
Physical and chemical changes of polystyrene nanospheres irradiated with laser
Upload
independentCategory
view
1download
0
End-Group Cleavage in MALDI of ATRP-Made Polystyrene: A Silver-Catalyzed Reaction during Sample PreparationAura Tintaru,† Christophe Chendo,‡ Trang N. T. Phan,† Marion Rollet,† Laurent Giordano,§
Stephane Viel,† Didier Gigmes,† and Laurence Charles*,†
†Institut de Chimie Radicalaire ICR, Aix-Marseille Universite−CNRS, UMR 7273, F-13397 Marseille, France‡Federation des Sciences Chimiques de Marseille, Aix-Marseille Universite−CNRS, Spectropole, FR 1739, F-13397 Marseille, France§Institut des Sciences Moleculaires de Marseille, Aix-Marseille Universite−CNRS, UMR 7313, & Ecole Centrale de Marseille,F-13397 Marseille, France
*S Supporting Information
ABSTRACT: Cleavage of the labile halide termination uponmatrix-assisted laser desorption/ionization (MALDI) hasalways been reported as a major concern in mass analysis ofpolystyrene prepared by atom transfer radical polymerization(ATRP). By studying this issue using nuclear magneticresonance (NMR) and electrospray ionization−mass spec-trometry, we evidence here that the ionization step is notinvolved in this deleterious process. Instead, removal of thehalogen was shown to readily occur upon interaction of thesilver salt (AgTFA) used as the cationizing agent in massspectrometry, either in solution or in the solid-state whenperforming solvent-free sample preparation. In solution, thissilver-induced reaction mostly consists of a nucleophilicsubstitution, leading to polystyrene molecules holding differentterminations, depending on relative nucleophilicity of speciespresent in the liquid-phase solution composition. In chloro-form supplemented with AgTFA, trifluoroacetate-terminatedPS were evidenced in ESI-MS spectra but experienced end-group cleavage in MALDI. In contrast, the major methoxy-terminatedPS macromolecules formed when the silver-catalyzed nucleophilic substitution was performed in methanol were generated asintact gas-phase ions using both ionization techniques. This controlled and fast modification could hence be advantageously usedas a rapid sample pretreatment for safe MALDI mass analysis of ATRP-made polystyrene.
Since the availability of soft ionization methods, massspectrometry (MS) has been increasingly employed for
synthetic polymer structural characterization.1−3 Transfer ofintact species into gas-phase ions is indeed mandatory forpolymer end-group analysis, since MS information about chainterminations mainly rely on the m/z values measured forpolymer adducts. As compared to electrospray ionization(ESI),4 the limited softness of matrix-assisted laser desorp-tion/ionization (MALDI)5,6 has often been pointed out as amajor issue for mass analysis of polymeric chains carrying labileend-groups.7 Such polymers are usually produced by controlledradical polymerization (CRP) processes,8 which are all basedon the low dissociation energy of the chemical bond betweenthe last monomer unit and the terminating group. As a result, ifmacromolecules are activated while being ionized, this chemicalbond is prone to cleavage, either in the source or during flighttime, and ions measured in the mass spectra no longer reflectthe original chain composition.However, using low laser fluence in MALDI, many CRP
polymers could be produced as intact gas-phase ions,
independent of the synthesis process from which they arose.9
Interestingly, when reported to occur during the ionizationprocess, structural alteration does not only depend on thenature of the labile end-group but also on the nature of thepolymer. This is the case of polystyrenes (PS) produced inatom transfer radical polymerization (ATRP), rarely reported asMALDI intact species, in great contrast to other ATRP-madepolymers (such as, for example, poly(acrylate)s).10−12 The onlystudy providing partial mass spectral evidence of intact silveradducts of PS-Br displayed poorly resolved signals, confined toa minor distribution with regard to signals corresponding to PSholding an unsaturated termination, arising from dehydrohalo-genation of the chain terminal group.13 Mechanistically, ATRPis based on a fast, dynamic equilibrium established between thedormant chains and active radical species, with transition-metal
Received: February 4, 2013Accepted: May 10, 2013Published: May 10, 2013
Article
pubs.acs.org/ac
© 2013 American Chemical Society 5454 dx.doi.org/10.1021/ac400375d | Anal. Chem. 2013, 85, 5454−5462

complexes acting as reversible halogen atom transfer reagent.14
The end-groups of ATRP polymers are thus defined by the RXalkyl halide (or a so-called pseudo-halide when X = SCN)initiator used, with R and the (pseudo)halide, respectively,expected at the α and ω position. Using a silver salt to promoteoptimal ionization of the PS chains, PS−CHCH−Phdegradation products were systematically observed as themajor species in MALDI mass spectra of ATRP-made PS,regardless of the matrix used for sample preparation(dithranol,15−19 2,5-dihydroxybenzoic acid,16,20 trans-2-[3-(4-tert-butyl-phenyl)-2-methyl-2-propenylidene]malononitrile(DCTB),21 or all-trans retinoic acid22,23). It was thus concludedthat the halogen termination was less stable upon laserionization when linked to PS, compared to other polymersprepared by ATRP,10 leading to the formation of a conjugated(hence, stable) terminal double bond. Some authors assumed acatalytic role of silver salt in the dehydrohalogenation processduring MALDI.13,17 A loss of HSCN was also reported to occurto a high extent from PS prepared by ATRP involving apseudohalide precursor, using dithranol and AgTFA to prepareMALDI samples.24 However, very low amounts of intact [PS−SCN+Ag]+ could be detected, suggesting that the eliminationof thiocyanate was not complete during MALDI, which wasconsistent with the bond strength argument (C−SCN > C−Br).25
Intriguingly, although silver adduction remains the bestcationization mode for polystyrenes,1 successful production ofsilverated ATRP PS has never been reported using electrospray.This result is particularly surprising, because the softness of ESIwas previously demonstrated for polymers with labile end-groups, such as nitroxides, allowing the production of intactgas-phase ions while MALDI did not.26−28 Intact PS−Br couldactually be electrosprayed only when using sodium as thecationization agent and providing that experimental settingshave been carefully optimized to prevent in-source dissocia-tion,29 while experiments performed at very high cone voltagesmainly conducted to the detection of dehydrohalogenated PSadducted with sodium.18,29 In contrast to ESI, MALDI of PS−Br prepared with sodium salts did not allow intact sodiatedoligomers to be observed.10,30−35 This end-group degradationwas attributed to the MALDI process and, more particularly, tothe usually high laser fluence required to circumvent the lowaffinity of polystyrene molecules toward alkali, leading to highlyactivated gas-phase ions prone to spontaneous dissociation.By combining nuclear magnetic resonance (NMR) and mass
spectrometry experiments to study an ATRP-made PS sampleat the different stages of a typical MALDI sample preparation,we evidence here that the halogen terminal group is readilyremoved from PS molecules upon mixing with a silver salt. Inother words, the MALDI process is not responsible for PS−Brend-group degradation but oligomers are actually chemicallymodified, prior to the ionization step, during a silver-catalyzedreaction.
