rather, gene splicing is a result of translational inhibition.
RNA interference (RNAi) is a biological process in which RNA molecules reduce gene expression, by causing the destruction of specific mRNA molecules. Two types of small ribonucleic acid (RNA) molecules – microRNA (miRNA) and small interfering RNA (siRNA) – are central to RNA interference. RNAs are the direct products of genes, and these small RNAs can bind to other specific messenger RNA (mRNA) molecules

16
and either increase or decrease their activity, for example by preventing an mRNA from producing a protein. RNA interference has an important role in defending cells against parasitic nucleotide sequences like viruses and transposons.
The RNAi pathway is found in many eukaryotes including animals and is initiated by the enzyme Dicer, which cleaves long double-stranded RNA (dsRNA) molecules into short fragments of ~20 nucleotides that are called siRNAs. Each siRNA is unwound into 2 single-stranded ssRNAs, namely the passenger strand and the guide strand. The passenger strand is degraded, and the guide strand is incorporated into the RNA-induced silencing complex (RISC). The most well-studied outcome is post-transcriptional gene silencing, which occurs when the guide strand base pairs with a complementary sequence in a messenger RNA molecule and induces cleavage by Argonaute, the catalytic component of the RISC complex.
RNAi has become a valuable research tool, both in cell culture and in living organisms, because synthetic dsRNA introduced into cells can selectively induce suppression of specific genes of interest. RNAi may be used for large-scale screens that systematically shut down each gene in the cell, which can help identify the components necessary for a particular cellular process or an event such as cell division. The pathway is also used as a practical tool in biotechnology and medicine for drug discovery and therapeutics.
Chapter 4: Prediction of microRNA targets
The prediction of microRNA targets is very crucial to study their interactions with mRNA and thus has a lot of use in diagnostics and therapeutics. In this project, I have used mainly 2 web-servers viz. miRanda and TargetScan for predicting the targets and find out certain important data useful for my results.
Identification of 3’UTRs
To identify miRNA targets in a given species, knowledge of the set of 3’UTRs for this species is a vital step. Despite accumulating genome sequences for many species, the location, extent or splice variation of 3’UTRs is still poorly characterized for many mammals. Some species-specific projects, such as the Berkeley Drosophila Genome Project (BDGP), produce really high-quality transcript info that makes possible the proper and accurate determination of a 3’UTR, from stop codon to polyadenylation site. The Ensembl database uses alignment of cDNAs and the expressed sequence tags to genomic sequences to extract 3’UTR regions. These regions can be estimated by selecting a downstream flanking sequence of the stop codon, corresponding to the length of an average human 3’UTR.
Conservation Analysis
Solutions to decrease the number of false positives in target prediction values include the filtering out of those binding sites that do not seem to be conserved across species. The use of orthologous 3’-UTRs in multiple species are considered

