
Intramembrane receptor–receptor interactions: a novel principle in molecular medicine
Upload
independentCategory
view
0download
0
www.elsevier.com/locate/yexcr
Experimental Cell Research 299 (2004) 91–100
DU145 human prostate carcinoma invasiveness is modulated by
urokinase receptor (uPAR) downstream of epidermal growth
factor receptor (EGFR) signaling
Asmaa Mamoune,a,b,1 Jareer Kassis,a,b,1,2 Sourabh Kharait,a,b Susanne Kloeker,c
Elisabeth Manos,c David A. Jones,c and Alan Wellsa,b,*
aDepartment of Pathology, University of Pittsburgh, Pittsburgh, PA 15261, USAbPittsburgh VAMC, Pittsburgh, PA 15261, USA
cDivision of Molecular Pharmacology, Huntsman Cancer Institute, University of Utah, Salt Lake City, UT 84112, USA
Received 16 January 2004, revised version recieved 6 May 2004
Available online 17 June 2004
Abstract
Tumor cell motility and invasion have been linked to upregulated signaling from both the epidermal growth factor receptor (EGFR) and
that for urokinase-type plasminogen activator (uPAR). However, we do not know whether these events are interdependent or unrelated,
despite the obvious diagnostic and therapeutic implications. Gene microarray analyses have suggested that EGFR signaling via
phospholipase C-g (PLCg) induces uPAR transcription. We utilized two sublines of the DU145 human prostate carcinoma cell line that are
genetically engineered to differentially activate the EGFR/PLCg cascade and are variously invasive in vitro and in vivo. uPAR protein levels
in these cells were found to be dependent on PLC signaling, pharmacologic inhibition of PLC signaling reduced uPAR expression. To
determine whether uPAR was a required element in EGFR-mediated invasion, we stably expressed uPAR cDNA in either sense or antisense
orientation in the two DU145 sublines. Interestingly, uPA production was modulated in parallel, although to a lesser degree, with uPAR in
these sublines. Antisense to uPAR significantly restricted invasion of the highly invasive DU145 WT cells through Matrigel and reduced
aggressiveness of tumors in nude mice. Up-regulation of uPAR significantly increased the invasiveness of the moderately invasive DU145
parental (DU145 P) cells through Matrigel, but this increased invasiveness was not seen in mice. uPA activity appears to contribute to
invasiveness at least through Matrigel, as antibody to uPA or amiloride limited the transmigration. These results support a model of tumor
invasion promoted by autocrine EGFR signaling involving reinforcing altered gene expression, of uPAR at least, that further induces cell
motility. Herein, a number of key molecules whose expression levels are interrelated, including both EGFR and uPAR, are required but none
are sufficient in the absence of other keys molecules in promoting tumor progression.
D 2004 Elsevier Inc. All rights reserved.
Keywords: Tumor progression; EGF receptor; Phospholipase C-g; Motility; Migration
Introduction have elucidated a variety of cellular pathways and mecha-
One of the rate-limiting steps of prostate tumor cell
invasion is its ability to breach an extracellular matrix
(ECM) [1]. An increasing amount of recent data not only
0014-4827/$ - see front matter D 2004 Elsevier Inc. All rights reserved.
doi:10.1016/j.yexcr.2004.05.008
* Corresponding author. Department of Pathology, University of
Pittsburgh, 713 Scaife Hall, Pittsburgh, PA 15261. Fax: +1-412-647-8567.
E-mail address: [email protected] (A. Wells).1 These authors contributed equally to this publication.2 Current address: Building 10, Room B1B40, National Cancer
Institute, NIH, Bethesda, MD 20892.
nisms involved, but also have alluded to the interdepen-
dence and cross-communication of many of these pathways.
However, one relatively common aspect of cellular invasion
mechanisms is their mediation by surface receptors which,
in transformed cell, are often overexpressed and upregu-
lated. Of these receptors, the epidermal growth factor
receptor (EGFR) is the most frequently upregulated in
tumors including prostate carcinomas [2,3]. EGFR signaling
promotes tumor progression via both epigenetic events such
as de-adhesion and cytoskeletal reorganization [4], as well
as altering the transcriptional profile of the cancer cell [5,6].

A. Mamoune et al. / Experimental Cell Research 299 (2004) 91–10092
A second, distinct class of receptor, the urokinase-type
plasminogen activator (uPAR) is similarly often found
upregulated in invasive tumor cells [7,8]; this also includes
prostate carcinomas [9–11]. uPAR is considered to promote
tumor invasion both via matrix degradation by its protease
ligand, uPA, and cellular signaling of migration via other
surface receptors including EGFR [12–14]. While both
EGFR and uPAR appear to promote tumor progression,
the extent of their interrelationship in this process is not well
characterized.
These two receptor systems are linked at many levels.
Part of cellular signaling from uPAR appears to occur via
EGFR transactivation [13,15]. Inhibition of EGFR kinase
activity blocks uPAR-initiated activation of ERK MAP
kinase but not of the small rho GTPases. As ERK is required
for EGFR induction of both motility and proliferation, it is
not surprising that abrogation of this secondary effector
blocks cell motility and proliferation induced by uPAR
[13,15]. In tumor model systems, abrogation of EGFR
signaling blocks uPAR-associated invasiveness through an
extracellular matrix [16] and growth of tumors in animals
[13,17]. Thus, EGFR appears to be a necessary element for
uPAR-mediated tumor progression.
Interestingly, transcription and production of both uPAR
and its ligand, uPA, are increased by EGFR signaling
[18,19]. Treatment of PC3 human prostate cells with genis-
tein, a tyrosine kinase inhibitor, reduces both uPAR and uPA
levels [20]. Furthermore, we found that transcription of
uPAR was increased in a highly invasive subline of
DU145 which presents upregulation of the motility-associ-
ated EGFR/phospholipase C-g (PLCg) signaling cascade
[6]. Thus, while EGFR activity increases the potential uPAR
signaling capacity, it is still not known if EGFR-mediated
tumor progression depends on uPAR upregulation and
activation.
