
Engagement of Transferrin Receptor by Polymeric IgA1: Evidence for a Positive Feedback Loop...
10
Engagement of Transferrin Receptor by Polymeric IgA1: Evidence for a Positive Feedback Loop Involving Increased Receptor Expression and Mesangial Cell Proliferation in IgA Nephropathy Ivan C. Moura,* Michelle Arcos-Fajardo,* Abdelaziz Gdoura,* Vale ´rie Leroy,* † Charlotte Sadaka,* Nizar Mahlaoui,* Yves Lepelletier, ‡ Franc ¸ois Vrtovsnik, § Elie Haddad,* † Marc Benhamou,* and Renato C. Monteiro* *INSERM U699, Bichat Medical School, † Pediatric Nephrology, Robert Debre ´ Hospital, ‡ CNRS UMR8147, Necker Hospital, and § Department of Nephrology, Bichat Hospital, Paris, France IgA nephropathy (IgAN), the most common primary glomerulonephritis in the world, is characterized by IgA immune complex–mediated mesangial cell proliferation. The transferrin receptor (TfR) was identified previously as an IgA1 receptor, and it was found that, in biopsies of patients with IgAN, TfR is overexpressed and co-localizes with IgA1 mesangial deposits. Here, it is shown that purified polymeric IgA1 (pIgA1) is a major inducer of TfR expression (three- to four-fold increase) in quiescent human mesangial cells (HMC). IgA-induced but not cytokine-induced HMC proliferation is dependent on TfR engagement as it is inhibited by both TfR1 and TfR2 ectodomains as well as by the anti-TfR mAb A24. It is dependent on the continued presence of IgA1 rather than on soluble factors released during IgA1-mediated activation. In addition, pIgA1- induced IL-6 and TGF- production from HMC was specifically inhibited by mAb A24, confirming that pIgA1 triggers a TfR-dependent HMC activation. Finally, upregulation of TfR expression induced by sera from patients with IgAN but not from healthy individuals was dependent on IgA. It is proposed that deposited pIgA1 or IgA1 immune complexes could initiate a process of auto-amplification involving hyperexpression of TfR, allowing increased IgA1 mesangial deposition. Altogether, these data unveil a functional cooperation between pIgA1 and TfR for IgA1 deposition and HMC proliferation and activation, features that are commonly implicated in the chronicity of mesangial injuries observed in IgAN and that could explain the recurrence of IgA1 deposits in the mesangium after renal transplantation. J Am Soc Nephrol 16: 2667–2676, 2005. doi: 10.1681/ASN.2004111006 G lomerular mesangium contains contractile specialized cells, the mesangial cells, located between the capil- lary endothelial cells and the basal membrane of the glomeruli. Mesangial cells are thought to regulate the blood flow through selected capillary loops and the uptake of mac- romolecules by phagocytosis (1–3). Because of their intracapil- lary location and their capacity to synthesize cytokines and other inflammatory molecules, mesangial cells are in a critical position to initiate and mediate glomerular damage. Thus, mes- angial cell proliferation is an early pathologic alteration char- acteristic of many forms of immunologically mediated glomer- ulonephritis, such as IgA nephropathy (IgAN), mesangial proliferative glomerulonephritis, and diabetic nephropathy (4). IgAN is a primary glomerulonephritis of common incidence worldwide. Clinical features include hematuria and protein- uria, and the disease is characterized histologically by the dep- osition of IgA1-containing complexes in glomerular mesan- gium, matrix expansion, and mesangial cell proliferation. IgA1 deposits are often associated with deposits of IgG, of the com- plement component C3, and less frequently of IgM. IgAN prog- nosis is unfavorable because 20 to 30% of patients develop progressive renal failure within 20 yr of disease activity (4). Recurrence of IgA1 deposits after transplantation indicates that circulating rather than local kidney abnormalities are crucial to the development of IgAN (5). Alterations in IgA circulating levels and structure are asso- ciated with IgAN. Serum IgA levels are two- to three-fold enhanced in one half of the patients. In addition, polymeric: monomeric IgA ratio is increased in their serum (6). Further- more, pathogenesis of IgAN involves IgA1 immune complex formation and alterations of IgA1 glycans composition (7). Indeed, patients with IgAN express a subpopulation of serum IgA1 that are hypogalactosylated (8 –10). Altered O-glycans present in IgA1 hinge region can be recognized by naturally occurring antiglycans IgG and IgA, thus forming immune com- plexes (7) that are deposited in the mesangium (11). Binding of IgA1 complexes on mesangial cells elicits cell activation involving an increase in intracellular Ca 2 , phospho- lipase (PLC)-1 activation, production of inositol trisphosphate Received November 25, 2004. Accepted May 31, 2005. Published online ahead of print. Publication date available at www.jasn.org. Address correspondence to: Dr. Renato C. Monteiro, INSERM U699, Bichat Medical School, 16 Rue Henri Huchard, 75870 Paris, Cedex 18, France. Phone: 33-1-4485-6261; Fax: 33-1-4485-6260; E-mail: [email protected] Copyright © 2005 by the American Society of Nephrology ISSN: 1046-6673/1609-2667
-
Upload
univ-paris7 -
Category
Documents
-
view
0 -
download
0
Transcript of Engagement of Transferrin Receptor by Polymeric IgA1: Evidence for a Positive Feedback Loop...

Engagement of Transferrin Receptor by Polymeric IgA1:Evidence for a Positive Feedback Loop Involving IncreasedReceptor Expression and Mesangial Cell Proliferation in IgANephropathy
Ivan C. Moura,* Michelle Arcos-Fajardo,* Abdelaziz Gdoura,* Valerie Leroy,*†
Charlotte Sadaka,* Nizar Mahlaoui,* Yves Lepelletier,‡ Francois Vrtovsnik,§ Elie Haddad,*†
Marc Benhamou,* and Renato C. Monteiro**INSERM U699, Bichat Medical School, †Pediatric Nephrology, Robert Debre Hospital, ‡CNRS UMR8147, NeckerHospital, and §Department of Nephrology, Bichat Hospital, Paris, France
IgA nephropathy (IgAN), the most common primary glomerulonephritis in the world, is characterized by IgA immunecomplex–mediated mesangial cell proliferation. The transferrin receptor (TfR) was identified previously as an IgA1 receptor,and it was found that, in biopsies of patients with IgAN, TfR is overexpressed and co-localizes with IgA1 mesangial deposits.Here, it is shown that purified polymeric IgA1 (pIgA1) is a major inducer of TfR expression (three- to four-fold increase) inquiescent human mesangial cells (HMC). IgA-induced but not cytokine-induced HMC proliferation is dependent on TfRengagement as it is inhibited by both TfR1 and TfR2 ectodomains as well as by the anti-TfR mAb A24. It is dependent on thecontinued presence of IgA1 rather than on soluble factors released during IgA1-mediated activation. In addition, pIgA1-induced IL-6 and TGF-� production from HMC was specifically inhibited by mAb A24, confirming that pIgA1 triggers aTfR-dependent HMC activation. Finally, upregulation of TfR expression induced by sera from patients with IgAN but notfrom healthy individuals was dependent on IgA. It is proposed that deposited pIgA1 or IgA1 immune complexes could initiatea process of auto-amplification involving hyperexpression of TfR, allowing increased IgA1 mesangial deposition. Altogether,these data unveil a functional cooperation between pIgA1 and TfR for IgA1 deposition and HMC proliferation and activation,features that are commonly implicated in the chronicity of mesangial injuries observed in IgAN and that could explain therecurrence of IgA1 deposits in the mesangium after renal transplantation.
J Am Soc Nephrol 16: 2667–2676, 2005. doi: 10.1681/ASN.2004111006
G lomerular mesangium contains contractile specializedcells, the mesangial cells, located between the capil-lary endothelial cells and the basal membrane of the
glomeruli. Mesangial cells are thought to regulate the bloodflow through selected capillary loops and the uptake of mac-romolecules by phagocytosis (1–3). Because of their intracapil-lary location and their capacity to synthesize cytokines andother inflammatory molecules, mesangial cells are in a criticalposition to initiate and mediate glomerular damage. Thus, mes-angial cell proliferation is an early pathologic alteration char-acteristic of many forms of immunologically mediated glomer-ulonephritis, such as IgA nephropathy (IgAN), mesangialproliferative glomerulonephritis, and diabetic nephropathy (4).
IgAN is a primary glomerulonephritis of common incidenceworldwide. Clinical features include hematuria and protein-uria, and the disease is characterized histologically by the dep-
osition of IgA1-containing complexes in glomerular mesan-gium, matrix expansion, and mesangial cell proliferation. IgA1deposits are often associated with deposits of IgG, of the com-plement component C3, and less frequently of IgM. IgAN prog-nosis is unfavorable because 20 to 30% of patients developprogressive renal failure within 20 yr of disease activity (4).Recurrence of IgA1 deposits after transplantation indicates thatcirculating rather than local kidney abnormalities are crucial tothe development of IgAN (5).
