
Amyloid Protofibrils of Lysozyme Nucleate and Grow Via Oligomer Fusion
Upload
independentCategory
view
1download
0
This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Chemical and nanomechanical analysis of rice husk modifiedby ATRP-grafted oligomer
Samir M. Morsi a,c,⇑, Anahita Pakzad b, Amal Amin c, Reza S. Yassar b, Patricia A. Heiden a,⇑a Department of Chemistry, Michigan Technological University, 1400 Townsend Ave., Houghton, MI 49931, USAb Department of Mechanical Engineering–Engineering Mechanics, Michigan Technological University, USAc National Research Centre, Chemical Industry Division, Polymer and Pigment Department, Cairo, Egypt
a r t i c l e i n f o
Article history:Received 16 January 2011Accepted 20 April 2011Available online 27 April 2011
Keywords:Rice huskNanomechanicalHeterogeneous ATRPMacroinitiatorSurface modificationGrafting
a b s t r a c t
Rice husk (RH), an abundant agricultural residue, was reacted with 2-bromoisobutyryl bromide, toconvert it to a heterogeneous polyfunctional macroinitiator for Atom Transfer Radical Polymerization(ATRP). The number of active sites placed on the RH surface was small, but they were ATRP active.Non-polar methyl methacrylate (MMA) and polar acrylonitrile (AN) were polymerized from the RH,and a sequential monomer addition was used to prepare an amphiphilic PMMA-b-PAN copolymer onRH surface. FTIR qualitatively confirmed the grafting. Gravimetric and XPS analysis of the different RHsurface compositions indicated thin layers of oligomeric PMMA, PAN, and PMMA-b-PAN. The modifiedsurfaces were mapped by nanomechanical AFM to measure surface roughness, and adhesion and moduliusing the Derjaguin–Muller–Toropov model. RH grafted with MMA possessed a roughness value of 7.92,and a hard and weakly adhering surface (13.1 GPa and 16.7 nN respectively) while RH grafted with ANyielded a roughness value of 29 with hardness and adhesion values of 4.0 GPa and 23.5 nN. ThePMMA-b-PAN modification afforded a surface with a roughness value of 51.5 nm, with hardness andadhesion values of 3.0 GPa and 75.3 nN.
� 2011 Elsevier Inc. All rights reserved.
1. Introduction
Utilization of biomass is of increasing importance because ofthe limited quantities of fossil feedstocks [1]. Cellulose rich bio-mass is increasingly important as a potential alternative fuel feed-stock, but also as a chemical feedstock. Cellulose, hemicellulosesand lignins can be broken down into smaller compounds or, giventheir high functionality, are suitable for chemical derivatization[2,3].
While wood resources are abundant in North America and havea long history of use as reinforcements, more recently it has be-come of great interest as a source of chemical feedstocks. But, forthose regions of the world that do not have abundant forest re-sources rice husk (RH) is a more convenient biomass. RH contains25–35% cellulose, 8–21% hemicelluloses, 26–31% lignin, 15–17%amorphous silica and waxes, and 2–5% of other soluble substances[4–7].
Like wood, RH can be used as a resource for chemical feedstocksor as reinforcement. For example, Silvia et al. [8] mercerized RHwith NaOH and then acetylated the RH surface using acetic acid.This was done to improve the RH compatibility with non-polarpolymers for use in composites. Emiliano et al. [9] modified ofRH with NaOH followed by H2O2 (bleaching) and found it to bean efficient treatment to partially eliminate hemicelluloses, ligninand silica from the RH. Rozman et al. [10] chemically modifiedRH with glycidyl methacrylate, maleic anhydride and succinicanhydride, and Wong et al. [11] reported the modification of RHusing different carboxylic acids (citric, salicylic, tartaric, oxalic,mandelic, maleic and nitrilotriacetic acid).
Grafting studies of oligomers or polymers onto RH surfaces donot appear to have been reported before, though polymers havebeen grafted to cellulose and even to wood fibers [12,13]. However,RH is a more complex material than wood, and is compositionally‘assymetric’ having one side that is silica enriched and an innersurface that is waxy. So, grafting to RH may be significantly lessefficient than grafting to wood.
Typical grafting strategies can be described as occurring by (1)the ‘grafting through’ process, (2) the ‘grafting onto’ process and(3) the ‘grafting from’ process. Common examples of the ‘graftingthrough’ technique include copolymerizing pre-made vinyl-functionalized cellulose with comonomers [13]. The ‘grafting onto’technique requires the pre-synthesis of end-functionalized linear
0021-9797/$ - see front matter � 2011 Elsevier Inc. All rights reserved.doi:10.1016/j.jcis.2011.04.065
⇑ Corresponding authors. Addresses: Department of Chemistry, Michigan Tech-nological University, 1400 Townsend Ave., Houghton, MI 49931, USA (P.A. Heiden).National Research Centre, Chemical Industry Division, Polymer and PigmentDepartment, Cairo, Egypt (S.M. Morsi).
E-mail addresses: [email protected] (S.M. Morsi), [email protected](A. Pakzad), [email protected] (A. Amin), [email protected] (R.S. Yassar),[email protected] (P.A. Heiden).
Journal of Colloid and Interface Science 360 (2011) 377–385
Contents lists available at ScienceDirect
Journal of Colloid and Interface Science
www.elsevier .com/locate / jc is

Author's personal copy
chains that are subsequently covalently bonded to the cellulose[12]. This strategy usually suffers from low grafting density dueto steric hindrance. The ‘grafting from’ technique, involves thegrowth of polymer grafts directly from the substrate surface, andhas been extensively investigated by step growth and by chaingrowth processes.
Radicals can be conveniently generated along cellulosic back-bones in the presence of chemical initiators or by applying irradi-ation. Both methods afford the straightforward preparation ofcellulosic-based graft copolymers through ‘grafting from’ free rad-ical polymerization. The irradiation route is simple but character-ized by cellulosic backbone degradation. Neither method givescontrol over the graft molecular weight, molecular weight distribu-tion (PDI), or the control needed to obtain block copolymer grafts.That level of control is possible using controlled radical polymeri-zation techniques (CRP), e.g. nitroxide-mediated polymerization[14] (NMP), atom transfer radical polymerization [15,16] (ATRP),or reversible addition–fragmentation chain transfer [17] (RAFT),These methods are also tolerant of moisture and compatible witha large range of functional groups.
