
A Hypothesis for DNA Viruses as the Origin of Eukaryotic Replication Proteins

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX10.1146/annurev.biochem.74.082803.133250
Annu. Rev. Biochem. 2005. 74:317–53doi: 10.1146/annurev.biochem.74.082803.133250
Copyright c© 2005 by Annual Reviews. All rights reservedFirst published online as a Review in Advance on March 4, 2005
EUKARYOTIC TRANSLESION SYNTHESIS
DNA POLYMERASES: Specificity ofStructure and Function
Satya Prakash, Robert E. Johnson, and Louise PrakashSealy Center for Molecular Science, University of Texas Medical Branch, Galveston,Texas 77555-1061; email: [email protected], [email protected],[email protected]
Key Words lesion bypass, Y-family DNA polymerases, DNA polymerasestructures, Rad6-Rad18 enzyme complex
■ Abstract This review focuses on eukaryotic translesion synthesis (TLS) DNApolymerases, and the emphasis is on Saccharomyces cerevisiae and human Y-familypolymerases (Pols) η, ι, κ , and Rev1, as well as on Polζ , which is a member of theB-family polymerases. The fidelity, mismatch extension ability, and lesion bypassefficiencies of these different polymerases are examined and evaluated in the contextof their structures. One major conclusion is that, despite the overall similarity of basicstructural features among the Y-family polymerases, there is a high degree of specificityin their lesion bypass properties. Some are able to bypass a particular DNA lesion,whereas others are efficient at only the insertion step or the extension step of lesionbypass. This functional divergence is related to the differences in their structures. Polζis a highly specialized polymerase specifically adapted for extending primer terminiopposite from a diverse array of DNA lesions, and depending upon the DNA lesion,it contributes to lesion bypass in a mutagenic or in an error-free manner. Proliferatingcell nuclear antigen (PCNA) provides the central scaffold to which TLS polymerasesbind for access to the replication ensemble stalled at a lesion site, and Rad6-Rad18-dependent protein ubiquitination is important for polymerase exchange.
CONTENTS
OVERVIEW . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 318GENERAL FEATURES OF Y-FAMILY DNA POLYMERASES . . . . . . . . . . . . . . . . 319
Conserved Motifs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 320Conserved Structural Features . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 321
DNA POLYMERASE η . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 322Fidelity and Mismatch Extension Ability of Polη . . . . . . . . . . . . . . . . . . . . . . . . . . . 323Lesion Bypass by Polη . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 323Polη Structure and Lesion Bypass . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 326Mechanism of Nucleotide Incorporation by Polη . . . . . . . . . . . . . . . . . . . . . . . . . . . 327
DNA POLYMERASE ι . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 329
0066-4154/05/0707-0317$20.00 317

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
318 PRAKASH � JOHNSON � PRAKASH
Fidelity and Mismatch Extension Ability . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 329Lesion Bypass by Polι . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 330Structure of Polι and its Relevance for Lesion Bypass . . . . . . . . . . . . . . . . . . . . . . . 331
REV1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 333Role of Rev1 in Lesion Bypass . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 334Possible Involvement of Rev1 in the Assembly of Other Y-Family
Polymerases for Polζ -Dependent TLS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 335DNA POLYMERASE κ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 335
Fidelity and Mismatch Extension Ability of Polκ . . . . . . . . . . . . . . . . . . . . . . . . . . . 336Lesion Bypass by Polκ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 338Polκ Structure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 339
DNA POLYMERASE ζ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 340Fidelity and Highly Proficient Mismatch Extension Ability of Polζ . . . . . . . . . . . . . 341Role of Polζ as an Extender in Lesion Bypass . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 341
LESION BYPASS BY ONE OR TWO DNA POLYMERASES . . . . . . . . . . . . . . . . . . 342TARGETING OF TLS POLYMERASES TO THE REPLICATION
ENSEMBLE STALLED AT A LESION SITE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 343Physical and Functional Interactions of Y-Family Polymerases with PCNA . . . . . . 344Rad6-Rad18-Dependent Lesion Bypass Pathways . . . . . . . . . . . . . . . . . . . . . . . . . . 345Ubiquitin and SUMO Modifications of PCNA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 345
CONCLUDING REMARKS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 347
OVERVIEW
Cellular DNA is continually exposed to a large variety of external and internalDNA-damaging agents. In the cell, DNA is subject to spontaneous hydrolysisof its phosphodiester, N-glycosylic, and amino bonds, and DNA is exposed toconstant attack by oxygen free radicals that damage the sugar and the bases. Ex-posure to sunlight and to chemical pollutants adds further to this load. AlthoughDNA lesions can be removed by nucleotide excision repair (NER) and by baseexcision repair processes, many lesions escape repair and present a block to con-tinued transcriptional elongation by RNA polymerases and to replication by DNApolymerases (Pols).
In eukaryotes, DNA polymerases, which belong to the Y family, and DNApolymerase ζ , which is a member of the B family, promote replication throughDNA lesions (1, 2). DNA polymerases of the Y family belong to one of fivesubfamilies, which include the UmuC branch (typified by Escherichia coli Pol Vand found exclusively in prokaryotes) and the DinB branch (present in prokaryotes,eukaryotes, and archaea and typified by E. coli Pol IV, human Polκ , and the archaeaDbh/Dpo4 enzymes). The other three branches, comprised of Rev1, Polη, andPolι, respectively, are found exclusively in eukaryotes. Polζ also is found only ineukaryotes.
Structural and biochemical studies of eukaryotic Y-family polymerases, as wellas genetic and biochemical studies with Polζ , have revealed striking differencesamong them in the roles they play in lesion bypass. This review focuses on these

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 319
translesion synthesis (TLS) polymerases of yeast and humans and highlights theimportant principles and conclusions that have emerged from their study, whichare as follows:
� In contrast to replicative DNA polymerases, which synthesize DNA with ahigh degree of accuracy and are blocked by lesions that significantly distortthe geometry of DNA, TLS DNA polymerases, particularly of the Y family,synthesize DNA with much higher error rates and are able to synthesize DNApast lesions that block replicative polymerases.
� In spite of the underlying similarities of the basic structure among the Y-family polymerases, a large degree of functional divergence has occurredamong them, rendering them highly specialized for the roles they play inlesion bypass. Thus, instead of being a jack-of-all-trades, they have becomeadapted to the specific roles they perform in lesion bypass.
� The functional divergence among the Y-family polymerases can be attributedto their specific structural features.
� Although a particular polymerase could carry out both steps of lesionbypass—nucleotide incorporation opposite the DNA lesion and the sub-sequent extension from the inserted nucleotide—for many DNA lesions,especially where the distortion of DNA to Watson-Crick geometry is quitesignificant, TLS through the DNA lesion is mediated by the action of twodifferent polymerases in which one inserts the nucleotide opposite the lesionand the other performs the subsequent extension step. This two-polymerasemechanism of lesion bypass is afforded by the high degree of structural andfunctional specificity of the different Y-family polymerases. Additionally,and importantly, Polζ , which incorporates nucleotides with a relatively highfidelity and is strongly inhibited at the nucleotide incorporation step of lesionbypass, is highly adept at carrying out the extension reaction opposite froma diverse array of DNA lesions.
� Proliferating cell nuclear antigen (PCNA) provides the central scaffold towhich the various TLS polymerases bind to gain access to the replicativeensemble stalled at the lesion site and to execute their roles in lesion bypass.
There is relatively little information available on how the replicative polymerasebecomes disengaged from the template-primer junction and on how the entriesand exits of different TLS polymerases at the replication fork are coordinatedand regulated. In this respect, Rad6-Rad18-dependent protein ubiquitination isexpected to have a major impact on the various lesion bypass processes.
GENERAL FEATURES OF Y-FAMILY DNA POLYMERASES
On the basis of phylogenetic relationships, DNA polymerases have been groupedinto six families—A, B, C, D, X, or Y. The Y-family polymerases differ fromothers in having a low fidelity and in their ability to replicate through DNA

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
320 PRAKASH � JOHNSON � PRAKASH
Figure 1 Schematic alignment of human Y-family DNA polymerases. The primary aminoacid sequences of human DNA Pols η, ι, and κ , and Rev1 are shown as gray bars. Regionscorresponding to motifs I–V, common to all Y-family polymerases, are shown in differentcolors. The colors for motifs I–V coincide with those used in the structures in Figures 2,5, and 10. Other shaded regions represent conserved sequences found within a subfamily.The pink regions in Rev1 are common to all members of this subfamily, which includes theBRCT domain present at the N terminus. The x, y, and z motifs in Polκ are shared among allmembers of the DinB subfamily. The C2H2 and C2HC potential zinc-binding motifs found inPolη and Polκ , respectively, are shown in blue, and their PCNA-binding domains are shownin yellow.
lesions. Even though the Y-family polymerases share five conserved sequencemotifs and a common structural architecture, they differ in remarkable ways inmany of their structural features, and these differences account for their functionalspecificity.
Conserved Motifs
All members of the Y family contain five conserved motifs, I–V (Figure 1).Motifs I and III harbor the invariant acidic residues in the consensus sequences(I/V)D(M/F/L) and (A/L)SIDE(V/A)(F/Y), respectively (3), which coordinate thetwo metal ions in the active site, necessary for catalysis (4). Motif II is character-ized by an invariant tyrosine (Y) and a basic residue in the consensus sequenceYxA(R/K), and motif IV has an invariant lysine residue embedded in a conservedsequence (5). The Y and R/K residues of motif II and the K residue of motif IVcontribute to dNTP binding and its incorporation into DNA (5, 6). Motif V is muchless conserved, but it is essential for DNA synthesis, as are all the other motifs.
In addition to these five conserved motifs, Y-family polymerases harbor otherconserved features that are specific to a particular subfamily or are shared onlyamong some of the members of a subfamily (Figure 1). For instance, the Rev1subfamily contains an N-terminal extension beyond motif I in which it harbors aBRCT domain. A second conserved sequence in the Rev1 subfamily, located justC-terminal to motif V and essential for DNA synthetic activity, is predicted to formthe polymerase-associated domain (PAD). In the DinB subfamily, which includeshuman Polκ , the conserved sequences x, y, and z form the PAD and are shared byall members, whereas an N-terminal extension with a conserved sequence motif is

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 321
found only in the eukaryotic members of this subfamily (Figure 1, see also Figure 7below).
Conserved Structural Features
Crystal structures of several Y-family DNA polymerases, Dpo4 and Dbh fromArcheae (7–9), as well as of yeast Polη (10), human Polι (11), and human Polκ (12)have been determined, and they all resemble one another in having a similar overallstructure. As exemplified from the structure of Polη (Figure 2), the palm domainis the catalytic epicenter of the protein, which harbors the three acidic residuesthat coordinate two metal ions required for catalyzing nucleotide transfer onto the3′-OH of the primer-terminal nucleotide (Figure 2b). This domain is formed by
Figure 2 Crystal structure of a Y-family DNA polymerase. (a) Structural features of yeastPolη and T7 DNA polymerase. The palm (red and blue), fingers (yellow), and thumb (orange)domains as well as the PAD domain (green), unique to Y-family DNA polymerases, are shown.(b) A close-up view of the active sites of yeast Polη and T7 polymerase. The incomingnucleotide in Polη was modeled. The acidic residues that bind magnesium, as well as theresidues that interact with the triphosphate moiety of the incoming nucleotide, are shown forboth polymerases (5, 10, 14).

