
Engineering of Large Numbers of Highly Specific Homing Endonucleases that Induce Recombination on...
16
Engineering of Large Numbers of Highly Specific Homing Endonucleases that Induce Recombination on Novel DNA Targets Sylvain Arnould 1 †, Patrick Chames 1 †, Christophe Perez 1 Emmanuel Lacroix 1 , Aymeric Duclert 1 , Jean-Charles Epinat 1 Franc ¸ ois Stricher 3 , Anne-Sophie Petit 1 , Ame ´ lie Patin 1 Sophie Guillier 1 , Sandra Rolland 1 , Jesu ´ s Prieto 2 , Francisco J. Blanco 2 Jero ´ nimo Bravo 2 , Guillermo Montoya 2 , Luis Serrano 3 Philippe Duchateau 1 and Fre ´de ´ric Pa ˆ ques 1 * 1 CELLECTIS S.A., 102 route de Noisy 93235 Romainville France 2 Structural Biology and Biocomputing Programme Centro Nacional de Investigaciones Oncologica C/ Melchor Fdez Almagro 28029 Madrid, Spain 3 European Molecular Biology Laboratory, Meyerhofstrasse D-62117 Heidelberg, Germany The last decade has seen the emergence of a universal method for precise and efficient genome engineering. This method relies on the use of sequence-specific endonucleases such as homing endonucleases. The structures of several of these proteins are known, allowing for site-directed mutagenesis of residues essential for DNA binding. Here, we show that a semi-rational approach can be used to derive hundreds of novel proteins from I-CreI, a homing endonuclease from the LAGLIDADG family. These novel endonucleases display a wide range of cleavage patterns in yeast and mammalian cells that in most cases are highly specific and distinct from I-CreI. Second, rules for protein/DNA interaction can be inferred from statistical analysis. Third, novel endonucleases can be combined to create heterodimeric protein species, thereby greatly enhancing the number of potential targets. These results describe a straightforward approach for engineering novel endonucleases with tailored specificities, while preserv- ing the activity and specificity of natural homing endonucleases, and thereby deliver new tools for genome engineering. q 2005 Published by Elsevier Ltd. Keywords: I-CreI; homing endonucleases; protein engineering; specificity; recombination *Corresponding author Introduction Meganucleases are by definition sequence- specific endonucleases with large ( O12 bp) cleavage sites and they can be used to achieve very high levels of gene targeting efficiencies in mammalian cells and plants, 1–6 making meganu- clease-induced recombination an efficient and robust method for genome engineering. The major limitation of the current technology is the require- ment for the prior introduction of a meganuclease cleavage site in the locus of interest. Thus, the generation of novel meganucleases with tailored specificities is under intense investigation. Such proteins could be used to cleave endogenous chromosomal sequences and lead to a wide range of applications, including the correction of mutations responsible for inherited monogenic diseases. Recently, fusion of Cys2-His2 type Zinc- Finger Proteins (ZFP) with the catalytic domain of the FokI nuclease were used to make functional sequence-specific endonucleases. 7,8 The binding specificity of ZFPs is relatively easy to manipulate, and a repertoire of novel artificial ZFPs able to bind many (G/A)NN(G/A)NN(G/A)NN sequences is now available. 9–11 Nevertheless, preserving a very narrow specificity is one of the major issues for genome engineering applications, and presently it is unclear whether ZFPs would fulfil the very strict requirements for therapeutic applications. 0022-2836/$ - see front matter q 2005 Published by Elsevier Ltd. † S.A. & P.C. contributed equally to this work. Abbreviations used: wt, wild-type; ORF, open reading frame; CHO, Chinese hamster ovary; HE, homing endonuclease. E-mail address of the corresponding author: [email protected] doi:10.1016/j.jmb.2005.10.065 J. Mol. Biol. (2005) xx, 1–16 DTD 5 ARTICLE IN PRESS
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Engineering of Large Numbers of Highly Specific Homing Endonucleases that Induce Recombination on...

doi:10.1016/j.jmb.2005.10.065 J. Mol. Biol. (2005) xx, 1–16
DTD 5 ARTICLE IN PRESS
Engineering of Large Numbers of Highly SpecificHoming Endonucleases that Induce Recombinationon Novel DNA Targets
Sylvain Arnould1†, Patrick Chames1†, Christophe Perez1
Emmanuel Lacroix1, Aymeric Duclert1, Jean-Charles Epinat1
Francois Stricher3, Anne-Sophie Petit1, Amelie Patin1
Sophie Guillier1, Sandra Rolland1, Jesus Prieto2, Francisco J. Blanco2
Jeronimo Bravo2, Guillermo Montoya2, Luis Serrano3
Philippe Duchateau1 and Frederic Paques1*
1CELLECTIS S.A., 102 route deNoisy 93235 RomainvilleFrance
2Structural Biology andBiocomputing ProgrammeCentro Nacional deInvestigaciones OncologicaC/ Melchor Fdez Almagro28029 Madrid, Spain
3European Molecular BiologyLaboratory, MeyerhofstrasseD-62117 Heidelberg, Germany
0022-2836/$ - see front matter q 2005 P
† S.A. & P.C. contributed equallyAbbreviations used: wt, wild-typ
frame; CHO, Chinese hamster ovarendonuclease.E-mail address of the correspond
The last decade has seen the emergence of a universal method for preciseand efficient genome engineering. This method relies on the use ofsequence-specific endonucleases such as homing endonucleases. Thestructures of several of these proteins are known, allowing for site-directedmutagenesis of residues essential for DNA binding. Here, we show that asemi-rational approach can be used to derive hundreds of novel proteinsfrom I-CreI, a homing endonuclease from the LAGLIDADG family. Thesenovel endonucleases display a wide range of cleavage patterns in yeast andmammalian cells that in most cases are highly specific and distinct fromI-CreI. Second, rules for protein/DNA interaction can be inferred fromstatistical analysis. Third, novel endonucleases can be combined to createheterodimeric protein species, thereby greatly enhancing the number ofpotential targets. These results describe a straightforward approach forengineering novel endonucleases with tailored specificities, while preserv-ing the activity and specificity of natural homing endonucleases, andthereby deliver new tools for genome engineering.
q 2005 Published by Elsevier Ltd.
Keywords: I-CreI; homing endonucleases; protein engineering; specificity;recombination
*Corresponding authorIntroduction
Meganucleases are by definition sequence-specific endonucleases with large (O12 bp)cleavage sites and they can be used to achievevery high levels of gene targeting efficiencies inmammalian cells and plants,1–6 making meganu-clease-induced recombination an efficient androbust method for genome engineering. The majorlimitation of the current technology is the require-ment for the prior introduction of a meganucleasecleavage site in the locus of interest. Thus, the
ublished by Elsevier Ltd.
to this work.e; ORF, open readingy; HE, homing
ing author:
generation of novel meganucleases with tailoredspecificities is under intense investigation. Suchproteins could be used to cleave endogenouschromosomal sequences and lead to a wide rangeof applications, including the correction ofmutations responsible for inherited monogenicdiseases. Recently, fusion of Cys2-His2 type Zinc-Finger Proteins (ZFP) with the catalytic domain ofthe FokI nuclease were used to make functionalsequence-specific endonucleases.7,8 The bindingspecificity of ZFPs is relatively easy to manipulate,and a repertoire of novel artificial ZFPs able tobindmany (G/A)NN(G/A)NN(G/A)NN sequencesis now available.9–11 Nevertheless, preserving avery narrow specificity is one of the major issuesfor genome engineering applications, andpresently it is unclear whether ZFPs would fulfilthe very strict requirements for therapeuticapplications.