ATRP has advantages in ‘‘grafting from’’ reactions because theinitiator can be anchored on the surface in advance, and its pres-ence can be confirmed. Also, as with other CRPs, chain terminationis reduced. Therefore, using CRP it is possible to tailor the surfaceproperties of cellulosic-based materials by grafting oligomers orpolymers with a variety of different monomers, including function-alized monomers, while exercising some control over graft length.
Therefore, through appropriate monomer selection and reactiondesign, many different surface compositions [18] and architectures[15,19] can be placed on surfaces. ATRP has already been employedto graft from surfaces such as glass [20], gold [21,22], magnetite[23], silica [24–26] and from macroscopic cellulose fibers [27–32], pulp [33], and powder [34].
Although several examples of ATRP polymerization from differ-ent forms of cellulose surfaces exist in the literature, no precedentexists for ATRP-grafting from RH. RH is 25–35% cellulose, so inprinciple the reaction should proceed in the same way as when itis performed on cellulose fiber. However, the surfaces of cellulosepulp or fibers, even in a suspension, are more readily available thanthe cellulose within the RH due to the asymmetric and composi-tionally heterogeneous RH anatomy [35]. The external surface ofthe RH is a thick 2–5 lm silica layer, covered by a very thin layerof cellulose. The other surface is waxy and shields additional cellu-loses [36]. The surfaces are designed to serve as a barrier to gasesand moisture to protect the rice kernel inside.
Therefore, the waxy layer needs to be at least partially removed toexpose the thicker layer of cellulose below it to expose its hydroxylsfor modification, but this is also likely to remove all or part of the thin(�50 nm) [37] cellulose layer on the external surface. Some of thesecellulose domains may even be sterically unavailable because of thepresence of silica nanodomains dispersed within the RH. The silicadomains are also expected to make the RH more dense and less proneto solvent swelling than wood cellulose, though for this work the sil-ica was considered desirable, despite the fact that it is abrasive tosome processing equipment. This is because it may allow compositesto maintain some of the barrier properties of RH. For all these rea-sons, RH cellulose is expected to be less available for modificationthan wood pulp cellulose.
In this work we describe an ATRP route to give controlled poly-merization from RH surfaces. While this route is unlikely to becommercially viable, the ultimate application for the modifiedRH is for use in fundamental studies of the effect of controlledinterface modifications on composite properties. Therefore, thesurface compositions must be controlled and thoroughly charac-terized before it is possible to understand the effect of variablessuch as interface thickness, composition, and rigidity on selected
composite properties, particularly moisture properties (to bedescribed in a paper to follow). The purpose of this paper is todescribe the manipulation and surface and nanomechanical char-acterizations of the RH surface. Gaining an understanding of the ef-fects of these kinds of modifications on the properties of thereinforcement, particularly biobased reinforcement that is proneto moisture uptake and biological attack, may give access to anew range of improved composites.
2. Experimental
2.1. Materials
Rice husk (RH, 80 mesh) was donated by Rice Hull SpecialtyProducts (Stuttgart, Arizona). 2-bromoisobutyryl bromide (BiBB)pyridine (99.5%, extra dry over molecular sieves), and dimethyl-formamide (DMF, 99.8%, extra dry over molecular sieve) werepurchased from Acros Organics (New Jersey), N,N,N0,N00,N00-penta-methyldiethylenetriamine (PMDETA, 99%) ethyl a-bromoisobuty-rate (EBiB), and copper (I) bromide (98%,) were from AldrichChemicals (Milwaukee, WI) were used as received. Methyl methac-rylate (MMA, 99%, Aldrich) and acrylonitrile (AN, 99%, Acros) weredistilled under vacuum just before use. Dichloromethane (DCM)and tetrahydrofuran (THF) were from Fisher Scientific (Pittsburgh,PA) and were degassed by bubbling nitrogen for 20 min just beforeuse.
2.2. Instrumentation
2.2.1. Fourier transform infra-red (FTIR)Fourier transform infra-red (FTIR) spectra were taken on a
Perkin–Elmer Spectrum One FTIR Spectrometer from 4000 to650 cm�1 (4.0 cm�1 resolution) were used to confirm changes infunctional groups on the chemically modified RH.
2.2.2. X-ray photon spectroscopy (XPS)X-ray photon spectroscopy (XPS) were performed in a Surface
Science Instruments S-probe spectrometer that uses monochroma-tized Al Ka X-ray and a low energy electron flood gun for chargeneutralization of non-conducting samples. Samples were preparedby lightly pressing samples of the material to be analyzed ontodouble-sided tape that had first been affixed to a silicon wafer.The silicon wafers were fixed to the sample stage by double-sidedtape. The samples were run as insulators and the X-ray spot size foreach acquisition was approximately 800 lm. Pressure in the ana-lytical chamber was maintained below 5 � 10�9 Torr. The passenergy for survey and detail spectra (to calculate composition)was 150 eV. Pass energy for high-resolution spectra was 50 eV.The take-off angle (the angle between the sample normal and theinput axis of the energy analyzer) was �55� (55� take-off angleat �50 Å sampling depth). Then Service Physics ESCA2000A Analy-sis Software was used to determine peak areas above a linear back-ground in order to calculate the elemental compositions usingelemental sensitivity factors, and to perform peak fitting on thehigh-resolution spectra. The binding energy scale of the spectrawas calibrated by assigning the hydrocarbon C1s peak to a bindingenergy of 285.0 eV.