Alterations in IgA circulating levels and structure are asso-ciated with IgAN. Serum IgA levels are two- to three-foldenhanced in one half of the patients. In addition, polymeric:monomeric IgA ratio is increased in their serum (6). Further-more, pathogenesis of IgAN involves IgA1 immune complexformation and alterations of IgA1 glycans composition (7).Indeed, patients with IgAN express a subpopulation of serumIgA1 that are hypogalactosylated (8–10). Altered O-glycanspresent in IgA1 hinge region can be recognized by naturallyoccurring antiglycans IgG and IgA, thus forming immune com-plexes (7) that are deposited in the mesangium (11).
Binding of IgA1 complexes on mesangial cells elicits cellactivation involving an increase in intracellular Ca2�, phospho-lipase (PLC)-�1 activation, production of inositol trisphosphate
Received November 25, 2004. Accepted May 31, 2005.
Published online ahead of print. Publication date available at www.jasn.org.
Address correspondence to: Dr. Renato C. Monteiro, INSERM U699, BichatMedical School, 16 Rue Henri Huchard, 75870 Paris, Cedex 18, France. Phone:33-1-4485-6261; Fax: 33-1-4485-6260; E-mail: [email protected]
Copyright © 2005 by the American Society of Nephrology ISSN: 1046-6673/1609-2667

(IP3), and protein tyrosine phosphorylation (12). As a conse-quence, mesangial cells proliferate, release cytokines, and syn-thesize extracellular matrix (13,14).
IgA1-mediated mesangial cell activation suggested the exis-tence of a receptor for IgA1 on these cells. However, none of theknown IgA receptors, such as Fc�RI, polymeric Ig receptor(pIgR), and asialoglycoprotein receptor (ASGPR), is expressedon mesangial cells (4). Recently, we identified the transferrin(Tf) receptor (TfR) as an IgA1 receptor (15). TfR binds poly-meric IgA1 (pIgA1) but neither monomeric IgA1 nor IgA2 andpreferentially IgA1 complexes and hypoglycosylated IgA1 (16).TfR mesangial expression is upregulated in IgAN and Henoch-Schonlein purpura nephritis but also in other nephritides in-volving IgA deposits (17). TfR co-localized with IgA1 depositsin the mesangium from these patients (17).
Our study was designed to characterize the cellular conse-quences of IgA1/TfR interaction. We show that pIgA1 inducesTfR expression in a time- and dose-dependent manner. In ad-dition, engagement of TfR by pIgA1 induces mesangial cellproliferation and IL-6 and TGF-� secretion, indicating an orig-inal TfR signaling pathway activated by pIgA1. Finally, serumfrom patients with IgAN but not from normal individualsinduces an IgA-dependent upregulation of TfR expression, sug-gesting a link among altered IgA glycosylation, TfR expression,and mesangial cell activation observed in IgAN.
Materials and MethodsAntibodies and Reagents
IL-1�, TNF-�, and IL-6 were from R&D Systems (Lille, France). LPSwas from Sigma (St. Louis, MO). The anti-human TfR (CD71) mAb A24(�2b�) has been previously described (15), and the isotype-matchedmAb 30.9 directed against the rat Fc�RI� chain was used as a control(18). The anti-IgA mAb CH-EB6–8 (�1�) was from American TypeCulture Collection (Manassas, VA). Human myeloma IgA1 (Dou) waspurified as described (15). pIgA1 fractions were separated by gel filtra-tion on Superdex 200 columns through HPLC (Amersham Biosciences,Uppsala, Sweden; �99% pure). Polyclonal anti-human transferrin an-tibody (Santa Cruz Biotechnology, Santa Cruz, CA) was coupled toactivated Sepharose 4B beads (Amersham) according to the manufac-turer’s recommendations. Tf-free IgA was purified by gel filtration andimmunoadsorption through anti-Tf Sepharose 4B columns as described(15). Human serum IgG was purified by ammonium sulfate precipita-tion and DEAE ion exchange chromatography.
IgA1 depletion of sera from normal individuals and from patientswith IgAN was performed using jacalin-coupled Sepharose beads.Briefly, jacalin was purified from jackfruits on DEAE column afterextraction in PBS and coupled to CNBr-activated Sepharose 4B beads(Pharmacia, St. Quentin, France). Sera (250 �l) were incubated threetimes for 30 min with 200-�l beads on a rotating wheel at roomtemperature to remove IgA1.
Expression and Purification of TfR1 and TfR2 EctodomainsSoluble (s) versions of human TfR1 and TfR2 (kindly provided by P.
Bjorkman, CalTech, CA) were expressed in a lytic baculovirus/insect cellexpression system as described previously (19). The construct for the controlsoluble form of the extracellular domain of CD89 (Fc�RI) that contained a6-His tag in the C-terminus was cloned in pFASTBAC1 baculovirus expres-sion system (Invitrogen, Carlsbad, CA) according to the manufacturer’s in-structions. Recombinant proteins were purified as described (19).
CellsHMC were purchased from Clonetics (San Diego, CA). Cells were
cultured in RPMI 1640, supplemented with l-glutamine (2 mM), 5�g/ml insulin, 20% FCS (Life Technologies, Gaithersburg, MD), 7 mMglucose, 50 U/ml penicillin, and 50 �g/ml streptomycin in a 5% CO2
atmosphere. Studies were performed between passages 4 and 8.
Proliferation AssayHMC were trypsinized, resuspended in RPMI 1640 with 0.5% FCS,
added in triplicate at the concentration of 1 � 104 cells/well in 96-welltissue culture plates (Falcon, Oxnard, CA), and starved for 24 h beforeexperiments. Proliferation of quiescent HMC was induced using IL-1�
or TNF-� at 10 ng/ml, IL-6 at 100 ng/ml (R&D Systems), LPS at 10�g/ml, or pIgA1 at 0.5 mg/ml for 48 h. For the inhibition experiments,mAb A24, mAb 30.9 (10 �g/ml), sTfR1, or sTfR2 (50 �g/ml) was added15 min before addition of IgA1 or cytokines for 24 h. Proliferation wasmeasured over 18 h, after pulses with 1 �Ci/well [3H]thymidine (Am-ersham Life Science, Buckinghamshire, UK). Cells then were washed,trypsinized for 2 h at 37°C, and harvested on filters with a 96-wellHarvester (Pharmacia), and the incorporation of [3H]thymidine wasmeasured with a �-plate microscintillation counter (LKB; Pharmacia).
For depletion experiments, culture supernatants of HMC that werestimulated for 24 h with pIgA1 were adsorbed three times with anexcess of either anti-IgA (CH-EB6-8) or irrelevant mAb (30.9) coupledto Sepharose beads. Depleted supernatants were used as a stimulatingagent on quiescent HMC for 24 h, and cell proliferation was measuredas described above.
Reverse Transcription–PCRTotal RNA was purified with Trizol (Invitrogen) and was reverse-tran-
scribed using Advantage RT-for-PCR kit (Clontech, Palo Alto, CA) witholigo-dT primers according to the manufacturer’s instructions. TfR cDNAwere amplified using primers (sense 5�-GCGTTGTATGTTGAAAAT-CAATTTC and anti-sense 5�-GCTTTTCTGCAGCAGCTCTGGA) on the ba-sis of the human TfR sequences obtained from GenBank (accession no.M11507). �-Actin cDNA were amplified using sense primer 5�-GGGGTAT-GCCCTCCCCCATGCCATCCTGCG and antisense primer 5�-TTGGCGTA-CAGGTCTTTGCGGATGTCCACG. Amplification was performed with ini-tial denaturation at 94°C for 3 min, followed by 25 cycles at 94°C for 45 s, 60°Cfor 45 s, 72°C for 2 min, and a single final extension at 72°C for 5 min. Thereaction mixture that lacked cDNA was used as a negative control. PCRproducts were analyzed in a 1.5% agarose gel.
Flow Cytometry AnalysisCells (0.25 � 106) were preincubated with 10 �l of human IgG (10
mg/ml) for 15 min on ice to mask Fc�R. TfR expression was examinedusing an indirect immunofluorescence assay in which cells that werepreincubated with human IgG were incubated with 10 �l of biotinyl-ated A24 (0.1 mg/ml) for 30 min on ice before washing and incubationwith streptavidin-PE for 20 min at 4°C. After washes, APC-labeledstreptavidin (Southern Biotechnology Associates, Birmingham, AL)was used as a developing reagent. Immunofluorescence was finallyanalyzed by flow cytometry (FACScalibur; Becton Dickinson, LePont-de Claix, France).
