
Platelet-Derived Growth Factor Receptor B-Mediated Phosphorylation of MUC1 Enhances Invasiveness in...
11
Platelet-Derived Growth Factor Receptor B–Mediated Phosphorylation of MUC1 Enhances Invasiveness in Pancreatic Adenocarcinoma Cells Pankaj K. Singh, 1,2 Yunfei Wen, 1,2 Benjamin J. Swanson, 1 Kandavel Shanmugam, 3 Andrius Kazlauskas, 4 Ronald L. Cerny, 5 Sandra J. Gendler, 3 and Michael A. Hollingsworth 1,2 1 Eppley Institute for Research in Cancer and Allied Diseases and 2 Department of Biochemistry and Molecular Biology, University of Nebraska Medical Center, Omaha, Nebraska; 3 Department of Biochemistry/Molecular Biology Program, Mayo Clinic College of Medicine, Scottsdale, Arizona; 4 Schepens Eye Research Institute, Harvard Medical School, Boston, Massachusetts; and 5 University of Nebraska-Lincoln, Lincoln, Nebraska Abstract MUC1 is a heterodimeric transmembrane glycoprotein that is overexpressed and aberrantly glycosylated in ductal adeno- carcinomas. Differential phosphorylation of the MUC1 cyto- plasmic tail (MUC1CT) has been associated with signaling events that influence the proliferation and metastasis of cancer cells. We identified a novel tyrosine phosphorylation site (HGRYVPP) in the MUC1CT by mass spectrometric analysis of MUC1 from human pancreatic adenocarcinoma cell lines. Analyses in vitro and in vivo showed that platelet- derived growth factor receptor B (PDGFRB) catalyzed phos- phorylation of this site and of tyrosine in the RDTYHPM site. Stimulation of S2-013.MUC1F cells with PDGF-BB increased nuclear colocalization of MUC1CT and B-catenin. PDGF-BB stimulation had no significant effect on cell proliferation rate; however, it enhanced invasion in vitro through Matrigel and in vivo tumor growth and metastases. Invasive properties of the cells were significantly altered on expression of phosphor- ylation-abrogating or phosphorylation-mimicking mutations at these sites. We propose that interactions of MUC1 and PDGFRB induce signal transduction events that influence the metastatic properties of pancreatic adenocarcinoma. [Cancer Res 2007;67(11):5201–10] Introduction Pancreatic cancer is associated with high mortality and poor prognosis, in part because it is known to metastasize early during disease progression. Several studies have shown that the MUC1 glycoprotein contributes to growth and metastasis of pancreatic adenocarcinomas. MUC1 protein has been detected in >90% of pancreatic tumors examined by immunohistochemistry (1), in the pancreatic juice of pancreatic adenocarcinoma patients by proteomic analysis, and in most pancreatic cancer cell lines (2, 3). Sialylated MUC1 is overexpressed by invading and metastatic pancreatic cancer cells but not by normal pancreas or in cases of chronic pancreatitis or pancreatic ductal hyperplasia (4). Previous studies from our laboratory have established a role for MUC1 in invasion and metastasis in pancreatic cancer (5, 6). MUC1 is a type I transmembrane protein that consists of a large extracellular subunit comprised by a mucin-type tandem repeat and a smaller subunit that includes a small extracellular domain and a transmembrane domain plus a 72–amino acid cytoplasmic tail (MUC1CT). These two subunits are generated from the intracellular autocatalytic proteolytic cleavage of a single precursor polypeptide chain (7–9). The larger extracellular subunit consists of a heavily O -glycosylated tandem repeat unit of 20 amino acids that is repeated from 18 to over 100 times in different alleles (10–12). The MUC1CT is phosphorylated and involved in different signaling pathways (13). Initial studies suggested that changes in phosphorylation of MUC1CT were correlated with differences in cell adhesion (14–17). An SXXXXXSSL motif in the MUC1CT interacts with h-catenin and may compete with E-cadherin for binding with h-catenin (18, 19). This interaction is abrogated by mutation of a tyrosine to phenylalanine at the DRSPYEKV sequence, a site that is phosphorylated by c-Src, epidermal growth factor receptor (EGFR), or Lyn kinases. A fragment of MUC1CT is translocated to the nucleus in association with h-catenin and may influence its activity as a transcriptional coactivator (19). Phosphorylated YEKV is believed to serve as a binding site for Src SH2 domains (20). Phosphorylation of tyrosine in the YTNP sequence in the MUC1CT is postulated to enhance binding to the adaptor protein Grb2 and its associated small G protein activator SOS, providing a potential activation mechanism for Ras and its effectors (21, 22). Thus, there is substantial evidence that phosphorylation of the MUC1CT modulates signaling related to proliferation and metastasis. Previous investigations into the phosphorylation of MUC1 have been based on expression of constructs harboring mutations at specific tyrosine residues and analysis of changes in degrees of phosphorylation of all tyrosine residues on the MUC1CT by Western blot analysis with anti-phosphotyrosine antibodies. Here, we directly evaluated the phosphorylation status of MUC1 in pancreatic cancer cell lines by mass spectrometry (MS)-based sequence analysis of the MUC1CT, which revealed a novel tyrosine phosphorylation site at the HGRYVPP sequence in MUC1CT. We showed that platelet-derived growth factor receptor h (PDGFRh) is a kinase for the site and that stimulation of the pancreatic cancer cell line S2-013.MUC1F with PDGF-BB enhanced phosphorylation of MUC1CT, increased nuclear localization of MUC1CT and its association partner, h-catenin, and enhanced the invasiveness of S2-013.MUC1F cells without significantly affecting their prolifera- tion rate. Nuclear localization of MUC1CT and invasiveness of the cells were significantly diminished by tyrosine to phenylalanine mutations at these sites, whereas mutations of the tyrosine Requests for reprints: Michael A. Hollingsworth, Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, 986805 Nebraska Medical Center, Omaha, NE 68198-6805. Phone: 402-559-8343; Fax: 402-559-3339; E-mail: [email protected]. I2007 American Association for Cancer Research. doi:10.1158/0008-5472.CAN-06-4647 www.aacrjournals.org 5201 Cancer Res 2007; 67: (11). June 1, 2007 Research Article Research. on June 19, 2015. © 2007 American Association for Cancer cancerres.aacrjournals.org Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Platelet-Derived Growth Factor Receptor B-Mediated Phosphorylation of MUC1 Enhances Invasiveness in...

Platelet-Derived Growth Factor Receptor B–Mediated
Phosphorylation of MUC1 Enhances Invasiveness
in Pancreatic Adenocarcinoma Cells
Pankaj K. Singh,1,2
Yunfei Wen,1,2
Benjamin J. Swanson,1Kandavel Shanmugam,
3
Andrius Kazlauskas,4Ronald L. Cerny,
5Sandra J. Gendler,
3
and Michael A. Hollingsworth1,2
1Eppley Institute for Research in Cancer and Allied Diseases and 2Department of Biochemistry and Molecular Biology, University ofNebraska Medical Center, Omaha, Nebraska; 3Department of Biochemistry/Molecular Biology Program, Mayo ClinicCollege of Medicine, Scottsdale, Arizona; 4Schepens Eye Research Institute, Harvard Medical School,Boston, Massachusetts; and 5University of Nebraska-Lincoln, Lincoln, Nebraska
Abstract
MUC1 is a heterodimeric transmembrane glycoprotein that isoverexpressed and aberrantly glycosylated in ductal adeno-carcinomas. Differential phosphorylation of the MUC1 cyto-plasmic tail (MUC1CT) has been associated with signalingevents that influence the proliferation and metastasis ofcancer cells. We identified a novel tyrosine phosphorylationsite (HGRYVPP) in the MUC1CT by mass spectrometricanalysis of MUC1 from human pancreatic adenocarcinomacell lines. Analyses in vitro and in vivo showed that platelet-derived growth factor receptor B (PDGFRB) catalyzed phos-phorylation of this site and of tyrosine in the RDTYHPM site.Stimulation of S2-013.MUC1F cells with PDGF-BB increasednuclear colocalization of MUC1CT and B-catenin. PDGF-BBstimulation had no significant effect on cell proliferation rate;however, it enhanced invasion in vitro through Matrigel andin vivo tumor growth and metastases. Invasive properties ofthe cells were significantly altered on expression of phosphor-ylation-abrogating or phosphorylation-mimicking mutationsat these sites. We propose that interactions of MUC1 andPDGFRB induce signal transduction events that influence themetastatic properties of pancreatic adenocarcinoma. [CancerRes 2007;67(11):5201–10]
Introduction
Pancreatic cancer is associated with high mortality and poorprognosis, in part because it is known to metastasize early duringdisease progression. Several studies have shown that the MUC1glycoprotein contributes to growth and metastasis of pancreaticadenocarcinomas. MUC1 protein has been detected in >90% ofpancreatic tumors examined by immunohistochemistry (1), in thepancreatic juice of pancreatic adenocarcinoma patients byproteomic analysis, and in most pancreatic cancer cell lines(2, 3). Sialylated MUC1 is overexpressed by invading and metastaticpancreatic cancer cells but not by normal pancreas or in cases ofchronic pancreatitis or pancreatic ductal hyperplasia (4). Previousstudies from our laboratory have established a role for MUC1 ininvasion and metastasis in pancreatic cancer (5, 6).