To this end, we constructed a full-length uPAR cDNA
fragment and inserted it in both sense and antisense posi-
tions into a vector carrying a constitutively active promoter
and introduced these constructs into our DU145 sublines
that show moderate and high invasiveness [21]. We dem-
onstrated that elevating uPAR expression in parental
DU145 (DU145 P) cells increased their invasiveness
through Matrigel to levels similar to the EGFR overexpress-
ing DU145 WT cells, while the highly invasive DU145 WT
cells’ invasiveness was retarded by the antisense uPAR
cDNA fragment. To establish whether this behavior is
reproducible in vivo, we injected the highly invasive
DU145 WT cells expressing antisense uPAR cDNA into
athymic nude mice, and while control DU145 WT cells
formed highly invasive tumors, the antisense-carrying cells
were significantly less aggressive and were reduced in
diaphragm invasiveness. However, upregulation of uPAR
in parental DU145 cells did not significantly increase
invasion in vivo. These results indicate that uPAR is
required, but not sufficient for EGFR-induced tumor cell
invasion.
Materials and methods
Animals
Male athymic BALB/c nu/nu mice, 6–8 weeks of age,
were purchased from the Animal Production Area of the
National Cancer Institute-Frederick Cancer Research and
Development (Frederick, MD) and housed in suitable con-
ditions. Mouse weights ranged from 20–30 g at onset of
experiments. All animal experimentation was approved by
the Institutional Animal Care and Use Committees of both
the University of Pittsburgh and the Pittsburgh VA Medical
Center.
Cell culture
DU145 human prostate carcinoma cells were derived as
described [22]; we designate these cells as parental
(DU145 P). DU145 WT prostate carcinoma cells were
generated as described previously by retroviral transduction
[23]. Two independent polyclonal isolates were used
throughout the experiments. WT cells overexpress EGFR
to levels that oversaturate the capacity of the degradation
pathway and therefore do not undergo autocrine-induced
downregulation. The DU145 WT cells present approxi-
mately one-third more EGFR when autocrine downregula-
tion is minimized and over twice the number of bindings in
the face of autocrine downregulation [23]. The cells were
maintained in DMEM (4.5 g/ml glucose) containing 10%
FBS and supplemented with L-glutamine (2 mM), penicil-
lin/streptomycin (100 units/ml), nonessential amino acids
(0.1 mM), and sodium pyruvate (1 mM). For stable
selection of WT EGFR, cells were grown in G418 (1000
Ag/ml). Before in vitro experimentation, cells were qui-
esced in DMEM containing 0.1% dialyzed FBS for 24 h.
Sense and antisense uPAR constructs were introduced
into the cells by electroporation. Briefly, uPAR was cloned
into the constitutively active pXf vector, which contains the
SV40 early promoter [24], in either sense or antisense
directions. The sense uPAR gene was cloned via PCR with
the XbaI restriction site at the 5V end and the HindIII site at
the 3V end, while the antisense gene was cloned by switch-
ing the positions of these two restriction sites. Primers used
for the sense constructs were as follows: leading strand
5VGGA TCT AGA CCA TGG GTC ACC CGC CGC,
lagging strand 5V GGA AGC TTC TAT CAG CGG TAC
TGG ACATCC AG. To append the GPI anchor to the sense
construct, two-step PCR was performed using the leading
strand primer above and 5V GGT GAT GGT GAG GCT
GAG ATG GGC AGG GCC AGG CTG AGG AGC AGC
CCC ACT GCG GTA CTG GAC ATC C for the lagging
strand, followed by PCR of the resulting template with the
same leading strand primer and 5V GGC AAG CTT TTA
GGT CCA GAG GAG AGT GCC TCC CCA CAG TCT
GGC AGT CAT TAG CAG GGT GAT GGT GAG GCT G
as the lagging strand primer. Primers for the antisense

A. Mamoune et al. / Experimental Cell Research 299 (2004) 91–100 93
construct were as follows: leading strand 5V GGA TCT
AGATCA GCG GTA CTG GAC ATC CAG, lagging strand
5V GGA AGC TTA CCA TGG GTC ACC CGC CGC.
Stable polyclonal transfectant cells, representing at least 20
initial colonies, were selected by additionally supplementing
the above media with 1.2 Ag/ml methotrexate. Two inde-
pendent polyclonal isolates were tested for each construct
with similar results.
Immunoblotting
Cells were grown in six-well plates to semiconfluence,
washed with PBS, and lysed with Laemmli buffer. The
lysate was separated by SDS–PAGE, transferred to a nylon
membrane, and immunoblotted. Primary antibodies used
were anti-uPAR, which binds to both uPAR and uPA/uPAR
complexes (American Diagnostica; catalog #3936), anti-
EGFR (Zymed; catalog #28-0005), anti-uPA (Oncogene,
catalog #IM13L), and antiactin (Sigma; catalog #A 2066).
The staining was visualized by a secondary antibody linked
to horseradish peroxidase and detected by ECL (Amer-
sham). Blots were scanned and quantitated using NIH
Image software.
Immunofluorescence
Cells, grown on coverslips, were fixed in 3% parafor-
maldehyde for 30 min and washed twice with PBS. In a
series of experiments, before fixation, the cells were treated
with or without bacterial PI-PLC, 0.5 U/ml for 1 h (Sigma;
catalog #8804) to remove any GPI-anchored uPAR. The
coverslips were blocked with 1% BSA for 1 h and then
stained with the anti-uPAR antibody (1:50 in PBS contain-
ing 1% BSA) for 2 h, washed twice in PBS/BSA, and
visualized with FITC-conjugated antimouse IgG (1:500 in
PBS/BSA) (Molecular Probes).
Transmigration assays
Cell invasiveness in vitro was determined by the ability
to transmigrate a layer of ECM, Matrigel, in a modified
Boyden chamber assay. Matrigel invasion chamber plates
were obtained from Becton Dickinson/Biocoat (catalog
#40480). For each experiment, 20,000 cells were plated
randomly and distributed among plates with different lot
numbers, with each experiment performed in duplicate or
triplicate. In one series of experiments, we incorporated into
the assay either antibody to uPA (4 Ag/ml; Oncogene,
catalog #IM13L) or amiloride (0.5 mM; Sigma) to block
the functioning of uPA. Despite possible variances in EGFR
ligand concentrations in Matrigel, these intrinsic concen-
trations are saturating since even ‘‘reduced growth factor
Matrigel’’ contains relatively high amounts of EGF (up to
0.5 ng/ml compared to 0.5–1.3 ng/ml in regular Matrigel)
(Becton Dickinson Labware 1997/98 catalog description,
page 128). Cells were kept in serum-free media containing
1% BSA for the first 24 h and then replaced with serum-free
media for the remaining 24 h. The bottom, targeting well
was filled with media containing 10% FBS. Enumeration of
the cells that invaded through the matrix over a 48-h period
was accomplished by removing uninvaded cells with a
cotton swab and then fixing, staining with crystal violet,
and visually counting cells on the bottom of the membrane.