2.2.3. Atomic force microscopy (AFM)Peak force-tapping mode was used in Dimension� Icon� AFM,
to generate nanomechanical maps of sample surfaces. In this mode,at each pixel, the AFM probe generates a small indentation on thesample surface and a force–separation curve is collected (Fig. 1)[38]. In this technique, by using the maximum indentation force(peak force) as the feedback signal, a smaller deformation can be
378 S.M. Morsi et al. / Journal of Colloid and Interface Science 360 (2011) 377–385

Author's personal copy
generated on the sample surface, which increases the imaging res-olution to a great extent.
On each curve, peak force and adhesion force are the maximumand minimum force values respectively. Deformation is the differ-ence of separation at zero force and peak force. The Derjaguin–Muller–Toropov (DMT) modulus is calculated by fitting the DMTmodel [39] to the initial part of the retrace curve:
F ¼ 43
E�ffiffiffiffiffiffiffiffiRd3
qþ Fadh ð1Þ
where F is the force, R is the tip radius, d is the separation, Fadh is theadhesion force, and E� is the reduced elastic modulus. By assuminginfinite elastic modulus for the tip (Etip) and knowing the Poisson’sratio of the sample ts, the Young’s modulus for the sample (Es) canbe calculated:
E� ¼ 1� t2s
Esþ
1� t2tip
Etip
!�1
ð2Þ
The generated force–separation curves are used to create quan-titative nanomechanical maps (DMT modulus, adhesion and defor-mation) simultaneously with a topography map. For this purpose,several calibration steps are completed before imaging the sample.First, the deflection sensitivity of the cantilever was measured byindenting a hard sapphire surface (56.67 nm/V). Then, the cantile-ver spring constant was measured using thermal tuning method(K = 38.9 N/m). And finally, the tip radius was measured by chang-ing the tip radius in the AFM software, while imaging a standardpolymer sample with known mechanical properties, until the de-sired value for the DMT modulus was obtained (R = 4.5 nm).
All AFM images were collected in air under ambient conditionand consisted of 512 � 512 pixels with scanning rates <1 Hz. Thepeak force was set such that the resultant average deformationin each scan line was not more than 3 nm. Other scanning param-eters include integral and proportional gains, and are automaticallyset by the AFM software. Although one set of AFM images fromeach sample is presented here, the same trend was observed dur-ing scanning other regions of the samples.
2.3. Alkali treatment of RH
The RH was soaked in 18 wt.% solution of NaOH. The volume ofthe solution was equivalent to 1.5 times the volume of the filler.Immersion time was 0.5 h � 2. Once this time was over, the fillers
were separated from the solution and washed with water to re-move the NaOH. The RH was dried at 75 �C for 48 h.
2.4. Immobilization of the ATRP initiator on the RH surface (RHABr)
An ATRP-initiating bromine (BIBB) was grafted to the surface ofRH by mixing RH (5.0 g), DMF (50 mL), and pyridine (10.0 mL,124 mmol) in a round bottom flask. The mixture was magneticallystirred at room temperature under nitrogen gas. Then BiBB wasadded dropwise (10.0 mL, 80.9 mmol) over a period of 25 min.After the addition was completed the reaction mixture was stirredan additional 24 h at 80 �C. The surface-initiated RH (RHABr) wasremoved from the reaction mixture, washed thoroughly with ace-tone and deionized water several times and dried at 50 �C for 48 h.By gravimetric analysis the moles of ATRP active Br groups intro-duced to the RH was found to be �0.2 mmol/5 g RH (0.04 mmol/1 g RH), or a 0.02% yield overall based on moles of starting BiBB.
2.5. Grafting of AN from RHABr (RH-g-PAN)
To a round bottom flask were added RHABr (1.0 g, �0.04 mmolactive Br sites), CuBr (118.8 mg, 0.828 mmol), PMDETA (0.35 mL,1.67 mmol) and DMF (50 mL). The mixture was magnetically stir-red for 30 min at room temperature and under nitrogen to allowcopper–ligand complex formation. AN (26.4 mL, 403 mmol) andEBiB initiator (0.132 mL, 0.899 mmol) were then added. The tem-perature of the reaction mixture was raised to 80 �C and main-tained at that temperature for 24 h. The RH with surface graftedPAN (RH-g-PAN) was removed from the solution, washed thor-oughly with deionized water and acetone and dried at 50 �C for48 h. Yield: 1.16 g. Theoretical Xn if all Br sites initiated at the timeand reacted for the same length of time is 10, 075. Calculated Xn
(gravimetrically) assuming all Br sites reacted equally is Xn � 76.
2.6. Grafting of MMA from RHABr (RH-g-PMMA)
To a round bottom flask were added RHABr (2.5 g, �0.1 mmolactive Br sites), CuBr (300 mg, 2.1 mmol) and PMDETA (0.9 mL,4.31 mmol) containing a 4:1 mixture solution of DMF (100 ml)and DCM (25 mL). The mixture was magnetically stirred for 0.5 hat room temperature and under nitrogen gas. MMA (45.0 mL,422.5 mmol) and the initiator, EBiB (0.3 mL, 2.044 mmol), werethen added. The temperature of the reaction mixture was raisedto 80 �C and kept for 24 h. The RH with surface grafted PMMA(RH-g-PMMA) was removed from the solution, washed thoroughlywith deionized water, acetone and THF several times and dried at50 �C for 48 h. Yield: 2.675 g. Theoretical Xn assuming all Br sitesinitiated and reacted at the time for the same length of time is4224. Calculated Xn (gravimetrically) assuming all Br sites reactedequally is Xn � 18.
2.7. Grafting of MMA and PAN from RHABr (RH-g-PMMA-b-PAN)
To a round bottom flask was added RH-g-PMMA (1.0 g,�0.04 mmol active Br sites), CuBr (118.8 mg, 0.828 mmol), PMDETA(0.35 mL, 1.67 mmol) and DMF (50 mL). The mixture was magneti-cally stirred for 30 min at room temperature and under nitrogengas. AN (26.4 mL, 403 mmol) and EBiB initiator (0.132 mL,0.899 mmol) were then added. The temperature of the reaction mix-ture was raised to 80 �C and kept at that temperature for 24 h. Theproduct, RH-g-PMMA-b-PAN was removed by filtration, washedthoroughly with deionized water and acetone and dried at 50 �Cfor 48 h. Yield: 1.22 g. The overall reaction steps of ATRP initiatorimmobilization and grafting of RH are shown in Fig. 2. For all graftedsamples, the increase in the mass of the extracted samples from thatof the original RH yielded the grafting percentage. Calculated (gravi-
Fig. 1. Analytical values obtained from a peak force-tapping mode in AFM. At eachpixel, the AFM tip generates a small indentation on the sample surface, force–separation curves are made, and quantitative nanomechanical properties of thesurface are measured.