MUC1 is a type I transmembrane protein that consists of a largeextracellular subunit comprised by a mucin-type tandem repeatand a smaller subunit that includes a small extracellular domainand a transmembrane domain plus a 72–amino acid cytoplasmictail (MUC1CT). These two subunits are generated from theintracellular autocatalytic proteolytic cleavage of a single precursorpolypeptide chain (7–9). The larger extracellular subunit consists ofa heavily O-glycosylated tandem repeat unit of 20 amino acids thatis repeated from 18 to over 100 times in different alleles (10–12).The MUC1CT is phosphorylated and involved in different
signaling pathways (13). Initial studies suggested that changes inphosphorylation of MUC1CT were correlated with differences incell adhesion (14–17). An SXXXXXSSL motif in the MUC1CTinteracts with h-catenin and may compete with E-cadherin forbinding with h-catenin (18, 19). This interaction is abrogated bymutation of a tyrosine to phenylalanine at the DRSPYEKVsequence, a site that is phosphorylated by c-Src, epidermal growthfactor receptor (EGFR), or Lyn kinases. A fragment of MUC1CT istranslocated to the nucleus in association with h-catenin and mayinfluence its activity as a transcriptional coactivator (19).Phosphorylated YEKV is believed to serve as a binding site forSrc SH2 domains (20). Phosphorylation of tyrosine in the YTNPsequence in the MUC1CT is postulated to enhance binding to theadaptor protein Grb2 and its associated small G protein activatorSOS, providing a potential activation mechanism for Ras and itseffectors (21, 22). Thus, there is substantial evidence thatphosphorylation of the MUC1CT modulates signaling related toproliferation and metastasis.Previous investigations into the phosphorylation of MUC1 have
been based on expression of constructs harboring mutations atspecific tyrosine residues and analysis of changes in degrees ofphosphorylation of all tyrosine residues on the MUC1CT byWestern blot analysis with anti-phosphotyrosine antibodies. Here,we directly evaluated the phosphorylation status of MUC1 inpancreatic cancer cell lines by mass spectrometry (MS)-basedsequence analysis of the MUC1CT, which revealed a novel tyrosinephosphorylation site at the HGRYVPP sequence in MUC1CT. Weshowed that platelet-derived growth factor receptor h (PDGFRh) isa kinase for the site and that stimulation of the pancreatic cancercell line S2-013.MUC1F with PDGF-BB enhanced phosphorylationof MUC1CT, increased nuclear localization of MUC1CT and itsassociation partner, h-catenin, and enhanced the invasiveness ofS2-013.MUC1F cells without significantly affecting their prolifera-tion rate. Nuclear localization of MUC1CT and invasiveness of thecells were significantly diminished by tyrosine to phenylalaninemutations at these sites, whereas mutations of the tyrosine
Requests for reprints: Michael A. Hollingsworth, Eppley Institute for Research inCancer and Allied Diseases, University of Nebraska Medical Center, 986805 NebraskaMedical Center, Omaha, NE 68198-6805. Phone: 402-559-8343; Fax: 402-559-3339;E-mail: [email protected].
I2007 American Association for Cancer Research.doi:10.1158/0008-5472.CAN-06-4647
www.aacrjournals.org 5201 Cancer Res 2007; 67: (11). June 1, 2007
Research Article
Research. on June 19, 2015. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

residues to glutamic acid enhanced nuclear localization andinvasiveness. These results provide compelling evidence thatPDGFRh-mediated phosphorylation of the MUC1CT regulatesinvasiveness of pancreatic adenocarcinoma cells.
Materials and Methods
Cells and reagents. Panc1 was obtained from the American Type
Culture Collection. S2-013 is a cloned subline of a human pancreatic tumor
cell line (SUIT-2) derived from a liver metastasis (23). Armenian Hamster
monoclonal antibody (mAb) CT2 against the MUC1CT was kindly providedby Dr. Sandra J. Gendler (Mayo Clinic, Scottsdale, AZ). The mAb anti-h-actin was purchased from Sigma. Horseradish peroxidase (HRP)-conjugated
AffiniPure Goat Anti-Armenian Hamster IgG(H+L) was purchased from
Jackson ImmunoResearch Laboratories, Inc. Rabbit polyclonal IgG againstthe kinase insert domain of PDGFRh has been described elsewhere (24).
HRP-conjugated rabbit anti-goat IgG was purchased from Pierce. Recom-
binant human PDGF-BB was purchased from PeproTech. Recombinantactive PDGFRh and c-Src were purchased from Upstate Cell Signaling
Solutions. MUC1F-wt, MUC1.FHPM (having tyrosine at YHPM mutated
to phenylalanine), MUC1.FVPP (having tyrosine at YVPP mutated to
phenylalanine), MUC1.EHPM (having tyrosine at YHPM site mutated toglutamate), and MUC1.EVPP (having tyrosine at YVPP site mutated
to glutamate) were created by using the QuikChange II XL Site-Directed
Mutagenesis kit (Stratagene) and cloned in pLNCX.1.
Cell culture. FLAG epitope-tagged MUC1 (MUC1F) transfectants of theS2-013 cell line (S2-013.MUC1F), Panc1 cell line (Panc1.MUC1F), and
cytoplasmic tail deleted MUC1 transfectants of the S2-013 cell line
(S2-013.CT3) were cultured as described previously (25). HPAF-2 cells weremaintained in Eagle’s MEM (Life Technologies, Inc.) supplemented with 10%
heat-inactivated fetal bovine serum (FBS), nonessential amino acids,
sodium pyruvate, and penicillin/streptomycin under similar conditions.
To express MUC1F-wt and the mutant MUC1 constructs, retroviraltransductions were done essentially as described previously (26).
Western blotting and immunoprecipitation. Cell lysate proteins wereresolved on 10% or 14% Novex Tris-glycine denaturing polyacrylamide gels
(Invitrogen) in a 1� SDS-PAGE buffer (1 g/L SDS, 3 g/L Tris base, 14.4 g/Lglycine). Western blotting and immunoprecipitations were done as
described previously (19).