Tumor cell inoculations
We utilized the intraperitoneal xenograft model for
invasiveness as determined by diaphragmatic invasion
[21,25]. This mode of invasion through the diaphragm
was chosen for reproducibility and quantitation; it correlates
strongly with invasion from orthotopic tumor growth. Cells
were suspended in culture media and injected (26.5-gauge
needle) into the peritoneal cavity (2 million cells in 200-Altotal volume). Mice were asphyxiated after 60 days with
halothane and necropsied with removal of the pancreas,
spleen, kidneys, diaphragm, lungs, and any tumorous tissue.
All tissues were fixed in 10% buffered formalin, paraffin-
embedded, serially sectioned, and stained with hematoxylin
and eosin (H&E). Invasion of the diaphragm was deter-
mined by microscopic quantitation. The protocol was ap-
proved by the Pittsburgh Veteran Administration Medical
Center Institutional Animal Care and Use Committee.
Results
uPAR protein levels are upregulated in invasive WT DU145
cells in relation to PLC signaling
Cell lysates of the two DU145 sublines were queried for
uPAR levels. As shown in Fig. 1A, uPAR protein levels
were about two times higher in DU145 WT overexpressing
EGFR receptor than parental DU145 cells (2.04 F0.49, N =
4, P < 0.01); the increased level of EGFR is consistent with
earlier studies, with the slight electrophoretic retardation
reflecting the reported greater stoichiometry of phosphory-
lation [23]. Surface location of the uPAR was confirmed by
treating the cells with bacterial PI-PLC to cleave the GPI
anchors of uPAR; this treatment removed much of the uPAR
staining (Fig. 1B). This level of increase was in the range
expected from an earlier gene microarray query that found
higher uPAR mRNA in DU145 WT compared to DU145 P
cells [6].
To investigate the causal link between the EGFR/PLCg
and uPAR signaling, we treated the highly invasive cell line
either with an EGFR tyrosine kinase inhibitor, PD153035, or
the specific PLC inhibitor, U73122. Exposure of DU145WT
cells to U73122 reduced significantly the level of uPAR
protein (68%F 2.5% decrease compared to nontreated cells,
n = 3), while no effect was seen using its inactive analogue,
U73343 (Fig. 1C). The EGFR kinase inhibitor PD153035
also decreased uPAR expression (37% F 12% decrease).

Fig. 1. Expression of endogenous uPAR protein in DU145 sublines. (A) Whole cell lysates from each of DU145 P and DU145 WT cells were collected and
quantitated using the BioRad protein assay. Identical concentrations of lysates were then separated in 10% SDS–PAGE and blotted with anti-uPAR antibody
under nonreducing conditions (band at 50 kDa). Concurrent blotting of an identical gel with antiactin (50 kDa) (for gel loading) or anti-EGFR (170 kDa) was
performed. Shown are representative of four independent blots in A. (B) DU145 sublines were examined for cell surface expression of uPAR by
immunofluorescence. Alternate coverslips were treated with bacterial PI-PLC to remove GPI anchors. Exposure times were preset to compare treated and
untreated sublines but were not constant between the DU145 P and DU145 WT sublines. Shown are representative of three independent analyses; uPAR is
green, red is nuclear staining. (C) Lysates from DU145 WT cells treated with 500 nM PD153035, 2 AM U73122, or 2 AM U73343 for 24 h were collected and
subjected to SDS–PAGE electrophoresis. Three different blots were scanned, the ratio uPAR/actin was determined using NIH Image software, and values were
normalized to untreated cells. Shown is a representative immunoblot of three.
A. Mamoune et al. / Experimental Cell Research 299 (2004) 91–10094
EGFR inhibition did not suppress uPAR as extensively as that
due to U73122, suggesting that other signals also are increas-
ing uPAR via PLC-mediated transcription.
Modulation of uPAR expression alters invasiveness through
Matrigel
The above studies suggested that uPAR expression may
either enhance invasion or be required for invasion. To
determine the requirement for uPAR in DU145 tumor cell
invasion, we selected DU145 WT cells since these cells are
significantly more invasive than parental cells both in vitro
and in vivo [21,23]. To determine if uPAR expression
enhances invasiveness, we utilized the moderately invasive
DU145 P line.
To study the effects of uPAR downregulation in aggres-
sively invasive cells, we cloned a full-length uPAR cDNA
fragment (including the GPI anchoring domain) downstream
from a constitutively active SV40 early promoter in the
eukaryotic expression vector pXf [25]. Stable selection of
the electroporated construct was accomplished using 1.2 Ag/ml methotrexate in the media. As shown in Fig. 2A, immu-
noblot analysis of cell lysates indicated increased expression
of uPAR protein in the sense construct-expressing cells (1.87
F 0.37, N = 4, P = 0.01). Similarly, we cloned the uPAR
cDNA fragment in the antisense direction into the pXf
promoter and stably selected the construct in the highly
invasive DU145WTcells; uPAR protein levels were reduced
significantly in the DU145 WTcells expressing the antisense
cDNA (0.38 F 0.11, N = 3, P < 0.01). Both DU145 P and
DU145WT lines were transfected and stably selected with an
empty pXf vector and ones containing irrelevant cDNA;
examination of lines demonstrated no change in uPAR levels
(data not shown). uPAR was visualized on the transfected
cells demonstrating altered levels in nearly all the cells and
that the majority of the staining was indicative of cell surface
uPAR. These findings are significant to our study since uPAR
is often actively cleaved at the GPI anchor and shed [26],
thereby reducing levels at the cell surface.
The second aspect of the uPAR system is its ligand, the
protease uPA. uPA has been reported as upregulated in
response to EGFR signaling, including in prostate carcino-
ma cells [16–19]. We found that uPA levels changed
qualitatively in the same manner as uPAR (Fig. 2B),
although to a lesser quantitative extent. It is possible that
the change in uPA levels might reflect the changes in surface
uPAR binding. Removal of the surface associated-uPAR
with bacterial PLC did not eliminate the differences noted

Fig. 2. Expression of sense or antisense uPAR cDNA constructs alters uPAR levels in DU145 sublines. (A) DU145 WT cells expressing antisense, DU145 P
overexpressing uPAR cDNA, or mock-transfected cells were lysed in equal protein contents after counting cells in separate identical wells and normalizing
protein concentrations. Lysates were separated by SDS–PAGE (10% gel) under nonreducing conditions and immunoblotted with anti-uPAR antibody. (B)
DU145 WT cells expressing antisense uPAR, DU145 P overexpressing uPAR cDNA or mock-transfected cells were lysed in equal protein contents after
counting cells in separate identical wells and normalizing protein concentrations. Lysates were separated by SDS–PAGE (10% gel) and immunoblotted with
anti-uPA antibody. (C) Protein expression of uPAwas determined in DU145 P and DU145 WT cells stably expressing the sense or antisense uPAR constructs.