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
322 PRAKASH � JOHNSON � PRAKASH
motifs I and III that harbor the invariant acidic residues. The structural similarityof the palm domain of Polη and other Y-family members with that in high-fidelityA/B-family DNA polymerases is remarkable given their sequence divergence, andthis similarity highlights the conserved catalytic mechanism utilized by all DNApolymerases (13–15). The fingers and thumb domains are strikingly different fromthose in A/B-family DNA polymerases (Figure 2a). The fingers of high-fidelityA/B-family DNA polymerases contain two α-helices, exemplified by the O andO1 helices in T7 polymerase (14), which close tightly on the incoming nucleotideand the templating residue, forming a snug active site that accommodates thecorrect nascent base pair by geometric selection. The thumb domain of A/B-family polymerases is also large, contributing to the large DNA-binding surfacearea in these polymerases (14). In the Y-family polymerases, the fingers and thumbdomains are small and stubby (Figure 2a). The conserved motif II comprises apart of the fingers domain, and the invariant tyrosine and the basic residue in itinteract with the triphosphate moiety of the incoming nucleotide, similar to whatis observed in A- and B-family DNA polymerases (Figure 2b). A unique featureof the palm in the Y-family polymerases, however, is the presence of a helix whichlies atop the six-stranded β-sheet, on the thumb side of the polymerase. This isformed by part of motif IV, and it contributes another conserved basic residuethat interacts with the triphosphate moiety of the incoming nucleotide (Figure 2b).Thus, despite the large degree of sequence divergence between the Y-family andA/B-family polymerases, the basic mechanisms of catalysis and dNTP bindinghave been conserved among them.
The active sites of Y-family polymerases, however, differ strikingly from that ofhigh-fidelity polymerases because they are much more open and sterically less con-strained around the nascent base pair. Lastly, but importantly, the DNA syntheticactivity of Y-family polymerases requires, in addition to motifs I–V, ∼120–140amino acids to form the PAD (Figure 2a). The PAD increases the potential DNA-binding surface area from ∼600–700 A, for just the fingers and thumb domains,to ∼1000–1100 A, which is comparable to that observed in A- and B-familypolymerases.
DNA POLYMERASE η
Polη was first identified in yeast and shown to replicate through a cis-syn TTdimer by inserting two As opposite the two Ts of the dimer (16). This observation,which implied that Polη could promote error-free replication through UV-inducedcyclobutane pyrimidine dimers (CPDs), was in concert with the observation thatdeletion of the RAD30 gene in yeast, which encodes Polη, conferred an enhance-ment of UV mutagenesis (17).
Unlike cells from patients belonging to the classical xeroderma pigmentosum(XP) complementation groups, which harbor a defect in NER, cells from thevariant form of XP (XPV) are proficient in NER but defective in the replication ofUV-damaged DNA (18), and unlike the extracts from normal cells, extracts from

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 323
XPV cells do not support replication through a cis-syn TT dimer (19, 20). XPV cellsare hypermutable with UV light, and the frequency of adenine (A) incorporationopposite the TT photoproducts is reduced in XPV cells compared to that in normalcells (21, 22). The ability of yeast Polη to replicate through the cis-syn TT dimerin an error-free manner had raised the strong possibility that defects in Polη arethe cause of XPV in humans (16), and this prediction was soon confirmed in twoseparate studies (23, 24).
Fidelity and Mismatch Extension Ability of Polη
The ability of Polη and other Y-family polymerases to replicate through DNAlesions implies that they are not inhibited by the geometric distortions imposedby the presence of lesions in DNA. A TLS polymerase, then, cannot be verysensitive to geometric distortions of DNA, including those conferred from mispairformation on undamaged DNA templates. The idea that TLS polymerases canbetter tolerate the geometric distortion of a mismatched base pair in their activesites was substantiated from steady-state kinetic analyses with yeast and humanPolη, indicating that they misincorporate nucleotides with a frequency of ∼10−2
to 10−3 (25, 26).The fact that Polη incorporates wrong nucleotides at a fairly high rate may
seem incongruous with its role in promoting error-free bypass of CPDs and insuppressing UV mutagenesis. Both yeast and human Polη, however, extend mis-matched primer termini with a frequency of ∼10−2 to 10−3 relative to extensionfrom matched primer termini (27). Consequently, Polη would have a much higherprobability of dissociating from the primer terminus following the incorporationof an incorrect nucleotide than of a correct one, and that could provide the proof-reading exonuclease of Polδ or another proofreading exonuclease an opportunityto remove the wrong nucleotide. Such an idea is supported from experiments donewith the SV40 origin-dependent replication system in human extracts, in whichit was shown that hPolη-induced replication errors are enhanced if the proofread-ing exonucleases are inhibited by the addition of high amounts of dGMP to thereplication reaction (28).
Lesion Bypass by Polη
In its proficient ability to replicate through CPDs, Polη stands apart from allother known DNA polymerases. This proficiency of Polη derives from its uniquestructural feature—the ability to accommodate both template nucleotides of a CPDin its active site (10). Although the proficient ability to replicate through CPDs isthe most prominent and biologically consequential feature of Polη, it can promotereplication through many other DNA lesions also.
CPDs Both yeast and human Polη replicate through a cis-syn TT dimer by insert-ing two As opposite the two Ts of the dimer, and steady-state kinetic analyses haveshown that the incorporation of an A opposite the 3′T and the 5′T of the dimer

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
324 PRAKASH � JOHNSON � PRAKASH
occurs with nearly the same efficiency and fidelity as opposite the two undam-aged Ts (26, 29). Moreover, the polymerase exhibits the same processivity on thedimer containing DNA as on undamaged DNA. For both undamaged and damagedDNAs, ∼50% of the yeast Polη molecules remain bound after the incorporationof two dNTPs opposite the 3′T and 5′Ts of the dimer or opposite the two Ts in theanalogous undamaged sequence, and ∼30% of the enzyme molecules incorporateat least three dNTPs on both DNAs (29).
From presteady-state kinetic analyses of the different steps of the nucleotideincorporation reaction, DNA binding (Kd
DNA), nucleotide binding (KddATP), and
the maximal first-order rate constant for nucleotide incorporation (kpol), oppositethe two Ts of the dimer, it has been determined that yeast Polη binds the DNAwhen opposite the 3′T of the dimer with about the same affinity as it binds theanalogous site in undamaged DNA, with a Kd
DNA of 40 nM versus 32 nM, respec-tively (30). Although the affinity for nucleotide binding (Kd
dATP) opposite the TTdimer was slightly lower than opposite undamaged Ts, being reduced ∼fourfoldopposite the 3′T and ∼twofold opposite the 5′T of the dimer, the kpol values fordATP incorporation of ∼1 to 2 per s opposite the damaged and undamaged Tswere remarkably similar (30). The nearly identical rate of nucleotide incorpora-tion opposite the damaged and undamaged Ts strengthens the inference derivedfrom structural studies (10) that this polymerase can well accommodate both theTs of the dimer in its active site.
The idea that both Ts of a TT dimer stay in the active site of Polη rather than thedimer being pushed out of the active site, as occurs in T7 polymerase (31–33), hasalso been examined in studies with N3-methyl derivatives of the 3′T and the 5′Tof a TT dimer (34). If the dimer is outside the active site, this would preclude thedirect insertion of an A opposite the 3′T, and methylation of N3 of the 3′T wouldhave no effect on the efficiency of A insertion. If, however, the 3′T of the dimerstays inside the active site and directs the insertion of an A, methylation of N3 ofthe 3′T should lead to a decrease in the efficiency of A insertion. The observationsthat methylation of the 3′T reduces the efficiency of A incorporation by ∼30-fold,and that methylation of the 5′T confers a reduction of ∼100-fold (34), adds furthersupport to the inference that both the Ts of the dimer remain in the active site anddirect the insertion of an A.
The ability of Polη to accommodate the two template nucleotides of the TTdimer and to incorporate an A directly opposite the two Ts via Watson-Crick hy-drogen bonding provides an elegant mechanism by which Polη can also replicateefficiently and accurately opposite CPDs formed at 5′-TC-3′ and 5′-CC-3′ dipyrim-idine sites. That Polη functions in the error-free bypass of CPDs formed at thesesites is supported by the observation that inactivation of Polη in both yeast andhumans leads to a large increase in the frequency of C→T transitions, which resultfrom the misincorporation of an A opposite the 3′C of CPDs formed at 5′TC-3′
and 5′-CC-3′ sites in the absence of Polη (35, 36).CPDs are responsible for the majority of UV-induced mutations in mammalian
cells because they are formed much more frequently than (6-4) photoproducts(37–42), and also because they are removed much less efficiently than the (6-4)

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 325
photoproducts by NER (43). Hence, the proficient ability of Polη to efficiently andaccurately replicate opposite CPDs formed at various dipyrimidine sites providesa large measure of protection from sunlight-induced skin cancers in humans.
(6-4) TT PHOTOPRODUCTS Whereas a cis-syn TT dimer has only a modest effecton DNA structure and retains the ability to form Watson-Crick base pairs with thecorrect nucleotides (44–47), a (6-4) TT photoproduct induces a large structuraldistortion in DNA, with the 3′T of the (6-4) lesion oriented perpendicular to the5′T (48, 49). NMR studies have indicated that the O2 carbonyl of the 3′T in the(6-4) TT lesion can hydrogen bond with the imino and amino protons of a guanine(G), whereas the 5′T of the lesion retains the ability to form normal Watson-Crick hydrogen bonding with an A (50). Although Polη is unable to replicate pasta (6-4) TT lesion, it can preferentially incorporate a G opposite the 3′T of thelesion. Both yeast and human Polη incorporate a G opposite the 3′T of the lesion∼eightfold better than an A, but still the efficiency of G insertion opposite the 3′Tof this lesion is reduced ∼50–100-fold compared to the insertion of an A oppositethe corresponding undamaged T residue (51). The subsequent extension step isperformed by Polζ (51).
In agreement with the incorporation of a G opposite the 3′T of a (6-4) TTlesion by Polη seen in biochemical experiments, studies with single-stranded andgapped duplex plasmids in yeast have indicated that a (6-4) TT photoproductis highly mutagenic, inducing a high frequency of 3′T→C substitutions (52).Also, in contrast to the enhanced frequency of UV-induced forward mutations atthe CAN1 locus in the rad30� strain of yeast (17, 53), attributable to a majorcontribution of Polη to the error-free bypass of cis-syn TT, TC, and CC dimers(35), at the arg4-17 ochre allele, inactivation of Polη in yeast results in an ∼20-foldreduction in the frequency of UV-induced reversion (54). This reversion occurs bya T→C transition of the 3′T of a potential TT photoproduct site, which presumablycould arise from the mutagenic bypass of (6-4) TT lesions by Polη. Confirmatoryevidence for this Polη role has come from studies in which replication througha site-specific (6-4) TT lesion in a double-stranded plasmid was examined andshown to be dependent upon Polη (55).
Because (6-4) photoproducts are formed more frequently at 5′-TC-3′ and CCsequences than at TT sites (37–42), the ability of Polη to insert a G opposite the 3′Tof a (6-4) TT lesion strongly suggests that Polη contributes also to the error-freebypass of (6-4) lesions formed at TC and CC sites; similar to the 3′T of (6-4) TT,the O2 carbonyl of the 3′C in the (6-4) TC and CC photoproducts can also formhydrogen bonds with the opposing G. The incorporation of a G opposite the 3′Cof TC and CC photoproducts, followed by extension by Polζ , provides for theerror-free bypass of (6-4) lesions formed at these sites (56).
OTHER LESIONS Polη plays a prominent role in efficient and accurate replica-tion through the 8-oxoguanine (8-oxoG) lesion (57). The efficiency with whichyeast Polη incorporates a C opposite the lesion and then extends from the insertednucleotide is remarkably similar to that at an undamaged G. Evidence for the

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
326 PRAKASH � JOHNSON � PRAKASH
in vivo role of Polη in the error-free bypass of the 8-oxoG lesion came from theobservation that the rate of spontaneous G:C to T:A transversions, a characteristicfeature of mutations resulting from the mutagenic bypass of 8-oxoG, is synergis-tically enhanced upon the simultaneous inactivation of Polη and the Ogg1 DNAglycosylase, which removes the 8-oxoG paired with a C. Human Polη also pre-dominantly incorporates a C opposite 8-oxoG; however, in this case, a low butsignificant level of A is also inserted (57).
Whereas the absorption of UVB and UVC radiation from sunlight by DNAresults in the formation of CPDs, (6-4) photoproducts, and other photolesions,the action of UVA radiation from sunlight on cellular chromophores generatesreactive oxygen species (58, 59). Hence, exposure of mammalian cells to sunlightproduces, in addition to CPDs and other photolesions, a significant level of 8-oxoG. By promoting error-free replication through the 8-oxoG lesion, Polη wouldcontribute to minimizing the incidence of skin cancers, as well as of internalcancers, that would have otherwise resulted from the mutagenic bypass of thislesion by replicative DNA polymerases.
Polη can replicate through an 6O-methyl guanine (m6G) lesion, but oppositethis lesion, it incorporates the C and T nucleotides nearly equally well. In contrastto the efficient bypass of CPDs and 8-oxoG, replication through the m6G lesionis inhibited ∼20-fold at the nucleotide incorporation step (60). An abasic sitepresents a severe block to replication by Polη (61). Even for a G or an A, the twonucleotides incorporated most often by Polη opposite an abasic site, the efficiencyof nucleotide incorporation opposite the lesion is reduced by almost 1000-fold,and the extension step is similarly affected (61). Polη is also inhibited by N2-deoxyguanosine adducts of benzo[a]pyrene 7,8-diol 9,10-epoxide (BPDE) (62),and butadiene epoxide (63), and by the acrolein-derived adduct γ -hydroxy-1,N2-propano-deoxyguanosine (γ -HOPdG) (64). Replication by Polη is also inhibitedacross from 1,N6-etheno-deoxyadenosine (65). Overall, it seems that DNA lesionsthat severely impinge upon the minor groove or affect the Watson-Crick hydrogenbonding of base pairs block synthesis by Polη.
Polη Structure and Lesion Bypass
Although the structure of the ternary complex of Polη has not been determined,the structure of yeast Polη modeled with a cis-syn TT dimer in the template DNAand an incoming dATP (10) has provided clear evidence that its active site isopen enough to accommodate both residues of a TT dimer (Figure 3). This isin contrast to most DNA polymerases, which include not only the high-fidelityreplicative/repair polymerases but also the other members of the Y family, inwhich only the templating residue can fit in the active site (Figure 3), whereas thenext 5′-template base along with the rest of the unpaired template is pushed out ofthe active site at a 90◦ angle (9, 11, 12, 14, 66–71). Because the two pyrimidinesof a CPD are covalently linked by the cyclobutane ring and therefore cannot beseparated, the active site of most polymerases is not equipped to handle this lesion;hence, they stop synthesis before the lesion.

