
Electrochemical investigation of Mn4O4-cubane water-oxidizing clusters
9
Electrochemical investigation of Mn 4 O 4 -cubane water-oxidizing clustersw Robin Brimblecombe, ab Alan M. Bond,* a G. Charles Dismukes,* b Gerhard F. Swiegers c and Leone Spiccia* a Received 22nd January 2009, Accepted 16th April 2009 First published as an Advance Article on the web 26th May 2009 DOI: 10.1039/b901419e High valence states in manganese clusters are a key feature of the function of one of the most important catalysts found in nature, the water-oxidizing complex of photosystem II. We describe a detailed electrochemical investigation of two bio-inspired manganese–oxo complexes, [Mn 4 O 4 L 6 ] (L = diphenylphosphinate (1) and bis(p-methoxyphenyl)phosphinate (2)), in solution, attached to an electrode surface and suspended within a Nafion film. These complexes contain a cubic [Mn 4 O 4 ] 6+ core stabilized by phosphinate ligands. They have previously been shown to be active and durable photocatalysts for the oxidation of water to dioxygen. A comparison of catalytic photocurrent generated by films deposited by two methods of electrode immobilization reveals that doping of the catalyst in Nafion results in higher photocurrent than was observed for a solid layer of cubane on an electrode surface. In dichloromethane solution, and under conditions of cyclic voltammetry, the one-electron oxidation processes 1/1 + and 2/2 + were found to be reversible and quasi-reversible, respectively. Some decomposition of 1 + and 2 + was detected on the longer timescale of bulk electrolysis. Both compounds also undergo a two-electron, chemically irreversible reduction in dichloromethane, with a mechanism that is dependent on scan rate and influenced by the presence of a proton donor. When immersed in aqueous electrolyte, the reduction process exhibits a limited level of chemical reversibility. These data provide insights into the catalytic operation of these molecules during photo-assisted electrolysis of water and highlight the importance of the strongly electron-donating ligand environment about the manganese ions in the ability of the cubanes to photocatalyze water oxidation at low overpotentials. Introduction The catalytic site of the O 2 -evolving water-oxidizing complex (WOC) in photosystem II (PSII) features an inorganic cluster composed of an Mn 4 Ca 1 core. This core is conserved in all known oxygen photosynthetic organisms. 1 A range of manganese complexes inspired by the PSII-WOC have been reported. A small number of these are capable of catalytically oxidizing water, although usually only at slow rates and with limited turnovers in the presence of sacrificial chemical oxidants like Ce IV (1.72 V vs. NHE). 2,3 Chemical oxidants are commonly used in lieu of more complex, light-driven photo-oxidants in such homogeneous ‘‘artificial photo- synthesis’’ systems. Such chemical oxidants have the dis- advantage that they complicate both the system and the interpretation of the results. Two approaches are available to eliminate the need for sacrificial chemical oxidants. These involve the attachment of the catalysts to conductive surfaces for direct electrochemical oxidation or to photoanodes for light-induced oxidation, as in a solar cell. In this work we consider only the former approach. In a recent study, we described the sustained, catalytic, light- driven electro-oxidation of water by a bio-inspired Mn complex containing a [Mn 4 O 4 ] 6+/7+ cubane core, 4 supported by six facially capping diarylphosphinate ligands, L, giving a [Mn 4 O 4 L 6 ] molecule (Fig. 1). 5 The formal oxidation states of the manganese ions in this cluster are [Mn III 2 Mn IV 2 ]. Two ligands have been employed: L = diphenylphosphinate, [Mn 4 O 4 (Ph 2 PO 2 ) 6 ](1), and bis(p-methoxyphenyl)phosphinate, [Mn 4 O 4 ((CH 3 OPh) 2 PO 2 ) 6 ](2). 5,6 Both complexes have been investigated in detail from both a structural and a chemical reactivity perspective. 5–12 The oxidized cubanes, [Mn III Mn 3 IV O 4 L 6 ] + (ClO 4 ), 1 + ClO 4 and 2 + ClO 4 , hereafter denoted the cubium oxidation state, have been previously isolated and characterized. 8,11 When trapped in a thin layer of Nafion cast onto an electrode surface, 2 + is an active and durable catalyst capable of photo-oxidizing water at an applied potential of 1.00 V vs. Ag/AgCl. 4 Chemical reactivity studies have revealed that the [Mn 4 O 4 L 6 ] 0/1+ cubane complexes are exceptionally strong oxidizing agents, with the core oxygen atoms capable of oxygenation and H-atom abstraction reactions. 7,8 The neutral cubane is capable of H-atom abstraction by cleaving RO–H a School of Chemistry, Monash University, Victoria 3800, Australia. E-mail: [email protected], [email protected] b Department of Chemistry & Princeton Environmental Institute, Princeton University, NJ 08544, USA. E-mail: [email protected] c Intelligent Polymer Research Institute, University of Wollongong, Wollongong, NSW 2522, Australia w Electronic supplementary information (ESI) available: Tables S1, S2 and S3 provide summaries of cyclic voltammetry data for solution oxidation and reduction processes. Cyclic voltammograms obtained at a glassy carbon electrode for the oxidation and reduction of 1 and 2 in dichloromethane (Fig. S1) and glassy carbon rotating disc voltammo- gram for the reduction of 1 in dichloromethane (Fig. S2). See DOI: 10.1039/b901419e This journal is c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 6441–6449 | 6441 PAPER www.rsc.org/pccp | Physical Chemistry Chemical Physics
-
Upload
eastanglia -
Category
Documents
-
view
0 -
download
0
Transcript of Electrochemical investigation of Mn4O4-cubane water-oxidizing clusters

Electrochemical investigation of Mn4O4-cubane water-oxidizing clustersw
Robin Brimblecombe,ab
Alan M. Bond,*aG. Charles Dismukes,*
b
Gerhard F. Swiegerscand Leone Spiccia*
a
Received 22nd January 2009, Accepted 16th April 2009
First published as an Advance Article on the web 26th May 2009
DOI: 10.1039/b901419e
High valence states in manganese clusters are a key feature of the function of one of the most
important catalysts found in nature, the water-oxidizing complex of photosystem II. We describe
a detailed electrochemical investigation of two bio-inspired manganese–oxo complexes, [Mn4O4L6]
(L = diphenylphosphinate (1) and bis(p-methoxyphenyl)phosphinate (2)), in solution, attached to
an electrode surface and suspended within a Nafion film. These complexes contain a cubic
[Mn4O4]6+ core stabilized by phosphinate ligands. They have previously been shown to be active
and durable photocatalysts for the oxidation of water to dioxygen. A comparison of catalytic
photocurrent generated by films deposited by two methods of electrode immobilization reveals
that doping of the catalyst in Nafion results in higher photocurrent than was observed for a solid
layer of cubane on an electrode surface. In dichloromethane solution, and under conditions of
cyclic voltammetry, the one-electron oxidation processes 1/1+ and 2/2+ were found to be
reversible and quasi-reversible, respectively. Some decomposition of 1+ and 2+ was detected on
the longer timescale of bulk electrolysis. Both compounds also undergo a two-electron, chemically
irreversible reduction in dichloromethane, with a mechanism that is dependent on scan rate and
influenced by the presence of a proton donor. When immersed in aqueous electrolyte, the reduction
process exhibits a limited level of chemical reversibility. These data provide insights into the
catalytic operation of these molecules during photo-assisted electrolysis of water and highlight
the importance of the strongly electron-donating ligand environment about the manganese ions in
the ability of the cubanes to photocatalyze water oxidation at low overpotentials.
