
A new view into prokaryotic cell biology from electron ... - CORE
Upload
independentCategory
view
1download
0
Characterization of Prokaryotic and Eukaryotic Promoters UsingHidden Markov Models
Anders Gorm Pedersen *, Pierre Baldi t, S0ren Brunak $ and Yves Chauvin §
Abstract
In this paper we utilize hidden Markov mod-els (HMMs) and information theory to analyzeprokaryotic and eukaryotic promoters. We per-form this analysis with special emphasis on thefact that promoters are divided into a number ofdifferent classes, depending on which polymerase-associated factors that bind to them. We findthat HMMs trained on such subclasses of Es-cherichia coli promoters (specifically, the so-called ar° and a54 classes) give an excellent clas-sification of unknown promoters with respect tosigma-class. HMMs trained on euka~yotic se-quences from human genes also model nicely allthe essential well known signals, in addition toa potentially new signal upstream of the TATA-box. We furthermore employ a novel techniquefor automatically discovering different classes inthe input data (the promoters) using a system selforganizing parallel HMMs. These selforganiz-ing HMMs have at the same time the ability tofind clusters and the ability to model the sequen-tial structure in the input data. This is highlyrelevant in situations where the variance in thedata is high, as is the case for the subclass struc-ture in for example promoter sequences.
Key words: hidden Markov models (HMMs), in-formation theory, DNA sequence analysis, Es-cherichia coli, Homo sapiens, promoters.
* Center for Biological Sequence Analysis, building 206,The Technical University of Denmark, DK-2800, Denmark,[email protected], (+45) 4525-2484, (+45) 4593-4808 (fax).
q)ivision of Biology, California Institute of Technology,Pasadena, CA 91125, pfbaldiC~cco.caltech.edu, (213) 222-6007, (213) 222-7742 (fax).
lCenter for Biological Sequence Analysis, building 206,The Technical University of Denmark, DK-2800, Den-mark, [email protected], (+45) 4525-2477, (+45) 4593-4808 (fax).
§ Net-ID, Inc., San Francisco, CA 94107,yves~netid.com, (415) 647-9402 (415) 642-9265 (fax).
IntroductionInitiation of transcription is the first step in gene ex-pression, and constitutes an important point of controlin prokaryotes as well as in eukaryotes (Reznikoff et al.1985). Transcription initiates when RNA-polymeraserecognizes and binds to certain DNA-sequences termedpromoters. Subsequent to binding, a short stretch ofthe DNA double helix is disrupted, and the polymerasestarts to synthesize RNA by the process of complemen-
tary basepairing. The sequence of the promoter deter-mines the position of the transcriptional start point,and is furthermore important for the frequency with
which the gene is transcribed (the strength of the pro-
moter).
Escherichia coli Promoters
In the prokaryote E.coli, the form of the RNA-polymerase that is responsible for recognizing pro-moter sequences, has the protein subunit composi-tion a.~/3/3Qr. This so-called holo-enzyme can be di-vided into two functional components: the core enzyme
(a2~q’, also designated E) and the sigma factor (~).The sigma factor plays an important role in recognizingpromoter sequences, and after successful initiation it is
released from the holoenzyme (Gross & Lonetto 1992;Loewen & ttengge-Aronis 1994). Several different sig-
mafactors exist, each recognizing a specific subset ofpromoters. These subsets have different nucleotide se-
quences. The biological significance of this is that eachpromoter group controls genes that are needed underphysiologically similar conditions, and that thereforeneed to be expressed simultaneously. E.g., all promot-ers recognized by the holo enzyme Ea32 control geneswhich are important for helping the bacterium surviveprolonged exposure to higher-than-normal tempera-tures (heat shock). Sigma factors derive their names
from the molecular weight of the proteins (thus, ~3.~has a M~ of 32 kDa). E.coli is known to contain thesigma factors ~70, ~54 ~32 ~F, and as. Briefly the
genes controlled by the different factors are:
182 ISMB-96
From: ISMB-96 Proceedings. Copyright © 1996, AAAI (www.aaai.org). All rights reserved.

¯ ~70 Primary sigma, majority of all E.coli genes.
¯ ~54 Nitrogen assimilation.
¯ ~3~ Heat shock response.
¯ crr Flagellum genes.
¯ crs Starvation stress response.