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 327
Figure 3 Accommodation of a cis-syn TT dimer in the Polη active site (10). A cis-synTT dimer with an incoming dATP was modeled into the active sites of Polη and T7 DNApolymerase. Whereas there is ample room in Polη to fit both residues of the TT dimer, the5′T of the dimer severely clashes with residues on the O helix of T7 polymerase.
The capacity of Polη to accommodate the two template residues in its active sitecould also be the basis for its proficient ability to insert a C opposite the first G of acisplatin-G intrastrand cross-link (72). Polη, however, is inhibited at incorporatinga nucleotide opposite the second G of the cross-link (72), which could result fromthe structural distortion of DNA at this site.
In its proficient ability to insert a C opposite an 8-oxoG lesion, Polη differs fromother polymerases, which in most cases incorporate an A opposite this lesion (57,73). Although in the 8-oxoG · C base pair the two bases form a normal Watson-Crick base pair, the phosphodiester backbone faces a steric clash with the oxygenat C-8. Polβ avoids this clash in the 8-oxoG · C base pair by flipping the phosphatebackbone of the 5′-templating residue by 180◦ instead of the normal 90◦ (74);however, there may be a considerable energetic barrier for adopting such a sharplykinked DNA structure. The accommodation of an additional 5′-templating residuein the Polη active site may be important for 8-oxoG bypass in two ways: first, bypreventing the flipping of 8-oxoG into the syn conformation, which is a prerequisitefor the Hoogsteen pairing with an A, and thereby avoiding the incorporation of anA; and second, by avoiding the clash of oxygen at C-8 with the phosphodiesterbackbone.
Mechanism of Nucleotide Incorporation by Polη
The tight fit of the active site of high-fidelity replicative/repair polymerases with thenascent base pair puts a strong premium on the geometric shape complementarity

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
328 PRAKASH � JOHNSON � PRAKASH
of the incoming nucleotide with the templating base (14, 66–70). As a result, ge-ometric selection makes a predominant contribution to the nucleotide insertionspecificities of these polymerases (75–77). DNA polymerases can check for thecorrect geometric alignment at the initial nucleotide-binding step, the rate-limitingdNTP-induced conformational change step, and at the chemical step of phospho-diester bond formation (78–81). T7 DNA polymerase checks for correct geometricalignment at all three steps, and that accounts for its almost 106-fold selectivityfor the correct nucleotide (80, 81).
Presteady-state kinetic studies with Polη have also provided evidence for asimilar three-step mechanism of nucleotide incorporation (82, 83). Both yeast andhuman Polη, however, discriminate poorly at the dNTP-binding step, as they bindthe correct nucleotide only three- to fivefold better than the incorrect one, whichcorresponds to a free energy difference (��G) of ∼1.0 kcal/mol. This small freeenergy difference can be accounted for in the stability provided by Watson-Crickhydrogen bonding of base pairs. At the nucleotide incorporation step, yeast andhuman Polη exhibit a selectivity of ∼100–150 for the correct nucleotide over theincorrect one, and that corresponds to a ��G‡ of ∼3.0 kcal/mol. The selectiv-ity at this step could primarily obtain from the geometric constraints within thePolη active site, so that the binding of a correct dNTP is more efficient at induc-ing the conformational change than is the binding of an incorrect one. Becausethe induced-fit conformational change step is rate-limiting for nucleotide incor-poration, whereas the chemical step occurs rapidly, Polη would check for propergeometric alignment primarily at the induced-fit step (82).
Studies with nucleotide analogs that have the same size and shape as the naturalones, but lack the hydrogen-bonding groups, have provided evidence that Watson-Crick hydrogen bonding is not needed for efficient replication by the high-fidelityreplicative/repair polymerases (84–86). For example, difluorotoluence (F), an isos-teric analog of T that cannot form hydrogen bonds with A, is replicated with nearnormal efficiency and fidelity by enzymes, such as Klenow and T7 DNA poly-merase, when F is present either as a template base on DNA or as a dFTP substrate.In sharp contrast, replication by Polη is strongly inhibited when F is present asa template base or dFTP is used as the incoming nucleotide, and the efficiencyof incorporating an A opposite template F or, conversely, of incorporating an Fopposite template A is reduced to almost the same level as the efficiency for incor-porating the wrong nucleotides (87). We have ascribed this strong dependence ofPolη upon Watson-Crick hydrogen bonding to its much less constrained active site.In the absence of any significant geometric selection imposed by the active site atthe dNTP-binding step, the stability provided by Watson-Crick hydrogen bondingmay become crucial for the polymerase to undertake the subsequent induced-fitconformational change and chemical steps.
Because the positions of minor-groove hydrogen-bonding acceptors, N3 forpurines and O2 for pyrimidines, are nearly identical in the correct Watson-CrickG:C and A:T base pairs but are displaced in mispairs, DNA polymerases canadditionally check for the correctness of a base pair via their hydrogen bonding

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 329
with the minor groove of bases in DNA. Ternary structures of high-fidelity DNApolymerases have indicated that they make several hydrogen bonds with the minorgroove of DNA (14, 66–70, 88), and biochemical studies with nucleotide analogslacking hydrogen-bonding acceptors in the minor groove have provided evidencethat they make a functionally important contact at the position of the primer-terminal base (89–91). Biochemical studies with 3-deazaguanine, which is a baseanalog of guanine that has a CH substituted for the N3 minor-groove hydrogenbond acceptor of guanine, however, have indicated a functional hydrogen-bondinginteraction of Polη with the base of the incoming nucleotide rather than with theprimer-terminal base, and this difference may also be a factor contributing to thenucleotide incorporation specificity and lesion bypass ability of Polη (92).
DNA POLYMERASE ι
The high-fidelity replicative/repair DNA polymerases form the four possible cor-rect base pairs with nearly equivalent catalytic efficiencies, and this results fromthe near geometric identity of the Watson-Crick A:T and G:C base pairs, whichfit snugly into their active sites. Moreover, in spite of their much less constrainedactive sites, polymerases η and κ also adhere to this axiom. Polι and Rev1 presentnotable exceptions to this rule.
Fidelity and Mismatch Extension Ability
Polι incorporates nucleotides opposite the four template bases with very differ-ent efficiencies and fidelities, and it incorporates nucleotides opposite templatepurines with a much higher efficiency and fidelity than opposite template pyrim-idines (93–97). Polι exhibits the highest efficiency and fidelity opposite templateA, where it misincorporates nucleotides with frequencies of ∼10−3 to 10−5; oppo-site template G, Polι incorporates the correct nucleotide with an at least 10-fold-reduced efficiency compared to that opposite template A, and it misincorporatesa T opposite template G with a frequency of ∼10−1 (93, 96, 97). Polι is highlyinefficient at incorporating the correct nucleotide opposite templates C and T. Op-posite template C, Polι misincorporates nucleotides with frequencies of ∼10−1 to10−2, whereas opposite template T, it misincorporates a G ∼10-fold better than anA and misincorporates a T as efficiently as an A (93, 96, 97).
Presteady-state kinetic analyses for nucleotide incorporation opposite templatesA and T have indicated that opposite template A, Polι discriminates against thewrong nucleotides at both the nucleotide binding (KD
dNTP) and the incorporationsteps (kPol) (97). Polι binds the correct nucleotide with a 40- to 100-fold-higheraffinity than the incorrect one (��G, 2.2 to 2.7 kcal/mol), and it incorporates thecorrect nucleotide 50 to over 300 times faster than the incorrect ones (��G‡, 2.2to 3.4 kcal/mol). These free energy differences have suggested that, for nucleotideincorporation opposite template A, steric constraints within the Polι active site do

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
330 PRAKASH � JOHNSON � PRAKASH
contribute to nucleotide selectivity at both these steps (97). Opposite template T,Polι binds a G approximately eightfold better than an A (��G, −1.2 kcal/mol),whereas the binding affinity for T is about the same as for an A (97). This and thevery similar kpol values for the incorporation of an A, a G, or a T opposite templateT have suggested that Polι exerts little geometric selectivity at either of thesesteps. Opposite template T then, Polι may hold the incoming nucleotides ratherloosely, so that the steric constraints within the active site make little contributionto selectivity.
For most mispairs, Polι extends from the mispaired primer terminus with aboutthe same efficiency as it forms the mispair. However, it extends the primer-templateG · T and T · G mispairs quite well, ∼3% and ∼8%, respectively, as efficiently asextension from the correct A:T and C:G pairs (98).
Lesion Bypass by Polι
Polι, unlike Polη, is unable to replicate through a cis-syn TT dimer (93). Polι,however, can incorporate nucleotides opposite an abasic site and also opposite the3′T of a (6-4) TT photoproduct, but it is unable to carry out the subsequent extensionstep (93). Opposite both the abasic site and the 3′T of the (6-4) TT photoproduct,Polι incorporates a G, an A, or a T about equally well, and incorporation of a C isthe least favored (93, 96). Interestingly, the nucleotide insertion pattern oppositeboth these lesions resembles that opposite template T, which raises the intriguingpossibility that nucleotide incorporation opposite these lesion sites entails the samemechanisms as opposite an undamaged template T. The rather low efficiency ofnucleotide incorporation opposite both these lesions, however, may preclude Polιfrom making a major contribution to their bypass.
Evidence for a more specific role of Polι in lesion bypass has emerged fromits proficient ability to incorporate nucleotides opposite an N2-adducted guanine(99). Because of its high reactivity, the N2 group of guanine can conjugate with avariety of endogenously formed adducts. Acrolein, an α,β-unsaturated aldehyde,is formed in vivo as the end product of lipid peroxidation and also from metabolicoxidation of polyamines. The reaction of acrolein with the N2 of a G followedby ring closure at N1 yields γ -HOPdG (Figure 4). Since γ -HOPdG exists pre-dominantly in the ring-closed form in the nucleoside and in single-stranded DNA(100, 101), it is expected to also be in the ring-closed form when it is a templatingbase. Because of the inability of the ring-closed form of γ -HOPdG to participatein normal Watson-Crick pairing with the complementary C, this adduct poses asevere block to synthesis by most DNA polymerases. NMR studies with an 1,N2-propanodeoxyguanosine (PdG) adduct, whose structure is similar to that of thering-closed γ -HOPdG, have shown that in duplex DNA it adopts a syn conforma-tion and forms a protonated Hoogsteen pair with the complementary C (102, 103).This observation has suggested that the ring-closed form of γ -HOPdG is simi-larly involved in Hoogsteen pairing with a C. Polι incorporates a C or a T residueopposite γ -HOPdG as efficiently as it incorporates them opposite an undamaged