17
more likely to reduce the number of false positives. In case of humans and chimpanzees’ conserved targets, 99% of the entire transcript will at least be conserved. The fact is genomes are not sequenced according to their evolutionary distances.
Algorithms for miRNA target prediction
The challenging task of predicting miRNA targets has resulted in the development of several methods, which fall into several categories. Roughly, we can describe 3 types of target sites: 5’-dominant canonical, 5’-dominant seed only and 3’-compensatory. These differ in the level of complementarity of miRNA sequences to the site sequences. The various algorithms can be broadly classified into 3 types:
1. Complementarity searching
2.
: These are oriented towards recovery of known targets and the subsequent detection of further targets for experimental validation so that knowledge of miRNA binding dynamics might be improved. Most of them use complementarity to identify potential targets. This is followed by iterative rounds of filtering based on thermodynamics, binding site structure and conservation. After filtering, a score is typically applied to each detected target, this score can be useful for target ranking. Initial attempts at false-positive rate estimation usually relied on comparing detection methods for real miRNAs and shuffled control miRNAs. Eg. Stark’s method, miRanda, TargetScan, PicTar.
Thermodynamic-based algorithms
3.
: These methods use thermodynamic as the initial indicator of miRNA-binding site potential. Eg. DIANA-microT and RNAHybrid
Motif-mining approaches
Performance of target-prediction methods
It is difficult to assess accurately the performance of many of the methods listed above. Traditionally, this is because few validated miRNA targets are known. Thus, although the methods published tend to be able to predict the few known targets, these constitute a small proportion of target predictions overall. Thus, the initial estimates of the false-positive rates tend to use sequence-shuffling approaches to approximate error rates. But, the problem remains that many methods are not available to be downloaded for inclusion in independent testing on a common dataset.
miRanda
: These approaches work from an opposite angle compared with previous sequence-based methods, by looking in genomic 3’UTR sequences for overrepresented motifs of 6,7 or 8 nucleotides. These motifs are then compared with miRNA seed regions to predict potential miRNA targets.
This algorithm identifies potential binding sites by looking for high-complementarity regions on the 3’-UTRs. The scoring matrix used by this algorithm is built so that complementary bases at the 5’ end of the miRNA are rewarded more than those at

18
the 3’ end. Hence, the binding sites exhibiting a perfect or almost-perfect match at the seed region of miRNAs display a better score. The resulting binding sites are then evaluated thermodynamically, using the Vienna RNA folding package.
TargetScan
This method requires perfect complementarity to the seed region of a miRNA and then extends these regions to unravel complementarity outside the region. This is aimed at filtering the many positives from the start of the prediction process. The conservation criteria are introduced early in the process by using groups of orthologous 3’UTRs as input data. The predicted binding sites are tested for their thermodynamic stability, in this case with RNAfold from the Vienna Package. This is the first method to be applied for human miRNA target prediction, using mouse, rat and fish genomes for conservation analysis. Shuffled sequences with maintained dinucleotide compositions that mimic real 3’UTRs, are used to determine the significance of binding sites. The estimated false-positive rate varies between 22% and 31%. The method predicts both miRNA binding sites and novel sites.
Methods For proper prediction of miRNA targets, the various responsible miRNA species and their respective genes were found out. This was done after consulting various research papers and work previously done.
Responsible miRNAs and genes
Schizophrenia is a broad spectrum disorder involving various genes and miRNAs. In my literature review, I came across 2 major miRNAs involved in pathways crucial to schizophrenia and autism. They are hsa-miR-132 and hsa-miR-29b respetively. The important genes responsible have already been talked about previously in another chapter.
Working on miRanda
miRanda is a webserver which works in tandem with www.microRNA.org. The sequence required is obtained from miRBase. We try to get the precursor miRNA sequence from the database, then obtain then 3’ and 5’ loop sequences (after Dicer splicing) from the same.
TargetScan
TargetScan comes as a user interface on the web for various species like human, mouse, worm, fly and fish. We choose the human server. Therein, we fill in the specific Entrez gene symbol for the gene is question. This leads us to a page where the 3’UTR of the species is illustrated with the locations of sites with higher and lower