ELISAIgA concentration in the various sera was determined by a sandwich
ELISA. Wells in 96-well plates were coated with 50 �l of anti-human IgAmouse mAb CH-EB6-8 at 5 �g/ml in borate buffer saline for 2 h at roomtemperature. Nonspecific sites then were saturated for 2 h at room tempera-ture in PBS that contained 1% BSA and 0.1% NaN3. Sera (between 1:1000 and
2668 Journal of the American Society of Nephrology J Am Soc Nephrol 16: 2667–2676, 2005
1:10,000 dilution) then were added for 16 h at 4°C in the wells, and wells werewashed and incubated with a polyclonal anti-IgA antibody coupled to alka-line phosphatase (at 1:1000 dilution). Optical density was measured at 405 nmafter addition of alkaline phosphatase substrate (Sigma), and the IgA concen-tration was determined by comparison with an IgA standard curve. IL-6 andTGF-� secretions were measured from culture cell supernatants using kitsfrom R&D Systems following the manufacturer’s instructions.
Statistical AnalysesThe results were analyzed by independent sample two-tailed t test,
and evaluation for correlation was conducted by linear regressionanalysis. Results are presented as means � SD.
ResultspIgA1 Induce TfR Expression on HMC
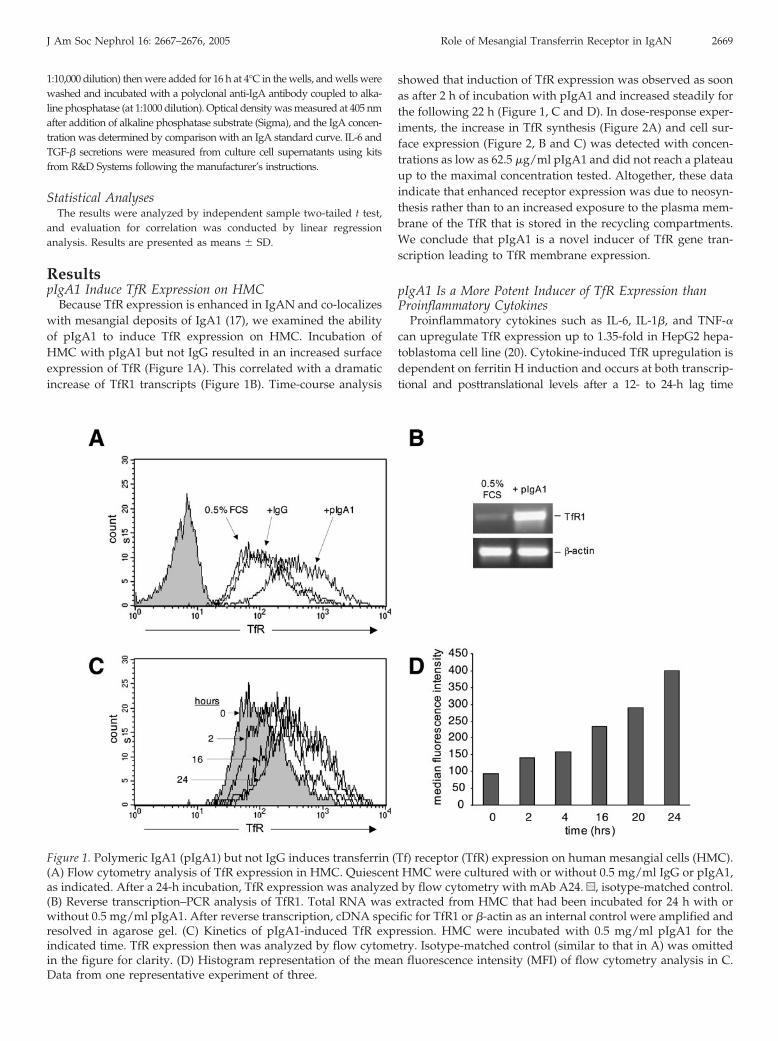
Because TfR expression is enhanced in IgAN and co-localizeswith mesangial deposits of IgA1 (17), we examined the abilityof pIgA1 to induce TfR expression on HMC. Incubation ofHMC with pIgA1 but not IgG resulted in an increased surfaceexpression of TfR (Figure 1A). This correlated with a dramaticincrease of TfR1 transcripts (Figure 1B). Time-course analysis
showed that induction of TfR expression was observed as soonas after 2 h of incubation with pIgA1 and increased steadily forthe following 22 h (Figure 1, C and D). In dose-response exper-iments, the increase in TfR synthesis (Figure 2A) and cell sur-face expression (Figure 2, B and C) was detected with concen-trations as low as 62.5 �g/ml pIgA1 and did not reach a plateauup to the maximal concentration tested. Altogether, these dataindicate that enhanced receptor expression was due to neosyn-thesis rather than to an increased exposure to the plasma mem-brane of the TfR that is stored in the recycling compartments.We conclude that pIgA1 is a novel inducer of TfR gene tran-scription leading to TfR membrane expression.
pIgA1 Is a More Potent Inducer of TfR Expression thanProinflammatory Cytokines
Proinflammatory cytokines such as IL-6, IL-1�, and TNF-�can upregulate TfR expression up to 1.35-fold in HepG2 hepa-toblastoma cell line (20). Cytokine-induced TfR upregulation isdependent on ferritin H induction and occurs at both transcrip-tional and posttranslational levels after a 12- to 24-h lag time
Figure 1. Polymeric IgA1 (pIgA1) but not IgG induces transferrin (Tf) receptor (TfR) expression on human mesangial cells (HMC).(A) Flow cytometry analysis of TfR expression in HMC. Quiescent HMC were cultured with or without 0.5 mg/ml IgG or pIgA1,as indicated. After a 24-h incubation, TfR expression was analyzed by flow cytometry with mAb A24. u, isotype-matched control.(B) Reverse transcription–PCR analysis of TfR1. Total RNA was extracted from HMC that had been incubated for 24 h with orwithout 0.5 mg/ml pIgA1. After reverse transcription, cDNA specific for TfR1 or �-actin as an internal control were amplified andresolved in agarose gel. (C) Kinetics of pIgA1-induced TfR expression. HMC were incubated with 0.5 mg/ml pIgA1 for theindicated time. TfR expression then was analyzed by flow cytometry. Isotype-matched control (similar to that in A) was omittedin the figure for clarity. (D) Histogram representation of the mean fluorescence intensity (MFI) of flow cytometry analysis in C.Data from one representative experiment of three.
J Am Soc Nephrol 16: 2667–2676, 2005 Role of Mesangial Transferrin Receptor in IgAN 2669
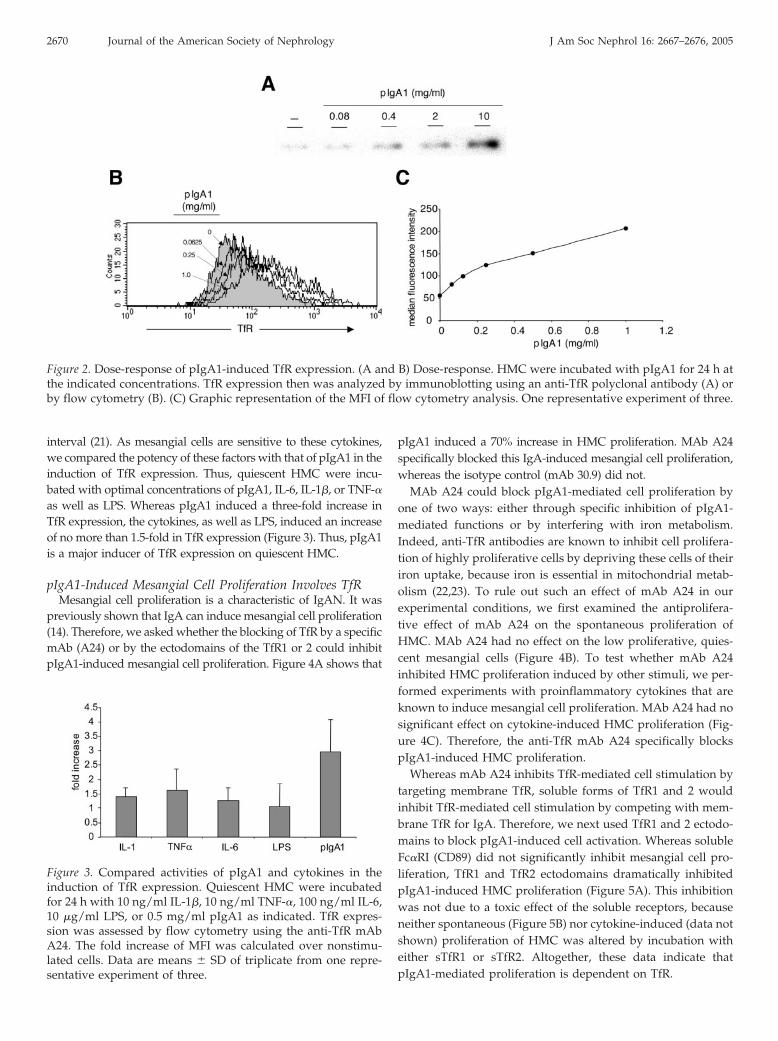
interval (21). As mesangial cells are sensitive to these cytokines,we compared the potency of these factors with that of pIgA1 in theinduction of TfR expression. Thus, quiescent HMC were incu-bated with optimal concentrations of pIgA1, IL-6, IL-1�, or TNF-�as well as LPS. Whereas pIgA1 induced a three-fold increase inTfR expression, the cytokines, as well as LPS, induced an increaseof no more than 1.5-fold in TfR expression (Figure 3). Thus, pIgA1is a major inducer of TfR expression on quiescent HMC.