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 331
Figure 4 Structure of acrolein and its deoxyguanosine adducts in ring-closed andring-open forms.
G. Subsequently, Polκ efficiently extends from the C · γ -HOPdG primer terminusbut not from the T · γ -HOPdG terminus. Efficient and accurate replication throughthis adduct could thus occur by the sequential action of Polι and Polκ (99).
Structure of Polι and its Relevance for Lesion Bypass
Although the basic structure of Polι resembles that of other Y-family polymerases(Figure 5a) with a palm, which harbors the active site acidic residues that cat-alyze the phosphodiester bond formation, a thumb, which contacts the DNA minorgroove, fingers, which lie over the incipient base pair, and a PAD, which contactsthe major groove (11), it differs in remarkable ways from Polη, Polκ , and Dpo4(Figure 5).
In contrast to all known DNA polymerases, which include the high-fidelityreplicative/repair polymerases, as well as the other Y-family polymerases, whichimpose Watson-Crick base pairing in their active site during normal DNA synthesis,Polι synthesizes DNA using Hoogsteen base pairing (Figure 5b,c). In the ternarystructure of Polι (11), the templating A adopts a syn conformation and forms twohydrogen bonds via its Hoogsteen edge (N7 and N6) with the Watson-Crick edge ofthe incoming dTTP (N3 and O4), which remains in the anti conformation (Figure5c). The templating A is driven to syn conformation primarily because of the Leu62and Gln59 residues in the fingers domain, which bear down upon it—then tilt(∼30◦) and rotate it to the syn conformation (χ = 46◦) (Figure 5b). This involvesa significant restructuring of the sugar-phosphate backbone. If the templating Awere to be in a trans configuration in this structure, it would severely clash withLeu62. Although the structure of Polι in a ternary complex with a templating G andan incoming dCTP or a dTTP residue has not yet been determined, in all likelihood,Polι replicates through this template base also via Hoogsteen base pairing. TheHoogsteen edge of guanine (N7 and O6) and the Watson-Crick edge of cytosine(N3 and N4), which would remain in the anti conformation, can also form two
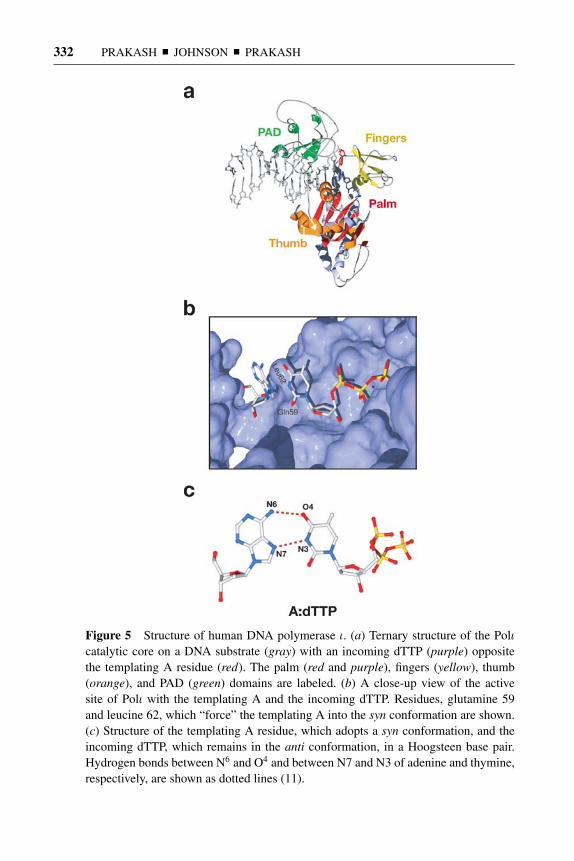
14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
332 PRAKASH � JOHNSON � PRAKASH
Figure 5 Structure of human DNA polymerase ι. (a) Ternary structure of the Polιcatalytic core on a DNA substrate (gray) with an incoming dTTP (purple) oppositethe templating A residue (red). The palm (red and purple), fingers (yellow), thumb(orange), and PAD (green) domains are labeled. (b) A close-up view of the activesite of Polι with the templating A and the incoming dTTP. Residues, glutamine 59and leucine 62, which “force” the templating A into the syn conformation are shown.(c) Structure of the templating A residue, which adopts a syn conformation, and theincoming dTTP, which remains in the anti conformation, in a Hoogsteen base pair.Hydrogen bonds between N6 and O4 and between N7 and N3 of adenine and thymine,respectively, are shown as dotted lines (11).

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 333
Figure 6 Hoogsteen base pair formation between guanine and cytosine and betweenguanine and thymine.
hydrogen bonds, one between O6 in G and N4 in C, and the other between N7 of Gand N3 of C; the latter hydrogen bonding, however, requires the protonation of N3in C (Figure 6). A geometrically similar Hoogsteen base pair can form between Gand T via the single hydrogen bond between the N7 of G and N3 of T (Figure 6).
The Hoogsteen base pairing of template purines with pyrimidines providesan elegant mechanism for the much higher efficiency and fidelity of nucleotideincorporation opposite template purines than that opposite template pyrimidines,as only the template purines can hydrogen bond via their Hoogsteen edge with thepyrimidines, whereas the lack of a Hoogsteen edge on the template pyrimidinesprecludes that from happening. Moreover, the different selectivities for nucleotideincorporation opposite templates A and G could then be attributed to the moreproficient Hoogsteen pairing of template A with a T than of template G with a Cor a T.
Hoogsteen base pairing can also serve as a mechanism to replicate throughminor-groove adducts of purines, such as those covalently attached to the N3of an A or the N2 group of G because the rotation of the purines from anti tosyn positions the adduct into the major groove, where there is much less sterichindrance. The proficient ability of Polι to incorporate nucleotides opposite γ -HOPdG supports this view (99). Hoogsteen base pairing can additionally serve topromote replication through purine adducts that impair the Watson-Crick pairingbut not the Hoogsteen pairing.
REV1
Rev1 functions in TLS together with Polζ , which is comprised of the Rev3 andRev7 proteins. Mutagenesis, induced by UV lesions (104–107) or from abasic sites(108), requires all three proteins. In the first reported biochemical study, yeast Rev1was shown to incorporate a C opposite template G and also opposite an abasic site.

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
334 PRAKASH � JOHNSON � PRAKASH
However, because of the apparent preferential incorporation of a C opposite anabasic site compared to that opposite an undamaged G, the Rev1 activity was thenreferred to as a deoxycytidyl transferase (109). Inherent in the nomenclature wasthe implication that Rev1 activity was not template specific. Subsequent kineticanalyses with yeast Rev1, however, have indicated that it preferentially incorpo-rates a C opposite template G, and it misincorporates nucleotides opposite thistemplate with a frequency of ∼10−3 to 10−4 (110). On a poly(dG) template, Rev1synthesizes DNA by C incorporation, and it incorporates ∼2 nucleotides per DNA-binding event. Rev1 can also incorporate a C opposite templates T, A, and C withfrequencies of ∼10−2 to 10−3 (110). Overall, the properties of Rev1 are consis-tent with its being a G-template-specific DNA polymerase, which also prefers toincorporate a C even at an abasic site. Thus, in addition to being specific for theG template, Rev1 is specific also for the incorporation of a C nucleotide. Rev1 isthe most extreme of DNA polymerases in its nucleotide incorporation specificity,much more so than even Polι.
Role of Rev1 in Lesion Bypass
Even though Rev1 is necessary for most base substitution mutations induced byUV light (104), its DNA polymerase activity seems to have no role in UV mu-tagenesis because C incorporation rarely occurs opposite UV lesions (52, 111)and inactivation of Rev1 DNA synthetic activity has no effect on UV mutagene-sis. And in accord with these genetic observations, Rev1 is unable to incorporatenucleotides opposite a cis-syn TT dimer or opposite a (6-4) TT photoproduct (L.Haracska and L. Prakash, unpublished observations). Rev1 is also indispensablefor mutagenesis resulting from TLS through abasic sites, but its DNA syntheticactivity has no significant effect on the bypass of this lesion, particularly whenthe lesion is in chromosomal DNA (112). Kinetic studies have indicated that Cincorporation by Rev1 opposite an abasic site is reduced ∼25-fold compared withthat opposite an undamaged G (110). Thus, although Rev1 could contribute to APbypass by C incorporation, genetic and biochemical evidence supports the sug-gestion that, in yeast as well as in humans, Polδ makes a major contribution to APbypass at the nucleotide incorporation step (112, 113).
N-2-acetyl aminofluorene (AAF) bound to the C8 of a G also presents a strongblock to nucleotide incorporation by Rev1. Genetic studies with a double-strandedplasmid harboring an AAF adduct have implicated a requirement for the Rev1protein to replicate also through this lesion, but its DNA polymerase activity isnot required (114). Mutagenic TLS occurring through UV lesions or abasic sitesor predominantly error-free TLS through AAF-adducted Gs, then, is dependenton the Rev1 protein but not on its DNA synthetic activity. Because Polζ is alsorequired for TLS through these various lesions at the extension step, Rev1 couldbe an important structural element for Polζ assembly at the replication fork.
Lesions, such as m6G and 8-oxoG, inhibit C incorporation by Rev1 ∼40- and400-fold, respectively (110), and genetic studies also have yielded no evidence

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 335
for a significant role of Rev1 or of Polζ in their bypass (S.L. Yu and L. Prakash,unpublished observations). Clear evidence for an important role of Rev1 DNAsynthetic activity in lesion bypass has emerged from biochemical studies indicatingthat it proficiently incorporates a C opposite an N2-γ -HOPdG (Figure 4), fromwhich Polζ extends (115). Error-free replication through a γ -HOPdG adduct couldthen occur by the sequential action of Rev1 and Polζ . In humans, in addition toRev1 and Polζ , Polι and Polκ would provide an alternate pathway to replicatethrough such lesions (99).
One major role of Rev1 DNA synthetic activity could then be to promote repli-cation through the variety of N2-guanine adducts that impinge upon the minorgroove. In this regard, Rev1 may resemble Polι, and as we have suggested for Polι,Rev1 also could proficiently incorporate a C opposite the various N2-adducted Gsby accommodating the adducted template G in the syn conformation and forminga Hoogsteen base pair with the incoming C.
Possible Involvement of Rev1 in the Assembly of OtherY-Family Polymerases for Polζ -Dependent TLS
In addition to its suggested role in the assembly of Polζ at the replication fork,recent evidence with mouse and human Rev1 has implicated this protein in theassembly of Polη, Polι, and Polκ , presumably with Polζ . Two hybrid analyses andcoimmunoprecipitation experiments have suggested that mouse and human Polη,Polι, and Polκ bind to the same ∼100 C-terminal amino acids of Rev1 (116–118).The requirement for the same region of Rev1 in the binding of these differentpolymerases might be to ensure that only one of them gains access to the Polζ -dependent TLS machinery at a time. Because human Rev1 interacts with Rev7 viathe same C-terminal portion (119, 120) as with the various Y-family polymerases,Rev7-bound Rev1 would then be inhibited from binding these polymerases.
The requirement for the activity of an inserter polymerase to be coordinatedwith that of Polζ necessitates the placement of a different inserter polymerasetogether with Polζ , depending upon the DNA lesion to be bypassed. Thus, Polηwould be needed at the insertion step for TLS through a (6-4) photoproduct orthrough an AAF-adducted G, and Polι would similarly promote Polζ -dependentlesion bypass of certain lesions by acting at the insertion step. Because of itspredominant role at the extension step, however, it is not clear how Polκ couldcontribute to Polζ -dependent lesion bypass. Perhaps, Polκ assists Polζ in someother, as yet unknown, way in lesion bypass.
DNA POLYMERASE κ
Polκ belongs to the DinB subfamily of Y-family DNA polymerases, which includesE. coli DinB (Pol IV) and archaeal Dbh and Dpo4 proteins. In addition to theconserved motifs I–V present in all Y-family polymerases, the DinB subfamily

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
336 PRAKASH � JOHNSON � PRAKASH
Figure 7 Schematic alignment of DinB subfamily members, human Polκ , E. coli DinB,and Sulfolobus solfataricus (S.s.) Dpo4. Amino acid sequences are depicted as gray boxes.Conserved motifs I–V, present in all Y-family polymerases, are shown. The C2HC and N-terminal regions unique to the eukaryotic members of this subfamily are shown in humanPolκ .
members have the conserved motifs x, y, and z, which form the PAD region.Human Polκ and other eukaryotic DinB homologs, however, differ from theirprokaryotic and archaeal counterparts by the presence of unique N-terminal andC-terminal extensions (Figure 7). The C-terminal region of Polκ , which is beyondthe PAD of the protein and contains two putative zinc-binding C2HC motifs, is notrequired for polymerase activity (12). The N-terminal region, however, is requiredfor activity, and Polκ , which is missing the N-terminal and C-terminal regionsand mimics the DinB and Dpo4 proteins, is completely inactive (12). Thus, thecatalytic core of eukaryotic Polκ , which includes the N-terminal extension of Polκ ,is larger than that of prokaryotic and archaeal DinB members.
Fidelity and Mismatch Extension Ability of Polκ
Polκ differs from other Y-family polymerases in nucleotide incorporation speci-ficity and mismatch extension ability. Polκ is the most faithful of all Y-familyDNA polymerases, misincorporating nucleotides with a frequency of ∼10−3 to10−4 opposite all four template bases (121). Polκ , however, is a proficient ex-tender of mispaired primer termini, extending them with a frequency of ∼10−1
to 10−2 (122). In its proficient mismatch extension ability, Polκ differs from theother members of the DinB subfamily because they extend the mispaired primerterminus with nearly the same frequency as they misincorporate the nucleotide. Forexample, E. coli Pol IV misincorporates nucleotides with a frequency of ∼10−3 to10−5, and it extends mispaired termini with nearly the same frequency (123). Ar-chael Dbh and Dpo4 polymerases misincorporate nucleotides with a frequency of∼10−2 to 10−3 (7, 124), and Dpo4 extends from mispaired termini with a frequencyof ∼10−3 (71).
Polκ shares with other DinB-family members the tendency to generate frame-shift errors, particularly single-base deletions. The mechanism of frameshifting,however, differs between Polκ and other members. Single-base deletions in nonit-erated sequences can be generated by two different mechanisms—dNTP-stabilizedmisalignment (125) and misinsertion misalignment (126). In dNTP-stabilized