In vitro kinase assay. Peptides (1 Ag) in 10 AL reaction buffer [PDGFRhreaction buffer: 40 mmol/L MOPS (pH 7.5), 1 mmol/L EGTA; Src reactionbuffer: 100 mmol/L Tris-HCl (pH 7.2), 125 mmol/L MgCl2, 25 mmol/L
MnCl2, 2 mmol/L EGTA, 0.25 mmol/L sodium orthovanadate, 2 mmol/L
DTT] were incubated at 37jC for 30 min along with 10 ACi [g-32P]ATP and500 ng of active human recombinant PDGFRh or 20 units of active humanrecombinant c-Src. The reaction was stopped by boiling the samples in
reducing SDS sample buffer. Samples were resolved on 16% Tricine gel. The
gel was then dried and by phosphorimager analysis.Tandem MS analysis of MUC1CT. Immunoprecipitated MUC1CT from
cell lysates or in vitro phosphorylated MUC1CT peptides were resolved on
14% Novex Tris-glycine denaturing polyacrylamide gels (Invitrogen) and
silver stained, and then bands corresponding MUC1CT were excised anddigested by a published method (27). Eluted peptides were analyzed using a
Q-TOF Ultima tandem mass spectrometer (Micromass/Waters) with
electrospray ionization. Analyses were done using data-dependent acqui-sition with the following variables: 1-s survey scan (380–1,900 Da) followed
by up to three 2.4-s MS/MS acquisitions (60–1,900 Da) at a mass resolution
of 8,000 Da. The instrument was calibrated using fragment ion masses of
doubly protonated Glu-fibrinopeptide.The MS/MS data were processed using MassLynx software (Micromass)
to produce peak lists that were input to the search engine MASCOT (Matrix
Science) and searched against the National Center for Biotechnology
Information nonredundant database. Search variables were as follows: massaccuracy 0.1 Da, enzyme specificity trypsin, fixed modification carboxya-
midomethylcysteine, and variable modification oxidized methionine.
Protein identifications were based on random probability scores with aminimum value of 25.
Immunofluorescence microscopy. Cells were cultured on glass cover-slips (Fisherbrand Microscope cover glass: 12-545-100 18CIR-1) at 4.5 � 105
per well for 12 h. Cells were rinsed once with serum-free medium, starved of
serum for 24 h, and then treated with PDGF-BB (50 ng/mL) for 2 h or were
left untreated in the serum-free medium. Immunofluorescence staining wasdone as described previously (19).
Matrigel invasion assay. In vitro invasive potential of cells under PDGF-
BB stimulation was assayed using the Biocoat Matrigel Invasion Chamber
(Becton Dickinson Labware) as described previously (6). Cells (5 � 104)were seeded onto the upper chamber of double-structured matrix gel
chamber. The lower chamber contained DMEM with or without 50 ng/mL
PDGF-BB or 1% FBS.
Tumor growth studies. Congenitally athymic female National CancerInstitute nude mice (NCr-nu/nu) were purchased from the National Cancer
Institute. Animals were maintained in pathogen-free conditions and fed
sterile water and food ad libitum . Mice were treated in accordance with theInstitutional Animal Care and Use Committee guidelines. Cells (1 � 105)
were used for orthotopic injections of cells into the pancreas of nude mice
as described previously (5). Mice (12–14) were used for each cell type. The
tumor size was measured postsurgery after the end of 4 weeks. Internalorgans, including lungs, liver, and all accessible mesenteric and mediastinal
lymph nodes, were dissected from sacrificed mice, fixed, and sectioned. The
identity of metastases within the dissected organs was confirmed by
staining the tissue sections with H&E before quantifying the numbers ofmetastases.
Statistical analysis. Results are expressed as mean F SE of three to five
independent experiments, each treatment done in triplicate. NonparametricKruskal-Wallis tests were used to compare differences between cell lines. If
the overall Kruskal-Wallis test indicated a difference in cell lines, a Wilcoxon
rank sum test was done for each pairwise comparison of interest. Analyses
were done using the Statistical Package for the Social Sciences or StatisticalAnalysis System for Windows version 8.2 (SAS Institute). Student’s t test was
used when appropriate. P < 0.05 was considered significant.
Results
Identification of a novel in vivo MUC1 phosphorylation site.Two human pancreatic adenocarcinoma cell lines, Panc1 andS2-013, stably expressing recombinant FLAG epitope-tagged full-length MUC1 (Panc1.MUC1F and S2-013.MUC1F, respectively) wereused for characterization of the phosphorylation status of MUC1.Panc1 is a poorly differentiated human pancreatic adenocarcinomacell line that expresses low levels of endogenous MUC1 andrelatively low levels of other glycoproteins (28). S2-013 is amoderately differentiated human pancreatic tumor cell line thatis known to express abundant O-glycosylated mucin-like proteins,including low levels of endogenous MUC1 (23). cDNA constructsencoding FLAG epitope-tagged forms of MUC1 have beendescribed previously (12).Cell lysates of S2-013.MUC1F and Panc1.MUC1F were immuno-
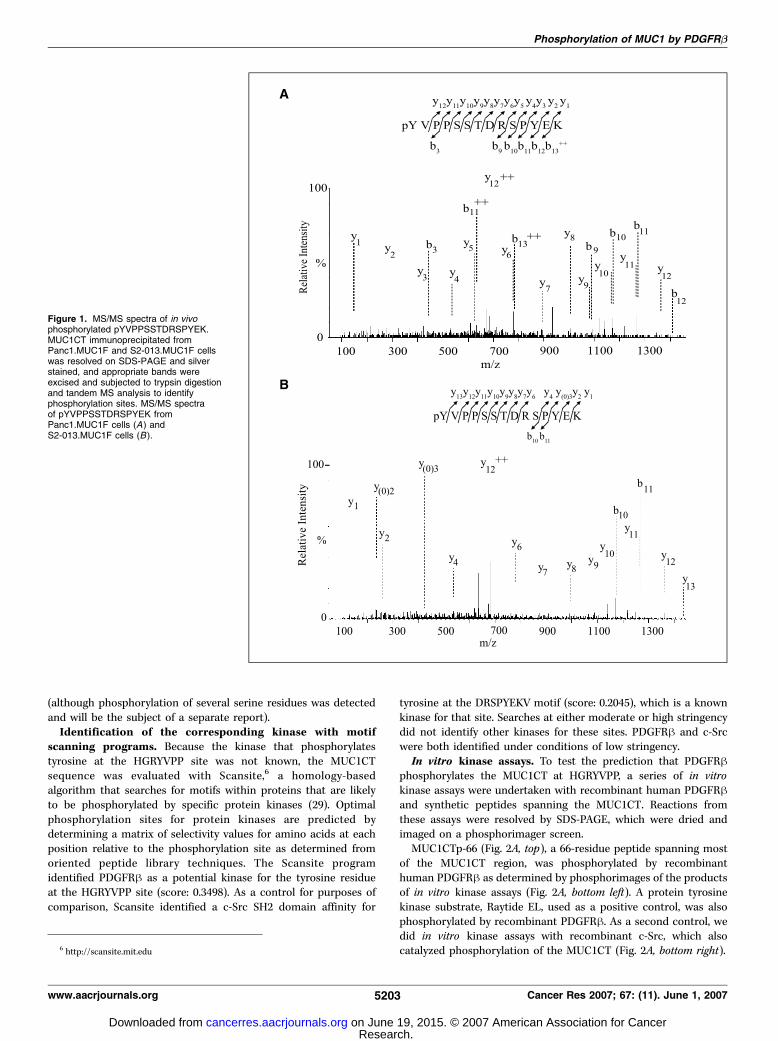
precipitated with CT2, a mAb raised against a peptide containingthe COOH-terminal 17 amino acids of MUC1CT, separated by SDS-PAGE, and silver stained with a MS-compatible silver stainprocedure. A duplicate gel was Western blotted and probed withCT2 antibody. Silver-stained bands of MUC1CT were identified atf30 kDa by comparison with bands obtained from the parallelWestern blotting experiment. These bands were sliced from the gel,subjected to in-gel trypsin digestion, and analyzed by tandem MSfor identification of phosphorylation sites. In repeated experiments,we detected tryptic fragments for the entire MUC1CT and observeda single and novel phosphorylation site at the first tyrosine residuein the YVPPSSTDRSPYEK peptide fragment in both Panc1.MUC1Fand S2-013.MUC1F cells (Fig. 1A and B). We did not detectphosphorylation of any other tyrosine residues in these analyses
Cancer Research
Cancer Res 2007; 67: (11). June 1, 2007 5202 www.aacrjournals.org
Research. on June 19, 2015. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from
(although phosphorylation of several serine residues was detectedand will be the subject of a separate report).Identification of the corresponding kinase with motif
scanning programs. Because the kinase that phosphorylatestyrosine at the HGRYVPP site was not known, the MUC1CTsequence was evaluated with Scansite,6 a homology-basedalgorithm that searches for motifs within proteins that are likelyto be phosphorylated by specific protein kinases (29). Optimalphosphorylation sites for protein kinases are predicted bydetermining a matrix of selectivity values for amino acids at eachposition relative to the phosphorylation site as determined fromoriented peptide library techniques. The Scansite programidentified PDGFRh as a potential kinase for the tyrosine residueat the HGRYVPP site (score: 0.3498). As a control for purposes ofcomparison, Scansite identified a c-Src SH2 domain affinity for
tyrosine at the DRSPYEKV motif (score: 0.2045), which is a knownkinase for that site. Searches at either moderate or high stringencydid not identify other kinases for these sites. PDGFRh and c-Srcwere both identified under conditions of low stringency.In vitro kinase assays. To test the prediction that PDGFRh
phosphorylates the MUC1CT at HGRYVPP, a series of in vitrokinase assays were undertaken with recombinant human PDGFRhand synthetic peptides spanning the MUC1CT. Reactions fromthese assays were resolved by SDS-PAGE, which were dried andimaged on a phosphorimager screen.MUC1CTp-66 (Fig. 2A, top), a 66-residue peptide spanning most
of the MUC1CT region, was phosphorylated by recombinanthuman PDGFRh as determined by phosphorimages of the productsof in vitro kinase assays (Fig. 2A, bottom left). A protein tyrosinekinase substrate, Raytide EL, used as a positive control, was alsophosphorylated by recombinant PDGFRh. As a second control, wedid in vitro kinase assays with recombinant c-Src, which alsocatalyzed phosphorylation of the MUC1CT (Fig. 2A, bottom right).6 http://scansite.mit.edu
Figure 1. MS/MS spectra of in vivophosphorylated pYVPPSSTDRSPYEK.MUC1CT immunoprecipitated fromPanc1.MUC1F and S2-013.MUC1F cellswas resolved on SDS-PAGE and silverstained, and appropriate bands wereexcised and subjected to trypsin digestionand tandem MS analysis to identifyphosphorylation sites. MS/MS spectraof pYVPPSSTDRSPYEK fromPanc1.MUC1F cells (A) andS2-013.MUC1F cells (B ).