2 Engineering of Specific Homing Endonucleases
DTD 5 ARTICLE IN PRESS
Homing endonucleases (HEs) are a widespreadfamily of natural meganucleases including hundredsof proteins.12 These proteins are encoded by mobilegenetic elements, which propagate by a processcalled “homing”: the endonuclease cleaves acognate allele from which the mobile element isabsent, thereby stimulating a homologous recom-bination event that duplicates the mobile DNA intothe recipient locus.13,14 Given their natural functionand their exceptional specificity, HEs provide idealscaffolds to derive novel endonucleases for genomeengineering. Data have been accumulated over thelast decade characterizing the LAGLIDADG family,the largest of the four HE families.12 LAGLIDADGrefers to the only sequence actually conservedthroughout the family and is found in one or(more often15) two copies in the protein. Proteinswith a single motif, such as I-CreI, form homo-dimers and cleave palindromic or pseudo-palin-dromic DNA sequences, whereas the larger, doublemotif proteins, such as I-SceI, are monomers andcleave non-palindromic targets. Seven differentLAGLIDADG proteins have been crystallized, andthey exhibit a very striking conservation of the corestructure that contrasts with the lack of similarity atthe primary sequence level.16–24 In this corestructure, two characteristic abbabba folds, con-tributed by two monomers, or by two domains indouble LAGLIDAG proteins, are facing each otherwith a 2-fold symmetry. DNA binding depends onthe four b strands from each domain, folded into anantiparallel b-sheet, and forming a saddle on theDNA helix major groove. The catalytic core iscentral, with a contribution of both symmetricmonomers/domains. In addition to this corestructure, other domains can be found: for example,PI-SceI, an intein, has a protein-splicing domain,and an additional DNA-binding domain.20,25 Thesestructural similarities prompted the construction ofchimeric and single-chain artificial HEs,26–28 show-ing that these proteins were robust enough towithstand extensive modifications. Recently,Seligman and co-workers used a rational approachto substitute specific individual residues of theI-CreI abbabba fold, and observed substantialcleavage of novel targets.29,30 In a similar way,Gimble et al. modified the additional DNA-bindingdomain of PI-SceI and obtained variant proteinswith altered binding specificity.31
Here, we used a rational approach along with acell-based high-throughput assay that directlymonitors endonuclease-induced recombinationin living cells, to identify functional variants ofthe I-CreI homodimeric protein. Hundreds ofhoming endonucleases with new specificities wereidentified, cleaving altogether 38 DNA targets.Many new proteins maintained their very narrowspecificity and displayed high levels of cleavage ofnew targets in living cells. At the same time,no detectable activity on the wild-type I-CreIcleavage site was detected, showing that engineer-ing can preserve efficacy and specificity. Second,statistical analysis revealed rules for protein/DNA
interaction. Finally, novel proteins could be com-bined in functional heterodimers to cleave chimerictargets. Our results describe a rational approachto rapidly evolve the specificity of homing endo-nucleases by collecting large samples of novelendonucleases, and combining them to achievetailored specificities.
Results
Screening for new functional endonucleases
I-CreI is a dimeric homing endonuclease thatcleaves a 22 bp pseudo-palindromic target.Analysis of I-CreI structure bound to its naturaltarget shows that in each monomer, eight residuesestablish direct interactions with seven bases.16
Residues Q44, R68, R70 contact three consecutivebase-pairs at position 3–5 (and K3 toK5, Figure 1).An exhaustive protein library versus target libraryapproach was undertaken to engineer locally thispart of the DNA-binding interface. First, the I-CreIscaffold was mutated from D75 to N to decreaselikely energetic strains caused by the replacement ofthe basic residues R68 and R70 in the library thatsatisfy the hydrogen-acceptor potential of theburied D75 in the I-CreI structure. The D75Nmutation did not affect the protein structure, butdecreased the toxicity of I-CreI in overexpressionexperiments (data not shown). Since I-CreI and itsN75 derivative display similar in vitro activities(data not shown) and levels of specificity (seebelow), further studies will be necessary todetermine the origin of I-CreI toxicity. Next,positions 44, 68 and 70 were randomized and 64palindromic targets resulting from substitutions inpositions G3,G4 and G5 of a palindromic targetcleaved by I-CreI18 were generated, as described inthe legend to Figure 1. Variant proteins, which allhave the D75N mutation, are named here after theresidues found in positions 44, 68 and 70 (forexample, the I-CreI N75 scaffold is QRR, and KTG isI-CreI K44 T68 G70 N75). Palindromic targets arenamed after the three bases found on the top strandatK5,K4 andK3 (for example, the CAT target hasC in K5, A in K4 and T in K3).
We have previously described an assay tomonitor homing endonuclease-induced recombina-tion in yeast cells.26 A modified assay, the principleof which is described in the legend to Figure 2(a),includes a mating step, and allows for the screeningof the same meganuclease with several differenttargets. We used a robot-assisted mating protocol toscreen a large number of endonucleases from ourlibrary. The general screening strategy is describedin the legend to Figure 2(b). A total of 13,824homing endonuclease-expressing clones (about2.3-fold the theoretical diversity) were spotted athigh density (20 spots/cm2) on nylon filters andindividually tested against each one of the 64 targetstrains (884,608 spots). A total of 2100 clones

Figure 1. Rationale of the experiment. (a) Localizationof the area of the binding interface (bottom view) chosenfor randomization (green) and interacting base-pairs (K3orange, K4 pink, K5 magenta). (b) Zoom showingresidues 44, 68, 70 chosen for randomization, D75 andinteracting base-pairs. R68 and R70 make classic inter-actions with two guanosine bases, in which the NH1 andNH2 groups of the arginine side-chain make a hydrogenbond with the N7 and O6 groups of the guanosine ring.Q44 makes a hydrogen bond to the side-chain N7 groupof an adenine. These residues are held in place by ahydrogen-bond network involving the side-chain OG group
Engineering of Specific Homing Endonucleases 3
DTD 5 ARTICLE IN PRESS
showing an activity against at least one target wereisolated (Figure 2(b)) and the open reading frame(ORF) encoding the homing endonuclease wasamplified by PCR and sequenced. A total of 410different sequences were identified and a similarnumber of corresponding clones were chosen toanalyse further. The spotting density was reducedto four spots/cm2 and each clone was tested againstthe 64 reporter strains in quadruplicate, therebycreating complete profiles (as in Figure 3(a)). A totalof 350 positives could be confirmed. Next, to avoidthe possibility of strains containing more than oneclone, mutant ORFs were amplified by PCR, andrecloned in the yeast vector. The resulting plasmidswere individually transformed back into yeast.A total of 294 such clones were obtained and testedat low density (four spots/cm2). Differences withprimary screening were observed mostly for weaksignals, with 28 weak cleavers appearing now asnegatives. Only one positive clone displayed apattern different from what was observed in theprimary profiling.The 350 validated clones showed very diverse
patterns. Some of these new profiles shared somesimilarity with the wild-type scaffold, whereasmany others were totally different. Variousexamples are shown in Figure 3(a). Homingendonucleases can usually accommodate somedegeneracy in their target sequences, and one ofour first findings was that the original I-CreI proteinitself cleaves seven different targets in yeast. In aformer study, Monnat and co-workers used anin vitro selection experiment to assess the degen-eracy of I-CreI cleavage, and had already shownthat GCC, TCC and ATC were cleavable, althoughthe impact of mutation in regions other than K5 toK3 was unclear in this study.32 Other sequencesfound by Monnat and co-workers, such as ATT andGGC, were not detected here, reflecting either thesensitivity of our yeast assay, or the impact of theadjacent mutations. Many of our mutants displayedcleavage degeneracy as well, with the numberof cleaved sequences ranging from one to 21 withan average of 5.0 sequences cleaved (standarddeviationZ3.6). Interestingly, in 50 mutants (14%),specificity was altered so that they cleaved exactlyone target; 37 (11%) cleaved two targets, 61 (17%)cleaved three targets and 58 (17%) cleaved fourtargets. For five targets and above, percentages
of T46 and the side-chain OD1 and OD2 groups of D75.)(c) Design of the library and targets. The interactions ofI-CreI residues Q44, R68 an R70 with DNA targets areindicated (top). The target described here is a palindromederived from the I-CreI natural target, and cleaved byI-CreI.18 Cleavage positions are indicated by arrowheads.In the library, residues 44, 68 and 70 are replaced withADEGHKNPQRST. Since I-CreI is a homodimer, thelibrary was screened with palindromic targets. A total of64 palindromic targets resulting from substitutions inpositions G3, G4 and G5 were generated. A fewexamples of such targets are shown (bottom).

Figure 2. Screening. (a) Yeast screening assay principle. A strain harbouring the expression vector encoding thevariants is mated with a strain harbouring a reporter plasmid. In the reporter plasmid, a LacZ reporter gene isinterrupted with an insert containing the site of interest, flanked by two direct repeats. Upon mating, the endonuclease(gray oval) performs a double-strand break on the site of interest, allowing restoration of a functional LacZ (white oval)gene by single-strand annealing (SSA) between the two flanking direct repeats. (b) Scheme of the experiment. A libraryof I-CreI variants is built using PCR, cloned into a replicative yeast expression vector and transformed in S. cerevisiaestrain FYC2-6A (MATa, trp1D63, leu2D1, his3D200). The 64 palindromic targets are cloned in the LacZ-based yeastreporter vector, and the resulting clones transformed into strain FYBL2-7B (MATa, ura3D851, trp1D63, leu2D1, lys2D202).Robot-assisted gridding on filter membrane is used to perform mating between individual clones expressing homingendonuclease variants and individual clones harbouring a reporter plasmid. After primary high-throughputscreening, the ORFs of positive clones are amplified by PCR and sequenced. A total of 410 different variants wereidentified among the 2100 positives, and tested at low density, to establish complete patterns, and 350 clones werevalidated. Also, 294 mutants were recloned in yeast vectors, and tested in a secondary screen, and results confirmedthose obtained without recloning. Chosen clones are then assayed for cleavage activity in a similar CHO-based assay andeventually in vitro.
4 Engineering of Specific Homing Endonucleases
DTD 5 ARTICLE IN PRESS
were lower than 10%. Altogether, 38 targets werecleaved by the mutants (Figure 4(a)). Interestingly,GTT has been identified in the cleavage site of twoI-CreI homologues, isolated from green algaechloroplastic introns.15 Cleavage was barelyobserved on targets with A in position G3, andnever with targets with TGN and CGN at positionsG5, G4, G3.