S.M. Morsi et al. / Journal of Colloid and Interface Science 360 (2011) 377–385 379

Author's personal copy
metrically, assuming all initial RHABr sites remained active) Xn AN� 103.
3. Results and discussion
ATRP is a useful technique for grafting from a complex cellulosicmaterial like RH because once the waxy inner layer is removed theprimary hydroxyl groups on the cellulose surface can be convertedto a-bromoesters, which are excellent initiators for ATRP polymer-
ization. Fig. 2 outlines the synthetic route used to produce thesurface grafted a-bromoester, RHABr. The RHABr functioned suc-cessfully as a heterogenous ATRP macroinitiator, being insolublein the solvent. Despite possessing a relatively low number of activeBr sites was then used to initiate three grafting from polymeriza-tions to give a hydrophobic RH-g-PMMA material, a hydrophilicRH-PAN material, and an amphiphilic RH-g-PMMA-b-PAN mate-rial. These modifications were made for use in studies of the effectsof surface-modified RH on the stability of composite interfaceswhen this reinforcement is used instead of unmodified RH. Before
OH
OH
Br
O
Br
Pyridine DMF, 80 oC
+
OH
O
OBr
OH
O
O Br
O O
n
MMA
CuBr
PMDETA
EBiB
DMF, DCM
80 oC
RH BiBB
RH-Br
RH-g-PMMA
OH
O
O Br
CNn
RH-g-PAN
OH
O
O
O O
n
Br
CNn
RH-g-PMMA-b-PAN
CuBr
PMDETA
EBiB
80 oC
AN
DMF
CuBr
PMDETA
EBiB
80oC
AN
DMF
(a )
(b) (c )
(d)
Fig. 2. The synthetic strategy used in the surface modification of RH: (a) conversion of hydroxyl groups to a-bromoesters, giving RHABr, (b) grafting of MMA on the surface,giving RH-g-MMA, (c) grafting of AN on RHABr giving RH-g-PAN, and (d) grafting AN onto RH-g-PMMA, giving RH-g-PMMA-b-PAN.
380 S.M. Morsi et al. / Journal of Colloid and Interface Science 360 (2011) 377–385

Author's personal copy
these studies can be done however, the surfaces of unmodified andmodified RH must be thoroughly characterized, which is the pur-pose of this work.
FTIR spectra (Fig. 3) qualitatively support the modification se-quence outlined in Fig. 2 to produce the RHABr macroinitiator,
and then give the RH-g-PAN, RH-g-PMMA, and RH-g-PMMA-b-PAN respectively through ATRP. The key features in the FTIR sup-porting the modification sequences are: a new band at 1730 cm�1
in the RHABr spectrum, which is attributed to the carbonyl vibra-tion of the ester group in RHABr (sequence 2a); an increase in the
Fig. 3. FTIR spectra of RH and the different modified RH species.
Fig. 4. AFM adhesion maps of: 500 � 500 nm2 ungrafted RH (a) and 1 � 1 lm2 RH-PMMA (b), RH PAN (c), and RH-g-PMMA-b-PAN (d).
S.M. Morsi et al. / Journal of Colloid and Interface Science 360 (2011) 377–385 381

Author's personal copy
intensity of the carbonyl band in the spectrum of RH-g-PMMAcompared to the RHABr attributed to the grafting of MMA(sequence 2b); the appearance of a new absorption band at2242.8 cm�1 in the RH-g-PAN spectrum, attributed to the nitrilegroup of AN (sequence 2c); and the presence of both the increasedcarbonyl band from the PMMA as well as the nitrile band of the ANin the spectrum of RH-g-PMMA-b-PAN (sequence 2d).
The FTIR data qualitatively support the successful modificationsproceeded as proposed, but do not quantify the modifications orthe distribution of the modifications. Therefore, AFM and XPS werealso used to characterize the different RH surfaces to understandhow well the RH surfaces are covered with grafted polymer, andif the RH-g-PMMA-b-PAN is appropriately layered to give a hydro-phobic PMMA layer at the RH and a PAN layer on the surface.
AFM gave both qualitative and quantitative information on thesurfaces of modified and unmodified RH. Figs. 4 and 5 show repre-sentative adhesion and DMT modulus maps of the samples. Fig. 4ashows the complex features and anatomy of unmodified RH. Thebasic anatomy is obscured in the images of the different graftedRH (Fig. 4b–d). The roughness of the samples was also measuredfrom the topography images (given in Supplemental information,Fig. S1). Average roughness (Ra) was equal to 7.92, 29.8 and51.5 nm on RH-PMMA, RH-PAN and RH-g-PMMA-b-PAN surfacesrespectively. This trend also correlates with the extent of graftingfrom the surface, indicating that the surfaces may not be uniformlycovered with grafted polymer. For example, the surface of unmod-ified RH is initially relatively rough (Fig. 4a), and grafting withMMA yielded oligomers with Xn � 18 (based on reacted mass andnumber of active grafting sites). This low molecular weight would
not have had a significant effect on the surface roughness. By con-trast, the AN modification yielded an Xn � 76, and the PMMA-b-ANcopolymer Xn � 103. The increase in oligomer chain length mayhave increased roughness without being sufficiently long to afforda uniform coating of the RH.
Fig. 5. AFM DMT modulus maps of: 500 � 500 nm2 ungrafted RH (a) and 1 � 1 lm2 RH-PMMA (b), RH-PAN (c), and RH-g-PMMA-b-PAN (d).