Comparison of E.coli o"T° promoters has led to theidentification of three major conserved features: the"-10 box", the "-35 box", and a pyrimidine (C orT) followed by a purine (A or G) at the initiation site(Rosenberg &: Court 1979; Hawley & McClure 1983).The -10 and -35 boxes are conserved hexanucleotideelements that are named according to the approximateposition of their central nucleotides relative to the tran-scriptional start point. The well known consensus se-quences are TTGACA for the -35 box, and TATAATfor the -10 box.
The sigma factors ~r32, ~F, and ~s are homolo-gous to crr° and all bind to promoters which have thesame overall architecture (signals at -10 and -35),but which differ at one or both sites (Lonetto, Grib-skov, & Gross 1992). However, "~4 i s n ot h omol-ogous to the ~r7°-family, and promoters recognizedby E~~4, have been found to contain two consensusboxes located at positions -12 and -24 (Merrick 1993;Morett & Segovia 1993).
Homo sapiens Promoters
Eukaryotes have three RNA-polymerases that are re-sponsible for transcribing different subsets of genes:RNA-polI transcribes ribosomal RNA, RNA-polII(which we will focus on in this paper) transcribesmRNA, while RNA-polIII transcribes tRNA and othersmall RNAs. RNA-polII consists of more than 10 sub-units, some of which are partly homologous to thebacterial subunits a, /3, and /?~. As with bacterialRNA-polymerase, the eukaryotic RNA-polII is depen-dent on additional factors for initiation. However, inthe case of RNA-polII the factors are more numer-ous and play a larger role in the determination of thestartpoint (Gill & Tjian 1992; Pugh & Tjian 1992;Eick, Wedel, & Heumann 1994). The factors that assistRNA-polII in initiating transcription, can be dividedinto three groups:
¯ The so-called basal factors are required for suc-cessful initiation at all promoters. Together withRNA-polII they form a complex surrounding thestartpoint, and they determine the transcriptionalstartpoint. The basal factors include the so-calledTFIIA, TFIIB, TFIID, TFIIE, TFIIF, TFIIH, andTFIIJ, many of which are multi-subunit factors.
¯ Upstream factors are DNA-binding proteins thatrecognize short sequence elements upstream of thestartpoint. They enhance the efficiency of initiation,presumably by protein-protein interactions with thebasal transcriptional apparatus. Different promot-ers may contain different combinations of bindingsites for these factors, in various distances from thestartpoint.
¯ Inducible factors are synthesized or activated un-der certain conditions or at certain times, but oth-erwise work like the upstream factors. They are re-sponsible for the control of transcription with regardto time and space.
Eukaryotic promoters are less similar to each otherthan bacterial promoters. Only two sequence elementsare reasonably conserved with respect to compositionand location: the TATA-box and the initiator element(Smale & Baltimore 1989; Guarente & Bermingham-McDonogh 1992; O’Shea-Greenfield & Smale 1992).The initiator is a sequence that is located at thestartpoint in some promoters. It has the consensusPy~CAPys, where Py is a pyrimidine (C or T). Mostpromoters have a TATA-box, which is a short sequencewith the consensus TATAAAA usually centered ap-proximately 25 bp upstream of the startpoint. TheTATA-box plays a crucial role when RNA-polII recog-nizes TATA-box containing promoters: the transcrip-tional apparatus is assembled factor by factor, start-ing with the TFIID-subunit TBP (the TATA BindingProtein). TBP arrives at the promoter and binds tothe TATA-box. During the subsequent steps, the tran-scriptional apparatus is assembled on the promoterby a process involving numerous protein-protein in-teractions. The so-called TAFs (TBP-associated fac-tors) are important in this respect (Gill & Tjian 1992;Pugh & Tjian 1992). In addition to the two elementsmentioned above, most promoters contain additionalupstream elements which bind activating factors, butthe sequence, number, orientation, and position ofthese is highly variable. However, there is an overallpreference for G’s and C’s in promoterregions.
Purpose
In this paper we have analyzed prokaryotic and eukary-otic promoters with statistical methods, and by usingpowerful hidden Markov models (HMMs). HMMs areexcellent for investigating conserved sequence signalsthat have variable spacing, and indeed we easily findthe known sequence signals in E.coli-genes, and themost conserved sequences in human genes. We findthat combinations of HMMs which have been trainedon sequences belonging to specific sigma-subclasses,
Pedersen 183

are able to classify unknown sequence with great suc-cess. Furthermore, we have developed a novel method,involving selforganizing parallel HMMs, for automaticclassification of the promoter sequences.