Introduction
The catalytic site of the O2-evolving water-oxidizing complex
(WOC) in photosystem II (PSII) features an inorganic cluster
composed of an Mn4Ca1 core. This core is conserved in
all known oxygen photosynthetic organisms.1 A range of
manganese complexes inspired by the PSII-WOC have been
reported. A small number of these are capable of catalytically
oxidizing water, although usually only at slow rates and with
limited turnovers in the presence of sacrificial chemical
oxidants like CeIV (1.72 V vs. NHE).2,3 Chemical oxidants
are commonly used in lieu of more complex, light-driven
photo-oxidants in such homogeneous ‘‘artificial photo-
synthesis’’ systems. Such chemical oxidants have the dis-
advantage that they complicate both the system and the
interpretation of the results. Two approaches are available
to eliminate the need for sacrificial chemical oxidants. These
involve the attachment of the catalysts to conductive surfaces
for direct electrochemical oxidation or to photoanodes for
light-induced oxidation, as in a solar cell. In this work we
consider only the former approach.
In a recent study, we described the sustained, catalytic, light-
driven electro-oxidation of water by a bio-inspired Mn
complex containing a [Mn4O4]6+/7+ cubane core,4 supported
by six facially capping diarylphosphinate ligands, L, giving a
[Mn4O4L6] molecule (Fig. 1).5 The formal oxidation states of
the manganese ions in this cluster are [MnIII2MnIV2]. Two
ligands have been employed: L = diphenylphosphinate,
[Mn4O4(Ph2PO2)6] (1), and bis(p-methoxyphenyl)phosphinate,
[Mn4O4((CH3OPh)2PO2)6] (2).5,6 Both complexes have been
investigated in detail from both a structural and a chemical
reactivity perspective.5–12 The oxidized cubanes,
[MnIIIMn3IVO4L6]
+(ClO4�), 1+ClO4
� and 2+ClO4
�, hereafter
denoted the cubium oxidation state, have been previously
isolated and characterized.8,11
When trapped in a thin layer of Nafion cast onto an
electrode surface, 2+ is an active and durable catalyst capable
of photo-oxidizing water at an applied potential of 1.00 V vs.
Ag/AgCl.4 Chemical reactivity studies have revealed that the
[Mn4O4L6]0/1+ cubane complexes are exceptionally strong
oxidizing agents, with the core oxygen atoms capable of
oxygenation and H-atom abstraction reactions.7,8 The neutral
cubane is capable of H-atom abstraction by cleaving RO–H
a School of Chemistry, Monash University, Victoria 3800, Australia.E-mail: [email protected],[email protected]
bDepartment of Chemistry & Princeton Environmental Institute,Princeton University, NJ 08544, USA.E-mail: [email protected]
c Intelligent Polymer Research Institute, University of Wollongong,Wollongong, NSW 2522, Australiaw Electronic supplementary information (ESI) available: Tables S1, S2and S3 provide summaries of cyclic voltammetry data for solutionoxidation and reduction processes. Cyclic voltammograms obtained ata glassy carbon electrode for the oxidation and reduction of 1 and 2 indichloromethane (Fig. S1) and glassy carbon rotating disc voltammo-gram for the reduction of 1 in dichloromethane (Fig. S2). SeeDOI: 10.1039/b901419e
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 6441–6449 | 6441
PAPER www.rsc.org/pccp | Physical Chemistry Chemical Physics

and RN–H bonds that are greater than 390 kJ mol�1 in
strength while the oxidized cubane (1+) can abstract hydride
anions, cleaving bonds of 530 kJ mol�1 (top line in Fig. 1).13
One striking comparison is that the O–H bond in the reduced
complex with the [Mn4O3(OH)]6+ core requires in excess of
390 kJ mol�1 to dissociate H and regenerate the [Mn4O4]6+
core vs. only 320 kJ mol�1 for the O–H bond in analogous
dimanganese complexes containing a rhombohedral core,
[Mn2O(OH)]3+ - [Mn2O2]3+.7 Thus, the oxo bridges of the
expanded [Mn4O4]6+ cubane cores form stronger O–H bonds
than those in the [Mn2O2]3+ rhombohedral core.13
The redox potentials of manganese complexes are strongly
influenced by the donor atoms and in particular the electron-
donating/withdrawing potential of the ligands.14 The presence
of a high charge of 7+ in the [Mn4O4] core elevates
the potential relative to monomeric metal ions. The higher
oxidation states of the cubane core can be stabilized by the
addition of electron-donating methoxy groups at the para-
position of the phosphate phenyl rings.6,11
Previous attempts to covalently attach the cubane to
conductive surfaces and to thereby form monolayers on gold
surfaces were successful using the iodine atom of p-iodo-
phenylphosphinate to bridge between the cubane core and a
gold surface.15 These monolayers were shown to be electro-
chemically active. However, the bridge was unstable and
shown to oxidatively cleave upon reaching the I�/IO3�
potential. Attempts to functionalize the phenyl rings at the
para-position with anchoring groups, such as carboxylic acid
and hydroxyl groups, have proven unsuccessful due to the
interference of these groups in cube formation.
In order to better understand the basis for the strong oxidative
capacity of the cubium state and to characterize the lower redox
states of these complexes, we have examined in detail, and describe
herein, their solution phase electrochemistry. We also compare
catalytic activity in aqueous electrolyte environments, when
the complexes are immobilized on electrode surfaces either by
deposition as a solid film or when ion-exchanged within thinNafion
membranes cast on the electrode surface.
Experimental section
Materials and methods
Diphenylphosphinic acid and bis(methoxyphenyl)phosphinic
acid were purchased from Lancaster and Aldrich, respectively,
and used without further purification. Tetrabutylammonium
hexafluorophosphate (Bu4NPF6) was obtained from GFS
Chemicals and was used as the electrolyte in organic solvents.