Novel homing endonucleases can cleave noveltargets while keeping high activity and narrowspecificity
Eight representative mutants (belonging to sixdifferent clusters, see below) were chosen forfurther characterization (Figure 3). We first deter-mined whether data in yeast could be confirmed in

Engineering of Specific Homing Endonucleases 5
DTD 5 ARTICLE IN PRESS
mammalian cells, by using an assay based on thetransient cotransfection of a homing endonucleaseexpressing vector and a target vector, as describedin previous reports.26,33 The eight mutant ORFs andthe 64 targets were cloned into appropriate vectors,and we used a robot-assisted microtitre-basedprotocol to co-transfect in Chinese hamster ovary(CHO) cells each selected variant with each one ofthe 64 different reporter plasmids. Homing endo-nuclease-induced recombination was measured bya standard, quantitative ONPG assay that monitorsthe restoration of a functional b-galactosidasegene. Profiles were found to be qualitatively andquantitatively reproducible in five independentexperiments (not shown). As shown in Figure 3(a),strong and medium signals were nearly alwaysobserved with both yeast and CHO cells (with theexception of ADK), thereby validating therelevance of our yeast HTS process. However,weak signals observed in yeast were often not
Figure 3(a) (legend o
detected in CHO cells, likely due to a difference inthe detection level (see QRR and targets GTG, GCT,and TTC).Four mutants were also produced in Escherichia
coli and purified by metal affinity chromatography.Their relative in vitro cleavage efficiencies againstthe wild-type site and their cognate sites weredetermined. The extent of cleavage under standar-dized conditions was assessed across a broad rangeof concentrations for the mutants (Figure 3(b)).Similarly, we analysed the activity of I-CreI wt onthese targets. In many case, 100% cleavage of thesubstrate could not be achieved, likely reflecting thefact that these proteins may have little or noturnover.34,35 In general, in vitro assays confirmedthe data obtained in yeast and CHO cells. Surpri-singly, the GTT target was efficiently cleaved byI-CreI, whereas no cleavage was observed in yeastin this study, nor in bacteria in a previous reportfrom Seligman and co-workers.30
n following page)
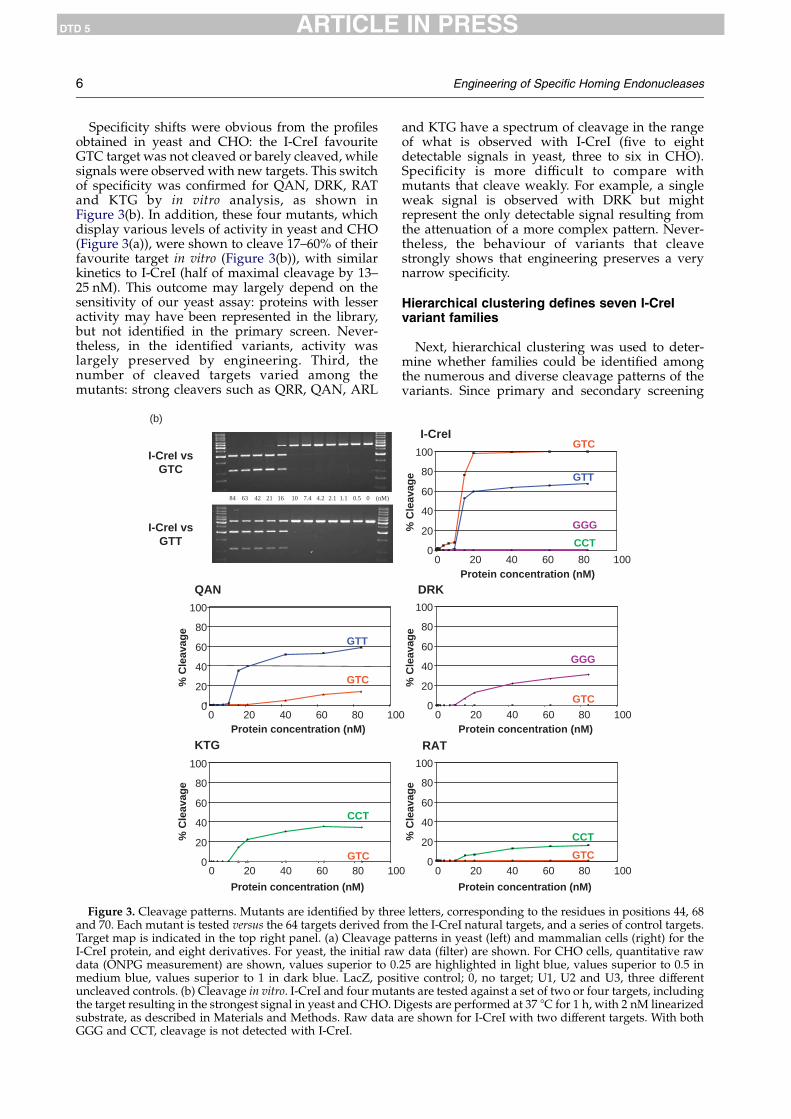
6 Engineering of Specific Homing Endonucleases
DTD 5 ARTICLE IN PRESS
Specificity shifts were obvious from the profilesobtained in yeast and CHO: the I-CreI favouriteGTC target was not cleaved or barely cleaved, whilesignals were observed with new targets. This switchof specificity was confirmed for QAN, DRK, RATand KTG by in vitro analysis, as shown inFigure 3(b). In addition, these four mutants, whichdisplay various levels of activity in yeast and CHO(Figure 3(a)), were shown to cleave 17–60% of theirfavourite target in vitro (Figure 3(b)), with similarkinetics to I-CreI (half of maximal cleavage by 13–25 nM). This outcome may largely depend on thesensitivity of our yeast assay: proteins with lesseractivity may have been represented in the library,but not identified in the primary screen. Never-theless, in the identified variants, activity waslargely preserved by engineering. Third, thenumber of cleaved targets varied among themutants: strong cleavers such as QRR, QAN, ARL
KTG
0
20
40
60
80
100
0 20 40 60 80 10
Protein concentration (nM)
% C
leav
age
I-CreI vsGTT
I-CreI vsGTC
84 63 42 21 16 10 7.4 4.2 2.1 1.1 0.5 0 (nM)
0
20
40
60
80
100
0 20 40 60 80 10
QAN
GTC
GTT
% C
leav
age
GTC
CCT
Protein concentration (nM)
(b)
Figure 3. Cleavage patterns. Mutants are identified by threand 70. Each mutant is tested versus the 64 targets derived froTarget map is indicated in the top right panel. (a) Cleavage pI-CreI protein, and eight derivatives. For yeast, the initial rawdata (ONPG measurement) are shown, values superior to 0.medium blue, values superior to 1 in dark blue. LacZ, posiuncleaved controls. (b) Cleavage in vitro. I-CreI and four mutathe target resulting in the strongest signal in yeast and CHO. Dsubstrate, as described in Materials and Methods. Raw data aGGG and CCT, cleavage is not detected with I-CreI.
and KTG have a spectrum of cleavage in the rangeof what is observed with I-CreI (five to eightdetectable signals in yeast, three to six in CHO).Specificity is more difficult to compare withmutants that cleave weakly. For example, a singleweak signal is observed with DRK but mightrepresent the only detectable signal resulting fromthe attenuation of a more complex pattern. Never-theless, the behaviour of variants that cleavestrongly shows that engineering preserves a verynarrow specificity.