Data
Prokaryotic Promoters
The E.coli promoter sequences were taken from thecompilation by Lisser and Margalit (Lisser & Margalit1993). This database, which contains 300 sequences,is superior to most other available E.coli promoterdatabases on two accounts:
¯ Each sequence has been compared to the original pa-per, minimizing the chance of database entry errors.
* For each sequence, the assignment of transcriptionalstart point(s) has been verified with the relevant pa-pers, and the most reliable have been chosen.
We processed the data in the following ways: first, weconcatenated the sequences that are partially overlap-ping (e.g., dnaK-P1 and dnaK-P2). This removed number of contradictions, since the nucleotide that ismarked as a transcriptional start point in one sequenceis not labeled as such in the partially overlapping se-quence, and vice versa. Concatenation resulted in asubset consisting of 248 sequences. Second, we dis-carded all the sequences that contain multiple startpoints, and all sequences not including at least 75 bpupstream and 25 bp downstream of the transcriptionalstartpoint. Then we cut out sequence surrounding thestartpoint, so that all sequences contain exactly 75bp upstream and 25 bp downstream of the transcrip-tional startpoint. The resulting set, which we use inthis study, contains 166 sequences. For the purpose oftraining HMMs that were able to analyze un-annotatedsequences with respect to sigma-class, we divided thedata into three sets: those sequences known to be rec-ognized by c~7° (38 sequences), those known to be rec-ognized by ~54 (3 sequences), and those where it wasnot known which sigma-factor is responsible for tran-scription (the remaining 125 sequences).
Eukaryotic Promoters
The human data was extracted from Genbank release90 (Benson et al. 1994). Specifically, all human se-quences containing the feature key "prim_transcript",were selected. This feature key indicates that the se-quence is an unprocessed transcript, and that it maytherefore contain one or more transcriptional start-points. From these sequences, those which had at least250 bp upstream and 250 bp downstream of the firsttranscriptional startpoint were selected, and the 501
184 ISMB-96
bp that symmetrically surrounded the startpoint werecut out, and kept for training. This resulted in a setof 340 sequences, of which 37 contained more than onetranscriptional startpoint.
Methods
Measures of Information Content
The Kullback Leibler distance (or relative entropy) foreach position in sequences aligned by the transcrip-tional startpoint, was calculated by the formula:
D(p, q) = Z Pi log2 P_.j_ii
qi
where pi and qi are the probabilities of occurrence fora particular nucleotide i (A, C, G,T) at the position(Kullback & Leibler 1951). Specifically, we took theprobability distribution from one sigma-subset (e.g.,6r7°) and compared it to the distribution from anothersubset. D(p,q) has values that range from 0 to oo.D(p,q) = indicates th at th e two di stributions ar eidentical at the given position, while larger values ofD(p, q) means that the distributions differ at that po-sition.
The traditional Shannon measure was also used andcalculated by the formula:
[(p) = H,,,a. - Pilogs Pii
where Hmax=log2(length of alphabet)=2, since the nu-cleotide alphabet contains 4 letters (Shannon 1948).
The results from both kinds of analysis were depictedby the use of sequence logos replacing the conven-tional numeric curves. The sequence logos wcre con-structed according to Schneider and Stephens (Schnei-der & Stephens 1990). Briefly, sequence logos combinethe information contained in consensus sequences witha quantitative measure of information, by representingeach position in an alignment by a stack of letters. Theheight of the stack is a measure of the non-randomnessat the position (here the Kullback Leibler distance orthe Shannon measure), while the height of a letter cor-responds to its frequency.