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 337
Figure 8 Model for generation of DNA polymerase-dependent single-base deletions. (a) IndNTP-stabilized misalignment, the templating base “loops out,” i.e., is extrahelical, and theresulting misalignment is stabilized by the pairing of the incoming dNTP with the next com-plementary template base. (b) In misinsertion misalignment, nucleotide misincorporation isfollowed by primer-template slippage and the repositioning of the misincorporated nucleotideopposite the next complementary template base. Reprinted by permission of publisher (127).
misalignment, the templating base is looped out in the polymerase active site, andthe misalignment is stabilized by the pairing of the incoming dNTP with the nextcomplementary template base (Figure 8a). E. coli Pol IV and archaeal Dpo4 utilizethis mechanism to generate frameshifting errors (9, 123). Polκ , instead, generatesframeshifting errors via misinsertion misalignment, wherein the template base atthe mispaired primer terminus is looped out and the primer-terminal nucleotideis then repositioned opposite the next complementary template base (Figure 8b).Polκ adopts this as a means to extend mispaired primer termini (127). The ability ofPolκ to extend mispaired primer termini by direct extension or via misalignment ofthe template and primer nucleotides (127) (Figure 9) could be used during normal
Figure 9 Two modes of mismatch extension by human Polκ . In direct mismatch ex-tension (left), following a primer-terminal mispair, the incoming dCTP (indicated bythe curved arrow) pairs with the correct templating base G. In extension by misalign-ment (right), the C · C mispair realigns so that the template C becomes extrahelicaland the primer-terminal C pairs with the next template base G. This is followed by thepairing of the incoming dTTP (indicated by the curved arrow) with the subsequentcomplementary template base. Reprinted by permission of publisher (127).

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
338 PRAKASH � JOHNSON � PRAKASH
DNA replication, thereby contributing to the continued and efficient progressionof the replication fork. Base substitution and single-base deletion mutations will,however, be generated as a result.
Lesion Bypass by Polκ
For most DNA lesions, Polκ is unable to insert nucleotides opposite the lesion site;it can, however, extend from nucleotides inserted by another DNA polymeraseopposite certain DNA lesions. For example, Polκ is unable to insert nucleotidesopposite the 3′T of a cis-syn T-T dimer (121), but it can extend from a G opposite the3′T of a dimer almost as efficiently as it extends from an A opposite a nondamagedT (122). Polκ , however, is unable to carry out either the nucleotide insertion orthe extension reaction opposite from a (6-4) TT photoproduct (121, 122). Polκis quite inefficient at inserting nucleotides opposite an abasic site (121, 128), andit also is very poor at extending from the nucleotide opposite an abasic site bymeans of direct incorporation of the next complementary nucleotide (127). It can,however, extend from an abasic site using the misalignment mechanism, whereinthe base opposite the abasic site pairs with the complementary 5′-template baseand is followed by the incorporation of the next complementary nucleotide (127).Polκ is also considerably inhibited at inserting nucleotides opposite an m6G lesion,but it can carry out the extension reaction opposite from this lesion. Opposite an8-oxoG lesion, Polκ inserts an A quite efficiently and a C less well, and it canextend with an ∼20–40-fold reduction in efficiency (129).
An AAF- or AF-adducted G presents a block to synthesis by Polκ at both thenucleotide incorporation and extension steps; the efficiency of incorporation of aC opposite both these lesions is reduced by almost 1000-fold, and the efficiency ofextension is reduced by over 10,000-fold (130). 1-N6-etheno dA is also inhibitoryat both these steps (65). dG-N2-BPDE adducts, too, pose a considerable block atboth the nucleotide incorporation and extension steps; the extension step, however,is less impaired (131). And, Polκ is inhibited at extending from an A insertedopposite a thymine glycol ∼100–200-fold (132).
This survey of the lesion bypass ability (or inability) of Polκ fails to yield anysatisfactory clues regarding the specificity of Polκ’s role in TLS. Although Polκcan efficiently extend opposite from the 3′T of a cis-syn TT dimer, and that mightaccount for the UV sensitivity of Polκ-deficient mouse cells (133), it seems ratherunlikely that the role of Polκ would be restricted to just promoting the mutagenicbypass of CPDs by direct extension, or to mediating the TLS through AP sites, andperhaps through other highly distorting lesions, via template-primer misalignment.
A role for Polκ in extending opposite from minor-groove-adducted templatenucleotides is indicated from its proficient ability to extend from a C oppositeγ -HOPdG (99), which is formed from the reaction of acrolein with the N2 of dG(Figure 4). Because of the high reactivity of the N2 group of dG, a variety ofDNA adducts would form at this position, and these include the propano adductsand malondialdehyde-derived adducts. The propano adducts are formed from

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 339
α,β-unsaturated aldehydes or enals, such as acrolein, crotonaldehyde, and trans-4-hydroxy-2-nonenal (HNE). Lipid peroxidation generates all these adducts, andthey are present in the DNA of human tissues at considerable levels. Recently, wehave observed that Polκ can also extend efficiently from the C incorporated oppo-site the N2-dG adduct of HNE by Polι (W.T. Wolfle and L. Prakash, unpublisheddata). The sequential action of Polι and Polκ , then, could provide an importantpathway for the efficient and error-free bypass of the various N2-dG adducts gen-erated from endogenous cellular reactions. The proficient ability of Polκ to extendopposite from lesions, such as γ -HOPdG and N2-dG HNE, suggests that its activesite can tolerate the distortions conferred by these adducts on the template base atthe template-primer junction.
Polκ Structure
Although deletion of the C-terminal 344 amino acids that lie beyond the PAD(Figure 7), as in Polκ (1–526), has no effect on DNA polymerase activity, Polκprotein lacking the first 91 amino acids shows no DNA polymerase activity. Dele-tion of the first 19 amino acids, however, has no effect on polymerase activity,whereas a protein deleted for the first 68 residues shows reduced activity. The Nterminus thus is indispensable for Polκ activity, and the region that lies betweenresidues 19–68 and that is conserved among all eukaryotic Polκ proteins (markedN in Figure 7) is required for complete activity (12).
The structure of Polκ containing residues 68–526 has been determined (12),and it reveals certain features that distinguish it from other Y-family polymerases(Figure 10). Even though the fingers domain of Polκ is smaller than that of Polη,the active site cleft at the junction of the fingers and palm domains is also smallerthan in Polη. Modeling of Polκ in a ternary complex bound to DNA and with anincoming nucleotide has indicated that Polκ’s active site is more constrained withrespect to the template base than that of Polη (12). The constrained active site cleftof Polκ may underlie its inability to incorporate nucleotides opposite the variousDNA lesions, such as a cis-syn TT dimer, a (6-4) TT photoproduct, and others andmay account also for its better fidelity on undamaged DNA templates.
The thumb domain of Polκ differs strikingly from that of other Y-family poly-merases. In addition to the five consecutive α-helices that are present betweenthe palm and PAD domains of Polκ and that correspond to the thumb domain ofother Y-family polymerases, Polκ has an additional sixth α-helix, which derivesfrom the unique N-terminal sequence (12). In the protein containing the entire Nterminus, this α-helix could potentially continue on, reach over, and interact withthe template primer; and in keeping with this suggestion, is the observation thatPolκ protein lacking the first 67 residues displays a reduced ability for bindingDNA (12).
Another striking feature of the Polκ structure is the position of the PAD relativeto the fingers domain. Compared to the other Y-family polymerases, Polη, Polι,and Dpo4, in which the PAD is located close to the fingers domain and binds

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
340 PRAKASH � JOHNSON � PRAKASH
Figure 10 Structure of human DNA polymerase κ from residues 68 to 526. Helix1, which forms part of the thumb domain and is unique to human Polκ , is labeledfrom Ser76 to Ser96. This helix is absent in Dpo4. The distance the PAD (green) musttraverse to bind DNA, akin to that in Dpo4, is shown by the green arrow (12).
duplex DNA, in the Polκ structure the PAD lies underneath the palm domainrather than juxtaposed next to the fingers domain (12). Even though the PAD isexpected to move upon DNA binding, still it may turn out that the Polκ PADmakes fewer contacts with the duplex DNA, and this contributes to its proficientability to extend mispaired primer termini on undamaged DNAs and to carry outthe extension reaction in lesion bypass.
DNA POLYMERASE ζ
Rev3, the catalytic subunit of Polζ , belongs to the B-family DNA polymerases,which include the replicative polymerases α, δ, and ε. Unlike Polδ or ε, however,Polζ lacks the proofreading exonuclease activity, and unlike the three replicative

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 341
polymerases, Rev3 is not essential for cell viability or for growth in yeast cells.Disruption of REV3 in mice, however, causes embryonic lethality (134–136).
Both subunits of Polζ , Rev3 and Rev7, are indispensable for UV mutagenesis(105–107) and for mutagenesis resulting from TLS occurring through abasic sites(108) or through bases damaged by certain chemical agents but not others (137).Polζ ’s action, however, is not limited to the mutagenic bypass of DNA lesions, asit also enables error-free bypass through a variety of DNA lesions.
Fidelity and Highly Proficient Mismatch ExtensionAbility of Polζ
Compared to the fidelity of Y-family polymerases, Polζ is a rather high-fidelityenzyme, since it misincorporates nucleotides opposite all four template bases witha frequency of ∼10−4 (93, 112, 138). The fidelity of Polζ is more akin to that ofPolα, which also synthesizes DNA with an error rate of ∼10−4 (139). But Polζdiffers strikingly from other DNA polymerases in its very proficient ability toextend from mispaired primer termini, which occurs with a frequency of ∼10−1 to10−2 (93, 112, 138). Although Polζ is more similar to Polκ in its high fidelity fornucleotide incorporation and in its proficient mismatch extension ability, overall,and in different sequence contexts, Polζ displays a higher fidelity and a bettermismatch extension ability than does Polκ .
Role of Polζ as an Extender in Lesion Bypass
Polζ is highly inefficient at replicating through DNA lesions, and this resultsfrom its inability to incorporate nucleotides opposite them. For example, Polζ isstrongly inhibited for nucleotide incorporation opposite the 3′T of a cis-syn TTdimer or a (6-4) TT photoproduct (93). An abasic site also presents a strong blockto nucleotide incorporation by Polζ , and moreover, even the incorporation of an Aopposite an abasic site, which even the replicative polymerases can accomplish, isreduced over 1000-fold (112). Also revealing in this respect is the greatly reducedefficiency of Polζ for nucleotide incorporation opposite lesions such as 8-oxoGand m6G (138).
Polζ is the most proficient extender of primer termini opposite from DNAlesions. In fact, in our studies, we have not yet encountered a DNA lesion fromwhich it could not extend. Opposite from the 3′T of a cis-syn TT dimer or a (6-4)TT photoproduct, Polζ extends from an A, a G, a T, or a C nucleotide almostas efficiently as when these nucleotides are opposite an undamaged T (93). Thus,neither of these lesions inhibits extension by Polζ . Likewise, Polζ extends from theA, T, or C nucleotides placed opposite an abasic site with nearly the same efficiencyas it does from the corresponding nucleotides placed opposite an undamaged C,and extension from a G opposite from an abasic site is only ∼10-fold reducedrelative to the extension from a G opposite an undamaged C (112).
Polζ can extend most efficiently from an A opposite an 8-oxoG, and it extendsabout fivefold less well from a C opposite an 8-oxoG. Polζ extends from a G,