Hierarchical clustering defines seven I-CreIvariant families
Next, hierarchical clustering was used to deter-mine whether families could be identified amongthe numerous and diverse cleavage patterns of thevariants. Since primary and secondary screening
0
0
0
20
40
60
80
100
0 20 40 60 80 100
RAT
CCT
GTC
Protein concentration (nM)
% C
leav
age
DRK
GTC0
20
40
60
80
100
0 20 40 60 80 100Protein concentration (nM)
% C
leav
age
GGG
I-CreI
0
20
40
60
80
100
0 20 40 60 80 100Protein concentration (nM)
% C
leav
age
GTC
CCT
GTT
GGG
e letters, corresponding to the residues in positions 44, 68m the I-CreI natural targets, and a series of control targets.atterns in yeast (left) and mammalian cells (right) for thedata (filter) are shown. For CHO cells, quantitative raw
25 are highlighted in light blue, values superior to 0.5 intive control; 0, no target; U1, U2 and U3, three differentnts are tested against a set of two or four targets, includingigests are performed at 37 8C for 1 h, with 2 nM linearizedre shown for I-CreI with two different targets. With both

Engineering of Specific Homing Endonucleases 7
DTD 5 ARTICLE IN PRESS
gave congruent results, we used quantitative datafrom the first round of yeast low-density screeningfor analysis to permit a larger sample size. Bothvariants and targets were clustered using standardhierarchical clustering with Euclidean distance andWard’s method,36 and seven clusters were defined(Figure 4(b)). Detailed analysis is shown for three of
7
(b)
5
6
1
3
4
2
GT
CG
TG
GT
TG
CT
GC
CT
TT
TT
CG
CG
AT
CA
TT
GT
AT
TG
AG
GG
GT
GG
C
GGG GGA(a)
GGT GGC
GAG GAA GAT GAC
GTG GTA GTT GTC
GCG GCA GCT GCC
TGG TGA TGT TGC
TAG TAA TAT TAC
TTG TTA TTT TTC
TCG TCA TCT TCC
C
C
C
C
AG
AA
A
AC
7
103
66
7
48
2
21
5
1
99109
146 95
3
53
58
7491
57
111
100 79
Figure 4(a) and (b) (lege
them (Figure 4(c)) and the results are summarizedin Table 1. For each cluster, a set of preferred targetscould be identified on the basis of the frequency andintensity of the signal (Figure 4(c)). The threepreferred targets for each cluster are indicated inTable 1, with their cleavage frequencies. The sum ofthese frequencies is a measurement of the specificity
GG
GC
AC
AA
CA
AT
AA
GC
AG
CA
TT
AG
TC
GC
CG
AC
TC
TC
AC
CC
TT
TA
CG
AC
GA
TG
AG
TA
TT
CT
CC
TT
CC
CC
C
GG CGA CGT CGC
AG CAA CAT CAC
TG CTA CTT CTC
CG CCA CCT CCC
G AGA AGT AGC
G AAA AAT AAC
TG ATA ATT ATC
G ACA ACT ACC
1
7 15 15
65
45 31
18
26
70105
39
38
4
6
nd on following page)

8 Engineering of Specific Homing Endonucleases
DTD 5 ARTICLE IN PRESS
of the cluster. For example, in cluster 1, the threepreferred targets (GTT/C/G), account for 78.1% ofthe observed cleavage, with 46.2% for GTT alone,revealing a very narrow specificity. Actually, thiscluster includes several proteins which, as QAN,cleave mostly GTT (Figure 3(a)). In contrast, the
Figure 4. Statistical analysis. (a) Cleaved target. Targets cleaproteins cleaving each target is shown below, and the levelintensity obtained with these cutters in yeast. (b) Hierarchicaland targets were clustered using hierarchical clustering withdone with hclust from the R package. Mutants and targets dclusters and the mutant dendrogram was cut at the height ofgreen. QRR mutant and GTC target are indicated by an arrowof three out of the seven clusters. For each mutant cluster (clwas computed and a bar plot (left column) shows, in decreasnumber of amino acid residue of each type at each position (4right column. The legend of amino acid colour code is at the
three preferred targets in cluster 2 represent only36.6% of all observed signals. In accordance withthe relatively broad and diverse patterns observedin this cluster, QRR cleaves five targets (Figure 3(a)),while other cluster members’ activity is notrestricted to these five targets.
ved by I-CreI variants are coloured in grey. The number ofof grey colouration is proportional to the average signalclustering of mutant and target data in yeast. Both mutantsEuclidean distance and Ward’s method.36 Clustering wasendrograms were reordered to optimize positions of the8 with deduced clusters alternately coloured in blue and. Grey levels reflect the intensity of the signal. (c) Analysisusters 1, 3 and 7), the cumulative intensity for each targeting order, the normalized intensities. For each cluster, the4, 68 and 70) is shown as a colour-coded histogram in thebottom of the Figure.

Table 1. Cluster analysis
Three preferred targetsa Preferred amino acidb
ClusterExamples
(Figure 3(a)) Sequence % CleavageNucleotide in position 4
(%)a 44 68 70
1 QAN GTT 46.2 G 0.5 QGTC 18.3 A 2.0 80.5%
77 proteins GTG 13.6 T 82.4 (62/77)SZ78.1 C 15.1
2 QRR GTT 13.4 G 0 Q RGTC 11.8 A 4.9 100.0% 100.0%
8 proteins TCT 11.4 T 56.9 (8/8) (8/8)SZ36.6 C 38.2
3 ARL GAT 27.9 G 2.4 A RTAT 23.2 A 88.9 63.0% 33.8%
65 proteins GAG 15.7 T 5.7 (41/65) (22/65)SZ66.8 C 3.0
4 AGR GAC 22.7 G 0.3 A&N R RTAC 14.5 A 91.9 51.6% & 35.
4%48.4% 67.7%
31 proteins GAT 13.4 T 6.6 (16&11/31) 15/31 21/31SZ50.6 C 1.2
5 ADK GAT 29.21 G 1.6DRK TAT 15.4 A 73.8
81 proteins GAC 11.4 T 13.4SZ56.05.9 C 11.2
6 KTG CCT 30.1 G 0 KRAT TCT 19.6 A 4.0 62.7%
51 proteins TCC 13.9 T 6.3 (32/51)SZ63.6 C 89.7
7 CCT 20.8 G 0 KTCT 19.6 A 0.2 91.9%
37 proteins TCC 15.3 T 14.4 (34/37)SZ55.7 C 85.4
a Frequencies according to the cleavage index, as described in the legend to Figure 4(c).b In each position, residues present in more than one-third of the cluster are indicated.
Engineering of Specific Homing Endonucleases 9
DTD 5 ARTICLE IN PRESS
Analysis of the residues found in each clustershowed strong biases for position 44: Q is over-whelmingly represented in clusters 1 and 2,whereas A and N are more frequent in clusters 3and 4, and K in clusters 6 and 7. Meanwhile, thesebiases were correlated with strong base preferencesfor DNA positions G4, with a large majority of T:Abase-pairs in cluster 1 and 2, A:T in clusters 3, 4 and5, and C:G in clusters 6 and 7 (see Table 1). Thestructure of I-CreI bound to its target shows thatresidue Q44 interacts with the bottom strand inposition K4 (and the top strand of position C4, seeFigure 1(b) and (c)). Our result suggests that thisinteraction is largely conserved in our mutants, andreveals a “code”, wherein Q44 would establishcontact with adenine, A44 (or less frequently N44)with thymine, and K44 with guanine. Such corre-lation was not observed for positions 68 and 70.
Structural perspective
To understand the reasons behind the change inspecificity of the new variants it is important to lookat the structure of the complex between I-CreI andthe DNA. DNA/protein interactions are summar-ized in Figure 1. Using a new version of theautomatic protein design algorithm FOLD-X,37–39
we have systematically mutated the base-pairs
in positions G3, G4 and G5 (a total of 64combinations) and calculated the interaction energyof the new protein DNA complexes after movingand relaxing the amino acid residues Q44, R68 andR70 and their neighbours (Table 2). The interactionof I-CreI with the 64 targets could be predictedrelatively well (compare the lowest value of Table 2with the I-CreI yeast pattern in Figure 3(a)).The first obvious conclusion from the FOLD-X
analysis and the modelled 64 complexes is that thebase-pair A:T in G3 is not tolerated at all with anyother DNAmutant combination due to a steric clashof the methyl group of the thymine with T46. Thisresult likely explains why nomutant was found thatrecognizes a template of the type NNA (compareTable 2 and Figure 4(a)). In addition, mutation inG3 automatically results in a loss of activity due tothe fact that to make a H-bond with another baseother than a G, R70 needs to break the double ion-pair with D75 and leave a buried unsatisfiedcharged group (Figure 1(b)). The D75N mutationallows more freedom to R70 (it is held by only onebond), so that it now can make a single hydrogenbond to the complementary base of TG3, explain-ing why QRR in the context of the N75 variantrecognizes the sequence GTT. For the base-pair T:Ain G4, the Q44 side-chain NH2 group makes ahydrogen bond to the N7 group of the A base

Table 2. Interaction energy for I-CreI in complex with the 64 variant targets
Difference in free energy of interaction between I-CreI and the DNAwith respect to the GTC sequence (in kcal/mol)a
GGG GGA GGT GGC AGG AGA AGT AGC3.9 8.3 4.3 1.6 4.4 8.6 4.7 3.5GAG GAA GAT GAC AAG AAA AAT AAC4.2 6.8 4.4 2.4 5.7 9.2 6.0 3.3GTG GTA GTT GTC ATG ATA ATT ATC3.8 6.2 4.3 0.0 2.9 5.6 3.3 1.3GCG GCA GCT GCC ACG ACA ACT ACC4.5 6.1 5.2 1.1 4.7 7.9 4.7 1.9TGG TGA TGT TGC CGG CGA CGT CGC3.6 8.6 4.1 3.5 4.7 7.9 5.3 4.0TAG TAA TAT TAC CAG CAA CAT CAC4.9 8.4 5.3 2.8 6.2 9.2 6.6 3.4TTG TTA TTT TTC CTG CTA CTT CTC3.5 7.2 4.1 0.7 4.8 6.5 5.3 1.0TCG TCA TCT TCC CCG CCA CCT CCC3.5 6.9 4.6 2.6 4.7 8.2 5.0 3.7
a All triplets predicted to bind with less than 1.5 kcal/mol difference with the I-CreI protein are in bold.