The cells were lysed and subjected to immunoblot detection before or after treatment with bacterial PI-PLC to remove GPI anchors and uPAR-complexed uPA.
uPAR was also visualized to demonstrate enzymatic removal of surface uPAR, denoted by decrease in total uPAR levels. Shown are representative experiments
of four (A) or two (B and C).
A. Mamoune et al. / Experimental Cell Research 299 (2004) 91–100 95
(Fig. 2C). More importantly than the change in uPA levels
which are slight compared to alterations in uPAR, these
immunoblots demonstrate that both uPAR and uPA are
produced by these cells, allowing for a human uPA/human
uPAR autocrine loop.
A hallmark of tumor progression is its ability to invade
through the ECM into surrounding tissue. We asked whether
lowering uPAR protein levels in the highly invasive DU145
WT cells reduced these cells invasiveness. To this end, we
compared transmigration of DU145 WT and DU145 WTAS
cells through Matrigel; transmigration by DU145 WT AS
was drastically reduced compared to the mock-transfected
cells (Fig. 3A). These results clearly indicated that down-
regulating uPAR reversed the invasive properties of upregu-
lated EGFR in these cells. Along the same lines, we
overexpressed uPAR in the less invasive parental DU145
cell lines (DU145 P SN). As shown in Fig. 3B, DU145 P
SN transmigration through Matrigel was dramatically in-
creased compared to DU145 P cells; uPAR expression
compensated for the relatively less PLCg signaling in these
cells [25]. Again, these results indicate that uPAR expres-
sion levels are both required for and contribute to invasion
at least in vitro and thus confirm a rate-limiting role for
uPAR functions.
uPAR initiates signals upon binding its ligand [7,8].
However, there have been suggestions of ligand-indepen-
dent uPAR modulation of cell behaviors. Addition of an
antibody against human uPA reduced DU145 WT invasive-
ness by about two thirds indicating that the invasiveness
noted herein is ligand-dependent (Fig. 3C). It is a further
question whether uPA protease activity is required for
uPAR-enhanced motility [27–29]. Amiloride binds to and
inhibits human uPA [30], and has been used to limit
invasiveness in vitro and in vivo of human carcinoma lines
[31,32]. We find that DU145 WT invasion of the Matrigel
barrier is partly reduced by amiloride (Fig. 3C), suggesting a
role for proteolytic activity.
uPAR downregulation reduces tumor formation and
invasiveness in mice
Our data so far showed a clear role for uPAR and uPA in
tumor cell invasion through Matrigel. In our previous
studies, we showed that DU145 invasiveness correlated
with PLCg activation via overexpressed EGFR. Inhibition
of PLCg pharmacologically with U73122 [21] or molecu-
larly via the dominant negative fragment PLCz [25] signif-
icantly reduced invasion of tumors formed by DU145 WT in
athymic nude mice. Because our recent data showed a clear
link between enhanced invasiveness and uPAR expression,
we asked whether downregulation of uPAR expression
would negate the effects of upregulated EGFR and PLCg
activity. Being able to demonstrate such an effect in mice
would be of paramount importance due to the fact that the
complex interactions of the uPA/uPAR system with various
integrins, growth factor receptors, and extracellular pro-
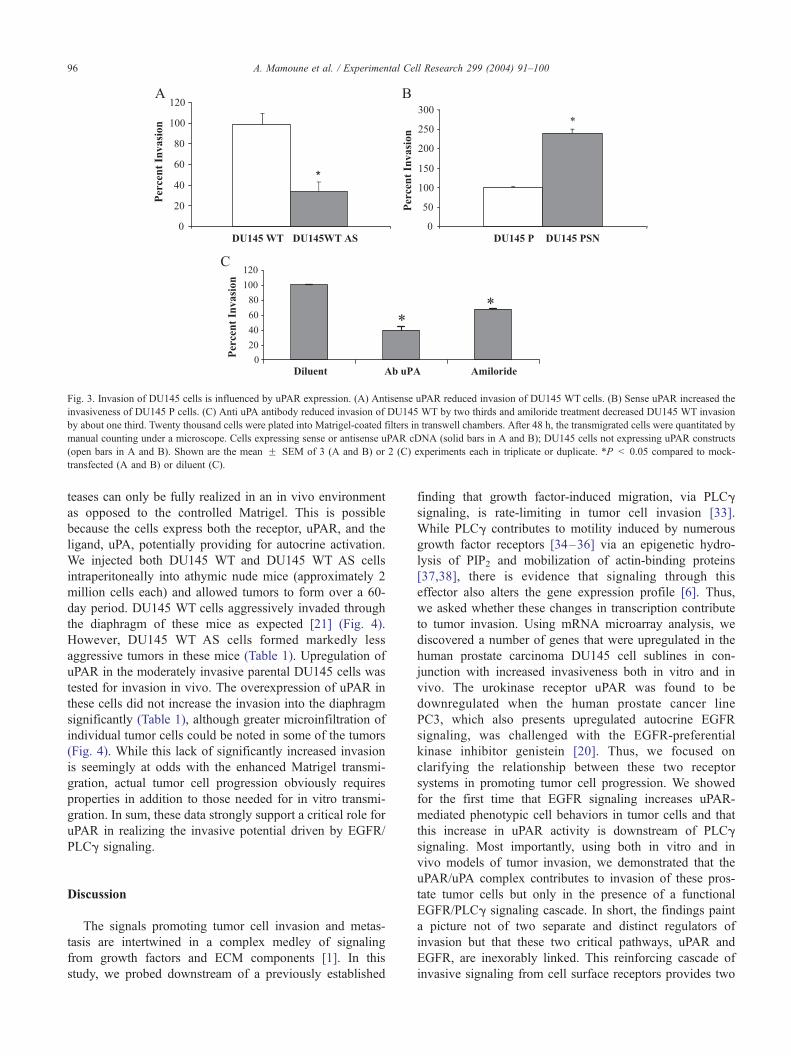
Fig. 3. Invasion of DU145 cells is influenced by uPAR expression. (A) Antisense uPAR reduced invasion of DU145 WT cells. (B) Sense uPAR increased the
invasiveness of DU145 P cells. (C) Anti uPA antibody reduced invasion of DU145 WT by two thirds and amiloride treatment decreased DU145 WT invasion
by about one third. Twenty thousand cells were plated into Matrigel-coated filters in transwell chambers. After 48 h, the transmigrated cells were quantitated by
manual counting under a microscope. Cells expressing sense or antisense uPAR cDNA (solid bars in A and B); DU145 cells not expressing uPAR constructs
(open bars in A and B). Shown are the mean F SEM of 3 (A and B) or 2 (C) experiments each in triplicate or duplicate. *P < 0.05 compared to mock-
transfected (A and B) or diluent (C).