pIgA1-Induced Mesangial Cell Proliferation Involves TfRMesangial cell proliferation is a characteristic of IgAN. It was
previously shown that IgA can induce mesangial cell proliferation(14). Therefore, we asked whether the blocking of TfR by a specificmAb (A24) or by the ectodomains of the TfR1 or 2 could inhibitpIgA1-induced mesangial cell proliferation. Figure 4A shows that
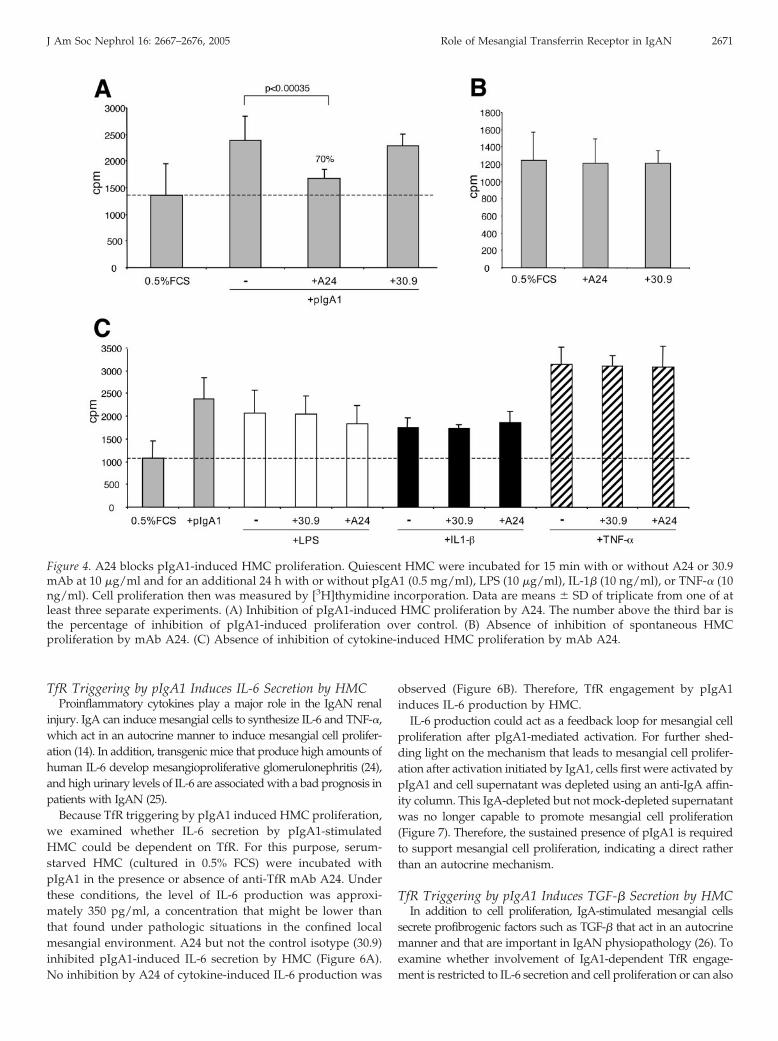
pIgA1 induced a 70% increase in HMC proliferation. MAb A24specifically blocked this IgA-induced mesangial cell proliferation,whereas the isotype control (mAb 30.9) did not.
MAb A24 could block pIgA1-mediated cell proliferation byone of two ways: either through specific inhibition of pIgA1-mediated functions or by interfering with iron metabolism.Indeed, anti-TfR antibodies are known to inhibit cell prolifera-tion of highly proliferative cells by depriving these cells of theiriron uptake, because iron is essential in mitochondrial metab-olism (22,23). To rule out such an effect of mAb A24 in ourexperimental conditions, we first examined the antiprolifera-tive effect of mAb A24 on the spontaneous proliferation ofHMC. MAb A24 had no effect on the low proliferative, quies-cent mesangial cells (Figure 4B). To test whether mAb A24inhibited HMC proliferation induced by other stimuli, we per-formed experiments with proinflammatory cytokines that areknown to induce mesangial cell proliferation. MAb A24 had nosignificant effect on cytokine-induced HMC proliferation (Fig-ure 4C). Therefore, the anti-TfR mAb A24 specifically blockspIgA1-induced HMC proliferation.
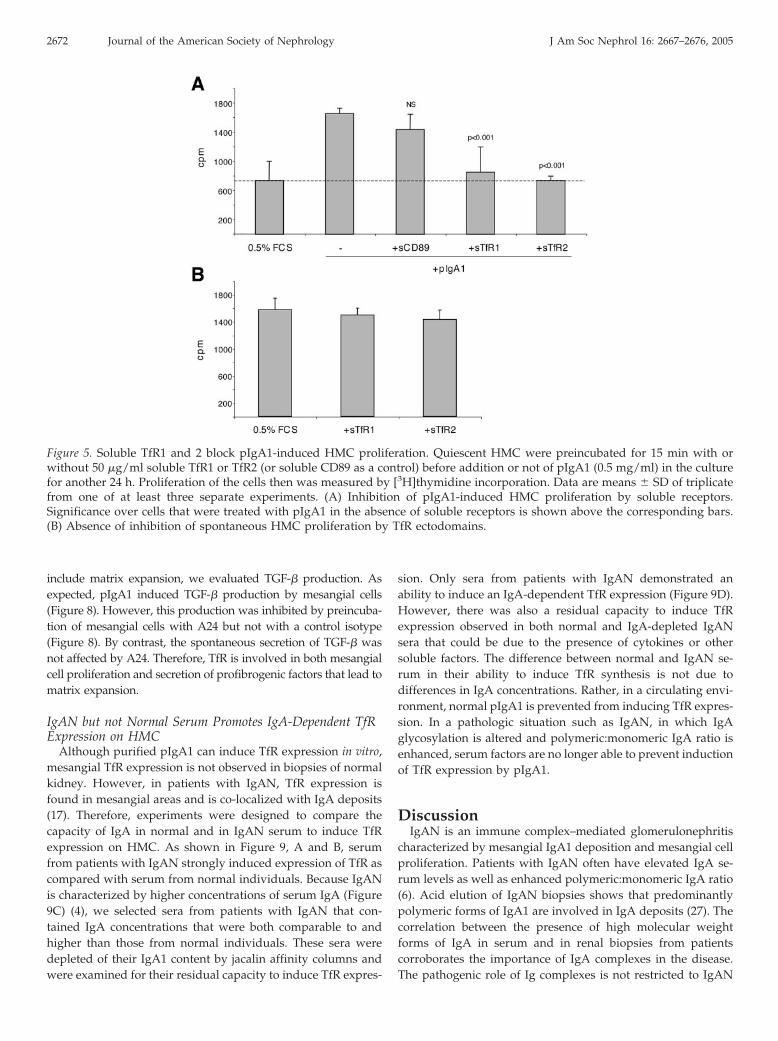
Whereas mAb A24 inhibits TfR-mediated cell stimulation bytargeting membrane TfR, soluble forms of TfR1 and 2 wouldinhibit TfR-mediated cell stimulation by competing with mem-brane TfR for IgA. Therefore, we next used TfR1 and 2 ectodo-mains to block pIgA1-induced cell activation. Whereas solubleFc�RI (CD89) did not significantly inhibit mesangial cell pro-liferation, TfR1 and TfR2 ectodomains dramatically inhibitedpIgA1-induced HMC proliferation (Figure 5A). This inhibitionwas not due to a toxic effect of the soluble receptors, becauseneither spontaneous (Figure 5B) nor cytokine-induced (data notshown) proliferation of HMC was altered by incubation witheither sTfR1 or sTfR2. Altogether, these data indicate thatpIgA1-mediated proliferation is dependent on TfR.
Figure 2. Dose-response of pIgA1-induced TfR expression. (A and B) Dose-response. HMC were incubated with pIgA1 for 24 h atthe indicated concentrations. TfR expression then was analyzed by immunoblotting using an anti-TfR polyclonal antibody (A) orby flow cytometry (B). (C) Graphic representation of the MFI of flow cytometry analysis. One representative experiment of three.