10 Engineering of Specific Homing Endonucleases
DTD 5 ARTICLE IN PRESS
(Figure 1(b)). In principle, a C:G pair also should beaccepted, since the same bond can be made with theequivalent group of a C base. This explains why a Qat position 44 creates a strong tendency to get a T:Apair in G4, or also a C:G, but no A:T or G:C pairs.Finally the guanine at position G5 that interactswith R68, cannot easily be replaced by an A, notonly because it will break the interaction with R68but also because it will introduce van der Waals’clashes with the side-chain of I24. This explainswhy few proteins cleave ANN targets (seeFigure 4(a)). A C:G pair will break the twoH-bonds with R68, and a T:A pair, in principle,could be tolerated, since still one H-bond could bemade (data not shown). Thus simple modeling andenergy calculation easily explains the experimentalpreferences found for the wt protein, as well as thegeneral trends observed for the mutants.
Variants can be assembled in functional het-erodimers to cleave new DNA target sequences
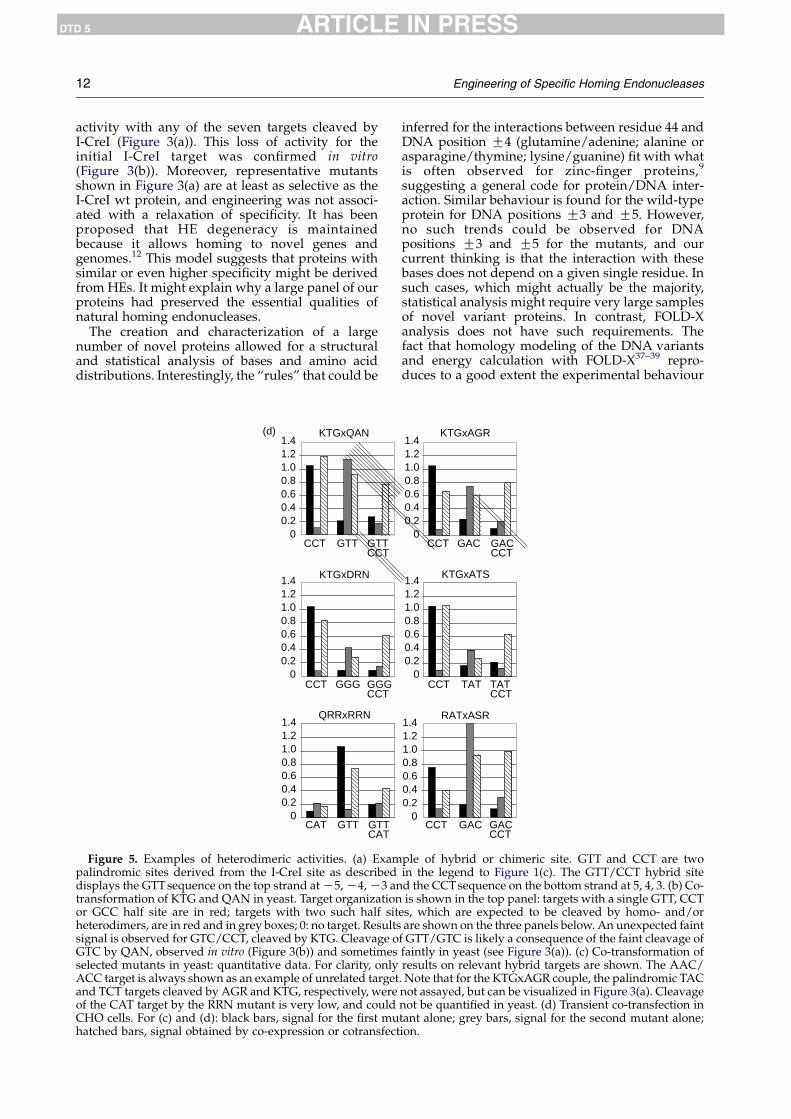
All selected variants are homodimers capable ofcleaving palindromic sites. To test whether the listof cleavable targets could be extended by creatingheterodimers that would cleave hybrid cleavagesites (as described in Figure 5(a)), we chose a subsetof mutants with distinct profiles and cloned them intwo different yeast vectors marked by LEU2 or KANgenes. We next co-expressed combinations ofmutants in yeast with a set of palindromic andnon-palindromic chimeric DNA targets. Anexample is shown in Figure 5(b): co-expression ofthe KTG and QANmutants resulted in the cleavageof two chimeric targets, GTT/GCC and GTT/CCT,that were not cleaved by either mutant alone. Thepalindromic GTT, CCT and GCC targets (and othertargets of KTG and QAN) were also cleaved, likelyresulting from homodimeric species formation, butunrelated targets were not. In addition, a GTT, CCTor GCC half-site was not sufficient to allowcleavage, since such targets were fully resistant(see GGG/GCC, GAT/GCC, GCC/TAC, and many
others, in Figure 5(b)). Unexpected cleavage wasobserved only with GTC/CCT, with KTG homo-dimers, but the signal remained very weak. Thus,efficient cleavage requires the cooperative bindingof two mutant monomers. These results demon-strate a good level of specificity for heterodimericspecies.
Altogether, a total of 112 combinations of 14different proteins were tested in yeast (not shown),and 37.5% of the combinations (42/112) revealed apositive signal on their predicted chimeric target.Quantitative data are shown for six examples inFigure 5(c), and for the same six combinations,results were confirmed in CHO cells in transient co-transfection experiments, with a subset of relevanttargets (Figure 5(d)). As a general rule, functionalheterodimers were always obtained when one ofthe two expressed proteins gave a strong signal ashomodimer. In these cases, unexpected cleavage ofan irrelevant target was rare, and always faint, asfor KTG (see Figure 5(b)). For example, DRN andRRN, two low-activity mutants, give functionalheterodimers with strong cutters such as KTG orQRR (Figure 5(c) and (d)), whereas no cleavage ofchimeric targets could be detected by co-expressionof the same weak mutants (not shown).
Discussion
We set out to determine whether a large numberof novel endonucleases could be derived from HEs,while keeping both high activity and high speci-ficity levels. The creation of a large number ofmutants with new specificities was clearly demon-strated. A large diversity of novel profiles wasfound, characterized by the cleavage of noveltargets. Based on the mutation of single I-CreIresidues, Seligman and co-workers29,30 found that afew I-CreI variants with altered specificity could beobtained. Here, we show that global alteration of awhole subdomain can yield hundreds of novelendonucleases. In our study, preservation of a verynarrow specificity was also achieved. In a former

Engineering of Specific Homing Endonucleases 11
DTD 5 ARTICLE IN PRESS
study, Gimble et al.31 derived several proteins fromPI-SceI, by mutating the DNA-binding residuesfrom the intein domain. However, most proteinsmaintained similar affinities for the wild-type target
5’ TCAAAACGTTGTACA3’ AGTTTTGCAACATGT
5’ TCAAAACCCTGTACA3’ AGTTTTGGGACATGT
5’ TCAAAACGTTGTACA3’ AGTTTTGCAACATGT
-12(a) -11-10 -9 -8 -7 -6 -5 -4 -3 -2 -1 21
GTT
CCT
GTT/CCT
AAC/ACT AAC/ACC AAC/CCT AAC/CCC ACT/CAC ACC
GGG/ACT GGG/ACC GGG/CAT GGG/CAC GGG/CCT GGG
GGG/GCT GGG/GCC GGG/TAT GGG/TAC GGG/TCT GGG
GAT/GTC GAT/GCC GAT/TAT GAC/ACT GAC/ACC GAC
GAC/GCT GAC/GCC GAC/TAT GAC/TCT GAC/TCC GTT
GTC/CAT GTC/CCT GTC/GCC GTC/TAT GCT/AAC GCT
GCC/CCT GCC/TAT GCC/TAC TAT/CAT TAT/CCT TAC
TAC/TCC TCT/AAC TCT/CAC TCC/AAC TCC/CAC GG
GCC
CCT
GCC/CCT
GTC/CCT
KTG
(b)
(c)
QAN
KTGxQAN
KTGxDRN
QRRxRRN
CCT GCC GTT GTTCCT
GTTGCC
AACACC
0
0.2
0.4
0.6
0.8
1.0
0
0.2
0.4
0.6
0.8
1.0
CCT GAC GAT GGG GGGCCT
GATCCT
GACCCT
AACACC
0
0.2
0.4
0.6
0.8
1.0
GTC GTT GCC CAT GTCCAT
GTTCAT
GCCCAT
AACACC
Figure 5(a)–(c) (legend
and the new targets in an in vitro binding test.Although it is difficult to compare results obtainedwith different assays and scaffolds, many of ourmutants cleaving a novel target had lost most or all
ACGTTTTGA 3’TGCAAAACT 5’
GGGTTTTGA 3’CCCAAAACT 5’
GGGTTTTGA 3’CCCAAAACT 5’
1211109876543
/CAC CAT/CCT CAC/CCT CAC/CCC GGG/AAC GCC
/CCC GGG/GAT GGG/GAC GGG/GTT GGG/GTC TAT
/TCC GAT/CAT GAT/CCT GAT/GAC GAT/GTT CAT
/CAT GAC/CCT GAC/CCC GAC/GTT GAC/GTC CCT
/CAT GTT/CCT GTT/GTC GTT/GCC GTT/TAT 0
/CAC GCT/TAC GCC/AAC GCC/CAT GCC/CAC 0
/ACT TAC/ACC TAC/CCT TAC/CCC TAC/TCT 0
G GAT GAC GTT GTC 0
KTGxQAN
GTT/CCTGTT/GCC
GTT
GTT/GTC
KTGxAGR
RATxASR
KTGxATS
0
0.2
0.4
0.6
0.8
1.0
CCT GAC GACCCT
GACTCT
TACCCT
TACTCT
AACACC
0
0.2
0.4
0.6
0.8
1.0
CCT CAT GAT TAT CATCCT
GATCCT
TATCCT
AACACC
0
0.2
0.4
0.6
0.8
1.0
CCT GAC GACCCT
AACACC
on following page)
12 Engineering of Specific Homing Endonucleases
DTD 5 ARTICLE IN PRESS
activity with any of the seven targets cleaved byI-CreI (Figure 3(a)). This loss of activity for theinitial I-CreI target was confirmed in vitro(Figure 3(b)). Moreover, representative mutantsshown in Figure 3(a) are at least as selective as theI-CreI wt protein, and engineering was not associ-ated with a relaxation of specificity. It has beenproposed that HE degeneracy is maintainedbecause it allows homing to novel genes andgenomes.12 This model suggests that proteins withsimilar or even higher specificity might be derivedfrom HEs. It might explain why a large panel of ourproteins had preserved the essential qualities ofnatural homing endonucleases.