A. Mamoune et al. / Experimental Cell Research 299 (2004) 91–10096
teases can only be fully realized in an in vivo environment
as opposed to the controlled Matrigel. This is possible
because the cells express both the receptor, uPAR, and the
ligand, uPA, potentially providing for autocrine activation.
We injected both DU145 WT and DU145 WT AS cells
intraperitoneally into athymic nude mice (approximately 2
million cells each) and allowed tumors to form over a 60-
day period. DU145 WT cells aggressively invaded through
the diaphragm of these mice as expected [21] (Fig. 4).
However, DU145 WT AS cells formed markedly less
aggressive tumors in these mice (Table 1). Upregulation of
uPAR in the moderately invasive parental DU145 cells was
tested for invasion in vivo. The overexpression of uPAR in
these cells did not increase the invasion into the diaphragm
significantly (Table 1), although greater microinfiltration of
individual tumor cells could be noted in some of the tumors
(Fig. 4). While this lack of significantly increased invasion
is seemingly at odds with the enhanced Matrigel transmi-
gration, actual tumor cell progression obviously requires
properties in addition to those needed for in vitro transmi-
gration. In sum, these data strongly support a critical role for
uPAR in realizing the invasive potential driven by EGFR/
PLCg signaling.
Discussion
The signals promoting tumor cell invasion and metas-
tasis are intertwined in a complex medley of signaling
from growth factors and ECM components [1]. In this
study, we probed downstream of a previously established
finding that growth factor-induced migration, via PLCg
signaling, is rate-limiting in tumor cell invasion [33].
While PLCg contributes to motility induced by numerous
growth factor receptors [34–36] via an epigenetic hydro-
lysis of PIP2 and mobilization of actin-binding proteins
[37,38], there is evidence that signaling through this
effector also alters the gene expression profile [6]. Thus,
we asked whether these changes in transcription contribute
to tumor invasion. Using mRNA microarray analysis, we
discovered a number of genes that were upregulated in the
human prostate carcinoma DU145 cell sublines in con-
junction with increased invasiveness both in vitro and in
vivo. The urokinase receptor uPAR was found to be
downregulated when the human prostate cancer line
PC3, which also presents upregulated autocrine EGFR
signaling, was challenged with the EGFR-preferential
kinase inhibitor genistein [20]. Thus, we focused on
clarifying the relationship between these two receptor
systems in promoting tumor cell progression. We showed
for the first time that EGFR signaling increases uPAR-
mediated phenotypic cell behaviors in tumor cells and that
this increase in uPAR activity is downstream of PLCg
signaling. Most importantly, using both in vitro and in
vivo models of tumor invasion, we demonstrated that the
uPAR/uPA complex contributes to invasion of these pros-
tate tumor cells but only in the presence of a functional
EGFR/PLCg signaling cascade. In short, the findings paint
a picture not of two separate and distinct regulators of
invasion but that these two critical pathways, uPAR and
EGFR, are inexorably linked. This reinforcing cascade of
invasive signaling from cell surface receptors provides two

Fig. 4. Diaphragm invasion of DU145 cells in athymic nude mice. Mice were injected in the peritoneal cavity with 2 million cells. (A) DU145 WT or DU145
WT antisense uPAR cells or (B) parental DU145 or parental DU145 expressing sense uPAR cells were inoculated. Mice were killed after 60 days, and organ/
tumor growth and metastasis were determined. Organs, including the diaphragm, were removed, fixed in 10% formalin and stained with H&E. The left column
shows a representative diaphragm taken from a mouse injected with DU145 WT or parental DU145, whereas the right column indicates diaphragms taken from
mice injected with DU145 WT antisense uPAR or parental DU145 sense uPAR cells.
A. Mamoune et al. / Experimental Cell Research 299 (2004) 91–100 97
new molecules that would be extracellularly accessible
targets for intervention.
Our previous studies demonstrated a rate-limiting role for
PLCg activity in tumor cell invasion. In prostate, breast and
bladder carcinomas, and glioblastoma, inhibition of PLCg
significantly retarded cell invasion in vitro and/or in vivo
[21,23,25,33]. In both breast and prostate cells, invasion
through PLCg activity was shown to be secondary to
autocrine EGFR activity, since (a) a truncated EGFR which
does not signal PLCg had reduced invasion [21] and (b)
Table 1
Invasion into the diaphragm of intraperitoneal tumors from DU145 sublines
Wild-type Wild-type
AS-uPAR
Parental Parental SN
Diaphragm
tumorsa24/24 17/17 16/16 14/15
Diaphragm
invasivenessb2.45 F 0.3 1.29* F 0.36 2.18 F 0.34 2.28 F 0.32
Data were collated from three different experiments of study with 5–8 mice
each.a Number of mice with tumors on diaphragm/mice inoculated.b On a scale of 0–4, with 0 being noninvasive with demonstrable fibrosis
capsule and 4 being transdiaphragmatic invasion.
*P < 0.05.
treating cells with the EGFR-specific kinase inhibitor
PD153035 likewise retarded cell invasion through Matrigel
[39]. These findings strongly suggest a pivotal role for
EGFR (or other growth factor receptors) promoting invasion
through the PLCg motility pathway. However, other signal-
ing pathways likely also contribute to invasion. Previous
studies have documented the role of upregulated uPAR in
invasion of various tumors, including glioblastomas, carci-
nomas of the bladder and breast, and highly metastatic
human lung cancer cells [10,40–42]. That this is linked to
tumor progression has been shown by interventional studies.
Antibodies against uPA or uPAR inhibit cancer invasion and
metastasis in vivo or in vitro in various tumors [11,43].