Phosphorylation of MUC1 by PDGFRb
www.aacrjournals.org 5203 Cancer Res 2007; 67: (11). June 1, 2007
Research. on June 19, 2015. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

To further evaluate the region of MUC1CT phosphorylated byrecombinant PDGFRh, in vitro kinase assays were done withthree overlapping peptides spanning the whole MUC1CT:MUC1CTp-1 (a 30-residue peptide), MUC1CTp-2 (a 32-residuepeptide), and MUC1CTp-3 (a 29-residue peptide). The sequencesencoded by these peptides are shown in Fig. 2A . The HGRYVPPsequence was included in only the MUC1CTp-2 fragment.MUC1CTp-1 and MUC1CTp-2 were phosphorylated by recombi-nant PDGFRh; however, MUC1CTp-3 was not phosphorylated byPDGFRh (Fig. 2A).MS analysis of in vitro phosphorylated MUC1CTp-66. MS
sequence analysis of in vitro PDGFRh-phosphorylated MUC1CTp-66 revealed two phosphorylation sites: tyrosines at RDTYHPM(Fig. 2B) and HGRYVPP (Fig. 2C) in the MUC1CT. In addition, thetyrosine at DRSPYEKV was phosphorylated by c-Src (Fig. 2D). Wealso conducted in vitro assays with recombinant human EGFRand c-Met (hepatocyte growth factor receptor), which did notphosphorylate MUC1 at the YVPP site but did phosphorylateother sites that will be the subject of a separate report (data notshown).
PDGFRB-mediated in vivo phosphorylation of MUC1CT.Labeling of S2-013.MUC1F cells with [32P]orthophosphate followingstimulation with 50 ng/mL PDGF-BB induced significant increasesin phosphorylation of MUC1CT compared with unstimulated cells(Fig. 3A). This enhancement in phosphorylation was abrogated bythe PDGFR-specific phosphorylation inhibitor AG1295 (10 Amol/L;Fig. 3A). The stimulation of phosphorylation was dependent on theduration of incubation with PDGF-BB, as there were increasingintensities of phosphorylated MUC1CT bands on the autoradio-grams when cells were treated for 15 min, 30 min, and 1 h,respectively (Fig. 3B and C).PDGF stimulation causes nuclear localization of MUC1CT
phosphorylated at tyrosines in the YVPP and YHPM motifs.We produced rabbit polyclonal antibodies specific for phosphory-lated tyrosines at the YVPPand YHPM sites, which were validated forspecificity by ELISA and Western blotting analysis with 66-residueMUC1CT peptides phosphorylated at the these sites (unphosphory-lated 66-residue MUC1CT peptide was used as a control; data notshown). These antibodies were used to evaluate the effect of PDGFstimulation on phosphorylation of these sites in the MUC1CT.
Figure 2. In vitro phosphorylation of MUC1CT by PDGFRh. A, MUC1CTp-66 is a 66-residue synthetic peptide corresponding to the cytoplasmic tail of human MUC1.MUC1CTp-1, MUC1CTp-2, and MUC1CTp-3 are three overlapping peptides, together spanning the whole cytoplasmic tail region of human MUC1. Each peptide wasdesigned to span sequences that included one or two tyrosines and to include one or two tyrosines in each overlap, thereby including all seven tyrosines from theMUC1CT in three peptides. Autoradiograms from 32P-labeled in vitro kinase reactions of MUC1CT peptides with PDGFRh (bottom left ) and c-Src (bottom right ).Phosphorylated substrate peptides were confirmed by MS analysis. A protein tyrosine kinase substrate, Raytide EL, was used as a control for tyrosine kinase activity.B toD, MUC1CTp-66 peptides phosphorylated by in vitro kinase assays with PDGFRh (B andC) or c-Src (D) were sequenced by tandemMS and showed phosphorylationat DTpYHPMSEYPTYHTHGR (B) and pYVPPSSTDRSPYEK by PDGFRh (C). MUC1CTp-66 was phosphorylated by c-Src at YVPPSSTDRSPpYEK (D).