19
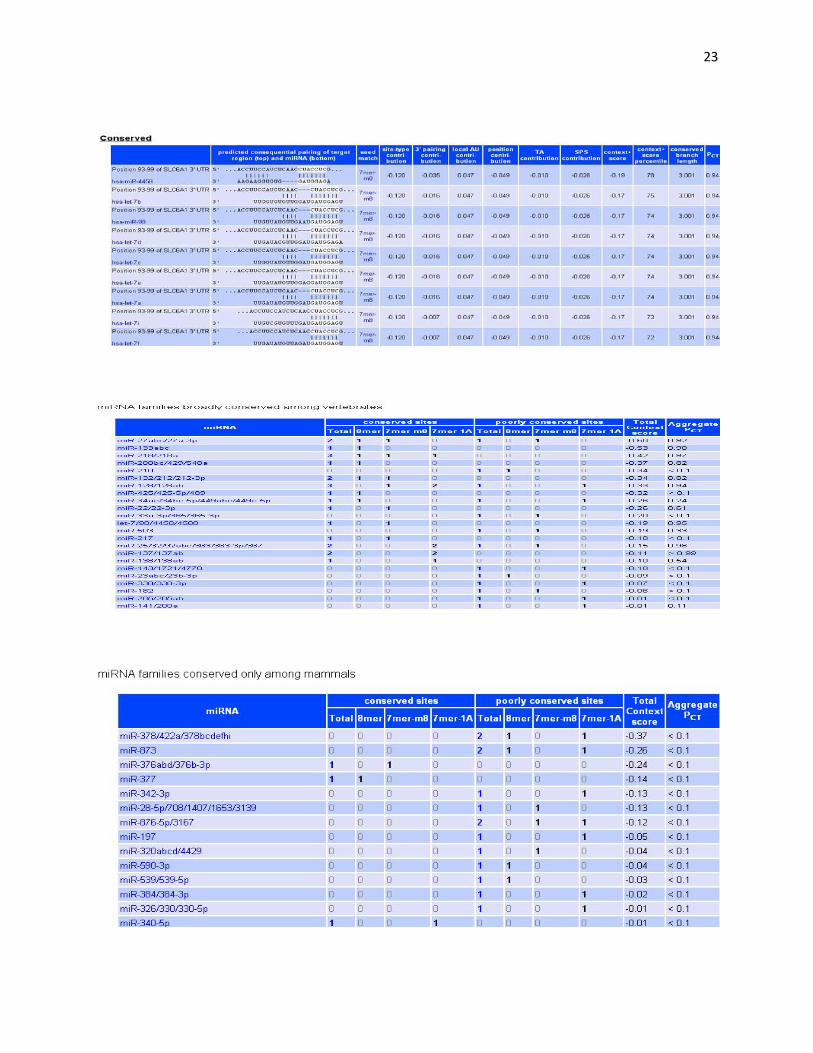
probabilities of preferential conservation. Essentially, it shows the conserved sites for miRNA families broadly conserved among vertebrates. The page also illustrates the nucleotide locations of conserved sites in different species listed in order. The conserved site positions alongwith the respective context scores and aggregate PCT scores are listed in a tabular format.
The TargetScan algorithm works like mentioned below:
1. Searches the UTRs in the first organism for segments of perfect Watson-Crick complementarity to bases 2-8 of the miRNA. The 7nt segment of the miRNA is the “miRNA seed”, whereas the UTR heptamers with perfect Watson-Crick complementarity to seed are the “seed matches”.
2. Extends each seed match with additional base pairs to the miRNA as far as possible in each direction, allowing G:U pairs but stopping at mismatches.
3. Optimizes base-pairing of the remaining 3’-portion of the miRNA to the 35 bases of the UTR immediately 5’ of each seed match using the RNA fold program.
4. Assigns a folding free energy G to each such miRNA:target site interaction (ignoring initiation free energy) using RNAeval.
5. Assigns a Z score to each UTR defined as , where n is the number of seed matches and Gk is the free energy of the miRNA target site interaction (kcal/mol) for the kth target site evaluated in the previous step.
6. Sorts the UTRs in this organism by Z score and assigns a rank Ri to each.
7. Repeats this process for the set of UTRs from each organism.
8. Predicts as targets those genes for which both both Zi ≥ZC and Ri ≤ RC for an orthologous UTR sequence in each organism, where ZC and RC are pre-chosen Z score and rank cutoffs.
Chapter 5: Results and Discussion The microRNA for our experiment, hsa-miR-132 is taken and filled in microRNA.org. a total of 7508 genes are targeted as shown by the server. The list of various targets is then seen after right clicking. The targets have been listed with respect to their mirSVR scores. The mirSVR score provided at mircrorna.org is the best tool for making predictions, as it utilizes the most recent miRanda prediction rules such as seed-site pairing, site context, free-energy, and conservation. A mirSVR cutoff of <= -1.2 is recommended. This value represents the top 5% of miRSVR scores, where the expected probability of observing a log expression change of <=.5 is ~50%, or of <=0.1 is 70%. The authors of the SVR score also suggest conservation as merely