HMMs of Promoter Sequences
A first order discrete HMM can be viewed as a stochas-tic generative model defined by a set of states S, analphabet ¢4 of m symbols, a probability transitionmatrix T = (tij), and a probability emission ma-trix E = (eix). The system randomly evolves fromstate to state, while emitting symbols from the alpha-bet. When the system is in a given state i, it has

a probability tij of moving to state j, and a prob-ability eix of emitting symbol X. As in the appli-cation of HMMs to speech recognition, a family ofDNA sequences can be seen as a set of different ut-terances of the same word, generated by a commonunderlying HMM. One of the standard HMM archi-tectures for molecular biology applications, first in-troduced in (Krogh et al. 1994), is the left-right ar-chitecture. The alphabet has m = 4 symbols, onefor each nucleotide (m = 20 for protein models, onesymbol per amino acids). In addition to the startand end states, there are three classes of states: themain states, the delete states and the insert states withS -- {s~ar~, ml .... , my, il, ..., iN+l, dl .... , dy, end}. Nis the length of the model, typically equal to the av-erage length of the sequences in the family. The mainand insert states always emit a nucleotide, whereas thedelete states are mute. The linear sequence of statetransitions start --* ml --~ m2 .... --+ mN --+ end is thebackbone of the model. For each main state, corre-sponding insert and delete states are needed to modelinsertions and deletions. The self-loop on the insertstates allows for multiple insertions at a given site.
Given a sample of K training sequences O1 .... , Ok,the parameters of an HMM can be iteratively modi-fied, in an unsupervised way, to optimize the data fitaccording to some measure, usually based on the like-lihood of the data. Since the sequences can be consid-ered as independent, the overall likelihood is equal tothe product of the individual likelihoods. Two targetfunctions, commonly used for training, are the negativelog-likelihood:
K K
Q=-EQk=-ElnP(Ok) (2.1)k=l k=l
and the negative log-likelihood based on the optimalpaths:
K K
Q = -~Qk = - ~ln P(Tr(O~)) (2.2)k=l k=l
where ~r(O) is the most likely HMM production pathfor sequence O. ~r(O) can be computed efficiently dynamic programming (the Viterbi algorithm). Whenpriors on the parameters are included, one can alsoadd regulariser terms to the objective functions forMAP (Maximum A Posteriori) estimation. Differentalgorithms are available for HMM training, includingthe Baum-Welch or EM (Expectation-Maximization)algorithm, and different forms of gradient descent andother GEM (Generalized EM) algorithms (Dempster,Laird, & Rubin 1977; Rabiner 1989; Baldi & Chauvin1994b). Regardless of the training method, once an
HMM has been successfully trained on a family of se-quences, it can be used in a number of different tasks.First, for any given sequence, one can compute its like-lihood according to the model, and also its most likelypath. A multiple alignment results immediately fromaligning all the optimal paths of the sequences in thefamily. The model can also be used for discrimina-tion tests, and data base searches (Krogh et al. 1994;Baldi ~: Chauvin 1994a), by comparing the likelihoodof any sequence to the likelihoods of the sequences inthe family. Finally the parameters of a model, such asthe emission distributions of the backbone states andtheir entropies, can be used to detect consensus pat-terns and other signals (see (Baldi et al. 1995) for anexample).
Another use of HMMs is in the classification of se-quences within a family, and the discovery of sub-classes. This may be particularly relevant for promotersequences, especially in eukaryotes, where a diverse ar-ray of sub-classes may exist, with different signals. Dif-ferent classification algorithms can be considered de-pending on the amount of prior knowledge available.One basic approach (Krogh et al. 1994) is to use "super-HMM", consisting of several basic sub-HMMsin parallel, one for each sub-class, see Figure 1. Thesuper-HMM can be trained using some form of compet-itive learning. For example, when a training sequenceis presented to the super-HMM, its Viterbi path is firstcomputed. Such a path goes through only one of thesub-HMMs, and only the parameters of this HMM areupdated during training, to increase the likelihood ofthe corresponding sequence. Thus, these selforganizingHMMs have at the same time the ability to find clustersand the ability to model the sequential structure in theinput data. Another more general approach to classi-fication, based on hybrid HMM/NN (Neural Network)architectures, is briefly described in (Baldi & Chauvin1995). In hybrid HMM/NN architectures, a NN is usedto calculate and nmdulate the parameters of an ttMM,so that a slightly different HMM is generated for eachsub-class.