The creation and characterization of a largenumber of novel proteins allowed for a structuraland statistical analysis of bases and amino aciddistributions. Interestingly, the “rules” that could be
QRRxRRN
CAT GTT GTTCAT
00.20.40.60.81.01.21.4
KTGxQAN
00.20.40.60.81.01.21.4
(d)
GTTCCT GTTCCT
KTGxDRN
CCT GGG GGGCCT
00.20.40.60.81.01.21.4
Figure 5. Examples of heterodimeric activities. (a) Exampalindromic sites derived from the I-CreI site as describeddisplays the GTTsequence on the top strand atK5,K4,K3 antransformation of KTG and QAN in yeast. Target organizationor GCC half site are in red; targets with two such half sitheterodimers, are in red and in grey boxes; 0: no target. Resultssignal is observed for GTC/CCT, cleaved by KTG. Cleavage oGTC by QAN, observed in vitro (Figure 3(b)) and sometimesselected mutants in yeast: quantitative data. For clarity, onlyACC target is always shown as an example of unrelated targetand TCT targets cleaved by AGR and KTG, respectively, wereof the CAT target by the RRN mutant is very low, and couldCHO cells. For (c) and (d): black bars, signal for the first muhatched bars, signal obtained by co-expression or cotransfect
inferred for the interactions between residue 44 andDNA position G4 (glutamine/adenine; alanine orasparagine/thymine; lysine/guanine) fit with whatis often observed for zinc-finger proteins,9
suggesting a general code for protein/DNA inter-action. Similar behaviour is found for the wild-typeprotein for DNA positions G3 and G5. However,no such trends could be observed for DNApositions G3 and G5 for the mutants, and ourcurrent thinking is that the interaction with thesebases does not depend on a given single residue. Insuch cases, which might actually be the majority,statistical analysis might require very large samplesof novel variant proteins. In contrast, FOLD-Xanalysis does not have such requirements. Thefact that homology modeling of the DNA variantsand energy calculation with FOLD-X37–39 repro-duces to a good extent the experimental behaviour
KTGxAGR
CCT GAC GACCCT
KTGxATS
TATCCT TATCCT
00.20.40.60.81.01.21.4
00.20.40.60.81.01.21.4
RATxASR
CCT GAC GACCCT
00.20.40.60.81.01.21.4
ple of hybrid or chimeric site. GTT and CCT are twoin the legend to Figure 1(c). The GTT/CCT hybrid sited the CCTsequence on the bottom strand at 5, 4, 3. (b) Co-is shown in the top panel: targets with a single GTT, CCT
es, which are expected to be cleaved by homo- and/orare shown on the three panels below. An unexpected faintf GTT/GTC is likely a consequence of the faint cleavage offaintly in yeast (see Figure 3(a)). (c) Co-transformation ofresults on relevant hybrid targets are shown. The AAC/. Note that for the KTGxAGR couple, the palindromic TACnot assayed, but can be visualized in Figure 3(a). Cleavagenot be quantified in yeast. (d) Transient co-transfection intant alone; grey bars, signal for the second mutant alone;ion.

Engineering of Specific Homing Endonucleases 13
DTD 5 ARTICLE IN PRESS
found for the I-CreI protein offers hope that acombination of protein design and screening willallow the generation of new, very specific homingendonucleases.
Recent work has shown that new homingendonucleases could be engineered by domainswapping of different monomers.26–28 These chi-meric proteins were able to cleave the hybrid targetcorresponding to the fusion of the two half parentDNA sequence targets. Here, we applied the samestrategy to the I-CreI mutants, by simple co-expression. The combinations were functional andstrictly predictable regarding their DNA specificityas long as the homodimeric activity was strong forat least one partner, which likely reflects thesensitivity of our assay. These results suggest thatfunctional heterodimers could be formed, andtherefore that the alteration of a precise area of theDNA-binding interface (positions 44, 68, 70) hasa limited influence on the protein/proteininterface constituted by the LAGLIDADG domains.However, co-expression leads to a mixture ofhomodimers and heterodimers, and the cleavageof palindromic cleavage sites by homodimersresults in a lesser specificity. We have demonstratedthe possibility to create a functional single chainprotein with I-CreI,26 and the same strategy can beused in this case to obtain a fusion protein that willcleave only non-palindromic chimeric sites.
The generation of collections of novel homingendonucleases, and the ability to combine them,considerably enriches the number of DNAsequences that can be targeted, but does not yetsaturate all potential sequences. However, there aremany ways to increase the number of targetsequences. First, by choosing the degenerate VVKcodon to create diversity, we excluded eight aminoacids from positions 44, 68 and 70, and theseresidues may be involved in new specificities.More importantly, we limited the scope of thisstudy to base-contacting residues, whereas anindirect but determinant influence of surroundingresidues is highly probable in terms of an inter-action with DNA and/or with DNA-bindingresidues. In the future, to target as many sequencesas possible, the binding interface has to beengineered more globally. Second, other areas ofthe DNA-binding domain need to be altered, tocreate new binding domains, which can ultimatelybe combined with the mutants described here. Thiswould expand further the potential targetsequences, with the proviso that a combinatorialapproach might be more difficult to apply within asingle monomer or domain than between mono-mers. Finally, we and others have shown thatLAGLIDADG domains from different proteins canbe assembled into functional chimeric molecules,26–28 and it should thus be possible to expand furtherthe number of combinations. Thus, combinations ofLAGLIDADG domains from a growing collection ofnovel proteins should eventually result in thegeneration of dedicated homing endonucleasesable to cleave sequences from many genes of
interest. Given the specificity of our proteins,potential applications include the cleavage of viralgenomes or the correction of genetic defects viadouble-strand break-induced recombination, bothof which will lead to future therapeutics.
Materials and Methods
Structure analyses
All analyses of protein structures were realized usingPymol. The structures from I-CreI correspond to ProteinData Bank accession number 1G9Y. Residue numbering inthe text always refers to these structures, except forresidues in the second I-CreI protein domain of thehomodimer, where residue numbers were set as for thefirst domain. Energies (Table 2) have been calculated forone half of the dimeric I-CreI complex. The FOLD-Xalgorithm37–39 modified to mutate DNAwas used with asofter version of the van der Waals’ clashes. Importantly,FOLD-X recognizes water molecules making two or morehydrogen bonds to the protein (either directly or throughanother water molecule). Also, FOLD-X can dress aprotein or DNA with predicted water-bridges.39 Thus,crystallographic and predicted water-bridges were con-sistent in the modelling.
Construction of mutant libraries
I-CreI wt and I-CreI D75N ORFs were synthesized asdescribed.26 Mutation D75N was introduced by replacingcodon 75 with AAC. The diversity of the homingendonuclease library was generated by PCR usingdegenerate primers from Sigma harbouring codon VVK(18 codons, amino acids ADEGHKNPQRST) at position44, 68 and 70, and as DNA template, the I-CreI gene. Thefinal PCR product was digested with specific restrictionenzymes, and cloned back into the I-CreI ORF digestedwith the same restriction enzymes (detailed protocol isavailable upon request), in pCLS542. In this 2m-basedreplicative vector marked with the LEU2 gene, I-CreIvariants are under the control of a galactose-induciblepromoter.26 After electroporation in E. coli, we obtained7!104 clones, representing 12 times the theoreticaldiversity at the DNA level (183Z5832). DNA wasextracted and transformed into Saccharomyces cerevisiaestrain FYC2-6A (MATa, trp1D63, leu2D1, his3D200).A total of 13,824 colonies were picked using a colonypicker (QpixII, genetix), and grown in 144 microtitreplates.