■ EXPERIMENTAL SECTION1. Chemicals. Styrene (99%), ethyl 2-bromoisobutyrate
(98%), copper(I) bromide (98%), and N,N,N′,N′′,N′′-pentam-ethyldiethylenetriamine (99%) (PMDETA), as well as tolueneand ethanol, used in ATRP were all obtained from Sigma−Aldrich (St. Louis, MO). Deuterated chloroform (CDCl3,99.8%) and deuterated methanol (CD3OD, 99.8%) were fromEuriso-Top (Saint-Aubin, France). Silver trifluoroacetate(AgTFA (98.0%)), silver nitrate (AgNO3 (≥99.0%)), and
sodium chloride (NaCl (99.5%)), as well as trans-2-[3-(4-tert-butyl-phenyl)-2-methyl-2-propenylidene]malononitrile(DCTB, ≥ 98.0%), which was used as the MALDI matrix, andPS and polypropylene glycol (PPG) standards, which wereused for mass calibration, were purchased from Sigma−Aldrich.High-performance liquid chromatography (HPLC)-gradetetrahydrofuran (THF) and methanol were obtained fromSDS (Peypin, France). All chemicals were used as received,without any further purification.
2. ATRP Synthesis of PS−Br. A three-neck round-bottomflask was charged with styrene, copper bromide, ethyl 2-bromoisobutyrate, and toluene. The system was fitted with awater-cooled condenser and rubber septum; the solution wasthen degassed under stirring with argon for 15 min. PMDETAwas mixed with 1 mL of toluene and then degassed by bubblingargon for 15 min. This ligand solution was transferred at roomtemperature, via cannula, to a monomer solution that wasprepurged with argon. The monomer concentration in thismixture was 60 wt %. The round-bottom flask was thenimmersed in a thermostatted oil bath that was heated at 105 °C.At the end of the polymerization, the reaction mixture wasquenched in cold water, dissolved with toluene, and filteredthrough a basic alumina column. The catalyst-free solution wasthen poured into a large excess of ethanol to precipitate PS.The solid was filtered and dried under vacuum to a constantmass. Two polystyrenes were prepared using an initiator/CuBr/PMDETA/monomer molar ratio of 1/1/1.5/25 and 1/2/1.5/144 and an initial initiator concentration of 187 mM and35 mM, respectively. The reaction time of these twopolymerizations was 45 and 120 min, respectively. Thenumber-average molecular weight (Mn) and dispersity (Ip) ofthe obtained PS were determined using size-exclusionchromatography (SEC). The used system was an EcoSEC(Tosoh, Japan) equipped with a PL Resipore Precolumn (4.6mm × 50 mm) and two PL Resipore columns (4.6 mm × 250mm) with gel particles 3 μm in diameter. These columns werethermostatted at 40 °C. The detection system was composed ofan UV/visible detector, operated at λ = 254 nm, and a dual-flowdifferential refractive index detector (both from Tosoh), and aviscometer (PSS, Model ETA2010). Measurements wereperformed in THF at a flow rate of 0.3 mL min−1. The Mnand Ip values derived from calibration curve based on PSstandards (ranging from 370 g mol−1 to 371 100 g mol−1) were1900 g mol−1 (Ip = 1.2) and 8000 g mol−1 (Ip = 1.1),respectively.
3. Nuclear Magnetic Resonance (NMR). NMR experi-ments were performed at 298 K on a Bruker Avance DRX 500NMR spectrometer (Karlsruhe, Germany) operating at 500.13MHz for the 1H Larmor frequency with a double-resonancebroadband fluorine observe (BBFO) probe head equipped witha z-magnetic-field gradient coil (maximum gradient strength ofGmax = 55 g cm−1). 1H and 19F diffusion-ordered NMRspectroscopy experiments were recorded using a double-stimulated echo pulse sequence incorporating bipolar gradientsand a longitudinal eddy current delay,36 as described else-where.37 In addition, so-called 1H diffusion-filtered NMRexperiments were recorded using the same pulse sequence(with a diffusion time of 100 ms and a gradient pulse durationand strength of 1.2 ms and 25 G cm−1, respectively). Suchexperiments allow the 1H signals due to low-molecular-weightcompounds (hence, exhibiting high translational molecularmobility) to be filtered out from the NMR spectrum, herebyevidencing preferentially the signals due to high-molecular-
Analytical Chemistry Article
dx.doi.org/10.1021/ac400375d | Anal. Chem. 2013, 85, 5454−54625455

weight compounds (see Figure S-1 in the SupportingInformation for an illustration with the PS-Br sample inCDCl3).