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
342 PRAKASH � JOHNSON � PRAKASH
an A, a T, or C nucleotide opposite from m6G with frequencies of ∼2 × 10−1–3 × 10−2 relative to extension from a C opposite undamaged G (138). Polζ ishighly proficient at extending from a C opposite γ -HOPdG (115).
Thymine glycol (Tg), a lesion formed from the oxidation of the 5,6 double bondof thymine, presents a strong block to synthesis by replicative DNA polymerases.Although a Tg does not block the incorporation of an A by these polymerases,synthesis is arrested opposite the lesion site, indicating a block at the extensionstep. Polζ extends from an A opposite Tg as efficiently as it extends from the Aopposite an undamaged T (140).
In its highly proficient ability to extend from nucleotides opposite a (6-4) TTphotoproduct, an abasic site, or a Tg, Polζ is strikingly different from other poly-merases because no other polymerase has such an ability. Even though a (6-4) TTphotoproduct induces a large structural distortion at the 3′T, it does not affect theability of the 5′T to form a normal Watson-Crick pair with an A; and an abasic sitealso does not affect the base pairing ability of the next 5′ template base. Conversely,a Tg destabilizes the stacking interactions with the 5′ base pair, as inferred fromenergy minimization and molecular dynamic simulation studies (141, 142). Thus,Polζ can extend not only from primer termini that are located opposite highly dis-torting DNA lesions or abasic sites, but it can extend also when the lesion stericallyinterferes with the next 5′ template base.
LESION BYPASS BY ONE OR TWO DNA POLYMERASES
Replication through certain DNA lesions can be accomplished by one polymerase,but replication through many DNA lesions requires the action of two different poly-merases, one for carrying out the nucleotide insertion reaction opposite the lesionsite and the other for the subsequent extension reaction (Table 1). Although Polηcan proficiently and accurately replicate through CPD and 8-oxoG lesions, oppo-site a (6-4) photoproduct, an abasic site, or an AAF- or AF-adducted G, it can onlycarry out the insertion reaction. The functions of Polι and Rev1 are largely restrictedto the insertion step, whereas Polκ and Polζ function primarily at the extensionstep. However, because Polκ extends only from a subset of lesions that are alsoproficiently handled by Polζ , the contribution of Polκ to the extension step seemsto be more limited than that of Polζ . That raises the possibility that Polκ contributesto lesion bypass in yet some other way and not just as a subsidiary to Polζ .
Mutations in REV3 were initially identified from their lack of UV mutagenesis,and the role of Polζ was thought to be primarily restricted to the mutagenic bypassof DNA lesions. However, it is becoming increasingly clear that Polζ promotes alsoerror-free TLS through a number of lesions. Thus, in addition to contributing to themutagenic bypass of CPDs and (6-4) photoproducts that would result followingextension from the wrong nucleotides incorporated by Polη or by some otherpolymerase opposite these lesions, Polζ carries out error-free bypass through the(6-4) TC and CC photoproducts by extending from the G nucleotide incorporated

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 343
TABLE 1 Lesion bypass by one or two DNA polymerases
Lesion bypass by
Two Pols
DNA lesion One Pol Inserter ExtenderError-free ormutagenic
8-oxoG Polη Error-free
CPDs at TT, TC, CC sites Polη Error-free
(6-4) photoproducts:at TT Polη Polζ Mutagenicat TC and CC Polη Polζ Error-freeat TT, TC, and CC Polι Polζ Mutagenic
Abasic sites Polδ Polζ MutagenicPolηRev1Polι
Tg Polδ Polζ Error-free
AAF- or AF-adducted G Polη Polζ Error-free
γ -HOPdG Rev1 Polζ Error-freePolι Polκ Error-free
by Polη opposite the 3′C of these photoproducts (51, 56, 93). By extending fromthe A nucleotide, inserted by Polδ or by another replicative polymerase oppositethe Tg lesion, Polζ provides a key pathway for error-free TLS through this lesion,and genetic observations in yeast support this conclusion (140). A role for Polζ inthe error-free bypass of an AAF- or AF-adducted G is also indicated from geneticstudies in yeast, where presumably it extends from the C inserted by Polη oppositethe lesion site (55, 114). And, error-free bypass through the γ -HOPdG adductcould occur by the sequential action of Rev1 and Polζ (115).
Proficient replication through the variety of DNA lesions that frequently formfrom endogenous cellular reactions could be important for ensuring that rapidcell divisions continue unabated during early embryonic development. The in-volvement of Polζ in promoting TLS through a wide array of DNA lesions thencould be the reason for its indispensability during early embryonic development inmice.
TARGETING OF TLS POLYMERASES TO THEREPLICATION ENSEMBLE STALLED AT A LESION SITE
How the action of various lesion bypass processes, including those of multipleTLS polymerases, in rescuing stalled replication forks is coordinated and regulatedremains enigmatic. Recent observations have indicated that PCNA plays a key role

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
344 PRAKASH � JOHNSON � PRAKASH
in promoting the access of TLS polymerases to the replication fork. The underlyingintricacies of protein-protein interactions, post-translational protein modifications,and of switching from the replicative mode to the lesion bypass mode, however,remain to be uncovered.
Physical and Functional Interactions of Y-FamilyPolymerases with PCNA
Both yeast and human Polη interact with PCNA physically and functionally (143,144). As determined from gel filtration studies with yeast Polη, in the Polη-PCNAcomplex, the two proteins are in a 1:3 molar ratio, respectively, consistent with thebinding of one Polη molecule to a PCNA trimer. PCNA, when loaded onto DNA byRFC in the presence of RPA, stimulates the DNA synthetic activity of both yeast andhuman Polη ∼ 10–15-fold (143, 144). Interestingly, this stimulation does not resultfrom an increase in processivity, as even in the presence of PCNA, RFC, and RPA,Polη processivity remains the same as in the absence of these accessory proteins,which is ∼three to four nucleotides per DNA-binding event. Instead, steady-statekinetic analyses have revealed that PCNA-binding stimulates the efficiency ofnucleotide incorporation by ∼15-fold, and importantly, this increase is achievedprimarily by a reduction in the apparent Km for the nucleotide (143, 144). The effectof PCNA on Polη in not affecting the processivity but in increasing the affinityfor the incoming nucleotide (as is suggested from the reduction of apparent Km)is remarkably different from its role of increasing the processivity of replicativeDNA polymerases δ and ε.
Mutations in the conserved PCNA-binding motif, I (L)–F F, which is presentat the C terminus of yeast and human Polη, inactivate the physical and functionalinteractions of these polymerases with PCNA (143, 144), and as has been deter-mined for the yeast protein, they inactivate the in vivo damage bypass ability ofPolη (143).
Human Pols ι and κ also interact physically with PCNA, and PCNA togetherwith RFC and RPA greatly stimulates their DNA synthetic activity (96, 128). Forboth polymerases, the efficiency of nucleotide incorporation is enhanced by ∼50–200-fold, and this is accompanied by a decrease in the apparent Km for dNTPsof nearly the same magnitude (96, 128). PCNA also stimulates the lesion bypassproficiency of these polymerases by a reduction in the Km for dNTPs, and themagnitude of stimulation in the efficiency of nucleotide incorporation is about thesame as that opposite undamaged template nucleotides (96).
In summary, although binding to PCNA does not alter the basic properties ofthese enzymes, such as their low processivity and low fidelity, or their abilityor inability to bypass a particular DNA lesion, PCNA stimulates their efficiencyfor nucleotide incorporation opposite both undamaged and damaged sites. Theincrease in the affinity for the nucleotide that obtains in the presence of PCNA raisesthe intriguing possibility that PCNA binding induces a conformational change inthese polymerases that is more conducive to dNTP incorporation.

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 345
Rad6-Rad18-Dependent Lesion Bypass Pathways
Rad6, a ubiquitin-conjugating enzyme (Ubc), exists in a complex with Rad18,a single-stranded DNA-binding protein (145, 146), which could target Rad6 tosingle-stranded DNA resulting from the stalled replication fork at a lesion site.There is extensive genetic evidence from yeast in support of a preeminent role forthis Ubc complex in the error-free as well as mutagenic bypass of DNA lesionsduring replication (147, 148).
Rad6-Rad18-mediated ubiquitin conjugation promotes replication throughDNA lesions via three different pathways: the Polζ - and Polη-dependent TLSpathways and a Rad5-Mms2-Ubc13-dependent pathway that promotes the repairof discontinuities that form in the newly synthesized strand opposite from DNAlesions (149). Although the mechanism of the Rad5-dependent pathway is notknown, it might involve a limited copy-choice type of DNA synthesis in which thenewly synthesized daughter strand of the undamaged complementary sequence isused as the template for bypassing the lesion.
The Polζ - and Polη-dependent TLS pathways promote mutagenic or error-freeTLS depending upon the DNA lesion (Table 1). The Rad5- and Mms2-Ubc13-dependent postreplication repair pathway operates primarily in an error-free man-ner. Rad5, a SWI/SNF-family DNA-dependent ATPase (150, 151), contains, inaddition to the helicase motifs, a C3HC4 ring finger motif that is found in many E3proteins that facilitate the assembly of the ubiquitin-conjugating enzyme, E2, withthe protein substrate. The Mms2 protein belongs to a class of Ubc enzyme variantproteins that lack Ubc activity because of the absence of an active site cysteineresidue (152). Mms2 forms a complex with Ubc13, and the Mms2-Ubc13 complexassembles polyubiquitin chains that are linked through lysine 63 of ubiquitin (153).As has been inferred from epistasis analyses, yeast cells carrying a mutated formof ubiquitin, ubiK63R, also confer a defect in the Rad5,Mms2-Ubc13-dependentlesion bypass pathway (153).
Although the Rad6-Rad18-dependent processes provide the major routes forlesion bypass during replication, the Rad52-dependent recombinational pathway,which presumably initiates from double-stranded DNA breaks, can also be usedas a back-up for lesion bypass (154). Because of the attendant risk of genomicrearrangements resulting from this double-strand break–initiated recombinationpathway, this pathway is kept suppressed in wild-type yeast cells by proteins suchas the Srs2 DNA helicase (155), which actively disrupts the Rad51-nucleoproteinfilament formation (156, 157), a necessary prerequisite for Rad52-dependent re-combinational repair to operate.
Ubiquitin and SUMO Modifications of PCNA
The evidence that PCNA interacts physically and functionally with the variousyeast and human TLS polymerases and that this interaction is important for theirrole in lesion bypass has indicated that TLS polymerases gain access to the repli-cation ensemble via their binding to PCNA (96, 128, 143, 144). Genetic studies

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
346 PRAKASH � JOHNSON � PRAKASH
Figure 11 Monoubiquitination (monoUb) of PCNA by Rad6-Rad18 at the lysine 164residue promotes TLS by DNA polymerases η and ζ , and polyubiquitination (polyUb)of lysine 164 of PCNA via a lysine 63-linked ubiquitin chain, which requires Rad5 andMms2-Ubc13, promotes the Rad5-dependent mode of lesion bypass. Ubc9-mediatedSUMO modification of lysine 164 of PCNA inhibits Rad52-dependent double-strandbreak–initiated recombinational repair.
in yeast have additionally provided evidence for the requirement of PCNA forRad6-Rad18-dependent error-free postreplication repair (158). Thus, it would ap-pear that all the Rad6-Rad18-dependent lesion bypass processes are coordinatedthrough PCNA.
In yeast cells, upon treatment with DNA-damaging agents, PCNA is monoubiq-uitinated at its lysine 164 residue in a Rad6-Rad18-dependent manner; subse-quently, this PCNA residue is polyubiquitinated via a lysine 63-linked ubiquitinchain in a Rad5-Mms2-Ubc13-dependent manner (159) (Figure 11). Presumably,in this reaction, Rad5 promotes PCNA polyubiquitination by connecting with bothMms2-Ubc13 and PCNA, thereby bringing the Ubc activity into close contact withPCNA (159–161).
PCNA is also modified by SUMO conjugation by Ubc9 at the lysine 164 andlysine 127 residues, with lysine 164 being the prominent site for this modification(159). SUMO conjugation of PCNA, which takes place in the S phase, remainsunaffected when yeast cells are treated with a low does of MMS (0.02%), con-ditions under which PCNA ubiquitination is stimulated. At a high dose of MMS(0.3%), however, SUMO conjugation of PCNA is markedly elevated, particularlyat the K164 residue; but under these conditions, PCNA ubiquitination is absent(159). SUMO conjugation of PCNA then seems to be important for normal DNAreplication, or when cells are exposed to a lethal dose of a DNA-damaging agent,whereas ubiquitin conjugation of PCNA seems to regulate the lesion bypass pro-cesses under conditions of low to moderate DNA damage.
Epistatic analysis of the PCNA mutation in which the lysine 164 residue hasbeen changed to arginine (pol 30 K164R) has suggested that this mutation inacti-vates all three Rad6-Rad18-dependent lesion bypass processes, and impairmentsof UV mutagenesis and postreplication repair occur (159, 162, 163). Even though