Construction of target clones
The C1221 24 bp palindrome (5 0 TCAAAACGTCGTACGACGTTTTGA 3 0) is a repeat of the half-site of thenearly palindromic natural I-CreI target (TCAAAACGTCGTGAGACAGTTTGG). C1221 is cleaved as efficiently asthe I-CreI natural target in vitro and ex vivo in both yeastand mammalian cells. The 64 palindromic targets werederived as follows: 64 pairs of oligonucleotides (5 0
GGCATACAAGTTTCAAAACNNNGTACNNNGTTTTGACAATCGTCTGTCA 3 0 and reverse complementarysequences) were ordered form Sigma, annealed andcloned into pGEM-T Easy (Promega). Next, a 400 bpPvuII fragment was excised and cloned into the yeastvector pFL39-ADH-LACURAZ and the mammalian

14 Engineering of Specific Homing Endonucleases
DTD 5 ARTICLE IN PRESS
vector pcDNA3.1-LACURAZ-DURA, both describedpreviously.26 The 75 hybrid target sequences were clonedas follows: oligonucleotides were designed that containedtwo different half sites of each mutant palindrome(Proligo). Double-stranded target DNA, generated byPCR amplification of the single-stranded oligonucleo-tides, was cloned using the Gateway protocol (Invitrogen)into yeast and mammalian reporter vectors. Yeastreporter vectors were transformed into S. cerevisiae strainFYBL2-7B (MAT a, ura3D851, trp1D63, leu2D1, lys2D202).
Mating of homing endonuclease-expressing clonesand screening in yeast
Mating was performed using a colony gridder (QpixII,Genetix). Mutants were gridded on nylon filters coveringYPD plates, using a high gridding density (about20 spots/cm2). A second gridding process was performedon the same filters to spot a second layer consisting of 64or 75 different reporter-harbouring yeast strains for eachvariant. Membranes were placed on solid agar YPD-richmedium, and incubated at 30 8C for one night, to allowmating. Next, filters were transferred to syntheticmedium, lacking leucine and tryptophan, with galactose(1%) as a carbon source (and with G418 for co-expressionexperiments), and incubated for five days at 37 8C, toselect for diploids carrying the expression and targetvectors. After five days, filters were placed on solidagarose medium with 0.02% X-Gal in 0.5 M sodiumphosphate buffer (pH 7.0), 0.1% (w/v) SDS, 6% dimethylformamide (DMF), 7 mM b-mercaptoethanol, 1% (w/v)agarose, and incubated at 37 8C, to monitor b-galacto-sidase activity. Results were analyzed by scanning andquantification was performed using a proprietary soft-ware.
Sequence and re-cloning of primary hits
The ORF of positive clones identified during theprimary screening in yeast was amplified by PCR andsequenced. Then, ORFs were recloned using the Gatewayprotocol (Invitrogen). ORFs were amplified by PCR onyeast colonies,40 using primers 5 0 GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAAGGAGATAGAACCATGGCCAATACCAAATATAACAAAGAGTTCC 3 0 and5 0 GGGGACCACTTTGTACAAGAAAGCTGGGTTTAGTCGGCCGCCGGGGAGGATTTCTTCTTCTCGC 3 0 fromProligo. PCR products were cloned either in (i) yeastgateway expression vectors harbouring a galactose-inducible promoter, LEU2 or KanR as selectable markerand a 2m origin of replication, (ii) a CHO gatewayexpression vector pCDNA6.2 from Invitrogen, (iii) apET 24d(C) vector from Novagen. Resulting cloneswere verified by sequencing (Millegen).
Mammalian cell assays
A total of 104 CHO cells were plated in each well of 96-well microplates. At 24 h after seeding, they weretransfected with Polyfect transfection reagent accordingto the supplier’s (Qiagen) protocol (175 ng of total DNAmix, 1 ml of polyfect, 150 ml of F12K medium per well).Cells were incubated at 37 8C, 5% CO2. At 72 h aftertransfection, culture medium was removed and 150 ml oflysis/revelation buffer added for b-galactosidase liquidassay. Typically, for 1 l of buffer, we used 100 ml of lysis
buffer (10 mM Tris–HCl (pH 7.5), 150 mM NaCl, 0.1%Triton X100, 0.1 mg/ml of BSA, protease inhibitors), 10 mlof Mg 100! buffer (100 mM MgCl2, 35% b-mercap-toethanol), 110 ml of ONPG (8 mg/ml) and 780 ml of0.1 M sodium phosphate (pH 7.5). After 3 h incubationat 37 8C, absorbance was measured at 420 nm. The entireprocess was performed on an automated Velocity11BioCel platform.
Protein expression and purification
His-tagged proteins were over-expressed in E. coli BL21(DE3)pLysS cells using pET-24d (C) vectors (Novagen).Induction with IPTG (0.3 mM), was performed at 25 8C.Cells were sonicated in a solution of 50 mM sodiumphosphate (pH 8), 300 mM sodium chloride containingprotease inhibitors (Complete EDTA-free tablets, Roche)and 5% (v/v) glycerol. Cell lysates were centrifuged at100,000g for 60 min. His-tagged proteins were thenaffinity-purified, using 5 ml Hi-Trap chelating HP col-umns (Amersham Biosciences) loaded with cobalt.Several fractions were collected during elution with alinear gradient of imidazole (up to 0.25 M imidazole,followed by plateau at 0.5 M imidazole, 0.3 M NaCl and50 mM sodium phosphate pH 8). Protein-rich fractions(determined by SDS-PAGE) were applied to the secondcolumn. The crude purified samples were taken to pH 6and applied to a 5 ml HiTrap Heparin HP column(Amersham Biosciences) equilibrated with 20 mMsodium phosphate (pH 6.0). Bound proteins were elutedwith a sodium chloride continuous gradient with 20 mMsodium phosphate and 1 M sodium chloride. The purifiedfractions were submitted to SDS-PAGE and concentrated(10 kDa cut-off Centriprep Amicon Ultra system), frozenin liquid nitrogen and stored at K80 8C. Purified proteinswere desalted using PD10 columns (Sephadex G-25M,Amersham Biosciences) in PBS or 10 mM Tris–HCl (pH 8)buffer.
In vitro cleavage assays
pGEM plasmids with single homing endonucleaseDNA target cut sites were first linearized with XmnI.Cleavage assays were performed at 37 8C in 10 mM Tris–HCl (pH 8), 50 mM NaCl, 10 mM MgCl2, 1 mM DTT,50 mg/ml of BSA. The target substrate concentration was2 nM. A dilution range between 0 nM and 85 nM wasused for each protein, in a 25 ml final volume reaction.Reactions were stopped after 1 h by addition of 5 ml of45% glycerol, 95 mM EDTA (pH 8), 1.5% (w/v) SDS,1.5 mg/ml of proteinase K and 0.048% (w/v) bromophe-nol blue (6! Buffer Stop) and incubated at 37 8C for30 min. Digests were run on agarose electrophoresis gel,and fragments quantified after ethidium bromide stain-ing, to calculate the percentage of cleavage.
Hierarchical clustering
Clustering was done using hclust from the R package.We used quantitative data from the primary, low-densityscreening. Both variants and targets were clustered usingstandard hierarchical clustering with Euclidean distanceand Ward’s method.36 Mutant and target dendrogramswere re-ordered to optimize positions of the clusters andthe mutant dendrogram was cut at the height of 8 todefine the cluster.

Engineering of Specific Homing Endonucleases 15
DTD 5 ARTICLE IN PRESS
Acknowledgements
We thank Rodney J. Rothstein, James E. Haberand Julianne Smith for critical reading of themanuscript, Daniel Padro for NMR analysis onthe mutants and Pilar Redondo for assistance inprotein purification and in vitro cleavage assays.This work was supported, in part, by grant0212508Q from The French Agency for Innovation(ANVAR), and grant 04W107 from the FrenchMinistry of Research, under label EUREKA S!3294.
References
1. Rouet, P., Smih, F. & Jasin, M. (1994). Introduction ofdouble-strand breaks into the genome of mouse cellsby expression of a rare-cutting endonuclease. Mol.Cell. Biol. 14, 8096–8106.
2. Choulika, A., Perrin, A., Dujon, B. & Nicolas, J. F.(1995). Induction of homologous recombination inmammalian chromosomes by using the I-SceI systemof Saccharomyces cerevisiae. Mol. Cell. Biol. 15,1968–1973.
3. Donoho, G., Jasin, M. & Berg, P. (1998). Analysis ofgene targeting and intrachromosomal homologousrecombination stimulated by genomic double-strandbreaks in mouse embryonic stem cells. Mol. Cell. Biol.18, 4070–4078.
4. Elliott, B., Richardson, C., Winderbaum, J., Nickoloff,J. A. & Jasin, M. (1998). Gene conversion tracts fromdouble-strand break repair in mammalian cells. Mol.Cell. Biol. 18, 93–101.