It was purified as follows prior to use: a sample of Bu4NPF6
was dissolved in acetone and KPF6 was added to precipitate
iodide impurities (as potassium iodide). The solution was
then filtered, evaporated to dryness and recrystallized from
ethanol. The resulting crystalline solid was dissolved in
dichloromethane. An insoluble white powder was removed
by filtration and the solvent evaporated to dryness to produce
electrochemically pure Bu4NPF6. Nafions was purchased
from Dupont as a Nafion-PFSA polymer dispersion DE
1020, which is an aqueous dispersion of 10–12% Nafion. All
other reagents were purchased from BDH or Aldrich and
used as received. [Mn4O4L16], compound 1 (L1 = diphenyl-
phosphinate),5 [Mn4O4L26], compound 2 (L2 = bis(p-methoxy-
phenyl)phosphinate),6 and 2+ClO4� were prepared11
as described in the literature. The pH of the aqueous solutions
and buffers used in the electrochemical studies were adjusted
by addition of either 0.1 M H2SO4 or 0.1 M NaOH to
aqueous 0.1 M Na2SO4 supporting electrolyte. A 10 mM
MOPS buffer was prepared by dissolving 3-(N-morpholino)-
propanesulfonic acid in water and adjusting the pH to 7 using
NaOH. The ionic strength of this buffer solution was adjusted
to 0.1 M using Na2SO4. pH values were measured with a
Metrohm 6.0234.100 pH electrode and 744 pH meter.
Electrochemistry
Electrochemical experiments were conducted at 22 (�2) 1C
with BAS (Bio Analytical Systems) Epsilon CS3 or BAS 100B
workstations. Cyclic voltammograms were obtained at scan
rates of 5 to 500 mV s�1 in a conventional three electrode
electrochemical cell containing inlet and outlet ports for
degassing solutions with nitrogen. Experiments in CH2Cl2 or
CH3CN (0.1 M Bu4NPF6) used an Ag/Ag+ (0.01 M AgNO3
in CH3CN) reference electrode, with a double glass frit
separating the electrode from the test solution. The ferrocene/
ferrocenium (Fc/Fc+) oxidation process was used to provide
an internal reference potential calibration system for
electrochemical studies in organic solvents, with potentials
adjusted to the Fc/Fc+ scale (0.400 V vs. NHE at 25 1C).
Aqueous experiments were conducted in distilled water
containing 0.1 M Na2SO4 as the electrolyte. In this case,
potentials were referenced against a BAS Ag/AgCl (3 MNaCl)
glass bodied reference electrode separated from the test solu-
tion by a Vycor frit, with a potential of 0.200 V vs. NHE at
25 1C (BAS reference electrode users manual). Potentials
(measured against Ag/AgCl (3 M NaCl)) are presented vs.
Fig. 1 Schematic representation of [Mn4O4L6], 1: L = diphenly-
phosphinate (PO2(Ph)2), and 2: L = bis(p-methoxyphenyl)phosphinate
(PO2(PhOMe)2), and previously observed oxidation states and
proposed reduced intermediates.
6442 | Phys. Chem. Chem. Phys., 2009, 11, 6441–6449 This journal is �c the Owner Societies 2009

Fc/Fc+ to allow comparison to organic solution measurements,
converted by adjusting the potential by�0.200 V. All voltammetric
experiments used Pt auxiliary electrodes. The working
electrodes used were 3 mm diameter glassy carbon and Pt disc
electrodes. The glassy carbon working electrode had an
electroactive area of 5.9 mm2 as determined by oxidation of
1.0 mM ferrocene in CH3CN (0.5 M Bu4NPF6) and use of the
Randles–Sevcik equation2 with a diffusion coefficient (D) of
1.7 � 10�5 cm�2 s�1 (CH3CN, 0.5 M Bu4NBF4).2
Bulk electrolysis experiments in CH2Cl2 (0.1 M Bu4NPF6)
were conducted under a nitrogen atmosphere with the same
Ag/Ag+ (0.01 M AgNO3 in CH3CN) reference electrode as
used in voltammetry, a large area glassy carbon working
electrode, and a platinum basket counter electrode. The bulk
electrolysis cell consisted of two compartments. The outer one
contained the counter electrode and a stirring rod immersed in
CH2Cl2 (0.1 M Bu4NPF6). This compartment was separated
by a glass frit from the inner compartment which contained 1
or 2 dissolved in CH2Cl2 (0.1 M Bu4NPF6) as well as the glassy
carbon working and the Ag/Ag+ reference electrode. Bulk
electrolysis experiments were stopped when the current
decayed to 1% of the initial value.
Solid film preparation
Electrodes were coated with a thin film of cubane by casting a
small volume of a CH2Cl2 solution of the complex onto the
electrode surface and allowing it to dry. Alternatively, films
were prepared by rubbing the face of the electrode over a small
amount of dry sample.
Nafion film deposition and Nafion doping
Nafion-modified electrodes were prepared by drop-casting
0.5 mL of 10% Nafion in ethanol onto a 3 mm diameter
electrode and allowing it to air dry before heating in a
laboratory oven at 120 1C for 20 min. Doping of the cast
Nafion membrane was achieved by immersion in a 2 mM
CH3CN solution of 2+ClO4�. 2+ in Nafion is reduced to 2, at
the initial potential used in cyclic voltammetry.
UV-visible spectrophotometry
The UV-visible spectra of 2+ in CH2Cl2 and 2+ supported in
Nafion in water were measured in 1 cm quartz cuvettes using a
Varian Cary 300 BIO spectrometer. Nafion membranes were
cast on the inside of quartz cuvettes, dried at 120 1C for
20 min, doped in situ with 2 mM solutions of 2+, rinsed with
acetonitrile, and then air-dried.
Light source
The xenon light source used for these experiments generated
white light with a stable output over the range 250–750 nm, from
a Rofin Australia-Polilight PL6, passed through a 1 m long
liquid light guide. Illumination experiments were conducted at a
light intensity of approximately 500 mW cm�2, measured at the
electrode surface.
Results and discussion
The cyclic voltammetry of 1 and 2 was investigated in three
different environments: (i) dissolved in CH2Cl2, (ii) as a solid
layer on the electrode surface in contact with water, and
(iii) when suspended in a Nafion membrane cast on the
electrode surface, again in contact with water. Using these
procedures, oxidation and reduction processes of the cubanes
are now described in detail.
Compound 1 has a very low solubility in many solvents
arising from the hydrophobic outer shell formed by the 12
phenyl rings of the phosphinate ligands. However, moderate
solubility is achieved in dichloromethane. As a consequence,
the solution electrochemistry for both compounds could be
studied over a concentration range of 0.4–1.0 mM in this
solvent. The nature of both oxidation and reduction processes
in dichloromethane was investigated by cyclic voltammetry,
rotating disc and bulk electrolysis; the reduction process was
studied in the presence of proton donors.
The electrochemistry of 1 was examined as a solid layer on
the surface of a glassy carbon electrode in contact with
aqueous electrolyte. Layers of this type were prepared by both
rubbing the electrode in solid cubane or casting a solution of
cubane dissolved in dichloromethane onto the electrode
surface and allowing the solvent to evaporate. Solid layers
were immersed in aqueous solutions of 0.1 M Na2SO4 and
were physically stable for at least 24 h.