Cancer Research
Cancer Res 2007; 67: (11). June 1, 2007 5204 www.aacrjournals.org
Research. on June 19, 2015. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

Western blotting of nuclear and cytoplasmic extracts from PDGF-BB–stimulated S2-013.MUC1F cells revealed a significant increase inphosphorylation of tyrosines in the YVPP and YHPMmotifs that wasconcordant with increasing duration of PDGF stimulation and wasmostly observed in the nuclear fraction (Fig. 3D). MS analysis ofindependent cell lysates confirmed phosphorylation but is not aquantitative method (data not shown). A single band was observedwith the anti-pYVPP antibody; however, two bands were observedwith the anti-pYHPM antibody, whose appearance was dependenton the duration of PDGF stimulation. The two bands could representdifferent MUC1CT fragments phosphorylated at one or bothtyrosines in the YHPM and YVPP motifs or tyrosine phosphorylatedonly in the YHPM motif but showing a distinct mobility perhapsbecause of other post-translational modifications that are notdefined at this time.PDGF-BB stimulation enhances nuclear localization of
B-catenin and MUC1CT. We evaluated the effects of PDGFRh-mediated phosphorylation of MUC1CT on colocalization withh-catenin. Immunofluorescence analysis with the CT2 mAb (raisedagainst the MUC1CT) and a mAb against h-catenin was done onS2-013.MUC1F cells treated with 50 ng/mL PDGF-BB for 2 h (30).The localization patterns of MUC1CT and h-catenin werecompared with serum-starved S2-013.MUC1F cells.Serum-starved S2-013.MUC1F cells showed localization of
MUC1CT and h-catenin at the periphery of cells (Fig. 4A, top).Some but not all MUC1 and h-catenin were colocalized at theperiphery; presumably, h-catenin at cell junctions is colocalizedwith E-cadherin (31), and MUC1 is found in areas of cell other thanat these junctions. PDGF-BB stimulation induced extensive nuclearlocalization of h-catenin and nuclear and diffuse cytoplasmicstaining of MUC1CT (Fig. 4A, bottom). These results show thatPDGFRh-mediated phosphorylation of MUC1CT enhanced nuclearlocalization of MUC1CT.To confirm that phosphotyrosines in the YHPM and YVPP motifs
contributed to the nuclear localization of MUC1CT, phosphoryla-tion-abrogating (Y to F) and phosphorylation-mimicking (Y to E)mutations were generated for the YHPM and YVPP motifs ofMUC1CT cloned into the pLNCX.1 retroviral expression vector.Retrovirally transduced S2-013 cells were tested for stable expres-sion of the recombinant MUC1 proteins by Western blotting withmAb CT2. These S2-013 transfectants, named S2-013.MUC1.FHPM,S2-013.MUC1.EHPM, S2-013.MUC1.FVPP, and S2-013.MUC1.EVPP,were used to evaluate the nuclear localization of MUC1CT underserum-starved and PDGF-BB–stimulated conditions. As expected,
the S2-013.MUC1.FHPM and S2-013.MUC1.FVPP cells expressingthe inactivating mutations showed significant localization ofMUC1CT to the cell periphery, which was not dramatically affectedby stimulation with PDGF-BB (Fig. 4B). S2-013.MUC1.EHPM andS2-013.MUC1.EVPP cells expressing the activating mutationsshowed significant constitutive nuclear localization of MUC1 evenunder serum-starved conditions (Fig. 4B).PDGFRB-mediated phosphorylation of MUC1 enhances inva-
siveness of pancreatic adenocarcinoma cells. Overexpression ofMUC1 and enhanced nuclear localization of MUC1CTand h-catenin
Figure 3. In vivo phosphorylation of MUC1 by PDGFRh. A, serum-starvedS2-013.MUC1F cells were incubated for 15 min with [32P]orthophosphate(250 ACi/mL) in the presence or absence of human recombinant PDGF-BB(50 ng/mL) or the PDGF inhibitor AG1295 (10 Amol/L). Immunoprecipitates (IP )with mAb CT2 were resolved on 14% SDS-PAGE and immunoblotted (IB )with CT2 (bottom ), and then the membrane was subjected to autoradiography(top ). B, autoradiography of CT2 immunoprecipitates from S2-013.MUC1F cellsincubated with PDGF-BB (50 ng/mL) and [32P]orthophosphate (250 ACi/mL)for increasing periods (15 min, 30 min, and 1 h), with unstimulated cells ascontrols (top ). Bottom, a Western blotting experiment was done in parallel todetermine the amounts of immunoprecipitated MUC1CT. Representative ofexperiments repeated thrice. C, average pixel intensity for each band wasnormalized for pixel intensity in MUC1CT bands and plotted against fold increasecompared with unstimulated cells. D, nuclear (N ) and cytoplasmic (C )localization of MUC1CT phosphorylated at tyrosines at the YVPP and YHPMsites determined by Western blotting with phosphorylation site-specificantibodies. The polyvinylidene difluoride membrane was reprobed withantibodies against MUC1CT (CT2 mAb), histone2B (H2B ), a marker for nuclearproteins, and h-actin as controls for protein loading.
Phosphorylation of MUC1 by PDGFRb
www.aacrjournals.org 5205 Cancer Res 2007; 67: (11). June 1, 2007
Research. on June 19, 2015. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

are associated with increased metastasis and poor prognosis fordifferent adenocarcinoma. Hence, Matrigel invasion assays weredone to evaluate the effect of PDGFRh-mediated phosphorylation ofMUC1 on invasiveness of pancreatic adenocarcinoma cells (Fig. 5).S2-013.MUC1F cells had significantly higher invasion rates com-pared with S2-013.CT3 (P < 0.0001) or S2-013.Neo cells (P < 0.0001;Fig. 5). The invasive potential of cytoplasmic tail-truncated MUC1containing S2-013 cells was not significantly different from control-
transfected cells (P > 0.3). Stimulation of S2-013.MUC1F with PDGF-BB (50 ng/mL) significantly enhanced the invasiveness of these cellscompared with unstimulated cells (P < 0.02). The PDGF-BB–mediated increase in invasive potential of S2-013.MUC1F cells wasablated by treatment of cells with a PDGFRh kinase inhibitor(10 Amol/L). There was no significant effect of PDGF-BB stimulationon the invasive potential of S2-013.CT3 and S2-013.Neo cells inresponse to the PDGF-BB stimulation (P = 0.3 and 0.7, respectively).
Figure 4. A, effect of PDGF-BB stimulation on the subcellular localization of h-catenin and MUC1CT in S2-013.MUC1F cells. S2-013.MUC1F cells were serum starvedfor 24 h, stimulated for 2 h or left unstimulated, fixed, and permeabilized before incubation with mAbs CT2, anti-h-catenin, and the nuclear dye propidium iodide.Fluorescence signals from each scan were acquired sequentially. mAb CT2 (anti-MUC1CT) was visualized as blue and detected by using a Cy5-conjugated secondaryantibody, mAb anti-h-catenin was identified with FITC-conjugated secondary antibody and visualized as green, and nuclei were visualized in red. White arrows,triple-merge of blue, green, and red (MUC1CT/b-catenin/Nucleus ) showed colocalization of MUC1CT and h-catenin at cell surfaces under serum-starved conditions.B, effect of tyrosine mutations on nuclear localization of MUC1. S2-013.MUC1.FHPM, S2-013.MUC1.EHPM, S2-013.MUC1.FVPP, and S2-013.MUC1.EVPPcells were stained for MUC1CT (mAb CT2) and nucleus [propidium iodide (PI )]. Bar, 25 Am. Magnification, �63. Results represent three to four individual cell scanningobservations.
Cancer Research
Cancer Res 2007; 67: (11). June 1, 2007 5206 www.aacrjournals.org
Research. on June 19, 2015. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

Role of tyrosines in the YHPM and YVPPmotifs of MUC1CT inthe invasive potential of S2-013.MUC1Fcells. After observing thatPDGF-BB stimulation increased invasiveness of MUC1-expressingcells, we sought to establish the role of phosphotyrosines in theYHPM and YVPP motifs of MUC1CT on the invasiveness of MUC1-expressing cells. Cell lines expressing inactivating and activatingmutations at these motifs (S2-013.MUC1.FHPM, S2-013.MUC1.EHPM, S2-013.MUC1.FVPP, and S2-013.MUC1.EVPP) and controlcells (S2-013.MUC1F and S2-013.Neo) were evaluated in Matrigelinvasion assays. S2-013 cells expressing either of the phosphory-lation-abrogating tyrosine mutations (S2-013.MUC1.FHPM or S2-013.MUC1.FVPP) showed reduced in vitro invasiveness comparedwith S2-013.MUC1F cells, suggesting that these residues contrib-ute to the invasiveness of S2-013.MUC1F cells (both P < 0.001;Fig. 6A). In contrast, increased in vitro invasiveness was observedby S2-013 cells expressing either of the phosphorylation-mimicking tyrosine mutations (S2-013.MUC1.EHPM or S2-013.MUC1.EVPP) compared with S2-013.MUC1F cells (P < 0.05 and0.001; Fig. 6A).We evaluated the effect of these MUC1 tyrosine mutations on
the tumorigenic and metastatic potential of S2-013.MUC1 cells byorthotopic injection into nude mice. Mice were sacrificed after4 weeks. Mean tumor size was analyzed postsurgery after the endof 4 weeks (Fig. 6B). S2-013.MUC1.EHPM and S2-013.MUC1.EVPPcells showed larger tumors than S2-013.MUC1F, whereas S2-013.MUC1.FHPM and S2-013.MUC1.FVPP showed slightly smallertumors; however, the differences did not achieve statisticalsignificance (Fig. 6B). Internal organs and lymph nodes fromsacrificed mice were dissected, fixed, sectioned, and stained withH&E to identify and quantify metastases. The incidence of lymphnode metastases was significantly lower in mice implanted withS2-013.MUC1.FHPM and S2-013.MUC1.FVPP cells compared withS2-013.MUC1F (P < 0.05 and 0.001, respectively; Fig. 6C). Miceimplanted with S2-013.MUC1.EHPM showed significantly higherrates of lymph node metastases compared with S2-013.MUC1F(P < 0.05). About lung metastases, only mice implanted withS2-013.MUC1.FHPM showed statistically significant differences(reduced; P < 0.05; Fig. 6D). There were no significant differencesin incidences of liver and diaphragm metastases (data notshown).