38 Importantly, because all the 1H NMR spectrareported in this study were obtained under these sameconditions, they will hereafter be simply referred to as 1Hspectra. Finally, 19F experiments were recorded with a standardone-pulse excitation pulse sequence (90° excitation pulse) with1H decoupling during signal acquisition (performed withWALTZ-16). 1H and 19F chemical shifts were externallyreferenced to TMS and CFCl3, respectively.4. Electrospray Mass Spectrometry. High-resolution
mass spectroscopy (MS) and tandem mass spectroscopy(MS/MS) experiments were performed using a QStar Elitemass spectrometer (Applied Biosystems SCIEX, Concord, ON,Canada) equipped with an electrospray ionization sourceoperated in the positive mode. The capillary voltage was set at+5500 V, and the cone voltage was set at +50 V. In this hybridinstrument, ions were measured using an orthogonalacceleration time-of-flight (oa-TOF) mass analyzer. A quadru-pole was used for selection of precursor ions to be furthersubmitted to collision-induced dissociation (CID) in MS/MSexperiments. In MS, accurate mass measurements wereperformed using reference ions from a PPG or a PS internalstandard. The precursor ion was used as the reference foraccurate measurements of product ions in MS/MS spectra. Inthis instrument, air was used as the nebulizing gas (10 psi)while nitrogen was used as the curtain gas (20 psi) as well asthe collision gas. Instrument control, data acquisition and dataprocessing of all experiments were achieved using Analystsoftware (QS 2.0) provided by Applied Biosystems. Samplesolutions were introduced in the ionization source at a 5 μLmin−1 flow rate, using a syringe pump.5. MALDI Mass Spectrometry. MALDI-TOF MS experi-
ments were carried out on a Bruker Autoflex I (BrukerDaltonics, Leipzig, Germany), equipped with a nitrogen laseremitting at 337 nm, a single-stage pulsed ion extraction sourceand dual microchannel plate detectors. Data acquisition wasperformed in reflectron mode. Positive-ion mode was used forall analyses with an accelerating voltage of 19 kV. The delaytime used in delayed extraction mode was optimized based onthe mass range of the polymer distributions and was generally∼300 ns. FlexControl software version 2.2 (Bruker Daltonics)was used for instrument control and data acquisition, andFlexAnalysis software version 2.2 (Bruker Daltonics) for dataprocessing. MALDI mass spectra result from averages over 100consecutive laser shots at a frequency of 10 Hz. All mass spectrawere acquired at threshold laser irradiance. External calibrationwas performed using PS standards.6. Sample Preparation. For NMR and ESI-MS analysis,
PS1900/AgTFA mixtures were prepared in a nearly 1:1 molarratio, with a slight excess of AgTFA (further reported as 1:1molar ratio, for the sake of clarity). A first-preparation protocolconsisted of mixing 500 μL of a 12.0 mg mL−1 PS1900 solutionin CDCl3 and 0.5 μL of a 1.55 g mL−1 AgTFA solution inCH3OH. This solution ([PS1900] ≈ 6.0 μmol mL−1 in CDCl3)was submitted to NMR analysis and, after dilution (1/10, v/v)with a methanol solution of AgTFA (0.155 mg mL−1), to ESI-MS analysis. A second type of solutions was obtained by mixing50 μL of the CDCl3 PS1900 solution (12.0 mg mL−1) with 450μL of a 0.155 mg mL−1 AgTFA solution in methanol (CD3ODor CH3OH). These latter methanol solutions ([PS1900] ≈ 0.6μmol mL−1) were subjected to both NMR and ESI-MS analysis
with no prior dilution. The same protocols were implementedwith NaCl as the salt.In solvent-based MALDI sample preparation, PS solutions
(in CDCl3 or CH3OH) were mixed with DCTB (in THF) andAgTFA (in CH3OH) at a matrix/polymer/salt molar ratio of1000:1:10 for PS1900 or 1000:1:100 for PS8000. PS standardsolutions used for mass calibration in MALDI were preparedusing the same protocol. MALDI samples were prepared usingthe dried droplet method, where a 1-μL aliquot of the liquidmixture was deposited onto the sample plate and allowed to air-dry. The solvent-free sample preparation39,40 consisted ofgrinding the matrix (130 mg) together with the polymer (21mg) and the salt (5 mg), that is a matrix/polymer/salt ratio of∼50:1:2, with a mortar and a pestle for ∼5 min at roomtemperature. A few grains of the solid mixture were applied tothe MALDI target and then pressed with a small spatula toform a thin film.
■ RESULTS AND DISCUSSION1. Evidence for Br Elimination in Solution. When
preparing MALDI samples using the dried droplet approach, aternary mixture of the matrix, the polymer and the cationizingagent has to be prepared in solution. To check whether mixingthe silver salt with the polymer in solution had any influence onthe integrity of the molecule halide end-group, 1H NMRspectra (see the Experimental Section) of the PS−Br (Mn =1900 g mol−1) sample were recorded in pure CDCl3 (Figure1a) and in a CDCl3 solution of AgTFA (Figure 1b), using apolymer/salt molar ratio of 1:1.The 1H spectral assignments shown in Figure 1 were
obtained by using two-dimensional (2D) correlation (1H−1HCOSY and 1H−13C HSQC) experiments (see Table S-1 in theSupporting Information). For this small-sized polymer, thepresence of the terminal bromine of PS−Br could be indirectlyevidenced in Figure 1a by the 1H signals observed at 4.5 ppm,
Figure 1. 1H NMR spectra (see the Experimental Section) of thePS1900 sample in CDCl3 (a) before and (b) after addition of AgTFA.Inset at the top of the image depicts the molecular structure of theATRP-made polystyrene (PS), along with the corresponding atomlabeling used for the spectral assignment.