Figure 3. Compared activities of pIgA1 and cytokines in theinduction of TfR expression. Quiescent HMC were incubatedfor 24 h with 10 ng/ml IL-1�, 10 ng/ml TNF-�, 100 ng/ml IL-6,10 �g/ml LPS, or 0.5 mg/ml pIgA1 as indicated. TfR expres-sion was assessed by flow cytometry using the anti-TfR mAbA24. The fold increase of MFI was calculated over nonstimu-lated cells. Data are means � SD of triplicate from one repre-sentative experiment of three.
2670 Journal of the American Society of Nephrology J Am Soc Nephrol 16: 2667–2676, 2005
TfR Triggering by pIgA1 Induces IL-6 Secretion by HMCProinflammatory cytokines play a major role in the IgAN renal
injury. IgA can induce mesangial cells to synthesize IL-6 and TNF-�,which act in an autocrine manner to induce mesangial cell prolifer-ation (14). In addition, transgenic mice that produce high amounts ofhuman IL-6 develop mesangioproliferative glomerulonephritis (24),and high urinary levels of IL-6 are associated with a bad prognosis inpatients with IgAN (25).
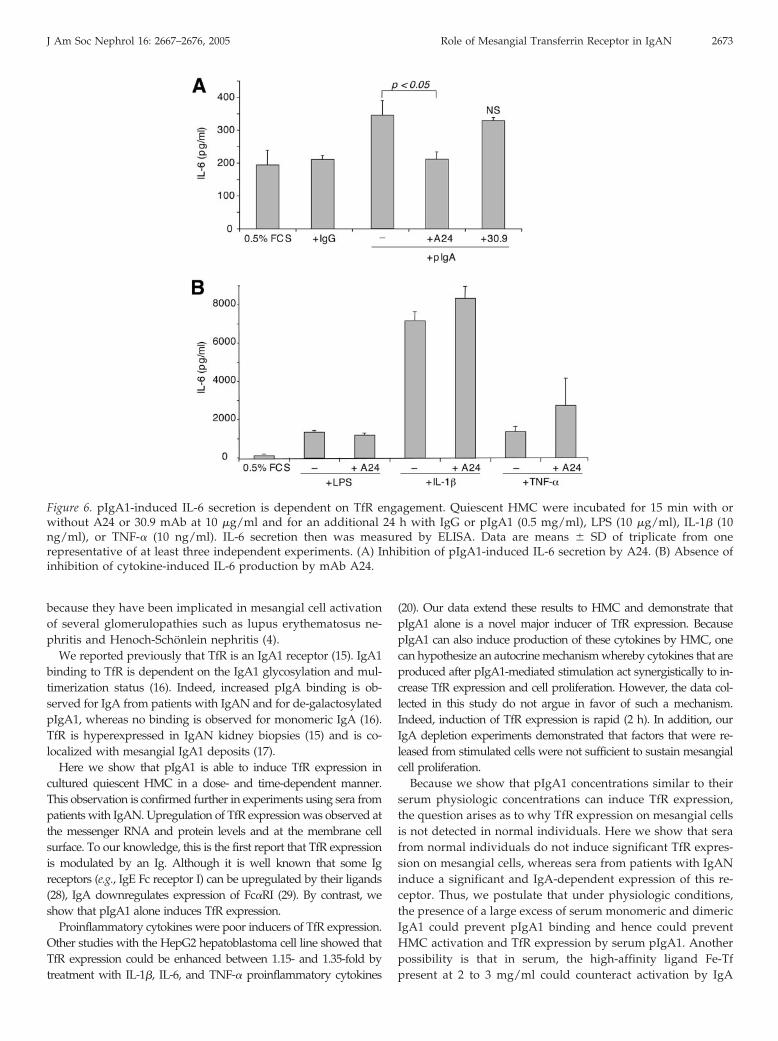
Because TfR triggering by pIgA1 induced HMC proliferation,we examined whether IL-6 secretion by pIgA1-stimulatedHMC could be dependent on TfR. For this purpose, serum-starved HMC (cultured in 0.5% FCS) were incubated withpIgA1 in the presence or absence of anti-TfR mAb A24. Underthese conditions, the level of IL-6 production was approxi-mately 350 pg/ml, a concentration that might be lower thanthat found under pathologic situations in the confined localmesangial environment. A24 but not the control isotype (30.9)inhibited pIgA1-induced IL-6 secretion by HMC (Figure 6A).No inhibition by A24 of cytokine-induced IL-6 production was
observed (Figure 6B). Therefore, TfR engagement by pIgA1induces IL-6 production by HMC.
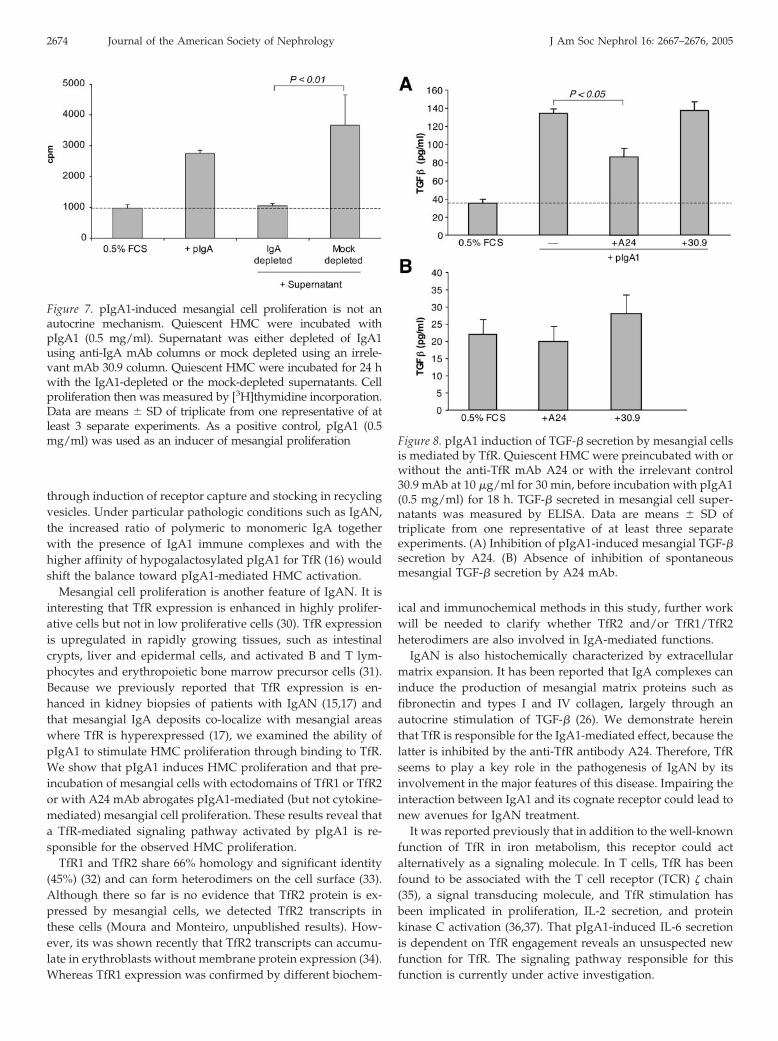
IL-6 production could act as a feedback loop for mesangial cellproliferation after pIgA1-mediated activation. For further shed-ding light on the mechanism that leads to mesangial cell prolifer-ation after activation initiated by IgA1, cells first were activated bypIgA1 and cell supernatant was depleted using an anti-IgA affin-ity column. This IgA-depleted but not mock-depleted supernatantwas no longer capable to promote mesangial cell proliferation(Figure 7). Therefore, the sustained presence of pIgA1 is requiredto support mesangial cell proliferation, indicating a direct ratherthan an autocrine mechanism.
TfR Triggering by pIgA1 Induces TGF-� Secretion by HMCIn addition to cell proliferation, IgA-stimulated mesangial cells
secrete profibrogenic factors such as TGF-� that act in an autocrinemanner and that are important in IgAN physiopathology (26). Toexamine whether involvement of IgA1-dependent TfR engage-ment is restricted to IL-6 secretion and cell proliferation or can also
Figure 4. A24 blocks pIgA1-induced HMC proliferation. Quiescent HMC were incubated for 15 min with or without A24 or 30.9mAb at 10 �g/ml and for an additional 24 h with or without pIgA1 (0.5 mg/ml), LPS (10 �g/ml), IL-1� (10 ng/ml), or TNF-� (10ng/ml). Cell proliferation then was measured by [3H]thymidine incorporation. Data are means � SD of triplicate from one of atleast three separate experiments. (A) Inhibition of pIgA1-induced HMC proliferation by A24. The number above the third bar isthe percentage of inhibition of pIgA1-induced proliferation over control. (B) Absence of inhibition of spontaneous HMCproliferation by mAb A24. (C) Absence of inhibition of cytokine-induced HMC proliferation by mAb A24.