Fig. 6. XPS survey spectra of alkali-treated RH (a, top) and RHABr (b, bottom). (Axesnumbers redrawn in Photoshop� for greater visibility.).
382 S.M. Morsi et al. / Journal of Colloid and Interface Science 360 (2011) 377–385
Author's personal copy
The adhesion test (Fig. 4a–d) shows that tip adhesion to the RH-PMMA surface (mean value 16.7 nN) is much less than to the sur-faces of RH-g-PAN or RH-g-PMMA-b-PAN (mean value 23.5 and75.3 nN respectively). This is because the PMMA surface is less po-lar and harder than the other surfaces and so adheres less to theAFM tip.
The DMT modulus values (Fig. 5a–d) show that RH-g-PMMA hasthe highest modulus (mean 13.1 GPa) so it is the hardest surface.The RH-g-PAN surface is much softer than the RH-PMMA surface(mean value of only 4.0 GPa). These data correlate well with obser-vations on the adhesion maps, since it is easier for the AFM tip toseparate from a harder surface (lower adhesion force) than a softerone. Interestingly, the modulus of the RH-g-PMMA-b-PAN surfaceis the softest, at a mean value of 3.0 GPa. The RH-g-PMMA-b-PANsurface also has the thickest oligomer-modified layer. It is possiblethat the thin modulus of the RH-g-PMMA layer was influenced by
silica, which exerts less influence as the thickness of the oligomerlayer increases.
These data also supported the successful grafting reactions andnon-uniform coverage of the RH surface. However, the sample sur-faces in general are quite rough for AFM analysis, which makesimaging larger scan areas and the ‘‘transition region’’ betweenphases troublesome. Addressing these kinds of issues and improv-ing quantitative analysis is the subject of ongoing research in theYassar lab.
XPS provided detailed quantitative information about thechanges in surface composition following grafting reactions.Fig. 6 shows representative XPS spectra of alkali-treated RH andRHABr. The alkali-treated RH (Fig. 6a) exhibits four distinct lines:C 1s (285 eV), O 1s (533 eV), N 1s (402 eV) and Si 2p (103 eV) lines.Both O 1s and C 1s arise from carbohydrate components (e.g. cel-luloses and lignin) while Si 2p arises from the silica in the outersurface of RH, and the N 1s may be a contaminant. The RHABr(Fig. 6b) spectrum shows the Br 3d (70 eV) line verifying theRHABr is available for use as a heterogeneous ATRP macroinitiator.
The quantitative results from these XPS measurements aresummarized in Table 1. The Br 3d signal, representing the surfaceactive sites for ATRP, rises from 0% to 2.8%. When the RH is con-verted to RHABr the C 1s electron signal increases and the O 1s sig-nal decreases, accounting for the increased C/O and C/Si ratios. Thedata verify the successful introduction of the ATRP initiator on thesurface and its attachment through an ester bond from exposed RHhydroxyl groups. The reduction in Si 2p on the surface with the in-crease in C 1s shows the modified surface of the RHABr is consid-erably more organic than the RH, even though the yield of activebromine sites introduced onto the surface is relatively small. Thelow yield of bromine sites is attributed to the non-uniform natureof the RH substrate. That is, the outside surface of RH possessessome thin cellulose layer on the surface that is likely to be at leastpartially degraded by the base washing. The inside layer is alsothought to be mixed silica and cellulose, so the availability of pri-mary hydroxyl units to be brominated on the RH is unlikely to behigh. Bromination efficiency might be increased by improvementsin conditions, but do not appear to be necessary to accomplish con-trolled modifications of the interface region between RH and apolymer matrix.
Fig. S1 (shown in Supplemental documents) shows a typicalhigh-resolution C 1s spectra of alkali-treated RH and RHABr. Anal-ysis of the different carbon types (three for RH at 250.0, 286.4 and288.7 eV, arising from (CAC, CAH), (CAO, CAOH) and (C@O),respectively and four from RHABr at 285.0, 286.6, 288 and289.4 eV, that are attributed to (CAC, CAH), (CABr, CAO, CAOH),(C@O) and (OAC@O) respectively). These data with their relative
Table 1Percentages of different elements, C/O and C/Si ratios of alkali-treated RH and RHABr.
Rice husk Element surface composition (%) Element ratio
C 1s N 1s O 1s Si 2p Br 3d C/O C/Si
Alkali-treated RH 55.5 2.0 37.0 5.5 – 1.50 10.09RHABr 63.7 3.1 28.2 2.2 2.8 2.26 28.95
Table 2Identity, quantity, and binding energy of RH and RHABr surface carbons.