20
another factor to consider when determining the overall confidence of the prediction, and not as an absolute cut off. We see later that the gene target we are trying to investigate i.e. CACNA1B has a very low mirSVR score of -0.7. But still it has an important role in schizophrenia since it carries the codes for proteins that use Calcium signals to help control how neurotransmitters are released in the brain.
Clicking on CACNA1B in the list of predicted gene targets takes us to a page which shows the miRNA-CACNA1B alignment alongwith the mirSVR and PhastCons score. PhastCons is a hidden Markov model-based method that estimates the probability that each nucleotide belongs to a conserved element, based on the multiple alignment. It considers not just each individual alignment column, but also it’s flanking columns. The phastCons scores represent probabilities of negative selection and range between 0 and 1. PhastCons treats alignment gaps and unaligned nucleotides as missing data, and it is run with the same parameters for each species set. The score in our hit is 0.3416.
Then we finally move to TargetScan. The specific gene name is filled in, which takes us to a page which shows the conserved sites for miRNA families broadly conserved among vertebrates. It also illustrates the nucleotide locations of the conserved sites. In our case, it is from 1090-1100. The positions of the conserved families along with the miRNA species is listed in a table which also includes the predicted consequential pairing of target region (top) and miRNA (bottom), seed matches, context score and aggregate PCT.
Fig. 3 It shows the conserved sites for miRNA families broadly conserved among vertebrates

21
Fig. 4 Positions of conserved sites alongwith context scores and aggregate PCT
The Context Score is the sum of the contribution of these 6 features:
• site-type contribution • 3' pairing contribution • local AU contribution • position contribution • TA contribution (Target-site abundance) • SPS contribution (Seed-pairing stability)
Fig. 5 miRNA families broadly conserved among vertebrates

22
Fig. 6 miRNA families conserved only among mammals
Fig. 7 SLC6A Results
23
24
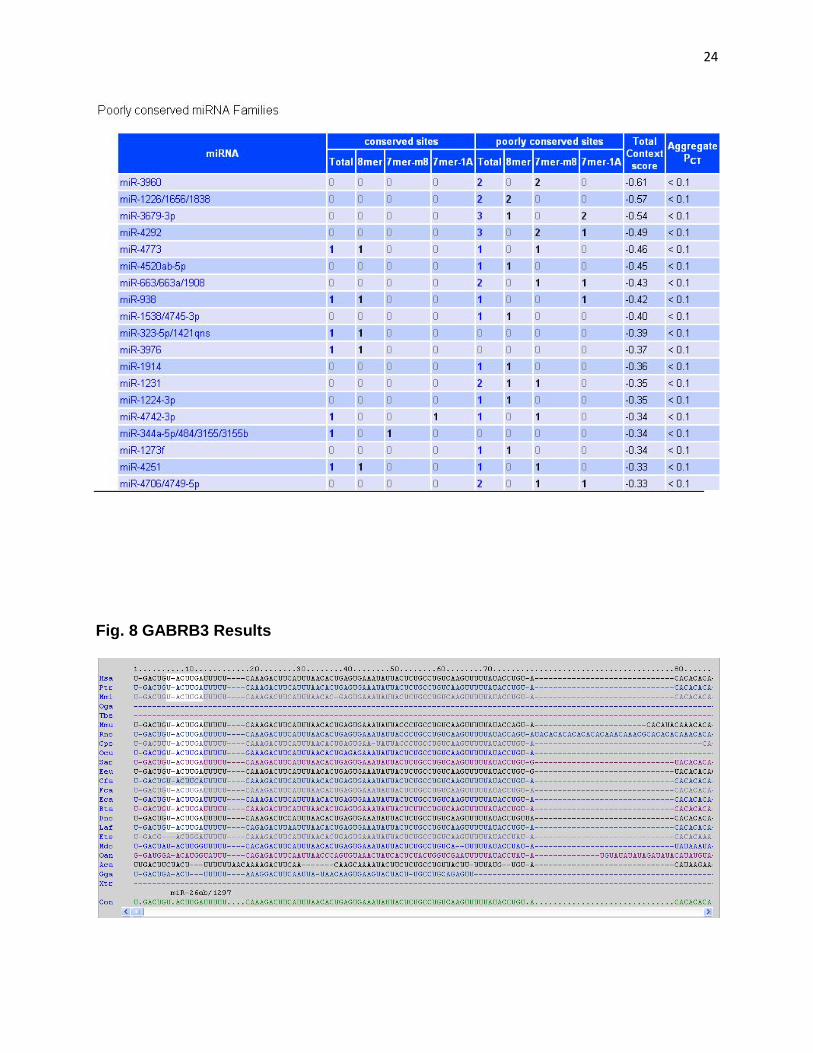
Fig. 8 GABRB3 Results