In practice, however, it is well known that selforga-nizing processes of this sort are far from trivial, espe-cially in the absence of any prior knowledge, and canbe plagued by local minima problems. The numberof classes, their definition and separation, the degreeto which each is represented in the available trainingset, are all crucial issues that impact the performanceof the algorithms. As an example, in one experiment,we tried a super-HMM consisting of two similar sub-HMMs, initialized randomly, against the entire subsetof E.coli promoter sequences with unique transcrip-tional starting point. Numerous training cycles always
Pedersen t85

Figure 1: A "super-HMM’, consisting of several basic sub-HMMs in parallel.
resulted in a disappointing result: essentially all thesequences were classified as a single sub-class, associ-ated with one of the two sub-HMMs. The underly-ing reason is as follows: with random initialisation,the initial sub-HMMs are far away, in sequence space,from the cloud of promoter sequences. The first train-ing sequence selects whichever sub-HMM happens tobe closer to the promoter cloud, and pulls it towardsthe cloud during the corresponding parameter update.This phenomenon is only repeated and reinforced bythe presentation of the following training sequences, sothat only one model is selected. Thus to produce a bi-furcation between the sub-models one must introducesome additional elements in the algorithm. One pos-sibility we have used is to take advantage of the priorknowledge gathered during the training of single HMMmodels, to initialise the sub-HMMs parameters close tothe promoter cloud, instead of randomly. More gener-ally, a bootstrap procedure of this sort can be usedanytime to go from n to n + 1 classification.
Results: Escherichia coil
Statistical analysis
The Shannon information measure was calculated forthree different subsets of the 166 sequences in theE.coli database: sequences known to be recognizedby sigma-70, sequences known to be recognized bysigma-54, and the rest (Figure 2). Not surprisingly,no strong and clear picture emerges from the analy-sis of the three sequences in the (r54-set. However,the well-known -10 box signal, and the CA-signalcan be seen in the subsets recognized by crr0, and thelarger subset of un-annotated sequences (Figure 2).
clear -35 signal can be seen in the aT°-subset, andonly a weak signal is visible in the larger subset ofun-annotated sequences. This can probably be ex-plained in part by the fact that the position of the-35 box, relative to the transcriptional start point, issomewhat flexible (Galas, Eggert, & Waterman 1985;Harley & Reynolds 1987). Consequently, the sequencesignal will not be clearly recognized without multip]ealignment.
In order to learn more about the differences betweenthe sequences in the different subsets, we used theKullback Leibler measure also in the analysis. Specif-ically, we calculated the Kullback Leibler distance be-tween sequences belonging to the aT°-subset and theentire set of sequences (data not shown). This analy-sis demonstrated that sequences from the (~7°-set werequite similar to the average sequence in the entire set.This is to be expected, since most of the sequencesin the un-annotated set probably do belong to theo’70-class. Nevertheless, a small overrepresentation ofT’s in the -10 box area was apparent, in good agree-ment with the fact that aZ°-sequences have the con-sensus TATAAT at this position. Furthermore, the(rZ°-sequences displayed some under-representation ofA’s and T’s in the area upstream of the -10 box.
HMMs
A hidden Markov model was trained on the entire set of166 E.coli-sequences. The main state emission proba-bilities of the resulting model are shown in figure 3. Asit can be seen, the HMM is very successful at modellingthe known sequence signals: around - 10 the TATAATconsensus is very clear. Further upstreana the most
186 ISMB-96

(b)
ATT
(c)
Figure 2: Shannon information content in the various subsets of the data depicted as sequence logos, a) Sequences known tobe recognized by as* (3 sequences), b) sequences known to be recognized by ~r0 (38 sequences), c) the remaining sequencesin our E.coli database (125 sequences). The sequences are aJigned by their transcriptionaJ initiation site (position
Pedersen 187

0.8
0.6
0.4
0.2
o
Figure 3: Emission probabifities of the main states in a hidden Markov model trained on the 166 E.coli-sequences. Noticethe clear pattern of the well-known consensus sequences at -35 (TTG), -10 (TATAAT), and at the transcriptional start.point(CA).
highly conserved part of the -35 box (TTG) can alsobe seen easily. Finally, there’s a reasonably clear CA-signal at the transcriptional startpoint.
Thus it can be seen that the HMM is very goodat handling the variably spaved E. colipromoter signalswithout a need for prior alignment, and certainly muchbetter than the simple statistical methods employedabove. This feature makes HMMs very strong toolsfor promoter analysis, since promoters are by naturemodular.