Discussion
Proteomics methods were used to identify sites of tyrosinephosphorylation on the MUC1CT produced by pancreatic tumorcells. Phosphorylation of tyrosine at the HGRYVPP sequence wasobserved in two different human pancreatic carcinoma cells,S2-013.MUC1F and Panc1.MUC1F, suggesting that this modificationis associated with pancreatic cancer. This is the first report ofphosphorylation of this site, the first report to directly show thesites of tyrosine phosphorylation on the MUC1CT by the use of MS,and the first report to confirm that these occur by the use of site-specific antibodies (previous studies used mutated constructs andshowed a decrease in total phosphorylation).We detected and sequenced peptides spanning the entire
cytoplasmic tail, which shows that the proteomics methods usedhere were sufficiently sensitive to detect multiple peptide speciesfrom the MUC1CT. Our failure to detect phosphorylation attyrosine sites other than HGRYVPP raises the possibility thatpancreatic tumor cells cultured under the growth conditionsanalyzed here predominantly phosphorylate the tyrosine atHGRYVPP and that phosphorylation of other sites is highlyregulated. It remains possible that there were low levels of othermodifications present in the extracts that were undetectable by thetechniques used here. Previous results reporting phosphorylationat DRSPYEKV were obtained with breast cancer cell lines or celllines derived from other organ sites, which raises anotherpossibility: that there are organ-specific or cell-specific differencesin the phosphorylation of the MUC1CT that reflect differences inthe expression or activity of receptor tyrosine kinases orphosphatases in these cells. In this regard, it is notable that wedetected phosphorylation of tyrosine in the DRSPYEKV site in vitroby c-Src (Fig. 2). In any case, there should be further definitivedetermination of specific sites on the MUC1CT that arephosphorylated by different normal and tumor cell types.Analysis by the Scansite algorithm revealed PDGFRh as a
putative kinase that catalyzed phosphorylation at the HGRYVPPsite. In vitro kinase assays with MUC1CTp-66, a 66-residuesynthetic peptide of the MUC1CT, clearly showed that PDGFRhphosphorylated MUC1CT (Fig. 2A) and subsequent analysisshowed that tyrosines at sequences RDTYHPM and HGRYVPP inthe MUC1CT were phosphorylated in vitro by PDGFRh (Fig. 2B–D).
Figure 5. Effect of PDGF-BB on Matrigelinvasion of S2-013 cells transfected withvector control (S2-013.Neo ), cytoplasmictail-truncated (S2-013.CT3 ), or full-lengthconstructs of MUC1 (S2-013.MUC1F ).Number of S2-013 cells invading throughMatrigel and an 8-Am-pored membraneafter 24 h of culture with or withoutPDGF-BB (50 ng/mL). Cells (5 � 104) wereseeded in the upper compartment. Thelower chamber contained DMEM with orwithout PDGF-BB (50 ng/mL). Columns,mean of invading cell number from threeindependent experiments done in triplicate;bars, SE.
Phosphorylation of MUC1 by PDGFRb
www.aacrjournals.org 5207 Cancer Res 2007; 67: (11). June 1, 2007
Research. on June 19, 2015. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

It should be noted that phosphorylation of tyrosine at theRDTYHPM sequence by PDGFR was predicted previously byWreschner et al. (32). Wang et al. (33) previously presentedevidence of phosphorylation of tyrosine at the RDTYHPM motif ina CD8/MUC1 chimeric protein synthesized from a recombinantchimeric construct expressed in Chinese hamster ovary cells, onstimulation with CD8 antibody, using phenylalanine substitutionmutants and anti-phosphotyrosine antibodies to detect phosphor-ylation. It is also notable that both of these sites are within a regionof the MUC1CT postulated to mediate association with p53 (34).We detected significant expression of PDGFRh in multiple
human pancreatic adenocarcinoma cell lines representing differentdifferentiation states of pancreatic adenocarcinoma and detectedphosphorylation of the tyrosine in the YVPP motif in FG, Colo357,and Capan-2 (data not shown). PDGFB, PDGFRa, and PDGFRh arehighly expressed in pancreatic cancer and involved in signalingcascades that regulate the proliferation of pancreatic acinar cells
and to be a key player in ductal cell carcinogenesis of pancreaticcancer as well as other cancers (35–38).We observed an enhancement in MUC1CT phosphorylation on
stimulation of S2-013.MUC1F cells with PDGF-BB, a ligand forPDGFRh, which supports the hypothesis that PDGFRh stimulationinfluences MUC1-mediated signaling. This enhancement of phos-phorylation was abolished by a PDGFR-specific kinase inhibitor,tyrphostin (AG1295), which has been used to specifically abrogatethe activation of PDGFR (39–42). Tyrphostin has shown promisingresults for the treatment of proliferative vitreoretinopathy, aorticallograft vasculopathy, and restenosis. Western blotting withantibodies specific for the phosphotyrosines at HGRYVPP andRDTYHPM confirmed PDGF-BB stimulation-dependent in vivophosphorylation of the MUC1CT at tyrosines in both these sites.Our studies showed that PDGF-BB stimulation induced
PDGFRh-mediated phosphorylation of the MUC1CT, which inturn enhanced the nuclear localization of MUC1CT and h-catenin
Figure 6. Effect of tyrosine mutations of MUC1CT on invasion and metastasis. A, effect of tyrosine mutations of MUC1CT on in vitro Matrigel invasion. Cells wereseeded onto the upper chamber in serum-free DMEM, whereas the lower chamber contained DMEMwith 1% FBS. B, effect of tyrosine mutations on in vivo tumor growthrate. Cells (1 � 105) were orthotopically implanted into athymic female nude mice, and the dimensions of the tumor were detected by excising and measuring thetumor after sacrificing mice at the end of 4th week.Columns, mean tumor diameter plotted in terms of area unit obtained by multiplication of the two largest cross sections;bars, SE. Effect of tyrosine mutations on lymph node (C ) and lung metastases (D ). The incidences of metastases were detected by microscopic observation of theH&E-stained tissue sections obtained from the orthotopically implanted athymic female nude mice. The mice were sacrificed after the 4th week of orthotopic implantationof tyrosine mutant or MUC1F or mock vector–expressing S2-013 cells. Columns, mean number of metastases as determined by counting of metastases by microscopicexamination of formalin-fixed and H&E-stained tissue sections; bars, SE. *, P < 0.05, compared with the S2-013.MUC1F cells; ***, P < 0.001, compared with theS2-013.MUC1F cells. Asterisk(s) over horizontal lines, statistical significance of the events for the cells represented by the vertical bars underneath the line.