The incorporation of 2+ into Nafion films was achieved by
immersing Nafion-modified glassy carbon electrodes into a
solution of 2+ClO4� (2 mM in CH3CN).4 Cubane in-
corporation was confirmed by the UV-visible and NMR
spectra of doped membranes on quartz,4 and by cyclic
voltammetry of doped films deposited on electrode surfaces
and immersed in an aqueous (0.1 M Na2SO4) electrolyte. The
voltammetry was also investigated when the Nafion-modified
electrode was in contact with buffered aqueous electrolyte.
Representative cyclic voltammograms obtained at slow scan
rates for each environment studied are displayed in Fig. 2.
Compounds 1 and 2 each exhibit one chemically reversible
oxidation and one chemically irreversible process in CH2Cl2(0.1 M Bu4NPF6). Voltammetry of 1 as a solid film on the
electrode surface in contact with aqueous 0.1 M Na2SO4
exhibits both an oxidation and a reduction process as well as
producing additional oxidation peaks at less positive potentials
after sweeping through the reduction step. Suspension of 2+ in
Nafion and immersion in aqueous electrolyte (0.1 M Na2SO4)
also reveal a well-defined process at positive potentials that is
known to generate a highly reactive 2+ species that acts as a
water-oxidation catalyst under conditions of bulk electrolysis.
For the Nafion system, the peak current magnitudes for the
reduction process are relatively weak compared to the waves
for the oxidation process (Fig. 2).
Oxidation in dichloromethane solution
Cyclic voltammograms for oxidation of 1 and 2 (scan rate of
10 mV s�1) are depicted in Fig. 3(A). A summary of
the electrochemical data derived from these experiments is
presented as a function of scan rate in Tables S1 and S2, ESIw.For compound 1, the reversible formal potential (E1f) derived
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 6441–6449 | 6443

from the average of the oxidation (Eoxp ) and reduction (Ered
p )
peak potentials [(Eoxp � Ered
p )/2] gives a value of 690 � 3 mV,
which, as expected, is almost unaffected by scan rate, whilst
the peak-to-peak separation (DEp) values observed for 1 are
slightly larger than the scan rate independent value of about
56 mV at 22 1C that is theoretically predicted for a reversible
one-electron process. They are equivalent to those obtained
for the known, reversible, one-electron Fc/Fc+ oxidation (for
the same Fc concentration and at the same scan rate in CH2Cl2(0.1 M Bu4NPF6)). Thus, oxidation of 1 is concluded to
represent an electrochemically and chemically reversible
one-electron process, with the higher than theoretically
expected DEp value predominantly attributed to un-
compensated ohmic IR drop in the high resistance CH2Cl2medium. The even larger DEp values for the oxidation of 2
imply that the rate of electron transfer for the 20/2+ process is
slower than for either 10/+ or Fc0/+. The oxidation of 2 is
therefore considered to be quasi-reversible at a glassy carbon
electrode. The E1f values determined by cyclic voltammetry for
the Mn4O4L6 " [Mn4O4L6]+ + e� processes are 690 � 3 mV
and 542 � 8 mV for 1 and 2, respectively, representing a
difference in E1f values of 150 mV.
In the initial brief report by Wu et al.,6 a difference in the E1fvalues of 109 mV was reported in CH2Cl2 (0.1 M Bu4NPF6)
(E1f values for 1 and 2 (0.4 mM) were 813 mV and 704 mV,
respectively (vs. Ag/Ag+ (0.1 M AgNO3 in CH3CN))). Also,
DEp values were reported to be much larger than those found
here (297 mV vs. 95 mV for 2 at a scan rate of 100 mV s�1),
suggesting that a higher level of uncompensated resistance was
present. This would have introduced considerable uncertainty
into the E1f values.6 The difference in the E1f values, for
1+ClO4�/1 and 2+ClO4
�/2, of 150 mV found in a more recent
study11 compares favorably with the result from the
current study.
The half-wave (E1/2) potentials obtained from rotating
disc voltammetry (Fig. 3(B)) were slightly more positive than
the E1f values, again consistent with the presence of un-
compensated IR drop and quasi-reversibility. For 1, E1/2
becomes slightly more positive with increasing rotation speed.
In contrast, E1/2 increased significantly with rotation rate for
2, as expected for a quasi-reversible system. E1/2 values range
from 696 to 709 mV for 1 and 542 to 607 mV for 2 (vs. Fc/Fc+)
when the scan rate changed from 500 to 5000 rpm. Diffusion
coefficients of 4.6 (�0.1) � 10�6 cm2 s�1 for 1 and 3.5 (�0.2) �10�6 cm2 s�1 for 2 were calculated from the rotation rate data
Fig. 2 Cyclic voltammograms obtained at 22 1C and with a scan rate
of 10 mV s�1 at a glassy carbon electrode for oxidation and reduction
of: (A) 0.4 mM CH2Cl2 (0.1 M Bu4NPF6) solutions of 1 (----) and 2
(—), 10 mV s�1; (B) solid layer of 1 adhered to the glassy carbon
electrode surface in contact with aqueous (0.1 M Na2SO4);
(C) 2+-doped in Nafion adhered to a glassy carbon electrode and
placed in contact with aqueous (0.1 M Na2SO4) electrolyte.
Fig. 3 Voltammograms obtained for oxidation of 1 and 2 at 22 1C at
a glassy carbon electrode: (A) Top: cyclic voltammetry with a scan rate
of 10 mV s�1 for 0.4 mM 1 (----) and 2 (—) in CH2Cl2 (0.1 M
Bu4NPF6); (B) Bottom: rotating disc voltammogram obtained for
1 mM 2 with a rotation rate of 500 rpm and a scan rate of 5 mV s�1.
6444 | Phys. Chem. Chem. Phys., 2009, 11, 6441–6449 This journal is �c the Owner Societies 2009

using the Levich equation. The slightly lower value for 2 may
be attributed to its higher molecular weight (1952 amu for 2 vs.
1587 amu for 1).
Bulk oxidation at a glassy carbon electrode of solutions of 1
(0.4 mM) and 2 and (1 mM) in CH2Cl2 was conducted at
a potential 100 mV more positive than the rotating disc
voltammetric E1/2 value for each complex. Coulometric
analysis of these experiments gave an n-value of 0.97 � 0.05
electrons, providing confirmation that the oxidation of 1 and 2
is a one-electron process (eqn (1)).
[MnIII2MnIV2O4L6] " [MnIIIMnIV3O4L6]+ + e� (1)
Reductive back electrolysis of the oxidized compounds yielded
an average of 67% of the charge collected in the oxidation
process, suggesting [Mn4O4L6]+ is not entirely stable in
CH2Cl2 over the time period of the experiment (ca. 1 hour).