Downregulation of uPAR mRNA using anti-uPAR oligonu-
cleotides or antisense uPAR cDNA reversed the invasive
phenotype of human fibroblasts and inhibited the invasion
of human colon cancer cell lines, respectively [44]. These
tumors are the same types in which upregulated EGFR
signaling strongly correlates with invasion and tumor pro-
gression [45–47]. However, it was not probed whether these
two pathways were upregulated separately or as a concor-
dant event. Our results herein strongly suggest a causal link
between EGFR-PLCg signaling and uPAR upregulation.
Thus, it appears that these two surface receptors are coor-

A. Mamoune et al. / Experimental Cell Research 299 (2004) 91–10098
dinately upregulated during progression of human tumors.
However, it was quite unknown if the contributions of
EGFR and uPAR signaling to tumor invasion were linked
or separate.
That uPAR and, to a lesser extent, uPA were found to be
induced by autocrine EGFR signaling through the PLCg
signaling pathway strongly suggests that the invasiveness
driven by these two molecules are interlinked. Particularly
intriguing is the finding that uPAR’s role in tumor invasion
likely occurs through increased cell migration. uPAR is
known to localize at the leading edge of the cell during
the migration process [48] and has been shown to be
required for migration of a number of cells. It also is vital
for promoting integrin signaling (reviewed in [8]), along
with various integrin-linked molecules such as focal adhe-
sion kinase (FAK) and mitogen-activated protein kinase
(MAPK) [49]. More recently, uPAR has been shown to
promote extravillous trophoblast migration via the ERK
MAP kinase and PLC pathways [29]. Furthermore, these
promotility signals from uPAR may occur, at least in part,
via transactivation of EGFR [50]. These same intracellular
effectors (ERK, PLC, etc.) are also required for signaling
from EGFR and other growth factor receptors [49,51]. Thus,
coordinate upregulation of uPAR in the face of an active
TGFa-EGFR autocrine signaling loop, present in practically
all prostate carcinomas [3], would serve either to reinforce
these signaling pathways or broaden the biological
responses contingent upon these.
We found that uPAR function was rate-limiting for
invasion of these cells that have active EGFR autocrine
signaling and activation of PLCg. In vitro transmigration of
the ECM Matrigel was modulated by uPAR levels in the
highly invasive DU145 WT and moderately invasive pa-
rental DU145 sublines (Table 1); abrogating uPAR by
antisense in DU145 WT (DU145 WT AS) reduced tumor
aggressiveness, whereas upregulation in parental DU145
(DU145 P SN) did not lead to significantly greater invasion
in vivo. Thus, invasion in vivo requires properties in
addition to those needed for ECM transmigration in vitro,
or uPAR may interact with tumor cell- or organismal-
derived ligands and inhibitors that modulate uPAR/uPA
functioning in animals. One suggestive hint is the finding
that HGF is also upregulated at the transcriptional level in
these same DU145 WT cells but not in the DU145 P cells
[6], particularly so since uPA proteolytically processes
HGF to an active form. Still, whatever the reason, uPAR
overexpression in itself was not capable of restoring the
increased invasiveness noted in the DU145 WT cells in
which endogenous uPAR expression is increased. This
demonstrates that the contribution of uPAR to tumor
invasion is not an independent event but linked to a larger
web of signals. That uPAR is required even for EGFR-
mediated tumor invasion presents obvious implications for
rational therapeutic interventions in that uPAR, being on
the cell surface, is targetable by noncell permeant agents.
Still, for this approach to be realized, the operative mech-
anism of uPAR signaling (i.e., protease, complexing with
integrins, synergizing with EGFR [50], or direct receptor
signaling) in this context needs to be determined.
One last point warrants further emphasis. While uPAR
gene expression levels correlated most strongly with inva-
siveness in our DU145 cells lines, a number of other
prominent receptors or ligands also were overexpressed in
DU145 WT, including HGF, IGF-1, and the integrin subunit
av. Not only have these been implicated in promoting tumor
cell invasion, but they are also known to interact with uPAR
in promoting cell growth as well as migration and invasion
[52,53]. This is particularly relevant for HGF, which is
activated from its proform by uPA-mediated cleavage
[54]. Thus, this initial study, in only prostate cancer that
will require experimental validation in other tumor types,
may only be the tip of the iceberg with a panoply of genes
being expressed in concert that further reinforce the motility,
scatter, and proliferative properties needed for successful
access to and growth in ectopic sites that are the essence of
metastasis.
Acknowledgments
We thank Wendy Mars for helpful discussions and
guidance and Diana Whaley and James Solava for critical
technical support. This study was supported by grants from
the Veterans Adminstration Merit Award Program and the
National Cancer Institute at NIH.
References
[1] A. Wells, Tumor invasion: role of growth factor-induced cell motility,
Adv. Cancer Res. 78 (2000) 31–101.
[2] S.A. Aaronson, Growth factors and cancer, Science 254 (1991)
1146–1153.
[3] H. Kim, T. Turner, J. Kassis, J. Souto, A.Wells, EGF receptor signaling
in prostate development, Histol. Histopathol. 14 (1999) 1175–1182.
[4] A. Glading, D.A. Lauffenburger, A. Wells, Cutting to the chase:
calpain proteases in cell migration, Trends Cell Biol. 12 (2002)
46–54.
[5] A. Malliri, M. Symons, R.F. Hennigan, A.F.L. Hurlstone, R.F. Lamb,
T. Wheeler, B.W. Ozanne, The transcriptional factor AP-1 is required
for EGF-induced activation of rho-like GTPases, cytoskeletal rear-
rangements, motility, and in vitro invasion of A431 cells, J. Cell Biol.
143 (1998) 1087–1099.
[6] E.J. Manos, M. Kim, J. Kassis, B. Chang, A. Wells, D.A. Jones,
Prostin-1, a novel phospholipase C-g regulated gene negatively asso-
ciated with prostate tumor invasion, Oncogene 20 (2001) 2781–2790.
[7] P.A. Andreasen, R. Egelund, H.H. Petersen, The plasminogen activa-
tion system in tumor growth, invasion, and metastasis, Cell. Mol. Life
Sci. 57 (2000) 25–40.
[8] F. Blasi, P. Carmeliet, uPAR: a versatile signalling orchestrator, Nat.
Rev., Mol. Cell Biol. 3 (2002) 932–943.