5. Sargent, R. G., Brenneman, M. A. & Wilson, J. H.(1997). Repair of site-specific double-strand breaks ina mammalian chromosome by homologous andillegitimate recombination.Mol. Cell. Biol. 17, 267–277.
6. Puchta, H., Dujon, B. & Hohn, B. (1996). Two differentbut related mechanisms are used in plants for therepair of genomic double-strand breaks by homolo-gous recombination. Proc. Natl Acad. Sci. USA, 93,5055–5060.
7. Smith, J., Berg, J. M. & Chandrasegaran, S. (1999).A detailed study of the substrate specificity of achimeric restriction enzyme. Nucl. Acids Res. 27,674–681.
8. Urnov, F. D., Miller, J. C., Lee, Y. L., Beausejour, C. M.,Rock, J. M., Augustus, S. et al. (2005). Highly efficientendogenous human gene correction using designedzinc-finger nucleases. Nature, 435, 646–651.
9. Pabo, C. O., Peisach, E. & Grant, R. A. (2001). Designand selection of novel Cys2His2 zinc finger proteins.Annu. Rev. Biochem. 70, 313–340.
10. Segal, D. J. & Barbas, C. F., 3rd (2001). Custom DNA-binding proteins come of age: polydactyl zinc-fingerproteins. Curr. Opin. Biotechnol. 12, 632–637.
11. Isalan, M., Klug, A. & Choo, Y. (2001). A rapid,generally applicable method to engineer zinc fingersillustrated by targeting the HIV-1 promoter. NatureBiotechnol. 19, 656–660.
12. Chevalier, B. S. & Stoddard, B. L. (2001). Homingendonucleases: structural and functional insight intothe catalysts of intron/intein mobility.Nucl. Acids Res.29, 3757–3774.
13. Kostriken, R., Strathern, J. N., Klar, A. J., Hicks, J. B. &
Heffron, F. (1983). A site-specific endonucleaseessential for mating-type switching in Saccharomycescerevisiae. Cell, 35, 167–174.
14. Jacquier, A. & Dujon, B. (1985). An intron-encodedprotein is active in a gene conversion process thatspreads an intron into a mitochondrial gene. Cell, 41,383–394.
15. Lucas, P., Otis, C., Mercier, J. P., Turmel, M. &Lemieux, C. (2001). Rapid evolution of the DNA-binding site in LAGLIDADG homing endonucleases.Nucl. Acids Res. 29, 960–969.
16. Jurica, M. S., Monnat, R. J., Jr & Stoddard, B. L. (1998).DNA recognition and cleavage by the LAGLIDADGhoming endonuclease I-CreI. Mol. Cell. 2, 469–476.
17. Chevalier, B. S., Monnat, R. J., Jr & Stoddard, B. L.(2001). The homing endonuclease I-CreI uses threemetals, one of which is shared between the two activesites. Nature Struct. Biol. 8, 312–316.
18. Chevalier, B., Turmel, M., Lemieux, C., Monnat, R. J.,Jr & Stoddard, B. L. (2003). Flexible DNA target siterecognition by divergent homing endonuclease iso-schizomers I-CreI and I-MsoI. J. Mol. Biol. 329,253–269.
19. Moure, C. M., Gimble, F. S. & Quiocho, F. A. (2003).The crystal structure of the gene targeting homingendonuclease I-SceI reveals the origins of its targetsite specificity. J. Mol. Biol. 334, 685–695.
20. Moure, C. M., Gimble, F. S. & Quiocho, F. A. (2002).Crystal structure of the intein homing endonucleasePI-SceI bound to its recognition sequence. NatureStruct. Biol. 9, 764–770.
21. Ichiyanagi, K., Ishino, Y., Ariyoshi, M., Komori, K. &Morikawa, K. (2000). Crystal structure of an archaealintein-encoded homing endonuclease PI-PfuI. J. Mol.Biol. 300, 889–901.
22. Duan, X., Gimble, F. S. & Quiocho, F. A. (1997). Crystalstructure of PI-SceI, a homing endonuclease withprotein splicing activity. Cell, 89, 555–564.
23. Bolduc, J. M., Spiegel, P. C., Chatterjee, P., Brady, K. L.,Downing, M. E., Caprara, M. G. et al. (2003). Structuraland biochemical analyses of DNA and RNA bindingby a bifunctional homing endonuclease and group Iintron splicing factor. Genes Dev. 17, 2875–2888.
24. Silva, G. H., Dalgaard, J. Z., Belfort, M. & Van Roey, P.(1999). Crystal structure of the thermostable archaealintron-encoded endonuclease I-DmoI. J. Mol. Biol. 286,1123–1136.
25. Grindl, W., Wende, W., Pingoud, V. & Pingoud, A.(1998). The protein splicing domain of the homingendonuclease PI-sceI is responsible for specific DNAbinding. Nucl. Acids Res. 26, 1857–1862.
26. Epinat, J. C., Arnould, S., Chames, P., Rochaix, P.,Desfontaines, D., Puzin, C. et al. (2003). A novelengineered meganuclease induces homologousrecombination in yeast and mammalian cells. Nucl.Acids Res. 31, 2952–2962.
27. Chevalier, B. S., Kortemme, T., Chadsey, M. S., Baker,D., Monnat, R. J. & Stoddard, B. L. (2002). Design,activity, and structure of a highly specific artificialendonuclease. Mol. Cell. 10, 895–905.
28. Steuer, S., Pingoud, V., Pingoud, A. & Wende, W.(2004). Chimeras of the homing endonuclease PI-SceIand the homologous Candida tropicalis intein: a studyto explore the possibility of exchanging DNA-bindingmodules to obtain highly specific endonucleases withaltered specificity. ChemBiochem, 5, 206–213.
29. Sussman, D., Chadsey, M., Fauce, S., Engel, A., Bruett,A., Monnat, R., Jr et al. (2004). Isolation and

16 Engineering of Specific Homing Endonucleases
DTD 5 ARTICLE IN PRESS
characterization of new homing endonuclease speci-ficities at individual target site positions. J. Mol. Biol.342, 31–41.
30. Seligman, L. M., Chisholm, K. M., Chevalier, B. S.,Chadsey, M. S., Edwards, S. T., Savage, J. H. & Veillet,A. L. (2002). Mutations altering the cleavage speci-ficity of a homing endonuclease. Nucl. Acids Res. 30,3870–3879.
31. Gimble, F. S., Moure, C. M. & Posey, K. L. (2003).Assessing the plasticity of DNA target site recognitionof the PI-SceI homing endonuclease using a bacterialtwo-hybrid selection system. J. Mol. Biol. 334,993–1008.
32. Argast, G. M., Stephens, K. M., Emond, M. J. &Monnat, R. J., Jr (1998). I-PpoI and I-CreI homing sitesequence degeneracy determined by random muta-genesis and sequential in vitro enrichment. J. Mol. Biol.280, 345–353.
33. Perez, C., Guyot, V., Cabaniols, J. P., Gouble, A.,Micheaux, B., Smith, J. et al. (2005). Factors affectingdouble-strand break-induced homologous recombi-nation in mammalian cells. Biotechniques, 39, 109–115.
34. Perrin, A., Buckle, M. & Dujon, B. (1993). Asymme-trical recognition and activity of the I-SceI endonu-clease on its site and on intron-exon junctions. EMBOJ. 12, 2939–2947.
35. Wang, J., Kim, H. H., Yuan, X. & Herrin, D. L. (1997).Purification, biochemical characterization andprotein-DNA interactions of the I-CreI endonucleaseproduced in Escherichia coli. Nucl. Acids Res. 25,3767–3776.
36. Ward, J. H. (1963). Hierarchical grouping to optimizean objective function. J. Am. Statist. Assoc. 58, 236–244.
37. Kiel, C., Wohlgemuth, S., Rousseau, F., Schymkowitz,J., Ferkinghoff-Borg, J., Wittinghofer, F. & Serrano, L.(2005). Recognizing and defining true Ras bindingdomains II: in silico prediction based on homologymodelling and energy calculations. J. Mol. Biol. 348,759–775.
38. Schymkowitz, J., Borg, J., Stricher, F., Nys, R.,Rousseau, F. & Serrano, L. (2005). The FoldX webserver: an online force field. Nucl. Acids Res. 33,W382–W388.
39. Schymkowitz, J. W., Rousseau, F., Martins, I. C.,Ferkinghoff-Borg, J., Stricher, F. & Serrano, L. (2005).Prediction of water and metal binding sites and theiraffinities by using the Fold-X force field. Proc. NatlAcad. Sci. USA, 102, 10147–10152.
40. Akada, R., Murakane, T. & Nishizawa, Y. (2000). DNAextraction method for screening yeast clones by PCR.Biotechniques, 28, 668–670, 672, 674.
Edited by M. Belfort
(Received 26 August 2005; received in revised form 19 October 2005; accepted 24 October 2005)




