J Am Soc Nephrol 16: 2667–2676, 2005 Role of Mesangial Transferrin Receptor in IgAN 2671
include matrix expansion, we evaluated TGF-� production. Asexpected, pIgA1 induced TGF-� production by mesangial cells(Figure 8). However, this production was inhibited by preincuba-tion of mesangial cells with A24 but not with a control isotype(Figure 8). By contrast, the spontaneous secretion of TGF-� wasnot affected by A24. Therefore, TfR is involved in both mesangialcell proliferation and secretion of profibrogenic factors that lead tomatrix expansion.
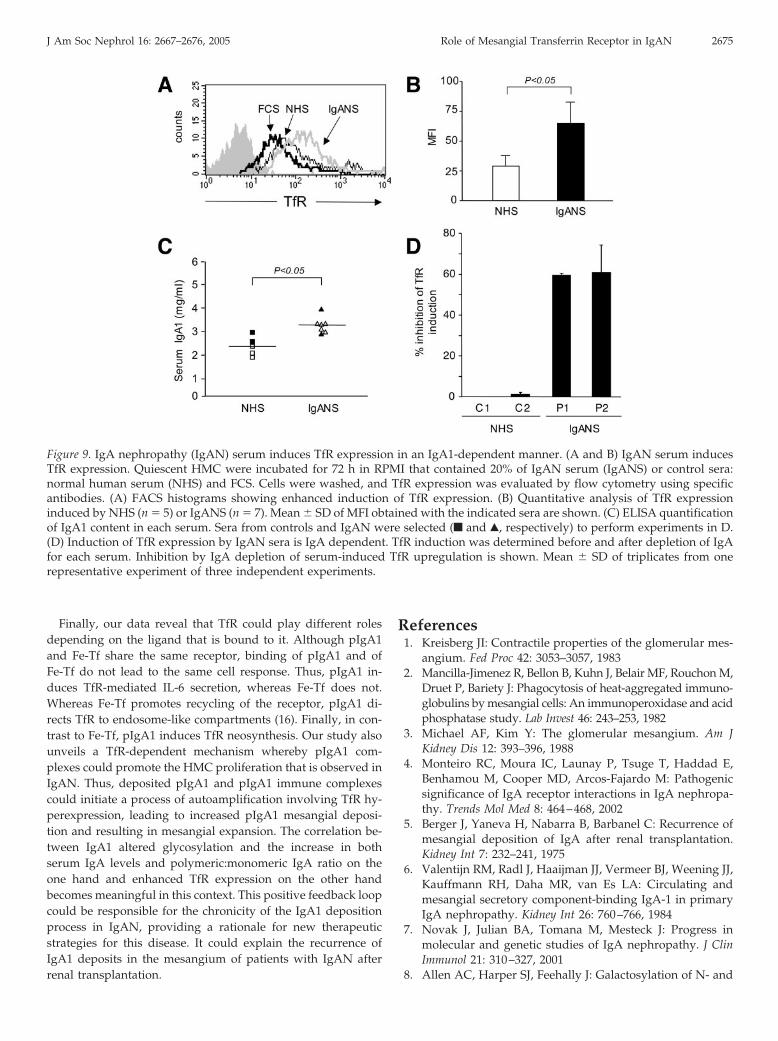
IgAN but not Normal Serum Promotes IgA-Dependent TfRExpression on HMC
Although purified pIgA1 can induce TfR expression in vitro,mesangial TfR expression is not observed in biopsies of normalkidney. However, in patients with IgAN, TfR expression isfound in mesangial areas and is co-localized with IgA deposits(17). Therefore, experiments were designed to compare thecapacity of IgA in normal and in IgAN serum to induce TfRexpression on HMC. As shown in Figure 9, A and B, serumfrom patients with IgAN strongly induced expression of TfR ascompared with serum from normal individuals. Because IgANis characterized by higher concentrations of serum IgA (Figure9C) (4), we selected sera from patients with IgAN that con-tained IgA concentrations that were both comparable to andhigher than those from normal individuals. These sera weredepleted of their IgA1 content by jacalin affinity columns andwere examined for their residual capacity to induce TfR expres-
sion. Only sera from patients with IgAN demonstrated anability to induce an IgA-dependent TfR expression (Figure 9D).However, there was also a residual capacity to induce TfRexpression observed in both normal and IgA-depleted IgANsera that could be due to the presence of cytokines or othersoluble factors. The difference between normal and IgAN se-rum in their ability to induce TfR synthesis is not due todifferences in IgA concentrations. Rather, in a circulating envi-ronment, normal pIgA1 is prevented from inducing TfR expres-sion. In a pathologic situation such as IgAN, in which IgAglycosylation is altered and polymeric:monomeric IgA ratio isenhanced, serum factors are no longer able to prevent inductionof TfR expression by pIgA1.
DiscussionIgAN is an immune complex–mediated glomerulonephritis
characterized by mesangial IgA1 deposition and mesangial cellproliferation. Patients with IgAN often have elevated IgA se-rum levels as well as enhanced polymeric:monomeric IgA ratio(6). Acid elution of IgAN biopsies shows that predominantlypolymeric forms of IgA1 are involved in IgA deposits (27). Thecorrelation between the presence of high molecular weightforms of IgA in serum and in renal biopsies from patientscorroborates the importance of IgA complexes in the disease.The pathogenic role of Ig complexes is not restricted to IgAN
Figure 5. Soluble TfR1 and 2 block pIgA1-induced HMC proliferation. Quiescent HMC were preincubated for 15 min with orwithout 50 �g/ml soluble TfR1 or TfR2 (or soluble CD89 as a control) before addition or not of pIgA1 (0.5 mg/ml) in the culturefor another 24 h. Proliferation of the cells then was measured by [3H]thymidine incorporation. Data are means � SD of triplicatefrom one of at least three separate experiments. (A) Inhibition of pIgA1-induced HMC proliferation by soluble receptors.Significance over cells that were treated with pIgA1 in the absence of soluble receptors is shown above the corresponding bars.(B) Absence of inhibition of spontaneous HMC proliferation by TfR ectodomains.
2672 Journal of the American Society of Nephrology J Am Soc Nephrol 16: 2667–2676, 2005
because they have been implicated in mesangial cell activationof several glomerulopathies such as lupus erythematosus ne-phritis and Henoch-Schonlein nephritis (4).
We reported previously that TfR is an IgA1 receptor (15). IgA1binding to TfR is dependent on the IgA1 glycosylation and mul-timerization status (16). Indeed, increased pIgA binding is ob-served for IgA from patients with IgAN and for de-galactosylatedpIgA1, whereas no binding is observed for monomeric IgA (16).TfR is hyperexpressed in IgAN kidney biopsies (15) and is co-localized with mesangial IgA1 deposits (17).
Here we show that pIgA1 is able to induce TfR expression incultured quiescent HMC in a dose- and time-dependent manner.This observation is confirmed further in experiments using sera frompatients with IgAN. Upregulation of TfR expression was observed atthe messenger RNA and protein levels and at the membrane cellsurface. To our knowledge, this is the first report that TfR expressionis modulated by an Ig. Although it is well known that some Igreceptors (e.g., IgE Fc receptor I) can be upregulated by their ligands(28), IgA downregulates expression of Fc�RI (29). By contrast, weshow that pIgA1 alone induces TfR expression.
Proinflammatory cytokines were poor inducers of TfR expression.Other studies with the HepG2 hepatoblastoma cell line showed thatTfR expression could be enhanced between 1.15- and 1.35-fold bytreatment with IL-1�, IL-6, and TNF-� proinflammatory cytokines
(20). Our data extend these results to HMC and demonstrate thatpIgA1 alone is a novel major inducer of TfR expression. BecausepIgA1 can also induce production of these cytokines by HMC, onecan hypothesize an autocrine mechanism whereby cytokines that areproduced after pIgA1-mediated stimulation act synergistically to in-crease TfR expression and cell proliferation. However, the data col-lected in this study do not argue in favor of such a mechanism.Indeed, induction of TfR expression is rapid (2 h). In addition, ourIgA depletion experiments demonstrated that factors that were re-leased from stimulated cells were not sufficient to sustain mesangialcell proliferation.