Cancer Research
Cancer Res 2007; 67: (11). June 1, 2007 5208 www.aacrjournals.org
Research. on June 19, 2015. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

and decreased localization of these proteins at cell surfaces.Activation of other receptor tyrosine kinases, such as EGFR,disrupts cadherin-h-catenin interactions at the cell surface andincreases nuclear signaling by h-catenin (43). PDGFRh activationcan lead to activation of a plethora of tyrosine kinases, includingc-Src, which directly phosphorylate h-catenin and disrupt itscell surface localization (44). Both MUC1CT and h-catenin can belocalized to the nucleus, and nuclear localization of h-catenin hasbeen correlated with increased proliferation and pathologic gradesof tumors. The increased nuclear localization of MUC1CT andh-catenin led us to investigate whether PDGFRh-mediated phos-phorylation of the MUC1CT influenced the proliferative andmetastatic activity of pancreatic adenocarcinoma cells. We observedno significant differences in proliferation in vitro among controlsor cells overexpressing recombinant MUC1 at any time pointsunder PDGF-BB stimulation compared with unstimulated cells asdetermined by flow cytometric cell cycle analysis (data not shown).These findings were confirmed by results of thymidine incorporationassays after 24 and 48 h of PDGF-BB stimulation (data not shown).In contrast, results of in vitro invasion assays clearly indicated
that PDGF-BB stimulation significantly enhanced the invasivepotential of MUC1-overexpressing S2-013 cells. The increasedinvasive potential was ablated by the PDGFRh kinase inhibitorAG1295, indicating that these effects were directly related to thekinase activity of PDGFRh. Independently, Hwang et al. (36) haveshown a reduction in growth and metastasis of a human pancreaticcarcinoma in an orthotopic nude mouse model by inhibitingPDGFR phosphorylation with STI571 (Gleevec). That there was nosignificant PDGF-BB–mediated increase in the invasive potential incontrol S2013.Neo or S2013.CT3 cells indicates that PDGFRh-mediated phosphorylation of MUC1CT is critical for the observedenhancement in invasive potential. Furthermore, expression ofphosphorylation-abrogating mutations in the MUC1CT diminishedthe invasive potential in both in vitro and in vivo assays. Incontrast, both invasion and metastasis were significantly enhancedby phosphorylation-mimicking mutations, supporting our hypoth-esis that phosphorylation at these sites regulates the tumorigenicand metastatic potential of pancreatic cancer cells.How is PDGFRh-mediated phosphorylation of the MUC1CT
regulated in normal pancreatic cells versus pancreatic adenocar-cinoma cells? One obvious factor is the requirement for tissue- orcell-specific coexpression of MUC1, PDGFRh, and PDGFB; however,simple coexpression of MUC1 and receptor tyrosine kinases, suchas PDGFRh or EGFR, in a given cell does not guarantee that theseproteins will interact and signal in a meaningful way. Differentialaccessibility of the substrate (MUC1CT) to the kinase (PDGFRh)can be regulated spatially in polarized epithelial cells and is likelyto contribute to regulation of these phosphorylation events, whichwe postulate are functional in different types of normal cells andtumor cells (13). We do not know what brings MUC1 and receptortyrosine kinases together. Normal epithelial cells express receptortyrosine kinases (such as EGFR and PDGFRh) at the basal surfaces,
whereas MUC1 is expressed at the apical surface (45). This spatialseparation would be predicted to preclude interaction of thesemolecules. One characteristic of oncogenic transformation is theloss of apical-basal polarity, which in theory would enablemembrane-associated mucins, such as MUC1, and growth factorreceptors to reside in physical proximity as has been shown in themammary gland (30) and thereby enable their association. Such amechanism may explain the phosphorylation of MUC1 by PDGFRhin pancreatic adenocarcinomas, which have lost some aspects ofapical-basal cell polarity and overexpress MUC1. Does such anevent represent aberrant signaling? Perhaps, but it is also possibleto envisage a model for the interaction of MUC1 and growth factorreceptors, such as PDGFR or EGFR, in normal epithelia that hadbeen injured or damaged and had lost some aspects of cellularpolarity or differentiated architecture. Damage or alterations inepithelia that resulted in disrupted cell polarity and enabled theassociation and phosphorylation of MUC1 by PDGFRh may sendsignals to the cell, informing it of this loss of differentiatedstructure or function. Such loss of cellular structure in normalepithelia would require activation of cellular repair mechanismsthat may involve cell proliferation and motility, both of which arefunctions previously associated with MUC1 in cancer. Thus,signaling mediated by MUC1, PDGFR, and other receptor tyrosinekinases in pancreatic cancer may be another example of tumorsappropriating a normal cellular function for purposes of survival,proliferation, and invasion.In summary, the findings presented here show that MUC1 is
phosphorylated by PDGFRh, which leads to increased nuclearlocalization of MUC1CT and h-catenin, and enhances the invasivepotential of pancreatic adenocarcinoma cells. These results alsosupport the hypothesis that phosphorylation events mediate MUC1-mediated tumor-associated signaling. Blockage of both MUC1 andPDGFRh signaling pathways might be of clinical relevance for thetreatment of pancreatic cancer and should be investigated further.The fact that we did not detect phosphorylation at other tyrosinesites in the MUC1CT raises the possibility that there are differencesamong tissues or tumors in site-specific phosphorylation of theMUC1CT or that phosphorylation of other sites is regulated byfactors not reflected in pancreatic tumor cell lines. In any case, it isimportant that additional studies be carried out to decipher the roleof phosphorylation of the MUC1CT in signal transduction.
Acknowledgments
Received 12/19/2006; revised 3/22/2007; accepted 3/28/2007.Grant support: Graduate Studies Assistantships (P.K. Singh, Y. Wen, and B.J.
Swanson) and NIH grants 5R01 CA57362 and T32 CA09476. The MS facility issupported in part by NIH grant P20 RR15635 from the COBRE Program of the NationalCenter for Research Resources, National Cancer Institute Cancer Center Support grantP30 CA36727, and the Nebraska Research Initiative.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.We thank Thomas Caffrey, Dr. Judith K. Christman, and Janice Taylor (Confocal
Laser Scanning Microscopy Core Facility) for their assistance.
References1. Qu CF, Li Y, Song YJ, et al. MUC1 expression in primaryand metastatic pancreatic cancer cells for in vitrotreatment by (213)Bi-C595 radioimmunoconjugate. Br JCancer 2004;91:2086–93.
2. Gronborg M, Bunkenborg J, Kristiansen TZ, et al.
Comprehensive proteomic analysis of human pancreaticjuice. J Proteome Res 2004;3:1042–55.3. Hollingsworth MA, Strawhecker JM, Caffrey TC, MackDR. Expression of MUC1, MUC2, MUC3 and MUC4mucin mRNAs in human pancreatic and intestinaltumor cell lines. Int J Cancer 1994;57:198–203.4. Masaki Y, Oka M, Ogura Y, et al. Sialylated MUC1
mucin expression in normal pancreas, benign pancre-atic lesions, and pancreatic ductal adenocarcinoma.Hepatogastroenterology 1999;46:2240–5.5. Tsutsumida H, Swanson BJ, Singh PK, et al. RNAinterference suppression of MUC1 reduces the growthrate and metastatic phenotype of human pancreaticcancer cells. Clin Cancer Res 2006;12:2976–87.