The HMM was also trained on random sequencesconstructed by shuffling the nucleotides within each ofthe sequences in the dataset. In this way the nucleotidecomposition is the same, but the sequential structureof the sequences is different. When one calculates thenegative log-likelihood of the sequences based on themodel trained on the E.coli-data, it is found that theshuffled sequences have much higher values than thenative sequences. Specifically, the average value of thenegative log-likelihood of the random sequences was144.0, while that of the E.coli-sequences was 137.6.This confirms that the E.coli-sequences have a lowerShannon information content than the randomly shuf-fled sequences (i.e., they contain conserved signals).
A super HMM was constructed by combining twosimple linear HMMs in parallel. One of the tlMMshad been trained on the c~7°-sequences (38 sequences),while the other HMM had been trained on the threecrS4-sequences. The super-model was subsequentlytrained using the entire set of 166 sequences. During allcycles of training (including cycle 0, i.e. prior to train-ing of the super-model), the classification induced on
the 166 sequences was completely constant: all the 166sequences are classified as belonging to the first sub-HMM (the one trained on sigma70), with the exceptionof 4 sequences. These 4 sequences are: glnA-P2, glnH-P2, fdhF, and lacI. The first three of these sequencesindeed belong to the sigma54 class, while lacI belongsto the o’7°-class. The lad promoter is associated witha weakly expressed gene (repressor for the lac-operon),and is known to be a ’non-consensus’ crr° representa-tive. It is therefore not. critical that the HMM doesn’tcharacterize it as belonging to the crT°-class Further-more, considering that the majority of E. coli-genes aretranscribed by the holoenzyme E~r7°, it is a very rea-sonable result, that most. sequences end up being stablyclassified as belonging to the J°-class. In conclusion,the HMM is able convincingly to characterize unknownsequences based on a relatively small number of train-ing sequences.
Results: Homo sapiensHMMs
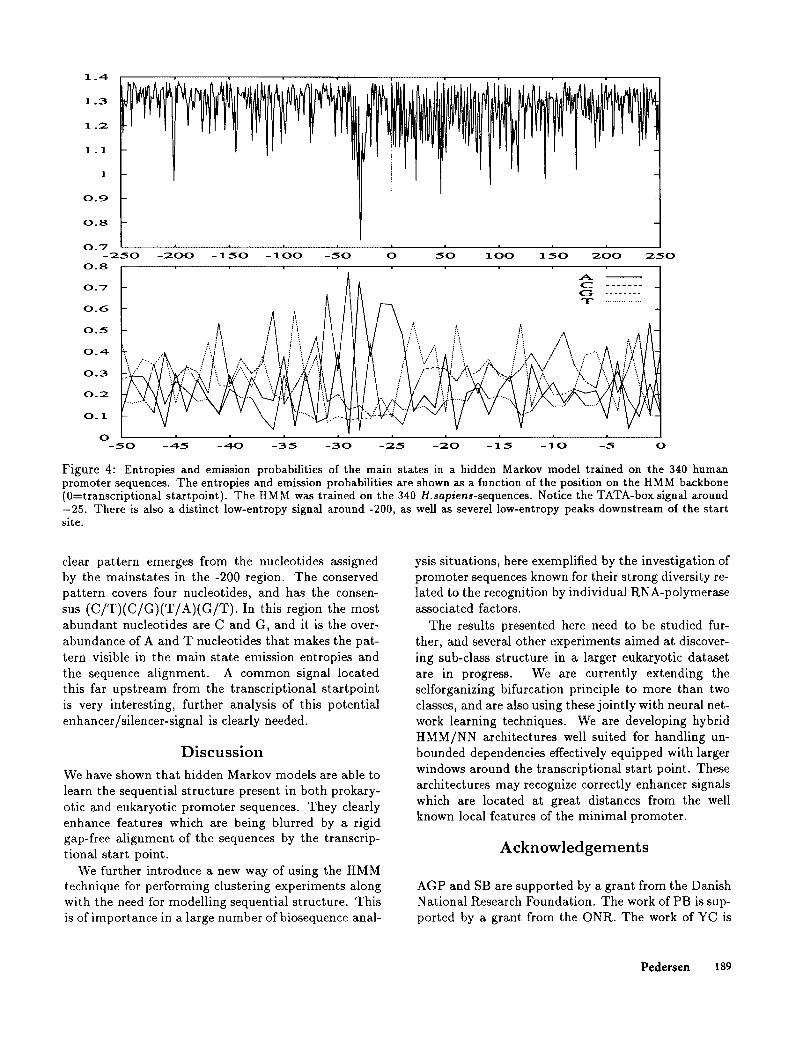
A hidden Markov model was trained on the 340H.sapiens-sequences (see Fig. 4 for the resulting mainstate entropies, and emission probabilities). Also inthis case the HMM can be seen to successfully modelthe most well-conserved part of the eukaryotic promot-ers, i.e.,the TATA-box. As it can be observed from theemission probability profiles, there is a clearly visibleoverabundance of A’s and T’s in the -25 region. In-terestingly, what appears to be an additional signalcan be seen around position -200. When analysing se-quence alignments made using the trained HMM, a
188 ISMB-96
1.4
1.3
1.2
1.1
]
0.9
0.8
0.7-250
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
i i i i i i i i i-200 -150 -100 -50 0 50 100 150 200 250
0 i | i i i i i i--50 --45 --ZI-O --35 --30 --25 --20 -- 15 -- I 0 --5 0
Figure 4: Entropies and emission probabilities of the main states in a hidden Maxkov model trained on the 340 humanpromoter sequences. The entropies and emission probabilities axe shown as a function of the position on the HMM backbone(0=transcriptional staxtpoint). The HMM was trained on the 340 H.sapiens-sequences. Notice the TATA-box signal around-25. There is also a distinct low-entropy signal around -200, as well as severel low-entropy peaks downstream of the startsite.