The limiting current recorded by rotating disc voltammetry,
after bulk oxidation followed by back electrolysis, gave an
average limiting current of 68% of the magnitude of the
pre-electrolysis limiting current value, consistent with the
coulometry. Analysis of the rotating disc voltammogram
obtained after bulk oxidation indicated that the E1/2 values
of 1 and 2, after allowing for the uncompensated IR drop,
were consistent with those obtained before electrolysis.
The UV-visible spectrum before and after bulk electrolysis
of 1 is shown in Fig. 4. The sharp absorption maxima at ca.
230 nm is attributed to a p - p* transition within the phenyl
rings of the ligands and, as expected, remains present when the
cubane is oxidized. There is a significant increase in absorption
in the range of 300–400 nm after oxidation. The intensity of
the bands in this region suggests that they are due to charge
transfer transitions. Thus, oxidation appears to reduce the
energy required for charge transfer within the complex which
agrees with the shortened metal–ligand bond lengths observed
in the crystal structure of 1+ClO4�.11
No other oxidation processes were observed for either
cubane between 1.0 and 2.0 V (vs. Fc/Fc+) (the latter potential
being the solvent/electrolyte limit). Thus, in CH2Cl2, even with
each manganese center coordinated to three oxo and three
phosphinate oxygens, it is not possible to reach a 4 MnIV state
at potentials less positive than 2.00 V.
Oxidation in Nafion
The oxidation process of the 2/2+ couple in Nafion at a scan
rate of 50 mV s�1 gave Eoxp = 738 mV, Ered
p = 517 mV,
and a mid-point potential of Em calculated as the value
[(Eoxp + Ered
p )/2] = 628 mV vs. Fc/Fc+ (Fig. 5(A)). Ep varied
with scan rate, suggesting significant resistance within the
system. The substantial level of chemical reversibility of the
2/2+ process suggests that the majority of the neutral cubane,
2 (generated electrochemically within the Nafion), is retained
within the membrane. This is likely due to an association
between the hydrophobic regions within the substructure of
the Nafion membrane and the hydrophobic aryl groups of 2,
which are insoluble in aqueous solution. Evidence in support
of this interpretation comes from the substantial red shift
(30 nm) in the ligand p - p* transition at 230 nm.4
In unbuffered aqueous electrolyte, the 2/2+ Nafion/GCE
oxidation process is initially well defined. However, with
potential cycling, the peak current deteriorates and the peak
potentials shift to less positive values (Fig. 5(A)). This pheno-
menon was also observed for oxidation of Mn(II) in Nafion/
GCE (Fig. 5(B)), revealing it to be a feature of the Nafion
support. Equivalent experiments in buffered solution at pH 3.3
lead to stable peak currents and potentials with smaller peak-
to-peak separations (Fig. 6(A)) than those observed in
unbuffered solution (Fig. 5(A)). In buffered solution at
pH 7, the peak current rapidly diminishes upon repetitive
cycling of the potential with the process becoming poorly
defined even after one sweep (Fig. 6(B)). After transferring
this electrode to a pH 3.3 solution environment, the oxidation
Fig. 4 UV-visible absorption spectra in CH2Cl2 (0.1 M Bu4NPF6) at
22 1C of 1 before (TT) and after (---) one-electron oxidative bulk
electrolysis with the potential of the glassy carbon working electrode
held at 950 mV (vs. Fc/Fc+) and after (—) two-electron bulk reduction
of 1, with the electrode potential held at �700 mV vs. Fc/Fc+, stirring
under N2.
Fig. 5 Cyclic voltammetry at 22 1C of 2+ (A) andMn2+ (B) doped in
Nafion on a glassy carbon electrode in H2O (0.1 MNa2SO4), scan rate:
50 mV s�1.
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 6441–6449 | 6445
process again became well defined. Thus, the voltammetric
response and conductivity of the system appear to be depen-
dent on the proton concentration within the Nafion.
Our findings imply that the Nafion support provides good
electrical contact between a fraction of the absorbed cubane
and the electrode, with some variability in peak current
magnitudes observed between different preparations. This
is presumably due to the variability of hand casting the
membranes onto the electrode surface. The success of this
system facilitated the investigation of the catalytic potential of
these complexes.4
Electro-photocatalytic oxidation of water
In a previous communication, we reported the photo-assisted
electrocatalytic oxidation of water using 2+–Nafion-coated
electrodes.4 In those experiments 2+–Nafion-modified electro-
des were poised at 0.8 V (vs. Fc/Fc+), above the oxidation
potential of the manganese cubanes, and illuminated with
light. The resulting photocurrent was shown to correlate with
the oxidation of water to oxygen and protons. In the present
study, we compare the photocurrent from the two methods of
electrode immobilization described above. Attempts to dope
1+ into Nafion were unsuccessful due to its insolubility
in polar solvents, which appears to be a pre-requisite for
achieving successful penetration into the Nafion membranes.
Photocurrent comparisons are thus made only for 2+, which is
readily soluble in acetonitrile.
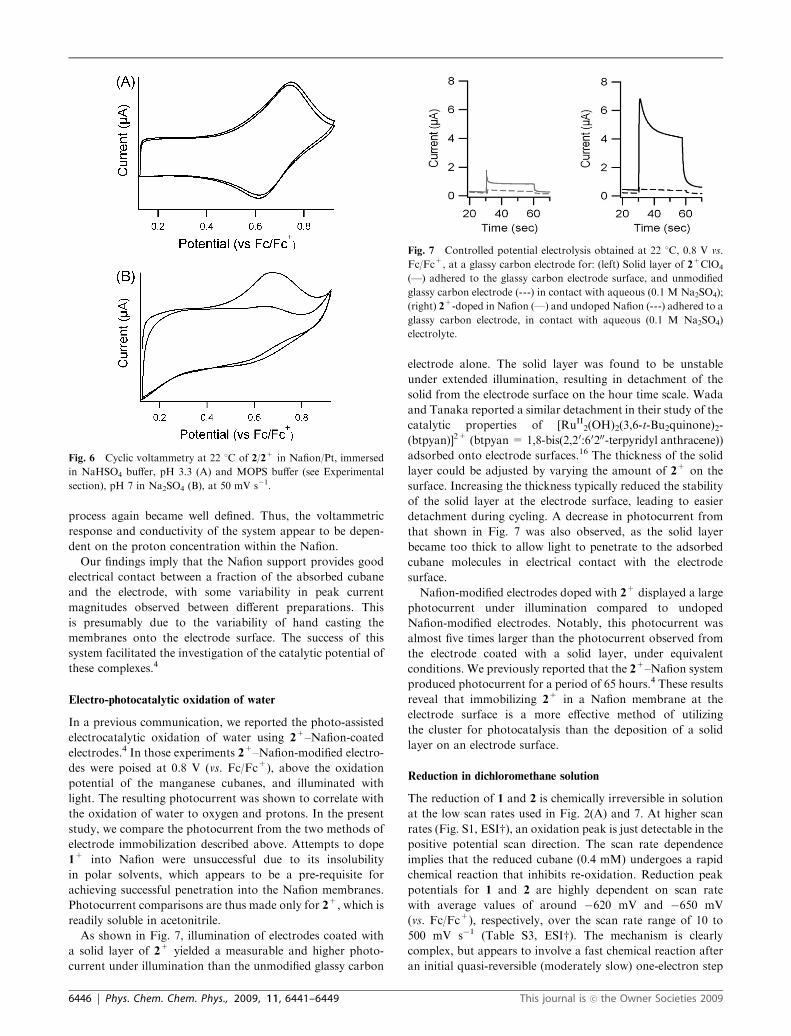
As shown in Fig. 7, illumination of electrodes coated with
a solid layer of 2+ yielded a measurable and higher photo-
current under illumination than the unmodified glassy carbon
electrode alone. The solid layer was found to be unstable
under extended illumination, resulting in detachment of the
solid from the electrode surface on the hour time scale. Wada
and Tanaka reported a similar detachment in their study of the
catalytic properties of [RuII2(OH)2(3,6-t-Bu2quinone)2-
(btpyan)]2+ (btpyan = 1,8-bis(2,20:60200-terpyridyl anthracene))
adsorbed onto electrode surfaces.16 The thickness of the solid
layer could be adjusted by varying the amount of 2+ on the
surface. Increasing the thickness typically reduced the stability
of the solid layer at the electrode surface, leading to easier
detachment during cycling. A decrease in photocurrent from
that shown in Fig. 7 was also observed, as the solid layer
became too thick to allow light to penetrate to the adsorbed
cubane molecules in electrical contact with the electrode
surface.