Phosphorylation of MUC1 by PDGFRb
www.aacrjournals.org 5209 Cancer Res 2007; 67: (11). June 1, 2007
Research. on June 19, 2015. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

6. Kohlgraf KG, Gawron AJ, Higashi M, et al. Contribu-tion of the MUC1 tandem repeat and cytoplasmic tail toinvasive and metastatic properties of a pancreaticcancer cell line. Cancer Res 2003;63:5011–20.7. Macao B, Johansson DG, Hansson GC, Hard T.Autoproteolysis coupled to protein folding in the SEAdomain of the membrane-bound MUC1 mucin. NatStruct Mol Biol 2006;13:71–6.8. Hollingsworth MA, Swanson BJ. Mucins in cancer:protection and control of the cell surface. Nat RevCancer 2004;4:45–60.9. Parry S, Silverman HS, McDermott K, Willis A,Hollingsworth MA, Harris A. Identification of MUC1proteolytic cleavage sites in vivo . Biochem Biophys ResCommun 2001;283:715–20.10. Silverman HS, Sutton-Smith M, McDermott K, et al.The contribution of tandem repeat number to the O-glycosylation of mucins. Glycobiology 2003;13:265–77.11. Silverman HS, Parry S, Sutton-Smith M, et al. In vivoglycosylation of mucin tandem repeats. Glycobiology2001;11:459–71.12. Burdick MD, Harris A, Reid CJ, Iwamura T,Hollingsworth MA. Oligosaccharides expressed onMUC1 produced by pancreatic and colon tumor celllines. J Biol Chem 1997;272:24198–202.13. Singh PK, Hollingsworth MA. Cell surface-associatedmucins in signal transduction. Trends Cell Biol 2006;16:467–76.14. Quin RJ, McGuckin MA. Phosphorylation of thecytoplasmic domain of the MUC1 mucin correlateswith changes in cell-cell adhesion. Int J Cancer 2000;87:499–506.15. Li Y, Bharti A, Chen D, Gong J, Kufe D. Interaction ofglycogen synthase kinase 3h with the DF3/MUC1carcinoma-associated antigen and h-catenin. Mol CellBiol 1998;18:7216–24.16. Kondo K, Kohno N, Yokoyama A, Hiwada K.Decreased MUC1 expression induces E-cadherin-medi-ated cell adhesion of breast cancer cell lines. Cancer Res1998;58:2014–9.17. Ren J, Li Y, Kufe D. Protein kinase Cy regulatesfunction of the DF3/MUC1 carcinoma antigen in h-catenin signaling. J Biol Chem 2002;277:17616–22.18. Schroeder JA, Adriance MC, Thompson MC, Came-nisch TD, Gendler SJ. MUC1 alters h-catenin-dependenttumor formation and promotes cellular invasion.Oncogene 2003;22:1324–32.19. Wen Y, Caffrey TC, Wheelock MJ, Johnson KR,Hollingsworth MA. Nuclear association of the cytoplas-
mic tail of MUC1 and h-catenin. J Biol Chem 2003;278:38029–39.20. Li Y, Kuwahara H, Ren J, Wen G, Kufe D. The c-Srctyrosine kinase regulates signaling of the human DF3/MUC1 carcinoma-associated antigen with GSK3h andh-catenin. J Biol Chem 2001;276:6061–4.21. Meerzaman D, Shapiro PS, Kim KC. Involvement ofthe MAP kinase ERK2 in MUC1 mucin signaling. Am JPhysiol Lung Cell Mol Physiol 2001;281:L86–91.22. Pandey P, Kharbanda S, Kufe D. Association of theDF3/MUC1 breast cancer antigen with Grb2 and theSos/Ras exchange protein. Cancer Res 1995;55:4000–3.23. Iwamura T, Taniguchi S, Kitamura N, et al. Correla-tion between CA19-9 production in vitro and histolog-ical grades of differentiation in vivo in clones isolatedfrom a human pancreatic cancer cell line (SUIT-2).J Gastroenterol Hepatol 1992;7:512–9.24. Tallquist MD, French WJ, Soriano P. Additive effectsof PDGF receptor h signaling pathways in vascularsmooth muscle cell development. PLoS Biol 2003;1:E52.25. McDermott KM, Crocker PR, Harris A, et al. Over-expression of MUC1 reconfigures the binding propertiesof tumor cells. Int J Cancer 2001;94:783–91.26. Thompson EJ, Shanmugam K, Hattrup CL, et al.Tyrosines in the MUC1 cytoplasmic tail modulatetranscription via the extracellular signal-regulatedkinase 1/2 and nuclear factor-nB pathways. Mol CancerRes 2006;4:489–97.27. Shevchenko A, Wilm M, Vorm O, Mann M. Massspectrometric sequencing of proteins silver-stainedpolyacrylamide gels. Anal Chem 1996;68:850–8.28. Yonezawa S, Byrd JC, Dahiya R, et al. Differentialmucin gene expression in human pancreatic and coloncancer cells. Biochem J 1991;276:599–605.29. Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0:proteome-wide prediction of cell signaling interactionsusing short sequence motifs. Nucleic Acids Res 2003;31:3635–41.30. Schroeder JA, Thompson MC, Gardner MM, GendlerSJ. Transgenic MUC1 interacts with epidermal growthfactor receptor and correlates with mitogen-activatedprotein kinase activation in the mouse mammary gland.J Biol Chem 2001;276:13057–64.31. Kurth T, Fesenko IV, Schneider S, et al. Immunocy-tochemical studies of the interactions of cadherins andcatenins in the early Xenopus embryo. Dev Dyn 1999;215:155–69.32. Wreschner DH, Zrihan-Licht S, Baruch A, et al. Doesa novel form of the breast cancer marker protein, MUC1,
act as a receptor molecule that modulates signaltransduction? Adv Exp Med Biol 1994;353:17–26.33. Wang H, Lillehoj EP, Kim KC. Identification of foursites of stimulated tyrosine phosphorylation in theMUC1 cytoplasmic tail. Biochem Biophys Res Commun2003;310:341–6.34. Wei X, Xu H, Kufe D. Human MUC1 oncoproteinregulates p53-responsive gene transcription in thegenotoxic stress response. Cancer Cell 2005;7:167–78.35. Ebert M, Yokoyama M, Friess H, Kobrin MS, BuchlerMW, Korc M. Induction of platelet-derived growth factorA and B chains and over-expression of their receptors inhuman pancreatic cancer. Int J Cancer 1995;62:529–35.36. Hwang RF, Yokoi K, Bucana CD, et al. Inhibition ofplatelet-derived growth factor receptor phosphorylationby STI571 (Gleevec) reduces growth and metastasis ofhuman pancreatic carcinoma in an orthotopic nudemouse model. Clin Cancer Res 2003;9:6534–44.37. Gotzmann J, Fischer AN, Zojer M, et al. A crucialfunction of PDGF in TGF-h-mediated cancer progres-sion of hepatocytes. Oncogene 2006;25:3170–85.38. Jechlinger M, Sommer A, Moriggl R, et al. AutocrinePDGFR signaling promotes mammary cancer metasta-sis. J Clin Invest 2006;116:1561–70.39. Iwamoto H, Nakamuta M, Tada S, Sugimoto R, EnjojiM, Nawata H. Platelet-derived growth factor receptortyrosine kinase inhibitor AG1295 attenuates rat hepaticstellate cell growth. J Lab Clin Med 2000;135:406–12.40. Kovalenko M, Gazit A, Bohmer A, et al. Selectiveplatelet-derived growth factor receptor kinase blockersreverse sis-transformation. Cancer Res 1994;54:6106–14.41. Twaddle GM, Turbov J, Liu N, Murthy S. Tyrosinekinase inhibitors as antiproliferative agents against anestrogen-dependent breast cancer cell line in vitro .J Surg Oncol 1999;70:83–90.42. Zheng Y, Ikuno Y, Ohj M, et al. Platelet-derivedgrowth factor receptor kinase inhibitor AG1295 andinhibition of experimental proliferative vitreoretinop-athy. Jpn J Ophthalmol 2003;47:158–65.43. Nelson WJ, Nusse R. Convergence of Wnt, h-catenin,and cadherin pathways. Science 2004;303:1483–7.44. Roura S, Miravet S, Piedra J, Garcia de Herreros A,Dunach M. Regulation of E-cadherin/catenin associa-tion by tyrosine phosphorylation. J Biol Chem 1999;274:36734–40.45. Ramsauer VP, Carraway CA, Salas PJ, Carraway KL.Muc4/sialomucin complex, the intramembrane ErbB2ligand, translocates ErbB2 to the apical surface inpolarized epithelial cells. J Biol Chem 2003;278:30142–7.
Cancer Research
Cancer Res 2007; 67: (11). June 1, 2007 5210 www.aacrjournals.org
Research. on June 19, 2015. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

2007;67:5201-5210. Cancer Res Pankaj K. Singh, Yunfei Wen, Benjamin J. Swanson, et al. Pancreatic Adenocarcinoma CellsPhosphorylation of MUC1 Enhances Invasiveness in
Mediated−βPlatelet-Derived Growth Factor Receptor
Updated version
http://cancerres.aacrjournals.org/content/67/11/5201
Access the most recent version of this article at:
Cited articles
http://cancerres.aacrjournals.org/content/67/11/5201.full.html#ref-list-1
This article cites 45 articles, 21 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/67/11/5201.full.html#related-urls
This article has been cited by 12 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
Research. on June 19, 2015. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from




