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 347
all the Rad6-Rad18-dependent lesion bypass pathways are apparently inactivatedby this PCNA mutation, it displays a much higher level of UV resistance thanthe rad6� and rad18� mutants. Genetic observations, indicating that the pol 30K164R mutation suppresses the UV sensitivity of the rad6� and rad18� mutantsand that this suppression occurs via Rad52-dependent recombinational repair pro-cesses, have suggested that this Rad52 pathway becomes activated in the PCNAmutant, and this accounts for its higher UV resistance (163). The observation thatactivation of the Rad52 pathway can occur in the absence of Rad6-Rad18 ubiquitinconjugation activity implies that the absence of SUMO modification in the pol 30K164R mutant PCNA is the cause of this activation. Overall, the various geneticobservations support the conclusion that monoubiquitination of K164 in PCNAactivates TLS by Polζ and Polη, whereas polyubiquintation of this PCNA residueactivates Rad5-dependent postreplication repair. SUMO modification of K164 ofPCNA, however, inactivates Rad52-dependent recombinational repair (Figure 11).
How ubiquitin conjugation of PCNA affects lesion bypass processes is notknown. One possibility, which we suggested before, is that PCNA monoubiquiti-nation disrupts the binding of the replicative polymerases to the template-primerjunction, and that in turn enables the binding of the TLS polymerase to PCNA andto the other components of the replication ensemble (163). Consistent with thisidea is the observation that, in UV-damaged human cells, Polη associates primarilywith monoubiquitinated PCNA (164).
CONCLUDING REMARKS
Biochemical and structural studies of yeast and human TLS polymerases haverevealed a surprisingly high degree of specificity in their roles in lesion bypass.The lesion bypass properties of Polη are affected by whether the lesion involvesone or two template nucleotides and whether the ability to form normal Watson-Crick base pairs is maintained. The proficiency and accuracy of Polη at replicatingthrough a CPD could only have been achieved from its ability to include bothtemplate residues of the lesion in its active site; otherwise, the reaction would nothave been as efficient. Polι is adept at incorporating nucleotides opposite minor-groove-adducted template purines that normally adopt a syn conformation in DNAand forms a Hoogsteen base pair with the incoming pyrimidine nucleotide. Theproficient ability of Polι to accommodate such a Hoogsteen base pair providesan exquisite mechanism for incorporating nucleotides opposite a large array ofminor-groove purine adducts that would otherwise be highly disruptive to thenormal progression of the replication fork. Rev1 is even more extreme than Polι inthis respect, and it remains to be seen whether it utilizes a mechanism similar to Polιfor incorporating a dCTP opposite the various N2-adducted guanines that adopt asyn conformation in DNA. Polκ functions at the extension step of lesion bypass.However, how the N-terminal extension, unique to Polκ and which constitutes theadditional helix of the thumb domain, and how the PAD, which remains far away

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
348 PRAKASH � JOHNSON � PRAKASH
from the fingers domain in the Polκ apo structure, contribute to this ability remainto be determined. Thus, even though all these polymerases share the same basicstructural plan, their specificity for lesion bypass is remarkably diverse, and thesedifferences emanate from their unique structural features.
Of all the eukaryotic TLS polymerases, Polζ is the most proficient at extendingprimer termini opposite from a diverse array of DNA lesions. Polζ , however, isstrongly inhibited at the nucleotide incorporation step of lesion bypass. Also, Polζis a very proficient extender of mispaired primer termini but exhibits a high speci-ficity for incorporating the correct nucleotide on undamaged DNAs. Polζ ’s activesite then is uniquely adapted for tolerating a high degree of geometric distortionat the template-primer junction but not at the site of the incoming nucleotide.
The observation that PCNA binding stimulates the efficiency of nucleotide in-corporation by Y-family polymerases opposite undamaged as well as damagedtemplates and the suggestion that this could be from a PCNA-induced conforma-tional change in the polymerase active site raise the possibility of protein-proteininteractions modifying the polymerase action, thereby providing for even morelatitude in their lesion bypass capabilities. Thus, in spite of the fact that nucleotideincorporation opposite the 3′T of a (6-4) TT lesion by Polη is reduced by 50–100-fold, genetic evidence from yeast documents an important role for this polymerasein its bypass. Perhaps, binding to other protein factors stimulates the proficiencyof Polη for dNTP incorporation opposite the (6-4) TT, TC, and CC photoproducts.There then may be lesions that are handled rather inefficiently when the polymeraseacts alone but are processed much more efficiently when it is in a complex withother proteins. Such a versatility in function, dependent upon the binding of otherprotein factors, might be more feasible for the Y-family polymerases because oftheir less rigid active sites.
Finally, conjugation of ubiquitin and SUMO not only to PCNA, but in all like-lihood also to other proteins, will be important for various lesion bypass processesand for polymerase exchange. Furthermore, checkpoint-induced phosphorylationsmay also have a significant role in these complex and diverse processes.
ACKNOWLEDGMENTS
We are most grateful to our colleagues Lajos Haracska, M. Todd Washington,Ildiko Unk, and William Wolfle for their contributions to the development andexecution of many of the ideas presented here. Without the structural studies thathave been done in the laboratory of our collaborator, Aneel Aggarwal, many ofthe conclusions of this review would not have been possible. We are grateful toAneel and his colleagues Deepak T. Nair, Sacha N. Uljon, Jose Trincao, and CarlosEscalante for their collaboration and insights. Collaborations with Jerry Hurwitzhave contributed much to the progress of our studies with human TLS polymerases,and Peter Burgers has helped through collaborations and by providing materi-als. We are also grateful to Patrick Sung for materials. Studies in our laboratorywere supported by NIH research grants GM19261, CA107650, and ES012411.

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 349
Structural studies in Aneel Aggarwal’s laboratory have been supported by NIHgrant CA094006.
The Annual Review of Biochemistry is online athttp://biochem.annualreviews.org
LITERATURE CITED
1. Ohmori H, Friedberg EC, Fuchs RPP,Goodman MF, Hanaoka F, et al. 2001.Mol. Cell. 8:7–8
2. Burgers PM, Koonin EV, Bruford E,Blanco L, Burtis KC, et al. 2001. J. Biol.Chem. 276:43487–90
3. Johnson RE, Washington MT, Prakash S,Prakash L. 1999. Proc. Natl. Acad. Sci.USA 96:12224–26
4. Kondratick CM, Washington MT, Pra-kash S, Prakash L. 2001. Mol. Cell Biol.21:2018–25
5. Johnson RE, Trincao J, Aggarwal AK,Prakash S, Prakash L. 2003. Mol. CellBiol. 23:3008–12
6. Glick E, Vigna KL, Loeb LA. 2001.EMBO J. 20:7303–12
7. Silvian LF, Toth EA, Pham P, GoodmanMF, Ellenberger T. 2001. Nat. Struct.Biol. 8:984–89
8. Zhou B-L, Pata JD, Steitz TA. 2001. Mol.Cell. 8:427–37
9. Ling H, Boudsocq F, Woodgate R, YangW. 2001. Cell 107:91–102
10. Trincao J, Johnson RE, Escalante CR,Prakash S, Prakash L, Aggarwal AK.2001. Mol. Cell. 8:417–26
11. Nair DT, Johnson RE, Prakash S, PrakashL, Aggarwal AK. 2004. Nature 430:377–80
12. Uljon SN, Johnson RE, Edwards TA,Prakash S, Prakash L, Aggarwal AK.2004. Structure 12:1395–404
13. Delarue M, Poch O, Tordo N, MorasD, Argos P. 1990. Protein Eng. 3:461–67
14. Doublie S, Tabor S, Long AM, Richard-son CC, Ellenberger T. 1998. Nature391:251–58
15. Ollis DL, Brick P, Hamlin R, Xuong NG,Steitz T. 1985. Nature 313:762–66
16. Johnson RE, Prakash S, Prakash L. 1999.Science 283:1001–4
17. McDonald JP, Levine AS, Woodgate R.1997. Genetics 147:1557–68
18. Lehmann AR, Kirk-Bell S, Arlett CF, Pa-terson MC, Lohman PHM, et al. 1975.Proc. Natl. Acad. Sci. USA 72:219–23
19. Cordeiro-Stone M, Zaritskaya LS, PriceLK, Kaufmann WK. 1997. J. Biol. Chem.272:13945–54
20. Nikolaishvili-Feinberg N, Cordeiro-Stone M. 2001. Biochemistry 40:15215–23
21. Wang Y-C, Maher VM, Mitchell DL,McCormick JJ. 1993. Mol. Cell Biol. 13:4276–83
22. Waters HL, Seetharam S, Seidman MM,Kraemer KH. 1993. J. Investig. Dermatol.101:744–48
23. Johnson RE, Kondratick CM, Prakash S,Prakash L. 1999. Science 285:263–65
24. Masutani C, Kusumoto R, Yamada A,Dohmae N, Yokoi M, et al. 1999. Nature399:700–4
25. Washington MT, Johnson RE, PrakashS, Prakash L. 1999. J. Biol. Chem. 274:36835–38
26. Johnson RE, Washington MT, PrakashS, Prakash L. 2000. J. Biol. Chem. 275:7447–50
27. Washington MT, Johnson RE, PrakashS, Prakash L. 2001. J. Biol. Chem. 276:2263–66
28. Bebenek K, Matsuda T, Masutani C,Hanaoka F, Kunkel TA. 2001. J. Biol.Chem. 276:2317–20

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
350 PRAKASH � JOHNSON � PRAKASH
29. Washington MT, Johnson RE, Prakash S,Prakash L. 2000. Proc. Natl. Acad. Sci.USA 97:3094–99
30. Washington MT, Prakash L, Prakash S.2003. Proc. Natl. Acad. Sci. USA 100:12093–98
31. Smith CA, Baeten J, Taylor J-S. 1998. J.Biol. Chem. 273:21933–40
32. Sun L, Wang M, Kool ET, Taylor J-S.2000. Biochemistry 39:14603–10
33. Li Y, Dutta S, Doublie S, Bdour HM, Tay-lor J-S, Ellenberger T. 2004. Nat. Struct.Mol. Biol. 11:784–90
34. Sun L, Zhang K, Zhou L, Hohler P, KoolET, et al. 2003. Biochemistry 42:9431–37
35. Yu S-L, Johnson RE, Prakash S, PrakashL. 2001. Mol. Cell Biol. 21:185–88
36. Stary A, Kannouche P, Lehmann AR,Sarasin A. 2003. J. Biol. Chem. 278:18767–75
37. Armstrong JD, Kunz BA. 1990. Proc.Natl. Acad. Sci. USA 87:9005–9
38. Bourre F, Renault G, Sarasin A. 1987. Nu-cleic Acids Res. 15:8861–75
39. Brash DE, Haseltine WA. 1982. Nature298:189–92
40. Brash DE, Seetharam S, Kraemer KH,Seidman MM, Bredberg A. 1987. Proc.Natl. Acad. Sci. USA 84:3782–86
41. Brash DE. 1997. Trends Genet. 13:410–14
42. Canella KA, Seidman MM. 2000. Mutat.Res. 450:61–73
43. Mitchell DL, Haipek CA, Clarkson JM.1985. Mutat. Res. 143:109–12
44. Ciarrocchi G, Pedrini AM. 1982. J. Mol.Biol. 155:177–83
45. Kemmink J, Boelens R, Koning T, vander Marel GA, van Boom JH, Kaptein R.1987. Nucleic Acids Res. 15:4645–53
46. Husain I, Griffith J, Sancar A. 1988. Proc.Natl. Acad. Sci. USA 85:2558–62
47. Park H, Zhang K, Ren Y, Nadji S, SinhaN, et al. 2002. Proc. Natl. Acad. Sci. USA99:15965–70
48. Kim J-K, Choi B-S. 1995. Eur. J.Biochem. 228:849–54
49. Kim J-K, Patel D, Choi B-S. 1995. Pho-tochem. Photobiol. 62:44–50
50. Lee J-H, Hwang G-S, Choi B-S. 1999.Proc. Natl. Acad. Sci. USA 96:6632–36
51. Johnson RE, Haracska L, Prakash S,Prakash L. 2001. Mol. Cell Biol. 21:3558–63
52. Gibbs PEM, Borden A, Lawrence CW.1995. Nucleic Acids Res. 23:1919–22
53. Johnson RE, Prakash S, Prakash L. 1999.J. Biol. Chem. 274:15975–77
54. Zhang H, Siede W. 2003. Nucleic AcidsRes. 30:1261–67
55. Bresson A, Fuchs RPP. 2002. EMBO J.21:3881–87
56. Prakash S, Prakash L. 2002. Genes Dev.16:1872–83
57. Haracska L, Yu S-L, Johnson RE, PrakashL, Prakash S. 2000. Nat. Genet. 25:458–61
58. Douki T, Perdiz D, Grof P, KuluncsicsZ, Moustacchi E, et al. 1999. Photochem.Photobiol. 70:184–90
59. Ravanat J-L, Douki T, Cadet J. 2001. J.Photochem. Photobiol. B 63:88–102
60. Haracska L, Prakash S, Prakash L. 2000.Mol. Cell Biol. 20:8001–7
61. Haracska L, Washington MT, Prakash S,Prakash L. 2001. J. Biol. Chem. 276:6861–66
62. Chiapperino D, Kroth H, Kramarczuk IH,Sayer JM, Masutani C, et al. 2002. J. Biol.Chem. 277:11765–71
63. Minko IG, Washington MT, Prakash L,Prakash S, Lloyd RS. 2001. J. Biol. Chem.276:2517–22
64. Minko IG, Washington MT, Kanuri M,Prakash L, Prakash S, Lloyd RS. 2003.J. Biol. Chem. 278:784–90
65. Levine RL, Miller H, Grollman A, OhashiE, Ohmori H, et al. 2001. J. Biol. Chem.276:18717–21
66. Kiefer JR, Mao C, Braman JC, Beese LS.1998. Nature 391:304–7
67. Li Y, Korolev S, Waksman G. 1998.EMBO J. 17:7514–25
68. Franklin MC, Wang J, Steitz TA. 2001.Cell 105:657–67