Analytical Chemistry Article
dx.doi.org/10.1021/ac400375d | Anal. Chem. 2013, 85, 5454−54625456

which is a chemical shift characteristic of methine protons in α-position to a halogen atom. In contrast, these signals were nolonger observed when PS−Br was in solution together with thesilver salt (Figure 1b), while a new resonance was detected at∼5.5 ppm (all other signals being unchanged). This new 1Hchemical shift value is characteristic of a proton in a methinegroup linked to an electron-withdrawing substituent. Formationof this new species, together with the white insolubleprecipitate observed to readily form upon the addition ofAgTFA in the CDCl3 polymer solution, suggest that brominehas been released as AgBr and replaced by a new X end-group.Importantly, when a sodium salt was supplemented to the PS−Br solution in CDCl3,
1H NMR data strictly identical to thosereported in Figure 1a were obtained (see Figure S-2 in theSupporting Information), which confirmed the key role of silverin this substitution reaction. These NMR experiments revealthat the terminal bromine end-group is totally released from thePS−Br polymer as soon as a silver salt is introduced into thesolution. Under such conditions, the production of intact PS−Br molecules using either ESI or MALDI is obviouslyimpossible. In contrast, NMR data indicate that intact PS−Brmolecules are still present in the MALDI sample to be laser-irradiated when using a sodium salt in the sample preparation.However, mass spectra obtained from such samples mainlyexhibited a distribution of PS oligomers with an unsaturatedtermination, as already reported by many other groups.10,30−35
This end-group degradation would, in this case, be due to theMALDI process in which the laser fluence must be increased,compared to experiments involving silver salts, in order toenable the production of sodiated molecules in the gas phase,despite the low affinity of PS toward this alkali.2. Structural Characterization of Modified PS End-
Groups. To further investigate the reaction occurring insolution between PS−Br and silver, the new polymeric speciesformed upon release of the bromide end-group was analyzed byelectrospray ionization−mass spectroscopy (ESI-MS). The
NMR sample solution was thus electrosprayed after beingdiluted (1:10, v/v) with methanol, which is a more ESI-friendlysolvent than chloroform. A major distribution of singly chargedPS oligomers, with peaks spaced by 104 m/z, was readilyobserved (Figure 2). Details of the isotopic pattern (the inset ofFigure 2) indicate that PS oligomers were adducted with silver,which is consistent with the salt present in solution and thecationization mode of PS.Knowing, from the 1H NMR results, that the α termination,
i.e., the CH3CH2O(CO)C(CH3)2 group, remains unchanged,with respect to the original polymer, and combining these MSdata with accurate mass measurements (see Table 1), the ωend-group of this major distribution was proposed to be atrifluoroacetate moiety. This assumption was further supportedby MS/MS experiments, which showed a product ion typicallyformed after the release of a trifluoroacetic acid (TFA)molecule (see Figure S-3 and Scheme S-1 in the SupportingInformation). This polymer is, hence, subsequently referred toas PS-TFA. Three other very minor distributions of singlycharged PS silver adducts were also detected, as designated byopen squares (□), open inverted triangles (▽), and opencircles (○) in Figure 2. None of these additional speciescorresponded to silver adducts of the investigated PS−Br.Alternatively, they could be assigned to PS oligomers holdingan unsaturated termination, a hydroxyl (PS−OH) and amethoxy (PS-OMe) end-group. Proposed end-groups for PSspecies observed in the ESI mass spectrum all exhibit anelectron-withdrawing character, consistent with results obtainedin 1H NMR, and these structural assumptions were supportedby ion elemental compositions derived from accurate massmeasurements (see Table 1).Assuming that relative ion abundances in the ESI mass
spectrum of Figure 2 actually reflect relative concentrations ofspecies in solution, the major PS−TFA distribution should giverise to specific signals in 19F NMR. Indeed, in addition to theunique resonance observed at −74.9 ppm due to AgTFA (i.e.,
Figure 2. Electrospray ionization (ESI) mass spectrum obtained after ESI analysis of the PS1900 sample previously analyzed by 1H NMR (Figure1b), after dilution with methanol. Peaks annotated with symbols were assigned to silver adducts of PS oligomers holding a trifluoroacetate (openstars, ☆), a hydroxyl (open inverted triangles, ▽), a methoxy (open circles, ○), or an unsaturated (open squares, □) termination. Asterisk symbol(*) designates silver adducts of PPG, used as an internal standard for accurate mass measurements.
Analytical Chemistry Article
dx.doi.org/10.1021/ac400375d | Anal. Chem. 2013, 85, 5454−54625457

CF3COO− ions), the 19F spectrum recorded on the 1:1
PS1900/AgTFA mixture exhibited a broad resonance atapproximately −75.2 ppm (see Figure S-4 in the SupportingInformation and the top spectrum in Figure 3b). To checkwhether or not this new resonance was characteristic of apolymer chain-end, 1H and 19F DOSY NMR experiments wererecorded in order to compare the self-diffusion coefficient (D)of the species present in solution. Results reported in Figure 3show that the D value measured for the 19F resonance at −75.2ppm is equal to the D value of the polymer (6.3 × 10−10 m2
s−1). This unambiguously indicates that, in agreement with MSdata, the main polymer holds a fluorinated termination. Inaddition, the −74.9 ppm resonance in the 19F DOSY spectrumcan be clearly attributed to a fast diffusing species (D = 2.5 ×
10−9 m2 s−1), consistent with the presence of residual free TFAin solution. Overall, these structural findings suggest thataddition of the silver salt in the PS−Br sample induces somesubstitution reactions of the terminal bromine by anionicspecies present in solution, the most abundant one (i.e.,trifluoroacetate) leading to the major PS−TFA product.The formation of PS−OMe species, revealed by a low-