[9] D.F. Liu, S.A. Rabbani, Induction of urinary plasminogen activator by
retinoic acid results in increased invasiveness of human prostate can-
cer cells PC-3, Prostate 27 (1995) 269–276.
[10] H. Miyake, I. Hara, K. Yamanaka, S. Arakawa, S. Kamidono, Eleva-
tion of urokinase-type plasminogen activator and its receptor densities

A. Mamoune et al. / Experimental Cell Research 299 (2004) 91–100 99
as new predictors of disease progression and prognosis in men with
prostate cancer, Int. J. Oncol. 14 (1999) 535–541.
[11] S.A. Rabbani, J. Gladu, Urokinase receptor antibody can reduce tu-
mor volume and detect the presence of occult tumor metastases in
vivo, Cancer Res. 62 (2002) 2390–2397.
[12] N. Sidenius, F. Blasi, The urokinase plasminogen activator system in
cancer: recent advances and implications for prognosis and therapy,
Cancer Metastasis Rev. 22 (2003) 205–222.
[13] D. Liu, J.A. Aguirre-Ghiso, Y. Estrada, L. Ossowski, EGFR is a
transducer of the urokinase receptor initiated signal that is required
for in vivo growth of a human carcinoma, Cancer Cell 1 (2002)
445–457.
[14] J.A. Aguirre-Ghiso, D. Liu, A. Mignatti, K. Kovalski, L. Ossowski,
Urokinase receptor and fibronectin regulate the ERKMAPK to
p38MAPK activity ratios that determine carcinoma cell proliferation
or dormancy in vivo, Mol. Biol. Cell 12 (2001) 863–879.
[15] M. Jo, K.S. Thomas, D.M. O’Donnell, S.L. Gonias, Epidermal
growth factor receptor-dependent and -independent cell-signaling
pathways originating from the urokinase receptor, J. Biol. Chem.
278 (2003) 1642–1646.
[16] A. Unlu, R.E. Leake, The effect of EGFR-related tyrosine kinase
activity inhibition on the growth and invasion mechanisms of prostate
carcinoma cell lines, Int. J. Biol. Markers 18 (2003) 139–146.
[17] T. Shiratsuchi, H. Ishibashi, K. Shirasuna, Inhibition of epidermal
growth factor-induced invasion by dexamethasone and AP-1 decoy
in human squamous cell carcinoma cell lines, J. Cell. Physiol. 193
(2002) 340–348.
[18] P.J. Jensen, U. Rodeck, Autocrine/paracrine regulation of keratinocyte
urokinase plasminogen activator through the TGF-alpha/EGF recep-
tor, J. Cell. Physiol. 155 (1993) 333–339.
[19] D.F. Jarrard, B.F. Blitz, R.C. Smith, B.L. Patai, D.B. Rukstalis,
Effects of epidermal growth factor on prostate cancer cell line PC3
growth and invasion, Prostate 24 (1994) 46–53.
[20] Y. Li, F. Sarkar, Down-regulation of invasion and angiogenesis-related
genes identified by cDNA microarray analysis of PC3 prostate cancer
cells treated with genistein, Cancer Lett. 186 (2002) 157–164.
[21] T. Turner, P. Chen, L.J. Goodly, A. Wells, EGF receptor signaling
enhances in vivo invasiveness of DU-145 human prostate carcinoma
cells, Clin. Exp. Metastasis 14 (1996) 409–418.
[22] K. Stone, D.D. Mickey, H. Wunderli, G.H. Mickey, D.F. Paulson,
Isolation of a human prostate carcinoma cell line (DU145), Int. J.
Cancer 21 (1978) 274–281.
[23] H. Xie, T. Turner, M.-H. Wang, R.K. Singh, G.P. Siegal, A. Wells, In
vitro invasiveness of DU-145 human prostate carcinoma cells is
modulated by EGF receptor-mediated signals, Clin. Exp. Metastasis
13 (1995) 407–419.
[24] P. Chen, K. Gupta, A. Wells, Cell movement elicited by epidermal
growth factor receptor requires kinase and autophosphorylation but is
separable from mitogenesis, J. Cell Biol. 124 (1994) 547–555.
[25] T. Turner, M. VanEpps-Fung, J. Kassis, A. Wells, Molecular inhibi-
tion of PLCg signaling abrogates DU-145 prostate tumor cell inva-
sion, Clin. Cancer Res. 3 (1997) 2275–2282.
[26] G. Hoyer-Hansen, E. Ronne, H. Solberg, N. Behrendt, M. Ploug,
L.R. Lund, V. Ellis, K. Dano, Urokinase plasminogen activator
cleaves its cell surface receptor releasing the ligand-binding domain,
J. Biol. Chem. 267 (1992) 18224–18229.
[27] D.H. Nguyen, I.M. Hussaini, S.L. Gonias, Binding of urokinase-type
plasminogen activator to its receptor in MCF-7 cells activates extra-
cellular signal-regulated kinase 1 and 2 which is required for in-
creased cellular motility, J. Biol. Chem. 273 (1998) 18268–18272.
[28] N. Busso, S.K. Masur, D. Lazega, S. Waxman, L. Ossowski, Induc-
tion of cell migration by pro-urokinase binding to its receptor:
possible mechanisms for signal transduction in human epithelial
cells, J. Cell Biol. 126 (1994) 259–270.
[29] J. Liu, C. Chakraborty, C.H. Graham, Y.P. Barbin, S.J. Dixon, P.K.
Lala, Noncatalytic domain of uPA stimulates human extravillous tro-
phoblast migration by using phospholipase C, phosphatidylinositol 3-
kinase and mitogen-activated protein kinase, Exp. Cell Res. 286
(2003) 138–151.
[30] J. Jankun, E. Skrzypczak-Jankun, Binding sites of amiloride to uro-
kinase plasminogen activator depends on species, Int. J. Mol. Med.
8 (2001) 365–371.
[31] N. Oka, Y. Okumura, H.O. Kanayama, H. Izaki, M. Okamoto, H.
Kido, S. Kagawa, Amiloride and urinary trypsin inhibitor inhibit
urothelial cancer invasion, Eur. Urol. 44 (2003) 737–741.
[32] J. Jankun, R.W. Keck, E. Skrzypczak-Jankun, R. Swiercz, Inhibitors
of urokinase reduce size of prostate cancer xenografts in severe com-
bined immunodeficient mice, Cancer Res. 57 (1997) 559–563.