clear pattern emerges from the nucleotides assignedby the mainstates in the -200 region. The conservedpattern covers four nucleotides, and has the consen-sus (C/T)(C/G)(T/A)(G/T). In this region abundant nucleotides are C and G, and it is the over-abundance of A and T nucleotides that makes the pat-tern visible in the main state emission entropies andthe sequence alignment. A common signal locatedthis far upstream from the transcriptional startpointis very interesting, further analysis of this potentialenhancer/silencer-signal is clearly needed.
Discussion
We have shown that hidden Markov models are able tolearn the sequential structure present in both prokary-otic and eukaryotic promoter sequences. They clearlyenhance features which are being blurred by a rigidgap-free alignment of the sequences by the transcrip-tional start point.
We further introduce a new way of using the HMMtechnique for performing clustering experiments alongwith the need for modelling sequential structure. Thisis of importance in a large number of biosequence anal-
ysis situations, here exemplified by the investigation ofpromoter sequences known for their strong diversity re-lated to the recognition by individual l~NA-polymeraseassociated factors.
The results presented here need to be studied fur-ther, and several other experiments aimed at discover-ing sub-class structure in a larger eukaryotic datasetare in progress. We are currently extending theselforganizing bifurcation principle to more than twoclasses, and are also using these jointly with neural net-work learning techniques. We are developing hybridHMM/NN architectures well suited for handling un-bounded dependencies effectively equipped with largerwindows around the transcriptional start point. Thesearchitectures may recognize correctly enhancer signalswhich are located at great distances from the wellknown local features of the minimal promoter.
Acknowledgements
AGP and SB are supported by a grant from the DanishNational P~esearch Foundation. The work of PB is sup-ported by a grant from the ONR. The work of YC is
Pedersen 189

supported in part by grant number R43 LM05780 fromthe National Library of Medicine. The contents of thispublication are solely the responsibility of the authorsand do not necessarily represent the official views ofthe National Library of Medicine.
References
Baldi, P., and Chauvin, Y. 1994a. Hidden markovmodels of the G-protein-coupled receptor family.Journal of Computational Biology 1 (4):311-335.
Baldi, P., and Chauvin, Y. 1994b. Smooth on-linelearning algorithms for hidden markov models. NeuralComputation 6(2):305-316.
Baldi, P., and Chauvin, Y. 1995. Protein mod-eling with hybrid hidden markov model/neural net-works architectures. In Proceedings of ¢he 1995 Con-fe.rence on Inlelligent Systems for Molecular Biology(ISMB95), in Cambridge (UK). Menlo Park, CA: TheAAI Press.
Baldi, P.; Brunak, S.; Chauvin, Y.; Engelbrecht, J.;and Krogh, A. 1995. Periodic sequence patterns in hu-man exons. In Proceedings of the 1995 Conference onIntelligent Systems for Molecular Biology (ISMB95),in Cambridge (UK). Menlo Park, CA: The AAI Press.