Alkali-treated RH RHABr
Binding energy (eV) Bond Carbon (%) Binding energy (eV) Bond Carbon (%)
250.0 CAC, CAH 50.6 285.0 CAC, CAH 39.0
286.4 CAO, CAOH 29.9 286.6 CABr, CAO, CAOH 37.9
288.7 C@O 19.6 288.0 C@O 13.6
289.4 OAC@O 9.5
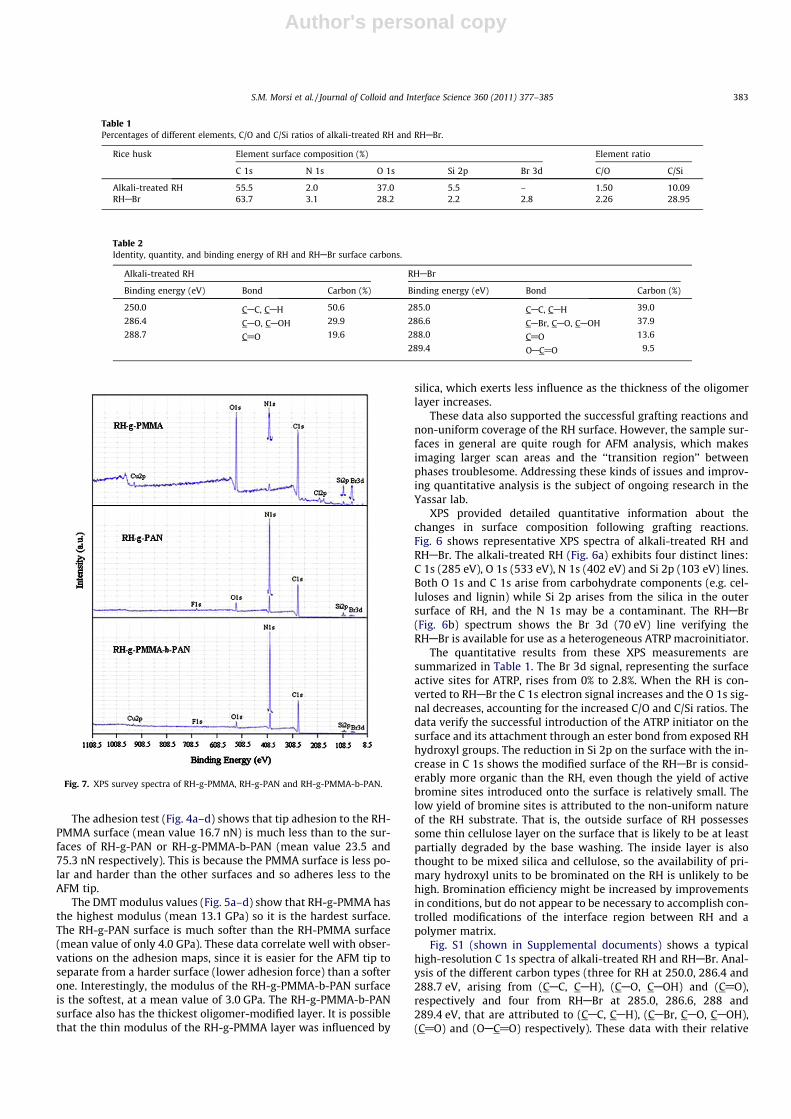
Fig. 7. XPS survey spectra of RH-g-PMMA, RH-g-PAN and RH-g-PMMA-b-PAN.
S.M. Morsi et al. / Journal of Colloid and Interface Science 360 (2011) 377–385 383

Author's personal copy
percentages and binding energies are summarized in Table 2. Thedata show that approximately 9.5% of the surface carbons are inthe a-bromoester groups that provide the ATRP active brominesites. The linking to the cellulose is through its primary hydroxylgroups, and the number of CAC and CAH groups on the surface de-creased (from 50.6% to 39.0%), while surface contributions fromdifferent CAO bonds increased.
The XPS survey spectra of RH-g-PMMA, RH-g-PAN and RH-g-PMMA-b-PAN are shown in Fig. 7. RH-g-PMMA shows strong C1s and O 1s peaks at 285 eV and 534 eV respectively that are attrib-uted to carbon and oxygen atoms of the grafted PMMA with possi-ble contribution from carbohydrate components that are very nearthe surface or are not coated by PMMA. Weak lines are also foundfrom Si 2p (100 eV) and Br 3d (69 eV). These peaks demonstrateincomplete surface coverage of the original silica surfaces of theRH. This indicates a low PMMA grafting percentage or a very thinPMMA layer, or the Si 2p regions would have been coated. Otherminor lines (Cu 2p, N 1s and Cl 2p) are likely to be residualcontaminants.
The RH-g-PAN and RH-g-PMMA-b-PAN spectra show a strong N1s line at 399 eV that is contributed by the AN nitrogen. The inten-sity of O 1s, Si 2p and Br 3d lines are less than in RH-PMMA. Thissuggests the PAN chains are higher molecular weight and coverthe RH surface more effectively than did the PMMA chains. Minorpeaks for Cu 2p and F 1s are contaminants. The quantitative surfacecomposition results are summarized in Table 3, along with per-centages of the different monomers grafted to RHABr, that werecalculated as:
G% ¼ ðWg �Wo=WoÞ � 100 ð3Þ
where Wg is the RH weight after grafting and Wo is the original RHweight.
Table 3 shows that the polymer-grafted samples possess a higherpercentage of the C 1s electron than either the alkali-washed RH orRHABr (Table 1). For RH-g-PMMA, the measured C/O ratio (2.78) is
close to the theoretical ratio (2.5). This suggests that the uppermostlayer of this structure is mostly PMMA, despite a low grafting effi-ciency (7%). The low MMA grafting efficiency may be due to its lowerreactivity compared to acrylonitrile. At 60 �C the kp � 10�3 of MMA isreported as 0.515 compared to 1.96 L mol�1 s�1, for AN [40]. Thislower reactivity is in part due to the large steric bulk of MMA (theArrhenius factor in propagation, Ap � 10�7, is given as 0.087) [40],reducing its ability to react with the ATRP active Br sites on the solidRH substrate. The XPS data, including the C/O ratio and the presenceof the Si 2p (2%), along with the low G% confirm that the surface of theRH-g-PMMA is either incompletely covered by the polymer or insome locations on the RH-g-PMMA there is a carbon contributionfrom components in the RH underlayer.
As expected, a comparison of the surface composition of RH-g-PAN and RH-g-PMMA (Table 3) shows a significant decrease in thenumber of surface oxygen atoms, along with the correspondingincrease in the number of observed nitrogen atoms. However, thelower Si 2p content (1.2, compared to 2.0 for RH-g-PMMA) sup-ports the evidence of higher molecular weight chains and higherG% (16%) leading to a more efficient coverage of the RH surface.The higher grafting of AN may be due to its lower steric bulk com-pared to MMA, which may have facilitated a more efficient initia-tion of active Br sites, and a faster propagation rate. Interestingly,the observed C/N ratio in the surface of the RH-g-PAN (4) is greaterthan the theoretical value (3). The reason is unknown, but suggeststhat the PAN chains have a conformation that gives an enrichednitrogen composition near the surface. This will be beneficial forinterfaces with some composite matrices.