intensity peak distribution in the ESI mass spectrum of Figure2, could be due to the introduction of an aliquot of methanol,used to ensure solubilization of AgTFA, in the CDCl3 polymersolution. Alternatively, this species could have been formedduring dilution of the CDCl3 sample solution with methanol forthe purpose of ESI: this would mean either that a small amountof PS−Br macromolecules (below the NMR detection limit)was still present after addition of the silver salt and that theterminal bromine has been substituted by MeO−, or thatmethanoate anions were able to displace the trifluoroacetatetermination in PS−TFA. Similarly, the formation of lowamounts of PS−OH molecules could reveal the presence ofwater traces in some of the employed solvents (in particular,CDCl3). A disproportionation reaction during polymerization(see Scheme S-2 in the Supporting Information) could accountfor the presence of PS chains with an unsaturated terminationin the sample. However, the absence of the complementaryPS−H adducts in the ESI mass spectrum strongly suggests thatthe detected PS−CHCH−Ph polymer would arise fromdehydrohalogenation of PS−Br in solution. This would meanthat, in addition to the substitution reactions of the terminalbromine by anionic species present in solution (proposed tolead to the formation of PS−TFA, PS−OMe, and PS−OH),elimination reactions could also occur in this PS−Br/silver saltmixture, accounting for the detection of low amounts of PSmolecules holding an unsaturated termination.The origin of the PS−OMe polymer observed with low
abundance in Figure 2 was further investigated using a differentsample preparation protocol. Instead of adding an aliquot of amethanolic solution of AgTFA to the polymer sample indeuterated chloroform, the CDCl3 PS−Br solution was diluted
Table 1. Accurate Mass Measurements of SilveratedOligomers in the Four PS Distributions Observed in Figure2a
m/z
n elemental composition theo exp error (ppm)
(☆) −O(CO)CF312 C104H107O4F3Ag
+ 1585.7194 1585.7205 +0.713 C112H115O4F3Ag
+ 1689.7822 1689.7795 −1.614 C120H123O4F3Ag
+ 1793.8451 1793.8415 −2.0(□) −CHCH−Ph
12 C110H114O2Ag+ 1575.7893 1575.7871 −1.4
13 C118H122O2Ag+ 1679.8521 1679.8569 +2.9
14 C126H130O2Ag+ 1783.9150 1783.9214 +3.6
(▽) −OH13 C110H116O3Ag
+ 1593.7999 1593.7916 −5.214 C118H124O3Ag
+ 1697.8627 1697.8634 +0.415 C126H132O3Ag
+ 1801.9255 1801.9189 −3.7(○) −OCH3
13 C111H118O3Ag+ 1607.8155 1607.8095 −3.7
14 C119H126O3Ag+ 1711.8784 1711.8787 +0.2
15 C127H134O3Ag+ 1815.9412 1815.9468 +3.1
aValues were measured at the maximum peak of the isotopic pattern.
Figure 3. (a) 1H-DOSY and (b) 19F-DOSY spectra of the PS1900/AgTFA mixture (molar ratio = 1:1) in CDCl3 at 298 K, showing NMR chemicalshifts (1H and 19F) and self-diffusion coefficients (D) on the horizontal and vertical axes, respectively. Diffusion coefficients are shown on alogarithmic scale. (In both cases, the corresponding 1H and 19F spectra are reported at the top of the panel.) The correlation peaks evidenced bydotted circles correspond to low molecular species present in the sample (e.g., 1, CHCl3; 2, H2O; 3, methanol (CH3OH) in panel (a); and 4,residual free TFA in panel (b)). Note that compounds 1−3 do not give rise to observable resonances in the 1H spectrum reported at top panel (a),because this spectrum was obtained using a diffusion-filtered pulse sequence (see the Experimental Section).
Analytical Chemistry Article
dx.doi.org/10.1021/ac400375d | Anal. Chem. 2013, 85, 5454−54625458

with methanol, supplemented or not with AgTFA, prior toanalysis. Both protonated (CH3OH) and fully deuterated(CD3OD) methanol were used. 1H NMR spectra recordedunder these conditions showed, once again, the disappearanceof the signal at 4.5 ppm (due to H-7 in the PS−Br sample)when AgTFA was present in the solution (see Figure S-5a inthe Supporting Information). Moreover, when using CH3OH,an additional 1H signal could be observed at ∼2.9 ppm, whereassuch signal was missing when using fully deuterated methanol(CD3OD). A two-dimensional (2D) 1H−13C HSQC experi-ment further revealed that the 13C resonance of thecorresponding carbon was close to 55 ppm, in agreementwith an OCH3 polymer termination (see Figure S5-b in theSupporting Information). This 2D experiment also evidencedthe presence of additional signals, at 3.5 ppm and 87 ppm for1H and 13C, respectively, which were assigned to the CH groupin position 7 (see atom numbering shown in Figure 1 with X =OCH3). In parallel, the ESI-MS spectrum obtained for thesample prepared using CD3OD exhibited a major distributionof silverated PS−OCD3 (see Figure 4a), which were observed
to shift to [PS−OCH3 + Ag]+ when CH3OH was used as thedilution solvent (Figure 4b). This last result unambiguouslyindicates that the methoxy termination is indeed caused by thesolvent, showing that efficient solvolysis of the PS−Brmolecules occurs when AgTFA is present in the medium. Itshould also be noted that no signal corresponding to the silveradduct of PS−TFA could be detected here, suggesting thatCH3O
− efficiently competes with trifluoroacetate anions insubstituting the bromine termination. Finally, structural
investigation of the two other very minor PS species observedbeside the major PS−OMe distribution in the ESI mass spectraof Figure 4 is detailed in the Supporting Information.In summary, as soon as the polymer interacts with silver in
solution, the halogen end-group is removed. While this reactionwas observed to occur regardless of the medium solvent, thenature of the substituting end-groups strongly depends on thesolution composition. To check whether or not this effect was asolution phenomenon that required the assistance of a solvent,MALDI-MS experiments were performed for solvent-freeprepared samples. As shown in Figure S-6 in the SupportingInformation, a major distribution of silverated PS holding anunsaturated termination was observed, together with the sameoligomers adducted with sodium as a minor species. This resultsuggests that the silver-catalyzed removal of the halogen end-group would also proceed in the solid phase, according to a so-called “mechano-induced reaction”, as previously evidenced byour group to occur for other systems during grinding thematrix/polymer/salt ternary mixture to prepare solvent-freeMALDI samples.41,42 A lack of any substitution products inthese samples would indicate low availability of the nucleophilicagents in the near environment of the reacting center, which isconsistent with the lower mobility of species in the solid phase,compared to solution.