Nafion-modified electrodes doped with 2+ displayed a large
photocurrent under illumination compared to undoped
Nafion-modified electrodes. Notably, this photocurrent was
almost five times larger than the photocurrent observed from
the electrode coated with a solid layer, under equivalent
conditions. We previously reported that the 2+–Nafion system
produced photocurrent for a period of 65 hours.4 These results
reveal that immobilizing 2+ in a Nafion membrane at the
electrode surface is a more effective method of utilizing
the cluster for photocatalysis than the deposition of a solid
layer on an electrode surface.
Reduction in dichloromethane solution
The reduction of 1 and 2 is chemically irreversible in solution
at the low scan rates used in Fig. 2(A) and 7. At higher scan
rates (Fig. S1, ESIw), an oxidation peak is just detectable in the
positive potential scan direction. The scan rate dependence
implies that the reduced cubane (0.4 mM) undergoes a rapid
chemical reaction that inhibits re-oxidation. Reduction peak
potentials for 1 and 2 are highly dependent on scan rate
with average values of around �620 mV and �650 mV
(vs. Fc/Fc+), respectively, over the scan rate range of 10 to
500 mV s�1 (Table S3, ESIw). The mechanism is clearly
complex, but appears to involve a fast chemical reaction after
an initial quasi-reversible (moderately slow) one-electron step
Fig. 6 Cyclic voltammetry at 22 1C of 2/2+ in Nafion/Pt, immersed
in NaHSO4 buffer, pH 3.3 (A) and MOPS buffer (see Experimental
section), pH 7 in Na2SO4 (B), at 50 mV s�1.
Fig. 7 Controlled potential electrolysis obtained at 22 1C, 0.8 V vs.
Fc/Fc+, at a glassy carbon electrode for: (left) Solid layer of 2+ClO4
(—) adhered to the glassy carbon electrode surface, and unmodified
glassy carbon electrode (---) in contact with aqueous (0.1 M Na2SO4);
(right) 2+-doped in Nafion (—) and undoped Nafion (---) adhered to a
glassy carbon electrode, in contact with aqueous (0.1 M Na2SO4)
electrolyte.
6446 | Phys. Chem. Chem. Phys., 2009, 11, 6441–6449 This journal is �c the Owner Societies 2009

(EC mechanism) perhaps followed on longer time scales (see
Fig. 1 (without acid) and equations below) by a further one-
electron reduction to give an ECEC type process of the kind:
[MnIII2MnIV2O4L6] + e� " [MnIII3MnIVO4L6]� (2a)
[MnIII3MnIVO4L6]� - product 1 (2b)
product 1 + e� " reduced product(s) 2 (2c)
The higher scan rate data imply that the reversible potentials
for the initial one-electron reduction processes are ca. 505 mV
and 530 mV for 1 and 2, respectively. This small difference of
only 25 mV is significantly less than that of about 150 mV
found for the oxidation process. Wu et al.6 previously reported
reduction peak potentials for 1 and 2 of �571 mV and
�884 mV, respectively (vs. Ag/AgNO3), with a difference of
over 300 mV. The reasons for this discrepancy are unclear but
as the reduction process is irreversible, significant uncertainty
is associated with values obtained at a single scan rate in a high
resistance medium.
Peak current magnitudes per unit concentration for
reduction and oxidation of 1 and 2 are similar, which may
imply that they are both one-electron processes (n = 1).
However, the peak heights for the reduction process
(eqn 2(a–c)) are a complex function of many variables in
addition to the n-value. In contrast, the limiting current (iL)
value at a rotating disc electrode directly reflects the n-value, if
the processes are mass transport limited in this potential
region.
Rotating disc experiments conducted on the reduction
processes (Fig. S2, ESIw) revealed limiting current magnitudes
(opposite sign) per unit concentration that were approximately
double that of the known one-electron oxidation process. For
example, in the case of 1, 0.4 mM in CH2Cl2 (0.1 M Bu4NPF6,
at a rotation rate of 1000 rpm and scan rate of 5 mV s�1) ioxL =
9.9 mA, iredL = �20 mA, suggesting the overall reduction step is
a two-electron process. Bulk reductive electrolysis of 0.4 mM 1
and 1 mM 2 in CH2Cl2 (0.1 M BuNPF6), at potentials 100 mV
more negative than Eredp , gave an n-value of 1.96 � 0.05
electrons, confirming that this is an overall two-electron
reduction process on long time scales. Oxidative bulk electro-
lysis of the reduction product yielded an insignificant amount
of charge, confirming that the process is chemically irreversible
on long time scales.
The intensity of the UV absorption at 250–400 nm became
significantly lower after a two-electron reduction (Fig. 4). This
may be due to a decrease in ligand to metal charge transfer
transitions, caused by an elongation of the core metal–ligand
bonds arising from the decrease in the manganese oxidation
state. However, it is likely that the two-electron reduced
species has undergone a change in structure, so the decrease
in intensity may also be due to the reduced cubane having
undergone a chemical change, such as the release of phosphinate
ligands.
Another fully irreversible reduction process is observed
at approximately �1500 mV in cyclic voltammograms.