Because we show that pIgA1 concentrations similar to theirserum physiologic concentrations can induce TfR expression,the question arises as to why TfR expression on mesangial cellsis not detected in normal individuals. Here we show that serafrom normal individuals do not induce significant TfR expres-sion on mesangial cells, whereas sera from patients with IgANinduce a significant and IgA-dependent expression of this re-ceptor. Thus, we postulate that under physiologic conditions,the presence of a large excess of serum monomeric and dimericIgA1 could prevent pIgA1 binding and hence could preventHMC activation and TfR expression by serum pIgA1. Anotherpossibility is that in serum, the high-affinity ligand Fe-Tfpresent at 2 to 3 mg/ml could counteract activation by IgA
Figure 6. pIgA1-induced IL-6 secretion is dependent on TfR engagement. Quiescent HMC were incubated for 15 min with orwithout A24 or 30.9 mAb at 10 �g/ml and for an additional 24 h with IgG or pIgA1 (0.5 mg/ml), LPS (10 �g/ml), IL-1� (10ng/ml), or TNF-� (10 ng/ml). IL-6 secretion then was measured by ELISA. Data are means � SD of triplicate from onerepresentative of at least three independent experiments. (A) Inhibition of pIgA1-induced IL-6 secretion by A24. (B) Absence ofinhibition of cytokine-induced IL-6 production by mAb A24.
J Am Soc Nephrol 16: 2667–2676, 2005 Role of Mesangial Transferrin Receptor in IgAN 2673
through induction of receptor capture and stocking in recyclingvesicles. Under particular pathologic conditions such as IgAN,the increased ratio of polymeric to monomeric IgA togetherwith the presence of IgA1 immune complexes and with thehigher affinity of hypogalactosylated pIgA1 for TfR (16) wouldshift the balance toward pIgA1-mediated HMC activation.
Mesangial cell proliferation is another feature of IgAN. It isinteresting that TfR expression is enhanced in highly prolifer-ative cells but not in low proliferative cells (30). TfR expressionis upregulated in rapidly growing tissues, such as intestinalcrypts, liver and epidermal cells, and activated B and T lym-phocytes and erythropoietic bone marrow precursor cells (31).Because we previously reported that TfR expression is en-hanced in kidney biopsies of patients with IgAN (15,17) andthat mesangial IgA deposits co-localize with mesangial areaswhere TfR is hyperexpressed (17), we examined the ability ofpIgA1 to stimulate HMC proliferation through binding to TfR.We show that pIgA1 induces HMC proliferation and that pre-incubation of mesangial cells with ectodomains of TfR1 or TfR2or with A24 mAb abrogates pIgA1-mediated (but not cytokine-mediated) mesangial cell proliferation. These results reveal thata TfR-mediated signaling pathway activated by pIgA1 is re-sponsible for the observed HMC proliferation.
TfR1 and TfR2 share 66% homology and significant identity(45%) (32) and can form heterodimers on the cell surface (33).Although there so far is no evidence that TfR2 protein is ex-pressed by mesangial cells, we detected TfR2 transcripts inthese cells (Moura and Monteiro, unpublished results). How-ever, its was shown recently that TfR2 transcripts can accumu-late in erythroblasts without membrane protein expression (34).Whereas TfR1 expression was confirmed by different biochem-
ical and immunochemical methods in this study, further workwill be needed to clarify whether TfR2 and/or TfR1/TfR2heterodimers are also involved in IgA-mediated functions.
IgAN is also histochemically characterized by extracellularmatrix expansion. It has been reported that IgA complexes caninduce the production of mesangial matrix proteins such asfibronectin and types I and IV collagen, largely through anautocrine stimulation of TGF-� (26). We demonstrate hereinthat TfR is responsible for the IgA1-mediated effect, because thelatter is inhibited by the anti-TfR antibody A24. Therefore, TfRseems to play a key role in the pathogenesis of IgAN by itsinvolvement in the major features of this disease. Impairing theinteraction between IgA1 and its cognate receptor could lead tonew avenues for IgAN treatment.
It was reported previously that in addition to the well-knownfunction of TfR in iron metabolism, this receptor could actalternatively as a signaling molecule. In T cells, TfR has beenfound to be associated with the T cell receptor (TCR) � chain(35), a signal transducing molecule, and TfR stimulation hasbeen implicated in proliferation, IL-2 secretion, and proteinkinase C activation (36,37). That pIgA1-induced IL-6 secretionis dependent on TfR engagement reveals an unsuspected newfunction for TfR. The signaling pathway responsible for thisfunction is currently under active investigation.
Figure 7. pIgA1-induced mesangial cell proliferation is not anautocrine mechanism. Quiescent HMC were incubated withpIgA1 (0.5 mg/ml). Supernatant was either depleted of IgA1using anti-IgA mAb columns or mock depleted using an irrele-vant mAb 30.9 column. Quiescent HMC were incubated for 24 hwith the IgA1-depleted or the mock-depleted supernatants. Cellproliferation then was measured by [3H]thymidine incorporation.Data are means � SD of triplicate from one representative of atleast 3 separate experiments. As a positive control, pIgA1 (0.5mg/ml) was used as an inducer of mesangial proliferation Figure 8. pIgA1 induction of TGF-� secretion by mesangial cells
is mediated by TfR. Quiescent HMC were preincubated with orwithout the anti-TfR mAb A24 or with the irrelevant control30.9 mAb at 10 �g/ml for 30 min, before incubation with pIgA1(0.5 mg/ml) for 18 h. TGF-� secreted in mesangial cell super-natants was measured by ELISA. Data are means � SD oftriplicate from one representative of at least three separateexperiments. (A) Inhibition of pIgA1-induced mesangial TGF-�secretion by A24. (B) Absence of inhibition of spontaneousmesangial TGF-� secretion by A24 mAb.
2674 Journal of the American Society of Nephrology J Am Soc Nephrol 16: 2667–2676, 2005
Finally, our data reveal that TfR could play different rolesdepending on the ligand that is bound to it. Although pIgA1and Fe-Tf share the same receptor, binding of pIgA1 and ofFe-Tf do not lead to the same cell response. Thus, pIgA1 in-duces TfR-mediated IL-6 secretion, whereas Fe-Tf does not.Whereas Fe-Tf promotes recycling of the receptor, pIgA1 di-rects TfR to endosome-like compartments (16). Finally, in con-trast to Fe-Tf, pIgA1 induces TfR neosynthesis. Our study alsounveils a TfR-dependent mechanism whereby pIgA1 com-plexes could promote the HMC proliferation that is observed inIgAN. Thus, deposited pIgA1 and pIgA1 immune complexescould initiate a process of autoamplification involving TfR hy-perexpression, leading to increased pIgA1 mesangial deposi-tion and resulting in mesangial expansion. The correlation be-tween IgA1 altered glycosylation and the increase in bothserum IgA levels and polymeric:monomeric IgA ratio on theone hand and enhanced TfR expression on the other handbecomes meaningful in this context. This positive feedback loopcould be responsible for the chronicity of the IgA1 depositionprocess in IgAN, providing a rationale for new therapeuticstrategies for this disease. It could explain the recurrence ofIgA1 deposits in the mesangium of patients with IgAN afterrenal transplantation.
References1. Kreisberg JI: Contractile properties of the glomerular mes-
angium. Fed Proc 42: 3053–3057, 19832. Mancilla-Jimenez R, Bellon B, Kuhn J, Belair MF, Rouchon M,
Druet P, Bariety J: Phagocytosis of heat-aggregated immuno-globulins by mesangial cells: An immunoperoxidase and acidphosphatase study. Lab Invest 46: 243–253, 1982
3. Michael AF, Kim Y: The glomerular mesangium. Am JKidney Dis 12: 393–396, 1988
4. Monteiro RC, Moura IC, Launay P, Tsuge T, Haddad E,Benhamou M, Cooper MD, Arcos-Fajardo M: Pathogenicsignificance of IgA receptor interactions in IgA nephropa-thy. Trends Mol Med 8: 464–468, 2002
5. Berger J, Yaneva H, Nabarra B, Barbanel C: Recurrence ofmesangial deposition of IgA after renal transplantation.Kidney Int 7: 232–241, 1975
6. Valentijn RM, Radl J, Haaijman JJ, Vermeer BJ, Weening JJ,Kauffmann RH, Daha MR, van Es LA: Circulating andmesangial secretory component-binding IgA-1 in primaryIgA nephropathy. Kidney Int 26: 760–766, 1984
7. Novak J, Julian BA, Tomana M, Mesteck J: Progress inmolecular and genetic studies of IgA nephropathy. J ClinImmunol 21: 310–327, 2001
8. Allen AC, Harper SJ, Feehally J: Galactosylation of N- and
Figure 9. IgA nephropathy (IgAN) serum induces TfR expression in an IgA1-dependent manner. (A and B) IgAN serum inducesTfR expression. Quiescent HMC were incubated for 72 h in RPMI that contained 20% of IgAN serum (IgANS) or control sera:normal human serum (NHS) and FCS. Cells were washed, and TfR expression was evaluated by flow cytometry using specificantibodies. (A) FACS histograms showing enhanced induction of TfR expression. (B) Quantitative analysis of TfR expressioninduced by NHS (n � 5) or IgANS (n � 7). Mean � SD of MFI obtained with the indicated sera are shown. (C) ELISA quantificationof IgA1 content in each serum. Sera from controls and IgAN were selected (f and Œ, respectively) to perform experiments in D.(D) Induction of TfR expression by IgAN sera is IgA dependent. TfR induction was determined before and after depletion of IgAfor each serum. Inhibition by IgA depletion of serum-induced TfR upregulation is shown. Mean � SD of triplicates from onerepresentative experiment of three independent experiments.