3. Nucleophilic Substitution for Reliable MALDIAnalysis of ATRP PS. Despite the low nucleophilicity oftrifluoroacetate anions, PS−TFA would readily be formed whenAgTFA is supplemented to the CDCl3 PS−Br solution(pathway (1) in Scheme 1), because of the halogenophiliccharacter of AgI.The reaction would be activated by specific interactions of
silver salts with the halogen and lead to the formation ofinsoluble silver halides.43 This nucleophilic substitution wouldmost probably occur via a SN1 mechanism, because of theresonance-stabilized carbocation produced upon Br− abstrac-tion. However, note that very few stoichiometric couplingreactions involving (organo)silver species (such as thatobserved to occur here) are known, and that they usuallyrequire the use of a co-catalyst.44 Exclusive formation of PS−OCH3 when methanol was the reaction medium (pathway (2)in Scheme 1) could be explained by the much-highernucleophilic character of CH3OH (also present in a muchlarger amount), compared to CF3COO−. Finally, sincenucleophiles also exhibit a basic character, an eliminationreaction was proposed to account for the presence of PSmolecules holding an unsaturated termination (pathway (3) inScheme 1). Because of the very low relative amount of thelatter species, this elimination would poorly compete with thesubstitution reactions.Nucleophilic displacement of the halogen termination of
ATRP polymers has already been used as an efficient way tointroduce several functional groups at the molecule chain-end,14,22,23,45−49 either to obtain intermediates for subsequentreactions, or to remove the terminal halogens which may bedetrimental to applications of polymers. Interestingly, in allcases, so-modified synthetic polymers could be readilyproduced as intact gas-phase ions upon MALDI. MALDI-MSexperiments were then performed for both PS−TFA and PS−OMe species (Figure 5). Mass spectra obtained for PS-TFAexhibited a main distribution corresponding to silver adducts ofPS molecules holding an unsaturated end-group (Figure 5a),suggesting that TFA has been released from the polymer duringthe ionization step. In contrast, silverated methoxy-terminated
Figure 4. ESI mass spectrum of the PS−Br sample (Mn = 1900 gmol−1) after dilution in AgTFA solutions in (a) deuterated methanol(CD3OD) and (b) methanol (CH3OH). Peaks marked by opensquares (□) were assigned to silver adducts of PS oligomers holdingan unsaturated termination, while open diamonds (◊) designate silveradducts of PS with an ethylisobutyrate moiety as both end-groups.
Analytical Chemistry Article
dx.doi.org/10.1021/ac400375d | Anal. Chem. 2013, 85, 5454−54625459
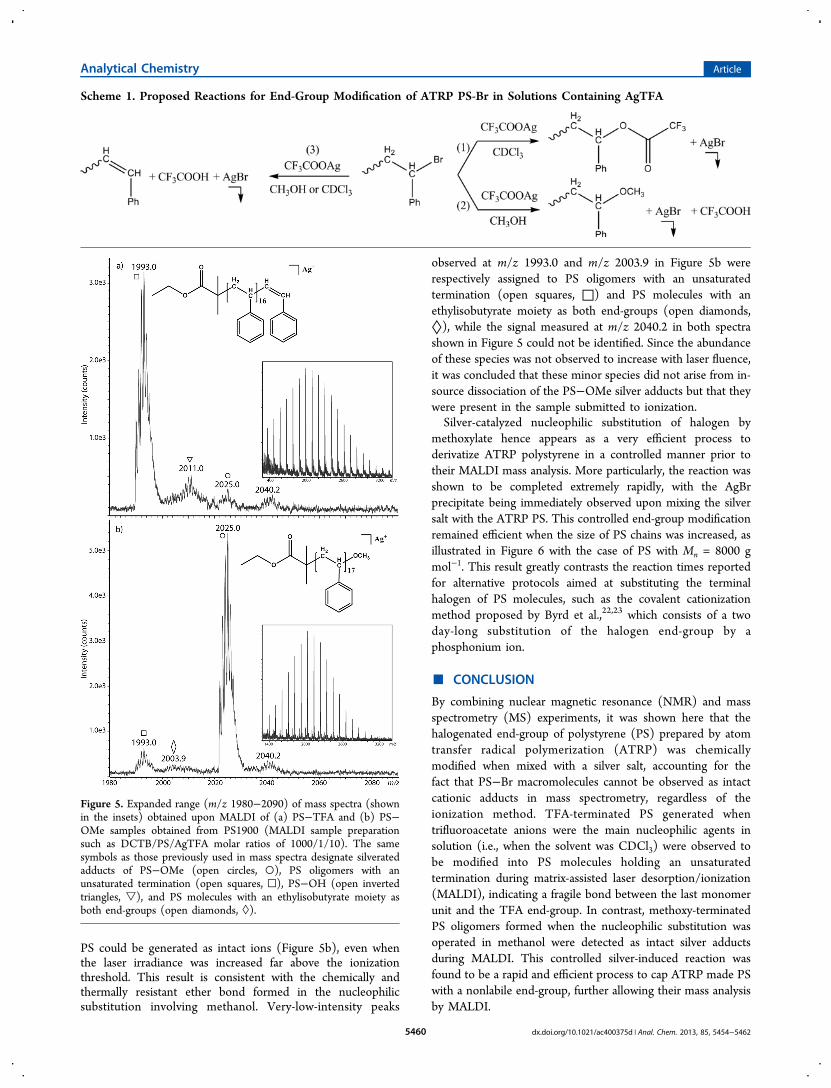
PS could be generated as intact ions (Figure 5b), even whenthe laser irradiance was increased far above the ionizationthreshold. This result is consistent with the chemically andthermally resistant ether bond formed in the nucleophilicsubstitution involving methanol. Very-low-intensity peaks
observed at m/z 1993.0 and m/z 2003.9 in Figure 5b wererespectively assigned to PS oligomers with an unsaturatedtermination (open squares, □) and PS molecules with anethylisobutyrate moiety as both end-groups (open diamonds,◊), while the signal measured at m/z 2040.2 in both spectrashown in Figure 5 could not be identified. Since the abundanceof these species was not observed to increase with laser fluence,it was concluded that these minor species did not arise from in-source dissociation of the PS−OMe silver adducts but that theywere present in the sample submitted to ionization.Silver-catalyzed nucleophilic substitution of halogen by
methoxylate hence appears as a very efficient process toderivatize ATRP polystyrene in a controlled manner prior totheir MALDI mass analysis. More particularly, the reaction wasshown to be completed extremely rapidly, with the AgBrprecipitate being immediately observed upon mixing the silversalt with the ATRP PS. This controlled end-group modificationremained efficient when the size of PS chains was increased, asillustrated in Figure 6 with the case of PS with Mn = 8000 gmol−1. This result greatly contrasts the reaction times reportedfor alternative protocols aimed at substituting the terminalhalogen of PS molecules, such as the covalent cationizationmethod proposed by Byrd et al.,22,23 which consists of a twoday-long substitution of the halogen end-group by aphosphonium ion.