Rotating disc experiments for this reaction give an iredL value
that is at least 10 times greater than the observed ioxL for
the reversible oxidation process, implying that this is a
multi-electron reduction process. A brown-black precipitate
is formed on the electrode surface which blocks the surface on
repeated cycling in protic media.
A 0.4 mM solution of 1 in CH2Cl2 (0.1 M BuNPF6)
was analyzed by cyclic voltammetry in the presence of
2.4 mM diphenylphosphinic acid. In the presence of the
proton source, the oxidation wave was slightly broadened
and two reduction processes were observed in the potential
range between 0 and �1.00 V (Fig. 8(A)). These processes are
also seen after addition of p-toluenesulfonic acid. A 2.4 mM
diphenylphosphinic acid solution in CH2Cl2 (0.1 M BuNPF6)
displays no faradaic current at potentials between�1.00 V and
1.00 V. Furthermore, splitting of the processes was not
observed when tetrabutylammonium diphenylphosphinate
was added to dichloromethane solutions of the cubane,
indicating that the presence of a proton donor is the origin
of the changes in the voltammetry. The first of the two
processes occurs at a less negative potential than observed
for the previously described cubane reduction (Fig. 5) and
the second at more negative potential than for the cubane
reduction. Analogous splitting into two processes was
observed in the solid film and Nafion supported systems when
these electrodes were immersed in pH 3.3 aqueous electrolyte
solutions (Fig. 8(B)).
Bulk reductive electrolysis was conducted at potentials
slightly more negative than those for the first reduction
Fig. 8 Cyclic voltammograms obtained at 22 1C at a glassy carbon
electrode with a scan rate of 10 mV s�1 for reduction of: (A) 0.4 mM 1
in CH2Cl2 (0.1 M Bu4NPF6), in the absence (---) and presence (—) of
the proton donor (2.4 mM diphenylphosphinic acid); (B) 2
(2+ converted to 2) doped in Nafion adhered to electrode in contact
with (---) aqueous 0.1 M Na2SO4 and (—) 0.1 M NaHSO4, pH 3.3
electrolyte.
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 6441–6449 | 6447

process (�450 mV vs. Fc, Fig. 8), on a 0.4 mM solution of 1
with 2.4 mM diphenylphosphinic acid. Coulombic analysis
again implied that an overall two-electron reduction occurred,
as in the absence of acid. After bulk electrolysis, the second
more negative process was absent. Proton assisted reduction
would explain the positive shift in potential for the first process
and the splitting into two processes. Thus, the initial step is
modified from that in eqn (2) to:
[Mn4O4]6+ + aH+ + e� - [Mn4O4Ha]
(5+a)+
- electroactive product(s) (3)
Multiple-process reduction is likely to be occurring on the
longer time scale of bulk electrolysis via an ECE type scheme.
This proton-assisted one-electron process is consistent with
our previous report describing the isolation and purification
of stable 1H via H atom transfer from phenothiazine (top line
in Fig. 1).7 This species was identified as having the core
composition of [Mn4O3(OH)]6+.
The addition of two electrons in this bulk electrolysis
experiment led to precipitation of a red-brown solid, leaving
a cloudy white solution. The electronic absorption spectra
of this solution reveal only the ligand absorption maxima
(B230 nm), implying that substantial decomposition of the
manganese core has occurred on long time scale experiments.
In addition to the solid product, water droplets formed on the
side of the glass cell, which was not observed in any of
the previously described bulk electrolysis experiments. The
red-brown solid obtained from the acidic bulk electrolysis
reduction experiment was insoluble in organic and aqueous
solvents and is proposed to be a reduced manganese oxide.
The cloudy white solution has been seen before in the course of
cubane reduction.10 Infrared data collected on this previous
solid indicated that it corresponded to a polymeric
Mn(II)(phosphinate)2.
Previous chemical reduction studies have revealed that the
cubane can undergo four-electron/four-proton reduction
process via proton-coupled electron transfer (PCET), yielding
two water molecules and the [Mn4O2L6] complex.7,10,17 PCET
was shown to occur via a sequential process with core
oxo-ligands being protonated to hydroxides and subsequently
water. This process goes via a reduced intermediate, Mn4O3L6,
that was detected by mass spectrometry after 2 reducing
equivalents were added. This intermediate forms when a
corner oxo is converted to a water molecule in the reduction
process. The presence of water after the reduction of 1 and 2 in
degassed CH2Cl2 solution suggests that the reduction of the
manganese atoms causes a weakening of the Mn–O bonds,
facilitating protonation of the core oxygens and their sub-
sequent release as water molecules (see Fig. 1 (with acid)).
Reduction of quasi-solid phases in contact with aqueous
environments
The electrochemistry of 1 as a solid (Fig. 2(B)) or 2 supported
in Nafion in aqueous 0.1 M Na2SO4 electrolyte yields new
oxidation processes in potential cycling experiments but only
after reduction of the cubane. Furthermore, these processes
are sensitive to the pH of the aqueous electrolyte solution.
Thus, a change of electrolyte to more acidic 0.1 M NaHSO4
causes the reduction component to split into two processes
(Fig. 2(B)), as also occurs when a weaker acid is added to
dichloromethane solutions subjected to solution phase
voltammetry. Clearly, there is a close relationship between
the voltammetry in the organic solvent and the solid phase
studies at chemically modified electrodes, even through
products of the reduction differ.
Conclusions
The high charge of the [Mn4O4]7+ core present in 1+ and 2+
means that the application of very positive electrode potentials
is needed for their generation. This provides a highly reactive
species, previously shown to be able to catalyze the oxidation
of water under illumination.4 We have confirmed that the
one-electron oxidation of the cubane to the cubium form is a
chemically reversible process on the voltammetric time scale
and displays significant stability under synthetic (bulk electro-
lysis) conditions (1 2 1+ transition in Fig. 1). The reversible
potentials for these 10/+ and 20/+ couples are ligand-dependent,
implying that the generation of a high valence state on a tetra-
manganese cluster is facilitated by strong electron donation
from the supporting ligands.6 In the [Mn2O2(bipyridine)4]3+
complex, charge transfer from two oxo-ligands and four
nitrogen lone pairs on the bipyridines gives rise to an electro-
chemical oxidation potential of the MnIIIMnIV core to 2MnIV
at 1.25 V vs. SCE.18 This potential is of the magnitude required
to oxidize aqueous MnII to MnIII (1.27 V vs. SCE). Increasing
electron donation from the ligands to the [Mn2O2] cluster, by
replacing two bipyridine ligands with two terminal dihydrogen
phosphates and one bridging hydrogen phosphate, means that
the 2 MnIV state in [Mn2O2(bpy)2(m-HPO4)(H2PO4)2] can be
achieved at 0.7 V vs. SCE.14
In the cubane cluster, each metal center is supported by
three oxo-ligands and three phosphinate oxygen donors to
achieve a large electron-donating field around the metals,
which causes oxidation of 2MnIII2MnIV to MnIII3MnIV to
occur at potentials as low as 0.73 V vs. SCE, yielding the
[Mn4O4]7+ species described above. The cubium state could be
achieved at even less positive potentials by developing cubanes
with still more strongly electron-donating phosphinate groups.