The RH-g-PMMA-b-PAN possessed the highest G% of measured(22%) and, the lowest quantity of Si 2p detected on the surface.(Table 3) indicating the most complete coverage of the husk. Thecopolymerization succeeded in covering both the MMA chain seg-ments and the silica domains that had remained uncovered in theRH-g-PMMA and the RH-g-PAN. The data in Table 3 also show thatwhile some Br 3d initiating sites remained in the RH-g-PAN, no ini-
Table 3Composition, grafting %, and C/O and C/N ratios of polymer-grafted RH surfaces.
Rice husk Elemental surface composition % and grafting % Elemental ratio
Observed Theoretical
C 1s N 1s O 1s F 1s Si 2p Cl 2p Cu 2p Br 3d G% C/O C/N C/O C/N
RH-g-PMMA 65.9 4.9 23.7 – 2.0 1.9 0.1 1.5 7 2.78 – 2.5 –RH-g-PAN 72.9 18.2 7.0 0.8 1.2 – – 0.2 16 – 4 – 3RH-g-PMMA-b-PAN 74.3 19.5 4.8 0.7 0.5 – 0.3 – 22 – 3.8 – 3
Table 4Binding energy, experimental percentage and theoretical value for the four carbon components of RH-g-PMMA.
Different carbons Binding energy (eV) Carbon bond Experimental (%) Theoretical (%)
C1 285.0 ACH2AC@ 37.1 40
C2 286.3 ACH2AC@ 30.8 20
C3 287.1 AOCH320.0 20
C4 289.0 >C@O 12.2 20
Table 5Binding energy, experimental percentage and theoretical value for different carbon components of PAN and PMMA-b-PAN grafted from the surface ofRH.
Carbon Binding energy (eV) Experimental (%) Theoretical (%)
Carbon bond RH-g-PAN RH-g-PMMA-b-PAN
C1 285.0 ACH2ACH(CN)A 22.6 28.1 33.33
C2 286.2–286.3 ACH2ACH(CN)A 74.7 71.9 66.66
C@O 288.4 >C@O 2.7 – –
384 S.M. Morsi et al. / Journal of Colloid and Interface Science 360 (2011) 377–385

Author's personal copy
tiating Br 3d sites were detected in the RH-g-PMMA-b-PAN, soeither these sites reacted or also were coated by copolymer.
Because the surface composition of the RH-g-PMMA-b-PAN isnearly identical to that of RH-g-PAN, this confirms that the amphi-philic block copolymer afforded a layered surface. The primary sig-nificance of this is that it proves that an amphiphilic copolymer canbe produced on the RH surface by ATRP, despite the heterogeneousRH anatomy and composition. This too is a significant benefit forthe intended application (biobased reinforcements in composites)because it demonstrates that the surface properties of modifiedRH can be modulated to impart a needed property or protectionat the surface of the biological component while the surfaceexposed to the matrix can be designed for a different purpose, tointeract effectively with a matrix material.
High-resolution C 1s XPS spectra for the RH-g-PMMA, RH-g-PAN, and RH-g-PMMA-b-PAN are given in the Supplemental infor-mation (Fig. S2). The data from those spectra are summarized inTable 4 (RH-g-PMMA) and Table 5 (RH-g-PAN and RH-g-PMMA-b-PAN). The four C 1s peaks centered at 285.0, 286.3, 287.1 and289.0 eV, correspond to the different bonding situations of the car-bon atoms in the PMMA repeat unit The RH-g-PAN spectrumshows a small third peak at 288.4 eV, which is consistent with a>C@O band seen in the underlying RH that is not seen in RH-g-PMMA-b-PAN. These data further confirms the coverage of thePMMA chain segments and proves both a thicker polymeric layeralmost completely coating both the substrate in RH-g-PMMA-b-PAN unlike the case with the RH-g-PAN, thus confirming layeringof the amphiphilic copolymer.
4. Conclusion
Commercial RH was reacted with BiBB to place ATRP activepolymerization sites on the RH surface. Despite a relatively lowyield of initiating groups on the RH surface, these groups were ac-tive, and able to polymerize MMA and AN. MMA gives a low graft-ing efficiency giving a low molecular weight graft oligomer. FTIR,XPS, and AFM all indicated successful modification of the surface,but AFM results suggest non-uniform coverage of the RH, particu-larly in the case of RH-g-PMMA. Nevertheless, XPS confirmed thesurface was enriched in carbon with decreased O and Si, confirm-ing a low-polarity surface was produced. Nanomechanical testingfound the new surface to be rough and hard. AN was found to bemore reactive in grafting from the RH surface, and gave a softerand highly nitrogen-enriched surface. Using sequential monomeraddition it was possibly to polymerize AN directly from the surfaceof the RH-g-PMMA, yielding an amphiphilic block copolymergrafted to the RH surface. This surface was compositionally verysimilar to that of the RH-g-PAN, and based on XPS analysis nearlycompletely covered the original RH surface as well as the PMMAchain segments. Composites prepared using RH bearing amphi-philic copolymers are expected to be able to blend well into polarmaterials and possess good interfacial adhesion with such materi-als while the inner non-polar PMMA component may help stabilizethe RH against biological decay or debonding from moisturecycling.
Given the increasing demands for the use of biomass in place ofpetroleum-derived materials, and given the relative worldwideabundance of RH compared to forest resources, their may be manyas-yet untapped applications for the use of RH biomass. Here forthe first time the RH has been shown to be able to be convertedinto a heterogenous and multifunctional ATRP initiator that canundergo controlled and sequential surface polymerizations. Also,the natural architecture of RH is intended to allow one surface tofunction as a barrier material to protect the rice kernel inside.Therefore, it may be possible to take advantage of the natural bar-
rier properties of one side of the RH and the ability to induce con-trolled organic modification of the surfaces for many differentapplications.
Acknowledgments
The authors gratefully acknowledge funding from the NSFthrough Grant Number OISE-0608608. R.S.Y and A.P. acknowledgethe funding support through the NSF-DMR Grant No. 0820884. Theauthors also express their gratitude to Gerry Hammer of NESAC/BIO for XPS analysis. NESAC/BIO is funded by an NIH NIBIB Grant(EB-002027).