■ CONCLUSION
By combining nuclear magnetic resonance (NMR) and massspectrometry (MS) experiments, it was shown here that thehalogenated end-group of polystyrene (PS) prepared by atomtransfer radical polymerization (ATRP) was chemicallymodified when mixed with a silver salt, accounting for thefact that PS−Br macromolecules cannot be observed as intactcationic adducts in mass spectrometry, regardless of theionization method. TFA-terminated PS generated whentrifluoroacetate anions were the main nucleophilic agents insolution (i.e., when the solvent was CDCl3) were observed tobe modified into PS molecules holding an unsaturatedtermination during matrix-assisted laser desorption/ionization(MALDI), indicating a fragile bond between the last monomerunit and the TFA end-group. In contrast, methoxy-terminatedPS oligomers formed when the nucleophilic substitution wasoperated in methanol were detected as intact silver adductsduring MALDI. This controlled silver-induced reaction wasfound to be a rapid and efficient process to cap ATRP made PSwith a nonlabile end-group, further allowing their mass analysisby MALDI.
Scheme 1. Proposed Reactions for End-Group Modification of ATRP PS-Br in Solutions Containing AgTFA
Figure 5. Expanded range (m/z 1980−2090) of mass spectra (shownin the insets) obtained upon MALDI of (a) PS−TFA and (b) PS−OMe samples obtained from PS1900 (MALDI sample preparationsuch as DCTB/PS/AgTFA molar ratios of 1000/1/10). The samesymbols as those previously used in mass spectra designate silveratedadducts of PS−OMe (open circles, ○), PS oligomers with anunsaturated termination (open squares, □), PS−OH (open invertedtriangles, ▽), and PS molecules with an ethylisobutyrate moiety asboth end-groups (open diamonds, ◊).
Analytical Chemistry Article
dx.doi.org/10.1021/ac400375d | Anal. Chem. 2013, 85, 5454−54625460

■ ASSOCIATED CONTENT
*S Supporting InformationThis material is available free of charge via the Internet athttp://pubs.acs.org.
■ AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected].
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTS
The authors acknowledge support from Spectropole, theAnalytical Facility of Aix-Marseille University, by allowing aspecial access to the instruments purchased with EuropeanFunding (No. FEDER OBJ2142-3341).
■ REFERENCES(1) Montaudo, M. S.; Lattimer, R. P. Mass Spectrometry of Polymers;CRC Press: Boca Raton, FL, 2001.(2) Weidner, S. M.; Trimpin, S. Anal. Chem. 2010, 82, 4811−4829.(3) Gruendling, T.; Weidner, S.; Falkenhagen, J.; Barner-Kowollik, C.Polym. Chem. 2010, 1, 599−617.(4) Fenn, J. B.; Mann, M.; Meng, C. K.; Wong, S. F.; Whitehouse, C.M. Mass Spectrom. Rev. 1990, 9, 37−70.(5) Karas, M.; Hillenkamp, F. Anal. Chem. 1988, 60, 2299−2301.(6) Tanaka, K.; Waki, H.; Ido, Y.; Akita, S.; Yoshida, Y.; Yoshida, T.;Matsuo, T. Rapid Commun. Mass Spectrom. 1988, 2, 151−153.(7) Barner-Kowollik, C.; Davis, T. P.; Stenzel, M. H. Polymer 2004,45, 7791−7805.(8) Matyjaszewski, K. In Controlled/Living Radical Polymerization:Progress in ATRP, NMP and RAFT; American Chemical Society:Washington, DC, 2000; Vol. 768.(9) Charles, L. Mass Spectrom. Rev. 2013, in press.