J Am Soc Nephrol 16: 2667–2676, 2005 Role of Mesangial Transferrin Receptor in IgAN 2675

O-linked carbohydrate moieties of IgA1 and IgG in IgAnephropathy. Clin Exp Immunol 100: 470–474, 1995
9. Hiki Y, Odani H, Takahashi M, Yasuda Y, Nishimoto A,Iwase H, Shinzato T, Kobayashi Y, Maeda K: Mass spec-trometry proves under-O-glycosylation of glomerularIgA1 in IgA nephropathy. Kidney Int 59: 1077–1085, 2001
10. Novak J, Tomana M, Kilian M, Coward L, Kulhavy R, BarnesS, Mestecky J: Heterogeneity of O-glycosylation in the hingeregion of human IgA1. Mol Immunol 37: 1047–1056, 2000
11. Allen AC, Bailey EM, Brenchley PE, Buck KS, Barratt J,Feehally J: Mesangial IgA1 in IgA nephropathy exhibitsaberrant O-glycosylation: Observations in three patients.Kidney Int 60: 969–973, 2001
12. Gomez-Guerrero C, Duque N, Egido J: Stimulation ofFc(alpha) receptors induces tyrosine phosphorylation ofphospholipase C-gamma (1), phosphatidylinositol phos-phate hydrolysis, and Ca2� mobilization in rat and humanmesangial cells. J Immunol 156: 4369–4376, 1996
13. Diven SC, Caflisch CR, Hammond DK, Weigel PH, Oka JA,Goldblum RM: IgA induced activation of human mesangialcells: Independent of FcalphaR1 (CD 89). Kidney Int 54: 837–847,1998
14. Gomez-Guerrero C, Lopez-Armada MJ, Gonzalez E, Egido J:Soluble IgA and IgG aggregates are catabolized by culturedrat mesangial cells and induce production of TNF-alpha andIL-6, and proliferation. J Immunol 153: 5247–5255, 1994
15. Moura IC, Centelles MN, Arcos-Fajardo M, Malheiros DM,Collawn JF, Cooper MD, Monteiro RC: Identification of thetransferrin receptor as a novel immunoglobulin (Ig)A1 re-ceptor and its enhanced expression on mesangial cells inIgA nephropathy. J Exp Med 194: 417–425, 2001
16. Moura IC, Arcos-Fajardo M, Sadaka C, Leroy V, BenhamouM, Novak J, Vrtovsnik F, Haddad E, Chintalacharuvu KR,Monteiro RC: Glycosylation and size of IgA1 are essentialfor interaction with mesangial transferrin receptor in IgAnephropathy. J Am Soc Nephrol 15: 622–634, 2004
17. Haddad E, Moura IC, Arcos-Fajardo M, Macher MA, BaudouinV, Alberti C, Loirat C, Monteiro RC, Peuchmaur M: Enhancedexpression of the CD71 mesangial IgA1 receptor in Berger dis-ease and Henoch-Schonlein nephritis: Association betweenCD71 expression and IgA deposits. J Am Soc Nephrol 14: 327–337,2003
18. Pastorelli C, Veiga J, Charles N, Voignier E, Moussu H, Mon-teiro R, Benhamou M: Phospholipid scramblase, a new effec-tor of FcepsilonRI signaling in mast cells. Mol Immunol 38:1235, 2002
19. Lebron JA, Bennett MJ, Vaughn DE, Chirino AJ, Snow PM,Mintier GA, Feder JN, Bjorkman PJ: Crystal structure of thehemochromatosis protein HFE and characterization of itsinteraction with transferrin receptor. Cell 93: 111–123, 1998
20. Hirayama M, Kohgo Y, Kondo H, Shintani N, Fujikawa K,Sasaki K, Kato J, Niitsu Y: Regulation of iron metabolism inHepG2 cells: A possible role for cytokines in the hepaticdeposition of iron. Hepatology 18: 874–880, 1993
21. Tsuji Y, Moran E, Torti SV, Torti FM: Transcriptional reg-ulation of the mouse ferritin H gene. Involvement of p300/CBP adaptor proteins in FER-1 enhancer activity. J BiolChem 274: 7501–7507, 1999
22. Kotamraju S, Chitambar CR, Kalivendi SV, Joseph J, Kaly-
anaraman B: Transferrin receptor-dependent iron uptake isresponsible for doxorubicin-mediated apoptosis in endo-thelial cells: Role of oxidant-induced iron signaling in ap-optosis. J Biol Chem 277: 17179–17187, 2002
23. Moura IC, Lepelletier Y, Arnulf B, England P, Baude C, Beau-mont C, Bazarbachi A, Benhamou M, Monteiro RC, Hermine O:A neutralizing monoclonal antibody (mAb A24) directedagainst the transferrin receptor induces apoptosis of tumor Tlymphocytes from ATL patients. Blood 103: 1838–1845, 2004
24. Suematsu S, Matsuda T, Aozasa K, Akira S, Nakano N,Ohno S, Miyazaki J, Yamamura K, Hirano T, Kishimoto T:IgG1 plasmacytosis in interleukin 6 transgenic mice. ProcNatl Acad Sci U S A 86: 7547–7551, 1989
25. Harada K, Akai Y, Kurumatani N, Iwano M, Saito Y: Prognosticvalue of urinary interleukin 6 in patients with IgA nephropathy:An 8-year follow-up study. Nephron 92: 824–826, 2002
26. Lopez-Armada MJ, Gomez-Guerrero C, Egido J: Receptorsfor immune complexes activate gene expression and synthe-sis of matrix proteins in cultured rat and human mesangialcells: Role of TGF-beta. J Immunol 157: 2136–2142, 1996
27. Monteiro RC, Halbwachs-Mecarelli L, Roque-Barreira MC,Noel LH, Berger J, Lesavre P: Charge and size of mesangialIgA in IgA nephropathy. Kidney Int 28: 666–671, 1985
28. Furuichi K, Rivera J, Isersky C: The receptor for immunoglobu-lin E on rat basophilic leukemia cells: Effect of ligand binding onreceptor expression. Proc Natl Acad Sci U S A 82: 1522–1525, 1985
29. Grossetete B, Launay P, Lehuen A, Jungers P, Bach JF, MonteiroRC: Down-regulation of Fcalpha receptors on blood cells of IgAnephropathy patients: Evidence for a negative regulatory role ofserum IgA. Kidney Int 53: 1321–1335, 1998
30. Trowbridge IS, Shackelford DA: Structure and function oftransferrin receptors and their relationship to cell growth.Biochem Soc Symp 51: 117–129, 1986
31. Greene WC, Robb RJ: Receptors for T-cell growth factor:Structure, function and expression on normal and neoplas-tic cells. Contemp Top Mol Immunol 10: 1–34, 1985
32. Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S,Gombart AF, Koeffler HP: Molecular cloning of transferrinreceptor 2. A new member of the transferrin receptor-likefamily. J Biol Chem 274: 20826–20832, 1999
33. Vogt TM, Blackwell AD, Giannetti AM, Bjorkman PJ, EnnsCA: Heterotypic interactions between transferrin receptorand transferrin receptor 2. Blood 101: 2008–2014, 2003
34. Calzolari A, Deaglio S, Sposi NM, Petrucci E, Morsilli O,Gabbianelli M, Malavasi F, Peschle C, Testa U: Transferrinreceptor 2 protein is not expressed in normal erythroidcells. Biochem J 381: 629–634, 2004
35. Salmeron A, Borroto A, Fresno M, Crumpton MJ, Ley SC,Alarcon B: Transferrin receptor induces tyrosine phos-phorylation in T cells and is physically associated with theTCR zeta-chain. J Immunol 154: 1675–1683, 1995
36. Manger B, Weiss A, Hardy KJ, Stobo JD: A transferrin receptorantibody represents one signal for the induction of IL 2 produc-tion by a human T cell line. J Immunol 136: 532–538, 1986
37. Cano E, Pizarro A, Redondo JM, Sanchez-Madrid F, Berna-beu C, Fresno M: Induction of T cell activation by mono-clonal antibodies specific for the transferrin receptor. EurJ Immunol 20: 765–770, 1990
2676 Journal of the American Society of Nephrology J Am Soc Nephrol 16: 2667–2676, 2005




