25

26
Fig. 9 DTNBP1 Results

27

28
Aggregate PCT
PCT is the probability of conserved targeting and is calculated for all highly conserved miRNA families.To control for site type and sequence features, calculate a signal-to-background ratio (S/B) for each site at each site's branch-length. For the purpose of evaluating individual sites, assessing controls at each branch length instead of at each branch-length cutoff is necessary in order to avoid crediting poorly conserved sites for having the same sequence as many highly conserved sites. Convert this S/B to a probability of preferentially conserved targeting (PCT), which is approximately equal to (S/B - 1)/(S/B) (or near zero, for sites with S/B < 1). This score reflects the estimate of the probability that a site is conserved due to selective maintenance of miRNA targeting rather than by chance or any other reason not pertinent to miRNA targeting, allowing for uncertainty in the S/B ratio. Predicted targets of a miRNA family can be sorted by decreasing aggregate PCT . Since PCT refers to a probability, the aggregate PCT is calculated as
1 - ( (1 - PCT)site1 x (1 - PCT)site2 x (1 - PCT)site3 ... )
Since overlapping sites cannot be occupied at the same time, some are removed from the table of predicted targets to create a set of non-overlapping sites while maximizing the total context score. These non-overlapping sites are used to calculate the aggregate PCT.
Conclusions miRNAs interact with their target mRNAs and inhibit translation and cleave the target mRNA. This interaction is guided by sequence complementarity and results in the reduction of mRNA and/or protein levels. miRNAs are involved in key biological processes and different diseases. Deciphering miRNA targets is crucial for diagnostics and therapeutics. In recent years, several computational methods based on sequence complementarity of the miRNA and the mRNAs have been developed. In this study, we found out that some mirSVR scores as predicted by TargetScan were not very satisfactory whereas some gave decent values. But, all the genes studied have been show to have important metabolic roles in autism and schizophrenia. Hence, it is difficult to say whether such quantitative studies and their results can be directly linked to the biological activities under observation. Moreover, neuro-developmental disorders are more of behavioral nature. Thus, it is not right to assume that miRNA-mRNA or other such interactions have a direct effect on the characterization of the disorders in patients.