Benson, D.; Boguski, M.; Lipman, D.; and Ostell, J.1994. Genbank. Nucl. Acids Res. 22:3441-3444.
Dempster, A. P.; Laird, N. M.; and Rubin, D. B.1977. Maximum likelihood from incomplete data viathe em algorithm. Journal Royal Statislical SocietyB39:1-22.
Eick, D.; Wedel, A.; and Heumann, H. 1994. From ini-tiation to elongation: Comparison of transcription byprokaryotic and eukaryotic RNA polymerases. Trendsin Gen. 10:292-296.
Galas, D. J.; Eggert, M.; and Waterman, M. S.1985. Rigorous pattern-recognition methods for DNAsequences, analysis of promoter sequences from Es-cherichia coli. J Mol Biol 186:117-28.
Gill, O., and Tjian, R. 1992. Eukaryotic coactivatorsassociated with the TATA box binding protein. Cur.Opin. Gen. Dev. 2:236-242.
Gross, C. A., and Lonetto, M. 1992. Bacterial sigmafactors. In Transcriptional regulation. Cold SpringHarbor Laboratory Press.
Guarente: L., and Bermingham-McDonogh, O. 1992.Conservation and evolution of transcriptional mecha-nisms in eukaryotes. Trends in Gen. 8:27-32.
190 ISMB-96
Harley, C. B., and Reynolds, R. P. 1987. Analy-sis of E. colt promoter sequences. Nucleic Acids Res15:2343-61.
Hawley, D. K., and McClure, W. R. 1983. Compila-tion and analysis of Escherichia colt promoter DNAsequences. Nucleic Acids Res 11:2237-55.
Krogh, A.; Brown, M.; Mian, I. S.; Sjolander, K.; andHaussler, D. 1994. Hidden Markov models in com-putational biology: Applications to protein modeling.Journal of Molecular Biology 235:1501-1531.
Kullback, S., and Leibler, R. A. 1951. On informationand sufficiency. Ann Math Stat 22:79-86.
Lisser, S., and Margalit, tI. 1993. Compilation of E.colt mRNA promoter sequences. Nucleic Acids Res21:1507-16.
Loewen, P., and Hengge-Aronis, R. 1994. The role ofthe sigma factor crs (katf) in bacterial global regula-tion. Annu. Re v. Microbiol. 48:53-80.
Lonetto, M.; Gribskov, M.; and Gross, C. 1992. Thea7° family: Sequence conservation and evolutionaryrelationships. J. Bact. 174:3843-3849.
Merrick, M. 1993. In a class of its own -- the RNApolymerase sigma factor cr54 (crN). Mol. Microbiol.10:903-909.
Morett, E., and Segovia, L. 1993. The (r 54 bacterialenhancer-binding protein family: Mechanism of ac-tion and phylogenetic relationship of their functionaldomains. J. Bact. 175:6067-6074.
O’Shea-Greenfield, A., and Smale, S. T. 1992. Rolesof TATA and initiator elements in determining thestart site location and direction of RNA polymeraseII transcription. J. Biol. Chem. 267:1391-1402.
Pugh, B., and Titan, R. 1992. Diverse transcriptionalfunctions of the multisubunit eukaryotic TFIID com-plex. J. Biol. Chem. 267:679-682.
Rabiner, L. R. 1989. A tutorial on hidden markovmodels and selected applications in speech recogni-tion. Proceedings of the IEEE 77(2):257-286.
Reznikoff, W. S.; Siegele, D. A.; Cowing, D. W.; andGross, C. A. 1985. The regulation of transcriptioninitiation in bacteria. Annu Rev Genet 19:355-87.
Rosenberg, M., and Court, D. 1979. Regulatory se-quences involved in the promotion and termination ofRNA transcription. Annu Rev Genet 13:319-53.
Schneider, T. D., and Stephens, R. M. 1990. Sequencelogos: A new way to display consensus sequences. Nu-cleic Acids Res. 18:6097-6100.

Shannon, C. E. 1948. A mathematical theory of com-munication. Bell System Tech. J. 27:379-423, 623-656.
Smale, S. T., and Baltimore, D. 1989. The "initiator"as a transcription control element. Cell 57:103-13.
Pedersen 191
Copyright © 2022 FDOKUMEN




