[33] J. Kassis, D.A. Lauffenburger, T. Turner, A. Wells, Tumor invasion as
dysregulated cell motility, Semin. Cancer Biol. 11 (2001) 105–118.
[34] K.E. Bornfeldt, E.W. Raines, T. Nakano, L.M. Graves, E.G. Krebs, R.
Ross, Insulin-like growth factor-1 and platelet-derived growth factor-
BB induce directed migration of human arterial smooth muscle cells
via signalling pathways that are distinct from those of proliferation,
J. Clin. Invest. 93 (1994) 1266–1274.
[35] V. Kundra, J.A. Escobedo, A. Kazlauskas, H.K. Kim, S.G. Rhee, L.T.
Williams, B.R. Zetter, Regulation of chemotaxis by the platelet-de-
rived growth factor receptor-h, Nature 367 (1994) 474–476.
[36] P. Chen, H. Xie, M.C. Sekar, K.B. Gupta, A. Wells, Epidermal growth
factor receptor-mediated cell motility: phospholipase C activity is
required, but MAP kinase activity is not sufficient for induced cell
movement, J. Cell Biol. 127 (1994) 847–857.
[37] P. Chen, J. Murphy-Ullrich, A. Wells, A role for gelsolin in actua-
ting EGF receptor-mediated cell motility, J. Cell Biol. 134 (1996)
689–698.
[38] J. Chou, D. Beer-Stolz, N. Burke, S.C. Watkins, A. Wells, Distribu-
tion of gelsolin and phosphoinositol 4,5-bisphosphate in lamellipodia
during EGF-induced motility, Int. J. Biochem. Cell Biol. 34 (2002)
776–790.
[39] J. Kassis, J. Moellinger, H. Lo, N. Greenberg, H.-G. Kim, A. Wells, A
role for phospholipase C-g-mediated signaling in tumor cell invasion,
Clin. Cancer Res. 5 (1999) 2251–2260.
[40] S.J. Gong, S.Y. Rha, H.C. Chung, N.C. Yoo, J.K. Roh, W.I. Yang,
K.S. Lee, J.S. Min, B.S. Kim, H.C. Chung, Tissue urokinase-type
plasminogen activator receptor levels in breast cancer, Int. J. Mol.
Med. 6 (2000) 301–305.
[41] M. Hudson, M. McReynolds, Urokinase and the urokinase receptor:
association with in vitro invasiveness of human bladder cancer cell
lines, J. Natl. Cancer Inst. 89 (1997) 709–717.
[42] T.J. MacDonald, Y.A. DeClerk, W.E. Laug, Urokinase induces recep-
tor mediated brain tumor cell migration and invasion, J. Neurooncol.
40 (1998) 215–226.
[43] S. Mohanam, R. Sawaya, I. McCutcheon, F. Ali-Osman, D. Boyd,
J.S. Rao, Modulation of in vitro invasion of human glioblastoma cells
by urokinase-type plasminogen activator receptor antibody, Cancer
Res. 53 (1993) 4143–4147.
[44] A. Quattrone, G. Fibbi, E. Anichini, M. Pucci, A. Zamperini, S.
Capaccioli, M.D. Rosso, Reversion of the invasive phenotype of
transformed human fibroblasts by anti-messenger ODNs inhibition
of urokinase receptor gene expression, Cancer Res. 55 (1995) 90–95.
[45] T.A. Libermann, H.R. Nusbaum, N. Razon, R. Kris, I. Lax, H. Soreq,
N. Whittle, M.D. Waterfield, A. Ullrich, J. Schlessinger, Amplifica-
tion, enhanced expression and possible rearrangement of EGF recep-
tor gene in primary human brain tumours of glial origin, Nature 313
(1985) 144–147.
[46] D.E. Neal, C. Marsh, M.K. Bennett, P.D. Abel, R.R. Hall, J.R.C.
Sainsbury, A.L. Harris, Epidermal-growth-factor receptors in human
blood cancer: comparison of invasive and superficial tumours, Lan-
ceti i (1985) 366–368.
[47] J.R.C. Sainsbury, J.R. Farndon, G.K. Needham, A.J. Malcolm,
A.L. Harris, Epidermal-growth-factor receptor status as predictor
of early recurrence of and death from breast cancer, Lanceti
(1987) 1398–1402.
[48] A. Estreicher, J. Muhlhauser, J. Carpentier, L. Orci, J. Vassalli, The

A. Mamoune et al. / Experimental Cell Research 299 (2004) 91–100100
receptor for urokinase type plasminogen activator polarizes expres-
sion of the protease to the leading edge of migrating edge of mono-
cytes and promotes degradation of enzyme inhibitor complexes, J.
Cell Biol. 111 (1990) 783–792.
[49] H. Tang, D.M. Kerins, Q. Hao, T. Inagami, D.E. Vaughan, The uro-
kinase-type plasminogen activator receptor mediates tyrosine phos-
phorylation of focal adhesion proteins and activation of mitogen-
activated protein kinase in cultured endothelial cells, J. Biol. Chem.
273 (1998) 18268–18272.
[50] M. Jo, K.S. Thomas, L. Wu, S.L. Gonias, Soluble urokinase-type
plasminogen activator receptor inhibits cancer cell growth and inva-
sion by direct urokinase-independent effects on cell signaling, J. Biol.
Chem. 278 (2003) 46692–46698.
[51] A. Wells, Molecule in focus: EGF receptor, Int. J. Biochem. Cell Biol.
31 (1999) 637–643.
[52] Y. Okusa, T. Ichikura, H. Mochizuki, N. Shinomiya, Urokinase type
plasminogen activator and its receptor regulates the invasive potential
of gastric cancer cell lines, Int. J. Oncol. 17 (2000) 1001–1005.
[53] Y. Wei, J.A. Eble, Z. Wang, J.A. Kreidberg, H.A. Chapman, Uroki-
nase receptors promote h1 integrin function through interactions with
integrin a3h1, Mol. Biol. Cell 12 (2001) 2975–2986.
[54] T. Moriyama, H. Kataoka, P. Hamasuna, E. Yoshida, T. Sameshima,
T. Iseda, K. Yokogami, S. Nakano, M. Koono, S. Wakisaka, Simul-
taneous up-regulation of urokinase-type plasminogen activator (uPA)
and uPA receptor by hepatocyte growth factor/scatter factor in human
glioma cells, Clin. Exp. Metastasis 17 (1999) 873–879.
Copyright © 2022 FDOKUMEN




