29
References
1. Sethupathy P, Corda B, Hatzigeorgiou AG. TarBase: A comprehensive database of experimentally supported animal microRNA
targets. RNA. 2006;12(2):192–197. [PMC free article] [PubMed]
2. Linsley P. S. S, Schelter J, Burchard J, Kibukawa M, Martin M. M. M, Bartz S. R. R, Johnson J. M. M, Cummins J. M. M,
Raymond C. K. K, Dai H, Chau N, Cleary M, Jackson A. L. L, Carleton M, Lim L. Transcripts targeted by the microRNA-16 family
cooperatively regulate cell cycle progression. Mol. Cell. Biol. 2007;27(6):2240. [PMC free article] [PubMed]
3. Bushati N, Cohen S. M. M. microRNA Functions. Annu. Rev. Cell Dev. Biol. 2007;23:175–205.[PubMed]
4. Ying SY, Chang DC, Lin SL. The MicroRNA (miRNA): Overview of the RNA Genes that Modulate Gene Function. Mol.
Biotechnol. 2008;38(3):257–268. [PubMed]
5. Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32 (Database issue), D109.
6. Griffiths-Jones S. miRBase: the microRNA sequence database. Methods Mol. Biol. 2006;342:129–138. [PubMed]
7. Baltimore D, Boldin MP, O'Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and
function. Nat. Immunol. 2008;9(8):839–845. [PubMed]
8. Song L, Tuan RS. MicroRNAs and cell differentiation in mammalian development. Birth Defects Res. C Embryo
Today. 2006;78(2):140–149. [PubMed]
9. Lu X, Huang X. Plant miRNAs and abiotic stress responses. Biochem. Biophys. Res. Commun.2008;368(3):458–462. [PubMed]
10. Stern-Ginossar N, Gur C, Biton M, Horwitz E, Elboim M, Stanietsky N, Mandelboim M, Mandelboim O. Human microRNAs
regulate stress-induced immune responses mediated by the receptor NKG2D. Nat. Immunol. 2008;9(9):1065–1073. [PubMed]
11. Sullivan C. S, Ganem D. MicroRNAs and Viral Infection. Mol. Cell. 2005;20(1):3–7. [PubMed]
12. Umbach JL, Kramer M F, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during
latent infection regulate viral mRNAs. Nat. Publ. Group. 2008;454:780–783.
13. Mcmanus MT. MicroRNAs and cancer. Semin. Cancer Biol. 2003;13(4):253–258. [PubMed]
14. Yang N, Coukos G, Zhang L. MicroRNA epigenetic alterations in human cancer: one step forward in diagnosis and
treatment. Int. J. Cancer. 2008;122(5):963–968. [PubMed]
15. Meltzer PS. Cancer genomics: small RNAs with big impacts. Nature-London. 2005;435(7043):745.[PubMed]
16. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska E A.
Requirement of bic/microRNA-155 for normal immune function. Sci. Signal.2007;316(5824):608.
17. Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Fren-dewey D, Valenzuela D, Kutok JL. Regulation of the
germinal center response by microRNA-155. Science. 2007;316(5824):604.[PubMed]

30
18. Metzler M, Wilda M, Busch K, Viehmann S, Borkhardt A. High expression of precursor microRNA-155/BIC RNA in children with
Burkitt lymphoma. Genes Chromosomes Cancer. 2004;39(2):167–169.[PubMed]
19. Kluiver J, Poppema S, Jong DD, Blokzijl T, Harms G, Ja-cobs S, Kroesen B, Berg AVD. BIC and miR-155 are highly expressed
in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J. Pathol. 2005;207(2):243–249. [PubMed]
20. Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, Stephens RM, Okamoto A, Yokota J, Tanaka T. Unique
microRNA molecular profiles in lung cancer diagnosis and prognosis.Cancer Cell. 2006;9(3):189–198. [PubMed]
21. Zhang B, Pan X, Cobb GP, Anderson TA. MicroRNAs as oncogenes and tumor suppressors. Dev. Biol. 2007;302(1):1–
12. [PubMed]
22. He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ. A
microRNA polycistron as a potential human oncogene. Nature-London.2005;435(7043):828. [PubMed]
23. Lanza G, Ferracin M, Gafµa R, Veronese A, Spizzo R, Pichiorri F, Liu C, Calin GA, Croce CM, Negrini M. mRNA/microRNA
gene expression profile in microsatellite unstable colorectal cancer. Mol. Cancer. 2007;6(1):54. [PubMed]




