Appendix A. Supplementary material
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.jcis.2011.04.065.
References
[1] C. Clasen, W-M. Kulicke, Prog. Polym. Sci. 26 (2001) 1839.[2] D.G. Barkalow, R.A. Young, J. Wood Chem. Technol. 5 (1985) 293.[3] R. Khullar, V.K. Varshney, S. Naithani, P.L. Soni, Express Polym. Lett. 2 (2008) 12.[4] V. Gerardi, F. Minelli, D. Viggiano, Biomass Bioenergy 14 (1998) 295.[5] G. Mansaray, A.E. Ghaly, Biores. Technol. 65 (1998) 13.[6] P.M. Stefani, D. Garcia, J. Lopez, A. Jimenez, J. Therm. Anal. Calorim. 81 (2005)
315.[7] S. Chandrasekhar, P.N. Pramada, L. Praveen, J. Mater. Sci. 40 (2005) 6535.[8] L.F. Silvia, S.L. Milena, G.V. Alberto, R. Ricardo, R. Eduardo, Composites Part A
41 (2010) 154.[9] M.C. Emiliano, M.S. Pablo, A.R. Roxana, Bioresour. Technol. 101 (2010) 818.
[10] H.D. Rozman, L. Musa, A. Abubakar, J. Appl. Polym. Sci. 97 (2005) 237.[11] K.K. Wong, C.K. Lee, K.S. Low, M.J. Haron, Chemosphere 50 (2003) 23.[12] P. Olivier, K. Mohammed, B. Julien, T. Wim, N.B. Mohamed, Acta Mater. 58
(2010) 792.[13] T. Morgan, C. Aurelia, F. Etienne, S. Martina, B. Julien, Macromol. Rapid
Commun. 31 (2010) 1751.[14] C.J. Hawker, A.W. Bosman, E. Harth, Chem. Rev. 101 (2001) 3661.[15] K. Matyjaszewski, J. Xia, Chem. Rev. 101 (2001) 2921.[16] M. Kato, M. Kamigaito, M. Sawamoto, T. Higashimura, Macromolecules 28
(1995) 1721.[17] J. Chiefari, Y.K. Chong, F. Ercole, J. Krstina, J. Jeffery, T.P. Le, R.T.A. Mayadunne,
G.F. Meijs, C.L. Moad, G. Moad, E. Rizzardo, S.H. Thang, Macromolecules 31(1998) 5559.
[18] G. Morandi, L. Heath, W. Thielemans, Langmuir 25 (2009) 8280.[19] M. Kamigaito, T. Ando, M. Sawamoto, Chem. Rev. 101 (2001) 3689.[20] Z. Zhang, T. Chao, S. Chen, S. Jiang, Langmuir 22 (2006) 10072.[21] D. Li, Y. Cui, K. Wang, Q. He, X. Yan, J. Li, Adv. Funct. Mater. 17 (2007) 3134.[22] Q. Wei, J. Ji, J. Shen, Macromol. Rapid Commun. 29 (2008) 645.[23] E. Marutani, S. Yamamoto, T. Ninjbadgar, Y. Tsujii, T. Fukuda, M. Takano,
Polymer 45 (2004) 2231.[24] K. Ohno, T. Morinaga, K. Koh, Y. Tsujii, T. Fukuda, Macromolecules 38 (2005)
2137.[25] A. Ramakrishnan, R. Dhamodharan, J. Ruehe, J. Polym. Sci., Part A: Polym.
Chem. 44 (2006) 1758.[26] J. Lindqvist, E. Malmstroem, J. Appl. Polym. Sci. 100 (2006) 4155.[27] J. Lindqvist, D. Nystroem, E. Oestmark, P. Antoni, A. Carlmark, M. Johansson, A.
Hult, E. Malmstroem, Biomacromolecules 9 (2008) 2139.[28] R. Westlund, A. Carlmark, A. Hult, E. Malmstroem, I.M. Saez, Soft Matter 3
(2007) 866.[29] A. Carlmark, E. Malmstrom, Biomacromolecules 4 (2003) 1740.[30] D. Plackett, K. Jankova, H. Egsgaard, S. Hvilsted, Biomacromolecules 6 (2005)
2474.[31] N. Singh, Z. Chen, N. Tomer, S.R. Wickramasinghe, N. Soice, S.M. Husson, J.
Membr. Sci. 311 (2008) 225.[32] M. Xiao, S. Li, W. Chanklin, A. Zheng, H. Xiaoa, Carbohydr. Polym. 83 (2011)
512–519.[33] G. Zampano, M. Bertoldo, S. Bronco, Carbohydr. Polym. 75 (2009) 22–31.[34] M. Coskun, M.M. Temuez, Polym. Int. 54 (2005) 342.[35] S.C. Byun, I.O. Jung, M.Y. Kim, et al., J. Nanosci. Nanotechnol. 11 (2) (2011)
1305–1309.[36] B.S. Ndazi, S. Karlsson, J.V. Tesha, Composites Part A 38 (2007) 925.[37] S.C. Byun, I.O. Jung, M.Y. Kim, S.J. So, C. Yang, C. Kim, G. Lei, C.S. Han, J. Nanosci.
Nanotechnol. 11 (2) (2011) 1305.[38] B. Pittenger, N. Erina, S. Chanmin, Quantitative Mechanical Mapping at
Nanoscale with Peak Force QNM, Bruker Application Note, Bruker, 2009,<www.briker-axs.de>.
[39] B.V. Derjaguin, V.M. Muller, Y.P. Toropov, J. Colloid Interface Sci. 53 (1975) 314.[40] George Odian (Ed.), in: Principles of Polymerization, fourth ed., vol. 270, Wiley
Interscience, Hoboken, NJ, USA, 2004
S.M. Morsi et al. / Journal of Colloid and Interface Science 360 (2011) 377–385 385
Copyright © 2022 FDOKUMEN




