In principle, this would lower the required energy in catalytic
applications of these clusters.
Catalytic utilization of the cubium state has been achieved
by immobilization of the cluster at electrode surfaces.
A comparison of two methods of catalyst immobilization,
solid layer adsorption16 and doping into electrode bound
Nafion membranes,19–23 revealed that Nafion membranes offer
superior catalyst support, higher photocurrents and greater
stability. The Nafion appears to create a more efficient
distribution of catalytic molecules over the electrode surface
and protects the catalysts from the bulk water electrolyte.
Further investigation of this system is currently underway.
We note that to achieve effective doping of the Nafion
membrane, the catalyst must be positively charged and soluble
in a polar solvent. The casting of solid layers onto an electrode
surface is, therefore, a useful technique for screening
the catalytic potential of molecules that do not meet these
requirements.
6448 | Phys. Chem. Chem. Phys., 2009, 11, 6441–6449 This journal is �c the Owner Societies 2009

Electrochemical investigation of the reduction has revealed it to
involve an overall two-electron process, with products and inter-
mediates being sensitive to the presence of protons (see Fig. 1,
proposed electrochemical reduction with acid and without acid).
Previous chemical reduction studies have revealed that the cubane
can undergo proton-coupled reduction, yielding two water
molecules and a [Mn4O2L6] complex.10 PCET was shown to be
a sequential process, with core oxo-ligands being protonated to
hydroxides and subsequently water. The observed resolution into
two reduction processes in this study, upon addition of protons,
demonstrates that reduction is more readily achieved when
coupled with protonation. The ability of the cubane to undergo
PCET may be an essential property in its ability to catalytically
oxidize water.7
The chemical irreversibility of the two-electron process
2MnIII2MnIV - 4MnIII in dichloromethane solution phase
suggests that a structural or chemical change prevents
re-oxidation of the cluster. A transient one-electron reduced
cluster is probably an intermediate. In the solid films, inter-
mediates are detected although it is unclear whether the original
complex may be reformed under conditions of cyclic voltammetry
or a new water-ligated product is generated. For the Nafion
supported cubane, the peak currents and potentials have been
shown to be sensitive to the pH of the supporting electrolyte, with
the reduction process appearing to undergo a PCET process
similar to that observed in dichloromethane solution. As reported
by Brimblecombe et al.,4 the development of the Nafion system
has provided a significant step forward in the use of these
complexes as photo-assisted oxidation catalysts.
Acknowledgements
This work was supported by the Australian Research Council
through the Discovery Program (LS/GCD/GFS/AMB) and
Federation Fellowship Scheme (AMB), the US National
Institutes of Health (GCD), a Lemberg Fellowship (GCD),
an Australian Academy of Sciences Travel Fellowship (GFS),
an Australian Postgraduate Award (RB), a Fullbright Post-
graduate Award (RB) and a Monash University Postgraduate
Publication Award (RB).
Notes and references
1 J. Barber and J. W. Murraya, Coord. Chem. Rev., 2008, 252,233–243.
2 A. J. Bard and L. R. Faulkner, Electrochemical Methods:Fundamentals and Applications, John Wiley & Sons Inc., New York,1990.
3 C. W. Cady, R. H. Crabtree and G. W. Brudvig, Coord. Chem.Rev., 2008, 252, 444–455.
4 R. Brimblecombe, G. F. Swiegers, G. C. Dismukes and L. Spiccia,Angew. Chem., Int. Ed., 2008, 47, 7335–7338.
5 W. Ruettinger and G. C. Dismukes, Chem. Rev., 1997, 97
1–24.6 J.-Z. Wu, E. Sellitto, G. P. A. Yap, J. Sheats and G. C. Dismukes,Inorg. Chem., 2004, 43, 5795–5797.
7 T. G. Carrell, E. Bourles, M. Lin and G. C. Dismukes, Inorg.Chem., 2003, 42, 2849–2858.
8 T. G. Carrell, S. Cohen and G. C. Dismukes, J. Mol. Catal. A:Chem., 2002, 187, 3–15.
9 W. Ruettinger, M. Yagi, K. Wolf, S. Bernasek andG. C. Dismukes, J. Am. Chem. Soc., 2000, 122, 10353–10357.
10 W. F. Ruettinger and G. C. Dismukes, Inorg. Chem., 2000, 39,1021–1027 (Inorg. Chem., 2000, 39, 4186).
11 J.-Z. Wu, F. D. Angelis, T. G. Carrell, G. P. A. Yap, J. Sheats,R. Car and G. C. Dismukes, Inorg. Chem., 2006, 45, 189–195.
12 M. Yagi, K. V. Wolf, P. J. Baesjou, S. L. Bernasek andG. C. Dismukes, Angew. Chem., Int. Ed., 2001, 40, 2925–2928.
13 W. F. Ruettinger, D. M. Ho and G. C. Dismukes, Inorg. Chem.,1999, 38, 1036–1037.
14 R. Manchanda, G. W. Brudvig and R. H. Crabtree, Coord. Chem.Rev., 1995, 144, 1–38.
15 Y. Yu, M. Dubey, S. L. Bernasek and G. C. Dismukes, Langmuir,2007, 23, 8257–8263.
16 T. Wada and K. Tanaka, Eur. J. Inorg. Chem.,2005, 3832–3839.
17 M. Maneiro, W. F. Ruettinger, E. Bourles, G. L. McLendon andG. C. Dismukes, Proc. Natl. Acad. Sci. U. S. A., 2003, 100,3707–3712.
18 S. R. Cooper and M. Calvin, J. Am. Chem. Soc., 1977, 99,6623–6630.
19 H. Hagiwara, N. Ono, T. Inoue, H. Matsumoto and T. Ishihara,Angew. Chem., Int. Ed., 2006, 45, 1420–1422.
20 M. A. Gondal, A. Hameed and Z. H. Yamani, Energy Sources,2005, 27, 1151–1165.
21 R. Bandyopadhyay, S. K. Maiti and R. Bhattacharyya, Inorg.Chem. Commun., 2002, 5, 452–452.
22 K. Sayama, K. Yase, H. Arakawa, K. Asakura, A. Tanaka,K. Domen and T. Onishi, J. Photochem. Photobiol., A, 1998,114, 125–135.
23 K. Hashimoto, H. Irie and A. Fujishima, Jpn. J. Appl. Phys., 2005,44, 8269–8285.
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 6441–6449 | 6449




