(10) Nonaka, H.; Ouchi, M.; Kamigaito, M.; Sawamoto, M.Macromolecules 2001, 34, 2083−2088.(11) Venkatesh, R.; Vergouwen, F.; Klumperman, B. J. Polym. Sci.,Part A: Polym. Chem. 2004, 42, 3271−3284.(12) Can, A.; Altuntas, E.; Hoogenboom, R.; Schubert, U. S. Eur.Polym. J. 2010, 46, 1932−1939.(13) Le Grognec, E.; Claverie, R.; Poli, R. J. Am. Chem. Soc. 2001,123, 9513−9524.(14) Matyjaszewski, K.; Xia, J. H. Chem. Rev. 2001, 101, 2921−2990.(15) Shen, X. F.; He, X. R.; Chen, G. Q.; Zhou, P.; Huang, L.Macromol. Rapid Commun. 2000, 21, 1162−1165.(16) Pascual, S.; Coutin, B.; Tardi, M.; Polton, A.; Vairon, J. P.;Chiarelli, R. Macromolecules 2001, 34, 5752−5758.(17) Stoffelbach, F.; Claverie, J.; Poli, R. C. R. Chim. 2002, 5, 37−42.(18) Ladaviere, C.; Lacroix-Desmazes, P.; Delolme, F.Macromolecules2009, 42, 70−84.(19) Li, Y.; Hoskins, J. N.; Sreerama, S. G.; Grayson, M. A.; Grayson,S. M. J. Mass Spectrom. 2010, 45, 587−611.(20) Cheng, G. L.; Boker, A.; Zhang, M. F.; Krausch, G.; Muller, A.H. E. Macromolecules 2001, 34, 6883−6888.(21) Kulis, J.; Bell, C. A.; Micallef, A. S.; Jia, Z.; Monteiro, M. J.Macromolecules 2009, 42, 8218−8227.(22) Lin-Gibson, S.; Bencherif, S. A.; Beers, K. L.; Byrd, H. C. M.Macromolecules 2003, 36, 4669−4671.(23) Byrd, H. C. M.; Bencherif, S. A.; Bauer, B. J.; Beers, K. L.; Brun,Y.; Lin-Gibson, S.; Sari, N. Macromolecules 2005, 38, 1564−1572.(24) Singha, N. K.; German, A. L. J. Appl. Polym. Sci. 2005, 98, 1418−1426.(25) Lovering, E. G.; Laidler, K. J. Can. J. Chem. 1960, 38, 2367−2372.(26) Mazarin, M.; Phan, T. N. T.; Charles, L. Rapid Commun. MassSpectrom. 2008, 22, 3776−3782.(27) Girod, M.; Phan, T. N. T.; Charles, L. J. Am. Soc. Mass Spectrom.2008, 19, 1163−1175.(28) Mazarin, M.; Girod, M.; Viel, S.; Phan, T. N. T.; Marque, S. R.A.; Humbel, S.; Charles, L. Macromolecules 2009, 42, 1849−1859.(29) Jia, Z.; Bell, C. A.; Monteiro, M. J. Macromolecules 2011, 44,1747−1751.
Figure 6. MALDI mass spectrum (DCTB/PS/AgTFA molar ratios = 1000/1/100) obtained after PS−Br (Mn = 8000 g mol−1) was dissolved in amethanolic solution of AgTFA. Inset shows details of the mass spectrum showing signals obtained for PSn−OMe molecules with n = 74 and n = 75(open circles, ○), respectively, expected at m/z 7960.7 and m/z 8064.7, together with minor peaks assigned to PS73−74 oligomers with an unsaturatedtermination (open squares, □) expected at m/z 7928.6 and m/z 8032.7, respectively.
Analytical Chemistry Article
dx.doi.org/10.1021/ac400375d | Anal. Chem. 2013, 85, 5454−54625461

(30) Francis, R.; Lepoittevin, B.; Taton, D.; Gnanou, Y. Macro-molecules 2002, 35, 9001−9008.(31) Bernaerts, K. V.; Schacht, E. H.; Goethals, E. J.; Du Prez, F. E. J.Polym. Sci., Part A: Polym. Chem. 2003, 41, 3206−3217.(32) Bernaerts, K. V.; Du Prez, F. E. Polymer 2005, 46, 8469−8482.(33) Pfeifer, S.; Lutz, J. F. Chem.Eur. J. 2008, 14, 10949−10957.(34) Pfeifer, S.; Lutz, J. F. Macromol. Chem. Phys. 2010, 211, 940−947.(35) Lepoittevin, B.; Elhiri, A.; Bech, L.; Belleney, J.; Baltaze, J. P.;Capron, I.; Planchot, V.; Roger, P. Carbohydr. Polym. 2011, 83, 1174−1179.(36) Jerschow, A.; Muller, N. J. Magn. Reson. 1997, 125, 372−375.(37) Barrere, C.; Mazarin, M.; Giordanengo, R.; Phan, T. N. T.;Thevand, A.; Viel, S.; Charles, L. Anal. Chem. 2009, 81, 8054−8060.(38) Jayawickrama, D. A.; Larive, C. K.; McFord, E. F.; Roe, D. C.Magn. Reson. Chem. 1998, 36, 755−760.(39) Skelton, R.; Dubois, F.; Zenobi, R. Anal. Chem. 2000, 72, 1707−1710.(40) Trimpin, S.; Rouhanipour, A.; Az, R.; Rader, H. J.; Mullen, K.Rapid Commun. Mass Spectrom. 2001, 15, 1364−1373.(41) Pizzala, H.; Barrere, C.; Mazarin, M.; Ziarelli, F.; Charles, L. J.Am. Soc. Mass Spectrom. 2009, 20, 1906−1911.(42) Major, Y.; Pizzala, H.; Ziarelli, F.; Phan, T. N. T.; Mollica, G.;Charles, L. Anal. Methods 2012, 4, 3118−3126.(43) Weibel, J.-M.; Blanc, A.; Pale, P. Chem. Rev. 2008, 108, 3149−3173.(44) Dillinger, S.; Bertus, P.; Pale, P. Org. Lett. 2001, 3, 1661−1664.(45) Matyjaszewski, K.; Nakagawa, Y.; Gaynor, S. G.Macromol. RapidCommun. 1997, 18, 1057−1066.(46) Coessens, V.; Nakagawa, Y.; Matyjaszewski, K. Polym. Bull.1998, 40, 135−142.(47) Weimer, M. W.; Frechet, J. M. J.; Gitsov, I. J. Polym. Sci., Part A:Polym. Chem. 1998, 36, 955−970.(48) Malz, H.; Komber, H.; Voigt, D.; Hopfe, I.; Pionteck, J.Macromol. Chem. Phys. 1999, 200, 642−651.(49) Chen, G. Q.; Wu, Z. Q.; Wu, J. R.; Li, Z. C.; Li, F. M.Macromolecules 2000, 33, 232−234.
Analytical Chemistry Article
dx.doi.org/10.1021/ac400375d | Anal. Chem. 2013, 85, 5454−54625462
Copyright © 2022 FDOKUMEN




