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 351
69. Pelletier H, Sawaya MR, Kumar A, Wil-son SH, Kraut J. 1994. Science 264:1891–903
70. Sawaya MR, Prasad R, Wilson SH,Kraut J, Pelletier H. 1997. Biochemistry36:11205–15
71. Trincao J, Johnson RE, Wolfle WT, Es-calante CR, Prakash S, et al. 2004. Nat.Struct. Mol. Biol. 11:457–62
72. Masutani C, Kusumoto R, Iwai S,Hanaoka F. 2000. EMBO J. 19:3100–9
73. Shibutani S, Takeshita M, Grollman AP.1991. Nature 349:431–34
74. Krahn JM, Beard WA, Miller H, GrollmanAP, Wilson SH. 2003. Structure 11:121–27
75. Echols H, Goodman MF. 1991. Annu. Rev.Biochem. 60:477–511
76. Goodman MF. 1997. Proc. Natl. Acad.Sci. USA 94:10493–95
77. Kool ET. 2002. Annu. Rev. Biochem. 71:191–219
78. Kuchta RD, Mizrahi V, Benkovic PA,Johnson KA, Benkovic SJ. 1987. Bio-chemistry 26:8410–17
79. Kuchta RD, Benkovic P, Benkovic SJ.1988. Biochemistry 27:6716–25
80. Patel SS, Wong I, Johnson KA. 1991. Bio-chemistry 30:511–25
81. Wong I, Patel SS, Johnson KA. 1991. Bio-chemistry 30:526–37
82. Washington MT, Prakash L, Prakash S.2001. Cell 107:917–27
83. Washington MT, Johnson RE, Prakash L,Prakash S. 2003. Mol. Cell Biol. 23:8316–22
84. Moran S, Ren RX-F, Kool ET. 1997. Proc.Natl. Acad. Sci. USA 94:10506–11
85. Morales JC, Kool ET. 1998. Nat. Struct.Biol. 5:950–54
86. Morales JC, Kool ET. 2000. J. Am. Chem.Soc. 122:1001–7
87. Washington MT, Helquist SA, Kool ET,Prakash L, Prakash S. 2003. Mol. CellBiol. 23:5107–12
88. Li Y, Mitaxov V, Waksman G. 1999. Proc.Natl. Acad. Sci. USA 96:9491–96
89. Morales JC, Kool ET. 1999. J. Am. Chem.Soc. 121:2323–24
90. Morales JC, Kool ET. 2000. Biochemistry39:12979–88
91. Spratt TE. 2001. Biochemistry 40:2647–52
92. Washington MT, Wolfle WT, Spratt TE,Prakash L, Prakash S. 2003. Proc. Natl.Acad. Sci. USA 100:5113–18
93. Johnson RE, Washington MT, HaracskaL, Prakash S, Prakash L. 2000. Nature406:1015–19
94. Tissier A, McDonald JP, Frank EG,Woodgate R. 2000. Genes Dev. 14:1642–50
95. Zhang Y, Yuan F, Wu X, Wang Z. 2000.Mol. Cell Biol. 20:7099–108
96. Haracska L, Johnson RE, Unk I, PhillipsBB, Hurwitz J, et al. 2001. Proc. Natl.Acad. Sci. USA 98:14256–61
97. Washington MT, Johnson RE, Prakash L,Prakash S. 2004. Mol. Cell Biol. 24:936–43
98. Vaisman A, Tissier A, Frank EG, Good-man MF, Woodgate R. 2001. J. Biol.Chem. 276:30615–22
99. Washington MT, Minko IG, Johnson RE,Wolfle WT, Harris TM, et al. 2004. Mol.Cell Biol. 24:5687–93
100. Nechev LV, Harris CM, Harris TM. 2000.Chem. Res. Toxicol. 13:421–29
101. Kurtz AJ, Lloyd RS. 2003. J. Biol. Chem.278:5970–76
102. Singh US, Moe JG, Reddy GR, Weis-enseel JP, Marnett LJ, Stone MP. 1993.Chem. Res. Toxicol. 6:825–36
103. Weisenseel JP, Reddy GR, Marnett LJ,Stone MP. 2002. Chem. Res. Toxicol. 15:127–39
104. Lawrence CW, Christensen RB. 1978. J.Mol. Biol. 122:1–21
105. Lawrence CW, Christensen RB. 1979.Genetics 92:397–408
106. Lawrence CW, O’Brien T, Bond J. 1984.Mol. Gen. Genet. 195:487–90
107. Lawrence CW, Nisson PE, ChristensenRB. 1985. Mol. Gen. Genet. 200:86–91
108. Johnson RE, Torres-Ramos CA, Izumi

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
352 PRAKASH � JOHNSON � PRAKASH
T, Mitra S, Prakash S, Prakash L. 1998.Genes Dev. 12:3137–43
109. Nelson JR, Lawrence CW, Hinkle DC.1996. Nature 382:729–31
110. Haracska L, Prakash S, Prakash L. 2002.J. Biol. Chem. 277:15546–51
111. Gibbs PEM, Kilbey BJ, Banerjee SK,Lawrence CW. 1993. J. Bacteriol. 175:2607–12
112. Haracska L, Unk I, Johnson RE, Johans-son E, Burgers PMJ, et al. 2001. GenesDev. 15:945–54
113. Avkin S, Adar S, Blander G, Livneh Z.2002. Proc. Natl. Acad. Sci. USA 99:3764–69
114. Baynton K, Bresson-Roy A, Fuchs RPP.1999. Mol. Microbiol. 34:124–33
115. Washington MT, Minko IG, Johnson RE,Haracska L, Harris TM, et al. 2004. Mol.Cell Biol. 24:6900–6
116. Guo C, Fischhaber PL, Luk-Paszyc MJ,Masuda Y, Zhou J, et al. 2003. EMBO J.22:6621–30
117. Ohashi E, Mukakumo Y, Kanjo N, AkagiJ-i, Masutani C, et al. 2004. Genes Cells9:523–31
118. Tissier A, Kannouche P, Reck M-P,Lehmann AR, Fuchs RPP, Cordonnier A.2004. DNA Repair 3:1503–14
119. Murakumo Y, Ogura Y, Ishii H, NumataS, Ichihara M, et al. 2001. J. Biol. Chem.276:35644–51
120. Masuda Y, Ohmae M, Masuda K, KamiyaK. 2003. J. Biol. Chem. 278:12356–60
121. Johnson RE, Prakash S, Prakash L. 2000.Proc. Natl. Acad. Sci. USA 97:3838–43
122. Washington MT, Johnson RE, Prakash L,Prakash S. 2002. Proc. Natl. Acad. Sci.USA 99:1910–14
123. Kobayashi S, Valentine MR, Pham P,O’Donnell M, Goodman MF. 2002. J.Biol. Chem. 277:34198–207
124. Boudsocq F, Iwai S, Hanaoka F, Wood-gate R. 2001. Nucleic Acids Res. 29:4607–16
125. Efrati E, Tucco G, Eritja R, Wilson SH,Goodman MF. 1997. J. Biol. Chem. 272:2559–69
126. Bebenek K, Kunkel TA. 1990. Proc. Natl.Acad. Sci. USA 87:4946–50
127. Wolfle WT, Washington MT, Prakash L,Prakash S. 2003. Genes Dev. 17:2191–99
128. Haracska L, Unk I, Johnson RE, PhillipsBB, Hurwitz J, et al. 2002. Mol. Cell Biol.22:784–91
129. Haracska L, Prakash L, Prakash S. 2002.Proc. Natl. Acad. Sci. USA 99:16000–5
130. Suzuki N, Ohashi E, Hayashi K, OhmoriH, Grollman AP, Shibutani S. 2001. Bio-chemistry 40:15176–83
131. Suzuki N, Ohashi E, Kolbanovskiy A,Geacintov NE, Grollman AP, et al. 2002.Biochemistry 41:6100–6
132. Fischhaber PL, Gerlach VL, Feaver WJ,Hatahet Z, Wallace SS, Friedberg EC.2002. J. Biol. Chem. 277:37604–11
133. Ogi T, Shinkai Y, Tanaka K, Ohmori H.2002. Proc. Natl. Acad. Sci. USA 99:15548–53
134. Bemark M, Khamlichi AA, Davies SL,Neuberger MS. 2000. Curr. Biol. 10:1213–16
135. Esposito G, Godin I, Klein U, Yaspo M-L, Cumano A, Rajewsky K. 2000. Curr.Biol. 10:1221–24
136. Wittschieben J, Shivji MKK, Lalani E,Jacobs MS, Marini F, et al. 2000. Curr.Biol. 10:1217–20
137. Prakash L. 1976. Genetics 83:285–301138. Haracska L, Prakash S, Prakash L. 2003.
Mol. Cell Biol. 23:1453–59139. Thomas DC, Roberts JD, Sabatino RD,
Myers TW, Tan C-K, et al. 1991. Bio-chemistry 30:11751–59
140. Johnson RE, Yu S-L, Prakash S, PrakashL. 2003. Genes Dev. 17:77–87
141. Clark JM, Pattabiraman N, Jarvis W,Beardsley GP. 1987. Biochemistry 26:5404–9
142. Miaskiewicz K, Miller J, Ornstein R, Os-man R. 1995. Biopolymers 35:113–24
143. Haracska L, Kondratick CM, Unk I,Prakash S, Prakash L. 2001. Mol. Cell.8:407–15

14 May 2005 14:58 AR AR261-BI74-12.tex XMLPublishSM(2004/02/24) P1: JRX
EUKARYOTIC LESION BYPASS POLYMERASES 353
144. Haracska L, Johnson RE, Unk I, PhillipsB, Hurwitz J, et al. 2001. Mol. Cell Biol.21:7199–206
145. Bailly V, Lamb J, Sung P, Prakash S,Prakash L. 1994. Genes Dev. 8:811–20
146. Bailly V, Lauder S, Prakash S, Prakash L.1997. J. Biol. Chem. 272:23360–65
147. Lawrence CW. 1982. Adv. Genet. 21:173–254
148. Prakash S, Sung P, Prakash L. 1993. Annu.Rev. Genet. 27:33–70
149. Torres-Ramos CA, Prakash S, Prakash L.2002. Mol. Cell Biol. 22:2419–26
150. Johnson RE, Henderson ST, Petes TD,Prakash S, Bankmann M, Prakash L.1992. Mol. Cell Biol. 12:3807–18
151. Johnson RE, Prakash S, Prakash L. 1994.J. Biol. Chem. 269:28259–62
152. Broomfield S, Chow BL, Xiao W. 1998.Proc. Natl. Acad. Sci. USA 95:5678–83
153. Hofmann RM, Pickart CM. 1999. Cell96:645–53
154. Prakash L. 1981. Mol. Gen. Genet. 184:471–78
155. Schiestl RH, Prakash S, Prakash L. 1990.Genetics 124:817–31
156. Krejci L, Van Komen S, Li Y, Villemain J,Reddy MS, et al. 2003. Nature 423:305–9
157. Veaute X, Jeusset J, Soustelle C, Kowal-czykowski SC, Le Cam E, Fabre F. 2003.Nature 423:309–12
158. Torres-Ramos CA, Yoder BL, BurgersPMJ, Prakash S, Prakash L. 1996. Proc.Natl. Acad. Sci. USA 93:9676–81
159. Hoege C, Pfander B, Moldovan G-L,Pyrowolakis G, Jentsch S. 2002. Nature419:135–41
160. Ulrich HD, Jentsch S. 2000. EMBO J.19:3388–97
161. Ulrich HD. 2003. J. Biol. Chem. 278:7051–58
162. Stelter P, Ulrich HD. 2003. Nature 425:188–91
163. Haracska L, Torres-Ramos CA, JohnsonRE, Prakash S, Prakash L. 2004. Mol. CellBiol. 24:4267–74
164. Kannouche PL, Wing J, Lehmann AR.2004. Mol. Cell. 14:491–500


Copyright © 2022 FDOKUMEN




















